the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 Sep 2019
| 25 Sep 2019
Diurnal variability, photochemical production and loss processes of hydrogen peroxide in the boundary layer over Europe
Horst Fischer
Raoul Axinte
Heiko Bozem
John N. Crowley
Cheryl Ernest
Stefan Gilge
Sascha Hafermann
Hartwig Harder
Korbinian Hens
Ruud H. H. Janssen
Rainer Königstedt
Dagmar Kubistin
Chinmay Mallik
Monica Martinez
Anna Novelli
Uwe Parchatka
Christian Plass-Dülmer
Andrea Pozzer
Eric Regelin
Andreas Reiffs
Torsten Schmidt
Jan Schuladen
Jos Lelieveld
Hydrogen peroxide (H2O2) plays a significant role in the oxidizing capacity of the atmosphere. It is an efficient oxidant in the liquid phase and serves as a temporary reservoir for the hydroxyl radical (OH), the most important oxidizing agent in the gas phase. Due to its high solubility, removal of H2O2 due to wet and dry deposition is efficient, being a sink of HOx (OH+HO2) radicals. In the continental boundary layer, the H2O2 budget is controlled by photochemistry, transport and deposition processes. Here we use in situ observations of H2O2 and account for chemical source and removal mechanisms to study the interplay between these processes. The data were obtained during five ground-based field campaigns across Europe from 2008 to 2014 and bring together observations in a boreal forest, two mountainous sites in Germany, and coastal sites in Spain and Cyprus. Most campaigns took place in the summer, while the measurements in the south-west of Spain took place in early winter. Diel variations in H2O2 are strongly site-dependent and indicate a significant altitude dependence. While boundary-layer mixing ratios of H2O2 at low-level sites show classical diel cycles with the lowest values in the early morning and maxima around local noon, diel profiles are reversed on mountainous sites due to transport from the nocturnal residual layer and the free troposphere. The concentration of hydrogen peroxide is largely governed by its main precursor, the hydroperoxy radical (HO2), and shows significant anti-correlation with nitrogen oxides (NOx) that remove HO2. A budget calculation indicates that in all campaigns, the noontime photochemical production rate through the self-reaction of HO2 radicals was much larger than photochemical loss due to reaction with OH and photolysis, and that dry deposition is the dominant loss mechanism. Estimated dry deposition velocities varied between approximately 1 and 6 cm s−1, with relatively high values observed during the day in forested regions, indicating enhanced uptake of H2O2 by vegetation. In order to reproduce the change in H2O2 mixing ratios between sunrise and midday, a variable contribution from transport (10 %–100 %) is required to balance net photochemical production and deposition loss. Transport is most likely related to entrainment from the residual layer above the nocturnal boundary layer during the growth of the boundary layer in the morning.
- Article
(1680 KB) - Full-text XML
-
Supplement
(594 KB) - BibTeX
- EndNote





Hydrogen peroxide (H2O2) plays a pivotal role in the oxidizing capacity of the atmosphere. In hydrometeors and aqueous particles it oxidizes dissolved inorganic trace gases, while in the gas phase it serves as a reservoir species for the atmosphere's most important oxidizing agent, the hydroxyl radical (OH). Thus, H2O2 has a dual role as a secondary source for OH radicals and an irreversible sink for HOx (OH+HO2) due to its physical removal by wet and dry deposition. The atmospheric chemistry, concentration levels in the troposphere and measurement techniques used to observe H2O2 have been discussed in a number of review articles (Gunz and Hoffmann, 1990; Jackson and Hewitt, 1999; Lee et al., 2000; Vione et al., 2003; Reeves and Penkett, 2003). The dominant photochemical source of H2O2 is the recombination of two hydroperoxy radicals (HO2):
Here M represents a collision partner, usually nitrogen (N2), oxygen (O2) or water vapour (H2O). Note that the rate coefficient for Reaction (R1) increases with increasing pressure (due to its dependence on M) and water vapour concentration [H2O] (Atkinson et al., 2004; http://iupac.pole-ether.fr, last access: 27 August 2019). Additional production of H2O2 can result from the ozonolysis of alkenes (Sauer et al., 1999), in particular biogenic alkenes emitted from forests.
The formation of H2O2 according to Reaction (R1) competes with the reaction of HO2 with NO,
one of the most important OH recycling reactions (Lelieveld et al., 2016) and an important step in photochemical ozone formation in the troposphere (Seinfeld and Pandis, 1997). Due to the competition for HO2 described in Reactions (R1) and (R2), it is expected that H2O2 mixing ratios will show a dependence on ambient NOx (NO+NO2) levels, with the highest levels expected at the lowest NOx.
Photochemical loss of H2O2 is due to either reaction with OH (Reaction R3) or photolysis (Reaction R4), partly reforming HOx radicals:
Since Reactions (3) and (4) recycle HOx, they do not constitute an irreversible loss mechanism for HOx or H2O2, the latter being due to physical removal of H2O2 by wet and dry deposition. Due to its high Henry's law coefficient (∼105 mol L−1 atm−1), H2O2 is highly soluble in water and will be efficiently removed by rain and the deposition of fog (Klippel et al., 2011). Additionally, dry deposition rates with deposition velocities of the order of 1–5 cm s−1 (see e.g. Table 6 in Stickler et al., 2007) lead to large losses of H2O2 in the boundary layer. Due to this strong surface sink, airborne observations often show increasing H2O2 mixing ratios with altitude, yielding a local maximum slightly above the boundary layer (Stickler et al., 2007; Klippel et al., 2011).
To understand the H2O2 budget and diurnal variability, one has to consider all chemical and physical processes. Along with net photochemical production (production minus loss) and deposition processes, horizontal and vertical transport have to be accounted for. In the absence of clouds, changes in the concentration of H2O2 can thus be described by Eq. (1):
Here Pchem is the photochemical production of H2O2 by Reaction (R1), neglecting additional contributions from the ozonolysis of alkenes, while Lchem is the loss due to Reactions (3) and (4). The third term on the right-hand side of Eq. (1) describes vertical transport due to entrainment across the top of the boundary layer ωeΔ[H2O2]/BLH (ωe is the entrainment velocity, Δ[H2O2] is the concentration difference between the boundary layer and the free troposphere, and BLH is the boundary layer height) and dry deposition to the surface vd [H2O2]/BLH (vd is the deposition velocity). The final term in Eq. (1) describes the horizontal advection of H2O2 due to a horizontal gradient in H2O2 mixing ratios. The relative strength of the individual terms in Eq. (1) strongly depends on local conditions. In the free troposphere, dry deposition can be neglected and horizontal and vertical transports are small due to small concentration gradients on a regional scale. Thus, net photochemical tendencies (Pchem–Lchem) and precipitation largely determine the H2O2 concentrations in the free troposphere (Klippel et al., 2011). In the boundary layer, both transport and dry deposition play a significant role. Due to a rather invariant boundary layer height over the oceans and small horizontal H2O2 concentration gradients in the marine boundary layer, wet and dry deposition and net photochemical tendencies are the dominant processes affecting the H2O2 concentrations (Fischer et al., 2015). In the continental boundary layer, the situation can be complex, since all processes described in Eq. (1) are expected to play a role. Following up on previous studies in the free troposphere (Klippel et al., 2011) and the marine boundary layer (Fischer et al., 2015), we examine the influence of chemical and physical processes on the H2O2 budget at various locations in the continental boundary layer at various locations in Europe. We use in situ observations of H2O2, its precursor (HO2), sinks (Reactions R2 and R3), as well as measurements of species that are expected to influence H2O2 photochemistry (i.e. nitrogen oxides (NOx) and ozone (O3)), together with meteorological and boundary layer height information to study the H2O2 budgets. Overall, we use observations from five measurement campaigns spanning a latitude range from 61.5 to 34.9∘ N between 2008 and 2014. With the exception of one campaign that was performed in early winter in southern Spain, all observations pertain to the summer. The main aim of this paper is to explore geographical differences in H2O2 mixing ratios and to what extent they are due to characteristics of the chemical environment, in particular with respect to HOx and NOx levels. Additionally, we investigate the role of transport and physical removal processes in H2O2 levels and diel variations. Rather than presenting individual time series, we will concentrate on diel variations, calculated from median values, being relatively less sensitive to individual events, e.g. precipitation or cloud processing. To illustrate atmospheric variability 25 %–75 % quartiles will be used. By using diel variations rather than time series, we neglect the influence of variability in air-mass origin and concentrate on the role of vertical transport due to boundary layer entrainment. Use of campaign-averaged diel profiles allows us to calculate median chemical tendencies and estimations of average deposition rates.
Section 2 describes the measurement sites, the techniques used for the in situ measurements of H2O2, OH, HO2, NOx, O3 and photolysis rates, and the derivation of the H2O2 photolysis rate that was not measured in all campaigns. In the results section (Sect. 3) we discuss H2O2 mixing ratios, their relation to NOx and HOx, and diel variations, and derive a H2O2 budget with respect to photochemical production and destruction, dry deposition and vertical entrainment. In the discussion (Sect. 4), the limitations of our approach are discussed and results are compared to literature values.
2.1 Campaigns and observation sites
Between winter 2008 and summer 2014, we performed five measurement campaigns at various locations across Europe. In Table 1 a summary of the different campaigns, their locations (latitude, longitude, height above sea level) and the time difference between UTC and local noon is given. The location of the different campaigns is documented in Fig. 1. The Diel Oxidant Mechanism in relation to Nitrogen Oxides (DOMINO) campaign was carried out at the El Arenosillo station (31.7∘ N, 6.7∘ W; 40 m a.s.l.) in the period between 21 November and 8 December 2008. El Arenosillo is located in the south-west of Spain approximately 200 m from the Atlantic Ocean. The site itself is situated in a national park. The city of Huelva, a large industrial complex, is situated 26 km to the north-west and the Seville metropolitan area is 75 km to the north-east. Back-trajectory calculations indicate air-mass origins from Huelva and Portugal, the continental north, Seville and the marine sectors, with an occurrence frequency of 42 %, 30 %, 7 % and 21 %, respectively. Maximum temperatures decreased from 23 ∘C in the beginning of the campaign to less than 16 ∘C towards the end. Typical wind speeds ranged between 1 and 4 m s−1. The boundary layer height varied between approximately 200 m during nighttime and 1400 m during the afternoon. Details of the site and an extensive discussion of the meteorological conditions, including a characterisation of air-mass origins, can be found in Adame et al. (2014).
