the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Jun 2019
| 04 Jun 2019
Atmospheric evolution of molecular-weight-separated brown carbon from biomass burning
Maria Tsagkaraki
Irini Tsiodra
Nikolaos Mihalopoulos
Kalliopi Violaki
Maria Kanakidou
Jean Sciare
Athanasios Nenes
Rodney J. Weber
Biomass burning is a major source of atmospheric brown carbon (BrC), and through its absorption of UV/VIS radiation, it can play an important role in the planetary radiative balance and atmospheric photochemistry. The considerable uncertainty of BrC impacts is associated with its poorly constrained sources, transformations, and atmospheric lifetime. Here we report laboratory experiments that examined changes in the optical properties of the water-soluble (WS) BrC fraction of laboratory-generated biomass burning particles from hardwood pyrolysis. Effects of direct UVB photolysis and OH oxidation in the aqueous phase on molecular-weight-separated BrC were studied. Results indicated that the majority of low-molecular-weight (MW) BrC (<400 Da) was rapidly photobleached by both direct photolysis and OH oxidation on an atmospheric timescale of approximately 1 h. High MW BrC (≥400 Da) underwent initial photoenhancement up to ∼15 h, followed by slow photobleaching over ∼10 h. The laboratory experiments were supported by observations from ambient BrC samples that were collected during the fire seasons in Greece. These samples, containing freshly emitted to aged biomass burning aerosol, were analyzed for both water- and methanol-soluble BrC. Consistent with the laboratory experiments, high-MW BrC dominated the total light absorption at 365 nm for both methanol and water-soluble fractions of ambient samples with atmospheric transport times of 1 to 68 h. These ambient observations indicate that overall, biomass burning BrC across all molecular weights has an atmospheric lifetime of 15 to 28 h, consistent with estimates from previous field studies – although the BrC associated with the high-MW fraction remains relatively stable and is responsible for light absorption properties of BrC throughout most of its atmospheric lifetime. For ambient samples of aged (>10 h) biomass burning emissions, poor linear correlations were found between light absorptivity and levoglucosan, consistent with other studies suggesting a short atmospheric lifetime for levoglucosan. However, a much stronger correlation between light absorptivity and total hydrous sugars was observed, suggesting that they may serve as more robust tracers for aged biomass burning emissions. Overall, the results from this study suggest that robust model estimates of BrC radiative impacts require consideration of the atmospheric aging of BrC and the stability of high-MW BrC.
- Article
(1817 KB) - Full-text XML
-
Supplement
(406 KB) - BibTeX
- EndNote





Brown carbon (BrC), the fraction of organic aerosol that absorbs solar radiation in the UV and near-visible wavelengths, may potentially be an important climate warmer, with estimated direct radiative forcing that varies from +0.03 to +0.60 W m−2 (e.g., Park et al., 2010; Feng et al., 2013; Lin et al., 2014; Wang et al., 2014; Saleh et al., 2015; Jo et al., 2016) and a vertical distribution that can be distinctly different from black carbon and other climate warmers (Zhang et al., 2017). In addition to climate impacts, the importance of UV solar radiation for photochemistry also implies that BrC may affect atmospheric chemistry (He and Carmichael, 1999; Mok et al., 2016).
Current knowledge on the sources, sinks, optical properties, and atmospheric lifetime of BrC is limited. Among the sources of BrC, combustion of biomass (Andreae and Gelencsér, 2006; Alexander et al., 2008; Hecobian et al., 2010; Kirchstetter and Thatcher, 2012; Saleh et al., 2014; Lack et al., 2012) and fossil fuels (Bond, 2001; Zhang et al., 2011) is thought to dominate; for example, a 1-year period in the southeastern USA, 50 % of BrC was attributed to biomass burning (Hecobian et al., 2010). Secondary processes that often involve carbonyls and nitrogen-containing compounds can also generate BrC in the atmosphere (Laskin et al., 2015). Molecular identification of the chromophores is a challenging task, as there may be a multitude of light-absorbing compounds. It remains unclear whether BrC is comprised of low concentrations of strongly absorbing chromophores or a large number of weakly absorbing chromophores in a complex organic matrix. To date, several classes of compounds have been identified as BrC in biomass burning organic aerosols (BBOA), such as nitroaromatic compounds (Desyaterik et al., 2013; Zhang et al., 2013; Mohr et al., 2013; Lin et al., 2016), humic-like substances (HULIS; Dinar et al., 2008; Hoffer et al., 2006; Fan et al., 2016; Wang et al., 2017), and other high-molecular-weight substances (i.e., compounds >400 Da; Di Lorenzo and Young, 2016; Di Lorenzo et al., 2017; Wong et al., 2017).
Studies have suggested that following the emission or formation of biomass burning BrC, their optical properties can be transformed by atmospheric aging processes. Laboratory studies, for both model compounds and complex mixtures of biomass burning BrC, suggested that initial stages of photochemical aging can increase light absorption (“photoenhancement”), followed by a subsequent decrease (“photobleaching”; Saleh et al., 2013; Zhong and Jang, 2014; Zhao et al., 2015; Sumlin et al., 2017; Hems and Abbatt, 2018). The atmospheric lifetime was constrained only for biomass burning BrC from nitrophenols, where photobleaching by aqueous OH oxidation in fog or cloud droplets was found to be its dominant atmospheric loss mechanism, with an estimated atmospheric lifetime of a few hours (Zhao et al., 2015; Hems and Abbatt, 2018). Field observations from wildfire emissions in the northwestern USA and the Amazon have suggested that most biomass burning BrC chromophores have an atmospheric lifetime in the range of 13 to 30 h (Forrister et al., 2015; Wang et al., 2016). It was observed, however, that the majority of light absorption in aged biomass burning BrC (approximately 2 d of atmospheric transport) from wildfire emissions in northeastern Canada was associated with compounds of 500 Da and larger (Di Lorenzo and Young, 2016; Di Lorenzo et al., 2017). Altogether, this indicated that a fraction of high-molecular-weight BrC is recalcitrant to atmospheric aging processes. While there is field evidence that aqueous-phase chemistry can transform the optical properties of BrC emitted from biomass burning (Gilardoni et al., 2016; Zhang et al., 2017), the specific aging processes leading to these field observations remain unknown. In addition, these contrasting laboratory and field observational constraints on the atmospheric lifetime of BrC are much shorter than the assumed atmospheric lifetime of BrC (approximately 4 d) utilized in models to estimate its impacts on aerosol direct radiative forcing (Jo et al., 2016).
Along with constraining the atmospheric lifetime of biomass burning BrC, accurate estimates of the contribution of biomass burning to global BrC are also critical for robust assessments of its climatic impacts. Levoglucosan, an anhydrous sugar emitted during biomass pyrolysis, is a molecular tracer widely used to estimate the contribution of biomass burning to ambient organic aerosol concentrations, as it was historically thought to be chemically inert (Simoneit et al., 1999). Yet a growing number of studies have demonstrated that levoglucosan is subject to significant atmospheric loss, with an estimated atmospheric lifetime of 0.7 to 2.2 d (Hennigan et al., 2010; Hoffmann et al., 2010; Kessler et al., 2010; May et al., 2012; Slade and Knopf, 2014; Zhao et al., 2014; Sang et al., 2016). Given that the average lifetime of atmospheric aerosols with respect to deposition is considerably longer, and recent field observations have demonstrated that aged BBOA had negligible concentrations of levoglucosan or of its aerosol mass spectrometric signatures (Bougiatioti et al., 2014; Zhou et al., 2017; Theodosi et al., 2018), these results suggest that levoglucosan or its mass spectral fragments cannot be used to estimate BrC levels in aged biomass burning emissions (>1 d).
In this study, we systematically investigated the photochemical aging of the molecular-weight-separated water-soluble (WS) BrC fraction of biomass burning particles produced by wood pyrolysis, by both aqueous OH oxidation and photolysis by UVB radiation. We build upon earlier laboratory experiments that examined the aging effects of photolysis by UVA radiation (Wong et al., 2017) to establish a more complete understanding of the effects of different photochemical aging processes. Based on the photobleaching rates determined from these laboratory experiments, we estimated the dominant photochemical pathway leading to the loss of biomass burning BrC in the atmosphere. To provide additional field evidence of the impacts of atmospheric aging on biomass burning BrC, and to further assess the atmospheric stability of high-molecular-weight BrC, the light absorptivity of the molecular-weight-separated water- and methanol-soluble BrC fraction from ambient samples of different atmospheric ages was determined. Finally, from these ambient samples, we assessed the use of levoglucosan and other proposed biomass burning species as robust tracers for aged biomass burning aerosol and BrC.
2.1 Laboratory experiments
2.1.1 Preparation of WS BrC
Wood smoke BrC was generated in the laboratory using the method described in Wong et al. (2017). Briefly, a small piece of dry cherry hardwood (5–10 g), placed on the bottom of a cylindrical electronically heated combustor, was pyrolyzed under an oxygen-free atmosphere at 210 ∘C to represent BrC emitted from smoldering combustion (Andreae and Gelencsér, 2006; Chen and Bond, 2010). The resulting smoke stream was subsequently diluted with filtered air by a factor of ∼30, and the BBOA was collected on polytetrafluoroethylene (PTFE) filters (47 mm, 2 µm pore size, Pall Corporation) at 6 L min−1 for 100 min and stored at −10 ∘C. Prior to each laboratory experiment, water-soluble (WS) BrC was extracted from the filter by adding 15 mL of purified water (18.2 mΩ) to a sealed glass vial and sonicated for 60 min. The extract was filtered using a 0.2 µm PTFE syringe filter (Fisher) to remove any insoluble materials that may damage the chromatography column or plug the waveguide used to characterize BrC properties (Sect. 2.1.4). Given that the WS BrC is dissolved in bulk aqueous solutions, the experimental conditions for the photochemical aging experiments, such as BrC concentrations and viscosity, most likely represent the aging of WS wood smoke BrC in fog and/or cloud droplets. For these laboratory studies, we only focused on the aging of WS BrC, as results from our previous work indicated that the majority of the light absorption of laboratory-generated BrC from wood smoke was contributed by the WS fraction (∼77 %) and that the trends in the evolution of light absorption of water-insoluble (i.e., methanol extracted) BrC due to photochemical aging are similar to those of the WS fraction (Wong et al., 2017).
2.1.2 Photolysis of WS BrC by UVB lights
Experiments examining the photolytic aging of WS BrC by UVB lights follow the same experimental procedure for UVA photolysis as described in Wong et al. (2017). All photochemical aging experiments were conducted in a photoreactor, with a slowly rotating vial rack (40 rpm) placed in the center and surrounded by 16 UVB lamps (Desert Series 50 T8, Zilla). With all UV lamps on, continuous ventilation by two fans maintained the temperature inside the photoreactor at 30±1 ∘C. The photon fluxes inside the photoreactor from UVB lamps were determined by chemical actinometry using 2-nitrobenzaldehyde (see Wong et al., 2017, for experimental details). The wavelength-dependent photon fluxes from both UVA and UVB lamps are shown in Fig. S1 in the Supplement, where the actinic flux at solar noon is provided for comparison. Most of the radiation fell in the range of 300–400 nm, with a maximum at 355 and 310 nm for UVA and UVB lamps, respectively. Note that while both UVB and UVA lamps have comparably similar photon fluxes for wavelengths lower than 320 nm, the UVA lamps have a correspondingly much higher photon flux at higher wavelengths. These spectral differences allowed for investigating the wavelength dependence of BrC aging by photolysis.
