the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Feb 2019
| 14 Feb 2019
Rate constant and secondary organic aerosol formation from the gas-phase reaction of eugenol with hydroxyl radicals
Changgeng Liu
Tianzeng Chen
Jun Liu
Methoxyphenols are an important organic component of wood-burning emissions and considered to be potential precursors of secondary organic aerosol (SOA). In this work, the rate constant and SOA formation potential for the OH-initiated reaction of 4-allyl-2-methoxyphenol (eugenol) were investigated for the first time in an oxidation flow reactor (OFR). The rate constant was cm3 molecule−1 s−1, determined by the relative rate method. The SOA yield first increased and then decreased as a function of OH exposure and was also dependent on eugenol concentration. The maximum SOA yields (0.11–0.31) obtained at different eugenol concentrations could be expressed well by a one-product model. The carbon oxidation state (OSC) increased linearly and significantly as OH exposure rose, indicating that a high oxidation degree was achieved for SOA. In addition, the presence of SO2 (0–198 ppbv) and NO2 (0–109 ppbv) was conducive to increasing SOA yield, for which the maximum enhancement values were 38.6 % and 19.2 %, respectively. The N∕C ratio (0.032–0.043) indicated that NO2 participated in the OH-initiated reaction, subsequently forming organic nitrates. The results could be helpful for further understanding the SOA formation potential from the atmospheric oxidation of methoxyphenols and the atmospheric aging process of smoke plumes from biomass burning emissions.
- Article
(1158 KB) - Full-text XML
-
Supplement
(819 KB) - BibTeX
- EndNote





Wood combustion is a major contributor to atmospheric fine particulate matter (PM) (Bruns et al., 2016), which could contribute approximately 10 %–50 % of the total organic fraction of atmospheric aerosols (Schauer and Cass, 2000). In some regions with cold climates, woodsmoke-associated aerosols are estimated to account for more than 70 % of PM2.5 in winter (Jeong et al., 2008; Ward et al., 2006). Recently, the significant potential for secondary organic aerosol (SOA) formation from woodsmoke emissions has been reported (Bruns et al., 2016; Gilardoni et al., 2016; Tiitta et al., 2016; Ciarelli et al., 2017; Ding et al., 2017). In addition, the organic compounds derived from wood combustion and their oxidation products may contribute significantly to global warming due to their light-absorbing properties (Chen and Bond, 2010). It has been reported that woodsmoke particles are predominant in the inhalable size range (Bari et al., 2010) and their extracts are mutagenic (Kleindienst et al., 1986). Exposure to woodsmoke can result in adverse health effects such as acute respiratory infections, tuberculosis, lung cancer, and cataracts (Bolling et al., 2009). Therefore, wood combustion has multifaceted impacts on climate, air quality, and human health.
Methoxyphenols produced by lignin pyrolysis are potential tracers for woodsmoke, and their emission rates are in the range of 900–4200 mg kg−1 wood (Schauer et al., 2001; Simpson et al., 2005; Nolte et al., 2001). The highest level of methoxyphenols in the atmosphere always appears during a woodsmoke-dominated period, with observed values up to several mg m−3 (Schauer and Cass, 2000; Schauer et al., 2001; Simpson et al., 2005). Methoxyphenols are semi-volatile aromatic compounds with low molecular weight, and many of them are found to mainly exist in the gas phase at typical ambient temperature (Schauer et al., 2001; Simpson et al., 2005). Thus, methoxyphenols can be chemically transformed through gas-phase reactions with atmospheric oxidants (Coeur-Tourneur et al., 2010a; Lauraguais et al., 2012, 2014a, b, 2015, 2016; Yang et al., 2016; Zhang et al., 2016; El Zein et al., 2015). The corresponding rate constants control their effectiveness as stable tracers for wood combustion and atmospheric lifetimes. In recent years, the rate constants for the gas-phase reactions of some methoxyphenols with hydroxyl (OH) radicals (Coeur-Tourneur et al., 2010a; Lauraguais et al., 2012, 2014b, 2015), nitrate (NO3) radicals (Lauraguais et al., 2016; Yang et al., 2016; Zhang et al., 2016), chlorine atoms (Cl) (Lauraguais et al., 2014a), and ozone (O3) (El Zein et al., 2015) have been determined. Some studies have indicated significant SOA formation from 2,6-dimethoxyphenol (syringol) and 2-methoxyphenol (guaiacol) with respect to their reactions with OH radicals (Sun et al., 2010; Lauraguais et al., 2012, 2014b; Ahmad et al., 2017; Yee et al., 2013; Ofner et al., 2011). Although biomass burning emissions have been indicated to have great SOA formation potential via atmospheric oxidation (Bruns et al., 2016; Gilardoni et al., 2016; Li et al., 2017; Ciarelli et al., 2017; Ding et al., 2017), SOA formation and growth from methoxyphenols are still poorly understood. Besides, the observed SOA levels in the atmosphere cannot be explained well by the present knowledge of SOA formation, which reflects the fact that a large number of precursors are not taken into account in the SOA formation reactions included in atmospheric models (Lauraguais et al., 2012).
4-Allyl-2-methoxyphenol (eugenol) is a typical methoxyphenol produced by lignin pyrolysis with a branched alkene group. It is widely detected in the atmosphere with a concentration of the order of ng m−3, which is comparable to that of other methoxyphenols (e.g., guaiacol and syringol) (Schauer et al., 2001; Simpson et al., 2005; Bari et al., 2009). Its average emission concentration and factor in beech burning are 0.032 µg m−3 and 1.52 µg g−1 PM, respectively, which are both higher than those (0.016 µg m−3 and 0.762 µg g−1 PM) of guaiacol (Bari et al., 2009). It has even been detected in human urine after exposure to woodsmoke (Dills et al., 2006). Eugenol has been observed to mainly distribute in the gas phase in woodsmoke emissions (Schauer et al., 2001), and its gas–particle partition coefficient is lower than 0.01 (Zhang et al., 2016), thus indicating the importance of its gas-phase reactions in the atmosphere. For this reason, the aim of this work was to determine the rate constant and explore the SOA formation potential for eugenol in gas-phase reactions with OH radicals using an oxidation flow reactor (OFR). In addition, the effects of SO2 and NO2 on SOA formation were investigated. To our knowledge, this work represents the first determination of the rate constant and SOA yield for the gas-phase reaction of eugenol with OH radicals.
The detailed schematic description of the experimental system used in this work is shown in Figs. S1 and S2 in the Supplement. The gas-phase reactions were conducted in the OFR, a detailed description of which has been presented elsewhere (Liu et al., 2014b). Before entering into the OFR, gas-phase species were mixed thoroughly in the mixing tube. The reaction time in the OFR was 26.7 s, calculated according to the illuminated volume (0.89 L) and the total flow rate (2 L min−1). OH radicals were generated by the photolysis of O3 in the presence of water vapor using a 254 nm UV lamp (Jelight Co., Inc.), and their formation reactions have been described elsewhere (Zhang et al., 2017). The concentration of OH radicals was governed by O3 concentration and relative humidity (RH). O3 concentration was controlled by changing the unshaded length of a 185 nm UV lamp (Jelight Co., Inc.). O3 with a concentration of 0.94–9.11 ppmv in the OFR was produced by passing zero air through an O3 generator (model 610-220, Jelight Co., Inc.), which was used to produce OH radicals. RH and temperature in the OFR were 44.0±2.0 % and 301±1 K, respectively, measured at the outlet of the OFR. The steady-state concentrations of OH radicals were determined using SO2 as the reference compound in separate calibration experiments. It is a widely used method for calculating OH exposure in the OFR, but could not sufficiently describe the potential OH suppression caused by the added external OH reactivity (Zhang et al., 2017; Lambe et al., 2015; Simonen et al., 2017; Li et al., 2015; Peng et al., 2015, 2016). The decay of SO2 from its reaction with OH radicals ( cm3 molecule−1 s−1) (Davis et al., 1979) was measured by an SO2 analyzer (model 43i, Thermo Fisher Scientific Inc.). The concentration of OH radicals ([OH]) in this work ranged from approximately 4.5×109 to 4.7×1010 molecules cm−3, and the corresponding OH exposures were in the range of 1.21–12.55×1011 molecules cm−3 s or approximately 0.93 to 9.68 days of equivalent atmospheric exposure, which was calculated using a typical [OH] of 1.5×106 molecules cm−3 in the atmosphere (Mao et al., 2009).
An Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) was applied to perform online measurements of the chemical composition of particles and the non-refractory submicron aerosol mass (DeCarlo et al., 2006). The size distribution and concentration of particles were monitored by a scanning mobility particle sizer (SMPS) consisting of a differential mobility analyzer (DMA) (model 3082, TSI Inc.) and a condensation particle counter (CPC) (model 3776, TSI Inc.). Assuming that particles are spherical and nonporous, the average effective particle density could be calculated as 1.5 g cm−3 using the equation (DeCarlo et al., 2004), where dva is the mean vacuum aerodynamic diameter measured by the HR-ToF-AMS and dm is the mean volume-weighted mobility diameter measured by the SMPS. The particle size for HR-ToF-AMS measurement was calibrated using NH4NO3 particles with a diameter between 60 and 700 nm selected by a DMA. The mass concentration of particles measured by HR-ToF-AMS was corrected by SMPS data in this work using the same method as in Gordon et al. (2014). Eugenol and reference compounds were measured by a high-resolution proton-transfer-reaction time-of-flight mass spectrometer (HR-ToF-PTRMS) (Ionicon Analytik GmbH). More experimental details are described in the Supplement.
