the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Jan 2019
| 22 Jan 2019
Organic peroxy radical chemistry in oxidation flow reactors and environmental chambers and their atmospheric relevance
Julia Lee-Taylor
John J. Orlando
Geoffrey S. Tyndall
Jose L. Jimenez
Oxidation flow reactors (OFRs) are a promising complement to environmental chambers for investigating atmospheric oxidation processes and secondary aerosol formation. However, questions have been raised about how representative the chemistry within OFRs is of that in the troposphere. We investigate the fates of organic peroxy radicals (RO2), which play a central role in atmospheric organic chemistry, in OFRs and environmental chambers by chemical kinetic modeling and compare to a variety of ambient conditions to help define a range of atmospherically relevant OFR operating conditions. For most types of RO2, their bimolecular fates in OFRs are mainly RO2+HO2 and RO2+NO, similar to chambers and atmospheric studies. For substituted primary RO2 and acyl RO2, RO2+RO2 can make a significant contribution to the fate of RO2 in OFRs, chambers and the atmosphere, but RO2+RO2 in OFRs is in general somewhat less important than in the atmosphere. At high NO, RO2+NO dominates RO2 fate in OFRs, as in the atmosphere. At a high UV lamp setting in OFRs, RO2+OH can be a major RO2 fate and RO2 isomerization can be negligible for common multifunctional RO2, both of which deviate from common atmospheric conditions. In the OFR254 operation mode (for which OH is generated only from the photolysis of added O3), we cannot identify any conditions that can simultaneously avoid significant organic photolysis at 254 nm and lead to RO2 lifetimes long enough (∼ 10 s) to allow atmospherically relevant RO2 isomerization. In the OFR185 mode (for which OH is generated from reactions initiated by 185 nm photons), high relative humidity, low UV intensity and low precursor concentrations are recommended for the atmospherically relevant gas-phase chemistry of both stable species and RO2. These conditions ensure minor or negligible RO2+OH and a relative importance of RO2 isomerization in RO2 fate in OFRs within of that in the atmosphere. Under these conditions, the photochemical age within OFR185 systems can reach a few equivalent days at most, encompassing the typical ages for maximum secondary organic aerosol (SOA) production. A small increase in OFR temperature may allow the relative importance of RO2 isomerization to approach the ambient values. To study the heterogeneous oxidation of SOA formed under atmospherically relevant OFR conditions, a different UV source with higher intensity is needed after the SOA formation stage, which can be done with another reactor in series. Finally, we recommend evaluating the atmospheric relevance of RO2 chemistry by always reporting measured and/or estimated OH, HO2, NO, NO2 and OH reactivity (or at least precursor composition and concentration) in all chamber and flow reactor experiments. An easy-to-use RO2 fate estimator program is included with this paper to facilitate the investigation of this topic in future studies.
- Article
(2323 KB) - Full-text XML
-
Supplement
(3817 KB) - BibTeX
- EndNote





Laboratory reactors are needed to isolate and study atmospheric chemical systems. Environmental chambers have been a major atmospheric chemistry research tool for decades (Cocker et al., 2001; Carter et al., 2005; Presto et al., 2005; Wang et al., 2011; Platt et al., 2013). Over the last few years, oxidation flow reactors (OFRs; see Appendix A for the meanings of the acronyms) (Kang et al., 2007) have emerged as a promising complement to chambers and are being used to investigate atmospheric oxidation processes, particularly volatile organic compound (VOC) oxidation and secondary organic aerosol (SOA) formation and aging (Kang et al., 2011; Lambe et al., 2015; Hu et al., 2016; Palm et al., 2016). These processes have air quality (Levy II, 1971), human health (Nel, 2005) and climate impacts (Stocker et al., 2014).
The most important advantage of OFRs is their ability to achieve relatively high photochemical ages (of the order of equivalent hours or days assuming an average ambient OH concentration of 1.5×106 molecules cm−3; Mao et al., 2009) in minutes instead of hours in chambers (Lambe et al., 2011). Rapid aging is usually achieved by highly active HOx radical chemistry initiated by low-pressure Hg lamp emissions (185 and 254 nm) (Li et al., 2015; Peng et al., 2015). This allows for shorter residence times in OFRs, thus reducing the relative importance of gas and particle losses to walls (Palm et al., 2016), which can be very important in Teflon chambers (Cocker et al., 2001; Matsunaga and Ziemann, 2010; Zhang et al., 2014; Krechmer et al., 2016). In addition, the lower costs and small size (volumes of the order of 10 L) of OFRs allow for better portability. These, together with the ability to rapidly achieve high photochemical ages, are advantageous for field applications. These advantages of OFRs have led a number of atmospheric chemistry research groups (Lambe and Jimenez, 2018) to deploy them in field (Hu et al., 2016; Ortega et al., 2016; Palm et al., 2016, 2017), source (Ortega et al., 2013; Tkacik et al., 2014; Karjalainen et al., 2016; Link et al., 2016) and laboratory studies (Kang et al., 2011; Lambe et al., 2013; Richards-Henderson et al., 2016; Lim et al., 2017).
While the use of oxidation flow reactors is growing rapidly in the atmospheric chemistry community, some researchers have raised two concerns with regard to OFRs: (1) the chemical regime of OFRs may be unrealistic compared to the atmosphere, and (2) OFRs are derivative of flow reactors with a long tradition in atmospheric chemistry, especially for chemical kinetic measurements, and thus there is not much new to be discussed or analyzed in their chemistry. While it is true that OFRs follow the tradition of flow tubes used in atmospheric chemistry, they attempt to simulate a much more complex system all at once and typically use much longer residence times, and thus many fundamental and practical issues arise that have not been addressed before. The need to achieve longer effective photochemical ages within a short residence time can, however, lead to the occurrence of undesirable oxidation pathways.
To clarify this issue, a series of chemical kinetic modeling studies have been performed: Li et al. (2015) and Peng et al. (2015) established a radical chemistry and oxidation model whose predictions compare well against laboratory experiments and found that OH can be substantially suppressed by external OH reactants (e.g., SO2, NOx and VOCs externally introduced into the reactor); Peng et al. (2016) identified a low water mixing ratio (H2O) and/or high external OH reactivity (OHRext, i.e., first-order OH loss rate constant contributed by external OH reactants) as conditions that can cause significant non-tropospheric VOC reactions (e.g., through photolysis at 185 and/or 254 nm); Peng and Jimenez (2017) studied NOy chemistry in OFRs and showed that high-NO conditions, under which organic peroxy radicals react more rapidly with NO than with HO2, can only be realized by simple NO injection in a very narrow range of physical conditions, whose application to investigating intermediate- and high-NO environments (e.g., urban area) is limited; Peng et al. (2018) thus evaluated a few new techniques to maintain high-NO conditions in OFRs and found the injection of percent-level N2O effective to achieve this goal.
While HOx and NOy chemistries have been extensively characterized in OFRs so far, organic peroxy radical (RO2) chemistry has yet to be considered in detail, as previous studies have only considered the balance between RO2+NO vs. RO2+HO2. There has been some speculation that due to high OH concentrations in OFRs, RO2 concentration and lifetime might be significantly different from ambient values, leading to the dominance of RO2 self- and cross-reactions and the elimination of RO2 isomerization pathways (Crounse et al., 2013; Praske et al., 2018). Given the central role RO2 plays in atmospheric chemistry (Orlando and Tyndall, 2012; Ziemann and Atkinson, 2012) and the rapidly increasing use of OFRs, RO2 chemistry in OFRs needs to be studied in detail to characterize the similarities and differences between their reaction conditions and those in the ambient atmosphere and traditional atmospheric reaction chambers.
In this paper, we address this need via modeling. All major known fates of RO2 in OFRs will be investigated and compared with those in typical chamber cases and in the atmosphere. This comparison will provide insights into the atmospheric relevance of RO2 chemistry in atmospheric simulation reactors and allow for the selection of experimental conditions with atmospherically relevant RO2 chemistry in experimental planning.
Due to a variety of loss pathways of RO2 and a myriad of RO2 types, RO2 chemistry is of enormous complexity. We detail the RO2 production and loss pathways of interest in this study, the approximations used to simplify this complex problem and the steps to investigate it methodically. We briefly introduce the base OFR design and the model, which are described in detail elsewhere (Kang et al., 2007; Peng et al., 2015, 2018).
2.1 Potential aerosol mass oxidation flow reactor (PAM OFR)
The concept of the base OFR design simulated in this study, the potential aerosol mass (PAM) reactor, was first introduced by Kang et al. (2007) The geometry of the most popular PAM OFR is a cylinder of ∼ 13 L volume. The PAM reactor we simulate is equipped with low-pressure Hg lamps (model no. 82-9304-03, BHK Inc.) emitting UV light at 185 and 254 nm. When both 185 and 254 nm photons are used to generate OH (termed “OFR185”), water vapor photolysis at 185 nm produces OH and HO2. Recombination of O2 and O(3P), formed by O2 photolysis at 185 nm, generates O3. O(1D), formed through O3 photolysis at 254 nm, reacts with water vapor and produces additional OH. 185 nm photons can be filtered by installing quartz sleeves around the lamps. This converts the reactor into “OFR254” mode, for which the photolysis of O3, which must be initially injected, is the only OH production route. The notation “OFR254-X” is used to specify the initial amount of injected O3 (X ppm) in OFR254. Lambe et al. (2017) and Peng et al. (2018) have shown that the initial injection of N2O is able to maintain up to tens of ppb NO in both OFR185 and OFR254. These modes are denoted “OFR185-iN2O” and “OFR254-X-iN2O”, or more generally “OFR-iN2O”. In OFR254-iN2O, O(1D) generated from O3 photolysis reacts with N2O to generate NO, while in OFR185-iN2O, O(1D) is mainly supplied by N2O photolysis at 185 nm (Peng et al., 2018).
2.2 RO2 production and loss pathways
A single generic RO2 is adopted for modeling purposes to avoid the huge number of RO2 types that would complicate effective modeling and analysis. In OH-initiated VOC oxidation, RO2 is primarily produced via VOC + OH → R (+H2O) followed by , where R is hydrocarbyl or oxygenated hydrocarbyl radical. Since the second step is extremely fast in air (Atkinson and Arey, 2003), the first step controls the RO2 production rate, which depends on OH concentration and OHRext due to VOCs (OHRVOC; see Appendix B for details). OHRVOC also includes the contribution from oxidation intermediates of primary VOCs (e.g., methyl vinyl ketone and pinonic acid). When the information about oxidation intermediates is insufficient to calculate OHRVOC, OHR due to primary VOCs is used instead as an approximant. RO2 production through other pathways, e.g., VOC ozonolysis and photolysis, is not considered, since all non-OH pathways of VOC destruction only become significant at low H2O and/or high OHRext (Peng et al., 2016). These conditions lead to significant non-tropospheric VOC photolysis and thus are of little experimental interest.
Table 1 lists all known RO2 loss pathways. Among those, RO2 photolysis, RO2+NO3 and RO2+O3 are not included in this study, since they are minor or negligible in OH-dominated atmospheres, chambers and OFRs for the following reasons.
Table 1Rate constants (in cm3 molecule−1 s−1 except for isomerization; in s−1), cross section (in cm2) and product(s) of RO2 loss pathways. Only organic species are listed for product(s).

a Ziemann and Atkinson (2012). b Orlando and Tyndall (2012); c typical value within the reported range in Orlando and Tyndall (2012); thermal decomposition rate constants of nitrates of acyl and non-acyl RO2 are assumed to be 0.0004 and 3 s−1, respectively, which are also typical values within the reported ranges in Orlando and Tyndall (2012). d Value used in the present work based on Bossolasco et al. (2014); Assaf et al. (2016, 2017a); Müller et al. (2016); Yan et al. (2016). e Müller et al. (2016); Yan et al. (2016); Assaf et al. (2017b, 2018). f Crounse et al. (2013). g Knap and Jørgensen (2017). h Burkholder et al. (2015). i Klems et al. (2015).
The first-order RO2 photolysis rate constant is of the order of 10−2 s−1 at the highest lamp setting in OFRs (Kalafut-Pettibone et al., 2013) and of the order of 10−5 s−1 in the troposphere under the assumption of unity quantum yield (Klems et al., 2015), while RO2 reacts with HO2 at > 1 s−1 at the highest lamp setting in OFRs and at s−1 in the troposphere. Note that in this study we assume an average ambient HO2 concentration of 1.5×108 molecules cm−3 (Mao et al., 2009; Stone et al., 2012) and RO2+HO2 rate constant of cm3 molecule−1 s−1 (Orlando and Tyndall, 2012).
When daytime photochemistry is active, NO3 is negligible in the atmosphere. In OFR-iN2O modes, RO2+NO3 is negligible unless at very low H2O and high UV intensity (abbreviated UV hereafter), which result in high O3 to oxidize NO2 to NO3 and keep HO2 minimized. However, very low H2O causes serious non-tropospheric organic photolysis (Peng et al., 2016) and thus these conditions are of no experimental interest.
In the atmosphere RO2+O3 is thought to play some role only at night (Orlando and Tyndall, 2012). Similar conditions may exist in some OFR254 cases if a very large amount of O3 is injected and H2O and UV are kept very low to limit HOx production. These conditions are obviously not OH dominated and not further investigated in this study.
