the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 29 May 2019
| 29 May 2019
Experimental budgets of OH, HO2, and RO2 radicals and implications for ozone formation in the Pearl River Delta in China 2014
Zhaofeng Tan
Keding Lu
Andreas Hofzumahaus
Hendrik Fuchs
Birger Bohn
Frank Holland
Yuhan Liu
Franz Rohrer
Min Shao
Kang Sun
Yusheng Wu
Limin Zeng
Yinsong Zhang
Astrid Kiendler-Scharr
Andreas Wahner
Yuanhang Zhang
Hydroxyl (OH) and peroxy radicals (HO2 and RO2) were measured in the Pearl River Delta, which is one of the most polluted areas in China, in autumn 2014. The radical observations were complemented by measurements of OH reactivity (inverse OH lifetime) and a comprehensive set of trace gases including carbon monoxide (CO), nitrogen oxides (NOx=NO, NO2) and volatile organic compounds (VOCs). OH reactivity was in the range from 15 to 80 s−1, of which about 50 % was unexplained by the measured OH reactants. In the 3 weeks of the campaign, maximum median radical concentrations were 4.5×106 cm−3 for OH at noon and 3×108 and 2.0×108 cm−3 for HO2 and RO2, respectively, in the early afternoon. The completeness of the daytime radical measurements made it possible to carry out experimental budget analyses for all radicals (OH, HO2, and RO2) and their sum (ROx). The maximum loss rates for OH, HO2, and RO2 reached values between 10 and 15 ppbv h−1 during the daytime. The largest fraction of this can be attributed to radical interconversion reactions while the real loss rate of ROx remained below 3 ppbv h−1. Within experimental uncertainties, the destruction rates of HO2 and the sum of OH, HO2, and RO2 are balanced by their respective production rates. In case of RO2, the budget could be closed by attributing the missing OH reactivity to unmeasured VOCs. Thus, the presumption of the existence of unmeasured VOCs is supported by RO2 measurements. Although the closure of the RO2 budget is greatly improved by the additional unmeasured VOCs, a significant imbalance in the afternoon remains, indicating a missing RO2 sink. In case of OH, the destruction in the morning is compensated by the quantified OH sources from photolysis (HONO and O3), ozonolysis of alkenes, and OH recycling (HO2+NO). In the afternoon, however, the OH budget indicates a missing OH source of 4 to 6 ppbv h−1. The diurnal variation of the missing OH source shows a similar pattern to that of the missing RO2 sink so that both largely compensate each other in the ROx budget. These observations suggest the existence of a chemical mechanism that converts RO2 to OH without the involvement of NO, increasing the RO2 loss rate during the daytime from 5.3 to 7.4 ppbv h−1 on average. The photochemical net ozone production rate calculated from the reaction of HO2 and RO2 with NO yields a daily integrated amount of 102 ppbv ozone, with daily integrated ROx primary sources being 22 ppbv in this campaign. The produced ozone can be attributed to the oxidation of measured (18 %) and unmeasured (60 %) hydrocarbons, formaldehyde (14 %), and CO (8 %). An even larger integrated net ozone production of 140 ppbv would be calculated from the oxidation rate of VOCs with OH if HO2 and all RO2 radicals react with NO. However, the unknown RO2 loss (evident in the RO2 budget) causes 30 ppbv less ozone production than would be expected from the VOC oxidation rate.
- Article
(3453 KB) - Full-text XML
-
Supplement
(1024 KB) - BibTeX
- EndNote





Hydroxyl radicals (OH) constitute the major atmospheric oxidant which is produced in the global troposphere by UV photolysis of ozone (Levy, 1971). The photolysis produces electronically excited O(1D) atoms which react with water molecules to form OH. In the polluted lower atmosphere, OH can also be produced efficiently by photolysis of nitrous acid (HONO). The reaction with OH initiates the degradation of most trace gases (e.g., CO and volatile organic compounds – VOCs), which leads in many cases to the formation of peroxy radicals, including hydroperoxy radicals (HO2) and organic peroxy radicals (RO2; R is alkyl group). In the presence of nitric oxide (NO), peroxy radicals recycle OH, an important mechanism that increases the oxidizing power of the atmosphere. The above reactions and other fundamental reactions controlling the formation and destruction of ROx radicals are listed in Table 1 (Ehhalt, 1999). The reaction of peroxy radicals with NO has another important implication. NO2 is formed as a product which can be photolyzed. The photodissociation produces ground-state oxygen atoms O(3P), which combine with O2 and form ozone. The combination of these reactions (Reactions R8, R9, and R11) establishes the basic mechanism of photochemical ozone formation in the troposphere (Fishman et al., 1979).
Table 1Chemical reactions considered in the radical budget analysis of OH, HO2, and RO2. The radical species are cyclically linked by chain reactions.
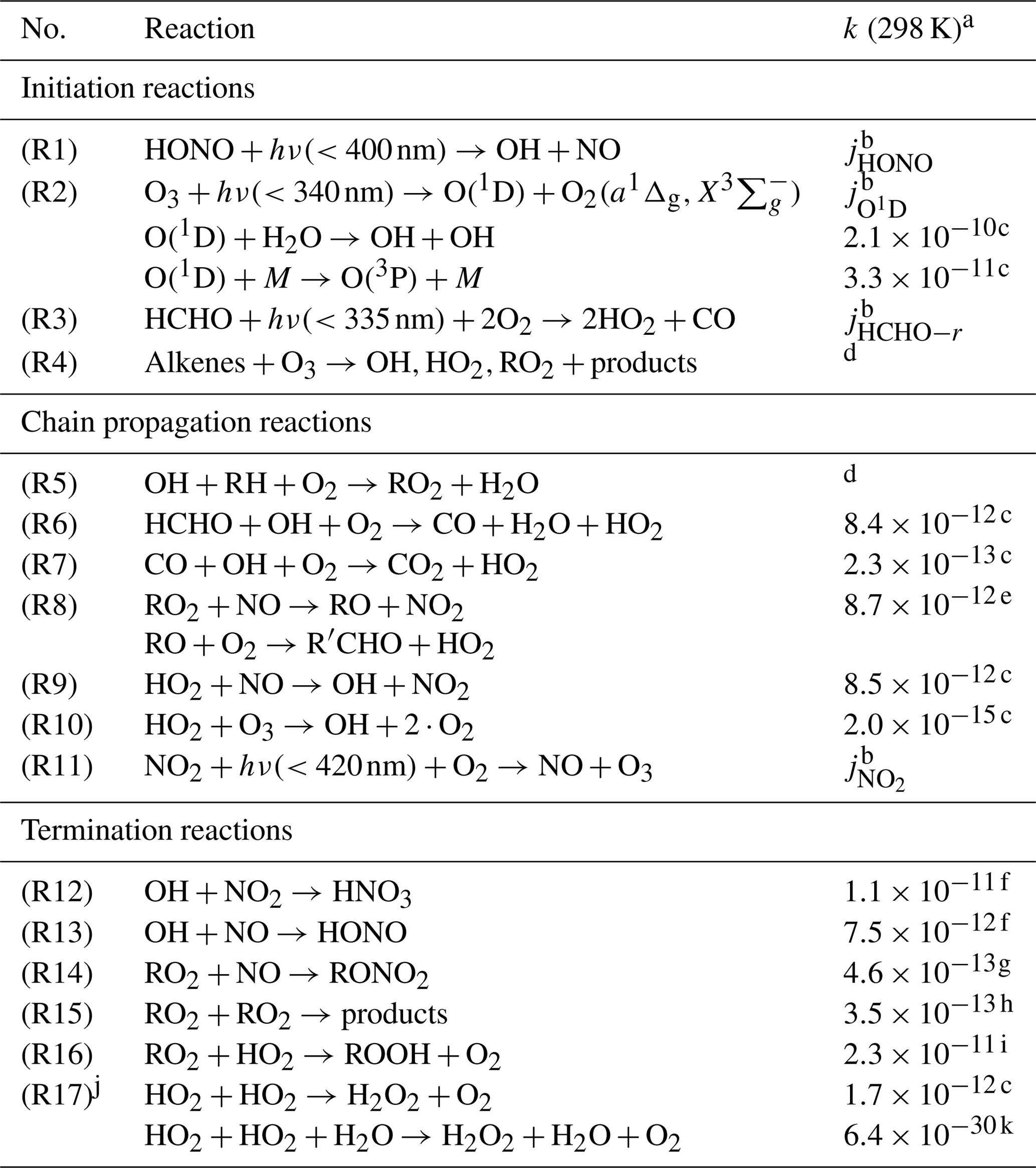
a Reaction rate coefficients (cm3 s−1) are shown in this
table for 298 K and 1 atm.
In the radical budget analysis (Figs. 2–4), the actual measured ambient
temperatures and pressures were used.
b Measured (see Table 2).
c MCM3.3.1.
d Specific kinetics data for each measured organic compound are taken
from MCM3.3.1.
e
(MCM3.3.1). The RO yield is assumed to be 0.95 (see text).
f NASA JPL (Burkholder et al., 2015).
g Reaction rate coefficient as for Reaction (R8). The yield of RONO2 is
assumed to be 0.05 (see text).
h (MCM3.3.1).
i (MCM3.3.1). f is a scaling factor which is
assumed
to be 1 (see text).
j The effective reaction rate k17 contains both reactions with and
without water.
k
(RACM2; Goliff et al., 2013). This reaction is a termolecular reaction
(with a unit of cm6 s−1).
China is a country with a large population and a fast-growing economy, which has caused increasing air pollution during the last decade (Chan and Yao, 2008). However, only a few field campaigns have been carried out so far studying photochemistry under polluted conditions in China with the support of OH and peroxy radical measurements. The campaigns were performed in the Pearl River Delta in southern China (Lu et al., 2012; Hofzumahaus et al., 2009) and in the North China Plain around Beijing (Tan et al., 2018a, b, 2017; Fuchs et al., 2017; Lu et al., 2013). Both large regions are densely populated and characterized by air pollution from energy production, traffic, industry, and farming. Chemical box model simulations of OH concentrations have shown general good agreement with measured OH in these regions at NO concentrations above 1 ppbv but a tendency to underpredict the measured OH at less than 1 ppbv NO. The largest underprediction of OH by a factor of 3–5 was observed in Backgarden near Guangzhou in the Pearl River Delta (PRD) in summer 2006 (Hofzumahaus et al., 2009). The PRIDE-PRD 2006 (Program of Regional Integrated Experiments of Air Quality over the Pearl River Delta, 2006 – Lu et al., 2012) campaign was characterized by high OH reactivities with mean daytime values of (20–50) s−1, where biogenic isoprene and its oxidation products made a contribution of 40 % in the afternoon. The results of this campaign showed similarities to those from studies in forested regions, where a model underprediction of OH by up to a factor of 10 was reported in isoprene-rich air (Tan et al., 2001; Ren et al., 2008; Lelieveld et al., 2008; Pugh et al., 2010; Whalley et al., 2011). The unexplained OH in forests and in the Pearl River Delta was generally assumed to be caused by an unknown OH recycling mechanism that is likely linked to the photochemical degradation of isoprene. In fact, subsequent research discovered previously unknown HOx regeneration reactions, which involve unimolecular isomerization and decomposition reactions of RO2 in the oxidation of isoprene (Peeters et al., 2014, 2009; Peeters and Muller, 2010; da Silva, 2010; Crounse et al., 2011; Fuchs et al., 2013; Teng et al., 2017) and methacrolein (Crounse et al., 2012; Fuchs et al., 2014). These reactions do not require NO and explain an OH enhancement by a factor of about 2 when the OH loss is dominated by isoprene (Fuchs et al., 2013). The mechanism is, however, not sufficient for resolving the large model–measurement discrepancies previously reported.
Another possible reason for the high OH observations could be an interference in the OH measurements. Artifacts of up to 80 % have been reported when OH was measured by laser-induced fluorescence (LIF) in forest environments (Mao et al., 2012; Hens et al., 2014; Novelli et al., 2014; Feiner et al., 2016). The interference was presumably caused by oxidation products of biogenic VOCs in the measuring system and was quantified by chemical modulation of ambient OH before the measured air enters the device. When the measurements were corrected for the artifact, the measured OH could be explained by a photochemical box model (Hens et al., 2014; Feiner et al., 2016). In other field studies, where different LIF instruments were used, chemical modulation resulted in only marginal unexplained OH signals at the instrumental limit of detection. One study was in an isoprene-rich forest (Griffith et al., 2016), and the other was in the polluted North China Plain (Tan et al., 2017). In both studies, the models could explain the OH measurements relatively well but showed a tendency to underpredict OH under low-NOx conditions (NO<300 pptv). These discrepancies could not be explained by OH measurement artifacts. Thus, while some of the previously reported high OH observations could have been affected by measurement interferences, there are also indications for an incomplete understanding of radical recycling in VOC rich atmosphere.
In the present paper, we report new radical measurements in the PRD during the campaign PRIDE-PRD2014 together with measurements of a large set of atmospheric trace gases. The campaign took place in a suburban area near Guangzhou and was carried out in autumn 2014, the enhanced pollution period for the PRD, to elucidate the radical chemistry and secondary pollution formation. Compared to the previous campaign PRIDE-PRD2006, RO2 was measured in addition to OH and HO2 radicals. Chemical modulation was applied to test the OH instrument with respect to possible OH interferences. In the present work, an experimental budget analysis approach is used for quantifying the main chemical reactions that control the radical abundances. This approach is possible, as the production and destruction rates of all radical species can be constrained by measurements. The concept has been applied before for OH, when its loss rate could be constrained by OH reactivity measurements (e.g., Shirley et al., 2006; Hofzumahaus et al., 2009; Whalley et al., 2011; Griffith et al., 2013; Hens et al., 2014). However, the budget analysis for HO2 and RO2 was generally done by chemical box models due to missing RO2 measurements. Here, the completeness of the radical measurements during the daytime is used for the first time to quantify the production and destruction processes for all radicals (OH, HO2, RO2) and their sum (ROx) and to analyze their role in photochemical ozone formation. New evidence is found for missing radical recycling under low-NOx conditions, which is a source of OH but not of ozone.
2.1 Measurement site
The field campaign took place at a long-term monitoring station, the Guangdong Atmospheric Supersite of China (22.728∘ N, 112.929∘ E), which is located about 6 km southwest of the city of Heshan. The site is situated on a 60 m high hill, surrounded by woods, small villages, and factories in the surrounding area. A closest highway is about 2 km to the northwest and showed moderate traffic during the measurement campaign. Two major cities, Guangzhou and Foshan, are located at a distance of 50 km to the northeast of the site. The measurement site is located about 140 km to the southwest of the measurement site (Backgarden) of the former PRIDE-PRD2006 campaign. The campaign was carried out from October to November to be representative of the photochemical polluted season in the PRD (Zhang et al., 2008).
2.2 Instrumentation
An overview of the instrumentation used for the radical budget analysis is given in Table S1 in the Supplement. The inlets of all instruments were located close to the radical measurement inlets at a height of 1.7 m above the roof of the building and 20 m above the ground.