3.1 Rate constant
The possible effect of O3 on the decay of eugenol and reference compounds was investigated in this work. As shown in Fig. S3, their concentrations were not affected by O3. Meanwhile, no SOA formation was observed by the SMPS and HR-ToF-AMS. In addition, in order to investigate the possible photolysis of eugenol and reference compounds at 254 nm UV light in the OFR, the comparative experiments were conducted with the UV lamp turned on and turned off when eugenol and reference compounds were introduced into the OFR. The normalized mass spectra of eugenol and reference compounds in the dark and light are shown in Fig. S4. The results showed that no significant decay (< 5 %) by photolysis was observed and could be neglected. According to the results reported by Peng et al. (2016), the photolysis of phenol and 1,3,5-trimethylbenzene can be ignored when the ratio of exposure to 254 nm and OH is lower than 1×106 cm s−1, a condition that the values (1.6×102 to 1.7×103 cm s−1) in this work also met. In addition, the initial concentration of eugenol was determined with the UV lamp turned on. Therefore, the effect of photolysis could be neglected in this work. However, it cannot be ruled out that photolysis under UV irradiation might have an influence on the evolution of oxidation products.
The rate constant for the gas-phase reaction of eugenol with OH radicals was determined by the relative rate method, which can be expressed by the following equation (Coeur-Tourneur et al., 2010a; Yang et al., 2016; Zhang et al., 2016):
where CE0 and CEt are the initial and real-time concentrations of eugenol, respectively. kE is the rate constant of the eugenol reaction with OH radicals. CR0 and CRt are the initial and real-time concentrations of the reference compound, respectively. kR is the rate constant of the reference compound with OH radicals, with values for m-xylene and 1,3,5-trimethylbenzene of and cm3 molecule−1 s−1, respectively (Kramp and Paulson, 1998; Coeur-Tourneur et al., 2010a).
Table 1Rate constant for the gas-phase reaction of eugenol with OH radicals and the associated atmospheric lifetime.
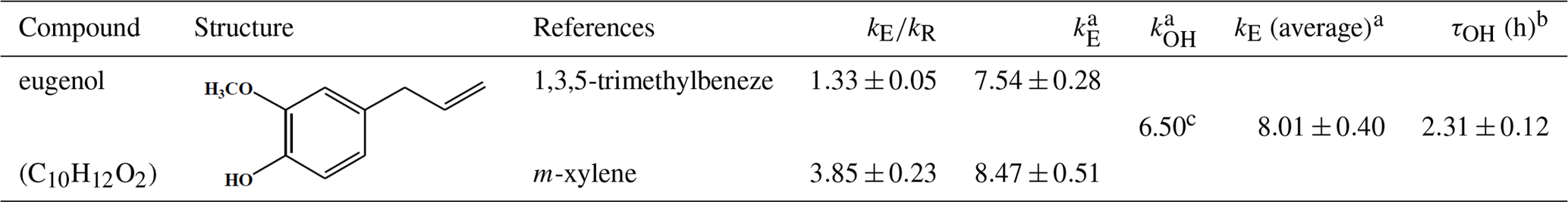
a Units of 10−11 cm3 molecule−1 s−1. b Atmospheric lifetime in hours. [OH], assuming a 24 h average [OH] = 1.5×106 molecules cm−3 (Mao et al., 2009). c Calculated using the US EPA AOP WIN model (US EPA, 2012).
Data obtained from the reactions were plotted in the form of Eq. (1) and were fitted well by linear regression (R2>0.97; Fig. 1). A summary of the slopes and the rate constants is listed in Table 1. The errors in kE∕kR are the standard deviations generated from the linear regression analysis and do not include the uncertainty in the rate constants of the reference compounds. The rate constants are and cm3 molecule−1 s−1, respectively, when using 1,3,5-trimethylbenzene and m-xylene as reference compounds. According to the US EPA AOP WIN model based on the structure–activity relationship (SAR) (US EPA, 2012), the rate constant was calculated to be cm3 molecule−1 s−1 (Table 1), which is lower than that obtained in this work. Inaccurate performance of the AOP WIN model has been observed for other multifunctional organics due to the inaccurate representation of the electronic effects of different functional groups on reactivity (Coeur-Tourneur et al., 2010a; Lauraguais et al., 2012). In addition, differences between density functional theory (DFT) calculations and lab studies have been also observed. For example, the DFT-predicted rate constant of 2-methoxyphenol with OH radicals ( cm3 molecule−1 s−1) is higher than that ( cm3 molecule−1 s−1) obtained by a lab study (Coeur-Tourneur et al., 2010a; Priya and Lakshmipathi, 2017). This suggests that it is necessary to determine the rate constants of multifunctional organics through lab experiments. The rate constant determined in this work can be used to calculate the atmospheric lifetime of eugenol with respect to its reaction with OH radicals. Assuming a typical [OH] for a 24 h average value to be 1.5×106 molecules cm−3 (Mao et al., 2009), the corresponding lifetime of eugenol was calculated to be 2.31±0.12 h with an average rate constant of cm3 molecule−1 s−1. This short lifetime indicates that eugenol is too reactive to be used as a tracer for woodsmoke emissions and also implies the possible fast conversion of eugenol from the gas phase to secondary aerosol during the transportation process.
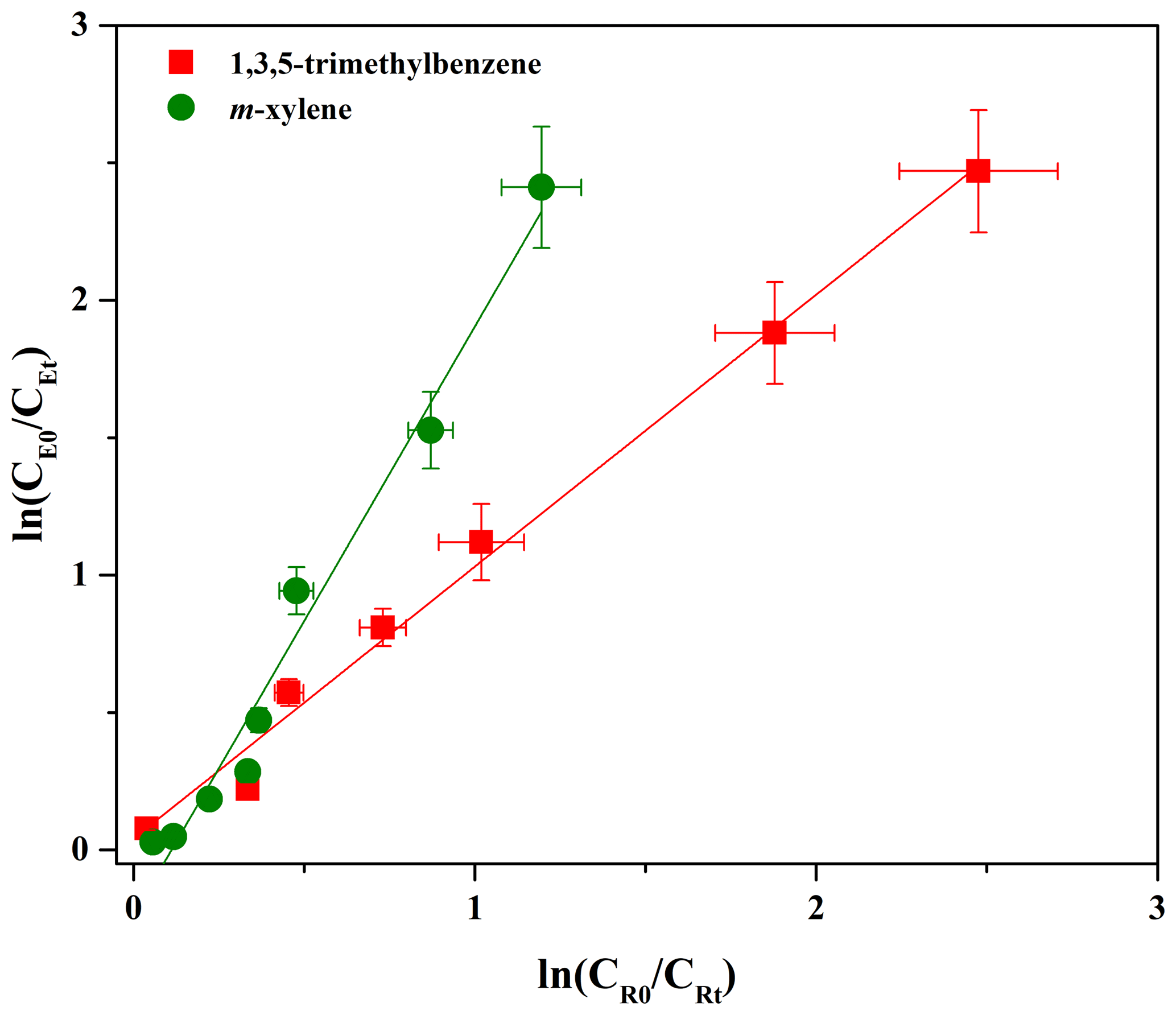
The rate constant obtained in this work is about 2 orders of magnitude faster than that for eugenol with NO3 radicals ( cm3 molecule−1 s−1) (Zhang et al., 2016), which suggests that the OH-initiated reaction of eugenol might be the main chemical transformation in the atmosphere. The rate constants of OH-initiated reactions of guaiacol, 2,6-dimethylphenol, and syringol were , , and cm3 molecule−1 s−1, respectively (Coeur-Tourneur et al., 2010a; Thuner et al., 2004; Lauraguais et al., 2012), while their corresponding rate constants were calculated to be , , and cm3 molecule−1 s−1 according to the US EPA AOP WIN model (US EPA, 2012). These differences among rate constants suggest that the rate constants of multifunctional organics should be necessarily determined via lab experiments. The reactivity of eugenol toward OH radicals is slightly higher than those of guaiacol and 2,6-dimethylphenol, but slightly slower than that of syringol. The presence of two methoxyl groups (–O–CH3) in syringol activates the electrophilic addition of OH radicals to the benzene ring by donating electron density through the resonance effect (Lauraguais et al., 2016). The activation effect of the methoxyl group is much larger than that of alkyl groups (McMurry, 2004). In a recent study, the reported energy barrier of NO3 electrophilic addition to eugenol was about 2-fold higher than that of 4-ethylguaiacol, indicating that the activation effect of the allyl group (–CH2CH=CH2) is lower than that of the ethyl group (–CH2CH3) (Zhang et al., 2016). These results are in accordance with the activation effects of the substituents toward the electrophilic addition of OH radicals (McMurry, 2004).