Of the RO2 fates considered in this study, RO2+HO2, RO2+NO and RO2+RO2 have long been known to play a role in the atmosphere (Orlando and Tyndall, 2012). Recommended general rate constants are available for RO2+HO2 and RO2+NO (Ziemann and Atkinson, 2012; Table 1), albeit with some small dependencies on the type of RO2 and a few deviations that are slightly larger but not important for the overall chemistry (e.g., CH3O2 and C2H5O2 for RO2+HO2). We use these recommended values for generic RO2 in this study. RO2+NO has two main product channels, i.e., RO+NO2 and RONO2, whose branching ratios are RO2 structure dependent (Ziemann and Atkinson, 2012). We do not include these product channels in this study, since they have negligible impacts on the chemical scheme described here. This feature results from two facts: (i) we focus on generic RO2 and do not explicitly consider the chemistry of the products of different RO2 loss pathways; and (ii) the channel producing RO and NO2 contributes little to NO2 production (Peng et al., 2018). However, RO2 self- and cross-reaction rate constants are highly dependent on the specific RO2 types and can vary over a very large range (10−17–10−10 cm3 molecule−1 s−1). Unsubstituted primary, secondary and tertiary RO2 radicals self-react at , and cm3 molecule−1 s−1, respectively (Ziemann and Atkinson, 2012). Rate constants of cross-reactions between these RO2 types also span this range (Orlando and Tyndall, 2012). Substituted RO2 types have higher self- and cross-reaction rate constants (Orlando and Tyndall, 2012). RO2+RO2 of highly substituted primary RO2 can be as high as cm3 molecule−1 s−1 (Orlando and Tyndall, 2012). Very recently, a few highly oxidized 1,3,5-trimethylbenzene-derived RO2s were reported to self- and cross-react at cm3 molecule−1 s−1 (Berndt et al., 2018). In the present work, we make a simplification to adapt to the generic RO2 treatment by assuming a single self- and cross-reaction rate constant for generic RO2 in each case. Three levels of RO2+RO2 rate constants, i.e., , and cm3 molecule−1 s−1, are studied in this paper. The first level is referred to as “medium RO2+RO2” as many other RO2 types can have self- and cross-reaction rate constants as low as 10−17 cm3 molecule−1 s−1; the second level is defined as “fast RO2+RO2”; and the last level is called “very fast RO2+RO2”. No RO2+RO2 rate constant lower than the medium level is investigated in the current work, although there are still a large variety of RO2 types whose self- and cross-reactions are at lower rate constants, since at the medium level, RO2+RO2 is already negligible in all the environments studied in this work, i.e., OFRs, chambers and the atmosphere (see Sect. 3.1.1). Since there are only a few very specific examples for very fast RO2+RO2 reported to date, we will not systematically explore this category but compare very fast RO2+RO2 as a sensitivity case with the other two types of RO2+RO2 reactions.
Acyl RO2 is considered as a separate RO2 type (neither medium nor fast RO2+RO2) in this study since its reaction with NO2 can be a major sink of RO2 in OFR (Peng and Jimenez, 2017). Thermal decomposition lifetimes of the product of RO2+NO2, i.e., acylperoxy nitrates, can be hours at laboratory temperatures (Orlando and Tyndall, 2012; also taken into account in the current work; see Table 1), while OFR residence times are typically minutes. Besides, acyl RO2 reacts with many RO2 types at cm3 molecule−1 s−1 (Orlando and Tyndall, 2012), similar to that of fast RO2+RO2. We thus assume the acyl RO2 self- and cross-reaction rate constant to also be cm3 molecule−1 s−1 to facilitate comparison with fast RO2+RO2 results.
In OFRs operated at room temperature, acylperoxy nitrates barely decompose, as their thermal decomposition lifetime is typically ∼ 1 h (Orlando and Tyndall, 2012), while OFR residence time is usually a few minutes. In contrast, peroxy nitrates of non-acyl RO2 do decompose on a timescale of 0.1 s (Orlando and Tyndall, 2012; Table 1). As a consequence, the production and decomposition of peroxy nitrates of non-acyl RO2 reach a steady state in OFRs, which can be greatly shifted toward the peroxy nitrate side in cases with very high NO2 (Peng and Jimenez, 2017; Peng et al., 2018).
RO2+OH (Fittschen et al., 2014) and RO2 isomerization (Crounse et al., 2013) have recently been identified as possible significant RO2 fates in the atmosphere. Reactions of the former type, according to several recent experimental and theoretical studies (Bossolasco et al., 2014; Assaf et al., 2016, 2017a, b; Müller et al., 2016; Yan et al., 2016), have similar rate constants ( cm3 molecule−1 s−1) regardless of RO2 type. Therefore, the reaction rate constant of generic RO2 with OH is assigned as cm3 molecule−1 s−1. RO2 isomerization reactivity is highly structure dependent (Crounse et al., 2013; Praske et al., 2018) and rate constant measurements are still scarce, preventing us from assigning a generic RO2 isomerization rate constant. However, for generic RO2, isomerization is generally not a sink but a conversion between two RO2 radicals (both encompassed by the generic one in this study), as RO2 isomerization usually generates an oxygenated hydrocarbyl radical, which rapidly recombines with O2 and forms another RO2. Therefore, RO2 isomerization is not explicitly taken into account in the modeling, but is considered in the RO2 fate analysis.
In summary, six pathways are included in the RO2 fate analysis of this study. The need to explore these six pathways for a high number of OFR, chamber and atmospheric conditions makes the presentation of results challenging. For clarity, we present the results in two steps. In the first step, only well-known RO2 fates (reaction with NO2, HO2, NO and RO2) will be included in the model. In the second step, the results of the first step will be used to guide the modeling and analysis of a more comprehensive set of significant RO2 fates.
2.3 Model description
The model used in the present work is a standard chemical kinetic box model implemented in the KinSim 3.4 solver in Igor Pro 7 (WaveMetrics, Lake Oswego, Oregon, USA) and has been described in detail elsewhere (Peng et al., 2015, 2018). Plug flow in the reactor with a residence time of 180 s is assumed, since the effects of non-plug flow are major only in a narrow range of conditions of little experimental interest, and the implementation of laminar flow or measured residence time distribution substantially increases computational cost (Peng et al., 2015; Peng and Jimenez, 2017). The reactions of RO2 discussed in Sect. 2.2 are added to the chemical mechanism. A generic slow-reacting VOC (with the same OH rate constant as SO2) is used as the external OH reactant. Its initial concentration is determined by the initial OHRext in each model case. Then as this proxy external OH reactant slowly reacts, OHRext slowly decays. This slow change in OHRext represents not only the decay of the initial reactant but also the generation and consumption of later-generation products that continue to react with OH. The reason for this approximation has been discussed in detail in previous OFR modeling papers (Peng and Jimenez, 2017; Peng et al., 2018). We exclude NOy species, which are explicitly modeled, from the calculation of OHRext; thus, OHRext only includes non-NOy OHRext hereafter. As OHRext is dominated by OHRVOC in most OFR experiments, we use OHRext to denote OHRVOC in OFRs (while for ambient and chamber cases OHRVOC is still used to exclude the contribution of CO, etc.). The outputs of our model (e.g., species concentrations and exposures) were estimated to be accurate to within a factor of 2–3 when compared with field OFR experiments; better agreement can generally be obtained for laboratory OFR experiments (Li et al., 2015; Peng et al., 2015).
Another key parameter in the model is the HOx recycling ratio (β), defined in this study as the number of HO2 molecule(s) produced per OH molecule destroyed by external OH reactants (Peng et al., 2015). This ratio depends on the products of RO2 loss pathways. The main product of RO2+HO2 is usually ROOH (Table 1), yielding no recycled HO2, while the main products of RO2+NO are RO and NO2, the former of which can often undergo extremely fast H abstraction by O2 to form a carbonyl and HO2. We used the fully chemically explicit (automated chemical mechanism generation based on available knowledge) box model GECKO-A (Aumont et al., 2005) to simulate OH oxidation of several simple VOCs (e.g., propane and decane) under various OFR conditions with zero NO. We consistently find that β∼ 0.3. At the other extreme, when RO2 is solely consumed by RO2+NO, the product RO yields HO2 at a branching ratio close to 1, β∼ 1. For intermediate cases, we assume that β may be interpolated as a linear function of , where r(RO2+NO) and r(RO2+HO2) are the local reactive fluxes of RO2+NO and RO2+HO2.
In the present work, we model OFR185, OFR254-70 and OFR254-7 (including their iN2O variants). We specify the same temperature and atmospheric pressure (295 K and 835 mbar, typical values in Boulder, Colorado, USA) as our previous OFR modeling studies (Li et al., 2015; Peng et al., 2015, 2016, 2018; Peng and Jimenez, 2017). The explored physical condition space follows that of our previous OFR-iN2O modeling work (Peng et al., 2018). The only differences are that in this study we also include cases without any N2O injected (OFR185 and OFR254 only) and exclude OHRext=0 conditions, which produce no RO2. In detail, the explored physical condition space covers the following: H2O of 0.07 %–2.3 % (relative humidity of 2 %–71 % at 295 K); UV photon flux at 185 nm (abbreviated F185) of 1.0×1011–1.0×1014 photons cm−2 s−1 (corresponding photon flux at 254 nm (F254) of 4.2×1013–8.5×1015 photons cm−2 s−1); OHRext of 1–1000 s−1; and N2O mixing ratio (abbreviated N2O hereafter) of 0 and 0.02 %–20 %. All model cases are logarithmically evenly distributed except for N2O =0 and F254. The latter is calculated based on the F185–F254 relationship for the lamps simulated here (Li et al., 2015).
For the classification of conditions, the same criteria as in the OFR-iN2O modeling study (Peng et al., 2018) are adopted. In detail, high- and low-NO conditions are classified by . In the current work, these reactive fluxes are explicitly tracked in the modeling instead of approximated as in previous studies (Peng and Jimenez, 2017; Peng et al., 2018). The terms “good,” “risky” and “bad” are used to describe OFR operating conditions in terms of non-tropospheric organic photolysis and are defined based on the ratios of F185 and F254 exposure (F185exp and F254exp, i.e., integrated photon fluxes over residence time) to OH exposure (OHexp), as presented previously (Peng and Jimenez, 2017; Peng et al., 2018). Briefly, under a given condition non-tropospheric photolysis is of different relative importance in the fate of each specific organic species: under good conditions, photolysis at 185 and/or 254 nm is unimportant for almost all VOCs; under bad conditions, non-tropospheric photolysis is problematic for most VOC precursors, since significant photolysis of their oxidation intermediates at 185 and/or 254 nm is almost inevitable; and risky conditions can be problematic for some but not all VOCs. Note that good, risky or bad conditions refer only to non-tropospheric organic photolysis and not to whether RO2 chemistry is atmospherically relevant. Table S1 summarizes our condition classification criteria.
In this section, the results are presented in two parts, i.e., first for the simulations with well-known pathways only and secondly with all significant pathways, as proposed in Sect. 2.2. Then based on the results and their comparison with the atmosphere and chamber experiments, we propose guidelines for OFR operation to ensure atmospherically relevant RO2 chemistry, as well as other chemistries already discussed in the previous studies (Peng et al., 2016, 2018), in OFRs.
3.1 Simulations with well-known pathways (RO2+HO2, RO2+RO2, RO2+NO and RO2+NO2)
Due to the significantly different reactivities of non-acyl and acyl RO2, the results of these two types of RO2 are shown separately.
3.1.1 Non-acyl RO2
In this case non-acyl RO2 radicals have only three fates, i.e., RO2+HO2, RO2+NO and RO2+RO2. The relative importance of these three fates can be shown in a triangle plot (Fig. 1). The figure includes data points of OFR185 (including OFR185-iN2O) and OFR254-70 (including OFR254-70-iN2O), as well as several typical ambient and chamber studies, including two pristine remote area cases (P1 and P2) from the ATom-1 study (Wofsy et al., 2018), two forested area cases (F1 and F2) from the BEACHON-RoMBAS and GoAmazon campaigns, respectively (Ortega et al., 2014; Martin et al., 2016, 2017), an urban area case (U) from the CalNex-LA campaign (Ryerson et al., 2013), and five typical chamber experiment cases (C1–C5) from the FIXCIT study (Nguyen et al., 2014). These typical cases shown in Fig. 1 bring to light several interesting points.
-
In all ambient and chamber cases, medium and slower RO2+RO2 contribute negligibly to the RO2 fate. This confirms a common impression that self- and cross-reactions of many RO2 radicals do not significantly affect RO2 fates.
-
However, if RO2 self- and cross-reacts rapidly, RO2+RO2 can be the most important loss pathway among RO2+RO2, RO2+HO2 and RO2+NO even in pristine regions with higher VOC (e.g., P1 in Fig. 1) compared to an average pristine region case (P2). Note that the P1 case is still very clean compared to typical forested and urban areas (Table 2).
-
Forested areas located in the same region as pollution sources are not as “low NO” as one may expect (points F1 and F2 in Fig. 1). RO2+NO contributes ∼ 20 %–50 % to RO2 loss, as NO and HO2 concentrations are of the same order of magnitude in these cases.
-
RO2+NO dominates over RO2+RO2 and RO2+HO2 in almost all urban areas. Even in relatively clean urban areas such as Los Angeles during CalNex-LA in 2010 (point U in Fig. 1), average NO is ∼ 1 ppb, still sufficiently high to ensure the dominance of RO2+NO among the three pathways.
-
Various chamber cases in the FIXCIT campaign (low to high OHRext; low to high NO; points Cx in Fig. 1) are able to represent specific RO2 fates that appear in different regions in the atmosphere.
On these plots, points for bad conditions (in terms of non-tropospheric photolysis) are not shown because of the lack of experimental interest. The triangle plots for OFR254-7 (including OFR254-7-iN2O) in the same form (Supplement Fig. S1a, b) show no qualitative differences from the results of OFR254-70, implying that initial O3 in OFR254 modes has only minor impacts on RO2 fate. We see this result not only for well-known non-acyl RO2 fate, but also for the aspects discussed in the following sections. The similarity between OFR254 modes can be explained by the minor effects of a lower O3 on HOx at relatively low OHRext (Peng et al., 2015). Cases at higher OHRext often have stronger non-tropospheric photolysis (Peng et al., 2016) and hence are more likely to be under bad conditions and are not shown in Figs. 1 and S1a, b. For simplicity, this similarity is not discussed further.
