the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 May 2019
| 07 May 2019
Impact of anthropogenic and biogenic sources on the seasonal variation in the molecular composition of urban organic aerosols: a field and laboratory study using ultra-high-resolution mass spectrometry
Kaspar R. Daellenbach
Ivan Kourtchev
Alexander L. Vogel
Emily A. Bruns
Jianhui Jiang
Tuukka Petäjä
Jean-Luc Jaffrezo
Sebnem Aksoyoglu
Markus Kalberer
Urs Baltensperger
Imad El Haddad
André S. H. Prévôt
This study presents the molecular composition of organic aerosol (OA) using ultra-high-resolution mass spectrometry (Orbitrap) at an urban site in Central Europe (Zurich, Switzerland). Specific source spectra were also analysed, including samples representative of wood-burning emissions from Alpine valleys during wood-burning pollution episodes and smog chamber investigations of woodsmoke, as well as samples from Hyytiälä, which were strongly influenced by biogenic secondary organic aerosol. While samples collected during winter in Alpine valleys have a molecular composition remarkably similar to fresh laboratory wood-burning emissions, winter samples from Zurich are influenced by more aged wood-burning emissions. In addition, other organic aerosol emissions or formation pathways seem to be important at the latter location in winter. Samples from Zurich during summer are similar to those collected in Hyytiälä and are predominantly impacted by oxygenated compounds with an H∕C ratio of 1.5, indicating the importance of biogenic precursors for secondary organic aerosol (SOA) formation at this location (summertime Zurich – carbon number 7.6, O:C 0.7; Hyytiälä – carbon number 10.5, O:C 0.57). We could explain the strong seasonality of the molecular composition at a typical European site by primary and aged wood-burning emissions and biogenic secondary organic aerosol formation during winter and summer, respectively. Results presented here likely explain the rather constant seasonal predominance of non-fossil organic carbon at European locations.
- Article
(4586 KB) - Full-text XML
-
Supplement
(580 KB) - BibTeX
- EndNote





Aerosols affect the Earth's climate and ecosystems, as well as human health. A main contributor to aerosol mass is organic aerosol (OA). OA can be directly emitted as primary particles (POA) or produced by oxidation and subsequent condensation of volatile organic compounds (VOCs) (secondary OA, SOA). Sources of both POA and SOA can be natural, such as plant debris, resuspension, and biogenic VOC (BVOC) oxidation, or anthropogenic, such as from traffic, cooking, or residential heating using wood or fossil fuels. The resulting SOA is typically a highly complex mixture of unknown compounds, the chemical characterization of which requires comprehensive analytical strategies (e.g. Noziere et al., 2015).
Field deployments of an aerosol mass spectrometer (AMS, Canagaratna et al., 2007) at several European stations have revealed a large impact of primary wood-burning emissions on OA (e.g. between 11 % and 59 % in Switzerland), while primary traffic emissions have a smaller contribution (4 %–14 %) (Lanz et al., 2010; Gilardoni et al., 2011; Daellenbach et al., 2017). The results are consistent with radiocarbon measurements (Zotter et al., 2014a), showing that during extreme winter pollution episodes non-fossil organic carbon may account for up to 97 % of organic carbon (OC) at Alpine valley sites (Magadino – 83 %, San Vittore – 97 %) and 74 % at an urban background site in Zurich and are also associated with levoglucosan, a pyrolysis product of cellulose. Similar results were obtained in French Alpine valleys such as in Chamonix (Bonvalot et al., 2016). Based on AMS measurements, a large fraction of OA, in addition to primary wood-burning emissions, is found to be SOA from unknown origins. Radiocarbon analyses suggest that a large fraction of SOA is also non-fossil, potentially arising from biomass smoke aging (Vlachou et al., 2018). Recent smog chamber studies were capable of clearly showing the significant SOA formation rates during the aging of wood-burning emissions, which could be largely attributed to phenolic derivatives, together with benzene and naphthalene (Bruns et al., 2016). During summer the dominant fraction of OA was reported to be SOA (Bozzetti et al., 2016, 2017a, b; Canonaco et al., 2015; Daellenbach et al., 2017; Lanz et al., 2010; Reyes-Villegas et al., 2016; Schlag et al., 2016) at many European sites even in large industrialized cities such as Marseille, France (El Haddad et al., 2011). Based on radiocarbon analyses, it is hypothesized that summertime SOA in Central Europe is largely produced from the oxidation of biogenic precursors (Vlachou et al., 2018; Bonvalot et al., 2016; Zotter et al., 2014a). While this is found to be the case in Europe, a strong contribution of fossil fuel emissions to SOA has been observed in the Los Angeles basin (Zotter et al., 2014b; Platt at el., 2017).
Despite these recent advancements of OA source apportionment using an AMS and radiocarbon analysis, direct links between observed SOA and its precursors are still missing. This remaining gap is strongly related to the extensive molecular fragmentation in the AMS caused by the use of electron ionization, which hinders retrieving detailed information on the chemical nature of wintertime and summertime SOA fractions. The use of soft ionization techniques, such as electrospray ionization coupled to ultra-high-resolution mass spectrometry (ESI-UHR-MS), is a powerful technique that may help in bridging such existing gaps (Nizkorodov et al., 2011). The technique provides the exact mass of molecular ions with minimal fragmentation, thus allowing the determination of the SOA molecular composition. The disadvantage of the technique is the relatively strong variability in the ionization efficiency of different compounds; i.e. the relative contribution of a compound cannot be directly linked to its concentration without using an authentic standard (Huffmann et al., 2012; Kruve et al., 2017; Noziere et al., 2015).
Despite this, laboratory studies using ESI-UHR-MS have deepened our understanding of the formation of SOA from various sources such as isoprene, monoterpenes, and vehicular exhaust (Bateman et al., 2009; Kourtchev et al., 2015; Mutzel et al., 2015; Nguyen et al., 2010, 2011; Romonosky et al., 2017; Walser et al., 2008). Studies focusing on the ESI-UHR-MS analysis of aerosol samples collected during field campaigns qualitatively revealed the influence of different sources at various sites (Dzepina et al., 2015; Kourtchev et al., 2013, 2014b; Lin et al., 2012; O'Brien et al., 2013, 2014; Rincón et al., 2012; Roach et al., 2010; Tao et al., 2014; Tong et al., 2016; Wang et al., 2017). However, the typical number of samples analysed remains very limited, with low temporal resolution in comparison to online measurement techniques. As a result, only limited knowledge is currently available about the changes of OA molecular characteristics throughout the seasons and especially during winter.
In this study, we examine the seasonal variability of the OA chemical composition at a molecular level at an urban background site in Central Europe (Zurich, Switzerland). In order to elucidate the influence of different sources on SOA chemical compositions, samples from Zurich are compared to those collected during wood-burning episodes and wood-burning smog chamber experiments, as well as samples dominated by biogenic SOA.
2.1 Aerosol sample collection
The field samples at the three sites in Switzerland were collected daily in 2013 using a high-volume sampler (500 L min−1, quartz-fibre filter, 14.7 cm filter diameter). Zurich is located on the northern Swiss plateau, and the site is classified as urban background. The sites in Magadino and San Vittore are in Alpine valleys in southern Switzerland. While the 15 samples collected in Zurich were analysed individually, the 4 samples from Magadino and 4 samples from San Vittore were grouped and analysed as composites (10, 14, 18, 22 December 2013).
Samples from smog chamber experiments were analysed to examine the composition of wood-burning emissions from the stable flaming phase and their evolution with aging (Bruns et al., 2016, 2017). During these experiments, fresh emissions were first injected into a 6 m3 Teflon smog chamber (Platt et al., 2013; Bruns et al., 2016, 2017). After 30 min of mixing, particles were sampled onto quartz-fibre filters (UV lights off and sampling time 30 min at ∼30 L min−1). Then, emissions were photochemically aged in the smog chamber by injecting HONO at a flow rate of 1–2 L min−1, which generates OH radicals upon photolysis. Samples were collected before aging (fresh) and at equivalent atmospheric aging times of 10 and 30 h (determined by the method of Barmet et al. (2012), assuming a wintertime OH concentration of 106 molec. cm−3). All samples were grouped and measured as three composites: fresh wood-burning emissions (16 samples), 10 h equivalent atmospheric aging (11 samples), and 30 h equivalent atmospheric aging (15 samples).
Further, we included two average spectra from the SMEAR-II (Station for Measuring Ecosystem–Atmosphere Relations, Hari and Kulmala 2005) at Hyytiälä (PM1, summer 2011, 12 h sampling time and summer 2014, 48 to 64 h sampling time, both using quartz-fibre filters) previously published in Kourtchev et al. (2016). The SMEAR-II (Hari and Kulmala, 2005) at Hyytiälä is a rural background site in Finland, strongly influenced by biogenic SOA (vegetation dominated by Scots pine and Norway spruce). The PM1 aerosol was collected between 16 and 25 August 2011 and between 7 July and 4 August 2014 using a low-volume sampler (35 L min−1). Since biogenic SOA is mostly found in PM1 (Vlachou et al., 2018), comparing PM1 samples from Hyytiälä to PM10 samples from Zurich does not affect the conclusions related to biogenic SOA.
In addition, we collected samples from Frauenfeld (PM10), Payerne (PM10), Bern (PM10), and Zurich (PM2.5) from the same year to complement the dataset with further chemical analyses (Sect. 2.3).
2.2 Direct-injection (-)ESI-UHR-MS
2.2.1 Analytical procedure
For each sample, a part of the quartz-fibre filter was extracted three times with 5 mL of methanol (Optima® grade, Fisher Scientific) under ultrasonic agitation in an ice-chilled bath for 30 min. The three extracts were combined, filtered through a Teflon® filter (0.2 µm), and reduced by volume to approximately 200 µL under a gentle stream of nitrogen. Extracts of field blanks were prepared analogously.
Recent results show that the positive-mode ESI, which is less selective, can provide additional valuable information, especially regarding fresh emissions (Lin et al., 2018). Here, to compare with previous results, and, as the main aim is to characterize SOA, we have focused on the negative-mode, (-)ESI. The aerosol extracts were analysed using an ultra-high-resolution LTQ Orbitrap Velos mass spectrometer in the negative mode (Thermo Fisher, Bremen, Germany) equipped with a TriVersa Nanomate robotic nanoflow chip-based ESI source (Advion Biosciences, Ithaca, NY, USA). The analyses were conducted with an ionization voltage of 1.51 kV, a vaporizer temperature of 230 ∘C for the Zurich samples and 254 ∘C for the other samples, and a capillary temperature of 200 ∘C (with only a small impact on measurements; see Fig. S8 in the Supplement). The Orbitrap MS instrument was calibrated using an Ultramark 1621 solution (Sigma-Aldrich, UK). The accuracy of the m∕z calibration was < 2 ppm. The mass resolution of the instrument was 100 000 at m∕z 400. Two or three replicate measurements were conducted for each extract (variability assessed in Fig. S9), and field blank extracts were analysed in the same way.
2.2.2 Data analysis
For each replicate measurement, approximately 38 mass spectral scans were averaged into one mass spectrum per measurement (representing 1 min measurement time). The gathered mass spectral information was processed with Xcalibur 2.1 (Thermo Scientific). In the first step, peaks exhibiting a signal below instrumental noise level were removed. We estimated the instrumental noise level as the 99.9th percentile of the signal recorded in regions of the mass spectrum where no signal is expected. In the second step, for all remaining peaks possible, molecular compositions were assigned to the signals using a tolerance level within ±5 ppm and the following constraints: 12C ≤100, 13C ≤1, 1H ≤200, 16O ≤50, 14N ≤5, 32S ≤2, and 34S ≤1. Among all the assigned molecular compositions, only molecular assignments in agreement with the following criteria described in detail in Kourtchev et al. (2013) were further considered.
Molecular assignments of a peak have to be within a defined mass difference from the measured mass (m∕z). The tolerated difference between the molecular assignments and the measured mass of a peak was assessed based on the respective difference for nine known compounds for every analysed sample (on average 0.5 ppm).
Molecular assignments have to be in agreement with the following elemental ratios: (i) O:C ≤1.5, (ii) 0.3≤ H∕C ≤2.5, (iii) 0≤ N∕C ≤0.5, and (iv) 0≤ S∕C ≤0.2.
Molecular assignments have to be consistent with a neutral formula with a positive-integer double-bond-equivalent (DBE, for any chemical formula , where NC, NH, NO, NN, and NS represent the number of carbon, hydrogen, oxygen, nitrogen, and sulfur atoms):
Nitrogen-containing compounds have to be in agreement with the nitrogen rule.
Formulae including either or both 13C and 34S were only further considered in the presence of a counterpart including only 12C and 32S, respectively.
Peaks were only further considered if their intensity in the sample was at least 3 times larger than in the blank.
2.2.3 Computation of bulk properties
All properties, molar ratios, and chemical formulae presented in this paper refer to neutral molecules. Literature data were additionally also filtered with criterion (2) for comparability. Bulk elemental ratios (H∕C, O:C, N∕C, and S∕C) and the number of carbons of the organic aerosol were computed as follows (Nizkorodov et al., 2011; Bateman et al., 2012):
O:C, H∕C, N∕C, S∕C, and carbon number, as well as the probability distribution function (pdf) are weighted by the number of O, H, N, S, and C atoms in a compound i (NO,i, NH,i, NN,i, NS,i, NC,i) by the respective peak intensity xi such that the probability p of, for example, an H∕C of h is defined as follows:
The compounds' aromaticity can be estimated by different parameterizations (e.g. aromaticity index, AI, and aromaticity equivalent, Xc). Xc (Yassine et al., 2014) has the advantage over AI (Koch and Dittmar, 2006) that (poly)aromatic compounds with significant alkylation are accurately classified. Therefore, we apply the widely used aromaticity equivalent (Xc) in this study, expressed as follows:
Here DBE is the double bond equivalent (Eq. 1), NO and NS are the number of oxygen and sulfur atoms present in the molecule, respectively, and m and n are the number of oxygen and sulfur atoms involved in π bonds, respectively (both assumed to be 0.5, Yassine et al., 2014). For molecular formulae with an odd number of sulfur or oxygen, the sum () was rounded down to the closest integer and for compounds with DBE was set to 0 (Yassine et al., 2014). Compounds with Xc < 2.5 were considered non-aromatic, with Xc≥2.5 aromatic and with Xc≥2.7143 condensed aromatic (Yassine et al., 2014).
2.3 Other chemical analyses
The chemical analyses were complemented with other filter-based analyses. Organic and elemental carbon (OC, EC) concentrations were determined for Zurich, Magadino, San Vittore, and Hyytiälä by a thermo-optical transmission (TOT) method with a Sunset OC/EC analyser (Birch and Cary, 1996), following the EUSAAR-2 thermal–optical transmission protocol (Cavalli et al., 2010). Water-soluble inorganic ions (K+, Na+, Mg2+, Ca2+, ; and , , Cl−) were measured by ion chromatography for Zurich, Magadino, and San Vittore (Piazzalunga et al., 2013; Jaffrezo et al., 1998). Pinic acid and 3-MBTCA (3-methyl-1,2,3-butanetricarboxylic acid) were quantified for the ambient filter samples from Switzerland, with analysis by liquid-chromatography mass spectrometry (LC-MS, (-)ESI LCQ-FLEET, Thermo Fisher), with chromatographic separation performed on a Synergi 4 µm Fusion-RP 80A (Phenomenex) with a water–acetonitrile–formic acid eluent. The calibration was performed with authentic standards. Online measurements of gas-phase compounds (NOx by chemiluminescence, SO2 by fluorescence and absorption) were performed and meteorological parameters were recorded at selected sites (for Hyytiälä no SO2). For the ambient samples from Switzerland, OA source apportionment contributions (presented in Daellenbach et al., 2017) were determined using an offline application of the HR-ToF-AMS, according to the protocol presented in Daellenbach et al. (2016). During the laboratory wood-burning experiments, the aerosol was monitored online by an HR-ToF AMS (Aerodyne, Canagaratna et al., 2007; Bruns et al., 2016).
