the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Mar 2019
| 12 Mar 2019
Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem
Claire E. Reeves
Alex R. Baker
Alfonso Saiz-Lopez
Rainer Volkamer
Theodore K. Koenig
Eric C. Apel
Rebecca S. Hornbrook
Lucy J. Carpenter
Stephen J. Andrews
Tomás Sherwen
Roland von Glasow
This study investigates the impact of reactive halogen species (RHS, containing chlorine (Cl), bromine (Br) or iodine (I)) on atmospheric chemistry in the tropical troposphere and explores the sensitivity to uncertainties in the fluxes of RHS to the atmosphere and their chemical processing. To do this, the regional chemistry transport model WRF-Chem has been extended to include Br and I, as well as Cl chemistry for the first time, including heterogeneous recycling reactions involving sea-salt aerosol and other particles, reactions of Br and Cl with volatile organic compounds (VOCs), along with oceanic emissions of halocarbons, VOCs and inorganic iodine. The study focuses on the tropical east Pacific using field observations from the Tropical Ocean tRoposphere Exchange of Reactive halogen species and Oxygenated VOC (TORERO) campaign (January–February 2012) to evaluate the model performance.
Including all the new processes, the model does a reasonable job reproducing the observed mixing ratios of bromine oxide (BrO) and iodine oxide (IO), albeit with some discrepancies, some of which can be attributed to difficulties in the model's ability to reproduce the observed halocarbons. This is somewhat expected given the large uncertainties in the air–sea fluxes of the halocarbons in a region where there are few observations of their seawater concentrations.
We see a considerable impact on the inorganic bromine (Bry) partitioning when heterogeneous chemistry is included, with a greater proportion of the Bry in active forms such as BrO, HOBr and dihalogens. Including debromination of sea salt increases BrO slightly throughout the free troposphere, but in the tropical marine boundary layer, where the sea-salt particles are plentiful and relatively acidic, debromination leads to overestimation of the observed BrO. However, it should be noted that the modelled BrO was extremely sensitive to the inclusion of reactions between Br and the oxygenated VOCs (OVOCs), which convert Br to HBr, a far less reactive form of Bry. Excluding these reactions leads to modelled BrO mixing ratios greater than observed. The reactions between Br and aldehydes were found to be particularly important, despite the model underestimating the amount of aldehydes observed in the atmosphere. There are only small changes to the inorganic iodine (Iy) partitioning and IO when the heterogeneous reactions, primarily on sea salt, are included.
Our model results show that tropospheric Ox loss due to halogens ranges between 25 % and 60 %. Uncertainties in the heterogeneous chemistry accounted for a small proportion of this range (25 % to 31 %). This range is in good agreement with other estimates from state-of-the-art atmospheric chemistry models. The upper bound is found when reactions between Br and Cl with VOCs are not included and, consequently, Ox loss by BrOx, ClOx and IOx cycles is high (60 %). With the inclusion of halogens in the troposphere, O3 is reduced by 7 ppbv on average. However, when reactions between Br and Cl with VOCs are not included, O3 is much lower than observed. Therefore, the tropospheric Ox budget is highly sensitive to the inclusion of halogen reactions with VOCs and to the uncertainties in current understanding of these reactions and the abundance of VOCs in the remote marine atmosphere.
- Article
(5641 KB) - Full-text XML
-
Supplement
(582 KB) - BibTeX
- EndNote




Reactive halogen species (RHS) cause ozone (O3) destruction, change the HOx (HO2+OH) and NOx (NO2+NO) partitioning, affect the oxidation of volatile organic compounds (VOCs) and mercury, and take part in new particle formation (Chameides and Davis, 1980; von Glasow et al., 2004; Saiz-Lopez and von Glasow, 2012). Moreover, reactive chlorine reduces the lifetime of methane (CH4). Halogen species are known to play an important role in the oxidizing capacity of the troposphere. The atmospheric oxidation capacity is to a large extent determined by budgets of the hydroxyl radical (OH) and O3; globally, most tropospheric OH is found in the tropics (Bloss et al., 2005). Therefore, a quantitative understanding of the composition and chemistry of the tropical marine atmosphere is essential to examine the atmospheric oxidative capacity and climate forcing.
In the troposphere, reactive halogen species catalyse ozone destruction cycles:
where X=Cl, Br, I.
In the past, tropospheric halogen chemistry has been studied using a number of box models and 1-D models (Sander and Crutzen, 1996; von Glasow et al., 2002a; Saiz-Lopez et al., 2006; Simpson et al., 2015; Lowe et al., 2009; Sommariva and von Glasow, 2012; Surl et al., 2015). Currently, there are several global models that have been used to study tropospheric halogens (Hossaini et al., 2010; Ordóñez et al., 2012; Saiz-Lopez et al., 2012a, 2015; Fernandez et al., 2014; Sherwen et al., 2016b; Schmidt et al., 2016). Numerical models predict that reactive halogen compounds account for 30 % of O3 destruction in the marine boundary layer (MBL) (von Glasow et al., 2002b, 2004; Saiz-Lopez et al., 2015; Sherwen et al., 2016b) and 5 %–20 % globally (Yang et al., 2005; Saiz-Lopez et al., 2015, 2012a; Sherwen et al., 2016b). Up to 34 % of O3 loss is calculated to be due to I and Br combined in the tropical east Pacific (Wang et al., 2015).
However, there are only a few regional models that have studied tropospheric halogens. Chlorine chemistry was implemented into the WRF-Chem model (Lowe et al., 2015; Li et al., 2016) and into the Community Multi-scale Air Quality (CMAQ) model (Sarwar et al., 2014) to study the formation of nitryl chloride (ClNO2) from the uptake of dinitrogen pentoxide (N2O5) on aerosols containing chloride. Moreover, bromine and iodine chemistry was implemented in CMAQ in Gantt et al. (2017) and Sarwar et al. (2015), where the impact of iodide-mediated O3 deposition on surface ozone concentrations was studied, and in the recent work of Muñiz-Unamunzaga et al. (2018), which concluded that oceanic halogens and dimethyl sulfide (DMS) emissions need to be included into the regional models to accurately reproduce the air quality in coastal cities.
Oceanic emissions provide a source of very-short-lived halocarbons (VSLHs) to the atmosphere, defined as trace gases with chemical lifetimes generally under 6 months, mainly in the form of bromoform (CHBr3), dibromomethane (CH2Br2) and methyl iodide (CH3I). Once in the atmosphere, VSLHs (and their degradation products) can ascend into the lower stratosphere (LS), where they can contribute to the Bry and lead to ozone depletion. Several emissions inventories for the VSLHs have been evaluated at a global scale (Bell et al., 2002; Ziska et al., 2013; Ordóñez et al., 2012; Hossaini et al., 2013; Lennartz et al., 2015; Wales et al., 2018). Recent measurements constrain the stratospheric injection of bromine from VSLHs as ∼5 pptv Bry (Wales et al., 2018), confirming recent WMO estimates. About 40 %–50 % of the bromine (2.1–2.6 pptv Bry) is injected into stratosphere as product gases (Koenig et al., 2017; Wales et al., 2018). Lennartz et al. (2015) presents a comparison of two simulations using the chemistry climate model EMAC. The first simulation computes the oceanic emissions online, mainly driven by the surface water concentrations and modelled meteorological variables, and the second uses prescribed emissions. These results reveal that calculating the air–sea fluxes online leads, in most cases, to more accurate atmospheric mixing ratios in the model in comparison with the simulation using prescribed emissions. Emissions of inorganic iodine compounds (HOI and I2) have been recognized as a significant source required to reproduce iodine oxide (IO) measurements over the open ocean (Mahajan et al., 2012; Carpenter et al., 2013) and have been included in some global models (Saiz-Lopez et al., 2014; Sherwen et al., 2016b).
There are indications that the chemistry of reactive halogens and that of oxygenated VOCs (OVOCs) in the tropics are interrelated. Model calculations suggest aldehydes are an important sink for bromine atoms and hence compete with the formation of bromine oxide (BrO) (Br+O3 → BrO). This illustrates a link between the cycles of halogens and OVOCs in the marine atmosphere (Sommariva and von Glasow, 2012; Toyota et al., 2004).
Recent studies have highlighted the key role that heterogeneous chemistry plays in explaining observations of BrO and IO abundances in the tropical troposphere. Cycling of Br and I through HOBr, BrNO3, HOI and INO3 is very slow in the gas phase, making it necessary to include heterogeneous reactions involving reactive halogen species to reproduce observed BrO and IO abundances (von Glasow et al., 2004; Saiz-Lopez et al., 2015; Sherwen et al., 2016a).
Another source of reactive inorganic bromine in the troposphere is the release of bromide (Br−) from sea-salt aerosols into the gas phase. This is known as debromination and occurs through the uptake of a gaseous species in sea salt and the subsequent reaction with Br−. Debromination has been included as a source of gas-phase bromine in several atmospheric models (Yang et al., 2005; Parrella et al., 2012; Ordóñez et al., 2012; Schmidt et al., 2016; Long et al., 2014). However, this process is poorly understood and its inclusion into the models can cause inconsistent high levels of bromine species (Schmidt et al., 2016).
Halogen chemistry in atmospheric models remains largely untested due to a lack of field observations of halogen species. However, during the last few years, there have been four campaigns that provided vertically resolved measurements of halogen radicals: the Tropical Ocean tRoposphere Exchange of Reactive halogen species and Oxygenated VOC (TORERO; Volkamer et al., 2015; Wang et al., 2015; Dix et al., 2016), the CONvective TRansport of Active Species in the Tropics (CONTRAST; Pan et al., 2017; Koenig et al., 2017), the Coordinated Airborne Studies in the Tropics (CAST; Harris et al., 2017) and the Airborne Tropical TRopopause EXperiment (ATTREX; Jensen et al., 2017).
The main objective of this study is to investigate the atmospheric chemistry in the tropical East Pacific with a focus on reactive halogens using the Weather Research and Forecasting model coupled with Chemistry (WRF-Chem; Grell et al., 2005) and field data from the TORERO campaign (Volkamer et al., 2015; Wang et al., 2015). Our reaction mechanism in WRF-Chem is based on the Model for OZone and Related chemical Tracers version 4 (MOZART-4) mechanism (Emmons et al., 2010; Knote et al., 2014) and has been extended to include halogen chemistry. Heterogeneous recycling reactions involving halogens have been included into the model, along with oceanic emissions of relevant VOCs and halocarbons. The observational data are described in Sect. 2. Model developments are described in Sect. 3. The model setup and the description of different sensitivity runs are in Sect. 4. The results of the model performance are discussed in Sect. 5. The last section summarizes the conclusions of this work.
The TORERO campaign (Volkamer et al., 2015; Wang et al., 2015), from 15 January to 1 March 2012, was used to evaluate the model. Data on halocarbons are available from the TORERO ship cruise (Andrews et al., 2015) and flights of the NSF/NCAR GV aircraft, whilst observations of O3, BrO, IO and OVOCs are available from the flights. The TORERO cruise aboard the NOAA RV Ka'imimoana (KA-12-01) took place from Honolulu, HI, to Puntarenas, Costa Rica, between 27 January and 1 March 2012. Air samples from the TORERO ship cruise were taken from a 10 m bow mast and surface water samples were taken from the underway supply. Halocarbons in air and water phases were measured using two automated online gas chromatography–mass spectrometry (GC-MS) systems (Andrews et al., 2015) and calibrated using NOAA standard SX-3570. Ozone was measured by UV absorption (Coburn et al., 2014), OVOCs were measured by the Trace Organic Gas Analyzer (TOGA) (Apel and UCAR/NCAR, 2016), and BrO and IO radicals were measured by the University of Colorado Airborne Multi-AXis Differential Optical Absorption Spectroscopy (CU AMAX-DOAS) instrument with typical detection limits of 0.5 pptv for BrO and 0.05 pptv for IO (Volkamer et al., 2015; Dix et al., 2016). A total of 13 flights provide O3 data and 16 flights provide BrO and IO data. Figure 1 displays the location of all the observational data with an orange line for the cruise track, red lines for the flights in the tropics and green lines for the flights in the subtropics.
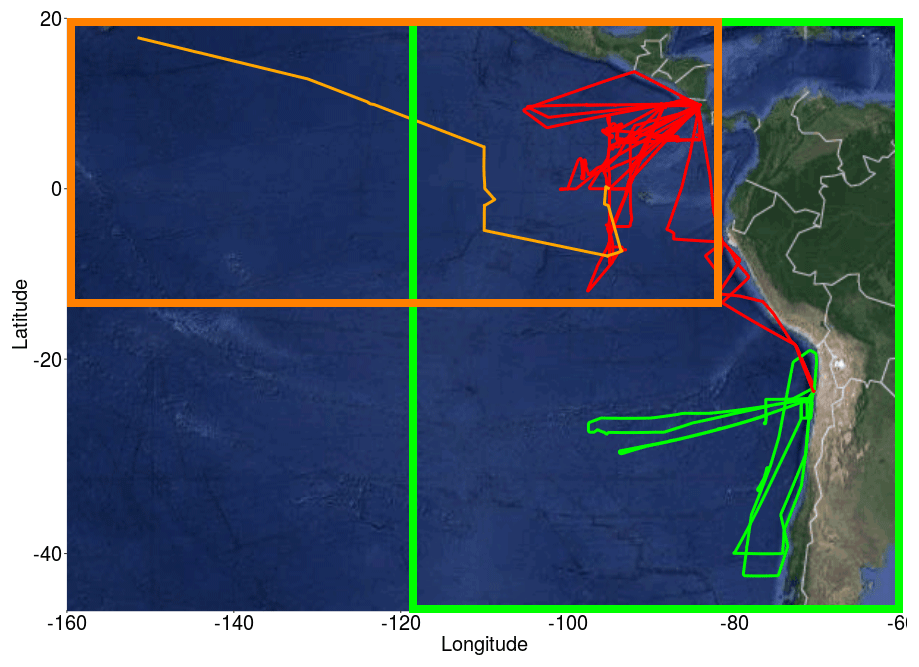
Figure 1Flight and cruise tracks from the TORERO campaign (January–February 2012). Cruise track is represented by a light orange line. Flights are grouped by the following regions: tropical (red lines) and subtropical (green lines). Two different domains were defined: domain to evaluate the cruises (dark orange square) and domain to evaluate aircrafts (green square).
WRF-Chem (Grell et al., 2005) is a highly flexible community model for atmospheric research where aerosol–radiation–cloud feedback processes are taken into account. Version 3.7.1 is used in this study.
The sources of halogen atoms considered in this study are inorganic I from the ocean (HOI and I2), oceanic source of organic halogens (CHBr3, CH2Br2, CH3I, CH2BrCl, CHBrCl2, CHBr2Cl, CH2I2, CH2IBr and CH2ICl) and debromination (Br2, IBr). These sources are explained in detail below in the following sections.
3.1 Oceanic fluxes
The oceanic emission of inorganic iodine (HOI and I2) follows the deposition of O3 to the surface ocean and reaction with iodide (I−) (Carpenter et al., 2013). We use Eqs. (19) and (20) in Carpenter et al. (2013) for the calculation of these emissions. Ocean surface I− is parameterized using MacDonald et al. (2014) (see Fig. S1 in the Supplement). Figure 2 shows the average oceanic emission for inorganic iodine (I2 in Fig. 2a and HOI in Fig. 2b) during January and February 2012. Higher emissions for inorganic iodine occur in the tropics with HOI being the dominant species.
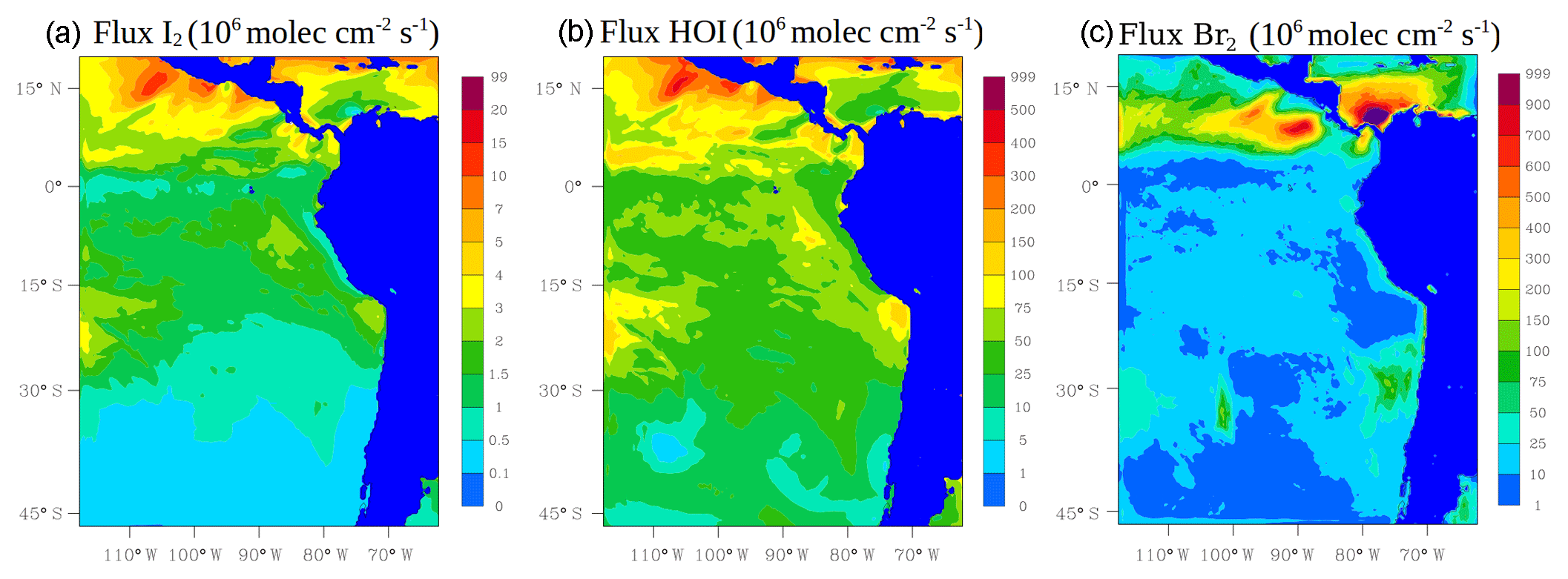
Figure 2Mean oceanic surface fluxes for inorganic iodine: I2 (a) and HOI (b). The column-integrated fluxes for inorganic bromine (Br2, c) from the debromination process during January and February 2012 are also shown. Values are given in 106 molec cm−2 s−1.