For each photolysis experiment, multiple pre-cleaned 2 mL borosilicate glass vials (sealed with Teflon-lined caps), each containing 1mL of the WS BrC extract solution, were placed on the rotating vial rack inside the photoreactor. Each extract solution was diluted by 5 % (with purified water) such that the final concentration of WS BrC used in the photolysis experiments is identical to that used in the aqueous OH oxidation experiments, where the WS BrC concentration was diluted by 5 % from the addition of H2O2 (Sect. 2.1.3). The vials were illuminated by UVB lights for up to approximately 100 h, where at different illumination times, one vial was removed for offline analysis (described in Sect. 2.1.4). Control experiments were conducted; no changes in WS BrC properties were observed when the vials containing the extract were completely covered by aluminium foil, where they were only exposed to the elevated temperatures inside the photoreactor and not UVB radiation. Photolysis experiments were repeated at least 3 times to ensure reproducibility.
2.1.3 Aqueous OH oxidation of WS BrC
Experiments for aqueous OH oxidation were conducted in the same experimental setup used in the photolysis experiments. H2O2 (30 %, Sigma Aldrich) was added to the WS BrC extract solution (final concentration of 1.5 mM) as a photolytic source of the OH radical upon irradiation with UVB lights (up to 18 h). Dark control experiments were conducted to confirm that the dark reaction of H2O2 with WS BrC did not change its optical or molecular-weight properties. Similar to photolysis experiments, at different illumination times, one vial was removed for offline analysis. The OH oxidation experiments were repeated 3 times to ensure reproducibility.
The steady-state OH concentration ([OH]ss) in these photochemical oxidation experiments was determined in order to estimate the atmospheric lifetime of WS BrC with respect to aqueous OH oxidation. This was performed by monitoring the formation of para-hydroxybenzoic acid (p-HBA) from the reaction of OH with benzoate, a commonly employed OH scavenger (Zhou and Mopper, 1990; Anastasio and McGregor, 2001; Badali et al., 2015). The OH quantification experiments are described in detail in Sect. S1. Briefly, sodium benzoate (0.1–1.0 µM) was added to WS wood smoke BrC extract solution with and without 1.5 mM H2O2. The resulting solutions were illuminated for up to 18 h, where at different times, a sample vial was removed to determine the concentration of p-HBA, using HPLC-UV/VIS absorption. The yield of p-HBA from the reaction of OH with benzoate (0.17) was used to convert the formation rate of p-HBA to an OH production rate from which the [OH]ss was estimated. p-HBA does not absorb radiation in the same wavelength regions as BrC, but given that other products formed from the reaction of OH + benzoate do, in order to avoid measurement interference from these products, the OH quantification experiments were conducted separately from the experiments where the effects of OH oxidation on BrC properties were examined. In these experiments, the relationship between OH photo-production and benzoate concentration was determined in order to quantify [OH]ss in the aqueous OH oxidation experiments where the BrC properties were monitored (i.e., no benzoate as an added OH scavenger). Figure S2 shows the relationship between the p-HBA formation rate and concentration of added benzoate, which was used to estimate the [OH]ss when [benzoate] = 0 (i.e., experiments where light absorptivity of BrC was monitored).
2.1.4 Offline WS BrC measurements
Following the removal of each sample vial from the photoreactor, the BrC solutions were divided to determine various BrC chemical and optical properties using the same procedures outlined in Wong et al. (2017). Briefly, changes in the water-soluble organic carbon (WSOC) concentration due to photochemical aging were monitored using a Sievers Total Organic Carbon (TOC) Analyzer (Model 900, GE Analytical Instruments). For these measurements, the WS BrC samples were diluted by a factor of 250 to ensure that the WSOC concentrations were in the linear range of the instrument, which was routinely calibrated using solutions of dissolved sucrose of known concentrations. Prior to photochemical aging, each sample vial contained 1400±178 ppb of WSOC. The light absorptivity of WS BrC of all molecular weights (i.e., bulk WS BrC) was monitored using the 250× diluted WS BrC solutions and an absorption spectrometer consisting of a liquid waveguide capillary (2.5 m optical path length, World Precision), a deuterium tungsten halogen light source (DT-Mini, Ocean Optics), and a light detector (USB4000, Ocean Optics) that can continuously monitor all wavelengths between 230 and 800 nm. The molecular-weight distributions of WS BrC were determined using size-exclusion chromatography (SEC), which separates analyte molecules due to differences in the extent of permeation into the column packing material, where larger molecules elute earlier than smaller molecules due to weaker interactions (Strigel et al., 2009). The technique was operated using high-performance liquid chromatography (HPLC; GP40 Dionex), equipped with an SEC column (Polysep GFC P-3000, Phenomenex) that was operated in isocratic mode using a 90:10 v∕v mixture of water and methanol with 25 mM ammonium acetate as the mobile phase at 1 mL min−1. The near-UV/VIS absorbance of the molecular-weight-separated BrC compounds was monitored using an absorbance spectrometer that was coupled in line with the SEC system. The spectrometer consisted of the same components as the one used for bulk WS BrC measurements, except a liquid waveguide capillary with a 1 m optical path length (World Precision) and a different model of the deuterium tungsten halogen light source (DT-Mini-2B, Ocean Optics) were used. This SEC approach was routinely calibrated using standards of known molecular weights from which the calibration methodology and the relationship between molecular weights and elution volumes were previously described (Wong et al., 2017). We note that the molecular weights reported using this SEC approach are only approximate, as the accuracy of the molecular-weight calibration depends on whether the molecular densities of calibration standards are representative of those of WS BrC molecules, which are currently unknown. Additionally, hydrophobic interactions of the sample molecules with the SEC column may affect the elution times (e.g., hydrophobic species elute at a later time compared to hydrophilic species of similar molecular weights; Sarah A. Styler, personal communication, 28 February 2019). The absorbance of the different molecular-weight fractions was determined by integrating the absorbance of a specific wavelength over the period of elution that corresponds to the molecular-weight fraction (PMW,λ). Since the coupling of the chromatographic technique to UV/VIS absorption measurements leads to the dilution of the BrC sample due to the use of the mobile phase, the absorbance of the molecular-weight-separated BrC (ABrC,λ) is related to absorbance of the injected BrC by accounting for the mobile phase flow rate (f) and the volume of the injected BrC sample (VBrC) using Eq. (1):
The measured light absorption for both bulk WS BrC were normalized by the WSOC concentration of the BrC extract to represent the light absorption per water-soluble organic carbon or the mass absorption coefficient (MAC) of the WS BrC (the calculation method described in Wong et al., 2017).
2.2 Field observations in Crete
2.2.1 Sampling site and identification of biomass burning events
Filter samples containing ambient BrC from biomass burning emissions were collected during the fire season (July to October) of 2016 and 2017 at the Heraklion station in Crete, Greece (Fig. 1). During the fire season, persistent northerly winds (the Etesians) move air masses across the Aegean Sea, where no further contribution from fire emissions can occur, transporting biomass burning emissions from continental eastern Europe towards the sampling site. The fire season in 2017 was more intense compared to 2016, due to extended droughts and high temperatures.
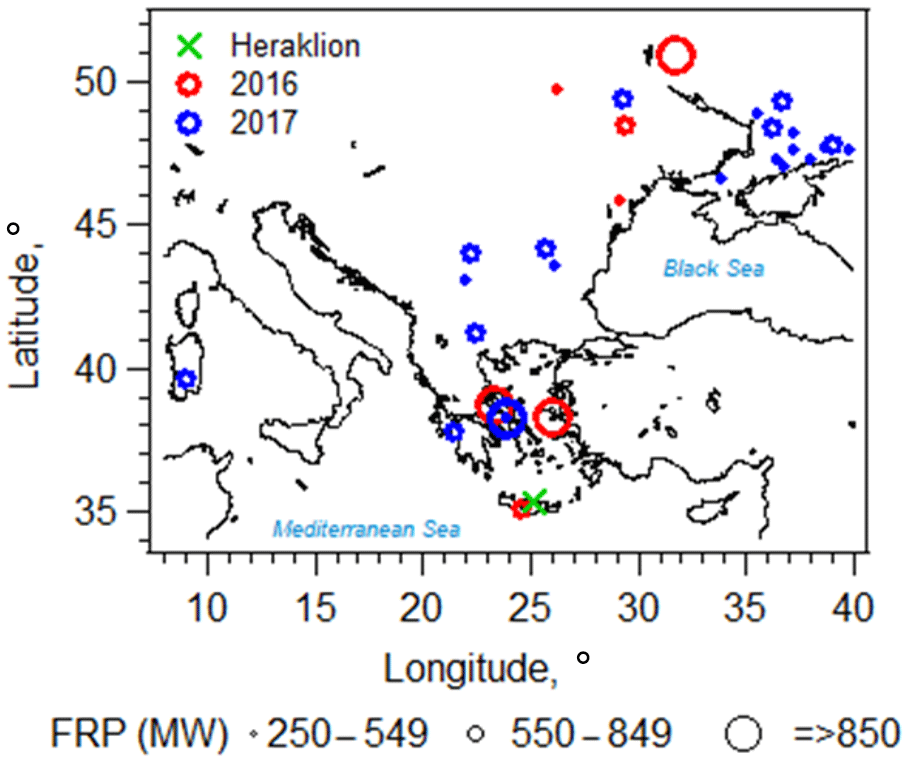
Figure 1Locations of biomass burning events relevant to the current study, as detected by the Moderate Resolution Imaging Spectroradiometer (MODIS) and the Fire Information for Resource Management System (FIRES; https://firms.modaps.eosdis.nasa.gov/map/, last access: 26 June 2018) during the fire seasons in 2016 (red circles) and 2017 (blue circles). The corresponding MODIS-measured fire radiative power (FRP; in megawatts) is represented by the size of the circle markers and is provided as a rough proxy of biomass burning emission rate (Wooster, 2002). Wildfires with FRP less than 100 MW are not shown here. The location of the sampling site (Heraklion, Crete, Greece) is indicated by the green cross.
