the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 May 2019
| 29 May 2019
Implication of strongly increased atmospheric methane concentrations for chemistry–climate connections
Franziska Winterstein
Fabian Tanalski
Patrick Jöckel
Martin Dameris
Michael Ponater
Methane (CH4) is the second-most important directly emitted greenhouse gas, the atmospheric concentration of which is influenced by human activities. In this study, numerical simulations with the chemistry–climate model (CCM) EMAC are performed, aiming to assess possible consequences of significantly enhanced CH4 concentrations in the Earth's atmosphere for the climate.
We analyse experiments with 2×CH4 and 5×CH4 present-day (2010) mixing ratio and its quasi-instantaneous chemical impact on the atmosphere. The massive increase in CH4 strongly influences the tropospheric chemistry by reducing the OH abundance and thereby extending the CH4 lifetime as well as the residence time of other chemical substances. The region above the tropopause is impacted by a substantial rise in stratospheric water vapour (SWV). The stratospheric ozone (O3) column increases overall, but SWV-induced stratospheric cooling also leads to a enhanced ozone depletion in the Antarctic lower stratosphere. Regional patterns of ozone change are affected by modification of stratospheric dynamics, i.e. increased tropical upwelling and stronger meridional transport towards the polar regions. We calculate the net radiative impact (RI) of the 2×CH4 experiment to be 0.69 W m−2, and for the 5×CH4 experiment to be 1.79 W m−2. A substantial part of the RH is contributed by chemically induced O3 and SWV changes, in line with previous radiative forcing estimates.
To our knowledge this is the first numerical study using a CCM with respect to 2- and 5-fold CH4 concentrations and it is therefore an overdue analysis as it emphasizes the impact of possible strong future CH4 emissions on atmospheric chemistry and its feedback on climate.
- Article
(4707 KB) - Full-text XML
-
Supplement
(1891 KB) - BibTeX
- EndNote





Methane (CH4) is a potent greenhouse gas (GHG), subject to strong anthropogenic emissions that contribute substantially to global warming. It is not just a radiatively active gas by itself but is chemically active as well, strongly influencing the chemical composition of the atmosphere. Beyond that, its sources are prone to temperature changes and it is generally expected that climate change (i.e. surface warming) will lead to enhanced CH4 emissions, accelerating the temperature rise. For instance, additional CH4 emissions are expected from wetlands due to climate-driven changes (Gedney et al., 2004; Zhang et al., 2017; Ma et al., 2017). Moreover, a large quantity of CH4 is stored as methane hydrate, not only in permafrost soil but also in the seafloor. Permafrost soil stores about a 100-fold of the current CH4 burden in the atmosphere, and oceanic methane hydrates store even a 1000-fold (IPCC, 2013). Current estimates indicate that GHG emissions from thawing permafrost soils could represent a major terrestrial biogeochemical feedback to climate change over the coming decades (Comyn-Platt et al., 2018).
At the same time, it is under debate whether a possible strong release of CH4 from thawing permafrost in the Arctic region could potentially force an abrupt climate change (as discussed by O'Connor et al., 2010). At present, the release of methane hydrate from reservoirs is highly uncertain as well as the magnitude of future natural and anthropogenic emissions of methane. Increasing surface temperatures cause enhanced CH4 emissions from thawing permafrost soils to the atmosphere, but the amount is currently poorly constrained (Hayes et al., 2014; Schaefer et al., 2014; Koven et al., 2015; Schuur et al., 2015). For instance, Dean et al. (2018) stated that there is basically no significant increase in Arctic methane emissions at the moment, though they may increase towards the end of the 21st century.
Nevertheless, permafrost thaw could potentially release trapped CH4 and transform frozen soil to wetland areas, which would then add to Arctic CH4 emissions. Moreover, ongoing heating of the Arctic sea surface temperature (SST) will also enhance future CH4 production in the ocean, and a reduction in sea ice concentration (SIC) may increase the direct transfer of CH4 from the ocean to the atmosphere. In particular, enhanced SST can increase the production of CH4, as permafrost underlying the continental shelf begins to thaw (Miller et al., 2018). How a changing climate will impact future CH4 emissions remains a topic of debate in atmospheric science, since emissions from the most climate-sensitive CH4 sources, i.e. wetlands, are difficult to quantify precisely.
Although there remain important knowledge gaps about the magnitude of CH4 emissions, it is important to improve our understanding of how strongly future CH4 emissions may impact our atmosphere and the environment. About 90 % of the emitted CH4 is removed in the troposphere. A change in tropospheric CH4 concentration affects the oxidizing capacity of the atmosphere, modifies ozone in the troposphere and influences the CH4 lifetime itself (e.g. Saunois et al., 2016; Frank, 2018; Holmes, 2018). Additionally, it affects the stratosphere. For example, enhanced CH4 emissions will lead to an abundance of stratospheric water vapour (SWV) and, as a consequence, will strongly influence stratospheric ozone (O3) (Stenke and Grewe, 2005; Revell et al., 2016).
To assess the direct and indirect effects of strongly enhanced CH4 emissions on atmospheric composition and Earth's climate, numerical model studies are able to support investigations such as identifying potential signatures impacting climate change. So far only a limited amount of numerical studies are available concerning the impact of very strong CH4 emissions. Exemplary, the effect of 2-fold CH4 was investigated in a 1-D radiative–convective climate model by Owens et al. (1982) and by MacKay and Khalil (1991). Shang et al. (2015) used a chemistry transport model (CTM) but doubled CH4 emission over China only. Other CTM studies have focused on recent changes and fluctuations in the atmospheric CH4 concentration (e.g. Dalsøren et al., 2016) or have tried to explain CH4 trends, which is a challenge because of important uncertainties in the global CH4 budget, i.e. the balance of surface sources and atmospheric and surface sinks (Saunois et al., 2016). Furthermore, CTMs are limited in assessing climate-change-related issues, because they do not include the feedback between chemistry and dynamics. Smith et al. (2018) investigated the fast radiative feedbacks (adjustments) in a model intercomparison using simulations with 3×CH4 without considering the chemical feedback effects. This investigation only includes physical components of the atmosphere, like direct cloud, water vapour and temperature adjustments, and the total radiative adjustment resulted in a value near zero. This example even more motivates an assessment of simulations that include chemically driven atmospheric adjustments to increases in CH4.
To our knowledge, studies using data derived from chemistry–climate model (CCM) simulations, including extreme CH4 emissions (i.e. beyond current and near-future amounts), are not available so far. A CCM is an atmospheric global circulation model that is interactively coupled to a detailed chemistry module. In contrast to CTMs, in CCMs the simulated concentrations of the radiatively active gases are used for the calculations of net heating rates. Changes in the abundance of these gases due to chemistry and advection influence heating rates and, consequently, variables describing atmospheric dynamics. This creates a dynamical–chemical coupling in which the chemistry influences the dynamics and vice versa. Since CH4 influences other trace gases due to its oxidation products as well as the removal of the hydroxyl radical (OH), a comprehensive chemistry module is necessary. In simulations with doubled carbon dioxide (CO2), in contrast, the feedback on climate and chemistry is induced only by its radiative impact. Apart from accounting for the direct radiative impact of CH4, the use of a CCM is strongly desired, since the atmospheric CH4 chemical feedback is a key process for understanding the variations in atmospheric CH4 and its effects on other chemical constituents of the atmosphere (Holmes, 2018).
The present work is the first study investigating atmospheric effects due to strong CH4 emissions with such a CCM. Idealized simulations of significantly enhanced CH4 concentrations are performed, i.e. 2-fold (2×CH4) and 5-fold (5×CH4) enhanced CH4 concentrations compared to present-day condition, allowing possible future consequences for atmospheric composition to be assessed while considering chemical feedback processes. In a first step, we conducted CCM simulations without interactive ocean coupling, i.e. the surface conditions regarding SST and SIC are prescribed (suppressed surface temperature feedback). Equivalent to the work of Smith et al. (2018), the results can be interpreted as rapid adjustments to a sudden CH4 enhancement before the ocean reacts to the perturbation, which would occur on a far larger timescale.
In this study we will use the ECHAM/MESSy Atmospheric Chemistry (EMAC) CCM (Jöckel et al., 2016), assessing the range of atmospheric responses by abrupt increases in CH4 concentrations. A short description of EMAC is given in Sect. 2, as well as an explanation of the simulation strategy. In Sect. 3 the reference simulation representing near-present-day condition is briefly evaluated with observations (Sect. 3.1) and a discussion of the impact of 2-fold and 5-fold increased CH4 concentrations in respective scenario simulations is presented in Sect. 3.2. In the final Sect. 4 we draw some conclusions from our investigation and give a brief outline of follow-up investigations.

Figure 1Annual zonal mean of absolute CH4 mixing ratios (in parts per billion volume (ppbv) of reference (REF) (a), S2 (b) and S5 (c). Note the different colour scales.
We use the EMAC model in version 2.52 (Jöckel et al., 2010) and operate it at a resolution of T42L90MA corresponding to a quadratic Gaussian grid of approx. in latitude and longitude with 90 levels up to 0.01 hPa. More details on the Modular Earth Submodel System (MESSy) can be found in Jöckel et al. (2016).