Two different approaches for the marine emissions of the halocarbons (CHBr3, CH2Br2, CH3I, CH2BrCl, CHBrCl2, CHBr2Cl, CH2I2, CH2IBr and CH2ICl) are examined in this model. The first approach uses prescribed monthly average oceanic fluxes from Ziska et al. (2013) and the second computes the oceanic fluxes online. Prescribed monthly average oceanic fluxes from Ziska et al. (2013) were calculated using 6-hourly means of wind speed and sea surface temperature from the ERA-Interim meteorological assimilation database (Dee et al., 2011) for the years 1989–2011 (). Computing the emissions online accounts for an interaction between the modelled atmosphere and the ocean at each time step. Thus, this approach can respond to changes in meteorological parameters, like surface temperature and surface wind speed. A two-layer model (Liss and Slater, 1974) is used to calculate the halocarbons air–sea fluxes:
where Ka is the transfer velocity of the gas (s−1), Cg (ppm) and Cl (nM) are the bulk gas and liquid-phase concentrations, and KH is Henry's law constant. Ka is parameterized following Johnson (2010) which is mainly a function of wind speed and sea surface temperature (SST) taken from the model at each time step. Cg is also taken from the model. Halocarbon seawater concentrations Cl are taken from Ziska et al. (2013). Figure 3 shows the average air–sea fluxes for CHBr3, CH2Br2 and CH3I during January and February 2012 for the two approaches. Note that the online calculation could increase, decrease or even reverse the fluxes in comparison with the prescribed emissions. This is the case for the online fluxes of CHBr3 over the tropics where the model calculates negative fluxes, whereas the prescribed fluxes are positive.
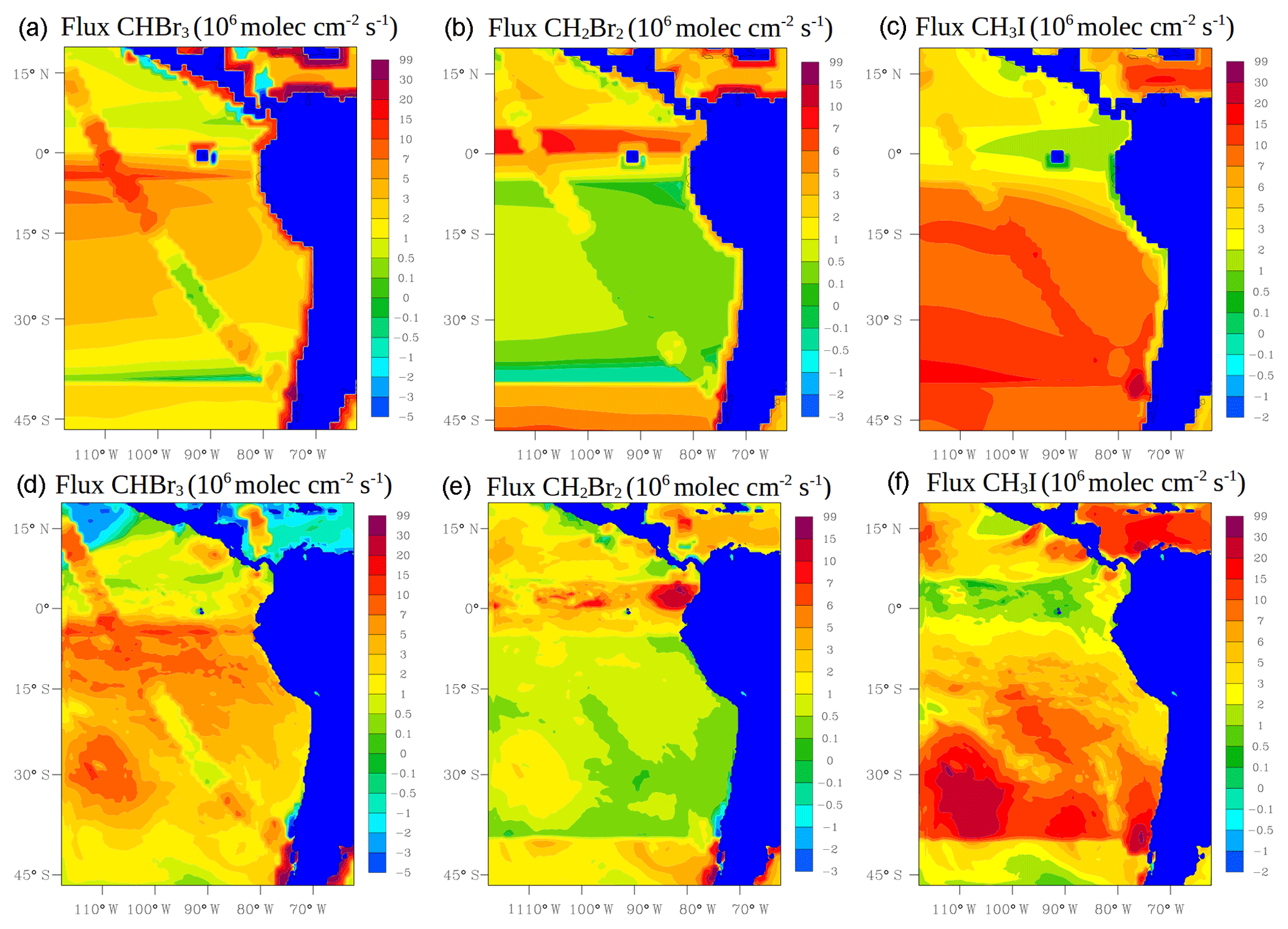
Figure 3Mean oceanic fluxes for halocarbons (CHBr3, CH2Br2 and CH3I) during January and February 2012. Prescribed fluxes are shown on the top (a, b, c) and online fluxes on the bottom (d, e, f). Values are given in 106 molec cm−2 s−1.
Recent studies suggest that the ocean is an important source of OVOCs such as acetaldehyde, ethanol and methanol (Coburn et al., 2014; Lawson et al., 2015; Mahajan et al., 2014; Myriokefalitakis et al., 2008; Sinreich et al., 2010; Volkamer et al., 2015; Yang et al., 2014; Fischer et al., 2012) that models do not generally consider or are not able to capture (Millet et al., 2010; Sherwen et al., 2016a). Oceanic fluxes of several VOCs have been included into WRF-Chem as part if this study. For the three OVOCs (acetaldehyde (CH3CHO), ethanol (C2H6O) and methanol (CH3OH)), the same online approach as for the VSLHs is used to calculate the marine fluxes where their seawater concentrations are taken from Yang et al. (2014).
Emissions for alkenes and alkanes (C2H4, C3H6, C2H6, C3H8) are prescribed and based on the POET (Granier et al., 2005) global inventory.
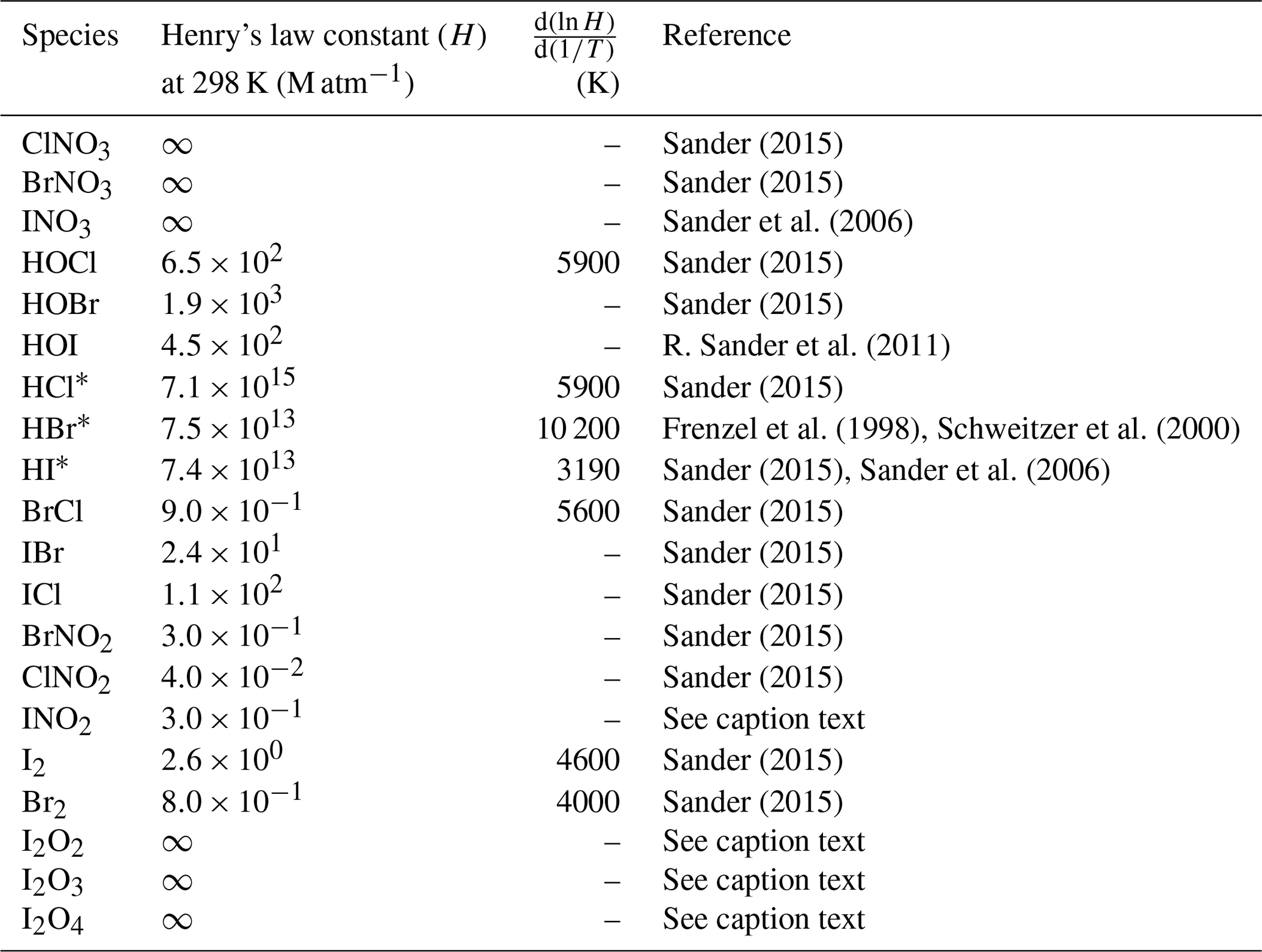
Deposition over the ocean for the halocarbons and OVOCs is included in the air–sea fluxes described above. For the rest of the species, dry deposition is calculated with the Wesely scheme (Wesely, 1989), which is used over land for several species. Washout of gases by precipitation is simulated using the scheme included in WRF-Chem (Grell and Dévényi, 2002; Zaveri et al., 2008) which was modified to include Henry's law constants for the RHS shown in Table 1.
Sander (2015)Sander (2015)Sander et al. (2006)Sander (2015)Sander (2015)R. Sander et al. (2011)Sander (2015)Frenzel et al. (1998)Schweitzer et al. (2000)Sander (2015)Sander et al. (2006)Sander (2015)Sander (2015)Sander (2015)Sander (2015)Sander (2015)Sander (2015)Sander (2015)Table 1Henry's law constant for relevant halogen species implemented in WRF-Chem. INO2 Henry's law constant is assumed equal to that of BrNO2. Iodine oxide (I2Ox) Henry's law constants are assumed to be infinity by analogy with INO3. Virtually infinity solubility is represented by using a very large number (2.69×1015).
The sea-salt aerosol emissions parameterization used in this study is described in Archer-Nicholls et al. (2014). This parameterization is mainly a function of wind speed from the model and uses the emissions scheme from Gong et al. (1997) for particles with dry diameters of 0.45 nm or more, and for smaller particles, it uses Fuentes et al. (2010).
3.2 Gas-phase chemistry scheme
Our reaction mechanism is based on the MOZART-4 mechanism (Emmons et al., 2010; Knote et al., 2014). This mechanism has been extended to include bromine, chlorine and iodine chemistry and has been coupled with the Model for Simulating Aerosol Interactions and Chemistry (MOSAIC) four-bin aerosol module (Zaveri et al., 2008). A total of 48 species and 159 halogen reactions have been included (see Tables 2, 3 and 4 for details). Inorganic, organic and inter-halogen reactions come from the 1-D model MISTRA (Sommariva and von Glasow, 2012). Production and loss reactions of the higher order of iodine oxides (I2Ox, where ) reactions have been included into the model. Photochemistry of I2Ox species is still an area of high uncertainty in atmospheric iodine chemistry (Sommariva et al., 2012; Saiz-Lopez et al., 2012b). Chemical loss of VSLHs through oxidation by the hydroxyl radical (OH) and by photolysis is included using data from S. P. Sander et al. (2011).
Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Burkholder et al. (2015)Atkinson et al. (2007)Atkinson et al. (2007)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)Atkinson et al. (2006)Atkinson et al. (2006)Atkinson et al. (2006)Atkinson et al. (2006)Atkinson et al. (2006)Burkholder et al. (2015)Atkinson et al. (2008)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)S. P. Sander et al. (2011)S. P. Sander et al. (2011)Atkinson et al. (2007)Orlando and Tyndall (1996)S. P. Sander et al. (2011)S. P. Sander et al. (2011)S. P. Sander et al. (2011)Atkinson et al. (2006)Atkinson et al. (2006)Atkinson et al. (2006)Toyota et al. (2004)Toyota et al. (2004)Toyota et al. (2004)Sander et al. (2006)Sander et al. (2006)Sander et al. (2006)Sander et al. (2006)Sander et al. (2006)Sander et al. (2006)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Burkholder et al. (2015)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Gómez Martín et al. (2007)Gómez Martín et al. (2007)Ordóñez et al. (2012)Ordóñez et al. (2012)Kaltsoyannis and Plane (2014)Atkinson et al. (2007)Riffault et al. (2005)McFiggans et al. (2000)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Kaltsoyannis and Plane (2014)Dillon et al. (2006)S. P. Sander et al. (2011)Bedjanian et al. (1997)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)Atkinson et al. (2007)S. P. Sander et al. (2011)Table 2Bimolecular and thermal decomposition halogen reactions included in WRF-Chem. These reactions are given in the Arrhenius form with the rate equal to .
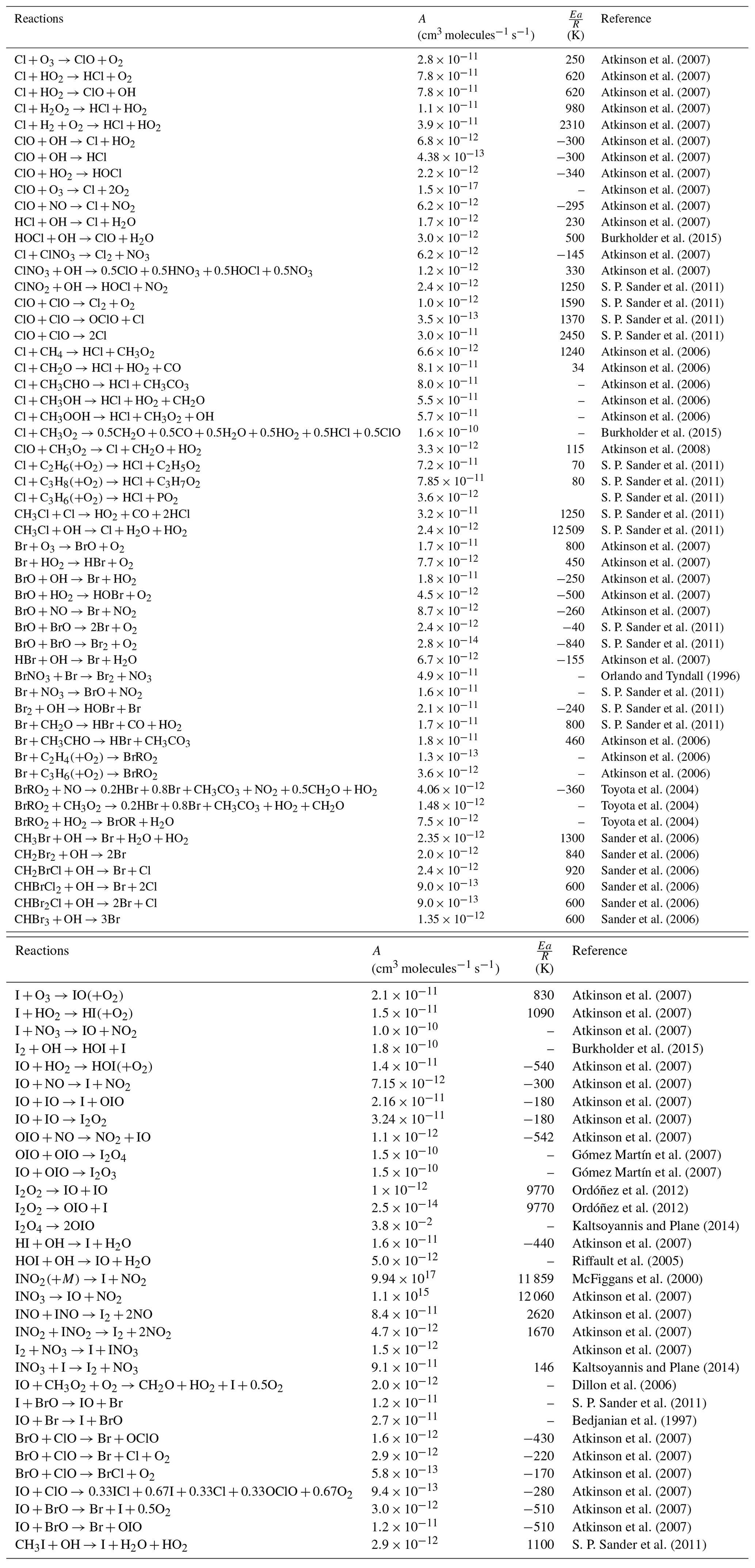
Table 3Termolecular reactions for halogens species included in WRF-Chem. The lower pressure limit rate (K0) is given by . The high pressure limit (K∞) is given by . Fc describes the fall of curve of the reaction described by Atkinson et al. (2007). Then the reaction rate (k) is defined as and n as .

A schematic representation of the main bromine and iodine chemistry implemented in the model is shown in Fig. 4. Chlorine chemistry is also included into the model; however, since our results are mainly focused on reactive bromine and iodine, we do not include chlorine chemistry in Fig. 4.