Information on location and date of fire events, along with fire radiative power, was obtained from the Fire Information for Resource Management System (FIRES; https://firms.modaps.eosdis.nasa.gov/map/, last access: 26 June 2018), which was detected by the Moderate Resolution Imaging Spectroradiometer (MODIS). Fire radiative power (in megawatts) was used as a rough proxy for the biomass burning emission rate (Wooster, 2002). Fires occurring 3 d prior to and within each filter sampling period, with fire radiative power over 100 megawatts, were included in the analysis. Air mass back trajectories were computed via HYSPLIT (Stein et al., 2015), using archived Global Data Assimilation System (GDAS) meteorology from which the vertical velocity was determined. New trajectories were computed every hour of each filter sampling interval (i.e., 22–24 h) for a total run time of 72 h. The information from FIRES and back trajectories was used together to identify filters samples that were influenced by fire events (i.e., intersection of back trajectories with the fire locations) and to estimate the corresponding atmospheric transport time from the fire location to the sampling site (example is shown in Fig. S3). This analysis approach was chosen to explore the stability of biomass burning tracers; however, it does not account for variability in BrC emissions from various fires. Out of the 65 field filter samples collected in the 2016 and 2017 fire seasons (method discussed below), 24 biomass burning filter samples of various atmospheric transport times were identified. We focus our following analysis on these identified biomass burning events, shown in Fig. 1. Note that several fires occurring near Athens and on the island of Chios were burn events that persisted for multiple days.
2.2.2 Filter collection, extraction, and analysis
Ambient BrC in PM2.5 were collected on prebaked 8 in×10 in quartz filters (2500QAT-UP, Pall) using a high-volume (Hi-Vol) sampler (TISCH) for 22–24 h at a flow rate of 1.4 and 2 m3 min−1 for 2016 and 2017, respectively. Immediately after collection, the filter samples were wrapped in prebaked aluminium foil and stored at −10 ∘C until analysis. In addition, multiple field blanks were collected during both fire seasons. Each quartz filter sample was divided into portions for the determination of various chemical properties. A 1.5 cm2 punch of the filter sample was analyzed for organic carbon (OC) and elemental carbon using an OCEC Analyzer (Sunset Laboratory Inc.) using the NIOSH Method. Another 1.5 cm2 filter punch was used to determine water-soluble components after extraction by sonication. This included analysis of carbohydrates (levoglucosan, mannosan, galactosan, glucose, mannose, and galactose) using high-performance anion exchange chromatography with pulsed amperometric detection (HPAEC-PAD; ICS-3000, Dionex), which is described in Fourtzio et al. (2017). Using a separate ion chromatographic system (Dionex), analysis of anions (Cl−, Br−, , , and ; CS12A column with CERS 500 suppressor) and cations (K+, Na+, Ca2+, Mg2+, and ; AS4A-SC column with AERS 500 suppressor) was also conducted. Potassium associated with biomass burning (i.e., non-sea-salt K+) was determined from the total potassium minus potassium associated with sea salt. This was calculated using the measured Na+ concentrations and a standard seawater mass ratio of K+ to Na+ of 0.0359 (Seinfeld and Pandis, 1998).
Two 1.5 cm2 punches were placed in a pre-cleaned 2 mL borosilicate glass vial, where either 1 mL of purified water (18.2 mΩ) or methanol (MeOH; HPLC grade, Merck) was added and sonicated for 1 h to extract either the WS- or MeOH-soluble (i.e., water-soluble and water-insoluble) BrC, respectively. Note that the extractions of MeOH BrC and WS BrC were done on separate sections of the same filter. Each extract was then filtered using a new 0.2 µm PTFE syringe filter (Fisher). For WS BrC, an aliquot of the filtered extract was used to determine the water-soluble organic carbon (WSOC) concentration using the TOC instrument discussed in Sect. 2.1.4. For both WS and MeOH BrC filtered extracts, aliquots of the solutions were used to determine the molecular-weight distributions of BrC using the HPLC-SEC-UV/VIS absorption technique discussed in Sect. 2.1.4. Additionally, an aliquot of both filtered solutions was used to measure the bulk light absorption properties (i.e., not molecular-weight-separated) of WS and MeOH BrC, using the absorption spectrometer described in Sect. 2.1.4. The MAC values of the WS- and MeOH-soluble BrC were determined through normalizing by the WSOC concentration (WS BrC and WSOC) or OC concentrations (MeOH BrC and OC), as determined using the TOC and OCEC analyzers, respectively. Note that for both TOC and bulk UV/VIS absorption measurements, the filtered extracts were diluted by a factor of 20 to ensure that the measured properties were in the linear response range of the instruments. The field filter blanks were analyzed using the same methodology as the BBOA filters, and all measurements were blank subtracted.
3.1 Laboratory experiments on WS BrC
3.1.1 Bulk WS BrC
Upon illumination by UVB lights, losses in WSOC were observed for both direct UVB photolysis and UVB + H2O2 experiments (Fig. 2a), with the majority of this loss occurring following 20 h of illumination. The effects of aqueous OH oxidation is taken to be the difference between the UVB and UVB + H2O2 experiments, assuming that the effects of direct UVB photolysis are identical in both types of experiments (i.e., the addition of H2O2 did not significantly alter the rate of direct photolysis). Here, aqueous OH oxidation did not lead to additional loss of WSOC, indicating that the loss of WSOC is only due to direct UVB photolysis.
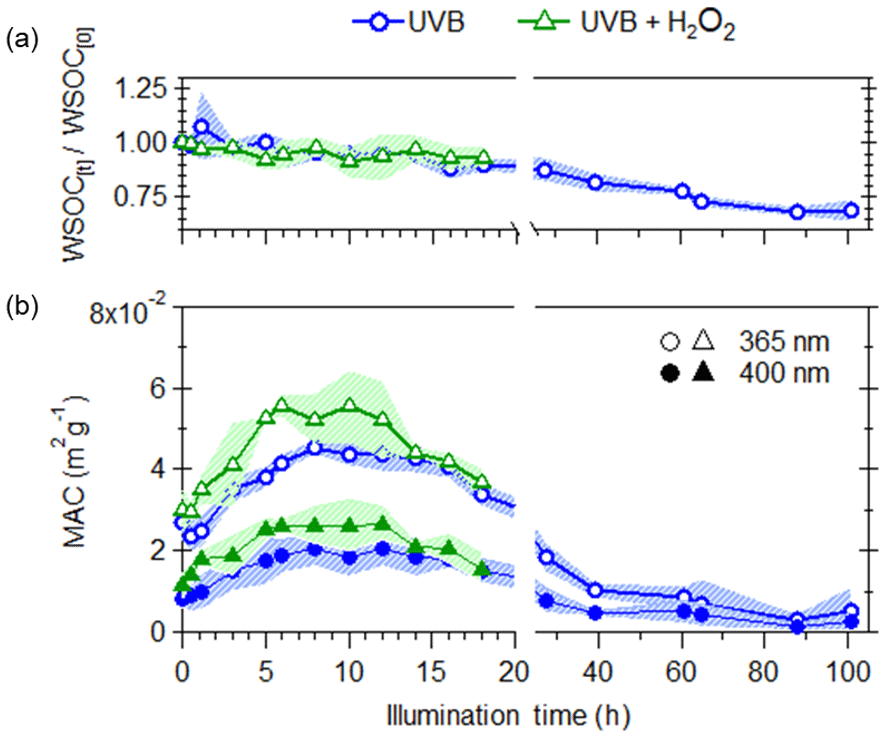
Figure 2Effects of UVB photolysis (blue circles) and aqueous OH oxidation (green triangles) on (a) changes in WSOC (b) WSOC mass normalized absorption coefficient (MAC) at 365 nm (open markers) and 400 nm (filled markers). The shaded areas represent the variability (±1σ) in multiple experiments (n=3).
The effects of photochemical aging on the light absorption per water-soluble organic carbon (mass absorption coefficient, MAC) of WS BrC are shown on Fig. 2b. The calculation method for the MAC at 365 and 400 nm is discussed previously in Wong et al. (2017). Note that these MAC values arise from light absorption measurements of water-extracted BrC and not from suspended BrC particles. The initial increase in MAC values was observed due to photochemical aging, suggesting that the WS BrC undergoes photoenhancement, leading to increased absorptivity of radiation at 365 and 400 nm, where the increase in MAC values at 365 nm was more significant compared to that at 400 nm. Given that a loss in WSOC was observed during this photoenhancement period, the increased MAC values may be driven by a loss in non-absorbing WSOC and/or the formation of more absorbing WS BrC. This initial increase in MAC values is likely driven by the formation of more absorbing WS BrC, as MAC values increased by a factor of ∼2, while WSOC decreased only by a factor of less than 1.1. During this initial period of photochemical aging (up to 10 h), the changes in WSOC and MAC365 are similar for UVB and UVB + H2O2 experiments, suggesting that the aging by UVB led to the observed photoenhancement. Following this period of initial photoenhancement (up to 12 h), photobleaching (i.e., decrease in MAC values) of WS BrC was observed. Here, a steeper slope for the decrease in MAC values was observed for the UVB + H2O2 compared to the UVB experiments, indicating that aqueous OH oxidation leads to enhanced decay in light absorptivity and that while BrC is susceptible to both degradation due to UVB photolysis and OH oxidation, certain BrC chromophores are more reactive towards OH radicals. Previous studies that have examined the photochemical aging of model biomass burning aromatic compounds (Gelencsér et al., 2003; Chang and Thompson, 2010; Ofner et al., 2011; Zhao et al., 2015; Smith et al., 2016; Hems and Abbatt, 2018), surrogate mixtures of biomass burning BrC (Schnitzler and Abbatt, 2018), and BrC emitted from the pyrolysis of various types of biomass (Zhao et al., 2015; Wong et al., 2017; Sumlin et al., 2017) have similarly observed initial photoenhancement, followed by photobleaching. It has been proposed that polymerization and/or functionalization of BrC leads to photoenhancement, while fragmentation results in photobleaching.
3.1.2 WS BrC separated by molecular weight
Molecular-weight-separated BrC measurements by the SEC provide additional insight into the reactivity of specific classes of WS BrC molecules leading to the observed bulk photoenhancement and photobleaching. This is illustrated in Figs. 3 and 4, where the Abs365 measurements were binned according to elution volumes where the high-molecular-weight fraction (high MW) is defined as the sum of light absorptivity (Abs365) for molecules with approximate molecular weights between 66 K Da and 401 Da (i.e., elution volumes between 8 and 15 mL) and a low-molecular-weight fraction (low MW) as the sum of light absorptivity (Abs365) for molecules with approximate molecular weights of 400 Da and less (i.e., elution volumes higher than 15 mL). Prior to photochemical aging, fresh WS BrC consisted of both high- and low-MW chromophores (Fig. 3a); however, the contribution of these two fractions to light absorption changes when photochemically aged. Figure 4 shows that both molecular-weight fractions exhibit dynamic changes in their light absorptivity due to photochemical aging, but to a different extent. High-MW WS BrC undergoes substantial initial photoenhancement, followed by photobleaching. Low-MW WS BrC initially decayed within the first hour; then there was an increase in light absorptivity between 1 and 5 h of illumination time, followed by another period of decreasing light absorptivity. The two separate phases of decreasing light absorptivity suggest that low-MW WS BrC contains chromophores of different photoreactivity. While the photobleaching of both molecular-weight fractions appeared largely to follow pseudo-first-order kinetics, the photoenhancement of high-MW WS BrC exhibited non-first-order behavior. This may be due to the presence of chromophores with different reactivity or because after the initial period of photoenhancement, further aging does not lead to further increases in light absorptivity. For simplicity, we treat this period of photoenhancement as pseudo-first-order. In order to isolate the effects of OH oxidation on light absorptivity, we follow the approach used by Zhao et al. (2015), where the pseudo-first-order OH oxidation rate constants () of photoenhancement and photobleaching were calculated by taking the difference between the observed first-order growth and decay rate constants due to UVB photolysis () and UVB + H2O2 photolysis experiments (), using Eq. (2). The second-order OH oxidation rate constant ( can be calculated from Eq. (3):
We note that for Eq. (2), [OH]ss represents the steady-state concentration of OH radicals due to the photolysis of 1.5 mM of H2O2 (i.e., difference in [OH]ss between UVB photolysis and UVB + H2O2 photolysis experiments). Additionally, for the high-MW BrC, the photoenhancement and photobleaching rate constants were determined by fitting first-order curves to the first 3 h and between 8 and 18 h of absorption data at 365 nm (Fig. S4a). For the low-MW WS BrC, to determine the decay rate constants of rapidly and slowly photobleached chromophores, a first-order decay curve was fitted to 0 to 1 h and 8 to 18 h of absorption data at 365 nm, respectively (Fig. S4b).