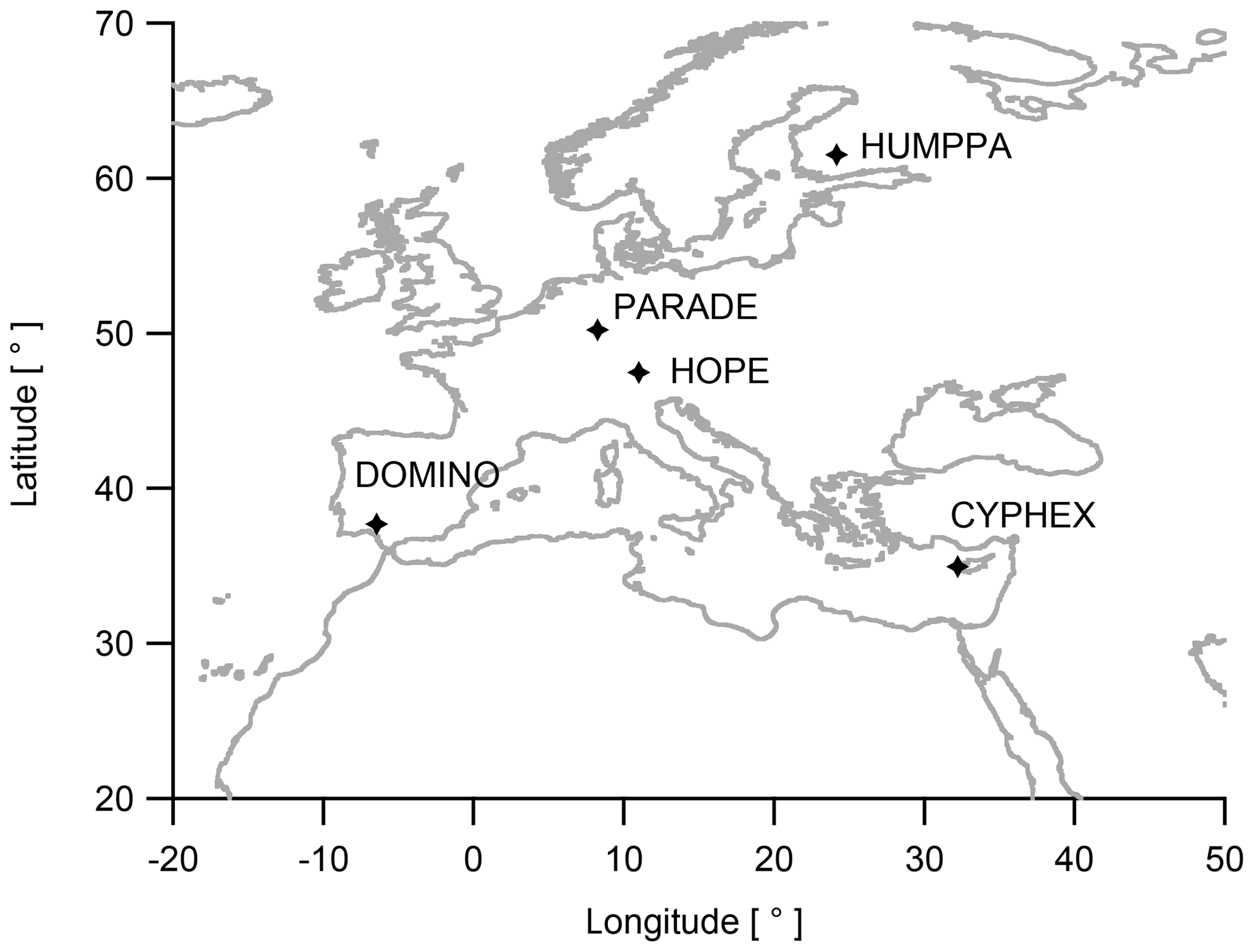
Figure 1Locations of the different campaigns performed between 2008 and 2014 in Europe.
The Hyytiälä United Measurements of Photochemistry and Particles – Comprehensive Organic Particle and Environmental Chemistry (HUMPPA-COPEC) campaign was conducted at the SMEAR II (Station for Measuring Ecosystem–Atmosphere Relation) boreal forest research station in Hyytiälä, Finland (61.5∘ N, 24.17∘ E; 181 m a.s.l.), from 12 July to 12 August 2010. The site is situated in a large boreal forest, with the next major urban setting being Tampere approximately 50 km to the south-west of the site. Back-trajectory calculations indicate air-mass origins from the Tampere (Finland) area (south-west) and the continental north and south-east (Russia) with occurrence frequencies of 53 %, 10 % and 21 %, respectively. Maximum temperatures varied between 16 and 30 ∘C. Precipitation was low with the exception of 3 days (15 and 27 July, 4 August). Typical wind speeds ranged between 1 and 3 m s−1. The boundary layer height varied between approximately 200 m during nighttime and 1500 m during the afternoon. Details of the site, the meteorology during HUMPPA and air-mass origins can be found in Williams et al. (2011).
The PArticles and RAdicals Diel observations of the impact of urban and biogenic Emissions (PARADE) campaign was conducted at the Taunus Observatory on the Kleiner Feldberg mountain (50.22∘ N, 8.45∘ E; 825 m a.s.l.) in south-western Germany between 15 August and 9 September 2011. The site is close to the Rhine–Main area, with the cities of Mainz 25 km to the south-south-west, Wiesbaden 20 km to the south-west and Frankfurt 30 km to the south-east. The first part of the campaign (15–26 August) was characterized by relatively high temperatures up to 25 ∘C, an air-mass origin from the south-west and a wind speed of the order of 5 m s−1. After the arrival of a series of cold fronts from the Atlantic temperatures dropped to maximum values between 10 and 22 ∘C, while wind speed increased to ∼10 m s−1. During the passage of the cold fronts on 27 August and 4 September rainfall occurred at the site. The boundary layer height varied between approximately 175 m during nighttime and 1300 m during the afternoon. Details of the site, the meteorology during PARADE and air-mass origins can be found in Li et al. (2015) and Sobanski et al. (2016).
The HOhenpeißenberg Photochemistry Experiment (HOPE 2012) was conducted at the Hohenpeißenberg Global Atmospheric Watch (GAW) Meteorological Observatory (47.48∘ N, 11∘ E; 988.8 m a.s.l.) in southern Germany between 11 June and 13 July 2012. This hilltop observatory operated by the German Weather Service is situated approximately 80 km south-west of the Bavarian capital of Munich in a rural area. Maximum temperatures during the campaign varied between 20 and 30 ∘C, while wind speeds between 2 and 5 m s−1 were measured. The main wind direction varied between south-east and south-west. Unfortunately, observations of the boundary layer height above the site were not made. Details of the site and trace gas measurements from HOPE 2012 can be found in Novelli et al. (2017).
The CYprus PHotochemistry EXperiment (CYPHEX) was conducted on a hilltop site in north-western Cyprus at Ineia (34.96∘ N, 32.37∘ E; 650 m a.s.l.) during the period between 7 July and 4 August 2014. The site is situated in a rural area with no major population centres upwind in the westerly and north-westerly directions. The distance to the Mediterranean Sea shoreline is approximately 10 km. Back-trajectory calculations indicate an air-mass origin from the western Mediterranean during the first half of the campaign, while the second half was characterized by an air-mass origin north of Cyprus over eastern Europe. Maximum temperatures varied between 21 and 28 ∘C. Typically, the wind speeds ranged between 2 and 6 m s−1. The boundary layer height varied between approximately 50 m during nighttime and 250 m during the afternoon. Details about the site, the meteorology during CYPHEX and air-mass origins can be found in Meusel et al. (2016), Hüser et al. (2017) and Derstroff et al. (2017).
2.2 Trace gas measurements
During the campaigns discussed here H2O2 was measured with a commercial analyser (AL2001 CA, Aero Laser, Garmisch Partenkirchen, Germany) based on the wet chemical dual-enzyme technique described by Lazarus et al. (1985, 1986). The instrument has been used previously for airborne (Stickler et al., 2007; Klippel et al., 2011) and ship-based (Fischer et al., 2015) measurements, and details of the instrument operation and performance are found in these publications. The detection limit of the instrument is of the order of 25 pptv at a time resolution (10 %–90 %) of 30 s. The total uncertainty is typically of the order of 12 %–15 % (Fischer et al., 2015).
Nitrogen oxides (NO and NO2) were measured with a highly sensitive two-channel chemiluminescence detector (CLD, ECO Physics CLD 790 SR, Duernten, Switzerland) during the DOMINO, HUMPPA, PARADE and CYPHEX campaigns. The instrument has been previously used in a number of airborne and ship-based campaigns and is described in detail in Hosaynali Beygi et al. (2011). The time resolution is 1 s and typical detection limits are in the low pptv range, with a total uncertainty of the order of 3 % and 5 % for NO and NO2, respectively. During HOPE NOx measurements were performed by the German Weather Service with a similar measurement technique.
Ozone was measured during all campaigns using a commercial UV Photometric O3-Analyzer (model 49, Thermo Environment Instruments, USA). The detection limit was typically 2 ppbv and the total uncertainty less than 5 %.
Measurements of OH and HO2 radicals were conducted with the Max Planck Institute for Chemistry HORUS instrument based on laser-induced fluorescence detection (Martinez et al., 2010; Hens et al., 2014). OH is detected directly, while HO2 is measured indirectly as OH following conversion with NO via Reaction (R2). Typical detection limits for OH and HO2 are 9×105 molec cm−3 and 0.4 pptv, respectively. The total uncertainty is of the order of 30 % (Hens et al., 2014). Note that since the HUMPPA campaign in 2010 an inlet pre-injector (IPI) has been used to determine the OH background signal via a chemical modulation (Novelli et al., 2014). This technique was not used during the DOMINO campaign so that OH measurements from this campaign are considered to be an upper limit. Moreover, during PARADE, HOPE and CYPHEX HO2 was measured using reduced amounts of NO sufficient to convert 10 %–30 % of the HO2, but low enough to avoid conversion of RO2 (Fuchs et al., 2011, Whalley et al., 2013). Previous measurements of HO2 reported for DOMINO and HUMPPA are therefore an upper limit as they are affected by a fractional measurement of RO2. Crowley et al. (2018) found in a data-constrained box model study that during HUMPPA at noon 30 % occurred due to RO2, confirming the finding by Hens et al. (2014). For the early morning and nighttime hours RO2 interference was significantly larger. During later campaigns (PARADE, HOPE, CYPHEX) the reduction of the amount of NO used to convert HO2 to OH resulted in RO2 interferences of the order 12 % to 15 % (Mallik et al., 2018).
During PARADE and CYPHEX photolysis rates for a large number of trace gases were measured with a commercial single monochromator spectroradiometer (Meteorologie Consult GmbH, Glashütten, Germany), while in all other campaigns J(NO2) was measured with a filter radiometer (Metorologie Consult GmbH, Glashütten, Germany). Based on a correlation analysis between measured J(NO2) and measured J(H2O2) during PARADE and CYPHEX, a second-order correlation function was determined (J(H2O2) =0.015 J(NO2)2+0.0004 J(NO2) , R2=0.99), which was used to calculate J(H2O2) from measured J(NO2) during DOMINO, HUMPPA and HOPE. We estimate the total uncertainty of the H2O2 photolysis rates obtained by this method to be of the order of 10 %.
