the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Sep 2020
| 01 Sep 2020
Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates
Iida Pullinen
Sebastian Schmitt
Sungah Kang
Mehrnaz Sarrafzadeh
Patrick Schlag
Stefanie Andres
Einhard Kleist
Franz Rohrer
Monika Springer
Ralf Tillmann
Jürgen Wildt
Defeng Zhao
Andreas Wahner
Astrid Kiendler-Scharr
The formation of organic nitrates (ONs) in the gas phase and their impact on mass formation of secondary organic aerosol (SOA) was investigated in a laboratory study for α-pinene and β-pinene photooxidation. Focus was the elucidation of those mechanisms that cause the often observed suppression of SOA mass formation by NOx, and therein the role of highly oxygenated multifunctional molecules (HOMs). We observed that with increasing NOx concentration (a) the portion of HOM organic nitrates (HOM-ONs) increased, (b) the fraction of accretion products (HOM-ACCs) decreased, and (c) HOM-ACCs contained on average smaller carbon numbers.
Specifically, we investigated HOM organic nitrates (HOM-ONs), arising from the termination reactions of HOM peroxy radicals with NOx, and HOM permutation products (HOM-PPs), such as ketones, alcohols, or hydroperoxides, formed by other termination reactions. Effective uptake coefficients γeff of HOMs on particles were determined. HOMs with more than six O atoms efficiently condensed on particles (γeff>0.5 on average), and for HOMs containing more than eight O atoms, every collision led to loss. There was no systematic difference in γeff for HOM-ONs and HOM-PPs arising from the same HOM peroxy radicals. This similarity is attributed to the multifunctional character of the HOMs: as functional groups in HOMs arising from the same precursor HOM peroxy radical are identical, vapor pressures should not strongly depend on the character of the final termination group. As a consequence, the suppressing effect of NOx on SOA formation cannot be simply explained by replacement of terminal functional groups by organic nitrate groups.
According to their γeff all HOM-ONs with more than six O atoms will contribute to organic bound nitrate (OrgNO3) in the particulate phase. However, the fraction of OrgNO3 stored in condensable HOMs with molecular masses > 230 Da appeared to be substantially higher than the fraction of particulate OrgNO3 observed by aerosol mass spectrometry. This result suggests losses of OrgNO3 for organic nitrates in particles, probably due to hydrolysis of OrgNO3 that releases HNO3 into the gas phase but leaves behind the organic rest in the particulate phase. However, the loss of HNO3 alone could not explain the observed suppressing effect of NOx on particle mass formation from α-pinene and β-pinene.
Instead we can attribute most of the reduction in SOA mass yields with increasing NOx to the significant suppression of gas phase HOM-ACCs, which have high molecular mass and are potentially important for SOA mass formation at low-NOx conditions.
- Article
(1933 KB) - Full-text XML
-
Supplement
(1720 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA) constitutes a substantial fraction of ambient aerosol. It is formed from oxidation products of volatile organic compounds (VOCs) and known to adversely affect visibility, climate, and human health (Hallquist et al., 2009). With annual emissions around 1100 Tg, the biosphere is the strongest source of tropospheric VOCs (Guenther et al., 2012), and thus, SOA formation from biogenic VOCs is of high importance. Despite the outstanding role of biogenic VOCs by amount and reactivity, anthropogenic trace gases affect SOA formation, and possible anthropogenic enhancement effects were found in laboratory and field studies (e.g., Carlton et al., 2010; De Gouw et al., 2005; Emanuelsson et al., 2013; Glasius et al., 2011; Hoyle et al., 2011; Shilling et al., 2013; Spracklen et al., 2011; Worton et al., 2011; Xu et al., 2015a). Examples of important anthropogenic trace gases are NO and NO2, which together form the NOx family. During nighttime NOx is converted to NO3 radicals, which oxidize biogenic VOCs, leading to organic nitrates and SOA formation (Boyd et al., 2015, 2017; Claflin and Ziemann, 2018; Faxon et al., 2018; Fry et al., 2013, 2014; Kiendler-Scharr et al., 2016; A. K. Y. Lee et al., 2016; Ng et al., 2017). During daytime, NOx controls the atmospheric HOx cycle and thus the oxidation cycle of VOCs by reaction with peroxy radicals. In this study we will focus on the role of NOx for SOA formation during daytime. In a number of studies the role of NOx in the formation of SOA mass was investigated (Eddingsaas et al., 2012; Han et al., 2016; Kim et al., 2012; Kroll et al., 2006; A. K. Y. Lee et al., 2016, 2020; Ng et al., 2007; Pandis et al., 1991; Presto et al., 2005; Rindelaub et al., 2015, 2016; Sarrafzadeh et al., 2016; Stirnweis et al., 2017; Zhang et al., 2006). In most cases it was observed that NOx decreased mass yields of SOA formation, and the effects were generally attributed to impacts of RO2•+NO reactions. Sarrafzadeh et al. (2016) show that parts of the apparent suppression of SOA yields from β-pinene by NOx were due to the role of NOx in the HOx cycle. As mass yields of SOA formation from α-pinene and β-pinene photooxidation depend on the actual OH concentrations, NOx also affects SOA formation via decreasing or increasing OH concentrations according to Reactions (R1) and (R2):
NOx inhibits new particle formation (NPF; Wildt et al., 2014); therefore, in absence of seed particles, NOx can prevent formation of sufficient particle surface where low-volatility compounds could condense on. In absence of particles other sinks gain in importance for low-volatility compounds as dry deposition in the environment or wall losses in chamber experiments. In order to circumvent these effects, Sarrafzadeh et al. (2016) used seed particles, always providing sufficient surface for the gas phase precursors of SOA mass to condense on, and kept the OH concentrations constant. As a result the remaining effect of NOx on SOA mass formation from β-pinene and on SOA yields was only moderate. Generally, in absence of NOx, peroxy radicals RO2•) mainly react with other peroxy radicals (including the HO2• radical) whereby termination products like hydroperoxides, ketones, alcohols, carboxylic acids, and percarboxylic acids are produced. In reactions between peroxy radicals alkoxy radicals are also formed, which continue the radical chain (R4b).
As observed in several studies with highly oxygenated multifunctional organic molecules (HOMs; e.g., Berndt et al., 2018a, b; Ehn et al., 2012, 2014; Mentel et al., 2015) accretion products can be formed in peroxy–peroxy radical reactions:
In laboratory studies HOM accretion products can contribute significantly to SOA yields (McFiggans et al., 2019).
The presence of NOx opens new pathways with large reaction rates and production of organic nitrates (Reaction R6a), including PAN-like compounds (Reaction R7). Furthermore, substantial amounts of alkoxy radicals (Reaction R6b) are formed.
From Reactions (R3) to (R7) it is obvious that the chemically stable products of peroxy radicals will have different termination groups under low- and high-NOx conditions: while hydroperoxides, ketones, carboxylic acids, etc. predominate at low-NOx conditions, organic nitrates (ONs), including PAN-like compounds, become more important at high-NOx conditions.
Recent studies demonstrated the dominant role of HOMs in SOA mass formation (Ehn et al., 2014; Jokinen et al., 2015; McFiggans et al., 2019; Mutzel et al., 2015; Zhang et al., 2017). HOMs are formed by addition of molecular oxygen to alkyl radicals that are formed after H migration in peroxy or in alkoxy radicals. Due to a relatively long lifetime of peroxy radicals, such H shifts with addition of molecular oxygen can appear several times in sequential steps and are therefore termed autoxidation (Crounse et al., 2011; Mentel et al., 2015; Rissanen et al., 2014). This process leads to highly oxygenated multifunctional peroxy radicals (HOM peroxy radicals). If the respective HOM is formed exclusively via autoxidation of peroxy radicals, the HOM moieties are very likely multiple hydroperoxides (Berndt et al., 2016; Rissanen et al., 2014). If there are intermediate steps via alkoxy radicals, there may also be alcohol groups (Mentel et al., 2015). Bianchi et al. (2019) suggested using the notation HOM when the autoxidation products carry six or more O atoms.
HOMs are low-volatility organic compounds (LVOCs) or even extremely low-volatility organic compounds (ELVOCs), and they substantially contribute to mass formation of particles and support NPF (Bianchi et al., 2016; Ehn et al., 2014; Kirkby et al., 2016; Lehtipalo et al., 2018; Tröstl et al., 2016). All experimental evidence shows that HOM peroxy radicals terminate with similar rates as less functionalized peroxy radicals (Berndt et al., 2016; Bianchi et al., 2019; Ehn et al., 2017). At low NOx levels, HOM hydroperoxides, HOM alcohols, HOM carboxylic acids, and HOM percarboxylic acids as well as HOM ketones are expected from the termination step and in addition HOM accretion products (HOM-ACCs).
At high NOx levels HOM-ONs become important termination products. In addition HOM-ACCs are suppressed (Lehtipalo et al., 2018; Rissanen, 2018) and shifted to smaller C numbers. The effect of NOx on HOM-RO2 chemistry is important for understanding the impact of NOx on SOA formation. In this paper we analyze two aspects of the effect of NOx which are important for SOA yield: the formation and the volatility of HOM-ONs as well as the suppression of HOM accretion products.
2.1 Description of the chamber setup and experiments
Experiments were conducted in the Jülich Plant Atmosphere Chamber (JPAC; Mentel et al., 2009, 2013). The actual setup of the chambers was already described in several recent publications (Ehn et al., 2014; Mentel et al., 2015; Sarrafzadeh et al., 2016; Wildt et al., 2014). A 1.45 m3 chamber made of borosilicate glass and set up in a climate-controlled housing was used for these experiments. The chamber was operated as a continuously stirred tank reactor. About 31 L of purified air per minute was pumped through the chamber, resulting in a residence time of approximately 46 min. Mixing was ensured by a fan, and the mixing time was about 2 min. The inlet flow was provided by two about equal purified air streams with one of them containing ozone and water vapor. In the second stream the VOC of interest was introduced from a diffusion source. Temperature (16±1 ∘C) and relative humidity (63±2 %) inside the chamber were held constant over the course of the experiments.
The chamber was equipped with several lamps. Two discharge lamps (HQI400 W/D, Osram) served to simulate the solar light spectrum. Twelve discharge lamps emitting UV light in the UV-A range (Philips, TL 60 W/10-R, 60 W, λmax=365 nm) provided the photolysis of NO2 with a photolysis frequency J(NO2) of s−1. A UV-C lamp (Philips, TUV 40 W, λmax=254 nm) was used to produce OH radicals by ozone photolysis and reaction of O1D atoms with water vapor. This lamp was housed in a quartz tube across the chamber diameter, and parts of the lamp were shielded by movable glass tubes. By altering the gap between these glass tubes, the photolysis frequency for ozone, J(O1D), and therewith the OH production rate could be adjusted. During most of the experiments described here, J(O1D) was about s−1. The photolysis frequencies J(NO2) and J(O1D) (as a function of the TUV gap) were determined experimentally by actinometry with NO2 and O3 in the chamber. Gas phase compounds such as ozone (O3, UV absorption, Thermo Environmental 49), nitrogen monoxide (NO, chemiluminescence, Eco Physics, CLD 770 AL ppt), and nitrogen dioxide (NO2, chemiluminescence after photolysis, Eco Physics, PLC 760) were measured. Water vapor concentrations were measured by dew point mirror (TP-2, Walz).
We used the monoterpenes α-pinene and β-pinene (both Aldrich, 95 % purity) as SOA precursors. The monoterpenes (MTs) were measured either by gas chromatography–mass spectrometry (GC–MS; Agilent GC–MSD system with HP6890 GC and 5973 MSD) or by proton-transfer-reaction mass spectrometry (PTR-MS; Ionicon, Innsbruck, Austria). Both devices were switched between the outlet and the inlet of the chamber in order to quantify concentrations in the chamber and to determine the VOC source strength.
To provide NOx to the photochemical system, we added NO2 (Linde, 100 ppm NO2 in nitrogen) in the β-pinene experiments or NO (90 ppm in nitrogen) in the α-pinene experiments to the VOC-containing air stream. In the case of NO2 addition, a fraction of the added NO2 was converted to NO due to the NO2 photolysis. In the case of NO addition, the major portion of the added NO was converted to NO2 by reaction with O3. We adjusted the O3 addition such that a steady state [O3] within a range of 60–90 ppb was achieved. We observed memory effects for NOx probably due to Teflon parts (tubing, fan) in the chamber. In particular after experiments at high NOx concentrations residual NOx appeared in the chamber on the next day when switching on the TUV lamp. To minimize the memory effect, the chamber was operated without NOx for 1 d between the NOx experiments. The background NOx concentration was around 300 ppt. When adding NOx, its initial concentration, [NOx]0, was between 5 and 150 ppb and thus substantially above the background levels.
The results presented here were obtained at steady-state conditions when all physical and chemical parameters were constant or in steady state, respectively. To indicate reference to steady state we mark the concentrations of MTs and NOx with the subscript “SS”. To allow better comparison to literature data, we refer in some instances to the initial concentrations, [NOx]0, [α-pinene]0, and [β-pinene]0, indicated by the subscript “0”, which are in our case input concentrations.
The OH concentrations were calculated from the decay of the respective MT in the chamber (Eq. 2).
Equation (1) describes mass balance of the MT in the chamber, and Eq. (2) results from Eq. (1) under steady-state conditions when d[VOC]∕dt=0. In Eqs. (1) and (2), V is the volume of the chamber, F is the total air flow through the chamber, and [VOC]0 and [VOC] are the concentrations of the VOC under investigation in the inlet air and in the chamber, respectively. kOH and are the respective rate coefficients for the reactions of the VOC with OH and with O3. In one case, where the concentration of β-pinene was altered during the experiment, m-xylene was added as a tracer for [OH].
Rate constants used for the determinations of [OH] were , , and cm−3 s−1 for α-pinene, β-pinene, and m-xylene, respectively (Atkinson, 1994, 1997). At typical conditions with [O3] ∼60–100 ppb and [OH] the consumption of α-pinene by O3 ( cm3 s−1) was about 5 % compared to that by OH. α-pinene losses due to ozonolysis were therefore neglected for the determination of [OH]. As β-pinene has an even lower ozonolysis rate constant ( cm3 s−1; Atkinson, 1997), and m-xylene does not react with O3 ( cm3 s−1; Atkinson et al., 1994), ozonolysis reactions were also neglected for OH estimations using these VOCs. The overall uncertainty in OH concentration was estimated to be about 20 % (Wildt et al., 2014).
Our results were obtained in different types of experiments. An overview of the performed experiments with their starting conditions is given in Table 1. Details of the procedures applied during individual experiments are described in the respective sections. Three experiment series (1–3) were conducted to characterize gas phase products, and experiment series 4 was conducted to characterize the particle phase. In the first experiment we estimated the molar yield of all organic nitrates (ONs) independent of their contribution to particle formation. This was made at the example of β-pinene at one NOx concentration (see Sect. 3.1). Experiment series 2 was performed to determine the fraction of HOM organic nitrates at the total amount of HOMs. Both monoterpenes were used with several NOx levels as indicated (see Sect. 3.3). In experiment series 3 we determined effective uptake coefficients for HOMs at a low and at a high NOx level for HOM-ONs and other HOM termination products (see Sect. 3.4). These experiments were performed with seed particles (ammonium sulfate), which were dried by a silica gel diffusion dryer to relative humidity (RH) < 30 % before they entered the chamber. In the last experiment series 4, we characterized the amount of nitrate bound to organics in particles for comparison with the amount of organic bound nitrate in those HOM-ONs that efficiently contribute to particle formation.
Table 1Overview of α-pinene and β-pinene experiments.