3.2 Effects of eugenol concentration and OH exposure on SOA formation
In this work, a series of experiments were conducted in the OFR with different eugenol concentrations. The SOA yield was determined as the ratio of the SOA mass concentration (Mo, µg m−3) to the reacted eugenol concentration ([eugenol], µg m−3) (Kang et al., 2007). The experimental conditions and maximum SOA yields are listed in Table 2. The wall loss of aerosol particles in the OFR could be ignored according to our previous results reported by Liu et al. (2014a). Figure S5 shows the plots of the SOA yield versus OH exposure at different eugenol concentrations. Higher concentrations resulted in higher amounts of condensable products and subsequently increased SOA yield (Lauraguais et al., 2012). SOA mass also directly influences gas–particle partitioning because SOA can serve as the adsorption medium for oxidation products, and higher SOA mass generally results in higher SOA yield (Lauraguais et al., 2012, 2014b). In the OFR, in all cases the SOA yield first increased and then decreased as a function of OH exposure (Fig. S5). This trend is the most common phenomenon observed in studies conducted in the OFR and a potential aerosol mass (PAM) reactor (Lambe et al., 2015; Ortega et al., 2016; Palm et al., 2016, 2018; Simonen et al., 2017). In this work, according to the OFR exposure estimator (v2.3) developed by Jimenez's group based on the estimation equations reported in previous work (Li et al., 2015; Peng et al., 2015, 2016), the maximum reduction of OH exposure by eugenol in the OFR was approximately 90 %. Its detailed calculation is shown in the Supplement. Although OH suppression by eugenol was not well determined in the OFR, OH radicals were expected to be the main oxidant due to the fast reaction rate constant of eugenol toward OH radicals obtained in this work. The decrease in SOA yield at high OH exposure is possibly contributed from the C–C bond scission of gas-phase species by further oxidation or heterogeneous reactions involving OH radicals, which would generate a large amount of fragmented molecules that subsequently volatilize out of aerosol particles (Lambe et al., 2015; Ortega et al., 2016; Simonen et al., 2017).
Table 2Experimental conditions and results for SOA formation.
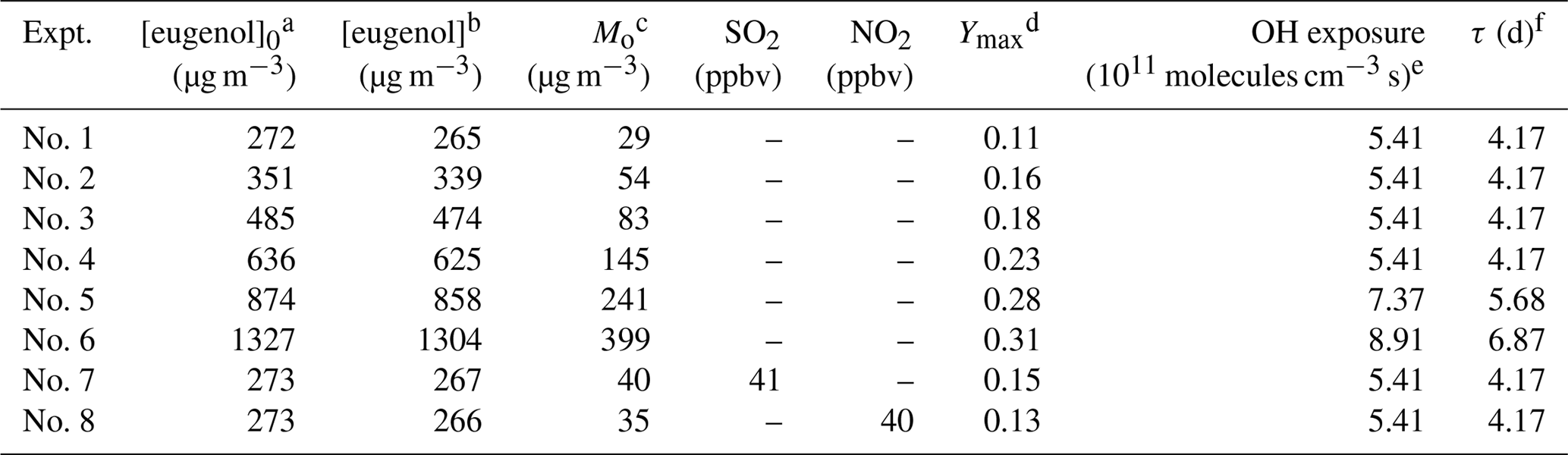
a Initial eugenol concentrations. b Reacted eugenol concentrations. c SOA concentrations. d Maximum SOA yields. e Corresponding OH exposure of maximum SOA yields. f Corresponding atmospheric aging time of maximum SOA yields, calculated using a typical [OH] in the atmosphere in this work (1.5×106 molecules cm−3) (Mao et al., 2009).
SOA yield can be described with a widely used semi-empirical model on the basis of the absorptive gas–particle partitioning of semi-volatile products, in which the overall SOA yield (Y) is given by Odum et al. (1996):
where αi is the mass-based stoichiometric coefficient for the reaction producing the semi-volatile product i, Kom,i is the gas–particle partitioning equilibrium constant, and Mo is the total aerosol mass concentration.
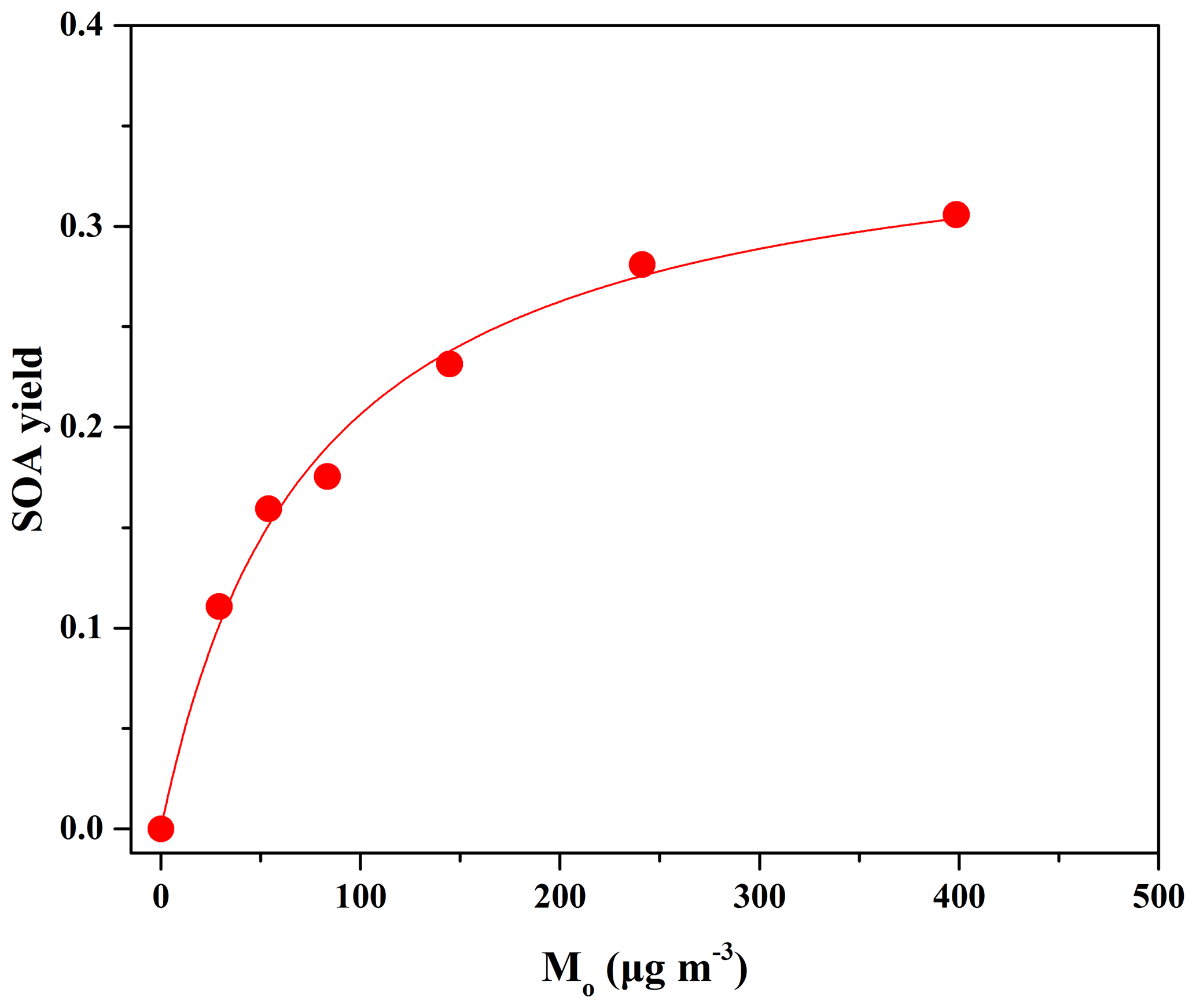
Figure 2Maximum SOA yields as a function of SOA mass concentration (Mo) formed from OH reactions at different eugenol concentrations. The solid line was fit to the experimental data using a one-product model. Values for αi and Kom,i used to generate the solid line are 0.36±0.02 and 0.013±0.002, respectively.