2.2.1 Radicals
OH and HO2 radicals were measured by the Peking University laser-induced fluorescence system (PKU-LIF), which was built by Forschungszentrum Jülich (FZJ). OH radicals are detected by laser-induced fluorescence (LIF) at 308 nm in a gas expansion inside a low-pressure (4 hPa) measurement cell (Hofzumahaus et al., 1996; Holland et al., 2003). In a second cell, HO2 radicals are chemically converted with NO, yielding OH, which is detected by LIF (Fuchs et al., 2011). The PKU-LIF instrument was applied first at Wangdu in the North China Plain in summer 2014 (Tan et al., 2017). It was then moved to the Heshan site for the present study. At both sites, the PKU-LIF system was complemented by devices from FZJ to measure RO2 (ROxLIF) and OH reactivity (kOH). In the ROx LIF system, the radicals RO2, HO2, and OH are quantitatively converted to HO2 in a pre-reactor by addition of 0.7 ppmv of NO and 0.11 % of CO at a total pressure of 25 hPa. In a second stage at lower pressure (4 hPa), the HO2 is further converted by a large excess of 0.5 % NO to OH, which is then detected by LIF (Fuchs et al., 2008). RO2 concentrations are calculated from the total sum of ROx (from ROxLIF) by subtracting the contributions of OH and HO2 measured in the other two detection chambers. A detailed description of the whole measurement system is given by Tan et al. (2017). Due to a technical problem, the integration time for the radical measurements in Heshan was increased to 5 min to achieve 1σ detection limits of 3.9×105 cm−3 for OH, 1.2×107 cm−3 for HO2, and 0.6×107 cm−3 for RO2 (Table S1). The accuracy of the calibrations depends on the uncertainty of the calibration source (10 %; 1σ) and the reproducibility of the calibrations, resulting in total accuracies of ±13 %, ±20 %, and ±26 % for OH, HO2, and RO2, respectively (Table S1).
As outlined by Tan et al. (2017), attention was paid to possible interferences in the measurement of HOx. Like in the Wangdu campaign, chemical modulation was applied on several occasions to test whether the OH measurements obtained normally by laser wavelength modulation are perturbed by artificial OH formation in the detection cell. Such kind of interference has been detected in some LIF instruments by applying chemical modulation in field campaigns (Mao et al., 2012; Novelli et al., 2014; Feiner et al., 2016). The chemical modulation system used at Wangdu and Heshan consisted of a flow tube in front of the OH measurement cell and allowed the scavenging of ambient OH by addition of propane in the sampled air flow. Switching between propane and nitrogen additions allows discriminating ambient OH from instrumental OH. For nitrogen, we used research-grade purity (>99.9990 %). A gas chromatograph (GC) analysis of the nitrogen showed no significant contamination by VOCs, which would scavenge OH in the chemical modulation system. In the OH detection cell, scavenging of artificially produced OH by the added propane is calculated to be less than 0.3 %. A description of the prototype chemical-modulation reactor used with PKU-LIF is given by Tan et al. (2017). Results of the chemical modulation experiments during the PRIDE-PRD2014 campaign are presented in Sect. 3.3.
The detection of HO2 by chemical conversion with NO has two known interferences. First, particular RO2 radicals (called ) from large alkanes (>C4), alkenes (including isoprene), and aromatics can be converted to OH in the HO2 detection cell, leading to a systematic positive bias of the HO2 measurement (Nehr et al., 2011; Fuchs et al., 2011; Whalley et al., 2013; Lew et al., 2018). The interference is most effective when the amount of added NO is high enough to convert most of the atmospheric HO2 to OH in the LIF cell. In the campaigns in Wangdu and Heshan, the concentration of the NO reagent was lowered by more than a factor of 10, thereby reducing the interference in the HO2 cell to less than 5 % (Tan et al., 2017).
On the other hand, the chemical conversion of can be used intentionally for concentration measurements (Whalley et al., 2013). For that purpose, ROxLIF was operated in an additional measurement mode where the NO addition in the pre-reactor was temporarily turned off and replaced by nitrogen (N2; Tan et al., 2017). In this mode, the sum of HO2 and is detected in the connected LIF cell, which is operated with a large amount of NO for chemical conversion. The concentration of is then determined as the difference of (ROxLIF) and HO2 (HO2 cell). The experimental error of the difference is quite large and occasionally exceeds 100 % in the present study because was much smaller than HO2 (see Fig. 1). Also, the calibration error of is larger than that of HO2. The detection sensitivity for individual species lies in the range of 0.8±0.2 times the detection sensitivity for HO2 (Fuchs et al., 2011; Lu et al., 2012). As the speciation of atmospheric is not exactly known, the possible range of sensitivities causes an additional error of 25 % that has to be added to the normal calibration error, yielding a total accuracy of ±32 % (1σ).
The second NO-related artifact, which is relevant for measurements in the HO2 cell and ROxLIF system, comes from spurious OH signals generated by the addition of NO in the LIF cells. The signal was regularly determined in humidified synthetic air during calibration and found to be stable over the campaign. It is equivalent to and cm−3 for HO2 and RO2, respectively, and is routinely subtracted from the measurements.
The total OH reactivity was measured by an instrument based on laser-flash photolysis laser-induced fluorescence (LP-LIF; Fuchs et al., 2017; Lou et al., 2010). Ambient air is sampled and pulled through a laminar flow tube at ambient conditions. Artificial OH is produced in the sampled air by pulsed laser photolysis (266 nm wavelength; 10 ns pulse duration) of ozone, which produces OH in nanoseconds according to Reaction (R2). The OH decay due to the reaction with atmospheric trace gases is then monitored in real time by LIF. kOH is determined as a pseudo-first-order rate coefficient from the decay curves. The precision of the measured kOH data is ±0.3 s−1 (1σ) and the accuracy is 10 % (Table S1) at an integration time of 180 s.
2.2.2 Trace gases and photolysis frequencies
As summarized in Table S1, the instrumentation used in the PRIDE-PRD2014 campaign was similar to that used in Wangdu (Tan et al., 2017; Fuchs et al., 2017). Photolysis frequencies were determined from spectral actinic photon-flux density measurements (Bohn et al., 2008). Meteorological parameters, including relative humidity, ambient pressure and temperature, and wind speed and direction, were also regularly measured at the site.
NO and NO2 were measured by a commercial chemiluminescence instrument (Thermo Scientific Model 42i). NO2 was converted to NO by a custom-built photolytic converter instead of an original molybdenum converter. O3, SO2, CO, and CO2 measurements were also measured by commercial instruments from Thermo Scientific (model 49i, 43i-TLE, 48i-TLE, and 410i). Greenhouse gases, including CO, CO2, CH4, and H2O, were measured by a cavity ring-down spectroscopy instrument (Picarro model G2401). The CO and CO2 measurements from the Thermo Scientific and Picarro instruments agreed within the instrumental accuracies. The Picarro measurements were used in this work due to the better data coverage. HONO measurements were performed using a custom-built long-path absorption photometer (LOPAP) from PKU (Liu et al., 2016). Measurements of non-methane hydrocarbons (NMHCs) were performed by a GC using a flame ionization detector (FID) and mass spectrometer (MS) for detection. The GC-FID–MS system provided measurements of C2–C6 alkanes, C2–C6 alkenes, and C6–C8 aromatics (Wang et al., 2014). Formaldehyde was measured by a commercial Hantzsch fluorimeter instrument (Aero-Laser GmbH model AL4021). A list of the measured VOCs is given in the Supplement (Table S2).
2.3 Experimental radical budget calculations
The radical budget analysis in this work is applied to OH, HO2, RO2, and the whole ROx family. The analysis is based on the chemical mechanism in Table 1, which describes fundamental reactions controlling the abundance of the radicals in the lower troposphere. The reactions include radical chain propagation reactions, which convert one radical species into another one, initiation reactions that produce radicals from closed-shell molecules, and chain termination reactions that destroy radicals. For the budget calculations, measurements for all relevant reactants and photolysis frequencies, together with published reaction rate coefficients (Table 1), are used. Unlike in model studies, the analysis does not include model-calculated species.
In the budget analysis, the total production and loss rates of each radical species are calculated and compared to each other. Since all radicals are short-lived (the OH lifetime is less than a second, and the lifetime of peroxy radicals is on the order of a minute), their concentrations are expected to be in a steady state, with total production and loss rates being balanced. In chemical box models, the balance is always enforced by the numerical solver of the rate equations, even if the chemical mechanism is incorrect. In the experimental budget analysis, however, imbalances are possible and indicate either unknown errors of the experimental input data (concentrations, photolysis frequencies, and rate coefficients) or an incorrect chemical mechanism.
The concept of an experimental radical budget analysis has been applied to atmospheric OH in previous studies, where the determination of the total OH loss rate was facilitated by the measurement of kOH. OH reactivity measurements avoid the problem that some relevant OH reactants may not be captured by direct measurements.
In the present work, the experimental budget analysis is extended to HO2 and RO2 radicals. In the case of peroxy radicals, no technique exists for the measurement of their atmospheric total reactivity. However, unlike for OH, the number of known reactant species removing peroxy radicals is relatively small. The main reactants are NO and the peroxy radicals themselves, all of which being measured, allowing the total loss rates from the individual reactions to be calculated.
2.3.1 ROx budget equations
The ROx budget is entirely controlled by initiation and termination reactions. ROx is primarily produced by photolysis of HONO (Reaction R1), O3 (Reaction R2), HCHO (Reaction R3), and ozonolysis of alkenes (Reaction R4). The total production rate is calculated as
In case of ozone photolysis, φOH is the yield of OH from the reaction of O(1D) with H2O, which competes with collisional deactivation of O(1D) with air molecules N2 and O2. In the ozonolysis reactions, the yields , , and are specific for each alkene species i. The summation of the radical production from ozonolysis is performed over all measured alkenes. This sum may be not complete if relevant alkenes are not measured (see discussion in Sect. 4.2).
The total destruction rate of ROx is given by the reactions of OH with NOx (Reactions R12, R13), RO2 with NO (Reaction R14), and self-reactions of peroxy radicals (Reactions R15–R17):
Since RO2 is measured as a sum of organic peroxy radicals, it is treated as a single species. The generalized rate coefficients are adopted from MCMv3.3.1 (see footnotes in Table 1). Reaction (R14), leading to the formation of organic nitrates, competes with Reaction (R8), which produces HO2 radicals. The branching ratio between Reactions (R8) and (R14) depends on the carbon chain lengths and structure (Atkinson et al., 1982; Lightfoot et al., 1992). The organic nitrate yield generally increases with carbon number and lies typically between 1 % for ethyl RO2 and 35 % for RO2 of C8 alkanes. Here, a nitrate yield of 5 % is assumed. The impact of larger nitrate yields is discussed in Sect. 4.
The thermal decomposition of HO2NO2 into HO2 and NO2 (an initiation reaction) and the back reaction to HO2NO2 (a termination reaction) are not explicitly considered in the budget equations. The two reactions reach a thermal equilibrium within seconds under the conditions of the campaign and have no net effect on the ROx balance. Likewise, equilibrium is assumed between thermal decomposition of PAN and its formation by the reaction of acetyl peroxy radicals with NO2. Also, this equilibrium is not explicitly considered in the budget Eqs. (1) and (2).
2.3.2 OH budget equations
As explained above, the total OH destruction rate can be directly quantified as the product of the OH concentration and the OH reactivity, both of which were measured during the PRIDE-PRD2014 campaign:
The total OH production rate is calculated from the primary (Reactions R1, R2, R4) and secondary (Reactions R9, R10) sources of OH. The primary sources are treated in the same way as in the ROx budget. The secondary sources include OH recycling from the reaction of HO2 with NO (Reaction R9) and O3 (Reaction R10):
2.3.3 HO2 budget equations
The total production rate of HO2 is calculated from primary sources, i.e. photolysis of HCHO (Reaction R3) and ozonolysis of alkenes (Reaction R4), and secondary sources which involve the conversion of OH and RO2 to HO2. The treatment of the primary production by Reactions (R3) and (R4) is explained in Sect. 2.3.1. In addition to the photolysis of HCHO, the photolysis of other oxygenated VOCs (OVOCs) could contribute to the HO2 production, but this is not considered here due to the absence of OVOC measurements.
Conversion from OH to HO2 can occur by reaction of OH with CO, HCHO, H2, and O3. Under the conditions of the campaign, the reaction rates for H2 and O3 were at least 2 orders of magnitude smaller than those for the reactions with HCHO (Reaction R6) and CO (Reaction R7). Therefore, only Reactions (R6) and (R7) are considered in the budget analysis. Also, the reaction of RO2 with NO (Reaction R8) constitutes an important secondary source of HO2. Reaction (R8) competes with the radical termination Reaction (R14), for which a nitrate yield of 5 % is assumed (see Sect. 2.3.1). Accordingly, an HO2 yield of 95 % is taken for Reaction (R8). The total HO2 production rate is then calculated as
HO2 is chemically removed by reaction with NO, O3, HO2, and RO2, all of which were measured in this campaign. It should be noted that the effective rate constant k17 for the self-recombination of HO2 has a water vapor dependence which is taken into account in k17 (Table 1). The total HO2 destruction rate is then given by
As explained in Sect. 2.3.1, the thermal equilibrium between HO2+NO2 and HO2NO2 is not explicitly considered in the budget equations.
2.3.4 RO2 budget equations
Primary RO2 production is possible by the ozonolysis of alkenes and the photolysis of OVOCs. Owing to the lack of OVOC measurements (except HCHO) in this study, only ozonolysis is considered to be a primary source. It is treated as described in Sect. 2.3.1.
In a broader sense, also reactions of hydrocarbons with NO3 radicals and chlorine atoms can be considered primary production processes because they do not consume ROx species. However, neither NO3 nor Cl were measured. NO3 is produced by reaction of NO2 with ozone. It is generally assumed that during the bright hours of the day, NO3 is predominantly destroyed by photolysis and reaction with NO. Recently, Liebmann et al. (2018) reported measurements in a forested environment in southern Germany, demonstrating that more than 25 % of daytime NO3 was removed by biogenic VOCs. The possible role of NO3 reactions with VOCs at Heshan is discussed in Sect. 3.
Cl atoms may play a role in the morning. Gaseous ClNO2 can be formed at night by heterogeneous reaction of N2O5 with Cl− ions and photolyze quickly after sunrise producing Cl atoms (Osthoff et al., 2008; Tham et al., 2016; Li et al., 2018). This mechanism, followed by the reaction of Cl with VOCs, made some contribution to the early morning RO2 production in a previous campaign in summertime in the North China Plain (Tan et al., 2017) and will be discussed in Sect. 4.2.
The main secondary source of RO2 is the reaction of OH with VOCs. As it is generally difficult to measure all reactive organic compounds in the atmosphere, we follow two different approaches to determine the RO2 production from OH reactions. The first approach calculates the RO2 production rate as the sum of the reaction rates of OH with all measured hydrocarbon species, denoted VOC(1). The resulting total RO2 production rate can be considered a lower limit:
Here, the first sum represents the primary production by ozonolysis, and the second term represents the RO2 production rate by OH reactions.