We conducted one reference simulation (REF) and two sensitivity simulations (S2 and S5) as 20-year time slice simulations, in general representing year 2010 conditions. Monthly SST and SIC are thereby repeatedly prescribed, representing a climatological annual cycle of the years 2000–2009 based on global analyses of Rayner et al. (2003). A spin-up of at least 10 years in length preceding each simulation (likewise time slice) ensures quasi-steady-state conditions but has been neglected in the evaluation. The spin-up started with initial conditions using a restart file representing the year 2010 of a reference simulation with specified dynamics (SD) of the Earth System Chemistry integrated Modelling (ESCiMo) project (Jöckel et al., 2016). To reduce the length of the spin up of the sensitivity simulations, we started those using 2-fold and 5-fold CH4 mixing ratios compared to the initialization of the reference.
The lower-boundary condition of CH4 in the reference simulation, i.e. the CH4 surface mixing ratio, is prescribed by Newtonian relaxation (i.e. nudged) following a zonal mean estimate from the Earth System Research Laboratory at the National Oceanic and Atmospheric Administration Earth System Research Laboratory (NOAA ESRL), based on observations of CH4 surface mixing ratios. These observations are provided by the Advanced Global Atmospheric Gases Experiment (AGAGE; http://agage.eas.gatech.edu, last access: 24 May 2019) as well as by NOAA ESRL (http://www.esrl.noaa.gov, last access: 24 May 2019). The mean surface mixing ratio is about 1.8 parts per million volume (ppmv). The two sensitivity simulations (S2 and S5) are carried out nudged at the surface to a 2-fold CH4 surface mixing ratio (compared to the present-day reference) of about 3.6 ppmv and a 5-fold CH4 surface mixing ratio of about 9.0 ppmv. Although this does not correspond to an equivalent increase in the surface fluxes, it scales the surface mixing ratio directly to the intended value, as has been done in similar studies (see Kirner et al., 2015; Forster et al., 2016; Smith et al., 2018). To put the chosen scaling factors into perspective, a surface mixing ratio of 3.6 ppmv (similar to the doubling above) will be reached according to the representative concentration pathway (RCP) 8.5 scenario towards the end of the 21st century (Riahi et al., 2007). The RCP 8.5 is the baseline climate change scenario, which does not employ any climate mitigation target (Riahi et al., 2011). Other prescribed conditions (SST, SIC, CO2, etc.) of the sensitivity simulations S2 and S5 are identical to REF. Particularly, all other GHG concentrations, as well as online simulated trace gas emissions, represent 2010 conditions.
In the following, changes and feedbacks are assessed by comparing the reference simulation REF with S2 and S5, with a focus on changes in the simulated chemically and radiatively active trace gases. To quantify the associated radiative impact (RI), the EMAC option for multiple radiation calls is used in submodel RAD (Dietmüller et al., 2016) in a separate additional simulation, which allows individual components of the total radiative impact to be estimated. This simulation is run for 1 year (plus 1-year spin-up) and uses climatological 20-year means of the species of interest (namely CH4, ozone and SWV from the corresponding reference or sensitivity simulation (REF, S2 and S5). The results are indicated hereafter by the associated simulation name and an asterisk (i.e. REF*, S2* and S5*). RAD performs multiple radiation calls with different inputs within one time step. Only the first call is used for providing the radiative heating feedback to the base model, while the other calls produce “perturbed” radiative fluxes and stratospheric temperature changes that are used diagnostically for calculating a stratospheric temperature-adjusted RI (Stuber et al., 2001; Dietmüller et al., 2016). In our set-up the first call receives the reference mixing ratios of the chemical species, while the other calls receive climatological means derived from the sensitivity simulations, replacing either all component species combined or each of the three species individually.
3.1 Evaluation of the reference simulation
The set-up of the reference simulation represents near-present atmospheric conditions of 2010. To ensure that this simulation is sufficiently realistic, the simulation results of CH4 mixing ratio in the troposphere and the stratosphere are compared to data derived from atmospheric observations indicated below. These observations are independent of the data sets used for the lower-boundary condition to ensure an objective evaluation.
For a detailed assessment of the performance of EMAC in general and how EMAC compares to observations (e.g. regarding temperature and ozone), we refer to Jöckel et al. (2016). This publication also includes an evaluation of transient simulations regarding CH4 in the upper troposphere and lower stratosphere (UTLS), using measurements of the Civil Aircraft for the Regular Investigation of the atmosphere Based on an Instrument Container (CARIBIC) project. That evaluation indicated a good CH4 representation with relative differences of less than 5 %.
In general, observed surface mixing ratios of CH4 indicate a north–south gradient with larger CH4 mixing ratios in the Northern Hemisphere (NH), mostly due to large wetland regions and anthropogenic sources on the northern continents. This north–south gradient is apparent by design (nudging to zonal mean surface mixing ratios based on observations) in our performed simulation. The simulated CH4 gradient of the REF simulation is compared to observations from a ship cruise of the research vessel Polarstern (Klappenbach et al., 2015, see Supplement Fig. S1). The simulation results reproduce the observed north–south gradient of the ship cruise qualitatively well, although an offset of about 0.055 ppmv exists. Note that the observations on the Polarstern were conducted in 2014, while the simulation represents 2010 conditions. Global CH4 surface mixing ratios have risen between 2010 and 2014 by about 0.030 ppmv, which explains some of the offset.
Additionally, the average vertical CH4 profile of the REF simulation is evaluated using balloon-borne measurements from Röckmann et al. (2011). The CH4 mixing ratio in the troposphere is approximately constant as a result of well-mixed tropospheric conditions. Above the tropopause the mixing ratio of CH4 decreases with altitude. This vertical gradient apparent in the balloon-borne observations is reasonably reproduced in REF (see Fig. S2).
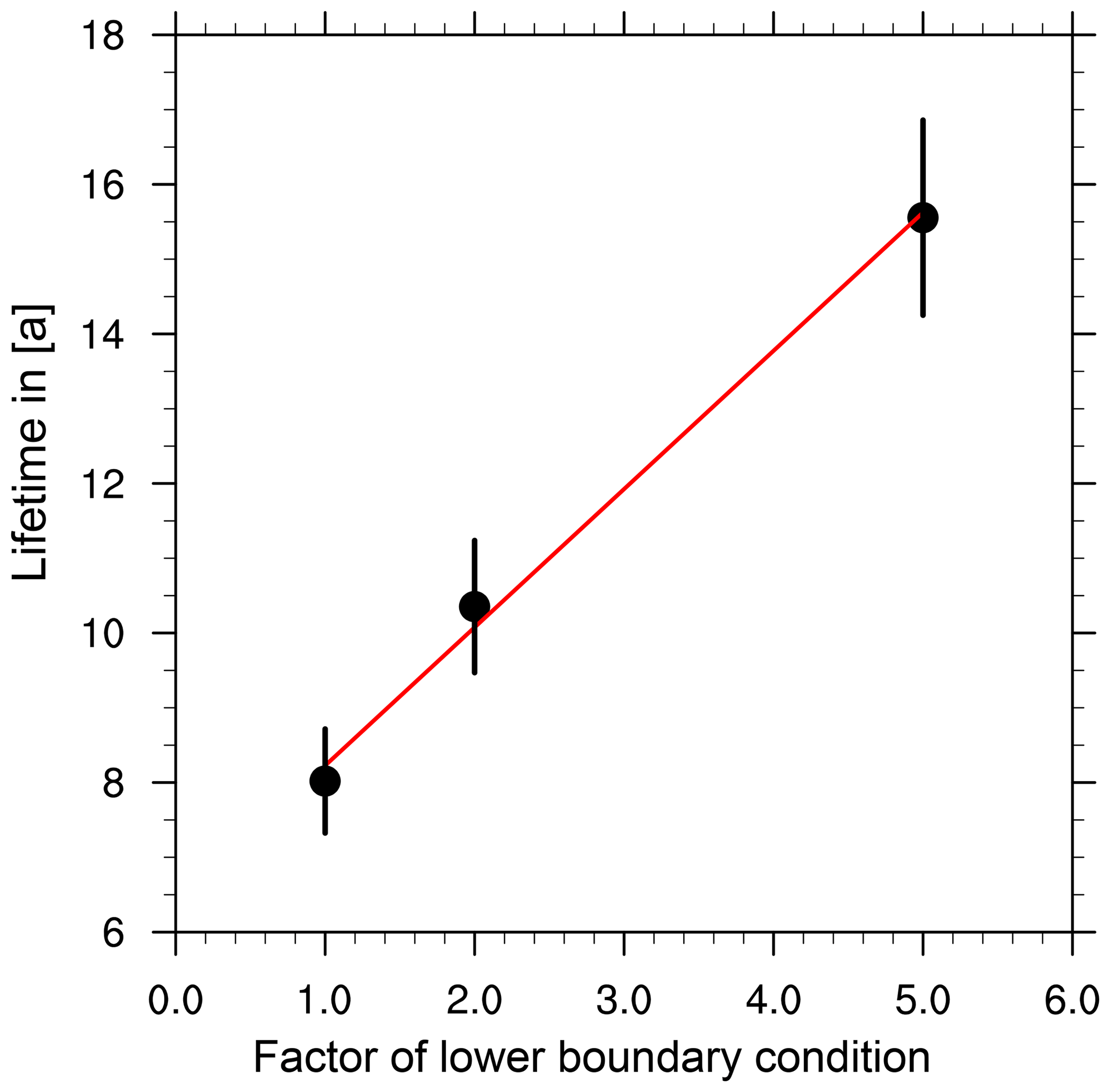
Figure 2CH4 lifetime calculated according to Eq. (1) vs. the corresponding scaling factor applied to the reference lower-boundary condition in the respective simulations: REF (1.0), S2 (2.0), S5 (5.0).
