the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Jul 2019
| 05 Jul 2019
An improved estimate for the δ13C and δ18O signatures of carbon monoxide produced from atmospheric oxidation of volatile organic compounds
Isaac J. Vimont
Jocelyn C. Turnbull
Vasilii V. Petrenko
Philip F. Place
Colm Sweeney
Natasha Miles
Scott Richardson
Bruce H. Vaughn
James W. C. White
Atmospheric carbon monoxide (CO) is a key player in global atmospheric chemistry and a regulated pollutant in urban areas. Oxidation of volatile organic compounds (VOCs) is an important component of the global CO budget and has also been hypothesized to contribute substantially to the summertime urban CO budget. In principle, stable isotopic analysis of CO could constrain the magnitude of this source. However, the isotopic signature of VOC-produced CO has not been well quantified, especially for the oxygen isotopes. We performed measurements of CO stable isotopes on air samples from two sites around Indianapolis, US, over three summers to investigate the isotopic signature of VOC-produced CO. One of the sites is located upwind of the city, allowing us to quantitatively remove the background air signal and isolate the urban CO enhancements. as well as the isotopic signature of these enhancements. In addition, we use measurements of Δ14CO2 in combination with the CO:CO2 emission ratio from fossil fuels to constrain the fossil-fuel-derived CO and thereby isolate the VOC-derived component of the CO enhancement. Combining these measurements and analyses, we are able to determine the carbon and oxygen isotopic signatures of CO derived from VOC oxidation as and 3.6 ‰±1.2 ‰, respectively. Additionally, we analyzed CO stable isotopes for 1 year at Beech Island, South Carolina, US, a site thought to have large VOC-derived contributions to the summertime CO budget. The Beech Island results are consistent with isotopic signatures of VOC-derived CO determined from the Indianapolis data. This study represents the first direct determination of the isotopic signatures of VOC-derived CO and will allow for improved use of isotopes in constraining the global and regional CO budgets.
- Article
(2277 KB) - Full-text XML
-
Supplement
(984 KB) - BibTeX
- EndNote





Table 1The four main CO sources and the OH sink listed with their isotopic signatures and uncertainties.
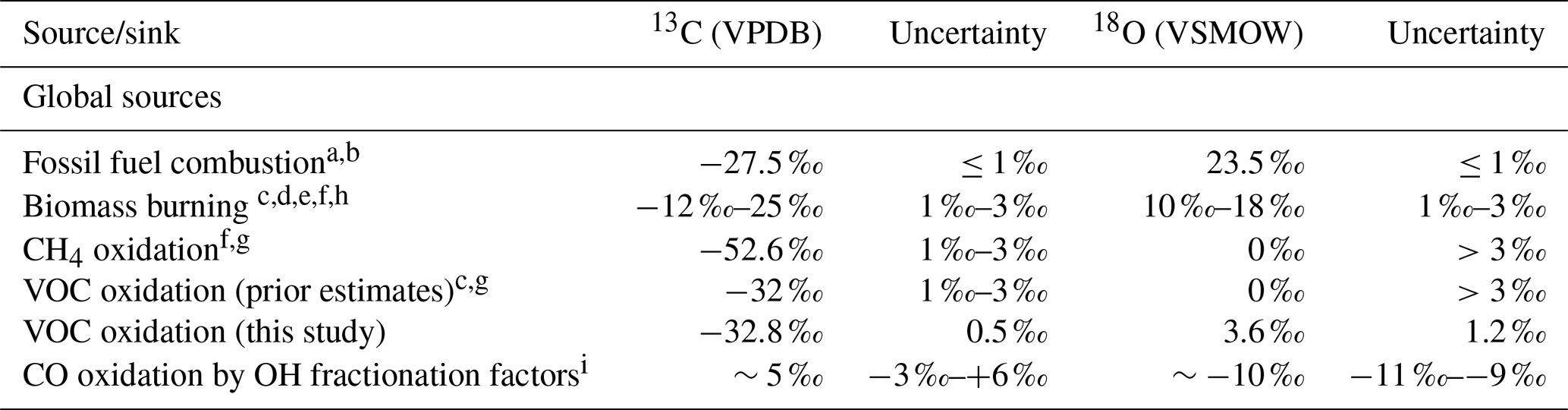
a Stevens et al. (1972). b Brenninkmeijer (1993). c Stevens and Wagner (1989). d Bergamaschi et al. (1998). e Saurer et al. (2009). f Manning et al. (1997). g Brenninkmeijer and Röckmann (1997). h Isotopic signatures vary based on type of vegetation burned (C3 ∕ C4) and temperature of fire. i These factors are the “best estimate” provided Brenninkmeijer et al. (1999). These are based on data from Röckmann et al. (1998) and Stevens et al. (1980). These studies report pressure-dependent fractionation factors for ε13C and very little pressure dependence for ε18O (pressure range ∼200 to 1100 mbar). The variability in the fractionation factors is reported here as the uncertainty.
The global carbon monoxide (CO) budget, along with regional and local CO budgets, remains uncertain (e.g., Holloway et al., 2000; Duncan et al., 2007; Granier et al., 2011; Zhou et al., 2017; Strode et al., 2018). CO stable isotope measurements can aid in the partitioning of the sources of CO, and hence improve global and regional budgets (e.g., Brenninkmeijer et al., 1999). Several studies have incorporated stable isotopes of CO to independently constrain the sources of CO (Manning et al., 1997; Bergamaschi et al., 2000; Park et al., 2015). On the global scale, CO has four major sources, which include biomass/biofuel burning, oxidation of methane (CH4), the incomplete combustion of fossil fuels, and the oxidation of volatile organic compounds (VOCs) (Logan et al., 1981; Duncan et al., 2007; Table 1). These sources are balanced by the oxidation of CO by the hydroxyl radical (OH) and a small soil sink, resulting in a residence time of CO in the atmosphere that is ≈ 2 months on average but varies by location and time of year (Logan et al., 1981; Duncan et al., 2007). Each CO source has a unique isotopic signature, which is determined by the isotopic signature of the source material (e.g., CH4) and the process(es) by which the CO is formed. The carbon isotopic signature of methane-derived CO is much more negative than that of the other sources, largely due to the depleted carbon isotopic signature of methane (Table 1, Brenninkmeijer et al., 1999). The oxygen isotopic signature can help distinguish between combustion (fossil fuel and biomass burning) and oxidation sources (methane and VOC-derived CO), with combustion sources having more positive isotopic values than oxidation sources (Table 1, Brenninkmeijer et al., 1999).
The isotopic signatures of CO from fossil fuel combustion and biomass burning have been relatively well quantified (Table 1). The 13CO produced by oxidation of methane has also been well quantified, although the C18O signature remains more uncertain (Brenninkmeijer et al., 1999). However, the isotopic signatures of CO produced by the oxidation of volatile organic compounds (VOCs) remain poorly known (Brenninkmeijer and Röckmann, 1997; Brenninkmeijer et al., 1999; Gros et al., 2001). The carbon isotopic signature of CO produced by oxidation of VOCs has been estimated to around −32 ‰ from atmospheric measurements (Stevens and Wagner, 1989) and through analysis of the isotopic signature of isoprene, accounting for fractionation during the oxidation reaction (Sharkey et al., 1991; Conny and Currie, 1996; Conny et al., 1997).
Only two prior studies have tried to estimate the oxygen isotopic signature of VOC-derived CO, yielding very different values: 0 ‰ (Brenninkmeijer and Röckmann, 1997) or 15 ‰ (Stevens and Wagner, 1989), with a reported uncertainty of “greater than 3 ‰” (e.g., Gros et al., 2001; Table 1). As VOC oxidation is a major source of CO on global and regional scales (e.g., Logan et al., 1981; Guenther et al., 1995; Duncan et al., 2007), the large uncertainty in the associated isotopic signatures presents a major obstacle to using isotopes in investigations of the atmospheric CO budget.
Our study uses a new set of measurements to evaluate the carbon and oxygen isotopic signatures of CO produced from VOCs by analyzing the urban CO isotopic enhancements at Indianapolis, Indiana, US. An urban setting for determining the isotopic signature of CO from oxidized VOCs may not seem like an obvious choice because of the large CO enhancements from fossil fuel burning (9; Vimont et al., 2017). However, previous literature suggests that during the summer months there may also be a large urban source of CO from the oxidation of VOCs, likely from biogenic sources (Guenther et al., 1993, 1995; Carter and Atkinson, 1996; Kanakidou and Crutzen, 1999; Cheng et al., 2017; Turnbull et al., 2006; Miller et al., 2012).
Some of these studies aimed to quantify fossil fuel CO2 enhancements (CO2FF) by using CO enhancements as a proxy measurement but noted that the ratio of CO:CO2FF enhancements was higher in the summer than the winter at several sites in the eastern United States (Turnbull et al., 2006; Miller et al., 2012). A higher CO:CO2FF ratio is inconsistent with a stronger sink process, such as an increase in OH during the summer months. Instead, a seasonal increase in a non-fossil fuel source provides the most likely explanation for the increase in the CO:CO2FF ratio. These studies hypothesized, but could not confirm, that oxidation of VOCs may be the source of this summertime increase in CO:CO2FF ratio.
Studies that model the effect of CO sources on the measured CO mole fraction have also indicated that oxidation of VOCs (particularly from biogenic sources) contributes significantly to the global and regional CO budget (e.g., Kanakidou and Crutzen, 1999). Isoprene and terpene emissions from broadleaf species have been shown to be a large source of VOCs (Guenther et al., 1995; Helmig et al., 1998; Harley et al., 1999), particularly in the southeastern United States (e.g., Chameides et al., 1988). Griffin et al. (2007) used the Caltech Atmospheric Chemistry Mechanism to investigate CO production by VOC oxidation at a regional scale in the United States. Their model determined that VOC oxidation could provide as much as 10 %–20 % of the CO observed in parts of New England, but, in a heavily polluted region such as the Los Angeles Basin, the percentage was much lower, on the order of 1 % or less. Cheng et al. (2017) measured O3 and CO mole fractions and then modeled CO production from the various sources using O3-to-CO ratios. Their model suggested the oxidation of isoprene might equal or exceed the total anthropogenic production of CO within the urban region of Baltimore, US.
This study focuses mainly on measurements from the Indianapolis FLUX project (INFLUX). INFLUX provides a sampling methodology that allows for quantitative removal of background air signals, which isolates the urban enhancement and simplifies the source and sink budget analysis (Turnbull et al., 2015, 2019; Vimont et al., 2017). Measurements are made not only at tower sites within and downwind of the city but also directly upwind of the city, so that the changes in CO mole fraction and isotopic values due to the urban influence can be isolated. The short transit time of air across the city means that removal of CO by OH (and the associated impact on the isotopic signature) can be ignored. Methane oxidation is similarly minimal in the short transit time, and biomass burning is known to be very small within the urban confines.