Another approach estimates the total atmospheric amount of organic reactants, here denoted VOC(2), from the measured OH reactivity (e.g., Shirley et al., 2006; Whalley et al., 2016). For this purpose, the reactivity of measured CO, NO, NO2, HCHO, SO2, and O3 is subtracted from the measured kOH to determine the total reactivity of VOCs that can potentially form RO2. This reactivity is called kOH(VOC(2)). This approach makes the implicit assumption that the missing OH reactivity found in the present study (see Sect. 3.2) is caused by unmeasured VOCs. The RO2 production rate is then given by
The RO2 destruction is determined by the reaction with NO (Reactions R8, R14) and with other peroxy radicals (Reactions R15, R16). These reactions and the thermal equilibrium of PAN are treated as in the ROx budget analysis (Sect. 2.3.1). Accordingly, the total destruction rate of RO2 can be calculated as
Equations (7) and (9) can be adapted for the budget analysis of radicals. In this case, the second term in Eq. (7) contains only OH reactions of VOCs that are known to produce . In Eq. (9), only the concentration of RO2 at the end of the equation has to be replaced by , assuming that all reacts with RO2.
3.1 Meteorological and chemical conditions
The complete suite of measurements (radicals, trace gases, and meteorological parameters) started on 22 October and ended on 14 November. The weather was generally cloudy, with temperatures in the 20 to 30 ∘C range, and water vapor volume mixing ratios were around 2 %. Solar UV radiation showed variability due to cloudy weather conditions, as can be seen from the photolysis-frequency variations of and (Fig. S1 in the Supplement). In this work, conditions with s−1 are referred to as daytime conditions, lasting from 06:00 to 18:00 LT (local time; all times listed in the text are in local time). After 6 November, the weather changed and became rainy, with little photochemical activity. Therefore, the current study was restricted to the time period from 22 October to 5 November. During this period, air transportation was dominated by northeasterly and easterly winds. The time dependence of measured trace gas concentrations is shown in Fig. S1, and median values are listed in Table 2. The chemical conditions are characterized by anthropogenic pollution. High concentrations of ozone were observed, with daily maxima reaching 100 ppbv on several days. In the morning and afternoon, median O3 values were 16 and 69 ppbv, respectively. NO mixing ratios reached maximum values of 10 ppbv, and median values were 3.7 ppbv in the morning and 0.4 ppbv in the afternoon. NO2 mixing ratios were 17 and 9 ppbv in the morning and afternoon, respectively.
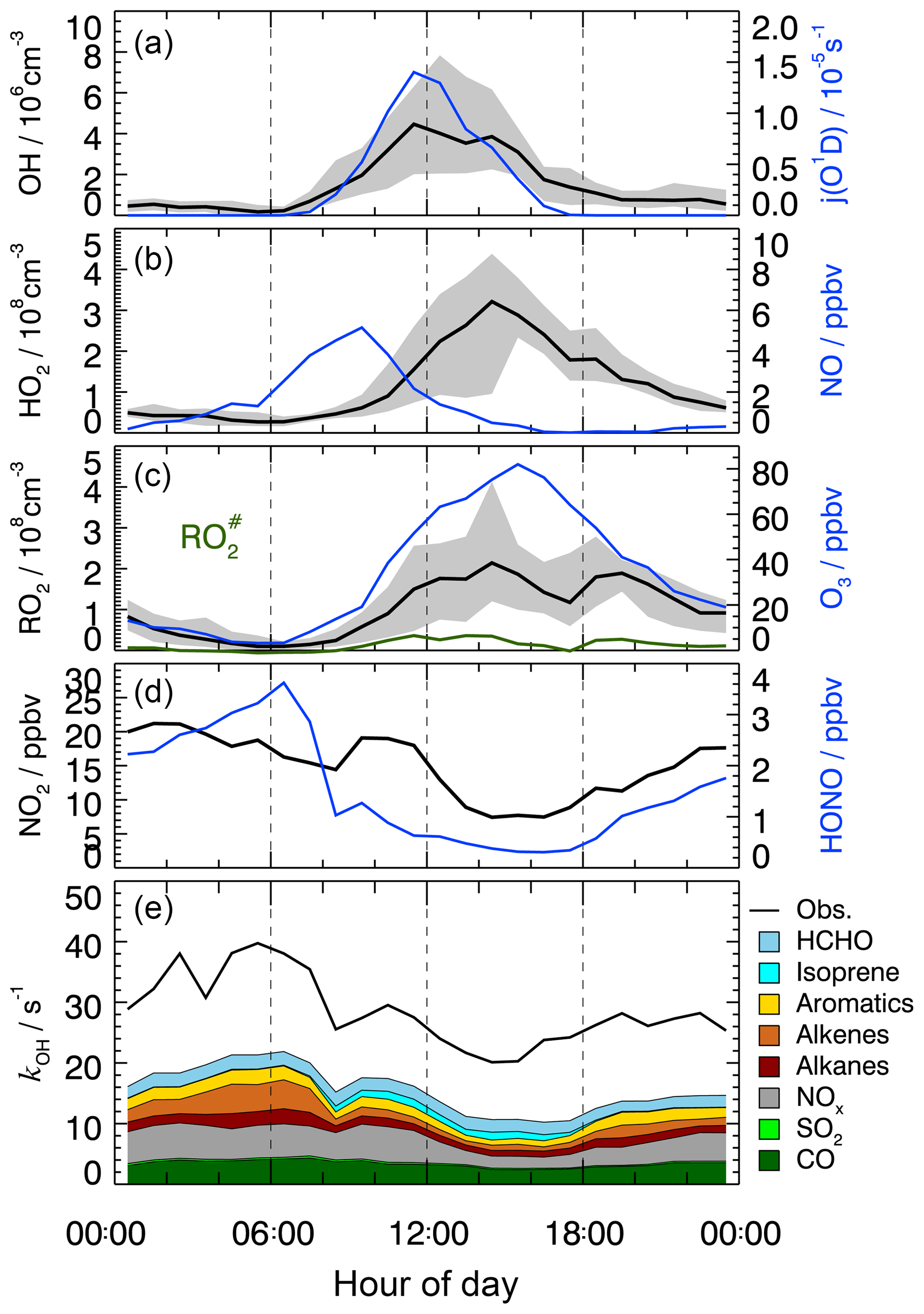
Figure 1Median diurnal profiles of measured OH, HO2, RO2, , kOH, j(O1D), NO, NO2, O3, and HONO. For OH, HO2, and RO2, the grey band around the median (black lines) denotes the 25 % and 75 % percentiles of the distributions. In the lowest panel, colored areas show the speciated reactivity contributions from measured OH reactants.
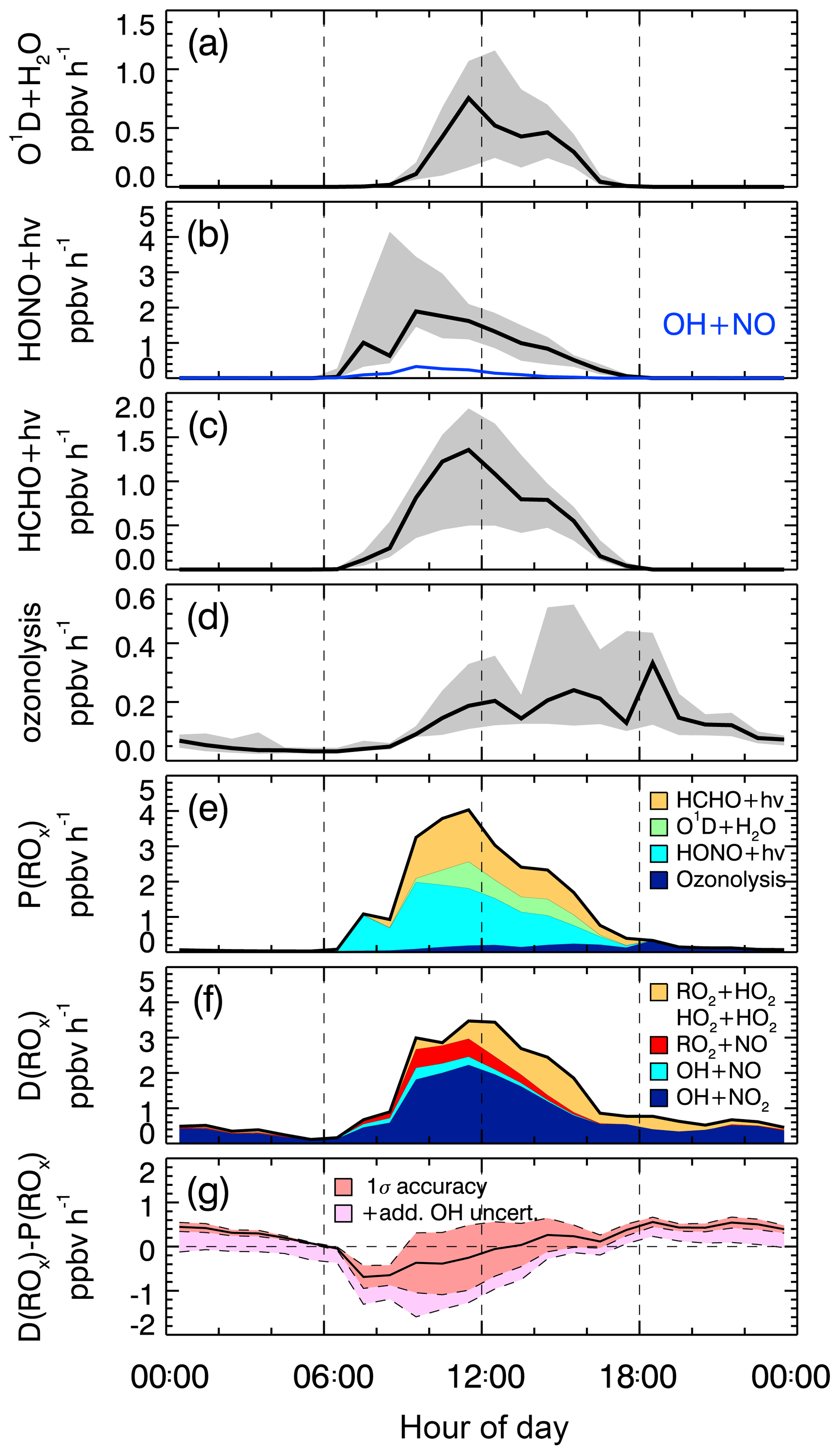
Figure 2Median production and destruction rates of ROx. (a)–(d) show production rates (black lines: median values) from the photolysis of ozone (a), HONO (b), HCHO (c), and ozonolysis of alkenes (d). Grey bands around the median denote 25 % and 75 % percentiles. The blue line in (b) represents the back reaction rate of OH+NO yielding HONO. (e) and (f) show cumulative plots of the production and destruction rates, respectively; (g) the solid black line is the difference between the total production and destruction rate. The red shaded band indicates the 1σ uncertainty due to experimental errors of the measured quantities (Table S1) and the reaction rate coefficients. The pink shaded area represents the maximum possible bias from a potential OH interference.
Table 2Median values of measured parameters in the morning and afternoon.
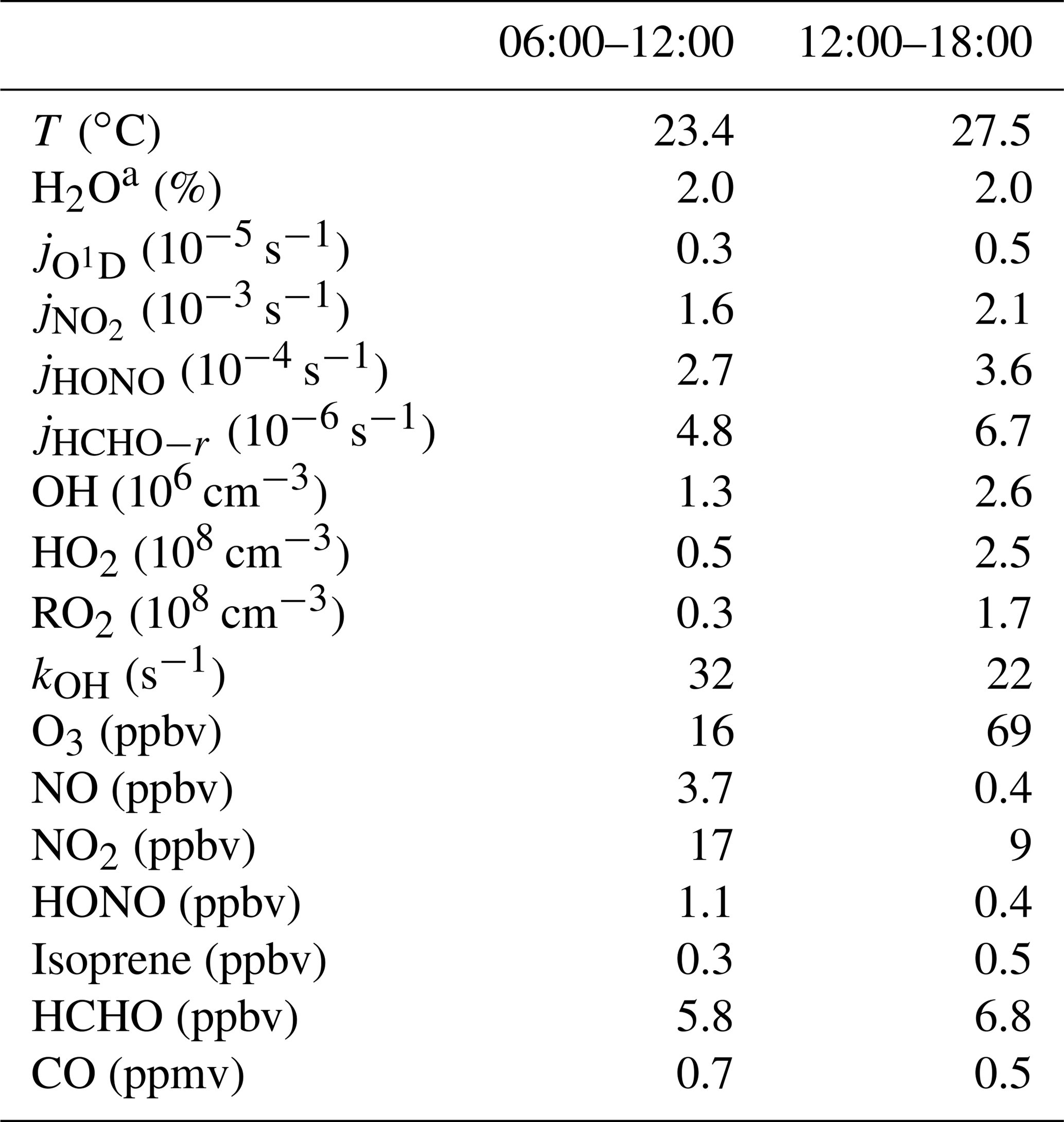
a Volume mixing ratio.
3.2 OH reactivity
The measured OH reactivity showed variations in the range from 15 to 80 s−1 (Fig. S2), with median values of 32 s−1 in the morning and 22 s−1 in the afternoon (Fig. 1). OH reactivities () that were calculated from measured trace gas concentrations, [i] (Table 2) and their OH reaction rate coefficients (ki+OH) show a similar temporal behavior to the measured kOH but underestimate its value systematically during the whole campaign (Fig. S2):
The comparison of measured and calculated OH reactivities (Fig. 1) indicates that the measured trace gases account for only half of the atmospheric OH reactivity. The contributions of CO, NOx, and measured NMHCs during the daytime were about 10 %, 14 %, and 20 %, respectively. Among the measured NMHCs, the groups of alkanes, alkenes (without isoprene), and aromatics had similar shares of reactivity during the day. In the night and in the morning, alkenes and aromatics were the dominating hydrocarbons, while isoprene made a contribution, with up to 6 % of the total kOH during the daytime. Formaldehyde was the only measured OVOC. It contributed 5 %–8 % during the day. The chemical nature of the missing reactivity (about 50 %) is not known but was likely caused by unmeasured VOCs (see Sect. 3.7).