Furthermore, a general comparison of the zonal mean of CH4 mixing ratio from REF above the tropopause is done with observations from the Michelson Interferometer for Passive Atmospheric Sounding (MIPAS) instrument mounted on the ENVISAT satellite (Fischer et al., 2008). The zonally averaged CH4 MIPAS climatology (2002–2012) from Plieninger (2017) corresponds qualitatively and quantitatively to our simulation results in Figure 1a. The REF simulation represents the observed CH4 mixing ratios in the stratosphere and mesosphere and also shows the apparent double bulge in the upper stratosphere with slightly higher values in the NH.
Overall, the agreement of the reference simulation results with observations is suitable for the purpose of our intended study. In the next section we compare the reference simulation with the two sensitivity simulations to present the general impact of strongly enhanced CH4 concentrations.
3.2 Impact of 2-fold and 5-fold increased CH4 concentrations
In this subsection we investigate the impact of 2-fold and 5-fold surface CH4 mixing ratios on the chemical composition of the atmosphere and analyse the new chemical equilibrium after a sufficient spin-up.
Since SST and SIC are prescribed, the larger part of the feedback on tropospheric temperature is suppressed in the present simulations. Therefore, in this study we are only considering rapidly evolving chemical feedback effects, including respective radiative adjustments and temperature adjustments in the stratosphere.

Figure 3Difference (in %) between the annual zonal mean CH4 of the simulations S2 relative to the 2-fold annual zonal mean and S5 relative to the 5-fold annual zonal mean of the reference in (%).
The oxidation capacity of the atmosphere is often measured in terms of the CH4 lifetime (Karlsdóttir and Isaksen, 2000; Dentener et al., 2003; Naik et al., 2013; Voulgarakis et al., 2013). In this study we calculate the tropospheric CH4 lifetime according to Jöckel et al. (2006) as
with being the mass of methane in kg, T the temperature, p the pressure and q the specific humidity, all depending on time and the specific box b∈B, with B being the set of all considered grid boxes, e.g. all boxes which lie below the tropopause. is the reaction coefficient of the reaction in [cm3 s−1]. is the concentration of air in [mol cm−3] and xOH is the mole fraction of OH in one mole of the chemical tracer per one mole of air (mol mol−1)dryair.
Our calculations yield a near-linear increase in tropospheric CH4 lifetime with respect to the CH4 scaling in the sensitivity simulations (see Fig. 2). It is known that the tropospheric CH4 lifetime is anti-correlated with OH concentration (Montzka et al., 2011). The strongly enhanced CH4 mixing ratios reduce the atmospheric OH mixing ratio, which leads to a longer (tropospheric) CH4 lifetime. It is not certain that the quasilinear behaviour will hold for even larger or smaller scaling factors, since the chemistry determining the OH abundance is highly non-linear. There are also not enough data points (sensitivity simulations) for a definite proposition on strictly linear dependence. Nevertheless, we assume that these results give evidence that in the troposphere CH4 and OH are almost linearly anti-correlated for the given range. The simulations clearly show that increasing CH4 emissions increase the residence time of CH4 in the atmosphere and therefore its global warming potential.
Next, we investigate the impact of CH4 concentration increases on changes in CH4 depletion, thus analysing possible non-linearities in the chemical cycles. For this purpose we compare both sensitivity simulation results with the reference CH4 mixing ratio multiplied by 2 and 5, as is shown in Fig. 3. This approach makes it possible to see where CH4 is impacted by non-linear processes, i.e. where the 2-folding (5-folding) at the surface does not lead to an equal increase in the upper layers in the steady state.
In both sensitivity simulations, the troposphere is largely controlled by the nudging at the lower boundary due to turbulent mixing. In this area the differences in the sensitivity simulations and the scaled reference simulation are near-zero (though slightly positive). Larger CH4 mixing ratios reduce its most important sink reactant in the troposphere, namely OH, which leads to a reduction in the CH4 depletion compared to the reference.
The n-fold methane concentrations at the surface do not generate a corresponding n-fold methane concentration throughout the upper stratosphere. In the sensitivity simulation with 2-fold (5-fold) CH4, lower CH4 values of about 5 % (10 %) are found between 50 and 1 hPa compared to the corresponding n-folded reference. Identically prescribed SST in all three model simulations determines the forcing of atmospheric dynamics to a large extent and also constrains the stratosphere to a large part (see Garny, 2010). Therefore, modified atmospheric circulation patterns are unlikely the cause of these changes in stratospheric CH4 mixing ratios.
An explanation for the relatively strong relative depletion in CH4 in the upper stratosphere could be the change in the reaction rates for the CH4 decomposition via OH and chlorine (Cl), which are both temperature dependent. However, since the stratosphere cools in the sensitivity simulations (as will be discussed below), this also cannot explain the simulated reduced CH4 content in the stratosphere. Nevertheless, the deviation of a linear signal in the stratospheric CH4 mixing ratio gives evidence that more CH4 in the upper stratosphere is destroyed due to secondary feedbacks caused by changes in the chemical composition of the stratosphere (in particular, O3, SWV and OH) and will be discussed in the paragraphs below.
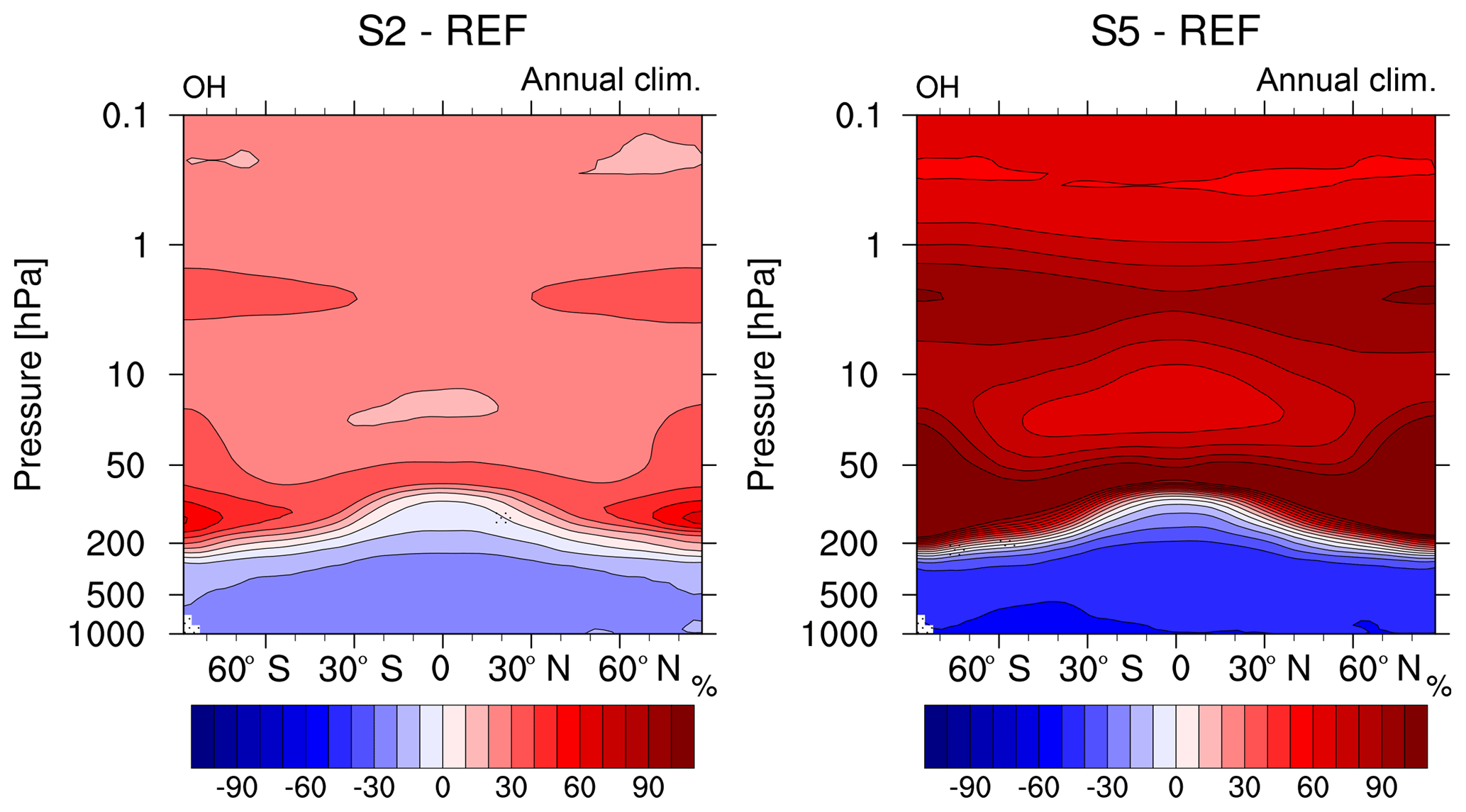
Figure 4Comparison of the relative changes (%) in annual zonal mean OH mixing ratio of the sensitivity simulations S2 and S5 (2- and 5-fold CH4) compared to the reference REF. Non-stippled areas are significant on a 95 % confidence level according to a two-sided Welch's test.
The OH concentration in the atmosphere is determined by its precursors, which are water vapour (H2O) and O3, and the photolysis rate of O3, as well as by its sinks, which are mostly CH4 and carbon monoxide (CO). The decline of OH in the troposphere by 20 %–30 % in the S2 simulation compared to REF (see Fig. 4) is caused, as stated above, by the increased sink via CH4. In the stratosphere, however, OH increases by about 30 % in the 2-fold CH4 case (S2) and by 60 %–80 % in the 5-fold CH4 case (S5; see Fig. 4). The mixing ratio of OH increases especially in the upper stratosphere at higher latitudes and fits to the decline in CH4 in the same regions (see Fig. 3). However, increases in CH4 mixing ratios influence the abundance of OH precursors, namely H2O and O3 via direct and secondary chemical effects and thereby feedback on the production of the CH4-associated sink OH.
