the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 01 Mar 2019
| 01 Mar 2019
Effect of salt seed particle surface area, composition and phase on secondary organic aerosol mass yields in oxidation flow reactors
Erik Ahlberg
Axel Eriksson
William H. Brune
Pontus Roldin
Birgitta Svenningsson
Atmospheric particulate water is ubiquitous, affecting particle transport and uptake of gases. Yet, research on the effect of water on secondary organic aerosol (SOA) mass yields is not consistent. In this study, the SOA mass yields of an α-pinene and m-xylene mixture, at a concentration of 60 µg m−3, were examined using an oxidation flow reactor operated at a relative humidity (RH) of 60 % and a residence time of 160 s. Wet or dried ammonium sulfate and ammonium nitrate seed particles were used. By varying the amount of seed particle surface area, the underestimation of SOA formation induced by the short residence time in flow reactors was confirmed. Starting at a SOA mass concentration of ∼5 µg m−3, the maximum yield increased by a factor of ∼2 with dry seed particles and on average a factor of 3.2 with wet seed particles. Hence, wet particles increased the SOA mass yield by ∼60 % compared to the dry experiment. Maximum yield in the reactor was achieved using a surface area concentration of ∼1600 µm2 cm−3. This corresponded to a condensational lifetime of 20 s for low-volatility organics. The O:C ratio of SOA on wet ammonium sulfate was significantly higher than when using ammonium nitrate or dry ammonium sulfate seed particles, probably due to differences in heterogeneous chemistry.
- Article
(1291 KB) - Full-text XML
-
Supplement
(311 KB) - BibTeX
- EndNote





The atmospheric aerosol is a dynamic mixture of organic and inorganic species. A large fraction of the organic aerosol is formed as a result of atmospheric processing of volatile organic compounds (VOCs), with products condensing onto pre-existing particles, forming secondary organic aerosol (SOA) (Hallquist et al., 2009). The partitioning of semivolatile organic species from gas to particles can be either adsorptive or absorptive depending on the chemical composition and phase of the particles (Pankow, 1994). Despite the complexity of the atmospheric aerosol, SOA mass yields (mass of formed particles divided by the mass of VOCs reacted) have traditionally been parameterized in models using simplified and relatively dry laboratory experiments.
Although not always measured, water is ubiquitous in the atmospheric aerosol, influencing particle size, scattering, transportation/deposition and uptake of gases, ultimately affecting both climate and health effects of particles (Pilinis et al., 1989; Nguyen et al., 2016). The aerosol liquid water content at subsaturation of water vapor depends on the relative humidity (RH), dry particle chemical composition and size. The most abundant inorganic aerosol compounds in submicron aerosol particles are the salts, ammonium sulfate (AS) and ammonium nitrate (AN), which are mostly of anthropogenic origin (e.g., Pöschl, 2005; Zhang et al., 2007). A portion of the water in particles can be regarded as anthropogenic water since both nitrate and sulfate generally increase particle hygroscopicity (Carlton and Turpin, 2013; Hodas et al., 2014), which in turn facilitates SOA formation of water-soluble organic compounds. This particle formation pathway is believed to be especially important for isoprene SOA, since isoprene's first-generation oxidation products are relatively small molecules with high-saturation vapor pressures compared to the oxidation products of other common SOA precursors (Carlton et al., 2009; Ervens et al., 2011; Sareen et al., 2017). Also, the electrolyte solution of ammonium sulfate and ammonium nitrate is acidic since ammonia is a weak base, which increases the reactive uptake of several SOA species (Jang et al., 2002; Gao et al., 2004).
SOA research has been substantial during the last two decades, and the effects of relative humidity and aerosol liquid water on particle yields have been investigated in numerous studies. From partitioning theory, it can be shown (Seinfeld et al., 2001; Hallquist et al., 2009; Pankow, 2010) that there should be a clear SOA mass yield dependence with RH, especially at low precursor concentrations, if an activity coefficient of 1 is assumed. Hennigan et al. (2008) showed that in an urban region dominated by biogenic emissions, partitioning to liquid water may be a significant contributor to SOA mass. However, adding water to laboratory oxidation experiments complicates the interpretation, since both gas- (Jonsson et al., 2006; Warren et al., 2009) and particle-phase (Ervens et al., 2011) chemistry may change. Furthermore, the yield variation with different RH can be NOx dependent (Ervens et al., 2011). For isoprene, both particulate water and acidity are believed to have strong effects on the SOA yield (Surratt et al., 2007; Carlton et al., 2009). Wong et al. (2015) showed that wet ammonium sulfate seed particles resulted in 60 % more isoprene SOA being formed compared to a system with dry seed particles at the same RH. However, laboratory studies using other SOA precursors are somewhat inconsistent. Prisle et al. (2010) saw no influence of RH (up to near 100 %) on α-pinene ozonolysis SOA yields with ammonium sulfate seeds. Cocker et al. (2001a), investigating the same system but using dry or wet seed particles, found that mass yields varied little with RH but decreased if the seed particles were wet. In similar studies, the SOA yields of m-xylene, 1,3,5-trimethylbenzene (Cocker et al., 2001b) and toluene (Edney et al., 2000) have been found to be unaffected by the aerosol liquid water content. Lu et al. (2009) found no effect on m-xylene SOA yields with wet or dry neutral seed particles, while the yield was increased with dry acidic seed particles. In contrast, other studies have found that higher RH significantly increases the SOA mass yield of toluene and xylenes (Kamens et al., 2011; Zhou et al., 2011). Also, more recently, Stirnweis et al. (2017) assessed the influence of NOx and RH on α-pinene SOA with different seed particles and concluded that particulate water significantly increases the organic mass yields. Further, Faust et al. (2017) found 13 % and 19 % increases in SOA yield from α-pinene and toluene, respectively, when SOA was formed on wet salt particles. It can be difficult to compare the results of different SOA oxidation experiments, since the conditions used are rarely the same. SOA yields can depend on, e.g., temperature, NOx concentrations, precursor concentration, oxidant exposure and type, seed particle concentration and composition.
In the following work, we report SOA yields from a mixture of α-pinene and m-xylene oxidized in a potential aerosol mass (PAM) oxidation flow reactor (OFR) (Kang et al., 2007; Lambe et al., 2011a), in the presence of wet or dry ammonium sulfate and ammonium nitrate seed particles. In contrast to many other studies looking into the effect of particulate water on SOA mass yields, the RH was held constant at 60 %, while the seed particles were either dried below an RH of 10 % or kept in their droplet state. Previous studies have shown that SOA produced in the reactor is similar to that produced in traditionally used smog chambers (Bruns et al., 2015; Lambe et al., 2015). The reactor can produce a more oxidized aerosol, which is strongly linked to the hygroscopicity of organic aerosols (Pang et al., 2006; Chang et al., 2010; Lambe et al., 2011b). Due to the fast processing in flow reactors, several studies have discussed the potential problem with low condensation sinks resulting in lower yields (Lambe et al., 2015; Palm et al., 2016; Ahlberg et al., 2017; Jathar et al., 2017; Simonen et al., 2017; Zhao et al., 2018). This effect was systematically investigated during the course of the experiments by using different seed particle concentrations.
2.1 Experimental setup
All experiments consisted of introducing a constant flow of SOA precursors with a varying concentration of seed particles into an oxidation flow reactor. The experimental setup is shown in Fig. 1. Seed particles were formed from atomization of a ∼1 g L−1 solution of ammonium sulfate (Sigma Aldrich, ≥99 %) or ammonium nitrate (Sigma Aldrich, ≥99.5 %) in molecular-grade water. The size distribution of seed particles had a maximum volume concentration at a mobility diameter between ∼150 and 200 nm. Although the molality of the solutions and pressure in the atomizer were similar in all experiments, the output number size distributions were not identical. The number of particles per volume unit, as measured by the scanning mobility particle sizer (SMPS), increased in the following order: dry AS > wet AS > dry AN > wet AN. The RH in the reactor was chosen between the deliquescence and efflorescence points of ammonium sulfate (Seinfeld and Pandis, 2006) and sodium chloride (experiments not reported here due to instrument failure) so that the hysteresis effect could be used to alternate between aqueous and dry particles. Ammonium nitrate has a reported deliquescence RH of 61.8 % (Tang and Munkelwitz, 1993) but efflorescence is not observed (Svenningsson, 1997; Lightstone et al., 2000). Seed particle mass concentrations of ∼0–100 µg m−3 were achieved by pulling a varying flow (0–0.7 L min−1) from the atomizer through the reactor.