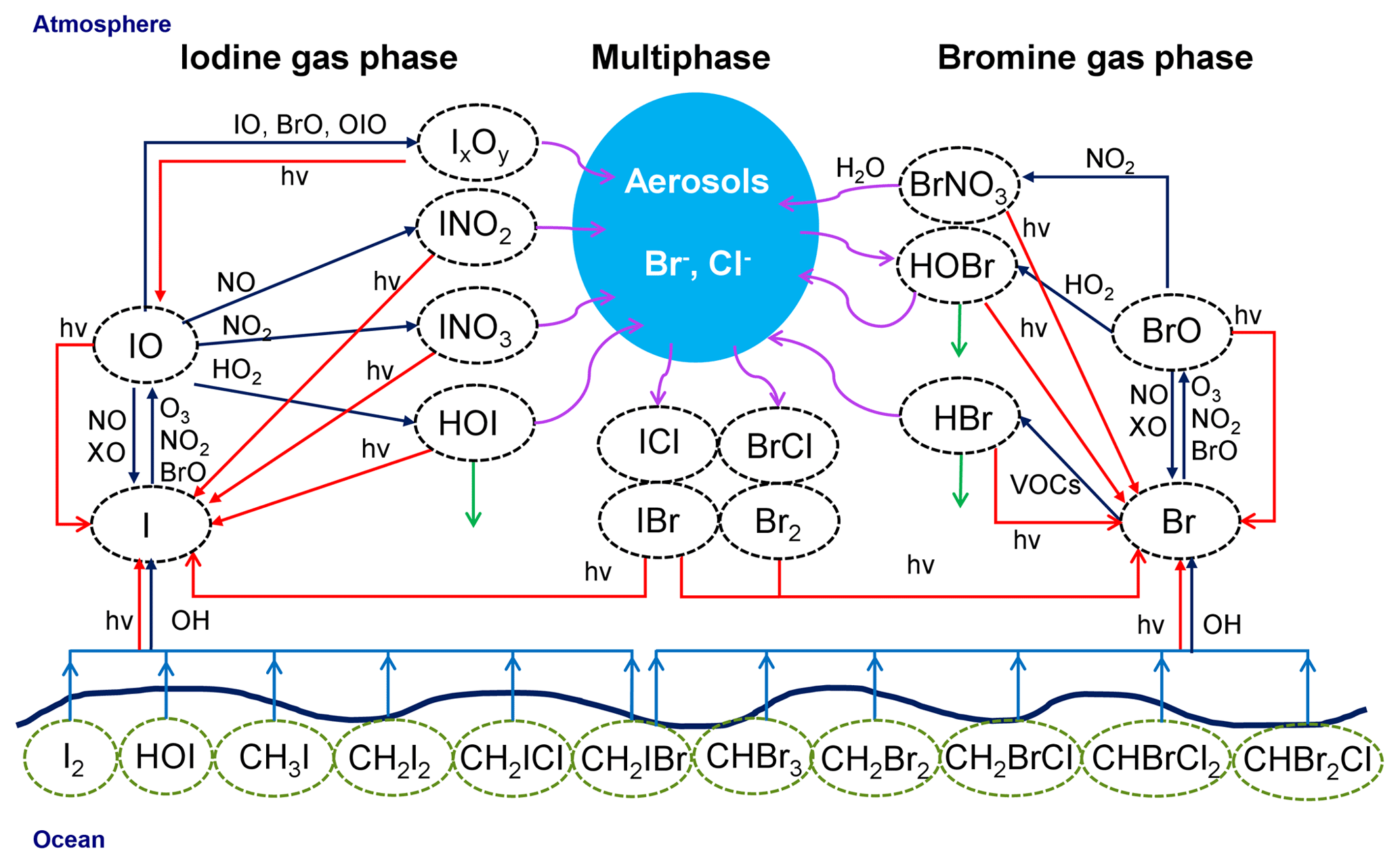
Figure 4Schematic representation of the implemented iodine and bromine chemistry in WRF-Chem. Chlorine chemistry has been included into the model; since our results are mainly focused on reactive bromine and iodine, we decided not to include chlorine chemistry in this figure. Red lines represent photolytic reactions, dark blue lines gas-phase pathways, light blue lines fluxes, green lines deposition and purple curved lines heterogeneous pathways.
Photolysis reactions included in the mechanism are listed in Table 4. To compute the photolysis rates the fast Tropospheric Ultraviolet–Visible (FTUV) online scheme (Tie et al., 2003) is used. The quantum yields and cross section for the photolytic reactions of halogens are from the Jet Propulsion Laboratory (JPL) 10-6 (S. P. Sander et al., 2011) and have been linearly interpolated onto the 17 wavelength bins used by FTUV. For I2Ox, we use the quantum yield and cross-section data from Gómez Martín et al. (2005).
3.2.1 Halogen and VOC reactions
Reactions between halogens and VOCs can be important for regulating reactive halogen chemistry in the MBL by promoting the conversion of Cl and Br atoms into HCl and HBr or more stable organic halogenated intermediates. The oxidation of methane (CH4), formaldehyde (CH2O), acetaldehyde (CH3CHO), methanol (CH3OH), methyl hydroperoxide (CH3OOH), methylperoxy (CH3O2), ethane (C2H6), ethene (C3H6) and propene (C3H6) by Cl is included in the chemical mechanism. In addition, the oxidation of CH2O, CH3CHO, C3H6 and C3H6 by bromine is also included in the chemical mechanism with a simplified version of the chemical scheme presented in Toyota et al. (2004) used for reactions of bromine with alkenes:
where BrRO2 is a brominated peroxy radical.
The loss of BrRO2 is represented by the following reactions:
The loss of BrOR is represented by the following reactions:
where BrOR is a brominated organic specie and x is a number between 0 and 1.
Reaction rates for these reactions and deposition velocities are taken from Toyota et al. (2004). Kinetic data for these reactions are poor, and the partitioning of the products (HBr:Br) is not clear. Based on the Toyota et al. (2004) description, it is assumed that x=0.2 such that the partitioning for HBr:Br is 1:4 (Kenjiro Toyota, personal communication, 2017).
3.3 Heterogeneous chemistry
Heterogeneous reactions on particle surfaces involving halogens are summarized in Table 5. The heterogeneous chemistry is assumed to take place between a gas-phase species and an adsorbed species. The bulk aqueous-phase chemistry in sea-salt aerosols is not treated. Uptake coefficients are used to calculate first-order rate constants for heterogeneous loss of the gas phase to the adsorbing surface (Jacob, 2000). This follows the approach used by McFiggans et al. (2000), which assumes a free molecular transfer regime approximation. The reaction rate constants, K (s−1), are given by
where γ is the uptake coefficient, S is the root mean square molecular speed (m s−1) and A is the total available aerosol surface area density (cm2 cm−3). Equation (2) does not take account of any diffusion limitation (i.e. the rate at which gases can diffuse towards the aerosol surface).
Table 5Halogen heterogeneous reactions added to WRF-Chem in this study.

a Uptake coefficient for moderate temperature.
b Uptake coefficient for cold temperatures.
We test the sensitivity of our results by adding the diffusion term, Dg (cm2 s−1), following Brasseur and Jacob (2017), where the reaction rate constant, K (s−1), is given by
where nbin is the number of particle-size bins, ri is the particle radius for bin i (cm), and Ai is the available aerosol surface area density for bin i (cm2 cm−3). K is integrated over the aerosol size distribution in order to resolve the dependence of the rate constant for the particle radius. Following Brasseur and Jacob (2017), Dg is given by
where mg is the mean molecular mass of the gas-phase specie (g mol−1), mair is the molecular mass of air (g mol−1), na is the total air number density (molecules cm−3), and T is the temperature (K).
Second-order reaction rate constants are calculated by dividing the first-order rate constant by the concentrations of the adsorbed species. Heterogeneous halogen activation is very efficient under cold or stratospheric conditions as compared to moderate temperatures. For this reason, we have made a distinction between moderate (>243.15 K) and cold temperatures (<243.15 K) in some reactions. Uptake coefficients for reactions in Table 5 are based on literature values where available (Jacob, 2000; Sander et al., 2006; Ordóñez et al., 2012).
There are six reactions implemented for sea-salt particles. The sea-salt surface area is calculated in the following way: (1) using the mass of Na and Cl and the associated H2O for each bin and the individual dry densities (for Na, Cl and H2O), the total volume of those particles for each bin is calculated and then, (2) assuming that sea-salt aerosols are spheres, the total surface area is calculated for each bin using this volume and the radius of aerosols in each bin.
It is known that the chemistry involving the release of bromine from the sea-salt aerosol (debromination) is strongly pH dependent, being more efficient for acidified aerosol especially with a pH <5.5 (Keene et al., 1998). Therefore, the pH value of the aerosol particles is calculated in the model for each size bin (see Zaveri et al., 2008 for further description of the pH calculation). We then apply a pH dependence to the heterogeneous reactions that occur on the surface of the sea salt. When the pH <5.5 debromination reactions occur with the release of Br2 and IBr resulting from the uptake of BrNO3, BrNO2, HOBr, INO3, INO2 and HOI (Reactions R11–R16). When the pH >5.5 no debromination reactions occur, although uptake of INO3, INO2 and HOI on the sea salt still occurs (Reactions R17–R19) leading to a change in iodine speciation but no release of Br. See also Table 5.
If the pH <5.5,
If the pH >5.5,
Due to the high uncertainty in the debromination process, the fraction of Br2 formed by Reactions (R11)–(R13) was chosen arbitrarily in order to add an extra bromine source in a simple way. A value of 0.6 was chosen. Figure 2 shows the column-integrated fluxes for inorganic bromine (Br2; Fig. 2c) during January and February 2012.
In addition, the heterogeneous uptake of N2O5 onto aerosol particles that contain Cl− to form ClNO2 is considered in the model. After uptake, N2O5 is taken up onto the particle; it reacts reversibly with liquid water to form protonated nitric acid intermediate (H2ONO+2). This then reacts with either liquid water, to form aqueous nitric acid (HNO3), or with chloride ions to form ClNO2. See Archer-Nicholls et al. (2014) for further description of this chemistry. In Archer-Nicholls et al. (2014), ClNO2 was considered as an inert specie; however, in our study, ClNO2 is not treated as an inert specie but is broken down via photolysis and reaction with OH (see Tables 2 and 4).
The model is set up with a horizontal grid spacing of 30 km×30 km and 30 vertical layers up to 50 hPa. Simulations that study the oxidation of VOCs by Br over the tropical area (described in Sect. 4.1) are performed with more vertical layers than the standard case in order to capture the vertical mixing in this area. Thus, 52 vertical layers up to 50 hPa are used in this case. The meteorological initial and lateral boundary conditions were determined using the ERA-Interim (Dee et al., 2011) data, and the meteorology was reinitialized every 3 days to reproduce the observed transport. Chemical initial and boundary conditions (ICs/BCs) are from the global atmospheric model GEOS-Chem described in Sherwen et al. (2016b). We conducted WRF-Chem simulations for January and February 2012 covering the TORERO domain (see Fig. 1). We performed a spin-up of 20 days. Table 6 describes the main configuration of the model.
(Emmons et al., 2010; Knote et al., 2014)(Sommariva and von Glasow, 2012)(Tie et al., 2003)Wesely (1989)Grell and Dévényi (2002)(Guenther et al., 2006)(Liss and Slater, 1974; Johnson, 2010)(Granier et al., 2005)Archer-Nicholls et al. (2014)Lowe et al. (2015)(Sherwen et al., 2016b)(Dee et al., 2011)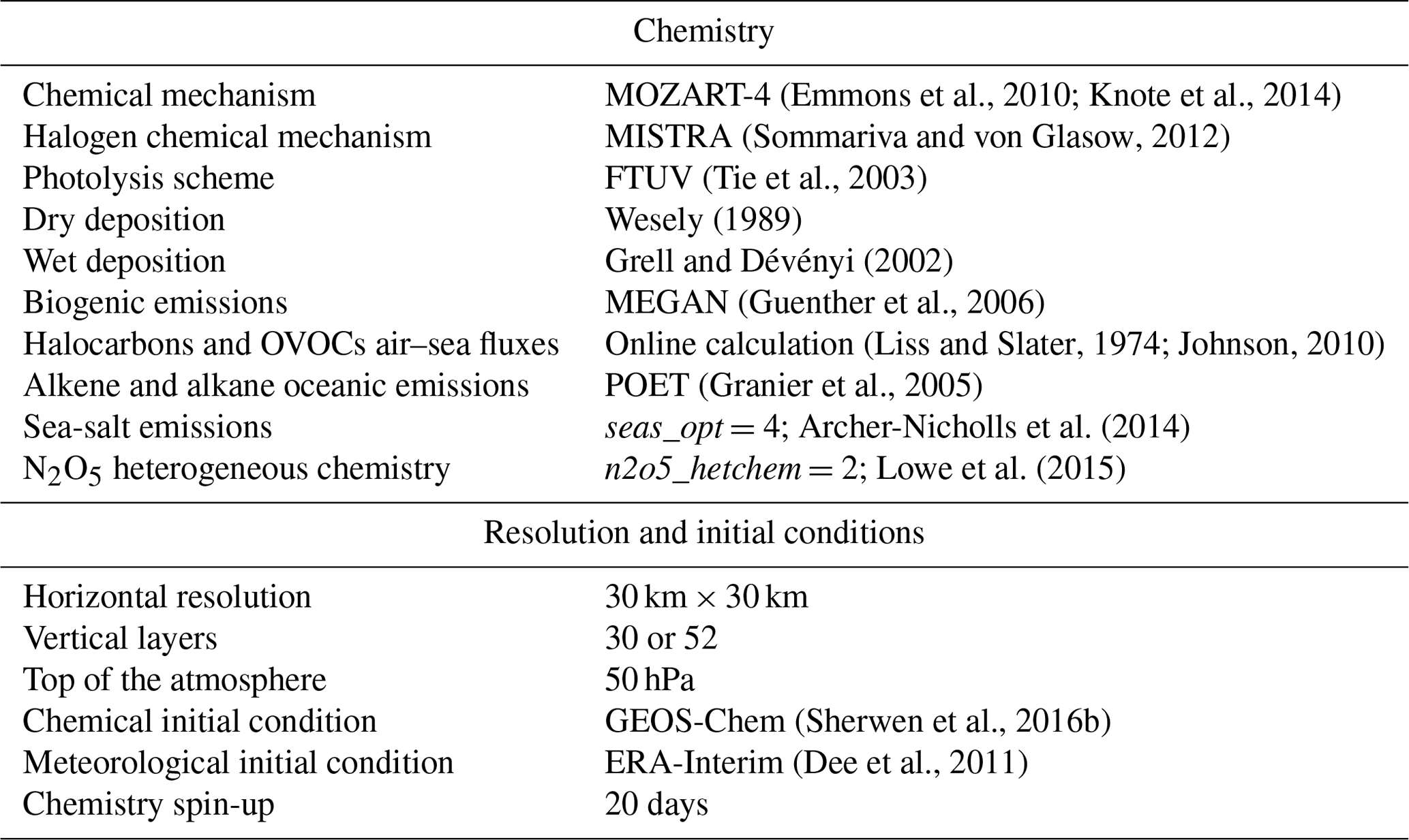
4.1 Sensitivity studies
A total of 11 different simulations were performed in this study. Our base simulation, WRF-DEBROM, considered all main processes involving halogen chemistry (sea-salt debromination, heterogeneous chemistry and reactions between halogens and VOCs) and computes the oceanic halocarbon fluxes online. The WRF-ZIS simulation is the same as WRF-DEBROM but uses prescribed oceanic emissions for the halocarbons. To test the sensitivity to the heterogeneous reaction rate constants, two runs were performed: the first one, in which the values for the uptake coefficient (γ from Eq. 2) from Table 5 have been divided by 2, WRF-GAMMADV2, and the second one, where Eq. (3) that has a diffusion term, is used, WRF-DIFF. To account for the importance of the debromination in sea-salt particles, we performed the simulation WRF-NODEBROM which is the same as the WRF-DEBROM simulation but without debromination. The WRF-NOHET simulation is the same as WRF-NODEBROM but without heterogeneous chemistry. A simulation with no halogen chemistry, WRF-NOHAL, is performed to study the effect of halogens on the tropospheric chemistry. All simulations except WRF-NOHAL use ICs/BCs from the GEOS-Chem model that include halogens. The WRF-NOHAL simulation uses ICs/BCs from the GEOS-Chem model with no halogen chemistry. Finally, to study the oxidation of VOCs by halogens, four simulations have been performed: (1) a simulation without the reactions of bromine reactions with alkenes (WRF-NOBRALKE), (2) a simulation without the reactions of bromine with aldehydes (WRF-NOBRALD), (3) a simulation without the reactions of bromine with VOCs, therefore neither alkenes nor aldehydes (WRF-NOBRVOCS), and (4) a simulation without reactions of bromine and chlorine with VOCs (WRF-NOHALVOCS). See Table 7 for a summary of all these simulations.
Table 7Summary of all the simulations to investigate the main processes involving reactions between halogen chemistry.

This section presents the model evaluation with observations of relevant trace gases. The model output is sampled at the nearest timestamp and grid box to the measurements. An ocean mask neglecting grid boxes above land was applied to compute all model results.
5.1 Oceanic emissions: halocarbons
Figure 5 shows the time series of CHBr3 (Fig. 5a), CH2Br2 (Fig. 5b) and CH3I (Fig. 5c) mixing ratios (in pptv) for the WRF-ZIS (green line) and WRF-DEBROM (black line) runs. In addition, the modelled wind speed (black line) is also shown in Fig. 5 (Fig. 5d). Measurements for the halocarbons and wind speed are represented by the solid red lines. Figure 6 presents the time series of CHBr3, CH2Br2 and CH3I water concentration (in pmol L−1) from the measurements (dashed red lines) and from the Ziska et al. (2013) climatology (dashed blue lines) used to compute both the prescribed and online fluxes.

Figure 5Time series of CHBr3 (a), CH2Br2 (b) and CH3I (c) mixing rations (in pptv) for the WRF-ZIS (green line) and WRF-DEBROM (black line) runs during the period of the TORERO campaign in 2012. In panel (d), the wind speed (m s−1) of the model is shown with a black line. Measurements during the TORERO campaign are depicted with red lines.