2.4 Hierarchical clustering analysis
Similarities in the mass spectral signatures were examined through hierarchical cluster analysis (HCA, Bar-Joseph et al., 2001) using the average linkage method. For a robust interpretation, we performed this analysis following two approaches, once considering the intensity of the peaks (approach A) and once based on the presence or absence of a peak (approach B). In approach A, we computed the Pearson correlation coefficient (R) between the mass spectral profiles of the samples in question and performed HCA based on the correlation matrix. In approach B, we computed the number of peaks k that a sample i had in common with another sample ii, normalized to the total number of peaks detected in sample i ().
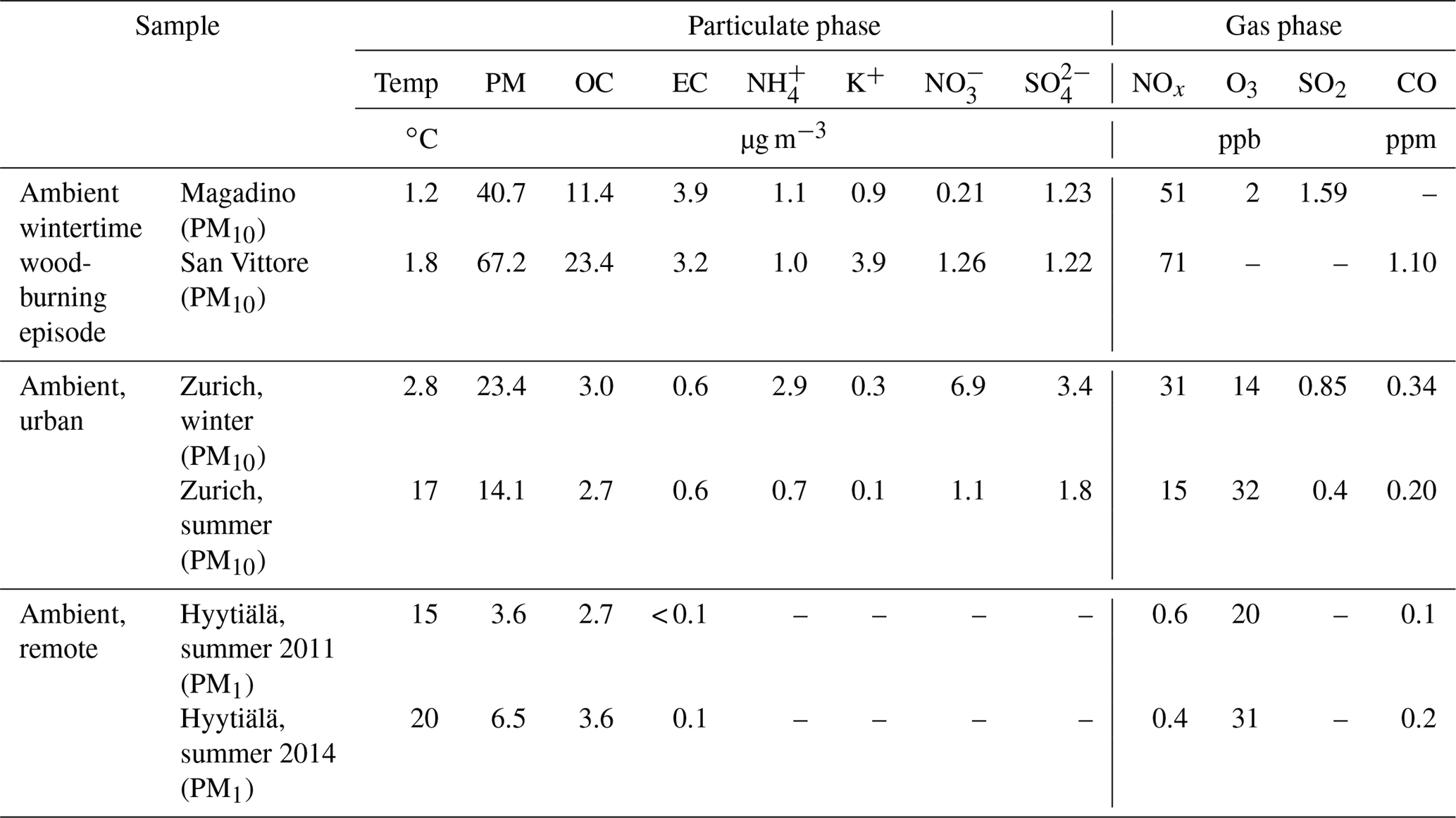
Table 1Concentrations of particulate and gaseous species and temperature.
3.1 Previous knowledge on aerosol composition and sources
The bulk aerosol composition for the different sites and periods is summarized in Table 1. On average, OC and EC loadings in Zurich were similar for the winter and summer samples. During the wood-burning pollution episode the concentrations were strongly elevated at the Alpine valley sites Magadino and San Vittore compared to the ones observed in Zurich. , , and concentrations exhibited a strong seasonality in Zurich, with higher concentrations during winter. The same is true for NOx and SO2, while higher O3 concentrations were observed during summer. While O3 concentrations at the remote site in Hyytiälä are comparable to Zurich, NOx and CO are strongly enhanced in Zurich, highlighting the anthropogenic influence at this location. The temperatures recorded in Zurich summer (17 ∘C) and winter (2 ∘C) were not significantly different from than those recorded during summer in Hyytiälä (between 15 and 20 ∘C for the measurement periods) and in Magadino (1.5 ∘C) during wood-burning episodes, respectively.
In earlier work, source apportionment analysis for the same samples (Daellenbach et al., 2017) quantified the contributions of POA from traffic (hydrocarbon-like OA, HOA), cooking (COA), biomass burning from residential heating (BBOA), as well as SOA (results for samples used in this study are presented in Fig. 1). These results showed that SOA was a main contributor to OA in the PM10 fraction in Zurich throughout the year. We further distinguished SOA into two seasonal components termed winter OOA (WOOA) and summer OOA (SOOA), which dominate SOA in winter and summer, respectively. WOOA correlated with anthropogenically influenced inorganic ions like and was, for this reason, interpreted as being formed from anthropogenic VOC emissions. SOOA in contrast showed a positive non-linear relation to temperature, consistent with the temperature-driven enhancement of biogenic terpene emissions (Daellenbach et al., 2017, for the entire dataset from Zurich (WOOA, , n=90) =0.79, Rs (SOOA, temp, n=91) =0.65). Therefore, we have hypothesized that summer SOA is formed from BVOC emissions (Daellenbach et al., 2017; Vlachou et al., 2018), consistent with previous 14C measurements at the same and other sites in Switzerland (Zotter et al., 2014a; Vlachou et al., 2018).
3.2 Seasonal differences in the OA chemical composition in Zurich
We present the average summer (T > 11 ∘C: 18 April, 12 May, 5 June, 29 June, 23 July, 16 August, 9 September, 3 October, 27 October 2013) and winter (T < 6 ∘C: 12 January, 5 February, 1 March, 25 March, 20 November, 14 December 2013) spectra from (-)ESI-UHR-MS at the urban background site in Zurich (mass spectral signature and van Krevelen diagrams in Fig. 2). We note that peak intensities, xi, are not directly linked to concentrations and only relative differences can be interpreted. The summer and winter average spectra exhibited a strong seasonal difference. During summer, peaks related to compounds only containing carbon, hydrogen, and oxygen (CHO) dominated the spectrum. The majority of these compounds had an H∕C around 1.5 and O:C between 0.4 and 1.4 (Fig. 2). These compounds were either absent or had a much lower intensity during winter.
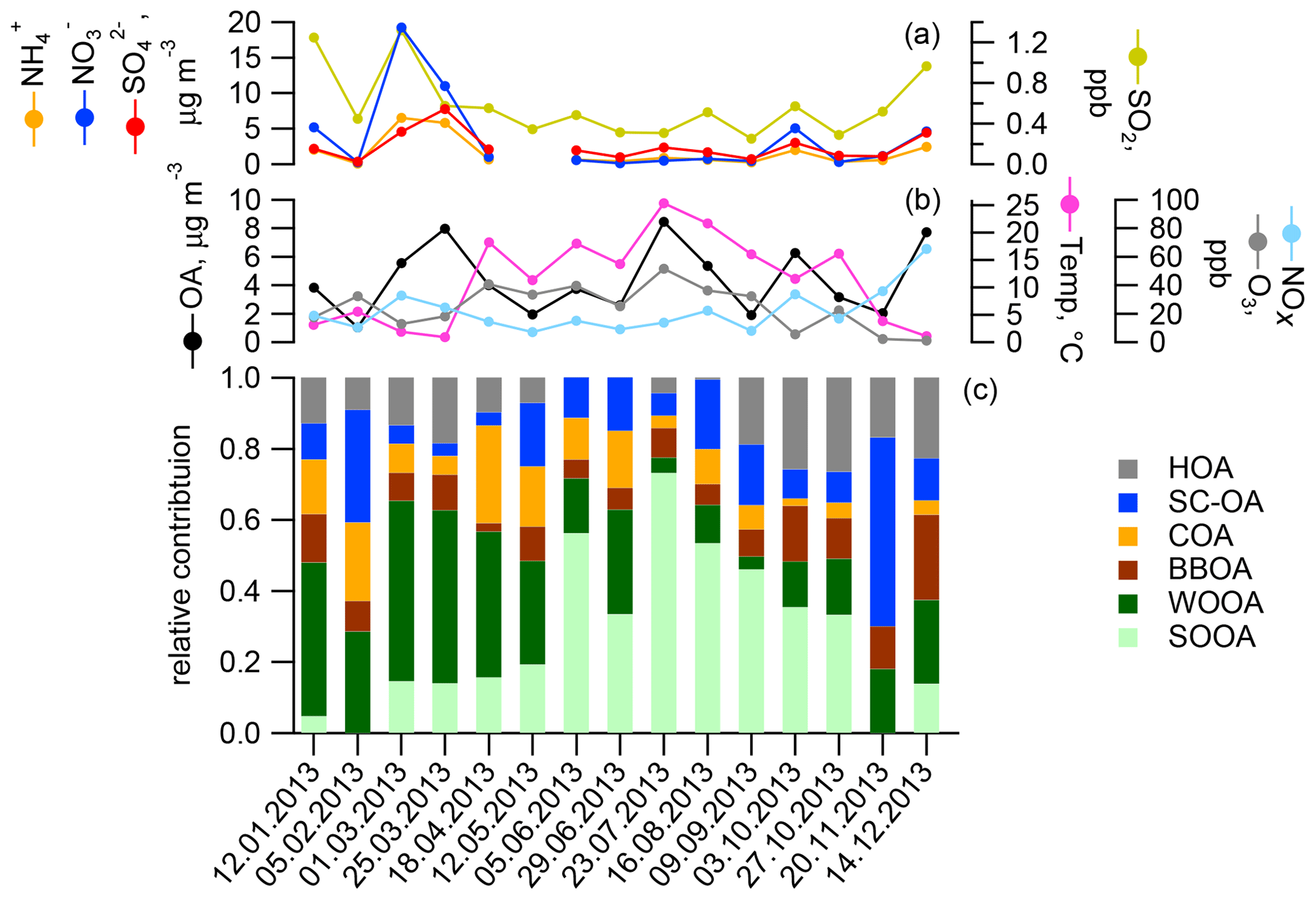
Figure 1Concentrations of particulate and gaseous species (a, b) and relative source contributions to the organic aerosol as determined by offline AMS in Zurich (c): traffic (HOA), cooking (COA), and wood-burning POA (BBOA), as well as a factor explaining sulfur-containing organic fragments (SC-OA) and SOA categories dominant in summer (SOOA) and winter (WOOA).
During winter, compounds also containing nitrogen (CHON) dominated the signal (Fig. 2). The largest intensity was assigned to C6H5O4N (possibly nitrocatechol), followed by C7H7O4N (possibly methyl-nitrocatechol). Some of these compounds were also present in summer with much lower contributions. Such compounds are formed through the oxidation of aromatic VOCs in the presence of NOx (Forstner et al., 1997; Jang and Kamens, 2001; Hamilton et al., 2005; Sato et al., 2012; Irei et al., 2015). In earlier work, these compounds were observed at urban and rural locations and were mainly associated with biomass-burning activities (Kitanovski et al., 2012; Iinuma et al., 2010; Claeys et al., 2012; Kourtchev et al., 2013, 2014b; Zhang et al., 2013; Mohr et al., 2013). The same peaks (C5H6O4N and C7H7O4N) were also abundant in measurements in the Pearl River Delta during the harvesting period (Lin et al., 2012). We did not observe nitrophenols (C6H5NO3,C7H7NO3, C8H9NO3), which were previously reported for measurements with the (-)ESI-Orbitrap in a road tunnel and related to vehicular emissions (Tong et al., 2016). However, nitrophenols were also reported to be influenced by biomass burning (Mohr et al., 2013). Some aromatic CHO compounds (generic formula C11H9O5) found during winter were either not detected or were only present at much lower concentrations during summer.

Figure 2(a) Average summer (T > 11 ∘C, mass-weighted OA, nine samples) and winter (T < 6 ∘C, mass-weighted OA, six samples) ultra-high-resolution mass spectra integrated into unit-mass resolution in the negative mode for the organic aerosol in PM10 sampled in Zurich during the year 2013 (weighed average with OA concentration from offline AMS analysis). The signal at a nominal mass is separated by ion family (CHO, CHON, CHOS, CHONS, or other). Peak assignments of dominant ions of selected unit mass resolution (UMR) peaks are labelled as neutral compounds. In the absence of a signal, the mass spectra are only plotted until m∕z 450 instead of 650. (b) Van Krevelen diagrams (neutral composition) of average summer (T > 11 ∘C, mass-weighted OA, nine samples) and winter (T < 6 ∘C, mass-weighted OA, six samples) ultra-high-resolution mass spectra in the negative mode for the organic aerosol in PM10 sampled in Zurich during the year 2013 (weighed average with OA concentration from offline AMS analysis). Peaks are displayed as circles with their size reflecting log(intensity) and colour code reflecting the molecular composition of the compound.
Both during summer and winter, CHOS and CHONS compounds characterized by elevated H∕C between 1.5 and 2 were observed, with little seasonal variability in (S∕C)bulk (0.03). A prominent CHONS compound observed both during summer and winter was C10H17O7NS. In early studies, this peak was linked to biogenic emissions, and recent publications observed this peak in bush fire plumes in Australia and linked it to outgassing of BVOCs during combustion (Iinuma et al., 2015). Additionally, this compound showed a high abundance during the harvest season in the Pearl River Delta (Lin et al., 2012), underlining possible additional links to human activities.
3.3 Comparison between Zurich and source samples
Hierarchical cluster analysis using R2 (approach A) as a measure of similarity of the recorded (-)ESI-UHR mass spectral profiles could distinguish the samples from summer and winter into two groups. On the one hand, the analysis revealed that the wintertime Zurich mass spectral signatures were similar to those recorded in Magadino and San Vittore during wood-burning episodes and laboratory wood-burning emissions (Fig. 3a). The spectra in San Vittore and Magadino were most similar to the fresh laboratory wood-burning emissions, suggesting the prevalence of primary wood-burning emissions during these pollution episodes. The wintertime Zurich spectrum was more similar to aged laboratory wood-burning emissions in the chamber under atmospherically relevant conditions (representing 10 and 30 h atmospheric aging). On the other hand, the summertime Zurich spectrum was most similar to that from Hyytiälä during summer (Kourtchev et al., 2014a, 2016). Since Hyytiälä is strongly influenced by biogenic SOA (e. g., Kourtchev et al., 2014a, 2016), the similarity between the mass spectral signatures for summertime Hyytiälä and summertime Zurich suggests that biogenic SOA also has a dominant influence at the urban background location in Zurich. However, the correlation between summertime Zurich and Hyytiälä was smaller than those between wintertime Zurich, Magadino, and San Vittore, indicating differences either in the SOA precursor emission patterns or in the SOA formation pathways.