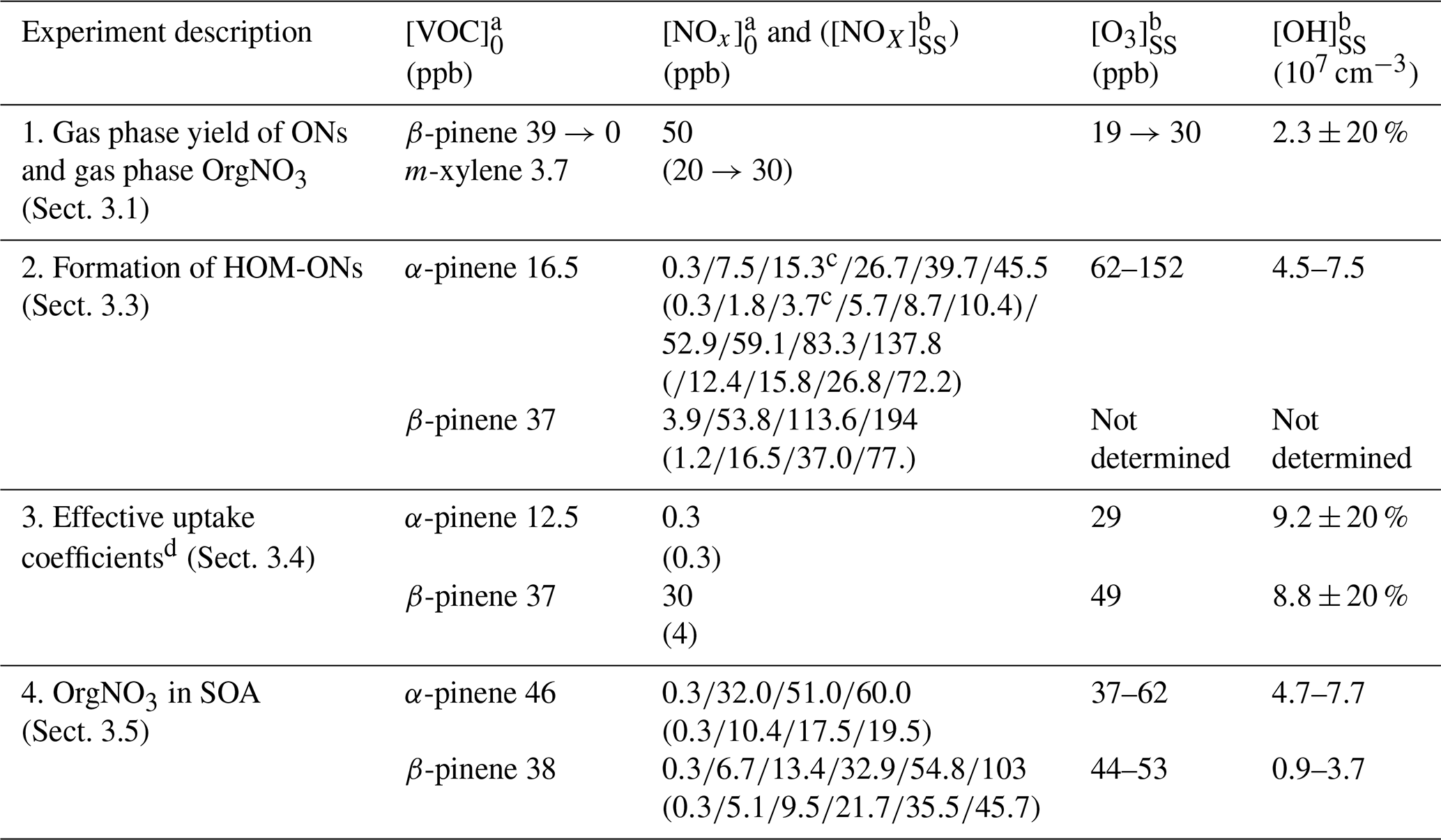
a The subscript 0 refers to mixing ratio in the inflow. b The subscript SS refers to mixing ratio at steady state. c Average of two experiments at [NOx]0 of 15 and 15.5 ppb ([NOx]SS of 3.6 and 3.75 ppb). d In the presence of ammonium sulfate seed aerosols.
2.2 Determination of highly oxygenated molecules (HOMs)
Highly oxygenated molecules (HOMs) were measured by a chemical ionization mass spectrometer (CIMS) operated with nitrate as reagent ion (-CIMS; Jokinen et al., 2012). First we determined that the relative transmission curve of our -CIMS was flat (Sect. S1.1 in the Supplement). This indicates that detection of HOMs within the mass range from ∼230 to ∼600 Da is nearly mass independent. So far, no absolute calibration method exists for HOMs. However, the charging efficiency for HOMs is close to the kinetic limit similar to sulfuric acid (Ehn et al., 2014; Kirkby et al., 2016). Thus, the sensitivity of the -CIMS to HOMs is supposedly similar to that of sulfuric acid. We therefore calibrated our -CIMS to sulfuric acid and used the calibration coefficient for the HOMs, too (see Sect. S1.2). Applying the sulfuric acid calibration coefficient to the HOMs, we investigated the mass balance between condensable HOMs and formed particle mass, which was closed within a factor of 2 (see Sect. S1.3). It is also likely that the sensitivity of the -CIMS does not depend much on the functional group formed in the final termination step. This assumption is reasonable as HOMs formed by autoxidation contain several hydroperoxy groups or a mixture of hydroxy and hydroperoxy groups, if they are formed via the alkoxy peroxy path (Mentel et al., 2015). Furthermore, a good agreement of the fraction of organic nitrates on the total reaction products of β-pinene (see Sect. 3.1) and the fraction of HOM-ONs of the total HOMs gives us confidence that the uncertainty induced by this assumption does not much affect the main conclusions.
This all together with the quantum mechanical results of the same sensitivity for HOMs containing six or more O atoms (Hyttinen et al., 2017) gave us confidence that concentrations of HOMs with six or more O atoms were determined with the same sensitivity to an uncertainty of less than a factor of 2.
In summary, we used the calibration coefficient for H2SO4 to calculate HOM concentrations. We further concluded a same sensitivity for the detection of all HOMs including accretion products when they contained six and more O atoms. These conclusions are supported by observations of Breitenlechner et al. (2017), who found that, once the HOM contains more than five O atoms, the sensitivity is to a good approximation independent of the number of O atoms. The sensitivity of the -CIMS is unclear for compounds containing fewer than five O atoms (Hyttinen et al., 2017; Rissanen et al., 2014; Riva et al., 2019). However, as will be shown in Sect. 3.4, HOMs with less than five O atoms are of minor importance for particle mass formation; hence we will neglect them, and this will not contribute much uncertainty to our results.
Identification of molecular formulas for individual HOMs was obtained using high-resolution spectra (resolution power ≈4000) as described in Sect. S2. In the case of HOM spectra from β-pinene photooxidation, we found many not fully resolved double peaks from the overlapping of C10, C9, and C8 progressions. The mass spectra of α-pinene HOMs in general consisted of singular peaks; i.e., they were quite well resolved. Figure S5 in Sect. S2 shows how HOM-RO2 and HOM-ONs were separated with increasing [NOx]. In the high-resolution analysis of α-pinene, we focus on the mass range 230 to 550 Da. The lower limit of 230 Da was chosen because of the equal sensitivity of the -CIMS towards HOMs as discussed before and because C10 compounds with six or more O atoms have molecular weights > 230 Da. As we will show in Sect. 4.2, they are LVOCs and ELVOCs and will contribute to SOA formation. The upper limit was set to 550 Da to reduce the influence of noise since not much signal is found for molecular masses > 550 Da.
The observed concentration of gas phase HOMs depends on the OH concentration and the condensation sink provided by newly forming particles. Adding to or removing NOx from a given photochemical system directly impacts [OH] by Reactions (R1) and (R2). NOx furthermore suppresses new particle formation (Wildt et al., 2014), which leads to a decreasing condensation sink for HOMs with increasing [NOx]. The actual OH concentration affects the actual turnover of the precursor, and thus the actual production of RO2• and HOMs, while the actual condensational sink leads to condensational loss of HOMs. Both factors change the observed HOM gas phase mixing ratio and can superimpose the impacts of NOx on peroxy radical chemistry itself. In order to separate the chemical impacts of NOx on HOM peroxy radical chemistry, we needed to take out the effects of [OH] and condensational sink as much as possible. This was achieved by normalizing the HOM mixing ratio to particle-free conditions and to a certain reference oxidation rate. The procedure is described in detail in Sect. S3.
2.3 Particle phase measurements
To characterize the particle phase, we used a condensation particle counter (CPC, TSI 3783), a scanning mobility particle sizer (Electrostatic classifier TSI 3080, including a differential mobility analyzer TSI 3081 and a CPC TSI 3025A), and an aerosol mass spectrometer (AMS, Aerodyne HR-ToF-MS, modified for application on a Zeppelin airship; Rubach, 2013). In the AMS the aerosol particles were vaporized at 600 ∘C and ionized by electron impact ionization at 70 eV. The AMS was routinely operated in V mode in two alternating modes: 1 min MS mode to measure the chemical composition and 2 min particle time of flight mode. Only MS mode data were analyzed here. In the following we will use the amount of nitrate bound to organics (OrgNO3) as a diagnostic to link observation of HOM-ONs in the gas phase to observations in the particulate phase. We separated organic and inorganic particulate nitrate and determined the amount of OrgNO3 by the method for AMS (Farmer et al., 2010; Kiendler-Scharr et al., 2016).
SOA yields were determined as described in Sarrafzadeh et al. (2016). For determining mass yields, the particle mass formed during steady-state conditions was divided by the mass of the consumed MT, which is the difference between inlet and outlet concentration:
During measurements of particle mass, the mean diameter of particles was above 100 nm. As the loss rates of such particles on the chamber walls were low (Mentel et al., 2009), they were neglected. Losses of oxidized SOA precursors to the chamber walls were considered by applying the correction function given by Sarrafzadeh et al. (2016). This function describes the ratio of wall losses over the sum of wall losses and losses on particles. For the data given here, the correction factors were between 1.5 and 2.1.
2.4 Determination of effective uptake coefficients
The experiments to determine effective uptake coefficients for HOMs, γeff, were performed as follows: signal intensities of the respective HOMs were measured at zero (α-pinene) or low particle load (β-pinene). Then we introduced dried seed particles into the reaction chamber by spraying ammonium sulfate solutions in two steps with concentrations of 4 and 40 g L−1. The particles were dried by passage through a silica gel diffusion tube and size selected at an electromobility diameter d=100 nm. Increasing amounts of ammonium sulfate seed particles instantaneously led to lowered HOM concentrations in the gas phase due to the additional loss by condensation on the seed particles. The decrease in signal intensity with increasing particle surface was used to evaluate γeff as described below.
We operate our chamber as a continuously stirred tank reactor (CSTR) in a flow-through mode with a well-mixed core of the chamber. HOMs lost at the chamber walls must diffuse through the laminar boundary layer at the chamber walls. In the chamber with no aerosols present, the walls constitute the major sink of HOMs. In this case the observed steady-state concentration is determined to a very good approximation only by the production rate and the wall loss rate. When seed aerosol is added or new particles are formed, the additional condensational sink provided by the particle surface lowers the steady-state concentration. Under conditions of unperturbed gas phase production and typical times for phase transfer smaller than the residence time of the air in the chamber, the lowered gas phase steady-state concentrations reflect the partitioning of HOMs, which is determined by the balance of condensation and evaporation. (Tröstl et al. (2016) noted that HOMs can be LVOCs; i.e., they have a very small but noticeable vapor pressure.) Since we are working in a steady-state system, we cannot easily separate between a kinetically slow uptake and a balance between (fast) uptake in steady state with a (fast) evaporation.
For molecules with noticeable volatility, steady state between condensation and evaporation is established on the timescales of less than 10 min in our CSTR; e.g., for molecules with molecular masses of 300 Da and at a particle surface of m2 m−3 the typical uptake time is about the same as the mixing time of 120 s. We express the net effect of condensation and evaporation by an effective uptake coefficient γeff and the gas kinetic collision rate of HOMs with the particle surface. The γeff can be determined by measurement of the ratio of steady-state HOM concentrations for the unseeded case and for seeded cases with the advantage that only signal intensities are required and hence no calibration is needed (Sarrafzadeh et al., 2016).
Size selection by electromobility produced bimodal size distributions in the chamber over the times of observation of 2 and 3.5 h with one mode around 100 nm and a second mode around 200 nm (≈25 % by number). The diameter of the median of the surface distributions was located in a range of 150–200 nm. Since it is likely that nearly every collision with the surface of particles will lead to phase transfer of HOMs, we considered the Fuchs–Sutugin correction factor (fFS) to calculate the collision rate (Fuchs and Sutugin, 1971) in order to correct for diffusion limitations. Taking into account the mean free path for a range of molecular compositions of and a median of the particle surface distribution in a range of 150–200 nm, we estimate fFS in a range of 0.65–0.75; diffusivity was calculated after Fuller et al. (1969).
In a first step wall loss rates of HOMs were measured. After stopping the OH production and thus photochemical HOM formation, we observe an exponential decay of HOM signals. The exponential decay of the signal intensity gives the lifetimes of those HOMs. In absence of particles the lifetimes reflect the wall loss rates. As shown in Sarrafzadeh et al. (2016) and Ehn et al. (2014) the lifetimes were in the range of 70 to 150 s, i.e., the loss rates on the walls of the chamber, LW(HOM), were in the range of to s−1. LW(HOM) values are more than an order of magnitude higher than those caused by the flush-out of the air in the chamber ( s−1 for the residence time of 46 min). Therefore we neglected flush-out as a sink for HOMs. In a second step, the chemical system was kept at the same steady-state conditions for [OH]SS, [O3]SS, and MT concentration.
Data evaluation was based on the following considerations: the concentration of any HOM, c(HOM), is determined by its production rate P(HOM) and the first-order loss rate, L(HOM), as given in Eq. (4):
In absence of particles, the total loss rate L(HOM) is given alone by the loss rate at the chamber walls, LW(HOM). In the presence of particles L(HOM) is the sum of loss rates at the walls and at the particle surface, LW(HOM) + LP(HOM). At constant production rate P(HOM) the ratio of concentrations is inversely proportional to their ratio of loss rates:
In Eq. (5), c(HOM)0 is the concentration of HOMs in the particle-free chamber, and c(HOM) is the concentration in the particle-containing chamber. Solving Eq. (5) for LP(HOM), we obtain Eq. (6):
We varied the surface area of seed particles (SP) and determined LP(HOM) by Eq. (6). We found a linear relationship between LP(HOM) and SP as expected from kinetic gas theory, Eq. (7):
In Eq. (7), fFS is the Fuchs–Sutugin correction, is the mean molecular velocity of the HOM, and γeff is an effective uptake coefficient. The coefficient γeff was obtained from the slope of such plots by dividing the values for slopes by with fFS=0.7, assuming a mean median of the surface size distribution of 175 nm. In the case of β-pinene some new particle formation was observed, which hindered measuring c(HOM)0 directly. Here, c(HOM)0 was calculated by linear extrapolation of 1∕c(HOM) to a zero-particle surface (compare to Sect. S3).
It has to be noted that γeff is only valid if SP is not too large for two reasons. First, in the presence of a strong condensational sink, many HOM signals come close to the detection limit (here for m2 m−3). Secondly, for large SP ( m2 m−3) distinct deviations from linearity were observed, likely due to the fact that the timescales of losses of peroxy radicals on particles become similar to the timescales of peroxy radical reactions (Pullinen, 2017). If so, the production rates P(HOM) of HOM termination products are not constant but decrease significantly with increasing particle load.
3.1 Yields of organic nitrates from β-pinene photooxidation
In the first step we determined the potential of organic nitrate (ON) formation in a β-pinene∕NOx mixture ([β-pinene]0∼39 ppb[NOx]0∼50 ppb). The β-pinene and NOx were added to the chamber that contained about 60 ppb O3. The OH production was started by switching on the UV lamp (time = −2.4 h in Fig. 1), inducing the photochemical oxidation of β-pinene and thereby the ON production. As shown in Fig. 1, concentrations of β-pinene and NOx decreased in the presence of OH. When the photochemical system was in a steady state after about 2 h (time t=0 h in Fig. 1), the β-pinene addition was stopped and β-pinene concentration decreased to zero. In parallel, [OH] increased, leading to a lower NOx concentration. At time t=1.7 h, the OH concentration was readjusted to the same OH level as before the removal of β-pinene by lowering J(O1D). The decrease in [OH] caused an increase in [NOx] by 15 to 32 ppb. Considering the NOx level of 20 ppb before the β-pinene had been removed, the net [NOx] increase amounts to 12 ppb. The inflow of NOx and the OH concentration were the same before and after removal of β-pinene, but now [NOx]SS was higher. Hence, with β-pinene we removed a strong NOx sink in the chamber. Most of this NOx sink is made up by reactions of NO and NO2 with peroxy radicals and peroxy acyl radicals that lead to ON formation (Reactions R6a and R7). Thus, the difference in [NOx]SS in the presence and in absence of β-pinene allowed us to calculate the fraction of ON formed from β-pinene. Defining the yield of ON formation as the molar amount of NOx “released” by not forming β-pinene ON over the molar amount of consumed β-pinene and with the assumption that one lost NOx molecule had produced one ON molecule, we derived a molar yield of ∼36 % for the ON formed from β-pinene. For later comparison with AMS results we calculated the mass concentration of nitrate bound to the organic moieties (OrgNO3), again with the assumption that one lost NOx molecule produces one OrgNO3. A total mass concentration of 33 µg m−3 OrgNO3 in the gas phase was obtained at the given condition in the chamber. The mass concentration of HNO3 formed during this time was about 24 µg m−3 (for details of these calculations see Sect. S3).