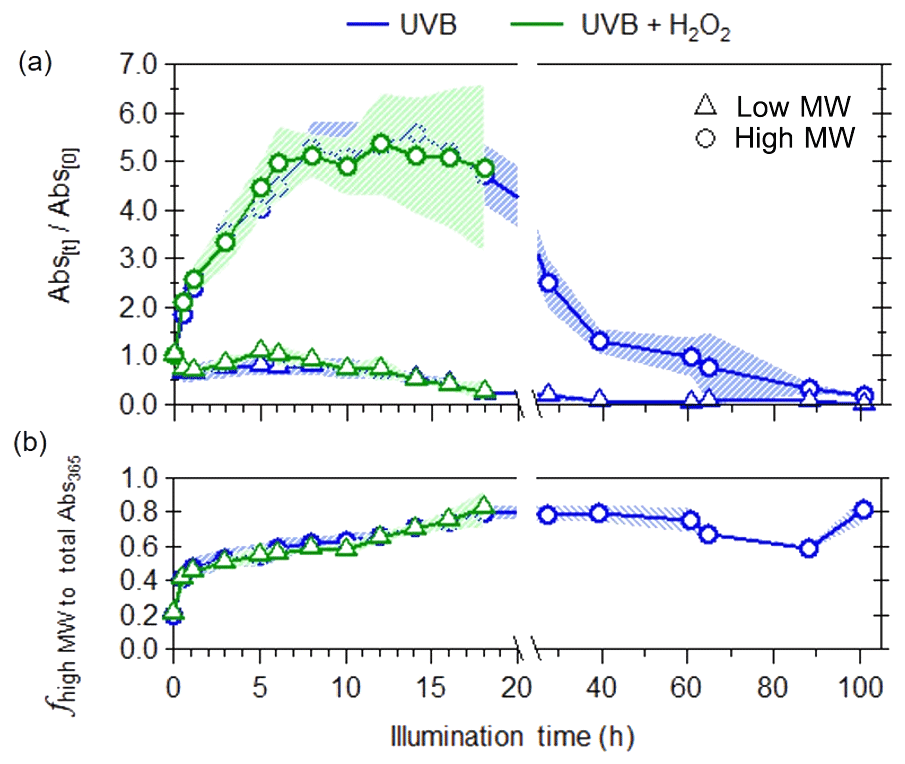
Figure 3Changes in (a) light absorption at a wavelength of 365 nm for low-MW (triangles) and high-MW (circles) BrC fractions and (b) contribution of high-MW fraction to total light absorbance at 365 nm in WS smoke BrC due to UVB photolysis (blue) and aqueous OH oxidation (green). The shaded areas represent the variability (±1σ) in multiple experiments (n=3).
The resulting observed rate constants for photoenhancement and photobleaching due to aqueous OH oxidation and UVB photolysis are shown in Table 1. Here, the corresponding rate constants due to UVA photolysis, which were determined by our previous work (Wong et al., 2017), are included for comparison. Considering the reaction rate constants for low-MW WS BrC, its initial photoenhancement was only observed in the presence of UVA radiation (i.e., enhanced photon flux at wavelengths above 310 nm), indicating that reactions leading to the increased light absorptivity are wavelength dependent, whereas photochemical aging of low-MW WS BrC by UVB photolysis and aqueous OH oxidation only leads to photobleaching. As mentioned previously, low-MW WS BrC of different reactivities with respect to photobleaching by aqueous OH oxidation and UVB photolysis was observed: chromophores that were rapidly photobleached due to exposure to UVB lights and chromophores that were slowly photobleached by both aqueous OH oxidation and UVB radiation. We note that the second-order rate constant for the decay in light absorptivity due to the OH reaction with slowly photobleached low-MW WS chromophores determined in this study [ M−1 s−1] is comparable to the range of the concentration-based rate constants for the OH reaction with three different nitrophenols ([(3.7–5.0)×109 M−1 s−1], as reported by Hems and Abbatt, 2018). Given that nitrophenols, a class of WS BrC that has been detected in BBOA in significant concentrations (Mohr et al., 2013; Lin et al., 2016), have molecular weights that are approximately less than 200 Da, it is reasonable that they govern the OH reactivity of low-MW WS BrC (i.e., ≤400 Da) observed in this study.
Table 1Rate constants for the photoenhancement and photobleaching of low- and high-MW WS BrC of wood smoke, with respect to photolysis (UVB and UVA) and OH oxidation. Note that for UVB photolysis and OH oxidation, the reported uncertainties represent the variability (±1σ) in multiple experiments (n=3) and that the rate constants for low-MW1 and low-MW2 BrC photobleaching correspond to chromophores that were rapidly and slowly photobleached, respectively. Rate constants for UVA photolysis were previously reported in Wong et al. (2017).

a High-MW BrC is defined as the sum of light absorptivity (Abs365) for molecules with approximate molecular weights between 66 kDa and 401 Da (i.e., SEC elution volumes between 8 and 15 mL), and low-molecular-weight fraction (low MW) is defined as the sum of light absorptivity for molecules with approximate molecular weights of 400 Da and less (i.e., SEC elution volumes higher than 15 mL).
For high-MW WS BrC, no differences in the evolution of their light absorption were observed between the two different photochemical aging experiments (Fig. 4a). This suggests that their initial photoenhancement (up to 15 h) and subsequent photobleaching were only due to exposure to UVB radiation. Given that the photoenhancement and photobleaching rates for high-MW WS chromophores due to UVB and UVA exposure (Table 1) are similar, and that both UVB and UVA lamps have similar photon fluxes for wavelengths lower than 310 nm, our results suggest that most of the photochemical aging was initiated by UVB radiation. Owing to its rapid photoenhancement and slow photobleaching, the contribution of high-MW WS BrC to total light absorptivity increases throughout photochemical aging, from 20 % when the WS BrC was freshly emitted up to 80 % after 100 h of UV exposure (Fig. 4b). Given that the integrated UVB photon flux in these laboratory experiments is roughly 92 % of the sun at solar noon (i.e., 1 h of UVB exposure in laboratory experiments is equivalent to 0.92 h in the atmosphere), these results further support earlier field observations where the light absorptivity of aged (up to 40 h) ambient biomass burning WS BrC was attributed to molecules larger than 500 Da (Di Lorenzo and Young, 2016; Di Lorenzo et al., 2017).
Changes in the absorption Ångström exponent (AAE) throughout UVB photolysis and OH oxidation were also determined from linear regression fits to log Abs vs. log λ (Fig. S5) in the wavelength ranges of 320–500 nm for both high and low-MW fractions of WS BrC molecules and 320–420 nm for high-MW BrC. Changes in AAE values that were determined from different wavelength ranges can provide insight into the effects of photochemical aging on the light absorption spectral properties of BrC in addition to monitoring the MAC values at different wavelengths. Shown in Fig. 5, prior to any photochemical aging, different AAE values for low-MW (10.0±0.4) and high-MW (6.4±0.7) WS BrC were observed, indicating that the low-MW WS BrC has a much stronger spectral dependence than that of high-MW WS BrC. We speculate that the lower AAE values of high-MW WS BrC are due to the highly conjugated nature of these molecules, as previous observations by Hopkins et al. (2007) have indicated that biomass burning BrC with lower AAE values has a higher extent of carbon sp2 hybridization compared to those with higher AAE values. As the low- and high-MW WS BrC was photochemically aged, changes in its AAE values were observed. For low-MW WS BrC, AAE values decreased due to decreased light absorptivity at lower wavelengths. For high-MW WS BrC, comparison of the temporal evolution of the AAE values determined in both wavelength ranges (Fig. 5b and c) shows that during initial photoenhancement (up to 15 h), a slight decrease in the AAE was only observed from 320–420 nm, suggesting that photoenhancement reactions, such as functionalization and polymerization, enhance the absorptivity of light of wavelengths only up to 500 nm. Following this period of initial photoenhancement, increasing AAE values were observed as the high-MW WS BrC was photobleached, where the increase in AAE values for 320–500 nm was more rapid compared to 320–420 nm, indicating a blue shift in the absorption spectra of high-MW WS BrC due to decreasing absorption in the 420 to 500 nm range. Collectively, the differences in evolution of AAE values further support that the reactivity and aging mechanisms of BrC are dependent on their molecular weight.

Figure 4Evolution of AAE due to UVB photolysis (blue) and aqueous OH oxidation (green) for (a) low-molecular-weight BrC (triangles), (b) high-molecular-weight BrC (circles) from 320–500 nm, and (c) high-molecular-weight BrC from 320–420 nm. The shaded areas represent the variability (±1σ) in multiple experiments (n=3).
3.1.3 Atmospheric fate of WS BrC from biomass burning
These experimentally determined rate constants indicate that ambient WS BrC is transformed within a day in the atmosphere (Table S1; calculation method discussed in Sect. S2), and considering that the average atmospheric lifetime of particles with respect to deposition is approximately 1 week, our laboratory results indicate that photochemical aging has an important effect on the optical properties of BBOA. For low-MW WS BrC, laboratory results suggest that there are two groups with different reactivities. A highly photolabile fraction of low-MW WS BrC is photobleached in the atmosphere, with UVB photolysis being their dominant atmospheric fate, with an estimated atmospheric lifetime of 0.7±0.2 h. A second, less reactive fraction of low-MW WS BrC has an estimated atmospheric lifetime of 9.8±1.6 h, with aqueous OH oxidation representing its dominant atmospheric loss mechanism. However, this less reactive low-MW WS BrC does not significantly contribute to total light absorptivity, as its decay only increased the fractional contribution of high MW to total light absorptivity for WS BrC by 0.2 (Fig. 4b). Conversely, for high-MW WS BrC, following its initial photoenhancement, where color formation occurs over a timescale of a few hours, this BrC is photobleached by UVB photolysis, with an atmospheric lifetime of 11±2.3 h.