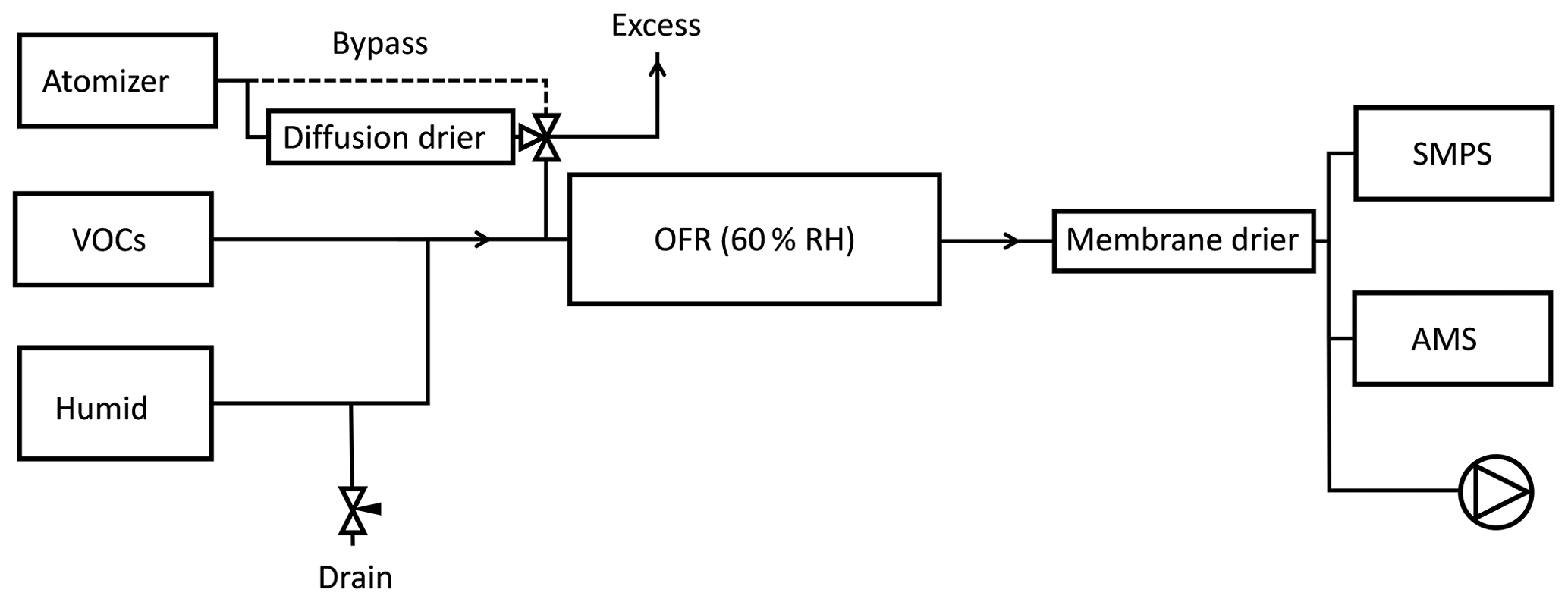
Figure 1Experimental setup. Seed particles were either dried or maintained in a liquid droplet. By changing the drain flow, the flow from the atomizer was varied without perturbing the VOC concentrations in the reactor. Total flow through the reactor was 5 L min−1 and RH was kept constant at 60 % by varying the RH of the humid flow.
VOCs were introduced into the reactor using a diffusion system with thin capillaries, described in Ahlberg et al. (2017). VOCs were chosen to get a mix of biogenic (α-pinene) and anthropogenic (m-xylene) SOA. The flow of VOCs was held constant throughout an experiment. A relatively low SOA mass concentration of ∼5 µg m−3 without seed particles was aimed for so that the nucleated particles would not be the dominant condensation sink. The VOC concentration was determined after the experiments by the liquid weight loss during 4 weeks. During these weeks, the evaporation rate declined, probably due to VOC oxidation or VOC condensation inside the capillaries. Therefore, the values of the first weighing were used. If the decline during the first week, prior to the first weighing, was the same as consecutive weeks, it would result in an overestimation of 13 % in the summed yield. Since the timescales of an experiment (∼8 h) were much shorter, oxidation or condensation inside capillaries is not expected to have taken place. The total concentration of VOCs at the reactor inlet was calculated to be 60 µg m−3 (5.2 ppb α-pinene and 6.7 ppb m-xylene); hence, the SOA mass yield with no seeds was ∼8 %, in agreement with previous measurements of the same mixture (Ahlberg et al., 2017), albeit at slightly different VOC ratios.
2.2 SOA formation
SOA was produced using a PAM oxidation flow reactor, which has been extensively used in laboratory and field measurements (https://sites.google.com/site/pamwiki/, last access: 24 January 2019). The reactor, which is a 13.2 L horizontal aluminium cylinder with passivated walls, produces very high concentrations of ozone and hydroxyl radicals (OH) from UV lights mounted inside (Kang et al., 2007; Lambe et al., 2011a). In recent years, measurements and modeling have significantly advanced the knowledge of the reactor and best practices during use have been developed (Ortega et al., 2013; Li et al., 2015; Peng et al., 2015, 2016; Palm et al., 2016). Briefly, the reactor should not be used with too-high OH reactivity (defined as the concentration of reactant multiplied by the OH reaction rate) input, since OH may be suppressed. The same problem may arise if the OH exposure is low due to low lamp voltage or low absolute humidity. In this work, the flow was set to 5 L min−1 and only one lamp was used. Lamp voltage was adjusted to reach an O3 concentration of 2.7–3 ppm. RH in the reactor was held constant at 60 % by proportional–integral–derivative (PID) regulation of a humidified flow. With these settings, the OH exposure, calibrated offline using 10 ppb of SO2 (for the detailed procedure, see Lambe et al., 2011a), was 7×1011 molec. cm−3 s, with an experimental uncertainty (1σ) of 5 %. The total OH reactivity was 9.4 s−1, which is not believed to have induced significant OH suppression (Peng et al., 2015, 2016). The temperature increase inside the reactor due to the lamp was measured to 1–2 ∘C prior to the experiments using a thermocouple inserted into the reactor. With an RH of 60 % at 22 ∘C (room temperature), RH inside the reactor is expected to be 53 %, which is above the efflorescence point of ammonium sulfate at the same temperature.
Particle losses depend on reactor settings and particle sizes but are generally lower than 10 % on a mass basis (Martinsson et al., 2015; Karjalainen et al., 2016; Ortega et al., 2016; Palm et al., 2016). Ortega et al. (2013) found that most of the particle losses take place at the inlet of the reactor. In this work, losses at the inlet are not of importance, since we look at SOA formed inside the reactor only. Palm et al. (2016) constructed a model for the fate of low-volatility organic compounds (LVOCs) in the reactor, in which four loss terms are competing: condensation onto particles, wall loss, fragmentation (assumed after reacting with OH five times) and outflow from the reactor. The model was compared with the SOA mass yields at different seed concentrations. For the reactor settings used, the modeled LVOC fate as a function of seed particle area is shown in Fig. 2. For model sensitivity tests and uncertainties, the reader is referred to the original paper.
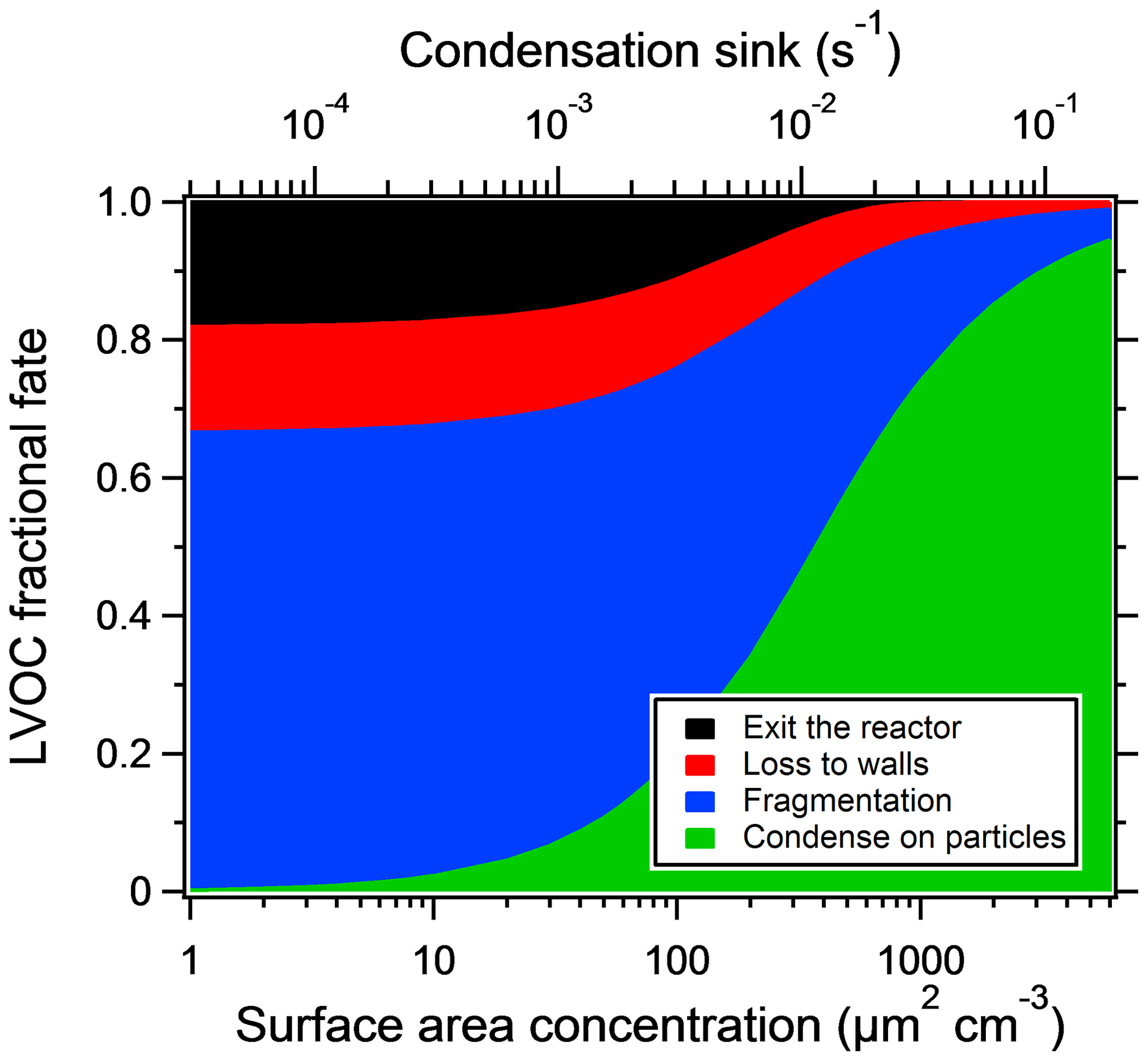
Figure 2The fractional fate of LVOCs as a function of particle surface area concentration, using the model of Palm et al. (2016), with the same OH reaction rate ( cm3 molec.−1 s−1) and assuming fragmentation after reaction with OH five times. For reactor settings, see text.