In addition to the CO mole fraction and stable isotopic measurements, 14CO2 measurements were also performed on the INFLUX samples, allowing for accurate quantification of CO2FF (Turnbull et al., 2015). This allowed us to partition the urban CO enhancement between fossil-fuel-derived and VOC-derived sources. We were then able to isolate the carbon and oxygen isotopic signatures of CO produced from VOC oxidation.
To further examine our estimates of the isotopic signatures of CO produced from oxidized VOCs, we analyzed bimonthly samples from a site at Beech Island, South Carolina, US. This site is heavily forested and the CO mole fraction at this site should be strongly influenced by isoprene oxidation during the summer. By analyzing the isotopic signatures at this site, we were able to compare the Beech Island isotopic signals to our estimates for VOC-derived CO.
2.1 Tower sampling at Indianapolis
Indianapolis, Indiana, is a metropolitan area of over 1 million people in the Midwest region of the United States. It is surrounded by mostly agricultural land, interspersed with trees and foliage. Broadleaf and deciduous foliage comprises approximately 25 %–100 % of the vegetative cover, both inside and outside of Indianapolis' borders (Fig. 1, Guenther et al., 2012; Fig. S1). It has hot summers (25–30 ∘C) and cold winters (−8 to 1 ∘C) that result in a distinct growing season, with the winter being relatively devoid of biogenic fluxes of CO and CO2 (Turnbull et al., 2015). INFLUX aims to develop and assess methods for determining urban greenhouse gas emissions. CO, though not a primary greenhouse gas, is measured and used as a tracer for fossil fuel CO2 emissions and to provide information for source attribution.

Figure 1Satellite image (image created using Google Earth (© Google 2018)) of INFLUX tower locations. Arrow indicates predominant wind direction during sampling. Samples from this study were taken from towers 1 and 2 (shown). Also note the vegetation cover between the two towers.
INFLUX has 12 instrumented towers within and around the urban boundary (Miles et al., 2017). The flask-sampling regime was described in detail by Vimont et al. (2017) and Turnbull et al. (2015). In brief, discrete hourly integrated air samples are collected at 6 of the towers, although the integrated samplers (Turnbull et al., 2012) are moved between the 12 towers occasionally. A total of 3 of the towers have had continuous flask samples and were sampled for CO isotopes (towers 1–3, Turnbull et al., 2015, 2019; Miles et al., 2017) approximately 6 d per month, during the early afternoon when the strongest boundary layer mixing occurs (19:00 UTC, 14:00 local time). Stable isotope measurements of CO were made on samples collected from July 2013 to July 2015. In this paper, we consider only the summer samples that were collected in July and August 2013, May–August 2014, and May–July 2015 (inclusive) from tower 1 (121 m above ground level, a.g.l., 39.5805∘ N, 86.4207∘ W) and tower 2 (136 m a.g.l., 39.7978∘ N, 86.0183∘ W) (Fig. 1). The winter samples were examined in a previous study (Vimont et al., 2017) that determined that in winter, CO enhancements in Indianapolis are primarily derived from fossil fuel combustion; the CO isotopic signature of the fossil fuel combustion source was also constrained. Though summer samples were also collected at tower 3 (39.7833∘ N, 86.1652∘ W), its proximity to Indianapolis' downtown district and its lower elevation above the ground (54 m a.g.l.) meant that the signals there were strongly dominated by fossil fuel combustion sources, even in summer. Tower 2, located to the east of the urban region, was the ideal candidate for determining the isotopic signature of the oxidized VOC source of CO. Tower 2 “sees” a more mixed signal of urban and suburban sources, including both fossil fuel sources and the influence of the substantial suburban vegetation (Turnbull et al., 2015, 2019).
For the samples in this study, collection was done when the wind was coming approximately from the west, so that tower 1 provides a clean-air background for the towers further to the east (Turnbull et al., 2012). Tower 2 is east of the city, with only a small residential influence and one major highway nearby and with significant foliage within its influence footprint (Turnbull et al., 2015). The distance between towers 1 and 2 is 51 km, and the average wind speed during the period sampled for this study was 4.4 m s−1, which results in an average transit time of air from tower 1 to tower 2 of 3.2 h.
The air samples were collected in portable flask packages (PFPs) provided by the National Oceanic and Atmospheric Administration (NOAA) Global Reference Network (GRN)(https://www.esrl.noaa.gov/gmd/ccgg/aircraft/sampling.html, last access: February 2019). A total of 1 h integrated samples were collected; this sampling regime allows for smoothing of very short-term variability that may be difficult to interpret (Turnbull et al., 2012). NOAA's Earth System Research Laboratory (ESRL) provides the infrastructure and logistical support for these PFPs and the CO mole fraction measurements used in this study (Novelli et al., 2003). 14CO2 measurements were performed at GNS Science with support from University of Colorado INSTAAR (Turnbull et al., 2015).
2.2 Tower sampling at Beech Island, South Carolina
Beech Island, South Carolina, US (33.4057∘ N, 81.8334∘ W), is a tall tower (305 m a.g.l.) site in the NOAA Global Greenhouse Gas Reference Network (GGGRN). The Beech Island sampling site is located in a sparsely populated region of South Carolina, approximately 5.5 km from the town of Beech Island. The climate is temperate with annual temperature varying between 6 and 28 ∘C (NOAA Center for Environmental Information, https://www.ncdc.noaa.gov/, last access: April 2019). The town of Beech Island has a population of approximately 8500, and the surrounding region population density is about 388 people per square kilometer (US Census Bureau, https://www.census.gov/, last access: April 2019). However, the sampling site is 25 km from Augusta, Georgia, a metropolitan center of approximately 200 000 (US Census Bureau, https://www.census.gov/, last access: April 2019). Deciduous, broadleaf trees and shrubs compose ∼80 % of the ground cover for much of the area surrounding the sampling site (Guenther et al., 2012, Fig. S2). Samples for CO stable isotopes were collected approximately bimonthly for 1 year (April 2015–March 2016) from this site. This site uses “grab sampling” rather than the integrating sampling used at the INFLUX towers. Flasks are flushed and then filled and pressurized over about a 2 min period. Flasks are measured by the same methods as the INFLUX samples. However, although 14CO2 measurements are made on some flasks from this site, limitations on the available air in each flask mean that the CO stable isotopes were measured on different flasks (collected on different dates) than the 14CO2 measurements.
2.3 Stable isotope analysis
The stable isotopic measurement procedure is described in detail in Vimont et al. (2017). Briefly, the air is extracted from the PFP by vacuum transfer through a cold loop trap at −70 ∘C that removes water vapor. Next, a mass flow controller is used to regulate the flow of the sample through a second cryogenic trap at −196 ∘C that removes CO2, N2O, and any other condensable species. The remaining air is passed through acidified I2O5 suspended on a silica gel matrix (Schutze's reagent, Schutze, 1944) that quantitatively oxidizes CO to CO2, adding oxygen with a consistent isotopic signature. The sample passes through a second cold loop trap (−70 ∘C) to remove any traces of sulfuric acid that has evolved from the reagent. Finally, the CO-derived CO2 is trapped on a third cryogenic trap (−196 ∘C) while the remaining gases are pumped away. The CO-derived CO2 is then transferred to a cryogenic focusing trap and finally released through a gas chromatographic (GC) column (PoraBond Q) to the isotope ratio mass spectrometer (GV Instruments IsoPrime 5 KeV).
Following convention, we use delta notation to report our isotopic results:
where Rs is the ratio of 13C to 12C in the sample and RVPDB is the ratio of 13C to 12C in the international standard Vienna Pee Dee Belemnite. The same notation describes δ18O except the international standard of reference is Vienna Standard Mean Ocean Water (VSMOW). Because we are oxidizing CO to CO2 in this analysis, we correct our CO2δ18O data to account for the added oxygen, as described in Stevens and Krout (1972), Brenninkmeijer (1993), and Mak and Yang (1998):
where the subscript CO indicates the original δ18O signature of the sample, CO2 indicates the δ18O of the CO2 measured in the mass spectrometer, CO2std indicates the δ18O of the CO2 measured on the standard gas, and COstd indicates the calibrated δ18O of the CO in the same standard gas (standard gas procedure was described in Vimont et al., 2017). Once the samples have been analyzed in the mass spectrometer, a correction for the 17O contribution to the δ13CO measurement is applied to the data based on the recommendations of Brand et al. (2009) (Vimont et al., 2017). This correction is required because 13CO and C17O are indistinguishable in our mass spectrometer. The 1σ repeatability over 2 years for our analysis system is 0.23 ‰ for δ13C and 0.46 ‰ for δ18O. For a more complete description of system performance; see Vimont et al. (2017).
We note that a significant deviation from the standard correction has been observed and quantified for CO (Röckmann and Brenninkmeijer, 1998; Röckmann et al., 1998). This so-called “17O excess”, or Δ17O, is a result of mass-independent fractionation (MIF) that arises in OH photolytic formation, which in turn affects CO during removal by OH (Röckmann et al., 1998b; Huff and Thiemens, 1998). Ozonolysis of VOCs also contributes to 17O excess (Röckmann et al., 1998a, b). The source of CO from ozonolysis of VOCs is discussed in more detail in Sect. 3.4. The combined Δ17O from these processes can introduce error of up to 0.35 ‰ in the corrected δ13C values, and the error is only quantifiable by measuring δ17O (Röckmann and Brenninkmeijer, 1998b). However, though we do not measure δ17O for our samples, our data analysis approach (Sect. 2.5) eliminates the need for this correction because both background and urban samples will see similar Δ17O effects. Additionally, because of the short transit time between our background and polluted tower sites (3.2 h, Sect. 2.1) and the long lifetime of most VOC ozonolysis relative to OH oxidation (Atkinson and Arey, 2003a), we expect any effect of ozonolysis-produced Δ17O error to our δ13C measurements to be insignificant relative to our measurement uncertainty.
2.4 Radiocarbon CO2 analysis
Each of the INFLUX samples analyzed for the stable isotopes of CO were also analyzed for 14CO2. 14CO2 is the best tracer for fossil-fuel-produced CO2 because fossil fuels contain no 14C (Levin et al., 2003; Turnbull et al., 2006). 14CO2 measurements were made by extracting CO2 from whole air in each flask at INSTAAR, University of Colorado, followed by graphitization and accelerator mass spectrometer (AMS) 14C measurement at GNS Science, New Zealand (Turnbull et al., 2015). CO2FF was determined for each sample using tower 1 as background, and the 14CO2 results for these and other INFLUX flask samples were reported in detail by Turnbull et al. (2015, 2019). 14C measurements of CO2 are reported as Δ14C, or the per mill deviation of the measured 14C from a standard material, corrected for fractionation effects and radioactive decay between sampling and measurement (Stuiver and Polach, 1977; Turnbull et al., 2015). The conversion of the 14CO2 measurements to CO2FF enhancements is done by the following equation (Turnbull et al., 2015):
is calculated using the observed (Δobs) and background (Δbg) Δ14C values and the observed CO2 mole fraction (). ΔFF is the Δ14C value of fossil fuel CO2 (by definition −1000 ‰). is a small correction that applied and consists primarily of sources from the nuclear industry and heterotrophic respiration typical values for are 0–0.5 ppm when a continental background is used (e.g., in Turnbull et al., 2006, 2015; Miller et al., 2012). The measurement precision of ∼ 1.8 ‰ results in uncertainties in CO2FF of better than 1 µmol : mol CO2FF for these samples.
