the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Apr 2019
| 11 Apr 2019
pH-dependent production of molecular chlorine, bromine, and iodine from frozen saline surfaces
Paul B. Shepson
Kerri A. Pratt
The mechanisms of molecular halogen production from frozen saline surfaces remain incompletely understood, limiting our ability to predict atmospheric oxidation and composition in polar regions. In this laboratory study, condensed-phase hydroxyl radicals (OH) were photochemically generated in frozen saltwater solutions that mimicked the ionic composition of ocean water. These hydroxyl radicals were found to oxidize Cl−, Br−, and I−, leading to the release of Cl2, Br2, I2, and IBr. At moderately acidic pH (buffered between 4.5 and 4.8), irradiation of ice containing OH precursors (either of hydrogen peroxide or nitrite ion) produced elevated amounts of I2. Subsequent addition of O3 produced additional I2, as well as small amounts of Br2. At lower pH (1.7–2.2) and in the presence of an OH precursor, rapid dark conversion of I− to I2 occurred from reactions with hydrogen peroxide or nitrite, followed by substantial photochemical production of Br2 upon irradiation. Exposure to O3 under these low pH conditions also increased production of Br2 and I2; this likely results from direct O3 reactions with halides, as well as the production of gas-phase HOBr and HOI that subsequently diffuse to frozen solution to react with Br− and I−. Photochemical production of Cl2 was only observed when the irradiated sample was composed of high-purity NaCl and hydrogen peroxide (acting as the OH precursor) at pH = 1.8. Though condensed-phase OH was shown to produce Cl2 in this study, kinetics calculations suggest that heterogeneous recycling chemistry may be equally or more important for Cl2 production in the Arctic atmosphere. The condensed-phase OH-mediated halogen production mechanisms demonstrated here are consistent with those proposed from recent Arctic field observations of molecular halogen production from snowpacks. These reactions, even if slow, may be important for providing seed halogens to the Arctic atmosphere. Our results suggest the observed molecular halogen products are dependent on the relative concentrations of halides at the ice surface, as we only observe what diffuses to the air–surface interface.
- Article
(978 KB) - Full-text XML
-
Supplement
(794 KB) - BibTeX
- EndNote





It is now well established that gas-phase halogen species influence atmospheric composition through reactions with ozone (O3), volatile organic compounds (VOCs), and gaseous elemental mercury (Hg0) (Barrie and Platt, 1997; Carpenter et al., 2013; Platt and Hönninger, 2003; Saiz-Lopez and von Glasow, 2012; Simpson et al., 2007, 2015; Steffen et al., 2008, 2014, and references therein). In polar regions, it is believed that halogens build up to effective concentrations through a heterogeneous reaction sequence known as the “halogen explosion” (Reactions R1–R4, where X represents Cl, Br, or I) (Garland and Curtis, 1981; Tang and McConnell, 1996; Vogt et al., 1996; Wennberg, 1999).
In this sequence, a molecular halogen (X2) is photolyzed to produce two reactive halogen radicals. These radicals can react with O3 to produce halogen oxides (XO). The XO produced in Reaction (R2) rapidly photolyzes (or reacts with NO) to regenerate O3 and X2 in a null cycle. To irreversibly remove ambient O3, XO must react with another halogen oxide or Hg0. Alternatively, XO can react with HO2 to form HOX (Reaction R3) or NO2 to form XONO2. Gas-phase HOX can heterogeneously react with salt-laden surfaces, including sea-salt aerosol particles (McConnell et al., 1992) and the “disordered interface” (often referred to as a quasi-liquid or quasi-brine layer) that exists on frozen saline surfaces (Bartels-Rausch et al., 2014; Cho et al., 2002) to produce X2, effectively returning two halogen radicals to the gas phase. Additionally, this mechanism is enhanced under acidic conditions, confirmed by laboratory studies of aqueous (Fickert et al., 1999) and frozen solutions (e.g., Abbatt et al., 2010; Sjostedt and Abbatt, 2008; Wren et al., 2013) and by field observations (Pratt et al., 2013).
While much has been learned about the atmospheric chemistry of reactive halogen species in the Arctic, knowledge gaps remain in the chemical mechanisms by which molecular halogens are produced from frozen surfaces (Liao et al., 2014; Pratt et al., 2013). Recently, in situ, light-induced production of Cl2 (Custard et al., 2016), Br2 (Pratt et al., 2013; Raso et al., 2017), and I2 (Raso et al., 2017) within snowpack interstitial air has been reported and was further demonstrated to be enhanced following the addition of O3. The Br2-producing snowpacks studied by Pratt et al. (2013) were characterized as having a larger surface area, lower pH (≤6.3), greater molar ratios (), and lower salinity relative to other frozen samples collected near Utqiaġvik, Alaska. The proposed mechanism for this chemistry is based on laboratory studies of condensed-phase, hydroxyl radical (OH)-mediated halogen oxidation (Reactions R5–R12), which is followed by partitioning of the molecular halogen to the gas phase (Abbatt et al., 2010; Knipping et al., 2000; Oum et al., 1998b).
Direct, light-induced halogen production from frozen surfaces in the presence of OH has been previously demonstrated in the laboratory for Br2 and possibly for I2 (Abbatt et al., 2010), but analogous chemistry for Cl2 has yet to be observed. Additionally, photochemical production of I2 has been directly observed in the absence of OH (Kim et al., 2016). Employing cavity ring-down spectroscopy, Kim et al. (2016) reported photochemical production of I2 from a frozen solution by known aqueous-phase chemistry (Reactions R13–R17). This proposed photochemical mechanism involves an (I−–O2) charge-transfer complex (Levanon and Navon, 1969).
Kim et al. (2016) also report enhanced photochemical production (determined spectrophotometrically) from sunlit frozen iodide solutions placed on Antarctic snowpack, as well as from refrozen field snow and glacier samples doped with iodide. A question is thus raised regarding the necessity of OH for I2 production under environmentally relevant conditions.
The role of O3 in halogen production on frozen surfaces is also unclear. Previous laboratory studies have demonstrated that halide-doped frozen surfaces exposed to O3 can lead to Br2 production (independent of radiation; Reactions R18–R19 and R4) (Oldridge and Abbatt, 2011; Oum et al., 1998a; Wren et al., 2013).
It has recently been shown that this process proceeds at the surface, through a water-stabilized ozonide, Br•OOO−, as shown in Reactions (R20)–(R22). Artiglia et al. (2017) observed this Br•OOO− intermediate via liquid-injection X-ray photoelectron spectroscopy.
Wren et al. (2013) found that Cl2 was produced primarily via heterogeneous recycling of HOCl, resulting from BrCl photolysis, on halide-rich artificial snow. However, the observation that O3 induces halogen production from natural frozen surfaces has yet to be confirmed by field observations of snowpack chemistry, in which exposure to only O3 in the absence of light has not been shown to produce molecular halogens (Custard et al., 2017; Pratt et al., 2013; Raso et al., 2017). This raises a question of whether O3 is more important for initial halogen release or in a gas-phase propagation/recycling capacity (i.e., per the halogen explosion).
In this study, we utilized a custom ice-coated-wall flow reactor in tandem with chemical ionization mass spectrometry to study Br2, Cl2, and I2 production from frozen surfaces with compositions mimicking sea ice. The effects of photochemically generated OH radicals, O3 addition, and pH are tested as they relate to the production of these halogens. Surface pH was controlled through the use of buffers.
2.1 Materials
Sample solutions were composed to mimic the halide composition of seawater. This was achieved using either dissolved Instant Ocean (Spectrum Brands) or commercially available halide salts at a composition that mimics Instant Ocean (for consistency) in solutions referred to hereafter as “saltwater”. The halide concentrations in these solutions were made to a final concentration of 0.56 M Cl−, 7.2 × 10−4 M Br−, and 1.9 × 10−6 M I−. Except for Instant Ocean, all chemicals were purchased from Sigma Aldrich. Halide salts include solid NaCl (puriss. p.a. grade, ≥99.5 % purity), NaBr (puriss. grade, > 99 % purity), and KI (puriss. p.a. grade, ≥ 99.5 % purity). We note that these halide concentrations are comparable to those in actual seawater (Herring and Liss, 1974; Luther et al., 1988; Tsunogai and Sase, 1969), which typically contains Cl−, Br−, and I− at ratios of . Solutes were dissolved in ultrapure water (Birck Nanotechnology Center). Dissolved organic carbon for Instant Ocean and halide salt solutions was analyzed using a Shimadzu TOC-VCSH Total Organic Carbon Analyzer and determined at approximately 70 mg L−1 for Instant Ocean solutions and less than 5 mg L−1 for saltwater solutions. No further characterization of carbon-containing compounds was performed.
While previous investigators have adjusted the pre-freezing pH of their samples, it is very difficult to know the pH in the surface brine (or disordered interface) of frozen samples (Bartels-Rausch et al., 2014), though there is evidence from laboratory studies suggesting that the pH of salt solutions remains largely unchanged after freezing (Wren and Donaldson, 2012b). To obviate this problem, the aqueous solutions used in this study were buffered so that the same pH should exist in the surface brine layer. All solutions were buffered by either a 20 mM acetic acid (ACS reagent grade, ≥99.7 % purity)/acetate (puriss. p.a. grade) buffer (pH ≈ 4.5–4.7) or a 20 mM bisulfate (ReagentPlus grade, 99 % purity)/sulfate (ReagentPlus grade, ≥99.0 % purity) buffer (pH ≈ 1.7–2.2). These buffer concentrations were chosen as a compromise between using as little buffer as possible, yet enough buffer to ensure adequate buffering ability, as buffer capacity rapidly decreases as constituent species concentrations approach the acid Ka value. pH values of sample solutions were determined before and after experiments with no significant changes observed, suggesting the buffer composition/buffering capacity does not appreciably change over the course of an experiment (discussed further in the Supplement). 100 µM of either hydrogen peroxide (trace analysis grade, ≥30 % purity) or sodium nitrite (ReagentPlus grade, ≥99.0 % purity) was included as photochemical hydroxyl radical precursors, via Reactions (R5)–(R7).