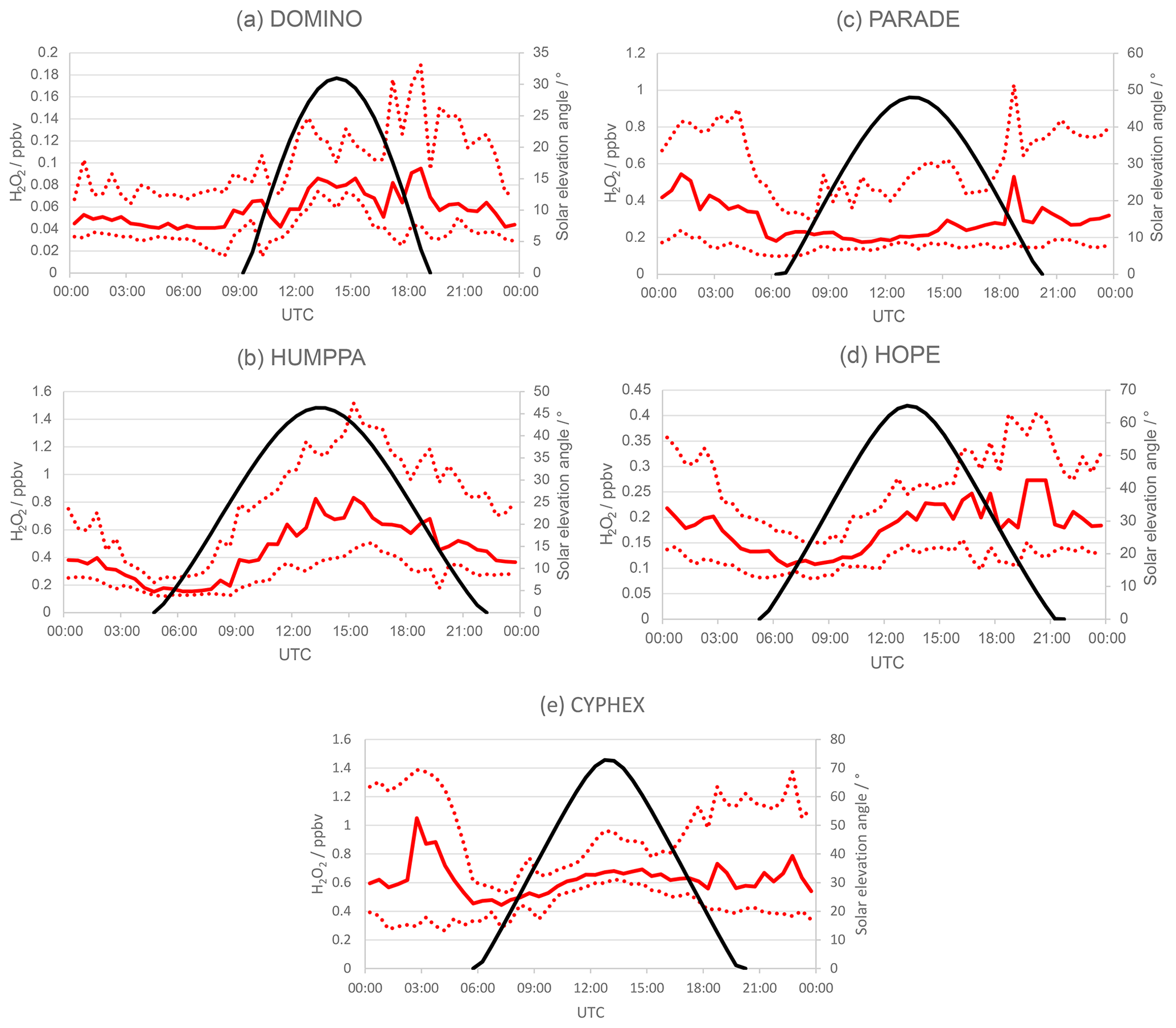
Figure 2Hydrogen peroxide diel variation of median mixing ratios (solid read line) and 25 % and 75 % quartiles (dashed red lines) for 30 min bins obtained for (a) DOMINO, (b) HUMPPA, (c) PARADE, (d) HOPE and (e) CYPHEX. Solar elevation angle is shown in black.
3.1 Diel variations
Diel variations have been calculated for NOx, O3, OH, HO2, H2O2, and J(NO2) by binning the data into 30 min bins and calculating median values, 25 % and 75 % quartiles and minimum and maximum values for each bin (Figs. S1–S5 in the Supplement). We use the medians instead of means to be less sensitive to outlier values, e.g. due to measurements below the detection limit or rain events. For the same reason, we use quartiles instead of standard deviations. Table S1 in the Supplement lists the data coverage (in %) for each species measured during the individual campaigns. Complete coverage (100 %) refers to uninterrupted measurements throughout the campaign time given in Table 1. In general, data coverage is less than 100 % due to calibrations, instrument maintenance and failure. Figure 2 shows the H2O2 diel variations and solar elevation angle for DOMINO, HUMPPA, PARADE, HOPE and CYPHEX, respectively. Visual inspection of the H2O2 diel variations indicates two groups with different behaviour: sites on flat terrain like those encountered during DOMINO (Fig. 2a) and HUMPPA (Fig. 2b) versus hilltop sites probed during PARADE (Fig. 2c), HOPE (Fig. 2d) and CYPHEX (Fig. 2e). The first group (flat terrain) exhibits local minima in the early morning hours between 05:30 and 08:30 UTC during DOMINO and between 04:30 and 07:30 UTC during HUMPPA, corresponding to local times between 06:00 and 09:00. Sunrise during DOMINO and HUMPPA was around 07:30 and 04:00 UTC, respectively. These minima are followed by steep increases in the H2O2 mixing ratios, reaching broad maxima between local noon and the early afternoon (DOMINO: 12:00–16:00 UTC; HUMPPA: 12:00–18:00 UTC), followed by a slow decrease during the late afternoon into the night. At these sites the daytime H2O2 mixing ratios are significantly higher than during the night, and the diel variations are similar to those observed for O3 (Figs. S1b and S2b). This is typical of the behaviour of a photochemically produced species in the continental boundary layer at a site with no significant orographic features. It is due to the interplay between net photochemical production during the day and strong deposition loss, scaling inversely with the variation of the boundary layer height.
The second group of sites is situated on hilltops and shows different characteristics. Although H2O2 mixing ratios during PARADE (Fig. 2c), HOPE (Fig. 2d) and CYPHEX (Fig. 2e) exhibit similar local minima in the early morning hours and increasing mixing ratios afterwards with maximum values between noon and the early afternoon, the nighttime mixing ratios are often higher than during the day. A similar evolution was observed for O3 (Figs. S3b–S5b) and is typical of mountainous sites with upslope air flow during the day due to local heating of the mountain slopes and descending air flow due to cooling during the night (Zaveri et al., 1995). Comparable H2O2 diel profiles have been described previously during observations at Mauna Loa, Hawaii (Heikes, 1992), and at Izana, Tenerife (de Reus et al., 2005). The higher mixing ratios of H2O2 and O3 during the night are generally due to sampling from higher altitudes (the nocturnal residual layer or the free troposphere), where mixing ratios for both species are expected to be higher as deposition losses are negligible.

Figure 3Relationship between H2O2 and HO2 for the five campaigns. Note that only daytime values (JNO2 > 10−3 s−1) have been used for the calculation of the median values and the 25 %–75 % quartiles.
3.2 Median values and dependence on HOx and NOx
Median H2O2 mixing ratios averaged across the diel cycle vastly differ from site to site. Here we investigate the causes of these differences by plotting campaign median (25–75 % quartiles) H2O2 mixing ratios versus median (and quartiles) HO2 and NOx, respectively (Figs. 3 and 4). The lowest H2O2 mixing ratios are observed for DOMINO, with median values (25 % to 75 % quartiles given in parentheses) of 58 pptv (37–91 pptv) for 24 h averages. Daytime mixing ratios (filtered by J(NO2) > 10−3 s−1) are slightly higher: 72 pptv (49–94 pptv). This was expected since DOMINO is the only campaign that took place in the early winter, when HOx levels and thus the oxidizing capacity of the atmosphere are generally lower. Higher mixing ratios are obtained during HOPE, 169 pptv (108–267 pptv), PARADE, 270 pptv (148–585 pptv), HUMPPA, 382 pptv (209–786 pptv), and CYPHEX, 601 pptv (420–936 pptv). Daytime-only values during HUMPPA are higher than the 24 h averages (473 pptv (227–907 pptv)), similar to observations at DOMINO. This is in line with the discussion of the diel variations in Sect. 3.1, where it was found that flat-terrain sites exhibit higher H2O2 mixing ratios during the day compared to the night. For the mountainous sites, there is no significant difference between the 24 h averages listed above and the daytime-only values: HOPE 155 pptv (103–241 pptv), PARADE 230 pptv (153–452), and CYPHEX 596 pptv (444–762 pptv).
These mixing ratios are consistent with previous observations over Europe, which indicated a general tendency for the highest mixing ratios in the summer season and lowest during winter; e.g. Morgan and Jackson (2002) observed a mean mixing ratio of 1.58 ppbv in June 1999 at Mace Head (Ireland) during the PARFORCE campaign and 0.23 ppbv in September 1998. This kind of seasonal variation is also observed at higher altitudes: Fels and Junkermann (1994) observed an average concentration of approximately 750 pptv of H2O2 in the summer of 1990 at an Alpine mountain station (Wank, Germany), while lower values of 185±233 pptv were reported for February/March 2006 at the neighbouring Jungfraujoch (Switzerland) (Walker et al., 2006). Airborne observations in the continental boundary layer (below 2 km) over Europe confirm this tendency, with mean (±1σ, standard deviation) mixing ratios of 0.55±0.37, 1.72±1.34, 1.74±0.75 and 0.92±0.47 ppbv during March 2004, July 2003, July 2007 and October 2006, respectively (Klippel et al., 2011).
With respect to diel variations, previous studies confirm the differences found here between mountainous sites and those at flat terrain. Fischer et al. (1998) reported higher H2O2 mixing ratios at night (∼2.4 ppbv) than during the day (2.1 ppbv) at high-altitude site Izana (Tenerife) during July/August 1993. This result was confirmed in July/August 2002 at the same side by de Reus et al. (2005). Average daytime mixing ratios were 1.24 ppbv±0.38 ppbv, increasing to 1.72±0.55 ppbv during the night. Contrary to mountainous and flat continental sites, coastal sites often exhibit no or only weak diel variations (e.g. Sauer et al., 1997; Morgan and Jackson, 2002) in line with the observations during DOMINO. Strong diel variations with daytime maxima have been reported for Tabua (Portugal) in June/July 1994 (night: < 15 pptv; day: 0.45±0.33 ppbv) (Sauer et al., 2001), Zagreb (Croatia) in the summer of 2004 (night: 0.2±0.35 ppbv; day: 0.4±0.56 ppbv) (Acker et al., 2008), and Waldstein (Germany) in July/August 2001 (night: ∼0.1 ppbv; day: ∼0.6 ppbv) (Ganzeveld et al., 2006; Valverde-Canossa et al., 2006). In general, the H2O2 mixing ratios and diel variations reported in this study are in good agreement with previous observations for similar locations.