While these estimated atmospheric lifetimes suggest that the majority of BrC is photobleached in the atmosphere, it is important to note that for high-MW WS BrC, the rate of decreasing light absorptivity slows down with time, where after 100 h of photochemical aging in the laboratory, approximately 20 % of the initial light absorptivity remained (Fig. 4a). These observations suggest that a fraction of high-MW WS BrC has an atmospheric lifetime longer than 11 h and is more persistent. It is critical to note that these atmospheric lifetimes were estimated from laboratory-generated BrC from one biomass fuel type, under a specific burn condition. In addition, the estimated atmospheric lifetimes were calculated assuming continuous exposure to solar radiation with an actinic flux corresponding to that at solar noon and [OH]ss of M, which represent the daily peak solar photon flux and the upper range of OH concentrations in cloud droplets (Herrmann et al., 2010; Arakaki et al., 2013), thus likely representing the lower range of BrC atmospheric lifetimes. We also stress that there are uncertainties in these estimates, as they assume that the photolysis quantum yield of BrC is wavelength independent. Also, estimates of [OH]ss in the condensed phase, which includes aqueous particles, cloud, and fog droplets, range over several orders of magnitude (Arakaki et al., 2013; Ervens, 2015). Under lower levels of [OH]ss, which better represent the oxidant concentrations of aerosol particles, photolysis by UVB becomes the dominant atmospheric loss mechanism for all molecular-weight fractions of BrC.
3.2 Field observations
3.2.1 Bulk- and molecular-weight-separated WS and MeOH BrC
Investigating the impacts of atmospheric aging on biomass burning BrC in the field and comparison to laboratory observations represent the other primary goals of this study. Both WS and MeOH (i.e., water-soluble and insoluble) BrC were analyzed for the field samples to assess the atmospheric evolution of BrC from ambient BBOA. Evolution of various properties of WS and MeOH BrC as a function of atmospheric transport time is shown in Fig. 6, where all individuals measurements (filled points), along with binned (every 5 h) median values (open points), are presented. We first discuss the bulk measurements (Fig. 6a–d).
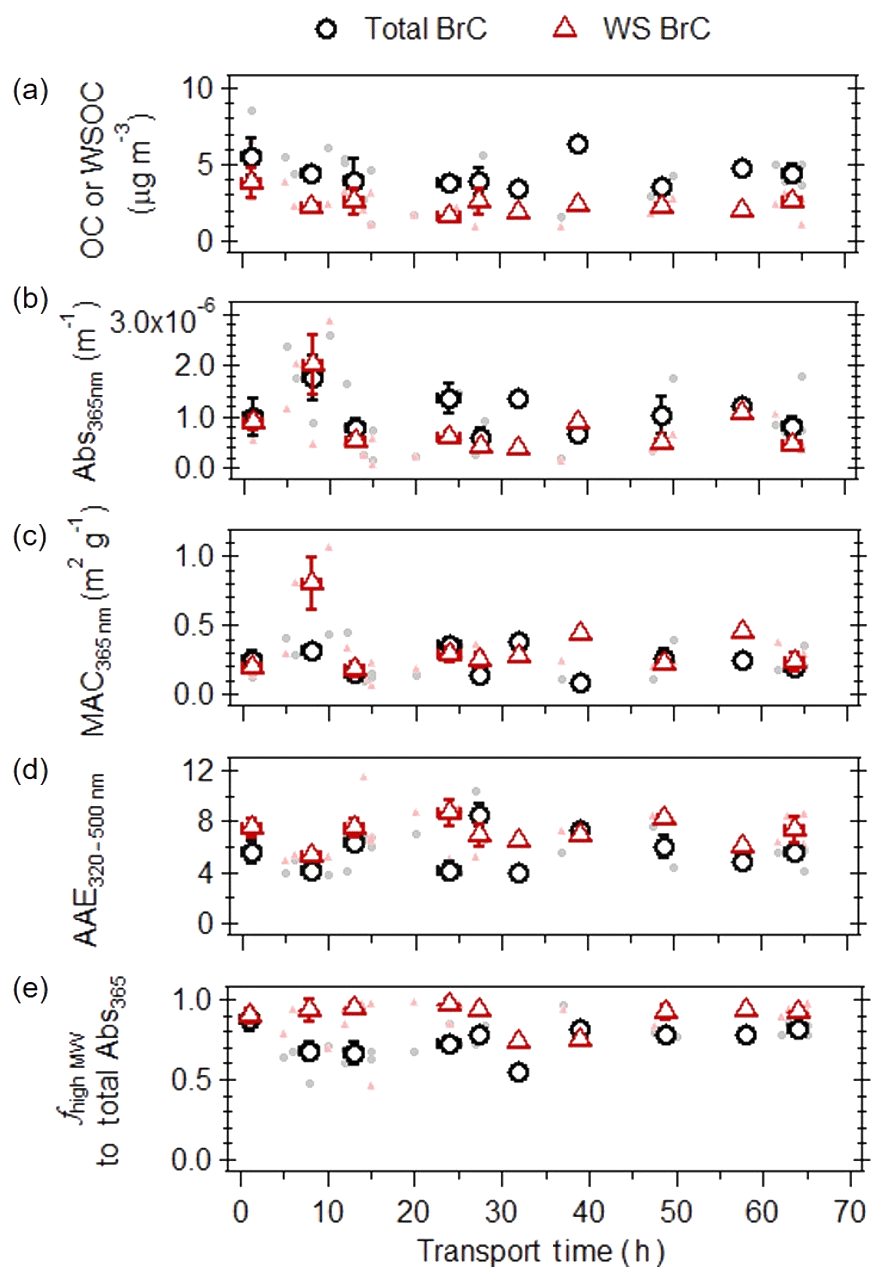
Figure 5(a) Organic carbon (OC) or water-soluble organic carbon (WSOC) concentrations, (b) light absorption at 365 nm, (c) mass absorption coefficients at 365 nm, (d) AAE (in the wavelength range of 320–500 nm), and (e) fractional contribution of high-molecular-weight fractions to total light absorption as a function of atmospheric transport time, for MeOH (black circles) and water (red triangles) extractable portion of the ambient filters collected on the island of Crete, Greece, during the 2016 and 2017 fire seasons. The filled points are individual filter measurements, the open points represent the binned median values, and the associated error bars represent the interquartile range.
Considering field samples with atmospheric transport times up to 10 h, an increase in bulk (i.e., not molecular-weight-separated) Abs365 nm (Fig. 6b) was observed for both MeOH and WS BrC, while a significant increase in MAC365 values (Fig. 6c) was only observed for the water-soluble portion. This is due to the more significant loss of WSOC compared to OC (Fig. 6a), suggesting that the non-absorbing water-soluble compounds in biomass burning are more rapidly lost in the atmosphere due to aging. For field samples with atmospheric transport times longer than 10 h, lower values of WSOC, OC and Abs365 values for MeOH and WS BrC were observed, suggesting that atmospheric aging processes led to a decay in the mass and light absorptivity of biomass burning BrC. These ambient observations of bulk WS BrC corroborate our earlier laboratory results, suggesting that atmospheric photochemical aging processes increase MAC values for WS BrC, at least in fresher biomass burning plumes, followed by photobleaching with further atmospheric aging. Despite the considerable scatter, the field data indicate that the light absorptivity of bulk MeOH BrC decreased at a slower rate compared to bulk WS BrC (see Fig. S6 for exponential decay). From these exponential decays in bulk MeOH and WS ambient BrC, the atmospheric lifetime of bulk MeOH and WS BrC is estimated to be between 15–28 h. We note that within the variability in all ambient observations, the MAC values for MeOH BrC did not change significantly. Lastly, from these bulk measurements, AAE values (Fig. 6d) did not change significantly throughout atmospheric transport, suggesting that aging processes do not greatly affect the wavelength dependence of BrC light absorption.
Molecular-weight-separated BrC measurements from the corresponding field samples (Fig. 6e) indicate that even for fresh biomass burning emissions (i.e., 1 h of atmospheric transport time), high-MW MeOH and WS BrC dominated the total light absorption at 365 nm. This is also highlighted in Fig. 3b, which shows the molecular-weight-separated absorption spectra of fresh ambient biomass burning WS BrC. Given that it remains unclear to which extent fuel type and burn conditions affect the molecular-weight distribution of BrC chromophores, the observed low contribution of low-MW BrC to total light absorptivity may either be due to their rapid photochemical removal in the atmosphere, as demonstrated from our laboratory experiments, or its low emission rate. In addition, the average contribution of high-MW chromophores to total BrC light absorptivity is lower compared to WS BrC, suggesting that some water-insoluble BrC is low-MW compounds.
Given that the atmospheric lifetime of ambient BrC from bulk measurements was estimated to be between 15–19 h, and that high-MW chromophores contributed on average 75–87 % of total light absorptivity for MeOH and WS ambient BrC, the atmospheric lifetime of ambient high-MW MeOH and WS BrC is likely to be at least 15–28 h as well, which is longer than the estimates from laboratory results for the WS BrC ( h, Table S1). We speculate that the difference in atmospheric lifetime may be due to several reasons. Firstly, the laboratory-constrained atmospheric lifetime represents BrC emitted from the combustion of one biomass fuel type under smoldering conditions, which may not represent ambient fire conditions, as the light absorption properties of BrC have been observed to be dependent on field and burn conditions (Chen and Bond, 2010). Secondly, the laboratory-constrained atmospheric lifetime represents high-MW WS BrC that was cloud processed, which may not apply for the ambient samples. Thirdly, high-MW WS BrC was continuously exposed to UV radiation in the laboratory, where this does not represent the diurnal cycle of solar radiation that ambient BrC is exposed to. This is particularly important for high-MW WS BrC emitted throughout the night, where its atmospheric lifetime will be longer relative to that emitted during the day, during which the high-MW WS BrC is photobleached. Fourthly, the lifetime obtained from laboratory results was based on the aging of BrC dissolved in bulk solutions, where parameters that may affect the reactivity of ambient BrC in suspended particles, such as the aerosol phase state, solute concentrations, and viscosity, were not accounted for. Similarly, ambient aerosols containing biomass burning BrC are likely to be more chemically complex than those studied in the laboratory (e.g., complex emissions from the combustion of multiple types of biomass), where the presence of other organic compounds that are more reactive may prolong the atmospheric lifetime of ambient high-MW BrC. Lastly, there may be uncertainties associated with the estimated atmospheric transport times for field samples, either due to unaccounted contributions from (a) fires occurring more than 3 d prior to filter collection (i.e., HYSPLIT runs times of 72 h were used), (b) small fires with fire radiative power less than 100 MW, which were not included in the current analysis, or (c) large fires that are not detected by MODIS due to interference effects by thick smoke (Schroeder et al., 2008).
3.2.2 Chemical tracers for aged BrC from biomass burning
Previous studies have suggested the limitations of using levoglucosan and non-sea-salt potassium (nss K+) as chemical tracers for biomass burning, due to levoglucosan's short atmospheric lifetime (e.g., Hennigan et al., 2010; May et al., 2012) and emission of nss K+ by non-biomass burning sources (Urban et al., 2012). More recently, Scaramboni et al. (2015) suggested the use of total hydrous sugars (e.g., glucose) as an alternative tracer for biomass burning, since the six-membered ring of the hydrated sugar molecule is potentially more stable than the five-membered ring of levoglucosan. While biological aerosols, such as microbes, are also a source of hydrated sugars (Graham et al., 2002) and may interfere with the signal from biomass burning, they are still useful for examining correlations of these types of sugars with BrC under known periods of biomass burning influence – especially given that high-MW BrC from biomass burning appears to be a relatively stable aerosol component, such that it can still be observed after significant periods of aging.