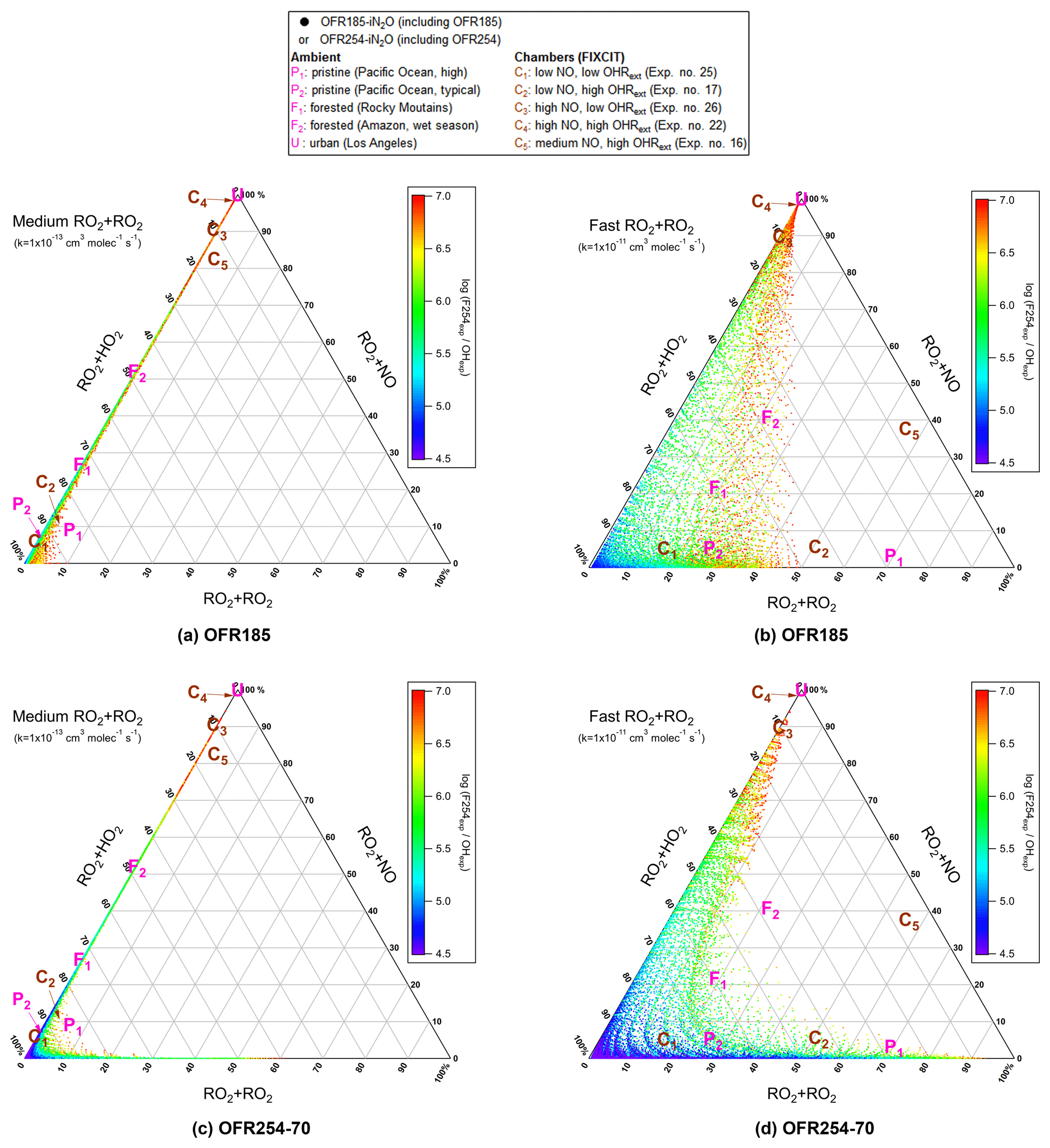
Figure 1Triangle plots of RO2 fate by RO2+HO2, RO2+RO2 and RO2+NO (without RO2+OH and RO2 isomerization considered in the model) for RO2 with the medium self- and cross-reaction rate constant ( cm−3 molecule−1 s−1) in (a) OFR185 (including OFR185-iN2O) and (c) OFR254-70 (including OFR254-70-iN2O) and for RO2 with the fast self- and cross-reaction rate constant ( cm−3 molecule−1 s−1) in (b) OFR185 (including OFR185-iN2O) and (d) OFR254-70 (including OFR254-70-iN2O). Inclined tick values on an axis indicate the grid lines that should be followed (in parallel to the inclination) to read the corresponding values on this axis. The OFR data points are colored by the logarithm of the exposure ratio between 254 nm photon flux and OH, a measure of badness of OFR conditions in terms of 254 nm organic photolysis. Several typical ambient and chamber cases (see Table 2 for details on these cases) are also shown for comparison.
Table 2Several typical ambient and chamber (the FIXCIT campaign) cases that are compared to OFR cases.

a Wofsy et al. (2018) for the Atom-1 campaign. b Fry et al. (2013) for the BEACHON-RoMBAS campaign. c RO2 concentration was given in Fry et al. (2013) (50 ppt) so that OHRVOC is not needed for RO2 fate estimation. d Daun Jeong and Saewung Kim, personal communication (2018), for the GoAmazon campaign (Martin et al., 2016, 2017). e Typical case in the CalNex-LA campaign (Ryerson et al., 2013). f Estimated (Peng et al., 2016). g Typical ambient value (Mao et al., 2009; Stone et al., 2012). h Data from Nguyen et al. (2014). i Initial value.
An important feature confirmed in Fig. 1 is that OFR-iN2O modes effectively realize conditions of experimental interest with variable relative importance of RO2+NO in RO2 fate (Lambe et al., 2017; Peng et al., 2018). Tuning initially injected N2O can achieve this goal (Fig. 2). While it is possible to reduce RO2+HO2 in OFR185-iN2O to negligible compared to RO2+NO by increasing N2O, this is not possible in OFR254-70-iN2O due to fast NO oxidation by the large amounts of O3 added in the reactor. Nevertheless, OFR254-70-iN2O can still make RO2+NO dominate over RO2+HO2 in RO2 fate. OFR and chamber cases span a range of ∼ 0 %–∼ 100 % in relative importance of RO2+NO in RO2 fate (Fig. 2), suggesting that both chambers and OFRs are able to ensure the atmospheric relevance of RO2+NO in RO2 fate.
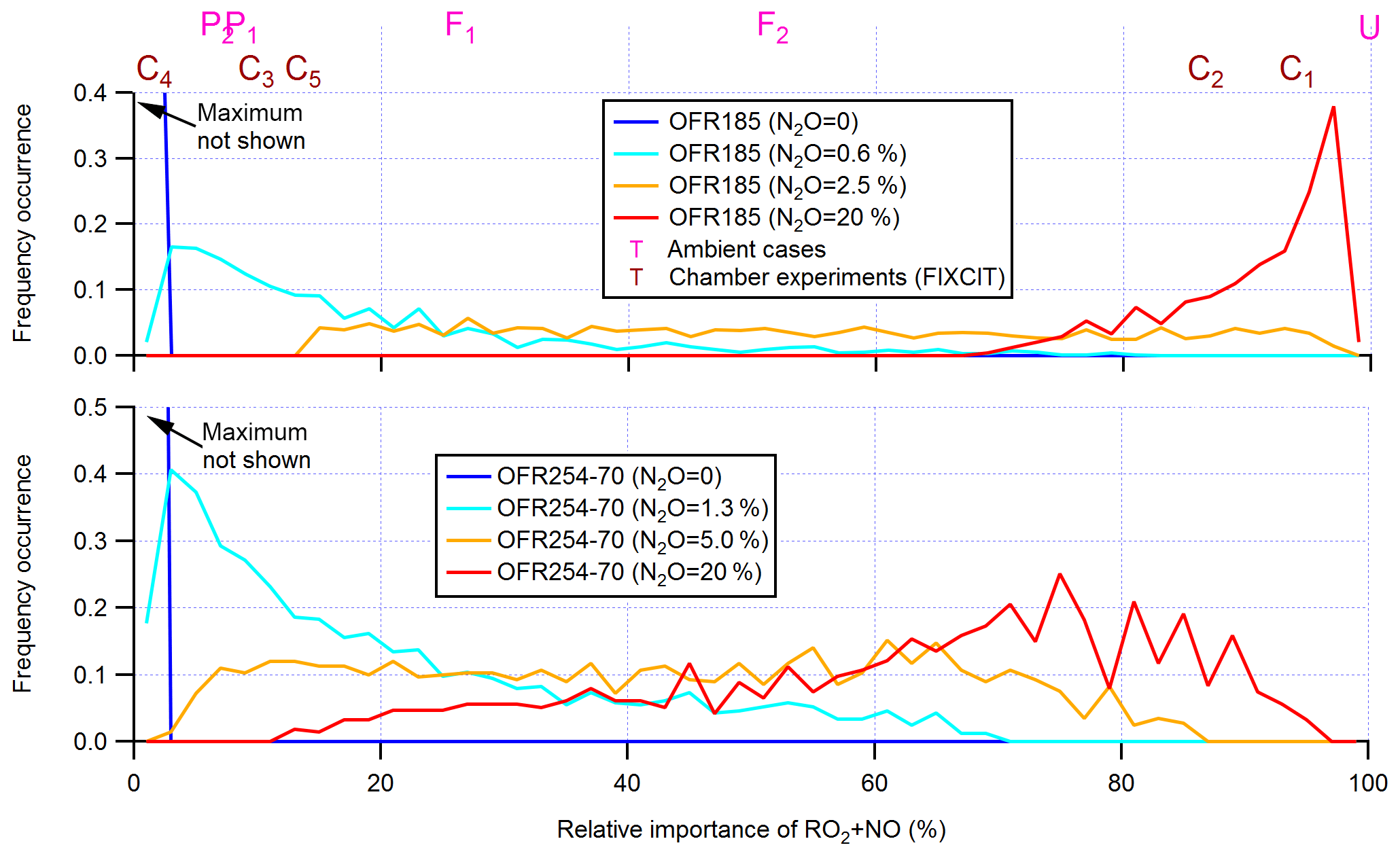
Figure 2Frequency distributions of the relative importance of RO2+NO in the fate of RO2 (with medium self- and cross-reaction rate constant and without RO2+OH and RO2 isomerization considered) for OFR185 (including OFR185-iN2O) and OFR254-70 (including OFR254-70-iN2O). Distributions for several different N2O levels are shown. Only good and risky conditions (in terms of non-tropospheric organic photolysis) are included in the distributions. Also shown is the relative importance of RO2+NO for several typical ambient and chamber cases (see Table 2 for details on these cases).
Another important feature that can be easily seen in Fig. 1 is that medium-rate RO2+RO2 (and hence also RO2+RO2 slower than 10−13 cm3 molecule−1 s−1) is of negligible importance in the fate of RO2 (Fig. 1a, c) in OFR185 (including OFR185-iN2O), OFR254-70 (under most conditions, including OFR254-70-iN2O), chambers and the atmosphere. Thus, a very large subset of RO2 types have only a minor or negligible contribution from RO2+RO2 to their fate. This is already known for ambient RO2 fate (Ziemann and Atkinson, 2012). The reason why this is also true in OFRs is that while OH is much higher than ambient levels, HO2 and NO (high-NO conditions only) are also higher. One can easily verify that steady-state RO2 concentrations (see Appendix B for details) would not deviate from ambient levels by orders of magnitude. The reactive fluxes of RO2+RO2 in OFRs are thus not substantially different than in the atmosphere, while RO2+HO2 and RO2+NO (high-NO conditions only) are both faster in OFRs because of higher HO2 and NO. The combined effect is a reduced relative importance of RO2+RO2 in RO2 fate in OFRs compared to the atmosphere. The only exception in OFRs occurs at very high VOC precursor concentrations (OHRext significantly > 100 s−1) in OFR254 (Fig. S2), in which OH levels are not substantially suppressed due to large amounts of O3 (Peng et al., 2015). As a result, RO2 concentration is remarkably increased by strong production, and RO2+RO2 relative importance increases roughly quadratically and becomes significant.
The generally lower relative importance of RO2+RO2 in OFRs than in the atmosphere is more obvious for the fate of RO2 with fast RO2+RO2 rate constants (Figs. 1b, d and 3). Although OFRs can reasonably reproduce RO2 fates in typical low- and moderate-OHRext ambient environments (e.g., typical pristine and forested areas; Figs. 1b, d and 3) and low-OHRext chambers, OFR185 cannot achieve a relative importance of RO2+RO2 significantly larger than 50 %, such as found in remote environments with higher VOC (e.g., P1 in Fig. 1) and high-OHRext chamber experiments (e.g., C2 and C5 in Fig. 1; the distribution for C2 is also shown in Fig. 3). In OFR254-70, a relative importance of RO2+RO2 as high as ∼ 90 % may be attained (Fig. S3). However, this requires very high OHRext, which leads to medium (and slower) RO2+RO2 showing higher-than-ambient relative importance. In reality, fast RO2+RO2 reactions all involve substituted RO2, which almost certainly arises from and coexists with unsubstituted RO2 (with slower self- and cross-reactions). Therefore, very high OHRext in OFR254 is not really suitable for attaining dominant RO2+RO2 conditions. In OFR185, a higher OHRext generally also results in a higher RO2+RO2 relative importance because of higher RO2 production (Fig. S3). Nevertheless, higher OHRext is more likely to lead to risky or bad conditions (Fig. 3; Peng et al., 2016). It should be noted that although it is difficult to reliably achieve RO2+RO2 with a relative importance larger than 50% in RO2 fate in OFRs, the distributions of RO2+RO2 relative importance in OFRs seem to be within a factor of 2 of those of field and aircraft campaigns (Fig. 3).
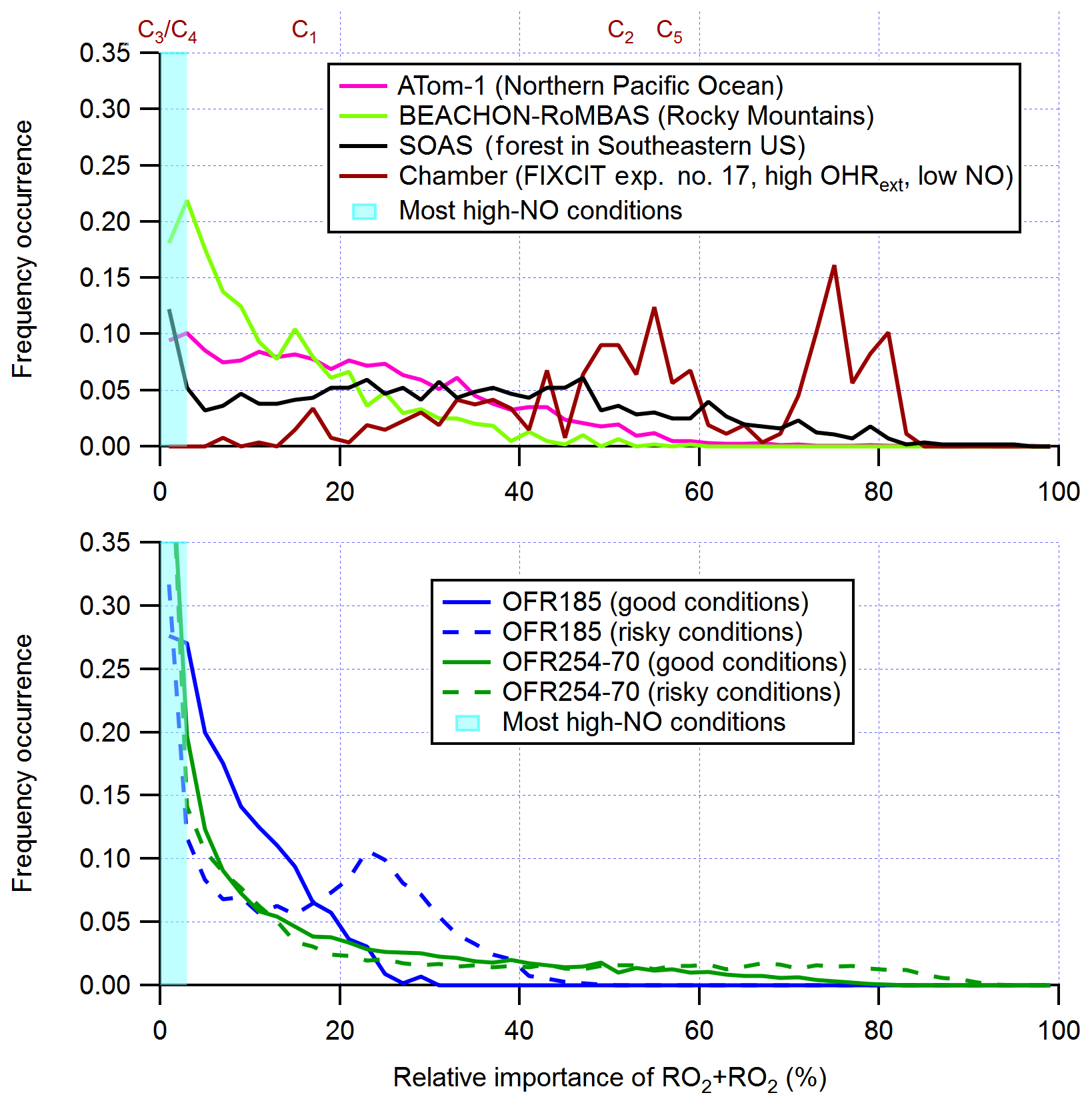
Figure 3Frequency distributions of the relative importance of RO2+RO2 in the fate of RO2 (with fast self- and cross-reaction rate constant and without RO2+OH and RO2 isomerization considered) for OFR185 (including OFR185-iN2O), OFR254-70 (including OFR254-70-iN2O), and a chamber experiment and in the atmosphere (a couple of different environments). The OFR distributions for good and risky conditions (in terms of 254 nm organic photolysis; see Table S1 for the definitions of these conditions) are shown separately. Also shown is the relative importance of RO2+RO2 for several typical chamber cases (see Table 2 for details on these cases). The range of the RO2+RO2 relative importance for most high-NO conditions is highlighted in cyan.