Based on airborne measurements in the continental boundary layer over Europe, Klippel et al. (2011) reported a significant latitudinal gradient of H2O2, with decreasing mixing ratios at increasing latitude, reflecting decreasing HOx and photochemical activity. This behaviour is only partly reproduced in the present study, indicating that other local effects have a strong influence on the mixing ratio of H2O2 at ground level. A suitable measure for the photochemical activity (or the oxidizing power of the lower troposphere) is the HO2 concentration during the day, which is also a precursor of H2O2 according to Reaction (R1). In Fig. 3 we therefore plot the daytime median H2O2 and HO2 mixing ratios against each other at the five measurement locations. The 25 % and 75 % percentiles are also plotted. In general, the range of mixing ratios for an individual site is too small to yield significant correlations, but by comparing different environments, this limitation is removed. Figure 3 indicates a strong positive correlation between H2O2 mixing ratios and its precursor HO2. Due to the quadratic dependency of the H2O2 production rate on [HO2] (Reaction R1), one expects that the mixing ratio of H2O2 will exhibit a quadratic relation as well. The data in Fig. 3 can be subdivided into two groups at median HO2 between 3 and 6 pptv (DOMINO, PARADE and HOPE) and those at higher HO2 levels (18–24 pptv during CYPHEX and HUMPPA). Visual inspection suggests a roughly linear relation between H2O2 and HO2. This is confirmed by a linear regression analysis based on median values, which yields a regression coefficient R2 of 0.73. Plotting H2O2 versus (HO2)2 (not shown) yields a smaller R2 of 0.61. This lower R2 is largely due to the HUMPPA data point whereby a lower H2O2 mixing ratio was measured at higher HO2 compared to CYPHEX. As reported by Hens et al. (2014), HO2 measurements with the HORUS instrument are prone to interferences from peroxy radicals, in particular from alkene-based peroxy radicals, which are expected to be most abundant in forest environments, e.g. during HUMPPA. In a recent modelling study, Crowley et al. (2018) determined the contribution of RO2 to the measured HO2 during the daylight hours to be of the order of 30 % around noon and close to 100 % around sunrise and sunset. If we correct the HUMPPA data by a weighted all-day value of 50 % for this potential interference, median daytime HO2 is reduced from 24 pptv (14 to 35 pptv for the 25 %–75 % percentiles) to 12 pptv (7–17.5 pptv). Thus, the data point for HUMPPA in Fig. 3 shifts to the left of the CYPHEX data point. A regression analysis of H2O2 versus (HO2)2 (not shown) with the reduced HUMPPA HO2 results in R2 of 0.9, yielding much better agreement with the hypothesis that the measured H2O2 follows a quadratic dependence on HO2. For further calculations on the HUMPPA data set, we have used the corrected HO2 data. For DOMINO the HO2 observations that were also measured at high NO additions were not corrected due to very low concentrations of biogenic VOCs (Sinha et al., 2012).
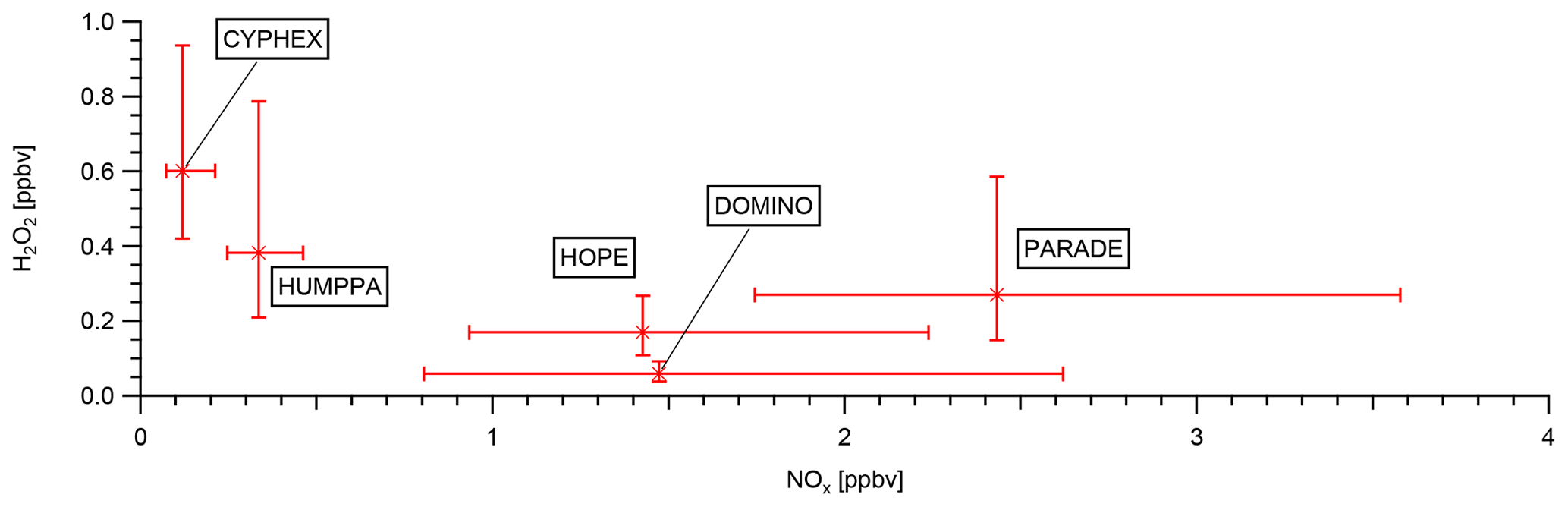
Figure 4Relationship between H2O2 and NOx for the five campaigns. Note that all data (day and night) have been used for the calculation of the median values and the 25 %–75 % quartiles.
Since the concentration of H2O2 according to Eq. (4) depends strongly on HO2, it is to be expected that the competing reaction of HO2 with NO (Reaction R3) will also have an effect. In Fig. 4, we therefore plot the median mixing ratio (25th–75th percentiles) of H2O2 versus the median mixing ratio (25th–75th percentiles) of NOx at the five measurement locations. Please note that, contrary to Fig. 3, we use data obtained during both day and night. Restriction of the analysis to daytime data only, as has been done in Fig. 3, will not change the results. As can be expected, there is a negative correlation between H2O2 and NOx, with the highest H2O2 mixing ratio observed at the lowest NOx values.
Besides being dependent on HO2 and NOx, H2O2 mixing ratios also show a positive (linear) correlation with O3 (Fig. S6), which is an indication of the dependence of H2O2 on photochemical activity. It is expected that higher O3 will be accompanied by higher HOx levels and thus an increasing H2O2 production rate.
Overall, it can be concluded that the H2O2 mixing ratios strongly depend on local oxidation rates represented by HOx and O3 levels. Higher photochemical activity leads to higher concentrations of H2O2. Nitrogen oxides play a key role in modulating the HOx partitioning and thus affect H2O2 levels indirectly by influencing the HO2 concentrations available. In order to study the role of physical processes (deposition and transport) in local H2O2 mixing ratio levels, we will next evaluate the H2O2 budget according to Eq. (1).
3.3 Hydrogen peroxide budgets
Equation (1) describes the temporal evolution of H2O2 neglecting scavenging by particles, cloud processing and wet deposition. Rain events and cloud processing that could result in total H2O2 removal were rare during the campaigns. Median values and 25 % and 75 % percentiles do not include such events. Therefore, we can neglect wet deposition in the analysis of Eq. (1). In the following, we concentrate on the observed increases in H2O2 during the period between sunrise and midday. During this period, net photochemical production, dry deposition and vertical entrainment associated with the growth of the boundary layer are expected to influence the observed change in H2O2. Based on a comparison of the mean observed change (dH2O2∕dt in pptv s−1) with calculations of the mean net production rate (Pchem–Lchem) and the deposition loss, we estimate potential entrainment rates during the growth of the boundary layer, neglecting horizontal advection. Please note that horizontal advection reflecting different air-mass origins will affect the absolute values of hydrogen peroxide, while the relative increase between sunrise and noon is mainly affected by local processes.
The chemical production (Eq. 2) and destruction (Eq. 3) rates for H2O2 in pptv s−1 are shown as a function of local time in Fig. 5a and b, respectively:
The calculation of the production term according to Eq. (2) is based on the rate coefficient k(R1) following the IUPAC recommendation, which takes into account an enhancement of the rate coefficient by water vapour (Atkinson et al., 2004; http://iupac.pole-ether.fr, last access: 27 August 2019). Measured water vapour concentrations varied between 0.9 % (DOMINO) and 2.2 % (HUMPPA), leading to enhancement factors of the reaction coefficient of Reaction (R)1 between 1.5 and 2.2. However, it is the difference in median HO2 concentrations that leads to the large variability in Pchem derived for the different campaigns (Fig. 5a). Maximum noontime H2O2 production rates were 0.0015 pptv s−1 (DOMINO), 0.004 pptv s−1 (PARADE), 0.017 pptv s−1 (HOPE), 0.04 pptv s−1 (HUMPPA) and 0.13 pptv s−1 (CYPHEX). This variation by 2 orders of magnitude reflects the dependence of Pchem on the HO2 precursor concentrations (Fig. 3), which are highest for those sites with the lowest NOx concentrations (Fig. 4). Median nighttime production was ∼0 pptv s−1 for DOMINO, PARADE, HOPE and CYPHEX, but between 0.01 and 0.02 pptv s−1 during HUMPPA, due to elevated HO2 concentrations during the night (∼20 pptv in Fig. S2d), which is most likely an artefact due to a RO2 interference of the HO2 measurements (Crowley et al. 2018). The total uncertainty is calculated by error propagation:
The largest contribution to the overall uncertainty is from atmospheric variability calculated from the 25 %–75 % percentiles of the input parameters H2O2 and HO2 used to calculate Pchem according to Eq. (2). The uncertainty in the rate constant is neglected, since it is much smaller that the atmospheric variability of the precursors. The uncertainty of Pchem is ±61 %, ±80 %, ±90 %, ±80 %, and ±35 % for PARADE, DOMINO, HUMPPA, HOPE and CYPHEX, respectively.

Figure 5Photochemical production and loss of H2O2 in pptv s−1. Note that the time used is local time.
Absolute differences in the photochemical destruction rates (Eq. 3) for the individual campaigns differ by an order of magnitude. Maximum Lchem values for DOMINO, PARADE, HOPE, HUMPPA and CYPHEX are −0.001, −0.001, −0.0025, −0.0046 and −0.015 pptv s−1, respectively. During the night, photochemical loss was zero during all the campaigns. Photolysis (Reaction R4) is the dominant photochemical H2O2 sink during CYPHEX (∼70 %), HUMPPA (∼75 %) and PARADE (∼90 %). During DOMINO and HOPE, photolysis and reaction with OH (Reaction R3) are of similar magnitude. Calculation of the uncertainty of Lchem according to Eq. (4) based on the atmospheric variability of the variables in Eq. (3) (i.e. H2O2, OH and J(H2O2)) yields ±86 %, ±62 %, ±76 %, ±72 %, and ±39 % for PARADE, DOMINO, HUMPPA, HOPE and CYPHEX, respectively.
Due to the much higher value of Pchem relative to Lchem (at least 1 order of magnitude), the net chemical production rate (NPR = Pchem−Lchem) is similar to Fig. 5a. The only exception is DOMINO, where the photochemical sources and sinks of H2O2 are almost balanced. It is interesting to evaluate observed H2O2 trends according to Eq. (1).
In order to calculate the effect of dry deposition we use two different approaches. The dry deposition of a trace gas depends on its loss at a surface (described by a surface resistance) and transport to the surface, mainly by turbulence. During the night the transport term is small due to low turbulence and thus we expect a low deposition velocity. In a first step, we therefore estimate the deposition loss rate constant from the decrease in H2O2 mixing ratios during the night, when photochemical production and loss as well as vertical entrainment are assumed to be negligible. This estimate of the dry deposition sink is a lower limit, since it neglects thermally driven turbulence and dry deposition due to stomatal uptake by vegetation, which occurs only during the day and does not take into account day–night changes in the rate of turbulent transport to the ground (e.g. Nguyen et al., 2015). In order to account for the contribution of enhanced turbulence and stomatal uptake, as a second step we also estimate dry deposition loss during local noontime. During this time of the day, associated with maximum H2O2 mixing ratios, it can be assumed that the daytime boundary layer is fully established and vertical intrusion is at minimum. Concentrating on periods with , only net chemical production, dry deposition and horizontal advection will influence the concentration of H2O2.