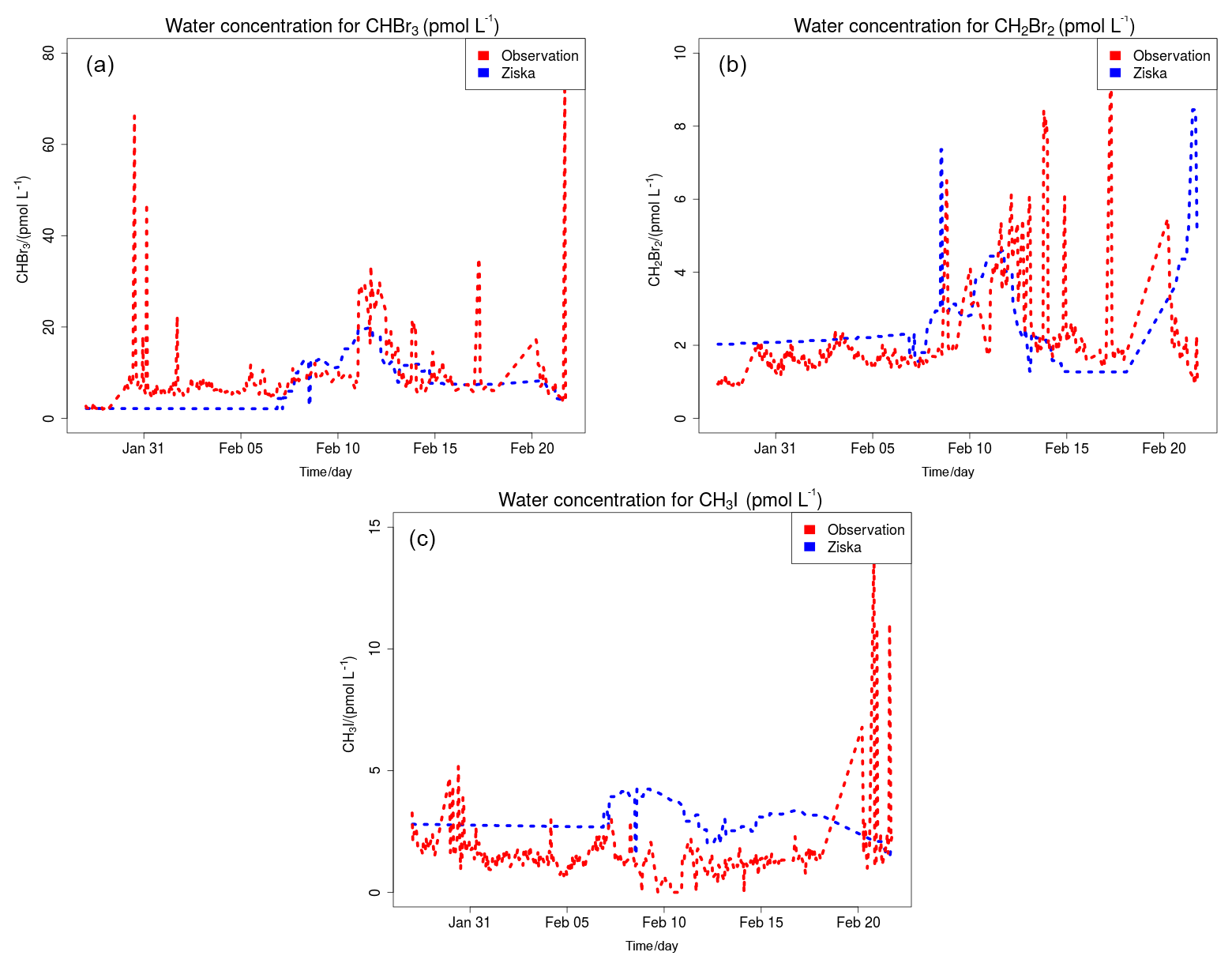
Figure 6Time series of measured CHBr3 (a), CH2Br2 (b) and CH3I (c) water concentration (in pmol L−1) during the TORERO campaign (red dashed line) and from the Ziska et al. (2013) climatology (blue dashed line).
In general, both simulations reproduce the concentrations of the halocarbons to the right order of magnitude, although there are specific periods with a negative bias. We see a tendency to underestimate CHBr3 for both model simulations during most of the period. This result is similar to the study of Hossaini et al. (2016), who compared 11 global models using different emissions inventories. The majority of the models do not reproduce the observed concentrations in the tropical marine boundary layer. Over the tropics, high emissions observed are associated with tropical upwelling and active planktonic production (Class and Ballschmiter, 1988; Atlas et al., 1993). One reason for low CHBr3 concentrations in our model simulations might be that the seawater concentrations are too low in this area (see Fig. 6 for CHBr3). The fluxes are also low (see Fig. S2). Note that Ziska et al. (2013) used only a very limited amount of data to derive the seawater concentration for the halocarbons in our domain, which leads to uncertainty in the calculated fluxes. The modelled CHBr3 is underestimated throughout the troposphere when it is compared with aircraft observations (see Fig. S3). Atmospheric concentrations of CH2Br2 are in good agreement with the observations although the model underestimates the observed values by ∼0.5 pptv during the periods 6–10 and 22–25 February. Bromocarbon concentrations agree better with the measurements when the oceanic fluxes are calculated online (WRF-DEBROM); in particular, the underestimation is less for specific periods (e.g. 20 February for CHBr3 and 10 and 22 February for CH2Br2) in comparison with WRF-ZIS. Moreover, the correlation coefficients between the observations and the simulations are better for the WRF-DEBROM compared to WRF-ZIS: 0.48 and 0.3 for CH2Br2 and CHBr3, respectively, in the case of WRF-ZIS, and 0.65 and 0.43 for CH2Br2 and CHBr3 in the case of WRF-DEBROM. Modelled CH3I concentrations show a similar trend to the observations, although, like the bromocarbons, both simulations underestimate the observations during specific periods (6–10 and 18–28 February). This underestimation is more prominent in the WRF-DEBROM simulation. One reason for that could be that the wind speed from WRF-Chem is lower than the wind speed used to calculate the prescribed emissions, producing lower online fluxes. Nevertheless, the correlation coefficients between the observed and simulated CH3I atmospheric concentrations are better for WRF-DEBROM than for WRF-ZIS: 0.19 is calculated for WRF-ZIS and 0.40 for the WRF-DEBROM simulation.
Specific periods of negative bias for both simulations demand further attention. A possible explanation for the underestimation in halocarbon atmospheric concentrations might be due to the input data (e.g. wind speed, SST, seawater concentration) that we used to compute these fluxes. In the case of the online fluxes, between 6 and 8 February, the model underestimates wind speed and this is directly accompanied by an underestimation for all three halocarbons' atmospheric concentrations. Ziska et al. (2013) demonstrate that changes in the input parameters, especially wind speed and SST, affect the fluxes calculation. The same study suggests that CH3I emissions are mainly influenced by variations of the wind speed. Moreover, the study of Lennartz et al. (2015), that uses the same seawater concentration as our study, suggests that the negative bias in the modelled atmospheric concentrations could indicate regions where the seawater concentration from the climatologies lacks hotspots; thus, it is missing an oceanic source region. This is clearly seen for the seawater concentrations of CHBr3 (during most of the period), CH2Br2 (peaks around 15 February) and CH3I (peaks around 20 February) used in this study that seem to be too low in comparison with the observations (see Fig. 6). More data on the seawater concentrations of these halocarbons in this region are required to better constrain the oceanic flux data sets available to models and so to improve the representation of these gases in the atmosphere.
5.2 Gas-phase and heterogeneous chemistry: bromine and iodine partitioning
Figure 7 compares model results sampled along 16 flight tracks with the observations for BrO (pptv) separating tropical from subtropical flights for the five simulations: WRF-NOHET, WRF-NODEBROM, WRF-GAMMADV2, WRF-DIFF and WRF-DEBROM. Results indicate that there is an improvement of the modelled BrO throughout the troposphere in both the tropics and subtropics when the heterogeneous chemistry is included in both tropics and subtropics.
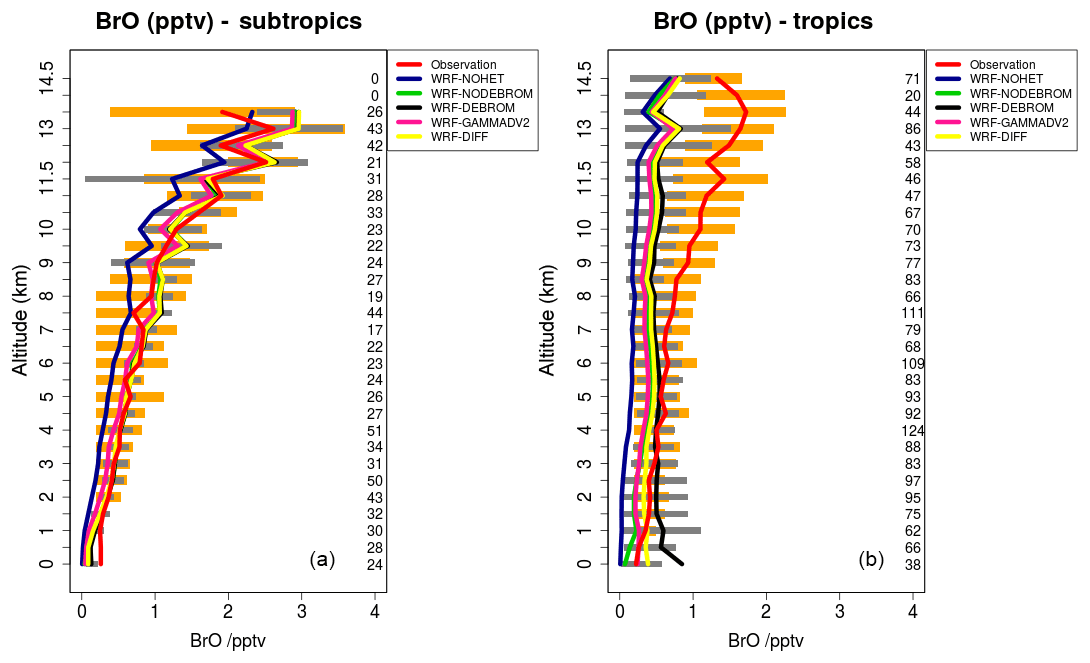
Figure 7Mean vertical profile of BrO (pptv) over the subtropics (a) and tropics (b). An average over 16 flights of the TORERO campaign (red line) are compared to the five different WRF-Chem simulations: WRF-NOHET (blue line), WRF-NODEBROM (green line), WRF-DEBROM (black line), WRF-GAMMADV2 (pink line) and WRF-DIFF (yellow line). Orange and grey horizontal bars indicate the 25th–75th quartile interval for the observations of the TORERO campaign and WRF-DEBROM simulation, respectively. Values are considered in 0.5 km bins and the number of aircraft measurement points for each altitude is given on the right side of each plot.
In the subtropics, higher values of BrO are found in the altitude range 11–13 km due to the lower altitude of the tropopause. Some data points in this altitude range will be in the lower stratosphere. There is really good agreement with the observations particularly in the middle and upper troposphere where the model is able to capture the higher values of BrO. Within the model, aerosols over the subtropical area tend to be alkaline; thus, BrO does not increase in this area when sea-salt debromination is included. Over the tropics, where the aerosol is more acidic and where the sea-salt aerosols are mostly located (see emissions of Br2 in Fig. 2), elevated BrO is seen with the inclusion of the debromination (WRF-DEBROM) in the MBL. Debromination improves the simulation of BrO concentrations in the middle troposphere although it excessively increases BrO levels up to 1 pptv in the MBL. Higher values are also seen in other modelling studies that include this process (Schmidt et al., 2016).
In areas, such as the tropics, where debromination dominates, the impact of halving gamma (WRF-GAMMADV2 run) is approximately half of the impact of including heterogeneous chemistry (i.e. the difference between the WRF-DEBROM run and the WRF-NOHET run) at least for the lower troposphere. Very little impact is seen in the upper troposphere (UT), a slight decrease in BrO, when gamma is halved (WRF-GAMMADV2). The simulation in which diffusion limitation is considered in the heterogeneous reactions (WRF-DIFF) gives values of BrO that are generally between the results from the WRF-GAMMADV2 and WRF-DEBROM simulations. They are similar to the WRF-GAMMADV2 values in the MBL, but over the subtropics, where debromination is lower, WRF-DIFF is very close to WRF-DEBROM values. Significant uncertainties still exist in the sea-salt debromination processes and the parameterizations used here might be too simple to represent them.
In addition, the conversion of BrO to HBr is dominated by the reaction between Br and OVOCs, such that the BrO overestimation seen in the MBL could be reduced if the modelled aldehyde concentrations were increased (discussed in Sect. 5.3). However, a reduction in the debromination would also reduce BrO concentrations. Thus, in order to capture the BrO concentrations in the MBL, the right balance between these two chemical processes is needed.
BrO is underestimated in the model by 1 pptv in the upper troposphere over the tropics. The breakdown of bromocarbons, such as CHBr3, contributes to BrO concentrations in the UT; thus, a good representation of bromocarbons is needed. CHBr3 is underestimated in the middle and upper troposphere especially over the tropics (see Fig. S3). The reason for that could be a combination of different factors: underestimation of the boundary conditions used in this study for CHBr3, underestimation in the oceanic fluxes (see Fig. S2) and overestimation of the loss rates. Moreover, an underestimation in the heterogeneous chemistry or uncertainties in the reactions between the halogens and VOCs (discussed in Sect. 5.3) can also contribute to the underestimation of BrO in the UT over the tropics.
Figure 8 shows the vertical profile distribution for inorganic bromine (Bry in pptv) for the three simulations – WRF-NOHET (Fig. 8a, d), WRF-NODEBROM (Fig. 8b, e) and WRF-DEBROM (Fig. 8c, f) – over the subtropics (Fig. 8a–c) and tropics (Fig. 8d–f). Inorganic bromine concentrations increase with altitude with a maximum of 8 pptv at 14 km in the subtropical area for all three simulations. This reflects the lifetime of the bromocarbon species that breakdown and release Br in the UT and LS. Over the tropical area, inorganic bromine concentrations have a peak in the middle troposphere at 6 km, then decrease until 12 km and then start to increase again. A big impact on the vertical Bry partitioning is seen between the three simulations. With the inclusion of the heterogeneous chemistry, there is a decrease of HBr and an increase of more reactive species: dihalogens (BrCl, Br2 and BrI) and BrO. HOBr increases and BrNO3 decreases in the UT due to BrNO3 hydrolysis. Over the tropics, Bry increases in the MBL (∼4 pptv) when debromination is included (WRF-DEBROM). This enhancement is seen for all inorganic species with a maximum in the surface where the concentration of sea-salt aerosols is highest. Over the subtropical area, little difference is seen between WRF-NODEBROM and WRF-DEBROM.
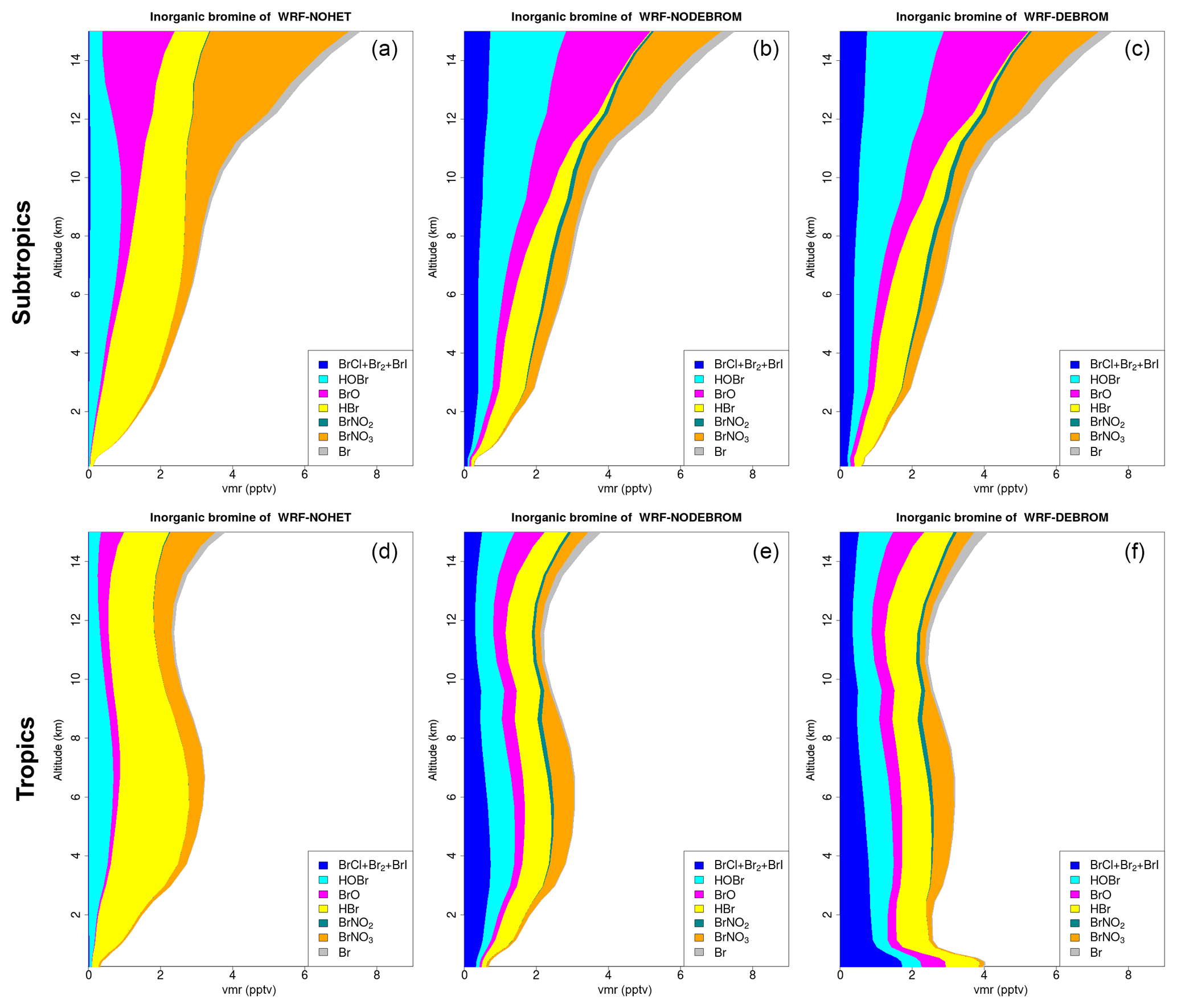
Figure 8Regional average vertical partitioning of inorganic bromine (Bry) for the three different simulations – WRF-NOHET (a, d), WRF-NODEBROM (b, e) and WRF-DEBROM (c, f) – during January and February 2012. Panels (a, b, c) are over the subtropical area and panels (d, e, f) over the tropical. Units are in pptv.
