the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Sep 2019
| 30 Sep 2019
Shipborne measurements of ClNO2 in the Mediterranean Sea and around the Arabian Peninsula during summer
Philipp G. Eger
Nils Friedrich
Jan Schuladen
Justin Shenolikar
Horst Fischer
Ivan Tadic
Hartwig Harder
Monica Martinez
Roland Rohloff
Sebastian Tauer
Frank Drewnick
Friederike Fachinger
James Brooks
Eoghan Darbyshire
Jean Sciare
Michael Pikridas
Jos Lelieveld
Shipborne measurements of nitryl chloride (ClNO2), hydrogen chloride (HCl) and sulfur dioxide (SO2) were made during the AQABA (Air Quality and climate change in the Arabian BAsin) ship campaign in summer 2017. The dataset includes measurements over the Mediterranean Sea, the Suez Canal, the Red Sea, the Gulf of Aden, the Arabian Sea, the Gulf of Oman, and the Arabian Gulf (also known as Persian Gulf) with observed ClNO2 mixing ratios ranging from the limit of detection to ≈600 pptv. We examined the regional variability in the generation of ClNO2 via the uptake of dinitrogen pentoxide (N2O5) to Cl-containing aerosol and its importance for Cl atom generation in a marine boundary layer under the (variable) influence of emissions from shipping and the oil industry. The yield of ClNO2 formation per NO3 radical generated was generally low (median of ≈1 %–5 % depending on the region), mainly as a result of gas-phase loss of NO3 dominating over heterogeneous loss of N2O5, the latter being disfavoured by the high temperatures found throughout the campaign. The contributions of ClNO2 photolysis and OH-induced HCl oxidation to Cl-radical formation were derived and their relative contributions over the diel cycle compared. The results indicate that over the northern Red Sea, the Gulf of Suez, and the Gulf of Oman the formation of Cl atoms will enhance the oxidation rates of some volatile organic compounds (VOCs), especially in the early morning.
- Article
(4794 KB) - Full-text XML
-
Supplement
(2117 KB) - BibTeX
- EndNote





The AQABA (Air Quality and climate change in the Arabian BAsin) campaign was designed to study air quality and climate in a region (eastern Mediterranean and Middle East) that is likely to be heavily impacted by future climate change with increasing frequency and intensity of droughts, heatwaves, and associated Aeolian dust and pollution emissions (Lelieveld et al., 2012). As the Arabian Gulf already suffers from some of the most polluted air on Earth with O3 levels regularly greater than 100 ppbv (Lelieveld et al., 2009), one aspect of the campaign was to investigate the factors that contribute to high levels of air pollution in the region. This includes the impact of reactive chlorine chemistry resulting from the interactions of pollutant emissions from ships and petrochemical activity with sea salt, under conditions influenced by intense photochemistry and high temperatures during summer.
The heterogeneous uptake of gaseous N2O5 to the aerosol phase represents an important atmospheric sink for NOx (NO+NO2) via conversion to nitric acid (HNO3), which is efficiently removed from the boundary layer via deposition (Lelieveld and Crutzen, 1990; Dentener and Crutzen, 1993; Macintyre and Evans, 2010). In the presence of aerosol chloride, nitryl chloride (ClNO2) can also be formed along with HNO3 () as shown in Reaction (R1) (Finlayson-Pitts et al., 1989; Behnke et al., 1997). ClNO2 has a lifetime of more than 30 h in the nocturnal marine boundary layer (Osthoff et al., 2008) but is rapidly photolysed after sunrise (Reaction R2), releasing nitrogen dioxide (NO2) and chlorine atoms.
The formation of ClNO2 can have a significant impact on regional NOx cycling and radical chemistry, especially in the polluted coastal and marine boundary layer (Simon et al., 2009; Riedel et al., 2014; Sarwar et al., 2014). The Cl atoms formed in Reaction (R2) can enhance oxidation rates of several volatile organic compounds (VOCs), especially during early morning hours (Phillips et al., 2012; Riedel et al., 2012a; Young et al., 2012) thus contributing to photochemical ozone production (Simon et al., 2009; Riedel et al., 2014; Sarwar et al., 2014; Faxon et al., 2015; Wang et al., 2019).
The chemical processes involved in the formation of ClNO2 are complex and, as outlined in Fig. 1, involve the sequential oxidation of NOx to N2O5 via NO3 (Reactions R3–R5). During the day NO3 is rapidly photolysed via Reaction (R6) or reacts with nitrogen oxide (NO) via Reaction (R7) so that N2O5 formation is suppressed. The heterogeneous reaction of N2O5 with particles is thus, to a good approximation, limited to the night-time. The equilibrium between NO3 and N2O5 (Reactions R4 and R5) is strongly temperature-dependent, with N2O5 formation favoured by high NO2 mixing ratios and low temperatures. NO3 can also react with volatile organic compounds (VOCs, Reaction R8) forming alkyl nitrates, which also reduces the rate of formation of N2O5.
The N2O5 loss rate via heterogeneous uptake to particles is described by Eq. (1), where is the average molecular velocity of N2O5, A is the particle surface area concentration and γ is the uptake coefficient.
The uptake coefficient, γ, has been characterized in several laboratory investigations (see Bertram and Thornton, 2009; Chang et al., 2011; Ammann et al., 2013, for summaries) and in numerous field studies where it has been found to be highly variable (between and 0.11) and dependent on temperature, relative humidity (RH), and aerosol composition (Brown et al., 2006, 2009, 2016; Bertram et al., 2009; Riedel et al., 2012b; Wagner et al., 2013; Morgan et al., 2015; Phillips et al., 2016). A value of ≈0.03 has been derived from measurements in the polluted marine environment (Aldener et al., 2006).
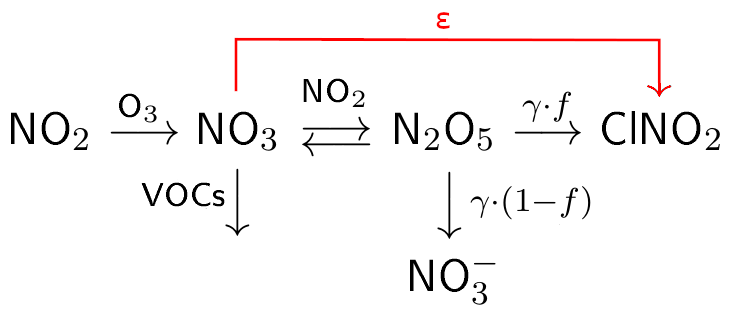
Figure 1Simplified scheme of chemical reactions and parameters involved in the formation of ClNO2. The ClNO2 production efficiency per NO3 formed is denoted as ε. The uptake coefficient to the particle phase is represented by γ and the ClNO2 yield (per N2O5 taken up) by f.
The ClNO2 yield, f, which controls the relative formation rates of and ClNO2 in Reaction (R1), is determined by the [Cl−]-to-[H2O] ratio in the aerosol phase (Behnke et al., 1997; Bertram and Thornton, 2009; Ammann et al., 2013), and can vary between zero and unity (Thornton et al., 2010; Wagner et al., 2012; Riedel et al., 2013; Phillips et al., 2016; Wang et al., 2016; McDuffie et al., 2018b). In Fig. 1 we introduce the ClNO2 production efficiency ε, which is the yield of ClNO2 per NO3 molecule formed in Reaction (R3) and will be discussed in detail in Sect. 3.2.
The established method to measure atmospheric ClNO2 mixing ratios from a few tens of parts per trillion by volume to several parts per billion by volume is chemical ionization mass spectrometry (CIMS) using iodide ions to generate , which can be detected at a mass-to-charge ratio (m∕z) of 208 and 210 (McNeill et al., 2006). The first measurement highlighting the importance of ClNO2 in the polluted marine boundary layer was performed by Osthoff et al. (2008), who detected mixing ratios exceeding 1 ppbv along the coast of Houston, Texas, originating from ship plumes and urban and industrial NOx sources. This was the starting point for numerous measurements of ClNO2 in various locations around the globe with an initial focus on coastal areas in the United States (US), e.g. the Los Angeles Basin in California (Riedel et al., 2012a; Wagner et al., 2012; Young et al., 2012). Other studies included coastal sites in Canada (Osthoff et al., 2018) and coastal/urban sites in the United Kingdom (Bannan et al., 2015, 2017; Priestley et al., 2018; Sommariva et al., 2018). Whereas ClNO2 was initially believed to play a significant role only in areas with marine influence (Behnke et al., 1997; Keene et al., 1999), mid-continental measurements in the US (Thornton et al., 2010; Riedel et al., 2013; Faxon et al., 2015) revealed the importance of anthropogenic sources (e.g. industrial combustion, cooling towers, natural gas extraction, and suspension of road salt) and sea salt chloride transported inland. Further studies reported significant mixing ratios of ClNO2 at a semi-rural site in continental Germany (Phillips et al., 2012, 2016) and at a mid-continental urban site in Canada (Mielke et al., 2011, 2016). Observations at continental sites could be reproduced by a global model (Wang et al., 2019) when considering the transport of HCl (aq), which had been initially formed in the gas phase through acid displacement in coastal regions. More recently, ClNO2 at the > 1 ppbv level has been observed in the heavily industrialized North China Plain (Tham et al., 2016, 2018; Liu et al., 2017; Wang et al., 2017), with even larger mixing ratios measured in Beijing (Le Breton et al., 2018; Zhou et al., 2018) and Hong Kong (Wang et al., 2016).
The great variability seen in ClNO2 mixing ratios in different locations reflects regional variability in its efficiency of production, which, as described above involves a complex set of chemical reactions, in both the gas and particle phases and which will vary over time and space. Most measurements of ClNO2 to date have been measurements at single locations, though some data from mobile platforms such as aircraft (Mielke et al., 2013; Lee et al., 2018; McDuffie et al., 2018a, b) and ships (Kercher et al., 2009; Riedel et al., 2012a) are available. With respect to understanding the formation and role of ClNO2, much of the atmospheric boundary layer remains unexplored.
Here we present shipborne measurements of ClNO2 in the marine boundary layer of the Mediterranean Sea and around the Arabian Peninsula, including the Red Sea and the Arabian Gulf. With a ship track from southern France to Kuwait we provide a unique marine ClNO2 dataset with a large spatial coverage. This allows us to investigate the ClNO2 production efficiency ε and its regional impact under various atmospheric conditions ranging from polluted marine and coastal environments to low-NOx conditions in chemically aged air masses.
2.1 AQABA campaign
The measurements presented in this study were performed during the AQABA campaign, which took place along the sea route between southern France and Kuwait in summer 2017. Five air-conditioned measurement containers with a variety of gas-phase and aerosol instrumentation were set up on board the research vessel Kommandor Iona, which departed from southern France on 24 June 2017 and passed various regions including the Mediterranean Sea, the Suez Canal, the Red Sea, the Gulf of Aden, the Arabian Sea, the Gulf of Oman, and the Arabian Gulf (see Fig. 2), reaching its destination Kuwait on 31 July 2017 (first leg) and covering a latitude–longitude span of 12–43∘ N and 6–60∘ E. After a short break in Kuwait the ship returned via the same route to southern France, arriving on 2 September 2017 (second leg). The trace gases described in this paper were sampled from the centre of a common, high-volume-flow inlet (10 m3 min−1, 0.15 m in diameter, 0.2 s residence time) made of stainless steel, which was located on a measurement container at the front of the ship at a height of approximately 5.5 m above the foredeck.
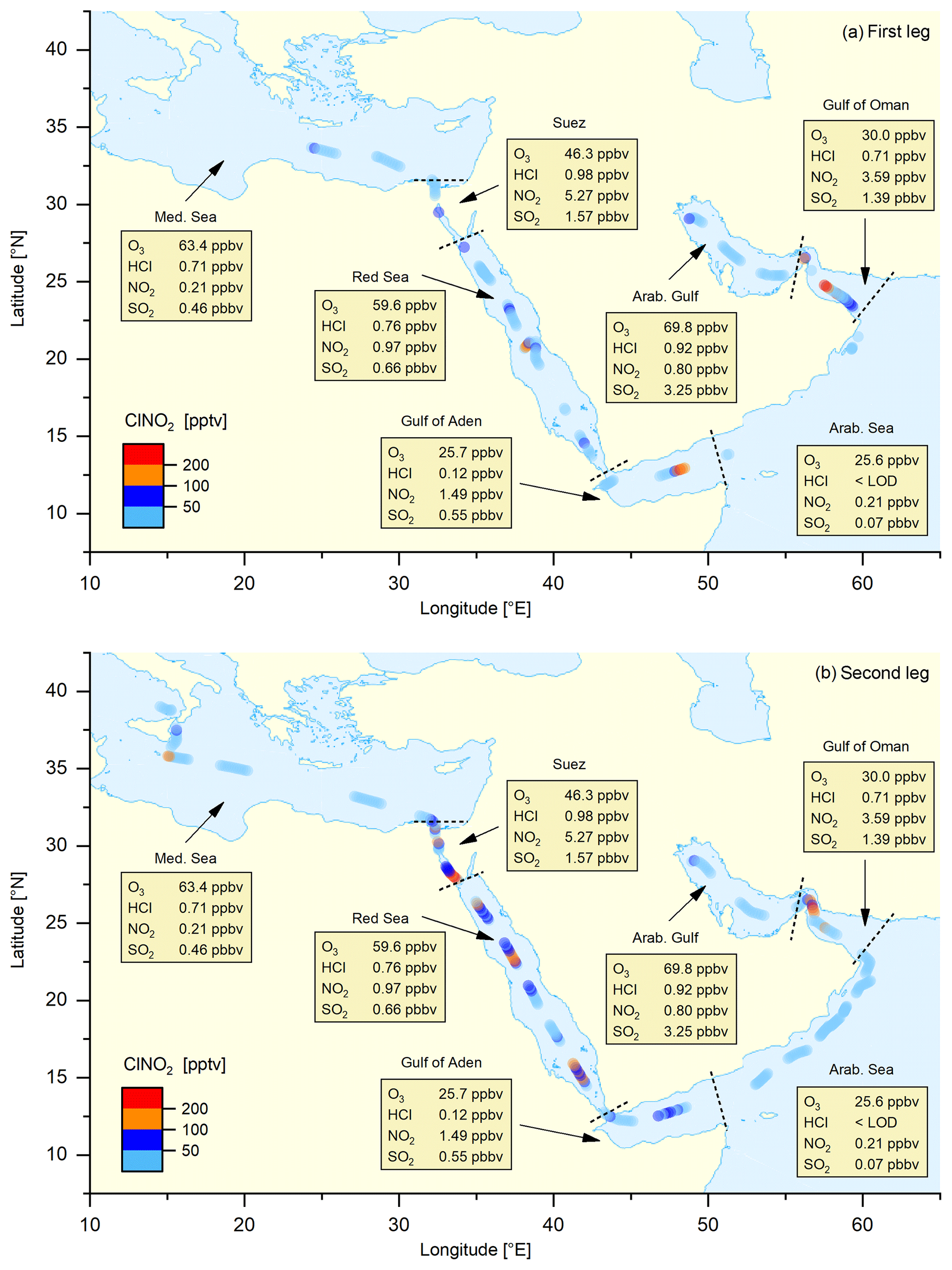
Figure 2Map of nocturnal ClNO2 mixing ratios (1 h averages) for the (a) first and (b) second legs, together with (median) night-time mixing ratios of O3, HCl, NO2, and SO2 for different regions (data from both legs combined) demarked by the dashed lines.