3.3 OH, HO2, and RO2 concentrations
Time series and median diurnal profiles of the measured radical concentrations are shown in Figs. S2 and 1. As in previous campaigns, the diurnal profile of OH shows a high correlation with , which is a proxy for the solar UV radiation driving much of the primary radical production during summertime (e.g., Rohrer et al., 2014; Ehhalt and Rohrer, 2000). It is noteworthy that the measured OH persisted even after sunset with median values 0.7×106 cm−3 until midnight (Fig. 1). Thereafter, OH concentrations dropped continuously until they reached values below the limit of detection shortly before sunrise.
Table 3Unexplained OH signal (mean±1σ) and chemical conditions during the OH chemical modulation test experiments. Given numbers are average values of multiple tests that were performed in the specified time period (see, for example, Fig. S3).
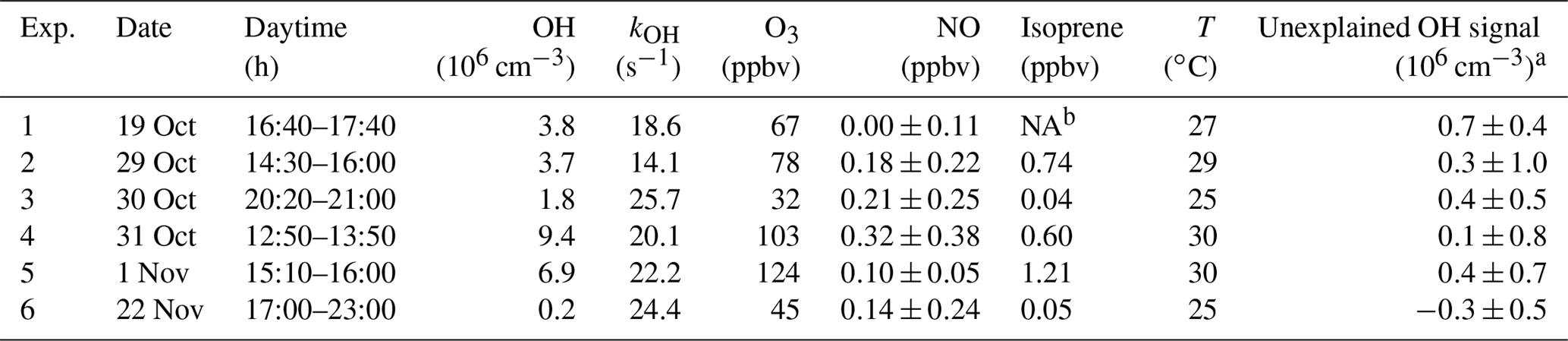
a Expressed as equivalent OH concentration. b Not available.
In order to test whether the OH measurements at Heshan could have been affected by an unknown interference, chemical modulation experiments were performed between noon and midnight on several days (Table 3). Within instrumental precision, no significant unexplained OH interference was detected. This result applies equally to day and night. Therefore, the OH concentration of 1.8×106 cm−3 measured on 30 October after sunset during the chemical modulation test (Table 3) must be considered real ambient OH. On the other hand, all unaccounted OH signals are slightly positively biased except the last one in Table 3. The mean value (±1σ) of the unaccounted OH signals is equivalent to an OH concentration of cm−3, which is at the limit of instrumental detection. In the further analysis, the mean plus 1σ is assumed to be the upper limit for a possible OH interference.
The peroxy radicals, HO2 and RO2, show qualitatively similar diurnal behavior to that which has been reported for other urban environments (e.g., Holland et al., 2003; Shirley et al., 2006; Kanaya et al., 2007; Emmerson et al., 2007). The photochemical build-up of peroxy radicals after sunrise is delayed due to their reaction with high NO concentrations in the morning. The peroxy radical concentrations reach a late maximum around 14:30 in the afternoon, when NO is decreasing (Fig. 1). Median ozone concentrations reach a maximum of 76 ppbv at the same time as the occurrence of the peroxy radical maxima. The concentration of represents about (15±15) % of the total organic peroxy radicals, RO2, during the daytime. This percentage is surprisingly small as measured precursors (alkenes, isoprene, aromatics and large alkanes) dominated the reactivity of the measured hydrocarbons (Fig. 1). A possible reason would be unmeasured VOCs which produce RO2 but not .
3.4 ROx budget
In the ROx budget analysis, only radical initiation and termination reactions play a role. Calculated median diurnal profiles for production (Eq. 1) and destruction (Eq. 2) rates of ROx are shown in Fig. 2. Both rates have maximum values on the order of 3–4 ppbv h−1 at noontime (Fig. 2e and f).
The observed daytime production of ROx (Fig. 2a–e) is dominated with 51 % by the photolysis of HONO (Reaction R1), followed by photolysis of formaldehyde (Reaction R3) and ozone (Reaction R2), which have contributions of 34 % and 15 %, respectively. The counteracting reaction forming HONO from OH and NO is comparatively slow (<5 %; Fig. 2b, blue line). Therefore, the net OH production from HONO photolysis is still the major primary source of ROx. Ozonolysis of measured alkenes (Reaction R4) contributes 7 % during the daytime and is the only primary source considered here at night.
ROx radical termination occurs via reactions with NOx (Reactions R12–R14) and by peroxy radical self-reactions (Reactions R15–R17). In the morning, when the median NOx concentration is about 21 ppbv (Table 2), the reaction of OH with NO2 (Reaction R15) is the dominating loss process (69 %), followed by the reaction of RO2 with NO (Reaction R14; 13 % on average) and the reaction of OH with NO (Reaction R13; 7 % on average). Radical self-reactions play a negligible role in the morning. They gain importance (12 %) in the afternoon, when the median NOx concentration decreases to 9.5 ppbv. Overall, ROx loss during the PRIDE-PRD2014 campaign was mainly controlled by reactions with NOx during the whole day.
The difference between the total ROx production and destruction rates is shown in Fig. 2g. During the daytime, there is an imbalance between production ( and destruction ( rates, which increase from −0.5 ppbv (08:00) to +0.5 ppbv (18:00). The deviations are small (<15 %) compared to the total ROx turnover rate at noon and can be explained by the experimental errors of the data between 08:30 and 17:30. After sunset until 2 h after midnight, remains about 0.5 ppbv h−1 larger than . The deviation may be caused, at least partly, by RO2 formation from reactions of VOCs with NO3 radicals, for which an upper limit of 0.7 ppbv h−1 can be estimated (see Sect. 4.2). The deviation can also largely be explained by experimental errors, when the upper limit of a possible OH interference is included in the total experimental uncertainty (Fig. 2g).
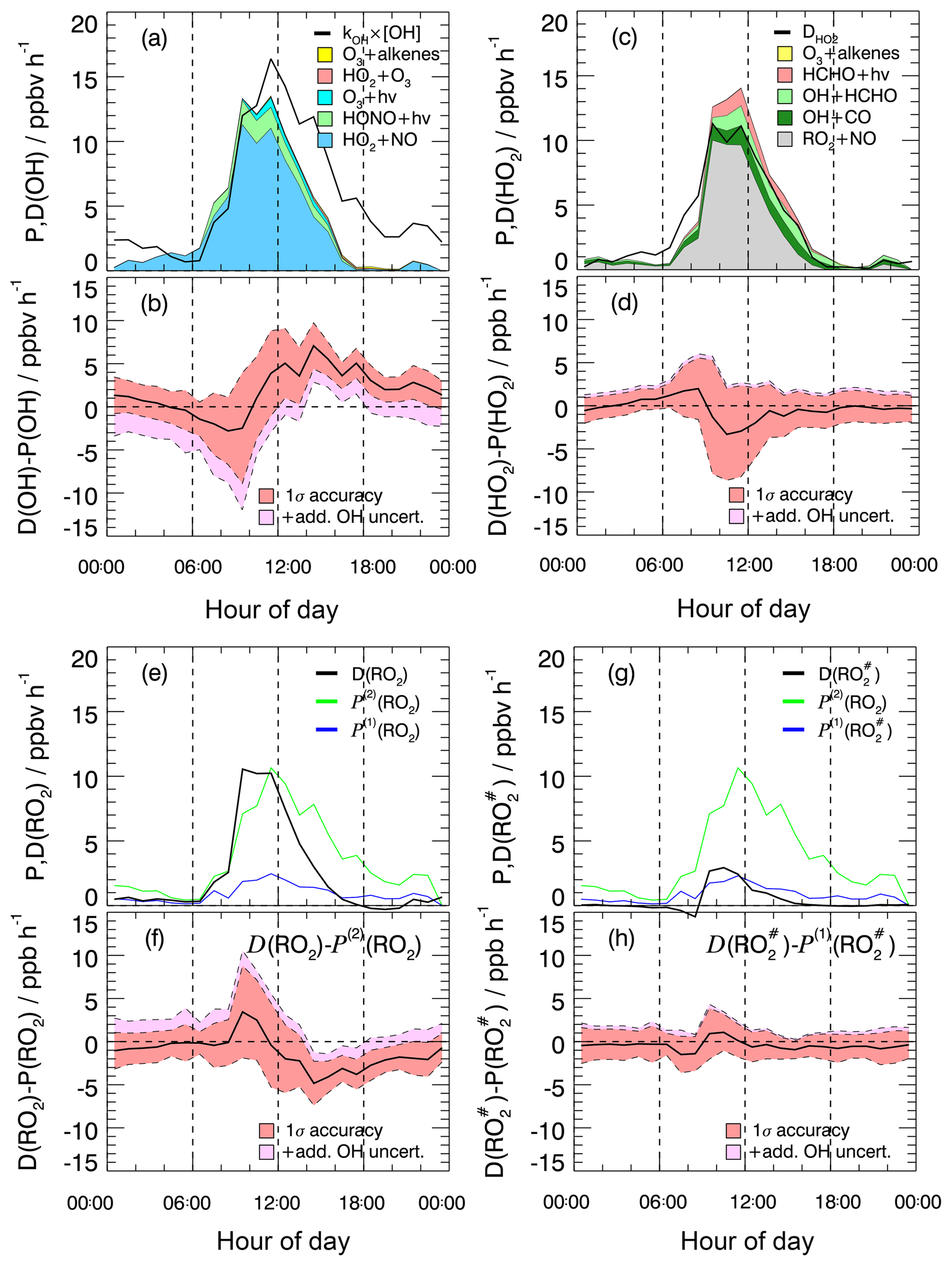
Figure 3Experimental budgets for OH (a, b), HO2 (c, d), RO2 (e, f), and (g, h). In the respective upper panels (a, c, e, g), solid black lines denote the median total destruction rates. The colored areas in (a) and (c) represent cumulative plots of the production rates from different reactions. The blue solid lines in (e) and (g) denote the production rates and , respectively, calculated from measured VOCs (Eq. 7). The green lines represent calculated from kOH(VOC(2)) (Eq. 8). In all four budgets (OH, HO2, RO2, ) the contribution of the radical production from ozonolysis is so small that it is hardly visible. The respective lower panels (b, d, f, h) show the difference between the total destruction and production rates. Red shaded bands indicate the 1σ uncertainty due to experimental errors of the measured quantities (Table S1) and the reaction rate coefficients. The pink shaded areas represent the maximum possible bias from a potential OH interference.
3.5 OH budget
The median OH production (Eq. 4) and destruction (Eq. 3) rates show diurnal profiles with noontime maxima of 13 and 16 ppbv h−1, respectively (Fig. 3a). The calculated OH production rate is dominated throughout the whole day by the recycling reaction of HO2 with NO, which contributes on average 79 % during the day. The most important primary daytime source is the photolysis of HONO. It contributes on average 13 % to the total OH production, whereas the photolysis of ozone and ozonolysis of alkenes add 4 % each. The OH destruction rate is balanced by the calculated production rate between sunrise and noon but is considerably larger than the production rate in the afternoon (Fig. 3b). The difference of (4–6) ppbv h−1 cannot be explained by the combined experimental uncertainties Fig. 3b, even if the upper limit of the potential OH interference is taken into account (Fig. 3b). Since the measured kOH provides a constraint for the total OH loss rate, the imbalance of POH being less than DOH indicates a significant missing OH source in the calculation of POH. Only after sunset would the remaining discrepancy be explainable by a potential OH artifact.
3.6 HO2 budget
The calculated HO2 production (Eq. 5) and destruction rates (Eq. 6) are in good agreement throughout the whole day. The maxima of the rates are on the order of (12–14) ppbv h−1 shortly before noontime (Fig. 3c). The major sources are the reactions of RO2 with NO (63 %) and the reactions of OH with CO (14 %) and formaldehyde (13 %). Primary production processes (photolysis of formaldehyde and ozonolysis of alkenes) contribute 10 % to the total HO2 production. Owing to the relatively high NO concentrations during the campaign, 96 % of the HO2 loss is caused by the reaction of HO2 with NO. The HO2 budget is closed within the experimental uncertainties (Fig. 3d). The magnitude of unexplained OH signals observed in the chemical modulation experiments has no noticeable influence on the closure of the budget (Fig. 3d).
3.7 RO2 budget
Like for HO2, the destruction rate of RO2 (Eq. 9) is almost completely (here, 98 %) due to the reaction with NO, and reaches a maximum shortly before noontime (Fig. 3e). In this case, the maximum has a value of (10–11) ppbv h−1. The production rate calculated by Eq. (7) from measured hydrocarbons (Fig. 3e) is far from being able to compensate for the loss of RO2 radicals. At noontime, the production rate is a factor of 4–5 too small. Once the missing OH reactivity is attributed to unmeasured VOCs, the resulting production rate calculated by Eq. (8) matches relatively well. From sunrise to noon, the budget becomes closed within the experimental uncertainties (Fig. 3f). This result strongly supports the hypothesis that the missing OH reactivity in the morning is caused by unmeasured VOCs.
Assuming that unmeasured VOCs are also responsible for the missing reactivity at other times of the day, an imbalance of (2–5) ppbv h−1 in the RO2 budget is left in the afternoon, where is greater than (Fig. 3f). Considering the experimental uncertainties in the budget equations, the difference is significant from noontime to midnight and indicates a missing RO2 sink. Even if the maximum potential OH interference is taken into account, the imbalance remains from the afternoon until 21:00, while it becomes insignificant later in the night (Fig. 3c).
The radical budget can be treated in a similar way as that for RO2 (see Sect. 2.3.4). The calculated destruction rate of shows a similar diurnal shape to RO2 and is also entirely determined by the reaction with NO (Fig. 3g). Within experimental uncertainty the destruction rate is balanced by the production rate calculated from the measured VOCs known to produce (Fig. 3h). Note that in Fig. 3g looks almost the same as in Fig. 3e, as most of the measured VOCs produce .
If the missing OH reactivity was caused entirely by precursor VOCs, the production rate of organic peroxy radicals would be up to factor of 5 higher than the loss rate (Fig. 3). This suggests that the missing OH reactivity is caused by chemical species that do not produce .
