the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Nov 2019
| 18 Nov 2019
Atmosphere–ocean exchange of heavy metals and polycyclic aromatic hydrocarbons in the Russian Arctic Ocean
Xiaowen Ji
Evgeny Abakumov
Xianchuan Xie
Heavy metals and polycyclic aromatic hydrocarbons (PAHs) can greatly influence biotic activities and organic sources in the ocean. However, fluxes of these compounds as well as their fate, transport, and net input to the Arctic Ocean have not been thoroughly assessed. During April–November of the 2016 “Russian High-Latitude Expedition”, 51 air (gases, aerosols, and wet deposition) and water samples were collected from the Russian Arctic within the Barents Sea, the Kara Sea, the Laptev Sea, and the East Siberian Sea. Here, we report on the Russian Arctic assessment of the occurrence of 35 PAHs and 9 metals (Pb, Cd, Cu, Co, Zn, Fe, Mn, Ni, and Hg) in dry and wet deposition as well as the atmosphere–ocean fluxes of 35 PAHs and Hg0. We observed that Hg was mainly in the gas phase and that Pb was most abundant in the gas phase compared with the aerosol and dissolved water phases. Mn, Fe, Pb, and Zn showed higher levels than the other metals in the three phases. The concentrations of PAHs in aerosols and the dissolved water phase were approximately 1 order of magnitude higher than those in the gas phase. The abundances of higher molecular weight PAHs were highest in the aerosols. Higher levels of both heavy metals and PAHs were observed in the Barents Sea, the Kara Sea, and the East Siberian Sea, which were close to areas with urban and industrial sites. Diagnostic ratios of phenanthrene/anthracene to fluoranthene/pyrene showed a pyrogenic source for the aerosols and gases, whereas the patterns for the dissolved water phase were indicative of both petrogenic and pyrogenic sources; pyrogenic sources were most prevalent in the Kara Sea and the Laptev Sea. These differences between air and seawater reflect the different sources of PAHs through atmospheric transport, which included anthropogenic sources for gases and aerosols and mixtures of anthropogenic and biogenic sources along the continent in the Russian Arctic. The average dry deposition of ∑9 metals and ∑35 PAHs was 1749 and 1108 ng m−2 d−1, respectively. The average wet deposition of ∑9 metals and ∑35 PAHs was 33.29 and 221.31 µg m−2 d−1, respectively. For the atmosphere–sea exchange, the monthly atmospheric input of ∑35 PAHs was estimated at 1040 t. The monthly atmospheric Hg input was approximately 530 t. These additional inputs of hazardous compounds may be disturbing the biochemical cycles in the Arctic Ocean.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(3261 KB)
- Corrigendum
-
Supplement
(1462 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(3261 KB) - Full-text XML
- Corrigendum
-
Supplement
(1462 KB) - BibTeX
- EndNote





The increasing anthropogenic activities associated with growing industries within boundary areas of the Arctic for economic reasons, including hydrocarbon exploration sites and mines in the Russian Arctic, represent potential pollution sources to Arctic ecosystems (Walker et al., 2003; Dahle et al., 2009; Ji et al., 2019). Additionally, the Arctic has long been contaminated by pollutants transported to polar areas from distant locations outside of this region (Hung et al., 2016). For example, anthropogenic sources of pollutants in the Arctic have been found to come from the Norilsk industrial area on the Taymyr Peninsula (Reimann et al., 1997; Zhulidov et al., 2011) and from the copper–nickel mining industry on the Kola Peninsula (Boyd et al., 2009; Jaffe et al., 1995). For pollutants transported from outside of the Arctic, reducing global emissions would be an ideal strategy to lessen the impacts of pollutants on Arctic ecosystems. For example, worldwide emissions of mercury will have increased by 25 % in 2020 over 2005 levels according to previous estimations (Pacyna et al., 2010). Mercury is a key problematic pollutant in the Arctic because it is a neurotoxic pollutant significantly influencing northern latitudes via human exposure from eating seafood and marine mammals (Stow et al., 2015). Thus, global emission reductions could help to alleviate problems associated with long-range mercury transport and contamination in the Arctic. In regard to sources close to the Arctic, these may inevitably cause localized ecological risks or risks over a wider regional range. For instance, Fernandes and Sicre (1999) showed that atmospheric transport of anthropogenic polycyclic aromatic hydrocarbons (PAHs) to the Eurasian Arctic mainly originated from eastern Europe and Russia. PAHs in aerosols from lower latitudes were deposited on soils and ice in winter and transported by rivers to the ocean by the occurrence of freshet (Fernandes and Sicre, 1999). The previous study also showed a strong net deposition in the marine transect from East Asia to the Arctic, and the controlling sources both contained East Asia as a potential continental source region and forest fires in the Arctic as a seasonal and regional source (Ma et al., 2013). In addition, high concentrations of heavy metals (Mn, Zn, Ni, Fe, and Cd) were observed in the west Arctic Ocean (Chukchi Sea); this enrichment was not only from Pacific-origin inflow water from the Bering Strait but also from additional sources such as melting sea ice and river water discharge (Kondo et al., 2016). Also of concern is the fact that the melting of contaminated ice may lead to more pollutant emission into the Arctic Ocean with rapid warming of the global climate, which could harm its fragile ecosystems.
Pollutants can be transported to the Arctic through both seawater and atmospheric pathways; the atmospheric pathway is the quickest and most direct way for long-range pollutant transportation, e.g., pollutants can be transported from distant sources to the Arctic within several days or weeks (Shevchenko et al., 2003). Reports have revealed that some pollutants such as heavy metals and polycyclic aromatic hydrocarbons (PAHs) can be transported with aerosols over thousands of kilometers to Arctic regions (Rahn and Lowenthal, 1984; Maenhaut et al., 1989; Shaw, 1991; Cheng et al., 1993). Approximately 100 t of airborne mercury originating from industrial sources is deposited in the Arctic Ocean annually (Valenti, 2006). While there is evidence that atmospheric inputs make large contributions to the chemical budgets in marine areas, the exact role of these inputs in the Arctic Ocean remains uncertain and may have been previously underestimated (Duce et al., 1991). Numerous studies have shown that aerosol transport is essential to transfer atmospheric compounds from air to ocean, and that this process is susceptible to changes in the climate of Arctic regions (Leck et al., 1996; Sirois and Barrie, 1999; Bigg and Leck, 2001). The compounds in aerosols over the Russian Arctic have been reported to show maximal concentrations during the winter/spring season; in addition, 50 % of the air pollutants were found to have originated from Russian Arctic pollution (Shevchenko et al., 2003). It has also been reported that the natural biodegradation rates of exogenous compounds in the Arctic Ocean could be lower than those in more temperate oceans such as the Atlantic and Pacific (Bagi et al., 2014). In addition, Vieira et al. (2019) found that Fe, Mn, and Co were predominantly controlled by reductive benthic inputs, and that their levels were affected by the biological processes of uptake and release in the Arctic Ocean. Due to their toxicity and persistence, high concentrations of heavy metals or other persistent pollutants such as PAHs may disturb the benthic fluxes in cross-shelf mixing in Arctic regions, which could result in adverse effects on marine life and, with the eventual biomagnification in the food web, on humans as well. However, the long-term influence of heavy metals and PAHs on biogeochemical cycles in the Arctic Ocean remains poorly understood.
Atmosphere–seawater exchange is the main process that controls the residence time and levels of chemical compounds in the Arctic Ocean. In particular, atmospheric deposition is a significant source for pollutants in seawater, and dry deposition in the ocean has been widely studied (Jickells and Baker, 2019; Wang et al., 2019; Park et al., 2019). Although wet deposition (precipitation scavenging) is regarded as playing a predominant role in eliminating pollutants in both gas and particulate phases, current reports on the spatial distribution of pollutants from wet (snow) deposition in high-latitude oceans are scarce (Custódio et al., 2014). Moreover, for volatile or semivolatile compounds, the volatilization process is an important pathway for atmosphere–seawater exchanges. Therefore, the atmosphere–water exchange of volatile or semivolatile compounds can be estimated by the net flux of pollutants either volatilizing from seawater to air or depositing from air to seawater (Rasiq et al., 2019; Cheng et al., 2013; Totten et al., 2001). Gonzalez-Gaya et al. (2016) reported on a global assessment of atmosphere–ocean fluxes of 64 PAHs; the net atmospheric PAH input to global ocean was 0.09 Tg per month. The atmosphere–seawater exchange rate is greatly influenced by atmospheric temperature variations, and the direction and magnitude of fluxes of compounds between air and seawater vary seasonally (Bamford et al., 1999; Hornbuckle et al., 1994). Additionally, inorganic salt ions can decrease the aqueous solubility of organic compounds such as PAHs (Rasiq et al., 2019). During the melting of sea ice in the Arctic Ocean, the magnitude and direction of atmosphere–seawater fluxes may be different from those in tropical and subtropical oceans (Gonzalez-Gaya et al., 2016; Rasiq et al., 2019). The Arctic Ocean is considered as a sink that receives global airborne pollutants (Environment Canada, Fisheries and Oceans Canada and Indian and Northern Affairs Canada Arctic, 2008); however, the fate of atmosphere–ocean exchange of trace metals and organic compounds remains unclear.
In this study, two categories of pollutants (i.e., 9 heavy metals and 35 PAHs) were measured in the Arctic Ocean, in aerosols, gas, and seawater, and atmosphere–ocean exchanges of Hg and PAHs were studied. We hypothesized about the relative equilibrium of chemical exchanges between seawater and air and calculated the net diffusion of atmosphere–ocean exchange of Hg and PAHs in the Arctic Ocean for an evaluation of the double-directional exchange. Meanwhile, the dry and wet deposition of heavy metals and PAHs in the Russian Arctic Ocean were determined. The distributions of heavy metals and PAHs in each sea of the Arctic Ocean and in various phases were also characterized to identify possible sources from the continents.
2.1 Study area and sample collection
All samples were collected during the period from 9 April to 10 November 2016 as part of the “Russian High-Latitude Expedition” carried out on the Mikhail Somov vessel (this vessel traveled from the city of Arkhangelsk to Wrangel Island). A total of 51 air and water samples, and 8 wet deposition samples were gathered from locations ranging from the southern inlet of the Barents Sea (from west sites to Vaygach Island) to across the Kara Sea (to Gerkules Island), Laptev Sea (to Bennett Island), and East Siberian Sea (to Wrangel Island) (Fig. 1).
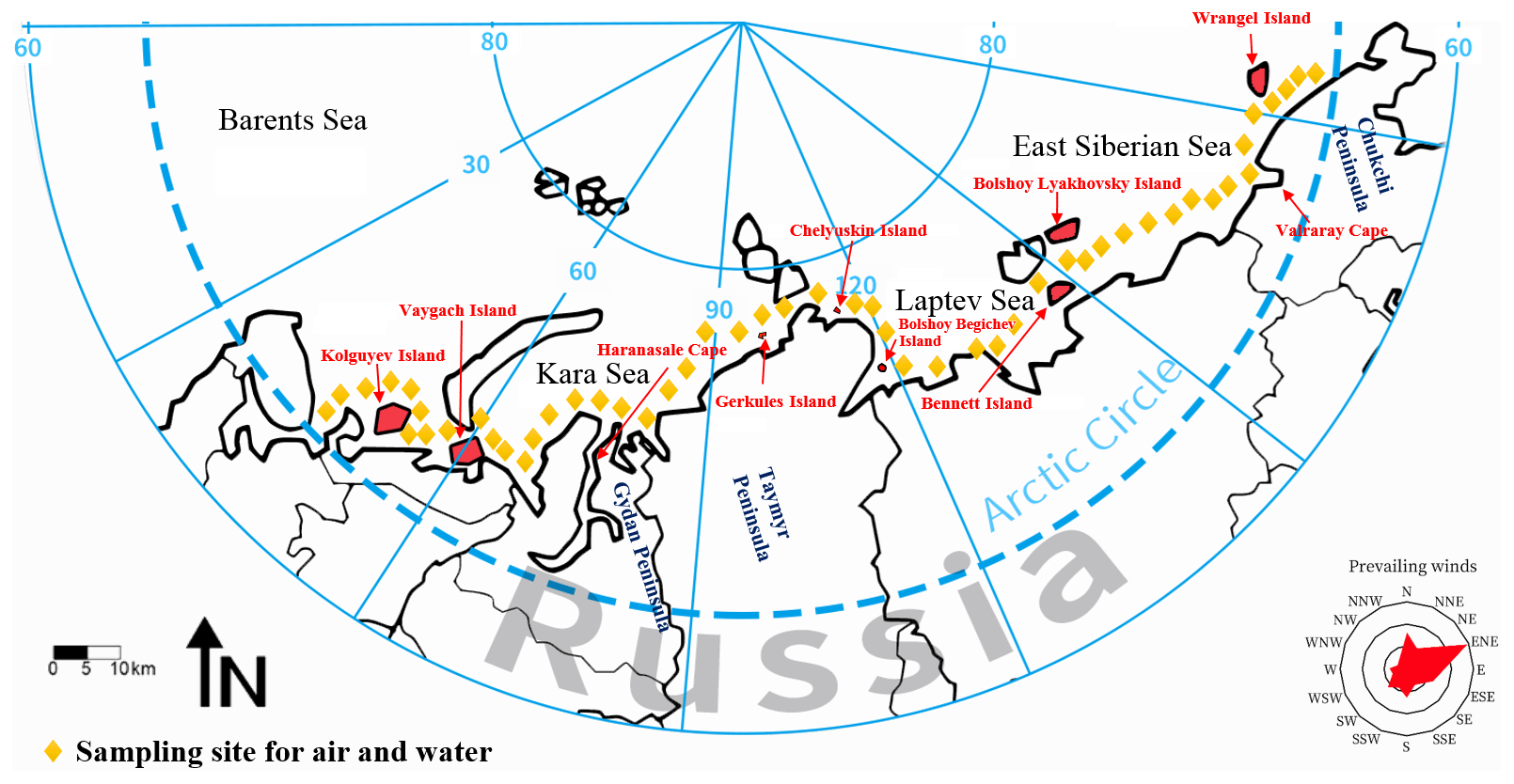
Figure 1Locations of investigated islands for soil sampling and trajectory of the vessel in the Russian Arctic.