Depending on the wind direction relative to the movement of the vessel, measurements were occasionally impacted by emissions from the stack of our own ship. Especially on the first leg, the relative wind direction was frequently from behind where the chimney was located. All datasets were filtered prior to analysis for periods where the measurements were contaminated by stack emissions to avoid a potential bias in the results. The filter is based on short-term variation in NO and SO2 signals and relative wind direction and reduces the useful data coverage to 58 % on the first leg and 95 % on the second leg.
2.2 Measurement of ClNO2, HCl, and SO2
Nitryl chloride (ClNO2), hydrogen chloride (HCl), and sulfur dioxide (SO2) were detected with a chemical ionization quadrupole mass spectrometer (CI-QMS) using an electrical, radio-frequency (RF) discharge ion source. The instrument and the ion–molecule reactions involved in the detection of the above-mentioned trace gases are described in detail by Eger et al. (2019). Briefly, ClNO2 was monitored as at a mass-to-charge ratio (m∕z) of 208 and 210 subsequent to the reaction of ClNO2 with I− (McNeill et al., 2006; Osthoff et al., 2008; Thornton et al., 2010). is more specific than ICl− (m∕z 162 and 164) and has a lower background signal, providing a sensitivity of 0.61 Hz pptv−1 per 106 Hz of I− at m∕z 208 (and 0.20 Hz pptv−1 at m∕z 210), a limit of detection (LOD) (2σ, 5 min) of 12 pptv, and a total measurement uncertainty of 30 % ± 6 pptv. We chose the signal at m∕z 208 for its higher signal-to-noise (S∕N) ratio to calculate the ClNO2 mixing ratios reported. For the whole campaign dataset, the ratio between m∕z 208 and 210 was 3.08 (with R2 = 0.96; see Fig. S1a of the Supplement), which is very close to the expected value of 3.13 derived from the natural abundance of the 35Cl and 37Cl isotopes. In addition, correlation plots from different regions (Fig. S1b–f) indicate the absence of significant interferences at either of the two m∕z.
HCl was observed as I(CN)Cl− (m∕z 188 and 190) (Eger et al., 2019) with a sensitivity of 0.17 Hz pptv−1 per 106 Hz of I− at m∕z 188 (and 0.05 Hz pptv−1 at m∕z 190), a detection limit of 98 pptv, and a total measurement uncertainty of 20 % ± 72 pptv. As m∕z 190 suffers from known interferences (e.g. ) and has a lower S∕N ratio, we used m∕z 188 to calculate the HCl mixing ratios reported. SO2 was detected as (m∕z 207) with a sensitivity of 0.10 Hz pptv−1 per 106 Hz of I−, a detection limit of 38 pptv and a total uncertainty of 20 % ± 23 pptv.
A flow of 2.5 slm (standard litres per minute) was drawn into the CI-QMS instrument via a ≈3 m long 6.35 mm (OD) PFA tubing while a 20 cm section of the inlet line in front of the IMR (ion–molecule reactor) was heated to 200 ∘C to enable detection of peroxyacetyl nitrate (PAN), which is not reported here. The IMR region was held at a pressure of (18.00±0.05) mbar by a dry vacuum scroll pump. The background signal was determined by periodically bypassing ambient air through a scrubber filled with steel wool where the trace gases of interest are efficiently destroyed at the hot surfaces (120 ∘C). To avoid condensation of water in the inlet lines in the containers, the pressure in the sampling line was reduced to ≈700–800 mbar with a bypass flow of ≈5 slm and by including an additional ≈50 cm long (coiled) piece of 3.18 mm (OD) PFA tubing. A 2 µm pore size membrane filter (Pall Teflo) was placed between the high-volume-flow inlet and CI-QMS sampling line to remove particles and was exchanged regularly to avoid accumulation of particulate matter. No indication for ClNO2 formation via N2O5 reactions on salty surfaces in the inlet line was observed during AQABA; i.e. whenever we changed the particle filter or the inlet line, no change in signal was observed. Further, the ClNO2-to-N2O5 ratio was highly variable during AQABA (range of 0.35–59 with a median of 3.2) and ClNO2 was occasionally measured in periods where no N2O5 was present.
ClNO2 was calibrated twice during the campaign by simultaneously sampling a source of ClNO2 via the CI-QMS and by a thermal dissociation cavity ring-down spectrometer (Sobanski et al., 2016). ClNO2 was generated by passing Cl2 over NaNO2 as described previously (Thaler et al., 2011; Eger et al., 2019). HCl was calibrated four times throughout the campaign by adding a small flow over a permeation source to the main flow and monitoring the CI-QMS signal at m∕z 188 and 190. SO2 calibrations were performed seven times during the AQABA campaign by addition of a known flow of SO2 from a gas cylinder (1 ppmv in synthetic air, Air Liquide). In contrast to ClNO2 and HCl, correction of the SO2 signal for its relative humidity (RH) dependence was necessary, which we derived from calibrations during AQABA where the RH was actively varied between 1 % and 80 %.
The CI-QMS was operated in selected ion monitoring mode measuring mainly ClNO2, HCl, SO2, peroxyacetyl nitrate (PAN), and peracetic–acetic acid with a temporal resolution of approximately 15 s for each molecule. Changes in sensitivity were captured by permanently monitoring the primary ion signal (I− and its water cluster) during ambient measurements and a background signal was recorded every 100 min. For further analysis, all datasets were averaged to 5 min temporal resolution. Our ClNO2, HCl, and SO2 datasets provide about 12 500 data points distributed over 61.4 d with interruptions due to background determinations, calibrations, filter and gas bottle changes, and instrument power-down at the harbours of Jeddah and Kuwait. For periods where the ship was in motion, the data coverage for all three trace gases was about 80 %.
2.2.1 Other trace gases
O3 was measured by a commercial ozone monitor (2B Technologies, model 202) based on optical absorption at 254 nm with a detection limit of 3 ppbv (10 s) and a total uncertainty of 2 % ± 1 ppbv. Mixing ratios of NOx and NOy (NOy=NOx + reactive nitrogen trace gases + particulate nitrate) were monitored via thermal dissociation cavity ring-down spectroscopy (TD-CRDS) using a modified version of the instrument described by Thieser et al. (2016). The difference between the NOy and the NOx signal is referred to as NOz, which includes organic nitrates (peroxyacetyl nitrates and alkyl nitrates), NO3, N2O5, ClNO2, HNO3, and particulate nitrate. In contrast to Thieser et al. (2016), the TD unit was operated at 850 ∘C to ensure detection of HNO3 and nitrate in the particle phase. The detection limits for NOx and NOy were 80 and 160 pptv, respectively, with total uncertainties of 9 % ± 30 pptv. NOz was calculated from measured NOx and NOy with a detection limit of 160 pptv and a total uncertainty of 13 % ± 42 pptv. NO2 (LOD = 52 pptv (1 s), total uncertainty = 7 %) and N2O5 (LOD = 6 pptv (1 s), total uncertainty = 15 %) were measured by a five-channel TD-CRDS described by Sobanski et al. (2016). NO and NO2 were measured by a chemiluminescence detector (CLD 790 SR, ECO Physics, Dürnten, Switzerland) (Fontijn et al., 1970; Li et al., 2015). The LOD (5 s) was 21 pptv for NO and 52 pptv for NO2 and the total uncertainty was 6 % and 7 %. The NO2 data were in good agreement with the CRDS dataset (R2 = 0.95) with a mean deviation of 6 %. The hydroxyl radical (OH) was measured using a laser-induced-fluorescence method (Martinez et al., 2010; Novelli et al., 2014).
2.2.2 Meteorological parameter and actinic flux
Photolysis rates (JO(1D), and ) were calculated from wavelength resolved actinic flux measured by a spectral radiometer (Metcon GmbH; Meusel et al., 2016) located close to the common trace-gas inlet. Cross sections and quantum yields were taken from Burkholder et al. (2015). J values were not corrected for upwelling UV radiation and are estimated to have an overall uncertainty of ≈10 %. A commercial Neptune weather station (Sterela) monitored various parameters such as temperature, relative humidity, wind speed and direction, speed of the vessel, and GPS position.
2.2.3 Aerosols
An aerosol mass spectrometer (Aerodyne HR-ToF-AMS; DeCarlo et al., 2006) measured PM1 non-refractory aerosol composition (30 s time resolution), including sulfate, nitrate, ammonium, chloride, and total organics with an overall uncertainty of 35 %. An optical particle spectrometer (OPC, Grimm model 1.109) measured the size distribution from 250 nm to 32 µm (6 s time resolution) with a total uncertainty of 25 %. A fast mobility particle spectrometer (FMPS, TSI model 3091) provided particle size distributions from 5.6 nm up to 560 nm (1 s time resolution). The particle surface area concentrations for PM1 and PM10 were calculated from the OPC and FMPS datasets; the overall uncertainty of these variables is estimated to be 30 %. The inlet for the aerosol instrumentation was located at the top of a measurement container at a distance of ≈5 m to the common trace-gas inlet described above. In order to avoid condensation in inlet lines, aerosol samples were passed through a drying system which reduced ambient relative humidity to an average value of ≈40 % in the measurement container. We calculated the ambient PM1 particle surface area concentration (A) from the measured surface area concentration using a hygroscopic growth factor (on average 1.32±0.24) based on ambient RH and aerosol composition. The calculation of the growth factor is described in the Supplement (Fig. S2–S5). The water-soluble fraction of total suspended particles (TSPs) was monitored with hourly resolution using a Monitor for AeRosols and Gases in Ambient Air, MARGA (Metrohm Applikon model S2), sampling at a distance of ≈5 m from the common gas-phase inlet. In this work only results from Na+ and Cl− measurements (TSP), with detection limits equal to 0.05 and 0.01 µg m−3, will be used.
In the following, we use only data (5 min averages) which were free from contamination by the ship's own exhaust (see Sect. 2.1).
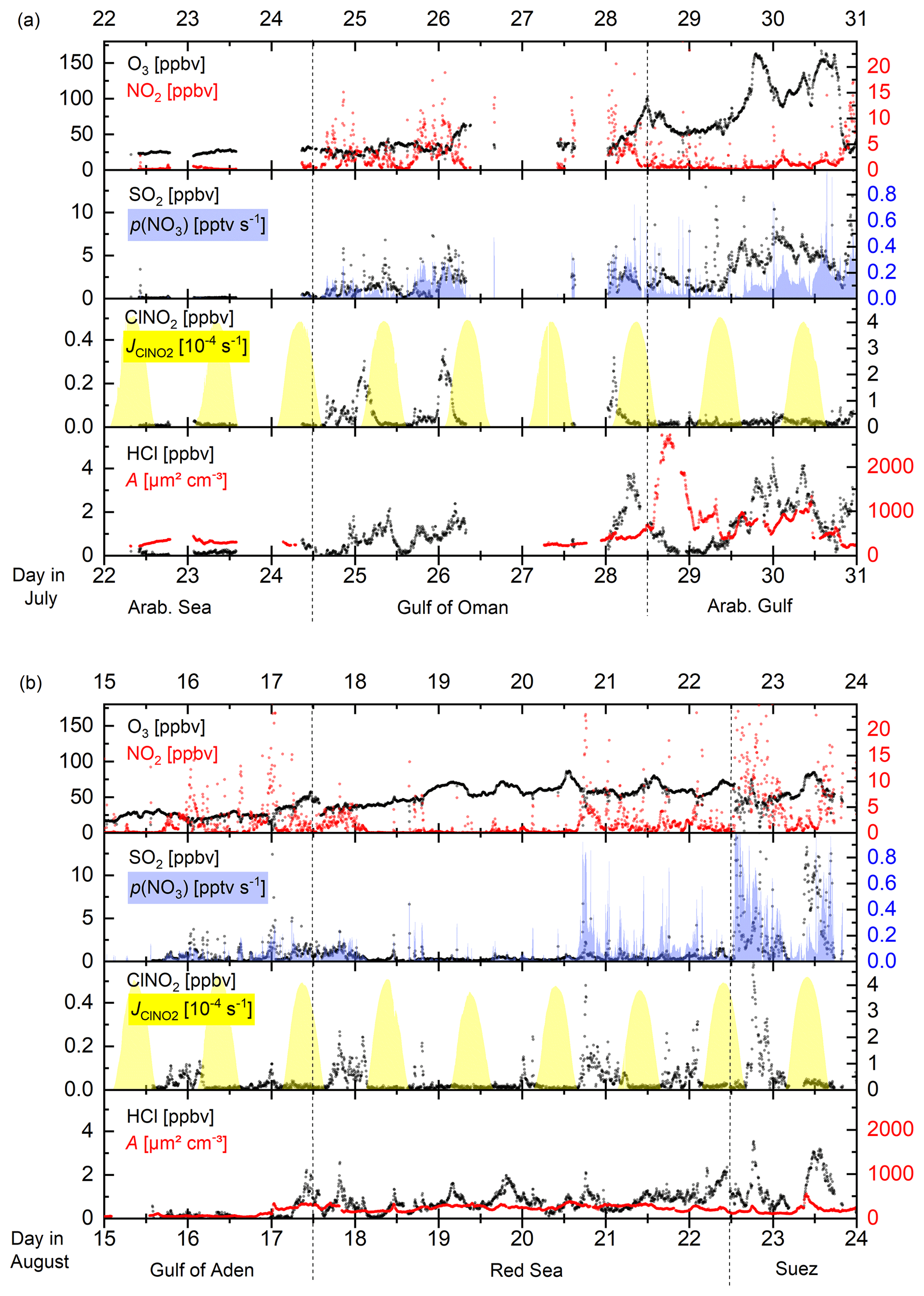
Figure 3Time series of ClNO2 and trace gases related to its production (NO2 and O3) as well as the NO3 production rate, p(NO3), ClNO2 photolysis rate (), PM1 particle surface area concentration (A), and HCl mixing ratio in different regions (separated by the dashed lines) for 9 d periods in the (a) first and (b) second legs.