The completeness of the radical measurements allows a budget analysis for all radicals (OH, HO2, and RO2) based on experimental data only, without application of a chemical box model, under the assumption that for the production and loss rates, all relevant species were measured. The ROx budget analysis compares whether the radical initiation reactions of ROx are balanced by the known radical termination reactions; the analysis of the OH, HO2, and RO2 budgets gives insight into the completeness of our understanding of the radical cycling. The interpretation of the budgets below will focus on daytime chemistry (06:00–18:00), as conclusions concerning the nighttime chemistry would be rather limited due to the possible OH interference and the lack of NO3 measurements.
4.1 Missing OH reactivity
Of the atmospheric OH reactivity measured at Heshan, approximately 25 % is explained by measured inorganic compounds (CO and NOx) and another 25 % is explained by measured NMHCs and formaldehyde. The missing reactivity of about 50 % indicates a considerable fraction by unmeasured reactants. Similar missing reactivities have also been observed in forests and other urban environments (Williams et al., 2016; Whalley et al., 2016; Ramasamy et al., 2016; Kaiser et al., 2016; Lu et al., 2013; Edwards et al., 2013; Dolgorouky et al., 2012; Lou et al., 2010; Sadanaga et al., 2005). Depending on the local conditions, missing kOH is generally attributed to unmeasured VOCs which have been emitted or produced by atmospheric oxidation. This hypothesis is plausible, since the atmosphere contains thousands of unknown organic species (Goldstein and Galbally, 2007), but the assumption of unmeasured VOCs is generally difficult to prove. In the present work, the existence of unmeasured atmospheric VOCs deduced from missing OH reactivity is independently confirmed by the analysis of the experimental RO2 budget presented in Sect. 3.7. Only if the missing reactivity is due to VOCs can the discrepancy of up to a factor of 5 between observed RO2 production and destruction rates be reconciled (Fig. 3e, f). In addition, the budget analysis for provides evidence that the unmeasured VOCs are mostly species that do not produce radicals (Fig. 3g, h). As such, they probably do not belong to the class of alkenes or aromatics.
Missing reactivity at the measurement site has also been reported by Yang et al. (2017), who analyzed kOH data measured from 20 October to 19 November 2014 using the comparative reactivity method (CRM) developed by Sinha et al. (2008). Although their time window encompasses the present study, there is little overlap of the data due to data gaps. As far as simultaneous data are available, the two instruments agreed within their combined errors (1 % for the CRM instrument and ±10 % for the LP-LIF instrument). For the time period analyzed by Yang et al. (2017) the fraction of missing reactivity to the total reactivity was reported to be 30 %. Although the percentage value is smaller than in the present paper (50 %), the absolute values for the OH reactivity from unmeasured reactants are comparable. The speciation of the missing reactivity, however, can be different because the higher NOx loading in the period analyzed by Yang et al. (2017) may lead to different photochemical products and may be correlated with different VOC emissions. Missing OH reactivity of about 50 % was also reported for the Backgarden site about 140 km northeast of Heshan, where daytime OH reactivities between 20 and 50 s−1 were measured in summer 2006 (Lou et al., 2010). In that study, the missing reactivity could be explained by unmeasured OVOCs (e.g., formaldehyde, acetaldehyde, methyl vinyl ketone – MVK, and methacrolein – MACR) which were simulated by a chemical box model as products from measured hydrocarbons. With a similar approach, Yang et al. (2017) explain one- to two-thirds of the missing reactivity at Heshan by organic oxidation products (e.g. aldehydes and dicarbonyl compounds) and suspect that the remaining missing reactivity was caused by unknown primary VOC emissions.
Many oxygenated VOCs produce RO2 when they react with OH. Examples are acetaldehyde and higher aldehydes, acetone and higher ketones, MACR, MVK, and methyl glyoxal, all of which were not measured in the present study. In the following discussion, we assume, based on the considerations given above, that the missing reactivity in the present study is entirely due to VOCs (including also OVOCs) which can produce RO2 by reaction with OH.
4.2 Radical budgets, their relationships, and uncertainties
The imbalances in the OH, HO2, and RO2 budgets (D-P) reach median values of up to (7±2.5), , and ppbv h−1, respectively, during the day. Interestingly, the imbalance in the ROx budget does not exceed ±0.5 ppbv h−1 (Fig. 2). This means that the largest uncertainties in the speciated radical budgets compensate each other in the ROx budget. The largest differences between destruction and production rates are found for OH (Fig. 3b) and RO2 (Fig. 3f). The respective diurnal profiles look similar in shape but with opposite signs. When added up in the ROx budget, their values largely compensate each other. The imbalances in the OH and RO2 budgets become large in the afternoon and show a growing trend when NO falls below 1 ppbv (Fig. 4). Above 1 ppbv NO (i.e., in the morning), however, both budgets are closed within their experimental uncertainties. In case of HO2, the destruction and production rates are balanced within experimental error during the whole day, independent of the NO mixing ratio (Figs. 3c and 4).

Figure 4Experimental production and destruction rates for OH, HO2, and RO2 as a function of NO. The circles represent median values for NO intervals of . Data are restricted to daytime conditions ( s−1). The number (no.) of data points included in each NO bin is given in (a). Vertical error bars (red and black) denote the 1σ uncertainties due to experimental errors of the measured quantities (Table S1) and the reaction rate coefficients. Vertical blue bars denote the maximum possible bias from a potential OH interference.
One possible explanation for the imbalances in the OH and RO2 radical budgets would be an unknown radical initiation reaction for OH and an unknown termination reaction for RO2, respectively, which fortuitously balance each other in time and quantity in the ROx budget. This coincidence also seems unlikely because this would mean a drastic increase in ROx production and destruction rates by a factor of 2.5 to 3 in the early afternoon. A more plausible explanation is a partially insufficient description of the radical chain propagation, which proceeds during the day at much higher rates of 10–16 ppbv h−1.
4.2.1 OH interference
The non-closure in the OH and RO2 budgets could be explained, for example, by experimental artifacts as recently reported for OH measurements in some LIF instruments (Mao et al., 2012; Novelli et al., 2014; Feiner et al., 2016). An experimental overestimation of the OH concentration would result in reaction rates that are too high being calculated for the OH destruction (kOH×[OH]) and the RO2 production from the reactions of VOCs with OH. However, the chemical modulation tests carried out under low NO conditions (<1 ppbv) do not show a significant interference. This result is consistent with other tests that were performed with our LIF technique in field campaigns in China (Tan et al., 2018b, 2017) and in laboratory and chamber experiments (Fuchs et al., 2016, 2012). For this reason, we consider OH interference to be an unlikely explanation here, although we cannot strictly exclude the possibility that there were OH interferences only at times when the chemical modulation system was not used.
4.2.2 OH regeneration mechanisms
An alternative explanation for the non-closure in the OH and RO2 budgets would be a chemical mechanism that effectively converts RO2 to OH. Unimolecular isomerization and decomposition reactions of RO2 can be such an OH source when the competing reaction with NO is slow. This chemistry has been known for a long time as autoxidation, for example, in low-temperature combustion (e.g., Cox and Cole, 1985; Glowacki and Pilling, 2010). Its potential relevance to atmospheric chemistry at ambient temperatures has only recently been recognized (e.g., Peeters et al., 2009; da Silva et al., 2010; Crounse et al., 2013; Praske et al., 2018). Autoxidation involves an intramolecular H shift in the RO2 molecule leading to a hydroperoxy alkyl radical, which is often named QOOH. This radical can generally undergo various types of reactions, such as a reaction with O2 producing an oxygenated VOC+HO2, or decomposition to an oxygenated VOC by elimination of OH from the -OOH group (e.g., Peeters et al., 2009; da Silva et al., 2010; Crounse et al., 2013; Praske et al., 2018). Another path is the addition of O2 forming a hydroperoxy peroxy radical (O2)QOOH. This new peroxy radical can then react with NO, HO2, and RO2 or undergo another internal H-shift reaction. Repetitive sequential H-shift reactions followed by O2 addition lead to highly oxidized RO2 radicals which produce highly oxidized molecules (HOMs) by radical termination reactions (e.g., Ehn et al., 2014, 2017; Jokinen et al., 2014). Due to their low vapor pressure, HOMs are efficient precursors for organic particles, which are produced from the original VOCs with yields in the low percent range (e.g., Ehn et al., 2014; Jokinen et al., 2015).
RO2 isomerization producing HOx radicals is known to occur in the oxidation of isoprene ((Peeters et al., 2014, 2009; Peeters and Muller, 2010; da Silva et al., 2010; Crounse et al., 2011; Fuchs et al., 2013; Teng et al., 2017) and methacrolein (Crounse et al., 2012; Fuchs et al., 2014). In Heshan, the production rate of isoprene peroxy radicals from the reaction of isoprene with OH never exceeded 0.5 ppbv h−1. Even if every isoprene-derived RO2 radical regenerated one OH molecule (which is not likely because of the competing reaction with NO), the process could explain only a small fraction of the missing OH production rate. The concentration of MACR was not measured but is generally not larger than that of isoprene (Karl et al., 2009; Shao et al., 2009). Since the OH rate constant is smaller than that for isoprene, OH regeneration by unimolecular reactions of MACR-derived RO2 is expected to be even less important.
Other than for isoprene and methacrolein, autoxidation of RO2 leading to HOx formation has been experimentally studied for only a few other VOCs, including 3-pentanone (Crounse et al., 2013), glyoxal (Lockhart et al., 2013), n-hexane and 2-hexanol (Praske et al., 2018), hydroxymethyl hydroperoxides (Allen et al., 2018), and 2-hydroperoxy-2-methylpentane (Praske et al., 2019). While isoprene and methacrolein chemistry is especially important in biogenically controlled environments, the new studies demonstrate that autoxidation can also be expected to play a role in urban atmospheres when NO concentrations are as low as 500 pptv (see Praske et al., 2018). Systematic theoretical studies have shown that the rates of H shifts in RO2 depend very much on their chemical structure (e.g., Crounse et al., 2013; Otkjær et al., 2018; Møller et al., 2019) and range from 10−4 to 10 s−1 at ambient temperatures. Low rate coefficients can be expected, for example, for 1,5-H or 1,6-H shift reactions in linear alkyl radicals, whereas the presence of (multiple) functional groups like -OH, -OOH, or -CHO may increase the H-shift rate by orders of magnitude. The possible yield of OH, HO2, or higher-oxidized RO2 radicals from a hydrogen shift depends on the chemical structure and functionality. Elimination of OH or HO2 is generally supported by the presence of functional groups (e.g., -OH, -OOH, or -CHO).
In the present campaign, the potential conversion of RO2 to OH by a unimolecular reaction would require a rate of about 0.08 s−1 to close the budgets of OH and RO2 if all measured RO2 radicals could produce OH from H-shift reactions (Fig. S4). Although the rate is in the possible range of H-shift reactions, it is questionable if a major fraction of RO2 was structurally capable of undergoing a fast H shift leading to OH formation. As approximately half of the measured OH reactivity is likely due to unmeasured VOCs with unknown speciation and owing to the general lack of kinetic and mechanistic studies for specific RO2 radicals, it is not possible to be more quantitative here. If the missing reactivity at Heshan was caused by photochemically aged, functionalized oxygenated VOCs, there is a chance that RO2 radicals from these compounds could have contributed significantly to the missing RO2 sink or missing OH source. If autoxidation played a significant role, additional HO2 formation from H-shift reactions would also be expected. However, the closed HO2 budget gives no indication for this.
Another possibility for converting RO2 to OH under low NO conditions is the reaction of RO2 with HO2. The reaction is generally considered to be chain terminating, but in the case of acyl peroxy and α-carbonyl peroxy radicals, a parallel reaction channel can produce OH with yields up to 80 % (Fuchs et al., 2018; Winiberg et al., 2016; Praske et al., 2015; Groß et al., 2014; Hasson et al., 2012, 2004; Dillon and Crowley, 2008; Jenkin et al., 2008). With reaction rate constants in the range of (1–2) , the OH production would be less than 0.1 ppbv h−1 even if all measured RO2 species produce OH. In conclusion, the mechanisms already known for conversion of RO2 to HO2 are not sufficient in explaining the missing RO2 sink and missing OH source.
The observations in Heshan resemble qualitatively those of the previous PRIDE-PRD 2006 campaign in Backgarden (Hofzumahaus et al., 2009), where the experimental OH budget indicated a missing OH source (28 ppbv h−1) in the afternoon, when NO was less than 1 ppbv. Box model simulations underestimated the measured OH concentrations by a factor 3–5 at low NO but agreed well with measured HO2 concentrations. In that study, the isoprene concentrations were considerably higher, reaching several parts per billion by volume. Isomerization reactions of isoprene peroxy radicals could explain only a small part (max 20 %) of the missing OH source (Lu et al., 2012; Fuchs et al., 2013). The observed behavior of OH and HO2 could be reproduced by assuming a hypothetical mechanism in which RO2 is converted to HO2 and HO2 is converted to OH by an unknown reactant X with a concentration equivalent to 0.8 ppbv NO. Owing to the lack of RO2 measurements, the mechanism could not be directly tested for RO2. In Heshan, the same mechanism would be able to close both the RO2 and OH budgets if an equivalent of 0.4 ppbv NO is assumed (Fig. S4). Here, the NO concentration of 0.4 ppbv corresponds to the rate coefficient of 0.08 s−1 discussed above. When using the X mechanism, the closure of the HO2 and ROx budgets remains unaffected.. Therefore, the X mechanism is one possibility for describing all radical budgets at Heshan consistently, although its chemical nature remains unresolved.
4.2.3 Additional uncertainties in the budget analyses
As pointed out in Sect. 4.1, unmeasured VOCs were most likely responsible for the observed missing OH reactivity. This not only considerably influences the radical chain propagation from OH to RO2 (Fig. 3e, g) but can also affect the primary production of radicals by ozonolysis and photolysis (see Supplement). Furthermore, Cl reactions with measured and unmeasured VOCs may have initiated ROx chain reactions in the early morning. These reactions could have slightly influenced the ROx budget but are of minor importance compared to the radical chain propagation reactions (Supplement).
Reactions of NO3 with VOCs are an additional RO2 source which are neglected in the budget calculation in Sect. 2.3.4. The relevance can be estimated from the production rate of NO3, which is calculated from the reaction of NO2 with O3 (); MCM3.3.1). In this campaign, the NO3 production rate was on the order of 1.4 and 0.7 ppbv h−1 during the daytime and nighttime, respectively. Because NO3 is efficiently photolyzed in the bright hours of the day, it is generally assumed that it plays a negligible role as an oxidant during the daytime. Liebmann et al. (2018) recently showed that this is not always the case. They reported measurements in a forested environment in southern Germany, demonstrating that more than 25 % of the daytime NO3 reacted with biogenic VOCs. Under the conditions at Heshan 2014, the main loss process during the daytime is the reaction with NO. If we neglect unmeasured VOCs, the percentage removal of NO3 in the morning is 96 % by NO, 3 % by photolysis, and 1 % by measured VOCs. In the afternoon, the corresponding values are 72 %, 21 %, and 7 %. Thus, the estimated RO2 production rate from NO3 reactions with known VOCs was probably not more than 0.1 ppbv h−1 during the daytime. It is conceivable that unmeasured VOCs, which probably accounted for 50 % of the OH reactivity, contributed by a similar magnitude. During the daytime, these contributions are relatively small compared to the total production rate of RO2. The tendency is to slightly increase the imbalance between the production and destruction rate of RO2 observed in the afternoon (Fig. 3). At sunset and in the night, the NO3 production rate of 0.7 ppbv h−1 can be considered an upper limit for the RO2 production. This value can possibly explain, at least partly, the imbalance of about 0.5 ppbv h−1 in the ROx budget after sunset (Fig. 2).