2.2 Flow tube
Experiments were performed in a custom-built 150 cm long, 2.5 cm ID frozen-walled Pyrex flow tube contained within a temperature-controlled cooling jacket. In each experiment, 80.0 mL of sample solution was poured into the tube in the presence of room air, which was subsequently sealed with vinyl caps (McMaster-Carr). The flow tube was then rotated on motorized rollers within a 170 cm × 50 cm × 50 cm insulated wooden cooling chamber. Crushed dry ice was placed along the bottom of the chamber, and fans were used to circulate the air throughout the chamber such that the flow tube was evenly cooled. After ∼30 min, the sample was evenly frozen (ice thickness of 0.9 mm). The flow tube was subsequently transferred to an enclosed 156 cm × 50 cm × 50 cm wooden Mylar-lined experiment chamber and connected to a recycling chiller set to 258 K (i.e., above the NaCl•2H2O eutectic point. At this temperature, the relevant chemical reactions are expected to occur with/in a brine on the ice surface; Cho et al., 2002; Oldridge and Abbatt, 2011). This conjecture is based on the work of Oldridge and Abbatt (2011), who reported from a series of similar experiments that when O3 is flowed over frozen NaCl∕NaBr solutions above the NaCl eutectic temperature, reaction kinetics were strongly consistent with chemistry occurring in a liquid brine. The cooling liquid used for the chiller was a mixture of 60 % ethylene glycol and 40 % distilled water. Six UVA-340 solar simulator lamps (Q-Lab, 295–400 nm with maximum wattage at 340 nm, irradiance spectrum in Fig. S1 in the Supplement) were installed in the experiment box (two on each side except bottom). Each side was lined with reflective Mylar sheets to evenly irradiate the flow tube when the lamps were powered.

Figure 1Experimental schematic. Purple bars represent powered solar simulator bulbs. The green shading around the flow tube (flow reactor) represents cooling liquid (60 % ethylene glycol, 40 % water) circulated through the chiller. The flow reactor region itself has an inner diameter of 2.5 cm.
A flow schematic representing typical experiments is shown in Fig. 1. The carrier gas (Air, Ultra Zero grade, Praxair) was scrubbed of volatile organic compounds using activated charcoal and water by traveling through coiled stainless-steel tubing surrounded by crushed dry ice (replaced throughout the course of an experiment). This gas was measured to contain ≤300–400 pmol mol−1 NO (experimentally determined limits of detection) using the Total REactive Nitrogen Instrument (TRENI) (Lockwood et al., 2010; Xiong et al., 2015). Though NO2 was not measured, it should have been removed by the charcoal trap. Before entering the coated-wall flow tube, the carrier gas flowed through a commercial O3 generator (2B Technologies model 306). Carrier gas air entered the tube near room temperature (20 ∘C). At the start of experiments, the O3 generator was set to 0 nmol mol−1. Carrier gas then entered the flow tube in the dark experiment chamber. In most experiments, the carrier gas was regulated to a volumetric flow rate of 4.0 L min−1, which yields a residence time in the flow tube of ∼12 s. On exiting the flow tube, sample air was characterized using a Thermo Environmental 49i O3 monitor (flow rate of ∼1.5 L min−1) and a chemical ionization mass spectrometer (CIMS; sampling flow rate of ∼1.7 L min−1; described below in Sect. 2.3). Excess flow air was vented away. At set times in an experiment, the solar simulator bulbs were activated, and O3 was added to the system by powering the O3 generator. At the end of each experiment, the ice was melted and the water collected for pH measurements. To clean the flow tube, its interior was washed three times with ultrapure water before a final rinse with wash acetone. The flow tube was then connected to a compressed nitrogen gas cylinder (Praxiar, > 99.99 % purity) to dry for at least 2 h. Once dry, the flow tube was disconnected and capped until the next experiment.
2.3 CIMS
Halogen species were detected using a chemical ionization mass spectrometer (CIMS), described previously by Liao et al. (2011) and Pratt et al. (2013). Chemical ionization is achieved by ion-molecule reactions that occur between iodide–water reagent clusters, in N2, and the gas-phase analytes in zero air. The iodide–water clusters are formed when gas-phase iodide ions, generated by flowing 5 ppm methyl iodide through a 210Po ionizer (NRD), combine with water in the humidified ion-molecule region of the CIMS. Ion were filtered using a quadrupole mass filter. The ice-coated flow tube was connected to the CIMS via approximately 50 cm of i.d. 1∕2 in. PFA Teflon tubing.
Table 1List of relevant species monitored by chemical ionization mass spectrometry ( as reagent ion) with corresponding m∕z values.
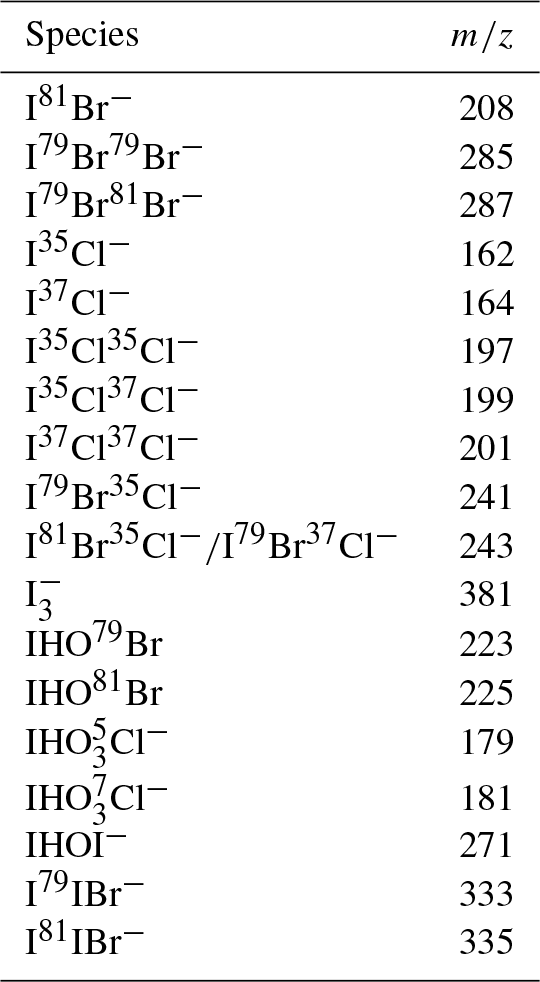
A typical CIMS sampling cycle consisted of an 8.35 s duty cycle. Dwell times for all monitored species were 250 ms except for the reagent ion (detected as m∕z 147, ), which was set to a dwell time of 100 ms. The 18 ions analyzed in this study are listed in Table 1, but we focus herein on results concerning masses related to Br2 (m∕z 285 and 287: I79Br79Br− and I81Br79Br−, respectively), Cl2 (m∕z 197, 199, and 201: I35Cl35Cl−, I37Cl35Cl−, and I37Cl37Cl−), and I2 (m∕z 381: ). In addition, IBr (m∕z 333 and 335: I79IBr−, I81IBr−) was unambiguously detected in some experiments. The presence of Br2, Cl2, and IBr was confirmed by measuring the ratios between the two isotope signals for each mass, compared to the natural abundances (i.e., 1.95 for m∕z 287:285; 1.54 for m∕z 197:199; and 1.03 for m∕z 333:335, respectively). Data outside ±25 % the expected isotope ratio were excluded from analysis. The signals for BrCl (m∕z 241 and 243: I79Br35Cl−, I81Br35Cl−, I79Br37Cl−) masses were never observed at the correct ratios (1.3 for m∕z 243:241), and so those data were not reported here. As the introduction of ∼60 nmol mol−1 O3 to the experimental system significantly increased the baseline signal of m∕z 197, but not m∕z 199 or 201, the presence of Cl2 could not be confirmed under elevated O3 conditions. In addition, background-subtracted, relative signals for m∕z 271 (IHOI−) and m∕z 225 (IHO81Br−) are discussed (signals are relative to that of the ionization gas (m∕z 147, ). According to isotope ratios, IHOBr− was not unambiguously observed, however, due to an interference at m∕z 223 (IHO79Br−), and our results here should be considered for only qualitative purposes as we only discuss relative changes in the signal.
CIMS calibrations were performed using I2, Br2, and Cl2 permeation devices (VICI) at the start and conclusion of each experiment. Br2 and Cl2 permeation outputs were quantified using the spectrophotometric method described by Liao et al. (2012). The I2 permeation output was quantified by flowing the I2 through an impinger containing a NaHCO3 (30 mM)/NaHSO3 (5 mM) reducing solution. This solution quantitatively reduces I2 to I−, which was then determined using a Dionex DX500 ion chromatography system. Permeation rates were calculated for each experiment and found to average (1.9±0.1) × 10−11, (5.5±0.1) × 10−10, and (8.6±0.1) × 10−10 mol min−1 of I2, Br2, and Cl2, respectively (uncertainties representing standard error of the mean). CIMS calibration factors were calculated for individual experiments. These factors are based on the average of the signal sensitivities, determined from the permeation sources, calculated at the start and completion of each experiment. Corresponding uncertainties for these calibration factors thus represent the 1σ standard deviation of the mean sensitivity. An approximate I79IBr− calibration factor was assumed to be the average of the sensitivities for m∕z 287 () and 381 (). Background measurements were performed before and after the experiment (minimum of 5 min) by passing the carrier gas through the experimental flow tube (without O3, in the dark) and subsequently through a glass wool scrubber, previously shown to remove molecular halogens with greater than 95 % efficiency (Liao et al., 2012; Neuman et al., 2010). Temporal variations in bromine-species signals while using the low pH sulfate/bisulfate buffer were observed in some experiments (Fig. S2) and are discussed in the Supplement.
