the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Feb 2019
| 07 Feb 2019
Free tropospheric aerosols at the Mt. Bachelor Observatory: more oxidized and higher sulfate content compared to boundary layer aerosols
Shan Zhou
Sonya Collier
Daniel A. Jaffe
Understanding the properties and life cycle processes of aerosol particles in regional air masses is crucial for constraining the climate impacts of aerosols on a global scale. In this study, characteristics of aerosols in the boundary layer (BL) and free troposphere (FT) of a remote continental region in the western US were studied using a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS) deployed at the Mount Bachelor Observatory (MBO; 2763 m a.s.l.) in central Oregon in summer 2013. In the absence of wildfire influence, the average (±1σ) concentration of non-refractory submicrometer particulate matter (NR-PM1) at MBO was 2.8 (±2.8) µg m−3 and 84 % of the mass was organic. The other NR-PM1 components were sulfate (11 %), ammonium (2.8 %), and nitrate (0.9 %). The organic aerosol (OA) at MBO from these clean periods showed clear diurnal variations driven by the boundary layer dynamics with significantly higher concentrations occurring during daytime, upslope conditions. NR-PM1 contained a higher mass fraction of sulfate and was frequently acidic when MBO resided in the FT. In addition, OA in the FT was found to be highly oxidized (average O∕C of 1.17) with low volatility while OA in BL-influenced air masses was moderately oxidized (average O∕C of 0.67) and semivolatile. There are indications that the BL-influenced OA observed at MBO was more enriched in organonitrates and organosulfur compounds (e.g., MSA) and appeared to be representative of biogenic secondary organic aerosol (SOA) originated in the BL. A summary of the chemical compositions of NR-PM1 measured at a number of other high-altitude locations in the world is presented and similar contrasts between FT and BL aerosols were observed. The significant compositional and physical differences observed between FT and BL aerosols may have important implications for understanding the climate effects of regional background aerosols.
- Article
(2676 KB) - Full-text XML
-
Supplement
(1650 KB) - BibTeX
- EndNote





Atmospheric aerosols can scatter and absorb incident sunlight, therefore altering the radiation budget of the earth directly. Depending on their chemical composition and microphysical properties, aerosol particles can also act as cloud condensation nuclei and or ice nuclei and affect climate indirectly by altering the lifetime and optical properties of clouds. Understanding the properties and the life cycle processes of atmospheric aerosols is important for reducing the uncertainties in aerosol climate forcing (Boucher, 2013).
Aerosols and their precursor gases are mostly emitted in the planetary boundary layer (PBL) but can be transported into the free troposphere (FT) through convection and frontal uplift. In the FT, aerosols are subjected to less efficient dry deposition and can have longer lifetimes than those at lower altitudes, facilitating regional recirculation or long-distance transport (Jaffe et al., 2005a; Dunlea et al., 2009; Sun et al., 2009). Under certain atmospheric conditions, aerosols in the FT can be entrained into the BL, affecting remote regions where local emissions may be minimal (Schroder et al., 2002; Timonen et al., 2013; Wang et al., 2016). A quantitative understanding of aerosol properties and processes in regional background air masses and in the FT would be useful for improving chemical transport models and global climate simulations.
High-altitude mountaintop observatories are important platforms for studying aerosols in regional and FT air masses without the added expense and difficulty of making airborne measurements. Another main advantage of mountaintop observatories is long-term continuous measurements, which are invaluable for statistics. Various mountaintop sites have been operated in North America and Europe to perform long-term measurements on aerosol optical properties, number, and size distributions and trace gases in continental background air masses (Jaffe et al., 2005b; Van Dingenen et al., 2005; Reidmiller et al., 2010; Fischer et al., 2011; Hallar et al., 2011, 2016; Rose et al., 2015; Bianchi et al., 2016; Tröstl et al., 2016; Zhang and Jaffe, 2017). Aerosol chemical composition has also been studied from high-elevation sites, through both filter collection followed by offline analysis (e.g., Takahama et al., 2011; Ahlm et al., 2013; Hallar et al., 2013; Dzepina et al., 2015) and real-time measurements using online aerosol mass spectrometers (e.g., Zhang et al., 2007; Cozic et al., 2008; Sun et al., 2009; Fröhlich et al., 2015; Rinaldi et al., 2015; Freney et al., 2016). These measurements have provided valuable information on the chemical and physical properties of remote aerosols in the FT and PBL as well as how they are influenced by various sources (e.g., biomass burning, BB; dust; and biogenic emissions) and atmospheric processes (e.g., new particle formation, NPF; long-range transport; and cloud processing).
The Mt. Bachelor Observatory (MBO) is a high-altitude atmospheric research site that has been utilized for studying atmospheric chemistry in the western US for more than a decade (Weiss-Penzias et al., 2006; Timonen et al., 2013). The observatory is located at 2763 m a.s.l. at the summit of Mt. Bachelor, a dormant volcano in the Deschutes National Forest in central Oregon (43.98∘ N, 121.69∘ W). Due to its elevation, MBO is situated in the FT at night and is under the influence of upslope flow from the PBL air during the daytime (McClure et al., 2016). The remote characteristics of the site make MBO an ideal location for studying transported plumes, such as biomass burning plumes from regional and distant sources (Jaffe et al., 2005b; Timonen et al., 2014; Briggs et al., 2016; Laing et al., 2016; Zhang and Jaffe, 2017; Zhang et al., 2018) and long-range transport of Asian pollution in the spring (Jaffe et al., 2005a; Weiss-Penzias et al., 2006; Fischer et al., 2010; Ambrose et al., 2011).
Continuous measurements of trace gases (e.g., ozone, carbon monoxide, carbon dioxide, mercury, nitrogen oxides) and aerosol optical properties have been made at MBO since 2004. In summer 2013, a high-resolution time-of-flight aerosol mass spectrometer (HR-AMS; Aerodyne Research, Inc.) was deployed at MBO as part of the US Department of Energy-sponsored Biomass Burning Observation Project (BBOP) (Collier et al., 2016; Zhou et al., 2017). This was the first real-time, highly time-resolved aerosol chemical measurement study performed at this site. MBO was frequently impacted by transported wildfire plumes during summer 2017 (Collier et al., 2016; Zhou et al., 2017), but during two periods, 25–30 July and 17–21 August, of this study the site was not influenced by wildfires and the concentrations of air pollutants remained low. Here, we focus on analyzing these clean periods in order to examine the chemical and physical properties of regional background aerosols and to investigate the differences of aerosol characteristics and processes in the PBL and the FT over the western US.
The HR-AMS was deployed at MBO from 25 July to 25 August 2013, as part of the BBOP campaign. Ambient aerosols were drawn through a PM2.5 cyclone inlet and dehumidified by a Nafion dryer to eliminate potential relative humidity (RH) effects on collection efficiency (CE). Treated particles then alternated between a heated thermodenuder (TD) line and an ambient bypass line every 5 min before entering the HR-AMS. Aerosol scattering (TSI nephelometer; 1 µm size cut), aerosol absorption (Tricolor Absorption Photometer, Brechtel; 1 µm size cut), CO and CO2 (Picarro Cavity Ring-Down Spectroscopy G2502), O3 (Dasibi), NOx (Air Quality Design two-channel chemiluminescence), NOy (chemiluminescence), and peroxyacetyl nitrate (PAN; custom gas chromatograph) were also measured. Water vapor mixing ratios were calculated from the measured temperature, relative humidity (Campbell Scientific HMP 45C), and pressure (Vaisala PTB101B) following Bolton (1980) and typically agreed to within ±15 % and ±0.3 g kg−1 (Ambrose et al., 2011). Additional details of the instrumentation and methodology can be found in previous publications (Briggs et al., 2016; Collier et al., 2016; Zhou et al., 2017).
HR-AMS data were analyzed using the established data analysis software tool Squirrel (v1.53) and Pika (v1.12; http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware, last access: 7 April 2017). A composition-dependent CE (ranging from 0.5 to 1; average = 0.66) was applied based on the algorithm by Middlebrook et al. (2012) to account for possible CE changes induced by changes in particle phase in the AMS. A time-dependent gas-phase subtraction (Collier and Zhang, 2013) was performed to improve the quantification of organic aerosol (OA), which is critical for low-aerosol-loading conditions (Setyan et al., 2012). Elemental analysis of high-resolution mass spectra (HRMS) utilized both the Aiken-ambient (AA) method (Aiken et al., 2008) and the improved-ambient (IA) method (Canagaratna et al., 2015).
Positive matrix factorization (PMF) was executed using the PMF2 algorithm (Paatero and Tapper, 1994) in the PET v2.05 program (Ulbrich et al., 2009) on the combined spectral matrices of organic and inorganic species (Sun et al., 2012; Zhou et al., 2017) during the clean periods without wildfire impact (i.e., 25–30 July and 17–21 August). Organic ions at m∕z 12–180 and major inorganic ions, i.e., SO+, , , , , and for sulfate; NO+ and for nitrate; NH+, , and for ammonium; and HCl+ for chloride were included. The error matrix was pretreated based on the procedures described in Ulbrich et al. (2009). After PMF analysis, the mass concentration of each OA factor was derived from the sum of organic signals in the corresponding mass spectrum after applying the default relative ionization efficiency (RIE = 1.4) for organics and the time-dependent CE. The solutions for two to five factors were explored with varying rotational parameters (−0.5 ≤ FPEAK ≤ 0.5, in increments of 0.1). Following the procedure listed in Table 1 in Zhang et al. (2011), PMF solutions were evaluated by investigating the key diagnostic plots, mass spectra, correlations with external tracers, and diurnal profiles. As shown in Fig. S1 in the Supplement, the two-factor solution showed relatively large residual while the four-factor solution showed signs of factor splitting. The three-factor solution resolved a less oxidized oxygenated OA (OOA) factor, a more oxidized OOA associated with some sulfate signals, and a sulfate-dominated OOA (Figs. S2 and S3 in the Supplement). As the sulfate-dominated OOA accounted for only 3 % of the total organic signal and its O∕C and HRMS highly resembled those in the more oxidized OOA factor (Fig. S3 in the Supplement), these two factors were combined to form a so-called “highly oxidized OOA” factor which has an O∕C of 1.17. Based on the chemical, physical characteristics, and the volatility properties (see detailed discussions in Sect. 3.3), the less oxidized OOA was found to be semivolatile OOA (SV-OOA) mainly associated with fresher air masses from the BL, whereas the highly oxidized OOA was comprised of low-volatility organic compounds (LV-OOA) representing regional background OA in the FT. Furthermore, the time series and mass spectra of the SV-OOA and LV-OOA derived here agreed well with the two background OOAs derived from PMF analysis of the whole dataset, including the clean periods discussed in this study and the periods influenced by wildfires (Zhou et al., 2017) (Figs. S4 and S5 in the Supplement; r2 > 0.9). This result suggests that the PMF results for the clean periods are statistically significant and robust. All aerosol data in this analysis are reported at ambient condition, except for aerosol light scattering, which is reported at standard temperature and pressure (STP; T=273 K and P=1013.25 hPa).