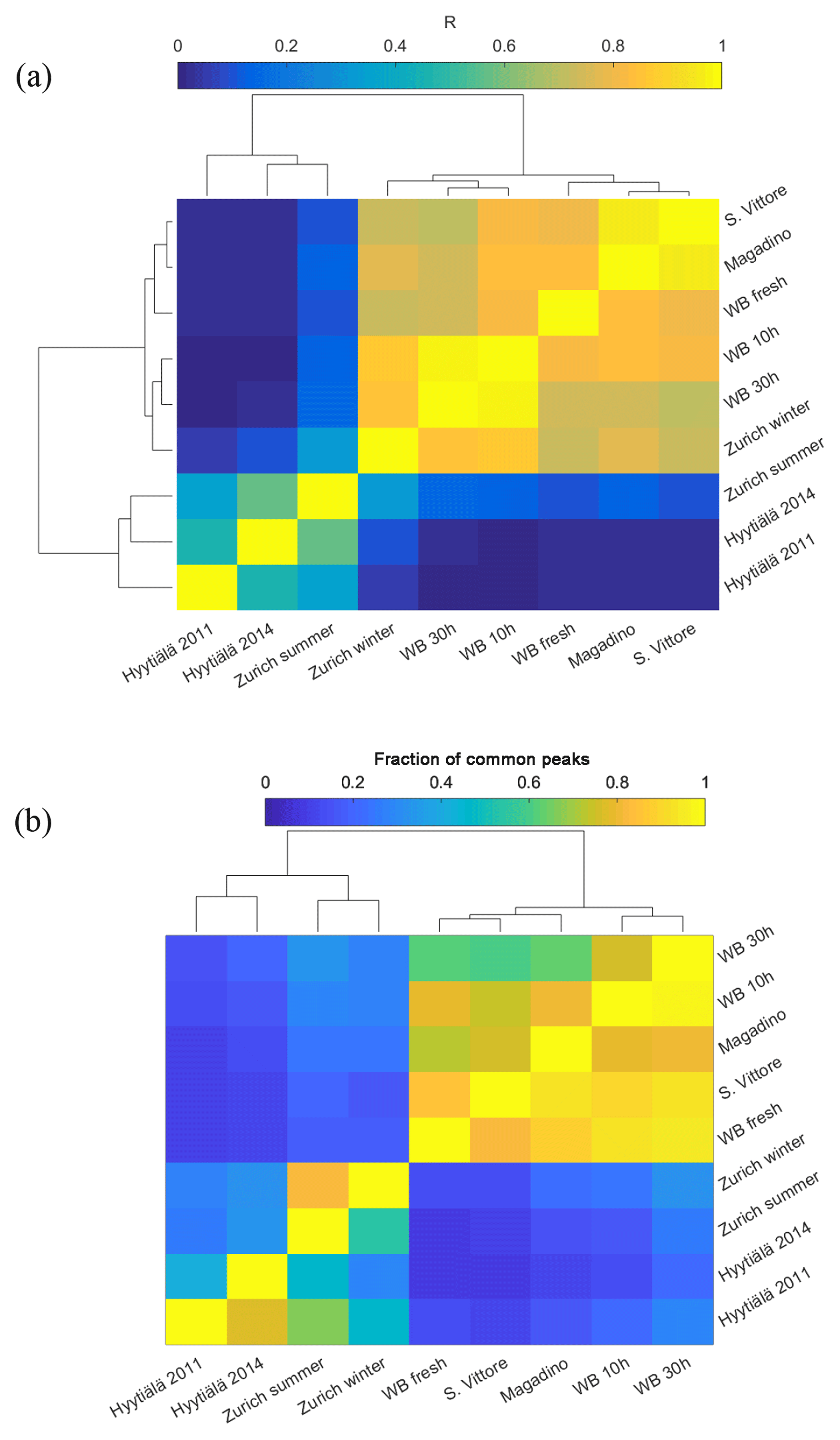
Figure 3(a) Correlation matrix of mass spectra sorted by hierarchical cluster analysis that also depicts the similarity as dendrograms. (b) Fraction of common peaks of a sample with the sample indicated on the y axis normalized to the total number of peaks of the respective sample sorted by hierarchical cluster analysis that also depicts the similarity as dendrograms.
As with any chemical ionization mass spectrometry, the spectral fingerprints are influenced by the variable relative ionization efficiencies of the different compounds. Therefore, we also performed hierarchical cluster analysis based on the normalized number of common peaks (approach B, Fig. 3b). Results confirmed the similarity of samples dominated by wood-burning emissions (from laboratory wood-burning experiments at San Vittore and Magadino), clearly distinguished from other samples. Among the wood-burning-dominated samples, fresh laboratory wood-burning emissions were most similar to San Vittore and to a lesser degree to Magadino. In addition, the analysis indicates a high number of peaks with relatively low intensity in common with the samples from Zurich during summer and winter. As will be discussed in more detail in the following sections, these peaks comprise CHONS compounds. Overall, the appearance of peaks related to biogenic SOA and wood-burning emissions in summer and winter, respectively, dominate the observed variability between the spectra. Therefore, samples from summer and winter will be discussed separately in the following.
3.4 Biogenic SOA: Zurich and Hyytiälä
3.4.1 Bulk chemical composition
In order to understand the spectral profiles recorded in summertime Zurich, we start by describing similar spectra from Hyytiälä. The largest fraction of signal in Hyytiälä can be attributed to CHO compounds with H∕C ∼1.5 and Cbulk 10.5 (H∕C 1.48, Cbulk 9.7 for 2011 and H∕C 1.50, Cbulk 11.3 for 2014; Figs. 4, 5, Table 2), characteristic of biogenic emissions. Compounds with 8 to 12 carbons (C8–C12), thought to arise from monoterpene oxidation, dominate the signals (Figs. 5, 6, 7). Additionally, CHO compounds with 13–16 carbons and 17–22 carbons significantly contribute to the signal. The C13–C16 compounds are thought to consist mainly of sesquiterpene oxidation products but may also be produced through reactions of monoterpene and isoprene RO2 radicals (Berndt et al., 2018), which is less probable in a boreal forest due to the low isoprene concentrations. Meanwhile, the latter class (C17–C22) of compounds is thought to consist mainly of dimeric oxidation products (e.g. Kristensen et al. 2016; Berndt et al., 2018; Frege et al., 2018). Larger oxidation products (C > 22) were detected but to a much lower extent.
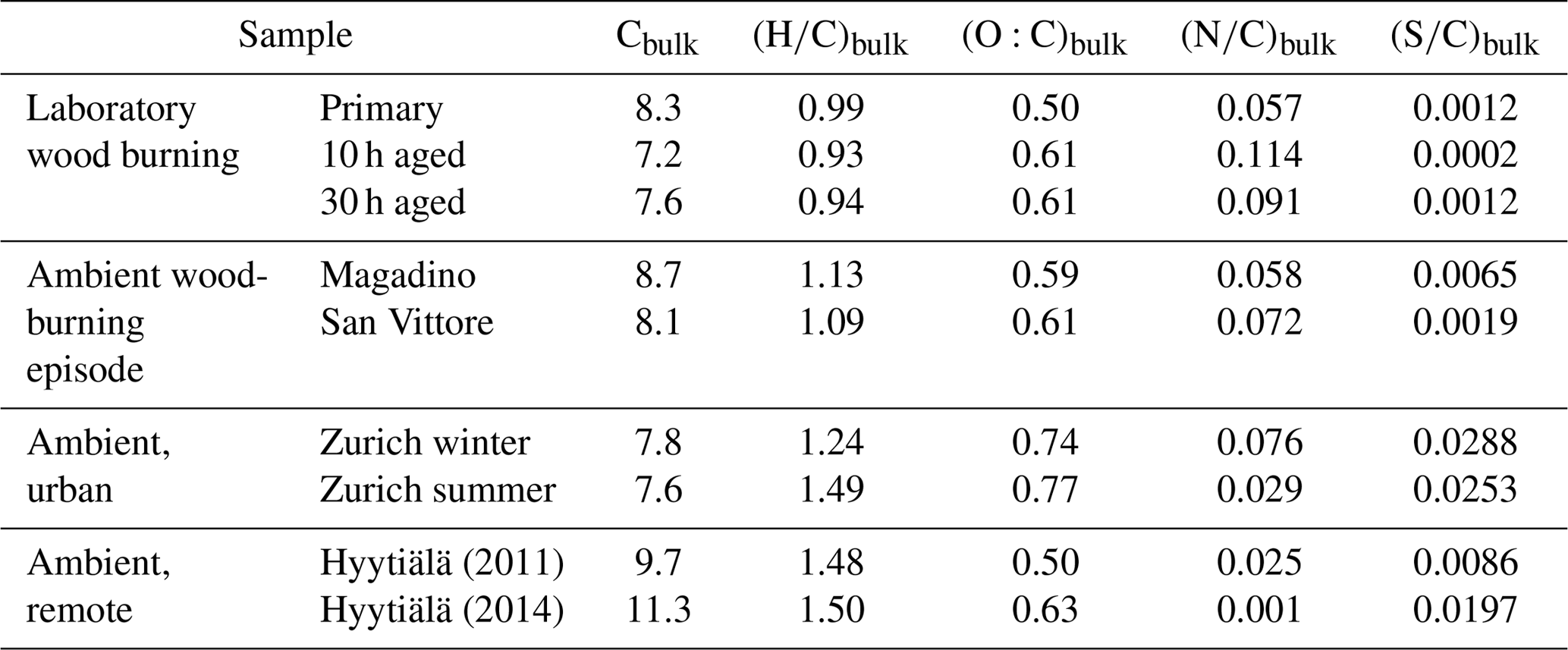
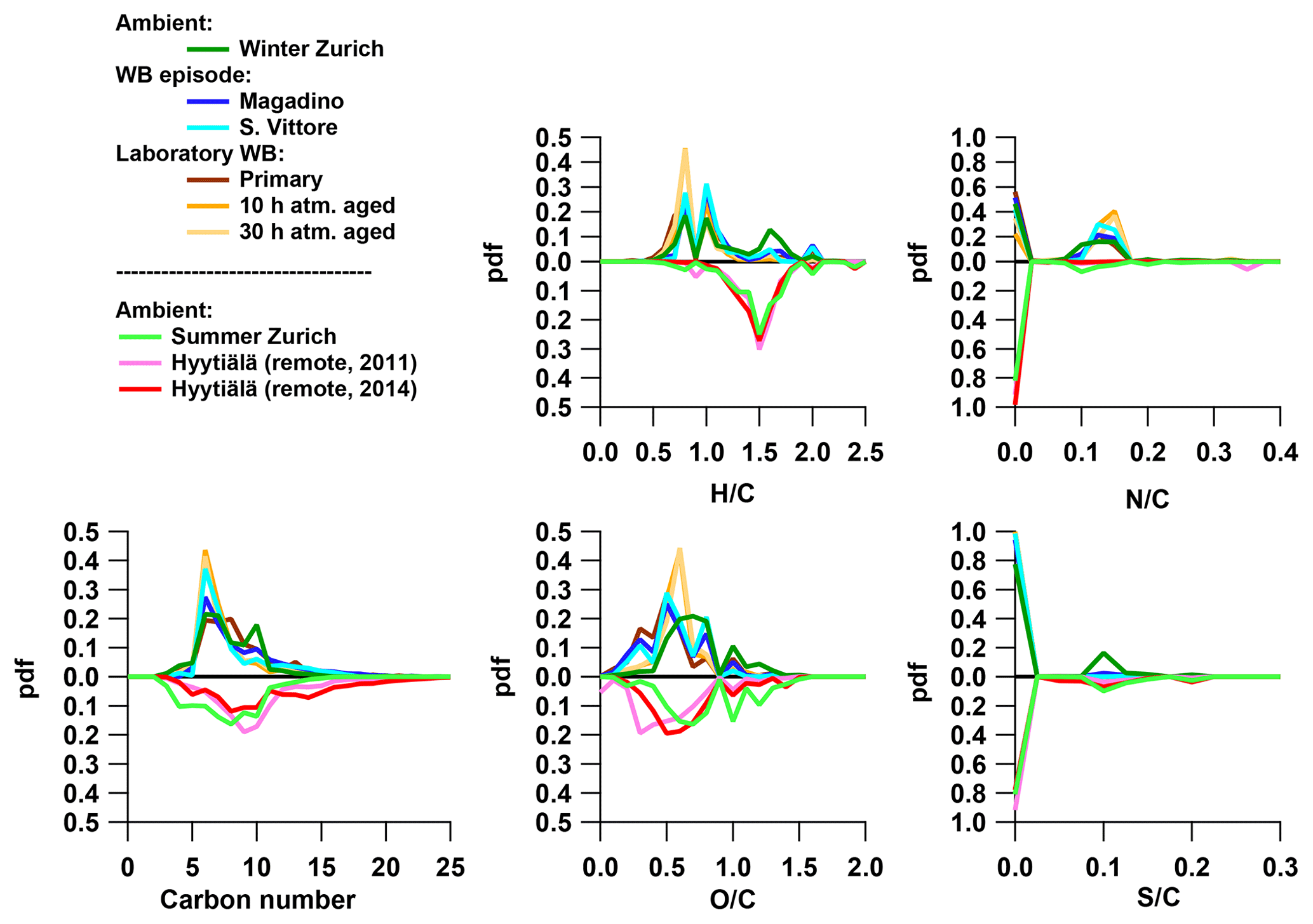
Figure 4The probability density function (pdf) of bulk properties of organic aerosol (neutral composition based on (-)ESI-ultra-high-resolution mass spectra) for different emission and aging conditions: the number of carbon (H∕C, O:C, N∕C, and S∕C) of ambient samples from Zurich in winter and summer, wood-burning episodes at the Alpine valley sites Magadino and San Vittore, and laboratory wood-burning experiments (fresh emissions and after simulated atmospheric aging of 10 and 30 h), as well as from boreal forest in Hyytiälä, Finland (Kourtchev et al., 2014a, 2016). The carbon number is binned in ranges of rounded masses: H∕C and O:C in ranges of 0.1 and N∕C and S∕C in ranges of 0.025.
The OA bulk composition in summertime Zurich was similar to that in Hyytiälä, especially in the C8–C12 range, and at both locations CHO compounds dominated the signal (Figs. 4, 5, 6). The (H∕C)bulk was 1.49 for Zurich samples during summertime, which is similar to those for samples from Hyytiälä and to the oxidation products of BVOCs (e.g. α-pinene, pinic acid: C9H14O4, MBTCA, C8H12O6). However, compared to Hyytiälä, signals in Zurich were clearly shifted toward smaller molecules (Cbulk 7.6: Figs. 4, 6, 7, 8, Table 2). While a high fractional contribution to the signal of C13–C16 and C17–C22 compounds was observed in Hyytiälä 2014, these compounds contributed much less in Zurich (Figs. 5, 6, 7). Meanwhile, small molecules such as C4H6O5 (possibly related to malonic acid) and C5H8O5 (possibly related to hydroxyglutaric acid) exhibited a higher fractional contribution in Zurich during summer than in Hyytiälä 2014 (Figs. 4, 6, 7, 8). Some of these compounds were related to OH-radical-induced atmospheric aging of monoterpene SOA, especially at high NOx conditions, in ambient as well as in laboratory experiments (Zhang et al., 2018; Mutzel et al., 2015) but could also originate from other biogenic precursors such as isoprene (see Sect. 3.4.2). In the following, we will discuss the possible reasons for the differences.
