the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 11 Dec 2019
| 11 Dec 2019
Secondary organic aerosol formation from OH-initiated oxidation of m-xylene: effects of relative humidity on yield and chemical composition
Qun Zhang
Yongfu Xu
The effect of relative humidity (RH) on secondary organic aerosol (SOA) formation from the photooxidation of m-xylene initiated by OH radicals in the absence of seed particles was investigated in a Teflon reactor. The SOA yields were determined based on the particle mass concentrations measured with a scanning mobility particle sizer (SMPS) and reacted m-xylene concentrations measured with a gas chromatograph–mass spectrometer (GC-MS). The SOA components were analyzed using a Fourier transform infrared (FTIR) spectrometer and an ultrahigh-performance liquid chromatograph–electrospray ionization–high-resolution mass spectrometer (UPLC-ESI-HRMS). A significant decrease was observed in SOA mass concentration and yield variation with the increasing RH conditions. The SOA yields are 14.0 %–16.5 % and 0.8 %–3.2 % at low RH (14 %) and high RH (74 %–79 %), respectively, with the difference being nearly 1 order of magnitude. Some of the reduction in the apparent yield may be due to the faster wall loss of semi-volatile products of oxidation at higher RH. The chemical mechanism for explaining the RH effects on SOA formation from m-xylene–OH system is proposed based on the analysis of both FTIR and HRMS measurements, and the Master Chemical Mechanism (MCM) prediction is used as the assistant. The FTIR analysis shows that the proportion of oligomers with C-O-C groups from carbonyl compounds in SOA at high RH is higher than that at low RH, but further information cannot be provided by the FTIR results to well explain the negative RH effect on SOA formation. In the HRMS spectra, it is found that C2H2O is one of the most frequent mass differences at low and high RHs, that the compounds with a lower carbon number in the formula at low RH account for a larger proportion than those at high RH and that the compounds at high RH have higher O : C ratios than those at low RH. The HRMS results suggest that the RH may suppress oligomerization where water is involved as a by-product and may influence the further particle-phase reaction of highly oxygenated organic molecules (HOMs) formed in the gas phase. In addition, the negative RH effect on SOA formation is enlarged based on the gas-to-particle partitioning rule.
- Article
(2250 KB) - Full-text XML
-
Supplement
(238 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA) is a significant component of atmospheric fine particulate matter in the troposphere (Hallquist et al., 2009; Spracklen et al., 2011; Huang et al., 2014), causing serious concern as it has a significant influence on air quality, the oxidative capacity of the troposphere, global climate change and human health (Jacobson et al., 2000; Hansen and Sato, 2001; Kanakidou et al., 2005; Zhang et al., 2014). In a previous study from a global model simulation, it has been found that SOA represents a large fraction, approximately 80 % of total organic aerosol sources (Spracklen et al., 2011).
The formation of SOA in the atmosphere is principally via the oxidation of volatile organic compounds (VOCs) by common atmospheric oxidants such as O3, OH and NO3 radicals (Seinfeld and Pandis, 2016). Aromatic compounds mainly from anthropogenic sources, including solvent usage, oil-fired vehicles and industrial emissions, contribute 20 %–30 % to the total VOCs in urban atmosphere, which play a significant role in the formation of ozone and SOA (Forstner et al., 1997; Odum et al., 1997; Calvert et al., 2002; Bloss et al., 2005; Offenberg et al., 2007; Ding et al., 2012; Zhao et al., 2017). Amongst aromatics, m-xylene is significant, and the mean concentration of which together with p-xylene in the daytime was determined to be up to 140.8 µg m−3 in the atmosphere of urban areas in developing countries (Khoder, 2007).
The oxidation of aromatics in the troposphere is mainly initiated through OH radicals, and is affected by many chemical and physical factors. The concentrations of oxidant species, VOCs and NOx concentrations, as well as the ratio of VOCs to NOx (Ge et al., 2017b) determine the main chemical mechanism. Light intensity (Warren et al., 2008), temperature (Qi et al., 2010) and relative humidity (RH) are the most significant physical parameters that affect the chemical process. RH governs the water concentration in the gas phase and the liquid water content (LWC) in the particle phase. Water plays a significant role as a reactant, product and solvent to directly participate in chemistry (Finlayson-Pitts and Pitts Jr., 2000) and indirectly affect the reaction environment such as the acidity of particles (Jang et al., 2002). The acid catalysis of heterogeneous reactions of atmospheric organic carbonyl species in particle phase can lead to a large increase in SOA mass, while this process can be suppressed by lower acidity at high RH (Czoschke et al., 2003). In addition, RH can change the viscosity of SOA and further affect the chemical processes of SOA formation (Kidd et al., 2014; Liu et al., 2017).
Investigations of the RH effects on aromatic SOA have been conducted in many previous works. In the presence of NOx, it was observed that RH significantly enhanced the yield of SOA from benzene, toluene, ethylbenzene and xylene photooxidation, which was explained by a higher formation of HONO, particle water, aqueous radical reactions and hydration from glyoxal (Healy et al., 2009; Kamens et al., 2011; Zhou et al., 2011; Jia and Xu, 2014, 2018; Wang et al., 2016). Meanwhile, under low-NOx conditions, wherein no NOx was introduced artificially and photolysis of H2O2 was as an OH radical source, it has been observed that deliquesced seed contributed to the enhancement of SOA yield from toluene (Faust et al., 2017; Liu et al., 2018). However, under low-NOx levels, it has been found that, in the study on toluene SOA formation, a moderate RH level (48 %) leads to a lower SOA yield than low-RH level (17 %–18 %; Cao and Jang, 2010). In a most recent study on SOA formation of toluene (Hinks et al., 2018), high RH led to a much lower SOA yield than low RH under low-NOx levels, which is attributed to condensation reactions that remove water, leading to less oligomerization at high RH. In a study on the chemical oxidative potential of SOA (Tuet et al., 2017) under low-NOx conditions, it was observed that the mass concentration of SOA from m-xylene irradiation under dry conditions was much larger than that under humid conditions, whereas the study did not focus on the mechanism of the RH effect on m-xylene SOA formation. These demonstrate that the RH effects on aromatic SOA yields, especially m-xylene, have not been fully understood, and RH effects are controversial under various NOx levels and seed particle conditions.
The chemical components of SOA are important, and are on which climate- and health-relevant properties of particles are dependent. Chemical composition of SOA from aromatics–NOx photooxidation has been investigated by gas chromatograph–mass spectrometer (GC-MS) analysis (Forstner et al., 1997). Nevertheless, this study was only performed at a limited RH range of 15 %–25 %. GC-MS in this study may not be the optimal technique for analysis of SOA components as high temperature at GC injection ports can easily decompose some low-volatility substances in SOA. FTIR was also used to study the chemical composition of SOA from aromatics–NOx photooxidation under different RH conditions, in which the information of functional groups in SOA was provided (Jia and Xu, 2014, 2018). In these studies, O-H, C=O, C-O and C-OH were found to be the main functional groups, intensities of which largely increased with increasing RH. Compounds in SOA with the O-H group mainly contributed to the increase of SOA, such as polyalcohols formed from aqueous reactions. A recent study on SOA components from toluene–OH system under both dry and humid conditions were analyzed via high-resolution mass spectrometry (HRMS) (Hinks et al., 2018). Although some chemical composition in SOA has been identified, the analysis and the mechanism of RH effects still need to be further studied. The RH effects on SOA formation from m-xylene under low-NOx conditions have not been studied well. In the present study, we present the results from the experiments about the SOA formation from the OH-initiated oxidation of m-xylene in the absence of seed particles in a Teflon bag. The SOA yields at different RHs and the chemical components under both low- and high-RH conditions will be reported. The underlying mechanism of SOA formation for these different conditions will be also discussed.
2.1 Equipment and reagents
Experiments on m-xylene photooxidation were performed in a 1 m3 air-tight Teflon FEP film reactor (DuPont 500A, USA), which is similar to our previous works (Jia and Xu, 2014, 2016, 2018; Ge et al., 2016, 2017c, b, a). A light source was provided by 96 lamps (F40BLB, GE; UVA-340, Q-Lab, USA) surrounding the Teflon bag to simulate the UV band of the solar spectrum in the troposphere. The NO2 photolysis rate was determined to be 0.23 min−1, which was used to reflect the light intensity in the reactor. To remove the electric charge on the surface of the FEP reactor, two ionizing air blowers were equipped outside the Teflon bag and were used throughout each experiment (McMurry and Rader, 2007).
The background gas was zero air, which was generated from Zero Air Supply and CO Reactor (Model 111 and 1150, Thermo Scientific, USA) and further purified by hydrocarbon traps (BHT-4, Agilent, USA). The humid zero air was obtained by bubbling dry zero air through ultrapure water (Milli Q, 18MU, Millipore Ltd., USA). To obtain the different desired RH in the reactor, a different ratio of dry and humid zero air was mixed. The RH and temperature in the reactor were measured by a hygrometer (Model 645, Testo AG, Germany).
Throughout each experiment, the background NOx concentration in the reactor was lower than 1 ppb and OH radicals were provided from H2O2 photolysis. Hydrogen peroxide was introduced into the reactor along with the zero air flow over a period of 30 min via an injection of H2O2 solution (30 wt %) into a three-way tube using a syringe to the desired concentration of 20 ppm. Though the H2O2 level was not measured, it was estimated through the measured volume of H2O2 solution evaporated. m-Xylene (99 %, Alfa Aesar) was introduced to the reactor subsequently using the same approach. No seed particles were introduced artificially. All reactants were introduced initially and then the lights were turned on and the reaction started. The experiments were conducted for 4 h. Thus, the “end” of the experiment in this study refers to the experiment at 4 h of reaction time.