2.3 Particle measurements
After oxidative aging in the reactor, the aerosol was dried below 30 % RH before size distribution and mass-based chemical composition was measured using a SMPS (Wiedensohler et al., 2012) and an Aerodyne high-resolution time-of-flight aerosol mass spectrometer (AMS; DeCarlo et al., 2006), respectively. The SMPS consisted of a custom-built differential mobility analyzer (DMA) and a TSI condensation particle counter (CPC) (model 3010). Silica gel driers decreased the RH of the sheath flow to below 10 %. The DMA voltages were calibrated prior to experiments and the number size concentration was checked using polystyrene latex (PSL) spheres. The AMS was calibrated using size-selected ammonium nitrate and ammonium sulfate particles.
2.4 Experimental procedure and data analysis
An overview of the experiments can be seen in Table 1. Before experiments, the reactor was run with the lamps on without seeds or VOCs until the volume concentration was below 0.2 µm3 cm−3, as measured by the SMPS. Before adding VOCs, two to five concentration levels of pure seed particles were measured to be able to parameterize organic impurities from the atomizer as a function of salt concentration. Despite using ultrapure water and zero air (Linde, GT30000), up to 6 % of the total mass of the seed particles were organic impurities and scaled roughly in a linear way with salt ion concentration. Pieber et al. (2016) found interferences in the m∕z 44 signal from reactions in the ionization region facilitated by inorganic salt particles. However, in our experiments, the m∕z 44 signal was only ∼15 % of the total organic impurity signal. Before calculating SOA yields, the impurities were removed from the organic signal using the linear relationship with the salt ions. After adding a constant concentration of VOCs, SOA was measured at five to eight different seed particle concentrations. For each seed type, experiments without seed particles were performed to get a base-level yield. This level was relatively stable between experiments, at 5.0±0.5 µg m−3 (1σ). Using data from a similar mixture in Ahlberg et al. (2017), the difference in base level corresponds to a difference in VOC concentrations of ±2 µg m−3 (±3.3 %). During the dry ammonium nitrate experiment, the base level drifted from 5 µg m−3 at the start to 6 µg m−3 at the end of the day. For this experiment, a time-adjusted base level was implemented. The adjustment translated to an increase in the SOA mass yield by at most 15 % for the lowest seed concentration, to 1 % at the highest seed concentration.
Table 1For each of the four experiments, the table shows dry seed surface area concentration, collection efficiency (CE) of the AMS for salts without SOA, initial SOA without seed particles (two replicates where available) and maximum SOA concentrations (with seed particles).

NA – not available.
The SMPS was used to determine the particle number size distribution and total particle volume and area concentrations. To calculate the input dry seed particle surface area concentration, a parameterization from pure salt measurements was made as a function of either sulfate or nitrate concentration as measured by the AMS. The size distribution was also used to calculate the condensation sink (CS) (Pirjola et al., 1999). However, we use area concentration when presenting our data, since this is a measurement more often used and in these experiments scaled linearly with CS.
AMS data were evaluated using standard AMS analysis programs (Squirrel v1.57 and Pika v1.16). Standard changes to the fragmentation table and high-resolution spectra were made, including corrections for zero-air CO2 concentrations and removal of organic peaks overlapping with either air or salt peaks (m∕z 14, 16, 32, 48, 64 for AS and 14, 16, 30, 46 for AN). The ammonium nitrate calibration of the AMS was used to calculate the relative ionization efficiency (RIE) of ammonium, which was subsequently used to calculate the RIE of sulfate. The RIE of ammonium was 4, which is the default value of the AMS, but the RIE of sulfate, at 1.96, was significantly higher than the default value of 1.2. Although this means that the measured sulfate mass was decreased during analysis, it does not affect the seed area calculations since the parameterization and seed mass changes cancel each other. For organics, the default RIE of 1.4 was used. To evaluate the AMS collection efficiency (CE) of the different experiments, the volume concentration as measured by the SMPS was multiplied by particle density calculated from the AMS chemical composition. A density of 1.4 g cm−3 was used for SOA from previous parameterizations of a similar mixture (Ahlberg et al., 2017). The collection efficiencies used as a function of SOA mass fraction can be seen in Fig. S1 in the Supplement. CEs for pure salts are listed in Table 1. CE of both wet and dry ammonium sulfate increased with increasing SOA mass fraction, likely due to decreased bounce. For ammonium nitrate, CE was roughly constant around 1. Pure SOA had a CE of 0.63±0.03 (1σ). The relatively low CE of SOA may not only be a bounce effect, since these particles were significantly smaller, with a number mode around 20 nm and a volume mode around 40–50 nm in mobility diameter, which to a higher degree are lost in the aerodynamic lens inlet of the AMS. An example of the volume size distributions during an experiment is shown in Fig. S2. The “improved ambient” parameterization was used to calculate elemental ratios (Canagaratna et al., 2015). However, the organic portion of the particles consisted of both SOA and salt impurities. To calculate the O:C and H:C ratios of SOA only, the elemental ratios of the impurities only and their fraction of the total organics were used. This correction increased SOA O:C by ∼8 % and decreased H:C by ∼0.5 % for AS, while for AN the change in O:C was below 1 % and H:C decreased 1 %–2 %.
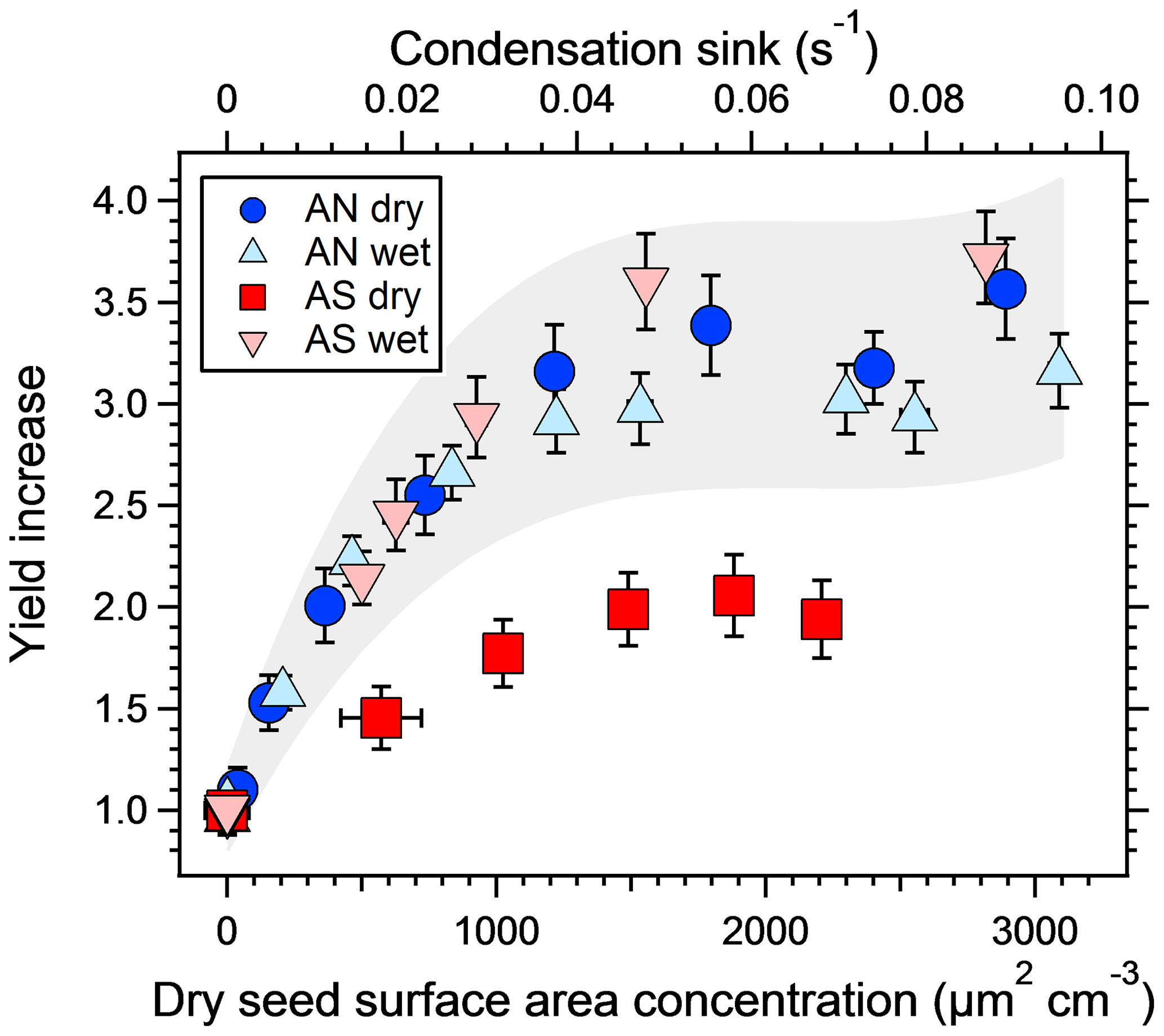
Figure 3The increase in SOA yield from a mixture of m-xylene and α-pinene at different dry salt seed surface areas, normalized to the yield from experiments with no seed particles. The corresponding condensation sink is shown on the top axis. Error bars denote 1σ of the measurements. The grey area represents ±20 % of the three experiments where the seeds are not effloresced to illustrate the expected repeatability of the experiments and the fact that the dry ammonium sulfate results are the only ones falling outside of this range.