3.1 Overview of measurement regions and ClNO2 mixing ratios observed
Figure 2 illustrates the ship's track during AQABA, divided into seven regions demarked by dashed black lines: the Mediterranean Sea, the Suez Canal including the Gulf of Suez, the Red Sea, the Gulf of Aden, the Arabian Sea, the Gulf of Oman, and the Arabian Gulf. On the first leg, the CI-QMS measurements started south of Crete; on the second leg measurements terminated close to Sicily after 2 months of almost continuous measurement. Maximum ClNO2 mixing ratios observed during each night ranged from the limit of detection to 586 pptv (see Fig. S6 for details). Figure 2 shows 1 h averaged ClNO2 mixing ratios along the ship track during the (a) first and (b) second legs. Text boxes indicate the median night-time mixing ratios of O3, HCl, NO2, and SO2 for the different regions where data from the first and second leg datasets have been combined. The night-time mean, median, and range of the mixing ratios of these trace gases (and also of NO2, temperature, relative humidity, NO3 production rate, and PM1 particle surface area concentration) are listed in Table 1; a time series of measured ClNO2, HCl, SO2, and O3 is provided in Fig. S7 of the Supplement. The predominant air mass origin for each night was derived from 48 h back trajectories calculated with HYSPLIT (Stein et al., 2015; Rolph et al., 2017) and is illustrated for both legs in Fig. S8 of the Supplement. While Fig. 2 provides an overview of the measurements during both legs, Fig. 3a and b highlight 9 d periods indicating features that characterized the transition from one region to the next. Based on these three figures, we will discuss observed ClNO2 mixing ratios and related parameters for the seven regions defined above.
Table 1Measured trace gases and other parameters (5 min, night-time only) for the different regions indicated in Fig. 2.
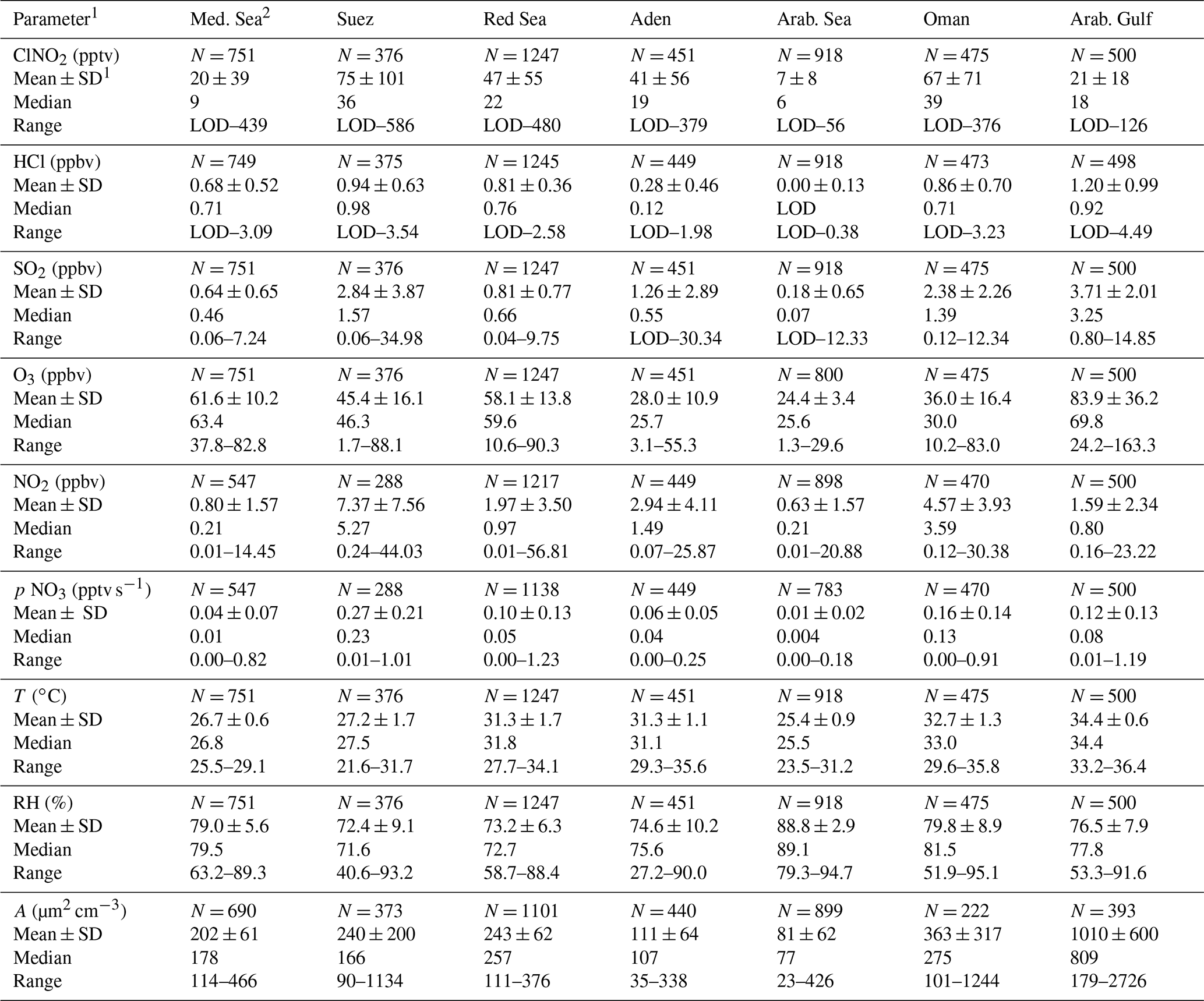
Notes: 1 Parameters are as follows: SD: standard deviation; RH: relative humidity; A: ambient PM1 particle surface area concentration (see Sect. 2.5); p denotes a production rate; N: number of data points; LOD: limit of detection (see Sect. 2.2). 2 Regions: Mediterranean Sea (Med. Sea): 30 June–1 July and 24–30 August. Suez Canal and Gulf of Suez (Suez): 2–3 July and 22–23 August. Red Sea: 3–15 July and 17–21 August. Gulf of Aden (Aden): 16–18 July and 15–16 August. Arabian Sea (Arab. Sea): 19–23 July and 7–14 August. Gulf of Oman (Oman): 24–27 July and 5–6 August. Arabian Gulf (Arab. Gulf): 28 July–4 August 2017.
Over the Mediterranean Sea, during periods when the CIMS was operational, we encountered mainly aged air masses which had passed over Italy, Greece, or Turkey (Fig. S8) characterized by relatively high O3 levels but low NO2-to-NOy ratios. As illustrated in Fig. 1 the formation of ClNO2 is initiated by NO3 production which will depend on O3 levels and availability of NO2. For the Mediterranean Sea the low NO2 mixing ratios resulted in a weak NO3 production term (Table 1) and low ClNO2 mixing ratios. The only exceptions are two nights south of Sicily on the second leg where ClNO2 mixing ratios up to 439 pptv were observed, which coincided with an increase in NO2 originating from industrial sources on the mainland (Sicily and Italy). There are no previous measurements of ClNO2 over the Mediterranean Sea but our data can be compared to the output of a regional model (Li et al., 2019), which predicts monthly average ClNO2 mixing ratios up to 100 pptv in the south-eastern Mediterranean Sea and around Sicily, in broad agreement with our observations.
The Suez Canal and the Gulf of Suez were impacted by fresh emissions from ships, industry, and urban centres with high NO2, SO2, HCl, and ClNO2 mixing ratios. On the night of 22–23 August 2017 we measured the highest ClNO2 mixing ratio of the whole campaign (586 pptv) due to exceptionally high NO2 levels and NO3 production rates.
Over the Red Sea, O3 levels were elevated with the highest NO3 production rates (up to 0.7 pptv s−1) observed when approaching the Gulf of Suez with NOx transported south from the region around the Suez Canal, the city of Cairo, and the Sinai Peninsula (see back trajectories in Fig. S8). ClNO2 mixing ratios exceeded 200 pptv on most of the nights (with a maximum of 480 pptv) whereby elevated PM1 particle surface area concentration and HCl mixing ratios indicated increased heterogeneous uptake and chloride availability.
Over the Gulf of Aden, with air mainly originating from Somalia, O3 levels were close to 25 ppbv and NO2 mixing ratios regularly exceeding 5 ppbv resulted in a NO3 production rate up to ≈0.2 pptv s−1. However, the PM1 particle surface area concentration remained low and ClNO2 was detected only occasionally (maximum of 379 pptv).
Over the Arabian Sea we experienced strong winds from the south with 48 h back trajectories touching the coast of Somalia. ClNO2 was generally below the detection limit (maximum 56 pptv) and NO2, HCl, and PM1 particle surface area concentrations were very low. Missing local sources of NOx and low O3 mixing ratios resulted in a weak NO3 production term, partially responsible for the lack of ClNO2. Low mixing ratios of ClNO2 were occasionally detected that originated from single ships or point sources on the mainland.
Upon entering the Gulf of Oman, which marks the transition between remote marine environment and increased emissions from petrochemical industry and shipping lanes, NO3 production rates increased significantly due to higher NOx and O3 levels. ClNO2 mixing ratios exceeded 200 pptv during two consecutive nights with a maximum value in this region of 376 pptv.
The Arabian Gulf was characterized by very high ozone levels (sometimes exceeding 150 ppbv) and SO2 mixing ratios that generally exceeded 5 ppbv. For the first leg (sailing into the Arabian Gulf), the air mass passed over Kuwait whereas for the second leg (sailing out of the Arabian Gulf) it mainly passed over Iran. In the gulf region, which was heavily polluted by emissions from shipping and the petrochemical industry, we also observed the highest HCl and PM1 particle surface area concentration throughout the whole campaign. However, despite high NO3 production rates (≈0.4 pptv s−1) due to NOx emissions from oil and gas refineries as well as emissions from shipping and urban areas, we only observed relatively low ClNO2 mixing ratios with a maximum value of 126 pptv close to Kuwait.
Consistent with Osthoff et al. (2008) we find significant amounts of nocturnal ClNO2 in aged ship plumes that could be identified by a defined peak shape (Fig. S9) and covariance of NO2 and SO2 indicative of upwind point sources. As SO2 and NO2 are co-emitted from the combustion of ship fuel, it is not surprising that they show a co-variance. The consequence of the co-emission of NO2 and SO2 is that ClNO2 is generally observed in the presence of both whereby high ClNO2 mixing ratios were associated with aged ship plumes.
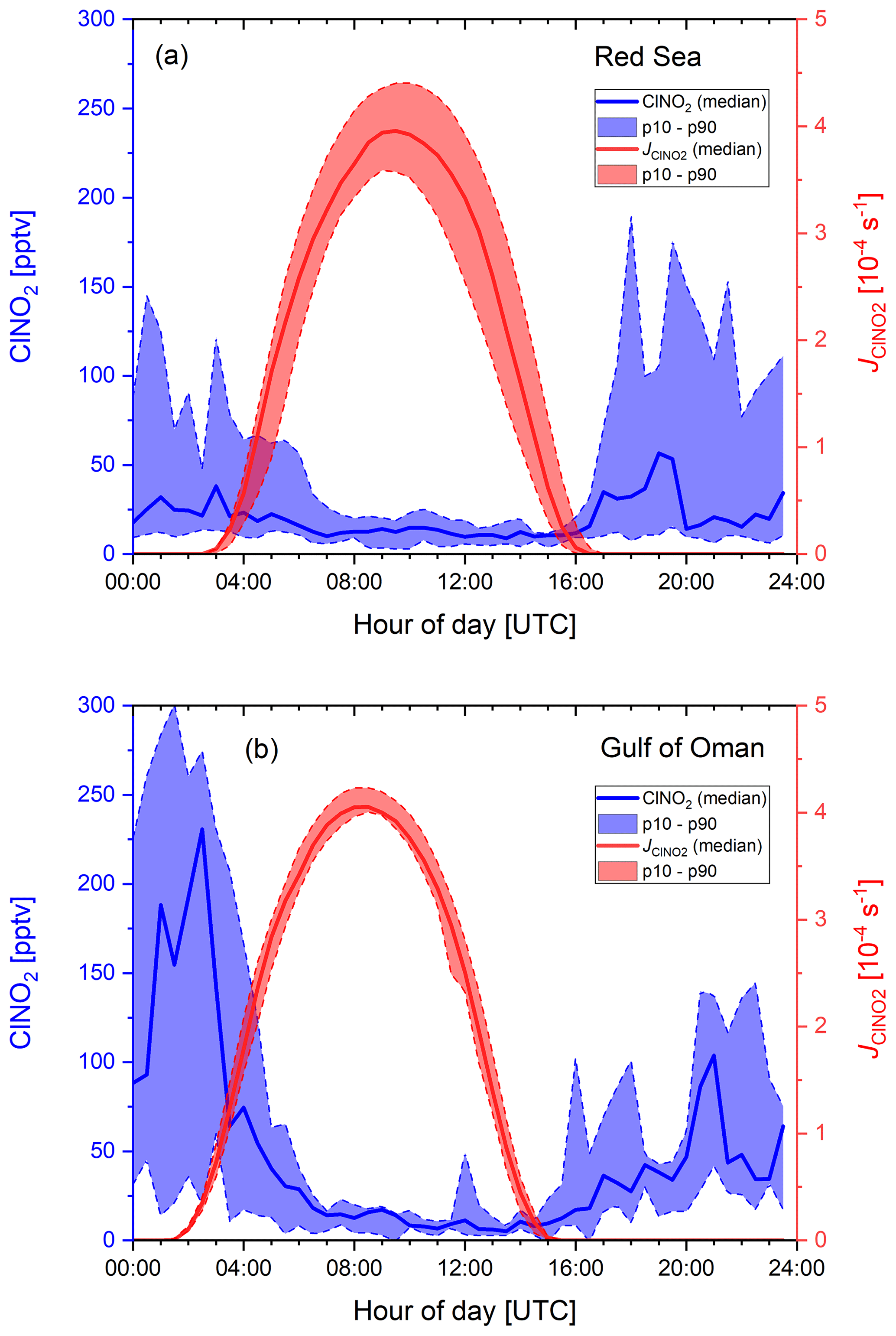
Figure 4Diurnal profiles of ClNO2 and its photolysis rate constant, , for (a) the Red Sea and (b) the Gulf of Oman. The solid lines represent the median values, the shaded areas correspond to the 10th and 90th percentiles.
Figure 4 shows diel profiles of nitryl chloride for the Red Sea and the Gulf of Oman together with the photolysis rate constant illustrating that mixing ratios generally decreased at sunrise with a ClNO2 lifetime of a few hours. Diel ClNO2 profiles for other regions (see Fig. S10 of the Supplement) look generally similar but with a varying maximum mixing ratio. Over the Red Sea ClNO2 was often observed in plumes, whereas mixing ratios over the Gulf of Oman increased continuously after sunset, indicating that we sampled a more homogeneously polluted air mass in which ClNO2 accumulated over the course of the night. The median mixing ratio in the afternoon was still around 10 pptv, which we attribute to an HCl interference at m∕z 208 and 210 described by Eger et al. (2019) rather than to the presence of ClNO2 during the day when its production rate is close to zero and its lifetime is short due to photolysis. The magnitude of the HCl interference at the m∕z used to monitor ClNO2 was derived during HCl calibrations on board the ship and found to be 0.006 Hz (pptv of HCl)−1, which is about 1 % of the ClNO2 count rate of 0.61 Hz (pptv of ClNO2)−1 at 106 Hz of I−. However, during ambient air measurements the interfering signal was variable with a campaign average of (0.008±0.005) Hz pptv−1, which implies that a correction based on the HCl signal alone is not sufficient. The variable offset at the ClNO2 mass contributes to the total measurement uncertainty and can be significant when analysing data close to the detection limit. Although on occasions several hundred parts per trillion by volume of ClNO2 were observed, below we show that the ClNO2 production efficiency was generally low. Reasons for this are examined in the following sections.