For these two cases we calculate an average loss rate kd constant according to
and the deposition velocity vd as
with BLH the boundary layer height in centimetres. The factor x takes into account a potential gradient in the mixing ratio profile. During the night x=2, assuming a linear increase in the mixing ratio with height in the nocturnal boundary layer (Shepson et al., 1992). During the day, we assume that the boundary layer is well mixed and x is equal to 1.
Table 2Calculation of the nighttime dry deposition loss rate kd and the deposition velocity vd. Uncertainties are reported in %.

Table 2 lists the time span over which d[H2O2]∕dt was analysed, the change in H2O2 mixing ratio ΔH2O2, the mean d[H2O2]∕dt, kd, BLH and vd. Values for the BLH during the night were taken from van Stratum et al. (2012) for DOMINO, Ouwersloot et al. (2012) for HUMPPA, and Berkes et al. (2016) for PARADE. For HOPE boundary layer height measurements are not available, so the nocturnal BLH was estimated to be 200 m, similar to measurements at the other sites. We assign an uncertainty of 20 % to all BLH values. Note that for CYPHEX this method cannot be applied, since the nighttime mixing ratios of H2O2 exhibit a tendency to increase while the hilltop extends into the free troposphere. Here, the observed decrease in H2O2 in the early morning occurs during sunlit hours. For the estimation of the nighttime deposition velocities we follow the approach of Shepson et al. (1992) and Hall and Claiborn (1997), assuming that the deposition loss is a first-order loss process resulting in an exponential decrease in H2O2 (Hall and Claiborn, 1997). Additionally, we assume a linear H2O2 gradient throughout the nocturnal boundary layer (Shepson et al., 1992). Please note that nighttime production of H2O2 due to the ozonolysis of alkenes is neglected in this approach, leading to a potential underestimation of the deposition velocities, in particular in environments with large biogenic emissions such as during HUMPPA. Estimated deposition velocities varied between 0.18 and 0.6 cm s−1 (Table 2) with a total uncertainty between ±53 and ±105 %. These values are similar to values for the H2O2 dry deposition velocity found in the literature. Walcek (1987) reported a value of 1 cm s−1 over the north-east of the USA, while Baer and Nester (1992) estimated an average vd of 1.5 cm s−1 for the upper Rhine Valley (Germany). From airborne measurements over the tropical rainforest in Suriname, Stickler et al. (2007) deduced a H2O2 deposition velocity of 1.35 cm s−1. Higher values of vd up to 5–10 cm s−1 are reported over forested regions due to enhanced uptake by stomatal openings (Hall and Claiborn, 1998; Valverde-Canossa et al., 2006; Nguyen et al., 2015). The nighttime vd values listed in Table 2 can thus be assumed to be lower limits of daytime values, since the effect of vegetation and enhanced turbulence is not accounted for.
Table 3Calculation of daytime dry deposition loss rate kd and the deposition velocity vd. Uncertainties are reported in %.
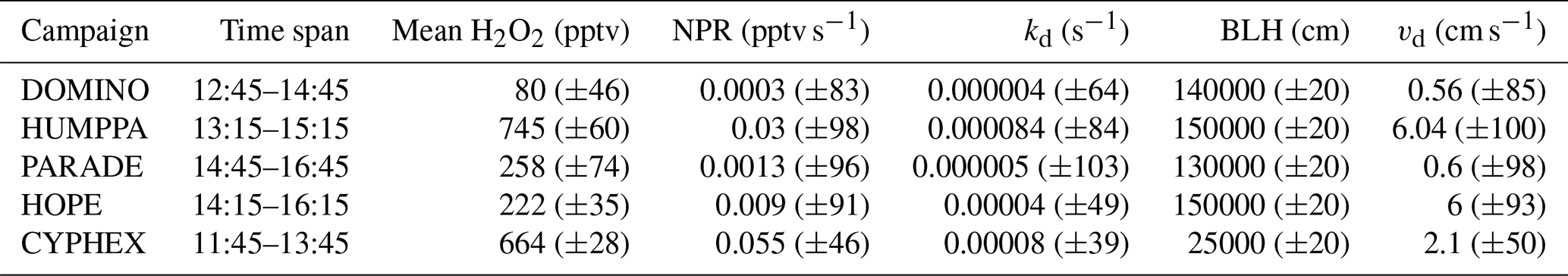
The daytime analysis of vd has been performed for periods of the day in which H2O2 can be assumed to be in a photostationary state (). This criterion is generally met between local noon and the early afternoon when the BLH is highest and vertical entrainment can be neglected. For the calculation of vd in Table 3 we assume that net H2O2 production (NPR =Pchem–Lchem) is balanced by dry deposition loss. The deposition velocities for DOMINO (0.56 cm s−1; uncertainty ±85 %) and PARADE (0.6 cm s−1; uncertainty ±98 %) are about a factor of 2 to 3 higher than the nighttime values for these sites documented in Table 2, while significantly higher daytime vd values (factor of 10 to 20) (Table 3) are calculated for HUMPPA and HOPE. The value of vd (day) for CYPHEX of 2.1 cm s−1 (uncertainty ±50 %) is within the range of observation at other sites both investigated here and those cited in the literature. The daytime vd value obtained for HOPE (6 cm s−1; uncertainty ±93 %) is also within the range of values reported in the literature for forested environments (Hall and Claiborn, 1997; Valverde-Canossa et al., 2006; Nguyen et al., 2015), while the value obtained for HUMPPA (6.04 cm s−1; uncertainty ±100 %) is comparable to the high values reported for a boreal forest in Canada (Hall and Claiborn, 1997). Note that uncertainties were calculated according to Eq. (4), taking into account the variability of all input variables to Eq. (1) derived from the 25 %–75 % range and an uncertainty of 20 % for the boundary layer height.
To evaluate Eq. (1) from sunrise to midday during the five campaigns we use the net photochemical production of H2O2 presented in Fig. 5 and calculate the deposition loss during the increase in the boundary layer height. For this calculation, we linearly interpolate the deposition velocity between the nighttime values presented in Table 2 and the noontime values presented in Table 3. For CYPHEX we use an average of all nighttime deposition velocities presented in Table 3. As mentioned before, during this period it is expected that vertical entrainment due to an increasing boundary layer height and horizontal advection will also have an effect on the temporal evolution of H2O2. The mean rate of d[H2O2]∕dt is derived from the observed H2O2 mixing ratio increase from the early morning minimum up to the maximum around local noon in Fig. 2. Average net photochemical production rates (Pchem–Lchem) and dry deposition loss rates over the periods for which d[H2O2]∕dt was analysed were derived from Eqs. (2), (3) and (7), respectively.
The residual (d[H2O2]∕dt–((Pchem–Lchem)–Ldep)) according to Eq. (1) is a measure of gain or loss of H2O2 due to the combination of vertical entrainment and horizontal advection. Table 4 lists the time periods over which d[H2O2]∕dt was analysed, the mean H2O2 mixing ratio (pptv), mean d[H2O2]∕dt (pptv h−1), mean net photochemical production rate (Pchem–Lchem) (pptv h−1), the mean boundary layer height (BLH) (cm), the deposition loss rate (Ldep) (pptv h−1) and the transport rate (Ptrans) (pptv h−1). Uncertainties in percentage were calculated from Eq. (4) based on the variabilities of the input variables. Positive residuals indicate vertical entrainment or advection of higher H2O2 mixing ratios, and negative values indicate dilution. The budget of net photochemical production, deposition loss, observed change in H2O2 mixing ratios from sunrise to noon and the inferred residual transport are graphically shown in Fig. 6. For DOMINO the calculated net photochemical production (1.3 pptv h−1) is of the same order of magnitude as the loss rate due to deposition (L pptv h−1), indicating a balance between sources and sinks of H2O2. Thus the observed increase in H2O2 (7.9 pptv −1h) during the morning is almost completely due to transport (7.6 pptv h−1).
Table 4Calculation of dry deposition loss Ldep and entrainment rate Pent. Uncertainties are reported in %.
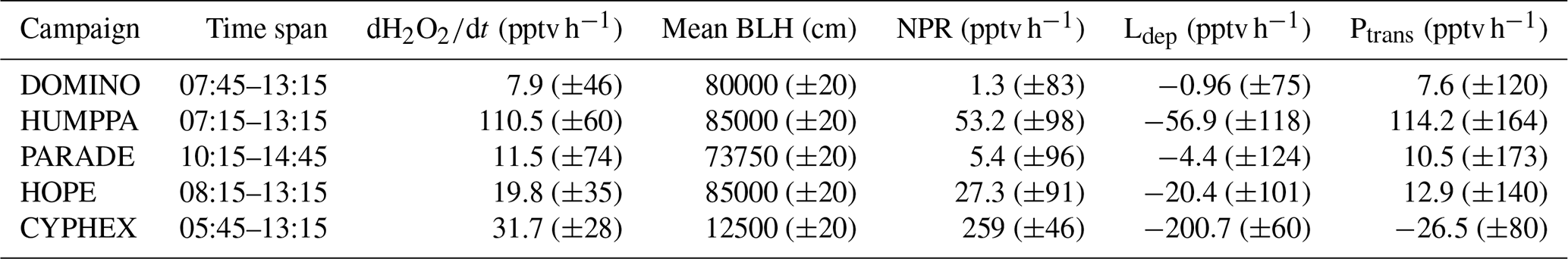
During PARADE the net production is 5.4 pptv h−1, which is also largely balanced by deposition loss (−4.4 pptv h−1), resulting in a positive residual indicating a missing source of the order of 10.5 pptv h−1. Since the PARADE site is on a hilltop it is likely that entrainment from the residual layer is responsible for this transport.
During HUMPPA the net photochemical production of 53.16 pptv h−1 is only slightly smaller than the deposition loss (−56.9 pptv h−1), resulting in a rather large entrainment rate of the order of 114.2 pptv h−1 required to explain the observed H2O2 increase. Since the surrounding area is rather homogeneous (Williams et al., 2011), we assume that this transport is due to vertical entrainment from the residual layer during the rise in the boundary layer height. The deduced entrainment rate of 118.3 pptv h−1 is an upper limit since we most likely underestimate the net production rate of H2O2. Axinte (2016) estimated that the ozonolysis of terpenes in the boreal forest would lead to an additional H2O2 production of the order of 8.3 pptv h−1, enhancing the net production by 7.5 %.
Slightly higher net production compared to deposition losses is observed for HOPE (net production =27.3 pptv h−1, deposition loss pptv h−1). This yields a contribution of 12.9 pptv h−1 from transport. Since HOPE was performed on a mountaintop, we assume that this increase is due to vertical entrainment during the growth of the boundary layer. Contrary to the other sites discussed above where close to 100 % of the morning increase in H2O2 was due to transport, this contribution is only 65 % during HOPE.