2.1.1 Aerosol and gas phase
Air samples, including aerosols and concurrent gases as described elsewhere (Reddy et al., 2012; Shoeib and Harner, 2002; Galarneau et al., 2017; Grosjean, 1983; Wu, 2014), were collected by a high-volume sampler set up at the top of a main rod. A wind vane was connected to the high-volume sampler so that samples could be collected only if the wind was derived from the bow to prevent contamination from ship emissions. The average sampled air volume was 632 m3 (412–963 m3) per sample. The aerosols were sampled on Teflon filters (P0325-100EA, Fluoropore, Darmstadt, Germany), and the compounds in the gas phase were then collected over pre-cleaned polyurethane foams (PUFs). After sampling, the filters and PUFs were tightly covered with aluminum foil for air-tightness, then immediately placed in polyethylene bags, and frozen at −20 ∘C prior to chemical analyses.
2.1.2 Wet deposition and water
Wet deposition samples were collected through a cleaned stainless steel funnel connected to a glass bottle during eight snow events. Snowfall samples were melted thoroughly at room temperature. Water samples were gathered continuously from surface seawater (at a depth of 5 m) along the vessel, and these samples were immediately filtered onto borosilicate microfiber glass filters (AP1504700, MilliporeSigma, Darmstadt, Germany). Then, the compounds in the dissolved phase were retained on XAD sorbent tubes subjected to controlled flows. The mean filtered water volume was 1239 mL (135–2876 mL). The XAD tubes were stored at 5 ∘C before their extraction in the laboratory.
2.2 Heavy metal extraction and analysis
For metal determinations in the aerosol, gas phase, wet deposition, and water samples, Teflon filters, PUFs, and dissolved phases were first Soxhlet-extracted for 8 h using HNO3. The samples were then diluted with deionized water to 23 mL and subjected to inductively coupled plasma mass spectrometry (ICP-MS) analysis. Specifically, the contents of Pb, Cd, Cu, Co, Zn, Fe, Mn, Ni, and Hg were analyzed on an ICP-MS instrument (Thermo Scientific ICE 3500, Waltham, MA, USA) while making use of rhodium (Rh) as an internal standard. High-resolution (10 000) data were collected to avoid any mass interference problems.
2.3 PAH extraction and analysis
For PAH determinations in the gas, aerosol, and dissolved phase samples, published procedures were used (Berrojalbiz et al., 2011; Castro-Jimenez et al., 2012; Gonzalez-Gaya et al., 2014). Snow-melt water was extracted by using solid phase Oasis HLB (3 cc/60 mg) cartridges on board. Briefly, cartridges were preconditioned with 5 mL methanol, 10 mL of a mixture of methanol:dichloromethane (1 : 2), and 10 mL deionized water. Afterward, each sample was combined with a recovery standard and concentrated by N2 until near dryness. Then, it was eluted with 5 mL hexane, 5 mL of a mixture of hexane:dichloromethane (1 : 2), and 10 mL deionized water.
A total of 35 PAH species were quantified, including naphthalene, methylnaphthalene (sum of two isomers), 1,4,5-trimethylnaphthalene, 1,2,5,6-tetramethylnaphthalene, acenaphthylene, acenaphthene, fluorene, dibenzothiophene, anthracene, 9-methylfluorene, 1,7-dimethylfluorene, 9-n-propylfluorene, 2-methyldibenzothiophene, 2,4-dimethyldibenzothiophene, 2,4,7-trimethyldibenzothiophene, 3-methylphenanthrene, 1,6-dimethylphenanthrene, 1,2,9-trimethylphenanthrene, 1,2,6,9-tetramethylphenanthrene, fluoranthene, pyrene, benzo[a]anthracene, chrysene, 3-methylchrysene, 6-ethylchrysene, 1,3,6-trimethylchrysene, benzo[b]fluoranthene, benzo[k]fluoranthene, benzo[a]pyrene, perylene, dibenzo[a,h]anthracene, indeno[1,2,3-cd]pyrene, dibenzo[a,h]anthracene, and benzo[]perylene. PAH quantification was performed by gas chromatography–mass spectrometry (GC-MS). Specifically, we used a gas chromatograph coupled with a triple quadrupole mass selective detector (GS-MS, ITQ 1100, Thermo Scientific, USA) equipped with a DB-5ms chromatographic capillary column (30 m × 0.25 mm i.d. and 0.25 µm film, Agilent Technologies, Santa Clara, CA, USA) operating in electron impact (EI) mode and with selected ion monitoring (SIM) as reported previously (Gonzalez-Gaya et al., 2014). Internal standards (anthracene-d10, p-terphenyl-d14, pyrene-d10, and benzo[b]fluoranthene-d12) were added before operating the GC-MS instrument for the quantification of PAHs, and the recovery of perdeuterated standards (acenaphthene-d10, chrysene-d12, phenanthrene-d10, and perylene-d12) was determined by addition prior to the procedures of extraction; these values were then used for the correction of measured concentrations.
2.4 Quality assurance and quality control
Analyses of every sample and phase were conducted in the laboratory with field blanks to determine the analytical limits and recoveries. Breakthroughs of aerosols and gas phases were checked for the Teflon filter and PUF samples. Approximately 90 % of the metals and PAHs were obtained during the first half of the sample analysis, while the remaining 10 % were obtained during the second half; for the PAHs, these mostly consisted of compounds with two to three rings. Six blanks (field and laboratory) were collected for the gas phase, while seven field banks and eight laboratory blanks were used for the dissolved phase, all of which were extracted along with the rest of the samples during the analytical procedure. For the gas phase, average ∑ metal values were approximately 0.049 and 0.052 ng per sample in the field and laboratory blanks, respectively, and average ∑ PAH values were approximately 2.44 and 2.06 ng per sample in the field and laboratory blanks, respectively (Tables S1 and S2 in the Supplement). For the aerosols, average ∑ metal values were 0.046 and 0.065 ng per sample in the field and laboratory blanks, respectively, and average ∑ PAH values were 2.95 and 2.96 ng per sample in the field and laboratory blanks, respectively. Similarly, for the dissolved phase, values of 0.053 and 0.052 ng per sample were obtained for the ∑ metals and values of 2 and 1.73 ng per sample were obtained for the ∑ PAHs. All measured PAHs from field samples exceeded the field and laboratory blank concentrations; therefore, the quantified compounds did not subtract the blank values. Mean recoveries of perdeuterated standards used as surrogates in dissolved samples were as follows: 63 % for acenaphthene-d10, 54 % for chrysene-d12, 73 % for phenanthrene-d10, and 82 % for perylene-d12.
All concentrations in each medium were corrected by the surrogate recovery for individual samples. The detection limit was used for the lowest limit of the calibration curve. The quantification limit was equivalent to the average blank concentration for each phase.
2.5 Data processing
Dry deposition fluxes (FDD, ng m−2 d−1) were calculated from field measurements of trace metals and PAHs collected during the expedition over eight time periods. Aerosol deposition fluxes for the metals were calculated as follows:
where Cd is the concentration of atmospheric aerosols and Vd is the velocity of deposition (m s−1). Vd was calculated as shown in Eq. (2), and the details have been described elsewhere (Zhang et al., 2001):
where ugrav is the gravitational settling velocity, and Ra and Rs are the aerodynamic resistance for gaseous species and the surface resistance, respectively. Rs can be calculated as follows:
where ε0 is an empirical constant (ε0=3) and u∗ is the friction velocity calculated for gases. EB is the collection efficiency of Brownian diffusion as a function of the Schmidt number Sc:
where γ is an empirical constant (γ=0.5). EIM is the collection efficiency from impaction based on the following formulas (Peters and Eiden, 1992):
where Va is the kinematic viscosity for air (m2 s−1). The correction factor (R1) is the fraction of particles close to the surface:
For PAHs, the specific compound deposition velocity (Vd, cm s−1) was derived from an empirical parameterization (Gonzalez-Gaya et al., 2014):
where PL is the subcooled liquid vapor pressure of each PAH, U10 is the 10 m height wind speed, and Chls is the concentration of surface chlorophyll. With Eq. (8), one can estimate the Vd for each PAH and sampling period by taking PL from references and using the field-measured U10 and Chls. In this study, FDD(PAH) values were estimated from the measured concentrations in the aerosol phase (CA, ng m−3) as follows:
where 864 is the unit conversion factor.
The wet deposition fluxes (FW, ng m−3 d−1) of metals/PAHs were estimated using the quantified concentrations of metals/PAHs from the collected snow and the precipitated volume of snow-melt water per surface and time period for each of the eight snow events during the expedition.
The air–water diffusive fluxes (FAW, ng m−2 d−1) for Hg/PAHs were calculated according to Fick's law:
where CG and CTW represent the concentration measured in the gas phase (ng m−3) and dissolved phase (ng L−1), respectively. H′ is the temperature dependence of Henry's law constant, and H′ values for PAHs were taken from Bamford et al. (1999); H′ for Hg was calculated from Eq. (11) for seawater (Andersson et al., 2008):
where T is the temperature of the surface water (K). H′ was corrected by the field-measured salinity. KAW represents the air–water mass transfer rate (m d−1) calculated by a two-film model (Singh and Xu, 1997) and while considering the nonlinear wind-speed effect. CTW for Hg was directly measured concentrations, and CTW values for PAHs were calculated using the measured concentrations in the dissolved phase (CW) as follows:
where KDOC was taken as the value of 10 % of the octanol–water partitioning coefficient (KOW) (Burkhard, 2000), and DOC represents the dissolved organic carbon (mg L−1).
KAW was calculated by the two-film model:
where KW and KA are the mass transfer coefficients (m d−1) of Hg and PAHs in the water and air films, respectively. The mass transfer coefficient of CO2 in the water phase (, m d−1) can be used to calculate KW (Gonzalez-Gaya et al., 2016), which is a wind-speed quadratic function at a height of 10 m (U10, m−1) (Nightingale et al., 2000). A Weibull distribution of wind speed was assumed to parameterize because average wind speeds were used during the sampling period as the gas and dissolved phase concentrations were averaged values for the sampling transects; was calculated using a previously reported method (Livingstone and Imboden, 1993):
where η and ξ are the constants of scale and shape in the Weibull distribution, respectively, and Γ represents a gamma function. ξ=2 (Rayleigh distribution) was used as recommended (Gonzalez-Gaya et al., 2016). η is related to wind speed and was calculated with (Livingstone and Imboden, 1993).
KW can be calculated as follows:
where SCPAH is the Hg ∕ PAH Schmidt number. The same applies for KA, which was also calculated from wind speed and the H2O mass transfer coefficient for the air phase (, cm s−1):
where Di,a and represent the Hg ∕ PAH and H2O diffusive coefficients in air, respectively.
The uncertainty was lower than a factor of 1–2 in these estimates for metals/PAHs. Most of the increasing uncertainty was associated with the Henry's law constants. The effect of uncertainty on the air–water exchange net direction was assessed by the ratios of air–water fugacity (fG∕fW) (Figs. S1 and S2 in the Supplement); moreover, the findings revealed that most metals and PAHs were not close to the equilibrium of air–water. Among the PAHs, net volatilization was detected only for dibenzothiophene, alkylated phenanthrenes, and fluoranthene. The details of the uncertainty analysis are shown in Sect. S1 (Supplement).