Figure 9 compares model results sampled along 16 flight tracks with the observations for IO separating tropical from subtropical flights for the five simulations: WRF-NOHET, WRF-NODEBROM, WRF-GAMMADV2, WRF-DIFF and WRF-DEBROM. No clear impact is seen with the inclusion of the heterogeneous chemistry. At the surface, simulations with heterogeneous chemistry (WRF-DEBROM, WRF-GAMMADV2, WRF-DIFF and WRF-NODEBROM) have slightly lower IO concentrations than the simulation without heterogeneous chemistry (WRF-NOHET). The main reason for that reduction is the sink for the iodine oxides (I2Ox, where ) included in the heterogeneous chemistry. Over the tropical region, the model overestimated surface IO. This overestimation might be explained by the large modelled inorganic iodine oceanic fluxes in this area. The biggest uncertainty in the inorganic iodine emissions parameterization is the calculation of the iodide concentration in the seawater. Over the subtropics, IO enhancements observed below 4 km are not captured by the model. Some studies suggest that there is abiotic CH3I production when dust contacts seawater containing iodide (Williams et al., 2007; Puentedura et al., 2012). Implementing this chemistry into the model is out of the scope of this paper and further investigation is needed to explain whether the production of CH3I enhances the IO concentration or if there are other missing IO precursors. Gómez Martín et al. (2013) presented an analysis of observations of several gas-phase iodine species made during a field campaign in the eastern Pacific marine boundary layer and suggested that the presence of elevated CH3I does not have a big impact on the IOx concentrations due to CH3I in the MBL having a long lifetime (∼2 days at the Equator). An overestimation of modelled IO in the UT needs further investigation. This overestimation is similar to other modelling studies (Sherwen et al., 2016a). Changing the heterogeneous rate constants (difference between the WRF-DEBROM, WRF-GAMMADV2 and WRF-DIFF runs) has very little impact on IO.
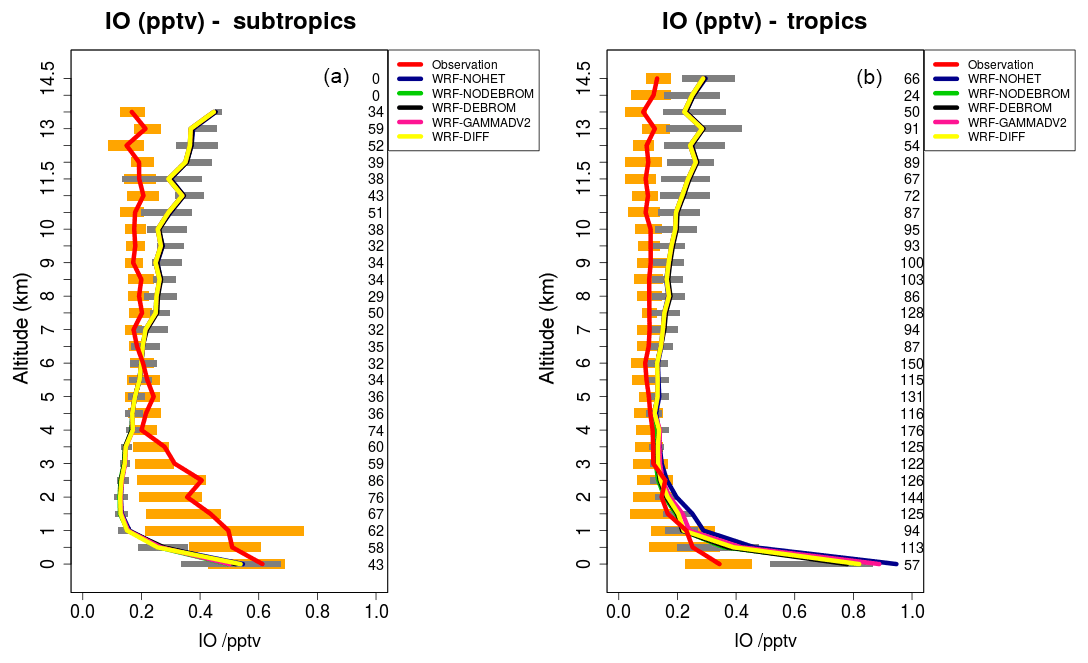
Figure 9Mean vertical profile of IO (pptv) over the subtropics (a) and tropics (b). An average over 16 flights of the TORERO campaign (red line) is compared to the five different WRF-Chem simulations: WRF-NOHET (blue line), WRF-NODEBROM (green line), WRF-DEBROM (black line), WRF-GAMMADV2 (pink line) and WRF-DIFF (yellow line). Orange and grey horizontal bars indicate the 25th–75th quartile interval for the observations of the TORERO campaign and WRF-DEBROM simulation, respectively. Values are considered in 0.5 km bins and the number of aircraft measurement points for each altitude is given on the right side of each plot.
Figure 10 shows the vertical profile distribution for inorganic iodine (Iy) for the three simulations – WRF-NOHET (Fig. 10a, d), WRF-NODEBROM (Fig. 10b, e) and WRF-DEBROM (Fig. 10c, f) – over the subtropics (Fig. 10a–c) and tropics (Fig. 10d–f). Iy is higher in the MBL where it is emitted, especially in the tropical region, with HOI being the dominant species. Concentrations start to decrease above the MBL due to the removal of soluble species by the wet deposition. Unlike Bry, we do not see a big impact on the vertical profile of Iy partitioning with the inclusion of the heterogeneous chemistry. The only differences are the Iy decreases in the surface with the inclusion of the heterogeneous chemistry, due to the removal of the iodine oxides, and the production of more dihalogens in the MBL, especially when debromination is included. Heterogenous iodine reactions (Reactions R11–R19) compete with the photolysis. Iodine species are more readily photolysed, so less is taken up into the aerosol and the impact of heterogeneous chemistry is lower.

Figure 10Regional average vertical partitioning of inorganic iodine (Iy) for the three different simulations – WRF-NOHET (a, d), WRF-NODEBROM (b, e) and WRF-DEBROM (c, f) – during January and February 2012. Panels (a, b, c) are over the subtropical area and panels (d, e, f) over the tropical. Units are in pptv.
5.3 Impact on VOCs
Several VOC oceanic fluxes have been included in the model (see Sect. 3.2.1) as well as the oxidation of VOCs by halogens. In order to see the impact of halogen reactions with the VOCs, average loss rates of all organic compounds due to the Cl and Br families are calculated as percent of the total tropospheric losses over the ocean for the WRF-DEBROM simulation. Bromine accounts for 9.2 % of the oxidation of CH3CHO, 1.4 % of CH2O, 0.8 % of C2H4 and 4.1 % of C3H6. Chlorine accounts for 0.6 % of the oxidation of CH3CHO, 0.3 % of CH2O, 7.7 % of CH3OH, 0.8 % of CH3OOH, 0.6 % of CH3O2, 35.5 % of C2H6 and 10.5 % of C3H8.
A subset of nine flights from the TORERO campaign over the tropics is compared with the WRF-DEBROM, WRF-NOBRVOCS, WRF-NOBRALKE and WRF-NOBRALD simulations for BrO (pptv) in Fig. 11. Comparisons between WRF-DEBROM and WRF-NOBRVOCS simulations show a clear difference (1–4 pptv) throughout the whole troposphere. VOCs play an important role in the MBL regulating the reactive halogens. Without the bromine reactions with the VOCs, BrO concentrations are higher than observed in the MBL. In the middle and upper troposphere, where VOCs emitted from the ocean and forests are transported by convection, the model underestimates the amounts of BrO when these reactions are considered. The results obtained indicate that BrO is highly sensitive to the conversion of reactive bromine into more stable species by these reactions. The partitioning of the products of these reactions (HBr∕Br), and thus the conversion of reactive bromine to more stable species, is highly uncertain (see Sect. 3.2.1) and the results suggest that it might be too effective in these upper layers of the model.
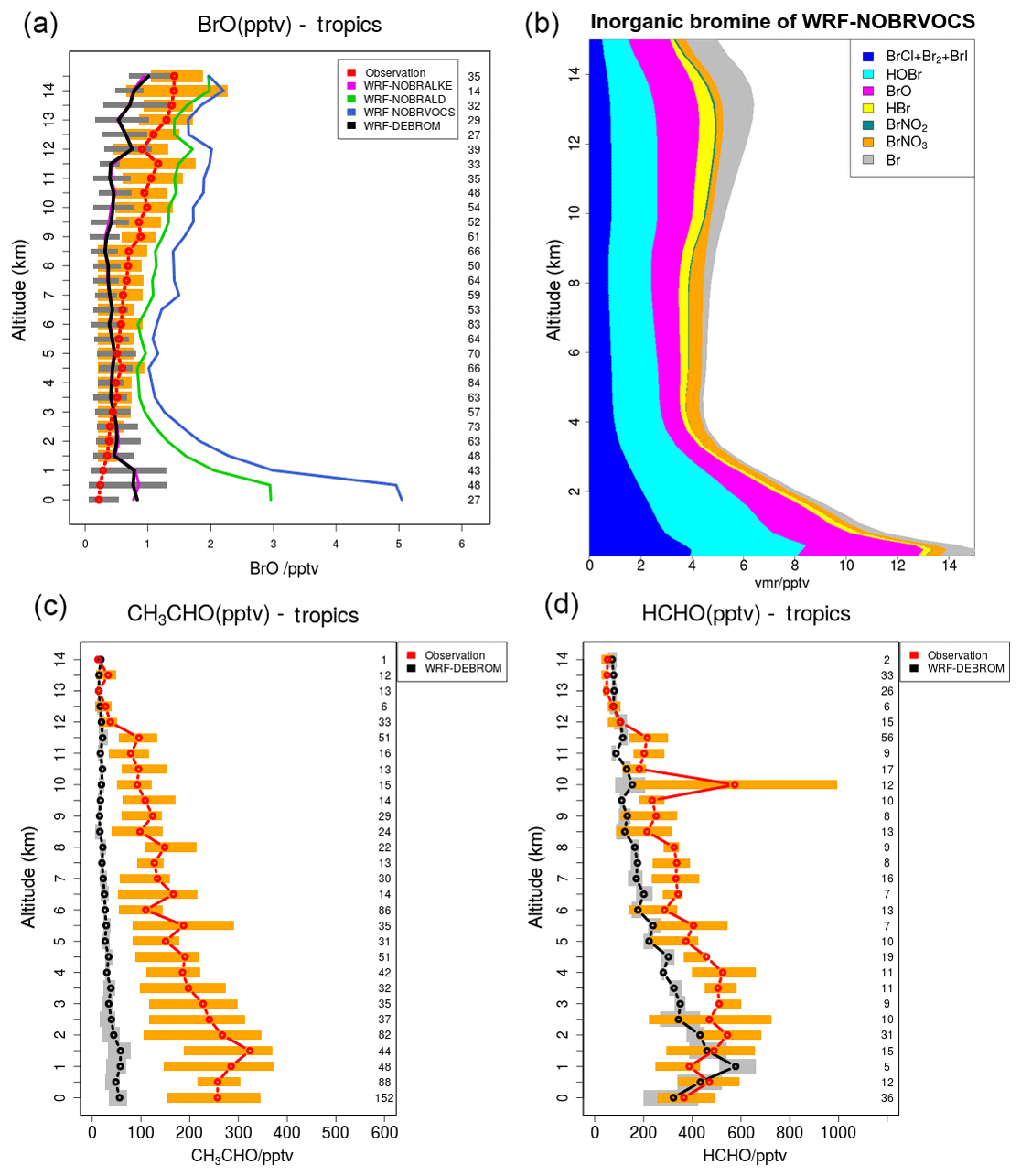
Figure 11(a) Mean vertical profile of BrO (pptv) over the tropics. A subset of nine flights from the TORERO campaign (red line) are compared to the four different WRF-Chem simulations: WRF-NOBRALKE (blue line), WRF-NOBRALD (green line), WRF-NOBRVOCS (blue line) and WRF-DEBROM (black line). (b) Regional average vertical partitioning of inorganic bromine (Bry) for the WRF-NOBRVOCS run over the tropical area during January and February 2012. (c, d) The WRF-DEBROM (black line) simulation is compared with acetaldehyde and formaldehyde TORERO observations for the same nine flights (red line). Orange and grey horizontal bars indicate the 25th–75th quartile interval for the observations of the TORERO campaign and WRF-DEBROM simulation, respectively. Values are considered in 0.5 km bin and the number of points for each altitude is given on the right side of each plot. Units are in pptv.
In order to understand which families of VOCs have a higher impact on the BrO concentrations, the oxidation of alkenes and aldehydes by Br has been studied separately in the WRF-NOBRALKE and WRF-NOBRALD simulations. Differences between WRF-DEBROM and WRF-NOBRALD are seen in the whole troposphere with higher differences up to 2 pptv in the MBL, where the concentrations of both bromine and aldehydes are high. The concentrations of the aldehydes are underestimated by the model, especially for CH3CHO, meaning that BrO-modelled concentrations would be even lower if the modelled concentrations of the aldehydes were reconciled with the observations. The model also seriously underestimates the observed glyoxal mixing ratios. The modelled values are typically ∼1 pptv, whilst the observed values are around 30–40 pptv in the MBL, decreasing to around 5–10 pptv in the upper troposphere (Volkamer et al., 2015; Sinreich et al., 2010). This illustrates that there are large gaps in our understanding of OVOCs in the remote marine atmosphere. Small differences are observed between WRF-DEBROM and WRF-NOBRALKE. However, differences up to 2 pptv between WRF-NOBRVOCS and WRF-NOBRALD are clearly seen especially in the MBL.
These findings suggest that when aldehyde oxidation by Br is included, reactive Br is reduced considerably, thus limiting the amount of alkene oxidation by Br (difference between WRF-DEBROM and WRF-NOBRALKE). However, when the oxidation of aldehydes is included, there is sufficient Bry present for the oxidation of alkenes by Br to have an impact on the BrO (difference between WRF-NOBRALD and WRF-NOBRVOCS).
Figure 11 also shows the vertical profile distribution for inorganic bromine (Bry in pptv) for the WRF-NOBRVOCS run over the tropics (Fig. 11b). When reactions of bromine with VOCs are not included, the amount of Bry increases considerably (difference between WRF-DEBROM and WRF-NOBRVOCS from Figs. 8 and 11), reaching values of 14 pptv in the MBL over the tropics. Moreover, when this chemistry is included, the partitioning of Bry shifts to more stable bromine species such as HBr.
Figure 12 shows the vertical profile distribution for inorganic chlorine (Cly in pptv) for the two simulations – WRF-NOHALVOCS (Fig. 12a, d) and WRF-DEBROM (Fig. 12b, e) – over the subtropics (Fig. 12a–c) and tropics (Fig. 12d–f). Regional average vertical partitioning of reactive chlorine species (Cl*) is also shown (Fig. 12c, f) where Cl* is defined as Cly gases other than HCl. When the VOCs react with Cl (WRF-DEBROM), almost all the inorganic Cl is in the form of HCl (see Fig. 12). When these reactions are not considered (WRF-NOHALVOCS), Cly increases and there is a shift in the partitioning to more reactive chlorine increases, in particular HOCl, but also ClO and the dihalogens.

Figure 12Regional average vertical partitioning of inorganic chlorine (Cly) for the two different simulations – WRF-NOHALVOCS (a, d) and WRF-DEBROM (b, e) – during January and February 2012. Regional average vertical partitioning of reactive chlorine species (Cl*) is also shown (c, f). Cl* is defined as Cly gases other than HCl. Panels (a, b, c) are over the subtropical area and panels (d, e, f) over the tropical. Units are in pptv.
From this, we concluded that VOCs play an important role in the reactive bromine and chlorine concentrations. Therefore, marine emissions of VOCs as well as halogen reactions with VOCs need to be included in models. However, large uncertainties still exist in some of these reactions (see Sect. 3.2.1).
5.4 Impact on O3 and Ox
Figure 13a presents a comparison of modelled O3 from seven simulations (WRF-DEBROM, WRF-GAMMADV2, WRF-DIFF, WRF-NODEBROM, WRF-NOHET, WRF-NOHALVOCS and WRF-NOHAL) sampled along 13 flight tracks with the observed O3 (ppbv). O3 is overestimated when halogens are not included (WRF-NOHAL) except in the upper troposphere. When halogens are included, the model (WRF-DEBROM) is in line with the observations, capturing the O3 gradient and variability of data throughout the troposphere. The average difference between WRF-DEBROM and WRF-NOHAL simulations throughout the troposphere is 7 ppbv. In the MBL, high concentrations of halogens due to ocean emissions destroy O3 and contribute to a negative bias up to 8 ppbv for WRF-DEBROM run. In the middle troposphere, the model results (WRF-DEBROM) improve with the inclusion of halogens, where the average underestimation is reduced from 4.0 to 2.4 ppbv. In the upper troposphere, where the differences between the simulations (WRF-DEBROM and WRF-NOHAL) are mainly driven by the boundary conditions used for each simulation, both simulations underestimate the ozone concentrations. The heterogeneous halogen chemistry has an impact on O3 concentrations where a difference of up to 3 ppbv of O3 is seen between the simulations with and without heterogeneous chemistry (WRF-DEBROM run WRF-NOHET run, respectively) mainly in the MBL. Dividing gamma by 2 (WRF-GAMMADV2) and considering the diffusion limitation (WRF-DIFF) reduces this difference to around 2 ppbv. The modelled O3 is highly sensitive to the inclusion of the reactions of the halogens with the VOCs (WRF-NOHALVOCS) where O3 concentrations are much lower (between 12 and 7 ppbv) than in the WRF-DEBROM run.