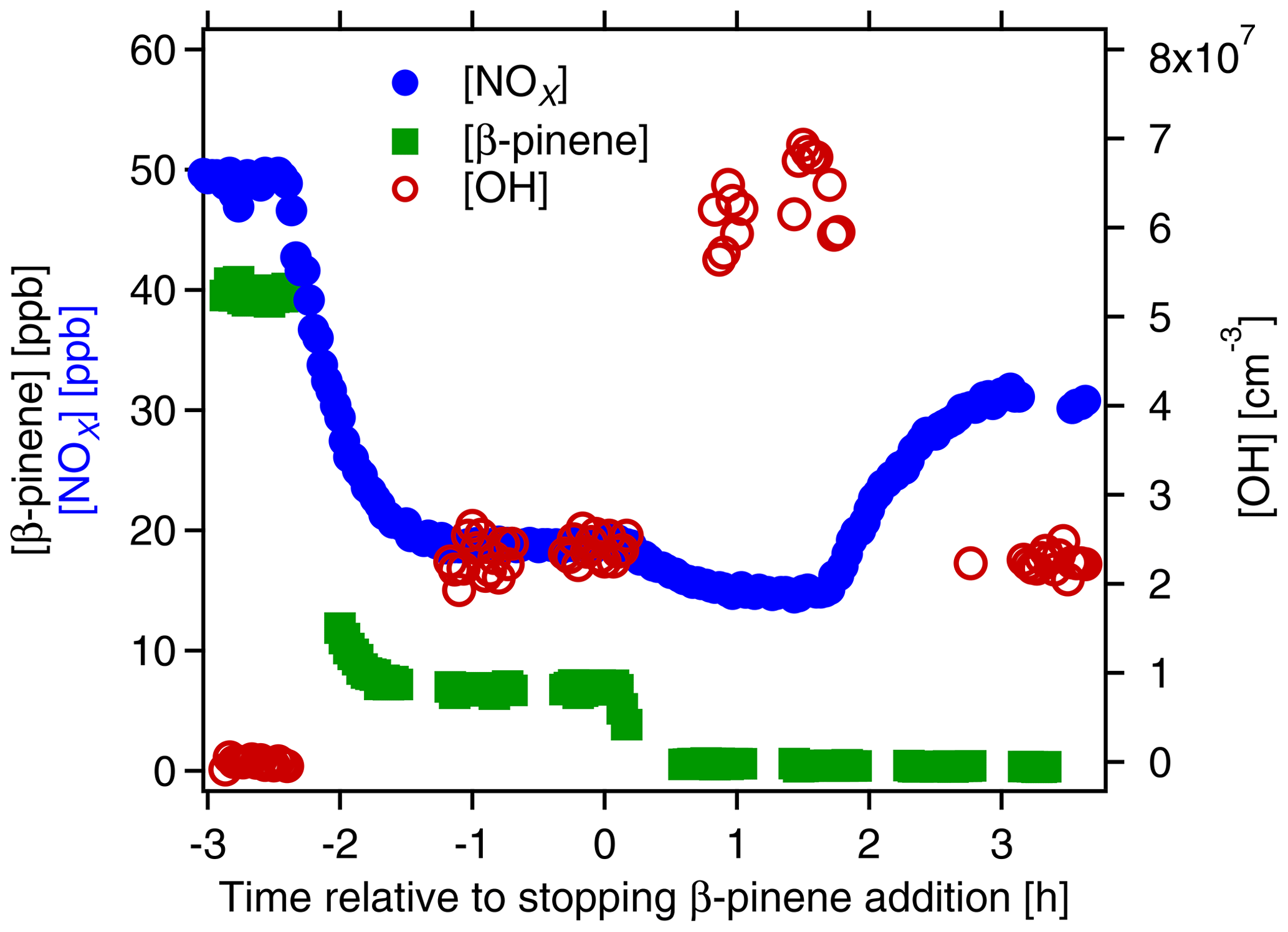
Figure 1Time series of [β-pinene] (green squares, left scale), [NOx] (blue circles, left scale) and [OH] (open brown circles, right scale). The experiment served to estimate the sum of organic nitrates (ON) formed in a mix of NOx and β-pinene. M-xylene ([m-xylene]0∼3.7 ppb) was added to the chamber as a tracer for OH. At time h OH formation was induced by O3 photolysis. At time t=0 h, β-pinene addition was stopped, and at time t=1.7 h J(O1D) was reduced to obtain the same [OH] as in the presence of β-pinene at time −1 h.
3.2 HOM formation from α-pinene and β-pinene photooxidation
We observed multifunctional peroxy radicals (HOM-RO2) as well as their termination products in the high-resolution mass spectra. The latter are formed in accordance with established pathways of peroxy radical chemistry (Bianchi et al., 2019). We will distinguish HOM-PPs, which arise from permutation reactions of HOM-RO2 with peroxy radicals, including HO2, and HOM-ONs, which are formed in the reaction of HOM peroxy and HOM acyl peroxy radicals with NO or NO2. In addition we found HOM accretion products with C>10 and C<20 (HOM-ACCs).
We observed two major differences in HOM formation and product patterns for α-pinene and β-pinene, which are both related to the position of their double bond.
-
For α-pinene, with an endocyclic double bond, addition of ozone is relatively fast, and at OH concentrations up to 1×107 cm−3 certain fractions of HOMs were produced by ozonolysis (compare Fig. S6). In contrast, ozonolysis of β-pinene, with an exocyclic bond, is slow and does not produce significant amounts of HOMs (Pullinen, 2017).
-
The HOM products of α-pinene in the monomer region mainly consisted of C10 molecules, as breaking of an endocyclic double bound will merely lead to ring opening. The breakage of the exocyclic double bond of β-pinene during the oxidation process will cause some fragmentation. We observed progressions of C10, C9, C8, and C7 HOMs leading to overlapping peaks in the mass spectra. For example, molecular masses where one C atom and four H atoms are replaced by one O atom were not fully resolved. However, by peak fitting we were able to attribute the contributing formula components in most cases (see Sect. S2 and peak list in Sect. S6). In addition, fragmented peroxy radicals in β-pinene form a larger variety of accretion products with fewer than 20 C atoms.
Besides these differences, the behavior with respect to NOx addition was very similar for α-pinene and β-pinene photooxidation: increase in HOM-ONs (increase in peaks with odd molecular masses), decrease in accretion products, and a shift of HOM monomers to the higher m∕z.
3.3 Accretion products and products from fragmentation
With increasing [NOx] we observed a strong decrease in HOM-ACCs relative to HOM monomers. These can be clearly seen in the HOM mass spectra obtained for α-pinene and β-pinene in Fig. 2 by comparing the ranges of and Da for low- and high-NOx conditions. To quantify the effect, the molecular-mass-weighted signals of HOM monomers (C5–C10, 230 Da < Da) and HOM-ACCs (C11–C20, 230 Da < Da) were converted to mass concentrations, summed up, and normalized as described in Sect. S3. Figure 3 shows the mass concentration of total HOMs and the fractions of HOM monomers and HOM-ACCs as a function of [NOx]. HOM-ACCs decrease with [NOx]: at the lowest and highest NOx levels of 0.3 and 72 ppb HOM-ACCs contribute 0.3 and 0.09 µg m−3, respectively, to total HOMs, whereas HOM monomers contribute about 0.4 µg m−3 over the whole range. At low-NOx conditions, accretion products contributed ≈40 % and monomers contributed ≈60 % to the total mass concentration of HOMs. At the highest NOx level the mass concentration of HOM-ACCs was suppressed by about 70 %. In comparison, the sum of HOM monomers was diminished only by less than 10 %. The decrease in HOM-ACC mixing ratio is attributed to the competition between HOM-ON formation channels (Reactions R6a and R7) and the HOM-ACC formation channel (Reaction R5). As we will show in the next section, in the presence of NOx more HOM-ON monomers were formed and thus fewer HOM accretion products.
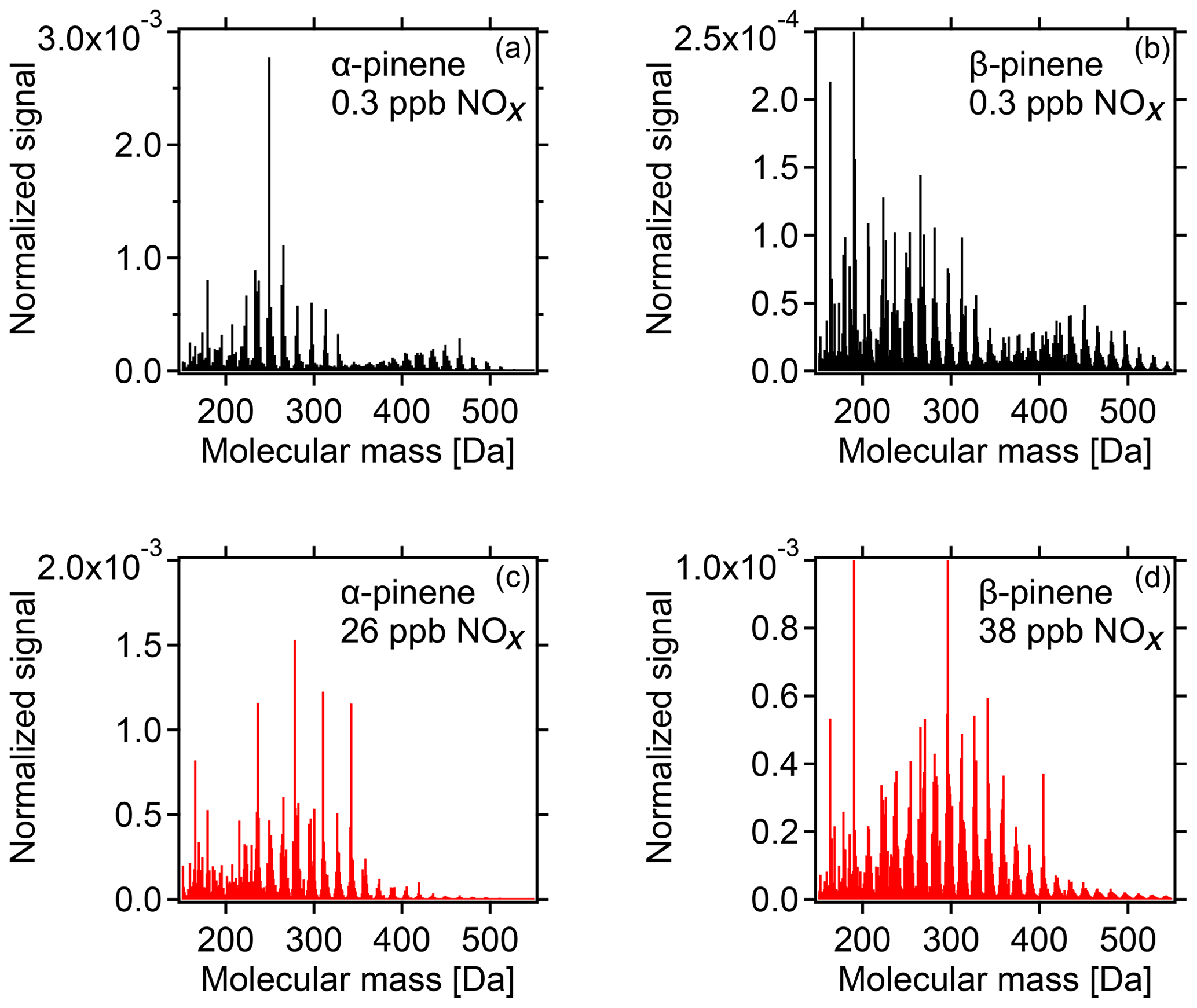
Figure 2HOM spectra from photooxidation of α-pinene (a, c) and β-pinene (b, d) without NOx addition (a, b) and with NOx addition (c, d). NOx concentrations in the α-pinene and β-pinene experiments were 26 and 38 ppb, respectively. Background NOx was 0.3 ppb. The signals were normalized to the sum over all detected ions. For the α-pinene example, in the low-NOx case HOM monomers contribute ≈0.4 µg m−3 and HOM-ACCs ≈0.3 µg m−3, whereas at 26 ppb NOx HOM monomers contribute ≈0.4 µg m−3 and HOM-ACCs less than 0.1 µg m−3 (compare Fig. 3).
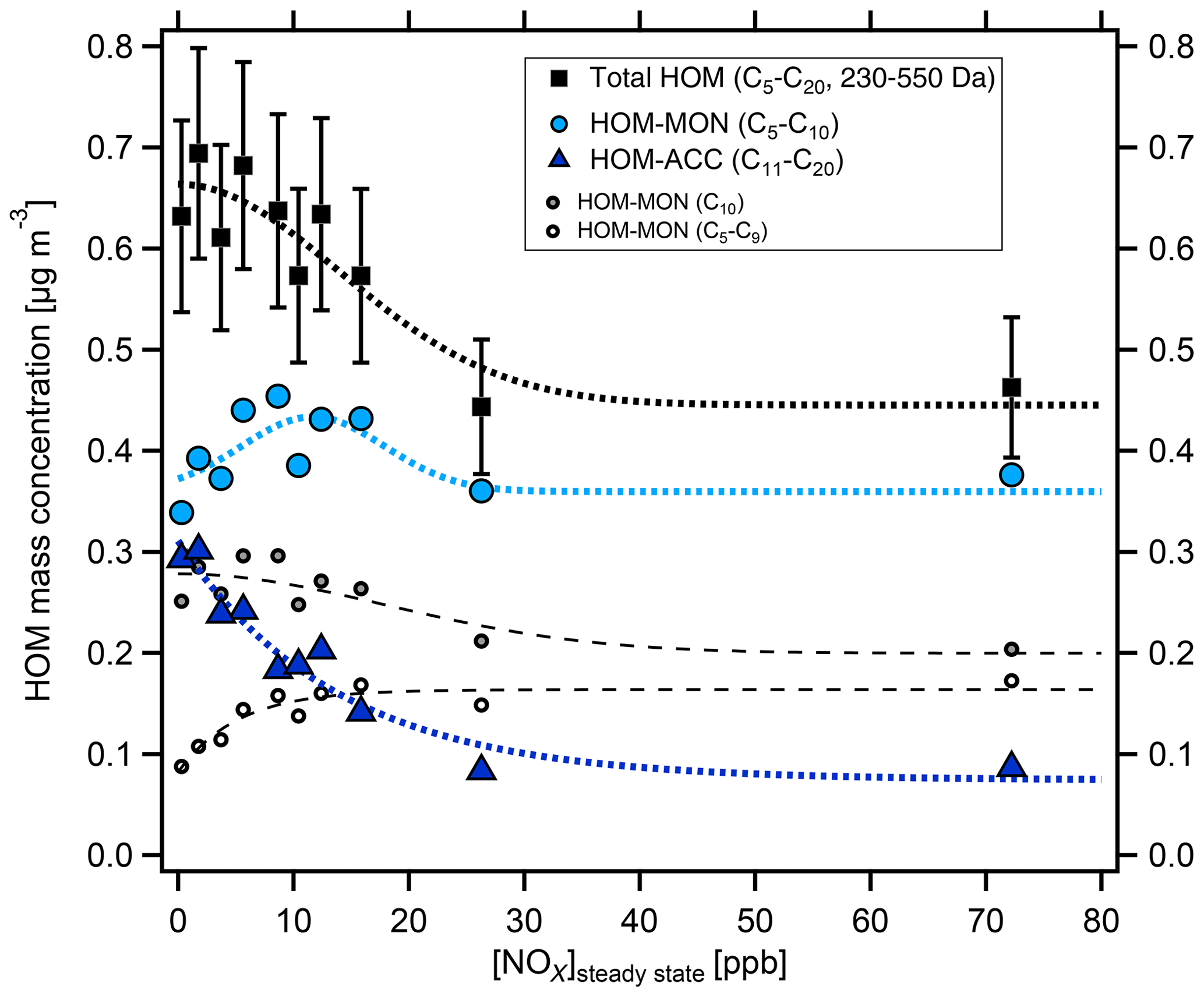
Figure 3Mass concentration of HOM products as a function of [NOx]SS in α-pinene photooxidation experiments. C5–C20 compounds with molecular masses 230–550 Da were added up for total HOMs (black squares) and divided into HOM monomers (light blue circles) and HOM accretion products (blue triangles). The analysis is based on the assigned peaks (>90 % of the total signal) and the sensitivity of 3.7×1010 molec. cm−3 nc−1 (Sect. S1.2). Dashed and dotted lines serve to guide the eye and have no further meaning. Concentrations were corrected as described in Sect. S1.2. Turnover ranged from 8.7×107 to 1.04×108 cm−3 s−1, leading to correction factors in the range of 1.1–0.8. The correction factors were close to 1 and thus did not add much uncertainty. Observed particle surface ranged from to m2 m−3, resulting in correction factors between 1.0 and 1.45, with the highest correction factors at lower [NOx]SS where new particle formation could not be suppressed.