Focusing on the ambient filter samples impacted by biomass burning with estimated atmospheric transport times over 10 h, we compare correlations of levoglucosan, nss K+, and total hydrous sugars (defined as the sum of glucose, mannose, and galactose mass concentration) to BrC to assess how these compounds compare to BrC as a biomass burning tracer for aged BBOA. Figure 7 shows their correlations to MeOH and WS BrC light absorption at 365 nm. The detailed results of these regression analyses are provided in Table S2. No correlation is seen between levoglucosan and aged MeOH and WS BrC (r2≤0.01), suggesting that levoglucosan is neither likely to be representative of absorbing organic components nor serves as an effective tracer of aged biomass burning. Given that we did not account for the fire conditions in this analysis, the lack of correlation can also arise due to varying relative emission ratios of BrC to levoglucosan, since it has been previously observed that the relative emission ratios of OA to levoglucosan and BrC to OA are not constant across burning conditions (i.e., smoldering vs. flaming; Mazzoleni et al., 2007; Kalogridis et al., 2018). In comparison, both WS and total BrC correlated moderately with nss K+ (), consistent with previous field measurements of BBOA from the boreal forest (Di Lorenzo et al., 2018) and Amazon rainforest (Fuzzi et al., 2007). The moderate correlations may potentially be due to differences in the dependence of potassium emissions and BrC light absorptivity on fire conditions (Chen and Bond, 2010; Lee et al., 2010). Additionally, contributions of nss K+ from other non-biomass burning sources, such as crustal material, could also diminish the correlation with BrC if BrC is more specific to biomass burning than nss K+. Moderate to strong correlations between light absorptivity of (MeOH and WS) BrC and total hydrous sugars were observed (), suggesting that both classes of compounds may serve as a robust tracer for aged biomass burning emissions.

Figure 6Correlations of light absorption at 365 nm for the MeOH (black circles) and water-soluble (red triangles) extractable portion of ambient filter samples that were impacted by biomass burning to biomass burning tracers: (a) levoglucosan, (b) nss K+, and (c) total hydrous sugars. For nss K+, outliers (circled points) were excluded from the linear regression analysis.
In this work, the effects of atmospheric aging on the light absorptivity of molecular-weight-separated BrC were demonstrated in both controlled laboratory experiments and ambient observations. The experimental conditions of the laboratory work focused on the aging of WS BrC most likely represent cloud processing of BBOA. The ambient data included analysis of both WS and MeOH BrC. In the laboratory experiments, photochemical aging processes led to significant changes in light absorptivity and molecular-weight distributions of BrC, where the reactivity of WS BrC was observed to be dependent on molecular weight. We found that low-MW WS BrC undergoes rapid photobleaching in the atmosphere on timescales of a few hours, whereas high-MW WS BrC likely persists in the atmosphere for up to a few days. These laboratory results bridge contrasting results from previous laboratory and field observations which demonstrated that low-MW WS BrC, such as nitrophenols, was rapidly photobleached within several hours, while high-MW WS BrC is more stable. Ambient BrC was largely composed of WS BrC species, and for both WS and MeOH BrC, their light absorptivity was dominated by high-MW BrC, consistent with our laboratory results. Ambient BrC was observed to undergo initial photoenhancement and total light absorption dominated by high-MW BrC at 365 nm, even for fresh (∼1 h of atmospheric transport) biomass burning emissions, further supporting that low-MW BrC is a short-lived component in atmospheric BBOA. Additionally, observations of initial photoenhancement due to atmospheric aging in both laboratory and field data support earlier ambient observations that secondary production of BrC from biomass burning emissions can be an important source of light-absorbing aerosol (Gilardoni et al., 2016), but only near fire emissions. From the observed decay of bulk WS light absorptivity, we estimate that ambient WS BrC have atmospheric lifetimes of approximately 15–28 h. This range of the atmospheric lifetime likely corresponds to that of high-MW BrC, as it contributed to over 75 % of total light absorptivity of ambient BrC of ages up to 68 h. It is important to note that while this estimated atmospheric lifetime likely represents a majority of high-MW BrC, the slope of the light-absorptivity decay curve (Fig. S6) decreased with atmospheric age, indicating that a fraction of the high-MW BrC is persistent in the atmosphere for at least up to 68 h. Collectively, this field-constrained lifetime of high-MW WS BrC is much larger than that obtained from laboratory results, which may be due to differences in the assumed versus ambient solar photon fluxes and oxidant concentrations. In addition, aging processes not investigated in the laboratory study, such as dilution, are also unlikely to result in more rapid removal of high-MW BrC from the atmosphere. Although the volatility of high-MW BrC compounds remains uncharacterized, given their large molecular weights and solubility in water, we expect them to exhibit very low volatility, and they are unlikely to be lost from biomass burning particles via volatilization during dilution with background air masses, which is consistent with the observation that the majority of wood smoke BrC is associated with extremely low-volatility organic compounds (Saleh et al., 2014).
Given that the average lifetime of particles in the atmosphere is approximately 1 week with respect to deposition, the estimated atmospheric lifetime of high-MW BrC continues to support earlier observations that it can be a persistent component in atmospheric BBOA (Di Lorenzo and Young, 2016; Wong et al., 2017; Di Lorenzo et al., 2017) and therefore has a larger impact on aerosol direct radiative forcing compared to low-MW BrC. The stability of high-MW BrC also suggests that it may be ubiquitous in the atmosphere and potentially undergo intercontinental transport. In addition, the estimated atmospheric lifetimes of ambient BrC from this study (15–28 h) are consistent with those from previous field studies of wildfires in the northwestern USA (τ: 13–22 h; Forrister et al., 2015) and in the Amazon (τ: 22–45 h; Wang et al., 2016). A recent modeling study indicated that incorporating a 1 d photobleaching e-folding time, which was constrained from these previous field studies, improved modeled-versus-observed BrC absorption and decreased the estimated positive direct radiative effect of organic aerosols (Wang et al., 2018). Further field data from different geographical regions are necessary for assessing the estimated 1 d BrC atmospheric lifetime and improving predictions of global BrC impacts.
Accurate predictions of the direct radiative forcing also require robust estimates of the contribution of biomass burning to global BrC. Our field observations suggest that total hydrous sugars and BrC may be more robust biomass burning markers compared to levoglucosan and nss K+. Other potential sources of hydrous sugars should also be assessed to better constrain their use as tracers for biomass burning.
From our field observations, the fractional contribution of high-MW MeOH and WS BrC to total (all molecular weights) light absorptivity remained relatively constant for up to 68 h of atmospheric aging, suggesting that this class of compounds may also provide an alternative marker for aged biomass burning emissions. However, it remains unclear whether fuel type and burn conditions influence the emission, chemical composition, and reactivity of high-MW BrC. For example, while the absorptivity of MeOH and WS BrC generated from wood smoke has been shown to be dependent on pyrolysis temperature and wood types (Chen and Bond, 2010), it remains unclear whether this is due to differences in the emission of low or high-MW BrC. Characterization of high-MW BrC generated from other commonly burned biomass, such as agriculture crop residues and biofuels used for residential heating under representative combustion and fuel conditions (dry or wet), is warranted.
SEC data can be requested from the corresponding author. All other data are uploaded and available at https://doi.pangaea.de/10.1594/PANGAEA.896731 (Wong et al., 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7319-2019-supplement.
JPSW, AN, and RJW designed the laboratory experiment. JPSW performed laboratory experiments and analyzed the data. NM, KV, MK, JS, and AN designed and organized the field campaign. MT and IT collected the field samples and conducted the OC–EC and ion chromatography measurements. JS carried out the carbohydrate analysis. JPSW conducted and analyzed light absorption and WSOC measurements for field samples. JPSW wrote the paper, with contributions from AN and RJW. All authors commented on the final paper.
The authors declare that they have no conflict of interest.
This work was supported by the Electric Power Research Institute (EPRI) through contract no. 00-10003806, by NASA through contract NNX14A974G, and by the project PyroTRACH (ERC-2016-COG) funded from H2020-EU.1.1. – Excellent Science – European Research Council (ERC), project ID 726165.
This paper was edited by Manvendra K. Dubey and reviewed by two anonymous referees.
Alexander, D. T. L., Crozier, P. A., and Anderson, J. R.: Brown Carbon Spheres in East Asian Outflow and Their Optical Properties, Science, 321, 833–836, https://doi.org/10.1126/science.1155296, 2008.
Anastasio, C. and McGregor, K. G.: Chemistry of fog waters in California's Central Valley: 1. In situ photoformation of hydroxyl radical and singlet molecular oxygen, Atmos. Environ., 35, 1079–1089, https://doi.org/10.1016/S1352-2310(00)00281-8, 2001.
Andreae, M. O. and Gelencsér, A.: Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols, Atmos. Chem. Phys., 6, 3131–3148, https://doi.org/10.5194/acp-6-3131-2006, 2006.
Arakaki, T., Anastasio, C., Kuroki, Y., Nakajima, H., Okada, K., Kotani, Y., Handa, D., Azechi, S., Kimura, T., Tsuhako, A., and Miyagi, Y.: A General Scavenging Rate Constant for Reaction of Hydroxyl Radical with Organic Carbon in Atmospheric Waters, Environ. Sci. Technol., 47, 8196–8203, https://doi.org/10.1021/es401927b, 2013.
Badali, K. M., Zhou, S., Aljawhary, D., Antiñolo, M., Chen, W. J., Lok, A., Mungall, E., Wong, J. P. S., Zhao, R., and Abbatt, J. P. D.: Formation of hydroxyl radicals from photolysis of secondary organic aerosol material, Atmos. Chem. Phys., 15, 7831–7840, https://doi.org/10.5194/acp-15-7831-2015, 2015.
Bond, T. C.: Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion, Geophys. Res. Lett., 28, 4075–4078, https://doi.org/10.1029/2001GL013652, 2001.
Bougiatioti, A., Stavroulas, I., Kostenidou, E., Zarmpas, P., Theodosi, C., Kouvarakis, G., Canonaco, F., Prévôt, A. S. H., Nenes, A., Pandis, S. N., and Mihalopoulos, N.: Processing of biomass-burning aerosol in the eastern Mediterranean during summertime, Atmos. Chem. Phys., 14, 4793–4807, https://doi.org/10.5194/acp-14-4793-2014, 2014.
Chang, J. L. and Thompson, J. E.: Characterization of colored products formed during irradiation of aqueous solutions containing H2O2 and phenolic compounds, Atmos. Environ., 44, 541–551, https://doi.org/10.1016/j.atmosenv.2009.10.042, 2010.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Desyaterik, Y., Sun, Y., Shen, X., Lee, T., Wang, X., Wang, T., and Collett, J. L.: Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China, J. Geophys. Res.-Atmos., 118, 7389–7399, https://doi.org/10.1002/jgrd.50561, 2013.