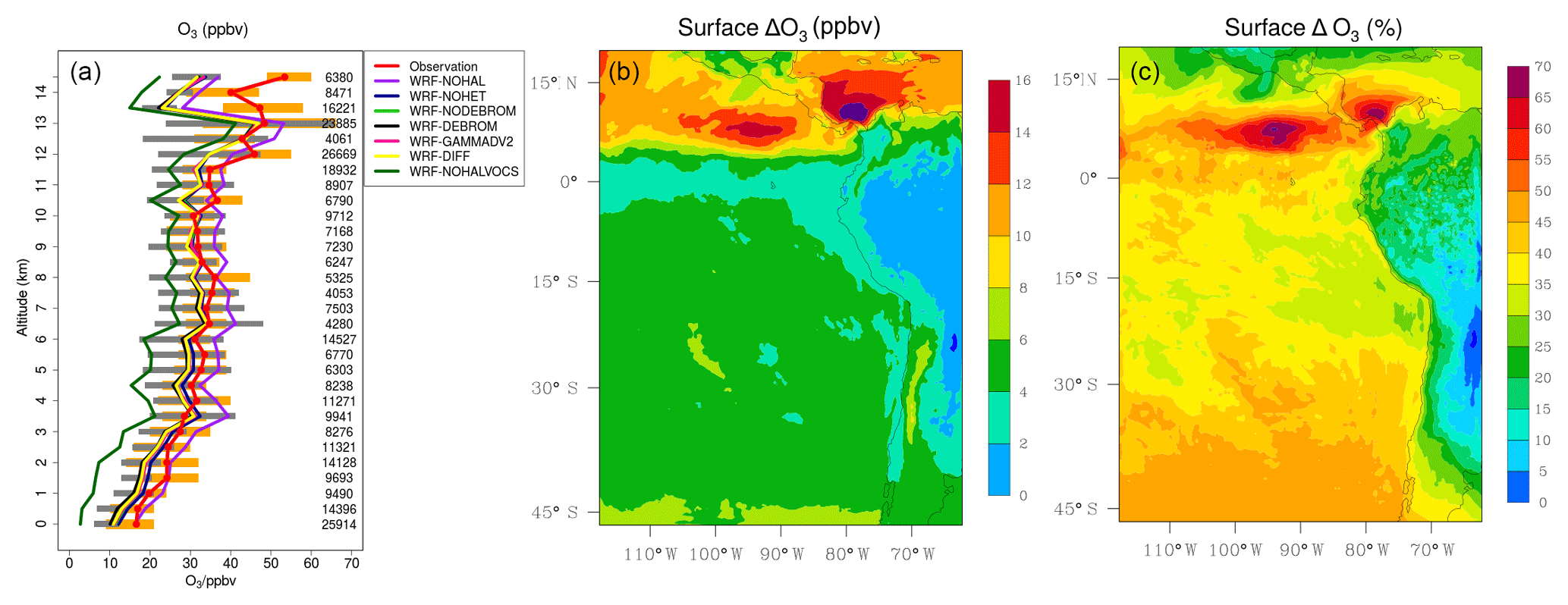
Figure 13(a) Mean vertical profile of O3 (ppbv) over the domain area using 13 flights from the TORERO campaign (red line) compared to the seven different WRF-Chem simulations: WRF-NOHAL (purple line), WRF-NOHET (blue line), WRF-NODEBROM (light green line), WRF-DEBROM (black line), WRF-GAMMADV2 (pink line), WRF-DIFF (yellow line) and WRF-NOHALVOCS (dark green line). Orange and grey horizontal bars indicate the 25th–75th quartile interval for the observations of the TORERO campaign and WRF-DEBROM simulation, respectively. Values are considered in 0.5 km bins and the number of aircraft measurement points for each altitude is given on the right side of each plot. (b, c) Mean O3 difference between the simulation with no halogen chemistry (WRF-NOHAL) and with halogen chemistry (WRF-DEBROM) for January and February 2012. Surface mean bias (ppbv) is shown in panel (b) and surface relative mean bias (%) in panel (c). Relative mean bias (%) is calculated as (WRF-NOHAL − WRF-DEBROM) ∕ WRF-NOHAL ×100.
Figure 13b–c show the regional effects of halogen chemistry on simulated O3 concentrations at the surface. Surface mean bias (ppbv) and relative mean bias (%) between the simulation with no halogen chemistry (WRF-NOHAL) and with halogen chemistry (WRF-DEBROM) for the simulation period are presented. We find that the regional O3 concentrations are reduced by 2–18 ppbv, corresponding to 25 %–70 %, with the inclusion of the halogens. Over the tropics, there is a substantial decrease of O3 (>8 ppbv, >40 %). As we see in Figs. 2 and 3, there are high iodine and bromocarbon emissions and especially large amounts of bromine produced from debromination over this area. These destroy ozone and contribute to higher difference in O3 concentrations in this area.
The odd oxygen Ox is defined as
where X is Cl, Br and I; PAN is peroxyacetyl nitrate, MPAN is methacryloyl peroxynitrate; ONIT is organic nitrate; ONITR is lumped isoprene nitrate; ISOPNO3 is peroxy radical from NO3+ISOP; PBZNIT is peroxybenzoyl nitrate; MBONO3O2 is peroxy radical from NO3 + 2-methyl-3-buten-2-ol.
The Ox loss resulting from reactions with each of the ozone-depleting families (Ox , HOx, NOy, VOCs, Br, Cl and I) is calculated. Note that to calculate the Ox loss due to the Ox depleting family we only consider reactions involving O(3P), O(1D) and O3. The average tropospheric vertical profile of Ox loss grouped by ozone-depleting families for the WRF-DEBROM simulation is given in Fig. 14. Figure 15 summarizes the relative contribution of each halogen family averaged at different altitude intervals for the WRF-DEBROM, WRF-GAMMADV2, WRF-DIFF, WRF-NODEBROM, WRF-NOHET and WRF-NOHALVOCS simulations.
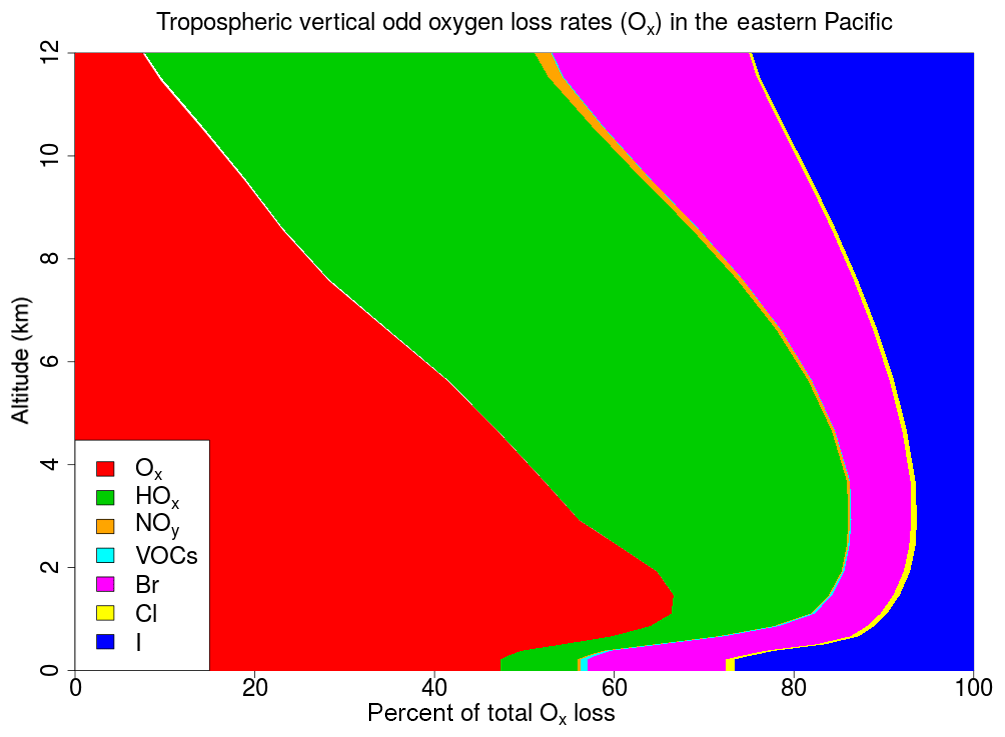
Figure 14Regional average percentage contribution of each ozone-depleting family to the total tropospheric vertical odd oxygen loss (Ox) for the WRF-DEBROM simulation.

Figure 15Integrated odd oxygen loss rates for each O3 depleting halogen family within the troposphere at different altitude levels: MBL (surface–900 hPa), FT (900–350 hPa), UT (350 hPa–tropopause) and troposphere (surface–tropopause) for the WRF-DEBROM, WRF-GAMMADV2, WRF-DIFF, WRF-NODEBROM, WRF-NOHET and WRF-NOHALVOCS simulations.
The regional average Ox percentage loss due to the halogens in our model domain is 34 %, 18 % and 40 % in the MBL (p>900 hPa), free troposphere (FT) ( hPa) and UT (), respectively, for the WRF-DEBROM simulation. The MBL Ox loss is in good agreement with Sherwen et al. (2016b), who reported 33 % and Prados-Roman et al. (2015) reported 31 %. The tropospheric Ox loss due to the BrOx, IOx and ClOx cycles is 14 %, 16 % and 1 % throughout the troposphere, respectively, for the WRF-DEBROM simulation. The very fast catalytic reactions of iodine species make the iodine loss higher than for bromine and chlorine, especially in the MBL for all simulations that include halogen–VOC reactions (19 %–23 %). With the inclusion of the sea-salt debromination, Ox loss due to the bromine is 14 % in the MBL. In the upper troposphere, iodine contributes 18 %–23 % and bromine 14 %–19 % to the total Ox loss. The impact of halogen chemistry on the tropospheric Ox loss is 31 % for the WRF-DEBROM simulation. This value is comparable with other studies that reported 28 % over the tropics (Saiz-Lopez et al., 2015) and 21.4 % at the global scale (Sherwen et al., 2016b). Moreover, our results are in agreement with Wang et al. (2015), who used a box model and concluded that bromine and iodine are responsible for 34 % of the column-integrated loss of tropospheric O3. The tropospheric Ox loss due to the iodine is higher than the box model study of Dix et al. (2013), which concluded that the fraction of iodine-induced ozone loss generally is around 10 %. When comparing different simulations with the WRF-DEBROM run, the biggest difference is seen with the WRF-NOHALVOCS simulation, where around 60 % of Ox is removed by halogens. BrO is much higher when the VOC reactions are not included (see Fig. 11), which explains why the amount of Ox loss by BrOx reactions is much larger (20.5 %). Moreover, the big change though is for the ClOx, which increases from <1 % to 26 %. Cl is very important in the oxidation of the alkanes. When this chemistry is not included the concentrations of Cly increases and there is an impact on the partitioning increasing reactive species (see Fig. 12); hence, the ClOx cycles play an important role in Ox loss. It should be noted that very little is known about the abundance and distribution of Cly, so this is a large uncertainty. Therefore, a large uncertainty in the impact of halogen cycling on the O3 budget is the reactions of halogens with VOCs. In the model runs performed, excluding these reactions doubled the percentage contribution of halogens to Ox loss (i.e. increase it from 31 % to 60 %) in the troposphere. Heterogeneous chemistry (including debromination) has the effect of increasing the Ox loss by halogen cycling from 25 % to 31 % for the whole troposphere (i.e. comparison between WRF-NOHET and WRF-DEBROM runs). For the UT, the equivalent values are 37 % to 40 %, for the FT 13 % to 18 % and for the MBL 23 % to 34 %. Hence, heterogeneous chemistry increases the percentage of the Ox loss that is attributable to the halogens by about 6 % for the troposphere, ranging from 3 % to 11 % depending on the region of the troposphere. Dividing gamma by 2 (WRF-GAMMADV2) and considering the diffusion limitation (WRF-DIFF) reduces the Ox loss in the troposphere by the halogens to 3 % and 2 %, respectively. Note that the gas-phase halogen chemistry makes a bigger contribution of around 25 % (WRF-NOHET run) to the Ox loss for the troposphere ranging from 13 % to 37 % depending on the region of the troposphere. Therefore, the overall impact of the halogen chemistry on Ox loss appears not to be very sensitive to the treatment of the heterogeneous chemistry.
We have presented a regional 3-D tropospheric model that includes halogen chemistry (bromine, iodine and chlorine). A comprehensive description has been provided for the halogen gas-phase chemistry, the heterogeneous recycling reactions in sea-salt aerosol and other particles, reactions of reactive halogens with volatile organic compounds (VOCs) and the oceanic emissions of halocarbons, inorganic iodine and several VOCs. It is the first time that a comprehensive halogen chemistry mechanism has been added into the online WRF-Chem model. Our results provide useful insight regarding the potential importance of reactive halogens in the tropical marine atmosphere and the many uncertainties that remain. Field data from the TORERO campaign (January–February 2012) have been used in the model evaluation.
Two different approaches to compute marine emissions, online and prescribed, for the VSLHs are discussed here. There is an improvement using online fluxes, WRF-DEBROM, in comparison with prescribed fluxes, WRF-ZIS, especially for CH2Br2 and CHBr3 atmospheric concentrations, where the overestimation seen for the model in comparison with ship measurements is decreased for specific periods. During the whole period, an underestimation is seen for both simulations for CHBr3. This underestimation is similar to other modelling studies, which indicates the oceanic fluxes for CHBr3 in this region are not well determined. Results indicate that the input data (especially wind speed and water concentrations) used in this study to calculate marine fluxes underestimate halocarbon concentrations. Large underestimation of CHBr3 and CH3I concentrations throughout the troposphere is seen when compared to the aircraft observations.
Five sensitivity studies are compared in order to understand the impact of the heterogeneous chemistry for bromine and iodine species. Results show that the inclusion of heterogeneous chemistry on marine aerosol has a considerable impact on the Bry partitioning, increasing reactive species like BrO. An increase of Bry is seen in the tropical MBL when debromination processes are included, due to the presence of relatively acidic particles.
The oxidation of alkenes and aldehydes by bromine has been studied in three different sensitivity runs. These runs suggest that reactions of bromine with OVOCs have a big impact on the BrO concentrations. The reactions between Br and aldehydes were found to be particularly important, despite the model underestimating the amount of aldehydes observed in the atmosphere.
The model shows an overall good agreement with the observed IO vertical profile. Higher modelled concentrations in the surface are seen over the tropics, indicating that inorganic iodine emissions might be too high in this area. The model is not able to capture the IO enhancements sometimes seen below 4 km over the subtropical area. Unlike Bry, the Iy partitioning is found to be relatively insensitive to inclusion of the heterogeneous chemistry.
The model captures the O3 vertical profile in the free troposphere. The simulation with halogens (WRF-DEBROM) underestimates the observed O3 values in the MBL, where the oceanic emissions of the halogenated species are higher. Over the tropics, the regional surface O3 concentrations are reduced between 2 and 18 ppbv with the inclusion of the halogens. When heterogeneous chemistry is included, O3 concentrations are reduced by up to 3 ppbv in the MBL. The biggest difference (7–12 ppbv) in O3 values is seen when reactions between Br and Cl and VOCs are not considered (WRF-NOHALVOCS run).
In our simulations, halogens constitute 25 %–60 % of the overall tropospheric Ox loss. This range of values is comparable with other studies. Uncertainties in the heterogeneous chemistry accounted for only a small proportion of this range (25 % to 31 % of the Ox loss). When reactions between Br and Cl with VOCs are not considered (WRF-NOHALVOCS), Ox loss by BrOx, ClOx and IOx cycles is high (60 %), which accounts for the upper limit of the overall range. The model results are clearly very sensitive to the VOCs and this is a large uncertainty given that their emissions over these remote areas are poorly known.
Our model results suggest that including halogen chemistry has a large affect on O3 (7 ppbv) and contributes typically about 25 %–30 % of Ox loss. Including heterogeneous halogen chemistry has a big impact on the Bry partitioning but not on the Iy partitioning. However, it does not have a large impact on the O3 concentrations or the percentage of Ox loss via halogen chemistry. Therefore, although the uncertainties in the heterogeneous chemistry are large, the Ox appears to be relatively insensitive to these uncertainties. However, the modelled O3 and Ox losses are very sensitive to the reactions between the halogens and the VOCs. Excluding these reactions leads to greater amounts of the reactive halogen species (Figs. 11 and 12), less O3 (Fig. 13) and greater Ox loss from halogens (60 %) (Fig. 15), in particular from ClOx. Very little is known about the abundance and distribution of Cly, so this is a large uncertainty. There are also large uncertainties in the degree to which Br is recycled or converted to the more stable product (HBr) in the reactions following Br reactions with the alkenes. Moreover, there is considerable uncertainty in the emissions and distributions of the VOCs in the remote marine atmosphere.
More data are required at the process level from laboratory studies along with field observations of, for example, more Bry, Iy and Cly species, to better constrain the modelled representation of these processes and to verify if halogens really do have such a large impact on Ox in the tropical troposphere. This is important given that the oxidizing capacity of this region of the atmosphere has a large impact on the lifetime of many pollutants including methane, a key greenhouse gas.
The WRF-Chem model code is available from http://www2.mmm.ucar.edu/wrf/users/download/get_sources.html (last access: 1 February 2019), with the specific code used in this study available from the authors upon request (alba.badia.moragas@gmail.com).
The TORERO data are available from the TORERO data archive: https://www.eol.ucar.edu/field_projects/torero, last access: 1 February 2019. The TORERO data set is open for use by the public, subject to the data policy: https://www.eol.ucar.edu/content/torero-data-policy, last access: 1 February 2019.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-3161-2019-supplement.
AB carried out all the model simulations and data analysis, and led the interpretation of the results and prepared the manuscript with contributions from all co-authors. CER contributed to the interpretation of the results and provided extensive comments on manuscript. ARB and AS made several comments and suggestions. RV, TKK, ECA, RSH, LJC and SJA conducted and provided the TORERO measurements. TS provided input data to run the model. RvG provided the initial motivation to this study, designed the research and secured the funding.
The authors declare that they have no conflict of interest.
This work is funded by the National Environmental Research Council (NERC)
grant NE/L005271/1. The authors wish to thank the TORERO team, especially
Barbara Dix and Theodore Konstantinos. TORERO was supported by the NSF under
award AGS-1104104 (PI: Rainer Volkamer). The involvement of the NSF-sponsored
Lower Atmospheric Observing Facilities, managed and operated by the National
Center for Atmospheric Research (NCAR) Earth Observing Laboratory (EOL), is
acknowledged. Rainer Volkamer acknowledges funding from NSF award
AGS-1620530. Lucy J. Carpenter acknowledges support from NERC (award
NE/J00619X/1). The authors also thank Carlos Cuevas, Douglas Lowe, Gordon McFiggans,
Kenjiro Toyota, Peter Braüer, Luke Surl, Deanna Donohoue and Roberto
Sommariva for their constructive suggestions and feedback during this study.
Finally, this work is specially dedicated to the friendship and memory of
Roland von Glasow.