Another uncertainty is caused by the measurement and incomplete representation of the RO2 chemistry. Due to the measurement principle of the ROxLIF instrument, only those RO2 species are measured which are converted in the instrument to HO2 by reaction with NO. This measurement is suitable to quantify the HO2 production rate (Eq. 5). Among the RO2 radicals which are not completely captured by ROxLIF are those species which produce a new RO2 radical when they react with NO. As these reactions are neutral with respect to the total RO2, the RO2 budget (D-P) is not sensitive to the RO2 measurement bias caused by these species (see Supplement).
Other uncertainties in the RO2 budget are caused by the rate constants for the reactions of RO2 with NO (Reactions R8, R14), RO2 (Reaction R15), and HO2 (Reaction R16) that are given in Table 1 as effective values for the lumped RO2 radicals (Jenkin et al., 2019). In this work, the uncertainty of the rate coefficients for Reactions (R15) and (R16) plays only a minor role because the daytime loss of the peroxy radicals was largely dominated by the reaction with NO (see Supplement). The relevant range for the reaction rate constants of different RO2 species with NO (Reactions R8, R14) is between and cm3 s−1 (see Supplement). As a sensitivity test, Figs. S5 and S6 show the budgets of ROx, RO2, and HO2 for a rate constant of cm3 s−1. The results are essentially the same as in Figs. 2 and 3, where a rate constant of cm3 s−1 was applied for Reactions (R8) and (R14). Thus, an increased rate constant cannot explain the missing RO2 sink in the RO2 budget.
Also the unknown branching ratio in the reaction of RO2 with NO, which can produce HO2 (Reaction R8, chain propagating) or organic nitrates (Reaction R14, chain terminating), is uncertain (see Sect. 2.3.1 and Supplement). Changing the yields for organic nitrates from 5 % to 20 % has a small but notable influence on the ROx budget, reversing the slightly negative bias (D<P) to a lightly positive one (D>P; Fig. S7). In both limits, the ROx budget remains closed within experimental errors. The influence of the different HO2 yields on the production rate of HO2 is small (Fig. S8). For the range of tested yields, the HO2 budget remains balanced within experimental uncertainties.
4.3 Photochemical ozone production
Photochemical ozone production in the troposphere is due to the oxidation of NO to NO2 by reaction with peroxy radicals (Reactions R8, R9), followed by NO2 photolysis yielding NO and O(3P) atoms. The O atoms combine with O2 and form ozone. The net ozone production can be estimated from the production rate of NO2 via Reactions (R8) and (R9), corrected for chemical loss of NO2 by reaction with OH (Reaction R12; e.g., Kanaya et al., 2007; Ren et al., 2013; Brune et al., 2016):
Chemical loss of ozone by photolysis (Reaction R2), ozonolysis reactions (Reaction R4), and dry deposition are negligible under the given conditions. Calculated losses of ozone by photolysis and ozonolysis are not larger than 0.2 ppbv h−1. The dry deposition rate during the daytime is estimated to be no more than 1 ppbv h−1 when assuming a mixed boundary layer height of 1 km and a maximum deposition velocity of 1 cm s−1 (e.g., Wesely et al., 2000). Using the rates shown in Fig. 2f (OH+NO2), Fig. 3c (), and Fig. 3e (), a daily integrated net ozone production of (102±31) ppbv is calculated (06:00 to 18:00). For comparison, the daily integrated OH+NO2 term is (14±3) ppbv. About 70 % of the ozone is produced in the morning (06:00–12:00), and 30 % is produced in the afternoon (12:00–18:00).
The radical budgets for OH, HO2, and RO2 allow tracing back the chemical processes that are driving the production of peroxy radicals and therefore ozone formation. The first term (k8[RO2][NO]) in Eq. (11) can be considered the contribution of the VOCs that form RO2, which continue to react with NO to HO2. As the HO2 budget is essentially balanced, the HO2 loss term (k9 [HO2] [NO]) can be replaced by the rate of HO2-producing processes (Reactions R3, R6, R7, R8), shown in Fig. 3c. This replacement implicitly assumes that other HO2 losses such as HO2+RO2 and HO2+HO2 are negligible, which is valid during this study. The ozone production from RO2 and HO2 can then be expressed as
Using Eq. (12) yields a daily net-ozone production of 112 ppbv (Fig. 5). This value is in close agreement with the result of Eq. (11), which uses different experimental input parameters. According to Eq. (12), a percentage of 78 % of the daily net-ozone production results from the oxidation of VOCs via Reactions (R5), (R8), and (R9); 14 % results from reactions of HCHO (Reactions R3 and R6, followed by Reaction R9); and 8 % results from CO oxidation (Reactions R7, R9). Measured VOCs that produce RO2 account for only 18 % of the total ozone production, while unmeasured VOCs contribute the dominant fraction of 60 %.
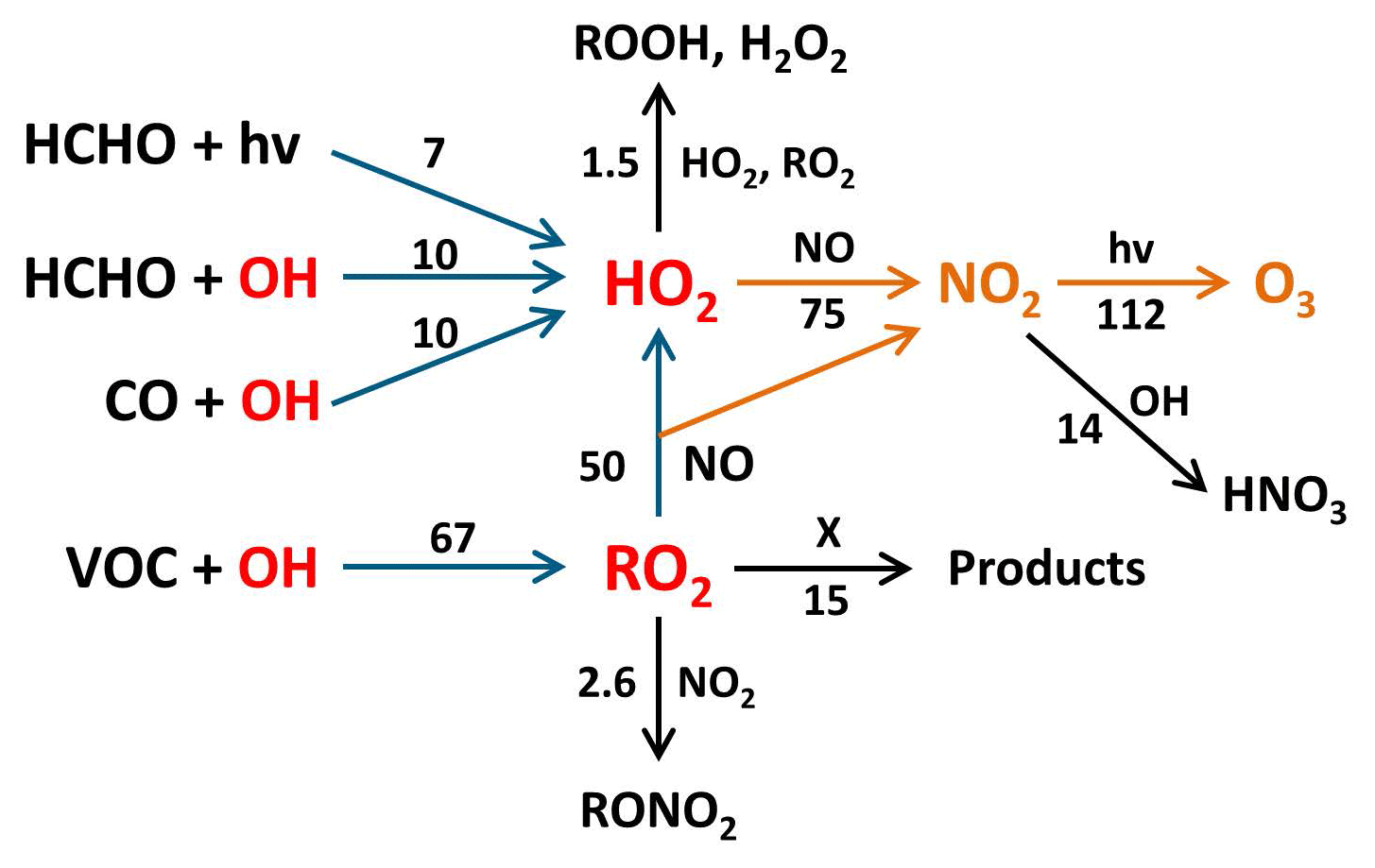
Figure 5Main reaction pathways leading to photochemical net ozone formation during the Heshan campaign. Blue arrows show the reaction paths leading to RO2 and HO2, and yellow arrows indicate the oxidation reactions of NO to NO2 by peroxy radicals with subsequent NO2 photolysis yielding ozone. Black arrows represent reactions that remove peroxy radicals or NO2 and thus reduce ozone production. Numbers represent median daily integrated reaction rates (ppbv) calculated by Eq. (12). The value for VOC+OH corresponds to kOH(VOC(2))×[OH] (see Eq. 8). The reaction with X indicates an unknown process that removes RO2 without producing NO2.
In principle, the first term (k8[RO2][NO]) in Eqs. (11) or (12) could be replaced by the production rate calculated from the total VOC reactivity (kOH(VOC(2))×[OH]) if the RO2 budget is balanced:
Using this equation, an integrated net-ozone production of 140 ppbv would be calculated. However, the RO2 budget is not balanced (Fig. 3e) and indicates a missing RO2 sink, which does not oxidize NO and therefore does not produce ozone. This possibility was first suggested when the conversion of RO2 to OH by X was proposed to explain a missing OH source in the PRD in 2006 (Hofzumahaus et al., 2009). In the present case, such an RO2 sink would remove 15 ppbv (22 %) of the daily produced RO2, resulting in an integrated ozone production that is 30 ppbv lower than expected from the rate of VOC oxidation.
The possible underprediction of the photochemical ozone production from unknown (unmeasured) atmospheric VOCs has been pointed out in previous studies, where RO2 concentrations have been modeled (e.g., Griffith et al., 2016) or estimated from OH reactivities (Whalley et al., 2016). In the ClearfLo campaign 2012 in central London, the ozone production calculated from the oxidation of C2-C8 VOC species (measured by a standard GC FID) was about 60 % smaller than calculated from the total organic OH reactivity (≤C12). The ozone underprediction for the case of using standard VOC measurements (C2-C8) alone is comparable to the present work. However, the calculated ozone production from the oxidation of VOCs may be overestimated if an unknown RO2 loss exists as is shown above. In a further study related to observations in the ClearfLo campaign, the comparison of measured and modeled radical concentrations (OH, HO2, , and RO2) points to a significant missing OH source and a missing sink for peroxy radicals at NO concentrations below 1 ppbv (Whalley et al., 2018), which is a similar trend to the Heshan campaign for OH and RO2. The results of both campaigns indicate significant gaps in the understanding of the radical chemistry and ozone formation in urban air at low NO conditions, which will require further investigations.
A field campaign was carried out near the city of Heshan in autumn 2014, studying the radical chemistry under anthropogenically polluted conditions in the Pearl River Delta in southern China. Measurements of radical concentrations (OH, HO2, RO2, and ), OH reactivity, and numerous other trace gases were performed. OH reactivity was in the range from 15 to 80 s−1, with median values of 32 s−1 in the morning and 22 s−1 in the afternoon. Approximately 25 % of the reactivity could be explained by measured CO and NOx, and another 25 % could be explained by measured hydrocarbons and formaldehyde, with a remainder of 50 % missing reactivity from unmeasured components. OH concentrations reached maximum median values of 4.5×106 cm−3 at noon. HO2 and RO2 reached their maximum concentrations later in the afternoon, with values of 3×108 and 2.0×108 cm−3, respectively. Measured (peroxy radicals mainly from alkenes and aromatics) made up only a small part (15 %) of the total RO2, although the fraction of -producing VOCs made the largest contribution (94 %) to the reactivity of measured VOCs. It suggests that at least part of the missing reactivity was due to unmeasured VOCs which produce RO2 but not .
The diurnal profile of OH was highly correlated with solar radiation, and a significant median OH concentration of 0.7×106 cm−3 remained after sunset until midnight. In the remaining night, the concentrations dropped below the limit of detection (0.4×106 cm−3; 1σ). Chemical modulation experiments were performed on several days between noon and midnight in order to test whether the OH measurements could be biased by artificially produced OH in the low-pressure LIF detection cell. The test experiments were performed at OH reactivities of (14–26) s−1, NO mixing ratios below 1 ppbv, relative high ozone concentrations (45–124 ppbv), and high air temperatures (25–30 ∘C). A possible OH interference equivalent to a concentration of cm−3 was found at the limit of detection.
In one of the test experiments, high OH nighttime values (around 1.8×106 cm−3) were measured after sunset. These relatively high values are significantly larger than the possible OH interference determined in that test, suggesting that the measured OH was real.
The completeness of the radical measurements during the daytime allowed, for the first time, performing experimental budget analyses for all radicals (OH, HO2, and RO2). There are differences between this method and the analysis often performed in model-based studies. In those studies, turnover rates are calculated from radicals and species that are simulated by a box model. Furthermore, balances between radical production and destruction rates are enforced even if the chemical mechanism is incorrect. In contrast, imbalances in a fully constrained experimental budget analysis, as applied here, indicate directly unknown experimental errors in the input data or an inconsistent chemical mechanism underlying the evaluation.
The balance between radical initiation and termination was studied in the ROx budget. ROx was mainly produced by photolysis of HONO (51 %), HCHO (34 %) and ozone (15 %), and ozonolysis of alkenes (7 %). The production with a maximum rate of 3–4 ppbv h−1 was essentially balanced within 0.5 ppbv h−1 by the destruction of ROx species with NO or NO2.
In case of RO2, the production rate calculated from measured VOCs is a factor of 4–5 too small to compensate the destruction rate of up to 11 ppbv h−1 in the afternoon, which is mainly determined by the loss reaction with NO. Only when the missing OH reactivity is explained by unmeasured VOCs can the RO2 loss rate be balanced. The general assumption that missing OH reactivity is equivalent to unmeasured VOCs is thus directly confirmed by RO2 measurements. Although the closure of the RO2 budget is greatly improved, a significant imbalance of (2–5) ppbv h−1 remains in the afternoon, indicating a missing RO2 sink under low NO conditions. As far as is concerned, the chemical budget can be quantitatively closed within relatively large experimental error margins if only measured VOCs are considered for RO2 production. This result implies that the unmeasured VOCs did not produce and therefore do not belong to the group of alkenes and aromatics.