3.2 ClNO2 yield per NO3 molecule formed
We define the ClNO2 production efficiency (ε) during AQABA as the number of ClNO2 molecules generated per NO3 molecule formed from the reaction of NO2 with O3 (Reaction R3). The instantaneous production rate of NO3 is given by k1[NO2][O3] and the total number of NO3 molecules formed over the course of the night is derived using a rate coefficient of k1=1.4 × 10−13 exp () cm3 molecule−1 s−1 (IUPAC, 2019) and integrating the NO3 production term from the beginning of the night (t0) to the time of the measurement (t) according to Eq. (2). In this calculation we assume that [NO2] changes over time but [O3] stays constant in good approximation.
In order to account for the pre-sunset production of NO3 at high solar zenith angles where N2O5 could already be detected, t0 was defined as the point in time at which was below 0.017 s−1 (about 10 % of maximum value during day). This was typically 30–50 min prior to sunset. All data points before sunset were however excluded from the analysis due to the increased uncertainty in the reaction time. The NO2 mixing ratio at the beginning of the night, [NO2]0, was derived from the measured NO2 mixing ratio at time t via Eq. (3) by assuming that NO2 had been consumed by reaction with O3 but the O3 mixing ratio did not change significantly. Consequently, the amount of NO3 produced along the air mass trajectory is equal to the difference between calculated [NO2]0 and measured [NO2] (t).
The ClNO2 production efficiency ε can be determined by inserting the integrated NO3 production over the course of the night and the measured ClNO2 mixing ratio (assuming no losses) into Eq. (4).
To account for fresh emissions of NO (e.g. by passing ships), the reaction time t′ was calculated from Eq. (5) according to McDuffie et al. (2018a):
where s represents the number of NO2 molecules required to make NOy and is 1 when NO3 reacts directly with VOCs and 2 when NO3 reacts with NO2 to form N2O5, which subsequently hydrolyses to HNO3. As discussed later, the direct NO3 losses are dominant throughout the campaign compared to the heterogeneous N2O5 production, so to a good approximation, s≈1. As discussed by McDuffie et al. (2018a) inherent to the use of this expression is the assumption that NOy is conserved during the night; any losses of NOy (e.g. via deposition of HNO3) lead to an underestimation of the true reaction time. As the calculated night-time air mass age depends on the ratio between [NO2] and [NOy], the calculation breaks down whenever a fresh NO emission (e.g. from a nearby ship) is injected into an air mass and unreacted NO is still present. To avoid this, we only analyse ClNO2 data when NO is below the detection limit. In addition, we only consider data points where the calculated age of the air mass is equal to or exceeds the time elapsed since sunset as these air masses are unlikely to have been impacted by recent emissions. As the loss of NOz via deposition will result in an air mass age that is shorter than the true one, we relax the criterion for equality of reaction times by also including calculated air mass ages that are up to 25 % shorter (i.e. ). The reduced dataset provides 1742 data points with a median value of ε=2.7 % for the whole campaign. The data reduction is described in more detail in the Supplement (all the data shown in the paper correspond to the application of method C), where the sensitivity of ε to these limitations and additional constraints is discussed. Here, we emphasize that even when limiting the dataset to ClNO2 mixing ratios exceeding 100 pptv (see Table S1 in the Supplement), the ClNO2 production efficiency still remains relatively low (ε=6.4 %, Fig. S11). Although the median values would be modified (Fig. S12), the relative differences between the regions and especially the low ε observed over the Arabian Gulf persist.
3.2.1 Temporal and regional variability in ε
To compare the efficiency of ClNO2 formation in different regions, a median value for ε was derived for each individual night with the results illustrated in Fig. 5 in which the size of the circles (first leg) and stars (second leg) scales with the median NO3 production rate (for a more detailed plot with 1 h averaged data points see Fig. S13). Despite high NO3 production rates (high O3 and NO2 levels), the lowest values of ε were observed over the Arabian Gulf, whereas elevated values of ε were found over the Arabian Sea, where NO3 production was lowest.
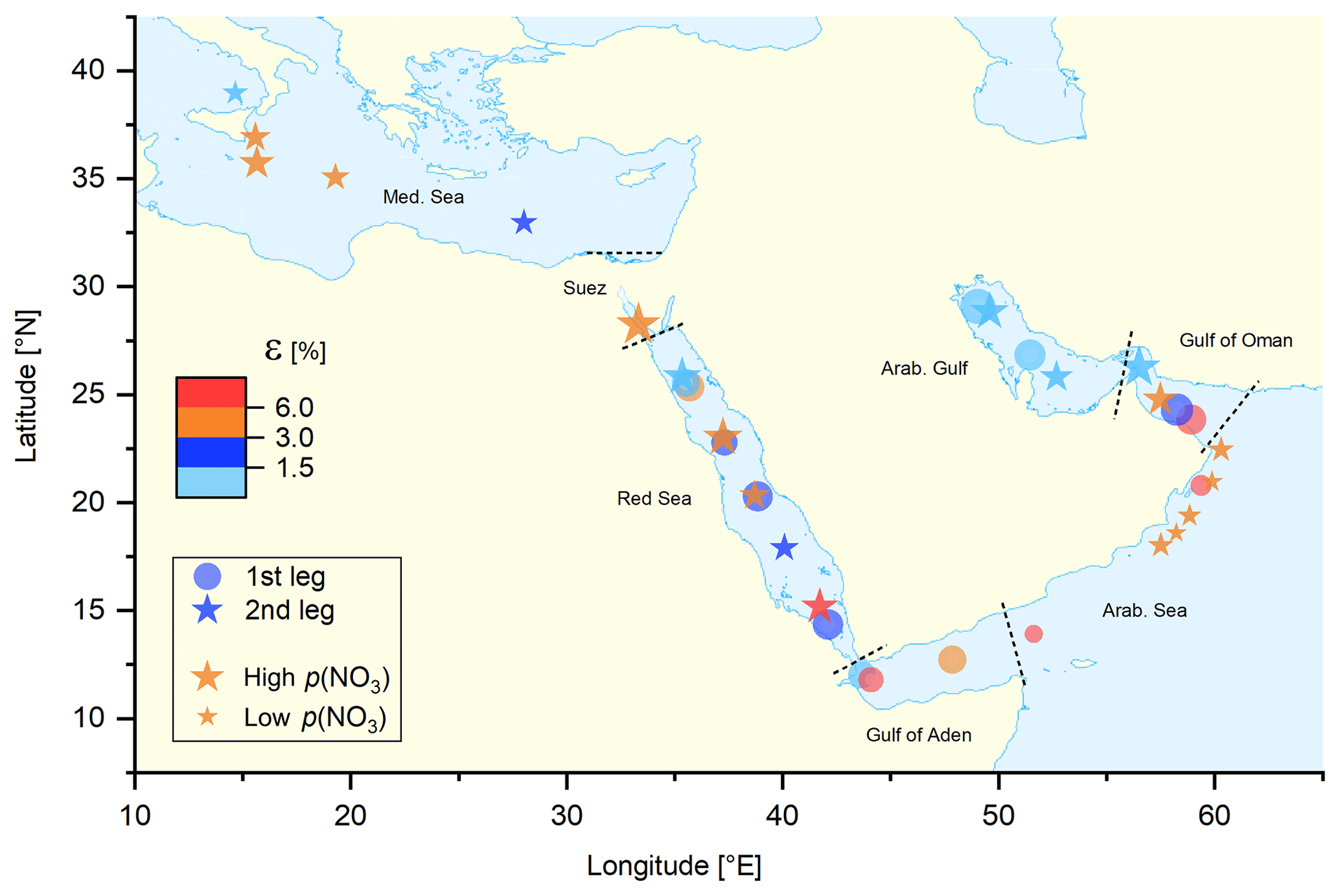
Figure 5Median night-time values of ε (the ClNO2 yield per NO3 molecule formed) calculated via Eq. (4). The size of the symbols represents (logarithmic scale) the median NO3 production rate, p(NO3), ranging from 0.001 pptv s−1 in the Arabian Sea to 1.2 pptv s−1 in the Suez Canal. Different regions are separated by the dashed black lines.
Figure 6 displays box plots of ε for each region, calculated from between 41 and 546 data points per region. The median ClNO2 production efficiency ε displays large night-to-night variability and interregional variability with the highest value found over the Gulf of Aden and the Arabian Sea (median = 4.7 %) and the lowest value found over the Arabian Gulf (median = 0.8 %). Median values of ε (in %) derived for the Mediterranean Sea, the Suez Canal, the Red Sea, and the Gulf of Oman were 2.9, 2.7, 2.1, and 2.0. In the following, we examine the factors that cause the generally low efficiency in ClNO2 production and also the regional variability in ε.

Figure 6Box plots of ε (ClNO2 production efficiency) for different regions, based on all corresponding individual night-time values calculated from Eq. (4).
3.3 Factors influencing the ClNO2 production efficiency
The uptake of N2O5 to aerosol particles can proceed via hydrolysis to HNO3 as well as formation of ClNO2, with yield f (see Fig. 1). Assuming no night-time losses, the concentration of ClNO2 is given by Eq. (6). The overall ClNO2 production efficiency ε as derived in Sect. 3.2 is dependent on f and on the relative rates of direct NO3 loss (kdir, Eq. 7) and indirect NO3 loss (khet, Eq. 8), where A is the particle surface area concentration, is the mean molecular velocity of N2O5 ((24 400±160) cm s−1 during AQABA), and cm3 molecule−1 (IUPAC, 2019) is the temperature-dependent equilibrium constant (Reactions R4 and R5). During night-time, kdir is determined by the NO3 reactivity towards VOCs and NO and khet by the rate of heterogeneous uptake of N2O5 to aerosol particles. In the following we calculate f and γ from our measurements, compare the values with the literature, and quantify the contributions of khet and kdir to the overall NO3 loss rate.
If no particulate chloride is available, f is zero and two ions are produced according to Reaction (R1), whereas if the particulate chloride concentration is large, fapproaches unity and one anion plus one ClNO2 molecule are formed. As particulate nitrate () can leave the particle as HNO3 we can write
where p signifies a production rate. Equation (9) assumes that N2O5 uptake is the sole mechanism for the night-time production of HNO3. In principal, as demonstrated by Phillips et al. (2016), f can be derived from field data using measured production rates (or concentrations) of inorganic nitrate () and ClNO2 according to Eq. (9). For AQABA, we derived the mixing ratio of total inorganic nitrate from measurements of NOz (corrected for ClNO2 and N2O5) assuming that, for this marine environment the contribution of organic nitrate in both gas and particle phases is small compared to inorganic nitrate. f could be derived using Eq. (9) whenever there was a significant correlation (over a period of several hours) between ClNO2 and inorganic nitrate, as illustrated in Fig. S14 for data obtained in the Gulf of Oman (25–26 July 2017) for which . The spatially and temporally variable sources of pollution during AQABA meant that requirement of a homogeneous fetch over periods of hours was rarely fulfilled and only a handful of values for f could be derived this way. Further values derived are 0.42±0.06 (Red Sea, 15 July 2017), 0.84±0.09 (Gulf of Oman, 24–25 July 2017), and 0.65±0.05 (Mediterranean Sea, 29 August 2017), indicating that the values of f were generally large whenever ClNO2 was observed.
f can also be calculated (Eq. 10) if the relative concentrations of particulate chloride and water are known (Behnke et al., 1997; Bertram and Thornton, 2009):
where is the ratio of rate constants (Ammann et al., 2013) for reaction of (formed along with upon dissociation of N2O5 in the aqueous phase) with either Cl− or H2O. The aerosol liquid water content [H2O] and chloride ion concentration [Cl−] were calculated using the E-AIM model (http://www.aim.env.uea.ac.uk/aim/model4/model4a.php, last access: 26 September 2019) (Clegg et al., 1998; Friese and Ebel, 2010) using the ambient temperature and relative humidity along with PM1 nitrate, sulfate, ammonium, and chloride mass concentrations (µg m−3) as reported by the AMS. From the median aerosol composition (PM1) in the seven different regions we calculated median values of f, which are shown in Table 2. The values of f obtained via Eq. (10) were variable between regions, with medians of 0.53 in the Mediterranean Sea, 0.90 in the Suez Canal, 0.86 in the Red Sea, 0.76 in the Gulf of Aden, 0.87 in the Arabian Sea, 0.50 in the Gulf of Oman, and 0.17 in the Arabian Gulf. To put these numbers in context, a value of f=0.9 corresponds to a ≈1.1 mol L−1 Cl− solution. MARGA measurements of Cl− and Na+ (total suspended particles, TSPs) also indicated sea salt concentrations up to 20 µg m−3 in the coarse mode over the Arabian Sea. A comparison of the [Cl−] (TSP) from the MARGA (which detects NaCl as well as NH4Cl) with the (largely) non-refractory [Cl−] reported by the AMS reveals a strong co-variance. The AMS concentrations (PM1) were on average ≈1 % of those reported by the MARGA (TSP) with the correlation between them indicating that the AMS chloride (PM1) is mainly due to sea salt rather than NH4Cl. If we assume that ≈10 % of the sea salt mass (TSP) is associated with the fine mode (PM1) as previously derived (Sommariva et al., 2018), we can use the MARGA (TSP) [Cl−] to estimate that the true PM1 [Cl−] would be about an order of magnitude higher than measured by the AMS. Under the assumption that this is true, f is > 0.67 in all seven regions, implying that a lack of Cl− is not the reason for low values of ε, as may be expected for a marine environment.
Table 2Regional variability in ClNO2 production efficiency (ε), ClNO2 yield (f), N2O5 uptake coefficient (γ), and heterogeneous NO3 loss rate (khet).