Analysis of experimental data was based on 1 min averages, with uncertainties representing the standard deviation of these averages. Subsequently, signals were converted to concentrations using the sensitivities calculated above, propagating the sensitivity uncertainty into the measurement uncertainty. Average limits of detection (3σ) across all experiments for the molecular halogens during background periods were 1.8±0.4, 1.2±0.3, and 9±2 pmol mol−1 for Br2, Cl2, and I2 respectively (uncertainties representing standard error of the mean). Additionally, reported uncertainties for integrated amounts of formed halogens are calculated as integrated halogen concentrations multiplied by the relative uncertainty in the CIMS signal sensitivity.
The experiments described here address the extent to which condensed-phase OH radicals in an ice surface brine (Cho et al., 2002) can produce I2, Br2, and Cl2 through condensed-phase reactions within frozen saline surfaces, as hypothesized by recent field (Custard et al., 2017; Pratt et al., 2013; Raso et al., 2017) and laboratory experiments (Abbatt et al., 2010). In addition, we test the pH dependence of this chemistry and whether gas-phase O3 enhances this production. We find the relative and absolute amounts of halogens produced from ice are a complex function of the relative concentrations of the precursor halide ions, pH, presence of oxidants, radiation, and O3.
Table 2Results for all experiments performed. The first line in an experiment represents the integrated totals of molecular halogen production after 1 h of irradiation (t=0 through t=1 h). The results on italicized lines are 1 h integrated production amounts beginning once additional ozone was introduced to the flow tube. Average LODs across experiments were 1.8±0.4, 1.2±0.3, and 9±2 pmol mol−1 for Br2, Cl2, and I2 respectively. “IO#” represents samples composed of Instant Ocean, and “SW#” represents saltwater samples, composed of reagent salts. “CL1” here represents the experiment performed using 0.56 M high-purity NaCl.
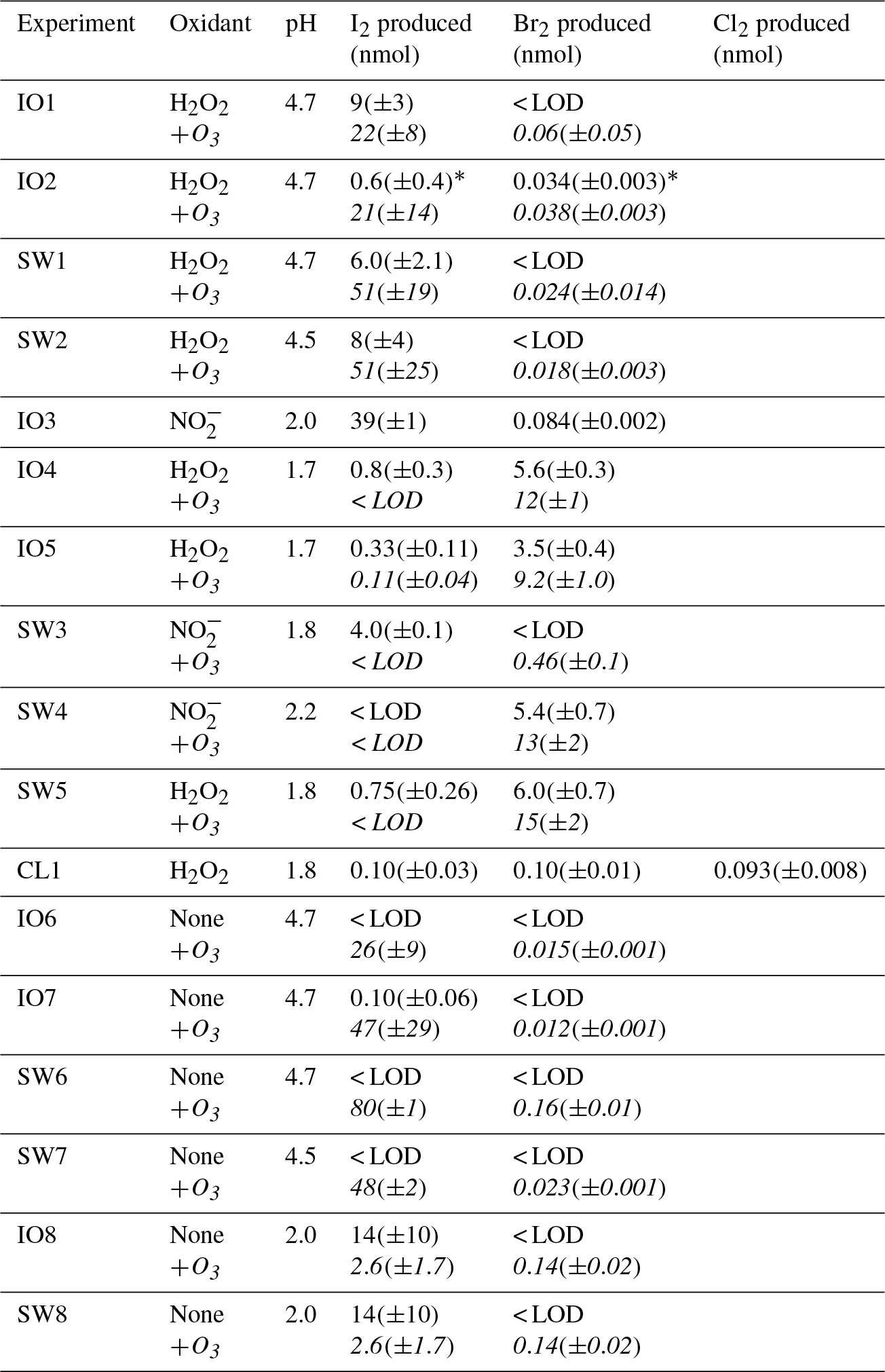
* The Br2 and I2 values presented for IO2 are discussed further in the Supplement.
The ice-coated flow tube experiments started under dark conditions and without addition of O3 (Sect. 3.1). Once signals stabilized, lights were activated (Sect. 3.2). After 1–2 h, ∼60 nmol mol−1 of O3 was introduced into the carrier gas (Sect. 3.3). Integrated amounts of produced molecular halogens are presented in Table 2 for all experiments. Unless otherwise specified, integrated amounts of produced halogens represent amounts produced over the course of 1 h of exposure to light (Sect. 3.2) and/or ozone (Sect. 3.3). Saline ices tested include frozen Instant Ocean (IO) solutions, saltwater (SW) solutions composed of dissolved reagent grade salts mimicking seawater composition, and 0.56 M high-purity NaCl (CL1). OH-radical precursors used include hydrogen peroxide (H2O2) or nitrite (), which have been estimated to account for 96 % of snowpack photochemical OH formation at Utqiaġvik, Alaska (France et al., 2012). Many of the salient features of our results are demonstrated by example experiments shown in Fig. 2, including the impact of irradiation in the presence of ice-phase OH-radical precursors, varied pH, and the presence of O3. Below we discuss the results and interpretations of our experiments, organized by the mechanism of halogen production and halogen products themselves.
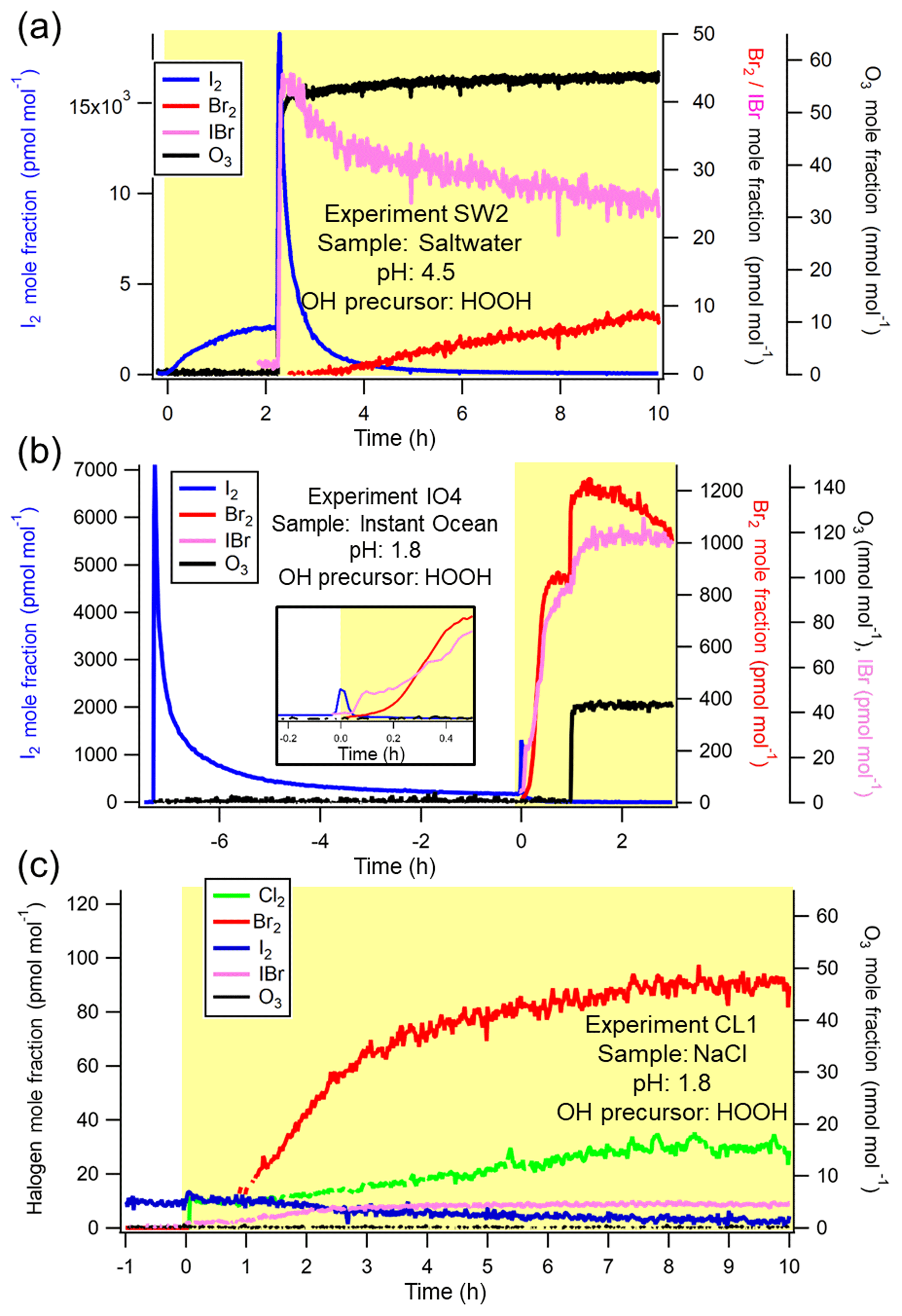
Figure 2Representative experiments of OH-mediated production of X2 and subsequent production of X2 from O3 addition. (a) Saltwater experiment (SW2) at pH = 4.5. (b) Instant Ocean experiment (IO4) at pH = 1.8. Time-varying Br2 and IBr signals before t=0 are shown in Fig. S2 in the Supplement. Inset more clearly shows the increase of I2 signal after irradiation. (c) NaCl experiment (CL1) at pH = 1.8. Timescale represents hours from the activation of the lights, and the yellow shading represents presence of radiation from solar simulator bulbs. Gaps in data represent periods when the isotopic ratios showed an interference.