The OH destruction is compensated in the morning by the known OH sources from photolysis (HONO and O3), ozonolysis of alkenes, and OH recycling (HO2+NO; Reaction R9). In the afternoon, however, the destruction rate is significantly higher than the calculated production rate, which indicates a considerable missing OH source of (4–6) ppbv h−1. The daily variation of the missing OH source looks similar to that of the missing RO2 sink, but with the opposite sign, so that both compensate each other largely in the ROx budget. Contrary to OH and RO2, the HO2 budget is essentially balanced over the whole day. The difference between production and destruction rates for OH and RO2 shows an increasing trend when NO falls below 1 ppbv and becomes insignificant above 1 ppbv of NO.
The observations indicate the existence of chemical processes that convert RO2 to OH without the involvement of NO. Such processes have been discovered in recent years in the photochemical degradation of isoprene and methacrolein, where OH is regenerated by unimolecular RO2 reactions. However, due to the low abundance of isoprene in the present campaign, these reactions account for only a small fraction (<10 %) of the missing RO2 sink and missing OH source. A generic mechanism has been postulated previously to explain a missing OH source in the PRD under low NO conditions (Hofzumahaus et al., 2009). It involves the successive conversion of RO2 to HO2 and then to OH by an unknown reactant X. A concentration of X equivalent to 0.4 ppbv NO would close the budgets of RO2 and OH in the present study and leave the budgets of HO2 and ROx unchanged. The X mechanism is, therefore, one possibility for describing all radical budgets at Heshan consistently, although its chemical nature remains unresolved.
The photochemical net ozone production rate was calculated from the reaction rates of HO2 and RO2 with NO, yielding a daily integrated amount of 102 ppbv O3. This amount is due to the oxidation of VOCs (78 %), formaldehyde (14 %), and CO (8 %). About 60 % of the ozone production is caused by unmeasured VOCs, which account for half of the measured OH reactivity. An even larger integrated net-ozone production would be calculated from the reaction rate of measured and unmeasured VOCs with OH if all RO2 radicals react with NO. However, the unknown RO2 loss reaction removes 22 % of the daily RO2 production and thus causes 30 ppbv less ozone production per day than would be expected from the VOC oxidation rate.
In summary, the current work provides new arguments for the existence of a missing OH source, which is most likely due to the conversion of RO2 radicals without the involvement of NO. Our line of argument depends critically on the assumption that the OH measurement technique is free of artifacts which would erroneously increase the calculated OH loss and RO2 production rates. The experimental tests that were performed in the campaign give no evidence for such an interference, but uncertainty remains because the tests have not covered the whole period of the campaign. Therefore, further field experiments with continuous control of the LIF measurements by chemical modulation are planned.
The data used in this study are available from the corresponding author upon request (a.hofzumahaus@fz-juelich.de).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-7129-2019-supplement.
YZ, AW, and KL organized the field campaign. AH and ZT analyzed the data and wrote the paper. All authors contributed to measurements, discussing results, and commenting on the paper.
The authors declare that they have no conflict of interest.
We thank the science teams of PRIDE-PRD2014 for support during the field campaign. We also thank the anonymous referees for insightful and constructive comments, which helped in improving the paper.
This research has been supported by the National Natural
Science Foundation of China (grant nos. 21522701, 91544225, 21190052, and 41375124),
the National Key R&D Plan of China (grant no. 2017YFC0213000), the National Science and
Technology Support Program of China (grant no. 2014BAC21B01), the
Strategic Priority Research Program of the Chinese Academy of Sciences (grant no. XDB05010500),
the Collaborative Innovation Center for Regional Environmental Quality,
the BMBF project ID-CLAR (grant no. 01DO17036), and the EU project AMIS
(Fate and Impact of Atmospheric Pollutants; grant no. PIRSES-GA-2011-295132).
The article
processing charges for this open-access
publication were
covered by a Research
Centre of the Helmholtz
Association.
This paper was edited by Dwayne Heard and reviewed by three anonymous referees.
Allen, H. M., Crounse, J. D., Bates, K. H., Teng, A. P., Krawiec-Thayer, M. P., Rivera-Rios, J. C., Keutsch, F. N., St. Clair, J. M., Hanisco, T. F., Møller, K. H., Kjaergaard, H. G., and Wennberg, P. O.: Kinetics and Product Yields of the OH Initiated Oxidation of Hydroxymethyl Hydroperoxide, J. Phys. Chem. A, 122, 6292–6302, https://doi.org/10.1021/acs.jpca.8b04577, 2018.
Atkinson, R., Lloyd, A. C., and Winges, L.: An updated chemical mechanism for hydrocarbon/NOx/SO2 photooxidations suitable for inclusion in atmospheric simulation models, Atmos. Environ., 16, 1341–1355, https://doi.org/10.1016/0004-6981(82)90055-5, 1982.
Bohn, B., Corlett, G. K., Gillmann, M., Sanghavi, S., Stange, G., Tensing, E., Vrekoussis, M., Bloss, W. J., Clapp, L. J., Kortner, M., Dorn, H.-P., Monks, P. S., Platt, U., Plass-Dülmer, C., Mihalopoulos, N., Heard, D. E., Clemitshaw, K. C., Meixner, F. X., Prevot, A. S. H., and Schmitt, R.: Photolysis frequency measurement techniques: results of a comparison within the ACCENT project, Atmos. Chem. Phys., 8, 5373–5391, https://doi.org/10.5194/acp-8-5373-2008, 2008.
Brune, W. H., Baier, B. C., Thomas, J., Ren, X., Cohen, R. C., Pusede, S. E., Browne, E. C., Goldstein, A. H., Gentner, D. R., Keutsch, F. N., Thornton, J. A., Harrold, S., Lopez-Hilfiker, F. D., and Wennberg, P. O.: Ozone production chemistry in the presence of urban plumes, Faraday Discuss., 189, 161–189, https://doi.org/10.1039/C5FD00204D, 2016.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov (last access: 27 May 2019), 2015.
Chan, C. K. and Yao, X.: Air pollution in mega cities in China, Atmos. Environ., 42, 1–42, https://doi.org/10.1016/j.atmosenv.2007.09.003, 2008.
Cox, R. A. and Cole, J. A.: Chemical aspects of the autoignition of hydrocarbon?air mixtures, Combust. Flame, 60, 109–123, https://doi.org/10.1016/0010-2180(85)90001-X, 1985.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011.
Crounse, J. D., Knap, H. C., Ornso, K. B., Jorgensen, S., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O-2, J. Phys. Chem. A, 116, 5756–5762, https://doi.org/10.1021/jp211560u, 2012.
Crounse, J. D., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
da Silva, G.: Hydroxyl radical regeneration in the photochemical oxidation of glyoxal: kinetics and mechanism of the HC(O)CO + O2 reaction, Phys. Chem. Chem. Phys., 12, 6698–6705, https://doi.org/10.1039/b927176g, 2010.
Dillon, T. J. and Crowley, J. N.: Direct detection of OH formation in the reactions of HO2 with CH3C(O)O2 and other substituted peroxy radicals, Atmos. Chem. Phys., 8, 4877–4889, https://doi.org/10.5194/acp-8-4877-2008, 2008.
Dolgorouky, C., Gros, V., Sarda-Esteve, R., Sinha, V., Williams, J., Marchand, N., Sauvage, S., Poulain, L., Sciare, J., and Bonsang, B.: Total OH reactivity measurements in Paris during the 2010 MEGAPOLI winter campaign, Atmos. Chem. Phys., 12, 9593–9612, https://doi.org/10.5194/acp-12-9593-2012, 2012.
Edwards, P. M., Evans, M. J., Furneaux, K. L., Hopkins, J., Ingham, T., Jones, C., Lee, J. D., Lewis, A. C., Moller, S. J., Stone, D., Whalley, L. K., and Heard, D. E.: OH reactivity in a South East Asian tropical rainforest during the Oxidant and Particle Photochemical Processes (OP3) project, Atmos. Chem. Phys., 13, 9497–9514, https://doi.org/10.5194/acp-13-9497-2013, 2013.
Ehhalt, D. H.: Photooxidation of trace gases in the troposphere, Phys. Chem. Chem. Phys., 1, 5401–5408, https://doi.org/10.1039/A905097C, 1999.
Ehhalt, D. H. and Rohrer, F.: Dependence of the OH concentration on solar UV, J. Geophys. Res., 105, 3565–3571, https://doi.org/10.1029/1999JD901070, 2000.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Ehn, M., Berndt, T., Wildt, J., and Mentel, T.: Highly Oxygenated Molecules from Atmospheric Autoxidation of Hydrocarbons: A Prominent Challenge for Chemical Kinetics Studies, Int. J. Chem. Kinet., 49, 821–831, https://doi.org/10.1002/kin.21130, 2017.
Emmerson, K. M., Carslaw, N., Carslaw, D. C., Lee, J. D., McFiggans, G., Bloss, W. J., Gravestock, T., Heard, D. E., Hopkins, J., Ingham, T., Pilling, M. J., Smith, S. C., Jacob, M., and Monks, P. S.: Free radical modelling studies during the UK TORCH Campaign in Summer 2003, Atmos. Chem. Phys., 7, 167–181, https://doi.org/10.5194/acp-7-167-2007, 2007.
Feiner, P. A., Brune, W. H., Miller, D. O., Zhang, L., Cohen, R. C., Romer, P. S., Goldstein, A. H., Keutsch, F. N., Skog, K. M., Wennberg, P. O., Nguyen, T. B., Teng, A. P., DeGouw, J., Koss, A., Wild, R. J., Brown, S. S., Guenther, A., Edgerton, E., Baumann, K., and Fry, J. L.: Testing Atmospheric Oxidation in an Alabama Forest, J. Atmos. Sci., 73, 4699–4710, https://doi.org/10.1175/jas-d-16-0044.1, 2016.
Fishman, J., Ramanathan, V., Crutzen, P. J., and Liu, S. C.: Tropospheric ozone and climate, Nature, 282, 818, https://doi.org/10.1038/282818a0, 1979.
Fuchs, H., Holland, F., and Hofzumahaus, A.: Measurement of tropospheric RO2 and HO2 radicals by a laser-induced fluorescence instrument, Rev. Sci. Instrum., 79, 084104, https://doi.org/10.1063/1.2968712, 2008.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Fuchs, H., Dorn, H.-P., Bachner, M., Bohn, B., Brauers, T., Gomm, S., Hofzumahaus, A., Holland, F., Nehr, S., Rohrer, F., Tillmann, R., and Wahner, A.: Comparison of OH concentration measurements by DOAS and LIF during SAPHIR chamber experiments at high OH reactivity and low NO concentration, Atmos. Meas. Tech., 5, 1611–1626, https://doi.org/10.5194/amt-5-1611-2012, 2012.
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H. P., Haseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/ngeo1964, 2013.
Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Holland, F., Kaminski, M., Li, X., Lu, K., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Wahner, A.: OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 14, 7895–7908, https://doi.org/10.5194/acp-14-7895-2014, 2014.
Fuchs, H., Tan, Z., Hofzumahaus, A., Broch, S., Dorn, H.-P., Holland, F., Künstler, C., Gomm, S., Rohrer, F., Schrade, S., Tillmann, R., and Wahner, A.: Investigation of potential interferences in the detection of atmospheric ROx radicals by laser-induced fluorescence under dark conditions, Atmos. Meas. Tech., 9, 1431–1447, https://doi.org/10.5194/amt-9-1431-2016, 2016.
Fuchs, H., Novelli, A., Rolletter, M., Hofzumahaus, A., Pfannerstill, E. Y., Kessel, S., Edtbauer, A., Williams, J., Michoud, V., Dusanter, S., Locoge, N., Zannoni, N., Gros, V., Truong, F., Sarda-Esteve, R., Cryer, D. R., Brumby, C. A., Whalley, L. K., Stone, D., Seakins, P. W., Heard, D. E., Schoemaecker, C., Blocquet, M., Coudert, S., Batut, S., Fittschen, C., Thames, A. B., Brune, W. H., Ernest, C., Harder, H., Muller, J. B. A., Elste, T., Kubistin, D., Andres, S., Bohn, B., Hohaus, T., Holland, F., Li, X., Rohrer, F., Kiendler-Scharr, A., Tillmann, R., Wegener, R., Yu, Z., Zou, Q., and Wahner, A.: Comparison of OH reactivity measurements in the atmospheric simulation chamber SAPHIR, Atmos. Meas. Tech., 10, 4023–4053, https://doi.org/10.5194/amt-10-4023-2017, 2017.
Fuchs, H., Albrecht, S., Acir, I., Bohn, B., Breitenlechner, M., Dorn, H.-P., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Kaminski, M., Keutsch, F. N., Novelli, A., Reimer, D., Rohrer, F., Tillmann, R., Vereecken, L., Wegener, R., Zaytsev, A., Kiendler-Scharr, A., and Wahner, A.: Investigation of the oxidation of methyl vinyl ketone (MVK) by OH radicals in the atmospheric simulation chamber SAPHIR, Atmos. Chem. Phys., 18, 8001–8016, https://doi.org/10.5194/acp-18-8001-2018, 2018.
Glowacki, D. R. and Pilling, M. J.: Unimolecular Reactions of Peroxy Radicals in Atmospheric Chemistry and Combustion, Chem. Phys. Chem., 11, 3836–3843, https://doi.org/10.1002/cphc.201000469, 2010.
Goldstein, A. H. and Galbally, I. E.: Known and Unexplored Organic Constituents in the Earth's Atmosphere, Environ. Sci. Technol., 41, 1514–1521, https://doi.org/10.1021/es072476p, 2007.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Stevens, P. S., Alaghmand, M., Bertman, S. B., Carroll, M. A., Erickson, M., Galloway, M., Grossberg, N., Hottle, J., Hou, J., Jobson, B. T., Kammrath, A., Keutsch, F. N., Lefer, B. L., Mielke, L. H., O'Brien, A., Shepson, P. B., Thurlow, M., Wallace, W., Zhang, N., and Zhou, X. L.: OH and HO2 radical chemistry during PROPHET 2008 and CABINEX 2009 – Part 1: Measurements and model comparison, Atmos. Chem. Phys., 13, 5403–5423, https://doi.org/10.5194/acp-13-5403-2013, 2013.
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of Hydroxyl and Hydroperoxy Radicals during CalNex-LA: Model Comparisons and Radical Budgets, J. Geophys. Res., 121, 4211–4232, https://doi.org/10.1002/2015JD024358, 2016.
Groß, C. B. M., Dillon, T. J., and Crowley, J. N.: Pressure dependent OH yields in the reactions of CH3CO and HOCH2CO with O2, Phys. Chem. Chem. Phys., 16, 10990–10998, https://doi.org/10.1039/C4CP01108B, 2014.
Hasson, A. S., Tyndall, G. S., and Orlando, J. J.: A product yield study of the reaction of HO2 radicals with ethyl peroxy (C2H5O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals, J. Phys. Chem. A, 108, 5979–5989, https://doi.org/10.1021/jp048873t, 2004.
Hasson, A. S., Tyndall, G. S., Orlando, J. J., Singh, S., Hernandez, S. Q., Campbell, S., and Ibarra, Y.: Branching Ratios for the Reaction of Selected Carbonyl-Containing Peroxy Radicals with Hydroperoxy Radicals, J. Phys. Chem. A, 116, 6264–6281, https://doi.org/10.1021/jp211799c, 2012.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Hofzumahaus, A., Aschmutat, U., Hessling, M., Holland, F., and Ehhalt, D. H.: The measurement of tropospheric OH radicals by laser-induced fluorescence spectroscopy during the POPCORN field campaign, Geophys. Res. Lett., 23, 2541–2544, https://doi.org/10.1029/96gl02205, 1996.