Gross fluxes of volatilization and absorption depend on the first and second terms of Eq. (10), respectively. The total accumulated fluxes for the Barents Sea, the Kara Sea, the Laptev Sea, and the East Siberian Sea were acquired by multiplying the mean basin flux with its standard deviation by the surface area of each basin.
The estimations of degradation fluxes of PAHs in the atmospheric ocean boundary were calculated as follows:
where CGf and CGi are the last concentration after a fixed time in a closed system (ng m−3) and the concentration in the gas phase at the initial time (ng m−3), respectively. t is the time period (average 5 h daytime per day), and ABL represents the average height of the atmospheric boundary layer (380 m). CGf can be calculated as follows:
where kOH is the rate constant for a PAH reaction with OH radicals (Keyte et al., 2013), and [OH] is the hydroxyl radical concentration in the mixed layer (1000–500 hPa) based on the monthly mean OH radical concentration (Spivakovsky et al., 2000). The mean concentrations of OH were calculated by Eq. (13). The OH concentrations ranged between 5.23 and 17.26×105 mol cm−3. In addition, only the PAHs in the gas phase were considered while the potential degradation of PAHs bound in aerosols was ignored. Considering the uncertainty of those sources, a relevant error factor of 2–3 was given for the degradative fluxes based on the individual PAHs. Because of the large uncertainties in kOH values, the degradation fluxes of PAHs in the atmosphere could not be calculated.
3.1 Heavy metals in the atmosphere and ocean
Nine heavy metals were measured, and the average concentration for each metal in each sea can be found in Table S3. The highest ∑9 metal concentrations in the Barents Sea were found in the gas phase (CG, ng m−3), where the average concentration was 0.418 ng m−3 (Fig. S3). The average values of CG showed no obvious differences among the four seas, whereas the oceanic area adjacent to the Chukchi Peninsula, Taymyr–Gydan peninsulas, and the Arkhangelsk region showed higher combined concentrations of the nine metals (Fig. 2a). High ∑9 metal concentrations in the aerosol phase (CA, ng m−3) were observed in the Barents Sea (p<0.05), where the average ∑9 metal concentration was 2.713 ng m−3 (Fig. 2b). These high levels may have been associated with the trajectories of air from Russian inland areas. The distributions of heavy metals in the Russian Arctic Ocean revealed that the concentrations of the ∑9 metals in seawater were lower than those in air. The concentrations of each metal in aerosols were comparable to those previously reported in the Russian Arctic, i.e., the Laptev Sea, the Kara Sea, the Barents Sea, Severnaya Zemlya, and Wrangel Island (Shevchenko et al., 2003; Vinogradova and Ivanova, 2017). For other parts of the Arctic Ocean, the average mass concentrations of each metal from Svalbard, the Fram Strait, the central Arctic, and Greenland (Ferrero et al., 2019; Maenhaut et al., 1979, 2002, 1989), were higher than those found in aerosols in our study. Metals' concentrations in aerosols in our study were lower than those in the Red Sea and the Mediterranean area (Chen et al., 2008). The average ∑9 metal concentrations in dissolved water (CW) ranged from 0.526 to 0.896 µg L−1 (the Laptev Sea to the Barents Sea). This is relatively lower than the concentrations of dissolved trace metals (Mn, Fe, Ni, Zn, and Cd) previously reported for the western Arctic Ocean (the Chukchi Sea and the Canada Basin, depth: 5–20 cm) (Kondo et al., 2016). Higher values of CW were observed in the Barents Sea–Kara Sea region (the Yamal Peninsula) and in the East Siberian Sea (close to the Chukchi Peninsula) in comparison with the CW values in other areas (Fig. 2c).
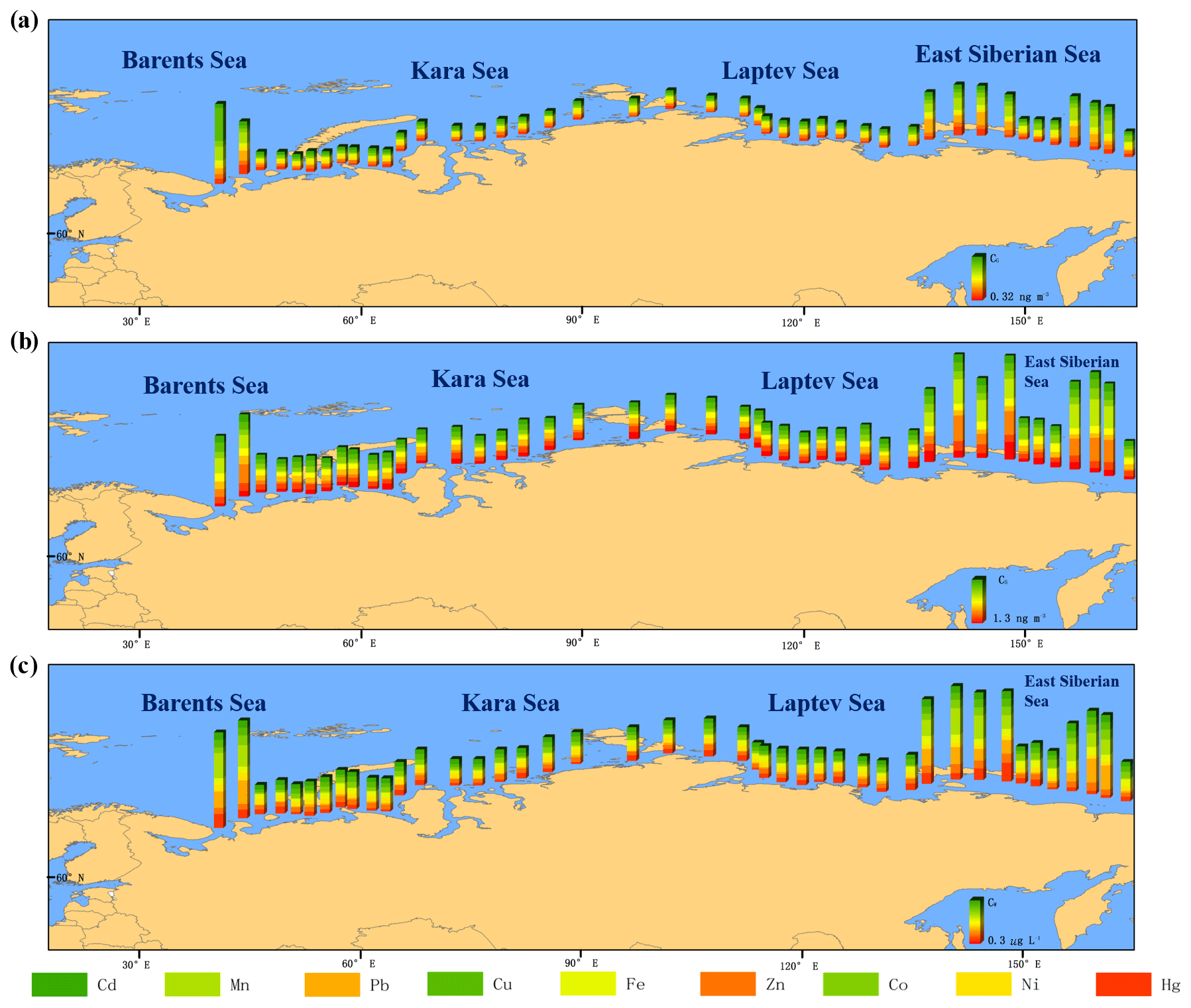
Figure 2Occurrence of heavy metals. Results show the concentrations of heavy metals in the (a) gas phase, (b) aerosol phase, and (c) dissolved water phase. Colored bars show the sum of nine quantified metals. The number at the bottom of the legend bars in the panels represent the concentration scale (and is the same for Figs. 3–7).
The abundance of each metal in gases, aerosols, and dissolved water is dependent on the emission sources. In this study, Fe and Zn were the most abundant metals detected in aerosols and dissolved water from the Russian Arctic Ocean, where the average ∑9 metal concentrations in aerosols and dissolved water were 0.64 ng m−3 and 0.91 ng L−1, respectively. Pb was the most abundant metal in the gas phase (the average concentration in the Russian Arctic Ocean =0.14 ng m−3). In comparison with aerosols and dissolved water, the gas phase contained higher levels of Hg, which is a finding consistent with the usual form of Hg in the atmosphere (>98 %) and the tendency for the remaining types of Hg to adsorb to particles during atmospheric transport (Poissant et al., 2008). In all phases, the proportions of Mn, Fe, Pb, and Zn were significantly higher than those of other heavy metals. Additionally, the metal distributions in the Barents Sea and the Kara Sea showed the highest proportions, followed by the metal distributions in the East Siberian Sea. On the Taymyr Peninsula (adjacent to the Kara Sea and the Laptev Sea), there is a mining and metallurgical factory operated by the Norilsk company that processes copper and nickel and is one of the biggest metallurgical factories in the world. This may be a likely source of metals in the Kara Sea region (Shevchenko et al., 2003). Due to the significant differences in the concentrations of metals in the marine boundary layer both temporally and spatially throughout the Russian Arctic Ocean (Vinogradova and Polissar, 1995; Shevchenko et al., 1999), as well as the scarcity of reported data on heavy metals in the atmosphere in this region, it was difficult to compare our data with historical findings. However, our data are similar to those reported for September 1993 in the Kara Sea (Rovinsky et al., 1995).
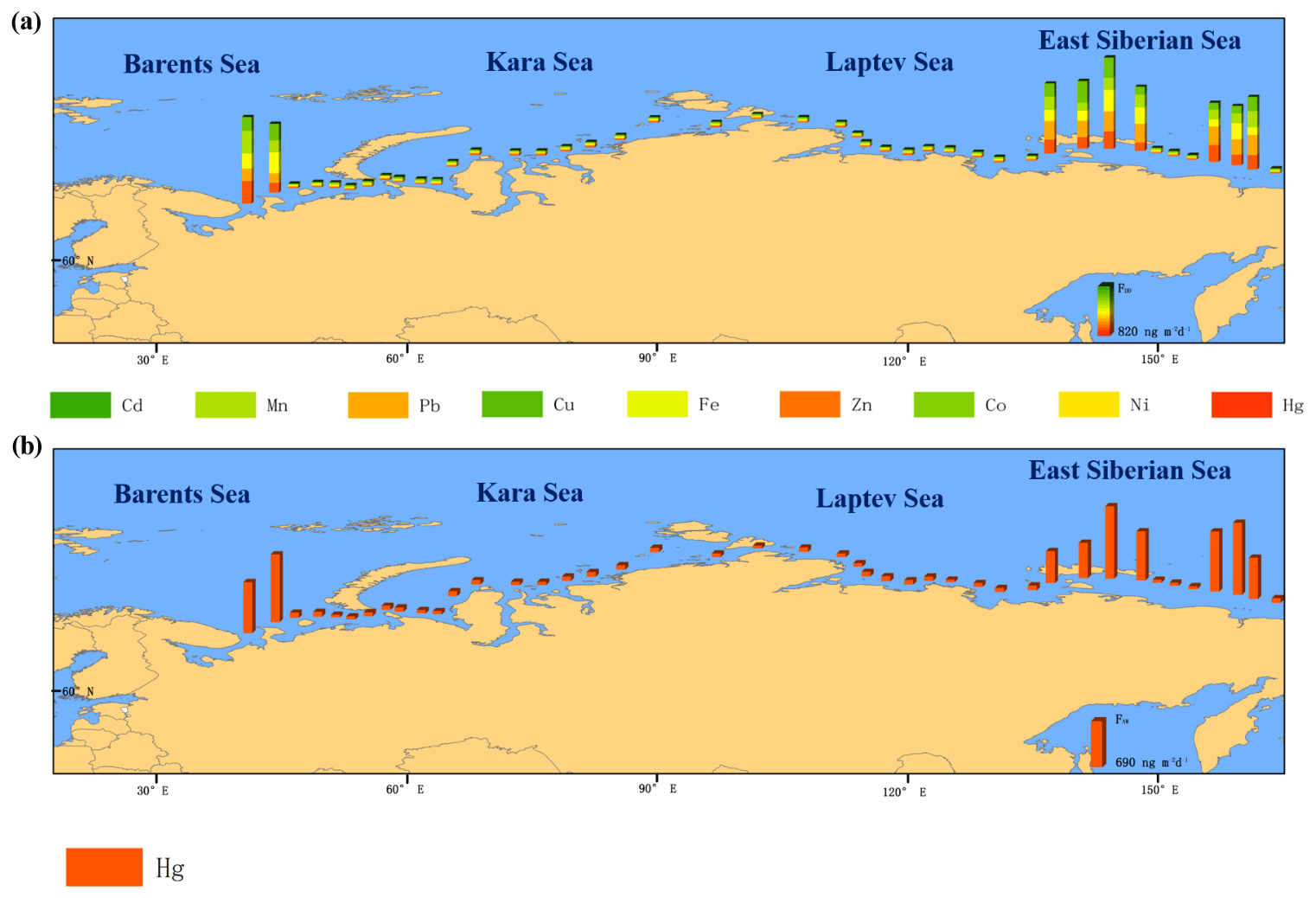
Figure 3Measured atmosphere–ocean exchange of heavy metals. (a) Fluxes of dry deposition for nine heavy metals; (b) fluxes of net diffusive air–water exchange for Hg. In panel (a), the colored bars represent the sum of nine heavy metals. In panel (b), the downward bars represent the net deposition into the ocean, and the upward bars represent the net volatilization of Hg.