Di Lorenzo, R. A. and Young, C. J.: Size separation method for absorption characterization in brown carbon: Application to an aged biomass burning sample, Geophys. Res. Lett., 43, 2015GL066954, https://doi.org/10.1002/2015GL066954, 2016.
Di Lorenzo, R. A., Washenfelder, R. A., Attwood, A. R., Guo, H., Xu, L., Ng, N. L., Weber, R. J., Baumann, K., Edgerton, E., and Young, C. J.: Molecular-Size-Separated Brown Carbon Absorption for Biomass-Burning Aerosol at Multiple Field Sites, Environ. Sci. Technol., 51, 3128–3137, https://doi.org/10.1021/acs.est.6b06160, 2017.
Di Lorenzo, R. A., Place, B. K., VandenBoer, T. C., and Young, C. J.: Composition of Size-Resolved Aged Boreal Fire Aerosols: Brown Carbon, Biomass Burning Tracers, and Reduced Nitrogen, ACS Earth Space Chem., 2, 278–285, https://doi.org/10.1021/acsearthspacechem.7b00137, 2018.
Dinar, E., Riziq, A. A., Spindler, C., Erlick, C., Kiss, G., and Rudich, Y.: The complex refractive index of atmospheric and model humic-like substances (HULIS) retrieved by a cavity ring down aerosol spectrometer (CRD-AS), Faraday Discuss., 137, 279–295, https://doi.org/10.1039/B703111D, 2008.
Ervens, B.: Modeling the Processing of Aerosol and Trace Gases in Clouds and Fogs, Chem. Rev., 115, 4157–4198, https://doi.org/10.1021/cr5005887, 2015.
Fan, X., Wei, S., Zhu, M., Song, J., and Peng, P.: Comprehensive characterization of humic-like substances in smoke PM2.5 emitted from the combustion of biomass materials and fossil fuels, Atmos. Chem. Phys., 16, 13321–13340, https://doi.org/10.5194/acp-16-13321-2016, 2016.
Feng, Y., Ramanathan, V., and Kotamarthi, V. R.: Brown carbon: a significant atmospheric absorber of solar radiation?, Atmos. Chem. Phys., 13, 8607–8621, https://doi.org/10.5194/acp-13-8607-2013, 2013.
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes: Brown Carbon in Biomass Burning Plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015GL063897, 2015.
Fourtziou, L., Liakakou, E., Stavroulas, I., Theodosi, C., Zarmpas, P., Psiloglou, B., Sciare, J., Maggos, T., Bairachtari, K., Bougiatioti, A., Gerasopoulos, E., Sarda-Estève, R., Bonnaire, N., and Mihalopoulos, N.: Multi-tracer approach to characterize domestic wood burning in Athens (Greece) during wintertime, Atmos. Environ., 148, 89–101, https://doi.org/10.1016/j.atmosenv.2016.10.011, 2017.
Fuzzi, S., Decesari, S., Facchini, M. C., Cavalli, F., Emblico, L., Mircea, M., Andreae, M. O., Trebs, I., Hoffer, A., Guyon, P., Artaxo, P., Rizzo, L. V., Lara, L. L., Pauliquevis, T., Maenhaut, W., Raes, N., Chi, X., Mayol-Bracero, O. L., Soto-García, L. L., Claeys, M., Kourtchev, I., Rissler, J., Swietlicki, E., Tagliavini, E., Schkolnik, G., Falkovich, A. H., Rudich, Y., Fisch, G., and Gatti, L. V.: Overview of the inorganic and organic composition of size-segregated aerosol in Rondônia, Brazil, from the biomass-burning period to the onset of the wet season, J. Geophys. Res.-Atmos., 112, D01201, https://doi.org/10.1029/2005JD006741, 2007.
Gelencsér, A., Hoffer, A., Kiss, G., Tombácz, E., Kurdi, R., and Bencze, L.: In-situ Formation of Light-Absorbing Organic Matter in Cloud Water, J. Atmos. Chem., 45, 25–33, https://doi.org/10.1023/A:1024060428172, 2003.
Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Graham, B., Mayol-Bracero, O. L., Guyon, P., Roberts, G. C., Decesari, S., Facchini, M. C., Artaxo, P., Maenhaut, W., Köll, P., and Andreae, M. O.: Water-soluble organic compounds in biomass burning aerosols over Amazonia 1. Characterization by NMR and GC-MS, J. Geophys. Res.-Atmos., 107, LBA14-1–LBA14-16, https://doi.org/10.1029/2001JD000336, 2002.
He, S. and Carmichael, G. R.: Sensitivity of photolysis rates and ozone production in the troposphere to aerosol properties, J. Geophys. Res.-Atmos., 104, 26307–26324, https://doi.org/10.1029/1999JD900789, 1999.
Hecobian, A., Zhang, X., Zheng, M., Frank, N., Edgerton, E. S., and Weber, R. J.: Water-Soluble Organic Aerosol material and the light-absorption characteristics of aqueous extracts measured over the Southeastern United States, Atmos. Chem. Phys., 10, 5965–5977, https://doi.org/10.5194/acp-10-5965-2010, 2010.
Hems, R. F. and Abbatt, J. P. D.: Aqueous Phase Photo-oxidation of Brown Carbon Nitrophenols: Reaction Kinetics, Mechanism, and Evolution of Light Absorption, ACS Earth Space Chem., 2, 225–234, https://doi.org/10.1021/acsearthspacechem.7b00123, 2018.
Hennigan, C. J., Sullivan, A. P., Collett, J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010.
Herrmann, H., Hoffmann, D., Schaefer, T., Bräuer, P., and Tilgner, A.: Tropospheric Aqueous-Phase Free-Radical Chemistry: Radical Sources, Spectra, Reaction Kinetics and Prediction Tools, Chem. Phys. Chem., 11, 3796–3822, https://doi.org/10.1002/cphc.201000533, 2010.
Hoffer, A., Gelencsér, A., Guyon, P., Kiss, G., Schmid, O., Frank, G. P., Artaxo, P., and Andreae, M. O.: Optical properties of humic-like substances (HULIS) in biomass-burning aerosols, Atmos. Chem. Phys., 6, 3563–3570, https://doi.org/10.5194/acp-6-3563-2006, 2006.
Hoffmann, D., Tilgner, A., Iinuma, Y., and Herrmann, H.: Atmospheric Stability of Levoglucosan: A Detailed Laboratory and Modeling Study, Environ. Sci. Technol., 44, 694–699, https://doi.org/10.1021/es902476f, 2010.
Hopkins, R. J., Lewis, K., Desyaterik, Y., Wang, Z., Tivanski, A. V., Arnott, W. P., Laskin, A., and Gilles, M. K.: Correlations between optical, chemical and physical properties of biomass burn aerosols, Geophys. Res. Lett., 34, L18806, https://doi.org/10.1029/2007GL030502, 2007.
Jo, D. S., Park, R. J., Lee, S., Kim, S.-W., and Zhang, X.: A global simulation of brown carbon: implications for photochemistry and direct radiative effect, Atmos. Chem. Phys., 16, 3413–3432, https://doi.org/10.5194/acp-16-3413-2016, 2016.
Kalogridis, A.-C., Popovicheva, O. B., Engling, G., Diapouli, E., Kawamura, K., Tachibana, E., Ono, K., Kozlov, V. S., and Eleftheriadis, K.: Smoke aerosol chemistry and aging of Siberian biomass burning emissions in a large aerosol chamber, Atmos. Environ., 185, 15–28, https://doi.org/10.1016/j.atmosenv.2018.04.033, 2018.
Kessler, S. H., Smith, J. D., Che, D. L., Worsnop, D. R., Wilson, K. R., and Kroll, J. H.: Chemical Sinks of Organic Aerosol: Kinetics and Products of the Heterogeneous Oxidation of Erythritol and Levoglucosan, Environ. Sci. Technol., 44, 7005–7010, https://doi.org/10.1021/es101465m, 2010.
Kirchstetter, T. W. and Thatcher, T. L.: Contribution of organic carbon to wood smoke particulate matter absorption of solar radiation, Atmos. Chem. Phys., 12, 6067–6072, https://doi.org/10.5194/acp-12-6067-2012, 2012.
Lack, D. A., Langridge, J. M., Bahreini, R., Cappa, C. D., Middlebrook, A. M., and Schwarz, J. P.: Brown carbon and internal mixing in biomass burning particles, P. Natl. Acad. Sci. USA, 109, 14802–14807, https://doi.org/10.1073/pnas.1206575109, 2012.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of Atmospheric Brown Carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lee, T., Sullivan, A. P., Mack, L., Jimenez, J. L., Kreidenweis, S. M., Onasch, T. B., Worsnop, D. R., Malm, W., Wold, C. E., Hao, W. M., and Collett Jr., J. L.: Chemical Smoke Marker Emissions During Flaming and Smoldering Phases of Laboratory Open Burning of Wildland Fuels, Aerosol Sci. Tech., 44, i–v, https://doi.org/10.1080/02786826.2010.499884, 2010.
Lin, G., Penner, J. E., Flanner, M. G., Sillman, S., Xu, L., and Zhou, C.: Radiative forcing of organic aerosol in the atmosphere and on snow: Effects of SOA and brown carbon, J. Geophys. Res.-Atmos., 119, 2013JD021186, https://doi.org/10.1002/2013JD021186, 2014.
Lin, P., Aiona, P. K., Li, Y., Shiraiwa, M., Laskin, J., Nizkorodov, S. A., and Laskin, A.: Molecular Characterization of Brown Carbon in Biomass Burning Aerosol Particles, Environ. Sci. Technol., 50, 11815–11824, https://doi.org/10.1021/acs.est.6b03024, 2016.
May, A. A., Saleh, R., Hennigan, C. J., Donahue, N. M., and Robinson, A. L.: Volatility of Organic Molecular Markers Used for Source Apportionment Analysis: Measurements and Implications for Atmospheric Lifetime, Environ. Sci. Technol., 46, 12435–12444, https://doi.org/10.1021/es302276t, 2012.
Mazzoleni, L. R., Zielinska, B., and Moosmüller, H.: Emissions of Levoglucosan, Methoxy Phenols, and Organic Acids from Prescribed Burns, Laboratory Combustion of Wildland Fuels, and Residential Wood Combustion, Environ. Sci. Technol., 41, 2115–2122, https://doi.org/10.1021/es061702c, 2007.
Mohr, C., Lopez-Hilfiker, F. D., Zotter, P., Prévôt, A. S. H., Xu, L., Ng, N. L., Herndon, S. C., Williams, L. R., Franklin, J. P., Zahniser, M. S., Worsnop, D. R., Knighton, W. B., Aiken, A. C., Gorkowski, K. J., Dubey, M. K., Allan, J. D., and Thornton, J. A.: Contribution of Nitrated Phenols to Wood Burning Brown Carbon Light Absorption in Detling, United Kingdom during Winter Time, Environ. Sci. Technol., 47, 6316–6324, https://doi.org/10.1021/es400683v, 2013.