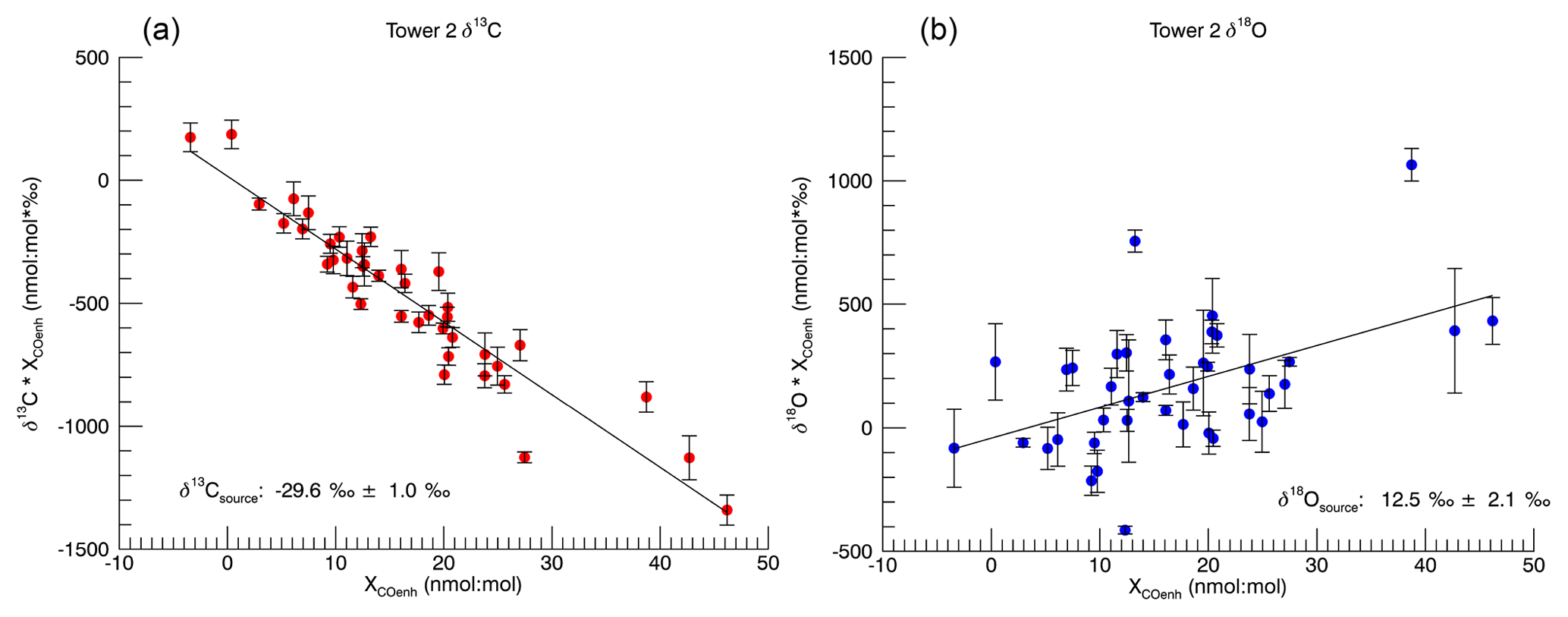
Figure 2Indianapolis Miller–Tans plots for late spring through summer (May, June, July, August, September). The error bars represent the propagated error for the calculation of the enhancements (see text for details).
2.5 Regression plot analysis
At Indianapolis, the CO measured at tower 2 is typically 20 nmol : mol higher than the background CO of ∼ 150 nmol : mol at tower 1. It is necessary to remove the background signal from the polluted tower to accurately constrain the urban CO signals. Using the method described by Miller and Tans (2003), we calculate the isotopic signature of the urban source:
where δs is the δ13C or δ18O of the urban source (Fig. 2), X indicates the mole fraction, and the subscript “meas” indicates the δ13C (or δ18O) and CO mole fraction measured at tower 2. The subscript “bkg” indicates the δ13C (or δ18O) and CO mole fraction measured at tower 1. In order to obtain a best-fit solution using Eq. (4) for all the data, we regressed the numerator against the denominator using an ordinary least-squares (model 1) Y|X approach (Isobe et al.,1990; Zobitz et al., 2006).
To account for uncertainty in our measurements, we used a Monte Carlo technique. Using the propagated measurement uncertainties, we assigned an error distribution to each point. We assumed a normally distributed error curve based on Q–Q plot analysis of our data against a synthetic normally distributed data set (not shown). This analysis allows us to assess if two data sets have the same distribution. A total of 10 000 regressions were run, randomly selecting values for each data point from that point's error distribution. The reported slopes are the median values from the 10 000 regressions. The reported errors on the slope are 1σ for the slopes of each simulation.
At the Beech Island measurement site, no local background measurement site with CO isotope measurements exists. Therefore, we performed a Keeling plot analysis, as well as a Miller–Tans plot analysis using monthly averaged CO mole-fraction, δ13C, and δ18O data from Izaña, Tenerife, in the Canary Islands (28∘ N, 16∘ W, 2370 m a.s.l.) as a background for Beech Island (Bräunlich, 2000, Table S4). The Beech Island Miller–Tans analysis was performed in the same manner as the Indianapolis source signatures described above.
In the Keeling plot approach, isotopic measurements are plotted against the reciprocal of the mole fraction (Keeling, 1958). This method uses the following relationship:
where δobs is the observed δ13C or δ18O at the measurement site, M is the slope determined from a regression of the data, and XCO is the observed CO mole fraction. δs is the intercept determined from a regression of the data. The intercept represents the isotopic signature of the sources influencing the measurement site (Keeling, 1958). The Keeling plot assumes that the background concentration and isotopic values are constant over the period of analysis, which is a reasonable but imperfect assumption for this data set measured over the summer season. The benefits and limitations of this approach are discussed more fully in Sect. 3.3.
To assess the uncertainty of our Keeling plot analysis, we perform a standard Monte Carlo analysis and additionally use a sampling with replacement Monte Carlo method (often referred to as a bootstrap Monte Carlo). Briefly, the boot strap Monte Carlo consists of calculating a linear regression for 1000 randomly chosen sample sets. These sets are chosen from the original data at random, such that the number of data points is always constant (n=7 for both summer and winter at Beech Island). However, in some sample sets, points may be selected more than once or not at all. In this way, any disproportionately large influence on the model by outlier points can be assessed, and the distribution of the model parameter of interest (in our case, the intercept) is representative of data as a whole. We report the mean of the 1000 intercepts, and both the 1σ standard deviation as well as the standard error of the mean are reported for the error on that value. The bootstrap Monte Carlo distributions are shown in the Supplement (Sect. S3).
2.6 Calculation of the VOC oxidation isotopic signatures using mass balance
The CH4 oxidation source, the biomass-burning source, and the OH oxidation sink have negligible impacts for the Indianapolis CO budget (detailed calculations can be found in the Supplement, Sect. S2). In order to constrain the remaining two sources (fossil fuel combustion and VOC oxidation), we use a simple isotope mass balance approach. We assume that the δs calculated at each polluted tower (Sect. 2.5, Eq. 4) can be represented by the following equations:
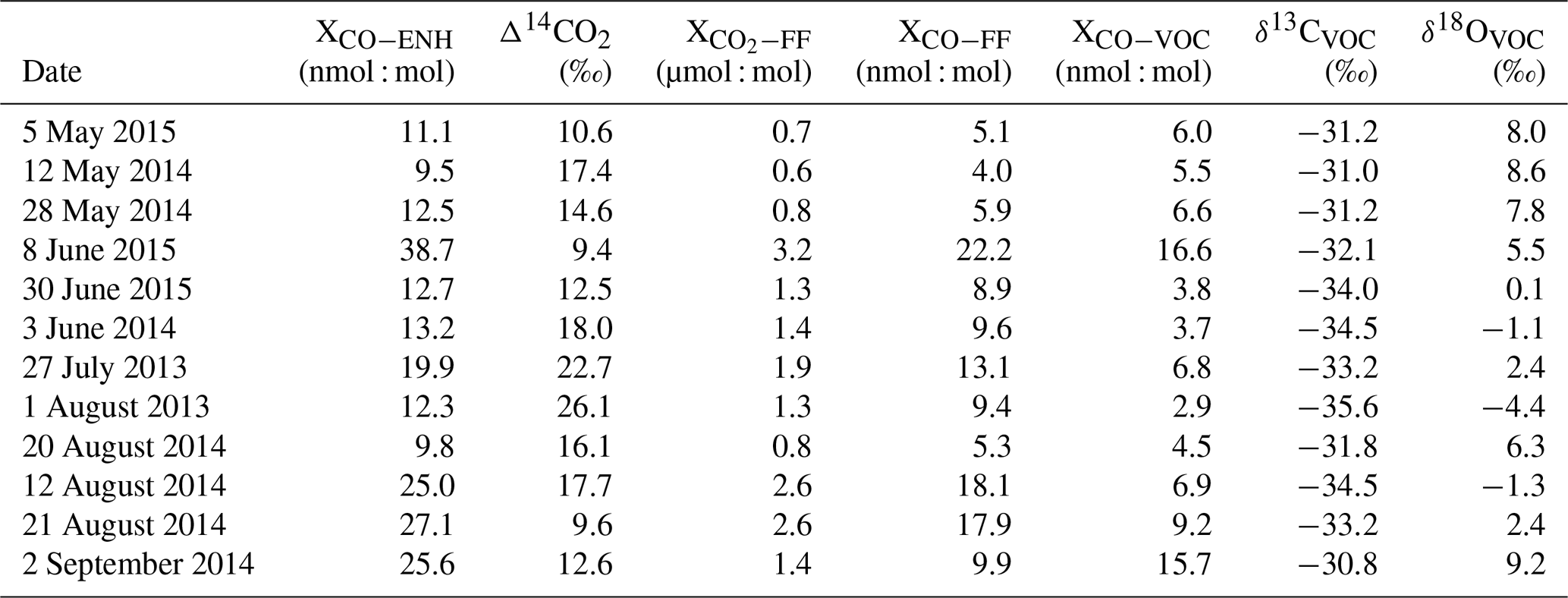
where fVOC and δVOC are the fraction (as compared to total urban CO enhancement) and isotopic signature of CO added from VOC oxidation, and fFF and δFF are the fraction and isotopic signature of CO added from fossil fuel combustion. XCO−VOC, XCO−FF, and XCO−ENH are the mole fractions for VOC-produced CO, the fossil-fuel-produced CO, and the total urban CO enhancement, respectively. The isotopic signatures of fossil fuel combustion at Indianapolis were previously determined from wintertime measurements when fossil fuel combustion is the only significant CO source in Indianapolis and are ‰ and 17.7±1.1 ‰ for δ13C and δ18O, respectively (Vimont et al., 2017). That study found that the isotopic signature in the winter did not vary significantly with temperature and that the primary source within the city was emissions from transportation (Vimont et al., 2017). Therefore, we use these values as the fossil-fuel-produced CO isotopic signatures for Indianapolis. Because we have only two sources (Supplement, Sect. S2), we can derive XCO−VOC as follows:
In order to determine XCO−FF we need to determine XCO−FF. This is done using the ratio of fossil fuel CO to CO2:
where is the fossil-fuel-produced enhancement in the CO2 mole fraction, determined by 14CO2 measurements (Sect. 2.4). is the ratio of COFF to CO2FF and was determined to be 7±1 nmol : µmol for Indianapolis in the winter, when nearly all CO produced is from fossil fuel combustion, primarily vehicles (Turnbull et al., 2019). We assume that this ratio holds across all seasons. We then solve Eqs. (8), (7), and (6a) to determine δVOC. In order to estimate a mean value for our limited sample set, we perform a bootstrap Monte Carlo approach, similar to that described in the previous section. We perform 10 000 calculations of the mean. We report the mean and standard deviation of the 10 000 individual mean values for our bootstrap Monte Carlo simulation as our estimate of the isotopic value and uncertainty of δVOC.