In the case of very fast RO2+RO2, all features for fast RO2+RO2 discussed above are still present (Fig. S1c, d). The only major difference between the results for fast RO2+RO2 and very fast RO2+RO2 is the significantly higher relative importance of RO2+RO2 in RO2 fate in the latter case, which is expected. In summary, fast RO2+RO2 is not perfectly reproduced in OFRs in terms of relative importance in RO2 fate, but it is significant when this pathway is also important in the atmosphere.
The HOx recycling ratio β (see Sect. 2.3) is one of the key factors determining HO2 in the OFR model, yet it is not well constrained. Although we make reasonable assumptions for it in the model input (see Sect. 2.3 for details), a sensitivity study to explore its effects is also performed here. For RO2 with the fast self- and cross-reaction rate constant, we perform simulations with the HOx recycling ratios fixed to a number of values from 0 (radical termination) to 2 (radical proliferation) in lieu of those calculated under the assumptions described in Sect. 2.3. As expected, the contribution of RO2+RO2 to RO2 fate increases monotonically between β=2 and β=0 (Fig. S4) as the recycling of the competing reactant HO2 decreases. Nevertheless, the change in the average RO2+RO2 relative importance from β=0 to β=2 is generally within a factor of 2. Thus, it still holds that the RO2+RO2 relative importance in OFRs is generally lower than in the atmosphere. Only at β∼ 0 may OFR185 theoretically attain a relative importance of RO2+RO2 of ∼ 70 %, as in the P1 case (pristine, but relatively high VOC; Fig. S5). Note that β=0 for all VOC oxidation (including oxidation of intermediates) is extremely unlikely. In OFR254, even if RO2+RO2 may contribute up to ∼ 100 % to RO2 fate at very high OHRext at β=0, these conditions still also lead to significant RO2+RO2 in the fate of RO2 that self- and cross-reacts more slowly, which is not atmospherically relevant.
3.1.2 Acyl RO2
As described in Sect. 2.1, the generic acyl RO2 modeled in this study has the same loss pathways as RO2 with the fast self- and cross-reaction rate constant, except for RO2+NO2, which can be a significant acyl RO2 loss pathway in OFRs as well as both chambers and the atmosphere. When this reaction is included in the simulations of acyl RO2, it is a minor or negligible loss pathway of RO2 at low N2O, while it can be the dominant fate of acyl RO2 at high N2O (Fig. 4). In general, the RO2+NO2 relative importance increases with initial N2O. This is always true in OFR254-70-iN2O between N2O = 0.02 % and N2O = 20 %, while in OFR185-iN2O, the average relative contribution of RO2+NO2 to RO2 fate starts to decrease at N2O ∼ 10 % because RO2+NO regains some importance. This results from the HOx suppression caused by high NOy and strong NO production at high N2O. Strong NO production increases its concentration and suppresses HOx under these conditions, limiting the conversion of NO to NO2. Because of the strong OH suppression by high NOy at N2O ≥10 %, these conditions are not desirable (Peng et al., 2018).

Figure 4Average relative importance of RO2+NO2 in acyl RO2 fate (RO2+OH and RO2 isomerization not considered) in OFR185 (including OFR185-iN2O) and OFR254-70 (including OFR254-70-iN2O). The averages are calculated based on good and risky conditions (in terms of non-tropospheric organic photolysis) only.
The only difference between the simulations of acyl RO2 and of the fast self- and cross-reacting non-acyl RO2 is the quasi-irreversible reaction at room temperature, whose effects are revealed by a comparison of the triangle plots of the RO2 fates in each case (Figs. 1b, d and S6). RO2+NO2 is clearly dominant in acyl RO2 fate in OFRs as long as RO2+NO plays some role (not necessarily under high-NO conditions). In OFR185-iN2O, the relative importance of RO2+RO2 in the sum of the HO2, NO and RO2 pathways is reduced (Fig. S6a) compared to that of non-acyl RO2 with fast RO2+RO2 (Fig. 1b) because RO2+NO2 decreases acyl RO2 concentration. Such a decrease is not significant in OFR254-70-iN2O (Fig. S6b, compared to Fig. 1d), since for non-acyl RO2, it is already stored in the form of RO2NO2 as an RO2 reservoir. In other words, the high initial O3 greatly accelerates NO-to-NO2 oxidation and shifts the equilibrium far to the right even for non-acyl RO2.
RO2+NO2 is an inevitable and dominant sink of most acyl RO2 in high-NOx OFRs, though the extent of this dominance differs substantially among the different OFR operation modes. In OFR254-70-iN2O, RO2+NO makes a minor or negligible contribution to acyl RO2 fate because the required high O3 very rapidly oxidizes NO to NO2 and leads to very low NO-to-NO2 ratios (e.g., ∼ 0.003–0.03; see Fig. S7). In OFR185-iN2O, the contribution of RO2+NO can be somewhat significant, with typical NO-to-NO2 of ∼ 0.03–0.4. (Fig. S7). Urban NO-to-NO2 ratios vary widely, for example (roughly, and excluding significant tails in the frequency distributions) 0.02–1 for Barcelona and 0.007–0.7 for Los Angeles and Pittsburgh (see Fig. S7). Given these variations among different urban areas, RO2+NO and RO2+NO2 for acyl RO2 in OFR185-iN2O can be regarded as relevant to urban atmospheres. Exceptions to the relevance of OFR185-iN2O occur during morning rush hours (e.g., see the high NO-to-NO2 tail for the Pittsburgh case in Fig. S7), near major NO sources and/or in urban atmospheres with stronger NO emission intensity (e.g., Beijing, especially in winter; Fig. S7). In these cases, NO-to-NO2 ratios may significantly exceed 1, and RO2+NO may be the dominant acyl RO2 loss pathway. Such high-NO conditions appear difficult to simulate in OFRs with the current range of techniques.
Acyl RO2 is not the dominant type among RO2 types under most conditions in OFRs, chambers and the atmosphere, since their formation usually requires multistep (at least 2 steps) oxidation via specific pathways leading to an oxidized end group (i.e., aldehyde and then acylperoxy). However, simulations using the GECKO-A model in urban (Mexico City) and forested (Rocky Mountains) atmospheres (Fig. S8) show that acyl RO2 can still be a major (very roughly 1∕3) component of RO2 at ages of several hours or more. Therefore, acyl RO2 chemistry in a high-NO OFR can significantly deviate from that in an urban atmosphere with NO dominating NOx and can be relevant to an urban atmosphere with NO2 dominating NOx. On the other hand, a few theoretical studies suggested that H abstraction by the acylperoxy radical site from hydroperoxy groups close to the acylperoxy site in multifunctional acyl RO2 may be extremely fast (Jørgensen et al., 2016; Knap and Jørgensen, 2017). If these theoretical predictions are sufficiently accurate, these acyl RO2 types may exclusively undergo an intramolecular H shift to form non-acyl RO2 or other radicals and prevent RO2+NO2 from occurring even at very high (ppm level) NO2. However, this type of RO2 is structurally specific and may not have strong impacts on the overall acyl RO2 chemistry.
3.2 Simulations with all significant pathways
Since RO2 isomerization does not significantly affect the generic RO2 concentration, the two RO2 fates that were recently found to be potentially important, i.e., RO2+OH and RO2 isomerization, can be discussed separately.
3.2.1 RO2+OH
In the troposphere, RO2+OH is a minor (at low NO) or negligible (at high NO) RO2 loss pathway (Fittschen et al., 2014; Assaf et al., 2016; Müller et al., 2016), as its rate constant is roughly an order of magnitude higher than that of RO2+HO2 (Table 1), while the ambient OH concentration is on average 2 orders of magnitude lower than that of HO2 (Mao et al., 2009; Stone et al., 2012; Fig. 5). We will not discuss RO2+OH in the high-NO cases in detail. Simply put, the relative importance of RO2+OH is generally negatively correlated with input N2O in OFR-iN2O, as NOx suppresses OH and the relative importance of RO2+NO increases. Below, we focus on low-NO (actually, for simplicity, zero-NO) conditions.
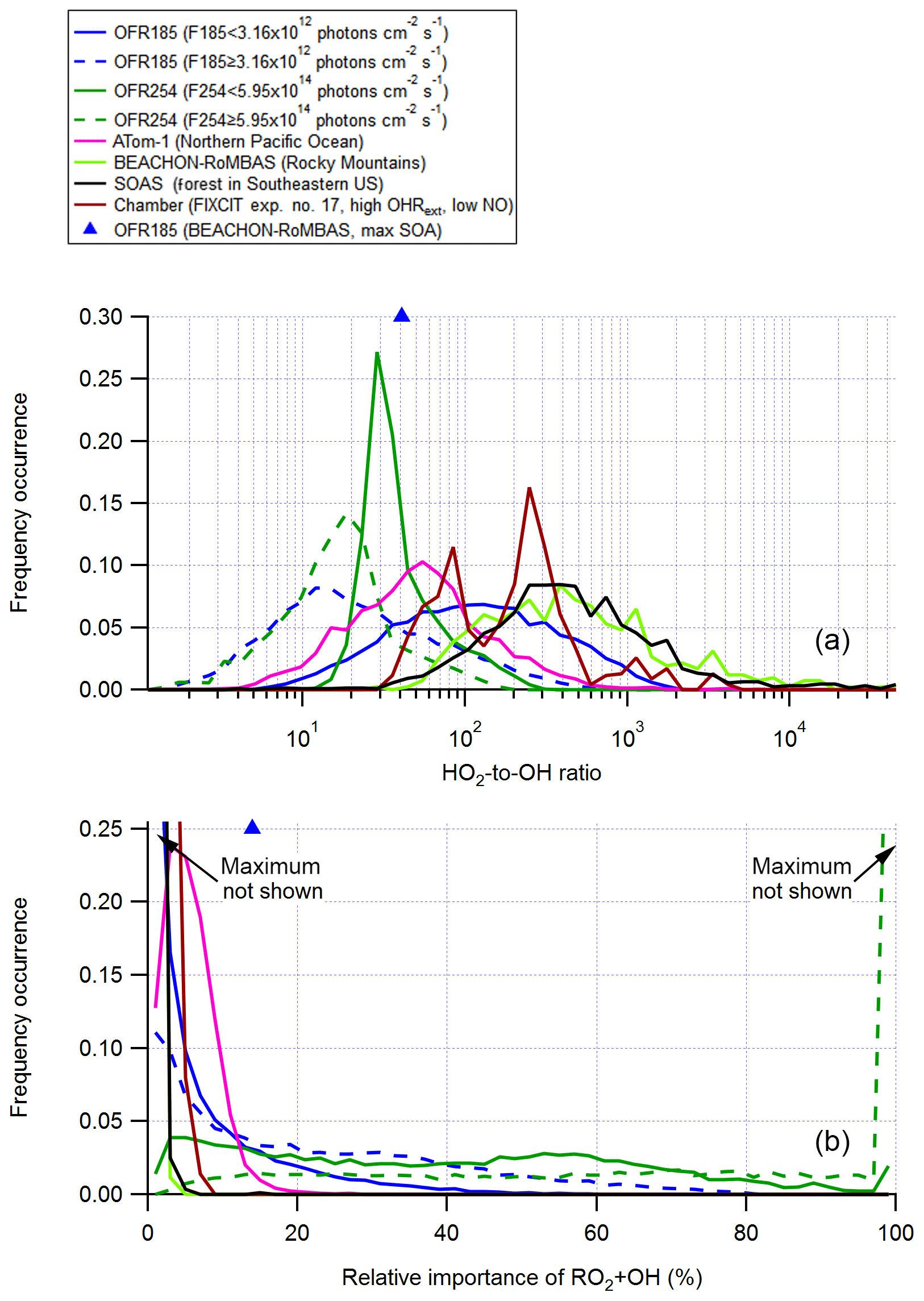
Figure 5Frequency distributions of (a) the HO2-to-OH ratio and (b) the relative importance of RO2+OH in the fate of RO2 (with medium self- and cross-reaction rate constant) for OFR185 (including OFR185-iN2O), OFR254-70 (including OFR254-70-iN2O), and a chamber experiment and in the atmosphere (a couple of different environments). The OFR distributions for lower (F185 < 3.16×1012 photons cm−2 s−1; F254 < 5.95×1014 photons cm−2 s−1) and higher UV (F185 photons cm−2 s−1; F254 photons cm−2 s−1) are shown separately. Only good and risky conditions (in terms of non-tropospheric organic photolysis) are included in the distributions for OFRs. Also shown are the HO2-to-OH ratio and the relative importance of RO2+OH for an OFR experiment with ambient air input in a field study (BEACHON-RoMBAS; Palm et al., 2016).
