the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Sep 2019
| 19 Sep 2019
Seasonal characteristics of organic aerosol chemical composition and volatility in Stuttgart, Germany
Wei Huang
Harald Saathoff
Xiaoli Shen
Ramakrishna Ramisetty
Thomas Leisner
The chemical composition and volatility of organic aerosol (OA) particles were investigated during July–August 2017 and February–March 2018 in the city of Stuttgart, one of the most polluted cities in Germany. Total non-refractory particle mass was measured with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; hereafter AMS). Aerosol particles were collected on filters and analyzed in the laboratory with a filter inlet for gases and aerosols coupled to a high-resolution time-of-flight chemical ionization mass spectrometer (FIGAERO-HR-ToF-CIMS; hereafter CIMS), yielding the molecular composition of oxygenated OA (OOA) compounds. While the average organic mass loadings are lower in the summer period (5.1±3.2 µg m−3) than in the winter period (8.4±5.6 µg m−3), we find relatively larger mass contributions of organics measured by AMS in summer (68.8±13.4 %) compared to winter (34.8±9.5 %). CIMS mass spectra show OOA compounds in summer have O : C of 0.82±0.02 and are more influenced by biogenic emissions, while OOA compounds in winter have O : C of 0.89±0.06 and are more influenced by biomass burning emissions. Volatility parametrization analysis shows that OOA in winter is less volatile with higher contributions of low-volatility organic compounds (LVOCs) and extremely low-volatility organic compounds (ELVOCs). We partially explain this by the higher contributions of compounds with shorter carbon chain lengths and a higher number of oxygen atoms, i.e., higher O : C in winter. Organic compounds desorbing from the particles deposited on the filter samples also exhibit a shift of signal to higher desorption temperatures (i.e., lower apparent volatility) in winter. This is consistent with the relatively higher O : C in winter but may also be related to higher particle viscosity due to the higher contributions of larger-molecular-weight LVOCs and ELVOCs, interactions between different species and/or particles (particle matrix), and/or thermal decomposition of larger molecules. The results suggest that whereas lower temperature in winter may lead to increased partitioning of semi-volatile organic compounds (SVOCs) into the particle phase, this does not result in a higher overall volatility of OOA in winter and that the difference in sources and/or chemistry between the seasons plays a more important role. Our study provides insights into the seasonal variation of the molecular composition and volatility of ambient OA particles and into their potential sources.
- Article
(5857 KB) - Full-text XML
-
Supplement
(2610 KB) - BibTeX
- EndNote





Air pollution has significant impacts on human health (D'Amato et al., 2014) and visibility (Majewski et al., 2014) and also interacts with climate change (Seinfeld and Pandis, 2016). Due to rapid urbanization, industrialization, and growing human population, air quality in urban environments has become a severe issue in more and more cities all over the world, particularly in densely populated megacities (Guttikunda et al., 2014; Chan and Yao, 2008; Mayer, 1999; Marlier et al., 2016). Air quality in urban environments is influenced by emissions, e.g., from sources such as industrial processes, automobile traffic, and domestic heating, and also by meteorological conditions (e.g., solar radiation, wind, temperature, precipitation), atmospheric dispersion, chemical transformation, location, and topography (D'Amato et al., 2014; Baumbach and Vogt, 2003; Kinney, 2018). Moreover, air pollution is not limited within the boundaries of urban areas but can be transported over long distances and contribute to background pollution on the regional to global scale (Baklanov et al., 2016).
The most abundant air pollutants are nitrogen dioxide (NO2), ozone (O3), sulfur dioxide (SO2), and particulate matter (PM; D'Amato et al., 2014). Despite its abundance and important impacts on climate and health, PM sources, physicochemical transformation, and fate in the atmosphere still remain to be fully understood in urban areas. This is especially true for the organic fraction (Hallquist et al., 2009; Fuzzi et al., 2015). Organic aerosol (OA) often makes up a significant fraction of submicron particulate mass in urban areas (Hallquist et al., 2009; Jimenez et al., 2009). OA can be directly emitted into the atmosphere from sources such as fossil fuel combustion and biomass burning (primary organic aerosol, POA), or it can be formed in the atmosphere from the oxidation of gas-phase precursors (secondary organic aerosol, SOA). POA is dominated by vehicular emissions in urban environments (Bhattu, 2018). SOA, which dominates the global budget of OA (Shrivastava et al., 2015), can be of biogenic and/or anthropogenic origin with biogenic sources dominating on a global scale (Heald et al., 2008). SOA also generally makes up the biggest mass fraction of OA in urban areas, as observed, e.g., in Mexico City (Volkamer et al., 2006; Kleinman et al., 2008), some heavily urbanized areas in the US (de Gouw et al., 2005; Zhang et al., 2005), and during severe haze pollution events in the big cities in China (Huang et al., 2014). In European cities, most of the OA mass observed consists of oxygenated compounds (oxygenated organic aerosol, OOA), and most OOA is of secondary origin and thus SOA (Lanz et al., 2007; Jimenez et al., 2009; Zhang et al., 2007; El Haddad et al., 2013). Robinson et al. (2007) suggest that semi-volatile and intermediate VOCs may play a dominant role in SOA formation in urban locations. In order to design effective mitigation strategies for urban air pollution, it is therefore of great importance to identify the sources of OA, and especially SOA, in urban areas.