The dry deposition that involves aerosols binding to heavy metals (FDD, ng m−2 d−1) is a major process for heavy metal deposition (Shevchenko et al., 2003). In the Russian Arctic Ocean, the average FDD of the ∑9 metals ranged from 392 to 8067 ng m−2 d−1 (mean of 1792 ng m−2 d−1). The largest FDD value was found close to the coast of the East Siberian Sea, where FDD values of 305 and 224 ng m−2 d−1 were observed for Hg and Pb, respectively, and were dominant (Fig. 3a). Our results seem to be 1 order of magnitude higher than those in the Red Sea (mean of 615 ng m−2 d−1) (Chen et al., 2008) and Mediterranean Sea (mean of 264 ng m−2 d−1; Chen et al., 2008). However, this comparison may not reflect the strength of the emission sources because dry deposition is highly dependent on the deposition velocity, which is affected by meteorological conditions such as the humidity, wind speed, and the stability of the air column (Mariraj Mohan, 2016). The relative humidity in the Arctic Ocean tends to be higher in coastal areas and, notably, we sampled during spring–winter when water vapor evaporates from the relatively warmer surfaces of seawater (Vihma et al., 2008). In addition, the wind over sampling sites in the Arctic Ocean was ∼7 m s−1 on average (the highest average wind speed was ∼9 m s−1 in the Barents Sea), which is significantly higher than the wind in the Red Sea and the Mediterranean Sea (0.36–1 m s−1) (Chen et al., 2008; Chester et al., 1999). During the eight snow events encountered in the course of the expedition, the wet deposition flux of the ∑9 metals (FWD, µg m−2 d−1) ranged from 23 to 32 µg m−2 d−1 (mean of 26 µg m−2 d−1) (Fig. 4a). Data relevant to the wet deposition flux of heavy metals in the Arctic region include results for Hg, which were estimated on land in Alaska; the highest deposition was detected along the southern and southeastern coasts (>0.05 µg m−2 d−1) (Pearson et al., 2019). The values were quite similar to the FWD for Hg in our study (0.05 to 0.09 µg m−2 d−1). Using analysis of variance tests, we did not find any significant difference in the FWD at different locations (p>0.05) for all heavy metals, whereas a relatively higher FWD for Hg was observed in coastal areas adjacent to the Taymyr Peninsula with industrial factories. Pearson et al. (2019) pointed out that there are larger contributions from Hg wet deposition in the Bering Sea and the Gulf of Alaska, which are influenced by the western Pacific winds from East Asia, where high Hg emissions from industrial activities and coal burning occur (Wong et al., 2006). The Russian Arctic Ocean is also affected by Pacific winds from East Asia, which could lead to a combination of heavy metal deposition from both local anthropogenic sources and long-range transport from Asia. Wet deposition is an important process for the transfer of heavy metals from gas and aerosol phases to ocean water. Snowfall in the Arctic is an important fraction of precipitation, but variations in measurements ranging from 20 % to 50 % can occur under windy conditions even with sampling equipment designed with wind protection (Rasmussen et al., 2012). However, snow events are quite sporadic in the Russian Arctic Ocean during spring–summer compared with the other deposition processes. Nevertheless, wet deposition in our study was under regional influences and had a relatively high uncertainty.
Figure 4Wet deposition of (a) heavy metals and (b) PAH fluxes. The measured wet deposition of heavy metals and PAHs occurred during the eight snow events encountered during the vessel's expedition.
For many heavy metals that form volatile species, there is additional evidence that their existence in water is strongly related to releases from terrestrial environments rather than internal cycling in aquatic systems (Robert, 2013). For example, following the deposition of atmospheric Fe, a nonvolatile species, the concentrations in water are influenced mainly by the particulate phase and its dissolution, whereas for Hg, a volatile species that predominantly exists in the atmosphere as a gas (Hg0), the concentrations of volatile Hg species in water are largely influenced by volatilization and deposition processes at the air–water interface; portions of the Hg in aquatic systems end up being converted to methylmercury (Mason and Sheu, 2002; Sunderland and Mason, 2007; Selin et al., 2007; Strode et al., 2007). Hg concentrations in the gas phase in the present study were significantly lower than those measured in 1996 in the Northern Hemisphere (1.5–1.7 ng m−3) and Southern Hemisphere (1.2–1.3 ng m−3) (Steffen et al., 2005; Slemr et al., 2003; Wängberg et al., 2007; Kim et al., 2005). Steffen et al. (2002) indicated that there has been increasing retention of Hg in the Arctic region based on analyses of long-term measurements of atmospheric Hg concentrations. Diffusive air–water exchange is the dominant process driving the exchange of Hg in the ocean. The net diffusive air–water exchange (FAW, ng m−2 d−1) was estimated by a two-film resistance model (Robert, 2013). The net input of Hg was calculated as shown in Fig. 3b and revealed that there was a net input from the atmosphere to the ocean at all stations, especially for the stations close to industrial/urban areas. The integrated monthly FAW fluxes (in tons per month) for Hg were of the same order of magnitude as the FDD fluxes for Hg and other heavy metals in the Russian Arctic Ocean (Fig. S4). For Hg, the gross volatilization and gross absorption in the Russian Arctic Ocean were 250 and 530 t per month, respectively. In consideration of previous studies of atmospheric mercury depletion events (AMDEs), during which the net input of Hg in the Arctic was evaluated (Brooks et al., 2006; Lindberg et al., 2001), we adjusted our sampling times to avoid sampling during sunrise: during this period the autocatalytic release of sea salt aerosols changes the oxidative photochemistry in the stratified planetary boundary layer where elemental and reactive Hg in the gas phase is oxidized by reactive halogens. It was estimated that ∼99–496 t of Hg are deposited annually in the Arctic during AMDEs (Skov et al., 2006; Ariya et al., 2004). The net input of Hg in the present study was 1 order of magnitude higher than that caused by the AMDEs; this discrepancy may result from the fact that the previous studies considered the terrestrial boundary between air masses and the snowpack, and that the different locations and seasons were affected by different meteorological conditions. In northern regions, it has been shown that Hg undergoes long-range transport from Eurasia, especially during the winter season (Poissant et al., 2008). These net amounts of Hg entering into the Arctic Ocean pose potential risks to marine biota because Hg is poorly mobile and can be retained by aquatic biota that are exposed to it during the deposition process (Harris et al., 2007).
3.2 PAHs in the atmosphere and ocean
A total of 35 individual PAHs, which included isomer groups such as alkylated PAHs, were measured. The average concentrations of PAHs in each sea of the Russian Arctic Ocean are shown in Table S4. The average values of CG showed no obvious differences in the Kara Sea, the Laptev Sea, and the East Siberian Sea (p>0.05), and no particularly high levels of CG were detected at any of the sampling sites (Fig. 5a). The range of ∑35 PAH CG is 19.87–22.14 ng m−3 for the Barents Sea, 19.01–22.34 ng m−3 for the Kara Sea, 19.23–21.70 ng m−3 for the Laptev Sea, and 19.28–22.61 ng m−3 for the East Siberian Sea. The highest CG of ∑35 PAH is observed in the Barents Sea, with a value of 22.61 ng m−3. ∑35 PAH CA in the Barents Sea (0.25–2.95 ng m−3) and the East Siberian Sea (0.24–3.32), with average values of 1.38 and 2.07 ng m−3 , respectively, were higher than those in the Laptev Sea (0.23–0.89 ng m−3) and the Kara Sea (0.23–0.27 ng m−3), with average values of 0.30 and 0.25 ng m−3, respectively (Fig. 5b). The average CA of ∑35 PAH in the present study is higher than the average CA of ∑64 PAH measured in the South Atlantic Ocean (0.93 ng m−3) and North Pacific Ocean (0.56 ng m−3), while it is much lower than the average CA of ∑64 PAH in the Indian Ocean (10 ng m−3) (Gonzalez-Gaya et al., 2016). The average ∑35 PAH CA of 1.02 ng m−3 in the Russian Arctic Ocean is comparable to the average ∑64 PAH CA observed in the South Atlantic Ocean and South Pacific Ocean (1.1 ng m−3) (Gonzalez-Gaya et al., 2016). ∑18 PAH CA was measured from the North Pacific towards the Arctic Ocean, ranging from 0.0002 to 0.36 ng m−3, with the highest concentration found in the coastal areas in East Asia (Ma et al., 2013). These concentrations were significantly lower than the average levels found in our study. In addition, Ma et al. (2013) observed relatively higher ∑18 PAH CA values in the most northern latitudes of the Arctic Ocean, which is associated with back trajectories of air masses from southern Asia. The higher levels of CA in our study could be attributed to the proximity of the coastline to larger areas of burning taiga forest and more industrial sources in the boreal regions of Russian continent. Similar to the pattern for heavy metals mentioned above, high levels of these chemicals may have been derived from atmospheric transport from the industrial areas of the Russian continent. Due to the various sampling methods and the differences in PAHs measured, and because not all studies separated gas and particles concentrations, it is quite difficult to compare PAH levels in aerosols. The average ∑35 PAH CW ranged from 13.07 ng L−1 (in the Laptev Sea) to 69.90 ng L−1 (in the Barents Sea), and its spatial variability was similar to that of PAH concentrations in aerosols. The ranges of ∑35 PAH CW for the Barents Sea, the Kara Sea, the Laptev Sea, and the East Siberian Sea are 12.36–162.05, 12.18–14.04, 11.21–15.82, and 11.40–129.60 ng L−1, respectively. Higher levels of CW were also found along the coast of the Yamal–Gydan peninsulas, where active petrol and natural gas industries sites are located (Fig. 5c).

Figure 5Occurrence of PAHs in the Russian Arctic Ocean. Concentrations of PAHs in the (a) gas phase, (b) aerosol phase, and (c) dissolved water phase. Color bars indicate the sum of 35 PAHs, where each PAH corresponds to the bottom legend (colors range from red for the heaviest molecular weight PAHs to green for the lightest molecular weight PAHs).
The contribution of each PAH in the gas, aerosol, and dissolved water phases is determined by its source, volatility, and hydrophobicity (Lima et al., 2005). The low-molecular-weight PAHs were dominant in gas and dissolved water (Fig. S5). In the gas phase, low-molecular-weight PAHs occupied more than 75 % of the ∑35 PAHs, which mainly contained methylated phenanthrenes, e.g., methylphenanthrene (mean of 1.31 ng m−3), dimethylphenanthrene (mean of 1.27 ng m−3), and trimethylphenanthrene (mean of 1.32 ng m−3), and methylated dibenzothiophenes, e.g., methyldibenzothiophene (mean of 1.29 ng m−3), dimethyldibenzothiophene (mean of 1.27 ng m−3), and trimethyldibenzothiophene (mean of 1.32 ng m−3). In dissolved water, methylnapthalene and tetramethylnaphthalene were the most abundant PAHs with average concentrations of 1.12 and 1.45 ng L−1, respectively. Measured values of CG, CA, and CW are known to vary with the changes in each PAH concentration in the marine environment (Berrojalbiz et al., 2011; Castro-Jimenez et al., 2012; Cabrerizo et al., 2014). However, there were no previous reports regarding the occurrence of PAHs in the Russian Arctic atmosphere and ocean.

Figure 6Dry deposition fluxes for the 35 measured PAHs. Color bars indicate the sum of the 35 quantified compounds, and each color represents the individual PAHs in the bottom legend (colors range from red for the heaviest molecular weight PAHs to green for the lightest molecular weight PAHs).