Simple filtering is applied to these data. Any samples with calculated XCO−VOC values that were near zero, were negative, or that exceeded the total urban enhancement were removed. XCO−VOC values that are negative or exceed the total enhancement are obviously nonphysical. Positive values of XCO−VOC that are extremely low (less than 15 % of the total enhancement), while physical, create extreme outliers when δ13CVOC or δ18OVOC are calculated (in one case, several hundred per mill). Likewise, in cases where XCO−VOC is calculated to be nearly the entire urban enhancement, our method will produce δCO−VOC estimates which approach or are equal to our urban enhancement δ values.
Large overestimates of XCO−VOC arise because the ratio method can produce unrealistically low calculated XCO−FF values if the enhancements are not significantly different from zero. enhancements near or below zero are a result of possible local contamination at or near the background tower, which violates the assumption of well-mixed background air flowing across the city. Conversely, the ratio method can overestimate XCO−FF and thereby underestimate XCO−VOC when is highly elevated without a corresponding elevation in XCO−ENH. One example of how this can occur is if the local power plant (the Harding Street Generating Station) plume is sampled by the polluted tower. In the period of this study, the Harding Street Generating Station contributed about 28 % of Indianapolis' CO2FF emissions and, while this source is often dispersed, the plume from this source is occasionally observed at tower 2. This source has a CO:CO2FF ratio of < 0.1 nmol : µmol, due to CO emissions controls fitted to the exhaust stack. Because we use a constant value for , any day where tower 2 samples contain power plant emissions will produce low or negative XCO−VOC values. We do not attempt to identify specific causes for high or low XCO−VOC values. For our sample set, we simply filter samples in which XCO−VOC was less than 15 % of the total enhancement, which produced strong outliers, and samples in which XCO−VOC was more than 85 % of the total enhancement, which produced values equal to our calculated urban enhancements. This filtering removed a total of six data points. The data used for calculating the isotopic signatures for VOC-derived CO are shown in Table 2.
Table 2VOC signature calculation table using data from Indianapolis, Indiana, US. Δ14CO2 and values are reported from Turnbull et al. (2015, 2019). XCO−ENH 1σ uncertainty is ±0.7 nmol : mol, Δ14CO2 1σ uncertainty is ‰ (Turnbull et al., 2015, 2019), and 1σ uncertainty is ±1 µmol : mol (Turnbull et al., 2015, 2019).
3.1 Determination of the urban enhancement CO isotopic signatures
The full time series from Indianapolis was published in Vimont et al. (2017). However, we have reproduced the data from towers 1 and 2 (Fig. 3) here to highlight the summertime data (not discussed in Vimont et al., 2017). The summertime mole fraction and isotopic data can be seen in Table S2 in the Supplement. One of the more salient features of the summer Indianapolis data as compared to the winter data is that, while tower 2 CO mole fraction remains enhanced over tower 1 throughout the year, the δ18O values at tower 2 tend to be much closer to those of tower 1 during the summer, yet are more positive during the winter. This is consistent with the hypothesis that the wintertime urban enhancement is dominated by a fossil source, while the summertime enhancement is a mixed source. Further, this mixed source must be more depleted in 18O than fossil-fuel-produced CO. The δ13C results are more difficult to interpret from the time series alone, which underscores the need for the Miller–Tans method at Indianapolis.
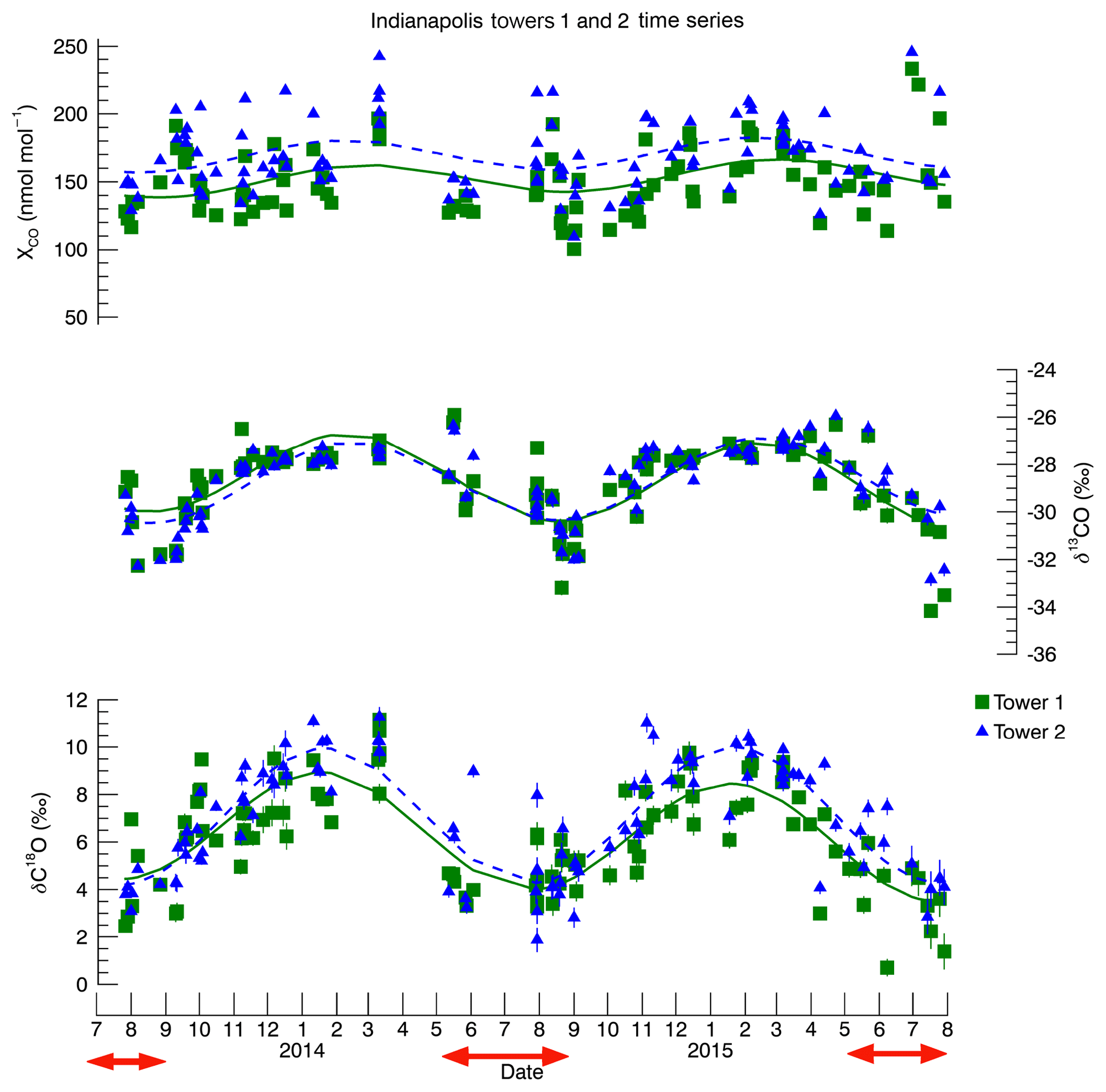
Figure 3Time series of towers 1 and 2 at Indianapolis. These data were previously shown in Vimont et al. (2017) but are reproduced here for the convenience of the reader. The curves shown are for sighting purposes only. They are a simple single harmonic polynomial smoothing and are meant to aid the reader in viewing the seasonal variability. The error bars represent 1σ uncertainty. CO mole fraction 1σ uncertainty is ±0.5 nmol : mol. The red arrows indicate the time periods used in this study, and these data, along with δ13C and δ18O 1σ uncertainty, are listed in the Supplement (Table S2).
The Miller–Tans Monte Carlo regression analysis produced isotopic results of ‰ for δ13C and 12.5 ‰±2.1 ‰ for δ18O (1σ) for the overall urban summertime CO source (Fig. 2). The δ13C source signature is very similar to that determined in winter (, Vimont et al., 2017). In contrast, the δ18O signature is substantially lower in summer than in winter (17.7 ‰±1.0 ‰ in winter, Vimont et al., 2017). These results are consistent with our hypothesized mixing of two sources of CO with different isotopic signatures contributing to the summertime CO enhancement. The determined δ13C of the urban CO source stays relatively consistent between winter and summer ( ‰ and ‰, respectively), suggesting that the VOC oxidation source must have a δ13C signature that is only slightly more negative than the fossil fuel source. In contrast, δ18O of the urban source changes substantially from winter to summer (17.7 ‰±1.0 ‰ and 12.5 ‰±2.1 ‰, respectively), indicating a VOC δ18O signature that is much more negative than the fossil fuel source. The increased scatter in the δ18O regression relative to δ13C is also consistent with this interpretation: variability in the relative contributions of fossil fuel and VOC CO sources for different samples will impart more variability in δ18O than δ13C.