3.1 Dark reaction production of I2
After the initial connection of the flow tube to the CIMS (i.e., before irradiation or addition of O3), large I2 signals (measured as , m∕z 381) were observed in several cases in which OH-radical precursors were utilized, especially when pH ≤ 2 (e.g., Figs. 2b and S2). Integrated calibrated sums of this dark I2 production are estimated in Table S1 in the Supplement and span from the time when the flow tube was connected to the CIMS until lights were activated. When pH ≤ 2, dark production of I2 sometimes caused significant depletion of reservoir I−. Experiment IO4 and SW5 (both using H2O2 as an OH precursor) only had, at most, ∼36 % of the initial 152 nmol of I− by the time lights were turned on (remaining I− was estimated by subtracting twice the observed I2 (i.e., two I− for every I2) from the initial 152 nmol of I− in the IO or SW solutions). Considerably less dark I2 production occurred using as an OH precursor (depleting I− by an average of 4.5 %; Table S1). However, the amounts in Table S1 represent lower limits of the dark-produced I2; it is impossible to accurately determine the extent of dark I2 production since some was lost from the flow tube during its connection to the CIMS after freezing (Fig. 1). At pH ≈ 4.7, this production was relatively modest. Only Experiment IO2 was noticeably affected, in which only ∼0.5 % of initial I− was removed by dark mechanisms (Table S1). Under both pH regimes (i.e., ∼4.7 and < 2), this signal subsequently decayed as I2 flushed out of the system until a low steady-state concentration was reached. No corresponding dark production of Br2 or Cl2 was observed for any experiments at any pH.
As previously reported, both H2O2 and can directly convert I− to I2 under dark acidic conditions. The oxidation of I− by H2O2 occurs through the condensed-phase reactions, Reactions (R23) and (R24) (Küpper et al., 1998):
Nitrite ions react with hydronium ions to form the nitroacidium ion, H2ONO+, which has been previously shown to produce I2 (Reactions R25–R27) (Hellebust et al., 2007; O'Driscoll et al., 2006, 2008; O'Sullivan and Sodeau, 2010):
Therefore, it is likely the I2 observed on connection of the flow tube to CIMS originated from the above reactions, Reactions (R23)–(R27), as the pH ≤ 2 experiments in this work (IO3-5, SW3-5) favor these forward reactions.
3.2 Hydroxyl radical-induced halogen production
3.2.1 pH ≈ 4.7
At pH ≈ 4.7, frozen solutions without OH-radical precursors produced no (IO6, SW6–SW7) or little (IO7, 0.10±0.06 nmol of I2) amounts of molecular halogens above their respective limits of detection (LODs) after activation of lights (Table 2). The small amount of I2 produced in IO7 possibly originates from the light and O2-mediated production mechanism proposed by Kim et al. (2016) as summarized within Reactions (R13)–(R17). However, as shown below, this mechanism of I2 production is of relatively minor importance at this pH.
In the presence of H2O2 at pH ≈ 4.7, I2 mole fractions increased rapidly upon irradiation, as shown in Fig. 2a. Of the four experiments performed in these conditions (IO1, IO2, SW1, SW2), three experiments (IO1, SW1, SW2) produced statistically similar amounts of I2 (mean: 8±2 nmol) after 1 h of irradiation (Table 2). The I2 signal behavior in Experiment IO2 qualitatively shared the same features as Experiment IO1, SW1, and SW2 (Fig. S3) but provided an apparently statistically different amount of I2 (0.6 (±0.4) nmol) based on the objectively chosen integration limits. This experiment is discussed further in the Supplement.
Regarding other molecular halogens, IBr was observed above the estimated limits of detection (3 pmol mol−1) upon irradiation during Experiment SW2 (Fig. 2a), starting approximately 20 min before the addition of O3. No photochemically produced (OH-induced) Br2 was unambiguously observed at this pH (note that the apparent IO2 Br2 production of 0.034±0.003 nmol is likely overestimated and is discussed in more detail in the Supplement). Cl2 mole fractions remained below limits of detection in all cases with OH precursors at this pH.
3.2.2 pH ≤ 2
In cases without OH precursors at pH ≤ 2, photochemical I2 production was observed (integrated production of 14±10 nmol for IO8, and 6±2 nmol for SW8) (Table 2), contrasting with experiments performed at pH ≈ 4.7 in which very little was produced. This production likely stems from the mechanisms outlined by Kim et al. (2016) (Reactions R13–R17), which require only light and oxygen to form a charge-transfer complex that results in I2 production (discussed in Sect. 1). Molecular Br2 and Cl2 concentrations remained below limits of detection, consistent with Abbatt et al. (2010), in which no Br2 or Cl2 was observed without an OH precursor.
As discussed in Sect. 3.1, inclusion of H2O2 or can result in direct oxidation of I− and reduce the available [I−] for photochemical OH oxidation when pH ≤ 2. Photochemical production of I2 across experiments yielded ≤0.82 nmol (IO4, IO5, and SW5) when H2O2 was used as an OH precursor. However, when instead was used (as in IO3, SW3, and SW4), initial observations of I2 on connection of the flow tube to CIMS were as much as 90 % less than when H2O2 was used (Table S1), thereby leaving more I− available for reaction. For Experiment IO3 (using ), the reduced pH led to an observed photochemical I2 production amount of 39±1 nmol, approximately 4 times larger than the largest amount observed at pH ≈ 4.7 (9±3 nmol; Table 2). That production would be enhanced at lower pH was expected based on the halogen activation reactions, Reactions (R4)–(R22). The corresponding saltwater experiments using were not as conclusive; Experiment SW3 only yielded 4.0±0.1 nmol of photochemical I2 (Fig. S5). Experiment SW4 (a repeat of SW3) did not produce any photochemical I2 and qualitatively resembles the H2O2 experiments performed at this pH. It is possible that, for SW3 and SW4, more I2 was produced by dark reactions and flushed out of the tube during connection with the CIMS and therefore would not have been measured.
Photochemical production of Br2 does not appear until I2 production decreases. The results shown in Fig. 2a and b demonstrate that when approximates the initial conditions of Instant Ocean (), OH-mediated I2 production precedes Br2 and IBr production (as in the pH ≈ 4.7 experiments and IO3, in which significant dark I2 production was not observed). After has sufficiently decreased, Br2 eventually becomes the dominant photochemical product. As demonstrated by Experiment IO4 (Fig. 2b and inset), there is a delay in Br2 production until I− was removed as I2, then as IBr. For experiments that used H2O2, photochemical Br2 yields averaged 4.5±0.5 nmol between IO4 and IO5 and 6.0±0.7 nmol from SW5. Experiment SW4 (using ) produced a comparable amount of Br2 (5.4±0.7 nmol). Given the initial depletion of I− from dark I2 production (Sect. 3.1), we can estimate at pH ≤ 2 in ice with H2O2 just before irradiation based on the remaining moles of I− in solution (Table S1) and the total moles of Br− in the solution. Averaging values from Experiment IO4-5 and SW5, was calculated as (compared to the initial ratio of ) and was sufficiently low to result in photochemical production of Br2.
Photochemical Cl2 production was only observed from a frozen solution of “pure” 0.56 M NaCl and H2O2 at pH = 1.8 (CL1), as shown in Fig. 2c. The initial Br− impurity of this CL1 solution was determined to be M via ion chromatography, while any I− impurity concentration could not be detected above the 3σ LOD of 90 nM. When the lights were turned on, slight increases in I2 and IBr were observed in concert with a rapid rise in Br2. After about 1 h of apparent equilibrium, I2 concentrations began decreasing, while Br2, IBr, and Cl2 continued rising. Over 1 h of illumination, 93±3 pmol of Cl2, 100±10 pmol of Br2, and 100±10 pmol of I2 were observed. However, as shown in Fig. 2c, the greatest rate of increase in Cl2 signal occurred just after this time. Integrating instead from t=0 until t=2 h, the amount of Cl2 produced was 190±10 pmol, while the amount of Br2 increased to 310±20 pmol. Utilizing the starting halide concentrations of Br− and Cl− for CL1, our results show Cl2 production was observed at of (1∕124 000), compared to the Instant Ocean of . Unfortunately, BrCl could not be observed due to an unknown interference at m∕z 241 and 243.