The SOA mass yield is defined as the amount of SOA formed divided by the amount of VOCs reacted. Assuming constant OH concentration, with our settings the VOC lifetimes for reaction with OH are short compared to the residence time (4.3 and 16.2 s for α-pinene and m-xylene, respectively). Therefore, we assume that all VOCs have reacted. However, comparing yields only would give a skewed result, since small differences in base SOA level between the experiments (Table 1) give large differences in yield. Instead, we compared the ratio of base-level SOA mass to SOA mass at different seed particle concentrations, which is equal to the relative increase in yield (unitless). This cancels out the VOC concentrations from the calculations. The uncertainty in the yield increase was calculated from error propagation of the standard deviations of the measurements. The fractional uncertainty with this method was between 6 % and 10 %. However, this only reflects the precision in one experiment. Although all flow, pressure and OFR settings were checked repeatedly, a variation larger than single experiment standard deviation is expected since the setup was highly sensitive to small perturbations. No experiment was repeated fully, but SOA levels without seeds were tested twice per day. Replicates of SOA yield with seeds gave a fractional error of 14 % compared to previous values. Therefore, a conservative expected repeatability of the experiments is within 20 %.
Figure 3 shows the increase in yield as a function of dry salt seed particle surface area concentration. In all experiments, the yield increased significantly with seed particle surface area, confirming previous findings (Lambe et al., 2015; Palm et al., 2016; Ahlberg et al., 2017; Jathar et al., 2017; Zhao et al., 2018). The extent of yield underestimation in these experiments can be calculated by normalizing the yield with the maximum yield. Doing this, all experiments follow the same trend with seed surface area seen in Fig. 4. Also seen in Fig. 4 is the modeled bias (fraction condensed on particles), following a similar trend but slightly lower, since the experimental data only consider the condensation sink of the seed particles, while in the model the total sink (seed plus SOA) is taken into account. The yield error decreases up to a condensation sink of ∼0.05 s−1, corresponding to a seed surface area of ∼1600 µm2 cm−3. Above this value, the LVOC fate model (Fig. 2) also indicates a slower increase. However, while the LVOC model yield continues to increase with increased seed area concentration, the increase in the experimental yields levels off. This could be due to increased fragmentation losses from heterogeneous oxidation, since a larger portion of the SOA will be exposed with higher seed particle concentration. The condensation sink at which the data level off corresponds to a lifetime (τCS) of 20 s, which is similar to the residence time of the reactor short circuit (Lambe et al., 2011a; Ahlberg et al., 2017).
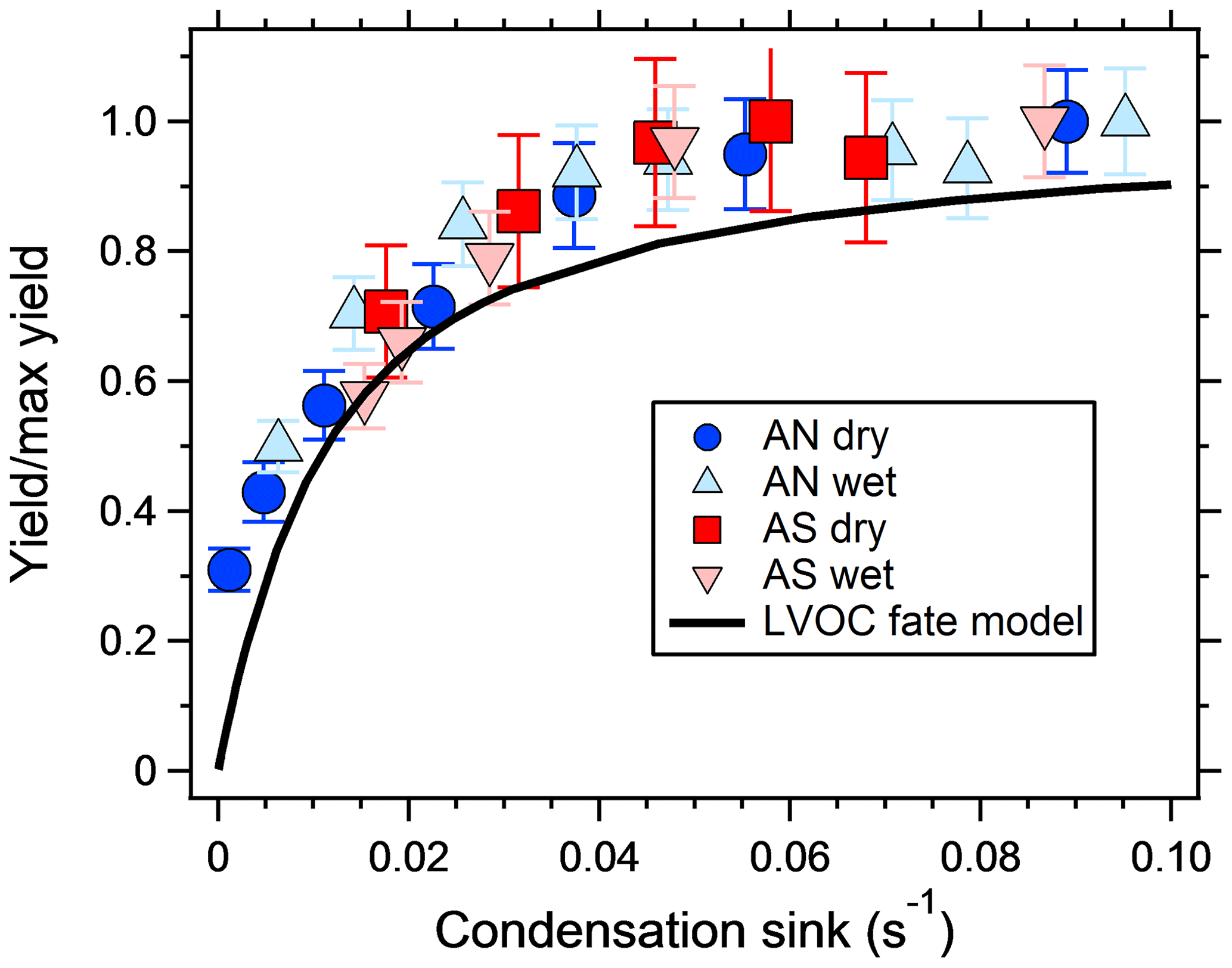
Figure 4SOA yield increase, from a mixture of m-xylene and α-pinene, normalized by the maximum SOA yield increase and plotted against the dried seed particle condensation sink. Error bars denote the propagated uncertainties (1σ). The black line shows the fraction of condensed LVOCs according the model of Palm et al. (2016).
The results suggest that previous measurements using similar reactors have underestimated the yield at low condensation sinks. Because the error is larger at low yields, the yield curves will have a steeper increase and reach a constant yield at lower mass concentrations. Applying corrections to previous reactor experiments relying on nucleated particles as the only condensation sink is not trivial, since the condensation sink varies with time in the reactor. However, at a similar SOA condensation sink as that used in this study (0.022 s−1), the yields should increase by a factor of 2–3 compared to when no seed is used (Fig. 3). Given the shape of the yield bias in Fig. 4, at lower concentrations (and condensation sinks) the increase should be even higher. According to the LVOC fate model a 3-fold increase in yield (yield/max yield of 0.33) corresponds to a condensation sink of ∼0.006 s−1, suggesting the effective CS at this mass concentration is approximately one-third (0.006/0.022) of the reactor outlet CS in nucleation experiments. At half of that condensation sink (0.003 s−1), the model predicts a 5-fold increase in yield, and at 1∕10 (0.0006 s−1) the increase could be as high as a factor of 45. In Fig. 5, we used the LVOC fate model to recalculate the yields of Ahlberg et al. (2017). The inverse of the fraction condensed on particles at one-third of the experimental CS was multiplied with the measured yields and mass concentrations. Because both x and y values increase (both SOA mass and yields change with the same factor), the change from the measurements is not as dramatic as when only looking at the absolute yield increase or if yields were plotted against reacted VOCs. At 10 µg m−3, the increase in yield is estimated, from linear regression between adjacent data points in Fig. 5, at 67 %, 80 %, 24 % and 94 % for α-pinene, m-xylene, myrcene and isoprene, respectively, with the differences arising from differences in the size distribution of each SOA precursor. It is likely that our assumption that the effective CS is one-third of the output underestimates the yield at low mass concentrations and overestimates the yield at high mass concentrations, since higher VOC concentrations also produce a condensation sink faster than a low-input VOC concentration. Although the calculations may be an oversimplification, it is clear that the SOA mass yields at low mass concentrations are biased low and that seed particles have a big impact in OFR experiments.