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified Trace Gas Removal in the Troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009.
Holland, F., Hofzumahaus, A., Schafer, R., Kraus, A., and Patz, H. W.: Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ, J. Geophys. Res., 108, 8246, https://doi.org/10.1029/2001jd001393, 2003.
Jenkin, M. E., Hurley, M. D., and Wallington, T. J.: Investigation of the radical product channel of the CH3C(O)CH2O2+HO2 reaction in the gas phase, Phys. Chem. Chem. Phys., 10, 4274–4280, https://doi.org/10.1039/b802898b, 2008.
Jenkin, M. E., Valorso, R., Aumont, B., and Rickard, A. R.: Estimation of rate coefficients and branching ratios for reactions of organic peroxy radicals for use in automated mechanism construction, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-44, in review, 2019.
Jokinen, T., Sipila, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angew. Chem., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016.
Kanaya, Y., Cao, R., Akimoto, H., Fukuda, M., Komazaki, Y., Yokouchi, Y., Koike, M., Tanimoto, H., Takegawa, N., and Kondo, Y.: Urban photochemistry in central Tokyo: 1. Observed and modeled OH and HO2 radical concentrations during the winter and summer of 2004, J. Geophys. Res., 112, 312, https://doi.org/10.1029/2007jd008670, 2007.
Karl, T., Guenther, A., Turnipseed, A., Tyndall, G., Artaxo, P., and Martin, S.: Rapid formation of isoprene photo-oxidation products observed in Amazonia, Atmos. Chem. Phys., 9, 7753–7767, https://doi.org/10.5194/acp-9-7753-2009, 2009.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, https://doi.org/10.1038/nature06870, 2008.
Levy, H.: Normal Atmosphere: Large Radical and Formaldehyde Concentrations Predicted, Science, 173, 141, https://doi.org/10.1126/science.173.3992.141, 1971.
Lew, M. M., Dusanter, S., and Stevens, P. S.: Measurement of interferences associated with the detection of the hydroperoxy radical in the atmosphere using laser-induced fluorescence, Atmos. Meas. Tech., 11, 95–109, https://doi.org/10.5194/amt-11-95-2018, 2018.
Li, Q., Zhang, L., Wang, T., Wang, Z., Fu, X., and Zhang, Q.: “New” Reactive Nitrogen Chemistry Reshapes the Relationship of Ozone to Its Precursors, Environ. Sci. Technol., 52, 2810–2818, https://doi.org/10.1021/acs.est.7b05771, 2018.
Liebmann, J., Karu, E., Sobanski, N., Schuladen, J., Ehn, M., Schallhart, S., Quéléver, L., Hellen, H., Hakola, H., Hoffmann, T., Williams, J., Fischer, H., Lelieveld, J., and Crowley, J. N.: Direct measurement of NO3 radical reactivity in a boreal forest, Atmos. Chem. Phys., 18, 3799–3815, https://doi.org/10.5194/acp-18-3799-2018, 2018.
Lightfoot, P. D., Cox, R. A., Crowley, J. N., Destriau, M., Hayman, G. D., Jenkin, M. E., Moortgat, G. K., and Zabel, F.: Organic peroxy radicals: Kinetics, spectroscopy and tropospheric chemistry, Atmos. Environ., 26, 1805–1961, https://doi.org/10.1016/0960-1686(92)90423-I, 1992.
Liu, Y., Lu, K., Dong, H., Li, X., Cheng, P., Zou, Q., Wu, Y., Liu, X., and Zhang, Y.: In situ monitoring of atmospheric nitrous acid based on multi-pumping flow system and liquid waveguide capillary cell, J. Environ. Sci., 43, 273–284, https://doi.org/10.1016/j.jes.2015.11.034, 2016.
Lockhart, J., Blitz, M., Heard, D., Seakins, P., and Shannon, R.: Kinetic Study of the OH + Glyoxal Reaction: Experimental Evidence and Quantification of Direct OH Recycling, J. Phys. Chem. A, 117, 11027–11037, https://doi.org/10.1021/jp4076806, 2013.
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Lu, K. D., Hofzumahaus, A., Holland, F., Bohn, B., Brauers, T., Fuchs, H., Hu, M., Häseler, R., Kita, K., Kondo, Y., Li, X., Lou, S. R., Oebel, A., Shao, M., Zeng, L. M., Wahner, A., Zhu, T., Zhang, Y. H., and Rohrer, F.: Missing OH source in a suburban environment near Beijing: observed and modelled OH and HO2 concentrations in summer 2006, Atmos. Chem. Phys., 13, 1057–1080, https://doi.org/10.5194/acp-13-1057-2013, 2013.
Mao, J., Ren, X., Zhang, L., Van Duin, D. M., Cohen, R. C., Park, J.-H., Goldstein, A. H., Paulot, F., Beaver, M. R., Crounse, J. D., Wennberg, P. O., DiGangi, J. P., Henry, S. B., Keutsch, F. N., Park, C., Schade, G. W., Wolfe, G. M., Thornton, J. A., and Brune, W. H.: Insights into hydroxyl measurements and atmospheric oxidation in a California forest, Atmos. Chem. Phys., 12, 8009–8020, https://doi.org/10.5194/acp-12-8009-2012, 2012.
Møller, K. H., Bates, K. H., and Kjaergaard, H. G.: The Importance of Peroxy Radical Hydrogen-Shift Reactions in Atmospheric Isoprene Oxidation, J. Phys. Chem. A, 123, 920–932, https://doi.org/10.1021/acs.jpca.8b10432, 2019.
Nehr, S., Bohn, B., Fuchs, H., Hofzumahaus, A., and Wahner, A.: HO2 formation from the OH + benzene reaction in the presence of O2, Phys. Chem. Chem. Phys., 13, 10699–10708, https://doi.org/10.1039/C1CP20334G, 2011.
Novelli, A., Vereecken, L., Lelieveld, J., and Harder, H.: Direct observation of OH formation from stabilised Criegee intermediates, Phys. Chem. Chem. Phys., 16, 19941–19951, https://doi.org/10.1039/C4CP02719A, 2014.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Otkjær, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 122, 8665–8673, https://doi.org/10.1021/acs.jpca.8b06223, 2018.
Peeters, J. and Muller, J. F.: HOx radical regeneration in isoprene oxidation via peroxy radical isomerisations. II: experimental evidence and global impact, Phys. Chem. Chem. Phys., 12, 14227–14235, https://doi.org/10.1039/c0cp00811g, 2010.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, https://doi.org/10.1039/b908511d, 2009.
Peeters, J., Muller, J.-F., Stavrakou, T., and Nguyen, V. S.: Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: The upgraded LIM1 mechanism, J. Phys. Chem. A, 118, 8625–8643, https://doi.org/10.1021/jp5033146, 2014.
Praske, E., Crounse, J. D., Bates, K. H., Kurten, T., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric Fate of Methyl Vinyl Ketone: Peroxy Radical Reactions with NO and HO2, J. Phys. Chem. A, 119, 4562–4572, https://doi.org/10.1021/jp5107058, 2015.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Intramolecular Hydrogen Shift Chemistry of Hydroperoxy-Substituted Peroxy Radicals, J. Phys. Chem. A, 123, 590–600, https://doi.org/10.1021/acs.jpca.8b09745, 2019.
Pugh, T. A. M., MacKenzie, A. R., Hewitt, C. N., Langford, B., Edwards, P. M., Furneaux, K. L., Heard, D. E., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J., Mills, G., Misztal, P., Moller, S., Monks, P. S., and Whalley, L. K.: Simulating atmospheric composition over a South-East Asian tropical rainforest: performance of a chemistry box model, Atmos. Chem. Phys., 10, 279–298, https://doi.org/10.5194/acp-10-279-2010, 2010.
Ramasamy, S., Ida, A., Jones, C., Kato, S., Tsurumaru, H., Kishimoto, I., Kawasaki, S., Sadanaga, Y., Nakashima, Y., Nakayama, T., Matsumi, Y., Mochida, M., Kagami, S., Deng, Y., Ogawa, S., Kawana, K., and Kajii, Y.: Total OH reactivity measurement in a BVOC dominated temperate forest during a summer campaign, 2014, Atmos. Environ., 131, 41–54, https://doi.org/10.1016/j.atmosenv.2016.01.039, 2016.
Ren, X., Olson, J. R., Crawford, J. H., Brune, W. H., Mao, J., Long, R. B., Chen, Z., Chen, G., Avery, M. A., Sachse, G. W., Barrick, J. D., Diskin, G. S., Huey, L. G., Fried, A., Cohen, R. C., Heikes, B., Wennberg, P. O., Singh, H. B., Blake, D. R., and Shetter, R. E.: HOx chemistry during INTEX-A 2004: Observation, model calculation, and comparison with previous studies, J. Geophys. Res., 113, 310, https://doi.org/10.1029/2007JD009166, 2008.
Ren, X., van Duin, D., Cazorla, M., Chen, S., Mao, J., Zhang, L., Brune, W. H., Flynn, J. H., Grossberg, N., Lefer, B. L., Rappenglück, B., Wong, K. W., Tsai, C., Stutz, J., Dibb, J. E., Jobson, B. T., Luke, W. T., and Kelley, P.: Atmospheric oxidation chemistry and ozone production: Results from SHARP 2009 in Houston, Texas, J. Geophys. Res.-Atmos., 118, 5770–5780, https://doi.org/10.1002/jgrd.50342, 2013.
Rohrer, F., Lu, K., Hofzumahaus, A., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Haseler, R., Holland, F., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S., Oebel, A., Shao, M., Zeng, L., Zhu, T., Zhang, Y., and Wahner, A.: Maximum efficiency in the hydroxyl-radical-based self-cleansing of the troposphere, Nat. Geosci., 7, 559–563, https://doi.org/10.1038/ngeo2199, 2014.
Sadanaga, Y., Yoshino, A., Kato, S., and Kajii, Y.: Measurements of OH reactivity and photochemical ozone production in the urban atmosphere, Environ. Sci. Technol., 39, 8847–8852, https://doi.org/10.1021/es049457p, 2005.
Shao, M., Lu, S., Liu, Y., Xie, X., Chang, C., Huang, S., and Chen, Z.: Volatile organic compounds measured in summer in Beijing and their role in ground-level ozone formation, J. Geophys. Res., 114, D00G06, https://doi.org/10.1029/2008JD010863, 2009.
Shirley, T. R., Brune, W. H., Ren, X., Mao, J., Lesher, R., Cardenas, B., Volkamer, R., Molina, L. T., Molina, M. J., Lamb, B., Velasco, E., Jobson, T., and Alexander, M.: Atmospheric oxidation in the Mexico City Metropolitan Area (MCMA) during April 2003, Atmos. Chem. Phys., 6, 2753–2765, https://doi.org/10.5194/acp-6-2753-2006, 2006.
Sinha, V., Williams, J., Crowley, J. N., and Lelieveld, J.: The Comparative Reactivity Method – a new tool to measure total OH Reactivity in ambient air, Atmos. Chem. Phys., 8, 2213–2227, https://doi.org/10.5194/acp-8-2213-2008, 2008.
Tan, D., Faloona, I., Simpas, J. B., Brune, W., Shepson, P. B., Couch, T. L., Sumner, A. L., Carroll, M. A., Thornberry, T., Apel, E., Riemer, D., and Stockwell, W.: HOx budgets in a deciduous forest: Results from the PROPHET summer 1998 campaign, J. Geophys. Res., 106, 24407–24427, https://doi.org/10.1029/2001jd900016, 2001.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Lu, K., Dong, H., Hu, M., Li, X., Liu, Y., Lu, S., Shao, M., Su, R., Wang, H., Wu, Y., Wahner, A., and Zhang, Y.: Explicit diagnosis of the local ozone production rate and the ozone-NOx-VOC sensitivities, Sci. Bull., 63, 1067–1076, https://doi.org/10.1016/j.scib.2018.07.001, 2018a.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018b.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Wang, M., Zeng, L., Lu, S., Shao, M., Liu, X., Yu, X., Chen, W., Yuan, B., Zhang, Q., and Hu, M.: Development and validation of a cryogen-free automatic gas chromatograph system (GC-MS/FID) for online measurements of volatile organic compounds, Anal. Methods, 6, 9424–9434, https://doi.org/10.1039/C4AY01855A, 2014.
Wesely, M. L. and Hicks, B. B.: A review of the current status of knowledge on dry deposition, Atmos. Environ., 34, 2261–2282, https://doi.org/10.1016/S1352-2310(99)00467-7, 2000.
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Whalley, L. K., Stone, D., Bandy, B., Dunmore, R., Hamilton, J. F., Hopkins, J., Lee, J. D., Lewis, A. C., and Heard, D. E.: Atmospheric OH reactivity in central London: observations, model predictions and estimates of in situ ozone production, Atmos. Chem. Phys., 16, 2109–2122, https://doi.org/10.5194/acp-16-2109-2016, 2016.
Whalley, L. K., Stone, D., Dunmore, R., Hamilton, J., Hopkins, J. R., Lee, J. D., Lewis, A. C., Williams, P., Kleffmann, J., Laufs, S., Woodward-Massey, R., and Heard, D. E.: Understanding in situ ozone production in the summertime through radical observations and modelling studies during the Clean air for London project (ClearfLo), Atmos. Chem. Phys., 18, 2547–2571, https://doi.org/10.5194/acp-18-2547-2018, 2018.
Williams, J., Kessel, S. U., Nolscher, A. C., Yang, Y. D., Lee, Y., Yanez-Serrano, A. M., Wolff, S., Kesselmeier, J., Klupfel, T., Lelieveld, J., and Shao, M.: Opposite OH reactivity and ozone cycles in the Amazon rainforest and megacity Beijing: Subversion of biospheric oxidant control by anthropogenic emissions, Atmos. Environ., 125, 112–118, https://doi.org/10.1016/j.atmosenv.2015.11.007, 2016.
Winiberg, F. A. F., Dillon, T. J., Orr, S. C., Groß, C. B. M., Bejan, I., Brumby, C. A., Evans, M. J., Smith, S. C., Heard, D. E., and Seakins, P. W.: Direct measurements of OH and other product yields from the HO2+CH3C(O)O2 reaction, Atmos. Chem. Phys., 16, 4023–4042, https://doi.org/10.5194/acp-16-4023-2016, 2016.
Yang, Y., Shao, M., Keßel, S., Li, Y., Lu, K., Lu, S., Williams, J., Zhang, Y., Zeng, L., Nölscher, A. C., Wu, Y., Wang, X., and Zheng, J.: How the OH reactivity affects the ozone production efficiency: case studies in Beijing and Heshan, China, Atmos. Chem. Phys., 17, 7127–7142, https://doi.org/10.5194/acp-17-7127-2017, 2017.
Zhang, Y. H., Su, H., Zhong, L. J., Cheng, Y. F., Zeng, L. M., Wang, X. S., Xiang, Y. R., Wang, J. L., Gao, D. F., Shao, M., Fan, S. J., and Liu, S. C.: Regional ozone pollution and observation-based approach for analyzing ozone–precursor relationship during the PRIDE-PRD2004 campaign, Atmos. Environ., 42, 6203–6218, https://doi.org/10.1016/j.atmosenv.2008.05.002, 2008.