Source apportionment of OA has been advanced by the application of positive matrix factorization (PMF) to aerosol mass spectrometer (AMS) or aerosol chemical speciation monitor (ACSM) data (Canonaco et al., 2015; Crippa et al., 2014; Ulbrich et al., 2009). However, the distinction of OOA sources (biogenic or anthropogenic) by AMS- or ACSM-PMF remains difficult due to excessive fragmentation and thus loss of molecular information in the AMS or ACSM. The recent advent of new methods provides more insights into the molecular composition of OA, such as linear trap quadrupole (LTQ) Orbitrap mass spectrometry (Daellenbach et al., 2019), filter inlet for gases and aerosols coupled to a high-resolution time-of-flight chemical ionization mass spectrometer (FIGAERO-HR-ToF-CIMS; Huang et al., 2019), and the newly developed extractive electrospray ionization time-of-flight mass spectrometer (EESI-ToF; Qi et al., 2019). Whereas AMS- and ACSM-PMF are not directly able to reveal OOA sources, they separate OOA into factors with different degrees of oxygenation and thus presumed volatility, such as semi-volatile oxygenated organic aerosol (SV-OOA) and low-volatility oxygenated organic aerosol (LV-OOA; Ulbrich et al., 2009; Jimenez et al., 2009; Lanz et al., 2007). The abovementioned state-of-the-art instruments (e.g., Orbitrap, FIGAERO-HR-ToF-CIMS, EESI-ToF) enable the link between the molecular composition of OA and its physicochemical properties through the use of molecular information in volatility parameterizations to calculate the effective saturation mass concentrations (Csat) of different compounds (Li et al., 2016; Donahue et al., 2011). This can be used to define, e.g., volatility basis sets (VBS), a framework that has been proposed and widely used for grouping the organic compounds into volatility classes (or bins) based on their Csat values (Donahue et al., 2006, 2011, 2012; Cappa and Jimenez, 2010).
Volatility determines whether an organic compound partitions into the particle phase and contributes to OA particulate mass. It is thus an important physicochemical property of OA that influences the lifetime of OA and with that air quality. As a consequence of the connection between a compound's molecular composition and structure with its volatility, different types of OA fall into different categories of volatility. For, e.g., OA measured in Mexico City, biomass burning OA (BBOA) was found to be the most volatile, followed by hydrocarbon-like OA (HOA), SV-OOA, and LV-OOA (Cappa and Jimenez, 2010). SOA derived from isoprene epoxydiol (IEPOX) was observed to have the highest volatility of the OA measured in the southeastern US (Lopez-Hilfiker et al., 2016). As ambient particles generally consist of a matrix of thousands of different compounds, OA apparent volatility can also be influenced by particle-phase diffusion limitations, e.g., due to amorphous phase state and/or the presence of a high mass fraction of oligomers (Vaden et al., 2011; Roldin et al., 2014; Cappa and Wilson, 2011; Yli-Juuti et al., 2017; Huang et al., 2018). Overall, the relationship between OA molecular composition and its volatility and how this relationship is influenced by environmental conditions and particle physicochemical properties are not well characterized, particularly for field data.