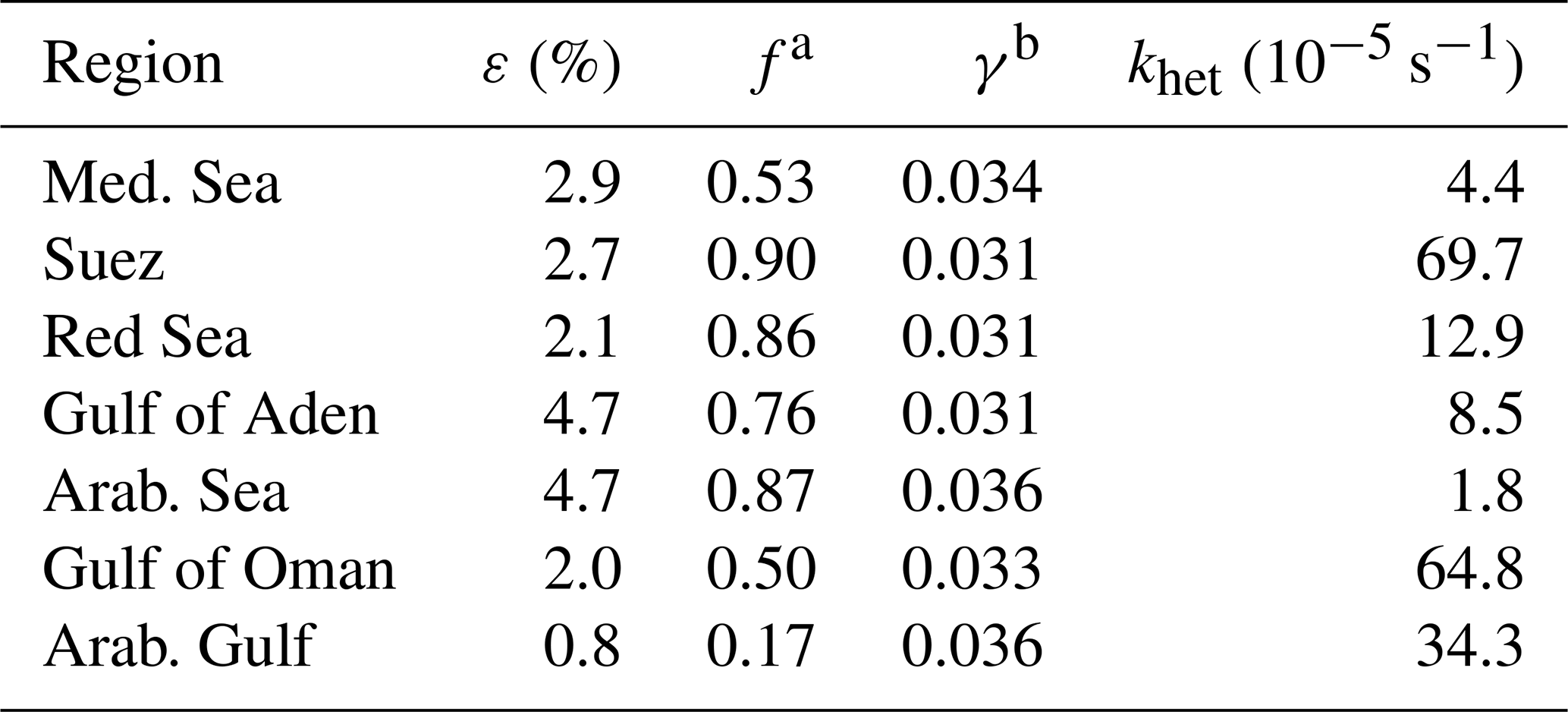
Notes: a Calculated from Eq. (10). b Calculated from Eq. (11).
Previous derivations of f in a marine environment (Texas coast) yield values between 0.1 and 0.65 (Osthoff et al., 2008) whereas even larger values (up to 0.9) have been reported for inland sites impacted by anthropogenic emissions (Young et al., 2013) or by long-range transport of sea salt (0.035<f<1) (Phillips et al., 2016). A median value of 0.138 (0.003<f<1) was derived for airborne measurements in a coastal region during winter (McDuffie et al., 2018b).
The uptake coefficient, γ, can be estimated using the parameterization in Eq. (11) (Bertram and Thornton, 2009):
where B=3.2 × 10−8 s, k=1.15 × 106−1.15 × 106 exp(−0.13 [H2O(l)]) s−1 is the rate constant for the reaction N2O5(aq) + H2O(l), a=0.06 denotes the ratio of rate constants for reactions (aq) + H2O(l) and + (aq), and b=29 denotes the ratio of rate constants for reactions and . Using AMS data for [] and [Cl−] we derive where the standard deviation encompasses the weak inter-regional variation (see Table 2). This value is consistent with reported by Aldener et al. (2006) for a polluted marine environment. The large values of γ reflect the low PM1 particulate nitrate concentrations observed during AQABA where the high temperatures favour the partitioning of particulate nitrate and HNO3(g) to the gas phase. A suppression of γ through the presence of organics in the particle phase has been reported (Bertram et al., 2009), though the low (generally < 2) organic-to-sulfate ratio in particles observed during AQABA suggests that this is not likely to be important for the present analysis.
The campaign-averaged fractional contribution of coarse-mode particles (PM10–PM1) to the total particle surface area concentration (A) was only (14±14) % so that the uncertainty incurred when using the PM1 particle surface area concentration to derive the heterogeneous NO3 loss rate (khet, Table 2) from Eq. (8) is negligible. However, during two periods (of 2–5 d duration) when the ship was sailing through the Gulf of Aden, Arabian Sea, and southern Red Sea and air masses originated from the deserts of Eritrea, Djibouti, and Ethiopia, the contribution from the coarse-mode particles to the aerosol surface area increased to about 60 % mainly due to dust (and sea salt). The relative contribution of dust and sea salt to the coarse mode was estimated using MARGA measurements of Ca2+ and Na+ in TSP. The dust loading was calculated by assuming that the dust aerosols of Saharan origin consist of 10 % calcium (Molinaroli et al., 1993). Freshly generated dust particles do not contain chloride and the uptake of N2O5 to them ( for Saharan dust, Tang et al., 2012) does not result in ClNO2 formation (f=0) but contributes to khet through the additional aerosol surface area, thus lowering the ClNO2 production efficiency. In contrast, the uptake of N2O5 to coarse-mode sea salt particles has a ClNO2 yield f close to unity (Ammann et al., 2013), and can therefore enhance formation of ClNO2. Throughout the whole campaign, the contribution of dust to the uptake of N2O5 to coarse-mode particles was much larger than that of sea salt (on average (13±10) % with a range of 0 %–40 %). A close examination of the periods (14–19 July and 15–17 August 2017) strongly influenced by dust particles did not reveal any (anti)correlation between elevated dust concentration and ClNO2 mixing ratios. This is also reflected by the relatively high median value of ε over the Gulf of Aden where the highest concentrations of coarse-mode particles were observed.
Using a campaign average of ε=2.7 % and a maximum value of f=1 we can show that, with 97.3 %, the direct (gas-phase) loss of NO3 (kdir) is much more important than indirect losses via N2O5 uptake (khet), which contribute the remaining 2.7 %. Assuming a very conservative estimate of f=0.5 would still result in a contribution of 94.6 and 5.4 %. To put this into context, over the Gulf of Oman we calculated a median value of khet=6.5 × 10−4 s−1 (Table 2), which would result (assuming ε=2.0 % and f=0.5) in a direct NO3 loss term towards VOCs of kdir=1.6 × 10−2 s−1. The relatively small contribution of the heterogeneous loss term is readily explained by high mean night-time temperatures of 25–35 ∘C during AQABA, which favour the existence of NO3 rather than N2O5. To illustrate this, we calculate that a nocturnal temperature of 20 ∘C (rather than 30 ∘C) would increase the contribution of khet to 21 % of the total NO3 loss and lead to 3 times higher ClNO2 mixing ratios assuming that the rate constants for reaction between NO3 and reactive trace gases are not strongly temperature-dependent. A further reduction in temperature to 1 ∘C would lead to equality in khet and kdir and result in a factor of 7 more ClNO2 than observed.
The direct (gas-phase) loss rate of NO3 can also be calculated from Eq. (7) if the concentrations of all VOCs contributing to its reactivity are known. However, for a large fraction of each night NO3 was below the detection limit (ca. 5 pptv) despite a high production rate (large mixing ratios of NO2 and O3). A steady-state analysis of NO3 production and loss indicated a high total reactivity which could not be attributed to measured trace gases (kdir) or heterogeneous losses of N2O5 (khet). A detailed analysis of the NO3 lifetime and the role of VOCs is beyond the scope of the present paper and will be described in detail in a separate publication.
3.3.1 Comparison with literature
In the following, we compare the generally low values of ε derived during AQABA with previous determinations. Mielke et al. (2013) report a yield of ClNO2 relative to the total amount of NO3 formed at night of 0.7 % to 62 % with a median of 12% in the polluted coastal boundary layer in Pasadena, California. In contrast, ClNO2 production efficiencies derived for the urban boundary layer of Calgary, Canada (Mielke et al., 2016), were significantly lower, ranging from 0.1 % to 4.5 % (10th and 90th percentiles, median 1.0 %). Osthoff et al. (2018) report very low efficiencies with a median of 0.17 % and a maximum of 5.4 % for the Lower Fraser Valley of British Columbia, Canada, potentially due to a lack of available aerosol chloride. For AQABA we derive a region-dependent median efficiency of 1 %–5 % with a campaign median of 2.8 %, despite similar conditions to Mielke et al. (2013), i.e. mostly polluted marine environment. The difference can be attributed to exceptionally high nocturnal temperatures during AQABA with a median of 25–35 ∘C for different regions, shifting the equilibrium from N2O5 towards NO3 and favouring direct NO3 losses. For comparison, daily minimum temperatures during the study reported by Mielke et al. (2013) were 10–20 ∘C (Ryerson et al., 2013), whereas the lowest temperature measured during the whole AQABA cruise was 22 ∘C. Based on the ClNO2 dataset reported by Phillips et al. (2012) for continental Germany, where ClNO2 mixing ratios up to 800 pptv were reported, we calculate values of ε that range from 0.4 % to 12.3 % (10th and 90th percentiles) with a median of 2.6 %. On nights where ClNO2 mixing ratios above 100 pptv were observed, range and median increase to 5.0 %–24.1 % and 10.6 %, respectively. Compared to AQABA the yield per NO3 molecule formed on nights where ClNO2 was present at levels > 100 pptv is about a factor of 2 higher for this dataset, again most likely a result of the lower nocturnal temperatures.
3.4 Cl atom generation from ClNO2 and HCl
In this section we assess the role of two gas-phase chlorine reservoirs, ClNO2 and HCl, as sources of Cl atoms during AQABA. Other potential Cl sources (e.g. Cl2 photolysis) are not considered here as we do not have experimental data to quantify their impact. Compared to the complex route to ClNO2 formation described above (Fig. 1), the formation of HCl in the polluted marine environment can be traced back to its displacement from sea salt particles by stronger acids, such as H2SO4 and HNO3 (Keene et al., 1999). The high emission rates of NOx and SO2 by ship traffic resulted in enhanced concentrations of both NO2 and SO2 (see Fig. S9) during parts of the AQABA campaign. Both NO2 and SO2 are oxidized via OH to form HNO3 and H2SO4, both of which can be taken up by sea salt-containing aerosol releasing HCl. The release of HCl through acid displacement leads to a deficit in particulate Cl− concentrations which can be expressed in terms of a chloride depletion factor (Eq. 12), where [Na+] and [Cl−] represent the concentrations in moles per cubic metre and 1.174 is the molar ratio of Cl− to Na+ found in seawater (Zhuang et al., 1999).
In Fig. 7 we present a time series (Gulf of Oman/Arabian Gulf) in which significant differences in particulate Na+ and Cl− concentrations coincide with high mixing ratios of NO2, SO2, and HCl (chloride depletion up to 90 %) indicating efficient HCl acid displacement by HNO3 and H2SO4 in this region.
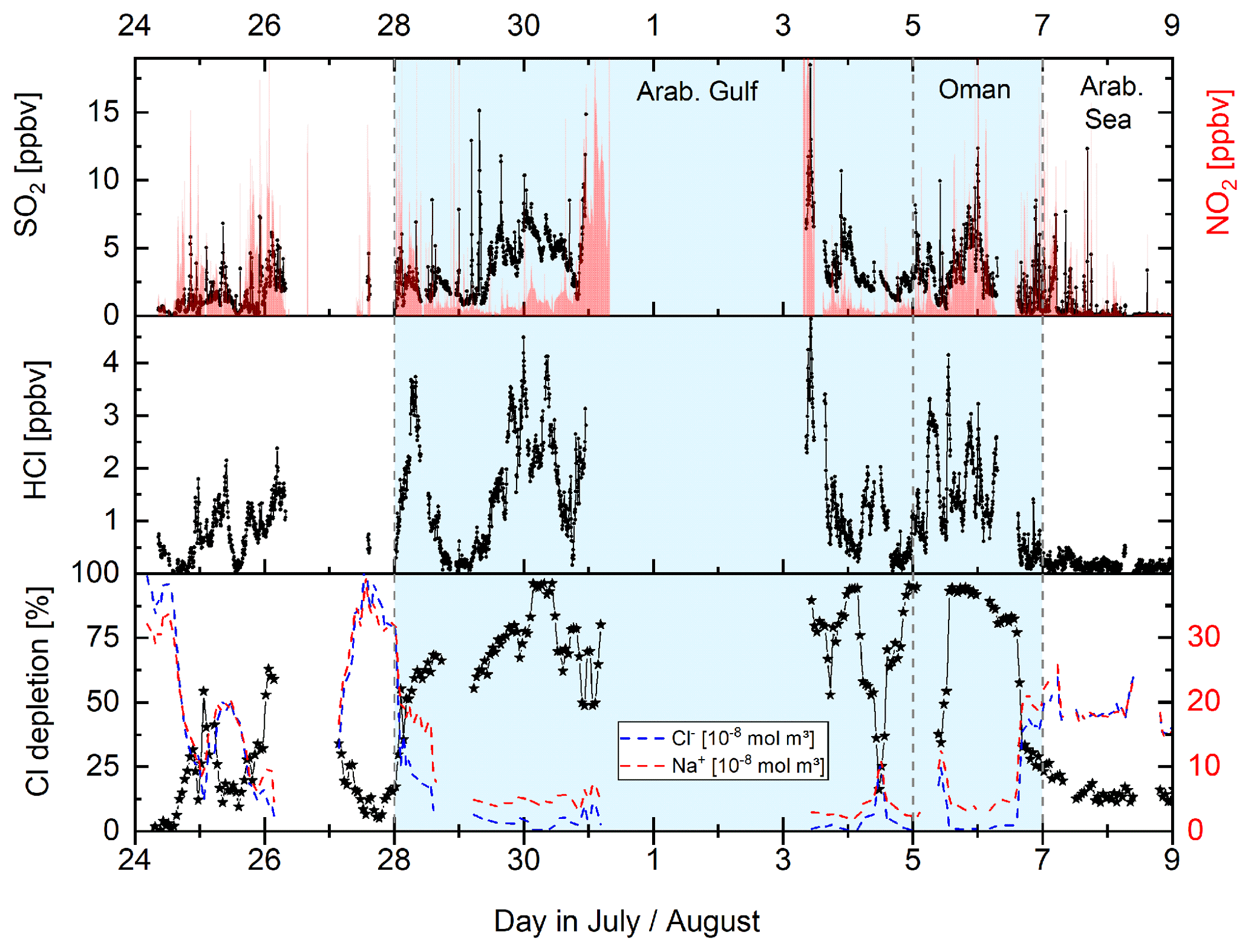
Figure 7Co-variance between mixing ratios of SO2, NO2, and HCl and particulate chloride depletion (calculated from Eq. 12) illustrated by the difference in Cl− and Na+ (PM1) measured. Chloride depletion of up to 90 % indicates effective acid displacement of HCl by HNO3 and H2SO4 in this region.