The SOA yield data in Table 2 can be plotted in the form of Eq. (2) to obtain the yield curve for eugenol (Fig. 2). The simulation of experimental data indicated that a one-product model could accurately reproduce the data (R2=0.98), while the use of two or more products in the model did not significantly improve the fitting quality. Odum et al. (1996) reported that the SOA yield data from the oxidation of aromatic compounds could be fitted well using a two-product model. However, a one-product model was also efficient for describing the SOA yields from the oxidation of aromatics including methoxyphenols (Coeur-Tourneur et al., 2010b; Lauraguais et al., 2012, 2014b). The success of simulations with a one-product model in this work is likely to indicate that the products in SOA have similar values of αi and Kom,i, i.e., that the obtained αi (0.36±0.02) and Kom,i (0.013±0.002 m3 µg−1) represent the average values. In this work, considering that the product composition of SOA was not determined, the volatility basis set (VBS) approach was not applied to simulate SOA yields. Figure S6 shows a plot of the SOA mass concentration (Mo) versus the reacted eugenol concentration ([eugenol]). Its slope was 0.37 as obtained using linear least-squares fitting, which is very close to the αi value (0.36). This suggests that the low-volatility products formed in the reaction almost completely disperse on the particle phase according to the theoretical partition model (Lauraguais et al., 2012, 2014b). In other words, SOA yield was approximately an upper limit for eugenol oxidation in the OFR. In view of the residence time in this work, it seems to be in contradiction to the recommendation of a longer residence time made by Ahlberg et al. (2017), who found that the condensation of low-volatility species on SOA in the OFR was often kinetically limited at low mass concentrations. In our recent experiments (not published), the SOA yields for guaiacol oxidation by OH radicals obtained under similar experimental conditions as this work could be comparable to those obtained in chamber studies conducted at low RH (Fig. S7) (Lauraguais et al., 2014b; Yee et al., 2013). This suggests that the effect of kinetic limitations on SOA condensation for the OH-initiated oxidation of methoxyphenols in this system might not be important.
Elemental ratios (H∕C and O∕C) could provide insights into SOA composition and chemical processes along with aging (Bruns et al., 2015). As shown in Fig. 3, the O∕C ratio of SOA increased and the H∕C ratio decreased with increasing OH exposure because oxygen-containing functional groups were formed in the oxidation products. In addition, the organic mass fractions of m∕z 44 () and m∕z 43 (mostly C2H3O+), named f44 and f43, respectively, could also provide information about the nature of SOA formation. Figure S8 shows the evolution of f44 and f43 versus OH exposure at low (272 µg m−3) and high (1328 µg m−3) concentrations of eugenol. The values of f44 were much higher than those of f43 and increased significantly as a function of OH exposure, suggesting that SOA formed in the experiments became more oxidized. The f44 value in this work ranged up to 0.26, which was consistent with that observed for ambient low-volatility OA (LV-OA) higher than 0.25 (Ng et al., 2010).
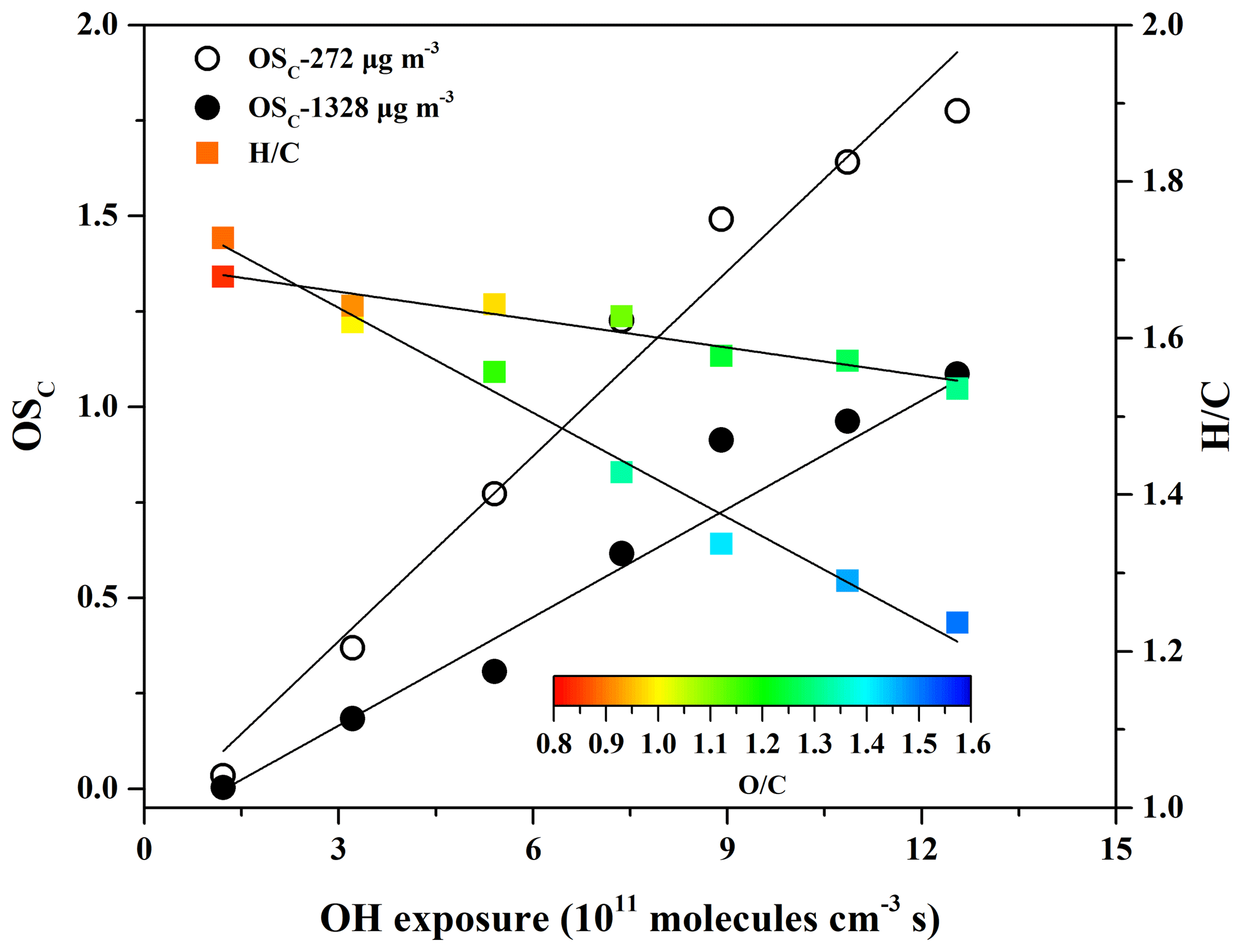
Figure 3OSC, H∕C, and O∕C vs. the OH exposure for SOA formed at two eugenol concentrations (272 and 1328 µg m−3).
The average carbon oxidation state (OSC) proposed by Kroll et al. (2011) is considered a more accurate indicator of the oxidation degree of atmospheric organic species than the O∕C ratio alone because it takes into account the saturation level of the carbon atoms in the SOA. OSC is defined as (Kroll et al., 2011), calculated according to the elemental composition of SOA measured by the HR-ToF-AMS. In this work, the OSC values obtained at low (272 µg m−3) and high (1328 µg m−3) concentrations of eugenol were compared. As shown in Fig. 3, OSC values for low concentration (0.035–1.78) were much larger than those for high concentration (0.0036–1.09) and increased linearly (R2>0.96) with OH exposure of molecules cm−3 s. The results were supported by the evolution of SOA mass spectra obtained by the HR-ToF-AMS at the same eugenol concentrations (Fig. S9). Similar trends have been observed in the smog chamber and PAM reactor (Simonen et al., 2017; Ortega et al., 2016). The OSC value in this work extended as high as 1.78, which was in good agreement with that observed for ambient LV-OA, up to 1.9 (Kroll et al., 2011). Recently, Ortega et al. (2016) reported that the OSC value for SOA formed from ambient air in an OFR ranged up to 2.0, and Simonen et al. (2017) determined a high OSC value (> 1.1) for SOA formed from the OH-initiated reaction of toluene in a PAM reactor with an OH exposure of 1.2×1012 molecules cm−3 s. In general, the OSC values for the PAM reactor are higher than those for smog chambers because OH exposure in the PAM reactor is about 1–3 orders of magnitude higher than in the smog chamber (Simonen et al., 2017; Ortega et al., 2016; Lambe et al., 2015). A higher OSC value indicates greater age, whereby the SOA components are further oxidized through heterogeneous oxidation, adding substantial oxygen and reducing hydrogen in the molecules in the particle phase to increase OSC values despite the overall loss of SOA mass (Ortega et al., 2016).