3.1 Temporal and diurnal variations of regional background aerosols observed at MBO
While observations at MBO were made continuously from 25 July to 25 August, for this work, we use only data from 25 to 30 July and 17 to 21 August 2013, which were classified as periods free of wildfire influence. The HR-AMS indicator for biomass burning influence, namely the fraction of (m∕z = 60.021) signal over total OA (f60), was used for differentiating wildfire influences. Periods with f60 below 0.3 % (Fig. S6 in the Supplement) likely received negligible influence from BB (Cubison et al., 2011) and thus were classified as clean periods. As shown in Fig. 1, throughout the clean periods, the CO mixing ratio and submicron aerosol light scattering at 550 nm (σ550 nm) were below 120 ppb and 25 Mm−1 at STP, respectively, similar to values previously observed at MBO under clean conditions (Fischer et al., 2011; Timonen et al., 2014). The site was influenced by transported wildfire plumes during the other periods of BBOP and air pollutant levels increased substantially; e.g., CO and σ550 nm increased by up to 8–10 times compared to the clean periods and NR-PM1 reached up to 140 µg m−3 (Zhou et al., 2017). Aerosol absorption data were available for the second clean period (17–21 August) and the average (±1σ) elemental carbon mass concentrations were estimated to be only 0.04 (±0.14) µgC m−3, further indicating a lack of BB influences. Additionally, although winds at MBO showed a persistent westerly component (Figs. 1a and S7b in the Supplement), the bivariate polar plot of NR-PM1 concentrations exhibited a dispersed profile (Fig. S7c in the Supplement), indicating regional sources of aerosols during the clean periods.
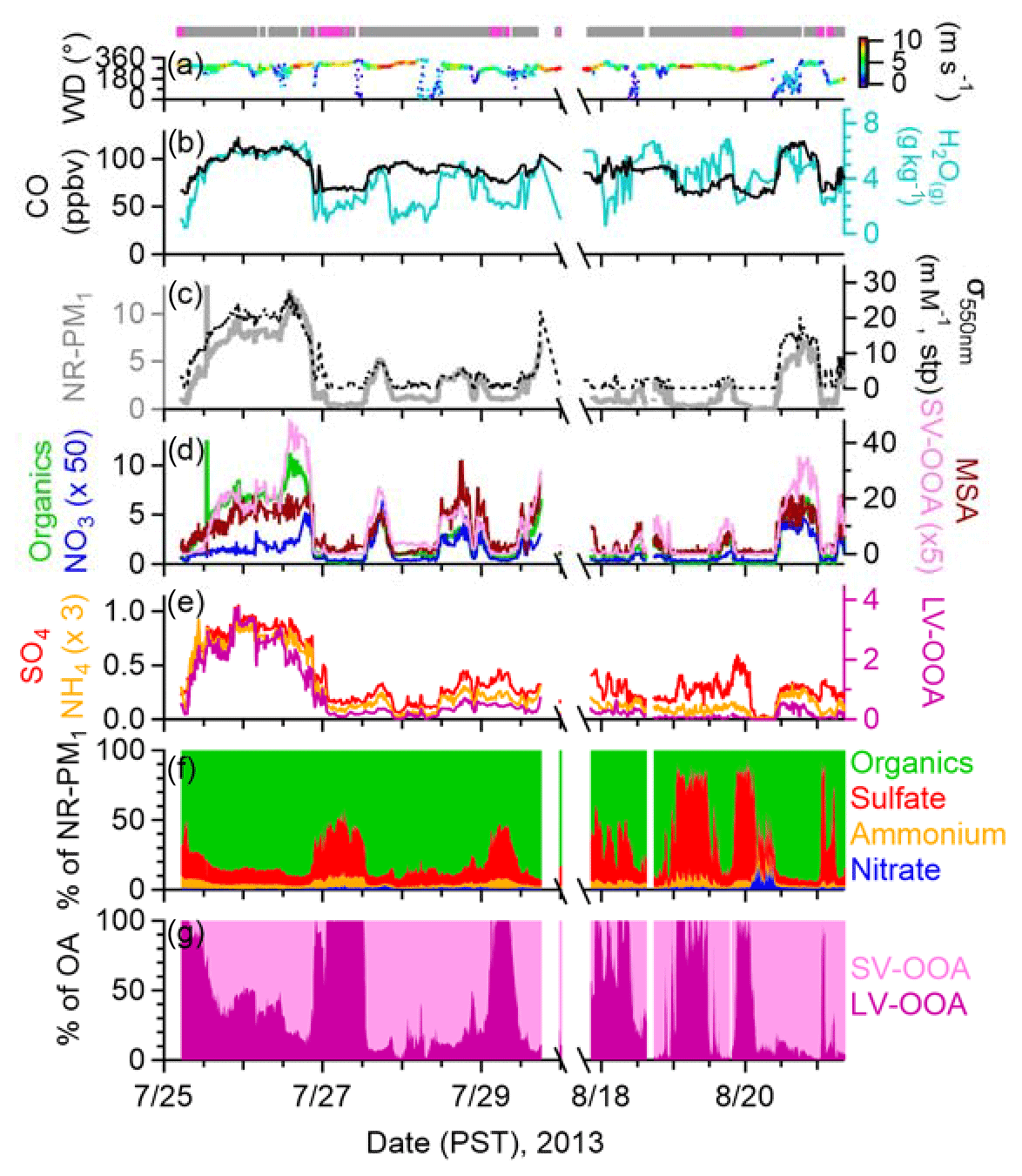
Figure 1Observations during two clean periods in summer 2013. Time series of (a) wind direction (WD) colored by wind speed (m s−1); (b) mixing ratios of CO and water vapor (H2O(g)); (c–e) mass concentrations of NR-PM1 species, OA factors (µg m−3) and MSA (ng m−3) at ambient conditions, and submicron aerosol light scattering at 550 nm (σ550 nm); (f) NR-PM1 composition; and (g) OA composition. The indicator bars at the top of the graph are colored by air mass types: free troposphere (pink) and boundary layer influenced (gray).
The average (±1σ) concentration of NR-PM1 (= sulfate + ammonium + nitrate + organics + chloride) during the clean periods was 2.8 (±2.8) µg m−3. OA was the largest PM1 component, contributing on average ∼84 % to the total NR-PM1 mass, followed by sulfate (11 %), ammonium (2.8 %), and nitrate (0.9 %) (Fig. S7a in the Supplement). Chloride was close or below detection limit for most of the time during the clean periods. Aerosol concentration and composition varied noticeably and showed diurnal changes that appeared to be mainly driven by BL dynamics. This is because MBO sits in the FT at night but is influenced by air masses transported from the PBL as the mixed layer height grows during the day. Indeed, the diurnal profile of the mixing-layer height retrieved from the HYbrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (Draxler and Hess, 1998) shows that the MBO is within the PBL between 12:00 and 20:00 PST (Pacific Standard Time) (Fig. 2). In addition, previous studies at MBO have shown that water vapor mixing ratio (H2O(g)) can be used to differentiate BL-influenced and FT air masses as FT conditions tend to be very dry (Weiss-Penzias et al., 2006; Reidmiller et al., 2010; McClure et al., 2016; Zhang and Jaffe, 2017). H2O(g) at MBO varied from as low as 0.42 g kg−1 at night to as high as 6.9 g kg−1 during the day (Fig. 1b) and showed a strong diurnal cycle similar to boundary layer height (BLH) and NOy∕CO (Fig. 2), another parameter for differentiating BL-influenced and FT air (Stohl et al., 2002).

Figure 2Diurnal cycles of the median values of water vapor (H2O(g)), NOy∕CO ratio (ppb ppb−1), estimated boundary layer height (BLH), mass concentrations of NR-PM1 species, elemental ratios of OA, and mixing ratios of gas species at MBO during two clean periods in summer 2013. Oxidation state of carbon (OS. The shaded areas indicate the 75th and 25th percentiles. The diurnal cycle of NR-PM1 composition displays the percent mass contributions, from top to bottom, of SV-OOA in light pink, LV-OOA in dark purple, sulfate in red, ammonium in orange, nitrate in blue, and chloride in purple. The diurnal cycle of NOy composition displays the percent mixing ratio contributions, from top to bottom, of NO in yellow, NO2 in light orange, nitrate in blue, PAN in dark orange, and NOz (= NOy − NO − NO2 − nitrate − PAN) in grey. Dashed line in the BLH plot indicates the altitude of MBO (2763 m).
NR-PM1 and σ550 nm generally followed the temporal trend of H2O(g) (Fig. 1b, c) and presented a pronounced diurnal profile with substantial daytime enhancements (Fig. 2). The median mass concentration of NR-PM1 was 0.5 µg m−3 at night and increased by more than 10 times to 5.6 µg m−3 in the afternoon. Similar temporal variations and substantial daytime increases were observed for OA, nitrate, and gaseous pollutants such as CO, NOy, and peroxyacetyl nitrate (PAN) (Figs. 1 and 2), indicating that these species are primarily emitted or formed within the BL and their concentrations at MBO are strongly influenced by BL dynamics. At night, the site is situated in the FT, above the shallow nocturnal BL formed over the surrounding lower areas and disconnected from aerosol and gas sources at the low altitudes. As the BL grows during the day, convective transport and thermal winds entrain pollution from lower altitudes and increase air pollutants at the site. In contrast, sulfate exhibited relatively constant concentrations (Fig. 1e) and a less pronounced diurnal pattern (Fig. 2). The weaker influence from BL evolution indicates similar sulfate concentrations in the BL and FT in the remote continental region of the western US. This is consistent with the relatively long atmospheric lifetime and the regional characteristics of sulfate particles. O3 and NO2 mixing ratios also showed flat diurnal patterns (Fig. 2). However, a previous study at MBO indicates that O3 is typically higher in FT air masses (Zhang and Jaffe, 2017) but this depends on the air mass origin and photochemical processing in both the BL and FT.