2.2 Monitoring and analysis
The concentration of m-xylene in the reactor was measured with a gas chromatograph–mass spectrometer (GC-MS, Model 7890A GC and Model 5975C mass selective detector, Agilent, USA), which was equipped with a thermal desorber (Master TD, Dani, Italy). The size distribution and concentrations of particles were monitored with a scanning mobility particle sizer (SMPS, Model 3936, TSI, USA). In our enclosed Teflon bag, our sampling instruments consumed 10 % of reactor volume during the photooxidation and we did not use make up air to dilute the bag. The particle wall loss constant has been determined to be s−1 and s−1 at low-RH and moist conditions, respectively. Though the particle wall loss constant is size dependent, it is not a strong function of particle size for the relatively narrow size distributions in smog chamber experiments (Park et al., 2001). Here we approximate the wall loss as size independent. In experiments under moist conditions, particles measured by SMPS consisted of liquid water content (LWC) and SOA. In low-RH experiments, as SOA hardly absorbs aerosol water, LWC can be negligible. Thus, the SOA mass can be directly measured by SMPS in low-RH experiments. To obtain the SOA mass in high-RH experiments, LWC should be excluded from total particle mass. The method for the measurement of LWC was already described in the previous study (Jia and Xu, 2018), so here only a brief introduction is provided. During each moist experiment, the SMPS measured the humid particles. After 4 h from the start of oxidation reaction in each moist experiment, the SMPS was modified to the dry mode. In the dry mode, a Nafion dryer (Perma Pure MD-700-12F-3) was added to the sampling flow and a Nafion dryer (Perma Pure PD-200T-24MPS) was added to the sheath flow. After the modification of SMPS, the humid air in SMPS was quickly replaced by dry air through venting the sheath air at 5 L min−1, so that the RH in the sheath air could decrease to 7 %. Then, SMPS at this dry mode measured dry particle concentrations as the RH in the sample air decreased to 10 % at this time. The LWC was determined by the difference of the particle mass concentrations before and after the SMPS modification to the dry mode.
The chemical composition of SOA originated from m-xylene–OH irradiation was investigated using a Fourier transform infrared (FTIR) spectrometer, which can provide information on the functional groups. The particles were collected on a ZnSe disk using a Dekati low-pressure impactor (DLPI, Dekati Ltd., Finland) at the end of each experiment (Ge et al., 2016; Jia and Xu, 2016). The duration of DLPI for FTIR was 15 min, and this sampling was taken just after 4 h of each experiment. Then, the ZnSe disk was directly put in a FTIR (Nicolet iS10, Thermos Fisher, USA) for the measurement of functional groups of the chemical composition in SOA samples.
To obtain the detailed information of chemical composition, SOA particles were sampled using the Particle into Liquid Sampler (PILS, model 4001, BMI, USA). The PILS samples water-soluble species in particles. As low-NOx m-xylene SOA is composed of almost all water-soluble species, it is reasonable and reliable to use PILS to sample SOA for the analysis of the chemical composition. The flow rate of the sample gas was around 11 L min−1, and the output flow rate of liquid sample was 0.05 mL min−1. Two denuders were used to remove the VOCs and acids in the sample gas. SOA liquid samples collected by PILS were finally transferred into vials for subsequent analysis of mass spectrometry. The duration of PILS was 5 min, and this sampling was taken just after 4 h of each experiment. Operatively, the blank measurements were obtained by replacing the sample gas with zero air collected in vials. It is well known that the PILS samples water-soluble species in the SOA with high efficiency. In addition, it is reported that the PILS can also sample slightly water-soluble organic compounds with average O : C ratios higher than 0.26 instead of the total SOA composition and the collection efficiency can exceed 0.6 (Zhang et al., 2016). Thus, the PILS can sample the overwhelming majority of the SOA system in our study, though PILS cannot sample water-insoluble species in the SOA.
The accurate mass of organic compounds in SOA was measured by a ultrahigh-performance liquid chromatograph (UPLC, Ultimate 3000, Thermo Scientific, USA) with a heated electrospray ionization high-resolution Orbitrap-based mass spectrometer (HESI-HRMS, Q Exactive, Thermo Scientific, USA). Methanol (Optima™ LC/MS Grade, Fisher Chemical, USA) was used as the eluent in UPLC system. The elution flow rate was 0.2 mL min−1, and the overall run time was 5 min. The injection volume was 20 µL. In this study, the UPLC was only used as the injection system of HRMS. The acquired mass spectrum of SOA was in the range of 80–1000 Da. The HESI source was conducted in positive and negative ion modes using the optimum method for characterization of organic compounds. We used the Thermo Scientific Xcalibur software (Thermo Fisher Scientific Inc., USA) to analyze the data from HRMS. To calculate the elemental composition of compounds, the accurate mass measurements were used. The reaction pathways and products of m-xylene–OH photooxidation in the Master Chemical Mechanism (MCM v3.3.1, the website at http://mcm.leeds.ac.uk/MCM; last access: 16 October 2017) was used for analysis of the products measured by HRMS (Jenkin et al., 2003; Jia and Xu, 2014).
Table 1Experimental conditions and results at 4 h of experiments in m-xylene–H2O2 photooxidation system.
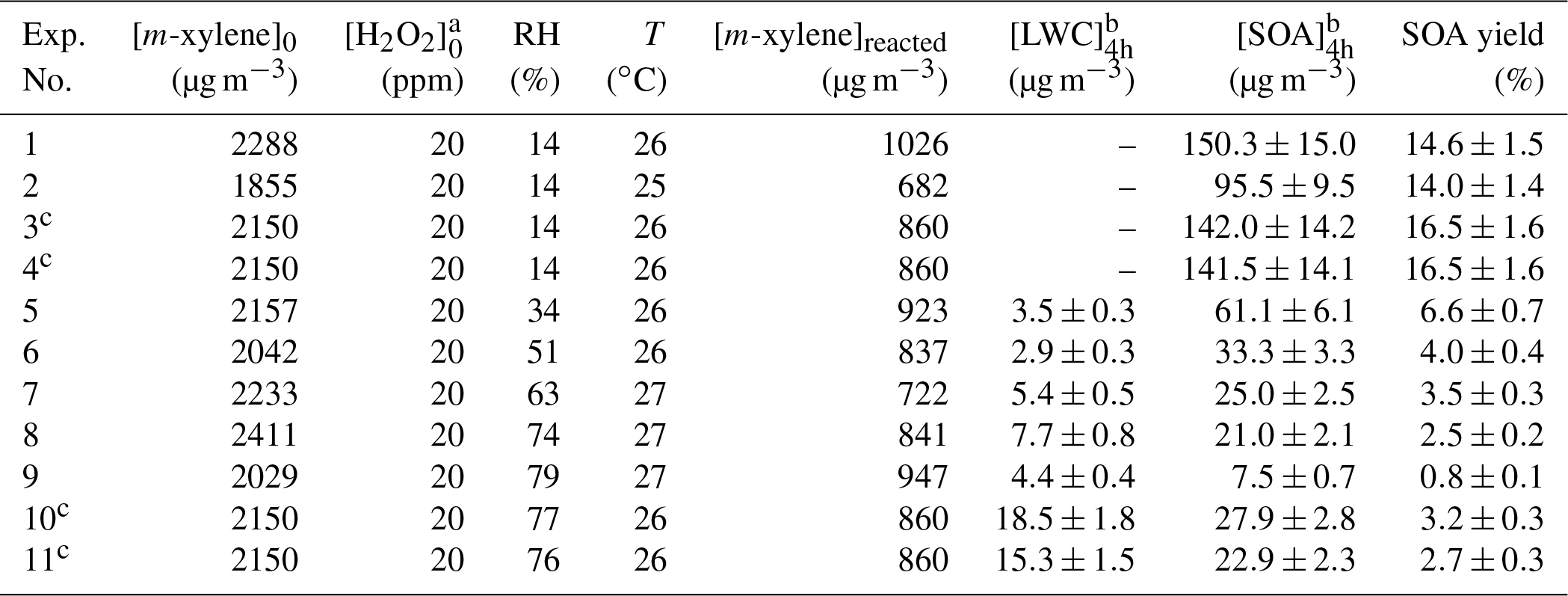
a Calculated using the density and mass concentration of the injected H2O2 solution, and the volume of the reactor. b The mass concentration at 4 h of reaction time with particle wall loss corrected. An SOA density of 1.4 g cm−3 was used to obtain the SOA mass concentrations (Song et al., 2007). c In Experiments. 3–4 and 10–11, the initial concentrations of m-xylene were calculated using the density and the volume of the injected m-xylene, and the volume of the reactor; the reacted concentrations of m-xylene were estimated using 40 % of the initial concentrations of m-xylene.
3.1 RH effects on SOA yields
Eleven experiments were conducted. Experimental conditions and results at 4 h of experiments in the m-xylene–H2O2 photooxidation system are summarized in Table 1. Experiments 1–4 were conducted in dry zero air, and are defined as the low-RH experiments. Experiments 8–11 were conducted in humid zero air, and are defined as the high-RH experiments. Experiments 5–7 were conducted in the mixed air of dry and humid zero air, and are defined as the intermediate-RH experiments. The initial concentrations of m-xylene and the consumed m-xylene proportion (typically ∼40 %) were approximately the same under different RH conditions. Under intermediate- and high-RH conditions, LWC accounts for a certain proportion of particles (Jia and Xu, 2018). To obtain the time evolution of SOA concentrations, the LWC has to be subtracted during the whole photooxidation period. Since LWC was only measured at the end of the reaction, the volume growth factor (VGF) was used to estimate the contribution of LWC in particles, which was defined as the ratio of the humid particle volume to the dry particle volume (Engelhart et al., 2011). It was assumed that the VGF did not change during the whole photooxidation period. The removal of aerosol water during the LWC measurement may cause dissolved species that are probably volatile/semi-volatile compounds to evaporate back into the gas phase (El-Sayed et al., 2015, 2016). Glyoxal is a typical semi-volatile compound with a high Henry's law constant, and is involved in SOA formation in m-xylene–OH system of our study. The Henry's law constant of glyoxal in pure water is as high as 4.19×105 M atm−1 at 298 K (Ip et al., 2009). Only 1 in 10 000 glyoxal molecules can dissolve in the LWC, whose concentration was obtained in our study. Thus, SOA concentrations for intermediate- and high-RH conditions were slightly underestimated, but the underestimation is extremely minimal and can be neglected. To obtain the mass concentrations of SOA, an SOA density of 1.4 g cm−3 was used (Song et al., 2007).