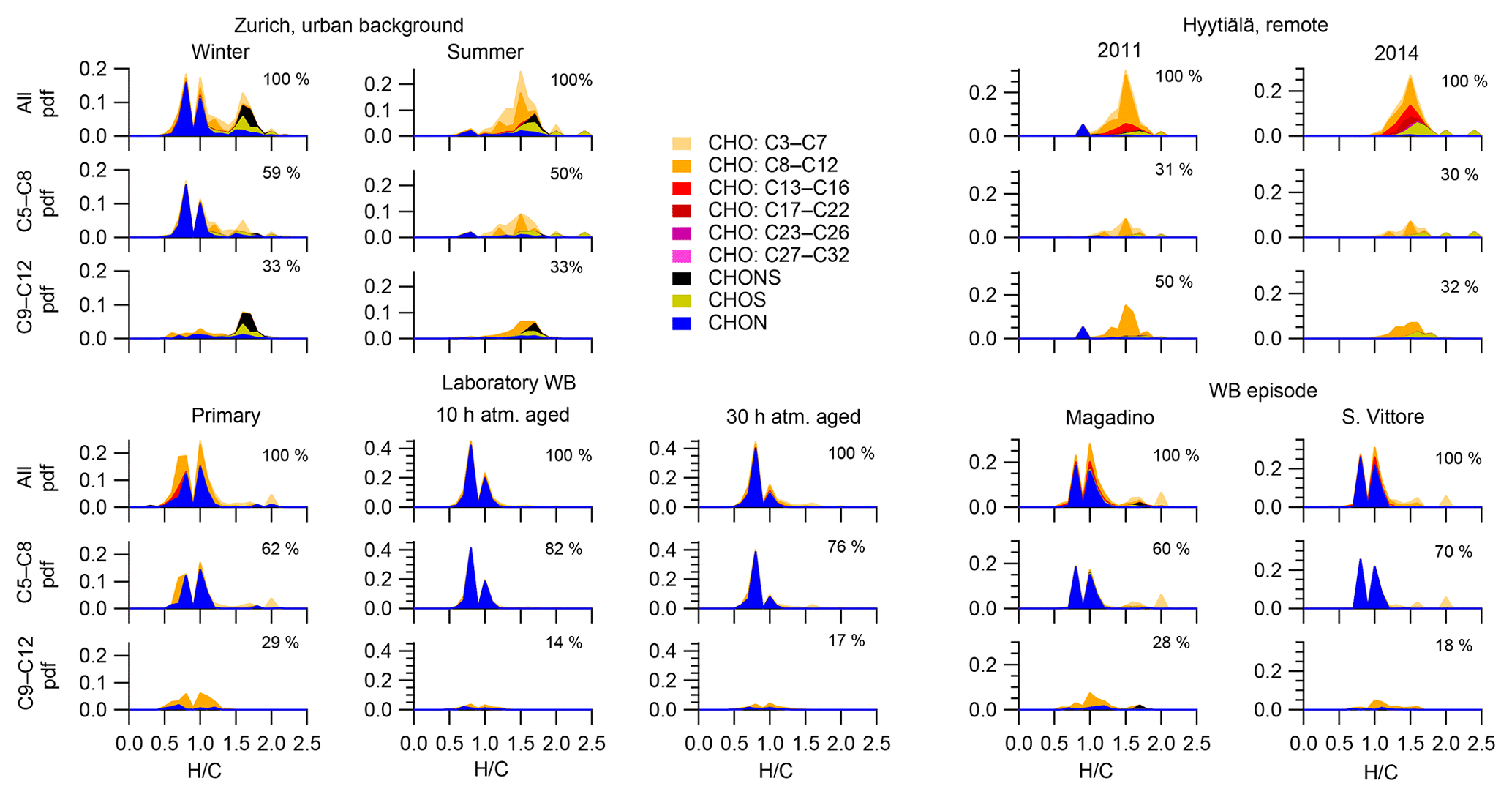
Figure 5Probability density functions (pdf) and contributions of different molecule families to H∕C for all molecules (neutral composition based on (-)ESI-ultra-high-resolution mass spectra), molecules with 5 to 8 carbon atoms, and 9 to 12 carbon atoms for the ambient samples collected in Zurich, Magadino, San Vittore, and wood-burning smog chamber experiments. The area of the histograms is proportional to the percentage of the total signal explained for each dataset.
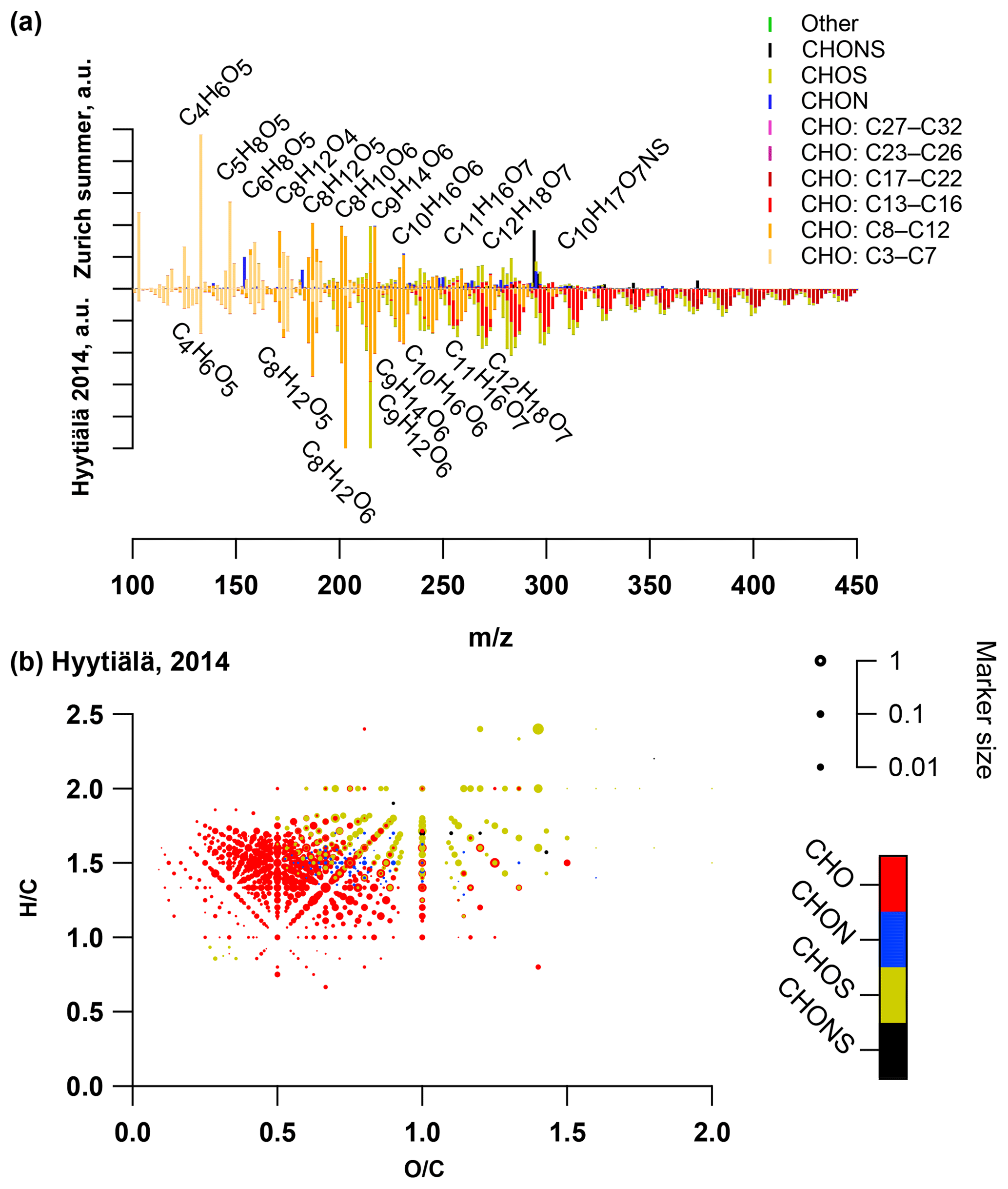
Figure 6(a) Average summer (T > 11 ∘C) ultra-high-resolution mass spectra integrated into unit-mass resolution in the negative mode for the organic aerosol in PM10 sampled in Zurich during the year 2013 (weighted average with OA concentration from offline AMS analysis) compared to the equivalent spectrum from Hyytiälä 2014. The signal at a nominal mass is separated by ion family (CHO, CHON, CHOS, CHONS, other). Peak assignments of dominant ions of selected UMR peaks are labelled as neutral compounds. (b) Van Krevelen diagram of negative ion mode spectra (neutral composition) of Hyytiälä 2014 sample peaks displayed as circles, with their size reflecting log(intensity) and the colour code reflecting the molecular composition of the compound (data from Kourtchev et al., 2016).
3.4.2 CHO and BVOC composition
The composition of BVOC emissions depends on various parameters, such as vegetation type and temperature. While many BVOCs lead to the formation of oxidation products characterized by H∕C ∼1.5, Cbulk depends on the size of the carbon backbone of the initially emitted precursor and the degree of accretion. Thus, the composition of the BVOC emissions has an impact on Cbulk. Modelled biogenic emissions showed a higher isoprene (ISO, C5H8) to monoterpene (MT, C10H16) ratio in Switzerland than in Finland (Fig. S2, Jiang et al., 2019). The higher ISO/MT ratio in BVOC emissions in Zurich could contribute to the higher C3–C7 CHO compound contribution at this site (see above, Figs. 4, 5, 7, 8). Sesquiterpene/monoterpene emission ratios (SQT/MT) did not show a clear difference between Finland and Switzerland and is therefore not expected to be the reason for the observed enhanced abundance of C13–C17 compounds in Hyytiälä compared to summertime Zurich (see the NOx discussion in the next section). In addition, increased temperatures lead to higher BVOC emissions and may induce some effects on the ratio of ISO/MT (mainly driven by photosynthetic activity) and of SQT/MT (Zhao et al., 2017). Biotic stress acting on plants may influence the SQT/MT emission ratio as well. Zhao et al. (2017) reported ratios of 0.15 for unstressed and 3.5 for stressed plants at 22–25 ∘C. These various effects could contribute to the variability in the abundance of C13–C17 CHO compounds observed between Zurich (17 ∘C, average Tmax=21 ∘C) and Hyytiälä in 2011 (15 ∘C, average Tmax=18 ∘C) and 2014 (20 ∘C, average Tmax=24 ∘C).
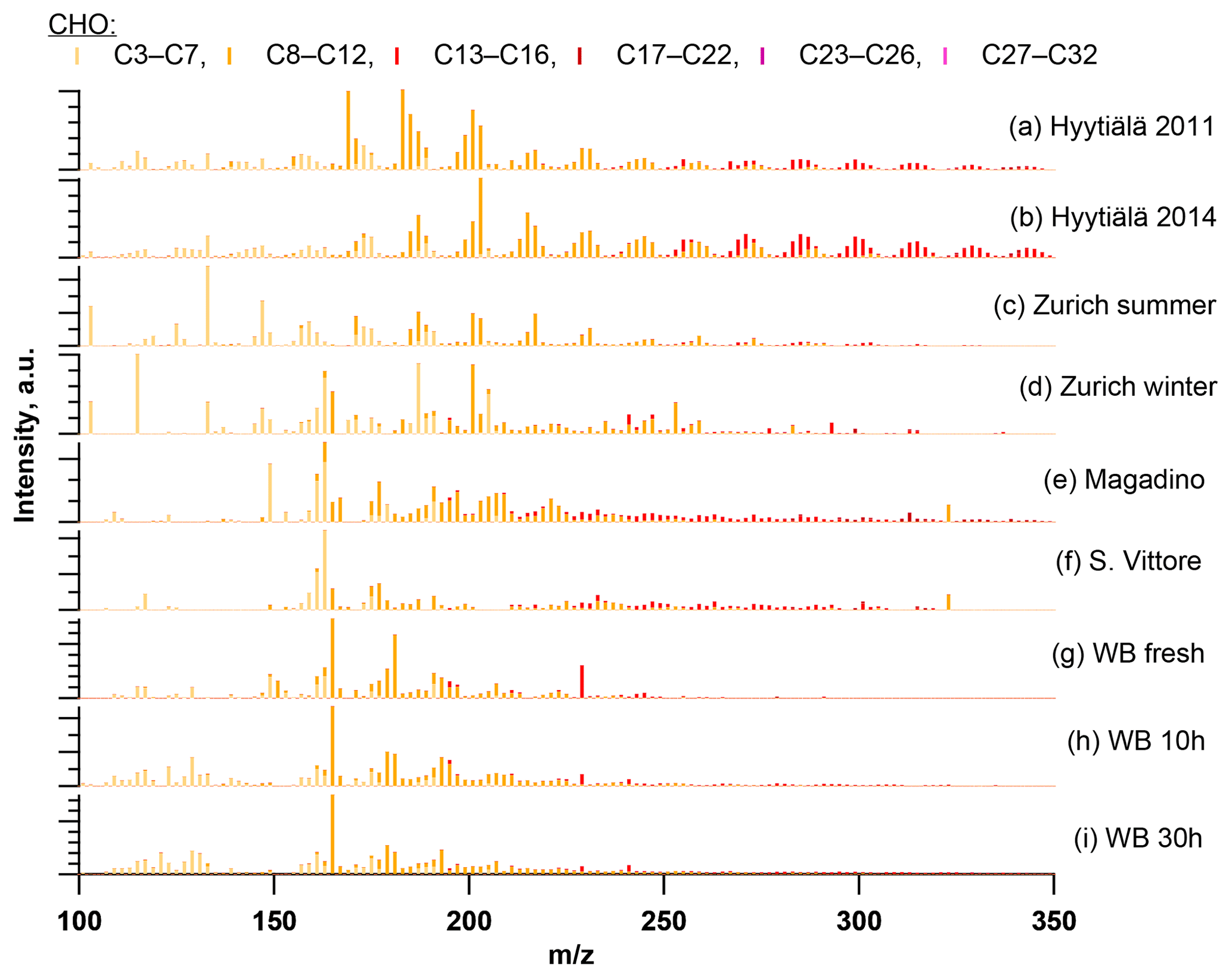
Figure 7Ultra-high-resolution mass spectra of CHO compounds integrated to unit-mass resolution in the negative mode for the organic aerosol in Zurich during summer (T > 11 ∘C) and winter (T < 6 ∘C) 2013 (PM10, OA-weighted average) in Hyytiälä during campaigns in 2011 and 2014, during wood-burning episodes in San Vittore and Magadino, and during laboratory wood-burning experiments (fresh emissions, 10 h, and 30 h atmospherically aged).
3.4.3 CHO and temperature
In 2014, higher daily average temperatures (20 ∘C, average Tmax=24 ∘C) were recorded compared to 2011 (15 ∘C, average Tmax=18 ∘C). The mass spectral signature recorded in Hyytiälä in 2011 showed distinct differences with the observations made in 2014 at the same place and was also less similar to summertime Zurich (Fig. 7). In 2011 (lower T), C8–C12 compounds dominated the signal, while larger compounds (C13–C16 and C17–C22) contributed significantly to the signal in 2014 (Fig. 5). As highlighted by Kourtchev et al. (2016), the higher contributions of dimeric and trimeric BVOC oxidation products in 2014 could be related to higher precursor and SOA mass, which is in agreement with laboratory experiments presented in the same study. Temperature differences affect not only the emissions from the biosphere but also the ratio between particle- and gas-phase concentration of compounds as a function of their volatility. This could lead to an enhancement of less volatile dimeric compared to more volatile monomeric BVOC oxidation products at higher T (Fig. 8, Hyytiälä 2014 vs. 2011). Temperature also affects the particle-phase contribution of first- (more volatile, such as pinic acid) and second-generation (less volatile, such as MBTCA) gas-phase oxidation products (Zhang et al., 2010; Vogel et al., 2013; Müller et al., 2012; Donahue et al., 2012). This is consistent with the observed enhancement of MBTCA compared to pinic acid with rising temperatures (Fig. 8). This phenomenon partially explains the variability in the observed composition of monomeric BVOC oxidation products.
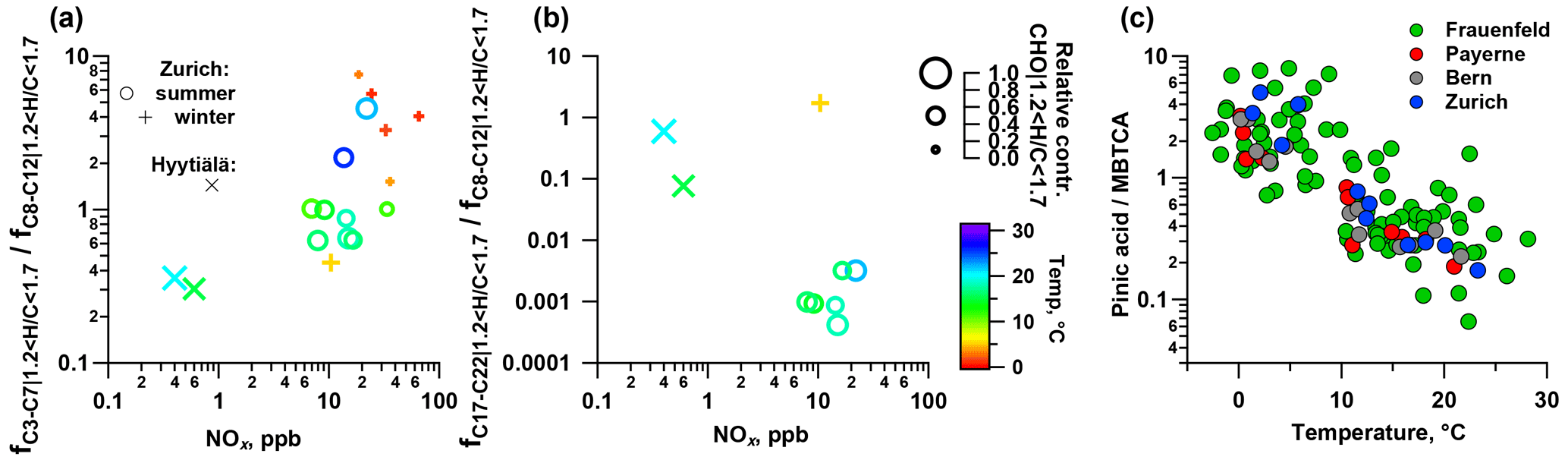
Figure 8Impact of NOx on the fraction of (a) C3–C7 and (b) C17–C22 relative to C8–C12 compounds and (c) the ratio of pinic acid to MBTCA from LC-MS as a function of temperature.