The average dry deposition flux (FDD) of the ∑35 PAHs was 1108 ng m−2 d−1. The increasing values of Vd may influence FDD in the marine environment due to the higher hydrophobicity of organic compounds, surface microlayer with reduced surface tension, and lipid floating (Del Vento and Dachs, 2007b). The higher average Vd was observed for 9-methylfluorene (1.01–10.02 cm s−1), followed by 1,7-dimethylfluorene (1.06–10.63 cm s−1) (Fig. S6). On a global scale, higher Vd values were found for heavier PAHs such as methylchrysene (0.17–13.30 cm s−1) and dibenzo[a,h]anthracene (0.29–1.38 cm s−1) (Gonzalez-Gaya et al., 2014). The Vd values reported previously ranged from 0.08 to 0.3 cm s−1 in the Atlantic Ocean (Del Vento and Dachs, 2007a) and from 0.01 to 0.8 cm s−1 in coastal areas (Holsen and Noll, 1992; Bozlaker et al., 2008; Esen et al., 2008; Eng et al., 2014); the higher values were observed in concentrated industrial and urban areas (Bozlaker et al., 2008). In our study, the highest Vd values were observed in the Barents Sea; the other three seas had similar Vd values (p>0.05) that were lower than in the Barents Sea except for 9-methylfluorene and 1,7-dimethylfluorene. The East Siberian Sea exhibited the lowest value of Vd, whereas the relatively higher Vd values were found for heavier PAHs (dibenzo[a,h]anthracene, indeno[1,2,3-cd]pyrene, dibenzo[a,h]anthracene and benzo[]perylene) in all seas (Fig. S7). This may be due to heavier PAHs being principally deposited via heavier aerosols with a higher Vd because they are bound to hydrophobic aerosols or because of gravity, e.g., soot carbon (Gonzalez-Gaya et al., 2014). Dry deposition is a major process for high-molecular-weight PAHs bound to aerosols (Figs. 6, S8). The deposition values varied mainly in accordance with the PAH concentrations of aerosols in suspension and the factors influencing the deposition velocities (wind speed, compound vapor pressure, etc.). The FWD of the ∑35 PAHs ranged from 14 to 19 µg m−2 d−1. Gonzalez-Gaya et al. (2014) found the highest FWD of ∑64 PAHs in the North Atlantic Ocean (24 µg m−2 d−1) with an average FWD value of ∼8 µg m−2 d−1 on the global scale based on rain samples. The higher FWD values of PAHs were found in urban areas in China (62.6 µg m−2 d−1) (Wang et al., 2016) from rain samples and the lower FWD values of PAHs (0.02–0.28 µg m−2 d−1) from both rain and snow samples were observed in high mountain areas in Europe (Arellano et al., 2018). Our FWD values were within the range of the previously reported global scale and the difference in wet deposition was mainly dependent on source distance and precipitation intensity. Wet deposition is an important purging process for semivolatile organic compounds such as PAHs in the gas and aerosol phase. Snow events are quite sporadic in the Arctic Ocean and, thus, these have lower relevance for wet deposition of PAHs in this region.
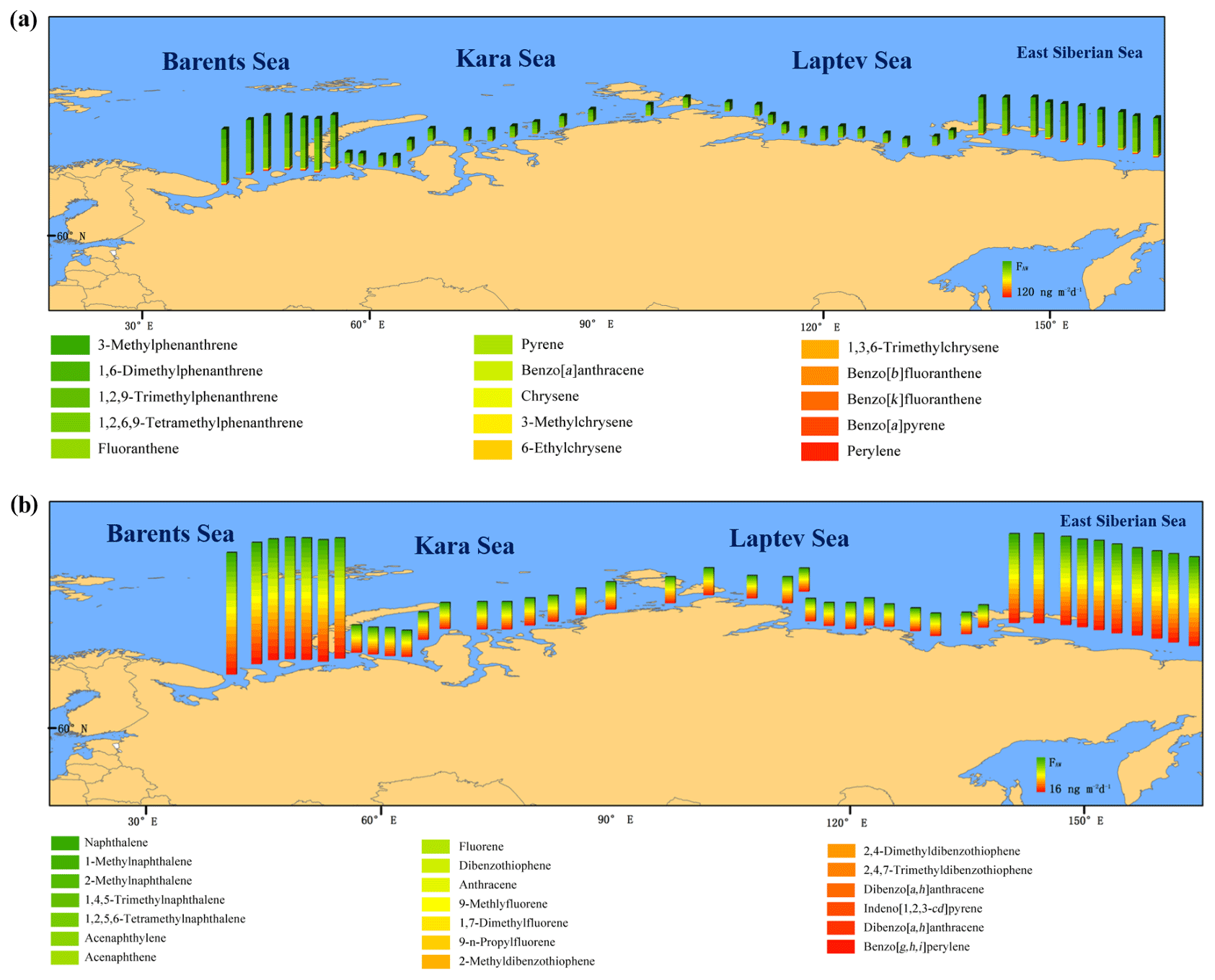
Figure 7Measured atmosphere–ocean exchange of PAHs. (a) Net diffusive air–water exchange fluxes (all net deposition into the ocean) and (b) volatilization air–water exchange fluxes. Color bars indicate the sum of the 35 quantified compounds, and each color represents the individual PAHs in the bottom legend (colors range from red for the heaviest molecular weight PAHs to green for the lightest molecular weight PAHs).
The estimated FAW values revealed that most PAHs had a net input from the atmosphere to the ocean except for the more volatile PAHs, such as two to three ring PAHs (Fig. 7). The lighter PAHs (two to three rings) appeared more volatile (978–4892 ng m−2 d−1), whereas heavier PAHs (four to six rings) showed net deposition (1561–7808 ng m−2 d−1), except dibenzo[a,h]anthracene (1322 ng m−2 d−1), indeno[1,2,3-cd]pyrene (1238 ng m−2 d−1), trimethylphenanthrene (1901 ng m−2 d−1), and benzo[]perylene (2708 ng m−2 d−1). A net deposition that was 3 orders of magnitude higher was observed for methylphenanthrene, dimethylphenanthrene, trimethylphenanthrene, and tetramethylphenanthrene (Fig. S9). Our results were similar to those observed in other PAH-affected areas such as the southeast Mediterranean (Castro-Jimenez et al., 2012), Narragansett Bay (Lohmann et al., 2011), and the North Atlantic Ocean (Lohmann et al., 2009). Ma et al. (2013) suggested that slight volatilization of lighter PAHs may exist from additional sources such as ship ballast and riverine runoff, which is consistent with our findings that volatilization occurred mainly in the East Siberian Sea and Barents Sea, where more industrial factories and urban areas are situated. Our study is also consistent with previous reports showing that diffusion during air–water exchange is the main process for the transfer of relatively light volatile organic compounds to the marine environment (Castro-Jimenez et al., 2012; Jurado et al., 2005). The integrated monthly FAW values (in tons per month) of five to six ring PAHs were comparable to FDD values in the Russian Arctic Ocean, whereas only the East Siberian Sea showed high levels of dry deposition (Fig. S9). In all four seas, FAW values of three to four ring PAHs were of the same magnitude as FDD values. The total volatilization and total adsorption of the ∑35 PAHs in the Russian Arctic Ocean amounted to 2600 and 3640 t per month, respectively. Therefore, there was a net input of the ∑35 PAHs from the atmosphere to the marine environment that reached 3276 t, which was 100 times higher than for the aerosol-bound ∑35 PAHs that underwent dry deposition (estimated at ∼30 t per month). In other reports, Gonzalez-Gaya et al. (2016) estimated the global input of PAHs from the atmosphere to the ocean to be of the order of 90 000 t per month, and Reddy et al. (2012) reported that the input of PAHs to the ocean in the Gulf of Mexico in 2010 after the Deepwater Horizon oil spill was 20 000 t. Such comparisons suggest that the diffusive fluxes in the Russian Arctic Ocean play an important role in the atmosphere–ocean exchange of PAHs, whereas there is a lower input of PAHs to the Russian Arctic Ocean on the global scale.
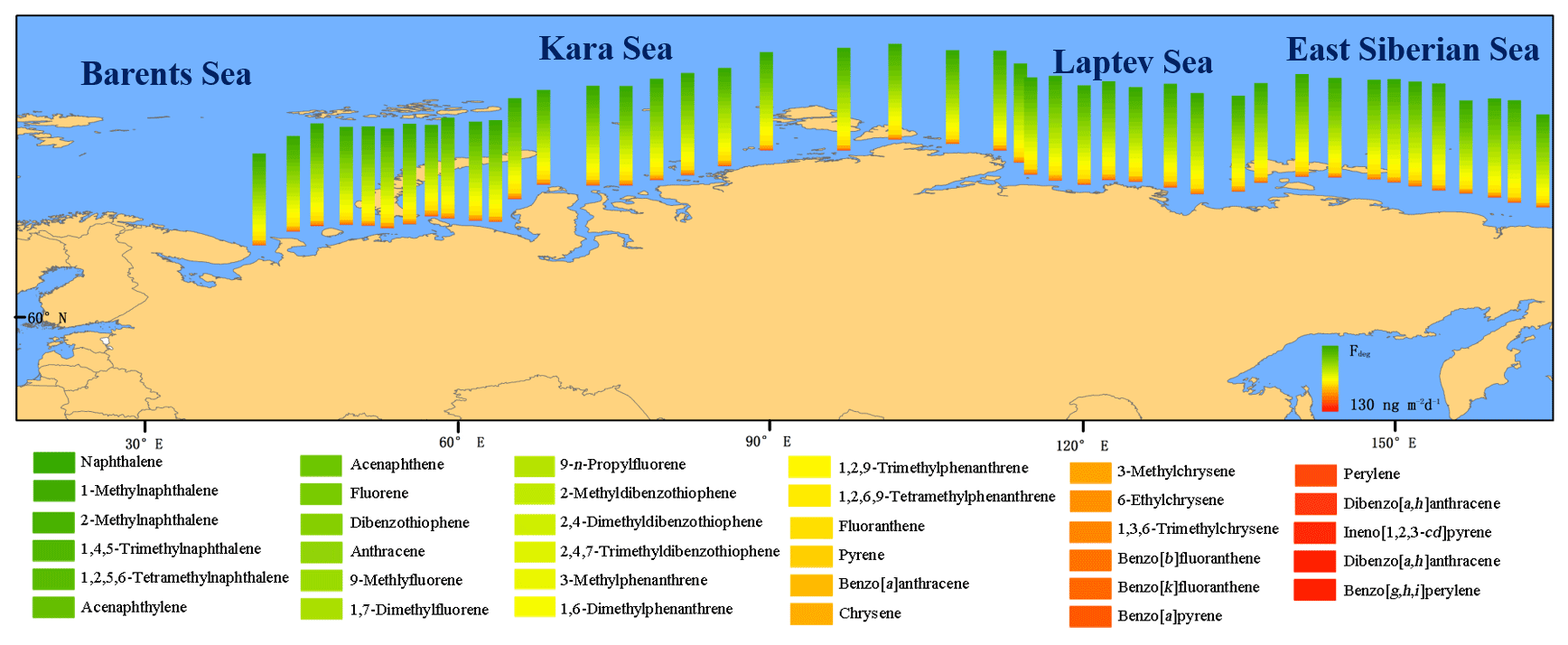
Figure 8Atmospheric degradation of PAHs. Estimated fluxes of degraded PAHs in the gas phase following reaction with OH radicals.