Day-to-day variability in the VOC oxidation source is expected and supports the hypothesis that secondary production of CO by VOCs strongly contributes to the urban enhancement. For example, isoprene has a short atmospheric lifetime in urban regions and rapidly forms CO (Atkinson and Arey, 2003a, b). Isoprene oxidation is highly variable because isoprene emissions depend exponentially on the ambient temperature, and the rate at which isoprene is oxidized will increase as NOx increases (Guenther et al., 1995; Carter and Atkinson, 1996). Additionally, boundary layer mixing will vary day to day, affecting the magnitude and transport of all sources within the tower domain.
3.2 Determination of the VOC-produced COδ13C and δ18O isotope signature
To determine the VOC-produced CO isotopic signature, we first determined the fossil-fuel-produced CO2 source (Sect. 2.4). The 14CO2, the derived CO2FF mole fractions, and the calculated COFF and COVOC mole fractions are presented in Table 2. The uncertainties reported are 1σ for CO2FF and Δ14CO2, while the uncertainties on the calculated COFF and COVOC values are the propagated errors for Eqs. (7) and (8). Using the mass balance approach and bootstrap Monte Carlo method described in Sect. 2.6, we use the isotopic source signatures determined in Sect. 3.1 to calculate the isotopic signatures of VOC-derived CO (Table 2) and the associated bootstrap Monte Carlo mean values: ‰ for δ13C and 3.6 ‰±1.2 ‰ for δ18O (1σ). The scatter in the VOC-derived CO isotopic signatures calculated for individual samples is relatively large (Table 2), likely due to a combination of uncertainties discussed in Sect. 2.6 and real day-to-day variability in the isotopic signatures. However, it is the mean values that are of most interest for CO budget studies, and these appear to be well constrained by the data set.
The δ13C results compare well to previously published estimates of the VOC oxidation signature: ‰ (e.g., Brenninkmeijer et al., 1999). This value is reasonable given the expected carbon isotopic ratio of isoprene and the fractionation processes associated with the isoprene oxidation reaction (e.g., Sharkey et al., 1991). Our δ18O result compares well with the previously published estimate from Brenninkmeijer and Röckmann (1997) (∼ 0 ‰) but contradicts Stevens and Wagner (1989) (∼ 15 ‰). We re-examine the methods and uncertainties of the previous studies to understand what might cause this discrepancy.
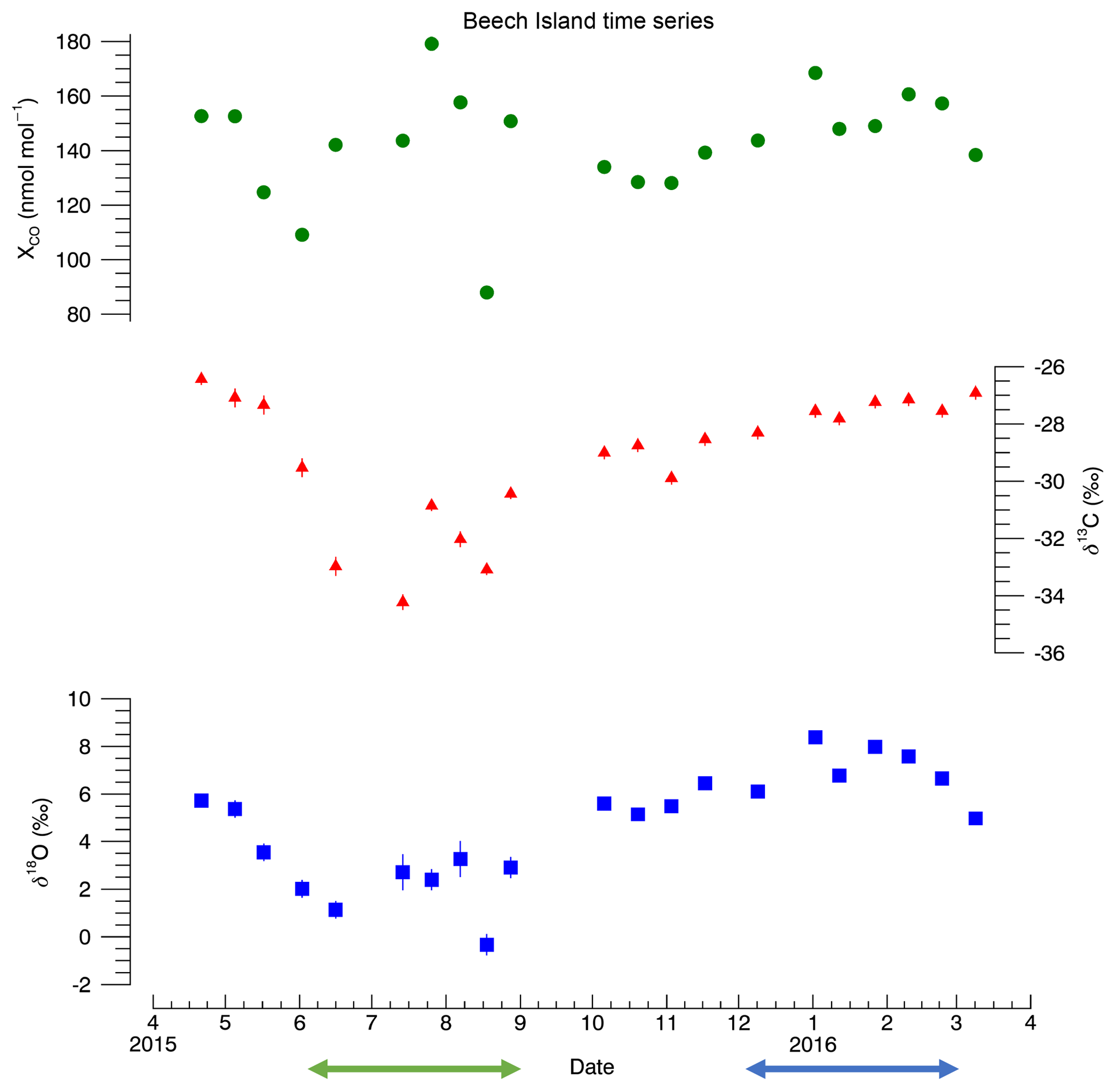
Figure 4Time series for Beech Island, South Carolina. No curves were fit to the data due to the short time frame for the measurements. The error bars represent 1σ analytical uncertainty. CO mole fraction 1σ uncertainty is ±0.5 nmol:mol. Uncertainty for δ13C and δ18O is listed in the Supplement (Table S3). The CO mole fraction data are taken from the NOAA GGGRN data set (Andrews et al., 2009). The green and blue arrows indicate the summer and winter periods used in this study, respectively.
Stevens and Wagner (1989) performed a Keeling plot analysis of samples collected in rural Illinois. They assumed a constant background, with VOC oxidation as the only added CO source, and performed a Keeling plot analysis. Their results indicated −32.2 ‰ for δ13C and 15 ‰ for δ18O of the added CO source. They also measured four samples from a coastal site in Australia and obtained an average δ18O of 5 ‰ for the atmospheric C18O signature. They did not perform a Keeling analysis on the Australian data. They reasoned that the effect of oxidation by OH on the Australia samples would reduce the δ18O by 10 ‰, which meant the source (assumed to be dominated by VOC and methane oxidation) must have been 15 ‰, in agreement with their rural Illinois samples.
The value of 0 ‰ suggested by Brenninkmeijer and Röckmann (1997) was based on a model-driven interpretation of CO isotope measurements in the Southern Hemisphere. Using mass balance, they were able to determine the oxidation of methane and VOCs should produce CO with an oxygen isotopic value near to 0 ‰, while the value of 15 ‰ suggested by Stevens and Wagner (1989) could not be consistent with the measurements. Bergamaschi et al. (2000) used an atmospheric inversion combined with CO mole fraction and isotopic measurements in an attempt to determine the isotopic signatures of CO sources at the global scale. However, their study resulted in wide ranges for δ13C (−17 ‰ to −31 ‰) and δ18O (−30 ‰ to +23 ‰) isotopic values, dependent on the input parameters of their model. Later studies using δ18O to partition the global budget generally use the 0 ‰ value for δ18O despite the lack of consensus (e.g., Park et al., 2015). By leveraging the INFLUX measurements, we are able to place a constraint on the VOC-produced CO isotopic signatures without relying on the uncertain assumptions of a constant background and VOCs as the only source, or on the use of a model to derive the CO mass balance.
3.3 Beech Island, South Carolina, isotopic data
The Beech Island results are shown in Fig. 4, while the data can be found in the Supplement (Table S3). One of the most striking features of this data set is that while the δ13C and δ18O both decrease from spring into summer and then increase into the fall and winter, the mole fraction values do not exhibit much seasonality. While any true seasonal cycles or trends are impossible to determine with only a single year of data, this nonetheless is consistent with a strong summer source of CO from VOC oxidation.
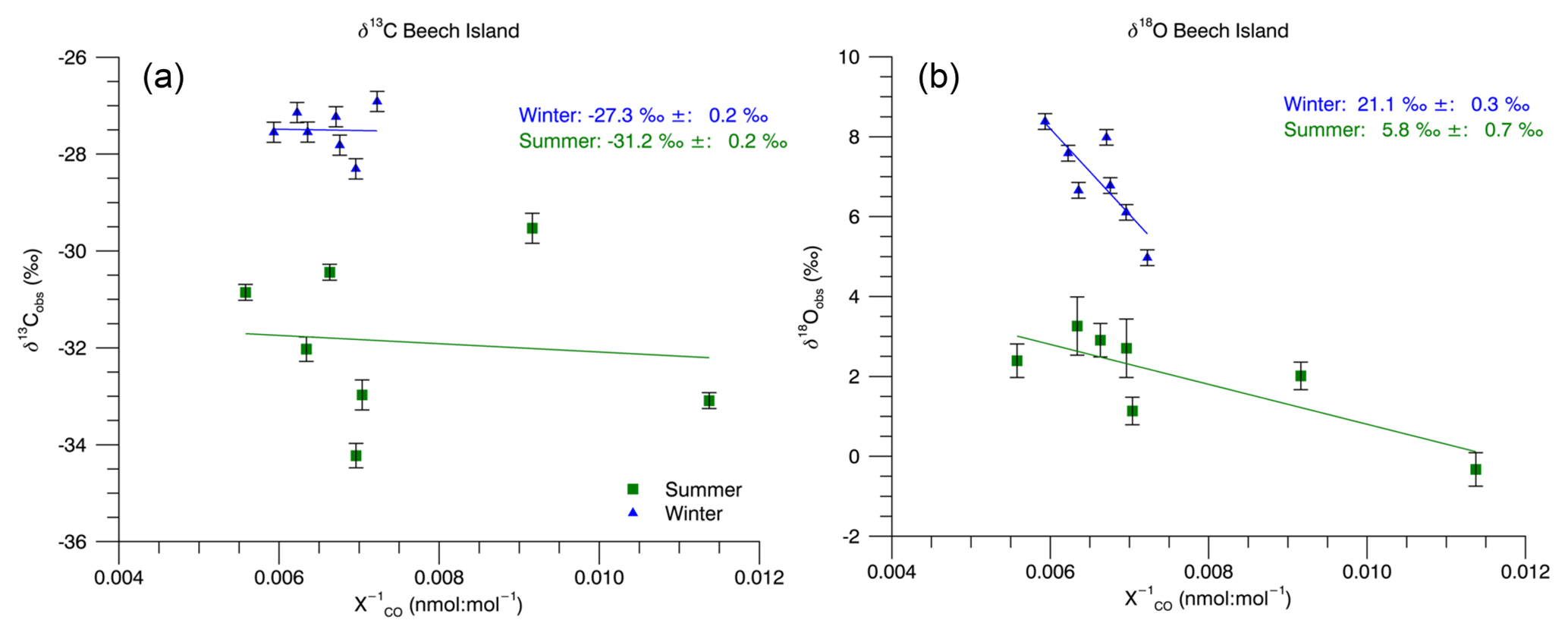
Figure 5Beech Island Keeling plots. The reported intercepts and uncertainties are the standard Monte Carlo simulation results. We also performed a bootstrap Monte Carlo. Those results are reported in the text.