The observations in this study indicate competition for the OH radical in which the most oxidizable halide is oxidized, and the corresponding molecular halogens are produced until that halide ion is depleted in the ice surface brine reaction environment. The trends in molecular halogen production confirm acid-enhanced mechanisms in which the dominant products are largely dependent on relative halide ratios. Here, Br2 and IBr were not observed until I2 production sufficiently decreased the ratio, and Cl2 was not observed unless the ratio was sufficiently low ( = , as discussed above). This observation is consistent with Sjostedt and Abbatt (2008), who exposed frozen salt solutions to gas-phase OH and found peak BrCl production occurred as Br− decreased from an initial of . Additionally, Abbatt et al. (2010) generated condensed-phase OH on frozen surfaces via the photolysis of nitrate and similarly found lower Br2 and IBr integrated amounts at lower when temperatures were higher than the eutectic point of sodium chloride. These halide ratios are also consistent with in situ snowpack observations of Br2, BrCl, and Cl2 formation (Custard et al., 2017; Pratt et al., 2013).
3.2.3 Relative reactivities of OH-induced halogen production
I2, Br2, and Cl2 have been previously observed at mole fractions within less than 2 orders of magnitude of each other in snowpack interstitial air at Utqiaġvik, Alaska (Custard et al., 2017; Raso et al., 2017). Custard et al. (2017) observed gas-phase [Br2]∕[Cl2] values for artificially irradiated, acidic snowpacks ranging from 2 to 95 for corresponding snowpack ratios of . Under similar conditions, Raso et al. (2017) observed [I2]∕[Br2] ranging from ∼0.4 to 0.8 from corresponding snowpack amounts of . Despite the large differences in relative halide abundance (i.e., [I−] ≪ [Br−] ≪ [Cl−]), it appears that halogen activation reaction kinetics favor the larger halide ions, effectively leveling the relative molecular halogen production rates. The observations herein provide an opportunity to explore the relative reactivities of OH-mediated halogen production.
If we assume that the observed X2 flux out of the ice is proportional to the production rate (i.e., X2 desorbs as it is produced, within the residence time of the flow tube) and that halogen production is limited by halide reaction with OH radicals, effective relative reactivities, , (where X and Y represent Br, Cl, or I) can be calculated using Eq. (1).
The initial molecular halogen flux is calculated as the integrated sum of X2 (in moles) divided by both integration time (t=0–3 min, starting from the beginning of irradiation) and the surface area of ice coverage in the flow tube. Because the surface area, as well as the [OH] and [H+] in the ice surface reaction environment, are identical within individual experiments and cancel in these calculations, the relative fluxes are simply equivalent to the relative outflow concentrations of halogens. The pre-freezing halide ion concentrations (defined in Sect. 2) thus allow us to solve for the effective relative reactivity, , by assuming the ratios of the halide ice concentrations are the same after freezing.
At pH = 1.8, was estimated to be from Experiment CL1; in other words, production of Br2 is 240 000 times more efficient than production of Cl2 via (OH + halide) in the surface layer. Across the six experiments performed at pH ≤ 2 (average of 1.85) using Instant Ocean (IO3, IO4, IO5) and saltwater (SW3, SW4, SW5), was calculated to average (reported uncertainty is the standard error of the mean and thus only represents the experiment repeatability). These relative reactivities are substantially larger than the corresponding relative aqueous OH + halide rate constants ( M−1 s−1; Buxton et al., 1988; Zehavi and Rabani, 1972; M−1 s−1; Grigor'ev et al., 1987), which are different by less than a factor of 4. However, these rate constants refer to the specific fundamental reaction of OH with X− to produce HOX−, as in Reaction (R8). Ultimately, X2 production would occur via Reactions (R8)–(R12), and this condensed-phase chemistry is much more complex when also considering interhalogen reactions, such as Reaction (R28), that involve combinations of the three molecular halogens, halides, and mixed molecular halogens (XY, where Y = Cl, Br, or I).
Thus, it must be the case that there exist competing reactions that make the production of the larger X2 more efficient. For example, may be faster than . Alternatively, the relative rates of the disproportionation reaction, Reaction (R11), are likely different, favoring the larger molecular halogens. We can thus only state from these observations that the apparent relative reactivities calculated are consistent with the overall reactivity of the larger ions compensating for their lower abundances. This may lead to comparable production rates in our laboratory experiments and comparable snowpack gas-phase concentrations.
The above relative reactivity calculations are considered upper limits since the halide ratios used represent those in the pre-freezing solution. In other words, it is assumed that the ions are excluded to the ice surface reaction environment–air interface in amounts proportional to their pre-freezing concentration. Malley et al. (2018) recently demonstrated that brine can be distributed throughout ice in channels, suggesting that only the solutes at the liquid–air interface (a fraction of the total pre-freezing solution) participate in heterogeneous chemistry. Indeed, we find evidence here suggesting not all ions are available for reaction at the ice brine surface, particularly for experiments for which little I− was lost from dark I2 production mechanisms (i.e., pH = 4.7 with OH precursors: IO1, IO2, SW1, SW2). Considering Experiment IO2 as an example (Fig. S5; pH = 4.7), integration of the I2 signal during ∼15 h of exposure to both light and O3 shows that 54 % (82 nmol) of the original 152 nmol of I− remained unreacted in the frozen solution despite the signal apparently stabilizing at its baseline. It is therefore probable that a significant number of the ions, as well as H2O2, exist within brine channels within the ice (Bartels-Rausch et al., 2014; Malley et al., 2018). Oxidation chemistry would then be occurring throughout the ice, but release of molecular halogens to the flow tube air would be determined by diffusion rates. The diffusion rates of the product molecular halogens through bulk ice are likely slow, such that only production occurring in the brine that is in the near-liquid–air interface is observed here (Abbatt et al., 2012). Of the halogens produced from frozen solutions here, it is expected that I2 is observed most readily given the high polarizability and surface affinity of I− in aqueous solutions (Gladich et al., 2011) and the relative ease of oxidation of I−. That is, surface concentrations will be relatively enhanced with larger, more polarizable anions (I− > Br− >Cl−) (Gladich et al., 2011), which favors production of I2 over Br2, and Br2 over Cl2. As the larger and more reactive ions are depleted through oxidation, the next largest ion then becomes more favorably oxidized. Thus, in addition to the impact of differential reactivities and competing reactions for Reactions (R9)–(R12), what we observe in the laboratory and in the field can also be influenced by the relative surface enhancements of the anions, especially with respect to O3 impacts as discussed below.
3.3 Effects of O3 on halogen production
In experiments without an OH source (IO6–IO8, SW6–SW8), I2 production was greatest when O3 was introduced to the irradiated tube for both pH regimes (Table 2). The amount of I2 produced over 60 min in these experiments was large, ranging from 26±9 to 80±1 nmol at pH = 4.7 and from 2.6±1.7 to 38±12 nmol at pH < 2. This production likely results from a combination of heterogeneous recycling and the surface and aqueous reactions between O3 and I− ( cm3 molecules−1 s−1; Liu et al., 2001). While the I2 produced when pH < 2 appears to be lower, I2 had already been produced in the presence of light prior to addition of O3 (Sect. 3.2.2), yielding a lower ratio when O3 was eventually added. Br2 production amounts ranged from 0.012±0.001 to 0.16 ±0.01 nmol at pH = 4.7 and took up to 6 h to rise above detection limits after O3 was added. At pH ≤ 2, Br2 production amounts ranged from 0.14±0.02 to 0.93±0.05 nmol. While O3-mediated halogen production has been observed directly from frozen surfaces in the absence of light in previous laboratory studies (Artiglia et al., 2017; Oldridge and Abbatt, 2011; Oum et al., 1998a; Wren et al., 2013), Br2 production has not been directly observed from the Arctic snowpack without irradiation (Pratt et al., 2013). This raises a question of the role of O3 in initial halogen release in the Arctic spring.
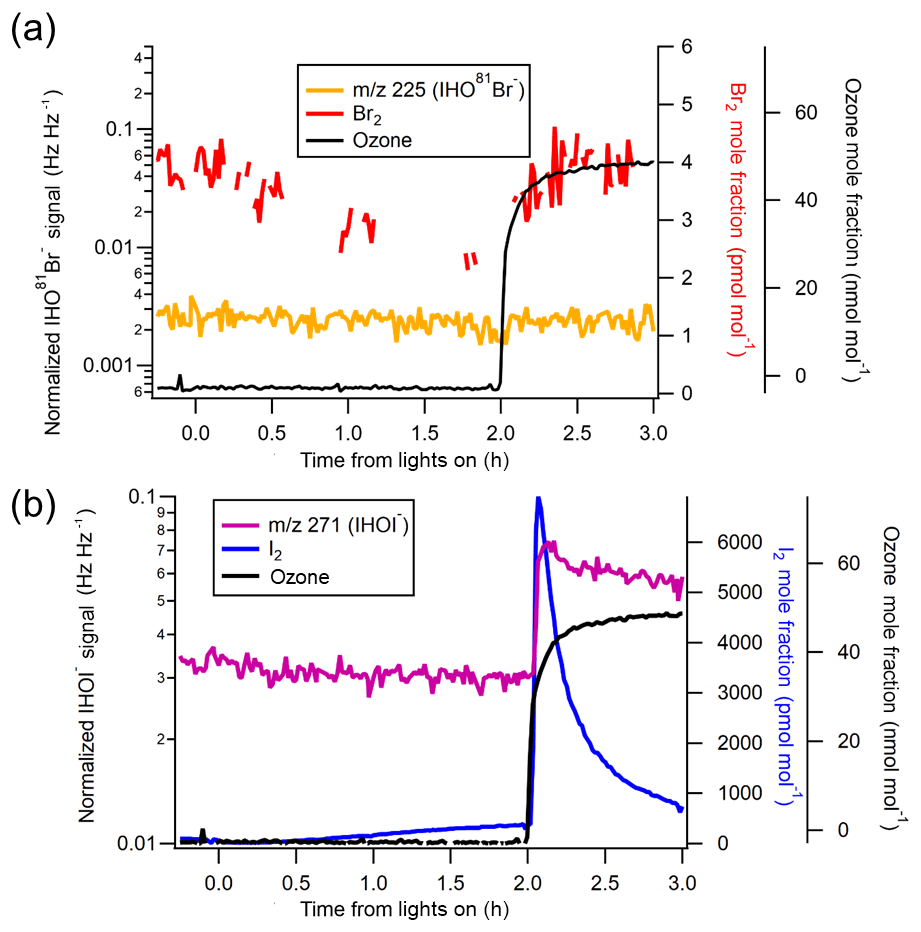
Figure 3Normalized, background-subtracted HOX signals from Experiment IO2, pH = 4.7. (a) Comparison of Br2 mole fractions to HOBr (m∕z 225). Note that the HOBr signal should be considered only qualitatively as its identity could not be confirmed using isotopic ratios with m∕z 223 due to its relatively large background signal. (b) Effect of O3 on I2 and HOI.