In addition to the transfers of PAHs to the ocean, PAHs can also be degraded during transport through the atmosphere due to reactions with OH radicals (Keyte et al., 2013). The degradation flux Datm of PAHs in the gas phase of the oceanic atmosphere (Fig. 8) was estimated at 3000 t per month for the ∑35 PAHs, which represents an additional PAH sink (see Sect. 2). In general, the large amounts of PAHs undergoing net deposition to the ocean and degradation during atmospheric transport are indicative of large source areas in the Russian Arctic. Notably, PAH concentrations have been increasing in the atmosphere owing to wildfires and fossil fuel use over the past century (Zhang and Tao, 2009). The high-molecular-weight PAHs were dominant in the aerosols (Fig. 5) originating from pyrolytic sources (Lima et al., 2005). In addition, high abundances of alkylated PAHs were observed in the gas and dissolved phases; along with the evaluations of the diagnostic ratios (Fig. S10), these results were suggestive of pyrogenic sources for PAHs in gases and aerosols, and mixtures of pyrogenic and petrogenic sources for PAHs in dissolved water (mostly for the Laptev Sea and the East Siberian Sea). Other sources contributing to the occurrence of PAHs may have involved both anthropogenic and biogenic sources on land (Cabrerizo et al., 2011). In the Russian Arctic Ocean, it can be assumed that PAHs in the atmosphere (gas and aerosol) originated from anthropogenic sources including industrial and urban activities, while PAHs in seawater, at sites with relatively fewer anthropogenic sources, i.e., the Laptev Sea and the East Siberian Sea, originated from a mixture of anthropogenic and biogenic sources. This indicates that the atmospheric transport of PAHs derived from anthropogenic activities occurs in all sectors of the Russian Arctic Ocean, while only the East Siberian Sea and the Laptev Sea have more anthropogenic PAHs in the water phase.
Because PAHs are toxic, these chemicals can have an adverse influence on food webs in marine ecosystems (Hylland, 2006). In particular, even though PAHs are present at natural background levels in the marine environment, the massive usage of fossil fuels has led to increases in PAH emissions and excessive PAH concentrations in many marine environments. The present study indicates that there are high contributions of diffusive atmospheric PAHs to the Arctic Ocean, and these chemicals are potentially perturbing the carbon cycle in the ocean and posing risks to the fragile Arctic marine food webs. Thus, further studies of the impacts of such chemicals are warranted.
This study presents the occurrence and atmosphere–ocean fluxes of 35 PAHs and 9 heavy metals in the Arctic Ocean. Dry deposition and wet deposition fluxes of nine heavy metals in aerosols were estimated at 2205 and 10.95 µg m−2 d−1, respectively. The net gross absorption of Hg in the Arctic Ocean was estimated at 280 t per month. A net input of PAHs from the atmosphere to the Arctic Ocean was observed for most of the PAHs, especially for the low-molecular-weight PAHs. The net atmospheric input of the 35 PAHs was estimated at 3276 t per month. The current occurrences of semivolatile aromatic hydrocarbons could have been derived from biogenic sources and anthropogenic sources from continental land masses, especially for the locations close to industrial areas. These inputs of Hg and PAHs may be causing adverse effects on the fragile Arctic marine ecosystems; this issue warrants further research. In addition, our study suggests that both PAHs and metals are affected by local depositional effects and changes of source emission; therefore, the spatial distribution of these compounds and source identification need to be further investigated.
All original data regarding the concentrations of PAHs and heavy metals in the gas, aerosol, and dissolved phases, as well as dry deposition velocity are shown in the Supplement. All other data utilized for calculations can be accessed by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-13789-2019-supplement.
XJ and EA set up the sampling equipment and analyzed the samples and the data. XX also helped to collect and analyze the data. XJ and XX wrote the paper.
The authors declare that they have no conflict of interest.
We would like to thank Yu Su from the School of Visual Arts at BFA Computer Art for helping with data visualization, and Kuznetsova Ekaterina for helping with the Russian translation.
This research has been supported by the National Key R&D Program of China (grant no. 2016YFE011230), the Russian Foundation for Basic Research (grant nos. 18-44-890003 and 16-34-60010), the Jiangsu Nature Science Fund (grant no. BK20151378), and the Fundamental Research Funds for the Central Universities (grant no. 090514380001).
This paper was edited by Ralf Ebinghaus and reviewed by two anonymous referees.
Andersson, M. E., Gårdfeldt, K., Wängberg, I., and Strömberg, D.: Determination of Henry's law constant for elemental mercury, Chemosphere, 73, 587–592, https://doi.org/10.1016/j.chemosphere.2008.05.067, 2008.
Arellano, L., Fernández, P., van Drooge, B. L., Rose, N. L., Nickus, U., Thies, H., Stuchlík, E., Camarero, L., Catalan, J., and Grimalt, J. O.: Drivers of atmospheric deposition of polycyclic aromatic hydrocarbons at European high-altitude sites, Atmos. Chem. Phys., 18, 16081–16097, https://doi.org/10.5194/acp-18-16081-2018, 2018.
Ariya, P. A., Dastroor, A. P., Amyot, M., Schroeder, W. H., Barrie, L., Anlauf, K., Raofie, F., Ryzhkov, A., Davignon, D., Lalonde, J., and Steffen, A.: The Arctic: a sink for mercury, Tellus B, 56, 397–403, https://doi.org/10.3402/tellusb.v56i5.16458, 2004.
Bagi, A., Pampanin, D. M., Lanzén, A., Bilstad, T., and Kommedal, R.: Naphthalene biodegradation in temperate and arctic marine microcosms, Biodegradation, 25, 111–125, https://doi.org/10.1007/s10532-013-9644-3, 2014.
Bamford, H. A., Poster, D. L., and Baker, J. E.: Temperature dependence of Henry's law constants of thirteen polycyclic aromatic hydrocarbons between 4 ∘C and 31 ∘C, Environ. Toxicol. Chem., 18, 1905–1912, https://doi.org/10.1897/1551-5028(1999)018<1905:Tdohsl>2.3.Co;2, 1999.
Berrojalbiz, N., Dachs, J., Jose Ojeda, M., Carmen Valle, M., Castro-Jimenez, J., Wollgast, J., Ghiani, M., Hanke, G., and Zaldivar, J. M.: Biogeochemical and physical controls on concentrations of polycyclic aromatic hydrocarbons in water and plankton of the Mediterranean and Black Seas, Global Biogeochem. Cy., 25, GB4003, https://doi.org/10.1029/2010gb003775, 2011.
Bigg, E. K. and Leck, C.: Properties of the aerosol over the central Arctic Ocean, J. Geophys. Res.-Atmos., 106, 32101–32109, https://doi.org/10.1029/1999JD901136, 2001.
Boyd, R., Barnes, S. J., De Caritat, P., Chekushin, V. A., Melezhik, V. A., Reimann, C., and Zientek, M. L.: Emissions from the copper-nickel industry on the Kola Peninsula and at Noril'sk, Russia, Atmos. Environ., 43, 1474–1480, https://doi.org/10.1016/j.atmosenv.2008.12.003, 2009.
Bozlaker, A., Muezzinoglu, A., and Odabasi, M.: Atmospheric concentrations, dry deposition and air-soil exchange of polycyclic aromatic hydrocarbons (PAHs) in an industrial region in Turkey, J. Hazard. Mater., 153, 1093–1102, https://doi.org/10.1016/j.jhazmat.2007.09.064, 2008.
Brooks, S. B., Saiz-Lopez, A., Skov, H., Lindberg, S. E., Plane, J. M. C., and Goodsite, M. E.: The mass balance of mercury in the springtime arctic environment, Geophys. Res. Lett., 33, L13812, https://doi.org/10.1029/2005GL025525, 2006.
Burkhard, L. P.: Estimating dissolved organic carbon partition coefficients for nonionic organic chemicals, Environ. Sci. Technol., 34, 4663–4668, https://doi.org/10.1021/es001269l, 2000.
Cabrerizo, A., Dachs, J., Moeckel, C., Ojeda, M.-J., Caballero, G., Barcelo, D., and Jones, K. C.: Ubiquitous Net Volatilization of Polycyclic Aromatic Hydrocarbons from Soils and Parameters Influencing Their Soil-Air Partitioning, Environ. Sci. Technol., 45, 4740–4747, https://doi.org/10.1021/es104131f, 2011.
Cabrerizo, A., Galban-Malagon, C., Del Vento, S., and Dachs, J.: Sources and fate of polycyclic aromatic hydrocarbons in the Antarctic and Southern Ocean atmosphere, Global Biogeochem. Cy., 28, 1424–1436, https://doi.org/10.1002/2014gb004910, 2014.
Castro-Jimenez, J., Berrojalbiz, N., Wollgast, J., and Dachs, J.: Polycyclic aromatic hydrocarbons (PAHs) in the Mediterranean Sea: Atmospheric occurrence, deposition and decoupling with settling fluxes in the water column, Environ. Pollut., 166, 40–47, https://doi.org/10.1016/j.envpol.2012.03.003, 2012.
Chen, Y., Paytan, A., Chase, Z., Measures, C., Beck, A. J., Sañudo-Wilhelmy, S. A., and Post, A. F.: Sources and fluxes of atmospheric trace elements to the Gulf of Aqaba, Red Sea, J. Geophys. Res.-Atmos., 113, D05306, https://doi.org/10.1029/2007JD009110, 2008.
Cheng, J.-O., Ko, F.-C., Lee, C.-L., and Fang, M.-D.: Air–water exchange fluxes of polycyclic aromatic hydrocarbons in the tropical coast, Taiwan, Chemosphere, 90, 2614–2622, https://doi.org/10.1016/j.chemosphere.2012.11.020, 2013.
Cheng, M. D., Hopke, P. K., Barrie, L., Rippe, A., Olson, M., and Landsberger, S.: Qualitative determination of source regions of aerosol in Canadian high Arctic, Environ. Sci. Technol., 27, 2063–2071, https://doi.org/10.1021/es00047a011, 1993.
Chester, R., Nimmo, M., and Preston, M. R.: The trace metal chemistry of atmospheric dry deposition samples collected at Cap Ferrat: a coastal site in the Western Mediterranean, Mar. Chem., 68, 15–30, https://doi.org/10.1016/S0304-4203(99)00062-6, 1999.
Custódio, D., Cerqueira, M., Fialho, P., Nunes, T., Pio, C., and Henriques, D.: Wet deposition of particulate carbon to the Central North Atlantic Ocean, Sci. Total Environ., 496, 92–99, https://doi.org/10.1016/j.scitotenv.2014.06.103, 2014.
Dahle, S., Savinov, V., Carroll, J., Vladimirov, M., Ivanov, G., Valetova, N., Gaziev, Y., Dunaev, G., Kirichenko, Z., Nikitin, A., Petrenko, G., Polukhina, A., Kalmykov, S., Aliev, R., and Sabodina, M.: A return to the nuclear waste dumping sites in the Bays of Novaya Zemlya, Radioprotection, 44, 281–284, https://doi.org/10.1051/radiopro/20095054, 2009.
Del Vento, S. and Dachs, J.: Atmospheric occurrence and deposition of polycyclic aromatic hydrocarbons in the northeast tropical and subtropical Atlantic Ocean, Environ. Sci. Technol., 41, 5608–5613, https://doi.org/10.1021/es0707660, 2007a.
Del Vento, S. and Dachs, J.: Influence of the surface microlayer on atmospheric deposition of aerosols and polycyclic aromatic hydrocarbons, Atmos. Environ., 41, 4920–4930, https://doi.org/10.1016/j.atmosenv.2007.01.062, 2007b.
Duce, R. A., Liss, P. S., Merrill, J. T., Atlas, E. L., Buat-Menard, P., Hicks, B. B., Miller, J. M., Prospero, J. M., Arimoto, R., Church, T. M., Ellis, W., Galloway, J. N., Hansen, L., Jickells, T. D., Knap, A. H., Reinhardt, K. H., Schneider, B., Soudine, A., Tokos, J. J., Tsunogai, S., Wollast, R., and Zhou, M.: The atmospheric input of trace species to the world ocean, Global Biogeochem. Cy., 5, 193–259, https://doi.org/10.1029/91GB01778, 1991.
Eng, A., Harner, T., and Pozo, K.: A Prototype Passive Air Sampler for Measuring Dry Deposition of Polycyclic Aromatic Hydrocarbons, Environ. Sci. Technol. Lett., 1, 77–81, https://doi.org/10.1021/ez400044z, 2014.