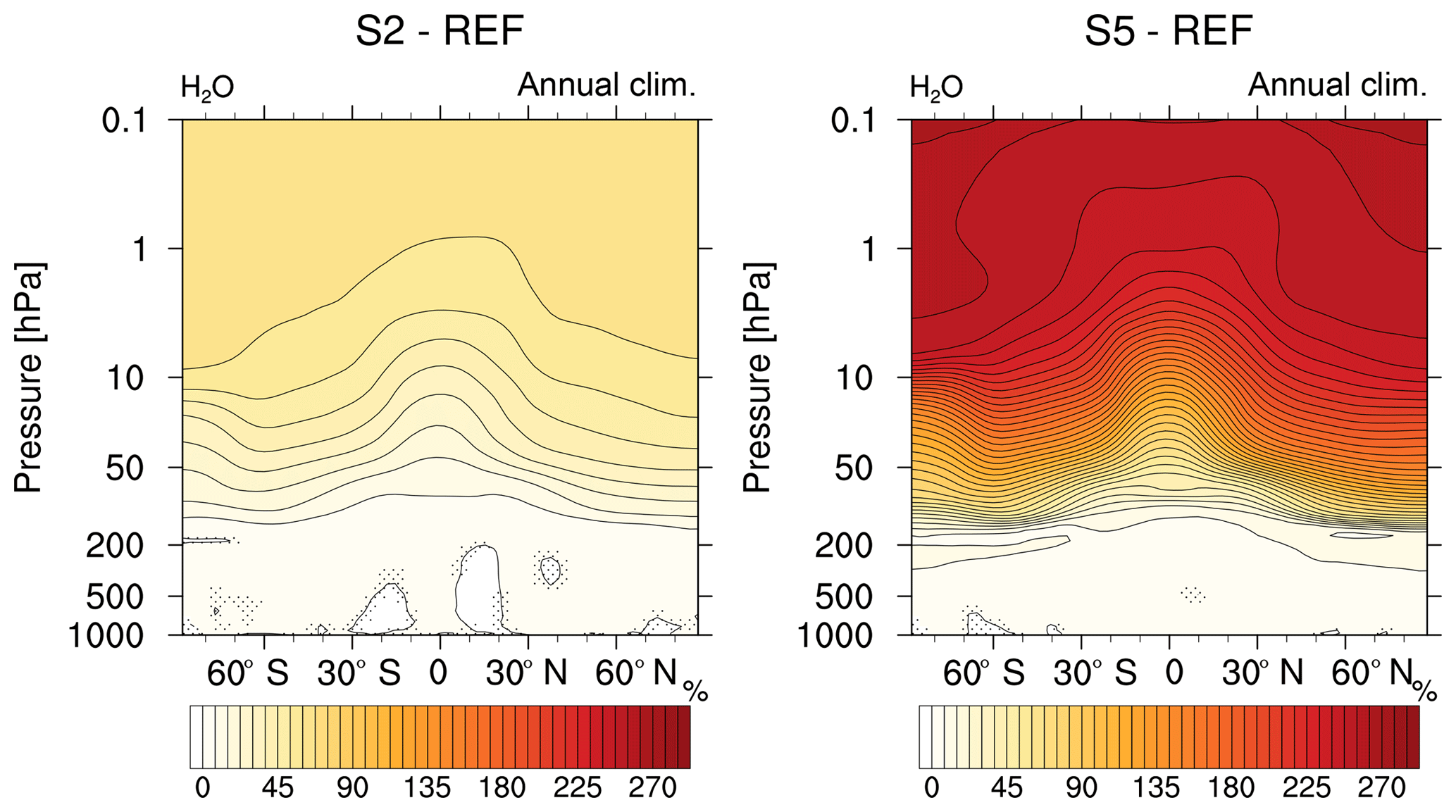
Figure 5Comparison of the relative changes (%) in annual zonal mean H2O mixing ratio of the sensitivity simulations S2 and S5 (2- and 5-fold CH4, respectively) compared to the reference REF. Non-stippled areas are significant on a 95 % confidence level according to a two-sided Welch's test.
Oxidation of CH4 in the stratosphere produces additional H2O and is therefore an important source for SWV (Hein et al., 2001; Rohs et al., 2006; Frank et al., 2018). The enhanced CH4 mixing ratios in the stratosphere cause a steady increase in SWV with height in both sensitivity simulations as indicated by Fig. 5. In the 2-fold CH4 case (S2) the amount of H2O is enhanced by up to 50 % in the middle and higher stratosphere, and in the 5-fold experiment (S5) the SWV increases by more than 250 %. The additional H2O leads to increasing OH in the upper stratosphere and lower mesosphere (Fig. 4).

Figure 6Comparison of the absolute changes (K) in annual zonal mean temperature of the sensitivity simulations S2 and S5 (2- and 5-fold CH4, respectively) compared to the reference REF. Non-stippled areas are significant on a 95 % confidence level according to a two-sided Welch's test.
The chemical changes indicated above influence the atmospheric temperature. However, since the SST is prescribed, the temperature response is largely suppressed in the troposphere. Confirmation is given in Fig. 6, where only a small change in tropospheric temperature is detected in both sensitivity simulations (S2 & S5). The stratosphere, however, shows larger changes in temperature. It can adjust to the perturbation since its temperature is mostly controlled by local radiative heating from trace gases and changing dynamics.
Around the tropopause there is a slight warming in the 2-fold CH4 case (S2), which reaches values of up to +3 K in the 5-fold case (S5; see Fig. 6). Elsewhere in the stratosphere, however, the higher abundance of CH4 induces a stratospheric cooling in our simulations. The 2-fold CH4 mixing ratios in S2 lead to a stratospheric cooling of about −1 to −2 K and a mesospheric cooling of up to −5 K. The results of the simulation with 5-fold CH4 mixing ratios indicate a cooling of about −3 K in the stratosphere and more than −10 K in the mesosphere. As will be discussed in detail later in this section, these temperature changes are induced by the radiative cooling from increasing CH4 and H2O in the stratosphere and mesosphere but in particular by the chemically induced O3 decrease and its associated radiative effect. However, this is clearly a matter of two-way interaction, as the cooling also impacts on chemical reaction rates affecting OH and O3 (see below).
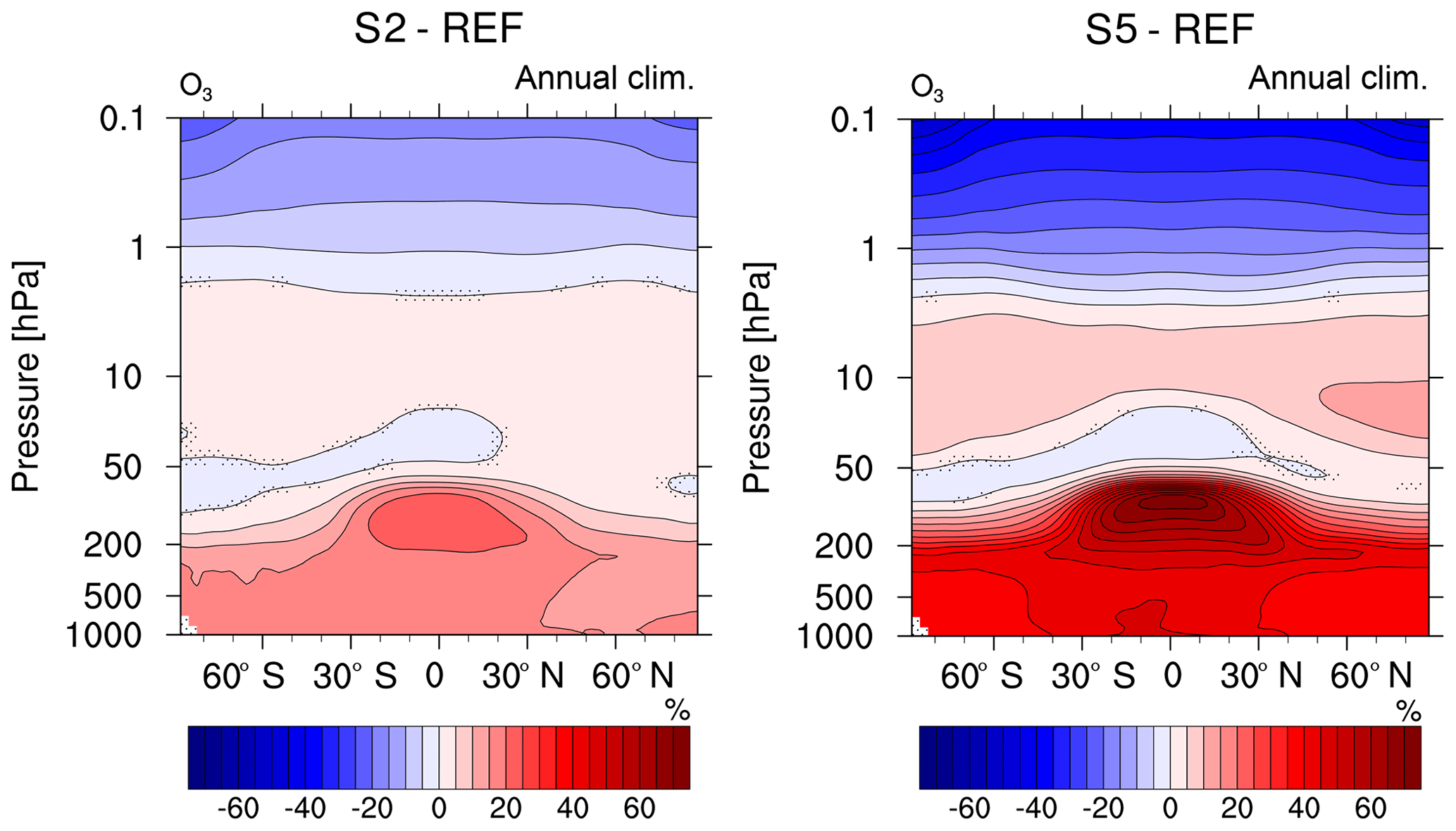
Figure 7Comparison of the relative changes (%) in annual zonal mean O3 mixing ratio of the sensitivity simulations S2 and S5 (2- and 5-fold CH4, respectively) compared to the reference REF. Non-stippled areas are significant on a 95 % confidence level according to a two-sided Welch's test.