Kourtchev et al. (2016) observed an increasing fraction of smaller molecules (C3–C7) in the total observed signal from biogenic SOA at higher temperatures. The increase in the proportion of smaller compounds (C3–C7) occurs despite their increasingly high evaporation rates. This could be related to a higher fraction of first-generation products residing in the gas phase where they are prone to further oxidation, possibly also promoting fragmentation. Since the average temperature in Zurich during summer is 17 ∘C (average Tmax=21 ∘C) this would partially explain the enhancement of the fraction of lower-molecular-weight compounds (C3–C7) compared to Hyytiälä.
3.4.4 CHO and NOx
While laboratory monoterpene experiments show a large contribution of functionalized monomeric oxidation products to SOA, ambient measurements have revealed an enhancement of fragmentation over functionalized products with increasing NOx concentrations (Zhang et al., 2018). Fragmentation products of RO2+NO reactions and subsequent autoxidation could explain such observations. Since we observe a similar behaviour (Fig. 8) in this study, the higher (C3–C7) ∕ (C8–C12) in summertime Zurich than in Hyytiälä can be related to enhanced NOx concentrations at urban sites (NOx summertime Zurich – 15 ppb; Hyytiälä – 0.5 ppb).
Dimeric monoterpene oxidation products (C17–C22) are mainly formed through RO2+RO2 reactions, while in the presence of NOx this reaction pathway is suppressed by radical termination reactions between RO2 and NO (Kristensen et al., 2016; Lehtipalo et al., 2018). This effect explains the considerable contribution of C17–C22 compounds in Hyytiälä, while they are largely absent in summertime Zurich (Figs. 5, 7, 8). Overall the enhanced NOx concentrations inhibit the formation of such dimeric C17–C22 compounds, leading to the smaller Cbulk in summertime Zurich than in Hyytiälä.
3.4.5 CHON, CHOS, and CHONS
A fraction of the signal is related to compounds also consisting of nitrogen and/or sulfur (CHON, CHOS, and CHON) in summertime Zurich (32 %) and Hyytiälä (19 %, 2011 – 15 %; 2014 – 23 %). Both in Hyytiälä and summertime Zurich CHOS compounds contribute to the signal (Zurich – 14 %; Hyytiälä 2011 – 6 %; Hyytiälä 2014 – 22 %), while the summertime Zurich SO2 concentration (average 0.4 ppb) exceeds a typical Hyytiälä concentration (June–July–August Q75=0.15 ppb, not available during sampling periods). Such compounds were also detected in wintertime Zurich. This group of compounds exhibits a similar composition at both sites (H∕C range of 1.3–1.9 and mostly C5–C12 compounds, though some larger compounds were also found in Hyytiälä 2014 with the 15 enhanced contributions of dimers, as expected). CHON compounds are enhanced in summertime Zurich (CHON: 13 %) compared to Hyytiälä (CHON – 4 %; 2011 – 7 %; 2014 – 1 %) but contribute clearly less than in wintertime Zurich. The CHON compounds have a similar H∕C (0.8–0.9) at both sites but a higher Cbulk in Hyytiälä (C9–C12) than in summertime Zurich (C5–C8). CHONS compounds are observed in summertime Zurich (4 %) and cover an H∕C range similar to CHOS compounds (1.4–1.9), but the signal can almost uniquely be explained by compounds with C9–C12 (most prominently C10H17O7NS). These compounds are largely absent in Hyytiälä (1 %), which might be explained by elevated NOx concentrations in Zurich.
3.5 Wood-burning emissions: laboratory experiments, ambient pollution episodes, and wintertime pollution
Chemical composition
Wood burning is an important wintertime source of OA in Central Europe (Herich et al., 2014; Lanz et al., 2010; Crippa et al., 2014; Zotter et al., 2014a; Daellenbach et al., 2017). In the following, filters from laboratory wood-burning experiments were used as a reference for understanding the influence of such emissions on wintertime pollution at different sites (Figs. 9, 10). During laboratory wood-burning experiments, aromatic CHON compounds with H∕C between 0.8 and 1.0 and 5 to 8 carbon atoms (C5–C8) contribute a large fraction of the signal (43 %, Figs. 4, 5). Additionally, CHO compounds, not present in summer samples with C8–C12 and H∕C 0.8–1.0, contribute significantly to fresh emissions (56 %). Since the contribution of CHON to biomass-burning aerosol increases at least initially during aging (65 %–78 % for aged and 43 % for fresh emissions), the relative contribution of CHO decreases (22 %–34 % for aged and 56 % for fresh emissions; Fig. 5). The composition during wood-burning episodes in Alpine valleys is similar to primary wood-burning emissions sampled in the laboratory (CHON – 47 %–58 %; CHO – 48 %–41 %; Fig. 5). In wintertime Zurich, the chemical composition is also characterized by a large contribution of CHON (43 %) and CHO (35 %) compounds to the signal but is smaller than at the Alpine valley sites (Fig. 5). However, additionally CHOS (12 %) and CHONS (10 %) compounds also contribute to the signal in wintertime Zurich, which is the case in neither the laboratory wood-burning experiments nor wood-burning episodes in the Alpine valleys (Figs. 5, 10). The composition of the laboratory wood-burning emissions, wood-burning episodes in Alpine valleys, and wintertime Zurich is clearly distinguishable from biogenic SOA by a higher contribution of CHON compounds (43 %–58 %) as well as a lower bulk H∕C (0.93–1.24) and Cbulk (7.2–8.7) than for biogenic SOA (CHON – 1 %–4 %; H∕C – 1.48–1.50; Cbulk – 9.7–11.3; Figs. 4, 5).
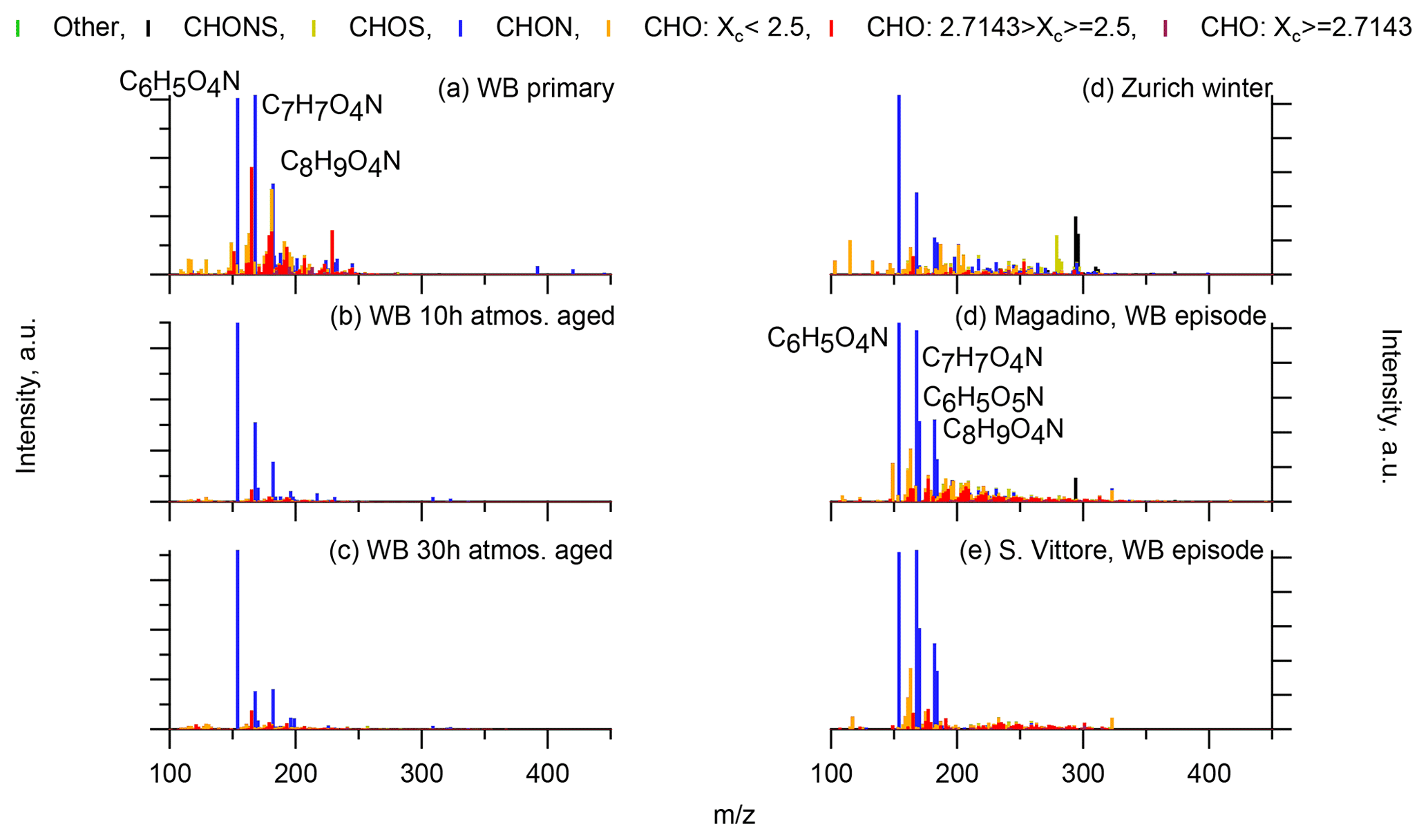
Figure 9(-)ESI-ultra-high-resolution mass spectra integrated to unit-mass resolution of wood-burning laboratory experiments (a primary emissions, b emissions after 10 h atmospheric aging, c emissions after 30 h atmospheric aging) and from winter wood-burning episodes at Alpine valley sites (d Magadino, e San Vittore). The signal at a nominal mass is separated by ion family (CHON, CHOS, CHONS, other), and the signal of CHO compounds is separated by aromaticity (non-aromatic, Xc > 2.5; aromatic, 2.5≤Xc < 2.7143; condensed aromatic, Xc≥2.7143). Peak assignments of dominant ions of selected UMR peaks are labelled as neutral compounds.
During the laboratory wood-burning experiments, compounds with H∕C of 0.8–1.0 and 5–8 carbon atoms dominate the CHON family. In fact, only a few compounds, such as nitrocatechols and similar compounds (C6H5O4N, C7H7O4N, C8H9O4N), contribute to the signal. Compounds detected in a road traffic tunnel, which were related to vehicular emissions (C7H7O3N and C8H9O3N, Tong et al., 2016) were also detected in the primary wood-burning emissions but with much lower intensities than C7H7O4N and C8H9O4N. While SOA formation from vehicular emissions might proceed via the oxidation of aromatic species, the most important precursors in biomass smoke are oxygenated aromatics such as phenol, cresol, and catechol (Harrison et al., 2005; Platt et al., 2013; Bruns, et al., 2015, 2016, 2017; Schauer et al., 2001, 2002). While CHON compounds were already present in the fresh emissions and thus were directly emitted, the increasing (N∕C)bulk indicates a strong additional secondary formation of such compounds (Figs. 4, 5, 9). In Magadino, San Vittore, and wintertime Zurich similar CHON compounds dominated the signal. Their composition suggests that the biomass-burning emissions observed in wintertime Zurich were further processed than in Magadino and San Vittore. During summer these compounds exhibited a much smaller contribution in Zurich (see more detail in Sect. 3.4 and Fig. 5).
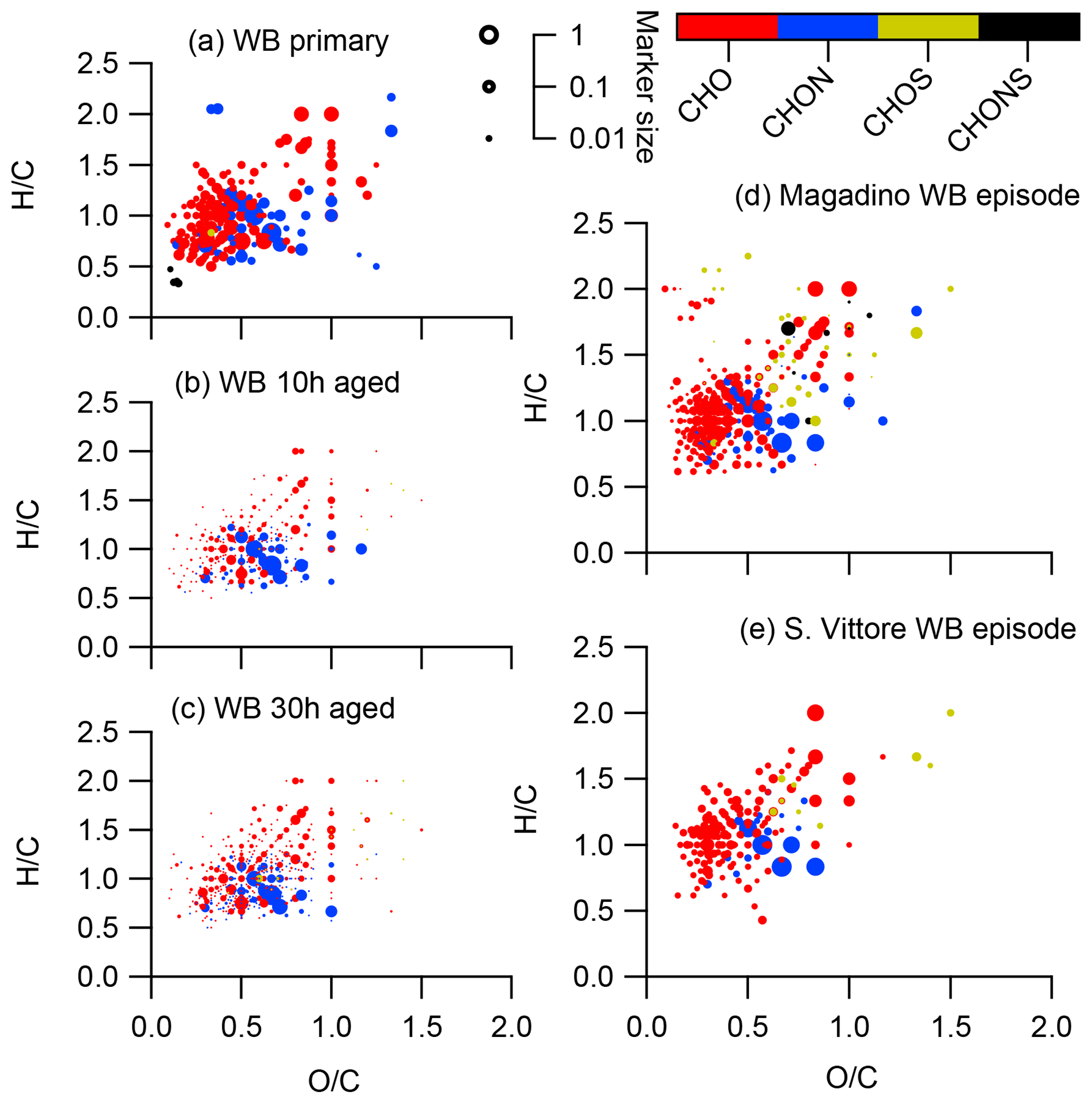
Figure 10Van Krevelen diagrams of negative ion mode spectra (neutral composition) of smog chamber wood-burning experiments (fresh emissions, after 10 h of simulated atmospheric aging, and after 30 h of simulated atmospheric aging) and of wood-burning episodes at the Alpine valley sites Magadino and San Vittore. Peaks are displayed as circles with their size reflecting log(intensity) and the colour code reflecting the molecular composition of the compound.