At N2O =0, it would be ideal if an HO2-to-OH ratio identical to the ambient values was realized in OFRs. In OFR185 cases with medium RO2+RO2, an HO2-to-OH ratio around 100 occurs at a combination of low H2O (of the order of 0.1%), low F185 (of the order of 1011 photons cm−2 s−1) and medium OHRext (10–100 s−1) and also at medium F185 (∼ 1012 photons cm−2 s−1) combined with very high OHRext (∼ 1000 s−1, Fig. S9). Under both sets of conditions, relatively high external OH reactants suppress OH, whose production is relatively weak, and convert some OH into HO2 through HOx recycling in organic oxidation (e.g., via alkoxy radical chemistry). The reason why such an OH-to-HO2 conversion is needed to attain an ambient-like HO2-to-OH ratio is that OFR185 is unable to achieve this via the internal (mainly assisted by O3) interconversion of HOx. This inability is most evident when F185 (1013–1014 photons cm−2 s−1) and H2O (of the order of 1 %) are high and OHRext is low (< ∼ 10 s−1; Fig. S9). Under these conditions, OH production by H2O photolysis is so strong that the HO2-to-OH ratio is lowered to ∼ 1, since OH and H (which recombines with O2 to form HO2) are produced in equal amounts from H2O photolysis. As the RO2+OH rate constant is only roughly 1 order of magnitude higher than that for RO2+HO2, slightly lower HO2-to-OH ratios (e.g., ∼ 30) suffice to keep RO2+OH minor in this case. A combination of UV and H2O that are not very high and a moderate OHRext that is able to convert some OH to HO2 and somewhat elevate the HO2-to-OH ratio results in minor relative importance for RO2+OH (Figs. S9 and S10).
In OFR254-70, it is more difficult to reach an HO2-to-OH ratio of ∼ 100, which can only be realized at a combination of very low H2O and F254 (∼ 0.07 % and photons cm−2 s−1, respectively) and very high OHRext (∼ 1000 s−1). This is mainly due to high O3 in OFR254-70, which controls the HOx interconversion through and and makes both OH and HO2 more resilient to changes due to OHRext (Peng et al., 2015). Even without H2O photolysis at 185 nm as a major HO2 source, the HOx interconversion controlled by O3 in OFR254-70 still brings the HO2-to-OH ratio to ∼ 1 in the case of minimal external perturbation (see the region at the highest H2O and UV and OHRext=0 in the OFR254-70 part of Fig. S9). This ratio cannot be easily elevated in OFR254-70 because of the resilience of OH to suppression for this mode (Peng et al., 2015). Thus, this ratio is relatively low (< 30) under most conditions (Fig. S9), and consequently (and undesirably) RO2+OH is a major RO2 fate in OFR254-70. There is an exception at relatively low H2O and UV with very high OHRext (Fig. S10); however, these conditions are undesirable in terms of non-tropospheric organic photolysis (Peng et al., 2016).
Only the results of RO2 with medium RO2+RO2 are discussed in this section. Those of RO2 with the fast RO2+RO2 are not shown as they are not qualitatively different. In OFR185, for the fast self- and cross-reacting RO2, RO2+RO2 is relatively important at high OHRext (> ∼ 100 s−1; Fig. S3), while RO2+OH is a major RO2 fate at low OHRext (generally of the order of 10 s−1 or lower) and relatively high H2O and UV (Fig. S10). These two ranges of conditions are relatively far away from each other, and hence there is no condition under which RO2+RO2 and RO2+OH are both major pathways that compete, which simplifies understanding RO2 fate. However, in OFR254-70, some conditions may lead to both significant RO2+RO2 (for the fast self- and cross-reacting RO2) and RO2+OH (e.g., H2O ∼ 0.5 %, F254 photons cm−2 s−1 and OHRext∼ 100 s−1). Nevertheless, as long as RO2+OH plays a major role, these conditions do not bear much experimental interest and thus do not need to be discussed in detail.
3.2.2 RO2 isomerization
RO2 isomerization is a first-order reaction. For this type of reaction to occur, RO2 does not need any other species but only a sufficiently long lifetime against all other reactants combined, as most RO2 isomerization rate constants are < 10 s−1. Radical (OH, HO2, NO, etc.) concentrations in OFRs are much higher than ambient levels and may shorten RO2 lifetimes compared to those in the troposphere. Possibly reduced RO2 lifetimes naturally raise concerns over the potentially diminished importance of RO2 isomerization in OFRs.
In this section we examine generic RO2 lifetimes against all reactions (calculated without RO2 isomerization taken into account) in OFR (including OFR-iN2O) cases (for the medium RO2+RO2 case) and compare them with the RO2 lifetimes in recent major field and aircraft campaigns in relatively clean environments and a field campaign in an urban area (CalNex-LA), as well as a low-NO chamber experiment (Fig. 6). Indeed, RO2 lifetimes in clean ambient cases and in chambers with near-ambient radical levels are generally much longer than those in OFRs. The RO2 lifetime distribution of the explored good and risky cases in OFR254-70 (including OFR254-70-iN2O) barely overlaps the ambient and chamber cases, while in OFR185 (including OFR185-iN2O), the RO2 lifetime can be as long as ∼ 10 s, which is longer than in urban areas and roughly at the lower end of the range of ambient RO2 lifetimes in clean environments (Fig. 6). The longest RO2 lifetime in OFR185 occurs at very low F185 (of the order of 1011 photons cm−2 s−1) and H2O (∼ 0.1 %; Fig. S11) when HOx is low. In OFR254-70, for RO2 to survive for ∼ 10 s, in addition to very low UV and H2O, high OHRext is also needed (Fig. S11). High-OHRext conditions in OFR254-70 cause OH suppression and a decrease in HOx concentration and hence result in relatively long RO2 lifetimes. However, the strong OH suppression is likely to create bad conditions (high contribution of non-tropospheric photolysis) (Peng et al., 2016). Low-OHRext conditions do not lead to long RO2 lifetimes in OFR254-70 even at very low F254 and H2O, since O3-assisted HOx recycling prevents a very low HOx level even if HOx primary production is low (Peng et al., 2015)
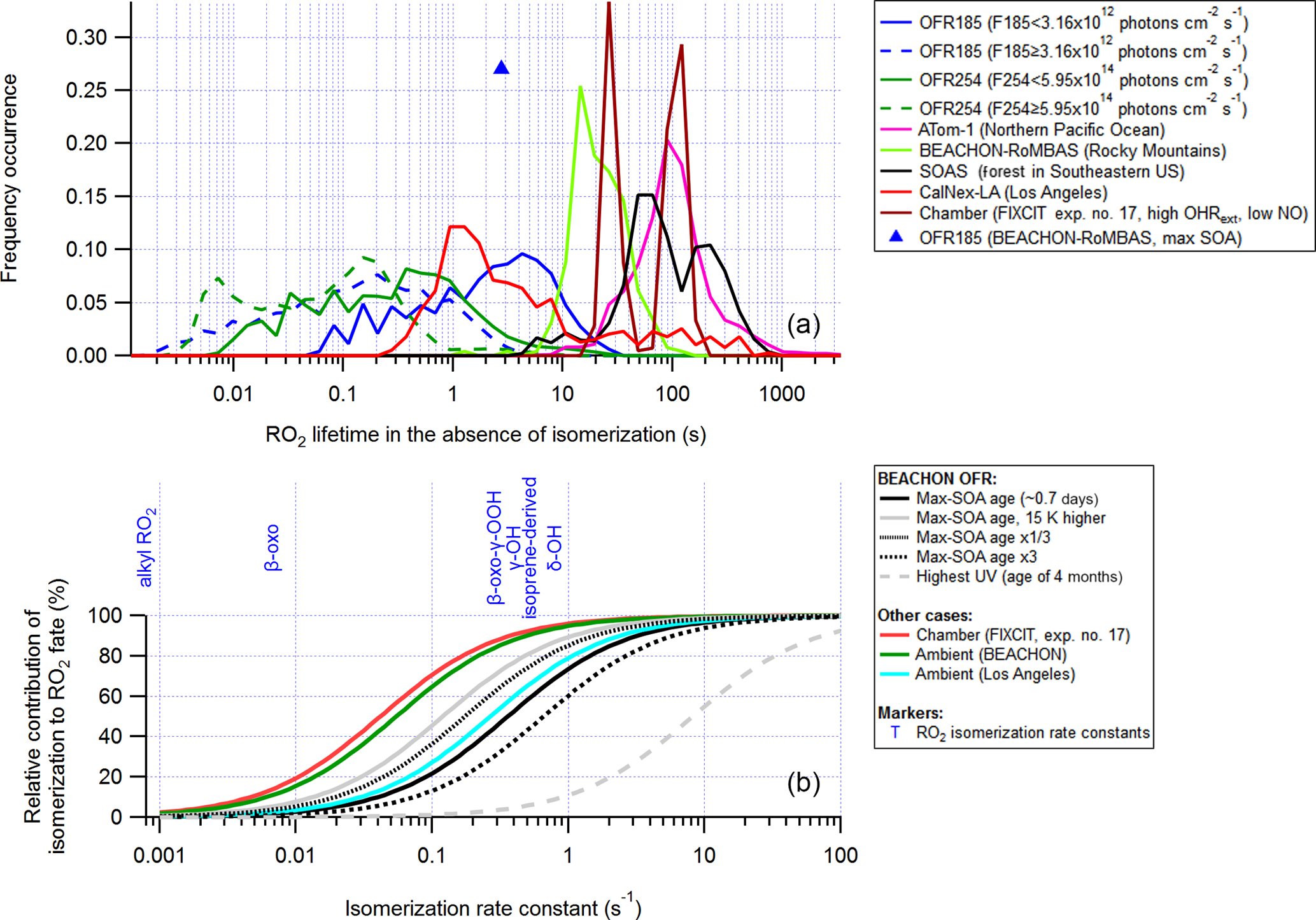
Figure 6(a) Same format as Fig. 5, but for RO2 lifetime (RO2 isomerization included in the model but excluded from lifetime calculation). (b) Relative contribution of isomerization to RO2 fate as a function of RO2 isomerization rate constant in several model cases for OFR experiments in the BEACHON-RoMBAS campaign (Palm et al., 2016), in a chamber experiment and in two ambient cases. Isomerization rate constants of several RO2 types (Crounse et al., 2013; Praske et al., 2018) are also shown.
An RO2 lifetime (without RO2 isomerization included) of 10 s leads to a relative importance of isomerization of 50 % in the total fate (including all loss pathways) of RO2 with an isomerization rate constant of 0.1 s−1, which is a typical order of magnitude for isomerization rate constants of multifunctional RO2 with hydroxyl and hydroperoxy substituents (Fig. 6; Crounse et al., 2013; D'Ambro et al., 2017; Praske et al., 2018). Although a 50 % relative importance of isomerization under some OFR conditions is still lower than those in relatively low-NO ambient environments and low-NO chambers, this relative importance should certainly be deemed major and far from negligible as some have speculated (Crounse et al., 2013). Other monofunctional RO2 (with peroxy radical site only) and bifunctional RO2 with a peroxy radical site and a carbonyl group isomerize so slowly (∼ 0.001–0.01 s−1) that their isomerizations are minor or negligible loss pathways in the atmosphere, chambers and OFRs with RO2 lifetimes around 10 s (Fig. 6). Isomerizations of other types of multifunctional RO2 (e.g., multifunctional acyl RO2 with hydroxyl and hydroperoxy substituents at favorable positions) are extremely fast (rate constants up to 106 s−1; Jørgensen et al., 2016; Knap and Jørgensen, 2017) and always dominate in their fates in the relatively low-NO atmosphere as well as in chambers and OFRs with RO2 lifetimes around 10 s.
In the discussion about RO2 isomerization above (as in the RO2+OH exploration in Sect. 3.2.1), we only examine low-NO (or zero-NO for simplicity) conditions with medium RO2+RO2. In high-NO environments, e.g., polluted urban atmospheres with NO of at least ∼ 10 ppb and high-NO OFRs in the iN2O modes, the RO2 lifetime is so short that isomerization is no longer a major fate for any but the most rapidly isomerizing multifunctional RO2 types discussed above. NO measured in Los Angeles during the CalNex-LA campaign (Ortega et al., 2016) was only ∼ 1 ppb, which would to allow RO2 to survive for a few seconds and isomerize (Fig. 6), even in an urban area.
The OFR simulations for the discussions about RO2 isomerization are the same as those conducted to study RO2+OH, i.e., the ones with medium RO2+RO2 and RO2+OH included. For fast RO2 self- and cross-reaction cases, RO2 lifetimes may be significantly shorter than for RO2 with the medium self- and cross-reaction rate constant at high OHRext (> ∼ 100 s−1) in OFR185 (Fig. S3). These high-OHRext conditions are likely to be risky or bad (of little experimental interest) (Peng et al., 2016) and thus do not need to be discussed further in detail. OFR254-70 (a zero-NO mode) does not generate good or risky (of at least some experimental interest in terms of non-tropospheric organic photolysis) conditions, also leading to low-NO-atmosphere-relevant RO2 lifetimes (Fig. 6). RO2 types with faster self- and cross-reaction rate constants have even shorter lifetimes in OFR254-70 and will not be discussed further.
3.3 Guidelines for OFR operation
In this section we discuss OFR operation guidelines for atmospherically relevant RO2 chemistry, with a focus on OFR185 and OFR254 (zero-NO modes). Since RO2+HO2 and RO2+NO both can vary from negligible to dominant RO2 fate in OFRs, chambers and the atmosphere (Figs. 1 and 2), these two pathways are not a concern in OFR atmospheric relevance considerations, and neither is RO2+RO2. Medium or slower RO2+RO2 is minor or negligible in the atmosphere and chambers, as well as in OFRs, as long as high OHRext is avoided in OFR254 (Fig. S2). Fast RO2+RO2 is somewhat less important in OFRs than in the atmosphere (Figs. 1b, d and 3), but is still qualitatively atmospherically relevant, given the uncertainties associated with the HOx recycling ratios of various reactive systems and the huge variety of RO2 types (and hence RO2+RO2 rate constants).