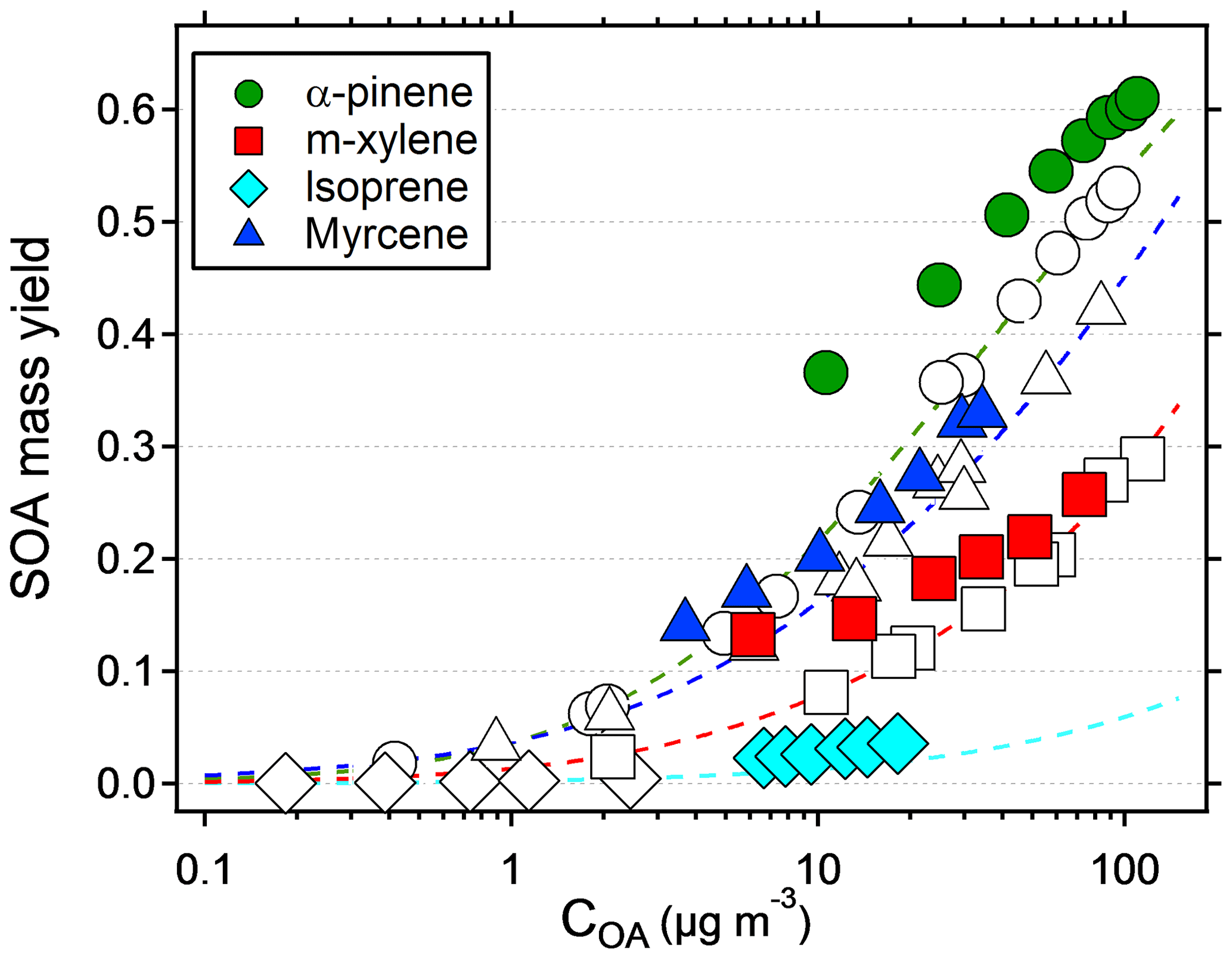
Figure 5Recalculated SOA mass yields from Ahlberg et al. (2017) as a function of organic aerosol mass concentration (COA). Assuming an effective condensation sink of one-third of the reactor output, the inverse of the fraction condensed in the LVOC fate model from Palm et al. (2016) was multiplied with the measured yields. Colored symbols are recalculated values, and original values are represented with the same symbol but no color. Dashed lines show the VBS models constructed from the original data.
The second main result, also seen in Fig. 3, is that the increase in yield with increased seed concentration is lower for the dry ammonium sulfate experiment. Since ammonium nitrate does not effloresce (Svenningsson, 1997; Lightstone et al., 2000), it is likely that both wet and dry AN adjusted to the RH of the reactor, and thus these experiments are essentially the same. Also, while the yield increase is highest for wet AS, this experiment had a lower base-level SOA mass concentration, making it harder to rank the three wet experiments. The grey area in the figure represents ±20 % of the three experiments where the seed particles did not effloresce and is added to emphasize the similarities between them. Dry ammonium sulfate was the only crystalline particle, with a yield bias of a factor of ∼2, while the other three experiments were similar given the experimental uncertainty, with a yield bias factor of 2.9–3.5. Hence, wet seed particles increased the yield by 45 %–75 %, with an average of 60 %, compared to the dry seed experiment.
The difference between wet and dry experiments can be due to either differences in partitioning, reactive uptake or both. Julin et al. (2014) showed that the mass accommodation coefficient of several different organic molecules is unity, regardless of the particle-phase state. In their study, the condensed and gaseous phases consisted of the same molecules, which is not the case in the present study. However, as soon as a layer of organics has condensed on the crystalline AS particles, the mass accommodation for uptake at the surface should approach unity. If an aqueous phase is to increase the yield by equilibrium partitioning, the organic molecules need to be water soluble and SOA mass concentration needs to be low enough to retain an appreciable amount in the gas phase (Hallquist et al., 2009; Pankow, 2010). Several studies have shown that organic aerosol particles may undergo liquid–liquid phase separation (Song et al., 2012b; You et al., 2012; Zuend and Seinfeld, 2012). However, the water solubility of organic molecules increases with decreasing molecular weight and increasing polarity (O:C) (Varutbangkul et al., 2006; Massoli et al., 2010; Duplissy et al., 2011), both of which are favored in OFR experiments compare to smog chambers. It has been shown (Song et al., 2012a, b) that liquid–liquid phase separation rarely occurs at O:C ratios higher than 0.7 in systems containing organics, water and AS. In the present study, O:C was always higher than 0.7, which is seen in Fig. 6 that shows the elemental ratios in Van Krevelen space as measured by the AMS.