We separated HOM monomers in compounds with C10 and compounds with C<10 (C5−9) shown by the small circles in Fig. 3. The mass concentration of C<10 HOMs increased with [NOx]SS (small open circles) whereas C10 HOMs decreased (small grey circles). Compounds with C5−9 double from ≈0.9 to ≈1.8 µg m−3 at the highest [NOx]SS, whereas the C10 compounds drop by only about 30 %. Since C5−9 compounds arise in large parts from fragmentation of alkoxy radicals, this indicated that a portion of C<10 arose from fragmentation of alkoxy radicals formed in R6b. Since we consider only molecular masses ≥230 Da, which is the molecular mass for formula C10H14O6, the C<10 compounds must still be highly functionalized; i.e., they must carry more oxygen than the respective C10 compounds to reach similar molecular masses, one O atom more per C lost.
We assume that compounds in the selected mass range will contribute to SOA formation. Since total HOMs decrease at higher [NOx]SS, but HOM monomers remain about stable over the whole NOx range, the suppression of the HOM-ACCs was the cause of the reduction of total HOMs and therewith of condensable mass. The fragmentation via alkoxy radicals played only a minor role.
3.4 Detection of termination permutation products of peroxy radical – peroxy radical reactions and HOM-organic nitrates
At low-NOx conditions, HOM-PPs with even molecular masses showed the highest concentrations. HOM-ACCs are also produced from peroxy–peroxy permutation reactions, but their intensity decreased strongly, and HOM-ACCs are barely observed at high-NOx conditions, as described in the previous Sect. 3.3. For that reason, we focus on monomer products in the following Sect. 3.5–3.7.
Because of the complexity of the product spectrum, the character of the functional group formed in the termination step of HOM-PPs cannot be derived unambiguously from the available elementary molecular formulas alone. As an example, HOM-PPs with the molecular formula C10H16Ox can be hydroperoxides formed from peroxy radicals with the molecular formula C10H15Ox in Reaction (R3a), they can be alcohols formed from peroxy radicals C10H15Ox+1 in Reaction (R4a), or they can be ketones formed from peroxy radicals with the molecular formula C10H17Ox+1 in Reaction (R4a). Dependent on the specific precursor peroxy radical, they can also be carboxylic acids or percarboxylic acids. We therefore lump the monomer HOMs with even masses (HOM-PPs) together independent of the chemical character of the termination group. In the case of β-pinene, we did not separate the contributions from different progressions (e.g., C10HyOx and ) but used the overall signal under the peak for further analysis and indicate the main components under the peak (e.g., Fig. 5). Separation between HOM-PPs and HOM-ONs was easier, because HOM-ONs have odd molecular masses. Peroxy radicals (without N) also contribute to the odd mass peaks, but in most cases they could be separated by the mass defect. Other contributors to odd mass peaks are HOMs containing 13C and clusters with the nitrate dimer () of HOM-PPs, but the latter contribution was small (Sect. S2). Analysis of the high-resolution mass spectra for α-pinene revealed that HOM-ON-peroxy radicals are rare. Peak lists for the HOMs measured in the absence and presence of NOx are given in Sect. S6. (Note that we cannot a priori distinguish between HOM-ONs formed in Reactions (R6a) and (R7).)
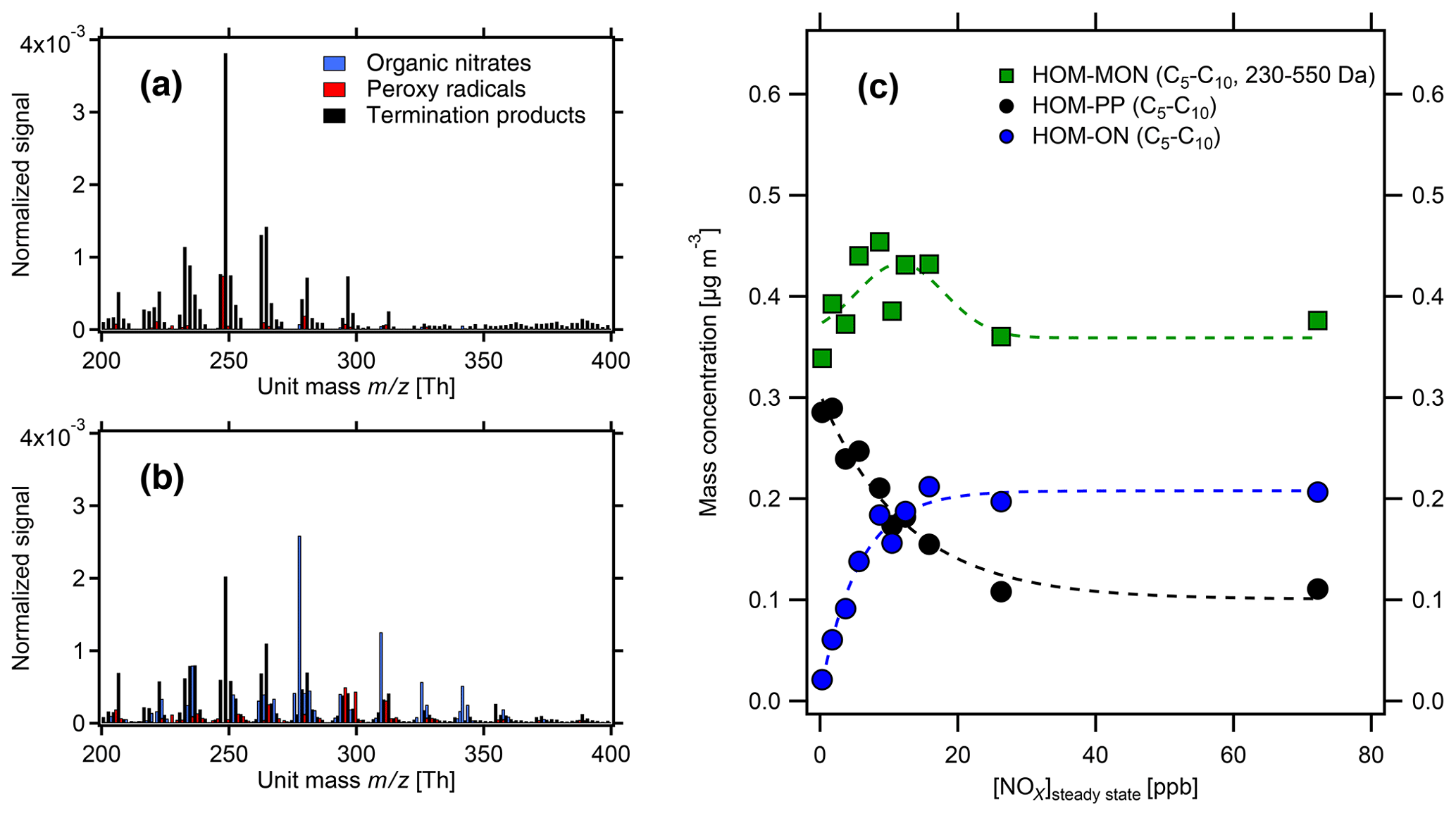
Figure 4HOM pattern from α-pinene photooxidation at two NOx levels in the monomer range. (a) Low-NOx conditions ([α-pinene]SS=1.7 ppb, [NOx]SS=0.3 ppb); (b) high-NOx conditions ([α-pinene]SS=1.0 ppb, [NOx]SS=8.7 ppb). Black bars: HOM-PP termination products of Reactions R3 and R4a. Blue bars: HOM-ONs (organic nitrates). Red bars: HOM-RO2 (peroxy radicals). The signals were normalized to the sum over all detected ions. (c) Mass concentrations of HOM monomers (green) in the molecular mass range 230–550 Da. HOM-ONs (blue) are increasing with increasing [NOx]SS, HOM-PPs (black) are decreasing, and the sum of all HOM monomers remains about the same. At about 10 ppb [NOx]SS HOM-ONs make up half of the HOM monomers, and at 26 ppb [NOx]SS they make up about 50 % of the total HOMs (shown in Fig. 3). Dashed lines serve only to guide the eye.
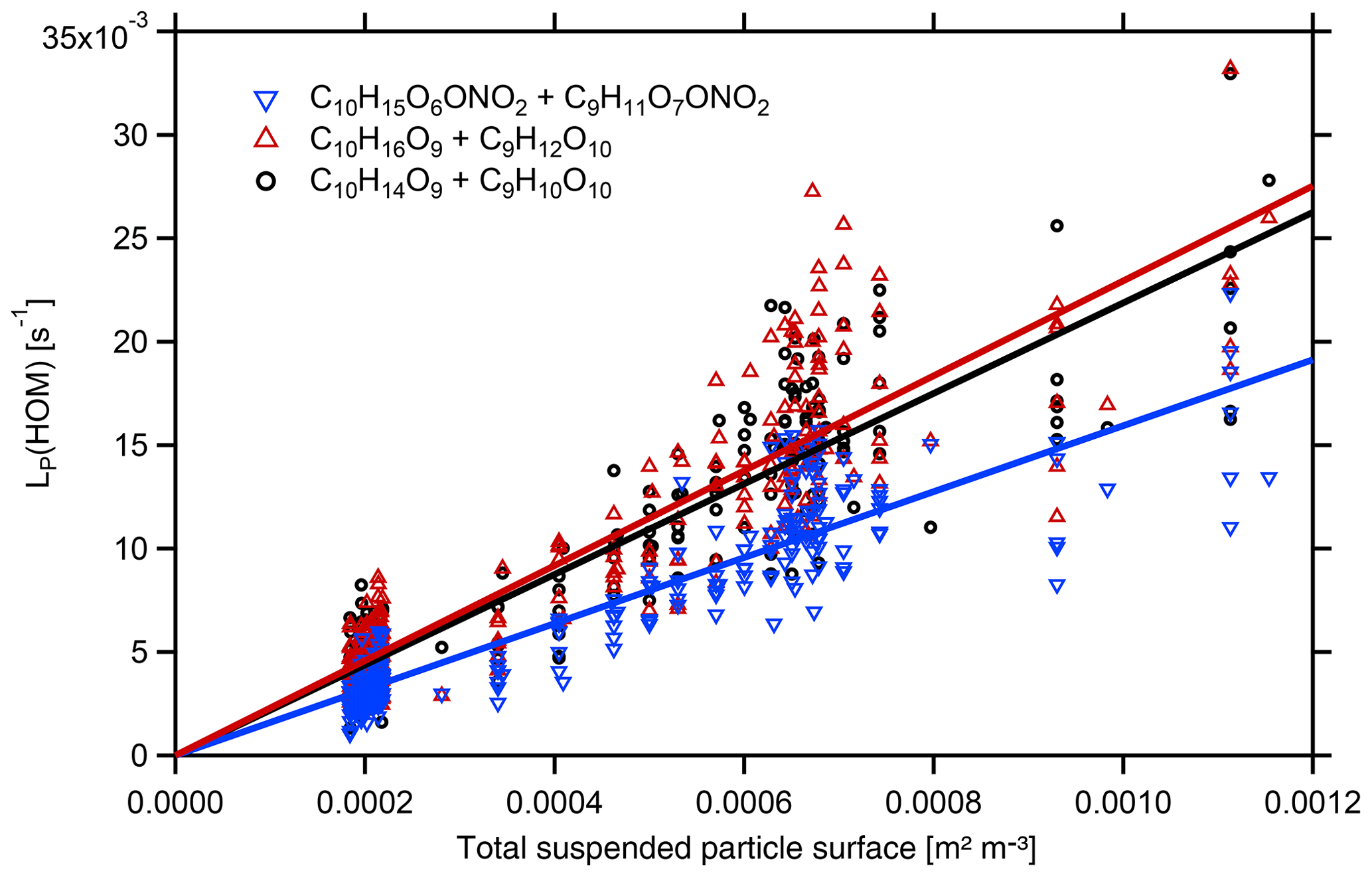
Figure 5Plot of LP(HOM) calculated by Eq. (6) versus particle surface area, SP, for the examples of HOM-ONs with a molecular mass of =293 Da (C10H15O6ONO2 and C9H11O7ONO2), HOM-PPs with a molecular mass of 280 Da (C10H16O9 and C9H12O10), and HOM-PPs with molecular mass of 278 (C10H14O9 and C9H10O10). HOMs from β-pinene photooxidation ([β-pinene]SS∼10 ppb, [NOx]SS∼4 ppb). Dividing the slopes by the respective led to for the example HOM-ONs and ∼0.6 in the latter cases. The main uncertainty arises from the scatter of LP(HOM) individual unit mass resolution data. Statistical errors of fFS×γeff were about ±5 %.
Figure 4 demonstrates the change in the HOM pattern when NOx is added to the reaction system. At low-NOx levels, HOM-PPs (black) were predominant, but already at the medium NOx levels the concentrations of HOM-ONs (blue) were similar to HOM-PPs (black). HOM-ON concentrations increased at the cost of HOM-PPs. The product spectra of β-pinene showed a similar shift from HOM-PPs to HOM-ONs.
With increasing NOx we observed a small but increasing fraction of highly oxidized nitrates with C<10. Their chemical formulas have a low H:C ratio compared to gas phase C10 HOM-ONs. Supposedly, they did not arise from gas phase chemistry but were likely formed at the walls. Their time series was not responding to the changes of experimental conditions like start of photochemistry or end of NOx addition. Instead these compounds increased steadily after NOx addition. For α-pinene, their maximum contribution appeared at 74 ppb [NOx]SS where they amounted up to 8 % of the total HOM concentration and 17 % of the HOM-ON concentration. Below 35 ppb NOx their contribution was less than 7 % and 12 %. For β-pinene, this was less distinct and the contribution was only 2 % to 3 %. Because these HOM-ONs had C<10 and appear at the lower end of the mass spectrum, their contribution to HOM mass (and therewith SOA mass) was small, and we did not correct for these products.