Accordingly, we focus on the atmospheric relevance of RO2+OH and RO2 isomerization, i.e., their relative contributions close to ambient values. Under typical high-NO conditions, RO2+NO dominates RO2 fate and RO2+OH is negligible. High NO also shortens the RO2 lifetime enough to effectively inhibit RO2 isomerization. Both the dominance of RO2+NO and the inhibition of RO2 isomerization also occur in the atmosphere and in chambers, so high-NO OFR operation (typically NO > 10 ppb) represents these pathways realistically. Some care is, however, required with the RO2+OH and RO2 isomerization pathways at low NO. Since RO2+HO2 in OFRs is always a major RO2 fate at low NO and RO2+RO2 is generally not problematic, RO2+OH and RO2+HO2 can be kept atmospherically relevant as long as the HO2-to-OH ratio is close to 100 (the ambient average). In addition, the RO2 lifetime (calculated without RO2 isomerization taken into account) should be at least around 10 s.
Practically, OH production should be limited to achieve this goal. Too-strong OH production at high H2O and UV can elevate OH and HO2 concentrations, which shortens RO2 lifetime and decreases the HO2-to-OH ratio to ∼ 1 (see Sect. 3.2.1). OH production is roughly proportional to both H2O and UV (Peng et al., 2015), so it can be limited by reducing either or both. However, H2O and UV have different effects on non-tropospheric organic photolysis. At a certain OHRext, the OH production rate roughly determines the OH concentration in OFRs. Reducing UV decreases both OH and UV roughly proportionally (Peng et al., 2015), and hence changes in F185exp ∕ OHexp and F254exp ∕ OHexp are small (Peng et al., 2016); i.e., non-tropospheric organic photolysis does not become significantly worse if UV is reduced. By contrast, if H2O is reduced without also decreasing UV, F185exp ∕ OHexp and F254exp ∕ OHexp both increase, signifying a stronger relative importance of non-tropospheric photolysis. Therefore, reducing UV is strongly preferred as an OH production limitation method and is effective in making both RO2+OH and RO2 isomerization more atmospherically relevant.
To further explore the effects of UV reduction on the RO2+OH (Fig. 5) and RO2 isomerization (Fig. 6) pathways, we divide our OFR case distributions into higher UV and lower UV classes, with the boundary being the midlevel (in logarithmic scale) UV in the explored range. The distributions for lower UV conditions (solid lines in Figs. 5 and 6) are clearly closer to the ambient cases (i.e., HO2-to-OH ratio closer to 100, smaller RO2+OH relative importance and longer RO2 lifetime).
Since OFR254 is unable to achieve both conditions with at least some experimental interest (i.e., with sufficiently low non-tropospheric photolysis) and an atmospherically relevant RO2 lifetime, we now discuss preferable conditions for OFR185 only. As F185 close to or lower than 1012 photons cm−2 s−1 is needed for the RO2 lifetime to be around 10 s or longer (Fig. S11), the OH concentration under preferable conditions for atmospherically relevant RO2 chemistry (∼ 109 molecules cm−3 or lower) is much lower than the maximum that OFR185 can physically reach (∼ 1010–1011 molecules cm−3). Furthermore, lower OH production leads to higher susceptibility to OH suppression by external OH reactants (Peng et al., 2015), which can create non-tropospheric photolysis problems (Peng et al., 2016). We thus recommend H2O as high as possible to maintain practically high OH while allowing lower UV to limit the importance of non-tropospheric organic photolysis.
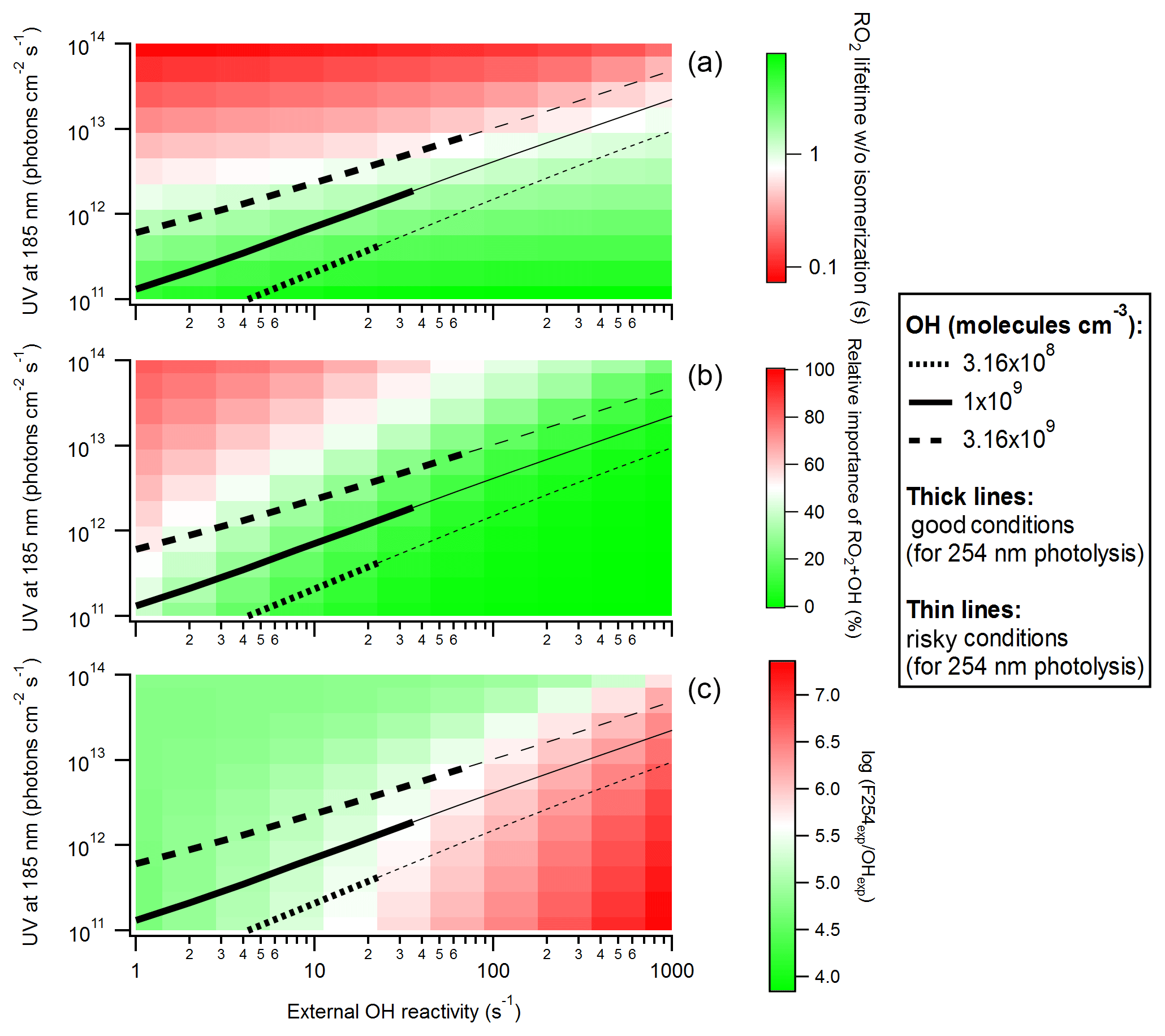
Figure 7(a) RO2 lifetime in the absence of isomerization, (b) relative importance of RO2+OH in RO2 fate, and (c) logarithm of the exposure ratio between 254 nm photon flux and OH as a function of 185 nm photon flux and external OH reactivity for OFR185 at N2O =0 and H2O =2.3 %. Three lines denoting conditions leading to OH of 3.16×108, 1×109 and 3.16×109 molecules cm−3 are added in each panel. The thick and thin parts of these lines correspond to good and risky conditions (in terms of 254 nm organic photolysis, which is usually worse than 185 nm organic photolysis; Peng et al., 2016), respectively.
The performance of various OFR185 conditions at high H2O (2.3 %) is illustrated in Fig. 7 as a function of F185 and OHRext. The three criteria for the performance, i.e., RO2 lifetime (calculated without RO2 isomerization considered), relative importance of RO2+OH and log(F254exp ∕ OHexp) (a measure of 254 nm non-tropospheric photolysis, which is usually worse than that at 185 nm; Peng et al., 2016), are shown. At F185 of ∼ 1011–1012 photons cm−2 s−1 and OHRext around or lower than 10 s−1, all three criteria are satisfied. Since UV (and hence OH production) is relatively low, a low OHRext (∼ 10 s−1) is required to avoid heavy OH suppression and keep conditions good (green area in Fig. 7c). Nevertheless, risky conditions (log(F254exp ∕ OHexp) < 7; light red area in Fig. 7c) may also bear some experimental conditions depending on the type of VOC precursors (specifically on their reactivity toward OH, their photolability at 185 and 254 nm, and the same quantities for their oxidation intermediates; Peng et al., 2016; Peng and Jimenez, 2017). Thus, higher OHRext (up to ∼ 100 s−1) may also be considered in OFR experiments with some precursors (e.g., alkanes). In practice, the preferred conditions may require F185 even lower than that our lowest simulated lamp setting (Li et al., 2015). Such a low F185 may be realized, e.g., by partially blocking 185 nm photons using nontransparent lamp sleeves with evenly placed holes that allow for some 185 nm transmission.
Under these preferred conditions, OH concentration in OFR185 is ∼ 109 molecules cm−3, equivalent to a photochemical age of ∼ 1 eq. days for a typical residence time of 180 s. This is much shorter than ages corresponding to the maximal oxidation capacity of OFRs (usually eq. weeks or months; Peng et al., 2015), but it is similar to the ages of the maximal organic aerosol formation in OFRs processing ambient air (Tkacik et al., 2014; Ortega et al., 2016; Palm et al., 2016). We show the maximal SOA formation case in the OFR185 experiments in the BEACHON-RoMBAS campaign in the Rocky Mountains (Palm et al., 2016) as an example (Figs. 5 and 6). During the campaign, relative humidity was high (> 60 % in most of the period), OHRext was estimated to be relatively low (∼ 15 s−1) in this forested area and UV in the OFR was limited in the case of the maximal SOA formation age (∼ 0.7 eq. days). All these physical conditions were favorable for atmospherically relevant RO2 fate (Figs. 5 and 6). RO2+OH was minor in this case and the relative importance of RO2 isomerization in RO2 fate in the OFR was within a factor of ∼ 2 of that in the atmosphere for all RO2 (regardless of the isomerization rate constant) during the BEACHON-RoMBAS campaign (Fig. 6). The effect of UV on the relative importance of RO2 isomerization for this example is also illustrated in Fig. 6. In the sensitivity case with a lower age, lower UV results in a larger contribution of isomerization to RO2 fate, while the relative importance of RO2 isomerization is lower in a sensitivity case with an age 3 times that of the maximal SOA formation. In an extreme sensitivity case with the highest UV in the range of this study (with an age of 4 eq. months), RO2 isomerization becomes minor or negligible for all RO2 except extremely rapidly isomerizing ones.
The discussions above indicate that the atmospheric relevance of gas-phase RO2 chemistry in OFRs deteriorates as the photochemical age over the whole residence time (180 s) increases. To reach longer ages, longer residence times (with UV still being low) can be adopted. However, OFR residence times > 10 min tend to be limited by the increasing importance of wall losses (Palm et al., 2016). As a result, longer residence times can only increase photochemical age in OFRs up to about a week. This implies that in OFR cases with ages much higher than that of maximal SOA formation (corresponding to the heterogeneous oxidation stage of SOA), the atmospheric relevance of gas-phase RO2 chemistry in the SOA formation stage (before the age of maximal SOA formation) often cannot be ensured. However, under those conditions new SOA formation is typically not observed, and the dominant process affecting OA is heterogeneous oxidation of the preexisting OA (Palm et al., 2016). If the heterogeneous oxidation of newly formed SOA is of interest, a two-stage solution may be required. Lower UV can be used in the SOA formation stage to keep the atmospheric relevance of the gas-phase chemistry, while high UV can be used in the heterogeneous aging stage to reach a high equivalent age. The latter approach is viable since heterogeneous oxidation of SOA by OH is slow and particle-phase chemistry is not strongly affected by gas-phase species except OH when OH is very high (Richards-Henderson et al., 2015, 2016; Hu et al., 2016). This two-stage solution may be realized through a cascade-OFR system or UV sources at different intensities within an OFR (e.g., spliced lamps).
Praske et al. (2018) measured RO2 isomerization rate constants at 296 and 318 K and observed an increase in the rate constants by a factor of ∼ 5 on average. A 15 K temperature increase in OFRs would lead to RO2 isomerization being accelerated by a factor of ∼ 3, while other major gas-phase radical reactions have weak or no temperature dependence (e.g., ∼ 7 %, ∼ 5 %, ∼ 6 % and ∼ 19 % slowdowns for isoprene+OH, toluene + OH, typical RO2+NO and RO2+HO2, respectively; Atkinson and Arey, 2003; Ziemann and Atkinson, 2012). As a consequence, the relative importance of RO2 isomerization in RO2 fate in OFRs can be elevated and closer to atmospheric values (Fig. 6). Nevertheless, a 15 K increase in temperature may also result in some OA evaporation (Huffman et al., 2009; Nault et al., 2018). Besides, the reduction of acylperoxy nitrate formation in OFRs, which may be useful to mimic some urban environments where NO plays a larger role in acyl RO2 fate (see Sect. 3.1.2), is unlikely to be achieved by increasing OFR temperature. The O–N bond energy of acylperoxy nitrates is ∼ 28 kcal mol−1 (Orlando and Tyndall, 2012), which can be taken as an approximate reaction energy of their decomposition. Then a 20 K temperature increase results in the equilibrium constant of acyl acyl RO2NO2 being shifted toward RO2+NO2 by a factor of ∼ 20. However, this shift is still too small relative to the equilibrium constant itself. It can be deduced by a simple calculation that for the generic acyl RO2 in this study in an OFR at 318 K (20 K higher than room temperature) with NO2 of 1012 molecules cm−3 (a relatively low level in typical OFR-iN2O experiments; Peng et al., 2018), ∼ 0.1 % of the total amount of acyl RO2+ acyl RO2NO2 will be present in the form of acyl RO2. Even if acylperoxy nitrate decomposition is 20 times faster than at room temperature and the formed acyl RO2 can irreversibly react with NO and decrease the acylperoxy nitrate concentration, this effect is small: typically up to an approximate 20 % decrease in acylperoxy nitrate and usually negligible changes in NO and NO2. The minor effect is due to (i) an acylperoxy concentration that is still very low, (ii) an NO concentration that is much lower than NO2 and (iii) an acylperoxy nitrate decomposition lifetime that is still of the order of minutes.