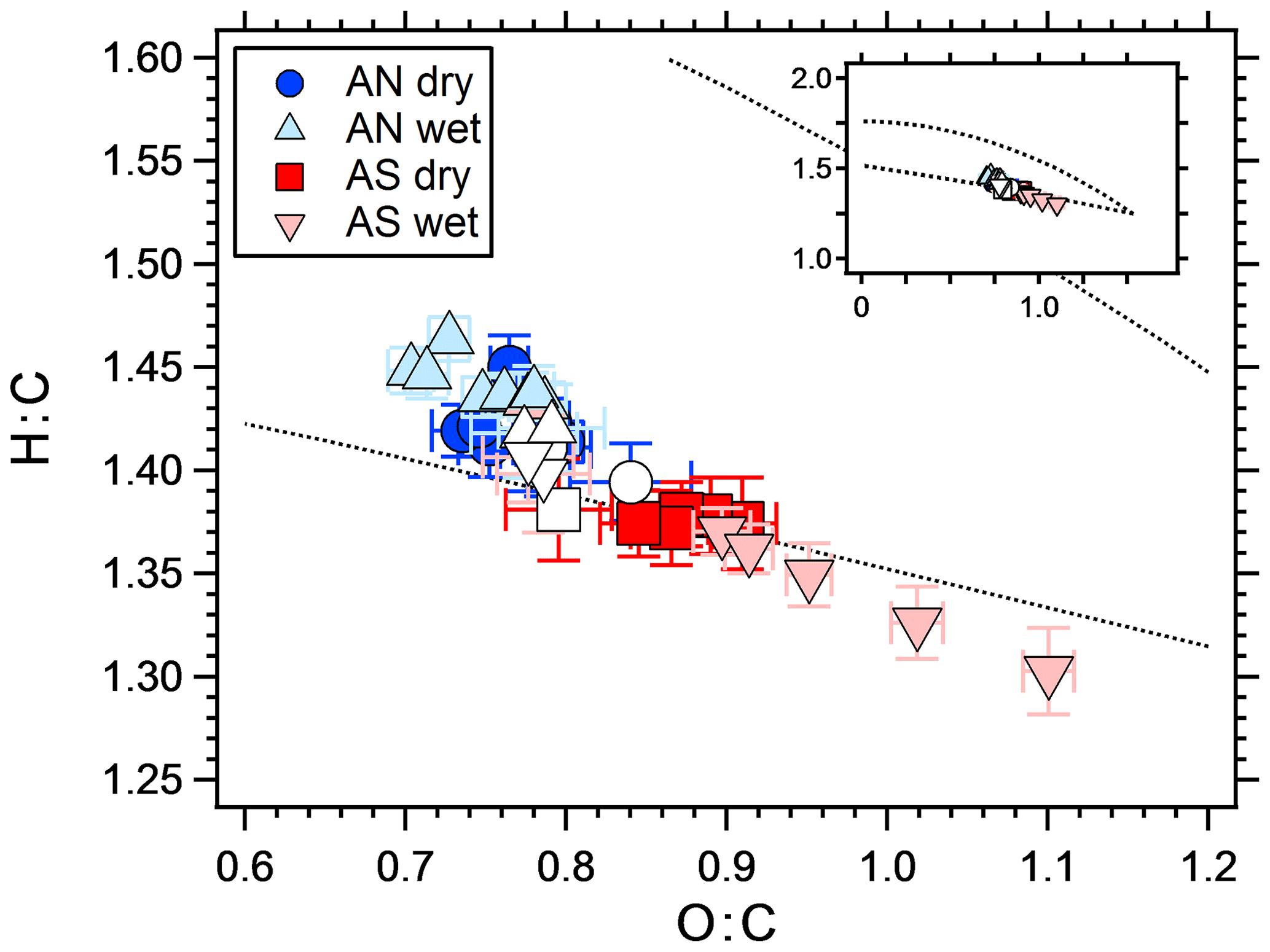
Figure 6Van Krevelen diagram showing the elemental ratios of SOA from a mixture of m-xylene and α-pinene, with different seed particles. The white markers represent SOA without seeds for corresponding symbol experiments. In general, O:C increased with increasing seed particle concentration. The dotted lines represent the Ng triangle (Ng et al., 2011a) translated to the improved ambient elemental ratio parameterization (Canagaratna et al., 2015), to orientate the reader. The insert shows the same figure with different axis ranges, illustrating the fact that all data are within a relatively narrow range compared to, e.g., ambient values.
The elemental ratios of all experiments fall within a relatively narrow range (Fig. 6). The difference between wet and dry AN and dry AS is similar to the difference between pure SOA on different days (white symbols), with O:C within 0.83±0.08 (1σ) and H:C within 1.33±0.05 (1σ). The wet AS experiment however reaches higher O:C values and spans a larger range. The O:C value increases with increasing SOA mass concentration (and seed particle concentration since these are connected), which is the opposite to what is expected since more oxidized molecules tend to be less volatile (Shilling et al., 2009). The increase is mostly due to the mass fragments with m∕z 28 and 44. Several acid-catalyzed oligomerization reactions change the elemental ratios of SOA but with lower O:C as a result (Jang et al., 2002; Chen et al., 2011). Also, saturated AN and AS solutions have similar pH (4.5 and 4.2, respectively, according to the E-AIM model; Clegg et al., 1998); hence, there should be no big difference between the wet experiments. A more likely explanation to the change in O:C with SOA mass is heterogeneous oxidation. Increasing the seed particle number concentration also decreases the SOA mass fraction per particle. It follows that SOA mass then is spread out on more particles, leaving less organics per particle, which enhances the area exposed to oxidation. Kroll et al. (2015) showed that heterogeneous OH oxidation of organic particles may increase the carbon oxidation state, however, at the expense of SOA mass. The water uptake (growth factor) of AN and AS is slightly different at 60 % RH, with an area increase of ∼1.4 and ∼1.7, respectively. However, the O:C increase with increasing area is much larger for wet AS than for any other experiment; hence, a chemical explanation is needed. To the best of our knowledge, there are no measurements on differences between OH radical uptake on different salt solution surfaces. Wang et al. (2016) found that the salting out effect (pushing dissolved molecules out of the water phase) of several different organic molecules is stronger in sulfate compared to nitrate solutions. However, the compounds used were not similar to the SOA used in this study and this effect should cause opposite results since the yield increase is slightly higher for wet AS than for the AN experiments. Takami et al. (1998) showed that below a pH of 7 the uptake coefficient of OH increases with acidity. Given the small difference in acidity between saturated AN and AS it is uncertain if this can explain the measurements. Wick and Dang (2006) found that solvation of OH was correlated with increasing NaCl salt concentration. A possible pathway for differences in reactive uptake of OH between the salt solutions is the reaction with , forming sulfate radicals (Jiang et al., 1992). Sulfate radicals have been shown to be an important source of organosulfates (Noziere et al., 2010; Schindelka et al., 2013). However, sulfate from organics is indistinguishable from inorganic sulfate in the AMS mass spectra since the fragmentation patterns are the same (Farmer et al., 2010) and thus should not affect the calculated O:C ratios. If sulfate aerosols affect the organic portion, this should be seen in ambient samples. Indeed, several studies have shown that the more oxidized SOA (LV-OOA) is correlated with sulfate (Ng et al., 2011b; Zhang et al., 2011; Crippa et al., 2013; Hao et al., 2014), but a more straightforward explanation to this is that both are secondary aerosol constituents.
Experiments were conducted with two aims: (i) to investigate the influence of an aerosol liquid water phase on SOA yields and (ii) verifying and quantifying the underestimation of SOA production in oxidation flow reactors due to limited time for condensation. It was found that in all cases there was a strong increase in yield with increased seed surface area concentration and that the yield with wet seed particles was 45 %–74 % higher compared to dry seed particles. The yield increase leveled off at a dry seed particle condensation sink of ∼0.05 s−1, corresponding to a surface area concentration of ∼1600 µm2 cm−3. This value will be different for different reactor geometries and settings (such as OH exposure and residence time) and implies that it is crucial that the condensation sink is evaluated in all OFR experiments where the absolute SOA mass is of interest.
If seed particles are used to drive the partitioning to the particle phase, the choice of seed may affect the results due to differences in heterogeneous chemistry and water uptake. This makes translation of lab results to atmospheric relevance more difficult since a much higher seed particle concentration is needed in a reactor than in the atmosphere to make the condensed fractions comparable. To further study and parameterize the effects of the condensation sink on OFR SOA, future experiments should focus on different VOC concentrations with varying seed particle surface area, as well as using different seeds or seed mixtures.
Using dry ammonium sulfate seed particles, the maximum yield increase was approximately a factor of 2, while all wet experiments were similar and induced an increase above a factor of 3. Hence, the wet particles produced around 60 % more SOA mass. The O:C ratio increased with decreasing SOA mass fraction. Also, O:C was higher with wet AS compared to other seeds, something which needs further research to fully explain but is likely due to heterogeneous chemistry. These results point to the importance of anthropogenic water as an important source of SOA.
Data are available upon request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-2701-2019-supplement.
EA designed the study with BS and PR. EA conducted the experiments with AE. EA analyzed the data and prepared the manuscript draft. AE, PR, BS and WHB critically reviewed the manuscript and conclusions.
The authors declare that they have no conflict of interest.
This work was financed by the Swedish research council Formas (grant 2011-00732).
Edited by: Barbara Ervens
Reviewed by: two
anonymous referees
Ahlberg, E., Falk, J., Eriksson, A., Holst, T., Brune, W. H., Kristensson, A., Roldin, P., and Svenningsson, B.: Secondary organic aerosol from VOC mixtures in an oxidation flow reactor, Atmos. Environ., 161, 210–220, 2017.
Bruns, E. A., El Haddad, I., Keller, A., Klein, F., Kumar, N. K., Pieber, S. M., Corbin, J. C., Slowik, J. G., Brune, W. H., Baltensperger, U., and Prévôt, A. S. H.: Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 8, 2315–2332, https://doi.org/10.5194/amt-8-2315-2015, 2015.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Carlton, A. G. and Turpin, B. J.: Particle partitioning potential of organic compounds is highest in the Eastern US and driven by anthropogenic water, Atmos. Chem. Phys., 13, 10203–10214, https://doi.org/10.5194/acp-13-10203-2013, 2013.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Chang, R. Y.-W., Slowik, J. G., Shantz, N. C., Vlasenko, A., Liggio, J., Sjostedt, S. J., Leaitch, W. R., and Abbatt, J. P. D.: The hygroscopicity parameter (κ) of ambient organic aerosol at a field site subject to biogenic and anthropogenic influences: relationship to degree of aerosol oxidation, Atmos. Chem. Phys., 10, 5047–5064, https://doi.org/10.5194/acp-10-5047-2010, 2010.
Chen, Q., Liu, Y. J., Donahue, N. M., Shilling, J. E., and Martin, S. T.: Particle-Phase Chemistry of Secondary Organic Material: Modeled Compared to Measured O:C and H:C Elemental Ratios Provide Constraints, Environ. Sci. Technol., 45, 4763–4770, 2011.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic model of the system H+----H2O at tropospheric temperatures, J. Phys. Chem. A, 102, 2137–2154, 1998.
Cocker, D. R., III, Clegg, S. L., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas-particle partitioning of secondary organic aerosol. Part I: alpha-pinene/ozone system, Atmos. Environ., 35, 6049–6072, 2001a.