The Keeling plot-derived CO source isotopic signatures at Beech Island, South Carolina, are shown in Fig. 5. During the summer months (June–July–August–September), the Keeling plot analysis (Sect. 2.5) produces a δ13C signature of ‰ and a δ18O signature of 5.8 ‰±0.7 ‰ (1σ) using a standard Monte Carlo simulation and a δ13C signature of ‰ and a δ18O signature of 5.6 ‰±2.4 ‰ (1σ) using the bootstrap Monte Carlo method. During the winter months (December–January–February–March), we obtain a δ13C signature of ‰ and a δ18O signature of 21.1 ‰±0.3 ‰ (1σ) using the standard Monte Carlo method. Using the bootstrap Monte Carlo, we obtain a δ13C of ‰ and a δ18O of 20.4 ‰±5.0 ‰ (1σ). The Keeling approach implicitly assumes constant background CO mole fraction and isotopic composition, which is unlikely to be correct for Beech Island for the entire duration of the summer. However, this approach is still useful for an approximate estimation of the CO source isotopic composition. This is particularly true for δ18O, where the difference between the inferred source isotopic signature and the measured δ18O values is larger than the scatter in the measured values.
In an alternative approach, we apply a background seasonal signal from data published by Bräunlich (2000) from Izaña, Tenerife, to allow for a Miller–Tans plot analysis. Tenerife is located in a similar latitudinal band to Beech Island (28∘ N vs. 33.4∘ N), and the amplitude of the background seasonal signal should be similar between the two sites. However, the Tenerife data set is from sampling done approximately 2 decades before our Beech Island sampling, and therefore global changes to the CO budget between the two studies will introduce error to this analysis that is not easily quantified. Figure 6 shows the isotopic source signatures derived from a Monte Carlo simulation for a Miller–Tans plot approach using monthly averaged data from Izaña, Tenerife (Bräunlich, 2000), as a background for Beech Island. This method produced summer (June–July–August–September) δ13C and δ18O source signatures of ‰ and 5.8 ‰±0.3 ‰ (1σ), respectively. During the winter months (December, January, February, March), we obtained δ13C and δ18O source signatures of ‰ and 20.5 ‰±1.7 ‰ (1σ), respectively. These results are in good agreement with our Keeling plot results.
While both the Keeling and the Miller–Tans approaches for analyzing Beech Island data have important weaknesses as discussed above, these weaknesses are different. The close agreement between the Keeling and Miller–Tans approaches for Beech Island therefore increases confidence in our findings and suggests that the primary drivers of the observed isotopic source signatures are local sources, rather than seasonal changes in background CO. The mean values (and standard deviations) of the isotopic signatures at Beech Island from our three analyses are ‰ and 5.7 ‰ ± 0.8 ‰ during the summer, and ‰ and 20.7 ‰ ± 1.7 ‰ during the winter for δ13C and δ18O, respectively.

Figure 6Miller–Tans analysis of Beech Island seasonal source signatures using monthly means from Izaña, Tenerife (Bräunlich, 2000), for background values. Green squares indicate summer data and blue triangles indicate winter data. The δ13C and δ18O values reported are the mean of 10 000 regression slopes from our Monte Carlo simulation (Sect. 2.5). The uncertainty is the standard deviation of the 10 000 slopes.
The wintertime source signatures derived at Beech Island are consistent with prior estimates of fossil fuel combustion sources (δ13C: ‰, δ18O: ∼23.5 ‰, Table 1). The Beech Island δ13C value is consistent with the wintertime value found at Indianapolis ( ‰, Vimont et al., 2017), while the δ18O value differs slightly from the value found at Indianapolis during the winter (17.7 ‰±1 ‰, Vimont et al., 2017). At Indianapolis, the winter CO urban enhancement was found to be primarily fossil fuel in origin, but it was noted that the oxygen isotopic signature was significantly different from prior estimates of fossil fuel combustion, possibly due to Indianapolis' emission regulation (Vimont et al., 2017). Nonetheless, this suggests that the main driver of CO variability during the winter at Beech Island is likewise fossil fuel combustion. In contrast, the summer CO source isotopic signatures at Beech Island (δ13C: −30.5 ‰, δ18O: 5.7 ‰) are lower than for Indianapolis (δ13C: −29.6 ‰, δ18O: 12.5 ‰), which is consistent with a larger relative contribution of CO from VOC oxidation. As stated above, the absence of a clear CO mole fraction summertime minimum at Beech Island (Fig. 4) is likely due to the large influence from CO produced by oxidation of VOCs during the summer, which offsets the expected summertime CO decline, such as is seen at Indianapolis (Fig. 3). The much higher contribution of CO produced by oxidized VOCs at Beech Island relative to Indianapolis is reasonable, given the more concentrated fossil fuel source in the Indianapolis urban area and the higher concentrations of biogenic VOCs expected at the densely forested and warmer Beech Island site.
While the small data set from Beech Island does not allow for a direct estimate of the isotopic signatures of VOC-produced CO, it is consistent with the values we obtained from Indianapolis and with values estimated by Brenninkmeijer and Röckmann (1997). Additionally, the Beech Island data are not consistent with the 15 ‰ value for δ18O of VOC-produced CO suggested by the prior Stevens and Wagner (1989) study. The Beech Island data suggest the dominant local CO wintertime source is fossil fuel combustion, with a δ18O isotopic signature of approximately 20 ‰. During the summer months, the addition of VOC-produced CO shifts the overall source δ18O to approximately 6 ‰. If the oxygen isotopic signature of CO produced by oxidation of VOCs was 15 ‰, as suggested by Stevens and Wagner (1989), this result would be impossible.
3.4 Discussion of the role of ozonolysis in the VOC-derived COδ18O signature
As noted above, Röckmann et al. (1998) suggested ozonolysis of VOCs may be a cause of significant Δ17O deviations, resulting from mass-independent fractionation (MIF) during the formation of O3 (see Röckmann et al., 1998a, b, for a more detailed explanation of the MIF process). Hatakeyama et al. (1991), Röckmann et al. (1998a), and Atkinson and Arey (2003a, b) have suggested that ozonolysis may be a large sink for terpenes in the atmosphere.
Röckmann et al. (1998a) found that O3, and subsequently the CO produced from ozonolysis of VOCs, had a substantially enriched δ18O signature relative to atmospheric oxygen and CO. The δ18O of O3 was shown to be around 80 ‰, and ethene, isoprene, and β-pinene-produced CO with a δ18O between 46 ‰ and 83 ‰ (relative to the original O2 used in the experiments) (Röckmann et al., 1998a). The δ18O of atmospheric O2 is around 23 ‰, and therefore the CO produced by ozonolysis of these VOCs in the atmosphere would have a δ18O of between 69 ‰ to 100 ‰. Röckmann et al. (1998a) acknowledge that a significant global source of CO with a δ18O of 69 ‰–100 ‰ is difficult to reconcile with the overall CO δ18O budget, and thus conclude that either (a) ozonolysis of VOCs is not the primary source of the observed mass-independent 17O deviations, or (b) a second source with sufficiently depleted δ18O and similar seasonal cycle to ozone, VOC emissions, and CO must be countering the ozonolysis δ18O contribution. Röckmann et al. (1998b) detail a second source of MIF from CO+OH and concluded that the ozonolysis source was a small contributor to the overall CO budget.
Our δ18O time series (Figs. 3 and 4) as well as summertime source isotopic signature analyses (Figs. 2, 5, 6) are not consistent with a summertime source with such a strong enrichment in 18O. Röckmann et al. (1998a) found no evidence for a seasonally covarying source that has depleted 18O of a similar magnitude to the ozonolysis source, which could obscure the impact of ozonolysis on CO−δ18O. Thus, we conclude that CO produced by the ozonolysis of VOCs is not a major component of the CO budget at both Indianapolis and Beech Island and that OH oxidation is the dominant source of VOC-produced CO in our study.
Nonetheless, our δ18O results do not preclude a minor source of CO from ozonolysis of VOCs and the VOC-produced CO δ13C and δ18O signatures calculated in this study cannot be separated between OH oxidation and ozonolysis. We note that, as discussed in Sect. 2.1, the mean transit time for air masses between our background and polluted sites is 3.2 h, which favors the oxidation of isoprene by OH (lifetime ∼ 1.4 h) relative to ozonolysis (lifetime ∼ 1.3 d), depending on the OH and O3 concentrations (Atkinson and Arey, 2003a). β-pinene (also tested by Röckmann et al., 1998a) has similar OH and O3 lifetimes (1.8 h vs. 1.1 d, respectively) (Atkinson and Arey, 2003a). Furthermore, Atkinson (2000) and Atkinson and Arey (2003a, b) have detailed the reaction schemes for VOCs and the OH oxidation and ozonolysis pathways, which are complex. Ozonolysis of isoprene, for example, produces an ozonide which is then destroyed via three possible reaction pathways (Atkinson, 2000; Atkinson and Arey, 2003a, b). Only one of these pathways produces formaldehyde, which is subsequently photolyzed and the only pathway by which the oxygen isotopic signature of ozone could be guaranteed to be preserved in the resultant CO (Atkinson, 2000; Atkinson and Arey, 2003a, b). Other reaction pathways involve further interaction with OH or other molecules (Atkinson, 2000; Atkinson and Arey, 2003a, b), which provides for possible fractionation or exchange of the oxygen isotopes. Other terpenes also form higher-order aldehydes, which primarily react with OH or NO3, but do not react further with O3 (Atkinson, 2000; Atkinson and Arey, 2003a, b). For reaction pathways other than photolysis of formaldehyde, the oxygen isotope fractionations or exchanges are difficult to trace and quantify and are beyond the scope of this study.