During CYPHEX, the net production of 259 pptv h−1 is only partly balanced by a dry deposition loss of −200.7 pptv h−1. Thus, the photochemical production of H2O2 minus deposition (58.3 pptv h−1) is slightly larger than the observed increase during the early morning (31.7 pptv h−1), yielding a negative residual of −26.5 pptv h−1, indicating a dilution. Since CYPHEX was performed at a high-altitude coastal site affected by a land–sea breeze, it is likely that the advection of marine air masses with slightly lower H2O2 mixing ratios is responsible for this dilution effect.
Please note that the error propagation according to Eq. (4) includes measurement uncertainties and atmospheric variability, with the latter being the dominant term. Exclusion of the atmospheric variability would result in much smaller errors. Therefore, the stated values for net production, deposition and transport are best estimates for the median values, while the error bars reflect atmospheric variability and are thus a very conservative measure of the uncertainty.
Besides the large uncertainty resulting from atmospheric variability affecting the median profiles (see Tables 2 to 4), this analysis is also influenced by uncertainties associated with respect to data coverage and limitations in the method to derive deposition velocities and subsequently transport rates, in particular during the day. These limitations will be discussed in the context of the HUMPPA campaign. This campaign is particularly suitable for this purpose since two other studies have been published that specifically address the temporal evolution of H2O2 using a box model (Crowley et al., 2018) and the contribution of entrainment to the early morning increase in O3 (Ouwersloot et al., 2012) for this particular campaign. Results from HUMPPA will also apply to the other campaigns discussed here.
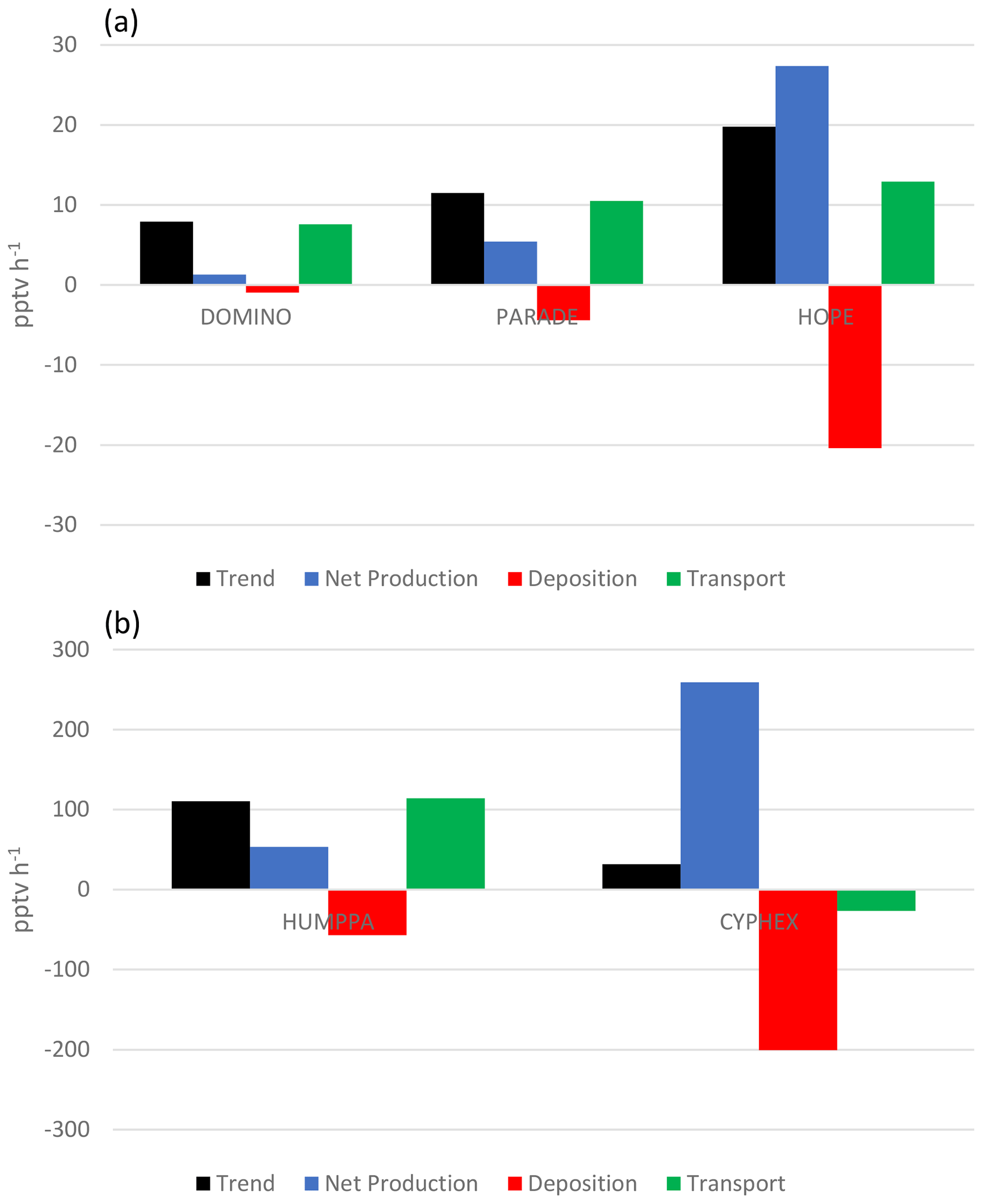
Figure 6Budget of the H2O change from the sunrise to midday for the individual campaigns. Trends, net production, deposition and transport are given in pptv h−1. Note the scale change from DOMINO, PARADE and HOPE (upper panel) to HUMPPA and CYPHEX (lower panel).
Uncertainties due to missing data are mainly due to gaps in measurements of radical species. Table S1 indicates that during HUMPPA data coverage for HO2 was only 14.7 %, so that it is questionable whether a median diel cycle based on this relatively small data set is representative of the whole campaign. Additionally, a potential interference in the HO2 observations by RO2 radicals (Hens et al., 2014) will also affect the H2O2 production rate. Crowley et al. (2018) used a box model to study PAA, PAN and H2O2 during HUMPPA, deriving HO2 concentrations that fit the temporal evolution of these species between 20 July and 12 August 2010. The limited measurements of HO2 during HUMPPA that cover approximately 12 days are in good agreement with the modelled data that cover a longer period (23 days). According to Eq. (2), using the modelled HO2 data from Crowley et al. (2018) leads to an approximately 23 % higher H2O2 production rate. The main effect of this higher production rate would be a higher deposition velocity derived from the steady-state assumption around noon, yielding a deposition velocity of 7.4 cm s−1 instead of 6 cm s−1 similar to the maximum value of 8.4 cm s−1 used by Crowley et al. (2018). The deposition velocity derived during the night by Crowley et al. (2018) is slightly larger than our estimate (0.8 cm s−1 versus 0.6 cm s−1), since Crowley et al. took the nighttime production of H2O2 via ozonolysis of terpenes into account, which was not considered in this study and which leads to an underestimation of the nocturnal deposition loss in this study. Since in our study the inferred entrainment rate strongly depends on the deposition sink, uncertainties in derived deposition velocities will linearly affect the entrainment flux needed to explain the morning rise in H2O2. Note that the deposition velocities used by us and Crowley et al. (2018) are in rather good agreement with observation-based estimates published in the literature (Hall and Claiburn, 1998; Valverde-Canossa et al., 2006; Nguyen et al., 2015), but are much higher than values used in the EMAC model for the boreal forest in Finland (∼0.2 cm s−1 at night and 0.8–1 cm s−1 during the day) (Jöckel et al., 2016). Using these low deposition velocities would yield a deposition loss of only 8.9 pptv h−1 instead of 118 pptv h−1 and thus a transport contribution of only 8.7 pptv h−1 (7.8 % of the morning increase). This illustrates that the H2O2 budget terms for deposition and transport in this study are highly coupled and depend strongly on the deposition velocities used.
Other potential error sources are trends in H2O2 mixing ratios over the campaign. While this study covers the whole period of the HUMPPA campaign (12 July until 12 August 2010), the model study by Crowley et al. (2018) started on 20 July 2010, missing the first week of the campaign. During this warm period, noontime H2O2 mixing ratios were higher than during the rest of the campaign, affecting the median increase after sunrise. Therefore, we obtained larger values of d[H2O2]∕dt over the whole campaign compared to the reduced period used by Crowley et al. (2018). Note that a smaller value for d[H2O2]∕dt at constant net-production and deposition loss yields a smaller residual, i.e. less transport. This is the reason that Crowley et al. (2018) did not include transport in their study.
In general, the results from our study are in good agreement with a H2O2 budget calculation for a coniferous forest in southern Germany based on a single-column chemistry-climate model made by Ganzeveld et al. (2006). They conclude that turbulent exchange is similar in magnitude to the deposition loss and much larger than net photochemical production. Since H2O2 and O3 have similar vertical profiles between the surface and the top of the boundary layer due to strong depositional sinks at the surface, they should behave similarly with respect to entrainment. Ouwersloot et al. (2012) simulated the O3 budget during HUMPPA with a single-column model, taking into account photochemical production, depositional loss and vertical transport, indicating that the rise in boundary layer height in the early morning and the subsequent in-mixing of residual layer air is responsible for the majority of the observed O3 increase.
The potential role of entrainment can also be illustrated by a simple scheme taking into account a two-box mixing process. If we assume that the H2O2 mixing ratio in the residual layer during the night is uniform and constant due to the absence of sinks (no photochemical production or loss and no deposition due to its isolation from the surface by the nocturnal inversion), this air will be mixed with air masses in the nocturnal boundary layer during the early morning rise in the BLH. The H2O2 mixing ratio in the nocturnal boundary layer just before sunrise is 260 pptv during HUMPPA. We further assume that the H2O2 mixing ratio in the residual layer is a remnant from the previous day with a mixing ratio of 600 pptv measured in the late afternoon at 16:15 UTC (17:45 local time). For simplicity we assume that the height of the nocturnal boundary layer is 200 m and the height of the residual layer corresponds to the top of the boundary layer (1500 m), yielding a depth of the residual layer of 1300 m. Mixing of these two boxes during the morning over a period of 6 h and taking into account the depth of both layers yields a H2O2 increase of 49 pptv h−1, which is a factor of 2 smaller than the value of 114 pptv h−1 derived in Table 4. This difference might be due to the restriction to two boxes, neglecting additional entrainment from the free troposphere.