NR-PM1 composition varied diurnally with a predominant organic composition during the day (up to 94 % of NR-PM1 mass; Figs. 1 and 2). However, at night when the site was situated in the FT, sulfate was a major component of aerosol (max = 83 % of NR-PM1; median = 37.6 %; mean = 33 %). OA during the clean periods at MBO was oxidized with an average (±1σ) O∕C of 0.85 (±0.36) and OM / OC of 2.26 (±0.46). The degree of oxidation was in agreement with regional background OA observed at other mountain sites such as Whistler Mountain in western Canada (Sun et al., 2009), Rocky Mountains in Colorado in the US (Schurman et al., 2015), and Mt. Cimone in Italy (Rinaldi et al., 2015). In addition, OA observed under the FT condition was overall more oxidized than those in the BL-influenced air masses. For example, O∕C peaked at night with a maximum value of 1.5 and reached a minimum of 0.7 in the afternoon (Fig. 2). H∕C anti-correlated with O∕C with a reversed diurnal trend that peaked during daytime. As a result, the average oxidation state of carbon (OSC; = 2 ; Kroll et al., 2011) of OA at MBO during clean periods differs by two units between day and night (Fig. 2). These trends highlight the different chemical properties as well as atmospheric ages of aerosols in the BL and the FT in this remote continental region in the western US. More discussions on the differences between aerosols in BL and FT air masses are given in Sect. 3.4.
3.2 Organonitrates and organosulfates in regional background aerosols
Particulate organonitrates have been shown to make a significant contribution to submicron aerosol mass, especially in rural and remote environments during summertime (Setyan et al., 2012; Fry et al., 2013; Kiendler-Scharr et al., 2016; Zhou et al., 2016). In this study, organonitrates were observed and appeared to account for most of the NO+ and (major ions of inorganic and organic nitrates in HR-AMS) signals detected in NR-PM1 during the clean periods. This is because the signal ratios of NO+ and measured for MBO aerosols, which ranged between 2.0 and 34.4 (average = 7.5; Fig. S9a in the Supplement), are substantially higher compared to the ratio for pure ammonium nitrate particles (RAN = 1.78 ± 0.07). Previous studies reported that the ratio for organonitrates (RON) is ∼2.25–3.7 times higher than RAN (Fry et al., 2009, 2013; Farmer et al., 2010). Based on this information and using Eq. (1) reported in Farmer et al. (2010), we estimated that nearly all the NO+ and signals measured during the clean periods were contributed by organonitrates (RONO2) from the fragmentation of the nitrate functional group (−ONO2). Assuming that organonitrate molecules on average contain one −ONO2 functional group per molecule and have an average molecular weight of 230 g mol−1 (Lee et al., 2006; Fry et al., 2009), we estimated that the average concentration of organonitrates was 0.13 (±0.12) µg m−3 (Fig. S9b in the Supplement) and accounted for ∼5 % of the total OA mass at MBO during the clean periods. Since MBO is situated in a forested region covered by coniferous trees at lower elevations, the reactions of monoterpenes with nitrate radicals were likely an important source of the observed particulate organonitrates (Fry et al., 2009, 2013; Boyd et al., 2015; Ng et al., 2017) in upslope daytime air. Similarly, Lee et al. (2016) observed that organonitrates made a significant contribution to the secondary OA (SOA) mass in the coniferous forested regions at Whistler – a mid-altitude site in western Canada.
The presence of organosulfur compounds in particles is also confirmed based on the unambiguous detection of sulfur-containing organic ions () such as CH3S+ (m∕z = 46.996), (m∕z = 77.978), (m∕z = 78.985), CH4 (m∕z = 95.988), (m∕z = 105.001), and (m∕z = 117.001). Previous studies have shown that , , and are HR-AMS signature ions for methanesulfonic acid (MSA) (Ge et al., 2012). In this study, the three ions correlate with each other (r=0.50–0.71; Fig. S10 in the Supplement) and their signal ratios are close to those observed for pure methanesulfonic acid (Ge et al., 2012). This indicates the presence of mesylate (, the deprotonated anion of MSA) in the regional background aerosols in the western US. Based on the fragmentation pattern of MSA, where contributed 8.7 % of the total major MSA fragments in the HR-AMS spectrum of MSA (Ge et al., 2012), we estimated that the average MSA mass concentration was 6.7 (±7.2) ng m−3, making up ∼0.3 % of the total OA mass during the clean periods. Sorooshian et al. (2015) measured MSA and organosulfates at inland ground sites near MBO and found broadly similar concentrations.
Oceans are generally considered a dominant source of dimethyl sulfide (DMS) and therefore its oxidation product MSA. However, the Pacific Ocean is 195 km to the west of MBO, whereas the bivariate polar plot of MSA revealed that high concentrations were associated with winds from the east and the south – the inland areas (Fig. S11 in the Supplement). In addition, MSA concentrations showed a clear diurnal cycle with a substantial daytime increase (Fig. 2), which suggests significant sources from the PBL. Aerosols in the PBL over this region likely have negligible oceanic influences since the Cascades mountain range lies between the Pacific Ocean and Mt. Bachelor and may obstruct surface wind bringing marine emissions inland. These results suggest that the sources of MSA at MBO were mostly continental, where a wide range of terrestrial sources including soil, vegetation, freshwater wetland, and paddy fields can emit DMS (Watts, 2000, and references therein). Furthermore, the maximum MSA∕SO4 ratio in this study was ∼0.081, much lower than those observed in marine aerosols (e.g., average = 0.23 in the sub-Arctic northeastern Pacific Ocean; Phinney et al., 2006). Similarly, lower MSA∕SO4 ratios were usually found in terrestrial regions, e.g., 0.01–0.17 in Fresno where MSA was mostly attributed to non-marine sources (Ge et al., 2012; Young et al., 2016), 0.007–0.15 along the Atlantic coast under continental influences (Zorn et al., 2008; Huang et al., 2017), and averages of 0.02–0.04 (maximum = 0.11) in California inland regions (Sorooshian et al., 2015).
3.3 Sources and processes of aerosols in the remote region of the western US
PMF analysis was performed on the NR-PM1 mass spectra acquired during the clean periods to further elucidate the sources and processes of the regional background aerosols observed at MBO. Two OA factors were identified, including an intermediately oxidized, semivolatile OOA (O∕C = 0.67; H∕C = 1.57) and a highly oxidized, low-volatility OOA (O∕C = 1.17 ± 0.08; H∕C = 1.18 ± 0.03). No hydrocarbon-like (HOA) factor was identified during the clean periods, which is consistent with a low abundance of (0.13 % of total OA signal), a tracer ion for primary OA from vehicle emissions (Collier et al., 2015). In addition, f60 was constantly lower than 0.3 % (Figs. S6 and S13b in the Supplement), indicating a lack of BB influence (Cubison et al., 2011). These results indicate the absence of primary aerosol sources at MBO during clean periods.
SV-OOA, which on average accounted for 70 % of total OA mass at MBO during clean periods (Fig. 3c), showed temporal features that indicate a strong influence from BL dynamics. Particularly, SV-OOA correlated well with CO, nitrate, and MSA (r=0.7–0.84) and exhibited a pronounced diurnal cycle that increases between 09:00 and 10:00 PST, peaks around 15:30 PST, and decreases to a very low concentration (∼0.1 µg m−3) at night (Fig. 2). The SV-OOA mass spectrum displayed the characteristics of secondary OA with two dominant oxygenated ions, C2H3O+ (m∕z = 43.018) and (m∕z = 43.989) (Fig. 3a). The signal intensity of C2H3O+ is similar to that of and the SV-OOA spectrum comprises relatively abundant and ions (Fig. 3a). These features, as well as an average O∕C of 0.67, indicate that SV-OOA was moderately oxidized and was likely not very aged.

Figure 3High-resolution mass spectra of (a) SV-OOA and (b) LV-OOA colored by eight ion families. The elemental ratios of OA determined using the IA method are shown in the legends. (c) Average OA composition. (d–e) The distribution of signals between SV-OOA and LV-OOA for 20 most abundant ions in SV-OOA (d) and LV-OOA (e). The nominal masses of the ions are shown on the top axes of (d) and (e).
The SV-OOA spectrum showed a significant signal at m∕z = 91.055 ( = 0.65 %) and a spectral pattern highly similar to biogenic SOA observed from a plant chamber (Kiendler-Scharr et al., 2009). was proposed as an indicator for the presence of β-pinene + NO3 reaction products (Boyd et al., 2015) and elevated was previously observed in the AMS spectra of biogenic SOA both in ambient air and in chamber experiments (Kiendler-Scharr et al., 2009; Sun et al., 2009; Robinson et al., 2011; Setyan et al., 2012; Budisulistiorini et al., 2015; Chen et al., 2015). In addition, as shown in Fig. S13a in the Supplement, the SV-OOA of this study situates along the right leg of the triangle defined by worldwide ambient OA in the f44 vs. f43 space. It has been illustrated previously that the f44 vs. f43 triangle plot could be used to indicate the source and type of the aerosols and that biogenic OAs usually situate on the right hand side of the triangle (Jimenez et al., 2009; Ng et al., 2010). These findings, together with the fact that organonitrates were predominantly associated with SV-OOA (e.g., 78 % of the aerosol nitrate signal was attributed to SV-OOA; Fig. S12 in the Supplement), indicate that the SV-OOA observed in this study likely represented biogenic SOA formed at lower altitudes in the region and transported upward to the site by thermal winds during the day.
LV-OOA, which accounted for an average of 30 % of total OA mass, likely represented more aged SOA in the regional background air. It exhibited a much less pronounced diurnal trend than SV-OOA (Fig. 2) and presented as a major OA component during most nights when the site was in the FT (Fig. 1g). These results suggest that LV-OOA likely represents OAs in the FT, which were transported over long distances and/or recirculated regionally due to longer aerosol lifetime and higher wind speed in the FT. LV-OOA was highly oxidized with an average O∕C of 1.17 (Fig. 3b) and contributed major fractions of highly oxygenated organic ions, e.g., (m∕z = 99.008), (m∕z = 89.024), and (m∕z = 125.024), as well as and (m∕z = 44.998) – HR-AMS signature ions for carboxylic acids (Fig. 3e). In contrast, nearly all the (m∕z = 107.086), C6H11O+ (m∕z = 99.081), (m∕z = 69.070), and (m∕z = 43.055) signals were attributed to SV-OOA, so were a majority of the (m∕z = 55.055; 91 %) and C2H3O+ (86 %) signals (Fig. 3d, e). In addition, LV-OOA was tightly associated with sulfate (Fig. 3b, e), a secondary aerosol species representative of aged, regional air masses. Furthermore, LV-OOA situates near the apex of the triangle region for ambient OAs in the f44 vs. f43 space (Fig. S13a in the Supplement), overlapping with the highly oxidized LV-OOA observed in various environments (Ng et al., 2010) as well as highly aged OOAs observed at high altitude (Sun et al., 2009; Fröhlich et al., 2015). These results together suggest that LV-OOAs likely represented free tropospheric SOA in the western US and were composed of highly oxidized organic compounds.