Edited by: Aurélien Dommergue
Reviewed by: two anonymous referees
Andrews, S. J., Hackenberg, S. C., and Carpenter, L. J.: Technical Note: A fully automated purge and trap GC-MS system for quantification of volatile organic compound (VOC) fluxes between the ocean and atmosphere, Ocean Sci., 11, 313–321, https://doi.org/10.5194/os-11-313-2015, 2015. a, b
Apel, E. and UCAR/NCAR: Earth Observing Laboratory, Trace Organic Gas Analyzer (TOGA) for HIAPER, UCAR/NCAR, Earth Observing Laboratory, Tech. rep., https://doi.org/10.5065/D6DF6P9Q, 2016. a
Archer-Nicholls, S., Lowe, D., Utembe, S., Allan, J., Zaveri, R. A., Fast, J. D., Hodnebrog, Ø., Denier van der Gon, H., and McFiggans, G.: Gaseous chemistry and aerosol mechanism developments for version 3.5.1 of the online regional model, WRF-Chem, Geosci. Model Dev., 7, 2557–2579, https://doi.org/10.5194/gmd-7-2557-2014, 2014. a, b, c, d
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a, b, c, d, e, f, g, h
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t, u, v, w, x, y, z, aa, ab, ac, ad, ae, af, ag, ah, ai, aj, ak, al, am, an, ao, ap
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species, Atmos. Chem. Phys., 8, 4141–4496, https://doi.org/10.5194/acp-8-4141-2008, 2008. a
Atlas, E., Pollock, W., Greenberg, J., Heidt, L., and Thompson, A. M.: Alkyl nitrates, nonmethane hydrocarbons, and halocarbon gases over the equatorial Pacific Ocean during SAGA 3, J. Geophys. Res., 98, 16933–16947, https://doi.org/10.1029/93JD01005, 1993. a
Bedjanian, Y., Le Bras, G., and Poulet, G.: Kinetic study of the BrCIO, ICBrO and BrCI2 reactions. Heat of formation of the BrO radical, Chem. Phys. Lett., 266, 233–238, https://doi.org/10.1016/S0009-2614(97)01530-3, 1997. a
Bell, N., Hsu, L., Jacob, D. J., Schultz, M. G., Blake, D. R., Butler, J. H., King, D. B., Lobert, J. M., and Maier-Reimer, E.: Methyl iodide: Atmospheric budget and use as a tracer of marine convection in global models, J. Geophys. Res., 107, 4340, https://doi.org/10.1029/2001JD001151, 2002. a
Bell, R. P.: The Proton in Chemistry, 2nd edn., Cornell University Press, Ithaca, N.Y., 1973. a
Bloss, W. J., Evans, M. J., Lee, J. D., Sommariva, R., Heard, D. E., and Pilling, M. J.: The oxidative capacity of the troposphere: Coupling of field measurements of OH and a global chemistry transport model, Faraday Discuss., 130, 425–436, https://doi.org/10.1039/B419090D, 2005. a
Brasseur, G. P. and Jacob, D. J.: Modeling of Atmospheric Chemistry, Cambridge University Press, Cambridge, https://doi.org/10.1017/9781316544754, 2017. a, b
Burkholder, B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and H., W. P.: Chemical kinetics and photochemical data for use in atmospheric studies, Evaluation number 18, Tech. rep., NASA, Jet Propulsion Laboratory, Pasadena, 2015. a, b, c
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013. a, b, c
Chameides, W. L. and Davis, D. D.: Iodine: Its possible role in tropospheric photochemistry, J. Geophys. Res., 85, 7383–7398, https://doi.org/10.1029/JC085iC12p07383, 1980. a
Class, T. and Ballschmiter, K.: Chemistry of organic traces in air, J. Atmos. Chem., 6, 35–46, https://doi.org/10.1007/BF00048330, 1988. a
Coburn, S., Ortega, I., Thalman, R., Blomquist, B., Fairall, C. W., and Volkamer, R.: Measurements of diurnal variations and eddy covariance (EC) fluxes of glyoxal in the tropical marine boundary layer: description of the Fast LED-CE-DOAS instrument, Atmos. Meas. Tech., 7, 3579–3595, https://doi.org/10.5194/amt-7-3579-2014, 2014. a, b
Dee, D. P., Uppala, S. M., Simmons, A. J., Berrisford, P., Poli, P., Kobayashi, S., Andrae, U., Balmaseda, M. A., Balsamo, G., Bauer, P., Bechtold, P., Beljaars, A. C. M., van de Berg, L., Bidlot, J., Bormann, N., Delsol, C., Dragani, R., Fuentes, M., Geer, A. J., Haimberger, L., Healy, S. B., Hersbach, H., Hólm, E. V., Isaksen, L., Kållberg, P., Köhler, M., Matricardi, M., McNally, A. P., Monge-Sanz, B. M., Morcrette, J.-J., Park, B.-K., Peubey, C., de Rosnay, P., Tavolato, C., Thépaut, J.-N., and Vitart, F.: The ERA-Interim reanalysis: configuration and performance of the data assimilation system, Q. J. Roy. Meteor. Soc., 137, 553–597, https://doi.org/10.1002/qj.828, 2011. a, b, c
Dillon, T. J., Tucceri, M. E., and Crowley, J. N.: Laser induced fluorescence studies of iodine oxide chemistry Part II. The reactions of IO with CH3O2, CF3O2 and O3, Phys. Chem. Chem. Phys., 8, 5185–5198, https://doi.org/10.1039/B611116E, 2006. a
Dix, B., Baidar, S., Bresch, J. F., Hall, S. R., Schmidt, K. S., Wang, S., and Volkamer, R.: Detection of iodine monoxide in the tropical free troposphere, P. Natl. Acad. Sci. USA, 110, 2035–2040, https://doi.org/10.1073/pnas.1212386110, 2013. a
Dix, B., Koenig, T. K., and Volkamer, R.: Parameterization retrieval of trace gas volume mixing ratios from Airborne MAX-DOAS, Atmos. Meas. Tech., 9, 5655–5675, https://doi.org/10.5194/amt-9-5655-2016, 2016. a, b
Emmons, L. K., Walters, S., Hess, P. G., Lamarque, J.-F., Pfister, G. G., Fillmore, D., Granier, C., Guenther, A., Kinnison, D., Laepple, T., Orlando, J., Tie, X., Tyndall, G., Wiedinmyer, C., Baughcum, S. L., and Kloster, S.: Description and evaluation of the Model for Ozone and Related chemical Tracers, version 4 (MOZART-4), Geosci. Model Dev., 3, 43–67, https://doi.org/10.5194/gmd-3-43-2010, 2010. a, b, c
Fernandez, R. P., Salawitch, R. J., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: Bromine partitioning in the tropical tropopause layer: implications for stratospheric injection, Atmos. Chem. Phys., 14, 13391–13410, https://doi.org/10.5194/acp-14-13391-2014, 2014. a
Fischer, E. V., Jacob, D. J., Millet, D. B., Yantosca, R. M., and Mao, J.: The role of the ocean in the global atmospheric budget of acetone, Geophys. Res. Lett., 39, L01807, https://doi.org/10.1029/2011GL050086, 2012. a
Frenzel, A., Scheer, V., Sikorski, R., George, C., Behnke, W., and Zetzsch, C.: Heterogeneous Interconversion Reactions of BrNO2, ClNO2, Br2, and Cl2, J. Phys. Chem. A, 102, 1329–1337, https://doi.org/10.1021/jp973044b, 1998. a
Fuentes, E., Coe, H., Green, D., de Leeuw, G., and McFiggans, G.: On the impacts of phytoplankton-derived organic matter on the properties of the primary marine aerosol – Part 1: Source fluxes, Atmos. Chem. Phys., 10, 9295–9317, https://doi.org/10.5194/acp-10-9295-2010, 2010. a
Gantt, B., Sarwar, G., Xing, J., Simon, H., Schwede, D., Hutzell, W. T., Mathur, R., and Saiz-Lopez, A.: The Impact of Iodide-Mediated Ozone Deposition and Halogen Chemistry on Surface Ozone Concentrations Across the Continental United States, Environ. Sci. Technol., 51, 1458–1466, https://doi.org/10.1021/acs.est.6b03556, 28051851, 2017. a
Gómez Martín, J. C., Spietz, P., and Burrows, J. P.: Spectroscopic studies of the I2/O3 photochemistry: Part 1: Determination of the absolute absorption cross sections of iodine oxides of atmospheric relevance, J. Photoch. Photobio. A, 176, 15–38, https://doi.org/10.1016/j.jphotochem.2005.09.024, 2005. a
Gómez Martín, J. C., Spietz, P., and Burrows, J. P.: Kinetic and Mechanistic Studies of the I2∕O3 Photochemistry, J. Phys. Chem. A, 111, 306–320, https://doi.org/10.1021/jp061186c, 2007. a, b
Gómez Martín, J. C., Mahajan, A. S., Hay, T. D., Prados-Román, C., Ordóñez, C., MacDonald, S. M., Plane, J. M., Sorribas, M., Gil, M., Paredes Mora, J. F., Agama Reyes, M. V., Oram, D. E., Leedham, E., and Saiz-Lopez, A.: Iodine chemistry in the eastern Pacific marine boundary layer, J. Geophys. Res.-Atmos., 118, 887–904, https://doi.org/10.1002/jgrd.50132, 2013. a
Gong, S. L., Barrie, L. A., and Blanchet, J.-P.: Modeling sea-salt aerosols in the atmosphere: 1. Model development, J. Geophys. Res., 102, 3805–3818, https://doi.org/10.1029/96JD02953, 1997. a
Granier, C., Lamarque, J., Mieville, A., Muller, J., Olivier, J., Orlando, J., Peters, J., Petron, G., Tyndall, G., and Wallens, S.: POET, a database of surface emissions of ozone precursors, available at: http://www.aero.jussieu.fr/projet/ACCENT/POET.php (last access: 1 February 2019), 2005. a, b
Grell, G. A. and Dévényi, D. o.: A generalized approach to parameterizing convection combining ensemble and data assimilation techniques, Geophys. Res. Lett., 29, 38-1–38-4, https://doi.org/10.1029/2002GL015311, 2002. a, b
Grell, G. A., Peckham, S. E., Schmitz, R., McKeen, S. A., Frost, G., Skamarock, W. C., and Eder, B.: Fully coupled “online” chemistry within the WRF model, Atmos. Environ., 39, 6957–6975, https://doi.org/10.1016/j.atmosenv.2005.04.027, 2005. a, b
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006. a
Harris, N. R. P., Carpenter, L. J., Lee, J. D., Vaughan, G., Filus, M. T., Jones, R. L., OuYang, B., Pyle, J. A., Robinson, A. D., Andrews, S. J., Lewis, A. C., Minaeian, J., Vaughan, A., Dorsey, J. R., Gallagher, M. W., Breton, M. L., Newton, R., Percival, C. J., Ricketts, H. M. A., Bauguitte, S. J.-B., Nott, G. J., Wellpott, A., Ashfold, M. J., Flemming, J., Butler, R., Palmer, P. I., Kaye, P. H., Stopford, C., Chemel, C., Boesch, H., Humpage, N., Vick, A., MacKenzie, A. R., Hyde, R., Angelov, P., Meneguz, E., and Manning, A. J.: Coordinated Airborne Studies in the Tropics (CAST), B. Am. Meteorol. Soc., 98, 145–162, https://doi.org/10.1175/BAMS-D-14-00290.1, 2017. a
Hossaini, R., Chipperfield, M. P., Monge-Sanz, B. M., Richards, N. A. D., Atlas, E., and Blake, D. R.: Bromoform and dibromomethane in the tropics: a 3-D model study of chemistry and transport, Atmos. Chem. Phys., 10, 719–735, https://doi.org/10.5194/acp-10-719-2010, 2010. a
Hossaini, R., Mantle, H., Chipperfield, M. P., Montzka, S. A., Hamer, P., Ziska, F., Quack, B., Krüger, K., Tegtmeier, S., Atlas, E., Sala, S., Engel, A., Bönisch, H., Keber, T., Oram, D., Mills, G., Ordóñez, C., Saiz-Lopez, A., Warwick, N., Liang, Q., Feng, W., Moore, F., Miller, B. R., Marécal, V., Richards, N. A. D., Dorf, M., and Pfeilsticker, K.: Evaluating global emission inventories of biogenic bromocarbons, Atmos. Chem. Phys., 13, 11819–11838, https://doi.org/10.5194/acp-13-11819-2013, 2013. a
Hossaini, R., Patra, P. K., Leeson, A. A., Krysztofiak, G., Abraham, N. L., Andrews, S. J., Archibald, A. T., Aschmann, J., Atlas, E. L., Belikov, D. A., Bönisch, H., Carpenter, L. J., Dhomse, S., Dorf, M., Engel, A., Feng, W., Fuhlbrügge, S., Griffiths, P. T., Harris, N. R. P., Hommel, R., Keber, T., Krüger, K., Lennartz, S. T., Maksyutov, S., Mantle, H., Mills, G. P., Miller, B., Montzka, S. A., Moore, F., Navarro, M. A., Oram, D. E., Pfeilsticker, K., Pyle, J. A., Quack, B., Robinson, A. D., Saikawa, E., Saiz-Lopez, A., Sala, S., Sinnhuber, B.-M., Taguchi, S., Tegtmeier, S., Lidster, R. T., Wilson, C., and Ziska, F.: A multi-model intercomparison of halogenated very short-lived substances (TransCom-VSLS): linking oceanic emissions and tropospheric transport for a reconciled estimate of the stratospheric source gas injection of bromine, Atmos. Chem. Phys., 16, 9163–9187, https://doi.org/10.5194/acp-16-9163-2016, 2016. a
Jacob, D. J.: Heterogeneous chemistry and tropospheric ozone, Atmos. Environ., 34, 2131–2159, 2000. a, b
Jensen, E. J., Pfister, L., Jordan, D. E., Bui, T. V., Ueyama, R., Singh, H. B., Thornberry, T. D., Rollins, A. W., Gao, R.-S., Fahey, D. W., Rosenlof, K. H., Elkins, J. W., Diskin, G. S., DiGangi, J. P., Lawson, R. P., Woods, S., Atlas, E. L., Rodriguez, M. A. N., Wofsy, S. C., Pittman, J., Bardeen, C. G., Toon, O. B., Kindel, B. C., Newman, P. A., McGill, M. J., Hlavka, D. L., Lait, L. R., Schoeberl, M. R., Bergman, J. W., Selkirk, H. B., Alexander, M. J., Kim, J.-E., Lim, B. H., Stutz, J., and Pfeilsticker, K.: The NASA Airborne Tropical Tropopause Experiment: High-Altitude Aircraft Measurements in the Tropical Western Pacific, Bulletin of the American Meteorol. Soc., 98, 129–143, https://doi.org/10.1175/BAMS-D-14-00263.1, 2017. a
Johnson, M. T.: A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas, Ocean Sci., 6, 913–932, https://doi.org/10.5194/os-6-913-2010, 2010. a, b
Kaltsoyannis, N. and Plane, J. M. C.: Quantum chemical calculations on a selection of iodine-containing species (IO, OIO, INO3, (IO)2, I2O3, I2O4 and I2O5) of importance in the atmosphere, Phys. Chem. Chem. Phys., 10, 1723–1733, https://doi.org/10.1039/B715687C, 2008. a, b
Keene, W. C., Sander, R., Pszenny, A. A., Vogt, R., Crutzen, P. J., and Galloway, J. N.: Aerosol pH in the marine boundary layer: A review and model evaluation, J. Aerosol Sci., 29, 339–356, https://doi.org/10.1016/S0021-8502(97)10011-8, 1998. a
Knote, C., Hodzic, A., Jimenez, J. L., Volkamer, R., Orlando, J. J., Baidar, S., Brioude, J., Fast, J., Gentner, D. R., Goldstein, A. H., Hayes, P. L., Knighton, W. B., Oetjen, H., Setyan, A., Stark, H., Thalman, R., Tyndall, G., Washenfelder, R., Waxman, E., and Zhang, Q.: Simulation of semi-explicit mechanisms of SOA formation from glyoxal in aerosol in a 3-D model, Atmos. Chem. Phys., 14, 6213–6239, https://doi.org/10.5194/acp-14-6213-2014, 2014. a, b, c
Koenig, T. K., Volkamer, R., Baidar, S., Dix, B., Wang, S., Anderson, D. C., Salawitch, R. J., Wales, P. A., Cuevas, C. A., Fernandez, R. P., Saiz-Lopez, A., Evans, M. J., Sherwen, T., Jacob, D. J., Schmidt, J., Kinnison, D., Lamarque, J.-F., Apel, E. C., Bresch, J. C., Campos, T., Flocke, F. M., Hall, S. R., Honomichl, S. B., Hornbrook, R., Jensen, J. B., Lueb, R., Montzka, D. D., Pan, L. L., Reeves, J. M., Schauffler, S. M., Ullmann, K., Weinheimer, A. J., Atlas, E. L., Donets, V., Navarro, M. A., Riemer, D., Blake, N. J., Chen, D., Huey, L. G., Tanner, D. J., Hanisco, T. F., and Wolfe, G. M.: BrO and inferred Bry profiles over the western Pacific: relevance of inorganic bromine sources and a Bry minimum in the aged tropical tropopause layer, Atmos. Chem. Phys., 17, 15245–15270, https://doi.org/10.5194/acp-17-15245-2017, 2017. a, b
Lawson, S. J., Selleck, P. W., Galbally, I. E., Keywood, M. D., Harvey, M. J., Lerot, C., Helmig, D., and Ristovski, Z.: Seasonal in situ observations of glyoxal and methylglyoxal over the temperate oceans of the Southern Hemisphere, Atmos. Chem. Phys., 15, 223–240, https://doi.org/10.5194/acp-15-223-2015, 2015. a
Lennartz, S. T., Krysztofiak, G., Marandino, C. A., Sinnhuber, B.-M., Tegtmeier, S., Ziska, F., Hossaini, R., Krüger, K., Montzka, S. A., Atlas, E., Oram, D. E., Keber, T., Bönisch, H., and Quack, B.: Modelling marine emissions and atmospheric distributions of halocarbons and dimethyl sulfide: the influence of prescribed water concentration vs. prescribed emissions, Atmos. Chem. Phys., 15, 11753–11772, https://doi.org/10.5194/acp-15-11753-2015, 2015. a, b, c
Li, Q., Zhang, L., Wang, T., Tham, Y. J., Ahmadov, R., Xue, L., Zhang, Q., and Zheng, J.: Impacts of heterogeneous uptake of dinitrogen pentoxide and chlorine activation on ozone and reactive nitrogen partitioning: improvement and application of the WRF-Chem model in southern China, Atmos. Chem. Phys., 16, 14875–14890, https://doi.org/10.5194/acp-16-14875-2016, 2016. a