Environment Canada, Fisheries and Oceans Canada and Indian and Northern Affairs Canada Arctic: Land-Based Pollution in the Arctic Ocean: Canadian Actions in a Regional and Global Context, Arctic, 61, 111–121, 2008.
Esen, F., Cindoruk, S. S., and Tasdemir, Y.: Bulk deposition of polycyclic aromatic hydrocarbons (PAHs) in an industrial site of Turkey, Environ. Pollut., 152, 461–467, https://doi.org/10.1016/j.envpol.2007.05.031, 2008.
Fernandes, M. B. and Sicre, M. A.: Polycyclic Aromatic Hydrocarbons in the Arctic: Ob and Yenisei Estuaries and Kara Sea Shelf, Estuar., Coast. Shelf Sci., 48, 725–737, https://doi.org/10.1006/ecss.1999.0472, 1999.
Ferrero, L., Sangiorgi, G., Perrone, M. G., Rizzi, C., Cataldi, M., Markuszewski, P., Pakszys, P., Makuch, P., Petelski, T., Becagli, S., Traversi, R., Bolzacchini, E., and Zielinski, T.: Chemical Composition of Aerosol over the Arctic Ocean from Summer ARctic EXpedition (AREX) 2011–2012 Cruises: Ions, Amines, Elemental Carbon, Organic Matter, Polycyclic Aromatic Hydrocarbons, n-Alkanes, Metals, and Rare Earth Elements, Atmosphere, 10, 54, https://doi.org/10.3390/atmos10020054, 2019.
Galarneau, E., Patel, M., Brook, J. R., Charland, J.-P., Glasius, M., Bossi, R., and Hung, H.: Artefacts in semivolatile organic compound sampling with polyurethane foam substrates in high volume cascade impactors, Aerosol Sci. Tech., 51, 247–257, https://doi.org/10.1080/02786826.2016.1267327, 2017.
Gonzalez-Gaya, B., Zuniga-Rival, J., Ojeda, M.-J., Jimenez, B., and Dachs, J.: Field Measurements of the Atmospheric Dry Deposition Fluxes and Velocities of Polycyclic Aromatic Hydrocarbons to the Global Oceans, Environ. Sci. Technol., 48, 5583–5592, https://doi.org/10.1021/es500846p, 2014.
Gonzalez-Gaya, B., Fernandez-Pinos, M.-C., Morales, L., Mejanelle, L., Abad, E., Pina, B., Duarte, C. M., Jimenez, B., and Dachs, J.: High atmosphere-ocean exchange of semivolatile aromatic hydrocarbons, Nat. Geosci., 9, 438–442, https://doi.org/10.1038/ngeo2714, 2016.
Grosjean, D.: Polycyclic aromatic hydrocarbons in Los Angeles air from samples collected on teflon, glass and quartz filters, Atmos. Environ., 17, 2565–2573, https://doi.org/10.1016/0004-6981(83)90084-7, 1983.
Harris, R. C., Rudd, J. W. M., Amyot, M., Babiarz, C. L., Beaty, K. G., Blanchfield, P. J., Bodaly, R. A., Branfireun, B. A., Gilmour, C. C., Graydon, J. A., Heyes, A., Hintelmann, H., Hurley, J. P., Kelly, C. A., Krabbenhoft, D. P., Lindberg, S. E., Mason, R. P., Paterson, M. J., Podemski, C. L., Robinson, A., Sandilands, K. A., Southworth, G. R., St. Louis, V. L., and Tate, M. T.: Whole-ecosystem study shows rapid fish-mercury response to changes in mercury deposition, P. Natl. Acad. Sci. USA, 104, 16586, https://doi.org/10.1073/pnas.0704186104, 2007.
Holsen, T. M. and Noll, K. E.: Dry deposition of atmospheric particles – application of current models to ambient data, Environ. Sci. Technol., 26, 1807–1815, https://doi.org/10.1021/es00033a015, 1992.
Hornbuckle, K. C., Jeremiason, J. D., Sweet, C. W., and Eisenreich, S. J.: Seasonal Variations in Air-Water Exchange of Polychlorinated Biphenyls in Lake Superior, Environ. Sci. Technol., 28, 1491–1501, https://doi.org/10.1021/es00057a018, 1994.
Hung, H., Katsoyiannis, A. A., Brorström-Lundén, E., Olafsdottir, K., Aas, W., Breivik, K., Bohlin-Nizzetto, P., Sigurdsson, A., Hakola, H., Bossi, R., Skov, H., Sverko, E., Barresi, E., Fellin, P., and Wilson, S.: Temporal trends of Persistent Organic Pollutants (POPs) in arctic air: 20 years of monitoring under the Arctic Monitoring and Assessment Programme (AMAP), Environ. Pollut., 217, 52–61, https://doi.org/10.1016/j.envpol.2016.01.079, 2016.
Hylland, K.: Polycyclic Aromatic Hydrocarbon (PAH) Ecotoxicology in Marine Ecosystems, J. Toxicol. Env. Heal. A, 69, 109–123, https://doi.org/10.1080/15287390500259327, 2006.
Jaffe, D., Cerundolo, B., Rickers, J., Stolzberg, R., and Baklanov, A.: Deposition of sulfate and heavy-metals on the Kola-Peninsula, Sci. Total Environ., 160–161, 127–134, https://doi.org/10.1016/0048-9697(95)04350-a, 1995.
Ji, X., Abakumov, E., and Polyakov, V.: Assessments of pollution status and human health risk of heavy metals in permafrost-affected soils and lichens: A case-study in Yamal Peninsula, Russia Arctic, Hum. Ecol. Risk Assess., 25, 2142–2159, https://doi.org/10.1080/10807039.2018.1490887, 2019.
Jickells, T. D. and Baker, A. R.: Atmospheric Transport and Deposition of Particulate Matter to the Oceans, in: Encyclopedia of Ocean Sciences (Third Edition), edited by: Cochran, J. K., Bokuniewicz, H. J., and Yager, P. L., Academic Press, Oxford, UK, 21–25, 2019.
Jurado, E., Jaward, F., Lohmarm, R., Jones, K. C., Simo, R., and Dachs, J.: Wet deposition of persistent organic pollutants to the global oceans (vol. 39, pg. 2426, 2005), Environ. Sci. Technol., 39, 4672–4672, https://doi.org/10.1021/es050660+, 2005.
Keyte, I. J., Harrison, R. M., and Lammel, G.: Chemical reactivity and long-range transport potential of polycyclic aromatic hydrocarbons – a review, Chem. Soc. Rev., 42, 9333–9391, https://doi.org/10.1039/C3CS60147A, 2013.
Kim, K.-H., Ebinghaus, R., Schroeder, W. H., Blanchard, P., Kock, H. H., Steffen, A., Froude, F. A., Kim, M.-Y., Hong, S., and Kim, J.-H.: Atmospheric Mercury Concentrations from Several Observatory Sites in the Northern Hemisphere, J. Atmos. Chem., 50, 1–24, https://doi.org/10.1007/s10874-005-9222-0, 2005.
Kondo, Y., Obata, H., Hioki, N., Ooki, A., Nishino, S., Kikuchi, T., and Kuma, K.: Transport of trace metals (Mn, Fe, Ni, Zn and Cd) in the western Arctic Ocean (Chukchi Sea and Canada Basin) in late summer 2012, Deep-Sea Res. Py. I, 116, 236–252, https://doi.org/10.1016/j.dsr.2016.08.010, 2016.
Leck, C., Bigg, E. K., Covert, D. S., Heintzenberg, J., Maenhaut, W., Nilsson, E. D., and Wiedensohler, A.: Overview of the atmospheric research program during the International Arctic Ocean Expedition of 1991 (IAOE-91) and its scientific results, Tellus B, 48, 136–155, https://doi.org/10.1034/j.1600-0889.1996.t01-1-00002.x, 1996.
Lima, A. L. C., Farrington, J. W., and Reddy, C. M.: Combustion-Derived Polycyclic Aromatic Hydrocarbons in the Environment – A Review, Environ. Forensics, 6, 109–131, https://doi.org/10.1080/15275920590952739, 2005.
Lindberg, S. E., Brooks, S., Lin, C. J., Scott, K., Meyers, T., Chambers, L., Landis, M., and Stevens, R.: Formation of Reactive Gaseous Mercury in the Arctic: Evidence of Oxidation of Hg0 to Gas-Phase Hg-II Compounds after Arctic Sunrise, Water Air Soil Poll., 1, 295–302, https://doi.org/10.1023/A:1013171509022, 2001.
Livingstone, D. M. and Imboden, D. M.: The nonlinear influence of wind-speed variability on gas transfer in lakes, Tellus B, 45, 275–295, https://doi.org/10.1034/j.1600-0889.1993.t01-2-00005.x, 1993.
Lohmann, R., Gioia, R., Jones, K. C., Nizzetto, L., Temme, C., Xie, Z., Schulz-Bull, D., Hand, I., Morgan, E., and Jantunen, L.: Organochlorine Pesticides and PAHs in the Surface Water and Atmosphere of the North Atlantic and Arctic Ocean, Environ. Sci. Technol., 43, 5633–5639, https://doi.org/10.1021/es901229k, 2009.
Lohmann, R., Dapsis, M., Morgan, E. J., Dekany, V., and Luey, P. J.: Determining Air-Water Exchange, Spatial and Temporal Trends of Freely Dissolved PAHs in an Urban Estuary Using Passive Polyethylene Samplers, Environ. Sci. Technol., 45, 2655–2662, https://doi.org/10.1021/es1025883, 2011.
Ma, Y., Xie, Z., Yang, H., Möller, A., Halsall, C., Cai, M., Sturm, R., and Ebinghaus, R.: Deposition of polycyclic aromatic hydrocarbons in the North Pacific and the Arctic, J. Geophys. Res.-Atmos., 118, 5822–5829, https://doi.org/10.1002/jgrd.50473, 2013.
Maenhaut, W., Zoller, W. H., Duce, R. A., and Hoffman, G. L.: Concentration and size distribution of particulate trace elements in the south polar atmosphere, J. Geophys. Res.-Oceans, 84, 2421–2431, https://doi.org/10.1029/JC084iC05p02421, 1979.
Maenhaut, W., Cornille, P., Pacyna, J. M., and Vitols, V.: Trace element composition and origin of the atmospheric aerosol in the Norwegian arctic, Atmos. Environ., 23, 2551–2569, https://doi.org/10.1016/0004-6981(89)90266-7, 1989.
Maenhaut, W., Ducastel, G., Leck, C., Nilsson, E., and Heintzenberg, J.: Multi-elemental composition and sources of the high Arctic atmospheric aerosol during summer and autumn, Tellus B, 48, 300–321, https://doi.org/10.1034/j.1600-0889.1996.t01-1-00011.x, 2002.
Mariraj Mohan, S.: An overview of particulate dry deposition: measuring methods, deposition velocity and controlling factors, Int. J. Environ. Sci. Te., 13, 387–402, https://doi.org/10.1007/s13762-015-0898-7, 2016.
Mason, R. P. and Sheu, G.-R.: Role of the ocean in the global mercury cycle, Global Biogeochem. Cy., 16, 40-1–40-14, https://doi.org/10.1029/2001GB001440, 2002.
Nightingale, P. D., Liss, P. S., and Schlosser, P.: Measurements of air-sea gas transfer during an open ocean algal bloom, Geophys. Res. Lett., 27, 2117–2120, https://doi.org/10.1029/2000gl011541, 2000.
Pacyna, E. G., Pacyna, J. M., Sundseth, K., Munthe, J., Kindbom, K., Wilson, S., Steenhuisen, F., and Maxson, P.: Global emission of mercury to the atmosphere from anthropogenic sources in 2005 and projections to 2020, Atmos. Environ., 44, 2487–2499, https://doi.org/10.1016/j.atmosenv.2009.06.009, 2010.
Park, G.-H., Lee, S.-E., Kim, Y.-I., Kim, D., Lee, K., Kang, J., Kim, Y.-H., Kim, H., Park, S., and Kim, T.-W.: Atmospheric deposition of anthropogenic inorganic nitrogen in airborne particles and precipitation in the East Sea in the northwestern Pacific Ocean, Sci. Total Environ., 681, 400–412, https://doi.org/10.1016/j.scitotenv.2019.05.135, 2019.
Pearson, C., Howard, D., Moore, C., and Obrist, D.: Mercury and trace metal wet deposition across five stations in Alaska: controlling factors, spatial patterns, and source regions, Atmos. Chem. Phys., 19, 6913–6929, https://doi.org/10.5194/acp-19-6913-2019, 2019.