3.3 Effect of SO2 on SOA formation
As shown in Fig. 4, the presence of SO2 favored SOA formation, and the sulfate concentration increased linearly (R2=0.99) as a function of OH exposure. The maximum SOA yield enhancement of 38.6 % was obtained at OH exposure of 5.41×1011 molecules cm−3 s and then decreased with the increase in OH exposure, possibly due to the fragmented molecules formed through the oxidation of gas-phase species by high OH exposure (Lambe et al., 2015; Ortega et al., 2016; Simonen et al., 2017). The SOA yield and sulfate concentration both increased linearly (R2>0.97) as the SO2 concentration increased from 0 to 198 ppbv at OH exposure of 1.21×1011 molecules cm−3 s (Fig. S10). Compared to the initial SOA yield (0.049) obtained in the absence of SO2, the SOA yield (0.066) obtained in the presence of 198 ppbv SO2 was enhanced by 34.7 %. In previous studies, Kleindienst et al. (2006) reported that the SOA yield from α-pinene photooxidation increased by 40 % in the presence of 252 ppbv SO2. T. Liu et al. (2016) recently found that the SOA yield from 5 h of photochemical aging for gasoline vehicle exhaust was enhanced by 60 %–200 % in the presence of ∼150 ppbv SO2.

Figure 4Evolution of the enhanced SOA yield and sulfate formation as a function of OH exposure in the presence of 41 ppbv SO2 at an average eugenol concentration of 273 µg m−3.
As shown in Figs. 4 and S10, the increase in sulfate concentration was favorable for SOA formation. In this system, it is difficult to completely remove trace NH3, and thus the formed sulfate was a mixture of sulfuric acid (H2SO4) and a small amount of ammonium sulfate ((NH4)2SO4). The in situ particle acidity was calculated as the H+ concentration ([H+], 40.23–648.39 nmol m−3) according to the AIM-II model for the H+––––H2O systems (http://www.aim.env.uea.ac.uk/aim/model2/model2a.php, last access: 18 June 2018; T. Liu et al., 2016). A detailed description of the calculation method has been represented elsewhere (T. Liu et al., 2016). The elevated concentration of sulfate in the particle phase with the increases in SO2 concentration and OH exposure was an important reason for the enhanced SOA yields (Kleindienst et al., 2006; T. Liu et al., 2016). Cao and Jang (2007) indicated that SOA yields from the oxidation of toluene and 1,3,5-trimethylbenzene increased by 14 %–36 % in the presence of acid seeds, with [H+] of 240–860 nmol m−3, compared to those obtained in the presence of nonacid seeds. Similar results concerning the effect of particle acidity on SOA yields were reported in other studies (Kleindienst et al., 2006; T. Liu et al., 2016; Jaoui et al., 2008; Xu et al., 2016). However, Ng et al. (2007b) found that particle acidity had a negligible effect on SOA yields from the photooxidation of aromatics, possibly due to the low RH (∼5 %) used in their work. The water content of aerosol plays an essential role in acidity effects (Cao and Jang, 2007). Under acidic conditions, the gas-phase oxidation products of eugenol partitioned onto the particle phase would be further oxidized into low-volatility products or produce oligomers by acid-catalyzed heterogeneous reactions, subsequently enhancing SOA yields (Cao and Jang, 2007; Jaoui et al., 2008; T. Liu et al., 2016; Xu et al., 2016). In addition, the formed sulfate not only serves as the substrate for product condensation and likely participates in new particle formation (NPF) (Jaoui et al., 2008; Wang et al., 2016), but also enhances the surface areas of particles to facilitate heterogeneous reactions on aerosols (Xu et al., 2016). These roles of sulfate are also favorable for increasing SOA yields. Recently, Friedman et al. (2016) have indicated that SO2 could participate in the oxidation reactions of α-and β-pinene and perturbs their oxidation in the OFR, but this possible effect could be ignored in this work due to the relatively high RH and the negligible S∕C ratio observed by the HR-ToF-AMS (Friedman et al., 2016).
3.4 Effect of NO2 on SOA formation
It is well known that a high NOx concentration almost always plays a negative role in NPF and SOA formation because the reaction of NO with RO2 radicals would result in the formation of more volatile products compared to the reaction of HO2 with RO2 radicals (Sarrafzadeh et al., 2016). Previous studies reported that nitro-substituted products were the main products for SOA formed from OH-initiated reactions of phenol precursors, including methoxyphenols, in the presence of NOx (Ahmad et al., 2017; Finewax et al., 2018; Lauraguais et al., 2012, 2014b). Thus, the effect of NO2 on SOA formation from eugenol oxidation by OH radicals was investigated. As shown in Fig. 5, the nitrate concentration measured by the HR-ToF-AMS increased as a function of OH exposure in the presence of 40 ppbv NO2, but it was much lower than the sulfate concentration (Fig. 4) even though the OH rate constant for NO2 was faster than that for SO2 (Atkinson et al., 1976; Davis et al., 1979). A possible explanation is that the formed HNO3 mainly existed in the gas phase, and the relatively high temperature (301±1 K) was not favorable for gaseous HNO3 distribution in the particle phase (Wang et al., 2016). It has been indicated that the temperature range for the greatest loss of nitrate is 293–298 K (Keck and Wittmaack, 2005). As illustrated in Fig. 5, the SOA yield enhancement and N∕C ratio both increased first and then decreased with rising OH exposure. The increase in NO2 concentration (40–109 ppbv) was beneficial to SOA yields (0.053–0.062), the N∕C ratio (0.032–0.041), and nitrate formation (4.29–6.30 µg m−3) (Fig. S11). Compared to the presence of 41 ppbv SO2, the maximum SOA yield enhancement (19.17 %) in the presence of 40 ppbv NO2 was lower. For most aromatic precursors, the addition of ppbv levels of NO2 should have a negligible effect on SOA formation because the rate constants of phenoxy radicals with O2 and NO2 are of the order of approximately 10−16 and 10−11 cm3 molecule−1 s−1, respectively (Atkinson and Arey, 2003). But, for phenol precursors, only about 0.5 ppbv NO2 is enough to compete with O2 for the reaction with phenoxy radicals (Finewax et al., 2018). Therefore, the enhancement effect of NO2 on SOA formation might be relevant to the special case of phenols or methoxyphenols but not for other aromatic precursors.

Figure 5Evolution of the enhanced SOA yields, nitrate formation, and N∕C ratio as a function of OH exposure in the presence of 40 ppbv NO2 at an average eugenol concentration of 273 µg m−3.
It is noteworthy that the N∕C ratio is in the range of 0.032–0.043, suggesting that NO2 participated in the OH reaction of eugenol through the addition of the phenoxy radical (Peng and Jimenez, 2017). Recently, Hunter et al. (2014) found that NO2 participated in OH reactions of cyclic alkanes, and the N∕C ratios were in the range of 0.031–0.064, higher than those obtained in this work. The nitro-substituted products were reported to be the main reaction products of the OH reactions of guaiacol and syringol in the presence of NO2 (Lauraguais et al., 2014b; Ahmad et al., 2017). N-containing products might be also formed through reactions involving NO3 radicals, which could be generated by the reaction between NO2 and O3 in this system (Atkinson, 1991). Using the box model (Peng et al., 2015) and the maximum O3 concentration (9.11 ppmv) in this work, the maximum NO3 exposure was calculated to be approximately 1.7×1011 molecules cm−3 s. Compared to the rate constant of eugenol with OH radicals obtained in this work, the rate constant ( cm3 molecule−1 s−1) of eugenol with NO3 radicals was about 2 orders of magnitude lower (Zhang et al., 2016). Thus, the contribution of NO3 radicals to the decay of eugenol was insignificant. The relative low volatility of N-containing products could reasonably contribute to SOA formation (Duporté et al., 2016; J. Liu et al., 2016). In addition, a higher NO2∕NO ratio favors the formation of nitro-substituted products, which are potentially involved in NPF and SOA growth (Pereira et al., 2015). Ng et al. (2007a) also indicated that NOx could be beneficial to SOA formation for sesquiterpenes due to the formation of low-volatility organic nitrates and the isomerization of large alkoxy radicals, resulting in less volatile products. The decrease in the N∕C ratio at high OH exposure suggested that more volatile products were generated through the oxidation of particle-phase species by OH radicals.
The ratios measured by the HR-ToF-AMS are widely used to identify inorganic and organic nitrates. The ratios for inorganic nitrates have been reported to be in the range of 1.08–2.81 (Farmer et al., 2010; Sato et al., 2010). The ratio ranged from 2.06 to 2.54 in this work as determined by the HR-ToF-AMS using ammonium nitrate as the calibration sample. However, the ratios during the oxidation of eugenol in the presence of 40 ppbv NO2 were 3.98–6.09. They were higher than those for inorganic nitrates and consistent with those for organic nitrates (3.82–5.84) from the photooxidation of aromatics (Sato et al., 2010). According to the method described by Fry et al. (2013) (shown in the Supplement), the fraction of organic nitrate was calculated to be in the range of 25.64 % to 82.05 % using the ratios (3.98–6.09) obtained at different OH exposure. The results were comparable to those reported in earlier studies (Liu et al., 2015; Hunter et al., 2014). Liu et al. (2015) reported that N-containing organic mass contributed 31.5±4.4 % to the total SOA derived from m-xylene oxidation by OH radicals. Hunter et al. (2014) estimated the organic nitrate yields of SOA to be 31 %–64 %, formed in OH-initiated reactions of acyclic, monocyclic, and polycyclic alkanes. This range obtained in this work should be the upper limit due to the possibility of C–C bond scission of gas- and particle-phase organics oxidized by high OH exposure. Besides, the maximum yield of nitrates for a single reaction step is expected to be approximately 30 % (Ziemann and Atkinson, 2012); this suggests that multiple reaction steps are needed.