3.4 Differences between aerosols in BL and FT air masses
To further examine the differences between aerosols in the free troposphere and boundary layer, we segregate periods using measurements of water vapor (H2O(g)). Extensive work has been done to differentiate free tropospheric air from boundary-layer-influenced air at MBO using water vapor chairlift soundings (Reidmiller et al., 2010) and other approaches (Weiss-Penzias et al., 2006; Fischer et al., 2010; Ambrose et al., 2011; McClure et al., 2016; Zhang and Jaffe, 2017), as discussed in more detail in Sect. 1 of the Supplement. Zhang and Jaffe (2017) established more accurate monthly H2O(g) criteria for FT air masses at MBO: 5.1 and 5.2 g kg−1 for July and August, respectively, and associated FT air masses with low H2O(g) values. However, since convection in summer enhances vertical transport and creates a thicker entrainment zone where BL mixes with FT, properly defining the top of BL is challenging (Wagner et al., 2015). To avoid the influences of the transition zone on FT, we used a more stringent H2O(g) criterion, 2.5 g kg−1, which is the lowest monthly cut point reported in Zhang and Jaffe (2017). In addition, we explored the usage of the estimated BL height from HYSPLIT back trajectory analysis as the segregation criteria. A comparison between these two methods can be found in Sect. 1 of the Supplement. After careful evaluation, we classify periods with H2O(g) < 2.5 g kg−1 and CO < 80 ppb as “FT air” and the rest as “BL-influenced air”.
The average concentration of NR-PM1 under BL influences was 3.16 µg m−3, approximately 4 times the average concentration in the FT (0.85 µg m−3). While OA concentration was on average 6 times higher in BL-influenced air than in FT air (2.7 vs. 0.34 µg m−3), sulfate mass concentrations in these two types of air masses were similar (0.35 vs. 0.33 µg m−3). The stoichiometric neutralization of the inorganic components of NR-PM1 was examined by comparing the molar equivalent ratio of ammonium () and sulfate () since inorganic nitrate and chloride concentrations were very low during clean periods. An ammonium-to-sulfate equivalent ratio of 1 suggests neutral particles, whereas a ratio significantly lower than 1 suggests acidic particles. This ratio varied between 0.005 and 1 for clean periods (Fig. 4a), indicating that remote aerosols in the western US were frequently acidic in the summer. Most significant is that a substantial amount of FT aerosols (∼78 % of the NR-PM1 mass in FT air vs. 16 % of the NR-PM1 mass in BL-influenced air) exhibited an ammonium-to-sulfate equivalent ratio lower than 0.3 (Fig. 4a), indicating the prevalence of very acidic particles in the free troposphere. Acidic FT particles were also observed at various high-altitude regional background sites, such as Jungfraujoch (Cozic et al., 2008; Fröhlich et al., 2015), Puy de Dôme station (Freney et al., 2016), Whistler Mountain (Sun et al., 2009), and Mauna Loa (Hawaii, US) (Johnson and Kumar, 1991), as well as during airborne measurements in the upper troposphere of the tropics (Froyd et al., 2009) and the Arctic (Brock et al., 2011; Fisher et al., 2011).
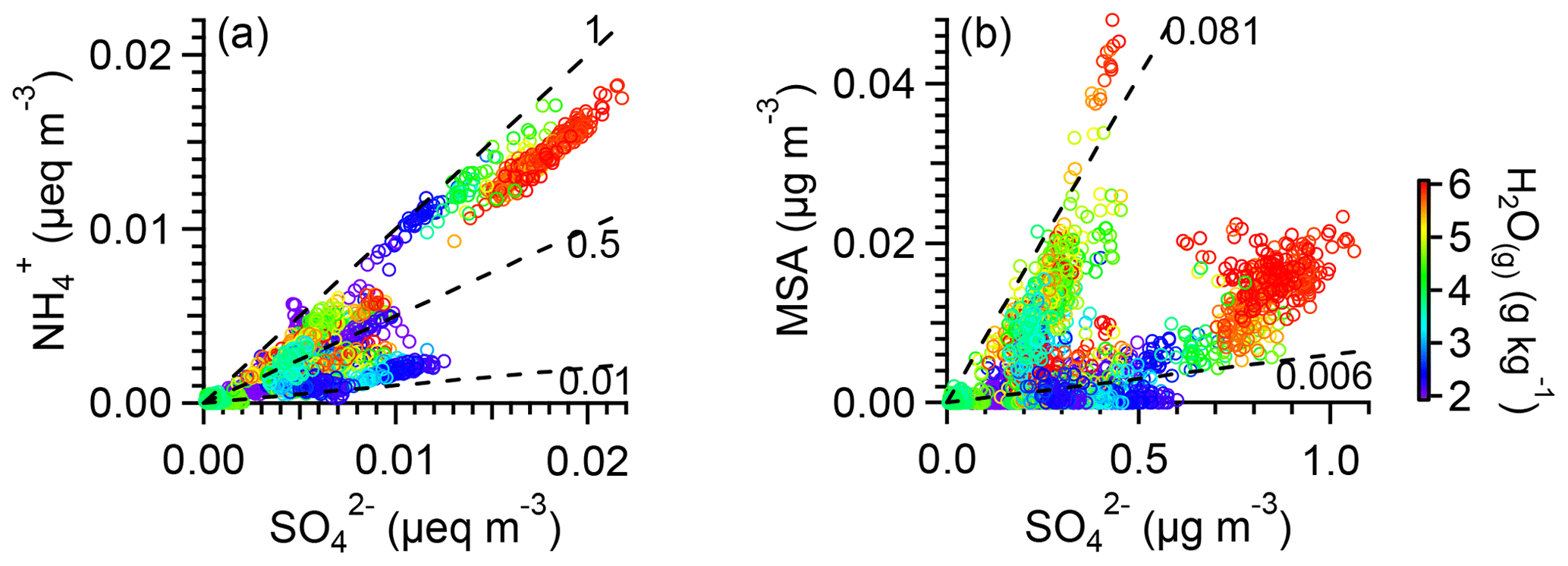
Figure 4Scatter plots that compare (a) ammonium molar equivalent concentration () vs. sulfate molar equivalent concentration () and (b) MSA mass concentration vs. sulfate mass concentration. Data points are colored by water vapor mixing ratio. Dashed lines with different slopes are added for reference.
MSA correlated with HR-AMS sulfate for different aerosol regimes with different slopes. As shown in Fig. 4b, BL-influenced aerosols showed a range of MSA∕SO4 ratios generally higher than FT aerosols. This may be attributed to higher MSA concentration near terrestrial sources in the BL. Indeed, airborne measurements of MSA in aerosol over the western US in summer 2013 have shown that MSA loading decreased with the increase in altitude (Sorooshian et al., 2015). Furthermore, as discussed later on, sulfate was likely produced in the FT during regional new particle formation and growth events, which may further contribute to a lower MSA∕SO4 ratio in FT aerosols.
In addition to aerosol chemical properties, the physical properties of MBO aerosols were examined as well. The average mass-based size distribution of NR-PM1 during the clean periods displayed a broad feature extending from 100 to 1000 nm in vacuum aerodynamic diameter (Dva, Fig. 5). Aerosol composition varied as a function of size with larger particles more enriched in sulfate than smaller particles (sulfate accounted for 12 % of the non-refractory aerosol mass in PM≥ 0.2 vs. 5 % in PM<0.2). Org43, the organic signal at m∕z = 43 (90 % of which was C2H3O+), presented a broad distribution peaking between 250 and 350 nm in Dva (Fig. 5b). In contrast, Org44, the organic signal at m∕z = 44 (95 % of which was ), and sulfate displayed distinctly narrower distributions peaking at a larger droplet accumulation mode close to 500 nm (Fig. 5b, c). The similar size distribution of Org44 and sulfate and the tight correlation between their concentrations (r2=0.61; Fig. S14 in the Supplement) suggest that highly oxidized organics and sulfate had similar sources and processes and are possibly internally mixed. In particular, the prominent droplet mode at 500 nm indicates an important influence of aqueous-phase reactions on the production of sulfate and highly oxidized organics. Indeed, previous studies have shown that aqueous-phase processing (i.e., fog and cloud droplets and aerosol-phase water) leads to production of more oxidized organics (Lee et al., 2011, 2012; Ervens et al., 2013; Kim et al., 2019) in the droplet mode (Ge et al., 2012) and that aqueous-phase production of sulfate is an important process in the atmosphere (e.g., Ervens et al., 2011). In addition, a similar sulfate size distribution was observed at the peak of Whistler Mountain, which had frequent cloud cover (Sun et al., 2009).