Figure 4Normalized, background-subtracted HOX signals from Experiment SW5, pH = 1.8. (a) Comparison of Br2 mole fractions to HOBr (m∕z 225). Note that the HOBr signal should be considered only qualitatively as its identity could not be confirmed using isotopic ratios with m∕z 223. (b) Effect of O3 on I2 and HOI.
When OH precursors were present, the addition of O3 to the zero-air flow over the irradiated frozen sample caused additional production of I2 and Br2, as shown in Fig. 2a and b, under both pH regimes (Table 2). In experiments at pH ≈ 4.7, in which remained sufficiently large due to minimal dark production of I2 (i.e., IO1-2, SW1-2), exposure to O3 caused a sharp increase in I2 (as in Fig. 2a). I2 production amounts for frozen Instant Ocean at pH ≈ 4.7 (IO1, IO2) averaged 22±10 nmol, about 2 times less than for frozen saltwater experiments SW1 and SW2 (average production amount of 51±25 nmol). As the I2 signal decayed, the corresponding Br2 signals gradually increased above detection limits, approximately 3 h after the introduction of O3 (Fig. 2a). The average integrated amounts of Br2 produced from these pH ≈ 4.7 experiments were very similar (0.05±0.01 nmol for IO experiments and 0.03±0.01 nmol for SW experiments).
When pH < 2, the effects of O3 addition varied according to the remaining availability of I−. When the surface I− reservoir had been reduced from dark reactions with H2O2 or (Reactions R17–R21; Sect. 3.1), exposure to O3 did not increase I2 above the LOD except in Experiment IO5, which exhibited a small spike before decaying below the LOD (0.11±0.06 nmol in IO5). However, O3 did cause additional Br2 production after 1 h (average of 10±2 nmol for IO4 (Fig. 2b) and IO5 (Fig. S4) and 14±2 nmol for SW4 and SW5). In contrast, for SW3 (using as an OH source), there was relatively little initial consumption of I− by dark reaction; therefore, when O3 was added, 1.1±0.1 nmol additional I2 was observed, comparable to what was observed with the higher pH experiments (Fig. S5). The amount of Br2 produced (0.46±0.01 nmol) was also significantly less than observed when I− was initially depleted, demonstrating the importance of the halide ratios.
This additional O3-induced halogen production could result from a combination of mechanisms. First, as discussed above, O3 can react with halides on frozen saline surfaces to produce Br2 or I2 per Reactions (R18)–(R19) and then Reaction (R4) (Artiglia et al., 2017; Carpenter et al., 2013; Gladich et al., 2015; Hayase et al., 2010; Oum et al., 1998a; Shaw and Carpenter, 2013; Wren et al., 2013). It is possible that Br2 (as well as other halogens) may have been produced via this mechanism at levels below the LOD in previous Arctic snowpack studies (Custard et al., 2017; Pratt et al., 2013; Raso et al., 2017).
The presence of O3 also yielded HOX compounds (Figs. 3–4), likely formed in the flow tube in part by O3 reactions with halides (Reactions R18–R19). Additionally, given a flow tube residence time of 12 s, gas-phase production of HOX is possible via Reactions (R1)–(R3) and could act as an additional X2 production source (via Reaction R4), given a timescale for molecular diffusion of 6.5 s for HOBr from the center of the tube to the ice surface. At this flow rate, there is enough time for one–two heterogeneous reaction cycles. Figure 3 shows HOX for IO2 (pH = 4.7 with H2O2 present, analogous to IO1, SW1, SW2). For each experiment in this series, increases in I2, HOI, and Br2 were readily observed when the O3 was introduced at hour 2 (Figs. 3 and S4). However, corresponding HOBr production was not observed, perhaps either due to a high LOD or the relatively low abundance of Br2 that would limit production of HOBr. Conversely, in pH ≤ 2 cases when substantial portions of I− had already reacted prior to irradiation (IO4, IO5, SW4, SW5), the addition of O3 produced negligible amounts of I2 and HOI (Fig. 4). But, in these cases, following the addition of O3, HOBr (m∕z 225 IHO81Br−), was observed together with Br2 (Figs. 4 and S4). We note in this case that m∕z 223, representative of IHO79Br−, does not appear to show an enhancement when O3 is added to the system. There was a much higher background signal for m∕z 223 compared with m∕z 225 (IHO81Br−) resulting from an unknown interference.
It was shown in this ice-coated-wall flow tube laboratory study that the hydroxyl radical can act as an effective condensed-phase halide oxidant, leading to I2, IBr, Br2, and Cl2 production under acidic conditions. Rates of molecular halogen production and release were dictated by both pH and relative halide concentrations. The identities of the molecular halogens produced appear to be highly influenced by which ions are enhanced at the ice surface, with I2 production occurring prior to Br2 production, which commenced as the was reduced. An opportunity exists to further explore this chemistry via surface-sensitive methods, for which recent developments have been shown to effectively enable characterization of the surface composition of frozen solutions of sodium chloride under near-atmospherically relevant conditions (Artiglia et al., 2017; Orlando et al., 2016). It would be useful to confirm the dominant ions involved in this surface-based chemistry over time. Further investigations into the effects of halide ratios on halogen production are also suggested, including measurements of how the ratios vary for different frozen Arctic surfaces, as well as how they vary spatially. While condensed-phase OH produces Br2 and I2 most rapidly in this study, it appears that other mechanisms, such as heterogeneous recycling of HOCl or ClONO2, could be a more dominant mechanism for in situ production of gas-phase Cl2 (Wang and Pratt, 2017). We find the addition of gas-phase O3 produces additional Br2 and I2, likely through aqueous reactions with halides and/or gas-phase production of HOX or possibly XONO2 (Deiber et al., 2004) and subsequent halogen explosion chemistry. These results lend support for the photochemical, condensed-phase molecular halogen production mechanisms proposed by the recent in situ snowpack experiments (Custard et al., 2017; Pratt et al., 2013; Raso et al., 2017).
Understanding the environmental pH dependence of halogen activation necessitates study of the pH on relevant Arctic frozen surfaces. Pratt et al. (2013) found that the frozen surfaces most conducive to in situ photochemical Br2 production had acidic pH after melting, while no production was observed from those with a well-buffered alkaline ice brine. Similarly, we find herein that condensed-phase OH-induced halogen production is enhanced at lower pH. Wren and Donaldson (2012a, b) found in laboratory studies that pH of acidic and basic solutions remains essentially unchanged after freezing and that saline solutions with buffers (i.e., seawater) maintain their buffering capacity following trace gas deposition, supporting the lack of observed Br2 production from the sea ice surface (Pratt et al., 2013). Therefore, it would be useful to test in situ production of halogens from Arctic frozen surfaces in tandem with measurement of the pH of said surfaces to determine the atmospherically relevant surface pH range required for halogen production.
Data used for this work are available for download from the NSF Arctic Data Center under the dataset “Laboratory experiments of the pH-dependent production of molecular chlorine, bromine, and iodine from frozen saline surfaces” (https://doi.org/10.18739/A22804Z17; Halfacre et al., 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-4917-2019-supplement.
JWH and PBS designed the research, and JWH performed the experiments and data analysis. All three authors contributed to the discussion and interpretation of the results and writing of the paper.
The authors declare that they have no conflict of interest.
We thank the National Science Foundation for their funding (PLR-1417668 and PLR-1417906, OPP-1417668). We also express thanks to Jonathan H. Slade, L. Gregory Huey, David J. Tanner, Fulizi Xiong, Angela R. W. Raso, and Kyle D. Custard for their assistance with CIMS operation and maintenance. Additionally, we thank the Purdue Chemistry Shop for helping build both the cooling and photolysis boxes, as well as the Jonathan Amy Facility for Chemical Instrumentation for their support in the fabrication of the experimental flow tube and setup of our experimental boxes. Thanks are also extended to Megan Haas and Marianne Bischoff for performing total organic carbon analysis of our samples and Angela R. W. Raso for confirmation of the iodide concentrations in our Instant Ocean samples. Finally, we thank Timothy Miller and the Purdue Birck Nanotechnology Center for the provision of the nanograde water used for our samples.
This paper was edited by James Roberts and reviewed by two anonymous referees.
Abbatt, J., Oldridge, N., Symington, A., Chukalovskiy, V., McWhinney, R. D., Sjostedt, S., and Cox, R. A.: Release of Gas-Phase Halogens by Photolytic Generation of OH in Frozen Halide-Nitrate Solutions: An Active Halogen Formation Mechanism, J. Phys. Chem. A, 114, 6527–6533, https://doi.org/10.1021/jp102072t, 2010.
Abbatt, J. P. D., Thomas, J. L., Abrahamsson, K., Boxe, C., Granfors, A., Jones, A. E., King, M. D., Saiz-Lopez, A., Shepson, P. B., Sodeau, J., Toohey, D. W., Toubin, C., von Glasow, R., Wren, S. N., and Yang, X.: Halogen activation via interactions with environmental ice and snow in the polar lower troposphere and other regions, Atmos. Chem. Phys., 12, 6237–6271, https://doi.org/10.5194/acp-12-6237-2012, 2012.