The instantaneous production rate of Cl atoms from the photolysis of ClNO2 () is given by the photolysis rate constant for ClNO2 () and its concentration (Eq. 13) whereas the instantaneous Cl production rate from HCl (pClHCl) requires knowledge of the OH concentration (Eq. 14) and the rate coefficient for reaction between OH and HCl ( × 10−12 exp() cm3 molecule−1 s−1; Atkinson et al., 2007; IUPAC, 2019).
In the following analysis, we focus on two consecutive nights in the Gulf of Oman region (Fig. 8) where we observed a monotonous increase in ClNO2 mixing ratios up to ≈300 pptv during the second half of the night followed by a decrease over a 4 h period starting at sunrise (Fig. 8 upper panel). The corresponding Cl production rate from ClNO2 photolysis reaches a maximum of 0.8×106 molecule cm−3 s−1 on the first night and 0.7×106 molecule cm−3 s−1 on the second night.
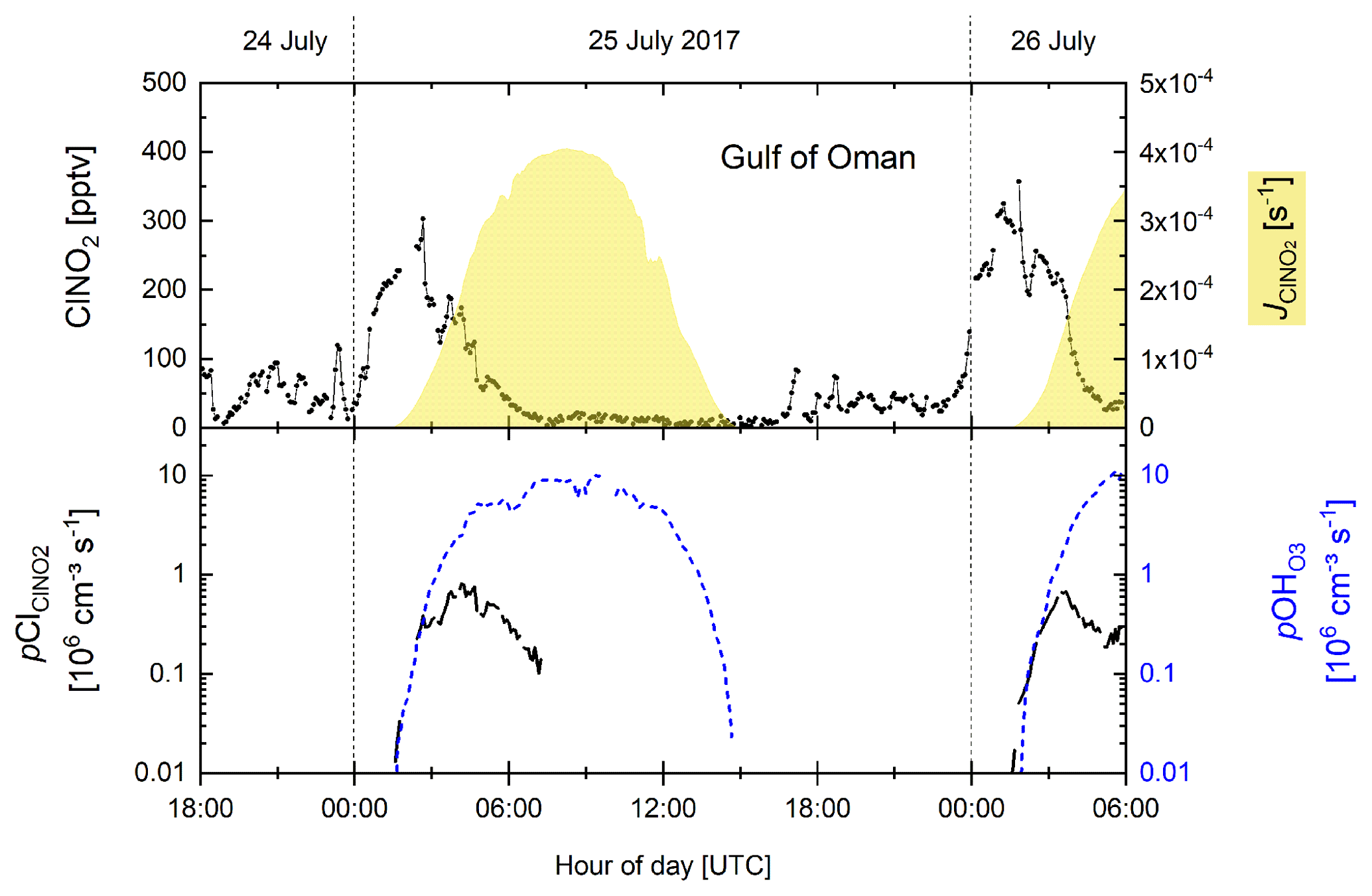
Figure 8Time series of ClNO2 mixing ratios, photolysis rates, and production of Cl radicals from ClNO2 photolysis (p) and OH radicals from O3 photolysis in the presence of H2O(p) for two consecutive nights in the Gulf of Oman.
To place this in context, we also make a rough estimate (lower limit) to the rate of OH radical production () from the photolysis of O3 in the presence of H2O (Eq. 15).
where is the photolysis rate constant for O3, and cm3 molecule−1 s−1, exp(110∕T) cm3 molecule−1 s−1, and exp(67∕T) cm3 molecule−1 s−1 (IUPAC, 2019) refer to reactions of O(1D) with H2O, N2, and O2, respectively. As we do not consider other OH production channels (e.g. photolysis of HONO or HO2+NO), which can be of importance under more polluted conditions, represents a lower limit of pOH. Although the maximum midday OH production rates from O3 photolysis ( OH molecule cm−3 s−1) are about an order of magnitude higher than Cl atom production rates, during the first ≈2 h after sunrise, and are roughly equal for this particular case study.
In order to examine the regional dependence of Cl formation we calculated the ClNO2 production rate from the regional median values of ε, the NO3 production rate, and the length of the night as described previously. As ClNO2 is completely photolysed to Cl during daytime, the total Cl production from ClNO2 photolysis (Σ) is equal to the median amount of ClNO2 formed during the night. The total Cl production from HCl+OH (ΣClHCl) was calculated by integrating the production rate pClHCl (based on [HCl] and [OH] measurements, Eq. 14) over the median diurnal profile. To calculate the contribution of O3 photolysis to the OH production (Σ), we integrated (calculated via Eq. 15) over the median diel profile. In Table 3 we summarize the average total daytime production of OH and Cl in the seven regions. The median radical production over one diurnal cycle calculated for the Suez region is exemplified in Fig. 9, whereby we assume an initial 363 pptv of ClNO2 at sunrise (Table 3), which is subsequently photolysed according to . Considering the entire campaign, we conclude that Cl atom formation via ClNO2 photolysis and OH-initiated HCl oxidation are of similar magnitude, though their relative contributions show large regional variability. For example, Cl formation from OH+HCl (ΣClHCl) is a factor ≈10 more important than ClNO2 photolysis (Σ) over the Mediterranean Sea where the NO3 (and thus ClNO2) production rate was low owing to low NOx levels and where the OH concentrations were highest. This is consistent with Li et al. (2019), who indicate the importance of HCl over the eastern Mediterranean Sea for which monthly average mixing ratios of 0.5–1.5 ppbv were predicted by a regional model. ΣClHCl is also a factor ≈2 higher than Σ over the Arabian Gulf where the ClNO2 production efficiency, ε, was low. In all other regions Σ was about equal to ΣClHCl or higher by factors between ≈1 and 4. This is in broad agreement with Riedel et al. (2012a), who report roughly equal importance of ClNO2 and HCl as chlorine atom sources in the polluted marine boundary layer of the Los Angeles region. They also report a maximum midday Cl production rate from ClNO2 photolysis of 0.6×106 molecule cm−3 s−1, which is similar to the production rates we obtained over the Gulf of Oman (Fig. 8).
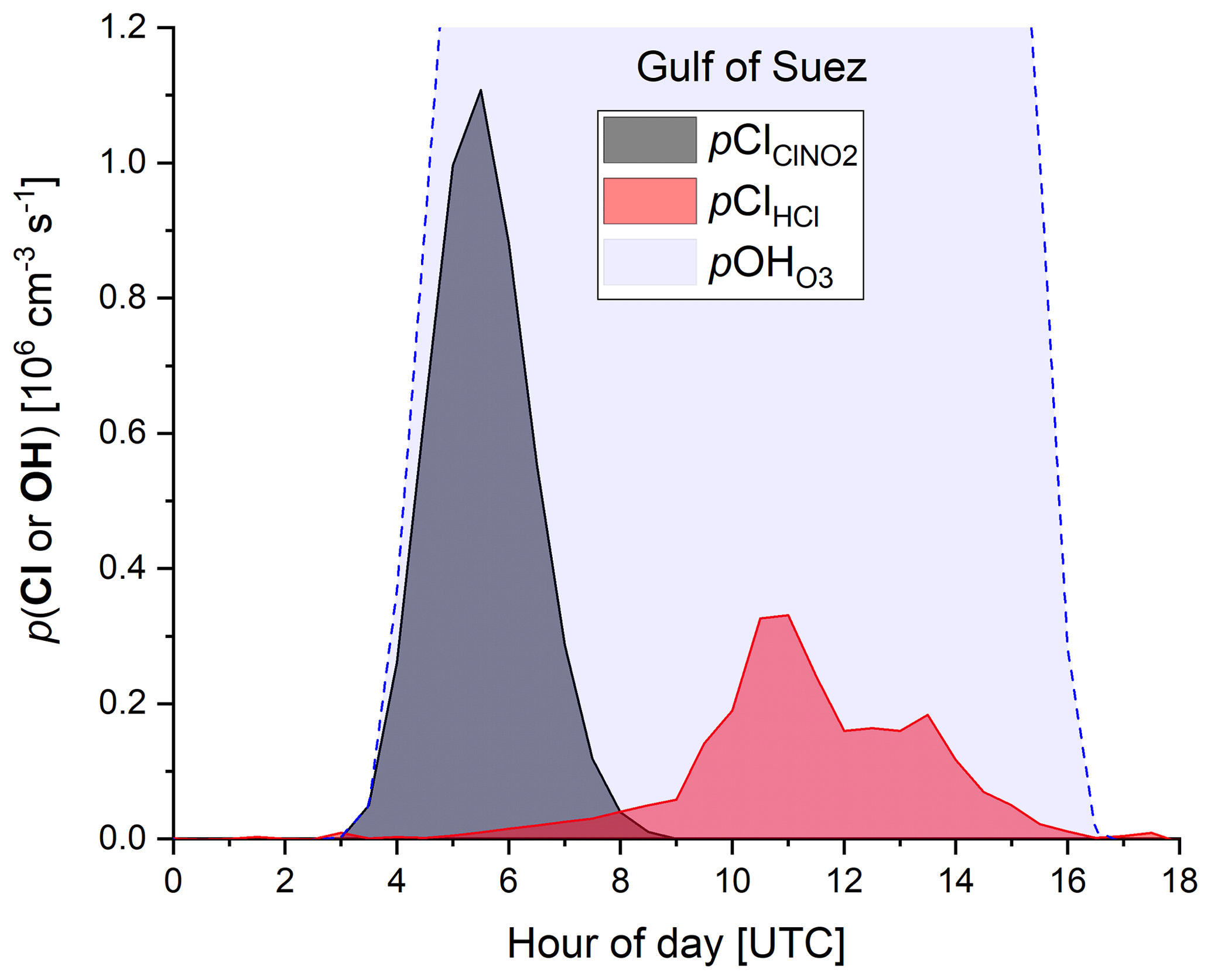
Figure 9Production of Cl atoms (from ClNO2 photolysis and HCl+OH) and OH radicals (from O3 photolysis) over one diurnal cycle in the Suez Canal and Gulf of Suez.
Table 3Regional variability in ε, pNO3, and OH and Cl radical production integrated over one diel cycle.
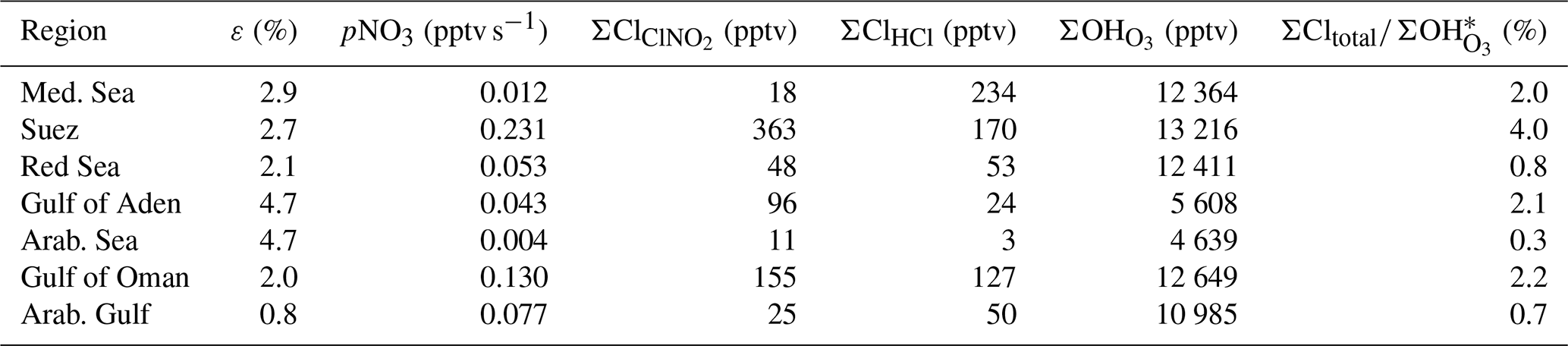
Notes: ΣCl and ΣClHCl are the integrated formation of Cl atoms from ClNO2 and HCl, respectively. ΣOH is the integrated formation of OH from O3 photolysis. * ΣCltotal=ΣClClHCl.