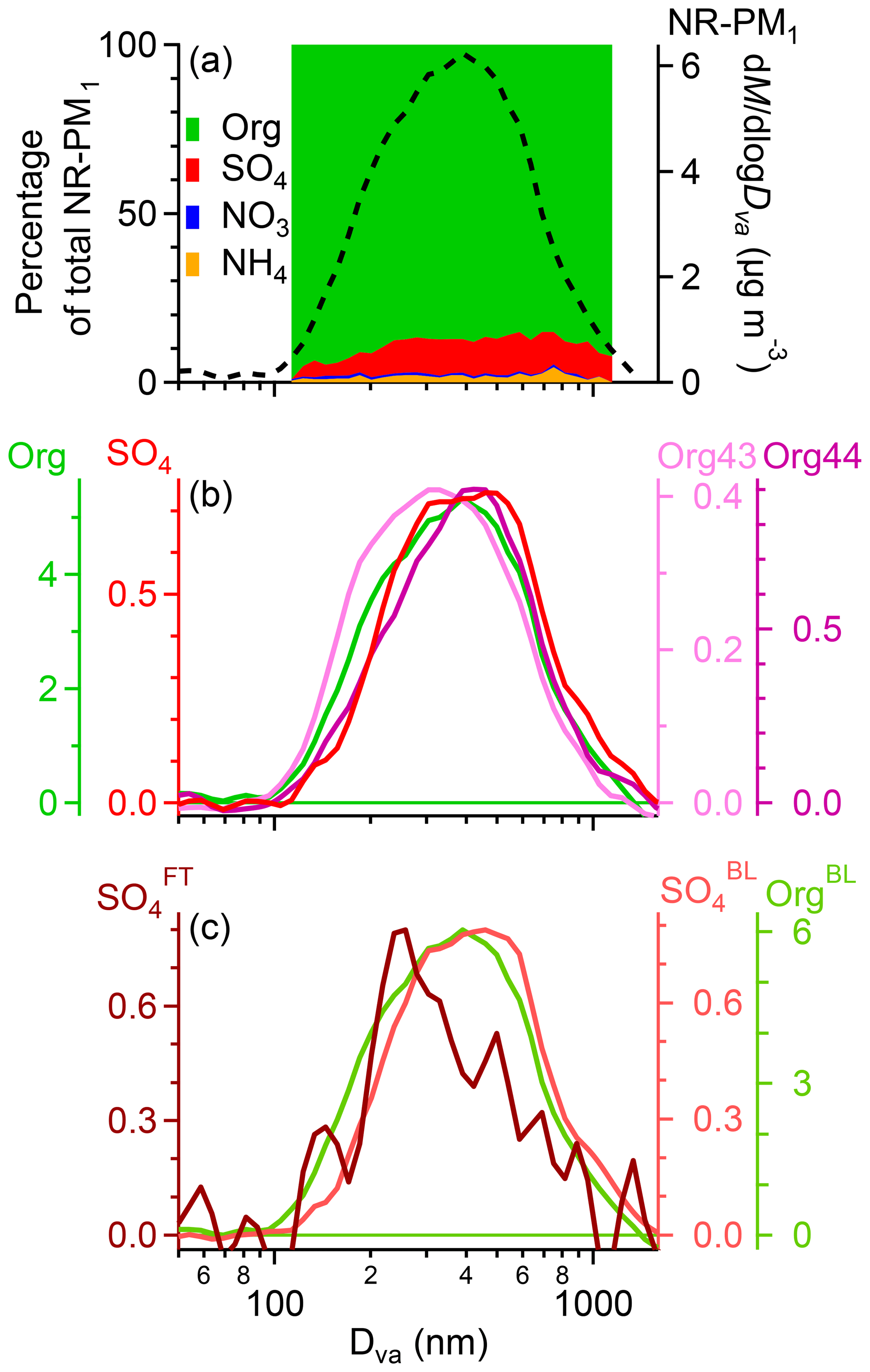
Figure 5(a) Size-resolved aerosol composition on the left axis and average size distributions of total NR-PM1 mass on the right. (b) Average mass-based size distributions of organics, sulfate, Org43, and Org44 during the clean periods. (c) Average mass-based size distributions of FT sulfate and BL-influenced sulfate and organics. The units for the y axes in (b) and (c) are µg m−3.
A distinctly different size distribution was observed for sulfate-containing particles in the FT (Fig. 5c), which exhibited a prominent mode at ∼250 nm. One possible explanation is preferential activation and/or washout of larger particles and thus the reduction of sulfate signal in larger size (droplet) modes. Condensational growth of newly nucleated particles in the FT may be another possibility. Scavenging could result in low particle surface area, which facilitates new particle formation in the FT. Although we did not observe NPF events in this study (due to instrumental limitations), in situ NPF events have been frequently observed in the FT (e.g., Hallar et al., 2011, 2013). Formation and growth of new particles have also been observed over broad regions in the FT (Tröstl et al., 2016). Condensation of gas-phase sulfate products on small FT particles could contribute to the observed condensation mode sulfate particles at MBO. These observations may shed light on the different sources and processes of aerosols in the BL and FT and suggest that sulfate and organic aerosols were likely present in both internal and external mixtures at MBO.
3.5 Comparisons with aerosols observed at other high-altitude locations
Figure 6 summarizes the average composition of NR-PM1 measured using AMS or aerosol chemical speciation monitors at various elevated regional background ground sites (Zhang et al., 2007; Sun et al., 2009; Worton et al., 2011; Fröhlich et al., 2015; Rinaldi et al., 2015; Ripoll et al., 2015; Schurman et al., 2015; Freney et al., 2016; Zhu et al., 2016; Xu et al., 2018) and by aircraft (Bahreini et al., 2003; Dunlea et al., 2009). All of these measurements were conducted under conditions absent of biomass burning influence and were representative of regional background aerosols in the Northern Hemisphere. Mountaintop studies separated FT air based on BLH calculated from lidar measurements (Freney et al., 2016) or tracers such as 222Rn concentrations and NOy∕CO and back trajectory analysis (Fröhlich et al., 2015). The average NR-PM1 mass concentration was 3.8 (±3.4) µg m−3 across all sites and was 2.6 (±1.6) µg m−3 in North America. NR-PM1 concentrations were, on average, substantially lower in FT air than in BL-influenced air (0.89 ± 0.43 µg m−3 vs. 4.7 ± 3.4 µg m−3), reflecting generally clean conditions in the FT.
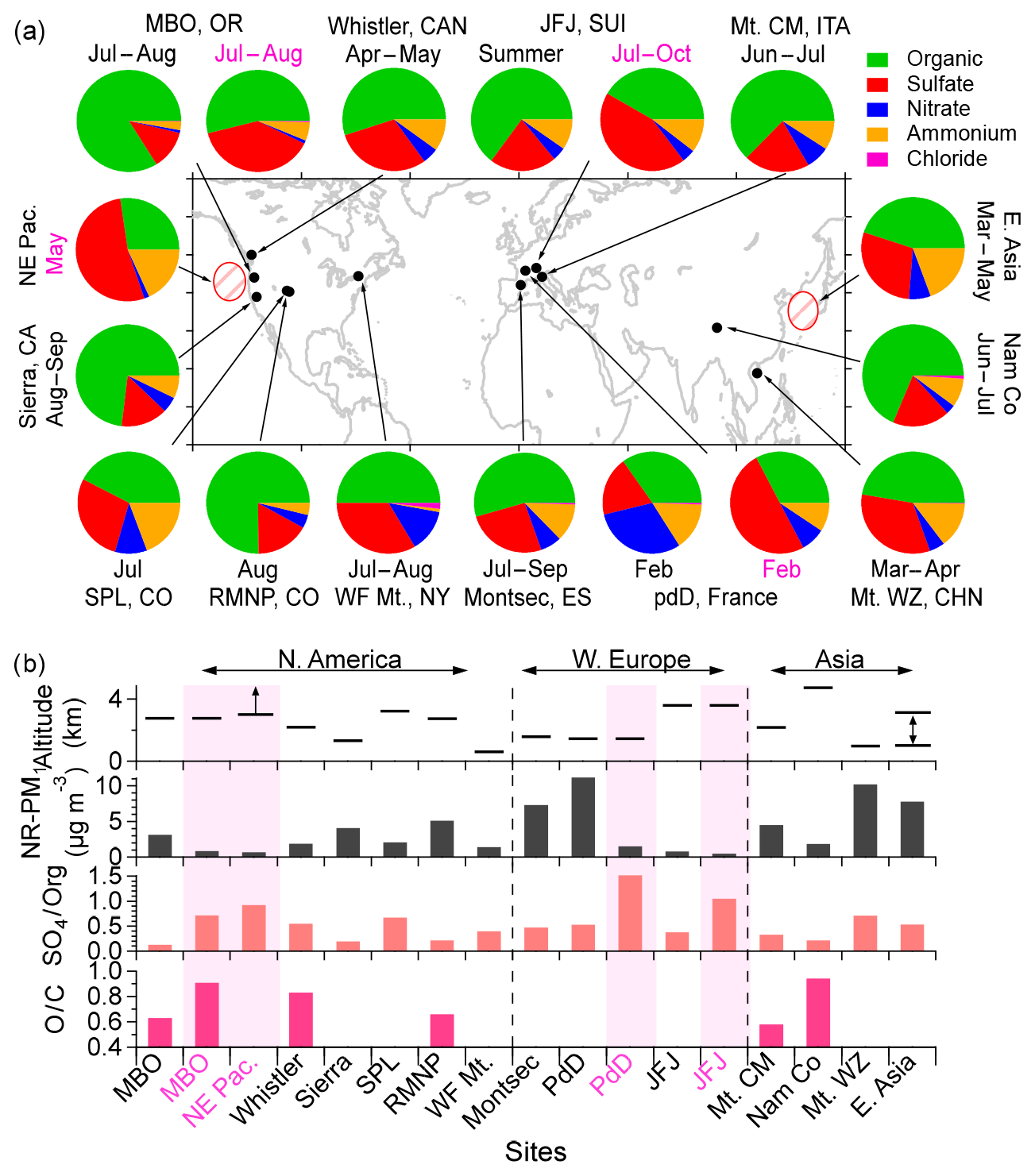
Figure 6(a) Location of selected high-altitude mountain sites and aircraft measurements of regional background aerosols in the world and the chemical composition of NR-PM1 (details are listed in Table S1 in the Supplement). Pie charts show the average chemical composition of NR-PM1. (b) Sampling altitude, average NR-PM1 mass concentration, sulfate-to-organic mass ratio (SO4∕Org), and average O∕C ratio of OA determined using the Aiken-Ambient method for each site. The sampling period labels for pie charts in (a) and the bottom axis labels for sites in (b) are colored according to air mass types: mixed BL–FT air (black) and FT air only (pink). Shaded pink bars in (b) highlight the FT data.
A major fraction (27 %–84 %; average = 51 %) of the NR-PM1 mass was organic matter at these remote high-altitude locations (Fig. 6a). OA in the FT air was generally more oxidized than that in the BL-influenced air (Fig. 6b). In addition, for the same site, marked chemical difference can be seen between aerosols in the FT and the BL. At all sites, FT aerosols contained a substantially higher mass fraction of sulfate (39 %–50 %) compared to the mixed BL–FT aerosols (11 %–35 %). Aircraft measurements also showed consistent results of higher sulfate content in aerosols at higher altitudes. For example, Bahreini et al. (2003) reported that the sulfate contribution to total NR-PM1 over east Asia increased from 17.4 % in the lower atmosphere (1–3 km) to 28.8 % in layers > 3 km. In the FT over the northeast Pacific, more than half of the background submicron mass was attributed to sulfate (Dunlea et al., 2009; Roberts et al., 2010). Elevated sulfate layers were also clearly observed in the higher altitudes above Mexico City (DeCarlo et al., 2008). As a result, the mass ratio of submicron sulfate to organics (SO4∕Org) showed significantly higher values (0.72 to 1.5) in the FT air masses than those in the mixed layers (0.13–0.7; Fig. 6b)
The extent to which sulfate particles are neutralized has major implications for aerosol radiative forcing. The average relative humidity at MBO was 25.6 (±8.9) % during the clean periods. Acidic sulfate aerosols are more hygroscopic than ammonium sulfate. The resulting increase in aerosol water content both increases the direct radiative forcing of sulfate (Adams et al., 2001; Jacobson, 2001) and promotes homogenous ice nucleation (Koop et al., 2000). In addition, while mineral dust particles coated with ammonium sulfate are efficient ice nuclei, those coated with sulfuric acid can lose their ice-nucleating ability (Eastwood et al., 2009).