The CHO compounds observed in the fresh wood-burning emissions during the laboratory experiments were characterized by a lower H∕C (0.7 to 1.0) than biogenic SOA (1.5) (Figs. 5, 10). Anhydrous sugars with high H∕C such as levoglucosan (C6H10O5, H∕C 1.67, O:C 0.83), mannosan, and galactosan, directly emitted from cellulose pyrolysis during biomass burning were detected in the laboratory wood-burning emissions as well as in Magadino and San Vittore but only contributed little to the signal. A considerable amount of the CHO compounds in wood-burning emissions could be considered aromatic (Xc≥2.5) or even condensed aromatic (Xc≥2.7143, Fig. S4). These compounds are consistent with products from lignin pyrolysis (Fig. S4, Bertrand et al., 2017, 2018) and contributed significantly to the CHO signal recorded for primary wood-burning emissions but less for aged emissions (57 %, 49 %, 44 % of CHO aromatic; 5 %, 3 %, 3 % of condensed aromatic; and 38 %, 48 %, 53 % of non-aromatic for fresh, 10 h, and 30 h atmospherically aged emissions, respectively). The ambient wood-burning pollution showed a similar distribution of the CHO signal as aged laboratory wood-burning emissions (Magadino – 46 % aromatic, 6 % condensed aromatic, 48 % non-aromatic; San Vittore – 44 % aromatic, 4 % condensed aromatic, 52 % non-aromatic), and the detailed chemical composition was similar to the fresh laboratory wood-burning emissions (Fig. S4). With proceeding aging during the wood combustion experiments, more oxygenated compounds dominated the signal during the wood combustion experiments (centre at H∕C of 1.0 and O:C of 0.5, Figs. 10, S4). These values are consistent with aqueous SOA from syringol–guaiacol–phenol formed through the reaction with hydroxyl radicals and excited states of organic compounds (Yu et al., 2014). Since wood burning is a known emitter of such compounds, it seems probable that the aged wood-burning emissions consisted of oxidation products of phenolic compounds (Schauer et al., 2001; Bruns et al., 2016, 2017). In wintertime Zurich, the CHO composition largely shows common features with aged laboratory wood-burning emissions (Figs. 10, S4). However, in wintertime Zurich the contribution of smaller CHO compounds (C3–C7) was higher (Figs. 5, 7, S3) and the contribution of aromatic compounds to the total CHO signal was clearly lower (22 % aromatic, 2 % condensed aromatic, 76 % non-aromatic) than for the laboratory wood-burning emissions (Figs. S3, S4, S5, S6). This indicates that additional processes leading to fragmentation play a role in the urban environment as already observed in summer. During summer the influence of aromatic compounds on the CHO signal was negligible (Zurich summer: condensed aromatic −0 %, aromatic −3 %, and non-aromatic −97 %).
In wintertime Zurich, CHOS and CHONS compounds contributed significantly to the signal as opposed to the laboratory wood-burning experiments and Alpine valleys. These compounds had between 9 and 12 carbons and were characterized by an H∕C between 1.5 and 2.0. During the laboratory wood-burning experiments (aging initialized with HONO) no SO2 was added during photochemical aging, which could explain the absence of these CHOS and CHONS compounds. On the other hand, a compound group with similar H∕C and Cbulk was also found in summertime Zurich but not in biogenic SOA. Thus, they can be linked to neither biogenic SOA nor wood-burning emissions. The presence of this compound group indicates the importance of additional sources and/or processes in the formation of urban SOA.
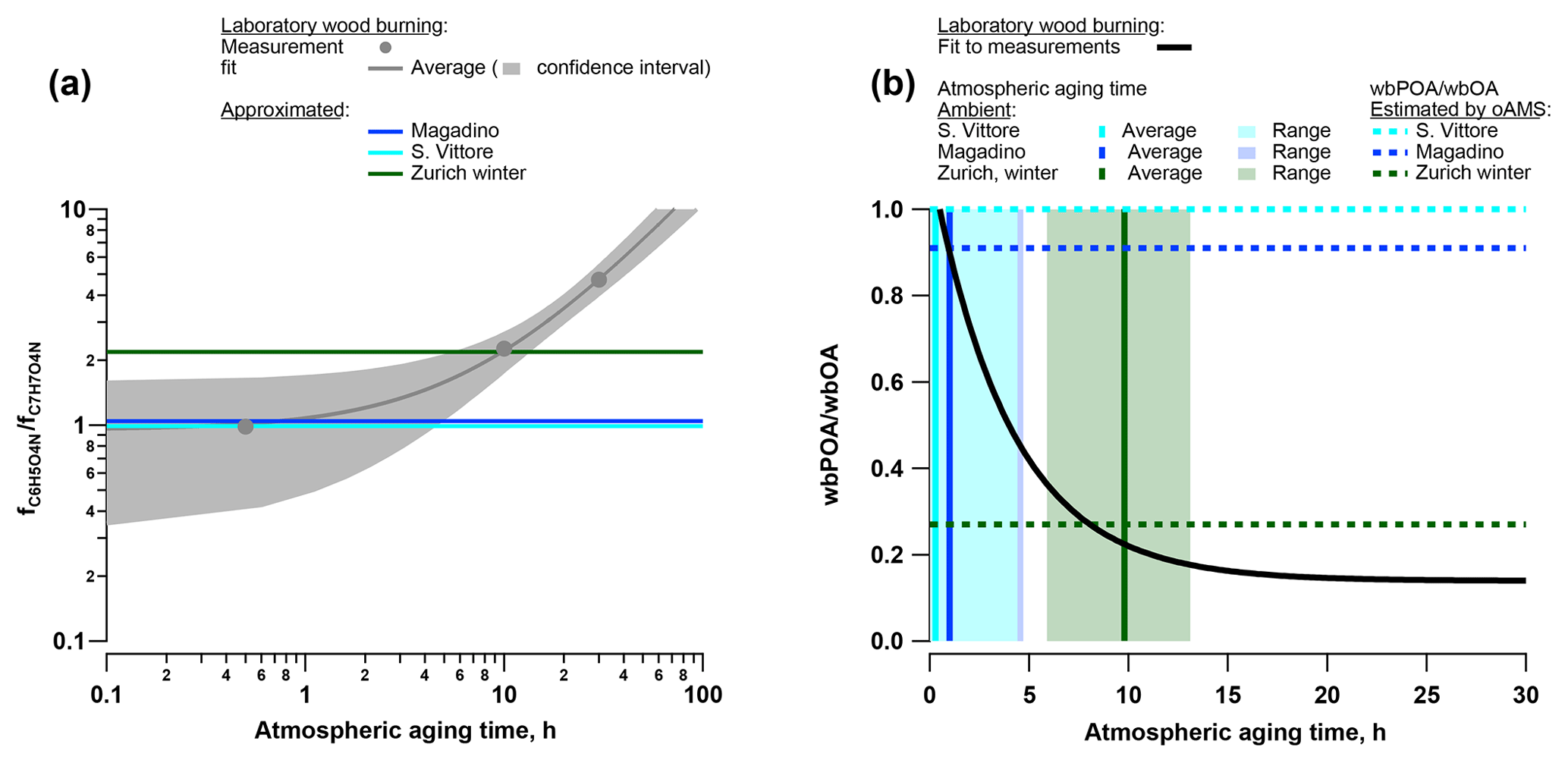
Figure 11(a) Atmospheric aging parameterized as a function of the ratio of the fraction of total signal related to C6H5O4N () and C7H7O4N (); for the wood-burning smog chamber experiments and estimated atmospheric aging time of the wood-burning emissions for the ambient analysis in Zurich (winter), Magadino, and San Vittore. (b) Fraction of primary wood-burning emissions to total wood-burning OA (wbPOA/wbOA) parameterized as a function of the atmospheric aging time of the wood-burning smog chamber experiments and estimated wbPOA/wbOA for the ambient analysis in Zurich (winter), Magadino, and San Vittore.
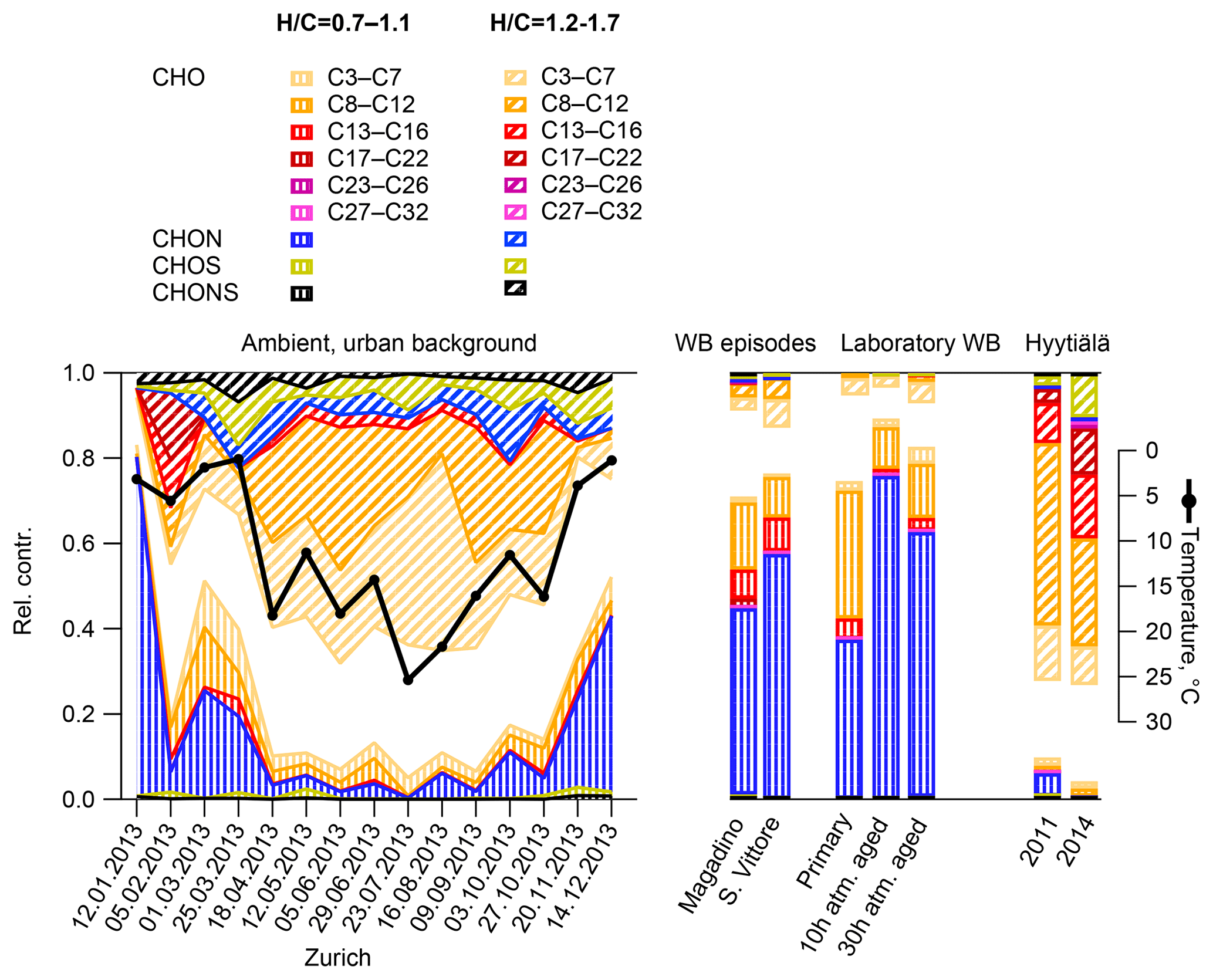
Figure 12Relative contributions of different compound classes to the signal of compounds, with as a surrogate for wood-burning emissions and as a surrogate for biogenic SOA.
3.6 Atmospheric aging of wood-burning OA
Nitrocatechol (C6H5O4N) and methyl-nitrocatechol (C7H7O4N) are two molecules commonly studied in ambient aerosol samples and used as markers for wood-burning SOA (Iinuma et al., 2010). These compounds were large contributors to the signal during the laboratory wood-burning experiments as well as in wintertime Zurich, Magadino, and San Vittore (Figs. 5, 9). Additionally, in the laboratory experiments the signal ratio of C6H5O4N to C7H7O4N increased with atmospheric aging (Fig. 9, H∕C 0.8 and 1.0). Based on this observation, we conclude that the wood-burning emissions analysed in San Vittore and Magadino are fresher than the ones in wintertime Zurich. In the following, we directly link the signal ratio C6H5O4N∕C7H7O4N to the atmospheric age of wood-burning emissions (Fig. 11). We derived a relation between the aging time in the smog chamber (10 and 30 h equivalent atmospheric aging) and the C6H5O4N∕C7H7O4N intensity ratio. Thereby, we estimated that in winter Zurich the wood-burning emissions were on average aged for 9.8 h (range 5.9–13.1 h). In contrast, at the Alpine valley sites the wood-burning emissions were only aged between 0.3 (San Vittore, range 0–4.4 h) and 1.0 h (Magadino, range 0–4.8 h). In the next step, we approximated the contribution of POA to OA during the smog chamber experiment using the OA (AMS) and BC (aethalometer) measurements and assuming a constant POA∕BC ratio during the measurement time yielding a POA∕OA of 0.22 after 10 h atmospheric aging and of 0.14 after 30 h. With this parameterization and the approximated aging time of the wood-burning emissions for the ambient samples, we estimated the contribution of primary wood-burning OA (wbPOA) to the total wood-burning OA (wbOA) for the ambient data (Fig. 11). The POA contribution to wbOA was 22 % in Zurich winter (best estimate by linear regression, range 18 %–36 % based on uncertainty of estimated atmospheric aging time) but much higher in Magadino (90 %, range 44 %–100 %) and San Vittore (100 %, range 47 %–100 %). Based on the offline AMS PMF, we use BBOA as an estimate of wbPOA and WOOA as an upper limit of wbSOA. At the Alpine valley sites, the influence of BBOA on the sum of BBOA and WOOA was higher than in wintertime Zurich (San Vittore – 100 % BBOA; Magadino – 90 % BBOA; wintertime Zurich – 27 % BBOA). These results are consistent with the findings in this study.
3.7 Temporal behaviour
The relative contribution of compounds with H∕C between 1.2 and 1.7 (characteristic range of BVOC SOA, Fig. 12) largely explained the signal in Hyytiälä (approximately 75 %) and contributed only little to the wintertime Alpine valley samples and laboratory wood-burning experiments (bulk composition in Fig. S7). The relative contribution of this compound class showed a seasonal behaviour similar to the local temperature in Zurich (R2=0.75, p < 10−4). The higher the temperature, the larger the contribution of small compounds was (C3–C7). Additionally, some CHON, CHOS, and CHONS molecules were also part of this compound class, which suggests a biogenic origin of these compounds. In Zurich, a good correlation was observed between the time series of the relative signal contribution of the compounds with an H∕C between 1.2 and 1.7 and a relative contribution of SOOA to OA (R2=0.57, p < 0.01).
On the other hand, a large part of the signal of the wintertime Alpine valley samples and the laboratory wood-burning samples could be explained by a compound class characterized with H∕C between 0.7 and 1.1 (characteristic range of wood-burning emissions, Fig. 12). The largest contribution to the relative signal of this group was from CHON molecules. In Zurich, the relative contribution of this compound class to the signal had a seasonal pattern consistent with the residential heating behaviour. In Zurich, a good correlation was observed between the time series of the relative signal contribution of compounds with H∕C between 0.7 and 1.1 and the relative contribution of the sum of BBOA and WOOA to OA (R2=0.59, p < 0.001), with lower correlations with either BBOA alone (R2=0.29, p=0.02) or WOOA alone (R2=0.37, p < 0.01).