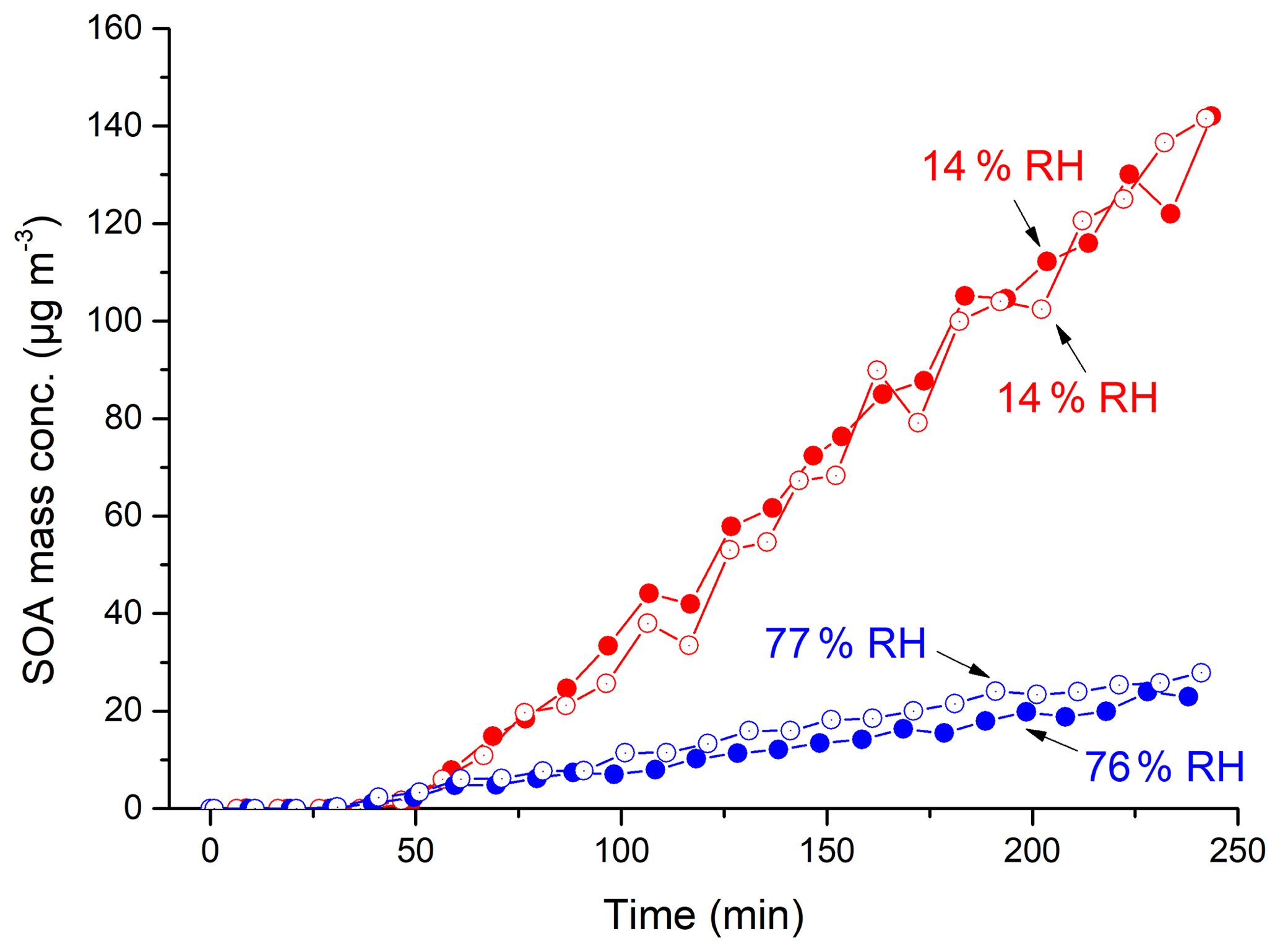
Figure 1Mass concentration time profiles of SOA from m-xylene–H2O2 photooxidation at 14 % (Exps. 3–4) and 76 %–77 % (Exps. 10–11) RH (corrected by particle wall loss and for the amount of LWC in particles).
In Fig. 1, the wall-loss-corrected SOA mass concentrations are plotted as a function of photooxidation reaction time for m-xylene–OH systems at low (Exps. 3–4) and high (Exps. 10–11) RHs. It can be clearly observed that there is a large difference in the maximum mass concentration between 14 % and 76 % RHs. In Table 1, the maximum mass concentrations are 95.5–150.3 µg m−3 at low RHs, whereas they are 7.5–27.9 µg m−3 at high RHs, with the largest difference being over a factor of 10. The RH effect was reproducible when the initial m-xylene concentration was slightly changed under similar conditions.
We used the definition of the ratio of the SOA mass to the consumed m-xylene mass to calculate the SOA yield at the end of each experiment. In Table 1, the SOA yields at low RH are 14.0 %–16.5 %, while those at high RH are only around 0.8 %–3.2 %. SOA yields at low RH are nearly 1 order of magnitude larger than those at high RH. Though temperatures at high RH are slightly higher than those at low RH as shown in Table 1, which can lead to a higher SOA yield, the difference of temperatures between low- and high-RH conditions is lower than 2 ∘C, which cannot lead to a significantly different SOA yield to affect the result (Qi et al., 2010).
Seed aerosols were not artificially introduced throughout all the experiments, which could lead to the underestimation of SOA, as SOA-forming vapors partly condense to the reactor walls instead of particles (Matsunaga and Ziemann, 2010; Zhang et al., 2014). The extent to which vapor wall deposition affects SOA mass yields depends on the specific parent hydrocarbon system (Zhang et al., 2014, 2015; Nah et al., 2016, 2017). Zhang et al. (2014) have estimated two m-xylene systems under low-NOx conditions and concluded that SOA mass yields were underestimated by factors of 1.8 (Ng et al., 2007) and 1.6 (Loza et al., 2012) under low-RH conditions. In addition, the excess OH radicals in our experimental system lead to less underestimation of SOA formation as the losses of SOA-forming vapors can be mitigated via the use of excess oxidant concentrations (Nah et al., 2016). Thus, the underestimation of SOA formation can be limited. In fact, the wall loss of m-xylene was not taken into consideration for the calculation of mass yields, which generally overestimates.
The wall loss of organic compounds that is sensitive to humidity can affect the RH effect on SOA yields, as the reduction of SOA yields at high humidity can be due to the loss to the wet reactor wall. There are thousands of SOA precursors from m-xylene–H2O2 photooxidation. However, as far as we know, there is no previous study that investigates the RH effect on the loss of these organic compounds to the wet reactor wall. Thus, we select three organic compounds that have relevant experimental data to estimate how much the wall loss of chemical species affects SOA formation at different RHs.
The first compound is glyoxal, a typical SOA precursor. Glyoxal can easily dissolve in the aqueous phase due to the large Henry's law constant of 4.19×105 M atm−1 at 298 K (Ip et al., 2009), and is very sensitive to humidity. Loza et al. (2010) found that the wall loss of glyoxal was minimal at 5 % RH, with s−1, whereas kW was s−1 at 61 % RH. Obviously, there is a large difference in wall loss between low and high RHs. We assume that kW linearly increases with RH, and the kW value is estimated to be s−1 at 80 % and at 13 % RH, with the difference being a factor of 8.2. According to the wall loss of glyoxal, glyoxal only decreased by 10 % at the end of our experiment at low RH, while glyoxal decreased by 59 % at high RH. This means that the SOA yield was underestimated by 59 % at high RH and by 10 % at low RH when the glyoxal lost to the wall was completely transformed into SOA. When this wall effect of SOA precursors was taken into consideration, the SOA yields at 74 % (Exp. 8) and 14 % (Exp. 2) RHs were 6.1 % and 15.5 %, respectively, still with a difference of a factor of nearly 3. Thus, the loss of organic vapor to the wet wall with a Henry's law constant as high as glyoxal's cannot completely explain the large difference of SOA formation at low and high RH in our study.
In a recent study, the signal decay of two compounds (C5H8O2 and C5H9O4N) generated from isoprene oxidation at RH = 5 %, 50 % and >90 % has been presented (Huang et al., 2018); these were selected as the second and third compounds. C5H8O2 decreased by 10 % after 8.3 h at 5 % and 50 % RH, while C5H9O4N decreased by 20 % and 40 % at 5 % and 50 % RH after 8.3 h, respectively. In our study, the SOA yield at the end of our experiment decrease by 71 % at 51 % RH (Exp. 6) relative to that at 14 % RH (Exp. 2). According to the wall loss of C5H8O2, the SOA yield would not decrease at intermediate RH, while taking C5H9O4N, as it only decreased by 10 % at low RH and decreased by 22 % at intermediate RH, the SOA yield at 51 % RH (Exp. 6) would decrease by 13 % relative to that at 14 % RH (Exp. 2). Obviously, the decay characteristic of the two compounds cannot explain the more than factor of 3 difference of SOA formation at 14 % and 51 % RH in our experiments. If we take an extreme case of >90 % RH to estimate the impact of the semi-volatile organic compound (SVOC) wall losses on SOA formation in our experiments, the results are indeed different. C5H8O2 and C5H9O4N decreased by 90 % and 70 % after 2 h ( and s−1), respectively and subsequently remained steady, indicating the saturation of the wet wall to absorb the organic vapor under humid conditions of >90 % RH. Taking the decay characteristics of C5H8O2, it is estimated that the SOA yield at the end of 2 h would decrease by 90 % and would not further decrease after 2 h. However, our experimental results show that the yield at the end of 2 h is 1.4 %, a decrease of 87 % relative to that at low RH, and then it further decreased by 82 % at the end of 4 h. It seems that for VOCs that generate intermediate SVOCs with a high-RH effects of wall losses, the RH effect of their SOA yields can be explained by SVOC wall losses during the first period. However, these SVOCs generally have saturation characteristics, which cannot explain our observed RH effect of SOA formation. In fact, there were many different SOA precursors from the m-xylene oxidation system that probably have a much smaller Henry's law constant relative to that of glyoxal, 4.19×105 M atm−1. Thus, we considered that other mechanisms might exist to explain the negative RH effect on SOA formation from m-xylene photooxidation.