Liss, P. S. and Slater, P. G.: Flux of Gases across the Air-Sea Interface, Nature, 247, 181–184, https://doi.org/10.1038/247181a0, 1974. a, b
Long, M. S., Keene, W. C., Easter, R. C., Sander, R., Liu, X., Kerkweg, A., and Erickson, D.: Sensitivity of tropospheric chemical composition to halogen-radical chemistry using a fully coupled size-resolved multiphase chemistry–global climate system: halogen distributions, aerosol composition, and sensitivity of climate-relevant gases, Atmos. Chem. Phys., 14, 3397–3425, https://doi.org/10.5194/acp-14-3397-2014, 2014. a
Lowe, D., Topping, D., and McFiggans, G.: Modelling multi-phase halogen chemistry in the remote marine boundary layer: investigation of the influence of aerosol size resolution on predicted gas- and condensed-phase chemistry, Atmos. Chem. Phys., 9, 4559–4573, https://doi.org/10.5194/acp-9-4559-2009, 2009. a
Lowe, D., Archer-Nicholls, S., Morgan, W., Allan, J., Utembe, S., Ouyang, B., Aruffo, E., Le Breton, M., Zaveri, R. A., Di Carlo, P., Percival, C., Coe, H., Jones, R., and McFiggans, G.: WRF-Chem model predictions of the regional impacts of N2O5 heterogeneous processes on night-time chemistry over north-western Europe, Atmos. Chem. Phys., 15, 1385–1409, https://doi.org/10.5194/acp-15-1385-2015, 2015. a, b
MacDonald, S. M., Gómez Martín, J. C., Chance, R., Warriner, S., Saiz-Lopez, A., Carpenter, L. J., and Plane, J. M. C.: A laboratory characterisation of inorganic iodine emissions from the sea surface: dependence on oceanic variables and parameterisation for global modelling, Atmos. Chem. Phys., 14, 5841–5852, https://doi.org/10.5194/acp-14-5841-2014, 2014. a
Mahajan, A. S., Gómez Martín, J. C., Hay, T. D., Royer, S.-J., Yvon-Lewis, S., Liu, Y., Hu, L., Prados-Roman, C., Ordóñez, C., Plane, J. M. C., and Saiz-Lopez, A.: Latitudinal distribution of reactive iodine in the Eastern Pacific and its link to open ocean sources, Atmos. Chem. Phys., 12, 11609–11617, https://doi.org/10.5194/acp-12-11609-2012, 2012. a
Mahajan, A. S., Prados-Roman, C., Hay, T. D., Lampel, J., Pöhler, D., Großmann, K., Tschritter, J., Frieß, U., Platt, U., Johnston, P., Kreher, K., Wittrock, F., Burrows, J. P., Plane, J. M., and Saiz-Lopez, A.: Glyoxal observations in the global marine boundary layer, J. Geophys. Res.-Atmos., 119, 6160–6169, https://doi.org/10.1002/2013JD021388, 2014. a
McFiggans, G., Plane, J. M. C., Allan, B. J., Carpenter, L. J., Coe, H., and O'Dowd, C.: A modeling study of iodine chemistry in the marine boundary layer, J. Geophys. Res., 105, 14371–14385, https://doi.org/10.1029/1999JD901187, 2000. a, b
Millet, D. B., Guenther, A., Siegel, D. A., Nelson, N. B., Singh, H. B., de Gouw, J. A., Warneke, C., Williams, J., Eerdekens, G., Sinha, V., Karl, T., Flocke, F., Apel, E., Riemer, D. D., Palmer, P. I., and Barkley, M.: Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations, Atmos. Chem. Phys., 10, 3405–3425, https://doi.org/10.5194/acp-10-3405-2010, 2010. a
Muñiz-Unamunzaga, M., Borge, R., Sarwar, G., Gantt, B., de la Paz, D., Cuevas, C. A., and Saiz-Lopez, A.: The influence of ocean halogen and sulfur emissions in the air quality of a coastal megacity: The case of Los Angeles, Sci. Total Environ., 610–611, 1536–1545, https://doi.org/10.1016/j.scitotenv.2017.06.098, 2018. a
Myriokefalitakis, S., Vrekoussis, M., Tsigaridis, K., Wittrock, F., Richter, A., Brühl, C., Volkamer, R., Burrows, J. P., and Kanakidou, M.: The influence of natural and anthropogenic secondary sources on the glyoxal global distribution, Atmos. Chem. Phys., 8, 4965-4981, https://doi.org/10.5194/acp-8-4965-2008, 2008. a
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012. a, b, c, d, e, f
Orlando, J. J. and Tyndall, G. S.: Rate Coefficients for the Thermal Decomposition of BrONO2 and the Heat of Formation of BrONO2, J. Phys. Chem., 100, 19398–19405, https://doi.org/10.1021/jp9620274, 1996. a
Pan, L. L., Atlas, E. L., Salawitch, R. J., Honomichl, S. B., Bresch, J. F., Randel, W. J., Apel, E. C., Hornbrook, R. S., Weinheimer, A. J., Anderson, D. C., Andrews, S. J., Baidar, S., Beaton, S. P., Campos, T. L., Carpenter, L. J., Chen, D., Dix, B., Donets, V., Hall, S. R., Hanisco, T. F., Homeyer, C. R., Huey, L. G., Jensen, J. B., Kaser, L., Kinnison, D. E., Koenig, T. K., Lamarque, J.-F., Liu, C., Luo, J., Luo, Z. J., Montzka, D. D., Nicely, J. M., Pierce, R. B., Riemer, D. D., Robinson, T., Romashkin, P., Saiz-Lopez, A., Schauffler, S., Shieh, O., Stell, M. H., Ullmann, K., Vaughan, G., Volkamer, R., and Wolfe, G.: The Convective Transport of Active Species in the Tropics (CONTRAST) Experiment, B. Am. Meteorol. Soc., 98, 106–128, https://doi.org/10.1175/BAMS-D-14-00272.1, 2017. a
Parrella, J. P., Jacob, D. J., Liang, Q., Zhang, Y., Mickley, L. J., Miller, B., Evans, M. J., Yang, X., Pyle, J. A., Theys, N., and Van Roozendael, M.: Tropospheric bromine chemistry: implications for present and pre-industrial ozone and mercury, Atmos. Chem. Phys., 12, 6723–6740, https://doi.org/10.5194/acp-12-6723-2012, 2012. a
Prados-Roman, C., Cuevas, C. A., Fernandez, R. P., Kinnison, D. E., Lamarque, J.-F., and Saiz-Lopez, A.: A negative feedback between anthropogenic ozone pollution and enhanced ocean emissions of iodine, Atmos. Chem. Phys., 15, 2215–2224, https://doi.org/10.5194/acp-15-2215-2015, 2015. a
Puentedura, O., Gil, M., Saiz-Lopez, A., Hay, T., Navarro-Comas, M., Gómez-Pelaez, A., Cuevas, E., Iglesias, J., and Gomez, L.: Iodine monoxide in the north subtropical free troposphere, Atmos. Chem. Phys., 12, 4909–4921, https://doi.org/10.5194/acp-12-4909-2012, 2012. a
Riffault, V., Bedjanian, Y., and Poulet, G.: Kinetic and mechanistic study of the reactions of OH with IBr and HOI, J. Photoch. Photobio. A, 176, 155–161, https://doi.org/10.1016/j.jphotochem.2005.09.002, 2005. a
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/C2CS35208G, 2012. a
Saiz-Lopez, A., Plane, J. M. C., McFiggans, G., Williams, P. I., Ball, S. M., Bitter, M., Jones, R. L., Hongwei, C., and Hoffmann, T.: Modelling molecular iodine emissions in a coastal marine environment: the link to new particle formation, Atmos. Chem. Phys., 6, 883–895, https://doi.org/10.5194/acp-6-883-2006, 2006. a
Saiz-Lopez, A., Lamarque, J.-F., Kinnison, D. E., Tilmes, S., Ordóñez, C., Orlando, J. J., Conley, A. J., Plane, J. M. C., Mahajan, A. S., Sousa Santos, G., Atlas, E. L., Blake, D. R., Sander, S. P., Schauffler, S., Thompson, A. M., and Brasseur, G.: Estimating the climate significance of halogen-driven ozone loss in the tropical marine troposphere, Atmos. Chem. Phys., 12, 3939–3949, https://doi.org/10.5194/acp-12-3939-2012, 2012, 2012a. a, b
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., von Glasow, R., Gómez Martín, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric Chemistry of Iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012b. a
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014. a
Saiz-Lopez, A., Baidar, S., Cuevas, C. A., Koenig, T. K., Fernandez, R. P., Dix, B., Kinnison, D. E., Lamarque, J.-F., Rodriguez-Lloveras, X., Campos, T. L., and Volkamer, R.: Injection of iodine to the stratosphere, Geophys. Res. Lett., 42, 6852–6859, https://doi.org/10.1002/2015GL064796, 2015. a, b, c, d, e
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015. a, b, c, d, e, f, g, h, i, j, k, l, m
Sander, R. and Crutzen, P. J.: Model study indicating halogen activation and ozone destruction in polluted air masses transported to the sea, J. Geophys. Res., 101, 9121–9138, https://doi.org/10.1029/95JD03793, 1996. a
Sander, R., Baumgaertner, A., Gromov, S., Harder, H., Jöckel, P., Kerkweg, A., Kubistin, D., Regelin, E., Riede, H., Sandu, A., Taraborrelli, D., Tost, H., and Xie, Z.-Q.: The atmospheric chemistry box model CAABA/MECCA-3.0, Geosci. Model Dev., 4, 373–380, https://doi.org/10.5194/gmd-4-373-2011, 2011. a
Sander, S. P., Golden, D. M., Kurylo, M. J., Moortgat, G. K., Wine, P. H., Ravishankara, A. R., Kolb, C. E., Molina, M. J., Finlayson-Pitts, B. J., Huie, R. E., and Orkin, V. L.: Chemical kinetics and photochemical data for use in atmospheric studies, Evaluation number 15, Tech. rep., NASA, Jet Propulsion Laboratory, Pasadena, 2006. a, b, c, d, e, f, g, h, i
Sander, S. P., Friedl, R. R., Abbatt, J. P. D., Barker, J. R., Burkholder, J. B., Golden, D. M., Kolb, C. E., Kurylo, M. J., Moortgat, G. K., Wine, P. H., Huie, R. E., and Orkin, V. L.: Chemical kinetics and photochemical data for use in atmospheric studies, Evaluation Number 17, Tech. rep., NASA, Jet Propulsion Laboratory, Pasadena, 2011. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t, u, v
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 41, 4050–4058, https://doi.org/10.1002/2014GL059962, 2014. a
Sarwar, G., Gantt, B., Schwede, D., Foley, K., Mathur, R., and Saiz-Lopez, A.: Impact of Enhanced Ozone Deposition and Halogen Chemistry on Tropospheric Ozone over the Northern Hemisphere, Environ. Sci. Technol., 49, 9203–9211, https://doi.org/10.1021/acs.est.5b01657, 2015. a
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 121, 11819–11835, https://doi.org/10.1002/2015JD024229, 2016. a, b, c, d
Schweitzer, F., Mirabel, P., and George, C.: Uptake of Hydrogen Halides by Water Droplets, J. Phys. Chem. A, 104, 72–76, https://doi.org/10.1021/jp992621o, 2000. a
Sherwen, T., Evans, M. J., Carpenter, L. J., Andrews, S. J., Lidster, R. T., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys., 16, 1161–1186, https://doi.org/10.5194/acp-16-1161-2016, 2016a. a, b, c
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016b. a, b, c, d, e, f, g, h
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and von Glasow, R.: Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015. a
Sinreich, R., Coburn, S., Dix, B., and Volkamer, R.: Ship-based detection of glyoxal over the remote tropical Pacific Ocean, Atmos. Chem. Phys., 10, 11359–11371, https://doi.org/10.5194/acp-10-11359-2010, 2010. a, b
Sommariva, R. and von Glasow, R.: Multiphase Halogen Chemistry in the Tropical Atlantic Ocean, Environ. Sci. Technol., 46, 10429–10437, https://doi.org/10.1021/es300209f, 2012. a, b, c, d
Sommariva, R., Bloss, W., and von Glasow, R.: Uncertainties in gas-phase atmospheric iodine chemistry, Atmos. Environ., 57, 219–232, https://doi.org/10.1016/j.atmosenv.2012.04.032, 2012. a
Surl, L., Donohoue, D., Aiuppa, A., Bobrowski, N., and von Glasow, R.: Quantification of the depletion of ozone in the plume of Mount Etna, Atmos. Chem. Phys., 15, 2613–2628, https://doi.org/10.5194/acp-15-2613-2015, 2015. a
Tie, X., Madronich, S., Walters, S., Zhang, R., Rasch, P., and Collins, W.: Effect of clouds on photolysis and oxidants in the troposphere, J. Geophys. Res., 108, 4642, https://doi.org/10.1029/2003JD003659, 2003. a, b
Toyota, K., Kanaya, Y., Takahashi, M., and Akimoto, H.: A box model study on photochemical interactions between VOCs and reactive halogen species in the marine boundary layer, Atmos. Chem. Phys., 4, 1961–1987, https://doi.org/10.5194/acp-4-1961-2004, 2004. a, b, c, d, e, f, g
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2−O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015. a, b, c, d, e, f
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer 1. Cloud-free MBL, J. Geophys. Res., 107, 4341, https://doi.org/10.1029/2001JD000942, 2002a. a
von Glasow, R., Sander, R., Bott, A., and Crutzen, P. J.: Modeling halogen chemistry in the marine boundary layer. 1. Cloud-free MBL, J. Geophys. Res., 107, 4341, https://doi.org/10.1029/2001JD000942, 2002b. a
von Glasow, R., von Kuhlmann, R., Lawrence, M. G., Platt, U., and Crutzen, P. J.: Impact of reactive bromine chemistry in the troposphere, Atmos. Chem. Phys., 4, 2481–2497, https://doi.org/10.5194/acp-4-2481-2004, 2004. a, b, c
Wales, P. A., Salawitch, R. J., Nicely, J. M., Anderson, D. C., Canty, T. P., Baidar, S., Dix, B., Koenig, T. K., Volkamer, R., Chen, D., Huey, L. G., Tanner, D. J., Cuevas, C. A., Fernandez, R. P., Kinnison, D. E., Lamarque, J.-F., Saiz-Lopez, A., Atlas, E. L., Hall, S. R., Navarro, M. A., Pan, L. L., Schauffler, S. M., Stell, M., Tilmes, S., Ullmann, K., Weinheimer, A. J., Akiyoshi, H., Chipperfield, M. P., Deushi, M., Dhomse, S. S., Feng, W., Graf, P., Hossaini, R., Jöckel, P., Mancini, E., Michou, M., Morgenstern, O., Oman, L. D., Pitari, G., Plummer, D. A., Revell, L. E., Rozanov, E., Saint-Martin, D., Schofield, R., Stenke, A., Stone, K. A., Visioni, D., Yamashita, Y., and Zeng, G.: Stratospheric Injection of Brominated Very Short-Lived Substances: Aircraft Observations in the Western Pacific and Representation in Global Models, J. Geophys. Res.-Atmos., 123, 5690–5719, https://doi.org/10.1029/2017JD027978, 2018. a, b, c
Wang, S., Schmidt, J. A., Baidar, S., Coburn, S., Dix, B., Koenig, T. K., Apel, E., Bowdalo, D., Campos, T. L., Eloranta, E., Evans, M. J., DiGangi, J. P., Zondlo, M. A., Gao, R.-S., Haggerty, J. A., Hall, S. R., Hornbrook, R. S., Jacob, D., Morley, B., Pierce, B., Reeves, M., Romashkin, P., ter Schure, A., and Volkamer, R.: Active and widespread halogen chemistry in the tropical and subtropical free troposphere, P. Natl. Acad. Sci. USA, 112, 9281–9286, https://doi.org/10.1073/pnas.1505142112, 2015. a, b, c, d, e
Wesely, M.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ., 23, 1293–1304, https://doi.org/10.1016/0004-6981(89)90153-4, 1989. a, b
Williams, J., Gros, V., Atlas, E., Maciejczyk, K., Batsaikhan, A., Schöler, H. F., Forster, C., Quack, B., Yassaa, N., Sander, R., and Van Dingenen, R.: Possible evidence for a connection between methyl iodide emissions and Saharan dust, J. Geophys. Res., 112, D07302, https://doi.org/10.1029/2005JD006702, 2007. a
Yang, M., Beale, R., Liss, P., Johnson, M., Blomquist, B., and Nightingale, P.: Air–sea fluxes of oxygenated volatile organic compounds across the Atlantic Ocean, Atmos. Chem. Phys., 14, 7499–7517, https://doi.org/10.5194/acp-14-7499-2014, 2014. a, b
Yang, X., Cox, R. A., Warwick, N. J., Pyle, J. A., Carver, G. D., O'Connor, F. M., and Savage, N. H.: Tropospheric bromine chemistry and its impacts on ozone: A model study, J. Geophys. Res., 110, D23311, https://doi.org/10.1029/2005JD006244, 2005. a, b
Zaveri, R. A., Easter, R. C., Fast, J. D., and Peters, L. K.: Model for Simulating Aerosol Interactions and Chemistry (MOSAIC), J. Geophys. Res., 113, D13204, https://doi.org/10.1029/2007JD008782, 2008. a, b, c
Ziska, F., Quack, B., Abrahamsson, K., Archer, S. D., Atlas, E., Bell, T., Butler, J. H., Carpenter, L. J., Jones, C. E., Harris, N. R. P., Hepach, H., Heumann, K. G., Hughes, C., Kuss, J., Krüger, K., Liss, P., Moore, R. M., Orlikowska, A., Raimund, S., Reeves, C. E., Reifenhäuser, W., Robinson, A. D., Schall, C., Tanhua, T., Tegtmeier, S., Turner, S., Wang, L., Wallace, D., Williams, J., Yamamoto, H., Yvon-Lewis, S., and Yokouchi, Y.: Global sea-to-air flux climatology for bromoform, dibromomethane and methyl iodide, Atmos. Chem. Phys., 13, 8915–8934, https://doi.org/10.5194/acp-13-8915-2013, 2013. a, b, c, d, e, f, g, h