Mok, J., Krotkov, N. A., Arola, A., Torres, O., Jethva, H., Andrade, M., Labow, G., Eck, T. F., Li, Z., Dickerson, R. R., Stenchikov, G. L., Osipov, S., and Ren, X.: Impacts of brown carbon from biomass burning on surface UV and ozone photochemistry in the Amazon Basin, Sci. Rep., 6, 36940, https://doi.org/10.1038/srep36940, 2016.
Ofner, J., Krüger, H.-U., Grothe, H., Schmitt-Kopplin, P., Whitmore, K., and Zetzsch, C.: Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS, Atmos. Chem. Phys., 11, 1–15, https://doi.org/10.5194/acp-11-1-2011, 2011.
Park, R. J., Kim, M. J., Jeong, J. I., Youn, D., and Kim, S.: A contribution of brown carbon aerosol to the aerosol light absorption and its radiative forcing in East Asia, Atmos. Environ., 44, 1414–1421, https://doi.org/10.1016/j.atmosenv.2010.01.042, 2010.
Saleh, R., Hennigan, C. J., McMeeking, G. R., Chuang, W. K., Robinson, E. S., Coe, H., Donahue, N. M., and Robinson, A. L.: Absorptivity of brown carbon in fresh and photo-chemically aged biomass-burning emissions, Atmos. Chem. Phys., 13, 7683–7693, https://doi.org/10.5194/acp-13-7683-2013, 2013.
Saleh, R., Robinson, E. S., Tkacik, D. S., Ahern, A. T., Liu, S., Aiken, A. C., Sullivan, R. C., Presto, A. A., Dubey, M. K., Yokelson, R. J., Donahue, N. M., and Robinson, A. L.: Brownness of organics in aerosols from biomass burning linked to their black carbon content, Nat. Geosci., 7, 647–650, https://doi.org/10.1038/ngeo2220, 2014.
Saleh, R., Marks, M., Heo, J., Adams, P. J., Donahue, N. M., and Robinson, A. L.: Contribution of brown carbon and lensing to the direct radiative effect of carbonaceous aerosols from biomass and biofuel burning emissions, J. Geophys. Res.-Atmos., 120, 2015JD023697, https://doi.org/10.1002/2015JD023697, 2015.
Sang, X. F., Gensch, I., Kammer, B., Khan, A., Kleist, E., Laumer, W., Schlag, P., Schmitt, S. H., Wildt, J., Zhao, R., Mungall, E. L., Abbatt, J. P. D., and Kiendler-Scharr, A.: Chemical stability of levoglucosan: An isotopic perspective, Geophys. Res. Lett., 43, 2016GL069179, https://doi.org/10.1002/2016GL069179, 2016.
Scaramboni, C., Urban, R. C., Lima-Souza, M., Nogueira, R. F. P., Cardoso, A. A., Allen, A. G., and Campos, M. L. A. M.: Total sugars in atmospheric aerosols: An alternative tracer for biomass burning, Atmos. Environ., 100, 185–192, https://doi.org/10.1016/j.atmosenv.2014.11.003, 2015.
Schnitzler, E. G. and Abbatt, J. P. D.: Heterogeneous OH oxidation of secondary brown carbon aerosol, Atmos. Chem. Phys., 18, 14539–14553, https://doi.org/10.5194/acp-18-14539, 2018.
Schroeder, W., Prins, E., Giglio, L., Csiszar, I., Schmidt, C., Morisette, J., and Morton, D.: Validation of GOES and MODIS active fire detection products using ASTER and ETM+ data, Remote Sens. Environ., 112, 2711–2726, https://doi.org/10.1016/j.rse.2008.01.005, 2008.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, Inc., New York, 1998.
Simoneit, B. R. T., Schauer, J. J., Nolte, C. G., Oros, D. R., Elias, V. O., Fraser, M. P., Rogge, W. F., and Cass, G. R.: Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles, Atmos. Environ., 33, 173–182, https://doi.org/10.1016/S1352-2310(98)00145-9, 1999.
Slade, J. H. and Knopf, D. A.: Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity, Geophys. Res. Lett., 41, 5297–5306, https://doi.org/10.1002/2014GL060582, 2014.
Smith, J. D., Kinney, H., and Anastasio, C.: Phenolic carbonyls undergo rapid aqueous photodegradation to form low-volatility, light-absorbing products, Atmos. Environ., 126, 36–44, https://doi.org/10.1016/j.atmosenv.2015.11.035, 2016.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: NOAA's HYSPLIT Atmospheric Transport and Dispersion Modeling System, B. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/BAMS-D-14-00110.1, 2015.
Strigel, A. M., Yau, W. W., Kirkland, J. J., and Bly, D. D.: Modern Size-Exclusion Liquid Chromatography, Second Edition, John Wiley & Sons, Inc., 2009.
Sumlin, B. J., Pandey, A., Walker, M. J., Pattison, R. S., Williams, B. J., and Chakrabarty, R. K.: Atmospheric Photooxidation Diminishes Light Absorption by Primary Brown Carbon Aerosol from Biomass Burning, Environ. Sci. Technol. Lett., 4, 540–545, https://doi.org/10.1021/acs.estlett.7b00393, 2017.
Theodosi, C., Panagiotopoulos, C., Nouara, A., Zarmpas, P., Nicolaou, P., Violaki, K., Kanakidou, M., Sempéré, R., and Mihalopoulos, N.: Sugars in atmospheric aerosols over the Eastern Mediterranean, Prog. Oceanogr., 163, 70–81, https://doi.org/10.1016/j.pocean.2017.09.001, 2018.
Urban, R. C., Lima-Souza, M., Caetano-Silva, L., Queiroz, M. E. C., Nogueira, R. F. P., Allen, A. G., Cardoso, A. A., Held, G., and Campos, M. L. A. M.: Use of levoglucosan, potassium, and water-soluble organic carbon to characterize the origins of biomass-burning aerosols, Atmos. Environ., 61, 562–569, https://doi.org/10.1016/j.atmosenv.2012.07.082, 2012.
Wang, X., Heald, C. L., Ridley, D. A., Schwarz, J. P., Spackman, J. R., Perring, A. E., Coe, H., Liu, D., and Clarke, A. D.: Exploiting simultaneous observational constraints on mass and absorption to estimate the global direct radiative forcing of black carbon and brown carbon, Atmos. Chem. Phys., 14, 10989–11010, https://doi.org/10.5194/acp-14-10989-2014, 2014.
Wang, X., Heald, C. L., Sedlacek, A. J., de Sá, S. S., Martin, S. T., Alexander, M. L., Watson, T. B., Aiken, A. C., Springston, S. R., and Artaxo, P.: Deriving brown carbon from multiwavelength absorption measurements: method and application to AERONET and Aethalometer observations, Atmos. Chem. Phys., 16, 12733–12752, https://doi.org/10.5194/acp-16-12733-2016, 2016.
Wang, X., Heald, C. L., Liu, J., Weber, R. J., Campuzano-Jost, P., Jimenez, J. L., Schwarz, J. P., and Perring, A. E.: Exploring the observational constraints on the simulation of brown carbon, Atmos. Chem. Phys., 18, 635–653, https://doi.org/10.5194/acp-18-635-2018, 2018.
Wang, Y., Hu, M., Lin, P., Guo, Q., Wu, Z., Li, M., Zeng, L., Song, Y., Zeng, L., Wu, Y., Guo, S., Huang, X., and He, L.: Molecular Characterization of Nitrogen-Containing Organic Compounds in Humic-like Substances Emitted from Straw Residue Burning, Environ. Sci. Technol., 51, 5951–5961, https://doi.org/10.1021/acs.est.7b00248, 2017.
Wong, J. P. S., Nenes, A., and Weber, R. J.: Changes in Light Absorptivity of Molecular Weight Separated Brown Carbon Due to Photolytic Aging, Environ. Sci. Technol., 51, 8414–8421, https://doi.org/10.1021/acs.est.7b01739, 2017.
Wong, J. P. S., Tsagaraki, M., Tsiodra, I., Mihalopoulos, N., Violaki, K., Kanakidou, M., Sciare, J., Nenes, A., and Weber, R. J. Atmospheric evolution of molecular weight separated brown carbon from biomass burning, PANGAEA, https://doi.org/10.1594/PANGAEA.896731, 2018.
Wooster, M. J.: Small-scale experimental testing of fire radiative energy for quantifying mass combusted in natural vegetation fires, Geophys. Res. Lett., 29, 2027, https://doi.org/10.1029/2002GL015487, 2002.
Zhang, X., Lin, Y.-H., Surratt, J. D., Zotter, P., Prévôt, A. S. H., and Weber, R. J.: Light-absorbing soluble organic aerosol in Los Angeles and Atlanta: A contrast in secondary organic aerosol, Geophys. Res. Lett., 38, L21810, https://doi.org/10.1029/2011GL049385, 2011.
Zhang, X., Lin, Y.-H., Surratt, J. D., and Weber, R. J.: Sources, Composition and Absorption Ångström Exponent of Light-absorbing Organic Components in Aerosol Extracts from the Los Angeles Basin, Environ. Sci. Technol., 47, 3685–3693, https://doi.org/10.1021/es305047b, 2013.
Zhang, Y., Forrister, H., Liu, J., Dibb, J., Anderson, B., Schwarz, J. P., Perring, A. E., Jimenez, J. L., Campuzano-Jost, P., Wang, Y., Nenes, A., and Weber, R. J.: Top-of-atmosphere radiative forcing affected by brown carbon in the upper troposphere, Nat. Geosci., 10, 486–489, https://doi.org/10.1038/ngeo2960, 2017.
Zhao, R., Mungall, E. L., Lee, A. K. Y., Aljawhary, D., and Abbatt, J. P. D.: Aqueous-phase photooxidation of levoglucosan – a mechanistic study using aerosol time-of-flight chemical ionization mass spectrometry (Aerosol ToF-CIMS), Atmos. Chem. Phys., 14, 9695–9706, https://doi.org/10.5194/acp-14-9695-2014, 2014.
Zhao, R., Lee, A. K. Y., Huang, L., Li, X., Yang, F., and Abbatt, J. P. D.: Photochemical processing of aqueous atmospheric brown carbon, Atmos. Chem. Phys., 15, 6087–6100, https://doi.org/10.5194/acp-15-6087-2015, 2015.
Zhong, M. and Jang, M.: Dynamic light absorption of biomass-burning organic carbon photochemically aged under natural sunlight, Atmos. Chem. Phys., 14, 1517–1525, https://doi.org/10.5194/acp-14-1517-2014, 2014.
Zhou, S., Collier, S., Jaffe, D. A., Briggs, N. L., Hee, J., Sedlacek III, A. J., Kleinman, L., Onasch, T. B., and Zhang, Q.: Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 17, 2477–2493, https://doi.org/10.5194/acp-17-2477-2017, 2017.
Zhou, X. and Mopper, K.: Determination of photochemically produced hydroxyl radicals in seawater and freshwater, Mar. Chem., 30, 71–88, https://doi.org/10.1016/0304-4203(90)90062-H, 1990.