3.5 Atmospheric implications
Biomass burning not only serves as a major contributor of atmospheric primary organic aerosol (POA), but also has great SOA formation potential through atmospheric oxidation (Bruns et al., 2016; Gilardoni et al., 2016; Li et al., 2017; Ciarelli et al., 2017; Ding et al., 2017). Recent studies have indicated that SOA formed from biomass burning plays an important role in haze pollution in China (Li et al., 2017; Ding et al., 2017). Residential combustion (mainly wood burning) could contribute approximately 60 %–70 % to SOA formation in winter at the European scale (Ciarelli et al., 2017). In addition, methoxyphenols are the major component of OA from biomass burning (Bruns et al., 2016; Schauer and Cass, 2000). Based on our results and those of previous studies (Sun et al., 2010; Lauraguais et al., 2012, 2014b; Ahmad et al., 2017; Yee et al., 2013; Ofner et al., 2011), more attention should be paid to SOA formation from the OH oxidation of biomass burning emissions and its subsequent effect on haze evolution, especially in China with nationwide biomass burning and high daytime average [OH] in the ambient atmosphere ( molecules cm−3) (Yang et al., 2017). Meanwhile, the potential contributions of SO2 and NO2 to SOA formation should also be taken into account because the concentrations of NOx and SO2 could be up to 200 ppbv in the severely polluted atmosphere in China (Li et al., 2017). Although eugenol concentrations in this work are higher than those in the ambient atmosphere, the results obtained in this work could provide new information for SOA formation from the atmospheric oxidation of methoxyphenols and might be useful for SOA modeling, especially for air quality simulation modeling of the specific regions experiencing serious pollution caused by fine particulate matter.
N-containing products formed from the oxidation of methoxyphenols could contribute to water-soluble organics in SOA (Lauraguais et al., 2014b; Yang et al., 2016; Zhang et al., 2016), which have been widely detected in atmospheric humic-like substances (HULIS) (Wang et al., 2017). Due to their surface-active and UV-light-absorbing properties, HULIS could influence the formation of cloud condensation nuclei (CCN), solar radiation balance, and photochemical processes in the atmosphere (Wang et al., 2017). In addition, the formation of oligomers in the particle phase via the OH-initiated reaction of methoxyphenols, which has been observed in the aqueous oxidation of phenolic species (Yu et al., 2014), might also enhance light absorption in the UV-visible region. The high reactivity of methoxyphenols toward atmospheric radicals suggests that SOA was formed from their oxidation processes with a relatively high oxidation level, subsequently leading to SOA with strong optical absorption and hygroscopic properties (Lambe et al., 2013; Massoli et al., 2010). Therefore, SOA formed from the reactions of methoxyphenols with atmospheric oxidants might have important effects on air quality and climate. In addition, the experimental results from this study could help to further the understanding of the atmospheric aging process of smoke plumes from biomass burning emissions.
For the first time, the rate constant and SOA formation from the gas-phase reaction of eugenol with OH radicals were investigated in an OFR. The second-order rate constant of eugenol with OH radicals was cm3 molecule−1 s−1, as measured by the relative rate method, and the corresponding atmospheric lifetime was 2.31±0.12 h. In addition, significant SOA formation of eugenol oxidation by OH radicals was observed. The maximum SOA yields (0.11–0.31) obtained at different eugenol concentrations could be expressed well by a one-product model. SOA yield was dependent on OH exposure and eugenol concentration, which first increased and then decreased as a function of OH exposure due to the possible C–C bond scission of gas-phase species by further oxidation or heterogeneous reactions involving OH radicals. The OSC and O∕C ratio both increased significantly as a function of OH exposure, suggesting that SOA became more oxidized. The presence of SO2 and NO2 was helpful to increase SOA yield, and the maximum enhanced yields were 38.6 % and 19.2 %, respectively. The observed N∕C ratio of SOA was in the range of 0.032–0.043, indicating that NO2 participated in the OH-initiated reaction of eugenol, consequently producing organic nitrates. The experimental results might be helpful to further understand the atmospheric chemical behavior of eugenol and its SOA formation potential from OH oxidation in the atmosphere.
The experimental data are available upon request to the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-2001-2019-supplement.
CL, YL, and HH designed the research and wrote the paper. CL, TC, and JL performed the experiments. CL, YL, TC, JL, and HH carried out the data analysis. All authors contributed to the final paper.
The authors declare that they have no conflict of interest.
This work was financially supported by the National Key R&D Program of
China (2016YFC0202700), the National Natural Science Foundation of China
(21607088 and 41877306), a China Postdoctoral Science Foundation funded
project (2017M620071), and the Applied Basic Research Project of Science and
Technology Department of Sichuan Province (2018JY0303). Yongchun Liu would like to
thank the Beijing University of Chemical Technology for financial support.
The authors would also like to acknowledge the experimental help provided by Xiaolei
Bao from the Hebei Provincial Academy of Environmental Sciences, Shijiazhuang,
China.
Edited by: Rainer Volkamer
Reviewed by: two anonymous referees
Ahlberg, E., Falk, J., Eriksson, A., Holst, T., Brune, W. H., Kristensson, A., Roldin, P., and Svenningsson, B.: Secondary organic aerosol from VOC mixtures in an oxidation flow reactor, Atmos. Environ., 161, 210–220, https://doi.org/10.1016/j.atmosenv.2017.05.005, 2017.
Ahmad, W., Coeur, C., Tomas, A., Fagniez, T., Brubach, J.-B., and Cuisset, A.: Infrared spectroscopy of secondary organic aerosol precursors and investigation of the hygroscopicity of SOA formed from the OH reaction with guaiacol and syringol, Appl. Opt., 56, 116–122, https://doi.org/10.1364/ao.56.00e116, 2017.
Atkinson, R., Perry, R. A., and Pitts, J. N.: Rate constants for the reactions of the OH radicals with NO2 (M = Ar and N2) and SO2 (M = Ar), J. Chem. Phys., 65, 306–310, https://doi.org/10.1063/1.432770, 1976.
Atkinson, R.: Kinetics and mechanisms of the gas-phase reactions of the NO3 radical with organic compounds, J. Phys. Chem. Ref. Data, 20, 459–507, https://doi.org/10.1063/1.555887, 1991.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bari, M. A., Baumbach, G., Kuch, B., and Scheffknecht, G.: Wood smoke as a source of particle-phase organic compounds in residential areas, Atmos. Environ., 43, 4722–4732, https://doi.org/10.1016/j.atmosenv.2008.09.006, 2009.
Bari, M. A., Baumbach, G., Kuch, B., and Scheffknecht, G.: Temporal variation and impact of wood smoke pollution on a residential area in southern Germany, Atmos. Environ., 44, 3823–3832, https://doi.org/10.1016/j.atmosenv.2010.06.031, 2010.
Bolling, A. K., Pagels, J., Yttri, K. E., Barregard, L., Sallsten, G., Schwarze, P. E., and Boman, C.: Health effects of residential wood smoke particles: the importance of combustion conditions and physicochemical particle properties, Part. Fibre Toxicol., 6, 29, https://doi.org/10.1186/1743-8977-6-29, 2009.
Bruns, E. A., El Haddad, I., Keller, A., Klein, F., Kumar, N. K., Pieber, S. M., Corbin, J. C., Slowik, J. G., Brune, W. H., Baltensperger, U., and Prévôt, A. S. H.: Inter-comparison of laboratory smog chamber and flow reactor systems on organic aerosol yield and composition, Atmos. Meas. Tech., 8, 2315–2332, https://doi.org/10.5194/amt-8-2315-2015, 2015.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prevot, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6., 27881, https://doi.org/10.1038/srep27881, 2016.
Cao, G. and Jang, M.: Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx, Atmos. Environ., 41, 7603–7613, https://doi.org/10.1016/j.atmosenv.2007.05.034, 2007.
Chen, Y. and Bond, T. C.: Light absorption by organic carbon from wood combustion, Atmos. Chem. Phys., 10, 1773–1787, https://doi.org/10.5194/acp-10-1773-2010, 2010.
Ciarelli, G., Aksoyoglu, S., El Haddad, I., Bruns, E. A., Crippa, M., Poulain, L., Äijälä, M., Carbone, S., Freney, E., O'Dowd, C., Baltensperger, U., and Prévôt, A. S. H.: Modelling winter organic aerosol at the European scale with CAMx: evaluation and source apportionment with a VBS parameterization based on novel wood burning smog chamber experiments, Atmos. Chem. Phys., 17, 7653–7669, https://doi.org/10.5194/acp-17-7653-2017, 2017.
Coeur-Tourneur, C., Cassez, A., and Wenger, J. C.: Rate Coefficients for the gas-phase reaction of hydroxyl radicals with 2-methoxyphenol (guaiacol) and related compounds, J. Phys. Chem. A, 114, 11645–11650, https://doi.org/10.1021/jp1071023, 2010a.
Coeur-Tourneur, C., Foulon, V., and Lareal, M.: Determination of aerosol yields from 3-methylcatechol and 4-methylcatechol ozonolysis in a simulation chamber, Atmos. Environ., 44, 852–857, https://doi.org/10.1016/j.atmosenv.2009.11.027, 2010b.
Davis, D. D., Ravishankara, A. R., and Fischer, S.: SO2 oxidation via the hydroxyl radical: Atmospheric fate of HSOx radicals, Geophys. Res. Lett., 6, 113–116, https://doi.org/10.1029/GL006i002p00113, 1979.
DeCarlo, P. F., Slowik, J. G., Worsnop, D. R., Davidovits, P., and Jimenez, J. L.: Particle morphology and density characterization by combined mobility and aerodynamic diameter measurements, Part 1: Theory, Aerosol Sci. Technol., 38, 1185–1205, https://doi.org/10.1080/027868290903907, 2004.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Dills, R. L., Paulsen, M., Ahmad, J., Kalman, D. A., Elias, F. N., and Simpson, C. D.: Evaluation of urinary methoxyphenols as biomarkers of woodsmoke exposure, Environ. Sci. Technol., 40, 2163–2170, https://doi.org/10.1021/es051886f, 2006.