As discussed above, high H2O, low UV and low OHRext are recommended for keeping the atmospheric relevance of RO2 chemistry in OFRs. These three requirements are also part of the requirements for attaining good high-NO conditions in OFR185-iNO (the OFR185 mode with initial NO injection; Peng and Jimenez, 2017). In addition to these three, an initial NO of several tens of ppb is also needed to obtain a good high-NO condition in OFR185-iNO. Under these conditions, RO2+NO dominates over RO2+HO2 and hence RO2+OH; UV is low, the photochemical age is typically ∼ 1 eq. days and the RO2 lifetime can be a few seconds. Therefore, these conditions are a good fit for studying the environments in relatively clean urban areas, such as Los Angeles during CalNex-LA (Ortega et al., 2016), where NO is high enough that the dominant bimolecular fate of RO2 is RO2+NO but low enough to maintain RO2 lifetimes that allow for the most common RO2 isomerizations.
As RO2 fate in OFRs is a highly complex problem and it can be tricky to find suitable physical conditions to simultaneously achieve experimental goals and keep the atmospheric relevance of the chemistry in OFRs, we provide here an OFR RO2 Fate Estimator (in the Supplement) to qualitatively aid experimental planning. The OFR RO2 Fate Estimator couples the OFR Exposure Estimator (Peng et al., 2016, 2018) to a general RO2 fate estimator (also in the Supplement; see Fig. S12 for a screenshot of its layout). The OFR Exposure Estimator updated in this study also contains estimation equations for the HO2-to-OH ratio in OFR185 (in OFR254, RO2 fate is always atmospherically irrelevant at low NO, while at high NO, RO2+NO dominates and a detailed RO2 fate analysis is no longer needed). In the general RO2 fate estimator, all RO2 reactant concentrations and all RO2 loss pathway rate constants can be specified. Thus, the general RO2 fate estimator can also be applied to the atmosphere and chamber experiments, in addition to OFRs. When applied to OFRs, the general RO2 fate estimator is provided by the OFR RO2 Fate Estimator with quantities estimated in the OFR Exposure Estimator (e.g., OH and NO). RO2 concentration and fate are calculated according to Appendix B in the RO2 fate estimators.
We investigated RO2 chemistry in OFRs with an emphasis on its atmospheric relevance. All potentially major loss pathways of RO2, i.e., reactions of RO2 with HO2, NO and OH, of acyl RO2 with NO2, and self- and cross-reactions of RO2 and RO2 isomerization, were studied and their relative importances in RO2 fate were compared to those in the atmosphere and chamber experiments. OFRs were shown to be able to tune the relative importance of RO2+HO2 vs. RO2+NO by injecting different amounts of N2O. For many RO2 types (including all unsubstituted non-acyl RO2 and substituted secondary and tertiary RO2), their self-reactions and the cross-reaction between them are minor or negligible in the atmosphere and chambers. This is also the case in OFR185 (including OFR185-iN2O) and OFR254-iN2O; however, those RO2 self- and cross-reactions can be important at high precursor concentrations (OHRext> 100 s−1) in OFR254. For substituted primary RO2 and acyl RO2, their self- and cross-reactions (including the ones with RO2 whose self-reaction rate constants are slower) can play an important role in RO2 fate in the atmosphere and chambers and may also be major RO2 loss pathways in OFRs, although they are somewhat less important in OFRs than in the atmosphere. Acylperoxy nitrates are the dominant sink of acyl RO2 at high NOx in OFRs (particularly in OFR254-iN2O for which RO2+NO is negligible for acylperoxy loss), while there is only a minor reservoir of acyl RO2 in the atmosphere under most conditions except in urban atmospheres, where RO2+NO and RO2+NO2 can both be the dominant acylperoxy loss pathway depending on conditions. In chambers, most acyl RO2 can be stored in the form of acylperoxy nitrates if NO2 is very high (hundreds of ppb to ppm level).
Besides the abovementioned well-known pathways, RO2+OH and RO2 isomerization may also play an important role in RO2 fate and sometimes results in atmospherically irrelevant RO2 chemistry in OFRs. Here we summarize the main findings about all the pathways and the related guidelines for OFR operation.
-
Under typical high-NO conditions, RO2+NO dominates RO2 fate and the RO2 lifetime is too short to allow for most RO2 isomerizations, regardless of whether in the atmosphere, chambers or OFRs, thus raising no concern about the atmospheric relevance of the OFR RO2 chemistry.
-
Under low-NO conditions, OFR254 cannot yield any physical conditions leading to a sufficiently long RO2 lifetime for its isomerization because of the high radical levels and their resilience to external perturbations in OFR254.
-
In OFR185 with strong OH production (and hence high OH), RO2+OH and RO2 isomerization may strongly deviate from that in the atmosphere (becoming important and negligible, respectively, for relatively rapidly isomerizing RO2; rate constants of the order of 0.1 s−1).
-
To attain both atmospherically relevant VOC and RO2 chemistries, OFR185 requires high H2O, low UV and low OHRext. These conditions ensure minor or negligible RO2+OH and a relative importance of RO2 isomerization in RO2 fate in OFRs within a factor of ∼ 2 of that in the atmosphere.
-
Under conditions allowing both VOC and RO2 chemistries to be atmospherically relevant, the maximal photochemical age that can be reached is limited to a few eq. days. This age roughly covers the period required for maximum SOA formation in ambient air.
-
To most realistically study much higher ages for SOA functionalization and fragmentation by heterogeneous oxidation, a sequence of low-UV SOA formation followed by a high UV condition (in the same reactor or in cascade reactors) may be needed.
-
High H2O, low UV and low OHRext in the OFR185-iNO mode can achieve conditions relevant to clean urban atmosphere, i.e., high NO but not sufficiently high to inhibit common RO2 isomerization.
Finally, RO2 chemistry is not only highly complex but also plays a central and instrumental role in atmospheric chemistry, in particular VOC oxidation and SOA formation. For all experiments conducted with an atmospheric chemistry simulation apparatus (chamber, flow reactor, etc.), an atmospherically relevant RO2 chemistry is crucial to meaningful experimental results. However, most literature studies have not published experimental data that are sufficient for estimating RO2 fate. The FIXCIT chamber experiment campaign is one of the few exceptions for which comprehensive data were reported (Nguyen et al., 2014) and used for the RO2 fate analysis in the present work. We recommend measuring and/or estimating and reporting OH, HO2, NO, NO2 and OHRVOC (or initial precursor composition at least) whenever possible for all future atmospheric laboratory and field experiments for organic oxidation to facilitate the analysis of RO2 fate and the evaluation of its atmospheric relevance.
The latest version of the general RO2 fate estimator and OFR RO2 Fate Estimator can be downloaded from https://sites.google.com/site/pamwiki/hardware/estimation-equations (last access: 17 January 2019). The model outputs in this study are available from the authors upon request (zhe.peng@colorado.edu or jose.jimenez@colorado.edu). All data shown in the figures in this paper (including the Supplement) can be downloaded from http://cires1.colorado.edu/jimenez/group_pubs.html (last access: 17 January 2019).
| OFR | oxidation flow reactor |
| VOC | volatile organic compound |
| SOA | secondary organic aerosol |
| H2O | water vapor mixing ratio |
| OHRext | external OH reactivity (due to CO, SO2, VOCs, etc.) |
| PAM | potential aerosol mass, a specific type of OFR |
| OFR185 | oxidation flow reactor using both 185 and 254 nm light |
| OFR254 | oxidation flow reactor using 254 nm light only |
| OFR254-X | OFR254 with X ppm O3 initially injected |
| OFR-iN2O | OFR with N2O initially injected |
| OFR185-iN2O | OFR185 with N2O initially injected |
| OFR254-iN2O | OFR254 with N2O initially injected |
| OFR254-X-iN2O | OFR254-X with N2O initially injected |
| OHRVOC | OH reactivity due to VOCs |
| F185, F254, etc. | UV photon flux at 185 nm, 254 nm, etc. |
| N2O | N2O mixing ratio |
| OHexp, F185exp, etc. | exposure (integral over time) to OH, F185, etc. |
The production rate of a generic RO2 is almost identical to the VOC consumption rate, since the second step of the conversion chain VOC is extremely fast. Therefore, the generic RO2 production rate, P, can be expressed as follows:
where OH is OH concentration and ci and ki are respectively the concentration and the reaction rate constant with OH of the ith VOC. OHRVOC is the total OHR due to VOC and equal to Σikici by definition.
For the generic RO2 loss rate, the reactions of RO2 with HO2, NO, RO2, NO2 (for acyl RO2 only) and OH are considered. Isomerization generally does not lead to a total RO2 concentration decrease and is thus not included in its loss rate. Then the RO2 loss rate is
where RO2, HO2, NO, NO2 and OH are the concentrations of corresponding species and kA (A= RO2, HO2, NO, NO2 and OH) is the reaction rate constant of RO2 with A. For non-acyl RO2, the term is not included; for cases with well-known pathways only (RO2+HO2, RO2+RO2, RO2+NO and RO2+NO2; see Sect. 3.1), the term kOHRO2⋅OH is excluded. needs to be given a value (which may be the main levels of RO2 self- and cross-reaction rate constants in this study, and cm3 molecule−1 s−1, or other values depending on the RO2 type).
At the steady state, P and L are equal. For an ambient and/or chamber setting, OH, HO2, NO, NO2 and OHRVOC are often measured or known. In this case, simultaneously considering Eqs. (B1) and (B2) yields a quadratic equation of RO2 concentration (the only unknown). Then the generic RO2 concentration can be easily obtained by solving this equation:
where .
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-813-2019-supplement.
ZP and JLJ designed the study. ZP performed most of the simulations, and JLT performed the GECKO-A simulations. JJO and GST provided advice on the organic peroxy radical chemistry. ZP, JJO, GST, and JLJ analyzed the results. ZP took the lead in writing the paper. All authors provided feedback on the paper.
The authors declare that they have no conflict of interest.
This work was partially supported by grants EPA STAR 83587701-0, NSF
AGS-1740610, NSF AGS-1822664, NASA NNX15AT96G and DOE(BER/ASR) DE-SC0016559.
We thank the following individuals for providing data from atmospheric field
studies: Tran Nguyen and Jordan Krechmer (FIXCIT), William Brune (SOAS and
ATom), Pedro Campuzano-Jost (ATom), Daun Jeong and Saewung Kim (GoAmazon),
and Weiwei Hu (Beijing). We are also grateful to John Crounse, Joel Thornton,
Paul Ziemann, Dwayne Heard, Paul Wennberg, Andrew Lambe and William Brune for
useful discussions and Donna Sueper for her assistance in the development of
the RO2 Fate Estimator. NCAR is sponsored by the National Science
Foundation. The EPA has not reviewed this paper and thus no endorsement
should be inferred.
Edited by: Dwayne Heard
Reviewed by: two anonymous referees
Assaf, E., Song, B., Tomas, A., Schoemaecker, C., and Fittschen, C.: Rate Constant of the Reaction between CH3O2 Radicals and OH Radicals Revisited, J. Phys. Chem. A, 120, 8923–8932, https://doi.org/10.1021/acs.jpca.6b07704, 2016.
Assaf, E., Tanaka, S., Kajii, Y., Schoemaecker, C., and Fittschen, C.: Rate constants of the reaction of C2–C4 peroxy radicals with OH radicals, Chem. Phys. Lett., 684, 245–249, https://doi.org/10.1016/j.cplett.2017.06.062, 2017a.
Assaf, E., Sheps, L., Whalley, L., Heard, D., Tomas, A., Schoemaecker, C., and Fittschen, C.: The Reaction between CH3O2 and OH Radicals: Product Yields and Atmospheric Implications, Environ. Sci. Technol., 51, 2170–2177, https://doi.org/10.1021/acs.est.6b06265, 2017b.
Assaf, E., Schoemaecker, C., Vereecken, L., and Fittschen, C.: Experimental and theoretical investigation of the reaction of RO2 radicals with OH radicals: Dependence of the HO2 yield on the size of the alkyl group, Int. J. Chem. Kinet., 50, 670–680, https://doi.org/10.1002/kin.21191, 2018.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Aumont, B., Szopa, S., and Madronich, S.: Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach, Atmos. Chem. Phys., 5, 2497–2517, https://doi.org/10.5194/acp-5-2497-2005, 2005.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bossolasco, A., Faragó, E. P., Schoemaecker, C., and Fittschen, C.: Rate constant of the reaction between CH3O2 and OH radicals, Chem. Phys. Lett., 593, 7–13, https://doi.org/10.1016/j.cplett.2013.12.052, 2014.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 18, Pasadena, CA, USA, available at: http://jpldataeval.jpl.nasa.gov/ (last access: 30 November 2018), 2015.
Carter, W. P. L., Cocker, D. R., Fitz, D. R., Malkina, I. L., Bumiller, K., Sauer, C. G., Pisano, J. T., Bufalino, C., and Song, C.: A new environmental chamber for evaluation of gas-phase chemical mechanisms and secondary aerosol formation, Atmos. Environ., 39, 7768–7788, https://doi.org/10.1016/j.atmosenv.2005.08.040, 2005.
Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: State-of-the-Art Chamber Facility for Studying Atmospheric Aerosol Chemistry, Environ. Sci. Technol., 35, 2594–2601, https://doi.org/10.1021/es0019169, 2001.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
D'Ambro, E. L., Møller, K. H., Lopez-Hilfiker, F. D., Schobesberger, S., Liu, J., Shilling, J. E., Lee, B. H., Kjaergaard, H. G., and Thornton, J. A.: Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields, Environ. Sci. Technol., 51, 4978–4987, https://doi.org/10.1021/acs.est.7b00460, 2017.
Fittschen, C., Whalley, L. K., and Heard, D. E.: The reaction of CH3O2 radicals with OH radicals: a neglected sink for CH3O2 in the remote atmosphere, Environ. Sci. Technol., 48, 7700–7701, https://doi.org/10.1021/es502481q, 2014.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Hu, W., Palm, B. B., Day, D. A., Campuzano-Jost, P., Krechmer, J. E., Peng, Z., de Sá, S. S., Martin, S. T., Alexander, M. L., Baumann, K., Hacker, L., Kiendler-Scharr, A., Koss, A. R., de Gouw, J. A., Goldstein, A. H., Seco, R., Sjostedt, S. J., Park, J.-H., Guenther, A. B., Kim, S., Canonaco, F., Prévôt, A. S. H., Brune, W. H., and Jimenez, J. L.: Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 16, 11563–11580, https://doi.org/10.5194/acp-16-11563-2016, 2016.
Huffman, J. A., Docherty, K. S., Aiken, A. C., Cubison, M. J., Ulbrich, I. M., DeCarlo, P. F., Sueper, D., Jayne, J. T., Worsnop, D. R., Ziemann, P. J., and Jimenez, J. L.: Chemically-resolved aerosol volatility measurements from two megacity field studies, Atmos. Chem. Phys., 9, 7161–7182, https://doi.org/10.5194/acp-9-7161-2009, 2009.