As evident from Fig. 7, the strongly enhanced CH4 concentrations prescribed in our sensitivity simulations have an impact on the vertical profile of O3. Concentrations of O3 increase between 50 and 5 hPa (see Fig. 7) and decrease above. There is also a decrease between 50 and 20 hPa in the tropics and between 100 and 50 hPa at the South Pole.
Table 1An estimation of separate RI [in Wm−2] of the changes in the chemical species CH4, SWV and O3. Values are calculated using the RAD submodel (Dietmüller et al., 2016) in a separate simulation using 20-year climatologies of the individual species. Solely values of the totals are directly calculated from the presented simulations R1, S2 and S5.
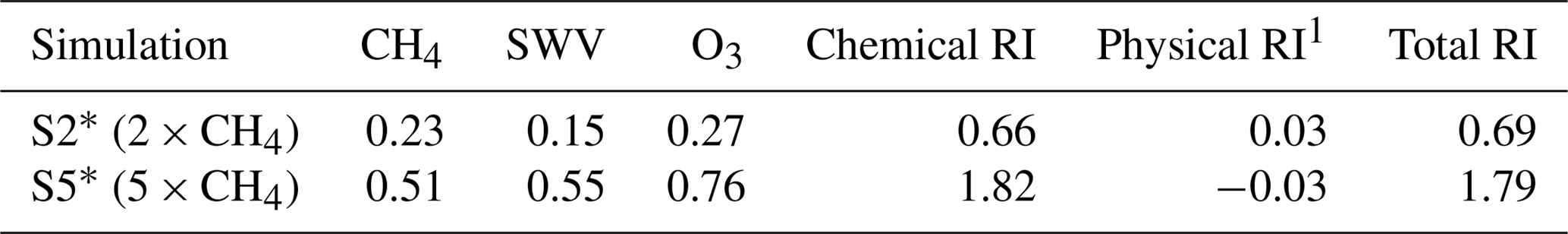
1 Total RI minus chemical RI.
The pattern of ozone reduction in the lowermost tropical stratosphere is typical of an enhanced tropical upwelling, which transports ozone depleted air from the upper troposphere to the lower stratosphere (Deckert and Dameris, 2008; Dietmüller et al., 2014). Although the main factor for such a strengthening, namely the SST, is prescribed, the increase in the GHG CH4 alone can also lead to an enhanced tropical upwelling (Garny et al., 2011). A similar pattern in stratospheric ozone changes due to CH4 increases (i.e. increase between 2000 and 2040–2049 according to the Intergovernmental Panel on Climate Change (IPCC) A1B greenhouse gas scenario) has been shown by Kirner et al. (2015). Nonetheless, we expect the impact on the tropospheric upwelling to intensify further in simulations where the SSTs are allowed to adjust to the CH4 radiative forcing.
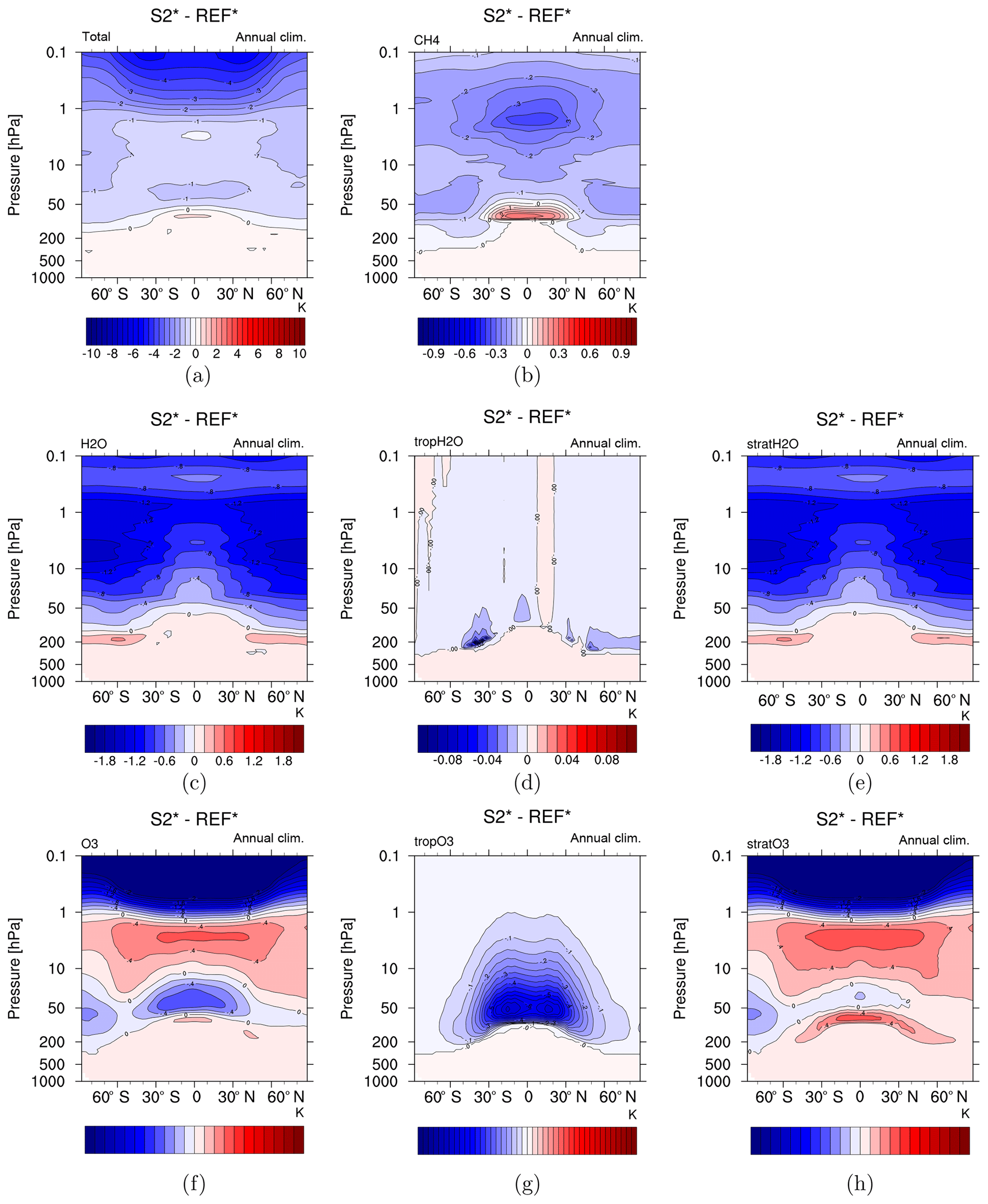
Figure 8Stratospheric adjusted temperature based on chemical changes in simulation S2* (2×CH4) in (a) CH4, H2O and O3 combined, (b) CH4, (c) H2O, (d) tropospheric H2O only, (e) stratospheric H2O only (SWV), (f) O3, (g) tropospheric O3 only and (h) stratospheric O3 only. Note the different colour bars in panels (a), (b), (d) and (g).
In the middle stratosphere the O3 production and depletion are influenced by the increased SWV and the corresponding stratospheric cooling, which restrains the reaction rates of O3-depleting catalytic cycles (Portmann and Solomon, 2007; Braesicke et al., 2013). Furthermore, excited oxygen (O(1D)) is depleted by increased abundances of H2O, which reduce the sink of O3 and eventually lead to increased O3 abundances. Above 2 hPa, increases in OH facilitate O3 destruction in the upper stratosphere and mesosphere (Kirner et al., 2015). Beyond that, there are interhemispheric differences in O3 mixing ratios in the polar regions (increased O3 at the polar middle stratosphere at about 30 hPa) that are an indication of a strengthening of the meridional transport towards the poles in the extreme case of 5×CH4 (see Fig. S4). In southern hemispheric winter (Fig. S4 upper right) this transport is suppressed by the polar vortex and is forming a corona of increased O3 mixing ratios outside the polar vortex. The stratospheric cooling also leads to enhanced forming of polar stratospheric clouds (PSCs) during the southern hemispheric winter and therefore to enhanced O3 depletion in the southern lower stratosphere (see Figs. S3c and S4c), as discussed in Dameris (2010), for example. The severity of the depletion barely increases from S2 to S5, which may be explained by a saturation effect reached with respect to additional PSCs. The same effect can be noticed in the total O3 column. Overall the total column of O3 (see Fig. S5) increases due to the rise of CH4 in the atmosphere, except in the Southern Hemisphere polar region, where S2 and S5 show about the same depletion in the total O3 column.