Based on field observations at a remote high-altitude atmospheric research station – the Mt. Bachelor Observatory (MBO; 43.98∘ N, 121.69∘ W; 2763 m a.s.l.) in central Oregon – we have characterized the chemical and physical properties of aerosols in the boundary layer and free troposphere air under clean conditions in the absence of wildfire influences in the western US. Water vapor mixing ratio, a tracer used to segregate FT and BL-influenced air masses at MBO, showed a strong diurnal cycle. Dry free tropospheric conditions were frequently observed at night, whereas more humid, boundary-layer-influenced air was often observed at MBO during daytime. The average (±1σ) NR-PM1 mass concentration during the entire clean period was 2.8 (±2.8) µg m−3, with OA dominating the NR-PM1 composition (∼84 %) followed by sulfate (11 %). OA, nitrate, and MSA displayed clear diurnal cycles with substantial daytime increases, suggesting significantly higher mass concentrations in the BL than in the FT.
Strong diurnal patterns driven by the boundary layer dynamics were also observed in aerosol chemical composition. NR-PM1 contained a significantly higher mass fraction of sulfate (up to 83 % of NR-PM1 mass) and was frequently acidic at night when MBO resided in the FT. In addition, nighttime free tropospheric OA was found to be more oxidized. PMF analysis identified two types of OOA that are present in the regional background air in the western US: a LV-OOA (30 % of OA mass) that was highly oxidized (O∕C = 1.17) and comprised of low-volatility organics, representative of SOA in the free troposphere; and an SV-OOA (70 % of OA mass) that was intermediately oxidized (O∕C = 0.67) and appeared to be semivolatile, representative of biogenic SOA originated in the BL. In addition, the chemical compositions of NR-PM1 observed at other high-altitude locations in the world under regional background conditions are summarized. These results highlight major differences between FT and BL aerosols, in that the FT aerosols are significantly more oxidized and contain a higher fraction of sulfate. The observed compositional difference suggests significant differences between FT and BL aerosols in microphysical and optical properties and may have important implications for understanding the climate effects of aerosols in remote regions.
Data presented in this paper are available upon request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-1571-2019-supplement.
All authors participated in the field campaign. SZ performed the data analysis. SZ and QZ wrote the manuscript with editing by all coauthors.
The authors declare that they have no conflict of interest.
This research was supported primarily by the US Department of Energy's
Atmospheric System Research, an Office of Science, Office of Biological and
Environmental Research, program, under grant no. DE-SC0014620. Shan Zhou also
acknowledges funding from the Chinese Scholarship Council (CSC) as well as the
Donald G. Crosby Fellowship and the Fumio Matsumura Memorial Fellowship from
the University of California at Davis. The Mt. Bachelor Observatory is
supported by the National Science Foundation (grant no. AGS-1447832) and the
National Oceanic and Atmospheric Administration (contract
no. RA-133R-16-SE-0758).
Edited by: Eleanor
Browne
Reviewed by: two anonymous referees
Adams, P. J., Seinfeld, J. H., Koch, D., Mickley, L., and Jacob, D.: General circulation model assessment of direct radiative forcing by the sulfate-nitrate-ammonium-water inorganic aerosol system, J. Geophys. Res.-Atmos., 106, 1097–1111, 2001.
Ahlm, L., Shakya, K. M., Russell, L. M., Schroder, J. C., Wong, J. P. S., Sjostedt, S. J., Hayden, K. L., Liggio, J., Wentzell, J. J. B., Wiebe, H. A., Mihele, C., Leaitch, W. R., and Macdonald, A. M.: Temperature-dependent accumulation mode particle and cloud nuclei concentrations from biogenic sources during WACS 2010, Atmos. Chem. Phys., 13, 3393–3407, https://doi.org/10.5194/acp-13-3393-2013, 2013.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O∕C and OM∕OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, 2008.
Ambrose, J. L., Reidmiller, D. R., and Jaffe, D. A.: Causes of high O3 in the lower free troposphere over the Pacific Northwest as observed at the Mt. Bachelor Observatory, Atmos. Environ., 45, 5302–5315, 2011.
Bahreini, R., Jimenez, J. L., Wang, J., Flagan, R. C., Seinfeld, J. H., Jayne, J. T., and Worsnop, D. R.: Aircraft-based aerosol size and composition measurements during ACE-Asia using an Aerodyne aerosol mass spectrometer, J. Geophys. Res.-Atmos., 108, 8645, https://doi.org/10.1029/2002JD003226, 2003.
Bianchi, F., Trostl, J., Junninen, H., Frege, C., Henne, S., Hoyle, C. R., Molteni, U., Herrmann, E., Adamov, A., Bukowiecki, N., Chen, X., Duplissy, J., Gysel, M., Hutterli, M., Kangasluoma, J., Kontkanen, J., Kurten, A., Manninen, H. E., Munch, S., Perakyla, O., Petaja, T., Rondo, L., Williamson, C., Weingartner, E., Curtius, J., Worsnop, D. R., Kulmala, M., Dommen, J., and Baltensperger, U.: New particle formation in the free troposphere: A question of chemistry and timing, Science, 352, 1109–1112, 2016.
Bolton, D.: The Computation of Equivalent Potential Temperature, Mon. Weather Rev., 108, 1046–1053, 1980.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., Lohmann, U., Rasch, P., Satheesh, S. K., Sherwood, S., Stevens, B., and Zhang, X. Y.: Clouds and Aerosols, in: Climate Change 2013: The Physical Science Basis, Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, UK and New York, NY, USA, 573–575, 2013.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Briggs, N. L., Jaffe, D. A., Gao, H., Hee, J. R., Baylon, P. M., Zhang, Q., Zhou, S., Collier, S. C., Sampson, P. D., and Cary, R. A.: Particulate Matter, Ozone, and Nitrogen Species in Aged Wildfire Plumes Observed at the Mount Bachelor Observatory, Aerosol Air Qual. Res., 16, 3075–3087, 2016.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Chen, Q., Farmer, D. K., Rizzo, L. V., Pauliquevis, T., Kuwata, M., Karl, T. G., Guenther, A., Allan, J. D., Coe, H., Andreae, M. O., Pöschl, U., Jimenez, J. L., Artaxo, P., and Martin, S. T.: Submicron particle mass concentrations and sources in the Amazonian wet season (AMAZE-08), Atmos. Chem. Phys., 15, 3687–3701, https://doi.org/10.5194/acp-15-3687-2015, 2015.
Collier, S. and Zhang, Q.: Gas-Phase CO2 Subtraction for Improved Measurements of the Organic Aerosol Mass Concentration and Oxidation Degree by an Aerosol Mass Spectrometer, Environ. Sci. Technol., 47, 14324–14331, 2013.
Collier, S., Zhou, S., Kuwayama, T., Forestieri, S., Brady, J., Zhang, M., Kleeman, M., Cappa, C., Bertram, T., and Zhang, Q.: Organic PM Emissions from Vehicles: Composition, O∕C Ratio, and Dependence on PM Concentration, Aerosol Sci. Tech., 49, 86–97, 2015.
Collier, S., Zhou, S., Onasch, T. B., Jaffe, D. A., Kleinman, L., Sedlacek, A. J., Briggs, N. L., Hee, J., Fortner, E., Shilling, J. E., Worsnop, D., Yokelson, R. J., Parworth, C., Ge, X., Xu, J., Butterfield, Z., Chand, D., Dubey, M. K., Pekour, M. S., Springston, S., and Zhang, Q.: Regional Influence of Aerosol Emissions from Wildfires Driven by Combustion Efficiency: Insights from the BBOP Campaign, Environ. Sci. Technol., 50, 8613–8622, 2016.
Cozic, J., Verheggen, B., Weingartner, E., Crosier, J., Bower, K. N., Flynn, M., Coe, H., Henning, S., Steinbacher, M., Henne, S., Collaud Coen, M., Petzold, A., and Baltensperger, U.: Chemical composition of free tropospheric aerosol for PM1 and coarse mode at the high alpine site Jungfraujoch, Atmos. Chem. Phys., 8, 407–423, https://doi.org/10.5194/acp-8-407-2008, 2008.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
DeCarlo, P. F., Dunlea, E. J., Kimmel, J. R., Aiken, A. C., Sueper, D., Crounse, J., Wennberg, P. O., Emmons, L., Shinozuka, Y., Clarke, A., Zhou, J., Tomlinson, J., Collins, D. R., Knapp, D., Weinheimer, A. J., Montzka, D. D., Campos, T., and Jimenez, J. L.: Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign, Atmos. Chem. Phys., 8, 4027–4048, https://doi.org/10.5194/acp-8-4027-2008, 2008.
Draxler, R. R. and Hess, G. D.: An overview of the Hysplit-4 modeling system for trajectories, dispersion, and deposition, Aust. Meteorol. Magn., 47, 295–308, 1998.
Dunlea, E. J., DeCarlo, P. F., Aiken, A. C., Kimmel, J. R., Peltier, R. E., Weber, R. J., Tomlinson, J., Collins, D. R., Shinozuka, Y., McNaughton, C. S., Howell, S. G., Clarke, A. D., Emmons, L. K., Apel, E. C., Pfister, G. G., van Donkelaar, A., Martin, R. V., Millet, D. B., Heald, C. L., and Jimenez, J. L.: Evolution of Asian aerosols during transpacific transport in INTEX-B, Atmos. Chem. Phys., 9, 7257–7287, https://doi.org/10.5194/acp-9-7257-2009, 2009.
Dzepina, K., Mazzoleni, C., Fialho, P., China, S., Zhang, B., Owen, R. C., Helmig, D., Hueber, J., Kumar, S., Perlinger, J. A., Kramer, L. J., Dziobak, M. P., Ampadu, M. T., Olsen, S., Wuebbles, D. J., and Mazzoleni, L. R.: Molecular characterization of free tropospheric aerosol collected at the Pico Mountain Observatory: a case study with a long-range transported biomass burning plume, Atmos. Chem. Phys., 15, 5047–5068, https://doi.org/10.5194/acp-15-5047-2015, 2015.
Eastwood, M. L., Cremel, S., Wheeler, M., Murray, B. J., Girard, E., and Bertram, A. K.: Effects of sulfuric acid and ammonium sulfate coatings on the ice nucleation properties of kaolinite particles, Geophys. Res. Lett., 36, L02811, https://doi.org/10.1029/2008GL035997, 2009.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Ervens, B., Wang, Y., Eagar, J., Leaitch, W. R., Macdonald, A. M., Valsaraj, K. T., and Herckes, P.: Dissolved organic carbon (DOC) and select aldehydes in cloud and fog water: the role of the aqueous phase in impacting trace gas budgets, Atmos. Chem. Phys., 13, 5117–5135, https://doi.org/10.5194/acp-13-5117-2013, 2013.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, 2010.