The budget of hydrogen peroxide in the continental boundary layer is defined by the balance between photochemical production and loss, physical removal by dry and wet deposition, as well as vertical entrainment into the boundary layer and horizontal advection. We used measurements of H2O2, its precursor HO2 and sinks (OH, J(H2O2)) at five European sites to calculate net photochemical production. Assuming horizontal homogeneity and negligible rainout, we estimated both the dry deposition loss and the entrainment rate. In general, absolute mixing ratios of H2O2 exhibit an inverse relation to local HOx levels. The net production is a strong function of HO2 and thus extremely sensitive to interferences in the measurement of this radical (Hens et al., 2014; Crowley et al., 2018). Calculated photochemical production rates generally exceed photochemical loss rates by at least an order of magnitude at all sites, except for one observation during the winter season (DOMINO) where production and loss are approximately equivalent. Estimates of deposition velocities during the night are of the order of 0.16–0.6 cm s−1 and thus at the lower end of values reported in the literature (Walcek 1987; Baer and Nester, 1992; Stickler et al., 2007; Hall and Claiborn, 1998; Valverde-Canossa et al., 2006; Nguyen et al., 2015). This is to be expected since deposition of H2O2 during the day is often enhanced by stomatal uptake (Hall and Claiborn, 1998; Valverde-Canossa et al., 2006; Nguyen et al., 2015). Daytime deposition rates during the five campaigns are consistently higher in forested areas and reach values of ∼6 cms−1, in good agreement with literature values (Hall and Claiborn, 1998; Valverde-Canossa et al., 2006; Nguyen et al., 2015). Using the individual terms for H2O2 photochemical production, photochemical loss and dry deposition, we could show that the early morning rise in H2O2 mixing ratios is influenced by dynamical processes. For DOMINO, HUMPPA and PARADE transport is responsible for almost all of the observed early morning increase in H2O2. Smaller contributions of transport are derived HOPE (65 %) and CYPHEX (10 %). This transport is most likely related to vertical entrainment from the residual layer during the early morning rise in the boundary layer height. As shown by aircraft measurements, strong deposition at the surface leads to increasing H2O2 mixing ratios with altitude up to the top of the boundary layer (Klippel et al., 2011), so that the entrainment during the early morning is a source of H2O2 (Fischer et al., 2015).
The findings of this study are in general agreement with previous studies of trace gas budgets for H2O2 (Ganzeveld et al., 2006; Stickler et al., 2007) and O3 (Ouwersloot et al., 2012; Kaser et al., 2017) in the continental boundary layer that emphasize the significant contribution of vertical entrainment in particular during the early morning hours. Nevertheless, the findings are rather qualitative since quantitative results strongly depend on the deposition velocity used in the budget calculations. In principle, the photochemical production and loss of H2O2 can be quantified by accurate local in situ measurements of precursors (mainly HO2) and losses due to photolysis and reaction with measured OH. The balance of net photochemical production, dry deposition and transport strongly depends on an accurate determination of the deposition velocity and its temporal evolution. Point measurements, as presented here, suffer from strong limitations in deriving deposition velocities and subsequently potential transport contributions to local budgets. Future studies should therefore include vertically resolved measurements, preferentially from the surface to the top of the boundary layer, and/or include flux measurements of the species of interest.
Readers who are interested in the data should contact Horst Fischer (horst.fischer@mpic.de).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-11953-2019-supplement.
HF, JNC, AP and JL designed the study. RA, HB, JNC, CE, SG, RJ, SH, HH, KH, RK, DK, CM, MM, AN, UW, CPD, ER, AR, TS, and JS conducted the measurements and processed the data. HF prepared the manuscript with contributions from all the co-authors.
The authors declare that they have no conflict of interest.
The article processing charges for this open-access publication were covered by the Max Planck Society.
This paper was edited by Markus Ammann and reviewed by two anonymous referees.
Acker, K., Kezele, N., Klasnic, L., Möller, D., Pehenec, G., Sorgo, G., Wieprecht, W., and Zuzul, S.: Atmospheric H2O2 measurement and modeling campaign during summer 2004 in Zagreb, Croatia, Atmos. Environ., 42, 2530–2542, https://doi.org/10.1016/j.atmosenv.2007.12.011, 2008.
Adame, J. A., Martínez, M., Sorribas, M., Hidalgo, P. J., Harder, H., Diesch, J.-M., Drewnick, F., Song, W., Williams, J., Sinha, V., Hernández-Ceballos, M. A., Vilà-Guerau de Arellano, J., Sander, R., Hosaynali-Beygi, Z., Fischer, H., Lelieveld, J., and De la Morena, B.: Meteorology during the DOMINO campaign and its connection with trace gases and aerosols, Atmos. Chem. Phys., 14, 2325–2342, https://doi.org/10.5194/acp-14-2325-2014, 2014.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Axinte, R.: The oxidation photochemistry and transport of hydrogen peroxide and formaldehyde at three site in Europe: trends budgets and 3D model simulations, PhD thesis, University of Mainz, Germany, 2016.
Baer, M. and Nester, K.: Parameterization of trace gas dry deposition velocities for a regional mesoscale diffusion model, Ann. Geophys., 10, 912–923, 1992.
Berkes, F., Hoor, P., Bozem, H., Kunkel, D., Sprenger, M., and Henne, S.: Airborne observation of mixing across the entrainment zone during PARADE 2011, Atmos. Chem. Phys., 16, 6011–6025, https://doi.org/10.5194/acp-16-6011-2016, 2016.
Crowley, J. N., Pouvesle, N., Phillips, G. J., Axinte, R., Fischer, H., Petäjä, T., Nölscher, A., Williams, J., Hens, K., Harder, H., Martinez-Harder, M., Novelli, A., Kubistin, D., Bohn, B., and Lelieveld, J.: Insights into HOx and ROx chemistry in the boreal forest via measurement of peroxyacetic acid, peroxyacetic nitric anhydride (PAN) and hydrogen peroxide, Atmos. Chem. Phys., 18, 13457–13479, https://doi.org/10.5194/acp-18-13457-2018, 2018.
Derstroff, B., Hüser, I., Bourtsoukidis, E., Crowley, J. N., Fischer, H., Gromov, S., Harder, H., Janssen, R. H. H., Kesselmeier, J., Lelieveld, J., Mallik, C., Martinez, M., Novelli, A., Parchatka, U., Phillips, G. J., Sander, R., Sauvage, C., Schuladen, J., Stönner, C., Tomsche, L., and Williams, J.: Volatile organic compounds (VOCs) in photochemically aged air from the eastern and western Mediterranean, Atmos. Chem. Phys., 17, 9547–9566, https://doi.org/10.5194/acp-17-9547-2017, 2017.
de Reus, M., Fischer, H., Sander, R., Gros, V., Kormann, R., Salisbury, G., Van Dingenen, R., Williams, J., Zöllner, M., and Lelieveld, J.: Observations and model calculations of trace gas scavenging in a dense Saharan dust plume during MINATROC, Atmos. Chem. Phys., 5, 1787–1803, https://doi.org/10.5194/acp-5-1787-2005, 2005.
Fels, M. and Junkermann, W.: The occurrence of organic peroxides in air at a mountain site, Geophys. Res. Lett., 21, 341–344, https://doi.org/10.1029/93GL01892, 1994.
Fischer, H., Nikitas, C., Parchatka, U., Zenker, T. Harris, G. W., Matuska, P., Schmitt, R., Mihelcic, D., Muesgen, P., Paetz, H.-W., Schultz, M., and Volz-Thomas, A.: Trace gas measurements during the Oxidizing Capacity of the Tropospheric Atmosphere campaign 1993 at Izana, J. Geophys. Res. 103, 13505–13518, https://doi.org/10.1029/97JD01497, 1998.
Fischer, H., Pozzer, A., Schmitt, T., Jöckel, P., Klippel, T., Taraborrelli, D., and Lelieveld, J.: Hydrogen peroxide in the marine boundary layer over the South Atlantic during the OOMPH cruise in March 2007, Atmos. Chem. Phys., 15, 6971–6980, https://doi.org/10.5194/acp-15-6971-2015, 2015.
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011.
Ganzeveld, L., Valverde-Canossa, J., Moortgat, G. K., and Steibrecher, R.: Evaluation of peroxide exchanges over a coniferous forest in a single-column chemistry-climate model, Atmos. Environm., 40, S68–S80, https://doi.org/10.1016/j.atmosenv.2006.01.062, 2006.
Gunz, D. W. and Hoffmann, M. R.: Atmospheric chemistry of peroxides: A review, Atmos. Environ., 24A, 1601–1633, https://doi.org/10.1016/0960-1686(90)90496-A, 1990.
Hall, B. D. and Claiborn, C. S.: Measurements of the dry deposition of peroxides to a Canadian boreal forest, J. Geophys. Res., 102, 29343–29353, https://doi.org/10.1029/97JD01113, 1997.
Heikes, B. G.: Formaldehyde and hydroperoxides at Mauna Loa Observatory, J. Geophys. Res., 97, 18001–10013, https://doi.org/10.1029/92JD00268, 1992.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Hosaynali Beygi, Z., Fischer, H., Harder, H. D., Martinez, M., Sander, R., Williams, J., Brookes, D. M., Monks, P. S., and Lelieveld, J.: Oxidation photochemistry in the Southern Atlantic boundary layer: unexpected deviations of photochemical steady state, Atmos. Chem. Phys., 11, 8497–8513, https://doi.org/10.5194/acp-11-8497-2011, 2011.
Hüser, I., Harder, H., Heil, A., and Kaiser, J. W.: Assumptions about footprint layer heights influence the quantification of emission sources: a case study for Cyprus, Atmos. Chem. Phys., 17, 10955–10967, https://doi.org/10.5194/acp-17-10955-2017, 2017.
Jackson, A. V. and Hewitt, C. N.: Atmospheric hydrogen peroxide and organic hydroperoxides: A review, Crit. Rev. Environ. Sci. Technol., 29, 175–228, https://doi.org/10.1080/10643389991259209, 1999.
Jöckel, P., Tost, H., Pozzer, A., Kunze, M., Kirner, O., Brenninkmeijer, C. A. M., Brinkop, S., Cai, D. S., Dyroff, C., Eckstein, J., Frank, F., Garny, H., Gottschaldt, K.-D., Graf, P., Grewe, V., Kerkweg, A., Kern, B., Matthes, S., Mertens, M., Meul, S., Neumaier, M., Nützel, M., Oberländer-Hayn, S., Ruhnke, R., Runde, T., Sander, R., Scharffe, D., and Zahn, A.: Earth System Chemistry integrated Modelling (ESCiMo) with the Modular Earth Submodel System (MESSy) version 2.51, Geosci. Model Dev., 9, 1153–1200, https://doi.org/10.5194/gmd-9-1153-2016, 2016.
Kaser, L., Patton, E. G., Pfister, G. G., Weinheimer, A. J., Montzka, D. D., Flocke, F., Thompson, A. M., Stauffer, R. M., and Halliday, H. S.: The effect of entrainment through atmospheric boundary layer growth on observed and model surface ozone in the Colorado Front Range, J. Geophys. Res.-Atmos., 122, 6075–6093, https://doi.org/10.1002/2016JD026245, 2017.
Klippel, T., Fischer, H., Bozem, H., Lawrence, M. G., Butler, T., Jöckel, P., Tost, H., Martinez, M., Harder, H., Regelin, E., Sander, R., Schiller, C. L., Stickler, A., and Lelieveld, J.: Distribution of hydrogen peroxide and formaldehyde over Central Europe during the HOOVER project, Atmos. Chem. Phys., 11, 4391–4410, https://doi.org/10.5194/acp-11-4391-2011, 2011.
Lazarus, A. I., Kok, G. L., Gitlin, S. N., and Lind, J. A.: Automated fluorometric method for hydrogen peroxide in atmospheric precipitation, Anal. Chem., 57, 917–922, https://doi.org/10.1021/ac00281a031, 1985.
Lazarus, A. I., Kok, G. L., Lind, J. A., Gitlin, S. N., Heikes, B. G., and Shetter, R. E.: Automated fluorometric method for hydrogen peroxide in air, Anal. Chem., 58, 594–597, https://doi.org/10.1021/ac00294a024, 1986.
Lee, M., Heikes, B. G., and O'Sullivan, D. W.: Hydrogen peroxide and organic hydroperoxide in the troposphere: a review, Atmos. Environ., 34, 3475–3494, https://doi.org/10.1016/S1352-2310(99)00432-X, 2000.