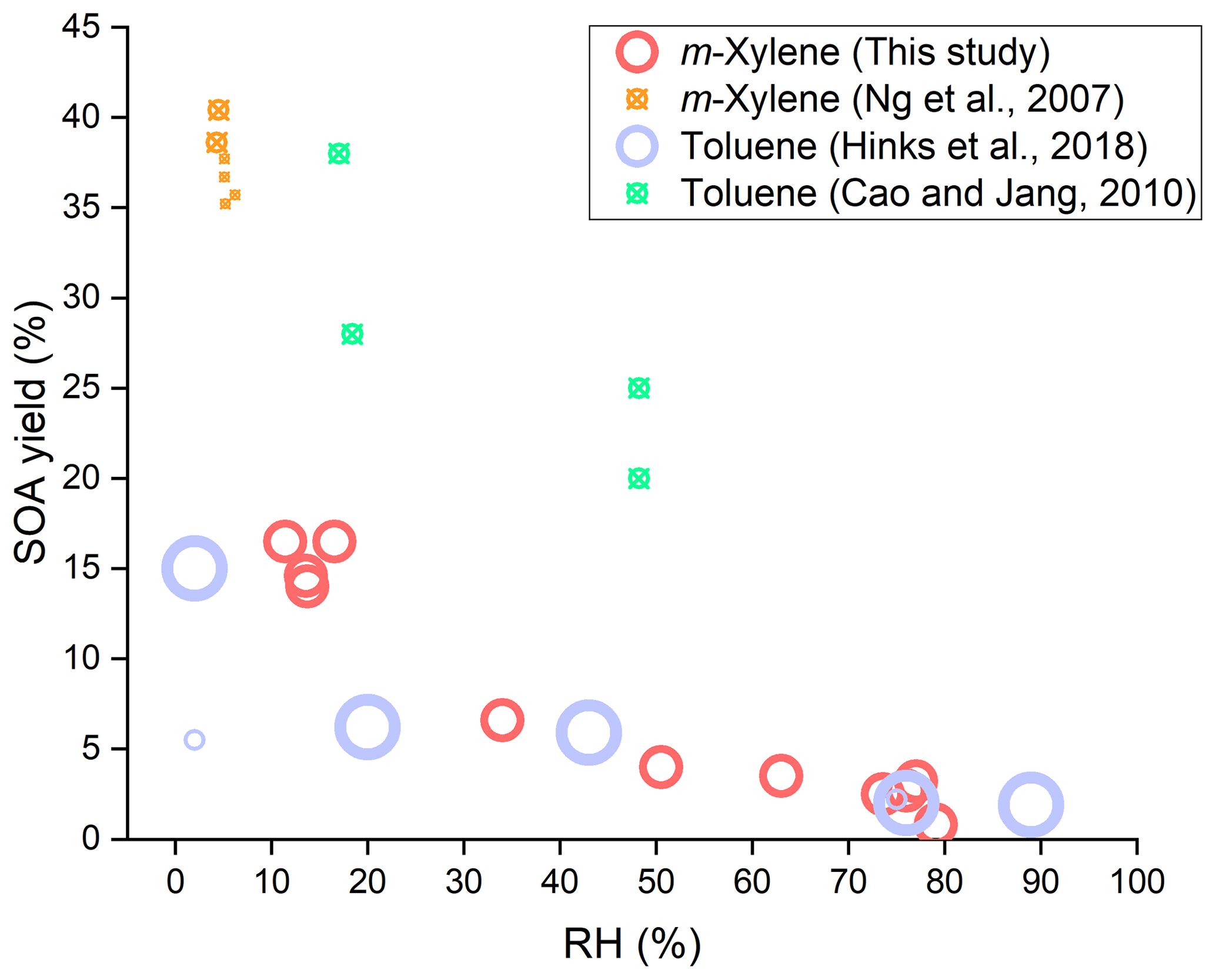
Figure 2SOA yields as a function of RH for different aromatic (toluene and m-xylene) oxidation under low-NOx conditions with photolysis of H2O2 as the OH source. The hollow circles represent where no seed particles were introduced and the circles with a cross where seed particles were introduced. The size of markers indicates the magnitude of the amount of reacted VOC.
For comparison and discussion of the results of SOA formation with other previous studies, Fig. 2 was plotted to show the SOA yields as a function of RH for the two aromatic compounds (toluene and m-xylene) oxidation under low-NOx conditions with the photolysis of H2O2 as the OH source. In Fig. 2, the hollow circles represent where no seed particles were introduced and the circles with a cross represent where seed particles were introduced, and the size of markers indicates the magnitude of the amount of reacted VOC. In the most recent study on toluene SOA formation conducted without seed particles (Hinks et al., 2018), the SOA yield at a low-NOx level was 15 % under dry conditions (<2 % RH) and 1.9 % under humid conditions (89 % RH), with the ratio of two yields of dry to humid conditions being over 7.5. The toluene SOA produced under high-RH conditions was significantly suppressed, in which the tendency of RH effects on SOA yield was very similar to our study, though the difference of SOA yield in the range of low- and high-RH conditions in Hinks et al. (2018) was slightly smaller than that in this study. The small difference of RH effects between Hinks et al. and our study is likely associated with the difference in experimental conditions, including RHs, initial and reacted VOCs and H2O2 concentrations, in addition to different species. This comparison demonstrates that different species of toluene and m-xylene of aromatics pose very similar RH effects under low-NOx conditions. Hinks et al. attributed the suppression of SOA yields by elevated RH to the lower level of oligomers generated by condensation reactions and the reduced mass loading at high RH.
In a study on an SOA model for toluene oxidation, the negative RH effect on SOA formation was also found in the presence of seed particles (Cao and Jang, 2010). In their study, the SOA yield at a low-NOx level was 28 %–30 % under low-RH conditions (17 %–18 % RH) and 20 %–25 % under moderate-RH conditions (48 % RH; Cao and Jang, 2010), but they did not focus on the RH effect to give an explanation. Furthermore, their RH only changed from 17 % to 48 %, the reacted parent VOC was smaller and the seed particles were present, so the RH effect on SOA yields was not as significant as that in Hinks et al. and our study. Ng et al. have investigated the yields of SOA formed from m-xylene–OH system at low RH (4 %–6 %) under low-NOx conditions (Ng et al., 2007). They obtained that the SOA yields were in the range of 35.2 %–40.4 % in the presence of seed particles. The SOA yields were larger than those of our study, as they conducted the experiments under a different irradiation time and with inorganic seed particles. These seed particles can provide not only a surface for chemical reactions, but also acidic and aqueous environments that can promote SOA formation (Jang et al., 2002; Liu et al., 2018; Faust et al., 2017). The reacted concentrations of the parent VOC was close between Cao and Jang and Ng et al. though the species were different. The results from these two studies can be considered together, since their experiments all had seed particles. As shown in Fig. 2, the negative RH effect on SOA yields can be found. In addition to these three previous studies shown in Fig. 2, a study on the chemical oxidative potential of SOA (Tuet et al., 2017) found that the concentration of SOA from m-xylene irradiation at a low-NOx level under dry conditions was much larger than that under humid conditions (89.3 µg m−3 at <5 % RH and 13.9 µg m−3 at 45 % RH), but they did not calculate the m-xylene SOA yields or give an explanation for the RH effect.

Figure 3FTIR spectra of particles from photooxidation of m-xylene–OH experiments under 14 % (Exp. 2) and 74 % RH (Exp. 8) conditions.
3.2 RH effects on functional groups of SOA
Figure 3 shows the FTIR spectra of particles from the photooxidation of m-xylene–OH experiments under both 14 % (Exp. 2) and 74 % (Exp. 8) RH conditions. The DLPI sample flow rate was 10 L min−1, and the sampling duration was 15 min. We used the same sampling flow rate and duration for both RH conditions. DLPI has 13 stages, and it can collect particles in the size range of 30 nm–10 µm. When we sampled using DLPI, the four plates for stages 4–7 were removed, so that particles in the range of 108–650 nm were collected on the third plate. As shown in Fig. S1 in the Supplement, the particles in the range of 108–650 nm can represent the total SOA from m-xylene oxidation in this study. The mean collection efficiency of the DLPI was 83 % for stages 4–7 (Durand et al., 2014). Thus, the SOA mass collected on the ZnSe window was 10.3 and 3.0 µg at 14 % RH (Exp. 2) and 74 % RH (Exp. 8), based on the SMPS measurement and the DLPI collection efficiency. As shown in Fig. 2, the SOA from m-xylene–OH experiments can be clearly observed under both RH conditions. The intensities of all functional groups from the 14 % RH experiment are much higher than those from the 74 % RH experiment, which is consistent with the reduced SOA yields under elevated RH conditions.
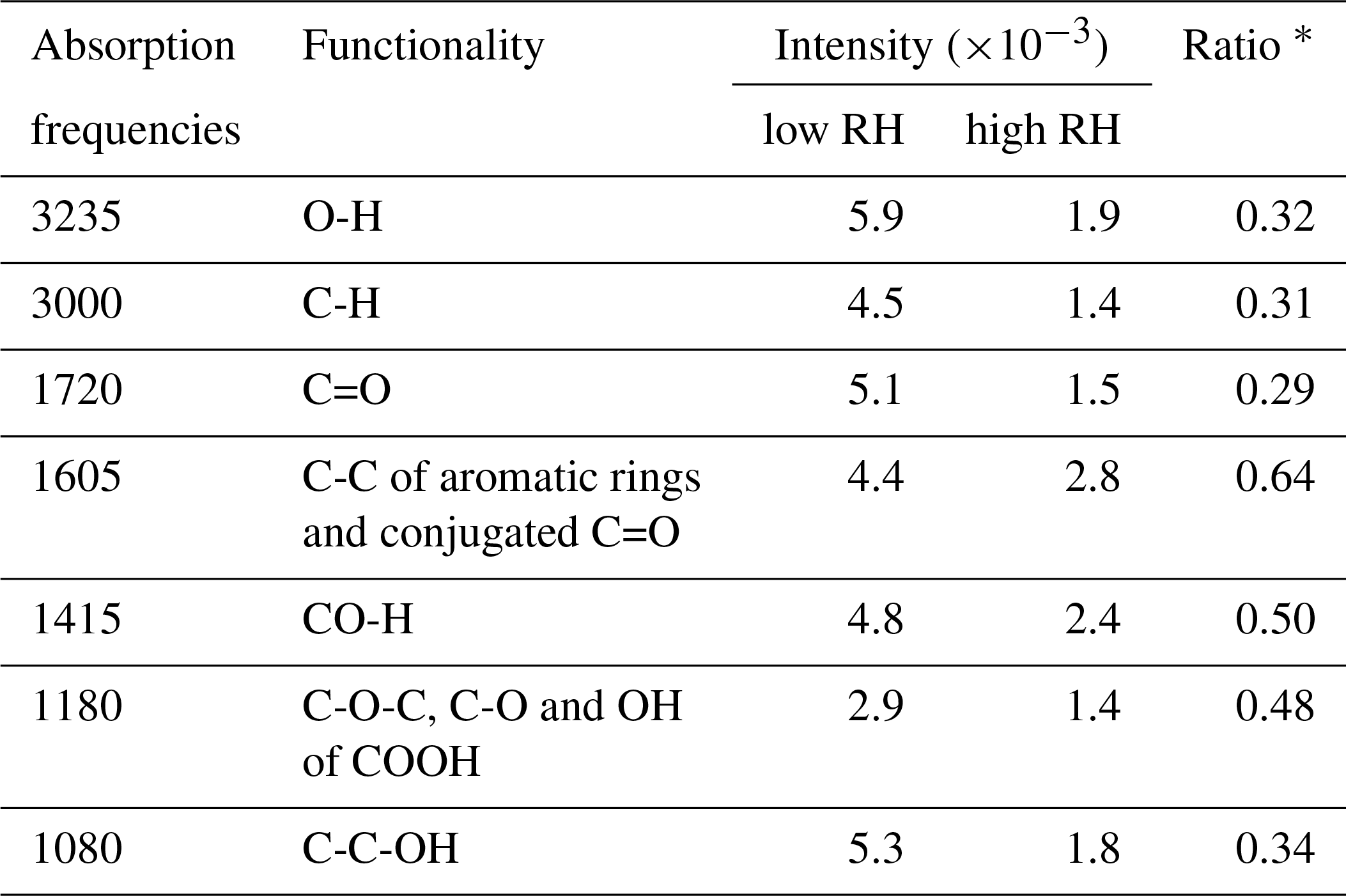
Table 2Absorbance positions of functional groups and the abundance at 14 % (Exp. 2) and 74 % (Exp. 8) RHs.