Artiglia, L., Edebeli, J., Orlando, F., Chen, S., Lee, M.-T., Corral Arroyo, P., Gilgen, A., Bartels-Rausch, T., Kleibert, A., Vazdar, M., Andres Carignano, M., Francisco, J. S., Shepson, P. B., Gladich, I., and Ammann, M.: A surface-stabilized ozonide triggers bromide oxidation at the aqueous solution-vapour interface, Nat. Commun., 8, 700, https://doi.org/10.1038/s41467-017-00823-x, 2017.
Barrie, L. and Platt, U.: Arctic tropospheric chemistry: an overview, Tellus B, 49, 450–454, https://doi.org/10.1034/j.1600-0889.49.issue5.2.x, 1997.
Bartels-Rausch, T., Jacobi, H.-W., Kahan, T. F., Thomas, J. L., Thomson, E. S., Abbatt, J. P. D., Ammann, M., Blackford, J. R., Bluhm, H., Boxe, C., Domine, F., Frey, M. M., Gladich, I., Guzmán, M. I., Heger, D., Huthwelker, Th., Klán, P., Kuhs, W. F., Kuo, M. H., Maus, S., Moussa, S. G., McNeill, V. F., Newberg, J. T., Pettersson, J. B. C., Roeselová, M., and Sodeau, J. R.: A review of air–ice chemical and physical interactions (AICI): liquids, quasi-liquids, and solids in snow, Atmos. Chem. Phys., 14, 1587–1633, https://doi.org/10.5194/acp-14-1587-2014, 2014.
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B.: Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals ( in Aqueous Solution, J. Phys. Chem. Ref. Data, 17, 513–886, https://doi.org/10.1063/1.555805, 1988.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Cho, H., Shepson, P. B., Barrie, L. A., Cowin, J. P., and Zaveri, R.: NMR Investigation of the Quasi-Brine Layer in Ice/Brine Mixtures, J. Phys. Chem. B, 106, 11226–11232, https://doi.org/10.1021/jp020449+, 2002.
Custard, K. D., Pratt, K. A., Wang, S., and Shepson, P. B.: Constraints on Arctic Atmospheric Chlorine Production through Measurements and Simulations of Cl2 and ClO, Environ. Sci. Technol., 50, 12394–12400, https://doi.org/10.1021/acs.est.6b03909, 2016.
Custard, K. D., Raso, A. R. W., Shepson, P. B., Staebler, R. M., and Pratt, K. A.: Production and Release of Molecular Bromine and Chlorine from the Arctic Coastal Snowpack, ACS Earth Space Chem., 1, 142–151, https://doi.org/10.1021/acsearthspacechem.7b00014, 2017.
Deiber, G., George, Ch., Le Calvé, S., Schweitzer, F., and Mirabel, Ph.: Uptake study of ClONO2 and BrONO2 by Halide containing droplets, Atmos. Chem. Phys., 4, 1291–1299, https://doi.org/10.5194/acp-4-1291-2004, 2004.
Fickert, S., Adams, J. W., and Crowley, J. N.: Activation of Br2 and BrCl via uptake of HOBr onto aqueous salt solutions, J. Geophys. Res.-Atmos., 104, 23719–23727, https://doi.org/10.1029/1999JD900359, 1999.
France, J. L., Reay, H. J., King, M. D., Voisin, D., Jacobi, H. W., Domine, F., Beine, H., Anastasio, C., MacArthur, A., and Lee-Taylor, J.: Hydroxyl radical and NOx production rates, black carbon concentrations and light-absorbing impurities in snow from field measurements of light penetration and nadir reflectivity of onshore and offshore coastal Alaskan snow, J. Geophys. Res., 117, D00R12, https://doi.org/10.1029/2011JD016639, 2012.
Garland, J. A. and Curtis, H.: Emission of iodine from the sea surface in the presence of ozone, J. Geophys. Res., 86, 3183, https://doi.org/10.1029/JC086iC04p03183, 1981.
Gladich, I., Shepson, P. B., Carignano, M. A., and Szleifer, I.: Halide Affinity for the Water-Air Interface in Aqueous Solutions of Mixtures of Sodium Salts, J. Phys. Chem. A, 115, 5895–5899, https://doi.org/10.1021/jp110208a, 2011.
Gladich, I., Francisco, J. S., Buszek, R. J., Vazdar, M., Carignano, M. A., and Shepson, P. B.: Ab Initio Study of the Reaction of Ozone with Bromide Ion, J. Phys. Chem. A, 119, 4482–4488, https://doi.org/10.1021/jp5101279, 2015.
Grigor'ev, A. E., Makarov, I. E., and Pikaev, A. K.: Formation of in the bulk of solution during radiolysis of concentrated aqueous solutions of chlorides, Khimiya Vysok. Ehnergij, 21, 123–126, 1987.
Halfacre, J. W., Shepson, P. B., and Pratt, K. A.: Laboratory experiments of the pH-dependent production of molecular chlorine, bromine, and iodine from frozen saline surfaces, NSF Arctic Data Center, https://doi.org/10.18739/A22804Z17, 2018.
Hayase, S., Yabushita, A., Kawasaki, M., Enami, S., Hoffmann, M. R., and Colussi, A. J.: Heterogeneous Reaction of Gaseous Ozone with Aqueous Iodide in the Presence of Aqueous Organic Species, J. Phys. Chem. A, 114, 6016–6021, https://doi.org/10.1021/jp101985f, 2010.
Hellebust, S., Roddis, T., and Sodeau, J. R.: Potential Role of the Nitroacidium Ion on HONO Emissions from the Snowpack, J. Phys. Chem. A, 111, 1167–1171, https://doi.org/10.1021/jp068264g, 2007.
Herring, J. R. and Liss, P. S.: A new method for the determination of iodine species in seawater, Deep-Sea Res., 21, 777–783, https://doi.org/10.1016/0011-7471(74)90085-0, 1974.
Kim, K., Yabushita, A., Okumura, M., Saiz-Lopez, A., Cuevas, C. A., Blaszczak-Boxe, C. S., Min, D. W., Yoon, H.-I., and Choi, W.: Production of Molecular Iodine and Tri-iodide in the Frozen Solution of Iodide: Implication for Polar Atmosphere, Environ. Sci. Technol., 50, 1280–1287, https://doi.org/10.1021/acs.est.5b05148, 2016.
Knipping, E. M., Lakin, M. J., Foster, K. L., Jungwirth, P., Tobias, D. J., Gerber, R. B., Dabdub, D., and Finlayson-Pitts, B. J.: Experiments and Simulations of Ion-Enhanced Interfacial Chemistry on Aqueous NaCl Aerosols, Science, 288, 301–306, https://doi.org/10.1126/science.288.5464.301, 2000.
Küpper, F. C., Schweigert, N., Gall, E. A., Legendre, J.-M., Vilter, H., and Kloareg, B.: Iodine uptake in Laminariales involves extracellular, haloperoxidase-mediated oxidation of iodide, Planta, 207, 163–171, https://doi.org/10.1007/s004250050469, 1998.
Levanon, H. and Navon, G.: Spectrum and stability of oxygen iodide charge-transfer complex, J. Phys. Chem., 73, 1861–1868, https://doi.org/10.1021/j100726a038, 1969.
Liao, J., Sihler, H., Huey, L. G., Neuman, J. A., Tanner, D. J., Friess, U., Platt, U., Flocke, F. M., Orlando, J. J., Shepson, P. B., Beine, H. J., Weinheimer, A. J., Sjostedt, S. J., Nowak, J. B., Knapp, D. J., Staebler, R. M., Zheng, W., Sander, R., Hall, S. R., and Ullmann, K.: A comparison of Arctic BrO measurements by chemical ionization mass spectrometry and long path-differential optical absorption spectroscopy, J. Geophys. Res.-Atmos, 116, D00R02, https://doi.org/10.1029/2010JD014788, 2011.
Liao, J., Huey, L. G., Tanner, D. J., Flocke, F. M., Orlando, J. J., Neuman, J. A., Nowak, J. B., Weinheimer, A. J., Hall, S. R., Smith, J. N., Fried, A., Staebler, R. M., Wang, Y., Koo, J.-H., Cantrell, C. A., Weibring, P., Walega, J., Knapp, D. J., Shepson, P. B., and Stephens, C. R.: Observations of inorganic bromine (HOBr, BrO, and Br2) speciation at Barrow, Alaska, in spring 2009, J. Geophys. Res.-Atmos., 117, D00R16, https://doi.org/10.1029/2011JD016641, 2012.
Liao, J., Huey, L. G., Liu, Z., Tanner, D. J., Cantrell, C. A., Orlando, J. J., Flocke, F. M., Shepson, P. B., Weinheimer, A. J., Hall, S. R., Ullmann, K., Beine, H. J., Wang, Y., Ingall, E. D., Stephens, C. R., Hornbrook, R. S., Apel, E. C., Riemer, D., Fried, A., Mauldin III, R. L., Smith, J. N., Staebler, R. M., Neuman, J. A., and Nowak, J. B.: High levels of molecular chlorine in the Arctic atmosphere, Nat. Geosci., 7, 91–94, https://doi.org/10.1038/ngeo2046, 2014.
Liu, Q., Schurter, L. M., Muller, C. E., Aloisio, S., Francisco, J. S., and Margerum, D. W.: Kinetics and Mechanisms of Aqueous Ozone Reactions with Bromide, Sulfite, Hydrogen Sulfite, Iodide, and Nitrite Ions, Inorg. Chem., 40, 4436–4442, https://doi.org/10.1021/ic000919j, 2001.