Here we present detailed chemical composition measurements of OA from July–August 2017 and February–March 2018 in the city of Stuttgart, Germany. We investigate the molecular composition and volatility of OA particles and discuss their seasonal variations as well as their potential sources. Stuttgart, a city located in southwest Germany with a population of more than 600 000 in a metropolitan area of 2.6 million inhabitants, is an important industrial center in Germany. It is situated in the steep valley of the Neckar river, in a “bowl” surrounded by a variety of hills, small mountains, and valleys. The complex topography can prevent the dispersion of air pollutants, and the location is characterized by low wind speeds and weak air circulation (Schwartz et al., 1991; Hebbert and Webb, 2012). Air quality has been a long-standing concern in Stuttgart, as it is one of the most polluted cities in Germany (Schwartz et al., 1991; Süddeutsche Zeitung, 2016; Office for Environmental Protection, 2016); however, few detailed studies are available. For the year 2017, the state environmental protection agency, LUBW (Landesanstalt für Umwelt Baden-Württemberg), attributes 58 % of the annual mean PM10 at their monitoring station “Am Neckartor” in downtown Stuttgart to road traffic (45 % abrasion, 7 % exhaust, 6 % secondary formation), 8 % to small- and medium-sized combustion sources, and 27 % to regional background (LUBW, 2019). Mayer (1999) showed the temporal variability of urban air pollutants (NO, NO2, O3, and Ox (sum of NO2 and O3)) caused by motor traffic in Stuttgart based on more than 10 years of observations, with higher NO concentrations in winter and higher Ox concentrations in summer. Bari et al. (2011) characterized air pollutants, such as polycyclic aromatic hydrocarbons (PAHs) and other woodsmoke tracer compounds (levoglucosan, methoxyphenols) from wood burning, in the residential areas of Dettenhausen (about 30 km south of Stuttgart) and attributed 57 % of the ambient PM10 pollution to hardwood combustion during wintertime. Our study therefore adds an important piece of information on air quality in Stuttgart by investigating the chemical composition, physicochemical properties, and potential sources of the OA particles in this city.
2.1 Measurement site
We performed particle and trace gas measurements from 5 July to 17 August 2017 and from 5 February to 5 March 2018 in the city of Stuttgart, Germany (48∘47′55.1′′ N, 9∘12′13.5′′ E). The measurement site was located near the park “Unterer Schlossgarten” of Stuttgart and can be classified as an urban background site. The only nearby source is a parcel distribution center with delivery trucks passing by with a low frequency during daytime. It was set up on a bridge over a train track about 2.2 km northeast of the Stuttgart main station with frequent train traffic (electric). The air quality monitoring station of LUBW, “Am Neckartor”, is 1.5 km southwest, and one of the busiest roads in Stuttgart, B14 (LUBW, 2019), is about 360 m southwest of the measurement location.
All instruments were set up in a temperature-controlled measurement container kept at ∼25 ∘C. The container has been described elsewhere (Huang et al., 2019; Shen et al., 2018). All sampling inlets were located 3.7 m a.g.l. and 1.5 m above the container roof. An overview of instruments and parameters measured is given in Table S1 in the Supplement.
2.2 Meteorological, particle, and trace gas measurements
Temperature, relative humidity, wind direction, wind speed, global radiation, pressure, and precipitation data were measured by a meteorological sensor (WS700, Lufft GmbH; see Table S1). The main wind directions during the campaign were southwest during the summer and northeast during the winter. Trace gases (O3, CO2, NO2, SO2) were measured with the corresponding sensors (Table S1). Particle number concentrations were recorded with two condensation particle counters (a CPC3022, measuring particles with mobility diameters larger than 7 nm, and a CPC3776, measuring particles with mobility diameters larger than 2.5 nm; both TSI Inc.). Particle size distributions were measured with a nanoscan scanning mobility particle sizer (NanoScan SMPS3910; measuring particles with mobility diameters between 10 and 420 nm; TSI Inc.). Black carbon (BC) concentrations were measured with an Aethalometer (AE51; Aethlabs Inc.).
A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc., hereafter AMS) equipped with an aerodynamic high-pressure lens (Williams et al., 2013) was deployed to continuously measure total non-refractory particle mass as a function of size (up to 2.5 µm particle aerodynamic diameter dva) at a time resolution of 0.5 min. The AMS inlet was connected to a PM2.5 head (flow rate 1 m3 h−1; Comde-Derenda GmbH) and a stainless-steel tube of 3.45 m length (flow rate 0.1 L min−1, residence time 0.9 s). AMS data were analyzed with the AMS data analysis software packages SQUIRREL (version 1.60C) and PIKA (version 1.20C).