∗ Ratio of the intensity at 74 % RH to that at 14 % RH.
The assignment and the intensity of the FTIR absorption frequencies at 14 % (Exp. 2) and 74 % (Exp. 8) RHs is summarized in Table 2. The broad absorption at 2400–3600 cm−1 is O-H stretching vibration in phenol, hydroxyl and carboxyl groups (Stevenson and Goh, 1971; Santos and Duarte, 1998; Duarte et al., 2005). The band at 3000 cm−1 is C-H stretching vibration (Stevenson and Goh, 1971; Santos and Duarte, 1998; Duarte et al., 2005). The sharp absorption at 1720 cm−1 is the C=O stretching vibration in carboxylic acids, formate esters, aldehydes and ketones (Stevenson and Goh, 1971; Santos and Duarte, 1998; Duarte et al., 2005). The absorptions at 1605 cm−1 match C-C stretching of aromatic rings and the C=O stretching of conjugated carbonyl groups. The absorptions at 1415 cm−1 match the deformation of CO-H, phenolic O-H and C-O (Coury and Dillner, 2008; Ofner et al., 2011). The absorptions at 1180 cm−1 match the C-O-C stretching of polymers, C-O and OH of COOH groups (Jang and Kamens, 2001; Jang et al., 2002; Duarte et al., 2005). The absorptions at 1080 cm−1 match the C-C-OH stretching of alcohols (Jang and Kamens, 2001; Jang et al., 2002).
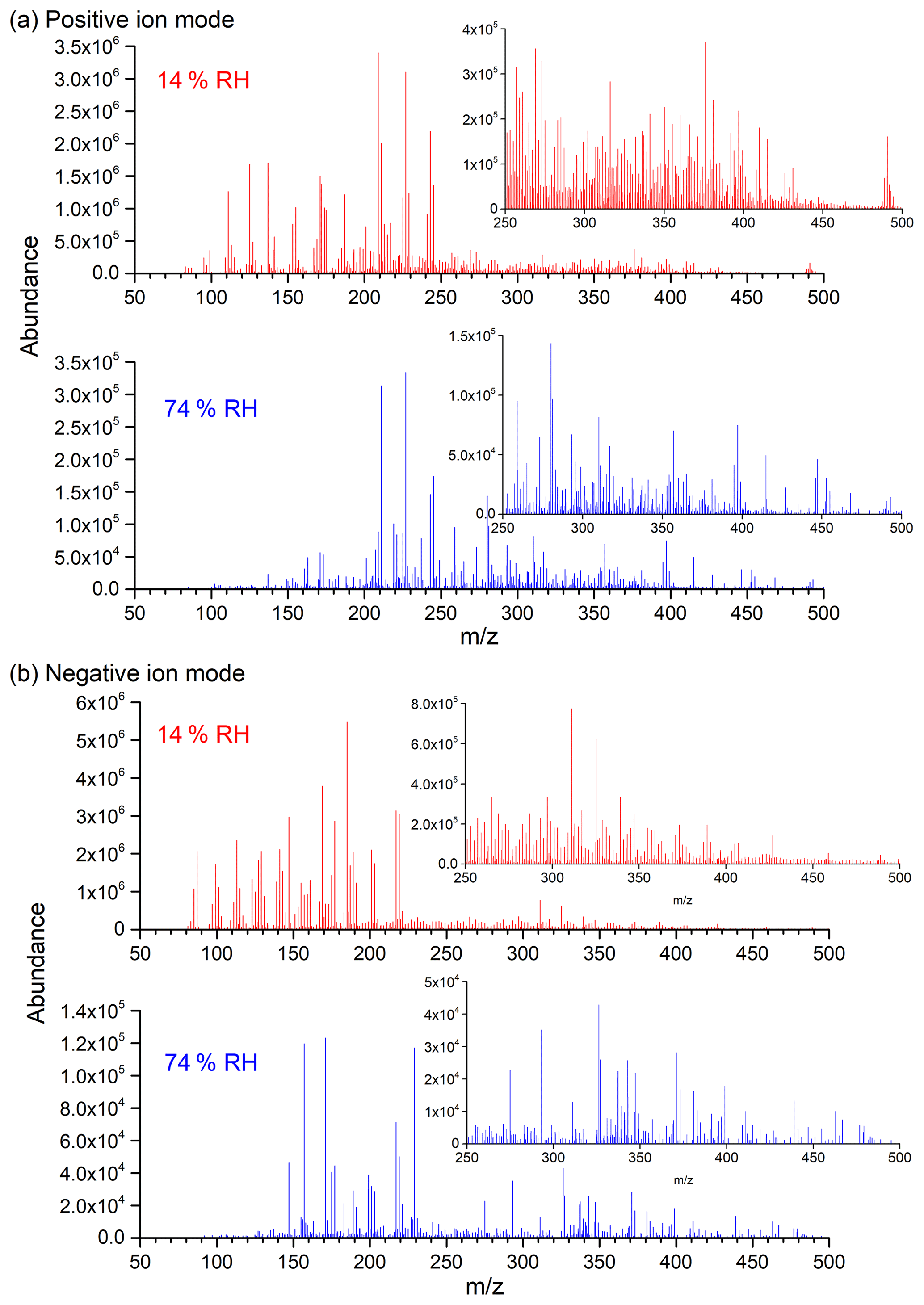
Figure 4Background-subtraction HESI-Q Exactive-Orbitrap MS results of SOA in positive (a) and negative (b) ion modes from the photooxidation of m-xylene–OH under both 14 % (Exp. 2) and 74 % (Exp. 8) RH conditions (note that the y axis scales for low and high RH are largely different: 106 at low RH and 105 at high RH).
The absorption intensity at ∼3200 cm−1 that is identified as the hydroxyl group is used as a representative of the reflection of the SOA formation. In addition, Table 2 gives the ratio of intensities at 74 % RH (Exp. 8) to those at 14 % RH (Exp. 2) to compare the differences of the relative intensities of functional groups. The intensities of functional groups are obviously suppressed at high RH, but the extents of the suppression for different functional groups are basically divided into two types. The ratios of O-H, C-H, C=O and C-C-OH groups are 0.29 to 0.34, which are close to the ratio of the SOA mass at 74 % RH to that at 14 % RH collected on the ZnSe disk, whereas the ratios of CO-H, C-O-C and C-O-H in COOH are above 0.48. The relative intensity of the C-O-C group is significantly higher than the C=O group, which can be explained by more oligomerization with the formation of C-O-C than other reactions at high RH. Nevertheless, the FTIR results cannot provide further information to well explain the differences of SOA yields between low and high RH, which will be further discussed in terms of mass spectra of SOA in the next section.
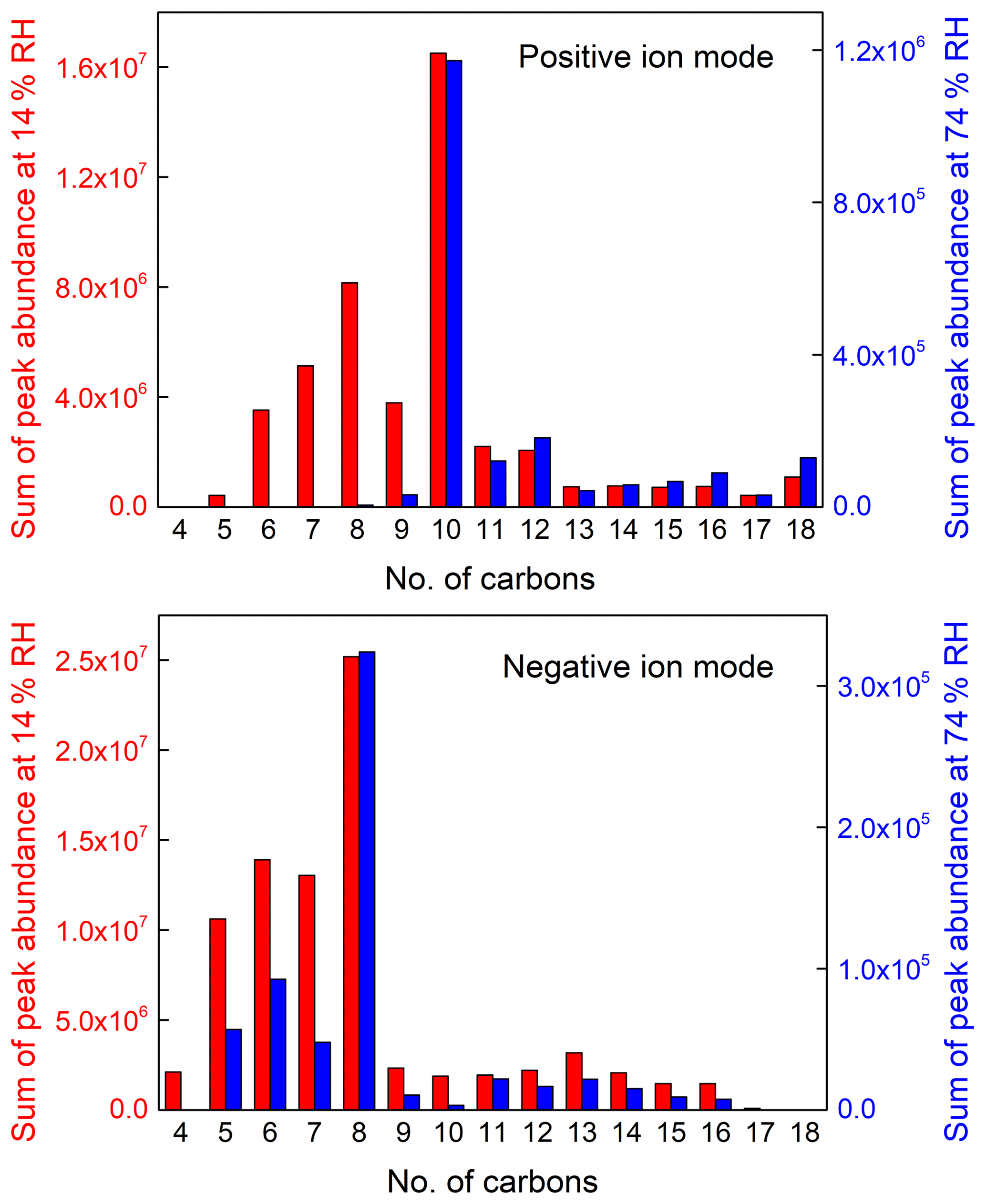
Figure 5Sum of peak abundance based on peaks selected in Fig. 4 as a function of the number of carbon atoms under the positive ion mode and negative ion mode (note that the y axis scale at low and high RH are largely different, with a label step of 4.0×106 at low RH and 4.0×105 at high RH in the positive ion mode, and 5.0×106 at low RH and 1.0×105 at high RH in the negative ion mode).