Overall, the enhanced CH4 mixing ratio with respect to 2010 is found to induce a radiative impact (RI) of about 0.69 Wm−2 in the 2-fold case (S2) and a RI of about 1.79 Wm−2 in the 5-fold case (S5) (see Table 1). The RI is calculated by the difference in the sum of long-wave and short-wave radiation at the top of the atmosphere between the reference and the respective sensitivity simulation. As the simulations are performed with prescribed SST, this net RI has the character of an effective radiative forcing (ERF), as pointed out by Forster et al. (2016). It includes the RI of CH4 itself, as well as rapid adjustments from physical and chemical processes. The chemical processes specifically include changes in SWV and O3 and have been quantified by estimating their individual RIs (including stratospheric temperature adjustments) with the EMAC submodel RAD (Dietmüller et al., 2016) in a separate simulation resulting in estimates corresponding to the reference simulation REF* and the sensitivity simulations S2* and S5* (see Sect. 2 for a detailed explanation of this simulation).
The 2-fold and 5-fold CH4 enhancements in the sensitivity simulations S2 and S5 correspond to net increases in surface CH4 of 1800 and 7200 ppbv, respectively. For example, the increase in the surface CH4 mixing ratio from 722 ppbv (pre-industrial) to 1803 ppbv (2011) has led to an ERF of 0.48 Wm−2 ± 0.1 Wm−2 (IPCC, 2013, chap. 8, Table 8.2), which corresponds to a net increase in surface CH4 of about 1100 ppbv. The net increase in 2-fold CH4 mixing ratios (S2) is larger than the increase since pre-industrial times. Thus, at first glance, the net RI calculated in this study seems consistent with the value in the IPCC AR5 and previous estimates from other models for a tripled CH4 concentration (assumed +3534 ppbv) ((Forster et al., 2016); (Smith et al., 2018)), which are, for example, for HadGEM2 1 Wm−2 and for CESM1 1.4 Wm−2.
However, those previous estimates do not account for contributions from O3 and stratospheric H2O changes (see also Smith et al., 2018). Only by looking at the direct RI simulations of individual species does a clearer picture emerge (see Table 1). The individual RIs of the chemical variations are once more reasonably compared with similar calculations of the IPCC (chap. 8, Fig. 8.17, derived from Shindell et al., 2009; Stevenson et al., 2013). We detect comparably low values for the RI of CH4, a feature which has been reported before for the ECHAM5 radiation module (Lohmann et al., 2010). A part of the underestimation can be attributed to a near-zero short-wave absorption contribution that is known from radiation schemes used in some other climate models (Etminan et al., 2016; Smith et al., 2018). As a residuum of the sum of chemically induced RI (Table 1) and the ERF, we estimate pure physical RIs of 0.03 Wm−2 (S2*, 2×CH4) and −0.03 Wm−2 (S5*, 5×CH4), i.e. small contributions that compare very well to the results of Smith et al. (2018).
The separation of the individual RI in the main contributing species allows further identification of the individual contribution to temperature adjustments in the stratosphere (see Fig. 8). The stratosphere cools in S2 by about −1 to −2 K, mostly due to SWV (up to −1.4 K). This cooling is amplified by the increased CH4 and tropospheric O3 but reduced by the radiative heating of increased stratospheric O3 in the altitude domain between roughly 20 and 1 hPa. This results in a quadrupole structure of the total temperature change pattern (warming in the troposphere with the maximum around the tropopause level, cooling in the lower and middle stratosphere, less cooling in the upper stratosphere and again stronger cooling in the mesosphere; see Figs. 8 and S6). Only a minor contribution to the stratospheric temperature change is made by tropospheric H2O, as to be expected. The difference between the pure RI of all trace gas changes (Figs. 8a and S6a) and the net stratospheric temperature change (Fig. 6) is small, indicating a dominating role of radiative effects over induced dynamical changes in forcing the temperature response in S2 and S5.
The present study summarizes the quasi-instantaneous chemical adjustments of the atmosphere in response to a very strong increase in atmospheric CH4. We emphasize that the applied doubling of present-day CH4 surface mixing ratios is not unrealistic as it is even part of the RCP 8.5 scenario. Considering further feedbacks with still uncertain quantitative consequences, it is indeed possible that the presented changes in the atmospheric chemistry will be faced by upcoming generations.
For the range of CH4 concentrations covered in this study, we find that the CH4 lifetime increases quasilinearly with enhanced surface mixing ratios. This is ascribed to a strong reduction in OH, which is the main sink of CH4 in the troposphere. We conclude that the strong reduction in OH will also influence other radiatively active, air-quality-relevant and ozone-depleting substances in the troposphere. The radical OH is the most important atmospheric detergent and its reduction will enhance the residence time of these substances, as well as of CH4, and thereby increase the global radiative burden.
Additionally, induced by CH4 oxidation, SWV will increase substantially by up to 50 % when CH4 is doubled and more than 250 % when CH4 is increased by a factor of five. This leads to a stratospheric cooling of several degrees, which in turn influences stratospheric chemistry and (to a smaller degree) dynamics. In particular it will lead to an increase in total O3 column (see Fig. S5) over nearly the whole globe. Only in the Antarctic spring does it cause a strengthening of the ozone depletion. We also detect an O3 reduction in the lowermost tropical stratosphere, typical of an enhanced tropical upwelling, which indicates small dynamical variations due to the strong increase in CH4, although more intense dynamical influences are suppressed by the predefined SST.
The rapid radiative adjustments of O3 and SWV are both positive and thus increase the radiative forcing directly induced by CH4, consistently with Fig. 8.17 of IPCC (2013). However, the direct CH4 radiative impact has a considerably low bias in the simulations, apparently through a systematic error in the radiation module. This bias remains masked if only the effective radiative forcing of the 2-fold and 5-fold CH4 simulations is considered. Individual radiative impact estimates also help to interpret the net stratospheric temperature change in the CH4 increase simulations. It reveals that the main part of the overall temperature pattern is controlled by cooling from SWV.
Since the SSTs are prescribed in the present simulations, tropospheric temperatures and atmospheric dynamics do not represent the situation after adaption of the ocean. This also prohibits the calculation of climate sensitivity parameters. In a future study, similar CCM simulations with a mixed layer ocean will be carried out and the contribution of feedbacks associated with SST changes will be investigated accordingly.
The Modular Earth Submodel System (MESSy) is continuously developed and applied by a consortium of institutions. The usage of MESSy and access to the source code is licensed to all affiliates of institutions, which are members of the MESSy Consortium. Institutions can become members of the MESSy Consortium by signing the MESSy Memorandum of Understanding. More information can be found on the MESSy Consortium website (http://www.messy-interface.org, last access: 24 May 2019, Jöckel and the MESSy Consortium, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7151-2019-supplement.
The simulations were set-up and carried out by PJ and FW. FT analysed the data and compiled the results. MD and FW structured and composed the manuscript. MP and FW contrived and carried out the radiative impact calculations. PJ and MP contributed to the text.
The authors declare that they have no conflict of interest.
This article is part of the special issue “The Modular Earth Submodel System (MESSy) (ACP/GMD inter-journal SI)”. It is not associated with a conference.
We acknowledge the DLR internal project KliSAW (Klimarelevanz von atmosphärischen Spurengasen, Aerosolen und Wolken), which provided the financial basis for the presented study. The model simulations have been performed at the German Climate Computing Centre (DKRZ) through support from the Bundesministerium für Bildung und Forschung (BMBF). We used the NCAR Command Language (NCL) for data analysis and to create some of the figures of this study. NCL is developed by UCAR/NCAR/CISL/TDD and available online: https://doi.org/10.5065/D6WD3XH5. We furthermore thank all contributors of the project ESCiMo (Earth System Chemistry integrated Modelling), which provides the model configuration and initial conditions, and Birgit Hassler for her internal review of the manuscript. Very constructive comments from the referees are gratefully acknowledged.
The article processing charges for this open-access publication were covered by a Research Centre of the Helmholtz Association.
This paper was edited by Andreas Hofzumahaus and reviewed by Christopher Smith and one anonymous referee.