To conclude, our results for the δ13C and δ18O signature of CO produced by oxidation of VOCs mainly represent OH oxidation processes with possible minor contributions from ozonolysis. Our atmospheric δ18O time series from Indianapolis and Beech Island are consistent with prior CO isotopic studies, for example, Mak et al. (2003) and Röckmann et al. (2002); i.e., they do not show evidence for a strong source of CO from ozonolysis of VOCs.
We analyzed carbon monoxide stable isotopes and Δ14CO2 during three summers at Indianapolis and determined the isotopic signature of the urban CO enhancement during the summer. Additionally, we analyzed CO stable isotopes approximately bimonthly for 1 year at Beech Island, South Carolina. Using the Δ14CO2 data and the ratio of CO:CO2FF, we calculated the fossil fuel component of the CO mole fraction enhancement at Indianapolis. We then used isotope mass balance and the Indianapolis COFF isotopic signatures from prior work to calculate the isotopic signature of CO produced from VOCs: ‰ for δ13C and 3.6 ‰±1.2 ‰ for δ18O. This result mainly reflects oxidation of VOCs by OH, with a possible minor contribution from ozonolysis of VOCs. Our measurements from Beech Island, South Carolina (a forest site strongly influenced by VOC-derived CO), are consistent with these results and confirm that VOC-derived CO is a large component of the summer Beech Island CO budget. Our estimate for the carbon isotopic signature of VOC-produced CO agrees well with and confirms prior estimates. Our oxygen isotopic result agrees well with estimates made by Brenninkmeijer and Röckmann (1997) but does not support prior work by Stevens and Wagner (1989).
This result is an important step to improving the constraints on global and regional CO budgets. Additional studies that quantify the isotopic signature of VOC-produced CO could confirm whether our result is valid regionally and globally, as well as attempt to better quantify the global importance of CO produced via ozonolysis of VOCs.
Data for this experiment are available in Table 2 in the main text and in the Supplement.
Please direct all requests for materials to Isaac J. Vimont (isaac.vimont@colorado.edu).
IJV performed the measurements, data analysis, and wrote the article. JCT assisted in data analysis and provided multiple coauthor revisions. VVP provided assistance with measurement issues, data analysis, and multiple coauthor revisions. PFP assisted in several of the measurements. CS provided several coauthor revisions. NM and SR provided logistical support for sample collection for the measurements. BHV and JWCW provided laboratory and equipment support.
The authors declare that they have no conflict of interest.
We thank NIST and NOAA for financial support, Sylvia Michel at INSTAAR, University of Colorado, for her advice and assistance during the sample analysis, and the Carbon Cycle and Climate Group at NOAA ESRL for their helpful suggestions.
This research has been supported by the National Institute of Standards and Technology, Physical Measurement Laboratory (grant no. 60 NANB10D023) and the National Oceanic and Atmospheric Administration, Climate Program Office (award nos. NA13OAR4310074 and RA-133R-15-CQ-0044).
This paper was edited by Jan Kaiser and reviewed by two anonymous referees.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/s1352-2310(99)00460-4, 2000.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003a.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003b.
Bergamaschi, P., Brenninkmeijer, C. A. M., Hahn, M., Röckmann, T., Scharffe, D. H., Crutzen, P. J., Elansky, N. F., Belikov, I. B., Trivett, N. B. A., and Worthy, D. E. J.: Isotope analysis based source identification for atmospheric CH4 and CO sampled across Russia using the Trans-Siberian railroad, J. Geophys. Res.-Atmos., 103, 8227–8235, https://doi.org/10.1029/97jd03738, 1998.
Bergamaschi, P., Hein, R., Brenninkmeijer, C. A. M., and Crutzen, P. J.: Inverse modeling of the global CO cycle 2, Inversion of 13C/12C and 18O/16O isotope ratios, J. Geophys. Res., 105, 1929–1945, 2000.
Brand, W. A., Assonov, S. S., and Coplen, T. B.: Correction for the 17O Interference in δ13C Measurements When Analyzing CO2 with Stable Isotope Mass Spectrometry, International Union of Pure and Applied Chemistry Inorganic Chemistry Division Commission on Isotopic Abundances and Atomic Weights, 2009.
Bräunlich, M.: Study of atmospheric carbon monoxide and methane using isotopic analysis, PhD, Institute of Environmental Physics, Rupertus Carola University, Heidelberg, Germany, 2000.
Brenninkmeijer, C. A. M.: Measurement of the Abundance of 14CO in the Atmosphere and the 13C/12C and 18O/16O Ratio of Atmospheric CO with Applications in New Zealand and Antarctica, J. Geophys. Res., 98, 10595–10614, 1993.
Brenninkmeijer, C. A. M. and Röckmann, T.: Principal factors determining the 18O/16O ratio of atmospheric CO as derived from observations in the southern hemispheric troposphere and lowermost stratosphere, J. Geophys. Res., 102, 25477, https://doi.org/10.1029/97jd02291, 1997.
Brenninkmeijer, C. A. M., Röckmann, T., Bräunlich, M., Jockel, P., and Bergamaschi, P.: Review of Progress in Isotope Sutdies of Atmospheric Carbon Monoxide, Chemosphere, 1, 33–52, 1999.
Carter, W. P. L. and Atkinson, R.: Development and evaluation of a detailed mechanism for the atmospheric reactions of isoprene and NOx, Int. J. Chem. Kinet., 28, 497–530, https://doi.org/10.1002/(sici)1097-4601(1996)28:7<497::aid-kin4>3.0.co;2-q, 1996.
Chameides, W. L., Lindsay, R. W., Richardson, J., and Kiang, C. S.: The role of biogenic hydrocarbons in urban photochemical smog – Atlanta as a case study, Science, 241, 1473–1475, https://doi.org/10.1126/science.3420404, 1988.
Cheng, Y., Wang, Y. H., Zhang, Y. Z., Chen, G., Crawford, J. H., Kleb, M. M., Diskin, G. S., and Weinheimer, A. J.: Large biogenic contribution to boundary layer O3-CO regression slope in summer, Geophys. Res. Lett., 44, 7061–7068, https://doi.org/10.1002/2017gl074405, 2017.
Conny, J. M. and Currie, L. A.: The isotopic characterization of methane, non-methane hydrocarbons and formaldehyde in the troposphere, Atmos. Environ., 30, 621–638, https://doi.org/10.1016/1352-2310(95)00305-3, 1996.
Conny, J. M., Verkouteren, R. M., and Currie, L. A.: Carbon 13 composition of tropospheric CO in Brazil: A model scenario during the biomass burn season, J. Geophys. Res.-Atmos., 102, 10683–10693, https://doi.org/10.1029/97jd00407, 1997.
Duncan, B. N., Logan, J. A., Bey, I., Megretskaia, I. A., Yantosca, R. M., Novelli, P. C., Jones, N. B., and Rinsland, C. P.: Global budget of CO, 1988–1997: Source estimates and validation with a global model, J. Geophys. Res.-Atmos., 112, D22301, https://doi.org/10.1029/2007jd008459, 2007.
Granier, C., Bessagnet, B., Bond, T., D'Angiola, A., van der Gon, H. D., Frost, G. J., Heil, A., Kaiser, J. W., Kinne, S., Klimont, Z., Kloster, S., Lamarque, J. F., Liousse, C., Masui, T., Meleux, F., Mieville, A., Ohara, T., Raut, J. C., Riahi, K., Schultz, M. G., Smith, S. J., Thompson, A., van Aardenne, J., van der Werf, G. R., and van Vuuren, D. P.: Evolution of anthropogenic and biomass burning emissions of air pollutants at global and regional scales during the 1980–2010 period, Clim. Change, 109, 163–190, https://doi.org/10.1007/s10584-011-0154-1, 2011.
Griffin, R. J., Chen, J. J., Carmody, K., Vutukuru, S., and Dabdub, D.: Contribution of gas phase oxidation of volatile organic compounds to atmospheric carbon monoxide levels in two areas of the United States, J. Geophys. Res.-Atmos., 112, D10S17, https://doi.org/10.1029/2006jd007602, 2007.
Gros, V., Braunlich, M., Röckmann, T., Jockel, P., Bergamaschi, P., Brenninkmeijer, C. A. M., Rom, W., Kutschera, W., Kaiser, A., Scheel, H. E., Mandl, M., van der Plicht, J., and Possnert, G.: Detailed analysis of the isotopic composition of CO and characterization of the air masses arriving at Mount Sonnblick (Austrian Alps), J. Geophys. Res., 106, 3179–3193, 2001.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., McKay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volitile organic compound emissions, J. Geophys. Res.-Atmos., 100, 8873–8892, https://doi.org/10.1029/94jd02950, 1995.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Guenther, A. B., Zimmerman, P. R., Harley, P. C., Monson, R. K., and Fall, R.: Isoprene and monoterpene emission rate variability – model evaluations and sensitivity analyses, J. Geophys. Res.-Atmos., 98, 12609–12617, https://doi.org/10.1029/93jd00527, 1993.
Harley, C. P., Monson, K. R., and Lerdau, T. M.: Ecological and evolutionary aspects of isoprene emission from plants, Oecologia, 118, 109–123, https://doi.org/10.1007/s004420050709, 1999.
Hatakeyama, S., Izumi, K., Fukuyama, T., Akimoto, H., and Washida, N.: Reactions of OH with alpha-pinene and beta-pinene in air-estimate of global CO production from the atmospheric oxidation of terpenes, J. Geophys. Res.-Atmos., 96, 947–958, https://doi.org/10.1029/90jd02341, 1991.
Helmig, D., Greenberg, J., Guenther, A., Zimmerman, P., and Geron, C.: Volatile organic compounds and isoprene oxidation products at a temperate deciduous forest site, J. Geophys. Res.-Atmos., 103, 22397–22414, https://doi.org/10.1029/98jd00969, 1998.
Holloway, T., Levy, H., and Kasibhatla, P.: Global distribution of carbon monoxide, J. Geophys. Res.-Atmos., 105, 12123–12147, https://doi.org/10.1029/1999jd901173, 2000.
Huff, A. K. and Thiemens, M. H.: O17/O16 and O18/O16 isotope measurements of atmospheric carbon monoxide and its sources, Geophys. Res. Lett., 25, 3509–3512, https://doi.org/10.1029/98gl02603, 1998.