3.3 RH effects on mass spectra of SOA
The blank-corrected mass spectra of the SOA sample formed from m-xylene–OH photooxidation under 14 % and 74 % RH conditions in positive and negative ion modes are presented in Fig. 4, which is plotted as a function of the mass-to-charge ratio. It should be noted that the y axis scales for low and high RH are largely different: 106 at low RH and 105 at high RH. In Fig. 4, a visible decrease in the overall peak abundance for both positive and negative ion modes can be clearly observed as the RH elevates, which is consistent with the result that the SOA mass concentration is lower at high RH. In addition, it is obvious that the number of peaks is less under high-RH conditions. As shown in Fig. 4, where the m∕z values of SOA samples are close for both low- and high-RH conditions, the absolute and relative peak abundance is much different, indicating that RH significantly affects the concentration of SOA components.
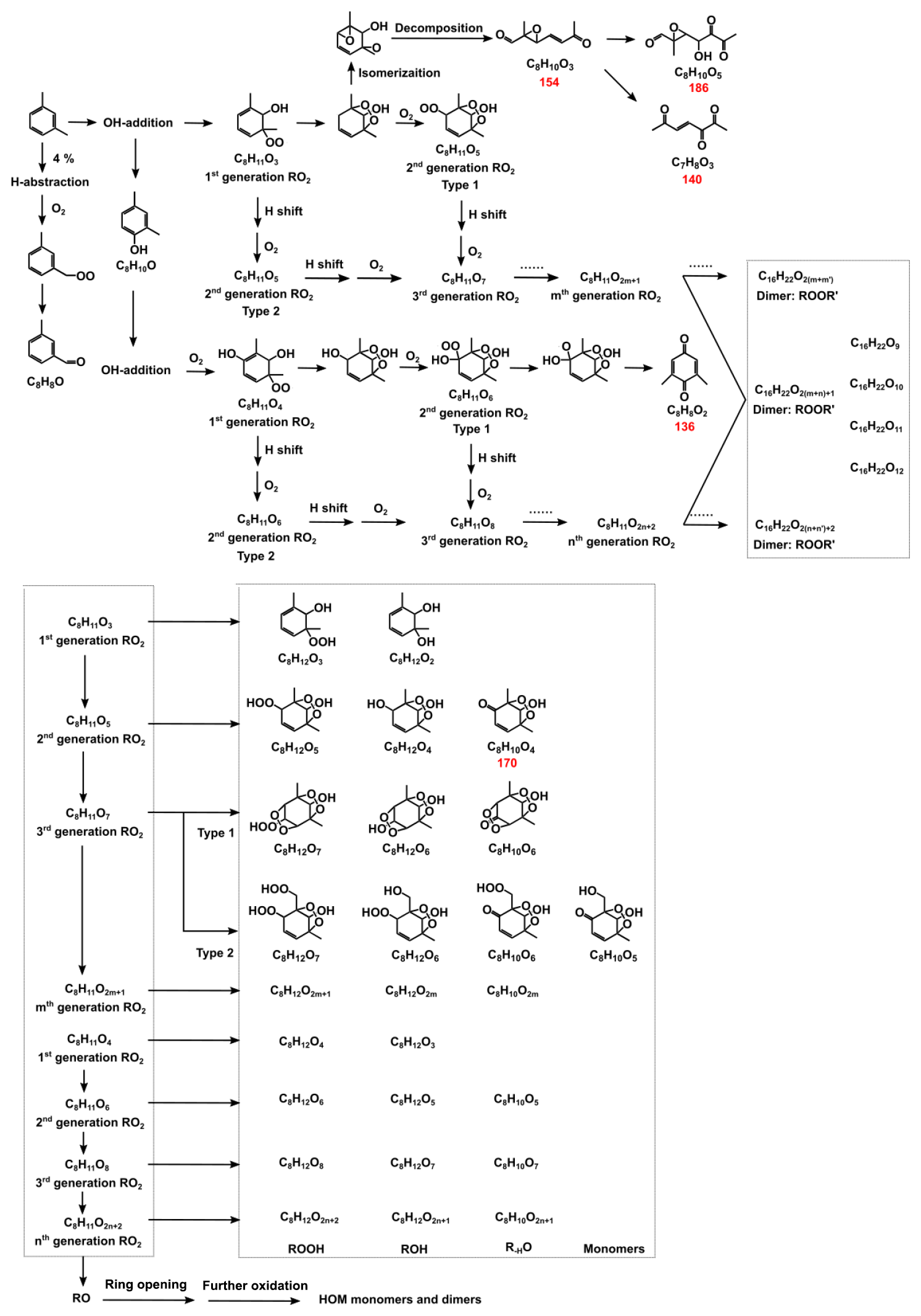
Scheme 1The route of OH-initiated m-xylene oxidation. The red number below the molecular formula is its molecular weight, which is determined by HRMS to exist in the particle phase.
For rough quantification of the RH effect, the blank-corrected mass peaks of SOA samples were selected according to whose abundance is larger than 105 under low-RH conditions and corresponding mass spectra under high-RH conditions, and then assigned with the number of carbon atoms. The peak abundance with the same number of carbon atoms (nC) is summed, which is presented in Fig. 5. It should be noted that the y axis scales at 14 % and 74 % RHs are largely different, with a label step of 4.0×106 at 14 % RH and 4.0×105 at 74 % RH in the positive ion mode, and 5.0×106 at 14 % RH and 1.0×105 at 74 % RH in the negative ion mode. The compounds with nC >8, a larger number of carbon atoms than m-xylene, are proposed to be oligomers that account for a large mass fraction of SOA due to their large molecular weights and lower volatilities, though their peak abundance is lower. As a result, the processes for formation of such compounds play an important role in the formation of SOA. It can be clearly observed that the peak abundance is much lower at high RH in the negative ion mode than that in the positive mode, indicating that the decrease of the compounds obtained in the negative ion mode account for a larger decrease at high RH.
3.4 Proposed mechanism of RH effects on SOA formation
The large difference of SOA yields and composition between low and high RHs suggests that water is directly involved in the chemical mechanism and further affects the SOA growth. In the particle-phase accretion equilibrium reactions, where water is involved as a by-product, the elevated RH alters the equilibrium of the reaction by moving toward reducing the fraction of oligomers with low volatility and increasing the fraction of monomers (Nguyen et al., 2011; Hinks et al., 2018). In this study and the previous study on toluene SOA formation, C2H2O was one of the most frequent mass differences at low and high RHs, but the peak abundance of its related compounds was much lower under elevated RH conditions (Hinks et al., 2018). C2H2O was proposed to be from the oligomerization reaction of glycolaldehyde (C2H4O2), which can react with carbonyl compounds by aldol condensation reactions with water as the by-product. This chemistry may dominantly affect the negative RH effect on the whole process of SOA formation.
Moreover, there may exist other processes that enlarge the difference of SOA formation under various RH conditions. Before we discuss the possible processes, the reaction pathway between m-xylene and OH radicals need to go through first. Reactions between m-xylene (C8H10) and OH radicals have two pathways, the H-abstraction from the methyl group and OH addition to the aromatic ring, which generates products such as methylbenzaldehyde (C8H8O) and methylbenzyl alcohol (C8H10O), as shown in Scheme 1. OH addition is the dominant pathway, as the branching ratio of H-abstraction only accounts for 4 % based on MCM. OH addition to the aromatic ring is followed by O2 adduct and isomerization to form a carbon-centered radical, which can form dimethylphenol (C8H10O) or is adducted by an O2 molecule forming a bicyclic peroxy radical (BPR, C8H11O5; Calvert et al., 2002; Birdsall et al., 2010; Wu et al., 2014). The BPR reacts with other RO2 radicals or HO2, forming the bicyclic oxy radical (C8H11O4). This RO radical can further react and finally form carbonylic products, such as (methyl) glyoxal and other SOA precursors (Jenkin et al., 2003; Hallquist et al., 2009; Carlton et al., 2010; Carter and Heo, 2013), react with HO2 radicals, forming bicyclic hydroxyhydroperoxides (ROOH, C8H12O5) or react with other RO2 radicals, forming ROH (C8H12O4) and R-HO (C8H10O4). The self- and cross-reactions of RO2 radicals also form ROOR (C16H22O10) or ROOR', which are the accretion products (Berndt et al., 2018; Molteni et al., 2018). The further O2 adduct of BPR can form highly oxygenated RO2 radicals and further react and finally form highly oxygenated organic molecules (HOMs; Types 1 and 2 in Scheme 1; Wang et al., 2017; Crounse et al., 2013; Ehn et al., 2014; Jokinen et al., 2015; Berndt et al., 2016). Dimethylphenol (C8H10O) and other products from the termination reaction with the benzene ring or double bond can react with OH radicals and further react to form HOMs as well.