When comparing the total number of chlorine atoms generated during the day (ΣCltotal = Σ +ΣClHCl) with the total number of OH formed from O3 photolysis (Σ) over the same period, we find the expected domination of OH for all regions. The largest contribution of Cl to the total radical production (4 %) was observed over the Suez Canal where the Cl production was highest (about 32 % from HCl and 68 % from ClNO2). The lowest ratio of Cl to OH production was observed over the Arabian Sea, reflecting the low levels of NOx and very low rates of NO3 generation. Although only (at maximum) ≈4 % of the radicals generated are Cl atoms, they react more rapidly than OH with some classes of hydrocarbons, especially saturated hydrocarbons and small oxygenates. For example, the relative room temperature rate coefficients (kCl ∕ kOH) are 16, 61, 127, and 242 for reaction with CH4, CH3OH, C3H8, and C2H6, respectively (Atkinson et al., 2004; IUPAC, 2019). The impact of chlorine atoms is thus mainly seen in the oxidation rates of unsaturated hydrocarbons, the relative concentrations of which may be modified according to the relative abundance of OH and Cl and the relative reaction rate constants. For the AQABA campaign, evidence for such effects has been reported by Bourtsoukidis et al. (2019), and for the global scale Wang et al. (2019) conclude that oxidation by Cl atoms accounts for 1.0 % of methane loss with larger impacts on ethane (20 %), propane (14 %), and methanol (4 %).
The AQABA campaign, which took place in summer 2017 along the sea route from southern France to Kuwait, provided the first ClNO2 measurements in the marine boundary layer of the Mediterranean Sea, the Suez Canal, the Red Sea, the Gulf of Aden, the Arabian Sea, the Gulf of Oman, and the Arabian Gulf. Along the ship track we observed a large variability in ClNO2 mixing ratios with nocturnal maxima ranging from below the detection limit over the Arabian Sea to a few hundred parts per trillion by volume over the Gulf of Oman, the northern part of the Red Sea, the Gulf of Suez, and the Mediterranean Sea close to Sicily, Italy, with a campaign maximum of ≈600 pptv observed over the Gulf of Suez.
The overall ClNO2 production efficiency, i.e. the yield of ClNO2 per NO3 molecule formed in the reaction of NO2 with O3, was generally low (median of 2.7 % for the whole campaign) and highly variable within individual nights and between different regions with values (in percent) of 2.9, 2.7, 2.1, 4.7, 4.7, 2.0, and 0.8 over the Mediterranean Sea, the Suez Canal, the Red Sea, the Gulf of Aden, the Arabian Sea, the Gulf of Oman, and the Arabian Gulf, respectively. The relatively low ClNO2 production efficiency compared to previous measurements in the polluted marine boundary layer or at continental sites was attributed to high nocturnal temperatures during AQABA (25–35 ∘C), which significantly shifted the equilibrium between NO3 and N2O5 towards NO3 and lowered the importance of N2O5 uptake to particles relative to direct NO3 losses. The low ClNO2 production efficiency in the Arabian Gulf (< 1 %) results from a combination of high temperatures, enhanced NO3 reactivity, and lowered chloride availability. The photolysis of ClNO2 was found to represent an important source of chlorine radicals in the early morning in areas where efficient night-time production was observed, and was augmented (and sometimes exceeded) by Cl atom formation from the reaction of OH with HCl, especially in areas where parts per billion by volume levels of HCl were observed such as the Mediterranean Sea or the Arabian Gulf. Although the number of Cl atoms generated was found to be a factor of 25 to 300 fewer than the number of OH molecules generated from O3 photolysis, the high rate coefficient ratio for Cl compared to OH reactions towards some hydrocarbons implies that Cl may enhance hydrocarbon oxidation, especially in the early morning.
The data are available upon request to all scientists agreeing to the AQABA protocol from August 2019 (https://doi.org/10.5281/zenodo.3460556) (Eger, 2019).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-12121-2019-supplement.
PGE performed the CI-QMS measurements of ClNO2, HCl, and SO2 during the AQABA campaign, evaluated the field data, and wrote the paper. JNC operated the CI-QMS and the CRDS instruments during parts of the first leg and supervised the study. NO2 and N2O5 data were provided by JSh. NOx, NOy, and NOz data were provided by NF. J values were measured by JSch. IT and HF contributed the NO and NO2 datasets. AMS, OPC, and FMPS measurements and analysis were performed by JB, ED, FF, and FD. MARGA data were provided by MP and JSci. RR, ST, MM, and HH provided the OH dataset. JL designed the AQABA campaign. All authors contributed to the paper.
The authors declare that they have no conflict of interest.
We acknowledge the co-operation with the Cyprus Institute (CyI), the King Abdullah University of Science and Technology (KAUST), and the Kuwait Institute for Scientific Research (KISR). We are grateful for the support of Hays Ships Ltd, the captain, and his crew on board Kommandor Iona. We would like to thank the whole AQABA team, particularly Marcel Dorf and Claus Koeppel, for logistical support.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Kyung-Eun Min and reviewed by two anonymous referees.
Aldener, M., Brown, S. S., Stark, H., Williams, E. J., Lerner, B. M., Kuster, W. C., Goldan, P. D., Quinn, P. K., Bates, T. S., Fehsenfeld, F. C., and Ravishankara, A. R.: Reactivity and loss mechanisms of NO3 and N2O5 in a polluted marine environment: Results from in situ measurements during New England Air Quality Study 2002, J. Geophys. Res.-Atmos., 111, D23S73, https://doi.org/10.1029/2006JD007252, 2006.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – gas phase reactions of inorganic halogens, Atmos. Chem. Phys., 7, 981–1191, https://doi.org/10.5194/acp-7-981-2007, 2007.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B. A., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prevot, A. S. H., Lee, J., Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp, T., Stone, D., Heard, D. E., Fleming, Z. L., Leigh, R., Shallcross, D. E., and Percival, C. J.: The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.-Atmos., 120, 5638–5657, https://doi.org/10.1002/2014JD022629, 2015.
Bannan, T. J., Bacak, A., Le Breton, M., Flynn, M., Ouyang, B., McLeod, M., Jones, R., Malkin, T. L., Whalley, L. K., and Heard, D. E.: Ground and airborne UK measurements of nitryl chloride: An investigation of the role of Cl atom oxidation at Weybourne Atmospheric Observatory, J. Geophys. Res.-Atmos., 122, 11154–11165, https://doi.org/10.1002/2017JD026624, 2017.
Behnke, W., George, C., Scheer, V., and Zetzsch, C.: Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments, J. Geophys. Res.-Atmos., 102, 3795–3804, https://doi.org/10.1029/96JD03057, 1997.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Bertram, T. H., Thornton, J. A., Riedel, T. P., Middlebrook, A. M., Bahreini, R., Bates, T. S., Quinn, P. K., and Coffman, D. J.: Direct observations of N2O5 reactivity on ambient aerosol particles, Geophys. Res. Lett., 36, L19803, https://doi.org/10.1029/2009GL040248, 2009.
Bourtsoukidis, E., Ernle, L., Crowley, J. N., Lelieveld, J., Paris, J.-D., Pozzer, A., Walter, D., and Williams, J.: Non-methane hydrocarbon (C2–C8) sources and sinks around the Arabian Peninsula, Atmos. Chem. Phys., 19, 7209–7232, https://doi.org/10.5194/acp-19-7209-2019, 2019.
Brown, S. S., Ryerson, T. B., Wollny, A. G., Brock, C. A., Peltier, R., Sullivan, A. P., Weber, R. J., Dube, W. P., Trainer, M., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. R.: Variability in nocturnal nitrogen oxide processing and its role in regional air quality, Science, 311, 67–70, https://doi.org/10.1126/science.1120120, 2006.
Brown, S. S., Dube, W. P., Fuchs, H., Ryerson, T. B., Wollny, A. G., Brock, C. A., Bahreini, R., Middlebrook, A. M., Neuman, J. A., Atlas, E., Roberts, J. M., Osthoff, H. D., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: Comparison to current model parameterizations, J. Geophys. Res.-Atmos., 114, D00F10, https://doi.org/10.1029/2008JD011679, 2009.
Brown, S. S., Dube, W. P., Tham, Y. J., Zha, Q. Z., Xue, L. K., Poon, S., Wang, Z., Blake, D. R., Tsui, W., Parrish, D. D., and Wang, T.: Nighttime chemistry at a high altitude site above Hong Kong, J. Geophys. Res.-Atmos., 121, 2457–2475, https://doi.org/10.1002/2015jd024566, 2016.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, http://jpldataeval.jpl.nasa.gov (last access: 26 September 2019), 2015.
Chang, W. L., Bhave, P. V., Brown, S. S., Riemer, N., Stutz, J., and Dabdub, D.: Heterogeneous atmosperic chemistry, ambient measurements and model calculatuons of N2O5: A review, Aerosol. Sci. Technol., 45, 655–685, https://doi.org/10.1080/02786826.2010.551672, 2011.
Clegg, S. L., Brimblecombe, P., and Wexler, A. S.: Thermodynamic model of the system at 298.15 K, J. Phys. Chem. A, 102, 2155–2171, https://doi.org/10.1021/jp973043j, 1998.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Dentener, F. J. and Crutzen, P. J.: Reaction of N2O5 on tropospheric aerosols – Impact on the global distributions of NOX, O3, and OH, J. Geophys. Res.-Atmos., 98, 7149–7163, https://doi.org/10.1029/92JD02979, 1993.
Eger, P. G.: Shipborne measurements of ClNO2 in the Mediterranean Sea and around the Arabian Peninsula during summer, https://doi.org/10.5281/zenodo.3460556, 2019.
Eger, P. G., Helleis, F., Schuster, G., Phillips, G. J., Lelieveld, J., and Crowley, J. N.: Chemical ionization quadrupole mass spectrometer with an electrical discharge ion source for atmospheric trace gas measurement, Atmos. Meas. Tech., 12, 1935–1954, https://doi.org/10.5194/amt-12-1935-2019, 2019.
Faxon, C. B., Bean, J. K., and Hildebrandt Ruiz, L.: Inland concentrations of Cl2 and ClNO2 in Southeast Texas suggest chlorine chemistry significantly contributes to atmospheric reactivity, Atmosphere, 6, 1487–1506, https://doi.org/10.3390/atmos6101487, 2015.
Finlayson-Pitts, B. J., Ezell, M. J., and Pitts, J. N. J.: Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2, Nature, 337, 241–244, https://doi.org/10.1038/337241a0, 1989.
Fontijn, A., Sabadell, A. J., and Ronco, R. J.: Homogeneous chemiluminescent measurement of nitric oxide with ozone - Implications for continuous selective monitoring of gaseous air pollutants, Anal. Chem., 42, 575–579, https://doi.org/10.1021/ac60288a034, 1970.
Friese, E. and Ebel, A.: Temperature dependent thermodynamic model of the system , The J. Phys. Chem. A, 114, 11595–11631, https://doi.org/10.1021/jp101041j, 2010.
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, edited by: Ammann, M., Cox, R. A., Crowley, J.N., Herrmann, H., Jenkin, M.E., McNeill, V.F., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J., http://iupac.pole-ether.fr/index.html, last access: 26 September 2019.
Keene, W. C., Khalil, M. A. K., Erickson, D. J., McCulloch, A., Graedel, T. E., Lobert, J. M., Aucott, M. L., Gong, S. L., Harper, D. B., Kleiman, G., Midgley, P., Moore, R. M., Seuzaret, C., Sturges, W. T., Benkovitz, C. M., Koropalov, V., Barrie, L. A., and Li, Y. F.: Composite global emissions of reactive chlorine from anthropogenic and natural sources: Reactive Chlorine Emissions Inventory, J. Geophys. Res.-Atmos., 104, 8429–8440, https://doi.org/10.1029/1998jd100084, 1999.
Kercher, J. P., Riedel, T. P., and Thornton, J. A.: Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry, Atmos. Meas. Tech., 2, 193–204, https://doi.org/10.5194/amt-2-193-2009, 2009.
Le Breton, M., Hallquist, Å. M., Pathak, R. K., Simpson, D., Wang, Y., Johansson, J., Zheng, J., Yang, Y., Shang, D., and Wang, H.: Chlorine oxidation of VOCs at a semi-rural site in Beijing: significant chlorine liberation from ClNO2 and subsequent gas-and particle-phase Cl–VOC production, Atmos. Chem. Phys., 18, 13013–13030, https://doi.org/10.5194/acp-18-13013-2018, 2018.
Lee, B. H., Lopez‐Hilfiker, F. D., Schroder, J. C., Campuzano‐Jost, P., Jimenez, J. L., McDuffie, E. E., and Weinheimer, A. J.: Airborne Observations of Reactive Inorganic Chlorine and Bromine Species in the Exhaust of Coal‐Fired Power Plants, J. Geophys. Res.-Atmos., 123, 11–225, 2018.
Lelieveld, J. and Crutzen, P. J.: Influences of Cloud Photochemical Processes on Tropospheric Ozone, Nature, 343, 227–233, https://doi.org/10.1038/343227a0, 1990.
Lelieveld, J., Hoor, P., Jöckel, P., Pozzer, A., Hadjinicolaou, P., Cammas, J.-P., and Beirle, S.: Severe ozone air pollution in the Persian Gulf region, Atmos. Chem. Phys., 9, 1393–1406, https://doi.org/10.5194/acp-9-1393-2009, 2009.
Lelieveld, J., Hadjinicolaou, P., Kostopoulou, E., Chenoweth, J., El Maayar, M., Giannakopoulos, C., Hannides, C., Lange, M. A., Tanarhte, M., Tyrlis, E., and Xoplaki, E.: Climate change and impacts in the Eastern Mediterranean and the Middle East, Climatic Change, 114, 667–687, https://doi.org/10.1007/s10584-012-0418-4, 2012.
Li, J., Reiffs, A., Parchatka, U., and Fischer, H.: In situ measurements of atmospheric CO and its correlation with NOx and O3 at a rural mountain site, Metrol. Meas. Sys. , XXII, 25–38, https://doi.org/10.1515/mms-2015-0001, 2015.
Li, Q., Borge, R., Sarwar, G., de la Paz, D., Gantt, B., Domingo, J., Cuevas, C. A., and Saiz-Lopez, A.: Impact of halogen chemistry on air quality in coastal and continental Europe: application of CMAQ model and implication for regulation, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2019-171, in review, 2019.
Liu, X., Qu, H., Huey, L. G., Wang, Y., Sjostedt, S., Zeng, L., Lu, K., Wu, Y., Hu, M., and Shao, M.: High levels of daytime molecular chlorine and nitryl chloride at a rural site on the North China Plain, Environ. Sci. Technol., 51, 9588–9595, 2017.
Macintyre, H. L. and Evans, M. J.: Sensitivity of a global model to the uptake of N2O5 by tropospheric aerosol, Atmos. Chem. Phys., 10, 7409–7414, https://doi.org/10.5194/acp-10-7409-2010, 2010.
Martinez, M., Harder, H., Kubistin, D., Rudolf, M., Bozem, H., Eerdekens, G., Fischer, H., Kluepfel, T., Gurk, C., Koenigstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: airborne measurements, Atmos. Chem. Phys., 10, 3759–3773, https://doi.org/10.5194/acp-10-3759-2010, 2010.