This work links seasonal variability in OA composition based on negative ESI-Orbitrap data from an urban background site in Central Europe (Zurich, Switzerland) to wood-burning emissions in winter and to biogenic SOA in summer. The mass spectral signatures observed for laboratory wood-burning emissions were dominated by CHON compounds (C6H5O4N, C7H7O4N). The influence of CHON increased during aerosol aging, while the relative contribution of CHO compounds decreased as the CHO content became less aromatic (with the aromatic fraction of the CHO signal accounting for 62 % in the fresh aerosol and 47 % in 30 h aged aerosol). Signatures from wintertime pollution episodes at two Alpine valley sites could be explained by the laboratory experiments. Wintertime Zurich signatures were also dominated by CHON compounds, but the CHO fraction showed some differences to the laboratory wood-burning experiments and ambient wood-burning pollution episodes (less aromatic and higher contribution of C3–C7 compounds in Zurich). Additionally, in Zurich a considerable influence of CHOS and CHONS was observed throughout the year, which suggests that additional sources and/or processes are important in Zurich. The summertime signature from Zurich was dominated by CHO compounds and showed a similar mass spectral signature as biogenic SOA observed in the boreal forest (Hyytiälä, Finland). C3–C7 compounds contributed a larger relative fraction to the signal in Zurich than in the boreal forest. While compounds related to sesquiterpenes (C13–C16) and dimers of α-pinene oxidation products (C17–C22) were prominent in the boreal forest, they were largely absent in Zurich during summer. These observations may be explained by differences in oxidant concentrations (mostly NOx) and in the composition of biogenic VOC emissions.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-5973-2019-supplement.
The data are available upon request from the corresponding author.
KRD, IEH, ASHP, IK, MK formulated the study. KRD, IK performed the Orbitrap analyses, JLJ measured MBTCA and pinic acid, EAB performed the smog chamber experiments, TP curated data related to Hyytiälä, JJ and SA performed the model calculations. KRD, IEH designed and performed the statistical analysis and data visualizations. Validation: KRD, IEH, IK, ALV. KRD, IEH, IK. KRD wrote the original draft, which was reviewed, commented and edited by all the authors. ASHP, UB, MK supported and supervised the research.
The authors declare that they have no conflict of interest.
This work was supported by the Swiss Federal Office of Environment; Liechtenstein; Ostluft; the Swiss cantons Basel, Graubünden, and Thurgau; the Competence Center Environment and Sustainability (CCES) (project OPTIWARES); and the Swiss National Science Foundation (SNF, WOOSHI grant 140590). Kaspar R. Daellenbach acknowledges the support of SNF grant P2EZP2_181599.
This paper was edited by Annele Virtanen and reviewed by two anonymous referees.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Bateman, A. P., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Applications of high-resolution electrospray ionization mass spectrometry to measurements of average oxygen to carbon ratios in secondary organic aerosols, Environ. Sci. Technol., 46, 8315–8324, https://doi.org/10.1021/es3017254, 2012.
Bar-Joseph, Z., Gifford, D. K., and Jaakkola, T. S.: Fast optimal leaf ordering for hierarchical clustering, Bioinformatics 17, S22–S29, https://doi.org/10.1093/bioinformatics/17.suppl_1.S22, 2001.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accertion product formation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bertrand, A., Stefenelli, G., Bruns, E. A., Pieber, S. M., Temime-Roussel, B., Slowik, J. G., Prévôt, A. S. H., Wortham, H., El Haddad, I., and Marchand, N.: Primary emissions and secondary aerosol production potential from woodstoves for residential heating: Influence of the stove technology and combustion efficiency, Atmos. Environ., 169, 65–79, https://doi.org/10.1016/j.atmosenv.2017.09.005, 2017.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018.
Birch, M. E. and Cary, R. A.: Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust, Aerosol Sci. Tech., 25, 221–241, https://doi.org/10.1080/02786829608965393, 1996.
Bonvalot, L., Tuna, T., Fagault, Y., Jaffrezo, J.-L., Jacob, V., Chevrier, F., and Bard, E.: Estimating contributions from biomass burning, fossil fuel combustion, and biogenic carbon to carbonaceous aerosols in the Valley of Chamonix: a dual approach based on radiocarbon and levoglucosan, Atmos. Chem. Phys., 16, 13753–13772, https://doi.org/10.5194/acp-16-13753-2016, 2016.
Bozzetti, C., Daellenbach, K., R., Hueglin, C., Fermo, P., Sciare, J., Kasper-Giebl, A., Mazar, Y., Abbaszade, G., El Kazzi, M., Gonzalez, R., Shuster Meiseles, T., Flasch, M., Wolf, R., Křepelová, A., Canonaco, F., Schnelle-Kreis, J., Slowik, J. G., Zimmermann, R., Rudich, Y., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: Size-resolved identification, characterization, and quantification of primary biological organic aerosol at a European rural site, Environ. Sci. Technol., 50, 3425–3434, https://doi.org/10.1021/acs.est.5b05960, 2016.
Bozzetti, C., Sosedova, Y., Xiao, M., Daellenbach, K. R., Ulevicius, V., Dudoitis, V., Mordas, G., Bycenkiene, S., Plauškaite, K., Vlachou, A., Golly, B., Chazeau, B., Besombes, J.-L., Baltensperger, U., Jaffrezo, J.-L., Slowik, J. G., El Haddad, I., and Prévôt, A. S. H.: Argon offline-AMS source apportionment of organic aerosol over yearly cycles for an urban, rural, and marine site in northern Europe, Atmos. Chem. Phys., 17, 117–141, https://doi.org/10.5194/acp-17-117-2017, 2017a.
Bozzetti, C., El Haddad, I., Salameh, D., Daellenbach, K. R., Fermo, P., Gonzalez, R., Minguillón, M. C., Iinuma, Y., Poulain, L., Elser, M., Müller, E., Slowik, J. G., Jaffrezo, J.-L., Baltensperger, U., Marchand, N., and Prévôt, A. S. H.: Organic aerosol source apportionment by offline-AMS over a full year in Marseille, Atmos. Chem. Phys., 17, 8247–8268, https://doi.org/10.5194/acp-17-8247-2017, 2017b.
Bruns, E. A., Krapf, M., Orasche, J., Huang, Y., Zimmermann, R., Drinovec, L., Mocnik, G., El-Haddad, I., Slowik, J. G., Dommen, J., Baltensperger, U., and Prévôt, A. S. H.: Characterization of primary and secondary wood combustion products generated under different burner loads, Atmos. Chem. Phys., 15, 2825–2841, https://doi.org/10.5194/acp-15-2825-2015, 2015.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27781, https://doi.org/10.1038/srep27881, 2016.
Bruns, E. A., Slowik, J. G., El Haddad, I., Kilic, D., Klein, F., Dommen, J., Temime-Roussel, B., Marchand, N., Baltensperger, U., and Prévôt, A. S. H.: Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry: fresh and aged residential wood combustion emissions, Atmos. Chem. Phys., 17, 705–720, https://doi.org/10.5194/acp-17-705-2017, 2017.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass. Spectrom. Rev., 26, 185–222, 2007.
Canonaco, F., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: Seasonal differences in oxygenated organic aerosol composition: implications for emissions sources and factor analysis, Atmos. Chem. Phys., 15, 6993–7002, https://doi.org/10.5194/acp-15-6993-2015, 2015.
Cavalli, F., Viana, M., Yttri, K. E., Genberg, J., and Putaud, J.-P.: Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol, Atmos. Meas. Tech., 3, 79–89, https://doi.org/10.5194/amt-3-79-2010, 2010.
Claeys, M., Vermeylen, R., Yasmeen, F., Gómez-González, Y., Chi, X., and Maenhaut, W.: Chemical characterisation of humic-like substances from urban, rural and tropical biomass burning environments using liquid chromatography with UV/vis photodiode array detection and electrospray ionisation mass spectrometry, Environ. Chem., 9, 273e284, https://doi.org/10.1071/EN11163, 2012.
Crippa, M., Canonaco, F., Lanz, V. A., Äijälä, M., Allan, J. D., Carbone, S., Capes, G., Ceburnis, D., Dall'Osto, M., Day, D. A., DeCarlo, P. F., Ehn, M., Eriksson, A., Freney, E., Hildebrandt Ruiz, L., Hillamo, R., Jimenez, J. L., Junninen, H., Kiendler-Scharr, A., Kortelainen, A.-M., Kulmala, M., Laaksonen, A., Mensah, A. A., Mohr, C., Nemitz, E., O'Dowd, C., Ovadnevaite, J., Pandis, S. N., Petäjä, T., Poulain, L., Saarikoski, S., Sellegri, K., Swietlicki, E., Tiitta, P., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach, Atmos. Chem. Phys., 14, 6159–6176, https://doi.org/10.5194/acp-14-6159-2014, 2014.
Daellenbach, K. R., Bozzetti, C., Krepelová, A., Canonaco, F., Wolf, R., Zotter, P., Fermo, P., Crippa, M., Slowik, J. G., Sosedova, Y., Zhang, Y., Huang, R.-J., Poulain, L., Szidat, S., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: Characterization and source apportionment of organic aerosol using offline aerosol mass spectrometry, Atmos. Meas. Tech., 9, 23–39, https://doi.org/10.5194/amt-9-23-2016, 2016.
Daellenbach, K. R., Stefenelli, G., Bozzetti, C., Vlachou, A., Fermo, P., Gonzalez, R., Piazzalunga, A., Colombi, C., Canonaco, F., Hueglin, C., Kasper-Giebl, A., Jaffrezo, J.-L., Bianchi, F., Slowik, J. G., Baltensperger, U., El-Haddad, I., and Prévôt, A. S. H.: Long-term chemical analysis and organic aerosol source apportionment at nine sites in central Europe: source identification and uncertainty assessment, Atmos. Chem. Phys., 17, 13265–13282, https://doi.org/10.5194/acp-17-13265-2017, 2017.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dom, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012.
Dzepina, K., Mazzoleni, C., Fialho, P., China, S., Zhang, B., Owen, R. C., Helmig, D., Hueber, J., Kumar, S., Perlinger, J. A., Kramer, L. J., Dziobak, M. P., Ampadu, M. T., Olsen, S., Wuebbles, D. J., and Mazzoleni, L. R.: Molecular characterization of free tropospheric aerosol collected at the Pico Mountain Observatory: a case study with a long-range transported biomass burning plume, Atmos. Chem. Phys., 15, 5047–5068, https://doi.org/10.5194/acp-15-5047-2015, 2015.
El Haddad, I., Marchand, N., Temime-Roussel, B., Wortham, H., Piot, C., Besombes, J.-L., Baduel, C., Voisin, D., Armengaud, A., and Jaffrezo, J.-L.: Insights into the secondary fraction of the organic aerosol in a Mediterranean urban area: Marseille, Atmos. Chem. Phys., 11, 2059–2079, https://doi.org/10.5194/acp-11-2059-2011, 2011.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: Molecular composition, Environ. Sci. Technol., 31, 1345–1358, https://doi.org/10.1021/es9605376, 1997.
Frege, C., Ortega, I. K., Rissanen, M. P., Praplan, A. P., Steiner, G., Heinritzi, M., Ahonen, L., Amorim, A., Bernhammer, A.-K., Bianchi, F., Brilke, S., Breitenlechner, M., Dada, L., Dias, A., Duplissy, J., Ehrhart, S., El-Haddad, I., Fischer, L., Fuchs, C., Garmash, O., Gonin, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kirkby, J., Kürten, A., Lehtipalo, K., Leiminger, M., Mauldin, R. L., Molteni, U., Nichman, L., Petäjä, T., Sarnela, N., Schobesberger, S., Simon, M., Sipilä, M., Stolzenburg, D., Tomé, A., Vogel, A. L., Wagner, A. C., Wagner, R., Xiao, M., Yan, C., Ye, P., Curtius, J., Donahue, N. M., Flagan, R. C., Kulmala, M., Worsnop, D. R., Winkler, P. M., Dommen, J., and Baltensperger, U.: Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation, Atmos. Chem. Phys., 18, 65–79, https://doi.org/10.5194/acp-18-65-2018, 2018.
Gilardoni, S., Vignati, E., Cavalli, F., Putaud, J. P., Larsen, B. R., Karl, M., Stenström, K., Genberg, J., Henne, S., and Dentener, F.: Better constraints on sources of carbonaceous aerosols using a combined 14C – macro tracer analysis in a European rural background site, Atmos. Chem. Phys., 11, 5685–5700, https://doi.org/10.5194/acp-11-5685-2011, 2011.
Hamilton, J. F., Webb, P. J., Lewis, A. C., and Reviejo, M. M.: Quantifying small molecules in secondary organic aerosol formed during the photo-oxidation of toluene with hydroxyl radicals, Atmos. Environ., 39, 7263–7275, https://doi.org/10.1016/j.atmosenv.2005.09.006, 2005.
Hari P. and Kulmala M.: Station for Measuring Ecosystem–Atmosphere Relations (SMEAR II), Boreal Environ. Res., 10, 315–322, 2005.
Harrison, M. A. J., Barra, S., Borghesi, D., Vione, D., Arsene, C., and Olariu, R. L.: Nitrated phenols in the atmosphere: a review, Atmos. Environ., 39, 231–248, https://doi.org/10.1016/j.atmosenv.2004.09.044, 2005.
Herich, H., M. F. D. Gianini, C. Piot, G. Mocnik, J. L. Jaffrezo, J. L. Besombes, A. S. H. Prevot, and C. Hueglin: Overview of the impact of wood burning emissions on carbonaceous aerosols and PM in large parts of the Alpine region, Atmos. Environ., 89, 64–75, https://doi.org/10.1016/j.atmosenv.2014.02.008, 2014.
Huffmann, B. A., Poltash, M. L., and Hughey, C. H.: Effect of polar protic and polar aprotic solvents on negative-ion electrospray ionization and chromatographic separation of small acidic molecules, Anal. Chem., 84, 9942–9950, https://doi.org/10.1021/ac302397b, 2012.
Iinuma, Y., Böge, O., Gräfe, R., and Herrmann, H.: Methylnitrocatechols: atmospheric tracer compounds for biomass burning secondary organic aerosols, Environ. Sci. Technol., 44, 8453e8459, https://doi.org/10.1021/es102938a, 2010.
Iinuma, Y., Keywood, M., and Herrmann, H.: Characterization of primary and secondary organic aerosols in Melbourne airshed: The influence of biogenic emissions, wood smoke and bushfires, Atmos. Environ., 130, 54–63, https://doi.org/10.1016/j.atmosenv.2015.12.014, 2015.
Irei, S., Rudolph, J., Huang, L., Auld, J., Collin, F., and Hastie, D.: Laboratory studies of carbon kinetic isotope effects on the production mechanism of particulate phenolic compounds formed by toluene photooxidation: A tool to constrain reaction pathways, J. Phys. Chem. A, 119, 5–13, https://doi.org/10.1021/jp5104609, 2015.
Jaffrezo, J. L., Calas, T., and Bouchet, M.: Carboxylic acids measurements with ionic chromatography, Atmos. Environ., 32, 2705–2708, https://doi.org/10.1016/S1352-2310(98)00026-0, 1998.
Jang, M. S. and Kamens, R. M.: Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene, Environ. Sci. Technol., 35, 3626–3639, https://doi.org/10.1021/es010676+, 2001.
Jiang, J., Aksoyoglu, S., Ciarelli, G., Oikonomakis, E., El-Haddad, I., Canonaco, F., O'Dowd, C., Ovadnevaite, J., Minguillón, M. C., Baltensperger, U., and Prévôt, A. S. H.: Effects of two different biogenic emission models on modelled ozone and aerosol concentrations in Europe, Atmos. Chem. Phys., 19, 3747-3768, https://doi.org/10.5194/acp-19-3747-2019, 2019.
Kitanovski, Z., Grgic, I., Vermeylen, R., Claeys, M., and Maenhaut, W.: Liquid chromatography tandem mass spectrometry method for characterization of monoaromatic nitro-compounds in atmospheric particulate matter, J. Chromatogr. A, 1268, 35e43, https://doi.org/10.1016/j.chroma.2012.10.021,2012.
Koch, B. P and Dittmar, T.: From mass to structure: an aromaticity index for high-resolution mass data of natural organic matter, Rapid. Commun. Mass. Spectrom., 20, 926–932, https://doi.org/10.1002/rcm.2386, 2006.