Cocker, D. R., III, Mader, B. T., Kalberer, M., Flagan, R. C., and Seinfeld, J. H.: The effect of water on gas-particle partitioning of secondary organic aerosol: II. m-xylene and 1,3,5-trimethylbenzene photooxidation systems, Atmos. Environ., 35, 6073–6085, 2001b.
Crippa, M., El Haddad, I., Slowik, J. G., DeCarlo, P. F., Mohr, C., Heringa, M. F., Chirico, R., Marchand, N., Sciare, J., Baltensperger, U., and Prévôt, A. S. H.: Identification of marine and continental aerosol sources in Paris using high resolution aerosol mass spectrometry, J. Geophys. Res.-Atmos., 118, 1950–1963, 2013.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, 2006.
Duplissy, J., DeCarlo, P. F., Dommen, J., Alfarra, M. R., Metzger, A., Barmpadimos, I., Prevot, A. S. H., Weingartner, E., Tritscher, T., Gysel, M., Aiken, A. C., Jimenez, J. L., Canagaratna, M. R., Worsnop, D. R., Collins, D. R., Tomlinson, J., and Baltensperger, U.: Relating hygroscopicity and composition of organic aerosol particulate matter, Atmos. Chem. Phys., 11, 1155–1165, https://doi.org/10.5194/acp-11-1155-2011, 2011.
Edney, E. O., Driscoll, D. J., Speer, R. E., Weathers, W. S., Kleindienst, T. E., Li, W., and Smith, D. F.: Impact of aerosol liquid water on secondary organic aerosol yields of irradiated toluene/propylene/NOx/(NH4)(2)SO4/air mixtures, Atmos. Environ., 34, 3907–3919, 2000.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, 2010.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 51, 1405–1413, 2017.
Gao, S., Ng, N. L., Keywood, M., Varutbangkul, V., Bahreini, R., Nenes, A., He, J. W., Yoo, K. Y., Beauchamp, J. L., Hodyss, R. P., Flagan, R. C., and Seinfeld, J. H.: Particle phase acidity and oligomer formation in secondary organic aerosol, Environ. Sci. Technol., 38, 6582–6589, 2004.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hao, L. Q., Kortelainen, A., Romakkaniemi, S., Portin, H., Jaatinen, A., Leskinen, A., Komppula, M., Miettinen, P., Sueper, D., Pajunoja, A., Smith, J. N., Lehtinen, K. E. J., Worsnop, D. R., Laaksonen, A., and Virtanen, A.: Atmospheric submicron aerosol composition and particulate organic nitrate formation in a boreal forestland–urban mixed region, Atmos. Chem. Phys., 14, 13483–13495, https://doi.org/10.5194/acp-14-13483-2014, 2014.
Hennigan, C. J., Bergin, M. H., Dibb, J. E., and Weber, R. J.: Enhanced secondary organic aerosol formation due to water uptake by fine particles, Geophys. Res. Lett., 35, L18801, https://doi.org/10.1029/2008gl035046, 2008.
Hodas, N., Sullivan, A. P., Skog, K., Keutsch, F. N., Collett, J. L., Decesari, S., Facchini, M. C., Carlton, A. G., Laaksonen, A., and Turpin, B. J.: Aerosol Liquid Water Driven by Anthropogenic Nitrate: Implications for Lifetimes of Water-Soluble Organic Gases and Potential for Secondary Organic Aerosol Formation, Environ. Sci. Technol., 48, 11127–11136, 2014.
Jang, M. S., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, 2002.
Jathar, S. H., Friedman, B., Galang, A. A., Link, M. F., Brophy, P., Volckens, J., Eluri, S., and Farmer, D. K.: Linking Load, Fuel, and Emission Controls to Photochemical Production of Secondary Organic Aerosol from a Diesel Engine, Environ. Sci. Technol., 51, 1377–1386, 2017.
Jiang, P. Y., Katsumura, Y., Nagaishi, R., Domae, M., Ishikawa, K., Ishigure, K., and Yoshida, Y.: Pulse-Radiolysis Study of Concentrated Sulfuric-Acid-Solutions – Formation Mechanism, Yield and Reactivity of Sulfate Radicals, J. Chem. Soc. Faraday T., 88, 1653-1658, 1992.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: Impact of humidity on the ozone initiated oxidation of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 40, 188–194, 2006.
Julin, J., Winkler, P. M., Donahue, N. M., Wagner, P. E., and Riipinent, I.: Near-Unity Mass Accommodation Coefficient of Organic Molecules of Varying Structure, Environ. Sci. Technol., 48, 12083–12089, 2014.
Kamens, R. M., Zhang, H. F., Chen, E. H., Zhou, Y., Parikh, H. M., Wilson, R. L., Galloway, K. E., and Rosen, E. P.: Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 45, 2324–2334, 2011.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Karjalainen, P., Timonen, H., Saukko, E., Kuuluvainen, H., Saarikoski, S., Aakko-Saksa, P., Murtonen, T., Bloss, M., Dal Maso, M., Simonen, P., Ahlberg, E., Svenningsson, B., Brune, W. H., Hillamo, R., Keskinen, J., and Rönkkö, T.: Time-resolved characterization of primary particle emissions and secondary particle formation from a modern gasoline passenger car, Atmos. Chem. Phys., 16, 8559–8570, https://doi.org/10.5194/acp-16-8559-2016, 2016.
Kroll, J. H., Lim, C. Y., Kessler, S. H., and Wilson, K. R.: Heterogeneous Oxidation of Atmospheric Organic Aerosol: Kinetics of Changes to the Amount and Oxidation State of Particle-Phase Organic Carbon, J. Phys. Chem. A, 119, 10767–10783, 2015.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011a.
Lambe, A. T., Onasch, T. B., Massoli, P., Croasdale, D. R., Wright, J. P., Ahern, A. T., Williams, L. R., Worsnop, D. R., Brune, W. H., and Davidovits, P.: Laboratory studies of the chemical composition and cloud condensation nuclei (CCN) activity of secondary organic aerosol (SOA) and oxidized primary organic aerosol (OPOA), Atmos. Chem. Phys., 11, 8913–8928, https://doi.org/10.5194/acp-11-8913-2011, 2011b.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W. W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J. A., and Jimenez, J. L.: Modeling the Radical Chemistry in an Oxidation Flow Reactor: Radical Formation and Recycling, Sensitivities, and the OH Exposure Estimation Equation, J. Phys. Chem. A, 119, 4418–4432, 2015.
Lightstone, J. M., Onasch, T. B., Imre, D., and Oatis, S.: Deliquescence, efflorescence, and water activity in ammonium nitrate and mixed ammonium nitrate/succinic acid microparticles, J. Phys. Chem. A, 104, 9337–9346, 2000.
Lu, Z. F., Hao, J. M., Takekawa, H., Hu, L. H., and Li, J. H.: Effect of high concentrations of inorganic seed aerosols on secondary organic aerosol formation in the m-xylene/NOx photooxidation system, Atmos. Environ., 43, 897–904, 2009.
Martinsson, J., Eriksson, A. C., Nielsen, I. E., Malmborg, V. B., Ahlberg, E., Andersen, C., Lindgren, R., Nystrom, R., Nordin, E. Z., Brune, W. H., Svenningsson, B., Swietlicki, E., Boman, C., and Pagels, J. H.: Impacts of Combustion Conditions and Photochemical Processing on the Light Absorption of Biomass Combustion Aerosol, Environ. Sci. Technol., 49, 14663–14671, 2015.
Massoli, P., Lambe, A. T., Ahern, A. T., Williams, L. R., Ehn, M., Mikkila, J., Canagaratna, M. R., Brune, W. H., Onasch, T. B., Jayne, J. T., Petaja, T., Kulmala, M., Laaksonen, A., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Relationship between aerosol oxidation level and hygroscopic properties of laboratory generated secondary organic aerosol (SOA) particles, Geophys. Res. Lett., 37, L24801, https://doi.org/10.1029/2010gl045258, 2010.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Chhabra, P. S., Seinfeld, J. H., and Worsnop, D. R.: Changes in organic aerosol composition with aging inferred from aerosol mass spectra, Atmos. Chem. Phys., 11, 6465–6474, https://doi.org/10.5194/acp-11-6465-2011, 2011a.
Ng, N. L., Canagaratna, M. R., Jimenez, J. L., Zhang, Q., Ulbrich, I. M., and Worsnop, D. R.: Real-Time Methods for Estimating Organic Component Mass Concentrations from Aerosol Mass Spectrometer Data, Environ. Sci. Technol., 45, 910–916, 2011b.
Nguyen, T. K. V., Zhang, Q., Jimenez, J. L., Pike, M., and Carlton, A. G.: Liquid Water: Ubiquitous Contributor to Aerosol Mass, Environ. Sci. Technol. Lett., 3, 257–263, 2016.
Noziere, B., Ekstrom, S., Alsberg, T., and Holmstrom, S.: Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 37, L05806, https://doi.org/10.1029/2009gl041683, 2010.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Ortega, A. M., Hayes, P. L., Peng, Z., Palm, B. B., Hu, W., Day, D. A., Li, R., Cubison, M. J., Brune, W. H., Graus, M., Warneke, C., Gilman, J. B., Kuster, W. C., de Gouw, J., Gutiérrez-Montes, C., and Jimenez, J. L.: Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area, Atmos. Chem. Phys., 16, 7411–7433, https://doi.org/10.5194/acp-16-7411-2016, 2016.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Pang, Y., Turpin, B. J., and Gundel, L. A.: On the importance of organic oxygen for understanding organic aerosol particles, Aerosol Sci. Tech., 40, 128–133, 2006.