Fischer, E. V., Jaffe, D. A., Reidmiller, D. R., and Jaegle, L.: Meteorological controls on observed peroxyacetyl nitrate at Mount Bachelor during the spring of 2008, J. Geophys. Res.-Atmos., 115, D03302, https://doi.org/10.1029/2009JD012776, 2010.
Fischer, E. V., Jaffe, D. A., and Weatherhead, E. C.: Free tropospheric peroxyacetyl nitrate (PAN) and ozone at Mount Bachelor: potential causes of variability and timescale for trend detection, Atmos. Chem. Phys., 11, 5641–5654, https://doi.org/10.5194/acp-11-5641-2011, 2011.
Fisher, J. A., Jacob, D. J., Wang, Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate–ammonium aerosol in the Arctic in winter–spring, Atmos. Environ., 45, 7301–7318, 2011.
Freney, E., Sellegri, K., Asmi, E., Rose, C., Chauvigne, A., Baray, J. L., Colomb, A., Hervo, M., Montoux, N., Bouvier, L., and Picard, D.: Experimental Evidence of the Feeding of the Free Troposphere with Aerosol Particles from the Mixing Layer, Aerosol Air Qual. Res., 16, 702–716, 2016.
Fröhlich, R., Cubison, M. J., Slowik, J. G., Bukowiecki, N., Canonaco, F., Croteau, P. L., Gysel, M., Henne, S., Herrmann, E., Jayne, J. T., Steinbacher, M., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Fourteen months of on-line measurements of the non-refractory submicron aerosol at the Jungfraujoch (3580 m a.s.l.) – chemical composition, origins and organic aerosol sources, Atmos. Chem. Phys., 15, 11373–11398, https://doi.org/10.5194/acp-15-11373-2015, 2015.
Froyd, K. D., Murphy, D. M., Sanford, T. J., Thomson, D. S., Wilson, J. C., Pfister, L., and Lait, L.: Aerosol composition of the tropical upper troposphere, Atmos. Chem. Phys., 9, 4363–4385, https://doi.org/10.5194/acp-9-4363-2009, 2009.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Fry, J. L., Draper, D. C., Zarzana, K. J., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Brown, S. S., Cohen, R. C., Kaser, L., Hansel, A., Cappellin, L., Karl, T., Hodzic Roux, A., Turnipseed, A., Cantrell, C., Lefer, B. L., and Grossberg, N.: Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 13, 8585–8605, https://doi.org/10.5194/acp-13-8585-2013, 2013.
Ge, X. L., Zhang, Q., Sun, Y. L., Ruehl, C. R., and Setyan, A.: Effect of aqueous-phase processing on aerosol chemistry and size distributions in Fresno, California, during wintertime, Environ. Chem., 9, 221–235, 2012.
Hallar, A. G., Lowenthal, D. H., Chirokova, G., Borys, R. D., and Wiedinmyer, C.: Persistent daily new particle formation at a mountain-top location, Atmos. Environ., 45, 4111–4115, 2011.
Hallar, A. G., Lowenthal, D. H., Clegg, S. L., Samburova, V., Taylor, N., Mazzoleni, L. R., Zielinska, B. K., Kristensen, T. B., Chirokova, G., McCubbin, I. B., Dodson, C., and Collins, D.: Chemical and hygroscopic properties of aerosol organics at Storm Peak Laboratory, J. Geophys. Res.-Atmos., 118, 4767–4779, 2013.
Hallar, A. G., Petersen, R., McCubbin, I. B., Lowenthal, D., Lee, S., Andrews, E., and Yu, F.: Climatology of New Particle Formation and Corresponding Precursors at Storm Peak Laboratory, Aerosol Air Qual. Res., 16, 816–826, 2016.
Huang, S., Poulain, L., van Pinxteren, D., van Pinxteren, M., Wu, Z., Herrmann, H., and Wiedensohler, A.: Latitudinal and Seasonal Distribution of Particulate MSA over the Atlantic using a Validated Quantification Method with HR-ToF-AMS, Environ. Sci. Technol., 51, 418–426, 2017.
Jacobson, M. Z.: Global direct radiative forcing due to multicomponent anthropogenic and natural aerosols, J. Geophys. Res.-Atmos., 106, 1551–1568, 2001.
Jaffe, D., Prestbo, E., Swartzendruber, P., Weiss-Penzias, P., Kato, S., Takami, A., Hatakeyama, S., and Kajii, Y.: Export of atmospheric mercury from Asia, Atmos. Environ., 39, 3029–3038, 2005a.
Jaffe, D., Tamura, S., and Harris, J.: Seasonal cycle and composition of background fine particles along the west coast of the US, Atmos. Environ., 39, 297–306, 2005b.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, 2009.
Johnson, S. A. and Kumar, R.: Composition and spectral characteristics of ambient aerosol at Mauna Loa Observatory, J. Geophys. Res.-Atmos., 96, 5379–5386, 1991.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Aijala, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, 2016.
Kiendler-Scharr, A., Zhang, Q., Hohaus, T., Kleist, E., Mensah, A., Mentel, T. F., Spindler, C., Uerlings, R., Tillmann, R., and Wildt, J.: Aerosol Mass Spectrometric Features of Biogenic SOA: Observations from a Plant Chamber and in Rural Atmospheric Environments, Environ. Sci. Technol., 43, 8166–8172, 2009.
Kim, H., Collier, S., Ge, X., Xu, J., Sun, Y., Jiang, W., Wang, Y., Herckes, P., and Zhang, Q.: Chemical processing of water-soluble species and formation of secondary organic aerosol in fogs, Atmos. Environ., 200, 158–166, 2019.
Koop, T., Luo, B., Tsias, A., and Peter, T.: Water activity as the determinant for homogeneous ice nucleation in aqueous solutions, Nature, 406, 611–614, 2000.
Kroll, J. H., Donahue, N. M., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R., Wilson, K. R., Altieri, K. E., Mazzoleni, L. R., Wozniak, A. S., Bluhm, H., Mysak, E. R., Smith, J. D., Kolb, C. E., and Worsnop, D. R.: Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 3, 133–139, 2011.
Laing, J. R., Jaffe, D. A., and Hee, J. R.: Physical and optical properties of aged biomass burning aerosol from wildfires in Siberia and the Western USA at the Mt. Bachelor Observatory, Atmos. Chem. Phys., 16, 15185–15197, https://doi.org/10.5194/acp-16-15185-2016, 2016.
Lee, A., Goldstein, A. H., Keywood, M. D., Gao, S., Varutbangkul, V., Bahreini, R., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res.-Atmos., 111, D07302, https://doi.org/10.1029/2005JD006437, 2006.
Lee, A. K. Y., Herckes, P., Leaitch, W. R., Macdonald, A. M., and Abbatt, J. P. D.: Aqueous OH oxidation of ambient organic aerosol and cloud water organics: Formation of highly oxidized products, Geophys. Res. Lett., 38, L11805, https://doi.org/10.1029/2011GL047439, 2011.
Lee, A. K. Y., Hayden, K. L., Herckes, P., Leaitch, W. R., Liggio, J., Macdonald, A. M., and Abbatt, J. P. D.: Characterization of aerosol and cloud water at a mountain site during WACS 2010: secondary organic aerosol formation through oxidative cloud processing, Atmos. Chem. Phys., 12, 7103–7116, https://doi.org/10.5194/acp-12-7103-2012, 2012.
Lee, A. K. Y., Abbatt, J. P. D., Leaitch, W. R., Li, S.-M., Sjostedt, S. J., Wentzell, J. J. B., Liggio, J., and Macdonald, A. M.: Substantial secondary organic aerosol formation in a coniferous forest: observations of both day- and nighttime chemistry, Atmos. Chem. Phys., 16, 6721–6733, https://doi.org/10.5194/acp-16-6721-2016, 2016.
McClure, C. D., Jaffe, D. A., and Gao, H.: Carbon Dioxide in the Free Troposphere and Boundary Layer at the Mt. Bachelor Observatory, Aerosol Air Qual. Res., 16, 717–728, 2016.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Tech., 46, 258–271, 2012.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Paatero, P. and Tapper, U.: Positive Matrix Factorization – a Nonnegative Factor Model with Optimal Utilization of Error-Estimates of Data Values, Environmetrics, 5, 111–126, 1994.
Phinney, L., Richard Leaitch, W., Lohmann, U., Boudries, H., Worsnop, D. R., Jayne, J. T., Toom-Sauntry, D., Wadleigh, M., Sharma, S., and Shantz, N.: Characterization of the aerosol over the sub-arctic north east Pacific Ocean, Deep Sea Res., 53, 2410–2433, 2006.
Reidmiller, D. R., Jaffe, D. A., Fischer, E. V., and Finley, B.: Nitrogen oxides in the boundary layer and free troposphere at the Mt. Bachelor Observatory, Atmos. Chem. Phys., 10, 6043–6062, https://doi.org/10.5194/acp-10-6043-2010, 2010.
Rinaldi, M., Gilardoni, S., Paglione, M., Sandrini, S., Fuzzi, S., Massoli, P., Bonasoni, P., Cristofanelli, P., Marinoni, A., Poluzzi, V., and Decesari, S.: Organic aerosol evolution and transport observed at Mt. Cimone (2165 m a.s.l.), Italy, during the PEGASOS campaign, Atmos. Chem. Phys., 15, 11327–11340, https://doi.org/10.5194/acp-15-11327-2015, 2015.
Ripoll, A., Minguillón, M. C., Pey, J., Jimenez, J. L., Day, D. A., Sosedova, Y., Canonaco, F., Prévôt, A. S. H., Querol, X., and Alastuey, A.: Long-term real-time chemical characterization of submicron aerosols at Montsec (southern Pyrenees, 1570 m a.s.l.), Atmos. Chem. Phys., 15, 2935–2951, https://doi.org/10.5194/acp-15-2935-2015, 2015.
Roberts, G. C., Day, D. A., Russell, L. M., Dunlea, E. J., Jimenez, J. L., Tomlinson, J. M., Collins, D. R., Shinozuka, Y., and Clarke, A. D.: Characterization of particle cloud droplet activity and composition in the free troposphere and the boundary layer during INTEX-B, Atmos. Chem. Phys., 10, 6627–6644, https://doi.org/10.5194/acp-10-6627-2010, 2010.
Robinson, N. H., Hamilton, J. F., Allan, J. D., Langford, B., Oram, D. E., Chen, Q., Docherty, K., Farmer, D. K., Jimenez, J. L., Ward, M. W., Hewitt, C. N., Barley, M. H., Jenkin, M. E., Rickard, A. R., Martin, S. T., McFiggans, G., and Coe, H.: Evidence for a significant proportion of Secondary Organic Aerosol from isoprene above a maritime tropical forest, Atmos. Chem. Phys., 11, 1039–1050, https://doi.org/10.5194/acp-11-1039-2011, 2011.