Ding, X., Zhang, Y.-Q., He, Q.-F., Yu, Q.-Q., Wang, J.-Q., Shen, R.-Q., Song, W., Wang, Y.-S., and Wang, X.-M.: Significant increase of aromatics-derived secondary organic aerosol during fall to winter in China, Environ. Sci. Technol., 51, 7432–7441, https://doi.org/10.1021/acs.est.6b06408, 2017.
Duporté, G., Parshintsev, J., Barreira, L. M. F., Hartonen, K., Kulmala, M., and Riekkola, M.-L.: Nitrogen-containing low volatile compounds from pinonaldehyde-dimethylamine reaction in the atmosphere: A laboratory and field study, Environ. Sci. Technol., 50, 4693–4700, https://doi.org/10.1021/acs.est.6b00270, 2016.
El Zein, A., Coeur, C., Obeid, E., Lauraguais, A., and Fagniez, T.: Reaction kinetics of catechol (1,2-benzenediol) and guaiacol (2-methoxyphenol) with ozone, J. Phys. Chem. A, 119, 6759–6765, https://doi.org/10.1021/acs.jpca.5b00174, 2015.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Finewax, Z., de Gouw, J. A., and Ziemann, P. J.: Identification and quantification of 4-nitrocatechol formed from OH and NO3 radical-initiated reactions of catechol in air in the presence of NOx: Implications for secondary organic aerosol formation from biomass burning, Environ. Sci. Technol., 52, 1981–1989, https://doi.org/10.1021/acs.est.7b05864, 2018.
Friedman, B., Brophy, P., Brune, W. H., and Farmer, D. K.: Anthropogenic sulfur perturbations on biogenic oxidation: SO2 additions impact gas-phase OH oxidation products of alpha- and beta-pinene, Environ. Sci. Technol., 50, 1269–1279, https://doi.org/10.1021/acs.est.5b05010, 2016.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Gordon, T. D., Presto, A. A., Nguyen, N. T., Robertson, W. H., Na, K., Sahay, K. N., Zhang, M., Maddox, C., Rieger, P., Chattopadhyay, S., Maldonado, H., Maricq, M. M., and Robinson, A. L.: Secondary organic aerosol production from diesel vehicle exhaust: impact of aftertreatment, fuel chemistry and driving cycle, Atmos. Chem. Phys., 14, 4643–4659, https://doi.org/10.5194/acp-14-4643-2014, 2014.
http://www.aim.env.uea.ac.uk/aim/model2/model2a.php, last access: 18 June 2018.
Hunter, J. F., Carrasquillo, A. J., Daumit, K. E., and Kroll, J. H.: Secondary organic aerosol formation from acyclic, monocyclic, and polycyclic alkanes, Environ. Sci. Technol., 48, 10227–10234, https://doi.org/10.1021/es502674s, 2014.
Jaoui, M., Edney, E. O., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Surratt, J. D., and Seinfeld, J. H.: Formation of secondary organic aerosol from irradiated alpha-pinene/toluene/NOx mixtures and the effect of isoprene and sulfur dioxide, J. Geophys. Res.-Atmos., 113, D09303, https://doi.org/10.1029/2007jd009426, 2008.
Jeong, C.-H., Evans, G. J., Dann, T., Graham, M., Herod, D., Dabek-Zlotorzynska, E., Mathieu, D., Ding, L., and Wang, D.: Influence of biomass burning on wintertime fine particulate matter: Source contribution at a valley site in rural British Columbia, Atmos. Environ., 42, 3684–3699, https://doi.org/10.1016/j.atmosenv.2008.01.006, 2008.
Kang, E., Root, M. J., Toohey, D. W., and Brune, W. H.: Introducing the concept of Potential Aerosol Mass (PAM), Atmos. Chem. Phys., 7, 5727–5744, https://doi.org/10.5194/acp-7-5727-2007, 2007.
Keck, L. and Wittmaack, K.: Effect of filter type and temperature on volatilisation losses from ammonium salts in aerosol matter, Atmos. Environ., 39, 4093–4100, https://doi.org/10.1016/j.atmosenv.2005.03.029, 2005.
Kleindienst, T. E., Shepson, P. B., Edney, E. O., Claxton, L. D., and Cupitt, L. T.: Wood smoke: Measurement of the mutagenic activities of its gas- and particulate-phase photooxidation products, Environ. Sci. Technol., 20, 493–501, https://doi.org/10.1021/es00147a009, 1986.
Kleindienst, T. E., Edney, E. O., Lewandowski, M., Offenberg, J. H., and Jaoui, M.: Secondary organic carbon and aerosol yields from the irradiations of isoprene and alpha-pinene in the presence of NOx and SO2, Environ. Sci. Technol., 40, 3807–3812, https://doi.org/10.1021/es052446r, 2006.
Kramp, F. and Paulson, S. E.: On the uncertainties in the rate coefficients for OH reactions with hydrocarbons, and the rate coefficients of the 1,3,5-trimethylbenzene and m-xylene reactions with OH radicals in the gas phase, J. Phys. Chem. A, 102, 2685–2690, https://doi.org/10.1021/jp973289o, 1998.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, https://doi.org/10.1038/nchem.948, 2011.
Lambe, A. T., Cappa, C. D., Massoli, P., Onasch, T. B., Forestieri, S. D., Martin, A. T., Cummings, M. J., Croasdale, D. R., Brune, W. H., Worsnop, D. R., and Davidovits, P.: Relationship between oxidation level and optical properties of secondary organic aerosol, Environ. Sci. Technol., 47, 6349–6357, https://doi.org/10.1021/es401043j, 2013.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Lauraguais, A., Coeur-Tourneur, C., Cassez, A., and Seydi, A.: Rate constant and secondary organic aerosol yields for the gas-phase reaction of hydroxyl radicals with syringol (2,6-dimethoxyphenol), Atmos. Environ., 55, 43–48, https://doi.org/10.1016/j.atmosenv.2012.02.027, 2012.
Lauraguais, A., Bejan, I., Barnes, I., Wiesen, P., Coeur-Tourneur, C., and Cassez, A.: Rate coefficients for the gas-phase reaction of chlorine atoms with a series of methoxylated aromatic compounds, J. Phys. Chem. A, 118, 1777–1784, https://doi.org/10.1021/jp4114877, 2014a.
Lauraguais, A., Coeur-Tourneur, C., Cassez, A., Deboudt, K., Fourmentin, M., and Choel, M.: Atmospheric reactivity of hydroxyl radicals with guaiacol (2-methoxyphenol), a biomass burning emitted compound: Secondary organic aerosol formation and gas-phase oxidation products, Atmos. Environ., 86, 155–163, https://doi.org/10.1016/j.atmosenv.2013.11.074, 2014b.
Lauraguais, A., Bejan, I., Barnes, I., Wiesen, P., and Coeur, C.: Rate coefficients for the gas-phase reactions of hydroxyl radicals with a series of methoxylated aromatic compounds, J. Phys. Chem. A, 119, 6179–6187, https://doi.org/10.1021/acs.jpca.5b03232, 2015.
Lauraguais, A., El Zein, A., Coeur, C., Obeid, E., Cassez, A., Rayez, M.-T., and Rayez, J.-C.: Kinetic study of the gas-phase reactions of nitrate radicals with methoxyphenol compounds: Experimental and theoretical approaches, J. Phys. Chem. A, 120, 2691–2699, https://doi.org/10.1021/acs.jpca.6b02729, 2016.
Li, H., Zhang, Q., Zhang, Q., Chen, C., Wang, L., Wei, Z., Zhou, S., Parworth, C., Zheng, B., Canonaco, F., Prévôt, A. S. H., Chen, P., Zhang, H., Wallington, T. J., and He, K.: Wintertime aerosol chemistry and haze evolution in an extremely polluted city of the North China Plain: significant contribution from coal and biomass combustion, Atmos. Chem. Phys., 17, 4751–4768, https://doi.org/10.5194/acp-17-4751-2017, 2017.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., de Gouw, J. A., and Jimenez, J. L.: Modeling the radical chemistry in an oxidation flow reactor: Radical formation and recycling, sensitivities, and the OH exposure estimation equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Liu, J., Lin, P., Laskin, A., Laskin, J., Kathmann, S. M., Wise, M., Caylor, R., Imholt, F., Selimovic, V., and Shilling, J. E.: Optical properties and aging of light-absorbing secondary organic aerosol, Atmos. Chem. Phys., 16, 12815–12827, https://doi.org/10.5194/acp-16-12815-2016, 2016.
Liu, T., Wang, X., Hu, Q., Deng, W., Zhang, Y., Ding, X., Fu, X., Bernard, F., Zhang, Z., Lu, S., He, Q., Bi, X., Chen, J., Sun, Y., Yu, J., Peng, P., Sheng, G., and Fu, J.: Formation of secondary aerosols from gasoline vehicle exhaust when mixing with SO2, Atmos. Chem. Phys., 16, 675-689, https://doi.org/10.5194/acp-16-675-2016, 2016.
Liu, Y., Huang, L., Li, S.-M., Harner, T., and Liggio, J.: OH-initiated heterogeneous oxidation of tris-2-butoxyethyl phosphate: implications for its fate in the atmosphere, Atmos. Chem. Phys., 14, 12195–12207, https://doi.org/10.5194/acp-14-12195-2014, 2014a.