Isobe, T., Feigelson, E. D., Akritas, M. G., and Babu, G. J.: Linear Regression in Astronomy, Astrophys. J., 364, 104–113, https://doi.org/10.1086/169390, 1990.
Kanakidou, M. and Crutzen, P. J.: The photochemical source of carbon monoxide: Importance, uncertainties and feedbacks, Chemosphere, 1, 91–109, 1999.
Keeling, C. D.: The concentration and isotopic abundances of atmospheric carbon dioxide in rural areas, Geochim. Cosmochim. Ac., 13, 322–334, https://doi.org/10.1016/0016-7037(58)90033-4, 1958.
Levin, I., Kromer, B., Schmidt, M., and Sartorius, H.: A novel approach for independent budgeting of fossil fuel CO2 over Europe by 14CO2 observations, Geophys. Res. Lett., 30, 2194, https://doi.org/10.1029/2003gl018477, 2003.
Logan, J. A., Prather, M. J., Wopsy, S. C., and McElroy, M. B.: Tropospheric Chemistry: A Global Perspective, J. Geophys. Res., 86, 7210–7254, 1981.
Mak, J. E. and Kra, G.: The isotopic composition of carbon monoxide at Montauk Point, Long Island, Chemosphere, 1, 205–218, 1999.
Mak, J. E., and Yang, W.: Technique for Analysis of Air Samples for 13C and 18O in Carbon Monoxide via Continuous-Flow Isotope Ratio Mass Spectrometry, Anal. Chem., 70, 5159–5161, 1998.
Mak, J. E., Kra, G., Sandomenico, T., and Bergamaschi, P.: The seasonally varying isotopic composition of the sources of carbon monoxide at Barbados, West Indies, J. Geophys. Res.-Atmos., 108, 4635, https://doi.org/10.1029/2003jd003419, 2003.
Manning, M. R., Brenninkmeijer, C. A. M., and Allan, W.: Atmospheric carbon monoxide budget of the southern hemisphere: Implications of 13C/12C measurements, J. Geophys. Res., 102, 10673, https://doi.org/10.1029/96jd02743, 1997.
Miles, N. L., Richardson, S. J., Lauvaux, T., Davis, K. J., Balashov, N. V., Deng, A., Turnbull, J. C., Sweeney, C., Gurney, K. R., Patarasuk, R., Razlivanov, I., Cambaliza, M. O. L., and Shepson, P. B.: Quantification of urban atmospheric boundary layer greenhouse gas dry mole fraction enhancements in the dormant season: Results from the Indianapolis Flux Experiment (INFLUX), Elem. Sci. Anth., 5, 27, https://doi.org/10.1525/elementa.127, 2017.
Miller, J. B. and Tans, P. P.: Calculating isotopic fractionation from atmospheric measurements at various scales, Tellus B, 55, 207–214, https://doi.org/10.1034/j.1600-0889.2003.00020.x, 2003.
Miller, J. B., Lehman, S. J., Montzka, S. A., Sweeney, C., Miller, B. R., Karion, A., Wolak, C., Dlugokencky, E. J., Southon, J., Turnbull, J. C., and Tans, P. P.: Linking emissions of fossil fuel CO2 and other anthropogenic trace gases using atmospheric 14CO2, J. Geophys. Res.-Atmos., 117, D08302, https://doi.org/10.1029/2011jd017048, 2012.
Park, K., Emmons, L. K., Wang, Z. H., and Mak, J. E.: Joint Application of Concentration and δ18O to Investigate the Global Atmospheric CO Budget, Atmosphere, 6, 547–578, https://doi.org/10.3390/atmos6050547, 2015.
Popa, M. E., Vollmer, M. K., Jordan, A., Brand, W. A., Pathirana, S. L., Rothe, M., and Röckmann, T.: Vehicle emissions of greenhouse gases and related tracers from a tunnel study: CO : CO2, N2O : CO2, CH4 : CO2, O2 : CO2 ratios, and the stable isotopes 13C and 18O in CO2 and CO, Atmos. Chem. Phys., 14, 2105–2123, https://doi.org/10.5194/acp-14-2105-2014, 2014.
Röckmann, T. and Brenninkmeijer, C. A. M.: The error in conventionally reported 13C/12C ratios of atmospheric CO due to the presence of mass independent oxygen isotope enrichment, Geophys. Res. Lett., 25, 3163–3166, 1998.
Röckmann, T., Brenninkmeijer, C. A. M., Neeb, P., and Crutzen, P. J.: Ozonolysis of nonmethane hydrocarbons as a source of the observed mass independent oxygen isotope enrichment in tropospheric CO, J. Geophys. Res.-Atmos., 103, 1463–1470, https://doi.org/10.1029/97jd02929, 1998a.
Röckmann, T., Brenninkmeijer, C. A. M., Saueressig, G., Bergamaschi, P., Crowley, J. N., Fischer, H., and Crutzen, P. J.: Mass-Independent Oxygen Isotope Fractionation in Atmospheric CO as a Result of the Reaction CO + OH, Science, 281, 544–546, https://doi.org/10.1126/science.281.5376.544, 1998b.
Röckmann, T., Jöckel, P., Gros, V., Bräunlich, M., Possnert, G., and Brenninkmeijer, C. A. M.: Using 14C, 13C, 18O and 17O isotopic variations to provide insights into the high northern latitude surface CO inventory, Atmos. Chem. Phys., 2, 147–159, https://doi.org/10.5194/acp-2-147-2002, 2002.
Saurer, M., Prévôt, A. S. H., Dommen, J., Sandradewi, J., Baltensperger, U., and Siegwolf, R. T. W.: The influence of traffic and wood combustion on the stable isotopic composition of carbon monoxide, Atmos. Chem. Phys., 9, 3147–3161, https://doi.org/10.5194/acp-9-3147-2009, 2009.
Schutze, M.: New Oxidation Means for the Quantitative Crossover from carboxide to carbon dioxide-Article on the chemsitry of iodine pentoxide, Ber. Dtsch. Chem. Ges., 77, 484–487, 1944.
Sharkey, T. D., Loreto, F., Delwiche, C. F., and Treichel, I. W.: Fractionation of carbon isotopes during biogenesis of atmospheric isoprene Plant Physiol., 97, 463–466, https://doi.org/10.1104/pp.97.1.463, 1991.
Stevens, C. M. and Krout, L.: Method for the Determination of the Concentration and of the Carbon and Oxygen Isotopic Composition of Atmospheric Carbon Monoxide, Int. J. Mass Spectrom., 8, 265–275, 1972.
Stevens, C. M. and Wagner, A. F.: The role of isotope fractionation effects in atmospheric chemsitry, Z. Naturforsch. A, 44, 376–384, 1989.
Stevens, C. M., Krout, L., Walling, D., and Venters, A.: The Isotopic Composition of Atmospheric Carbon Monoxide, Earth Planet. Sc. Lett., 16, 147–165, 1972.
Stevens, C. M., Kaplan, L., Gorse, R., Durkee, S., Compton, M., Cohen, S., and Bielling, K.: The kinetic isotope effect for carbon and oxygen in the reaction CO + OH, Int. J. Chem. Kinet., 12, 935–948, https://doi.org/10.1002/kin.550121205, 1980.
Strode, S. A., Liu, J., Lait, L., Commane, R., Daube, B., Wofsy, S., Conaty, A., Newman, P., and Prather, M.: Forecasting carbon monoxide on a global scale for the ATom-1 aircraft mission: insights from airborne and satellite observations and modeling, Atmos. Chem. Phys., 18, 10955–10971, https://doi.org/10.5194/acp-18-10955-2018, 2018.
Stuiver, M. and Polach, H. A.: Reporting of C-14 data – discussion, Radiocarbon, 19, 355–363, 1977.
Turnbull, J. C., Miller, J. B., Lehman, S. J., Tans, P. P., Sparks, R. J., and Southon, J.: Comparison of 14CO2, CO, and SF6 as tracers for recently added fossil fuel CO2 in the atmosphere and implications for biological CO2 exchange, Geophys. Res. Lett., 33, L01817, https://doi.org/10.1029/2005gl024213, 2006.
Turnbull, J., Guenther, D., Karion, A., Sweeney, C., Anderson, E., Andrews, A., Kofler, J., Miles, N., Newberger, T., Richardson, S., and Tans, P.: An integrated flask sample collection system for greenhouse gas measurements, Atmos. Meas. Tech., 5, 2321–2327, https://doi.org/10.5194/amt-5-2321-2012, 2012.
Turnbull, J. C., Sweeney, C., Karion, A., Newberger, T., Lehman, S. J., Tans, P. P., Davis, K. J., Lauvaux, T., Miles, N. L., and Richardson, S. J.: Toward quantification and source sector identification of fossil fuel CO2 emissions from an urban area: Results from the INFLUX experiment, J. Geophys. Res.-Atmos., 120, 292–312, https://doi.org/10.1002/2014JD022555, 2015.
Turnbull, J. C., Karion, A., Davis, K. J., Lauvaux, T., Miles, N. L., Richardson, S. J., Sweeney, C., McKain, K., Lehman, S. J., Gurney, K. R., Patarasuk, R., Liang, J. M., Shepson, P. B., Heimburger, A., Harvey, R., and Whetstone, J.: Synthesis of Urban CO2 Emission Estimates from Multiple Methods from the Indianapolis Flux Project (INFLUX), Environ. Sci. Technol., 53, 287–295, https://doi.org/10.1021/acs.est.8b05552, 2019.
U.S. Environmental Protection Agency (EPA): National Emissions Inventory, available at: https://www.epa.gov/air-emissions-inventories/2014-national-emissions-inventory-nei-data (last access: June 2016), 2014.
Vimont, I. J., Turnbull, J. C., Petrenko, V. V., Place, P. F., Karion, A., Miles, N. L., Richardson, S. J., Gurney, K. R., Patarasuk, R., Sweeney, C., Vaughn, B., and White, J. W. C.: Carbon monoxide isotopic measurements in Indianapolis constrain urban source isotopic signatures and support mobile fossil fuel emissions as the dominant wintertime CO source, Elem. Sci. Anth., 5, 63, https://doi.org/10.1525/elementa.136, 2017.
Zhou, Y., Mao, H., Demerjian, K., Hogrefe, C., and Liu, J.: Regional and hemispheric influences on temporal variability in baseline carbon monoxide and ozone over the Northeast US, Atmos. Environ., 164, 309–324, https://doi.org/10.1016/j.atmosenv.2017.06.017, 2017.
Zobitz, J. M., Keener, J. P., Schnyder, H., and Bowling, D. R.: Sensitivity analysis and quantification of uncertainty for isotopic mixing relationships in carbon cycle research, Agricult. Forest Meteorol., 136, 56–75, https://doi.org/10.1016/j.agrformet.2006.01.003, 2006.