Lelieveld, J., Gromov, S., Pozzer, A., and Taraborrelli, D.: Global tropospheric hydroxyl distribution, budget and reactivity, Atmos. Chem. Phys., 16, 12477–12493, https://doi.org/10.5194/acp-16-12477-2016, 2016.
Li, J., Reiffs, A., Parchatka, U., and Fischer, H.: In situ measurements of atmospheric CO and its correlation with NOx and O3 at a rural mountain site, Metrol. Meas. Syst., XXII, 25–28, https://doi.org/10.1515/mms-2015-0001, 2015.
Mallik, C., Tomsche, L., Bourtsoukidis, E., Crowley, J. N., Derstroff, B., Fischer, H., Hafermann, S., Hüser, I., Javed, U., Keßel, S., Lelieveld, J., Martinez, M., Meusel, H., Novelli, A., Phillips, G. J., Pozzer, A., Reiffs, A., Sander, R., Taraborrelli, D., Sauvage, C., Schuladen, J., Su, H., Williams, J., and Harder, H.: Oxidation processes in the eastern Mediterranean atmosphere: evidence from the modelling of HOx measurements over Cyprus, Atmos. Chem. Phys., 18, 10825–10847, https://doi.org/10.5194/acp-18-10825-2018, 2018.
Martinez, M., Harder, H., Kubistin, D., Rudolf, M., Bozem, H., Eerdekens, G., Fischer, H., Klüpfel, T., Gurk, C., Königstedt, R., Parchatka, U., Schiller, C. L., Stickler, A., Williams, J., and Lelieveld, J.: Hydroxyl radicals in the tropical troposphere over the Suriname rainforest: airborne measurements, Atmos. Chem. Phys., 10, 3759–3773, https://doi.org/10.5194/acp-10-3759-2010, 2010.
Meusel, H., Kuhn, U., Reiffs, A., Mallik, C., Harder, H., Martinez, M., Schuladen, J., Bohn, B., Parchatka, U., Crowley, J. N., Fischer, H., Tomsche, L., Novelli, A., Hoffmann, T., Janssen, R. H. H., Hartogensis, O., Pikridas, M., Vrekoussis, M., Bourtsoukidis, E., Weber, B., Lelieveld, J., Williams, J., Pöschl, U., Cheng, Y., and Su, H.: Daytime formation of nitrous acid at a coastal remote site in Cyprus indicating a common ground source of atmospheric HONO and NO, Atmos. Chem. Phys., 16, 14475–14493, https://doi.org/10.5194/acp-16-14475-2016, 2016.
Morgan, R. B. and Jackson, A. V.: Measurement of gas-phase hydrogen peroxide and methyl hydroperoxide in the coastel environment during the PARFORCE project, J. Geophys. Res., 107, 8109, https://doi.org/10.1029/2000JD000257, 2002.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Novelli, A., Hens, K., Tatum Ernest, C., Kubistin, D., Regelin, E., Elste, T., Plass-Dülmer, C., Martinez, M., Lelieveld, J., and Harder, H.: Characterisation of an inlet pre-injector laser-induced fluorescence instrument for the measurement of atmospheric hydroxyl radicals, Atmos. Meas. Tech., 7, 3413–3430, https://doi.org/10.5194/amt-7-3413-2014, 2014.
Novelli, A., Hens, K., Tatum Ernest, C., Martinez, M., Nölscher, A. C., Sinha, V., Paasonen, P., Petäjä, T., Sipilä, M., Elste, T., Plass-Dülmer, C., Phillips, G. J., Kubistin, D., Williams, J., Vereecken, L., Lelieveld, J., and Harder, H.: Estimating the atmospheric concentration of Criegee intermediates and their possible interference in a FAGE-LIF instrument, Atmos. Chem. Phys., 17, 7807–7826, https://doi.org/10.5194/acp-17-7807-2017, 2017.
Ouwersloot, H. G., Vilà-Guerau de Arellano, J., Nölscher, A. C., Krol, M. C., Ganzeveld, L. N., Breitenberger, C., Mammarella, I., Williams, J., and Lelieveld, J.: Characterization of a boreal convective boundary layer and its impact on atmospheric chemistry during HUMPPA-COPEC-2010, Atmos. Chem. Phys., 12, 9335–9353, https://doi.org/10.5194/acp-12-9335-2012, 2012.
Reeves, C. E. and Penkett, S. A.: Measurements of peroxides and what they tell us, Chem. Rev. 103, 5199–5218, https://doi.org/10.1021/cr0205053, 2003.
Sauer, F., Limbach, S., and Moortgat, G. K.: Measurements of hydrogen peroxide and individual organic peroxides in the marine troposphere, Atmos. Environ., 31, 1173–1184, https://doi.org/10.1016/S1352-2310(96)00289-0, 1997.
Sauer, F., Schäfer, Ch., Neeb, P., Horie, O., and Moortgat, G. K.: Formation of hydrogen peroxide in the ozonolysis of isoprene and simple alkenes under humid conditions, Atmos. Environ., 33, 229–241, https://doi.org/10.1016/S1352-2310(98)00152-6, 1999.
Sauer, F., Beck, J. Schuster, G., and Moortgat, G. K.: Hydrogen peroxide, organic peroxides and organic acids in a forested area during FIELDVOC'94, Chemosphere – Global Change Science, 3, 309–326, https://doi.org/10.1016/S1465-9972(01)00013-7, 2001.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics, John Wiley & Sons, New York, USA, 299–302, 1998.
Sinha, V., Williams, J., Diesch, J. M., Drewnick, F., Martinez, M., Harder, H., Regelin, E., Kubistin, D., Bozem, H., Hosaynali-Beygi, Z., Fischer, H., Andrés-Hernández, M. D., Kartal, D., Adame, J. A., and Lelieveld, J.: Constraints on instantaneous ozone production rates and regimes during DOMINO derived using in-situ OH reactivity measurements, Atmos. Chem. Phys., 12, 7269–7283, https://doi.org/10.5194/acp-12-7269-2012, 2012.
Shepson, P. B., Bottenheim, J. W., Hastie, D. R., and Venkatram, A.: Determination of the relative ozone and PAN deposition velocities at night, Geophys. Res. Lett., 19, 1121–1124, https://doi.org/10.1029/92GL01118, 1992.
Sobanski, N., Tang, M. J., Thieser, J., Schuster, G., Pöhler, D., Fischer, H., Song, W., Sauvage, C., Williams, J., Fachinger, J., Berkes, F., Hoor, P., Platt, U., Lelieveld, J., and Crowley, J. N.: Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE, Atmos. Chem. Phys., 16, 4867–4883, https://doi.org/10.5194/acp-16-4867-2016, 2016.
Stickler, A., Fischer, H., Bozem, H., Gurk, C., Schiller, C., Martinez-Harder, M., Kubistin, D., Harder, H., Williams, J., Eerdekens, G., Yassaa, N., Ganzeveld, L., Sander, R., and Lelieveld, J.: Chemistry, transport and dry deposition of trace gases in the boundary layer over the tropical Atlantic Ocean and the Guyanas during the GABRIEL field campaign, Atmos. Chem. Phys., 7, 3933–956, https://doi.org/10.5194/acp-7-3933-2007, 2007.
Valverde-Canossa, J., Ganzeveld, L., Rappenglück, B., Steinbrecher, R., Klemm, O., Schuster, G., and Moortgat, G. K.: First measurements of H2O2 and organic peroxides surface fluxes by the relaxed eddy-accumulation technique, Atmos. Environ., 40, S55–S767, https://doi.org/10.1016/j.atmosenv.2006.03.038, 2006.
van Stratum, B. J. H., Vilà-Guerau de Arellano, J., Ouwersloot, H. G., van den Dries, K., van Laar, T. W., Martinez, M., Lelieveld, J., Diesch, J.-M., Drewnick, F., Fischer, H., Hosaynali Beygi, Z., Harder, H., Regelin, E., Sinha, V., Adame, J. A., Sörgel, M., Sander, R., Bozem, H., Song, W., Williams, J., and Yassaa, N.: Case study of the diurnal variability of chemically active species with respect to boundary layer dynamics during DOMINO, Atmos. Chem. Phys., 12, 5329–5341, https://doi.org/10.5194/acp-12-5329-2012, 2012.
Vione, D., Maurino, V., Minero, C., and Pelizzetti, E.: The atmospheric chemistry of hydrogen peroxide: A review, Ann. Chim., 93, 477–488, 2003.
Walcek, C. J.: A theoretical estimate of O3 and H2O2 dry deposition over the northeast United States, Atmos. Environ., 21, 2649–2659, https://doi.org/10.1016/0004-6981(87)90196-X, 1987.
Walker, S. J., Evans, M. J., Jackson, A. V., Steinbacher, M., Zellweger, C., and McQuaid, J. B.: Processes controlling the concentration of hydroperoxides at Jungfraujoch Observatory, Switzerland, Atmos. Chem. Phys., 6, 5525–5536, https://doi.org/10.5194/acp-6-5525-2006, 2006.
Whalley, L. K., Blitz, M. A., Desservettaz, M., Seakins, P. W., and Heard, D. E.: Reporting the sensitivity of laser-induced fluorescence instruments used for HO2 detection to an interference from RO2 radicals and introducing a novel approach that enables HO2 and certain RO2 types to be selectively measured, Atmos. Meas. Tech., 6, 3425–3440, https://doi.org/10.5194/amt-6-3425-2013, 2013.
Williams, J., Crowley, J., Fischer, H., Harder, H., Martinez, M., Petäjä, T., Rinne, J., Bäck, J., Boy, M., Dal Maso, M., Hakala, J., Kajos, M., Keronen, P., Rantala, P., Aalto, J., Aaltonen, H., Paatero, J., Vesala, T., Hakola, H., Levula, J., Pohja, T., Herrmann, F., Auld, J., Mesarchaki, E., Song, W., Yassaa, N., Nölscher, A., Johnson, A. M., Custer, T., Sinha, V., Thieser, J., Pouvesle, N., Taraborrelli, D., Tang, M. J., Bozem, H., Hosaynali-Beygi, Z., Axinte, R., Oswald, R., Novelli, A., Kubistin, D., Hens, K., Javed, U., Trawny, K., Breitenberger, C., Hidalgo, P. J., Ebben, C. J., Geiger, F. M., Corrigan, A. L., Russell, L. M., Ouwersloot, H. G., Vilà-Guerau de Arellano, J., Ganzeveld, L., Vogel, A., Beck, M., Bayerle, A., Kampf, C. J., Bertelmann, M., Köllner, F., Hoffmann, T., Valverde, J., González, D., Riekkola, M.-L., Kulmala, M., and Lelieveld, J.: The summertime Boreal forest field measurement intensive (HUMPPA-COPEC-2010): an overview of meteorological and chemical influences, Atmos. Chem. Phys., 11, 10599–10618, https://doi.org/10.5194/acp-11-10599-2011, 2011.
Zaveri, R. A., Saylor, R. D., Peters, L. K., McNider, R., and Song, A.: A model investigation of summertime diurnal ozone bahavior in rural mountainous locations, Atmos. Environ., 29, 1043–1065, https://doi.org/10.1016/1352-2310(94)00319-G, 1995.