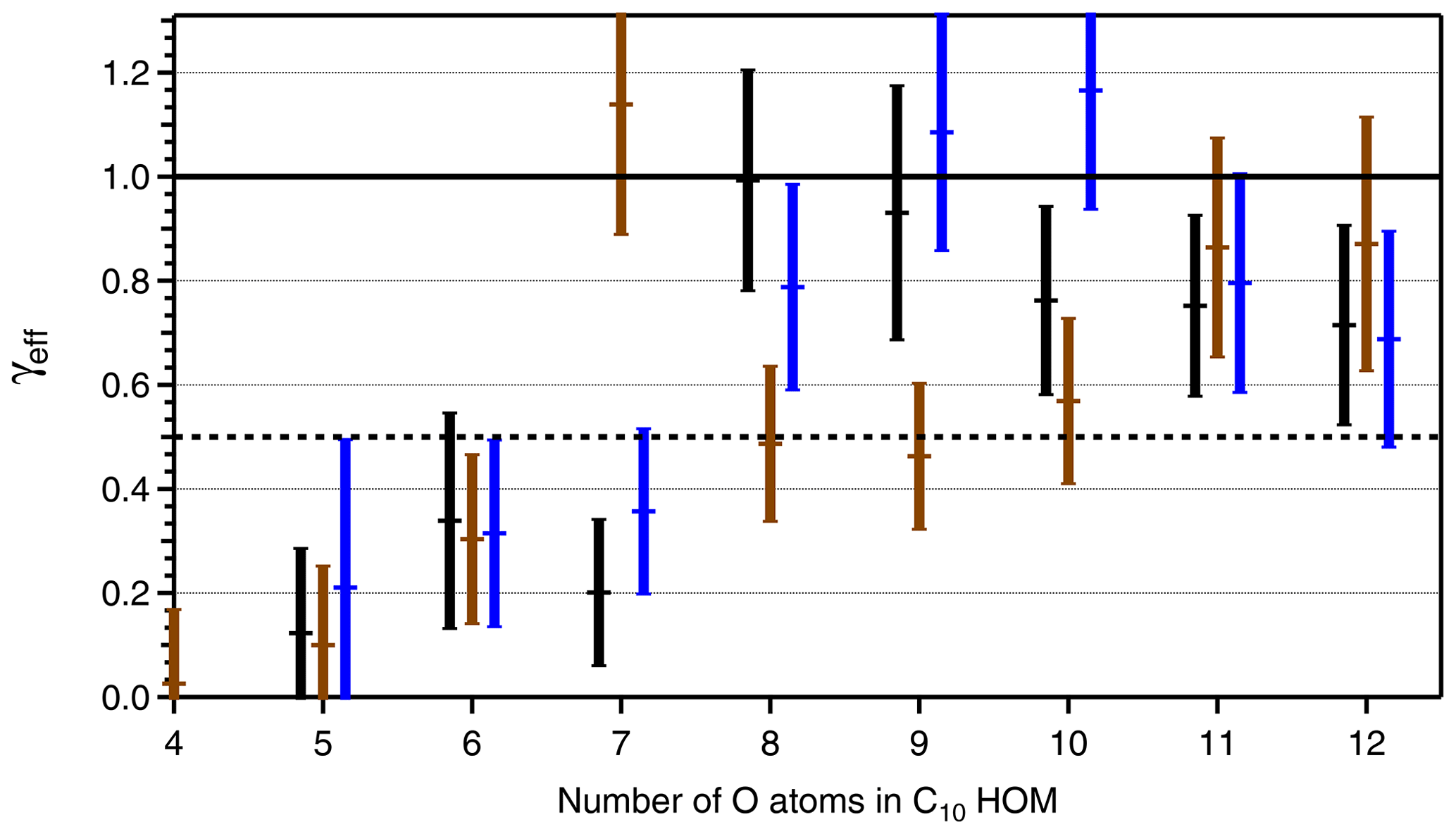
Figure 6Effective uptake coefficients γeff for HOM-PPs (C10H14Ox black bars, C10H16Ox, brown bars) and HOM-ONs (C10H15OxNO2, blue bars) as a function of the number of O atoms in the respective HOM. HOMs with different numbers of C, H, and O atoms, e.g., C10HyOx and HOM-PPs, are treated together, and the number of O atoms is given for the C10-HOM-PPs. The second component, C9-HOM-PPs, has one O atom more. Data were taken from the β-pinene photooxidation experiment with [β-pinene]SS∼10 ppb, [NOx]SS∼4 ppb. The signal intensity for the C10H14O4 and HOM-ONs with four O atoms was too low to allow reliable determination of γeff, and the respective data are left out. Uncertainties in γeff arise from the determination procedure as shown in Fig. 5. The black line indicates γeff=1 and 0.5. An average Fuchs–Sutugin correction factor of 0.70 (dp=175 nm) was applied to calculate γeff.
3.5 Effective uptake coefficients for HOM-PPs and HOM-ONs
Based on identified HOM-ONs and HOM-PPs, we characterized their potential contribution to SOA formation by determining their loss rates on seed particles LP(HOM), Eq. (6). According to Eq. (7), plots of LP(HOM) versus particle surface, SP, should exhibit a linear dependence between LP(HOM) and SP, allowing for the determination of effective uptake coefficients, γeff. However, for conditions with sufficient HOM production, we could not fully suppress new particle formation in absence of seed particles. As a result c(HOM)0 in Eqs. (5) and (6) had to be determined by extrapolating 1∕c(HOM) as f(SP) to SP=0. The wall loss coefficient LW(HOM) was determined to 150 s in independent experiments at lower concentrations and in absence of new particle formation. With c(HOM)0 and LW(HOM) LP(HOM) was calculated, allowing the derivation of γeff from Eq. (7) by applying fFS=0.7 to the slope of LP(HOM) as a function of particle surface SP (Fig. 5). Figure 5 is based on HR data, which show substantial scatter. The unit mass resolution (UMR) data showed a better signal-to-noise ratio than the individual HR peaks under the same UMR signal. In order to reduce the scatter, we thus used the respective UMR data for the evaluation of γeff. In Fig. 6 we compare γeff for HOMs with the same number of O atoms. Note that compared to their chemical sum formula, ONs were shifted by one O to lower O in order to account for the addition of NO. When comparing data for HOMs with the same numbers of O atoms (in the precursor peroxy moiety), no significant and systematic differences were found for γeff within the uncertainty limits; i.e., the potential to condense on particles was about the same for HOM-PPs and HOM-ONs. For HOM moieties with eight and more O atoms, γeff approaches 1 independent of whether they were HOM-PPs or HOM-ONs. HOM-PPs and HOM-ONs with six and more O atoms with upper limits of γeff near 0.5 will still reside to a large degree in the particle phase; therefore they should also contribute significantly to SOA mass.
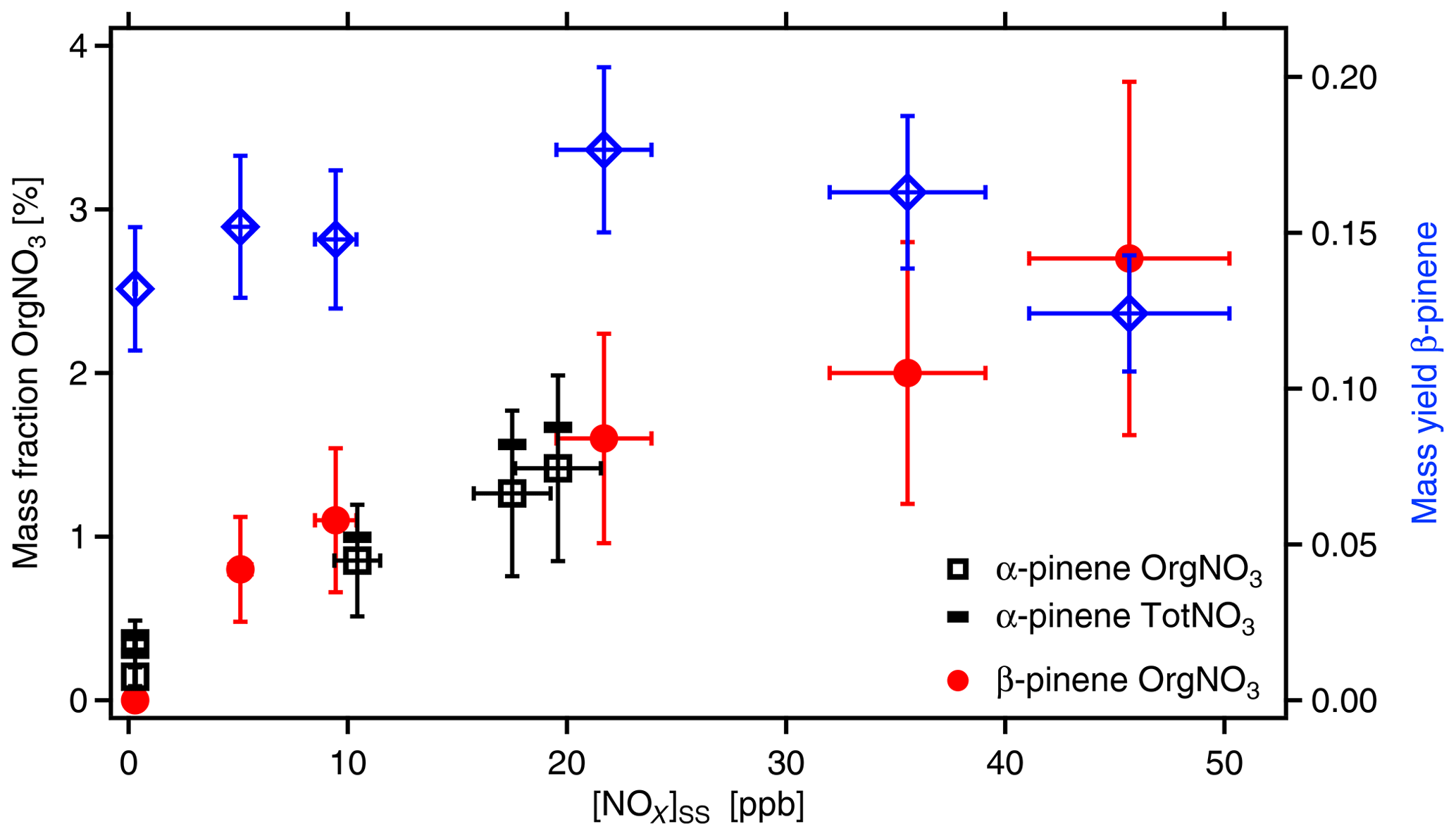
Figure 7Mass fraction of organic-bound nitrate (OrgNO3) in SOA as a function of [NOx]SS (left y axis). Black squares and red circles show data measured from α-pinene and β-pinene, respectively. SOA mass yields are shown for the respective experiment with β-pinene (blue diamonds). The SOA load ranged from 11 to 23 µg m−3 with an average of 16±5 µg m−3. The data are corrected for wall losses of HOMs. In absence of OH, [α-pinene]0 was around 46 ppb, and [β-pinene]0 was around 37 ppb. NOx was added at different amounts with [NOx]0 up to 103 ppb. Due to losses in reactions with OH and formation of organic nitrates, [NOx] decreased to the [NOx]SS levels shown here. Uncertainties in NOx data are estimated to ±10 %, uncertainties in SOA masses to ±10 %, and uncertainties in the content of OrgNO3 to ±40 %. The black bars indicate the fraction of total nitrate (TotNO3, left scale) for the example of α-pinene, which is dominated by organic nitrate.
3.6 Organic-bound nitrate in SOA and in gas phase HOM-ONs
SOA yields for α-pinene and β-pinene (wall loss corrected) were between 0.08 and 0.18, thus in the same range as those reported by Sarrafzadeh et al. (2016). The amount of formed SOA mass and the SOA yields changed with the addition of NOx to the photochemical system because NOx affects [OH]SS and thereby also the formation of higher-generation products. The mass fractions of OrgNO3 in the SOA particles are shown in Fig. 7. They were calculated for the different NOx concentrations by dividing the mass concentration of OrgNO3 by the mass concentration of the respective organic as measured with the AMS. The fraction of OrgNO3 in particles was dependent on the NOx concentrations. It was negligible when no NOx was added and increased steadily with increasing [NOx]. At the same time, the fraction of inorganic nitrate also increased with increasing [NOx] but was a factor of about 3 lower than that of OrgNO3 (Fig. 7). Calculating [HNO3]SS in the gas phase from [NO2] and [OH] shows that, at the highest NOx concentrations up to 24 µg m−3 of HNO3 was formed in the gas phase, but less than 0.1 µg m−3 of inorganic nitrate was found in the particle phase. For the same [NOx]SS, the mass fractions of organic or inorganic nitrate in SOA were about the same for α-pinene and β-pinene, indicating that the formation of condensable OrgNO3 was similar. The determination of OrgNO3 comprises some uncertainty, but even if we count all inorganic nitrate as OrgNO3 we get an upper limit of less than 4 %.
We compared the amount of OrgNO3 in particles to that in the gas phase. As shown in Sect. 3.1, at [NOx]SS∼20 ppb about 33 µg m−3 OrgNO3 was formed in the gas phase. At similar [NOx]SS and similar β-pinene concentrations([NOx]SS∼22 ppb, [β-pinene]SS∼6 ppb) the fraction of OrgNO3 in the particle phase was only 1.6±0.64 % (Fig. 7); i.e., less than 0.4 µg m−3 OrgNO3 was bound in particles. We conclude that many ONs are too volatile to significantly contribute to the particulate phase. However, considering the low-volatility HOM-ONs (Sect. 3.5), HOM-ONs should contribute to particle mass, eventually providing the particulate OrgNO3. We estimated the mass fraction of OrgNO3 bound in HOM-ONs. For this we considered all HOMs with six and more O atoms in the HOM moiety (molecular mass > 230 Da) because their γeff is large enough to partition significantly into the particle phase and to contribute efficiently to SOA mass. As shown in Fig. 6, the HOMs partitioned without preference, independent of being HOM-PPs or HOM-ONs. We first determined the fraction on a molecular base by using the ratios of signal intensities, which is identical to using concentrations c:
In Eq. (8), the left-hand term represents the molar fraction of HOM-ONs. We summed all HOMs with O≥6, which included all HOMs with γeff>0.5 and provides a lower limit. Signals at Da were not taken into account, since they were very low at high NOx levels and thus uncertain. In a second step we calculated from Eq. (8) the mass ratio of OrgNO3. We split all HOMs in the denominator of Eq. (8) in HOM-ONs and other termination products and multiplied the concentrations with the respective molar weight (Eq. 9). The numerator was multiplied with the molecular weight of the nitrate termination group:
In words, the left-hand term in Eq. (9) gives the ratio of the total mass of OrgNO3 over the total organic mass of HOMs with γeff>0.5 (O≥6). This value can be compared with the direct AMS observation of OrgNO3. Figure 8 shows the molecular ratios (calculated by Eq. 8) and the mass fractions (Eq. 9) as a function of NOx. For α-pinene we were able to separate HOM-ONs and HOM-RO2 unambiguously (see Supplement). For β-pinene we give lower and upper limits of the molecular and mass fractions, because of uncertainties in the HR analysis that were induced by the stronger fragmentation and overlapping progressions of compounds with a different number of C atoms but the same unit molecular mass. For the lower limit shown in Fig. 8, we applied a peak list with all identified signals at low and high NOx concentrations for fitting, whereas the upper limit was achieved by using the peak list optimized for high-NOx cases. (The reason for the spread can be explained as follows: the approach with the peak list with all identified peaks attributes some HOM-RO2 to the HON-ON signal, independent of whether the specific HOM-RO2 exists in the chemical system or not, while the approach with the high-NOx peak list has the tendency to falsely attribute HOM-ONs to existing HOM-RO2 missing in the high-NOx peak list.)