Rose, C., Sellegri, K., Asmi, E., Hervo, M., Freney, E., Colomb, A., Junninen, H., Duplissy, J., Sipilä, M., Kontkanen, J., Lehtipalo, K., and Kulmala, M.: Major contribution of neutral clusters to new particle formation at the interface between the boundary layer and the free troposphere, Atmos. Chem. Phys., 15, 3413–3428, https://doi.org/10.5194/acp-15-3413-2015, 2015.
Schroder, F., Karcher, B., Fiebig, M., and Petzold, A.: Aerosol states in the free troposphere at northern midlatitudes, J. Geophys. Res.-Atmos., 107, 8126–8133, 2002.
Schurman, M. I., Lee, T., Sun, Y., Schichtel, B. A., Kreidenweis, S. M., and Collett Jr., J. L.: Investigating types and sources of organic aerosol in Rocky Mountain National Park using aerosol mass spectrometry, Atmos. Chem. Phys., 15, 737–752, https://doi.org/10.5194/acp-15-737-2015, 2015.
Setyan, A., Zhang, Q., Merkel, M., Knighton, W. B., Sun, Y., Song, C., Shilling, J. E., Onasch, T. B., Herndon, S. C., Worsnop, D. R., Fast, J. D., Zaveri, R. A., Berg, L. K., Wiedensohler, A., Flowers, B. A., Dubey, M. K., and Subramanian, R.: Characterization of submicron particles influenced by mixed biogenic and anthropogenic emissions using high-resolution aerosol mass spectrometry: results from CARES, Atmos. Chem. Phys., 12, 8131–8156, https://doi.org/10.5194/acp-12-8131-2012, 2012.
Sorooshian, A., Crosbie, E., Maudlin, L. C., Youn, J.-S., Wang, Z., Shingler, T., Ortega, A. M., Hersey, S., and Woods, R. K.: Surface and airborne measurements of organosulfur and methanesulfonate over the western United States and coastal areas, J. Geophys. Res.-Atmos., 120, 8535–8548, 2015.
Stohl, A., Trainer, M., Ryerson, T. B., Holloway, J. S., and Parrish, D. D.: Export of NOy from the North American boundary layer during 1996 and 1997 North Atlantic Regional Experiments, J. Geophys. Res.-Atmos., 107, 4131–4139, 2002.
Sun, Y., Zhang, Q., Macdonald, A. M., Hayden, K., Li, S. M., Liggio, J., Liu, P. S. K., Anlauf, K. G., Leaitch, W. R., Steffen, A., Cubison, M., Worsnop, D. R., van Donkelaar, A., and Martin, R. V.: Size-resolved aerosol chemistry on Whistler Mountain, Canada with a high-resolution aerosol mass spectrometer during INTEX-B, Atmos. Chem. Phys., 9, 3095–3111, https://doi.org/10.5194/acp-9-3095-2009, 2009.
Sun, Y. L., Zhang, Q., Schwab, J. J., Yang, T., Ng, N. L., and Demerjian, K. L.: Factor analysis of combined organic and inorganic aerosol mass spectra from high resolution aerosol mass spectrometer measurements, Atmos. Chem. Phys., 12, 8537–8551, https://doi.org/10.5194/acp-12-8537-2012, 2012.
Takahama, S., Schwartz, R. E., Russell, L. M., Macdonald, A. M., Sharma, S., and Leaitch, W. R.: Organic functional groups in aerosol particles from burning and non-burning forest emissions at a high-elevation mountain site, Atmos. Chem. Phys., 11, 6367–6386, https://doi.org/10.5194/acp-11-6367-2011, 2011.
Timonen, H., Wigder, N., and Jaffe, D.: Influence of background particulate matter (PM) on urban air quality in the Pacific Northwest, J. Environ. Manage., 129, 333–340, 2013.
Timonen, H., Jaffe, D. A., Wigder, N., Hee, J., Gao, H., Pitzman, L., and Cary, R. A.: Sources of carbonaceous aerosol in the free troposphere, Atmos. Environ., 92, 146–153, 2014.
Tröstl, J., Herrmann, E., Frege, C., Bianchi, F., Molteni, U., Bukowiecki, N., Hoyle, C. R., Steinbacher, M., Weingartner, E., Dommen, J., Gysel, M., and Baltensperger, U.: Contribution of new particle formation to the total aerosol concentration at the high-altitude site Jungfraujoch (3580 m a.s.l., Switzerland), J. Geophys. Res.-Atmos., 121, 11692–11711, 2016.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Van Dingenen, R., Putaud, J.-P., Martins-Dos Santos, S., and Raes, F.: Physical aerosol properties and their relation to air mass origin at Monte Cimone (Italy) during the first MINATROC campaign, Atmos. Chem. Phys., 5, 2203–2226, https://doi.org/10.5194/acp-5-2203-2005, 2005.
Wagner, N. L., Brock, C. A., Angevine, W. M., Beyersdorf, A., Campuzano-Jost, P., Day, D., de Gouw, J. A., Diskin, G. S., Gordon, T. D., Graus, M. G., Holloway, J. S., Huey, G., Jimenez, J. L., Lack, D. A., Liao, J., Liu, X., Markovic, M. Z., Middlebrook, A. M., Mikoviny, T., Peischl, J., Perring, A. E., Richardson, M. S., Ryerson, T. B., Schwarz, J. P., Warneke, C., Welti, A., Wisthaler, A., Ziemba, L. D., and Murphy, D. M.: In situ vertical profiles of aerosol extinction, mass, and composition over the southeast United States during SENEX and SEAC4RS: observations of a modest aerosol enhancement aloft, Atmos. Chem. Phys., 15, 7085–7102, https://doi.org/10.5194/acp-15-7085-2015, 2015.
Wang, J., Krejci, R., Giangrande, S., Kuang, C., Barbosa, H. M., Brito, J., Carbone, S., Chi, X., Comstock, J., Ditas, F., Lavric, J., Manninen, H. E., Mei, F., Moran-Zuloaga, D., Pohlker, C., Pohlker, M. L., Saturno, J., Schmid, B., Souza, R. A., Springston, S. R., Tomlinson, J. M., Toto, T., Walter, D., Wimmer, D., Smith, J. N., Kulmala, M., Machado, L. A., Artaxo, P., Andreae, M. O., Petaja, T., and Martin, S. T.: Amazon boundary layer aerosol concentration sustained by vertical transport during rainfall, Nature, 539, 416–419, 2016.
Watts, S. F.: The mass budgets of carbonyl sulfide, dimethyl sulfide, carbon disulfide and hydrogen sulfide, Atmos. Environ., 34, 761–779, 2000.
Weiss-Penzias, P., Jaffe, D. A., Swartzendruber, P., Dennison, J. B., Chand, D., Hafner, W., and Prestbo, E.: Observations of Asian air pollution in the free troposphere at Mount Bachelor Observatory during the spring of 2004, J. Geophys. Res.-Atmos., 111, D10304, https://doi.org/10.1029/2005JD006522, 2006.
Worton, D. R., Goldstein, A. H., Farmer, D. K., Docherty, K. S., Jimenez, J. L., Gilman, J. B., Kuster, W. C., de Gouw, J., Williams, B. J., Kreisberg, N. M., Hering, S. V., Bench, G., McKay, M., Kristensen, K., Glasius, M., Surratt, J. D., and Seinfeld, J. H.: Origins and composition of fine atmospheric carbonaceous aerosol in the Sierra Nevada Mountains, California, Atmos. Chem. Phys., 11, 10219–10241, https://doi.org/10.5194/acp-11-10219-2011, 2011.
Xu, J., Zhang, Q., Shi, J., Ge, X., Xie, C., Wang, J., Kang, S., Zhang, R., and Wang, Y.: Chemical characteristics of submicron particles at the central Tibetan Plateau: insights from aerosol mass spectrometry, Atmos. Chem. Phys., 18, 427–443, https://doi.org/10.5194/acp-18-427-2018, 2018.
Young, D. E., Kim, H., Parworth, C., Zhou, S., Zhang, X., Cappa, C. D., Seco, R., Kim, S., and Zhang, Q.: Influences of emission sources and meteorology on aerosol chemistry in a polluted urban environment: results from DISCOVER-AQ California, Atmos. Chem. Phys., 16, 5427–5451, https://doi.org/10.5194/acp-16-5427-2016, 2016.
Zhang, L. and Jaffe, D. A.: Trends and sources of ozone and sub-micron aerosols at the Mt. Bachelor Observatory (MBO) during 2004–2015, Atmos. Environ., 165, 143–154, 2017.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y. L.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, 2011.
Zhang, Q., Zhou, S., Collier, S., Jaffe, D., Onasch, T., Shilling, J., Kleinman, L., and Sedlacek, A.: Understanding Composition, Formation, and Aging of Organic Aerosols in Wildfire Emissions via Combined Mountain Top and Airborne Measurements. In Multiphase Environmental Chemistry in the Atmosphere, Am. Chem. Soc., 1299, 363–385, 2018.
Zhou, S., Collier, S., Jaffe, D. A., Briggs, N. L., Hee, J., Sedlacek III, A. J., Kleinman, L., Onasch, T. B., and Zhang, Q.: Regional influence of wildfires on aerosol chemistry in the western US and insights into atmospheric aging of biomass burning organic aerosol, Atmos. Chem. Phys., 17, 2477–2493, https://doi.org/10.5194/acp-17-2477-2017, 2017.
Zhou, S., Collier, S., Xu, J., Mei, F., Wang, J., Lee, Y.-N., Sedlacek, A. J., Springston, S. R., Sun, Y., and Zhang, Q.: Influences of upwind emission sources and atmospheric processing on aerosol chemistry and properties at a rural location in the Northeastern U.S., J. Geophys. Res.-Atmos., 121, 6049–6065, 2016.
Zhu, Q., He, L.-Y., Huang, X.-F., Cao, L.-M., Gong, Z.-H., Wang, C., Zhuang, X., and Hu, M.: Atmospheric aerosol compositions and sources at two national background sites in northern and southern China, Atmos. Chem. Phys., 16, 10283–10297, https://doi.org/10.5194/acp-16-10283-2016, 2016.
Zorn, S. R., Drewnick, F., Schott, M., Hoffmann, T., and Borrmann, S.: Characterization of the South Atlantic marine boundary layer aerosol using an aerodyne aerosol mass spectrometer, Atmos. Chem. Phys., 8, 4711–4728, https://doi.org/10.5194/acp-8-4711-2008, 2008.