Pankow, J. F.: An Absorption-Model of Gas-Particle Partitioning of Organic-Compounds in the Atmosphere, Atmos. Environ., 28, 185–188, 1994.
Pankow, J. F.: Organic particulate material levels in the atmosphere: Conditions favoring sensitivity to varying relative humidity and temperature, P. Natl. Acad. Sci. USA, 107, 6682–6686, 2010.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Frohlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jimenez, J. L., Worsnop, D. R., Baltensperger, U., and Prevot, A. S. H.: Inorganic Salt Interference on CO2+ in Aerodyne AMS and ACSM Organic Aerosol Composition Studies, Environ. Sci. Technol., 50, 10494–10503, 2016.
Pilinis, C., Seinfeld, J. H., and Grosjean, D.: Water-Content of Atmospheric Aerosols, Atmos. Environ., 23, 1601–1606, 1989.
Pirjola, L., Kulmala, M., Wilck, M., Bischoff, A., Stratmann, F., and Otto, E.: Formation of sulphuric acid aerosols and cloud condensation nuclei: An expression for significant nucleation and model comparison, J. Aerosol Sci., 30, 1079–1094, 1999.
Pöschl, U.: Atmospheric aerosols: Composition, transformation, climate and health effects, Angew. Chem. Int. Edit., 44, 7520–7540, 2005.
Prisle, N. L., Engelhart, G. J., Bilde, M., and Donahue, N. M.: Humidity influence on gas-particle phase partitioning of alpha-pinene + O-3 secondary organic aerosol, Geophys. Res. Lett., 37, L01802, https://doi.org/10.1029/2009gl041402, 2010.
Sareen, N., Waxman, E. M., Turpin, B. J., Volkamer, R., and Carlton, A. G.: Potential of Aerosol Liquid Water to Facilitate Organic Aerosol Formation: Assessing Knowledge Gaps about Precursors and Partitioning, Environ. Sci. Technol., 51, 3327–3335, 2017.
Schindelka, J., Iinuma, Y., Hoffmann, D., and Herrmann, H.: Sulfate radical-initiated formation of isoprene-derived organosulfates in atmospheric aerosols, Faraday Discuss., 165, 237–259, 2013.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, John Wiley & Sons, Hoboken, New Jersey, 2006.
Seinfeld, J. H., Erdakos, G. B., Asher, W. E., and Pankow, J. F.: Modeling the formation of secondary organic aerosol (SOA). 2. The predicted effects of relative humidity on aerosol formation in the alpha-pinene-, beta-pinene-, sabinene-, Delta(3)-Carene-, and cyclohexene-ozone systems, Environ. Sci. Technol., 35, 1806–1817, 2001.
Shilling, J. E., Chen, Q., King, S. M., Rosenoern, T., Kroll, J. H., Worsnop, D. R., DeCarlo, P. F., Aiken, A. C., Sueper, D., Jimenez, J. L., and Martin, S. T.: Loading-dependent elemental composition of α-pinene SOA particles, Atmos. Chem. Phys., 9, 771–782, https://doi.org/10.5194/acp-9-771-2009, 2009.
Simonen, P., Saukko, E., Karjalainen, P., Timonen, H., Bloss, M., Aakko-Saksa, P., Rönkkö, T., Keskinen, J., and Dal Maso, M.: A new oxidation flow reactor for measuring secondary aerosol formation of rapidly changing emission sources, Atmos. Meas. Tech., 10, 1519–1537, https://doi.org/10.5194/amt-10-1519-2017, 2017.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid-liquid phase separation and morphology of internally mixed dicarboxylic acids/ammonium sulfate/water particles, Atmos. Chem. Phys., 12, 2691–2712, https://doi.org/10.5194/acp-12-2691-2012, 2012a.
Song, M., Marcolli, C., Krieger, U. K., Zuend, A., and Peter, T.: Liquid-liquid phase separation in aerosol particles: Dependence on O:C, organic functionalities, and compositional complexity, Geophys. Res. Lett., 39, L19801, https://doi.org/10.1029/2012gl052807, 2012b.
Stirnweis, L., Marcolli, C., Dommen, J., Barmet, P., Frege, C., Platt, S. M., Bruns, E. A., Krapf, M., Slowik, J. G., Wolf, R., Prévôt, A. S. H., Baltensperger, U., and El-Haddad, I.: Assessing the influence of NOx concentrations and relative humidity on secondary organic aerosol yields from a-pinene photo-oxidation through smog chamber experiments and modelling calculations, Atmos. Chem. Phys., 17, 5035–5061, https://doi.org/10.5194/acp-17-5035-2017, 2017.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, 2007.
Svenningsson, B.: Hygroscopic growth of atmospheric aerosol particles and its relation to nucleation scavenging in clouds, Division of Nuclear Physics, Lund University, Box 118, 221 00 Lund, Sweden, 1997.
Takami, A., Kato, S., Shimono, A., and Koda, S.: Uptake coefficient of OH radical on aqueous surface, Chem. Phys., 231, 215–227, 1998.
Tang, I. N. and Munkelwitz, H. R.: Composition and Temperature-Dependence of the Deliquescence Properties of Hygroscopic Aerosols, Atmos. Environ. A-Gen., 27, 467–473, 1993.
Varutbangkul, V., Brechtel, F. J., Bahreini, R., Ng, N. L., Keywood, M. D., Kroll, J. H., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Hygroscopicity of secondary organic aerosols formed by oxidation of cycloalkenes, monoterpenes, sesquiterpenes, and related compounds, Atmos. Chem. Phys., 6, 2367–2388, https://doi.org/10.5194/acp-6-2367-2006, 2006.
Wang, C., Lei, Y. D., and Wania, F.: Effect of Sodium Sulfate, Ammonium Chloride, Ammonium Nitrate, and Salt Mixtures on Aqueous Phase Partitioning of Organic Compounds, Environ. Sci. Technol., 50, 12742–12749, 2016.
Warren, B., Malloy, Q. G. J., Yee, L. D., and Cocker, D. R.: Secondary organic aerosol formation from cyclohexene ozonolysis in the presence of water vapor and dissolved salts, Atmos. Environ., 43, 1789–1795, 2009.
Wick, C. D. and Dang, L. X.: Computational observation of enhanced solvation of the hydroxyl radical with increased NaCl concentration, J. Phys. Chem. B, 110, 8917–8920, 2006.
Wiedensohler, A., Birmili, W., Nowak, A., Sonntag, A., Weinhold, K., Merkel, M., Wehner, B., Tuch, T., Pfeifer, S., Fiebig, M., Fjäraa, A. M., Asmi, E., Sellegri, K., Depuy, R., Venzac, H., Villani, P., Laj, P., Aalto, P., Ogren, J. A., Swietlicki, E., Williams, P., Roldin, P., Quincey, P., Hüglin, C., Fierz-Schmidhauser, R., Gysel, M., Weingartner, E., Riccobono, F., Santos, S., Grüning, C., Faloon, K., Beddows, D., Harrison, R., Monahan, C., Jennings, S. G., O'Dowd, C. D., Marinoni, A., Horn, H.-G., Keck, L., Jiang, J., Scheckman, J., McMurry, P. H., Deng, Z., Zhao, C. S., Moerman, M., Henzing, B., de Leeuw, G., Löschau, G., and Bastian, S.: Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high quality long-term observations of atmospheric particle number size distributions, Atmos. Meas. Tech., 5, 657–685, https://doi.org/10.5194/amt-5-657-2012, 2012.
Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Impacts of Sulfate Seed Acidity and Water Content on Isoprene Secondary Organic Aerosol Formation, Environ. Sci. Technol., 49, 13215–13221, 2015.
You, Y., Renbaum-Wolff, L., Carreras-Sospedra, M., Hanna, S. J., Hiranuma, N., Kamal, S., Smith, M. L., Zhang, X. L., Weber, R. J., Shilling, J. E., Dabdub, D., Martin, S. T., and Bertram, A. K.: Images reveal that atmospheric particles can undergo liquid-liquid phase separations, P. Natl. Acad. Sci. USA, 109, 13188–13193, 2012.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007gl029979, 2007.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y. L.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, 2011.
Zhao, Y., Lambe, A. T., Saleh, R., Saliba, G., and Robinson, A. L.: Secondary Organic Aerosol Production from Gasoline Vehicle Exhaust: Effects of Engine Technology, Cold Start, and Emission Certification Standard, Environ. Sci. Technol., 52, 1253–1261, 2018.
Zhou, Y., Zhang, H. F., Parikh, H. M., Chen, E. H., Rattanavaraha, W., Rosen, E. P., Wang, W. X., and Kamens, R. M.: Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: Water and particle seed effects (II), Atmos. Environ., 45, 3882–3890, 2011.
Zuend, A. and Seinfeld, J. H.: Modeling the gas-particle partitioning of secondary organic aerosol: the importance of liquid-liquid phase separation, Atmos. Chem. Phys., 12, 3857–3882, https://doi.org/10.5194/acp-12-3857-2012, 2012.