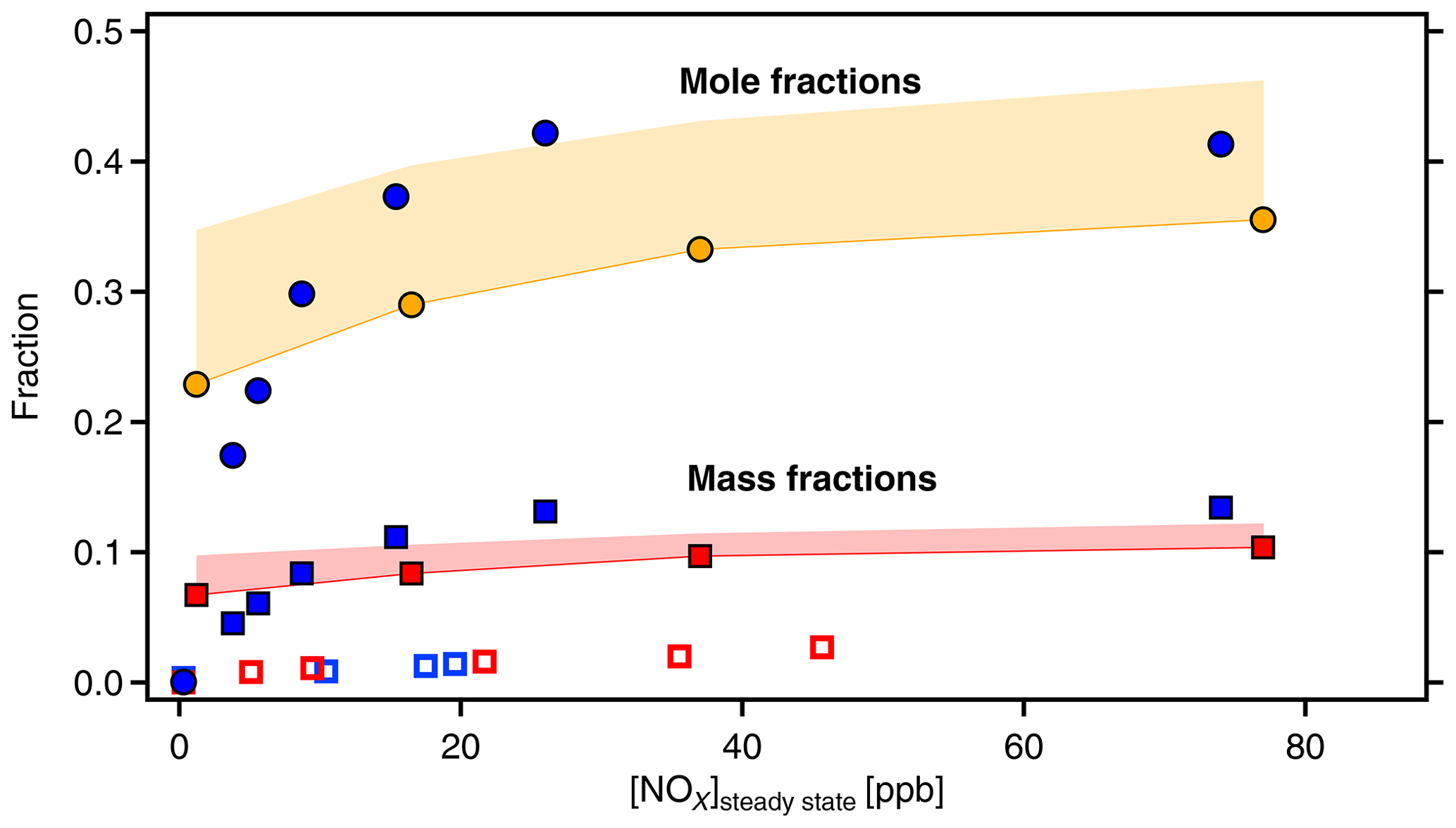
Figure 8Molecular fractions of organic-bound nitrate (OrgNO3, filled circles) and mass fractions of OrgNO3 (squares) as a function of [NOx]SS. Data from α-pinene (blue symbols) and β-pinene (orange and red symbols and areas). Molecular fractions of OrgNO3 and HOM-ONs are the same by definition. The mass fraction of OrgNO3 in the gas phase HOMs is significantly higher than in the particulate phase as determined by AMS (open blue and red squares). The areas in orange and red give the potential error for β-pinene due to unresolved progressions and overlap of organic nitrates with peroxy radicals (as explained in text).
Both HOM-ON and OrgNO3 mass fraction increased with [NOx], similarly for both MTs. For α-pinene about 40 % of the detected HOMs were HOM-ONs and more than 10 % of the HOM mass was OrgNO3 once [NOx] was larger than 30 ppb ([BVOC] ppbC ppb−1). For the upper limit case of β-pinene we achieved about the same fractions as observed for α-pinene (50 ppb NOx: [BVOC] ppbC ppb−1). Since we considered only HOMs that efficiently condense on particles, one would expect that OrgNO3 brought by HOM-ONs alone should contribute about 10 % of the SOA mass. This was not the case, as the direct comparison in Fig. 8 shows. The maximum contribution of particulate OrgNO3 was about 3 %; i.e., the measured OrgNO3 was a factor of 3 to 4 lower than expected: the OrgNO3 bound in low-volatility HOM-ONs which could potentially contribute to SOA mass was significantly higher than OrgNO3 directly observed in the particle phase.
3.7 Mass concentration of HOMs
To estimate the possible effect of hydrolysis of OrgNO3 and re-evaporation of HNO3, potential condensable mass concentration (cmass) was derived by weighing the concentration () of each HOMi by its molecular mass (Mi) in the range of 230 to 550 Da:
Herein cN is the concentration that was corrected according to the method described in Sect. S2. Figure 9 shows the calculated mass concentrations with and without hydrolysis as a function of NOx. The relative uncertainty limits were estimated to be 19 %. The uncertainty was estimated from the standard deviation of the data obtained at low-[NOx] conditions (nine measurements). Uncertainties of [NOx]SS were estimated to be ±10 %. The uncertainty of absolute concentrations caused by the uncertainty of the calibration factor (see Sect. S1) is much higher than the uncertainty limits shown in Fig. 9. However, as the systematic error of the calibration factor is the same for each data point it does not affect the observed trend of only somewhat decreasing mass concentrations of HOMs with increasing NOx.
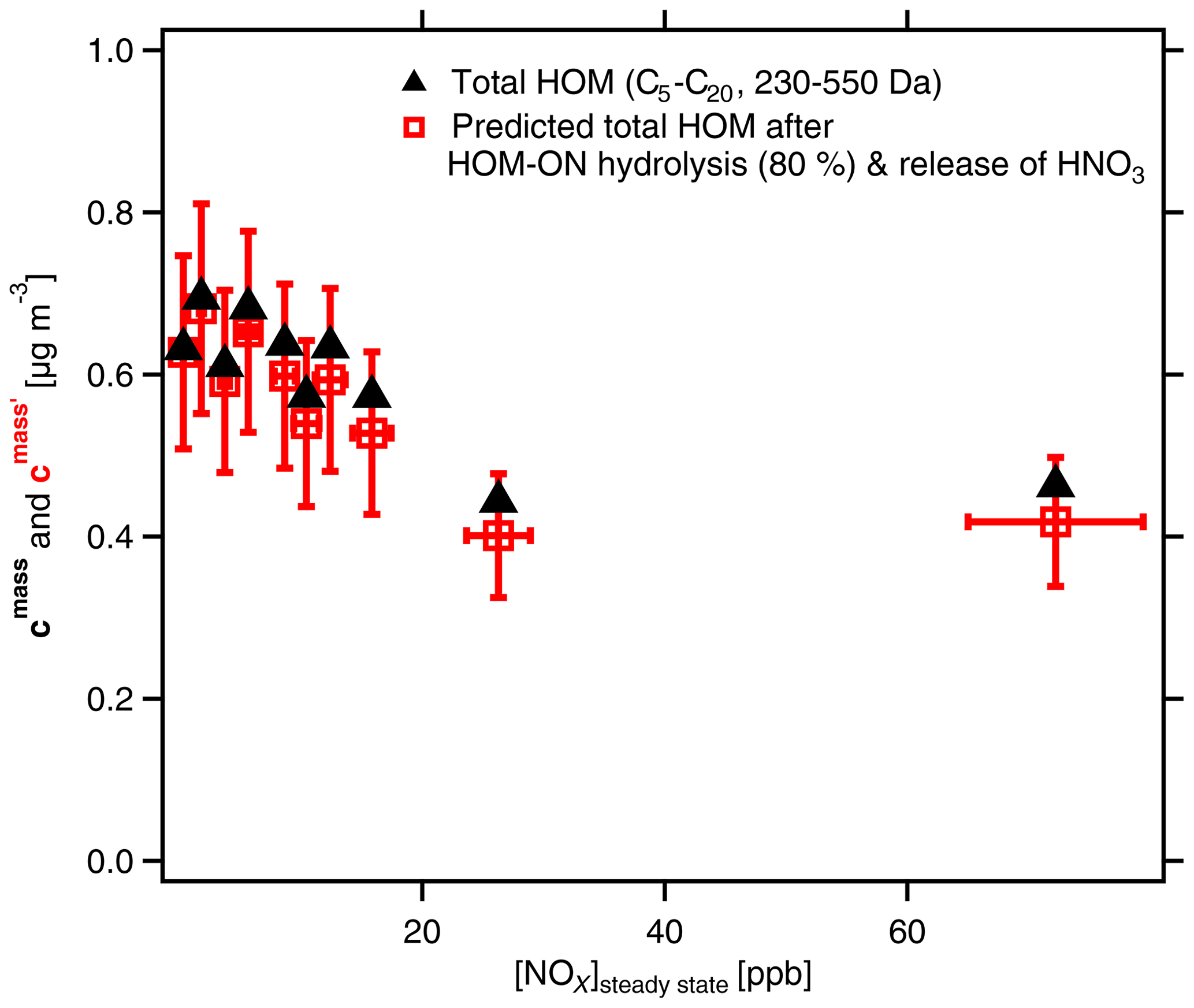
Figure 9Mass concentrations of total HOMs (C5–C20) with molar masses between 230 and 550 Da. Black triangles show mass concentrations cmass as determined. Red squares show , i.e., the resulting SOA mass after considering OrgNO3 loss by hydrolysis and evaporation of HNO3. [α-pinene]SS=0.9 to 2.2 ppb; [NOx]0 up to 125 ppb; [NOx]SS=0.3 to 74 ppb. The effect of hydrolysis of 80 % of the organic-bound nitrate has no substantial effect on the SOA mass. Analysis is based on assigned molecular formulas (>90 % of the total signal) applying the sensitivity of 3.7×1010 molec. cm−3 nc−1 (Sect. S1.2).
In the case of HOM-ONs, cmass includes the mass of OrgNO3. According to many studies particulate ON is undergoing hydrolysis, leading to loss of HNO3 (Bean and Hildebrandt Ruiz, 2016; Boyd et al., 2015; Rindelaub et al., 2015; Takeuchi and Ng, 2019). It is not clear whether organic material is lost from the particulate phase besides OrgNO3 (Fisher et al., 2016; Takeuchi and Ng, 2019; Zare et al., 2019). The efficiency of the hydrolysis of OrgNO3 depends on RH and particle acidity, and several studies report fractions of hydrolyzed particulate ON in a range of 10 %–60 % (Bean and Hildebrandt Ruiz, 2016; Boyd et al., 2015; Browne et al., 2013; Rindelaub et al., 2015; Takeuchi and Ng, 2019).
We indicate the resulting SOA mass after considering OrgNO3 loss by hydrolysis and evaporation of HNO3 by a prime as in Fig. 9 (details for the calculations of ; see Sect. 4.3). It is obvious from Fig. 9 that the mass concentration of condensable HOMs is about 30 % lower at the highest-NOx conditions compared to those at low-NOx conditions. It is furthermore evident that the differences between cmass and are quite low, which shows re-evaporation of HNO3 is of minor importance for explaining the SOA mass suppression with increasing [NOx] in the system. An explanation must then be the observed strong decrease in the accretion products with increasing [NOx] as shown in Fig. 2.
4.1 Organic nitrates and SOA formation
Several studies on organic nitrates (ONs) or organic-bound nitrate (OrgNO3) in SOA refer to reactions of unsaturated volatile organic compounds with NO3 (Claflin and Ziemann, 2018; Faxon et al., 2018; Fry et al., 2013, 2014; Kiendler-Scharr et al., 2016; A. K. Y. Lee et al., 2016; Ng et al., 2017, and references therein). The pathway of forming ON by NO3 was negligible in our experiments as we applied quite high light intensity, humidity, and also high NO concentrations during our experiments. These experimental conditions inhibited formation of NO3 at relevant concentrations because NO3 was efficiently destroyed by photolysis, by reactions with NO, and by scavenging of N2O5 at the humid surfaces of the chamber walls. The HOM-ONs measured during our experiments were formed in Reactions (R6a) and (R7).
There are some studies with respect to the SOA content of ON formed by photooxidation (Berkemeier et al., 2016; B. H. Lee et al., 2016; Nozière et al., 1999; Rollins et al., 2010; Takeuchi and Ng, 2019; Xu et al., 2015b; Zhao et al., 2018) wherein in most cases mass fraction of ONs of the total SOA mass is reported. According to literature data, ONs produced during photooxidation or ozonolysis contribute between 3 % (B. H. Lee et al., 2016) and 40 % (Berkemeier et al., 2016) to the total SOA mass. This compares well with the mass fraction of HOM-ONs with molecular masses > 230 Da, which varied from 0 % to 50 % with increasing NOx (Fig. 4). HOM-ONs (>230 Da) provide a measure of the expected contribution of ON to SOA as HOM-ONs and all other HOMs should condense with the same efficiency as shown by their γeff in Fig. 6.
We determined OrgNO3 by the AMS as a diagnostic for ONs in the particulate phase. The OrgNO3 mass fractions ranged from 0 % (no NOx addition) to 2.7 %, which is within the range 0.6 %–8 % of most literature data but at the lower end (Nozière et al., 1999; Rollins et al., 2010; Xu et al., 2015b; Berkemeier et al., 2016; B. H. Lee et al., 2016; Zhao et al., 2018). (When mass fractions of particulate ON were given, we estimated OrgNO3 assuming an average molecular mass of 300 Da and one nitrate group per ON; see Sect. S5.) In our study we showed that OrgNO3 depends on the NOx level ([VOC]∕[NOx] level) as expected from established peroxy radical chemistry in the presence of NOx. This finding can probably explain the wide range of ON and OrgNO3 fractions reported for SOA formation in the presence of NOx. Detailed and meaningful comparison of our data to those reported in literature requires knowing the [VOC]∕[NOx] ratios during SOA formation in the respective experiments. The [VOC]∕[NOx] ratio is known for experiments made by us in the SAPHIR chamber in Jülich (Zhao et al., 2018). Zhao et al. (2018) achieved a mass fraction of 11 % for OrgNO3, which is significantly higher than the OrgNO3 mass fractions in this study. Interestingly, the 11 % found by Zhao et al. (2018) is close to the 10 % mass fraction of OrgNO3 determined for the total HOMs in this study. These findings will be further discussed in Sect. 4.3.
4.2 Effective uptake coefficients
We provided data on effective uptake coefficients, γeff, which allowed differentiation between semivolatile organic compounds (SVOCs), LVOCs, and ELVOCs. The dependence of γeff on the number of O atoms in the HOM (Fig. 6) suggests that the OrgNO3 found in SOA predominantly originates from HOM-ONs with six and more O atoms (without −NO). We conclude that ONs with fewer than five O atoms are not so important for the formation of SOA, at least at atmospheric loads of SOA > 10 µg m−3. This conclusion is confirmed by our observation of small mass fractions of OrgNO3 in HOM monomers (0 %–10 %) and in the particle phase (0 %–3 %) despite the large ON fractions produced overall in the gas phase (molecular yield > 30 %, Fig. 1 in Sect. 3.1). From all ONs only a few percent – the HOM-ONs – made it into the particulate phase (Sect. 3.1 and 3.6).
Our findings are in agreement with observations by B. H. Lee et al. (2016) in a field study. They also show that the distribution of signal intensities for HOM-ONs in the gas phase is different from that in the particle phase. Comparing the signal intensities for HOM-ONs with the same number of O atoms in the gas phase and the particle phase (Fig. 2 in B. H. Lee et al., 2016), it seems that the higher the number of O atoms, the more the ON partition in the particle phase. As there was no calibration for gas phase HOM-ONs, absolute numbers for partitioning coefficients or effective uptake coefficients were not obtained. Our data are qualitatively consistent with those of B. H. Lee et al. (2016), suggesting that the basics of HOM-ON condensation in our laboratory studies are similar to those in the environment.
Comparison between γeff determined for HOM-PPs and HOM-ONs indicated that there were no significant and systematic differences at least for the HOM moieties with more than six O atoms. The variability in γeff in Fig. 6 is probably caused by the fact that more than one compound with different structure and functionalization contributes to each data point. For example, C10H16Ox showed a large γeff at an O number of 7 (Fig. 6). The signal assigned to C10H16O7 could be dominated by a compound with extremely low vapor pressure.
The independence of the overall uptake behavior from the termination groups can be understood from the basics of group contribution models (Capouet and Müller, 2006; Pankow and Asher, 2008; Compernolle et al., 2011): HOM peroxy radicals with more than six O atoms already carry protic functional groups – OH, −OOH, C(= O)OH, or C(= O)OOH – from the several autoxidation steps. Thus their vapor pressure is low because of the ability to form (multiple) hydrogen bonds. The termination Reactions (R3), (R4a), (R6a), and (R7) only form one more functional group of the respective HOM. Except for the functional group added by the termination reaction, distributions of functional groups are the same for all monomer termination products originating from the same HOM peroxy radical. This also includes HOM-ONs. Hence, no substantial differences should be expected for the vapor pressures of all monomer termination products produced originating from the same HOM peroxy radical.
Considering the molecular mass instead of O atoms of the HOM moiety, HOM-ONs have higher vapor pressure compared to HOM-PPs (Peräkylä et al., 2020), despite the heavier termination group −ONO2 compared to −OOH, = O, or −OH. Note that the number of functional groups cannot be inferred one to one from the number of O atoms. HOM peroxy radicals can be formed via autoxidation of peroxy radicals or via H shifts and O2 addition in alkoxy radicals, and thus there may be different numbers of O atoms per functional group. In addition, peroxy radicals may have the same molar weights but may have different molecular structures. With this limitation in mind, we will exploit the relationship between γeff and O atoms as a proxy for the number of functional groups in the next step of interpretation.