McDuffie, E. E., Fibiger, D. L., Dubé, W. P., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., Shah, V., Jaeglé, L., Guo, H., and Weber, R. J.: Heterogeneous N2O5 uptake during winter: Aircraft measurements during the 2015 WINTER campaign and critical evaluation of current parameterizations, J. Geophys. Res.-Atmos., 123, 4345–4372, https://doi.org/10.1002/2018JD028336, 2018a.
McDuffie, E. E., Fibiger, D. L., Dubé, W. P., Lopez Hilfiker, F., Lee, B. H., Jaeglé, L., Guo, H., Weber, R. J., Reeves, J. M., and Weinheimer, A. J.: ClNO2 yields from aircraft measurements during the 2015 WINTER campaign and critical evaluation of the current parameterization, J. Geophys. Res.-Atmos., 123, 12994–13015, https://doi.org/10.1029/2018JD029358, 2018b.
McNeill, V. F., Patterson, J., Wolfe, G. M., and Thornton, J. A.: The effect of varying levels of surfactant on the reactive uptake of N2O5 to aqueous aerosol, Atmos. Chem. Phys., 6, 1635–1644, https://doi.org/10.5194/acp-6-1635-2006, 2006.
Meusel, H., Kuhn, U., Reiffs, A., Mallik, C., Harder, H., Martinez, M., Schuladen, J., Bohn, B., Parchatka, U., Crowley, J. N., Fischer, H., Tomsche, L., Novelli, A., Hoffmann, T., Janssen, R. H. H., Hartogensis, O., Pikridas, M., Vrekoussis, M., Bourtsoukidis, E., Weber, B., Lelieveld, J., Williams, J., Pöschl, U., Cheng, Y., and Su, H.: Daytime formation of nitrous acid at a coastal remote site in Cyprus indicating a common ground source of atmospheric HONO and NO, Atmos. Chem. Phys., 16, 14475–14493, https://doi.org/10.5194/acp-16-14475-2016, 2016.
Mielke, L. H., Furgeson, A., and Osthoff, H. D.: Observation of CINO2 in a mid-continental urban environment, Environ. Sci. Technol., 45, 8889–8896, https://doi.org/10.1021/es201955u, 2011.
Mielke, L. H., Stutz, J., Tsai, C., Hurlock, S. C., Roberts, J. M., Veres, P. R., Froyd, K. D., Hayes, P. L., Cubison, M. J., Jimenez, J. L., Washenfelder, R. A., Young, C. J., Gilman, J. B., de Gouw, J. A., Flynn, J. H., Grossberg, N., Lefer, B. L., Liu, J., Weber, R. J., and Osthoff, H. D.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res.-Atmos., 118, 10638–10652, https://doi.org/10.1002/jgrd.50783, 2013.
Mielke, L. H., Furgeson, A., Odame-Ankrah, C. A., and Osthoff, H. D.: Ubiquity of ClNO2 in the urban boundary layer of Calgary, Alberta, Canada, Can. J. Chem., 94, 414–423, https://doi.org/10.1139/cjc-2015-0426, 2016.
Molinaroli, E., Guerzoni, S., and Rampazzo, G.: Contribution of Saharan dust to the Central Mediterranean Basin, Geological Society of America, Special Paper, 284, 303–312, 1993.
Morgan, W. T., Ouyang, B., Allan, J. D., Aruffo, E., Di Carlo, P., Kennedy, O. J., Lowe, D., Flynn, M. J., Rosenberg, P. D., Williams, P. I., Jones, R., McFiggans, G. B., and Coe, H.: Influence of aerosol chemical composition on N2O5 uptake: airborne regional measurements in northwestern Europe, Atmos. Chem. Phys., 15, 973–990, https://doi.org/10.5194/acp-15-973-2015, 2015.
Novelli, A., Hens, K., Ernest, C. T., Kubistin, D., Regelin, E., Elste, T., Plass-Duelmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413-3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/ngeo177, 2008.
Osthoff, H. D., Odame-Ankrah, C. A., Taha, Y. M., Tokarek, T. W., Schiller, C. L., Haga, D., Jones, K., and Vingarzan, R.: Low levels of nitryl chloride at ground level: nocturnal nitrogen oxides in the Lower Fraser Valley of British Columbia, Atmos. Chem. Phys., 18, 6293–6315, https://doi.org/10.5194/acp-18-6293-2018, 2018.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012GL051912, 2012.
Phillips, G. J., Thieser, J., Tang, M. J., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Priestley, M., le Breton, M., Bannan, T. J., Worrall, S. D., Bacak, A., Smedley, A. R. D., Reyes-Villegas, E., Mehra, A., Allan, J., Webb, A. R., Shallcross, D. E., Coe, H., and Percival, C. J.: Observations of organic and inorganic chlorinated compounds and their contribution to chlorine radical concentrations in an urban environment in northern Europe during the wintertime, Atmos. Chem. Phys., 18, 13481–13493, https://doi.org/10.5194/acp-18-13481-2018, 2018.
Riedel, T. P., Bertram, T. H., Crisp, T. A., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Gilman, J., de Gouw, J., Bon, D. M., Wagner, N. L., Brown, S. S., and Thornton, J. A.: Nitryl chloride and molecular chlorine in the coastal marine boundary layer, Environ. Sci. Technol., 46, 10463–10470, https://doi.org/10.1021/es204632r, 2012a.
Riedel, T. P., Bertram, T. H., Ryder, O. S., Liu, S., Day, D. A., Russell, L. M., Gaston, C. J., Prather, K. A., and Thornton, J. A.: Direct N2O5 reactivity measurements at a polluted coastal site, Atmos. Chem. Phys., 12, 2959–2968, https://doi.org/10.5194/acp-12-2959-2012, 2012b.
Riedel, T. P., Wagner, N. L., Dube, W. P., Middlebrook, A. M., Young, C. J., Ozturk, F., Bahreini, R., VandenBoer, T. C., Wolfe, D. E., Williams, E. J., Roberts, J. M., Brown, S. S., and Thornton, J. A.: Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl-2 measurements from a tall tower in a polluted continental setting, J. Geophys. Res.-Atmos., 118, 8702–8715, https://doi.org/10.1002/jgrd.50637, 2013.
Riedel, T. P., Wolfe, G. M., Danas, K. T., Gilman, J. B., Kuster, W. C., Bon, D. M., Vlasenko, A., Li, S. M., Williams, E. J., Lerner, B. M., Veres, P. R., Roberts, J. M., Holloway, J. S., Lefer, B., Brown, S. S., and Thornton, J. A.: An MCM modeling study of nitryl chloride (ClNO2) impacts on oxidation, ozone production and nitrogen oxide partitioning in polluted continental outflow, Atmos. Chem. Phys., 14, 3789–3800, https://doi.org/10.5194/acp-14-3789-2014, 2014.
Rolph, G., Stein, A., and Stunder, B.: Real-time environmental applications and display system: READY, Environ. Model. Softw., 95, 210–228, https://doi.org/10.1016/j.envsoft.2017.06.025, 2017.
Ryerson, T. B., Andrews, A. E., Angevine, W. M., Bates, T. S., Brock, C. A., Cairns, B., Cohen, R. C., Cooper, O. R., de Gouw, J. A., Fehsenfeld, F. C., Ferrare, R. A., Fischer, M. L., Flagan, R. C., Goldstein, A. H., Hair, J. W., Hardesty, R. M., Hostetler, C. A., Jimenez, J. L., Langford, A. O., McCauley, E., McKeen, S. A., Molina, L. T., Nenes, A., Oltmans, S. J., Parrish, D. D., Pederson, J. R., Pierce, R. B., Prather, K., Quinn, P. K., Seinfeld, J. H., Senff, C. J., Sorooshian, A., Stutz, J., Surratt, J. D., Trainer, M., Volkamer, R., Williams, E. J., and Wofsy, S. C.: The 2010 California Research at the Nexus of Air Quality and Climate Change (CalNex) field study, J. Geophys. Res.-Atmos., 118, 5830–5866, https://doi.org/10.1002/jgrd.50331, 2013.
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 41, 4050–4058, https://doi.org/10.1002/2014GL059962, 2014.
Simon, H., Kimura, Y., McGaughey, G., Allen, D. T., Brown, S. S., Osthoff, H. D., Roberts, J. M., Byun, D., and Lee, D.: Modeling the impact of ClNO2 on ozone formation in the Houston area, J. Geophys. Res.-Atmos., 114, D00F03, https://doi.org/10.1029/2008JD010732, 2009.
Sobanski, N., Schuladen, J., Schuster, G., Lelieveld, J., and Crowley, J. N.: A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 9, 5103–5118, https://doi.org/10.5194/amt-9-5103-2016, 2016.
Sommariva, R., Hollis, L. D., Sherwen, T., Baker, A. R., Ball, S. M., Bandy, B. J., Bell, T. G., Chowdhury, M. N., Cordell, R. L., and Evans, M. J.: Seasonal and geographical variability of nitryl chloride and its precursors in Northern Europe, Atmos. Sci. Lett., 19, e844, https://doi.org/10.1002/asl.844, 2018.
Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B., Cohen, M. D., and Ngan, F.: NOAA'S HYSPLIT atmospheric transport and dispersion modeling system, Bull. Am. Meteorol. Soc., 96, 2059–2077, https://doi.org/10.1175/bams-d-14-00110.1, 2015.
Tang, M. J., Thieser, J., Schuster, G., and Crowley, J. N.: Kinetics and mechanism of the heterogeneous reaction of N2O5 with mineral dust particles, Phys. Chem. Chem. Phys, 14, 8551–8561, https://doi.org/10.1039/C2CP40805H, 2012.
Thaler, R. D., Mielke, L. H., and Osthoff, H. D.: Quantification of nitryl chloride at part per trillion mixing ratios by thermal dissociation cavity ring-down spectroscopy, Anal. Chem., 83, 2761–2766, https://doi.org/10.1021/ac200055z, 2011.
Tham, Y. J., Wang, Z., Li, Q. Y., Yun, H., Wang, W. H., Wang, X. F., Xue, L. K., Lu, K. D., Ma, N., Bohn, B., Li, X., Kecorius, S., Gross, J., Shao, M., Wiedensohler, A., Zhang, Y. H., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Tham, Y. J., Wang, Z., Li, Q., Wang, W., Wang, X., Lu, K., Ma, N., Yan, C., Kecorius, S., and Wiedensohler, A.: Heterogeneous N2O5 uptake coefficient and production yield of ClNO2 in polluted northern China: roles of aerosol water content and chemical composition, Atmos. Chem. Phys., 18, 13155–13171, https://doi.org/10.5194/acp-18-13155-2018, 2018.
Thieser, J., Schuster, G., Phillips, G. J., Reiffs, A., Parchatka, U., Pöhler, D., Lelieveld, J., and Crowley, J. N.: A two-channel, thermal dissociation cavity-ringdown spectrometer for the detection of ambient NO2, RO2NO2 and RONO2, Atmos. Meas. Tech., 9, 553–576, https://doi.org/10.5194/amt-9-553-2016, 2016.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
Wagner, N. L., Riedel, T. P., Roberts, J. M., Thornton, J. A., Angevine, W. M., Williams, E. J., Lerner, B. M., Vlasenko, A., Li, S. M., Dube, W. P., Coffman, D. J., Bon, D. M., de Gouw, J. A., Kuster, W. C., Gilman, J. B., and Brown, S. S.: The sea breeze/land breeze circulation in Los Angeles and its influence on nitryl chloride production in this region, J. Geophys. Res.-Atmos., 117, D00V24, https://doi.org/10.1029/2012jd017810, 2012.
Wagner, N. L., Riedel, T. P., Young, C. J., Bahreini, R., Brock, C. A., Dube, W. P., Kim, S., Middlebrook, A. M., Ozturk, F., Roberts, J. M., Russo, R., Sive, B., Swarthout, R., Thornton, J. A., VandenBoer, T. C., Zhou, Y., and Brown, S. S.: N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements, J. Geophys. Res.-Atmos., 118, 9331–9350, https://doi.org/10.1002/jgrd.50653, 2013.
Wang, T., Tham, Y. J., Xue, L., Li, Q., Zha, Q., Wang, Z., Poon, S. C. N., Dube, W. P., Blake, D. R., Louie, P. K. K., Luk, C. W. Y., Tsui, W., and Brown, S. S.: Observations of nitryl chloride and modeling its source and effect on ozone in the planetary boundary layer of southern China, J. Geophys. Res.-Atmos., 121, 2476–2489, https://doi.org/10.1002/2015jd024556, 2016.
Wang, X., Wang, H., Xue, L., Wang, T., Wang, L., Gu, R., Wang, W., Tham, Y. J., Wang, Z., and Yang, L.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Env., 156, 125–134, https://doi.org/10.1016/j.atmosenv.2017.02.035, 2017.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Young, A. H., Keene, W. C., Pszenny, A. A. P., Sander, R., Thornton, J. A., Riedel, T. P., and Maben, J. R.: Phase partitioning of soluble trace gases with size-resolved aerosols in near-surface continental air over northern Colorado, USA, during winter, J. Geophys. Res.-Atmos., 118, 9414–9427, https://doi.org/10.1002/jgrd.50655, 2013.
Young, C. J., Washenfelder, R. A., Roberts, J. M., Mielke, L. H., Osthoff, H. D., Tsai, C., Pikelnaya, O., Stutz, J., Veres, P. R., Cochran, A. K., VandenBoer, T. C., Flynn, J., Grossberg, N., Haman, C. L., Lefer, B., Stark, H., Graus, M., de Gouw, J., Gilman, J. B., Kuster, W. C., and Brown, S. S.: Vertically resolved measurements of nighttime radical reservoirs; in Los Angeles and their contribution to the urban radical budget, Environ. Sci. Technol., 46, 10965–10973, https://doi.org/10.1021/es302206a, 2012.
Zhou, W., Zhao, J., Ouyang, B., Mehra, A., Xu, W., Wang, Y., Bannan, T. J., Worrall, S. D., Priestley, M., Bacak, A., Chen, Q., Xie, C., Wang, Q., Wang, J., Du, W., Zhang, Y., Ge, X., Ye, P., Lee, J. D., Fu, P., Wang, Z., Worsnop, D., Jones, R., Percival, C. J., Coe, H., and Sun, Y.: Production of N2O5 and ClNO2 in summer in urban Beijing, China, Atmos. Chem. Phys., 18, 11581–11597, https://doi.org/10.5194/acp-18-11581-2018, 2018.
Zhuang, H., Chan, C. K., Fang, M., and Wexler, A. S.: Formation of ntrate and non-sea-salt sulfate on coarse particles, Atmos. Environ., 33, 4223–4233, https://doi.org/10.1016/s1352-2310(99)00186-7, 1999.