Kourtchev, I., Fuller, S., Aalto, J., Ruuskanen, T. M., McLeod, M. W., Maenhaut, W., Jones, R., Kulmala, M., and Kalberer, M.: Molecular composition of boreal forest aerosol from Hyytiälä, Finland, using ultrahigh resolution mass spectrometry, Environ. Sci. Technol., 4, 4069–4079, https://doi.org/10.1021/es3051636, 2013.
Kourtchev, I., Fuller, S. J., Giorio, C., Healy, R. M., Wilson, E., O'Connor, I., Wenger, J. C., McLeod, M., Aalto, J., Ruuskanen, T. M., Maenhaut, W., Jones, R., Venables, D. S., Sodeau, J. R., Kulmala, M., and Kalberer, M.: Molecular composition of biogenic secondary organic aerosols using ultrahigh-resolution mass spectrometry: comparing laboratory and field studies, Atmos. Chem. Phys., 14, 2155–2167, https://doi.org/10.5194/acp-14-2155-2014, 2014a.
Kourtchev, I., O'Connor, I. P., Giorio, C., Fuller, S. J., Kristensen, K., Maenhaut, W., Wenger, J. C., Sodeau, J. R., Glasius, M., and Kalberer, M.: Effects of anthropogenic emissions on the molecular composition of urban organic aerosols: An ultrahigh resolution mass spectrometry study, Atmos. Environ., 89, 525–532, https://doi.org/10.1016/j.atmosenv.2014.02.051, 2014b.
Kourtchev, I., Giorio, C., Manninen, A., Wilson, E., Mahon, B., Aalto, J., Kajos, M., Venables, D., Ruuskanen, T., Levula, J., Loponen, M., Connors, S., Harris, N., Zhao, D., Kiendler-Scharr, A., Mentel, T., Rudich, Y., Hallquist, M., Doussin, J.-F., Maenhaut, W., Bäck, J., Petäjä, T., Wenger, J., Kulmala, M., and Kalberer, M.: Enhanced volatile organic compounds emissions and organic aerosol mass increase the oligomer content of atmospheric aerosols, Sci. Rep., 6, 35038, https://doi.org/10.1038/srep35038, 2016.
Kourtchev, I., Doussin, J.-F., Giorio, C., Mahon, B., Wilson, E. M., Maurin, N., Pangui, E., Venables, D. S., Wenger, J. C., and Kalberer, M.: Molecular composition of fresh and aged secondary organic aerosol from a mixture of biogenic volatile compounds: a high-resolution mass spectrometry study, Atmos. Chem. Phys., 15, 5683–5695, https://doi.org/10.5194/acp-15-5683-2015, 2015.
Kristensen, K., Watne, A. K., Hammer, J., Lutz, A., Petäjä, T., Hallquist, M., Bilde, M., and Glasius, M.: High-molecular weight dimer esters are major products in aerosols from alpha-pinene ozonolysis and the boreal forest, Environ. Sci. Technol. Lett., 3, 280–285, https://doi.org/10.1021/acs.estlett.6b00152, 2016.
Kruve, A. and Kaupmees, K.: Predicting ESI/MS signal change for anions in different solvents, Anal. Chem. 89, 5079–5086, https://doi.org/10.1021/acs.analchem.7b00595, 2017.
Lanz, V. A., Prévôt, A. S. H., Alfarra, M. R., Weimer, S., Mohr, C., DeCarlo, P. F., Gianini, M. F. D., Hueglin, C., Schneider, J., F avez, O., D'Anna, B., George, C., and Baltensperger, U.: Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: an overview, Atmos. Chem. Phys., 10, 10453–10471, https://doi.org/10.5194/acp-10-10453-2010, 2010.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F. Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A.-K., Breitenlechner, M., Brilke, S., Buchholz, A., Buenrostro Mazon, S., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fucks, C., Garmash, O., Gordon, H., Hakala, J., He, X., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V.-M., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin III, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipilä, M., Tauber, C., Tomé, A., Tröstl, J., Väisänen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation acid, ammonia, and biogenic vapors, Sci. Adv., 4, eaau5363, https://doi.org/10.1126/sciadv.aau5363, 2018.
Lin, P., Rincon, A. G., Kalberer, M., and Yu, J. Z.: Elemental composition of HULIS in the Pearl river delta region, China: Results inferred from positive and negative electrospray high resolution mass spectrometry data, Environ. Sci. Technol., 46, 7454–7462, https://doi.org/10.1021/es300285d, 2012.
Lin, P., Fleming, L. T., Nizkorodov, S., Laskin, J., and Laskin, A.: Comprehensive Molecular Characterization of Atmospheric Brown Carbon by High Resolution Mass Spectrometry with Electrospray and Atmospheric Pressure Photoionization, Anal. Chem., 90, 12493–12502, https://doi.org/10.1021/acs.analchem.8b02177, 2018.
Mohr, C., Lopez-Hilfiker, F., Zotter, P., Prévôt, A. S. H., Xu, L., Ng, N. L., Herndon, S. C., Williams, L. R., Franklin, J. P., Zahniser, M. S., Worsnop, D. R., Knighton, W. B., Aiken, A. C., Gorkowski, K. J., Dubey, M. K., Allan, J. D., and Thornton, J. A.: Contribution of nitrated phenols to wood burning brown carbon light absorption in Detling, United Kingdom during winter time, Environ. Sci. Technol., 47, 6316–6324, https://doi.org/10.1021/es400683v, 2013.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Mutzel, A., Poulain, L., Berndt, T., Iinuma, Y., Rodigast, M. Böge, O., Richters, S., Spindler, G., Sipilä, M., Jokinen, T., Kulmala, M., and Herrmann, H.: Highly oxidized multifunctional organic compounds observed in tropospheric particles: a field and laboratory study, Environ. Sci. Technol., 49, 7754–7761, https://doi.org/10.1021/acs.est.5b00885, 2015.
Nguyen, T. B., Bateman, A. P., Bones, D. L., Nizkorodov, S. A., Laskin, J., and Laskin, A.: High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 44, 1032–1042, https://doi.org/10.1016/j.atmosenv.2009.12.019, 2010.
Nguyen, T. B., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Nitrogen-Containing Organic Compounds and Oligomers in Secondary Organic Aerosol Formed by Photooxidation of Isoprene, Environ. Sci. Technol., 45, 6908–6918, https://doi.org/10.1021/es201611n, 2011.
Noziere, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decessari, S., Finessi, E., Glasius, M., Grgic, I., Hamilton., J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The molecular identification of organic compounds in the atmosphere: State of the art and challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Nizkorodov, S. A., Laskin, J., and Laskin, A.: Molecular chemistry of organic aerosols through the application of high resolution mass spectrometry, Phys. Chem. Chem. Phys., 13, 3612–3629, https://doi.org/10.1039/c0cp02032j, 2011.
O'Brien, R. E., Laskin, A., Laskin, J., Liu, S., Weber, R., Russell, L. M., and Goldstein, A. H.: Molecular characterization of organic aerosol using nanospray desorption/electrospray ionization mass spectrometry: CalNex 2010 field study, Atmos. Environ., 68, 265–272, https://doi.org/10.1016/j.atmosenv.2012.11.056, 2013.
O'Brien, R. E., Laskin, A., Laskin, J., Rubitschun, C. L., Surratt, J. D., and Goldstein, A. H.: Molecular characterization of S- and N-containing organic constituents in ambient aerosols by negative ion mode high-resolution Nanospray Desorption Electrospray Ionization Mass spectrometry: CalNex 2010 field study, J. Geophys. Res.-Atmos., 119, 12706–12720, https://doi.org/10.1002/2014JD021955 , 2014.
Piazzalunga, A., Bernardoni, V., Fermo, P., and Vecchi, R.: Optimisation of analytical procedures for the quantification of ionic and carbonaceous fractions in the atmospheric aerosol and applications to ambient samples, Anal. Bioanal. Chem., 56, 30–40, https://doi.org/10.1007/s00216-012-6433-5, 2013.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Mocnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Platt, S. M., El Haddad, I., Pieber, S. M., Zardini, A. A., Suarez-Bertoa, R., Clairotte, M., Daellenbach, K. R., Huang, R.-J., Slowik, J. G., Hellebust, S., Temime-Roussel, B., Marchand, N., de Gouw, J., Jimenez, J. L., Hayes, P. L., Robinson, A. L., Baltensperger, U., Astorga, C., and Prévôt, A. S. H.: Gasoline cars produce more carbonaceous particulate matter than modern filter-equipped diesel cars, Sci. Rep., 7, 4926, https://doi.org/10.1038/s41598-017-03714-9, 2017.
Reyes-Villegas, E., Green, D. C., Priestman, M., Canonaco, F., Coe, H., Prévôt, A. S. H., and Allan, J. D.: Organic aerosol source apportionment in London 2013 with ME-2: exploring the solution space with annual and seasonal analysis, Atmos. Chem. Phys., 16, 15545–15559, https://doi.org/10.5194/acp-16-15545-2016, 2016.
Rincón, A. G., Calvo, A. I., Dietzel, M., and Kalberer, M.: Seasonal differences of urban organic aerosol composition – An ultra-high resolution mass spectrometry study, Environ. Chem., 9, 298–319, https://doi.org/10.1071/EN12016, 2012.
Roach, P. J., Laskin, J., and Laskin, A.: Molecular characterization of organic aerosols using nanospray-desorption/electrospray ionization-mass spectrometry, Anal. Chem., 82, 7979–7986, https://doi.org/10.1021/ac101449p, 2010.
Romonosky, D. E., Li, Y., Shiraiwa, M., Laskin, A., Laskin, J., and Nizkorodov, S. A.: Aqueous photochemistry of secondary organic aerosol of alpha-pinene and alpha-humulene oxidized with ozone, hydroxyl radical, and nitrate radical, J. Phys. Chem. A, 121, 1298–1309, https://doi.org/10.1021/acs.jpca.6b10900, 2017.
Sato, K., Takami, A., Kato, Y., Seta, T., Fujitani, Y., Hikida, T., Shimono, A., and Imamura, T.: AMS and LC/MS analyses of SOA from the photooxidation of benzene and 1,3,5-trimethylbenzene in the presence of NOx: effects of chemical structure on SOA aging, Atmos. Chem. Phys., 12, 4667–4682, https://doi.org/10.5194/acp-12-4667-2012, 2012.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources, 3. C1-C29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources, 5. C-1-C-32 organic compounds from gasoline-powered motor vehicles, Environ. Sci. Technol., 36, 1169–1180, https://doi.org/10.1021/es0108077, 2002.
Schlag, P., Kiendler-Scharr, A., Blom, M. J., Canonaco, F., Henzing, J. S., Moerman, M., Prévôt, A. S. H., and Holzinger, R.: Aerosol source apportionment from 1-year measurements at the CESAR tower in Cabauw, the Netherlands, Atmos. Chem. Phys., 16, 8831–8847, https://doi.org/10.5194/acp-16-8831-2016, 2016.
Tao, S., Lu, X., Levac, N., Bateman, A. P., Nguyen, T. B., Bones, D. L., Nizkorodov, S. A., Laskin, J., Laskin, A., and Yang, X.: Molecular characterization of organosulfates in organic aerosols from shanghai and Los Angeles urban areas by nanospray-desorption electrospray ionization high-resolution mass spectrometry, Environ. Sci. Technol., 48, 10993–11001, https://doi.org/10.1021/es5024674, 2014.
Tong, H., Kourtchev, I., Pant, P., Keyte, I. J., O'Connoer, I. P., Wenger, J. C., Pope, F. D., Harrison, R. M., and Kalberer, M.: Molecular composition of organic aerosols at urban background and road tunnel sites using ultra-high resolution mass spectrometry, Faraday Discuss., 189, 51–68, https://doi.org/10.1039/C5FD00206K, 2016.
Vogel, A. L., Äijälä, M., Corrigan, A. L., Junninen, H., Ehn, M., Petäjä, T., Worsnop, D. R., Kulmala, M., Russell, L. M., Williams, J., and Hoffmann, T.: In situ submicron organic aerosol characterization at a boreal forest research station during HUMPPA-COPEC 2010 using soft and hard ionization mass spectrometry, Atmos. Chem. Phys., 13, 10933–10950, https://doi.org/10.5194/acp-13-10933-2013, 2013.
Vlachou, A., Daellenbach, K. R., Bozzetti, C., Chazeau, B., Salazar, G. A., Szidat, S., Jaffrezo, J.-L., Hueglin, C., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: Advanced source apportionment of carbonaceous aerosols by coupling offline AMS and radiocarbon size-segregated measurements over a nearly 2-year period, Atmos. Chem. Phys., 18, 6187–6206, https://doi.org/10.5194/acp-18-6187-2018, 2018.
Wang, X., Hayeck, N., Brüggemann, M., Yao, L., Chen, H., Zhang, C., Emmelin, C., Chen, J., George, C., and Wang, L.: Chemical characteristics of organic aerosols in Shanghai: a study by ultra-high-performance liquid chromatography coupled with orbitrap mass spectrometry, J. Geophys. Res., 122, 11703–11722, https://doi.org/10.1002/2017JD026930, 2017.
Walser, M. L., Desyaterik, Y., Laskin, J., Laskin, A., and Nizkorodov, S. A.: High-resolution mass spectrometric analysis of secondary organic aerosol produced by ozonation of limonene, Phys. Chem. Chem. Phys., 10, 1009–1022, https://doi.org/10.1039/B712620D, 2008.
Yassine, M. M., Harir, M., Dabek-Zlotorzynska, E., and Schmitt-Kopplin, P.: Structural characterization of organic aerosol using Fourier transform cyclotron resonance mass spectrometry: Aromaticity equivalent approach, Rapid. Commun. Mass Spectrom., 28, 2445–2454, https://doi.org/10.1002/rcm.7038, 2014.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., Beaver, M. R., Holder, A. L., Lonneman, W. A., Docherty, K. S., Jaoui, M., Pye, H. O. T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Guo, H., Weber, R. J., de Gouw, J., Koss, A. R., Edgerton, E. S., Brune, W., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Kreisberg, N. M., Spielman, S. R., Hering, S. V., Wilson, K. R., Thornton, J. A., and Goldstein, A. H.: Monoterpens are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, https://doi.org/10.1073/pnas.1717513115, 2018.
Zhang, X., Lin, Y. H., Surratt, J. D., and Weber, R. J.: Sources, composition and absorption Ångström exponent of light-absorbing organic components in aerosol extracts from the Los Angeles basin, Environ. Sci. Technol., 47, 3685e3693, https://doi.org/10.1021/es305047b, 2013.
Zhang, Y. Y., Müller, L., Winterhalter, R., Moortgat, G. K., Hoffmann, T., and Pöschl, U.: Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter, Atmos. Chem. Phys., 10, 7859–7873, https://doi.org/10.5194/acp-10-7859-2010, 2010.
Zhao, D. F., Buchholz, A., Tillmann, R., Kleist, E., Wu, C., Rubach, F., Kiendler-Scharr, A., Rudich, Y., Wildt, J., and Mentel, T. F.: Environmental conditions regulate the impact of plants on cloud formation, Nat. Commun., 8, 14067, https://doi.org/10.1038/ncomms14067, 2017.
Zotter, P., Ciobanu, V. G., Zhang, Y. L., El-Haddad, I., Macchia, M., Daellenbach, K. R., Salazar, G. A., Huang, R.-J., Wacker, L., Hueglin, C., Piazzalunga, A., Fermo, P., Schwikowski, M., Baltensperger, U., Szidat, S., and Prévôt, A. S. H.: Radiocarbon analysis of elemental and organic carbon in Switzerland during winter-smog episodes from 2008 to 2012 – Part 1: Source apportionment and spatial variability, Atmos. Chem. Phys., 14, 13551–13570, https://doi.org/10.5194/acp-14-13551-2014, 2014a.
Zotter, P., El-Haddad, I., Zhang, Y., Hayes, P. L., Zhang, X., Lin, Y.-H., Wacker, L., Schnelle-Kreis, J., Abbaszade, G., Zimmermann, R., Surratt, J. D., Weber, R., Jimenez, J. L., Szidat, S., Baltensperger, U., and Prévôt, A. S. H.: Diurnal cycle of fossil and nonfossil carbon using radiocarbon analyses during CalNex, J. Geophys. Res., 119, 6818–6835, https://doi.org/10.1002/2013JD021114, 2014b.