Starting from a given HOM peroxy radical, the number of functional groups in the termination products is the same, independent of whether a HOM-PP or a HOM-ON is being formed. However, the masses of HOM-PPs and HOM-ONs differ. If a HOM-ON is formed, it contains one N and one or two O atoms more than the parent HOM peroxy radical (depending on NO or NO2 being added). If a HOM-PP is formed in a reaction of the same HOM peroxy radical with another peroxy radical, HO2 or RO2•, the number of O atoms stays constant in the case of hydroperoxide formation or decreases by 1 in the case of ketone or alcohol formation. This means that the formation of HOM-ONs generally increases the molecular mass compared to a HOM-PPs (as was considered in Fig. 6). Since the γeff values are similar for all monomer HOMs originating from the same HOM peroxy radical (i.e., HOM-PP and HOM-ON have similar vapor pressures) a gain of SOA mass could be expected if HOM-ONs are produced instead of HOM-PPs (≈10 % gain per HOM-ON for NO and at a molecular mass of 300 Da). However, whether this potential mass gain can be realized at all in a long-term net increase in SOA mass in NOx-containing systems will depend on the fate of the ON in the particle phase.
4.3 Comparison of OrgNO3 in HOMs to OrgNO3 in particles
Comparing the data shown in Fig. 8, it is obvious that the mass fraction of OrgNO3 in gas phase HOMs is 3 to 4 times higher than that found in the particulate phase. As all considered HOMs (molecular mass > 230 Da) have a high efficiency for SOA formation, a fraction of about 10 % OrgNO3 would be expected in the particle phase. Zhao et al. (2018) performed experiments of α-pinene photooxidation in the presence of 20 ppb NOx at a lower relative humidity of ≈30 % in the SAPHIR chamber in Jülich. Of course there are differences between steady-state experiments in JPAC and time-dependent experiments in the batch reactor SAPHIR. However, applying the same instrumentation and using the same evaluation schemes as here, Zhao et al. (2018) find a mass fraction of 11 % for OrgNO3 in the particulate phase, i.e., mass closure as expected if all HOMs with more than six O atoms contribute to SOA formation. This is more than the 2 %–3 % OrgNO3 that we realized in the particulate phase. A difference between the experiments here in JPAC and Zhao et al. (2018) in SAPHIR was the relative humidity, which was about 30 % by Zhao et al. (2018) and 63 % in the study here. We suggest that the RH may be the key to bring this study and the results by Zhao et al. (2018) in agreement, although we cannot provide further experimental proof. However, there are several studies implying that ONs undergo hydrolysis in the condensed phase (Bean and Hildebrandt Ruiz, 2016; Boyd et al., 2015; Browne et al., 2013; Day et al., 2010; Fisher et al., 2016; Jacobs et al., 2014; B. H. Lee et al., 2016; Liu et al., 2012; Rindelaub et al., 2016, 2015). Thus, hydrolysis can be suspected as a mechanism explaining both the lower fraction of OrgNO3 in the particle phase compared to that stored in HOM organic nitrates and the lower amount or OrgNO3 found in this study compared to that found by Zhao et al. (2018).
In addition, hydrolysis of OrgNO3 could contribute to SOA mass suppression. We estimated the effect of ON hydrolysis on the remaining SOA mass under the assumption that all HOM-ONs with γeff>0.5 had condensed on SOA. By hydrolysis of organic nitrates HNO3 is formed, and the nitrate functionality at the organic rest is replaced by an OH group (Hu et al., 2011). HNO3 is too volatile to stay in the particle phase (e.g., Browne et al., 2013; Romer et al., 2016), and we found only negligibly small amounts of inorganic nitrate in particles despite the high HNO3 production in the gas phase (Sect. 3.1). As a consequence, on average one-fifth of the mass of the HOM-ONs that originally condensed on particles might re-evaporate and indeed reduce the amount of condensed mass. The question is what happens to the remaining organic moiety. As long as hydrolysis does not lead to fragmentation of the organic rest, hydrolysis just replaces the nitrate group by the protic OH group. The number of functional groups remains the same, and no strong changes in the vapor pressures are expected for the organic rest. As a consequence the organic rest of the former organic nitrate would stay in the particle phase. If so, hydrolysis and re-evaporation of HNO3 should not lead to a mass loss high enough to explain the often observed suppressing effect of NOx on SOA mass formation in laboratory studies. The mass loss per evaporated HNO3 is 63 Da. The water molecule driving the hydrolysis can be from the gas phase or particulate phase, and exchange of water between the phases is fast. The de facto mass loss therefore should be 45 Da per evaporating HNO3. A part of this loss is “compensated”, depending on the HOM-ON being formed by NO or NO2 and depending on the HOM-PP not produced instead of the organic nitrate, this mass gain is between 29 and 63 Da per formed HOM-ON. Thus, the net effect of mass gain by the formation of a HOM-ON instead of a HOM-PP in the gas phase and the possible mass loss due to evaporation of HNO3 from the particle phase should be very low or negligible.
4.4 Suppression of accretion products and SOA yield
In the previous section we have shown that the formation of organic nitrates instead of other HOM monomer termination products cannot explain the suppressing effect of NOx even if we consider hydrolysis and loss of HNO3. Also, increasing fragmentation via alkoxy radicals from Reaction (R6b) seemed to play only a minor role (see Sect. 3.3, Fig. 3). Reasons are that fragments formed in α-scission can still be highly functionalized, and thus are simply HOMs with fewer C atoms. In addition bond scission in alkoxy radicals of α- and β-pinene can lead to ring opening retaining the carbon number. Moreover, isomerization of alkoxy radicals is another pathway (unimolecular or bimolecular) that leads eventually to peroxy radicals with the same number of carbon atoms that could undergo further autoxidation and/or terminate to highly functionalized HOMs (Vereecken and Peeters, 2009, 2010).
The most probable explanation for the NOx-induced suppression of SOA formation in laboratory studies is the suppression of HOM-ACC formation (compare Rissanen, 2018). At low-NOx conditions, the mass fraction of HOM-ACCs is similar to that of monomers and it dropped to less than 30 % at high [NOx] (Fig. 3). Hence, besides reduction of [OH] at high [NOx], suppression of HOM-ACC formation by NOx might lead to a strong suppression of SOA formation in laboratory studies.
The precursors of the respective HOM-ACCs are the key to understand how the suppression of HOM-ACC formation could lead to a suppression of SOA formation. As we showed in Fig. 3 HOM-ACCs decreased with NOx, while HOM monomers remained about constant. We therefore analyze how a reduction of SOA could be realized when HOM monomers (here HOM-ONs) are formed instead of HOM-ACCs. According to Reaction (R5), HOM accretion products are produced from two peroxy radicals (Berndt et al., 2018a, b). The other product of this reaction is molecular oxygen, and thus the molecular mass of the HOM accretion product is lower by 32 Da than the sum of the molecular masses of both monomer peroxy radicals. If two HOM-ONs are formed from two HOM-RO2 by Reactions (R6a) and (R7) instead of one HOM-ACC, each of them gains molecular mass due to the addition of NO (30 Da) or NO2 (46 Da). Comparing the molecular mass of two HOM-ONs to that of the HOM-ACCs formed from the same HOM-RO2, there may even be a gain of 92 to 124 Da when two HOM-ONs are formed at the cost of one HOM-ACC. This gain considers the addition of two ONs or two NO2 particles in the formation of HOM-ONs and the loss of O2 in HOM-ACC formation. Hence, if the respective HOM-ACC is formed by HOM-RO2 radicals that anyhow would form low-volatility HOM-ONs, the suppression of HOM-ACCs should actually lead to an increase in total condensable mass.
The situation is different if we assume that classical (“non-SOA-forming”) RO2• with a lower O:C ratio were involved in HOM-ACC formation in addition to HOM-RO2. If the RO2 is not terminated by a HOM-RO2 to HOM-ACC, the volatility of its classical termination products may be too high to allow for effective condensation and contribution to SOA formation. In such cases, HOM-ACC formation would lead to a gain of condensable mass by scavenging the non-SOA-forming peroxy radical. In turn, suppression of HOM-ACC formation would indeed lead to a net loss of condensable mass. This effect is de facto the same as accretion product suppression by isoprene peroxy radicals described by McFiggans et al. (2019).
The loss could be even stronger when two intermediate-level oxidized (functionalized) peroxy radicals are involved in the accretion product formation. If both form volatile termination products otherwise, the whole accretion product accounts for loss. Involvement of non-SOA-forming RO2• in HOM-ACC formation can be verified by looking at average O:C ratios derived from high-resolution peak identification. For α-pinene at the background level of NOx the average O:C is 0.97 for the monomers and 0.68 for the accretion products. The lower O:C ratios of HOM-ACCs indicate a substantial contribution of RO2• with smaller numbers of O atoms.
Since HOM-ACCs can be formed from many permutations of HOM-RO2 and non-SOA-forming RO2•, clear identification of the respective precursors is not possible. Referring to rate coefficients reported by Berndt et al. (2018b), which decrease with the degree of functionalization by 2 orders of magnitude, we propose that there may be three types of pathways to accretion products: HOM-RO2• + HOM-RO2•, HOM-RO2• + RO2•, and RO2• + RO2•. Formation of accretion products by reaction HOM-RO2• + RO2• was observed for cyclopentene by Mentel et al. (2015). For illustration we simply assume that accretion product formation involves a pre-stabilized adduct (in analogy to the Lindemann–Hinshelwood mechanism). Then HOM-RO2• + HOM-RO2• would form a relatively long-lived and relatively stable adduct because of the high functionalization of the reactants (with protic functional groups). Such an adduct would live long enough to react to the accretion products, e.g., as proposed by Valiev et al. (2019). The reaction HOM-RO2• + RO2• forms weaker adducts with shorter lifetimes, but there are more collisions to form adducts as RO2• concentrations are higher than HOM-RO2• concentrations. RO2• + RO2• may still take place, driven by the bare number of collisions. All involved RO2• species must have a certain degree of functionalization (Berndt et al., 2018a, b). First-generation RO2• contain only three O atoms, but are by far the most abundant. Reactions of first-generation peroxy radicals could therefore still make a contribution to accretion products.
We conclude that suppression of HOM accretion product formation is a mechanism that leads to lower amounts of condensable mass because of involvement of non-SOA-forming RO2• and therefore can explain the suppressing effect of NOx on SOA formation. Note that in the experiments here RO2• dominated over HO2•. This is often the case in laboratory studies with enhanced VOC and oxidant levels. Thus, suppression of accretion products may well explain the dependence of SOA formation on [NOx] (and the variability) observed in laboratory studies. In the atmosphere, photochemical accretion product formation at low [NOx] can often be less important because termination reactions with HO2 are more important for HOM formation than termination reactions with RO2• (compare Berndt et al. (2018a) for the example of isoprene).
We characterized the role of ON in SOA mass formation. One finding was that low functionalized ONs do not contribute much to particle formation. Only HOM-ONs with more than six O atoms at the HOM moiety can efficiently contribute to SOA mass formation at least at mass loads as investigated here. Thereby HOM-ONs with six to seven O atoms showed partitioning with γeff of about 0.5, i.e., about 50 % staying in the particulate phase. Once the HOM-ONs contained more than eight O atoms their loss on particles was collision limited, and nearly 100 % resided in the particulate phase. This supports expectations that HOM-ONs with more than eight O atoms will have extreme low volatility. No significant and systematic differences in γeff were found between HOM-ONs and HOM-PPs when they have the same number of O atoms in the moiety, i.e., when they arise from the same HOM-RO2. Hence, different volatility of HOM-ONs and HOM-PPs from different termination reactions can be discarded as the reason for the suppressing effect of NOx on SOA mass formation. Hydrolysis of HOM-ONs in the particle phase and re-evaporation of HNO3 also seems insufficient to explain the suppressing impacts of NOx on SOA mass formation. Re-evaporation of HNO3 more or less just compensates for the mass gain due to the formation of a HOM-ON instead of a HOM-PP. Thus we conclude that formation of HOM-ONs instead of HOM-PPs (i.e., hydroperoxides, alcohols, ketones, carboxylic, or percarboxylic acids) cannot be the main reason for the often observed suppressing effect of NOx on SOA formation in photochemical systems. Since a suppressing effect of NOx on SOA mass formation is well documented in the literature (Presto et al., 2005; Kroll et al., 2006; Ng et al., 2007; Eddingsaas et al., 2012; Sarrafzadeh et al., 2016; Stirnweis et al., 2017), there must be other mechanisms causing this suppression. One effect is the lowering of the OH level by NOx (e.g., Sarrafzadeh et al., 2016; Lee et al. 2020). If OH is kept constant, we observed strong suppression of HOM-ACCs with increasing [NOx]. Formation of HOM-ONs via fast Reaction (R6a) and Reaction (R7) in competition with Reaction (R4a) are probably the reason for the observed phenomenon, especially if Reactions (R6a) and (R7) prevent less functionalized RO2• from being trapped in low-volatility accretion products. Without forming accretion products, there is no chance for them to participate in SOA mass formation because of the high volatility of their other termination products. Keeping less functionalized HOM-RO2 from forming accretion products leads to loss of one molecular mass unit of less functionalized RO2• or even the whole accretion product if it was formed by two intermediate-level oxidized peroxy radicals. This effect may be less expressed in the atmosphere as RO2•–RO2• interactions in low-NOx cases are less important than in our laboratory study.
There is another contribution left for an explanation of SOA mass suppression by NOx: the decomposition of alkoxy radicals that are formed in Reaction (R6b). We showed in Fig. 3 that fragmentation of alkoxy radicals led to C<10 compounds that are still HOMs that contribute to SOA. At the current stage the overall impact of alkoxy radicals on HOM and SOA formation is difficult to address and needs closer study (in preparation).
Note that we considered the photochemistry of NOx to SOA contribution for two major MTs, α-pinene and β-pinene. We find that SOA yields are fairly independent of [NOx] but drop significantly at the highest NOx levels. Model studies show that an increase in NOx emissions may also lead to more SOA, when NO3 is the oxidant (e.g., Pye et al., 2015) or when isoprene is involved (Marais et al., 2016). In the latter case NO directs the gas phase mechanism toward isoprene products with reactive uptake, while for compounds like α-pinene and β-pinene, investigated here, condensation is more important for SOA formation, and thus vapor pressures control SOA yields.
All data given in figures can be displayed in tables or in digital form. This includes the data given in the Supplement where we describe methodological issues including calibrations, peak separation in CIMS, and peak lists. Please send all requests for data to t.mentel@fz-juelich.de.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-20-10125-2020-supplement.
TFM, JW, EK, IP, AW, and AKS designed the experiments. Instrument deployment and operation were carried out by IP, SS, MS, PS, SA, EK, FR, MS, RT, JW, CW, and DZ. Data analysis was done by IP, SK, SS, FR, PS, and RT. IP, SK, SS, JW, and TFM interpreted the compiled data set. IP, SK, JW, TFM, and AKS wrote the paper. All co-authors discussed the results and commented on the paper.
Sebastian Schmitt works for TSI GmbH. The authors declare that they have no conflict of interest.
We would like to thank the anonymous reviewers for their helpful comments; in particular we acknowledge anonymous reviewer 2, who helped a lot to improve the manuscript in the second round.
This research has been supported by the European Commission (grant no. PIMMS (287382)).
The article processing charges for this open-access
publication were covered by a Research
Centre of the Helmholtz Association.
This paper was edited by Hinrich Grothe and reviewed by three anonymous referees.
Atkinson, R.: Gas-Phase Tropospheric Chemistry of Organic Compounds, J. Phys. Chem. Ref. Data, 2, 1–216, 1994.
Atkinson, R.: Gas-Phase Tropospheric Chemistry of Volatile Organic Compounds: 1. Alkanes and Alkenes, J. Phys. Chem. Ref. Data, 26, 215–290, https://doi.org/10.1063/1.556012, 1997.
Bean, J. K. and Hildebrandt Ruiz, L.: Gas–particle partitioning and hydrolysis of organic nitrates formed from the oxidation of α-pinene in environmental chamber experiments, Atmos. Chem. Phys., 16, 2175–2184, https://doi.org/10.5194/acp-16-2175-2016, 2016.