Jørgensen, S., Knap, H. C., Otkjær, R. V, Jensen, A. M., Kjeldsen, M. L. H., Wennberg, P. O., and Kjaergaard, H. G.: Rapid Hydrogen Shift Scrambling in Hydroperoxy-Substituted Organic Peroxy Radicals, J. Phys. Chem. A, 120, 266–275, https://doi.org/10.1021/acs.jpca.5b06768, 2016.
Kalafut-Pettibone, A. J., Klems, J. P., Burgess, D. R., and McGivern, W. S.: Alkylperoxy radical photochemistry in organic aerosol formation processes, J. Phys. Chem. A, 117, 14141–14150, https://doi.org/10.1021/jp4094996, 2013.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Kang, E., Toohey, D. W., and Brune, W. H.: Dependence of SOA oxidation on organic aerosol mass concentration and OH exposure: experimental PAM chamber studies, Atmos. Chem. Phys., 11, 1837–1852, https://doi.org/10.5194/acp-11-1837-2011, 2011.
Karjalainen, P., Timonen, H., Saukko, E., Kuuluvainen, H., Saarikoski, S., Aakko-Saksa, P., Murtonen, T., Bloss, M., Dal Maso, M., Simonen, P., Ahlberg, E., Svenningsson, B., Brune, W. H., Hillamo, R., Keskinen, J., and Rönkkö, T.: Time-resolved characterization of primary particle emissions and secondary particle formation from a modern gasoline passenger car, Atmos. Chem. Phys., 16, 8559–8570, https://doi.org/10.5194/acp-16-8559-2016, 2016.
Klems, J. P., Lippa, K. A., and McGivern, W. S.: Quantitative Evidence for Organic Peroxy Radical Photochemistry at 254 nm, J. Phys. Chem. A, 119, 344–351, https://doi.org/10.1021/jp509165x, 2015.
Knap, H. C. and Jørgensen, S.: Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals, J. Phys. Chem. A, 121, 1470–1479, https://doi.org/10.1021/acs.jpca.6b12787, 2017.
Krechmer, J. E., Pagonis, D., Ziemann, P. J., and Jimenez, J. L.: Quantification of Gas-Wall Partitioning in Teflon Environmental Chambers Using Rapid Bursts of Low-Volatility Oxidized Species Generated in Situ, Environ. Sci. Technol., 50, 5757–5765, https://doi.org/10.1021/acs.est.6b00606, 2016.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D)+N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T. and Jimenez, J. L.: PAM Wiki: User Groups, available at: https://sites.google.com/site/pamwiki/user-groups, last access: 2 July 2018.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between Oxidation Level and Optical Properties of Secondary Organic Aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Levy II, H.: Normal atmosphere: large radical and formaldehyde concentrations predicted., Science, 173, 141–143, https://doi.org/10.1126/science.173.3992.141, 1971.
Li, R., Palm, B. B., Ortega, A. M., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J., and Jimenez, J. L.: Modeling the radical chemistry in an Oxidation Flow Reactor (OFR): radical formation and recycling, sensitivities, and OH exposure estimation equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Lim, C. Y., Browne, E. C., Sugrue, R. A., and Kroll, J. H.: Rapid heterogeneous oxidation of organic coatings on submicron aerosols, Geophys. Res. Lett., 44, 2949–2957, https://doi.org/10.1002/2017GL072585, 2017.
Link, M. F., Friedman, B., Fulgham, R., Brophy, P., Galang, A., Jathar, S. H., Veres, P., Roberts, J. M., and Farmer, D. K.: Photochemical processing of diesel fuel emissions as a large secondary source of isocyanic acid (HNCO), Geophys. Res. Lett., 43, 4033–4041, https://doi.org/10.1002/2016GL068207, 2016.
Mao, J., Ren, X., Brune, W. H., Olson, J. R., Crawford, J. H., Fried, A., Huey, L. G., Cohen, R. C., Heikes, B., Singh, H. B., Blake, D. R., Sachse, G. W., Diskin, G. S., Hall, S. R., and Shetter, R. E.: Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 9, 163–173, https://doi.org/10.5194/acp-9-163-2009, 2009.
Martin, S. T., Artaxo, P., Machado, L. A. T., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Andreae, M. O., Barbosa, H. M. J., Fan, J., Fisch, G., Goldstein, A. H., Guenther, A., Jimenez, J. L., Pöschl, U., Silva Dias, M. A., Smith, J. N., and Wendisch, M.: Introduction: Observations and Modeling of the Green Ocean Amazon (GoAmazon2014/5), Atmos. Chem. Phys., 16, 4785–4797, https://doi.org/10.5194/acp-16-4785-2016, 2016.
Martin, S. T., Artaxo, P., Machado, L., Manzi, A. O., Souza, R. A. F., Schumacher, C., Wang, J., Biscaro, T., Brito, J., Calheiros, A., Jardine, K., Medeiros, A., Portela, B., De Sá, S. S., Adachi, K., Aiken, A. C., Alblbrecht, R., Alexander, L., Andreae, M. O., Barbosa, H. M. J., Buseck, P., Chand, D., Comstmstmstock, J. M., Day, D. A., Dubey, M., Fan, J., Fastst, J., Fisch, G., Fortner, E., Giangrande, S., Gilllles, M., Goldststein, A. H., Guenther, A., Hubbbbe, J., Jensen, M., Jimenez, J. L., Keutstsch, F. N., Kim, S., Kuang, C., Laskskin, A., McKinney, K., Mei, F., Millller, M., Nascimento, R., Pauliquevis, T., Pekour, M., Peres, J., Petäjä, T., Pöhlklker, C., Pöschl, U., Rizzo, L., Schmid, B., Shilllling, J. E., Silva Dias, M. A., Smith, J. N., Tomlmlinson, J. M., Tóta, J., and Wendisch, M.: The green ocean amazon experiment (GOAMAZON2014/5) observes pollution affecting gases, aerosols, clouds, and rainfall over the rain forest, B. Am. Meteorol. Soc., 98, 981–997, https://doi.org/10.1175/BAMS-D-15-00221.1, 2017.
Matsunaga, A. and Ziemann, P. J.: Gas-Wall Partitioning of Organic Compounds in a Teflon Film Chamber and Potential Effects on Reaction Product and Aerosol Yield Measurements, Aerosol Sci. Tech., 44, 881–892, https://doi.org/10.1080/02786826.2010.501044, 2010.
Müller, J.-F., Liu, Z., Nguyen, V. S., Stavrakou, T., Harvey, J. N., and Peeters, J.: The reaction of methyl peroxy and hydroxyl radicals as a major source of atmospheric methanol, Nat. Commun., 7, 13213, https://doi.org/10.1038/ncomms13213, 2016.
Nault, B. A., Campuzano-Jost, P., Day, D. A., Schroder, J. C., Anderson, B., Beyersdorf, A. J., Blake, D. R., Brune, W. H., Choi, Y., Corr, C. A., de Gouw, J. A., Dibb, J., DiGangi, J. P., Diskin, G. S., Fried, A., Huey, L. G., Kim, M. J., Knote, C. J., Lamb, K. D., Lee, T., Park, T., Pusede, S. E., Scheuer, E., Thornhill, K. L., Woo, J.-H., and Jimenez, J. L.: Secondary organic aerosol production from local emissions dominates the organic aerosol budget over Seoul, South Korea, during KORUS-AQ, Atmos. Chem. Phys., 18, 17769–17800, https://doi.org/10.5194/acp-18-17769-2018, 2018.
Nel, A.: Air Pollution-Related Illness: Effects of Particles, Science, 308, 804–806, https://doi.org/10.1126/science.1108752, 2005.
Nguyen, T. B., Crounse, J. D., Schwantes, R. H., Teng, A. P., Bates, K. H., Zhang, X., St. Clair, J. M., Brune, W. H., Tyndall, G. S., Keutsch, F. N., Seinfeld, J. H., and Wennberg, P. O.: Overview of the Focused Isoprene eXperiment at the California Institute of Technology (FIXCIT): mechanistic chamber studies on the oxidation of biogenic compounds, Atmos. Chem. Phys., 14, 13531–13549, https://doi.org/10.5194/acp-14-13531-2014, 2014.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Ortega, A. M., Hayes, P. L., Peng, Z., Palm, B. B., Hu, W., Day, D. A., Li, R., Cubison, M. J., Brune, W. H., Graus, M., Warneke, C., Gilman, J. B., Kuster, W. C., de Gouw, J., Gutiérrez-Montes, C., and Jimenez, J. L.: Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area, Atmos. Chem. Phys., 16, 7411–7433, https://doi.org/10.5194/acp-16-7411-2016, 2016.
Ortega, J., Turnipseed, A., Guenther, A. B., Karl, T. G., Day, D. A., Gochis, D., Huffman, J. A., Prenni, A. J., Levin, E. J. T., Kreidenweis, S. M., DeMott, P. J., Tobo, Y., Patton, E. G., Hodzic, A., Cui, Y. Y., Harley, P. C., Hornbrook, R. S., Apel, E. C., Monson, R. K., Eller, A. S. D., Greenberg, J. P., Barth, M. C., Campuzano-Jost, P., Palm, B. B., Jimenez, J. L., Aiken, A. C., Dubey, M. K., Geron, C., Offenberg, J., Ryan, M. G., Fornwalt, P. J., Pryor, S. C., Keutsch, F. N., DiGangi, J. P., Chan, A. W. H., Goldstein, A. H., Wolfe, G. M., Kim, S., Kaser, L., Schnitzhofer, R., Hansel, A., Cantrell, C. A., Mauldin, R. L., and Smith, J. N.: Overview of the Manitou Experimental Forest Observatory: site description and selected science results from 2008 to 2013, Atmos. Chem. Phys., 14, 6345–6367, https://doi.org/10.5194/acp-14-6345-2014, 2014.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Palm, B. B., Campuzano-Jost, P., Day, D. A., Ortega, A. M., Fry, J. L., Brown, S. S., Zarzana, K. J., Dube, W., Wagner, N. L., Draper, D. C., Kaser, L., Jud, W., Karl, T., Hansel, A., Gutiérrez-Montes, C., and Jimenez, J. L.: Secondary organic aerosol formation from in situ OH, O3, and NO3 oxidation of ambient forest air in an oxidation flow reactor, Atmos. Chem. Phys., 17, 5331-5354, https://doi.org/10.5194/acp-17-5331-2017, 2017.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z., Palm, B. B., Day, D. A., Talukdar, R. K., Hu, W., Lambe, A. T., Brune, W. H., and Jimenez, J. L.: Model Evaluation of New Techniques for Maintaining High-NO Conditions in Oxidation Flow Reactors for the Study of OH-Initiated Atmospheric Chemistry, ACS Earth Sp. Chem., 2, 72–86, https://doi.org/10.1021/acsearthspacechem.7b00070, 2018.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Mocnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary Organic Aerosol Production from Terpene Ozonolysis. 1. Effect of UV Radiation, Environ. Sci. Technol., 39, 7036–7045, https://doi.org/10.1021/es050174m, 2005.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Large Enhancement in the Heterogeneous Oxidation Rate of Organic Aerosols by Hydroxyl Radicals in the Presence of Nitric Oxide, J. Phys. Chem. Lett., 6, 4451–4455, https://doi.org/10.1021/acs.jpclett.5b02121, 2015.
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Sulfur Dioxide Accelerates the Heterogeneous Oxidation Rate of Organic Aerosol by Hydroxyl Radicals, Environ. Sci. Technol., 50, 3554–3561, https://doi.org/10.1021/acs.est.5b05369, 2016.
Ryerson, T. B., Andrews, A. E., Angevine, W. M., Bates, T. S., Brock, C. A., Cairns, B., Cohen, R. C., Cooper, O. R., De Gouw, J. A., Fehsenfeld, F. C., Ferrare, R. A., Fischer, M. L., Flagan, R. C., Goldstein, A. H., Hair, J. W., Hardesty, R. M., Hostetler, C. A., Jimenez, J. L., Langford, A. O., McCauley, E., McKeen, S. A., Molina, L. T., Nenes, A., Oltmans, S. J., Parrish, D. D., Pederson, J. R., Pierce, R. B., Prather, K., Quinn, P. K., Seinfeld, J. H., Senff, C. J., Sorooshian, A., Stutz, J., Surratt, J. D., Trainer, M., Volkamer, R., Williams, E. J., and Wofsy, S. C.: The 2010 California Research at the Nexus of Air Quality and Climate Change (CalNex) field study, J. Geophys. Res.-Atmos., 118, 5830–5866, https://doi.org/10.1002/jgrd.50331, 2013.
Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M.: Climate Change 2013 – The Physical Science Basis, edited by Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, 2014.
Stone, D., Whalley, L. K., and Heard, D. E.: Tropospheric OH and HO2 radicals: field measurements and model comparisons, Chem. Soc. Rev., 41, 6348–6404, https://doi.org/10.1039/c2cs35140d, 2012.
Tkacik, D. S., Lambe, A. T., Jathar, S., Li, X., Presto, A. A., Zhao, Y., Blake, D., Meinardi, S., Jayne, J. T., Croteau, P. L., and Robinson, A. L.: Secondary Organic Aerosol Formation from in-Use Motor Vehicle Emissions Using a Potential Aerosol Mass Reactor, Environ. Sci. Technol., 48, 11235–11242, https://doi.org/10.1021/es502239v, 2014.
Wang, J., Doussin, J. F., Perrier, S., Perraudin, E., Katrib, Y., Pangui, E., and Picquet-Varrault, B.: Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research, Atmos. Meas. Tech., 4, 2465–2494, https://doi.org/10.5194/amt-4-2465-2011, 2011.
Wofsy, S. C., Apel, E., Blake, D. R., Brock, C. A., Brune, W. H., Bui, T. P., Daube, B. C., Dibb, J. E., Diskin, G. S., Elkiins, J. W., Froyd, K., Hall, S. R., Hanisco, T. F., Huey, L. G., Jimenez, J. L., McKain, K., Montzka, S. A., Ryerson, T. B., Schwarz, J. P., Stephens, B. B., Weinzierl, B., and Wennberg, P.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, Oak Ridge, Tennessee, USA, 2018.
Yan, C., Kocevska, S., and Krasnoperov, L. N.: Kinetics of the Reaction of CH3O2 Radicals with OH Studied over the 292–526 K Temperature Range, J. Phys. Chem. A, 120, 6111–6121, https://doi.org/10.1021/acs.jpca.6b04213, 2016.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.