Peters, K. and Eiden, R.: Modelling the dry deposition velocity of aerosol particles to a spruce forest, Atmos. Environ. A-Gen., 26, 2555–2564, https://doi.org/10.1016/0960-1686(92)90108-W, 1992.
Poissant, L., Zhang, H. H., Canário, J., and Constant, P.: Critical review of mercury fates and contamination in the arctic tundra ecosystem, Sci. Total Environ., 400, 173–211, https://doi.org/10.1016/j.scitotenv.2008.06.050, 2008.
Rahn, K. A. and Lowenthal, D. H.: Elemental traces of distant regional pollution aerosols, Science, 223, 132–139, https://doi.org/10.1126/science.223.4632.132, 1984.
Rasiq, K. T., El-Maradny, A., Orif, M., Bashir, M. E., and Turki, A. J.: Polycyclic aromatic hydrocarbons in two polluted lagoons, eastern coast of the Red Sea: Levels, probable sources, dry deposition fluxes and air-water exchange, Atmos. Pollut. Res., 10, 880–888, https://doi.org/10.1016/j.apr.2018.12.016, 2019.
Rasmussen, R., Baker, B., Kochendorfer, J., Myers, T., Landolt, S., Fischer, A., Black, J., Thériault, J., Kucera, P., Gochis, D., Smith, C., Nitu, R., Hall, M., Cristanelli, S., and Gutmann, A.: How well are we measuring snow: the NOAA/FAA/NCAR winter precipitation test bed, B. Am. Meteorol. Soc., 93, 811–829, https://doi.org/10.1175/BAMS-D-11-00052.1, 2012.
Reddy, C. M., Arey, J. S., Seewald, J. S., Sylva, S. P., Lemkau, K. L., Nelson, R. K., Carmichael, C. A., McIntyre, C. P., Fenwick, J., Ventura, G. T., Van Mooy, B. A. S., and Camilli, R.: Composition and fate of gas and oil released to the water column during the Deepwater Horizon oil spill, P. Natl. Acad. Sci. USA, 109, 20229–20234, https://doi.org/10.1073/pnas.1101242108, 2012.
Reimann, C., Boyd, R., deCaritat, P., Halleraker, J. H., Kashulina, G., Niskavaara, H., and Bogatyrev, I.: Topsoil (0–5 cm) composition in eight arctic catchments in northern Europe (Finland, Norway and Russia), Environ. Pollut., 95, 45–56, https://doi.org/10.1016/s0269-7491(96)00102-9, 1997.
Robert, P. M.: Trace Metals in Freshwaters, in: Trace Metals in Aquatic Systems, Wiley-Blackwell, Chichester, UK, 2013.
Rovinsky, F., Pastukhov, B., Bouyvolov, Y., and Burtseva, L.: Present day state of background pollution of the natural environment in the Russian Arctic in the region of the Ust-Lena Reserve, Sci. Total Environ., 160–161, 193–199, https://doi.org/10.1016/0048-9697(95)04356-6, 1995.
Selin, N. E., Jacob, D. J., Park, R. J., Yantosca, R. M., Strode, S., Jaeglé, L., and Jaffe, D.: Chemical cycling and deposition of atmospheric mercury: Global constraints from observations, J. Geophys. Res.-Atmos., 112, D02308, https://doi.org/10.1029/2006JD007450, 2007.
Shaw, G. E.: Aerosol chemical components in Alaska air masses 1. Aged pollution, J. Geophys. Res.-Atmos., 96, 22357–22368, https://doi.org/10.1029/91jd02058, 1991.
Shevchenko, V., Lisitzin, A., Vinogradova, A., and Stein, R.: Heavy metals in aerosols over the seas of the Russian Arctic, Sci. Total Environ., 306, 11–25, https://doi.org/10.1016/S0048-9697(02)00481-3, 2003.
Shevchenko, V. P., Lisitzin, A. P., Stein, R., Serova, V. V., Isaeva, A. B., and Politova, N. V.: The Composition of the Coarse Fraction of Aerosols in the Marine Boundary Layer over the Laptev, Kara and Barents Seas, in: Land-Ocean Systems in the Siberian Arctic: Dynamics and History, edited by: Kassens, H., Bauch, H. A., Dmitrenko, I. A., Eicken, H., Hubberten, H.-W., Melles, M., Thiede, J., and Timokhov, L. A., Springer Berlin Heidelberg, Berlin, Heidelberg, Germany, 53–58, 1999.
Shoeib, M. and Harner, T.: Characterization and Comparison of Three Passive Air Samplers for Persistent Organic Pollutants, Environ. Sci. Technol., 36, 4142–4151, https://doi.org/10.1021/es020635t, 2002.
Singh, V. P. and Xu, C. Y.: Evaluation and generalization of 13 mass-transfer equations for determining free water evaporation, Hydrol. Process., 11, 311–323, https://doi.org/10.1002/(SICI)1099-1085(19970315)11:3<311::AID-HYP446>3.0.CO;2-Y, 1997.
Sirois, A. and Barrie, L. A.: Arctic lower tropospheric aerosol trends and composition at Alert, Canada: 1980–1995, J. Geophys. Res.-Atmos., 104, 11599–11618, https://doi.org/10.1029/1999JD900077, 1999.
Skov, H., Brooks, S. B., Goodsite, M. E., Lindberg, S. E., Meyers, T. P., Landis, M. S., Larsen, M. R. B., Jensen, B., McConville, G., and Christensen, J.: Fluxes of reactive gaseous mercury measured with a newly developed method using relaxed eddy accumulation, Atmos. Environ., 40, 5452–5463, https://doi.org/10.1016/j.atmosenv.2006.04.061, 2006.
Slemr, F., Brunke, E.-G., Ebinghaus, R., Temme, C., Munthe, J., Wängberg, I., Schroeder, W., Steffen, A., and Berg, T.: Worldwide trend of atmospheric mercury since 1977, Geophys. Res. Lett., 30, 1516, https://doi.org/10.1029/2003GL016954, 2003.
Spivakovsky, C. M., Logan, J. A., Montzka, S. A., Balkanski, Y. J., Foreman-Fowler, M., Jones, D. B. A., Horowitz, L. W., Fusco, A. C., Brenninkmeijer, C. A. M., Prather, M. J., Wofsy, S. C., and McElroy, M. B.: Three-dimensional climatological distribution of tropospheric OH: Update and evaluation, J. Geophys. Res.-Atmos., 105, 8931–8980, https://doi.org/10.1029/1999jd901006, 2000.
Steffen, A., Schroeder, W., Bottenheim, J., Narayan, J., and Fuentes, J. D.: Atmospheric mercury concentrations: measurements and profiles near snow and ice surfaces in the Canadian Arctic during Alert 2000, Atmos. Environ., 36, 2653–2661, https://doi.org/10.1016/S1352-2310(02)00112-7, 2002.
Steffen, A., Schroeder, W., Macdonald, R., Poissant, L., and Konoplev, A.: Mercury in the Arctic atmosphere: An analysis of eight years of measurements of GEM at Alert (Canada) and a comparison with observations at Amderma (Russia) and Kuujjuarapik (Canada), Sci. Total Environ., 342, 185–198, https://doi.org/10.1016/j.scitotenv.2004.12.048, 2005.
Stow, J., Krümmel, E., Leech, T., Donaldson, S., Hansen, J. C., and Van Oostdam, J.: What is the impact of mercury contamination on human health in the Arctic?, AMAP Assessment 2011: Mercury in the Arctic, Arctic Council, Copenhagen, Denmark, 159–169, 2015.
Strode, S. A., Jaeglé, L., Selin, N. E., Jacob, D. J., Park, R. J., Yantosca, R. M., Mason, R. P., and Slemr, F.: Air-sea exchange in the global mercury cycle, Global Biogeochem. Cy., 21, GB1017, https://doi.org/10.1029/2006GB002766, 2007.
Sunderland, E. M. and Mason, R. P.: Human impacts on open ocean mercury concentrations, Global Biogeochem. Cy., 21, GB4022, https://doi.org/10.1029/2006GB002876, 2007.
Totten, L. A., Brunciak, P. A., Gigliotti, C. L., Dachs, J., Glenn, Nelson, E. D., and Eisenreich, S. J.: Dynamic Air-Water Exchange of Polychlorinated Biphenyls in the New York-New Jersey Harbor Estuary, Environ. Sci. Technol., 35, 3834–3840, https://doi.org/10.1021/es010791k, 2001.
Valenti, J. M.: Book Review: Silent Snow: The Slow Poisoning of the Arctic, Sci. Commun., 27, 574–576, https://doi.org/10.1177/1075547006288605, 2006.
Vieira, L. H., Achterberg, E. P., Scholten, J., Beck, A. J., Liebetrau, V., Mills, M. M., and Arrigo, K. R.: Benthic fluxes of trace metals in the Chukchi Sea and their transport into the Arctic Ocean, Mar. Chem., 208, 43–55, https://doi.org/10.1016/j.marchem.2018.11.001, 2019.
Vihma, T., Jaagus, J., Jakobson, E., and Palo, T.: Meteorological conditions in the Arctic Ocean in spring and summer 2007 as recorded on the drifting ice station Tara, Geophys. Res. Lett., 35, L18706, https://doi.org/10.1029/2008GL034681, 2008.
Vinogradova, A. A. and Ivanova, Y. A.: Heavy Metals in the Atmosphere over the Northern Coast of Eurasia: Interannual Variations in Winter and Summer, Izv. Atmos. Ocean. Phy.+, 53, 711–718, https://doi.org/10.1134/S000143381707009X, 2017.
Vinogradova, A. A. and Polissar, A.: Elemental composition of the aerosol in the atmosphere of the central Russian Arctic (in Russian), Izv. Atmos. Ocean. Phy.+, 31, 248–257, 1995.
Walker, T. R., Young, S. D., Crittenden, P. D., and Zhang, H.: Anthropogenic metal enrichment of snow and soil in north-eastern European Russia, Environ. Pollut., 121, 11–21, https://doi.org/10.1016/s0269-7491(02)00212-9, 2003.
Wang, F., Feng, T., Guo, Z., Li, Y., Lin, T., and Rose, N. L.: Sources and dry deposition of carbonaceous aerosols over the coastal East China Sea: Implications for anthropogenic pollutant pathways and deposition, Environ. Pollut., 245, 771–779, https://doi.org/10.1016/j.envpol.2018.11.059, 2019.
Wang, Q., Liu, M., Li, Y., Liu, Y., Li, S., and Ge, R.: Dry and wet deposition of polycyclic aromatic hydrocarbons and comparison with typical media in urban system of Shanghai, China, Atmos. Environ., 144, 175–181, https://doi.org/10.1016/j.atmosenv.2016.08.079, 2016.
Wängberg, I., Munthe, J., Berg, T., Ebinghaus, R., Kock, H. H., Temme, C., Bieber, E., Spain, T. G., and Stolk, A.: Trends in air concentration and deposition of mercury in the coastal environment of the North Sea Area, Atmos. Environ., 41, 2612–2619, https://doi.org/10.1016/j.atmosenv.2006.11.024, 2007.
Wong, C. S. C., Duzgoren-Aydin, N. S., Aydin, A., and Wong, M. H.: Sources and trends of environmental mercury emissions in Asia, Sci. Total Environ., 368, 649–662, https://doi.org/10.1016/j.scitotenv.2005.11.024, 2006.
Wu, S.: Polycyclic Aromatic Hydrocarbons in the Atmosphere of Two Subtropical Cities in Southeast China: Seasonal Variation and Gas/Particle Partitioning, Aerosol Air Qual. Res., 14, 1232–1246, https://doi.org/10.4209/aaqr.2013.01.0015, 2014.
Zhang, L., Gong, S., Padro, J., and Barrie, L.: A size-segregated particle dry deposition scheme for an atmospheric aerosol module, Atmos. Environ., 35, 549–560, https://doi.org/10.1016/S1352-2310(00)00326-5, 2001.
Zhang, Y. and Tao, S.: Global atmospheric emission inventory of polycyclic aromatic hydrocarbons (PAHs) for 2004, Atmos. Environ., 43, 812–819, https://doi.org/10.1016/j.atmosenv.2008.10.050, 2009.
Zhulidov, A. V., Robarts, R. D., Pavlov, D. F., Kamari, J., Gurtovaya, T. Y., Merilainen, J. J., and Pospelov, I. N.: Long-term changes of heavy metal and sulphur concentrations in ecosystems of the Taymyr Peninsula (Russian Federation) North of the Norilsk Industrial Complex, Environ. Monit. Assess., 181, 539–553, https://doi.org/10.1007/s10661-010-1848-y, 2011.
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(3261 KB) - Full-text XML
- Corrigendum
-
Supplement
(1462 KB) - BibTeX
- EndNote