Berkemeier, T., Ammann, M., Mentel, T. F., Poschl, U., and Shiraiwa, M.: Organic nitrate contribution to new particle formation and growth in secondary organic aerosols from α-pinene ozonolysis, Environ. Sci. Technol., 50, 6334–6342, https://doi.org/10.1021/acs.est.6b00961, 2016.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurten, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Tröstl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kürten, A., Manninen, H. E., Munch, S., Perakyla, O., Petaja, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, https://doi.org/10.1126/science.aad5456, 2016.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Boyd, C. M., Nah, T., Xu, L., Berkemeier, T., and Ng, N. L.: Secondary Organic Aerosol (SOA) from Nitrate Radical Oxidation of Monoterpenes: Effects of Temperature, Dilution, and Humidity on Aerosol Formation, Mixing, and Evaporation, Environ. Sci. Technol., 51, 7831–7841, https://doi.org/10.1021/acs.est.7b01460, 2017.
Breitenlechner, M., Fischer, L., Hainer, M., Heinritzi, M., Curtius, J., and Hansel, A.: PTR3: An Instrument for Studying the Lifecycle of Reactive Organic Carbon in the Atmosphere, Anal. Chem., 89, 5825–5832, https://doi.org/10.1021/acs.analchem.6b05110, 2017.
Browne, E. C., Min, K.-E., Wooldridge, P. J., Apel, E., Blake, D. R., Brune, W. H., Cantrell, C. A., Cubison, M. J., Diskin, G. S., Jimenez, J. L., Weinheimer, A. J., Wennberg, P. O., Wisthaler, A., and Cohen, R. C.: Observations of total RONO2 over the boreal forest: NOx sinks and HNO3 sources, Atmos. Chem. Phys., 13, 4543–4562, https://doi.org/10.5194/acp-13-4543-2013, 2013.
Capouet, M. and Müller, J.-F.: A group contribution method for estimating the vapour pressures of α-pinene oxidation products, Atmos. Chem. Phys., 6, 1455–1467, https://doi.org/10.5194/acp-6-1455-2006, 2006.
Carlton, A. G., Pinder, R. W., Bhave, P. V., and Pouliot, G. A.: To What Extent Can Biogenic SOA be Controlled?, Environ. Sci. Technol., 44, 3376–3380, https://doi.org/10.1021/es903506b, 2010.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of beta-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
Compernolle, S., Ceulemans, K., and Müller, J.-F.: EVAPORATION: a new vapour pressure estimation methodfor organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/c1cp21330j, 2011.
Day, D. A., Liu, S., Russell, L. M., and Ziemann, P. J.: Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California, Atmos. Environ., 44, 1970–1979, https://doi.org/10.1016/j.atmosenv.2010.02.045, 2010.
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, D16305, https://doi.org/10.1029/2004jd005623, 2005.
Eddingsaas, N. C., Loza, C. L., Yee, L. D., Chan, M., Schilling, K. A., Chhabra, P. S., Seinfeld, J. H., and Wennberg, P. O.: α-pinene photooxidation under controlled chemical conditions – Part 2: SOA yield and composition in low- and high-NOx environments, Atmos. Chem. Phys., 12, 7413–7427, https://doi.org/10.5194/acp-12-7413-2012, 2012.
Ehn, M., Kleist, E., Junninen, H., Petäjá, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Emanuelsson, E. U., Hallquist, M., Kristensen, K., Glasius, M., Bohn, B., Fuchs, H., Kammer, B., Kiendler-Scharr, A., Nehr, S., Rubach, F., Tillmann, R., Wahner, A., Wu, H.-C., and Mentel, Th. F.: Formation of anthropogenic secondary organic aerosol (SOA) and its influence on biogenic SOA properties, Atmos. Chem. Phys., 13, 2837–2855, https://doi.org/10.5194/acp-13-2837-2013, 2013.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Faxon, C., Hammes, J., Le Breton, M., Pathak, R. K., and Hallquist, M.: Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry, Atmos. Chem. Phys., 18, 5467–5481, https://doi.org/10.5194/acp-18-5467-2018, 2018.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkle, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Fuchs, N. A. and Sutugin, A. G.: High-dispersed aerosols, in: International Reviews in Aerosol Physics and Chemistry (Vol. 2): Topics in current aerosol research, edited by: Hidy, G. M. and Brock, J. R., Pergamon Press, Oxford, New York, 1–60, https://doi.org/10.1016/B978-0-08-016674-2.50006-6, 1971.
Fuller, E. N., Ensley, K., and Giddings, J. C.: Diffusion of halogenated hydrocarbons in helium. Effect of structure on collision cross sections, J. Phys. Chem., 73, 3679–3685, https://doi.org/10.1021/j100845a020, 1969.
Glasius, M., la Cour, A., and Lohse, C.: Fossil and nonfossil carbon in fine particulate matter: A study of five European cities, J. Geophys. Res.-Atmos., 116, D11302, https://doi.org/10.1029/2011JD015646, 2011.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Han, Y., Stroud, C. A., Liggio, J., and Li, S.-M.: The effect of particle acidity on secondary organic aerosol formation from α-pinene photooxidation under atmospherically relevant conditions, Atmos. Chem. Phys., 16, 13929–13944, https://doi.org/10.5194/acp-16-13929-2016, 2016.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., Petäjä, T., Rosenoern, T., and Sullivan, A. P.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Hyttinen, N., Rissanen, M. P., and Kurten, T.: Computational Comparison of Acetate and Nitrate Chemical Ionization of Highly Oxidized Cyclohexene Ozonolysis Intermediates and Products, J. Phys. Chem. A, 121, 2172–2179, https://doi.org/10.1021/acs.jpca.6b12654, 2017.
Jacobs, M. I., Burke, W. J., and Elrod, M. J.: Kinetics of the reactions of isoprene-derived hydroxynitrates: gas phase epoxide formation and solution phase hydrolysis, Atmos. Chem. Phys., 14, 8933–8946, https://doi.org/10.5194/acp-14-8933-2014, 2014.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V. M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipila, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kim, H., Barkey, B., and Paulson, S. E.: Real Refractive Indices and Formation Yields of Secondary Organic Aerosol Generated from Photooxidation of Limonene and alpha-Pinene: The Effect of the HC/NOx Ratio, J. Phys. Chem. A, 116, 6059–6067, doi:10.1021/jp301302z, 2012.
Kirkby, J., Duplissy, J., Sengupta, K., Frege, C., Gordon, H., Williamson, C., Heinritzi, M., Simon, M., Yan, C., Almeida, J., Tröstl, J., Nieminen, T., Ortega, I. K., Wagner, R., Adamov, A., Amorim, A., Bernhammer, A. K., Bianchi, F., Breitenlechner, M., Brilke, S., Chen, X. M., Craven, J., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Hakala, J., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lehtipalo, K., Makhmutov, V., Mathot, S., Molteni, U., Onnela, A., Perakyla, O., Piel, F., Petaja, T., Praplan, A. P., Pringle, K., Rap, A., Richards, N. A. D., Riipinen, I., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Scott, C. E., Seinfeld, J. H., Sipila, M., Steiner, G., Stozhkov, Y., Stratmann, F., Tome, A., Virtanen, A., Vogel, A. L., Wagner, A. C., Wagner, P. E., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Zhang, X., Hansel, A., Dommen, J., Donahue, N. M., Worsnop, D. R., Baltensperger, U., Kulmala, M., Carslaw, K. S., and Curtius, J.: Ion-induced nucleation of pure biogenic particles, Nature, 533, 521–526, https://doi.org/10.1038/nature17953, 2016.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photo-oxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Lee, A. K. Y., Abbatt, J. P. D., Leaitch, W. R., Li, S.-M., Sjostedt, S. J., Wentzell, J. J. B., Liggio, J., and Macdonald, A. M.: Substantial secondary organic aerosol formation in a coniferous forest: observations of both day- and nighttime chemistry, Atmos. Chem. Phys., 16, 6721–6733, https://doi.org/10.5194/acp-16-6721-2016, 2016.
Lee, B., D'Ambro, E. L., Lopez-Hilfiker, F. D., Schobesberger, S., Mohr, C., Zawadowicz, M. A., Liu, J. M., Shilling, J. E., Hu, W. W., Palm, B. B., Jimenez, J. L., Hao, L. Q., Virtanen, A., Zhang, H. F., Goldstein, A. H., Pye, H. O. T., and Thornton, J. A.: Resolving Ambient Organic Aerosol Formation and Aging Pathways with Simultaneous Molecular Composition and Volatility Observations, ACS Earth Space Chem., 4, 391–402, https://doi.org/10.1021/acsearthspacechem.9b00302, 2020.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurten, T., Hu, W. W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H. Y., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J. M., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D. X., Chen, X. M., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X. C., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V. M., Kim, C., Kirkby, J., Kontkanen, J., Kurten, A., Lawler, M. J., Mai, H. J., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petaja, T., Piel, F., Pospisilova, V., Quelever, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipila, M., Tauber, C., Tome, A., Trostl, J., Vaisanen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M. Y., Weitz, L., Wimmer, D., Ye, P. L., Ylisirnio, A., Zha, Q. Z., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018.
Liu, S., Shilling, J. E., Song, C., Hiranuma, N., Zaveri, R. A., and Russell, L. M.: Hydrolysis of organonitrate functional groups in aerosol particles, Aerosol Sci. Tech., 46, 1359–1369, https://doi.org/10.1080/02786826.2012.716175, 2012.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mentel, Th. F., Wildt, J., Kiendler-Scharr, A., Kleist, E., Tillmann, R., Dal Maso, M., Fisseha, R., Hohaus, Th., Spahn, H., Uerlings, R., Wegener, R., Griffiths, P. T., Dinar, E., Rudich, Y., and Wahner, A.: Photochemical production of aerosols from real plant emissions, Atmos. Chem. Phys., 9, 4387–4406, https://doi.org/10.5194/acp-9-4387-2009, 2009.
Mentel, Th. F., Kleist, E., Andres, S., Dal Maso, M., Hohaus, T., Kiendler-Scharr, A., Rudich, Y., Springer, M., Tillmann, R., Uerlings, R., Wahner, A., and Wildt, J.: Secondary aerosol formation from stress-induced biogenic emissions and possible climate feedbacks, Atmos. Chem. Phys., 13, 8755–8770, https://doi.org/10.5194/acp-13-8755-2013, 2013.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M., Boge, O., Richters, S., Spindler, G., Sipila, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nozière, B., Barnes, I., and Becker, K. H.: Product study and mechanisms of the reactions of alpha-pinene and of pinonaldehyde with OH radicals. J. Geophys. Res.-Atmos., 104, 23645–23656, https://doi.org/10.1029/1999JD900778, 1999.
Pandis, S. N., Paulson, S. E., Seinfeld, J. H., and Flagan, R. C.: Aerosol formation in the photo-oxidation of isoprene and β-pinene, Atmos. Environ. A, 25, 997–1008, https://doi.org/10.1016/0960-1686(91)90141-S, 1991.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020.
Presto, A. A., Hartz, K. E. H., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 2. Effect of NOx concentration, Environ. Sci. Technol., 39, 7046–7054, https://doi.org/10.1021/es050400s, 2005.
Pullinen, I.: Photochemistry of Highly Oxidized Multifunctional Organic Molecules: a Chamber Study, Schriften des Forschungszentrums Jülich Reihe Energie & Umwelt/Energy & Environment, Mathematisch-Naturwissenschaftliche Fakultät, Köln, 2017.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016.
Rissanen, M. P.: NO2 Suppression of Autoxidation–Inhibition of Gas-Phase Highly Oxidized Dimer Product Formation, ACS Earth Space Chem., 2, 1211–1219, https://doi.org/10.1021/acsearthspacechem.8b00123, 2018.
Rissanen, M. P., Kurten, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jorgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petaja, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Rollins, A. W., Smith, J. D., Wilson, K. R., and Cohen, R. C.: Real time in situ detection of organic nitrates in atmospheric aerosols, Environ. Sci. Technol., 44, 5540–5545, https://doi.org/10.1021/es100926x, 2010.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., de Gouw, J., Draper, D. C., Feiner, P. A., Fry, J. L., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K., Teng, A. P., Wennberg, P. O., Wild, R. J., Zhang, L., and Cohen, R. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016.
Rubach, F.: Aerosol processes in the Planetary Boundary Layer: High resolution Aerosol Mass Spectrometry on a Zeppelin NT Airship, Fachgruppe Chemie und Biologie, Physikalische und Theoretische Chemie, in: Reihe Energie &Umwelt/Energy &Environment, Band 196, Schriften des Forschungszentrums Jülich, Universität Wuppertal, Wuppertal, 2013.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248,https://doi.org/10.5194/acp-16-11237-2016, 2016.
Shilling, J. E., Zaveri, R. A., Fast, J. D., Kleinman, L., Alexander, M. L., Canagaratna, M. R., Fortner, E., Hubbe, J. M., Jayne, J. T., Sedlacek, A., Setyan, A., Springston, S., Worsnop, D. R., and Zhang, Q.: Enhanced SOA formation from mixed anthropogenic and biogenic emissions during the CARES campaign, Atmos. Chem. Phys., 13, 2091–2113, https://doi.org/10.5194/acp-13-2091-2013, 2013.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Stirnweis, L., Marcolli, C., Dommen, J., Barmet, P., Frege, C., Platt, S. M., Bruns, E. A., Krapf, M., Slowik, J. G., Wolf, R., Prévôt, A. S. H., Baltensperger, U., and El-Haddad, I.: Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from α-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 17, 5035–5061, https://doi.org/10.5194/acp-17-5035-2017, 2017.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kurten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Mohler, O., Nieminen, T., Onnela, A., Petaja, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipila, M., Smith, J. N., Steiner, G., Tome, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P. L., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Valiev, R. R., Hasan, G., Salo, V. T., Kubecka, J., and Kurten, T.: Intersystem Crossings Drive Atmospheric Gas-Phase Dimer Formation, J. Phys. Chem. A, 123, 6596–6604, https://doi.org/10.1021/acs.jpca.9b02559, 2019.
Vereecken, L. and Peeters, J.: Decomposition of substituted alkoxy radicals – part I: a generalized structure-activity relationship for reaction barrier heights, Phys. Chem. Chem. Phys., 11, 9062–9074, https://doi.org/10.1039/b909712k, 2009.
Vereecken, L. and Peeters, J.: A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals, Phys. Chem. Chem. Phys., 12, 12608–12620, https://doi.org/10.1039/c0cp00387e, 2010.
Wildt, J., Mentel, T. F., Kiendler-Scharr, A., Hoffmann, T., Andres, S., Ehn, M., Kleist, E., Müsgen, P., Rohrer, F., Rudich, Y., Springer, M., Tillmann, R., and Wahner, A.: Suppression of new particle formation from monoterpene oxidation by NOx, Atmos. Chem. Phys., 14, 2789–2804, https://doi.org/10.5194/acp-14-2789-2014, 2014.
Worton, D. R., Goldstein, A. H., Farmer, D. K., Docherty, K. S., Jimenez, J. L., Gilman, J. B., Kuster, W. C., de Gouw, J., Williams, B. J., Kreisberg, N. M., Hering, S. V., Bench, G., McKay, M., Kristensen, K., Glasius, M., Surratt, J. D., and Seinfeld, J. H.: Origins and composition of fine atmospheric carbonaceous aerosol in the Sierra Nevada Mountains, California, Atmos. Chem. Phys., 11, 10219–10241, https://doi.org/10.5194/acp-11-10219-2011, 2011.
Xu, L., Guo, H. Y., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S. H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015a.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015b.
Zhang, J. Y., Hartz, K. E. H., Pandis, S. N., and Donahue, N. M.: Secondary organic aerosol formation from limonene ozonolysis: Homogeneous and heterogeneous influences as a function of NOx, J. Phys. Chem. A, 110, 11053–11063, https://doi.org/10.1021/jp062836f, 2006.
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Be, A. G., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z. F., Gold, A., Graf, S., Cubison, M. J., Groessl, M., Jayne, J. T., Worsnop, D. R., and Canagaratna, M. R.: Highly Oxygenated Multifunctional Compounds in alpha-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 51, 5932–5940, https://doi.org/10.1021/acs.est.6b06588, 2017.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zare, A., Fahey, K. M., Sarwar, G., Cohen, R. C., and Pye, H. O. T.: Vapor-Pressure Pathways Initiate but Hydrolysis Products Dominate the Aerosol Estimated from Organic Nitrates, ACS Earth Space Chem., 3, 1426–1437, https://doi.org/10.1021/acsearthspacechem.9b00067, 2019.