Braesicke, P., Keeble, J., Yang, X., Stiller, G., Kellmann, S., Abraham, N. L., Archibald, A., Telford, P., and Pyle, J. A.: Circulation anomalies in the Southern Hemisphere and ozone changes, Atmos. Chem. Phys., 13, 10677–10688, https://doi.org/10.5194/acp-13-10677-2013, 2013. a
Comyn-Platt, E., Hayman, G., Huntingford, C., Chadburn, S. E., Burke, E. J., Harper, A. B., Collins, W. J., Webber, C. P., Powell, T., Cox, P. M., Gedney, N., and Sitch, S.: Carbon budgets for 1.5 and 2 ∘C targets lowered by natural wetland and permafrost feedbacks, Nat. Geosci., 11, 568–573, https://doi.org/10.1038/s41561-018-0174-9, 2018. a
Dalsøren, S. B., Myhre, C. L., Myhre, G., Gomez-Pelaez, A. J., Søvde, O. A., Isaksen, I. S. A., Weiss, R. F., and Harth, C. M.: Atmospheric methane evolution the last 40 years, Atmos. Chem. Phys., 16, 3099–3126, https://doi.org/10.5194/acp-16-3099-2016, 2016. a
Dameris, M.: Climate Change and Atmospheric Chemistry: How Will the Stratospheric Ozone Layer Develop?, Angewandte Chemie International Edition, 49, 8092–8102, https://doi.org/10.1002/anie.201001643, 2010. a
Dean, J. F., Middelburg, J. J., Röckmann, T., Aerts, R., Blauw, L. G., Egger, M., Jetten, M. S. M., Jong, A. E. E., Meisel, O. H., Rasigraf, O., Slomp, C. P., Zandt, M. H., and J., D. A.: Methane Feedbacks to the Global Climate System in a Warmer World, Rev. Geophys., 56, 207–250, 2018. a
Deckert, R. and Dameris, M.: Higher tropical SSTs strengthen the tropical upwelling via deep convection, Geophys. Res. Lett., 35, L10813, https://doi.org/10.1029/2008GL033719, 2008. a
Dentener, F., Peters, W., Krol, M., van Weele, M., Bergamaschi, P., and Lelieveld, J.: Interannual variability and trend of CH4 lifetime as a measure for OH changes in the 1979–1993 time period, J. Geophys. Res., 108, 4442, https://doi.org/10.1029/2002JD002916, 2003. a
Dietmüller, S., Ponater, M., and Sausen, R.: Interactive ozone induces a negative feedback in CO2-driven climate change simulations, J. Geophys. Res.-Atmos., 119, 1796–1805, https://doi.org/10.1002/2013JD020575, 2014. a
Dietmüller, S., Jöckel, P., Tost, H., Kunze, M., Gellhorn, C., Brinkop, S., Frömming, C., Ponater, M., Steil, B., Lauer, A., and Hendricks, J.: A new radiation infrastructure for the Modular Earth Submodel System (MESSy, based on version 2.51), Geosci. Model Dev., 9, 2209–2222, https://doi.org/10.5194/gmd-9-2209-2016, 2016. a, b, c, d
Etminan, M., Myhre, G., Highwood, E., and Shine, K.: Radiative forcing of carbon dioxide, methane, and nitrous oxide: A significant revision of the methane radiative forcing, Geophys. Res. Lett., 43, 12614–12623, https://doi.org/10.1002/2016GL071930, 2016. a
Fischer, H., Birk, M., Blom, C., Carli, B., Carlotti, M., von Clarmann, T., Delbouille, L., Dudhia, A., Ehhalt, D., Endemann, M., Flaud, J. M., Gessner, R., Kleinert, A., Koopman, R., Langen, J., López-Puertas, M., Mosner, P., Nett, H., Oelhaf, H., Perron, G., Remedios, J., Ridolfi, M., Stiller, G., and Zander, R.: MIPAS: an instrument for atmospheric and climate research, Atmos. Chem. Phys., 8, 2151–2188, https://doi.org/10.5194/acp-8-2151-2008, 2008. a
Forster, P. M., Richardson, T., Maycock, A. C., Smith, C. J., Samset, B. H., Myhre, G., Andrews, T., Pincus, R., and Schulz, M.: Recommendations for diagnosing effective radiative forcing from climate models for CMIP6, J. Geophys. Res.-Atmos., 121, 12460–12475, https://doi.org/10.1002/2016JD025320, 2016. a, b, c
Frank, F.: Atmospheric methane and its isotopic composition in a changing climate: A modelling study, Ph.D. thesis, Ludwigs Maximillian Universität München, 2018. a
Frank, F., Jöckel, P., Gromov, S., and Dameris, M.: Investigating the yield of H2O and H2 from methane oxidation in the stratosphere, Atmos. Chem. Phys., 18, 9955–9973, https://doi.org/10.5194/acp-18-9955-2018, 2018. a
Garny, H.: Causes and impacts of changes in the stratospheric meridional circulation in a chemistry-climate model, Ph.D. thesis, Ludwig-Maximilians-Universität München, https://edoc.ub.uni-muenchen.de/12215/ (last access: 24 May 2019), 2010. a
Garny, H., Dameris, M., Randel, W., Bodeker, G. E., and Deckert, R.: Dynamically Forced Increase of Tropical Upwelling in the Lower Stratosphere, J. Atmos. Sci., 68, 1214–1233, https://doi.org/10.1175/2011JAS3701.1, 2011. a
Gedney, N., Cox, P. M., and Huntingford, C.: Climate feedback from wetland methane emissions, Geophys. Res. Lett., 31, L20503, https://doi.org/10.1029/2004GL020919, 2004. a
Hayes, D. J., Kicklighter, D. W., McGuire, A. D., Chen, M., Zhuang, Q., Yuan, F., Melillo, J. M., and Wullschleger, S. D.: The impacts of recent permafrost thaw on land-atmosphere greenhouse gas exchange, Environ. Res. Lett., 9, 045005, http://stacks.iop.org/1748-9326/9/i=4/a=045005 (last access: 24 May 2019), 2014. a
Hein, R., Dameris, M., Schnadt, C., Land, C., Grewe, V., Köhler, I., Ponater, M., Sausen, R., B. Steil, B., Landgraf, J., and Brühl, C.: Results of an interactively coupled atmospheric chemistry – general circulation model: Comparison with observations, Ann. Geophys., 19, 435–457, https://doi.org/10.5194/angeo-19-435-2001, 2001. a
Holmes, C. D.: Methane Feedback on Atmospheric Chemistry: Methods, Models, and Mechanisms, J. Adv. Model. Earth Sy., 10, 1087–1099, https://doi.org/10.1002/2017MS001196, 2018. a, b
IPCC: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, https://doi.org/10.1017/CBO9781107415324, 2013. a, b, c
Jöckel, P., Tost, H., Pozzer, A., Brühl, C., Buchholz, J., Ganzeveld, L., Hoor, P., Kerkweg, A., Lawrence, M. G., Sander, R., Steil, B., Stiller, G., Tanarhte, M., Taraborrelli, D., van Aardenne, J., and Lelieveld, J.: The atmospheric chemistry general circulation model ECHAM5/MESSy1: consistent simulation of ozone from the surface to the mesosphere, Atmos. Chem. Phys., 6, 5067–5104, https://doi.org/10.5194/acp-6-5067-2006, 2006. a
Jöckel, P., Kerkweg, A., Pozzer, A., Sander, R., Tost, H., Riede, H., Baumgaertner, A., Gromov, S., and Kern, B.: Development cycle 2 of the Modular Earth Submodel System (MESSy2), Geosci. Model Dev., 3, 717–752, https://doi.org/10.5194/gmd-3-717-2010, 2010. a
Jöckel, P., Tost, H., Pozzer, A., Kunze, M., Kirner, O., Brenninkmeijer, C. A. M., Brinkop, S., Cai, D. S., Dyroff, C., Eckstein, J., Frank, F., Garny, H., Gottschaldt, K.-D., Graf, P., Grewe, V., Kerkweg, A., Kern, B., Matthes, S., Mertens, M., Meul, S., Neumaier, M., Nützel, M., Oberländer-Hayn, S., Ruhnke, R., Runde, T., Sander, R., Scharffe, D., and Zahn, A.: Earth System Chemistry integrated Modelling (ESCiMo) with the Modular Earth Submodel System (MESSy) version 2.51, Geosci. Model Dev., 9, 1153–1200, https://doi.org/10.5194/gmd-9-1153-2016, 2016. a, b, c, d
Jöckel, P. and the MESSy Consortium: The highly structured Modular Earth Submodel System (MESSy), available at: http://www.messy-interface.org, last access: 24 May 2019.