Most HOMs can fall into the category of extremely low or low volatility organic compounds, and a small number of HOMs are semi-volatile organic compounds (SVOC; Bianchi et al., 2019). Extremely low volatility organic compounds can condense onto particles but SVOCs exist in significant fractions in the condensed and gas phases at equilibrium. As SMPS measured, at the end of the experiment the number concentrations (not corrected) of Exp. 1 (14 % RH) and Exp. 9 (79 % RH) were 1.9×103 and 5.8×102 particles cm−3, with a factor of 3, while the mass concentrations (not corrected) of Exp. 1 (14 % RH) and Exp. 9 (79 % RH) were 116.9 and 8.7 µg m−3, with a factor of 13. This indicates that the size of particles at low RH are higher than that at high RH. The O : C ratios in positive and negative ion modes under low- and high-RH conditions were roughly calculated using the carbon and oxygen atom numbers multiplied by the relative abundance obtained by HRMS. The O : C ratio in the positive ion mode was close to each other, 0.56 and 0.58 at low and high RHs, respectively, while the O : C ratio in the negative ion mode was different, 0.66 and 0.77 at low and high RHs, respectively. Based on the gas-to-particle partitioning rule, more-volatile compounds in the gas phase can condense to the particles of a larger size (Li et al., 2018). It can be deduced that particles of a larger size in reduced RH result in more SVOC in the gas phase condensing, leading to the difference of SOA mass at various RHs. As shown in Fig. 5, more compounds with less nC (nC < 8) are present under the low-RH experiment, also indicating that more SVOCs in the gas phase condense onto the particles. SVOCs tend to escape to the wet reactor wall as we discussed in Sect. 3.1, which interprets a certain proportion of SOA reduction at high RH. The wall process of the reactor enlarges the difference of SOA mass between low and high RH.
The higher O : C ratio in the negative ion mode demonstrates that the compounds in the negative ion mode are much more oxygenated than those in the positive ion mode. As shown in Fig. 5, the peak abundance at high RH is much lower in the negative ion mode than in the positive mode, indicating that the decrease of the more oxygenated compounds accounts for the larger fraction at high RH. These high O : C ratios cannot be explained by any of the formerly known oxidization pathways, except that the formation of HOMs from RO2 autoxidation is taken into consideration (Crounse et al., 2013; Barsanti et al., 2017). To our knowledge, RH does not directly impact the formation of HOMs (Li et al., 2019). It is possible that HOMs undergo further particle-phase reactions, as suggested in a previous study (Bianchi et al., 2019), which may be influenced by RH, but this process need to be further investigated in future studies.
The current study investigates the effect of RH on SOA formation from the oxidation of m-xylene under low-NOx conditions in the absence of seed particles. The elevated RH can significantly obstruct the SOA formation from the m-xylene–OH system, so that the SOA yield decrease from 14.0 %–16.5 % at low RH to 0.8 %–0.8 % at high RH, with a significant discrepancy of nearly 1 order of magnitude. Some of the reduction in the apparent yield may be due to the faster wall loss of semi-volatile products of oxidation at higher RH. The FTIR analysis shows that the proportion of oligomers with C-O-C groups from carbonyl compounds in SOA at high RH is higher than that at low RH, but the negative RH effect on SOA formation cannot be well explained as the FTIR results cannot provide further information. From the analysis of the HRMS spectra, it is found that C2H2O is one of the most common mass differences at low and high RHs, that the compounds with a lower carbon number in the formula at low RH account for a larger proportion than those at high RH, and that the compounds at high RH have higher O : C ratios than those at low RH. The HRMS results suggest that the RH may suppress oligomerization where water is involved as a by-product and may influence the further particle-phase reaction of highly oxygenated organic molecules (HOMs) formed in the gas phase. In addition to the chemical processes, the negative RH effect on SOA formation is enlarged based on the gas-to-particle partitioning rule. Together with the previous study on toluene SOA, it is conceivable that the effect of RH on SOA yield is a common feature of SOA formation from monocyclic aromatics oxidation under low-NOx conditions and using H2O2 as the OH radical source. Our results indicate that the production of SOA from aromatics in low-NOx environments can be modulated by the ambient RH. Our study highlights the role of water in SOA formation, which is particularly related to chemical mechanisms used to explain observed air quality and to predict chemistry in air quality models and climate models. The clear pathway of the influence of H2O on the particle-phase reaction of HOMs formed in the gas phase needs to be further studied in the future.
Data are available by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-15007-2019-supplement.
QZ and YX designed the research. QZ carried out the experiments and analyzed the data. LJ provided valuable advice on the experiment operations. YX and LJ provided advice on the analysis of results. QZ prepared the manuscript with contributions from all co-authors.
The authors declare that they have no conflict of interest.
The authors are very grateful to the four anonymous reviewers for useful discussions.
This research has been supported by the National Key R&D Program of China (2017YFC0210005) and National Natural Science Foundation of China (no. 41375129).
This paper was edited by Sergey A. Nizkorodov and reviewed by four anonymous referees.
Barsanti, K. C., Kroll, J. H., and Thornton, J. A.: Formation of low-volatility organic compounds in the atmosphere: Recent advancements and insights, J. Phys. Chem. Lett., 8, 1503–1511, https://doi.org/10.1021/acs.jpclett.6b02969, 2017.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurten, T., Otkjaer, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipila, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion product formation from self- and cross-reactions of RO2 radicals in the atmosphere, Angew. Chem. Int. Ed., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: a key contributor to atmospheric aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Birdsall, A. W., Andreoni, J. F., and Elrod, M. J.: Investigation of the role of bicyclic peroxy radicals in the oxidation mechanism of toluene, J. Phys. Chem. A, 114, 10655–10663, https://doi.org/10.1021/jp105467e, 2010.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Calvert, J. G., Atkinson, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. H., and Yarwood, G.: The mechanisms of atmospheric oxidation of the aromatic hydrocarbons, Oxford University Press, 1–566, 2002.
Cao, G. and Jang, M.: An SOA model for toluene oxidation in the presence of inorganic aerosols, Environ. Sci. Technol., 44, 727–733, https://doi.org/10.1021/es901682r, 2010.
Carlton, A. G., Bhave, P. V., Napelenok, S. L., Edney, E. O., Golam, S., Pinder, R. W., Pouliot, G. A., and Marc, H.: Model representation of secondary organic aerosol in CMAQv4.7, Environ. Sci. Technol., 44, 8553–8560, https://doi.org/10.1021/es100636q, 2010.
Carter, W. P. L. and Heo, G.: Development of revised SAPRC aromatics mechanisms, Atmos. Environ., 77, 404–414, https://doi.org/10.1016/j.atmosenv.2013.05.021, 2013.
Coury, C. and Dillner, A. M.: A method to quantify organic functional groups and inorganic compounds in ambient aerosols using attenuated total reflectance FTIR spectroscopy and multivariate chemometric techniques, Atmos. Environ., 42, 5923–5932, https://doi.org/10.1016/j.atmosenv.2008.03.026, 2008.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Czoschke, N. M., Jang, M., and Kamens, R. M.: Effect of acidic seed on biogenic secondary organic aerosol growth, Atmos. Environ., 37, 4287–4299, 10.1016/s1352-2310(03)00511-9, 2003.
Ding, X., Wang, X.-M., Gao, B., Fu, X.-X., He, Q.-F., Zhao, X.-Y., Yu, J.-Z., and Zheng, M.: Tracer-based estimation of secondary organic carbon in the Pearl River Delta, south China, J. Geophys. Res., 117, D05313, https://doi.org/10.1029/2011jd016596, 2012.
Duarte, R. M. B. O., Pio, C. A., and Duarte, A. C.: Spectroscopic study of the water-soluble organic matter isolated from atmospheric aerosols collected under different atmospheric conditions, Anal. Chim. Ac., 530, 7–14, https://doi.org/10.1016/j.aca.2004.08.049, 2005.
Durand, T., Bau, S., Morele, Y., Matera, V., Bémer, D., and Rousset, D.: Quantification of low pressure impactor wall deposits during zinc nanoparticle sampling, Aerosol Air Qual. Res., 14, 1812–1821, https://doi.org/10.4209/aaqr.2013.10.0304, 2014.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., and Lee, B.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476-479, 10.1038/nature13032, 2014.
El-Sayed, M. M. H., Wang, Y., and Hennigan, C. J.: Direct atmospheric evidence for the irreversible formation of aqueous secondary organic aerosol, Geophys. Res. Lett., 42, 5577–5586, https://doi.org/10.1002/2015gl064556, 2015.
El-Sayed, M. M. H., Amenumey, D., and Hennigan, C. J.: Drying-Induced evaporation of secondary organic aerosol during summer, Environ. Sci. Technol., 50, 3626–3633, https://doi.org/10.1021/acs.est.5b06002, 2016.
Engelhart, G. J., Hildebrandt, L., Kostenidou, E., Mihalopoulos, N., Donahue, N. M., and Pandis, S. N.: Water content of aged aerosol, Atmos. Chem. Phys., 11, 911–920, https://doi.org/10.5194/acp-11-911-2011, 2011.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of aerosol liquid water in secondary organic aerosol formation from volatile organic compounds, Environ. Sci. Technol., 51, 1405–1413, 10.1021/acs.est.6b04700, 2017.
Finlayson-Pitts, B. J. and Pitts Jr., J. N.: Chapter 6-Rates and mechanisms of gas-phase reactions in irradiated organic-NOx-air mixtures, in: Chemistry of the Upper and Lower Atmosphere, Academic Press, San Diego, 179–263, 2000.
Forstner, H. J. L., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol from the photooxidation of aromatic hydrocarbons: Molecular composition, Environ. Sci. Technol., 31, 1345–1358, https://doi.org/10.1021/es9605376, 1997.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from ethyne in the presence of NaCl in a smog chamber, Environ. Chem., 13, 699–710, https://doi.org/10.1071/en15155, 2016.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from ethylene ozonolysis in the presence of sodium chloride, J. Aerosol Sci., 106, 120–131, https://doi.org/10.1016/j.jaerosci.2017.01.009, 2017a.
Ge, S., Xu, Y., and Jia, L.: Secondary organic aerosol formation from propylene irradiations in a chamber study, Atmos. Environ., 157, 146–155, https://doi.org/10.1016/j.atmosenv.2017.03.019, 2017b.
Ge, S., Xu, Y., and Jia, L.: Effects of inorganic seeds on secondary organic aerosol formation from photochemical oxidation of acetone in a chamber, Atmos. Environ., 170, 205–215, https://doi.org/10.1016/j.atmosenv.2017.09.036, 2017c.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hansen, J. E. and Sato, M.: Trends of measured climate forcing agents, P. Natl. Acad. Sci. USA, 98, 14778–14783, https://doi.org/10.1073/pnas.261553698, 2001.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of relative humidity on gas/particle partitioning and aerosol mass yield in the photooxidation of p-xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Hinks, M. L., Montoya-Aguilera, J., Ellison, L., Lin, P., Laskin, A., Laskin, J., Shiraiwa, M., Dabdub, D., and Nizkorodov, S. A.: Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 18, 1643–1652, https://doi.org/10.5194/acp-18-1643-2018, 2018.