Liu, Y., Liggio, J., Harner, T., Jantunen, L., Shoeib, M., and Li, S.-M.: Heterogeneous OH initiated oxidation: A possible explanation for the persistence of organophosphate flame retardants in air, Environ. Sci. Technol., 48, 1041–1048, https://doi.org/10.1021/es404515k, 2014b.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Mao, J., Ren, X., Brune, W. H., Olson, J. R., Crawford, J. H., Fried, A., Huey, L. G., Cohen, R. C., Heikes, B., Singh, H. B., Blake, D. R., Sachse, G. W., Diskin, G. S., Hall, S. R., and Shetter, R. E.: Airborne measurement of OH reactivity during INTEX-B, Atmos. Chem. Phys., 9, 163–173, https://doi.org/10.5194/acp-9-163-2009, 2009.
Massoli, P., Lambe, A. T., Ahern, A. T., Williams, L. R., Ehn, M., Mikkila, J., Canagaratna, M. R., Brune, W. H., Onasch, T. B., Jayne, J. T., Petaja, T., Kulmala, M., Laaksonen, A., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Relationship between aerosol oxidation level and hygroscopic properties of laboratory generated secondary organic aerosol (SOA) particles, Geophys. Res. Lett., 37, L24801, https://doi.org/10.1029/2010gl045258, 2010.
McMurry, J. E.: Organic Chemistry, 6th edn., Brooks/Cole, Belmont, CA, 2004.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007a.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007b.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Nolte, C. G., Schauer, J. J., Cass, G. R., and Simoneit, B. R. T.: Highly polar organic compounds present in wood smoke and in the ambient atmosphere, Environ. Sci. Technol., 35, 1912–1919, https://doi.org/10.1021/es001420r, 2001.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943, 1996.
Ofner, J., Krüger, H.-U., Grothe, H., Schmitt-Kopplin, P., Whitmore, K., and Zetzsch, C.: Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS, Atmos. Chem. Phys., 11, 1–15, https://doi.org/10.5194/acp-11-1-2011, 2011.
Ortega, A. M., Hayes, P. L., Peng, Z., Palm, B. B., Hu, W., Day, D. A., Li, R., Cubison, M. J., Brune, W. H., Graus, M., Warneke, C., Gilman, J. B., Kuster, W. C., de Gouw, J., Gutiérrez-Montes, C., and Jimenez, J. L.: Real-time measurements of secondary organic aerosol formation and aging from ambient air in an oxidation flow reactor in the Los Angeles area, Atmos. Chem. Phys., 16, 7411–7433, https://doi.org/10.5194/acp-16-7411-2016, 2016.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Palm, B. B., de Sá, S. S., Day, D. A., Campuzano-Jost, P., Hu, W., Seco, R., Sjostedt, S. J., Park, J.-H., Guenther, A. B., Kim, S., Brito, J., Wurm, F., Artaxo, P., Thalman, R., Wang, J., Yee, L. D., Wernis, R., Isaacman-VanWertz, G., Goldstein, A. H., Liu, Y., Springston, S. R., Souza, R., Newburn, M. K., Alexander, M. L., Martin, S. T., and Jimenez, J. L.: Secondary organic aerosol formation from ambient air in an oxidation flow reactor in central Amazonia, Atmos. Chem. Phys., 18, 467–493, https://doi.org/10.5194/acp-18-467-2018, 2018.
Peng, Z., Day, D. A., Stark, H., Li, R., Lee-Taylor, J., Palm, B. B., Brune, W. H., and Jimenez, J. L.: HOx radical chemistry in oxidation flow reactors with low-pressure mercury lamps systematically examined by modeling, Atmos. Meas. Tech., 8, 4863–4890, https://doi.org/10.5194/amt-8-4863-2015, 2015.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Peng, Z. and Jimenez, J. L.: Modeling of the chemistry in oxidation flow reactors with high initial NO, Atmos. Chem. Phys., 17, 11991–12010, https://doi.org/10.5194/acp-17-11991-2017, 2017.
Pereira, K. L., Hamilton, J. F., Rickard, A. R., Bloss, W. J., Alam, M. S., Camredon, M., Ward, M. W., Wyche, K. P., Munoz, A., Vera, T., Vazquez, M., Borras, E., and Rodenas, M.: Insights into the formation and evolution of individual compounds in the particulate phase during aromatic photo-oxidation, Environ. Sci. Technol., 49, 13168–13178, https://doi.org/10.1021/acs.est.5b03377, 2015.
Priya, A. M. and Lakshmipathi, S.: DFT study on abstraction reaction mechanism of oh radical with 2-methoxyphenol, J. Phys. Org. Chem., 30, e3713, https://doi.org/10.1002/poc.3713, 2017.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Sato, K., Takami, A., Isozaki, T., Hikida, T., Shimono, A., and Imamura, T.: Mass spectrometric study of secondary organic aerosol formed from the photo-oxidation of aromatic hydrocarbons, Atmos. Environ., 44, 1080–1087, https://doi.org/10.1016/j.atmosenv.2009.12.013, 2010.
Schauer, J. J. and Cass, G. R.: Source apportionment of wintertime gas-phase and particle-phase air pollutants using organic compounds as tracers, Environ. Sci. Technol., 34, 1821–1832, https://doi.org/10.1021/es981312t, 2000.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of emissions from air pollution sources, 3. C-1-C-29 organic compounds from fireplace combustion of wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Simonen, P., Saukko, E., Karjalainen, P., Timonen, H., Bloss, M., Aakko-Saksa, P., Rönkkö, T., Keskinen, J., and Dal Maso, M.: A new oxidation flow reactor for measuring secondary aerosol formation of rapidly changing emission sources, Atmos. Meas. Tech., 10, 1519–1537, https://doi.org/10.5194/amt-10-1519-2017, 2017.
Simpson, C. D., Paulsen, M., Dills, R. L., Liu, L. J. S., and Kalman, D. A.: Determination of methoxyphenols in ambient atmospheric particulate matter: Tracers for wood combustion, Environ. Sci. Technol., 39, 631–637, https://doi.org/10.1021/es0486871, 2005.
Sun, Y. L., Zhang, Q., Anastasio, C., and Sun, J.: Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 10, 4809–4822, https://doi.org/10.5194/acp-10-4809-2010, 2010.
Thuner, L. P., Bardini, P., Rea, G. J., and Wenger, J. C.: Kinetics of the gas-phase reactions of OH and NO3 radicals with dimethylphenols, J. Phys. Chem. A, 108, 11019–11025, https://doi.org/10.1021/jp046358p, 2004.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
US EPA: Estimation Programs Interface Suite™ for Microsoft® Windows, v 4.11, United States Environmental Protection Agency, Washington, DC, USA, 2012.
Wang, D., Zhou, B., Fu, Q., Zhao, Q., Zhang, Q., Chen, J., Yang, X., Duan, Y., and Li, J.: Intense secondary aerosol formation due to strong atmospheric photochemical reactions in summer: observations at a rural site in eastern Yangtze River Delta of China, Sci. Total Environ., 571, 1454–1466, https://doi.org/10.1016/j.scitotenv.2016.06.212, 2016.
Wang, Y., Hu, M., Lin, P., Guo, Q., Wu, Z., Li, M., Zeng, L., Song, Y., Zeng, L., Wu, Y., Guo, S., Huang, X., and He, L.: Molecular characterization of nitrogen-containing organic compounds in humic-like substances emitted from straw residue burning, Environ. Sci. Technol., 51, 5951–5961, https://doi.org/10.1021/acs.est.7b00248, 2017.
Ward, T. J., Rinehart, L. R., and Lange, T.: The 2003/2004 Libby, Montana PM2.5 source apportionment research study, Aerosol Sci. Technol., 40, 166–177, https://doi.org/10.1080/02786820500494536, 2006.
Xu, L., Middlebrook, A. M., Liao, J., de Gouw, J. A., Guo, H., Weber, R. J., Nenes, A., Lopez-Hilfiker, F. D., Lee, B. H., Thornton, J. A., Brock, C. A., Neuman, J. A., Nowak, J. B., Pollack, I. B., Welti, A., Graus, M., Warneke, C., and Ng, N. L.: Enhanced formation of isoprene-derived organic aerosol in sulfur-rich power plant plumes during Southeast Nexus, J. Geophys. Res.-Atmos., 121, 11137–11153, https://doi.org/10.1002/2016jd025156, 2016.
Yang, B., Zhang, H., Wang, Y., Zhang, P., Shu, J., Sun, W., and Ma, P.: Experimental and theoretical studies on gas-phase reactions of NO3 radicals with three methoxyphenols: Guaiacol, creosol, and syringol, Atmos. Environ., 125, 243–251, https://doi.org/10.1016/j.atmosenv.2015.11.028, 2016.
Yang, Y., Shao, M., Keßel, S., Li, Y., Lu, K., Lu, S., Williams, J., Zhang, Y., Zeng, L., Nölscher, A. C., Wu, Y., Wang, X., and Zheng, J.: How the OH reactivity affects the ozone production efficiency: case studies in Beijing and Heshan, China, Atmos. Chem. Phys., 17, 7127–7142, https://doi.org/10.5194/acp-17-7127-2017, 2017.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Zhang, H., Yang, B., Wang, Y., Shu, J., Zhang, P., Ma, P., and Li, Z.: Gas-phase reactions of methoxyphenols with NO3 radicals: Kinetics, products, and mechanisms, J. Phys. Chem. A, 120, 1213–1221, https://doi.org/10.1021/acs.jpca.5b10406, 2016.
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Be, A. G., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z., Gold, A., Graf, S., Cubison, M. J., Groessl, M., Jayne, J. T., Worsnop, D. R., and Canagaratna, M. R.: Highly oxygenated multifunctional compounds in alpha-pinene secondary organic aerosol, Environ. Sci. Technol., 51, 5932–5940, https://doi.org/10.1021/acs.est.6b06588, 2017.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.