Karlsdóttir, S. and Isaksen, I. S. A.: Changing methane lifetime: Possible cause for reduced growth, Geophys. Res. Lett., 27, 93–96, https://doi.org/10.1029/1999GL010860, 2000. a
Kirner, O., Ruhnke, R., and Sinnhuber, B.-M.: Chemistry–Climate Interactions of Stratospheric and Mesospheric Ozone in EMAC Long-Term Simulations with Different Boundary Conditions for CO2, CH4, N2O, and ODS, Atmos.-Ocean, 53, 140–152, https://doi.org/10.1080/07055900.2014.980718, 2015. a, b, c
Klappenbach, F., Bertleff, M., Kostinek, J., Hase, F., Blumenstock, T., Agusti-Panareda, A., Razinger, M., and Butz, A.: Accurate mobile remote sensing of XCO2 and XCH4 latitudinal transects from aboard a research vessel, Atmos. Meas. Tech., 8, 5023–5038, https://doi.org/10.5194/amt-8-5023-2015, 2015. a
Koven, C. D., Lawrence, D. M., and Riley, W. J.: Permafrost carbon-climate feedback is sensitive to deep soil carbon decomposability but not deep soil nitrogen dynamics, P. Natl. Acad. Sci. USA, 112, 3752–3757, https://doi.org/10.1073/pnas.1415123112, 2015. a
Lohmann, U., Rotstayn, L., Storelvmo, T., Jones, A., Menon, S., Quaas, J., Ekman, A. M. L., Koch, D., and Ruedy, R.: Total aerosol effect: radiative forcing or radiative flux perturbation?, Atmos. Chem. Phys., 10, 3235–3246, https://doi.org/10.5194/acp-10-3235-2010, 2010. a
Ma, S., Jiang, J., Huang, Y., Shi, Z., Wilson, R. M., Ricciuto, D., Sebestyen, S. D., Hanson, P. J., and Luo, Y.: Data-Constrained Projections of Methane Fluxes in a Northern Minnesota Peatland in Response to Elevated CO2 and Warming, J. Geophys. Res.-Biogeo., 122, 2841–2861, https://doi.org/10.1002/2017JG003932, 2017. a
MacKay, R. and Khalil, M.: Theory and development of a one dimensional time dependent radiative convective climate model, Chemosphere, 22, 383–417, https://doi.org/10.1016/0045-6535(91)90326-9, 1991. a
Miller, S. M., Taylor, M. A., and Watts, J. D.: Understanding high-latitude methane in a warming climate, Earth and Space Science News, 99, https://doi.org/10.1029/2018EO091947, 2018. a
Montzka, S. A., Krol, M., Dlugokencky, E., Hall, B., Lelieveld, P. J., and Jöckel, P.: Small Interannual Variability of Global Atmospheric Hydroxyl, Science, 331, 67–69, 2011. a
Naik, V., Voulgarakis, A., Fiore, A. M., Horowitz, L. W., Lamarque, J.-F., Lin, M., Prather, M. J., Young, P. J., Bergmann, D., Cameron-Smith, P. J., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R., Eyring, V., Faluvegi, G., Folberth, G. A., Josse, B., Lee, Y. H., MacKenzie, I. A., Nagashima, T., van Noije, T. P. C., Plummer, D. A., Righi, M., Rumbold, S. T., Skeie, R., Shindell, D. T., Stevenson, D. S., Strode, S., Sudo, K., Szopa, S., and Zeng, G.: Preindustrial to present-day changes in tropospheric hydroxyl radical and methane lifetime from the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 5277–5298, https://doi.org/10.5194/acp-13-5277-2013, 2013. a
O'Connor, F. M., Boucher, O., Gedney, N., Jones, C. D., Folberth, G. A., Coppell, R., Friedlingstein, P., Collins, W. J., Chappellaz, J., Ridley, J., and Johnson, C. E.: Possible role of wetlands, permafrost, and methane hydrates in the methane cycle under future climate change: A review, Rev. Geophys., 48, RG4005, https://doi.org/10.1029/2010RG000326, rG4005, 2010. a
Owens, A. J., Steed, J. M., Filkin, D. L., Miller, C., and Jesson, J. P.: The Potential effects of increased methane on atmospheric ozone, Geophys. Res. Lett., 9, 1105–1108, https://doi.org/10.1029/GL009i009p01105, 1982. a
Plieninger, J. F.: Ableitung von CH4 und N2O aus MIPAS – ENVISAT-Beobachtungen, Ph.D. thesis, Karlsruher Institut für Technologie Fakultät Physik, 2017. a
Portmann, R. W. and Solomon, S.: Indirect radiative forcing of the ozone layer during the 21st century, Geophys. Res. Lett., 34, L02813, https://doi.org/10.1029/2006GL028252, 2007. a
Rayner, N. A., Parker, D. E., Horton, E. B., Folland, C. K., Alexander, L. V., Rowell, D. P., Kent, E. C., and Kaplan, A.: Global analyses of sea surface temperature, sea ice, and night marine air temperature since the late nineteenth century, J. Geophys. Res.-Atmos., 108, 4407, https://doi.org/10.1029/2002JD002670, 4407, 2003. a
Revell, L., Stenke, A., Rozanov, E., Ball, W., Lossow, S., and Peter, T.: The role of methane in projections of 21st century stratospheric water vapour, Atmos. Chem. Phys., 16, 13067–13080, https://doi.org/10.5194/acp-16-13067-2016, 2016. a
Riahi, K., Grübler, A., and Nakicenovic, N.: Scenarios of long-term socio-economic and environmental development under climate stabilization, Technol. Forecast. Soc., 74, 887–935, https://doi.org/10.1016/j.techfore.2006.05.026, 2007. a
Riahi, K., Rao, S., Krey, V., Cho, C., Chirkov, V., Fischer, G., Kindermann, G., Nakicenovic, N., and Rafaj, P.: RCP 8.5 – A scenario of comparatively high greenhouse gas emissions, Climatic Change, 109, 33–57, https://doi.org/10.1007/s10584-011-0149-y, 2011. a
Röckmann, T., Brass, M., Borchers, R., and Engel, A.: The isotopic composition of methane in the stratosphere: high-altitude balloon sample measurements, Atmos. Chem. Phys., 11, 13287–13304, https://doi.org/10.5194/acp-11-13287-2011, 2011. a
Rohs, S., Schiller, C., Riese, M., Engel, A., Schmidt, U., Wetter, T., Levin, I., Nakazawa, T., and Aoki, S.: Long-term changes of methane and hydrogen in the stratosphere in the period 1978–2003 and their impact on the abundance of stratospheric water vapor, J. Geophys. Res.-Atmos., 111, D14315, https://doi.org/10.1029/2005JD006877, 2006. a
Saunois, M., Jackson, R. B., Bousquet, P., Poulter, B., and Canadell, J. G.: The growing role of methane in anthropogenic climate change, Environ. Res. Lett., 11, 120207, https://doi.org/10.1088/1748-9326/11/12/120207, 2016. a, b
Schaefer, K., Lantuit, H., Romanovsky, V. E., Schuur, E. A. G., and Witt, R.: The impact of the permafrost carbon feedback on global climate, Environ. Res. Lett., 9, 085003, https://doi.org/10.1088/1648-9326/9/8/085003, 2014. a
Schuur, E. A. G., McGuire, A. D., Schädel, C., Grosse, G., Harden, J. W., Hayes, D. J., Hugelius, G., Koven, C. D., Kuhry, P., Lawrence, D. M., Natali, S. M., Olefeldt, D., Romanovsky, V. E., Schaefer, K., Turetsky, M. R., Treat, C. C., and Vonk, J. E.: Climate change and the permafrost carbon feedback, Nature, 520, 171–179, https://doi.org/10.1038/nature14338, 2015. a
Shang, L., Liu, Y., Tian, W., and Zhang, Y.: Effect of methane emission increases in East Asia on atmospheric circulation and ozone, Adv. Atmos. Sci., 32, 1617–1627, https://doi.org/10.1007/s00376-015-5028-4, 2015. a
Shindell, T. D., Faluvegi, G., Koch, D., Schmidt, G., Unger, N., and Bauer, S.: Improved Attribution of Climate Forcing to Emissions, Science, 326, 716–718, https://doi.org/10.1126/science.1174760, 2009. a
Smith, C. J., Kramer, R. J., Myhre, G., Forster, P. M., Soden, B., Andrews, T., Boucher, O., Faluvegi, G., Fläschner, D., Hodnebrog, O. ., Kasoar, M., Kharin, V., Kirkevå g, A., Lamarque, J.-F., Mülmenstädt, J., Olivié, D., Richardson, T., Samset, B. H., Shindell, D., Stier, P., Takemura, T., Voulgarakis, A., and Watson-Parris, D.: Understanding Rapid Adjustments to Diverse Forcing Agents, Geophys. Res. Lett., 45, 12023–12031, https://doi.org/10.1029/2018GL079826, 2018. a, b, c, d, e, f, g
Stenke, A. and Grewe, V.: Simulation of stratospheric water vapor trends: impact on stratospheric ozone chemistry, Atmos. Chem. Phys., 5, 1257–1272, https://doi.org/10.5194/acp-5-1257-2005, 2005. a
Stevenson, D. S., Young, P. J., Naik, V., Lamarque, J.-F., Shindell, D. T., Voulgarakis, A., Skeie, R. B., Dalsoren, S. B., Myhre, G., Berntsen, T. K., Folberth, G. A., Rumbold, S. T., Collins, W. J., MacKenzie, I. A., Doherty, R. M., Zeng, G., van Noije, T. P. C., Strunk, A., Bergmann, D., Cameron-Smith, P., Plummer, D. A., Strode, S. A., Horowitz, L., Lee, Y. H., Szopa, S., Sudo, K., Nagashima, T., Josse, B., Cionni, I., Righi, M., Eyring, V., Conley, A., Bowman, K. W., Wild, O., and Archibald, A.: Tropospheric ozone changes, radiative forcing and attribution to emissions in the Atmospheric Chemistry and Climate Model Intercomparison Project (ACCMIP), Atmos. Chem. Phys., 13, 3063–3085, https://doi.org/10.5194/acp-13-3063-2013, 2013. a
Stuber, N., Sausen, R., and Ponater, M.: Stratosphere Adjusted Radiative Forcing Calculations in a comprehensive climate model, Theor. Appl. Climatol., 68, 125–135, https://doi.org/10.1007/s007040170041, 2001. a
Voulgarakis, A., Naik, V., Lamarque, J.-F., Shindell, D. T., Young, P. J., Prather, M. J., Wild, O., Field, R. D., Bergmann, D., Cameron-Smith, P., Cionni, I., Collins, W. J., Dalsøren, S. B., Doherty, R. M., Eyring, V., Faluvegi, G., Folberth, G. A., Horowitz, L. W., Josse, B., MacKenzie, I. A., Nagashima, T., Plummer, D. A., Righi, M., Rumbold, S. T., Stevenson, D. S., Strode, S. A., Sudo, K., Szopa, S., and Zeng, G.: Analysis of present day and future OH and methane lifetime in the ACCMIP simulations, Atmos. Chem. Phys., 13, 2563–2587, https://doi.org/10.5194/acp-13-2563-2013, 2013. a
Zhang, Z., Zimmermann, N. E., Stenke, A., Li, X., Hodson, E. L., Zhu, G., Huang, C., and Poulter, B.: Emerging role of wetland methane emissions in driving 21st century climate change, P. Natl. Acad. Sci. USA, 114, 9647–9652, https://doi.org/10.1073/pnas.1618765114, 2017. a