Huang, R. J., Zhang, Y., Bozzetti, C., Ho, K. F., Cao, J. J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., El Haddad, I., and Prevot, A. S.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Ip, H. S. S., Huang, X. H. H., and Yu, J. Z.: Effective Henry's law constants of glyoxal, glyoxylic acid, and glycolic acid, Geophys. Res. Lett., 36, L01802, https://doi.org/10.1029/2008gl036212, 2009.
Jacobson, M. C., Hansson, H. C., Noone, K. J., and Charlson, R. J.: Organic atmospheric aerosols: Review and state of the science, Rev. Geophys., 38, 267–294, https://doi.org/10.1029/1998rg000045, 2000.
Jang, M. and Kamens, R. M.: Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene, Environ. Sci. Technol., 35, 3626–3639, https://doi.org/10.1021/es010676+, 2001.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions, Science, 298, 814–817, https://doi.org/10.1126/science.1075798, 2002.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jia, L. and Xu, Y.: Effects of relative humidity on ozone and secondary organic aerosol formation from the photooxidation of benzene and ethylbenzene, Aerosol Sci. Technol., 48, 1–12, https://doi.org/10.1080/02786826.2013.847269, 2014.
Jia, L. and Xu, Y.: Ozone and secondary organic aerosol formation from Ethylene-NOx-NaCl irradiations under different relative humidity conditions, J. Atmos. Chem., 73, 81–100, https://doi.org/10.1007/s10874-015-9317-1, 2016.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V. M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., and Herrmann, H.: Rapid autoxidation forms highly oxidized RO2 radicals in the atmosphere, Angew. Chem. Int. Ed., 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2015.
Kamens, R. M., Zhang, H., Chen, E. H., Zhou, Y., Parikh, H. M., Wilson, R. L., Galloway, K. E., and Rosen, E. P.: Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: Water and particle seed effects, Atmos. Environ., 45, 2324–2334, https://doi.org/10.1016/j.atmosenv.2010.11.007, 2011.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Khoder, M. I.: Ambient levels of volatile organic compounds in the atmosphere of Greater Cairo, Atmos. Environ., 41, 554–566, https://doi.org/10.1016/j.atmosenv.2006.08.051, 2007.
Kidd, C., Perraud, V., Wingen, L. M., and Finlayson-Pitts, B. J.: Integrating phase and composition of secondary organic aerosol from the ozonolysis of α-pinene, P. Natl. Acad. Sci. USA, 111, 7552–7557, https://doi.org/10.1073/pnas.1322558111, 2014.
Li, K., Li, J., Wang, W., Li, J., Peng, C., Wang, D., and Ge, M.: Effects of gas-particle partitioning on refractive index and chemical composition of m-xylene secondary organic aerosol, J. Phys. Chem. A, 122, 3250–3260, https://doi.org/10.1021/acs.jpca.7b12792, 2018.
Li, X., Chee, S., Hao, J., Abbatt, J. P. D., Jiang, J., and Smith, J. N.: Relative humidity effect on the formation of highly oxidized molecules and new particles during monoterpene oxidation, Atmos. Chem. Phys., 19, 1555–1570, https://doi.org/10.5194/acp-19-1555-2019, 2019.
Liu, T., Huang, D. D., Li, Z., Liu, Q., Chan, M., and Chan, C. K.: Comparison of secondary organic aerosol formation from toluene on initially wet and dry ammonium sulfate particles at moderate relative humidity, Atmos. Chem. Phys., 18, 5677–5689, https://doi.org/10.5194/acp-18-5677-2018, 2018.
Liu, Y., Wu, Z., and Hu, M.: Advances in the phase state of secondary organic aerosol, China Environ. Sci., 37, 1637–1645, 2017 (in Chinese).
Loza, C. L., Chhabra, P. S., Yee, L. D., Craven, J. S., Flagan, R. C., and Seinfeld, J. H.: Chemical aging of m-xylene secondary organic aerosol: laboratory chamber study, Atmos. Chem. Phys., 12, 151–167, https://doi.org/10.5194/acp-12-151-2012, 2012.
Matsunaga, A. and Ziemann, P. J.: Gas-wall partitioning of organic compounds in a Teflon film chamber and potential effects on reaction product and aerosol yield measurements, Aerosol Sci. Technol., 44, 881–892, https://doi.org/10.1080/02786826.2010.501044, 2010.
McMurry, P. H. and Rader, D. J.: Aerosol wall losses in electrically charged chambers, Aerosol Sci. Technol., 4, 249–268, https://doi.org/10.1080/02786828508959054, 2007.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Nah, T., McVay, R. C., Zhang, X., Boyd, C. M., Seinfeld, J. H., and Ng, N. L.: Influence of seed aerosol surface area and oxidation rate on vapor wall deposition and SOA mass yields: a case study with α-pinene ozonolysis, Atmos. Chem. Phys., 16, 9361–9379, https://doi.org/10.5194/acp-16-9361-2016, 2016.
Nah, T., McVay, R. C., Pierce, J. R., Seinfeld, J. H., and Ng, N. L.: Constraining uncertainties in particle-wall deposition correction during SOA formation in chamber experiments, Atmos. Chem. Phys., 17, 2297–2310, https://doi.org/10.5194/acp-17-2297-2017, 2017.
Ng, N. L., Kroll, J. H., Chan, A. W. H., Chhabra, P. S., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from m-xylene, toluene, and benzene, Atmos. Chem. Phys., 7, 3909–3922, https://doi.org/10.5194/acp-7-3909-2007, 2007.
Nguyen, T. B., Roach, P. J., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Effect of humidity on the composition of isoprene photooxidation secondary organic aerosol, Atmos. Chem. Phys., 11, 6931–6944, https://doi.org/10.5194/acp-11-6931-2011, 2011.
Odum, J. R., Jungkamp, T. P., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: The atmospheric aerosol-forming potential of whole gasoline vapor, Science, 276, 96–99, https://doi.org/10.1126/science.276.5309.96, 1997.
Offenberg, J. H., Lewis, C. W., Lewandowski, M., Jaoui, M., Kleindienst, T. E., and Edney, E. O.: Contributions of toluene and α-pinene to SOA formed in an irradiated toluene/α-pinene/NOx/air mixture: comparison of results using 14C content and SOA organic tracer methods, Environ. Sci. Technol., 41, 3972–3976, https://doi.org/10.1021/es070089+, 2007.
Ofner, J., Krüger, H.-U., Grothe, H., Schmitt-Kopplin, P., Whitmore, K., and Zetzsch, C.: Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS, Atmos. Chem. Phys., 11, 1–15, https://doi.org/10.5194/acp-11-1-2011, 2011.
Park, S. H., Kim, H. O., Han, Y. T., Kwon, S. B., and Lee, K. W.: Wall loss rate of polydispersed aerosols, Aerosol Sci. Technol., 35, 710–717, https://doi.org/10.1080/02786820152546752, 2001.
Qi, L., Nakao, S., Tang, P., and Cocker III, D. R.: Temperature effect on physical and chemical properties of secondary organic aerosol from m-xylene photooxidation, Atmos. Chem. Phys., 10, 3847–3854, https://doi.org/10.5194/acp-10-3847-2010, 2010.
Santos, E. B. H. and Duarte, A. C.: The influence of pulp and paper mill effluents on the composition of the humic fraction of aquatic organic matter, Water Res., 32, 597–608, https://doi.org/10.1016/S0043-1354(97)00301-1, 1998.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: From air pollution to climate change, 3 Edn., Wiley, Hoboken, 2016.
Song, C., Na, K., Warren, B., Malloy, Q., and Cocker, D. R., III: Secondary organic aerosol formation from m-xylene in the absence of NOx, Environ. Sci. Technol., 41, 7409–7416, https://doi.org/10.1021/es070429r, 2007.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Stevenson, F. J., and Goh, K. M.: Infrared spectra of humic acids and related substances, Geochim. Cosmochim. Ac., 35, 471–483, https://doi.org/10.1016/0016-7037(71)90044-5, 1971.
Tuet, W. Y., Chen, Y., Xu, L., Fok, S., Gao, D., Weber, R. J., and Ng, N. L.: Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 17, 839–853, https://doi.org/10.5194/acp-17-839-2017, 2017.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, Y., Luo, H., Jia, L., and Ge, S.: Effect of particle water on ozone and secondary organic aerosol formation from benzene-NO2-NaCl irradiations, Atmos. Environ., 140, 386–394, https://doi.org/10.1016/j.atmosenv.2016.06.022, 2016.
Warren, B., Song, C., and Cocker III, D. R.: Light intensity and light source influence on secondary organic aerosol formation for the m-xylene/NOx photooxidation system, Environ. Sci. Technol., 42, 5461–5466, https://doi.org/10.1021/es702985n, 2008.
Wu, R., Pan, S., Li, Y., and Wang, L.: Atmospheric oxidation mechanism of toluene, J. Phys. Chem. A, 118, 4533–4547, https://doi.org/10.1021/jp500077f, 2014.
Zhang, X., Cappa, C. D., Jathar, S. H., McVay, R. C., Ensberg, J. J., Kleeman, M. J., and Seinfeld, J. H.: Influence of vapor wall loss in laboratory chambers on yields of secondary organic aerosol, P. Natl. Acad. Sci. USA, 111, 5802–5807, https://doi.org/10.1073/pnas.1404727111, 2014.
Zhang, X., Schwantes, R. H., McVay, R. C., Lignell, H., Coggon, M. M., Flagan, R. C., and Seinfeld, J. H.: Vapor wall deposition in Teflon chambers, Atmos. Chem. Phys., 15, 4197–4214, https://doi.org/10.5194/acp-15-4197-2015, 2015.
Zhang, X., Dalleska, N. F., Huang, D. D., Bates, K. H., Sorooshian, A., Flagan, R. C., and Seinfeld, J. H.: Time-resolved molecular characterization of organic aerosols by PILS + UPLC/ESI-Q-TOFMS, Atmos. Environ., 130, 180–189, https://doi.org/10.1016/j.atmosenv.2015.08.049, 2016.
Zhao, Y., Saleh, R., Saliba, G., Presto, A. A., Gordon, T. D., Drozd, G. T., Goldstein, A. H., Donahue, N. M., and Robinson, A. L.: Reducing secondary organic aerosol formation from gasoline vehicle exhaust, P. Natl. Acad. Sci. USA, 114, 6984–6989, https://doi.org/10.1073/pnas.1620911114, 2017.
Zhou, Y., Zhang, H., Parikh, H. M., Chen, E. H., Rattanavaraha, W., Rosen, E. P., Wang, W., and Kamens, R. M.: Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: Water and particle seed effects (II), Atmos. Environ., 45, 3882–3890, https://doi.org/10.1016/j.atmosenv.2010.12.048, 2011.