Lockwood, A. L., Shepson, P. B., Fiddler, M. N., and Alaghmand, M.: Isoprene nitrates: preparation, separation, identification, yields, and atmospheric chemistry, Atmos. Chem. Phys., 10, 6169–6178, https://doi.org/10.5194/acp-10-6169-2010, 2010.
Luther, G. W., Swartz, C. B., and Ullman, W. J.: Direct determination of iodide in seawater by cathodic stripping square wave voltammetry, Anal. Chem., 60, 1721–1724, https://doi.org/10.1021/ac00168a017, 1988.
Malley, P. P. A., Chakraborty, S., and Kahan, T. F.: Physical Characterization of Frozen Saltwater Solutions Using Raman Microscopy, ACS Earth Space Chem., 2, 702–710, https://doi.org/10.1021/acsearthspacechem.8b00045, 2018.
McConnell, J. C., Henderson, G. S., Barrie, L., Bottenheim, J., Niki, H., Langford, C. H., and Templeton, E. M. J.: Photochemical bromine production implicated in Arctic boundary-layer ozone depletion, Nature, 355, 150–152, https://doi.org/10.1038/355150a0, 1992.
Neuman, J. A., Nowak, J. B., Huey, L. G., Burkholder, J. B., Dibb, J. E., Holloway, J. S., Liao, J., Peischl, J., Roberts, J. M., Ryerson, T. B., Scheuer, E., Stark, H., Stickel, R. E., Tanner, D. J., and Weinheimer, A.: Bromine measurements in ozone depleted air over the Arctic Ocean, Atmos. Chem. Phys., 10, 6503–6514, https://doi.org/10.5194/acp-10-6503-2010, 2010.
O'Driscoll, P., Lang, K., Minogue, N., and Sodeau, J.: Freezing Halide Ion Solutions and the Release of Interhalogens to the Atmosphere, J. Phys. Chem. A, 110, 4615–4618, https://doi.org/10.1021/jp060491v, 2006.
O'Driscoll, P., Minogue, N., Takenaka, N., and Sodeau, J.: Release of Nitric Oxide and Iodine to the Atmosphere from the Freezing of Sea-Salt Aerosol Components, J. Phys. Chem. A, 112, 1677–1682, https://doi.org/10.1021/jp710464c, 2008.
Oldridge, N. W. and Abbatt, J. P. D.: Formation of Gas-Phase Bromine from Interaction of Ozone with Frozen and Liquid NaCl∕NaBr Solutions: Quantitative Separation of Surficial Chemistry from Bulk-Phase Reaction, J. Phys. Chem. A, 115, 2590–2598, https://doi.org/10.1021/jp200074u, 2011.
Orlando, F., Waldner, A., Bartels-Rausch, T., Birrer, M., Kato, S., Lee, M.-T., Proff, C., Huthwelker, T., Kleibert, A., van Bokhoven, J., and Ammann, M.: The Environmental Photochemistry of Oxide Surfaces and the Nature of Frozen Salt Solutions: A New in Situ XPS Approach, Top. Catal., 59, 591–604, https://doi.org/10.1007/s11244-015-0515-5, 2016.
O'Sullivan, D. and Sodeau, J. R.: Freeze-Induced Reactions: Formation of Iodine-Bromine Interhalogen Species from Aqueous Halide Ion Solutions, J. Phys. Chem. A, 114, 12208–12215, https://doi.org/10.1021/jp104910p, 2010.
Oum, K. W., Lakin, M. J., and Finlayson-Pitts, B. J.: Bromine activation in the troposphere by the dark reaction of O3 with seawater ice, Geophys. Res. Lett., 25, 3923–3926, https://doi.org/10.1029/1998GL900078, 1998a.
Oum, K. W., Lakin, M. J., DeHaan, D. O., Brauers, T., and Finlayson-Pitts, B. J.: Formation of Molecular Chlorine from the Photolysis of Ozone and Aqueous Sea-Salt Particles, Science, 279, 74–76, https://doi.org/10.1126/science.279.5347.74, 1998b.
Platt, U. and Hönninger, G.: The role of halogen species in the troposphere, Chemosphere, 52, 325–338, https://doi.org/10.1016/S0045-6535(03)00216-9, 2003.
Pratt, K. A., Custard, K. D., Shepson, P. B., Douglas, T. A., Pöhler, D., General, S., Zielcke, J., Simpson, W. R., Platt, U., Tanner, D. J., Gregory Huey, L., Carlsen, M., and Stirm, B. H.: Photochemical production of molecular bromine in Arctic surface snowpacks, Nat. Geosci., 6, 351–356, https://doi.org/10.1038/ngeo1779, 2013.
Raso, A. R. W., Custard, K. D., May, N. W., Tanner, D., Newburn, M. K., Walker, L., Moore, R. J., Huey, L. G., Alexander, L., Shepson, P. B., and Pratt, K. A.: Active molecular iodine photochemistry in the Arctic, P. Natl. Acad. Sci. USA, 114, 10053–10058, https://doi.org/10.1073/pnas.1702803114, 2017.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/C2CS35208G, 2012.
Shaw, M. D. and Carpenter, L. J.: Modification of Ozone Deposition and I2 Emissions at the Air–Aqueous Interface by Dissolved Organic Carbon of Marine Origin, Environ. Sci. Technol., 47, 10947–10954, https://doi.org/10.1021/es4011459, 2013.
Simpson, W. R., von Glasow, R., Riedel, K., Anderson, P., Ariya, P., Bottenheim, J., Burrows, J., Carpenter, L. J., Frieß, U., Goodsite, M. E., Heard, D., Hutterli, M., Jacobi, H.-W., Kaleschke, L., Neff, B., Plane, J., Platt, U., Richter, A., Roscoe, H., Sander, R., Shepson, P., Sodeau, J., Steffen, A., Wagner, T., and Wolff, E.: Halogens and their role in polar boundary-layer ozone depletion, Atmos. Chem. Phys., 7, 4375–4418, https://doi.org/10.5194/acp-7-4375-2007, 2007.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and Glasow, R. von: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Sjostedt, S. J. and Abbatt, J. P. D.: Release of gas-phase halogens from sodium halide substrates: heterogeneous oxidation of frozen solutions and desiccated salts by hydroxyl radicals, Environ. Res. Lett., 3, 045007, https://doi.org/10.1088/1748-9326/3/4/045007, 2008.
Steffen, A., Douglas, T., Amyot, M., Ariya, P., Aspmo, K., Berg, T., Bottenheim, J., Brooks, S., Cobbett, F., Dastoor, A., Dommergue, A., Ebinghaus, R., Ferrari, C., Gardfeldt, K., Goodsite, M. E., Lean, D., Poulain, A. J., Scherz, C., Skov, H., Sommar, J., and Temme, C.: A synthesis of atmospheric mercury depletion event chemistry in the atmosphere and snow, Atmos. Chem. Phys., 8, 1445–1482, https://doi.org/10.5194/acp-8-1445-2008, 2008.
Steffen, A., Bottenheim, J., Cole, A., Ebinghaus, R., Lawson, G., and Leaitch, W. R.: Atmospheric mercury speciation and mercury in snow over time at Alert, Canada, Atmos. Chem. Phys., 14, 2219–2231, https://doi.org/10.5194/acp-14-2219-2014, 2014.
Tang, T. and McConnell, J. C.: Autocatalytic release of bromine from Arctic snow pack during polar sunrise, Geophys. Res. Lett., 23, 2633–2636, https://doi.org/10.1029/96GL02572, 1996.
Tsunogai, S. and Sase, T.: Formation of iodide-iodine in the ocean, Deep-Sea Res., 16, 489–496, https://doi.org/10.1016/0011-7471(69)90037-0, 1969.
Vogt, R., Crutzen, P. J., and Sander, R.: A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer, Nature, 383, 327–330, https://doi.org/10.1038/383327a0, 1996.
Wang, S. and Pratt, K. A.: Molecular Halogens Above the Arctic Snowpack: Emissions, Diurnal Variations, and Recycling Mechanisms, J. Geophys. Res.-Atmos., 122, 11991–12007, https://doi.org/10.1002/2017JD027175, 2017.
Wennberg, P.: Atmospheric chemistry: Bromine explosion, Nature, 397, 299–301, https://doi.org/10.1038/16805, 1999.
Wren, S. N. and Donaldson, D. J.: How does deposition of gas phase species affect pH at frozen salty interfaces?, Atmos. Chem. Phys., 12, 10065–10073, https://doi.org/10.5194/acp-12-10065-2012, 2012a.
Wren, S. N. and Donaldson, D. J.: Laboratory Study of pH at the Air–Ice Interface, J. Phys. Chem. C, 116, 10171–10180, https://doi.org/10.1021/jp3021936, 2012b.
Wren, S. N., Donaldson, D. J., and Abbatt, J. P. D.: Photochemical chlorine and bromine activation from artificial saline snow, Atmos. Chem. Phys., 13, 9789–9800, https://doi.org/10.5194/acp-13-9789-2013, 2013.
Xiong, F., McAvey, K. M., Pratt, K. A., Groff, C. J., Hostetler, M. A., Lipton, M. A., Starn, T. K., Seeley, J. V., Bertman, S. B., Teng, A. P., Crounse, J. D., Nguyen, T. B., Wennberg, P. O., Misztal, P. K., Goldstein, A. H., Guenther, A. B., Koss, A. R., Olson, K. F., de Gouw, J. A., Baumann, K., Edgerton, E. S., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., and Shepson, P. B.: Observation of isoprene hydroxynitrates in the southeastern United States and implications for the fate of NOx, Atmos. Chem. Phys., 15, 11257–11272, https://doi.org/10.5194/acp-15-11257-2015, 2015.
Zehavi, D. and Rabani, J.: Oxidation of aqueous bromide ions by hydroxyl radicals. Pulse radiolytic investigation, J. Phys. Chem., 76, 312–319, https://doi.org/10.1021/j100647a006, 1972.