Aerosol particles were deposited during daytime (between 10:00 and 16:00 UTC+2) on polytetrafluoroethylene (PTFE) filters (Zefluor PTFE membrane, 2 µm pore size, 25 mm diameter; Pall Corp.) that were prebaked at 200 ∘C in an oven overnight and stored in clean filter slides, using a stainless-steel filter holder connected to a PM10 head (flow rate 1 m3 h−1; Comde-Derenda GmbH) via a stainless-steel tube and conductive tubing of 2.85 m length (flow rate 8.7 L min−1 in summer and 10 L min−1 in winter, residence time 0.75 s in summer and 0.72 s in winter). Deposition times were varied (20–260 min) based on ambient organic mass concentrations in order to achieve similar organic mass loadings on the filter (summer: 3.5±1.4 µg; winter: 4.0±1.0 µg based on concurrent AMS measurements during the deposition period) and to avoid mass loading effects (Huang et al., 2018; Wang and Ruiz, 2018). A total of 21 filter samples were collected in the summer and 10 in the winter. After deposition, each filter sample was stored in a filter slide, wrapped in aluminum foil, and then stored in a freezer at −20 ∘C until analysis in the laboratory by a filter inlet for gases and aerosols coupled to a high-resolution time-of-flight chemical ionization mass spectrometer (FIGAERO-HR-ToF-CIMS, Aerodyne Research Inc.; hereafter CIMS) deploying iodide (I−) as the reagent ion. Particles collected on the filter were thermally desorbed by a flow of ultra-high-purity (99.999 %) nitrogen heated to 200 ∘C over the course of 35 min. The resulting mass spectral desorption signals are termed thermograms (Lopez-Hilfiker et al., 2014). For individual compounds, signals that peak at distinct desorption temperatures (Tmax) correlate with their saturation vapor pressure (Lopez-Hilfiker et al., 2015; Mohr et al., 2017); however, interference from isomers with different vapor pressures or thermal fragmentation of larger oligomeric molecules can lead to more complex, multimodal thermograms (Lopez-Hilfiker et al., 2015). Integration of the thermograms of individual compounds yields their total particle-phase signal. We assume the sensitivity to be the same for all compounds measured by CIMS (Huang et al., 2019) and convert the signal to mass so that the molecular weight of a compound is taken into account. In this study we do not attempt to derive any atmospheric mass concentrations from these filter measurements, since the actual deposited area of aerosol particles on the filter was larger than the area of the desorption flow, and the deposition was not evenly distributed across the filter. We therefore focus on the molecular composition and volatility distributions of OA particles, their seasonal variations, and the interpretation of these observations for potential sources. In order to correct for filter backgrounds, we collected prebaked clean filters from the measurement site without deposition flow for both winter and summer. Field blank samples for winter and summer were analyzed by CIMS in the laboratory and used for background subtraction.
3.1 Particulate OA mass loadings
We observe higher total non-refractory PM2.5 mass concentrations measured by AMS in winter (27.0±11.9 µg m−3, average ±1 standard deviation) than in summer (7.1±3.3 µg m−3) at this measurement site (Fig. S1 in the Supplement). Similar observations were also made for other central European locations, e.g., Zurich, Switzerland (Jimenez et al., 2009; Zhang et al., 2007). The reasons for this observation are differences in emission sources between the seasons, boundary layer height dynamics, and/or meteorological conditions (Canonaco et al., 2015; Daellenbach et al., 2019; Baumbach and Vogt, 2003). The surface temperature inversion, which develops by radiative cooling of the ground and is dissolved from the bottom up by solar radiation and heating of the ground (Baumbach and Vogt, 2003), is expected to be stronger in winter due to lower ambient temperature and global radiation (Fig. S2), weaker air circulation (i.e., wind speed; Fig. S3), and less precipitation. Air pollutants are therefore more likely to be kept beneath this inversion and have a longer local residence time in the atmosphere in winter. While the average organic mass loadings measured by AMS are lower in summer (5.1±3.2 µg m−3) than in winter (8.4±5.6 µg m−3; see Fig. S1), organics contribute relatively more mass to total non-refractory PM2.5 measured by AMS in summer (68.8±13.4 %) compared to winter (34.8±9.5 %; see Fig. S1). Contributions of fragments containing only C and H atoms (CH), or also one oxygen atom (CHO1), or more than one oxygen atom (CHO>1) to total OA measured by AMS are similar for both seasons (CH: 29.4±3.9 % for summer and 27.9±4.6 % for winter; CHO1: 415.7±1.6 % for summer and 15.3±1.9 % for winter; CHO>1 14.0±2.6 % for summer and 15.8±2.8 % for winter). Higher elemental oxygen-to-carbon ratios (O : C) measured by AMS were observed in winter (0.61±0.12) than in summer (0.55±0.10), implying that OA is more oxygenated in winter. Due to fragmentation of organic molecules during the ionization process in the AMS, molecular information of OA is lost. Molecular formulae can be retrieved using CIMS analysis of filter samples. Due to the fact that the iodide CIMS is selective towards polarizable and thus oxygenated compounds (Lee et al., 2014), the organic compounds measured by CIMS are oxygenated organic aerosol (OOA). In the next section we will discuss the molecular composition of OOA measured by CIMS.
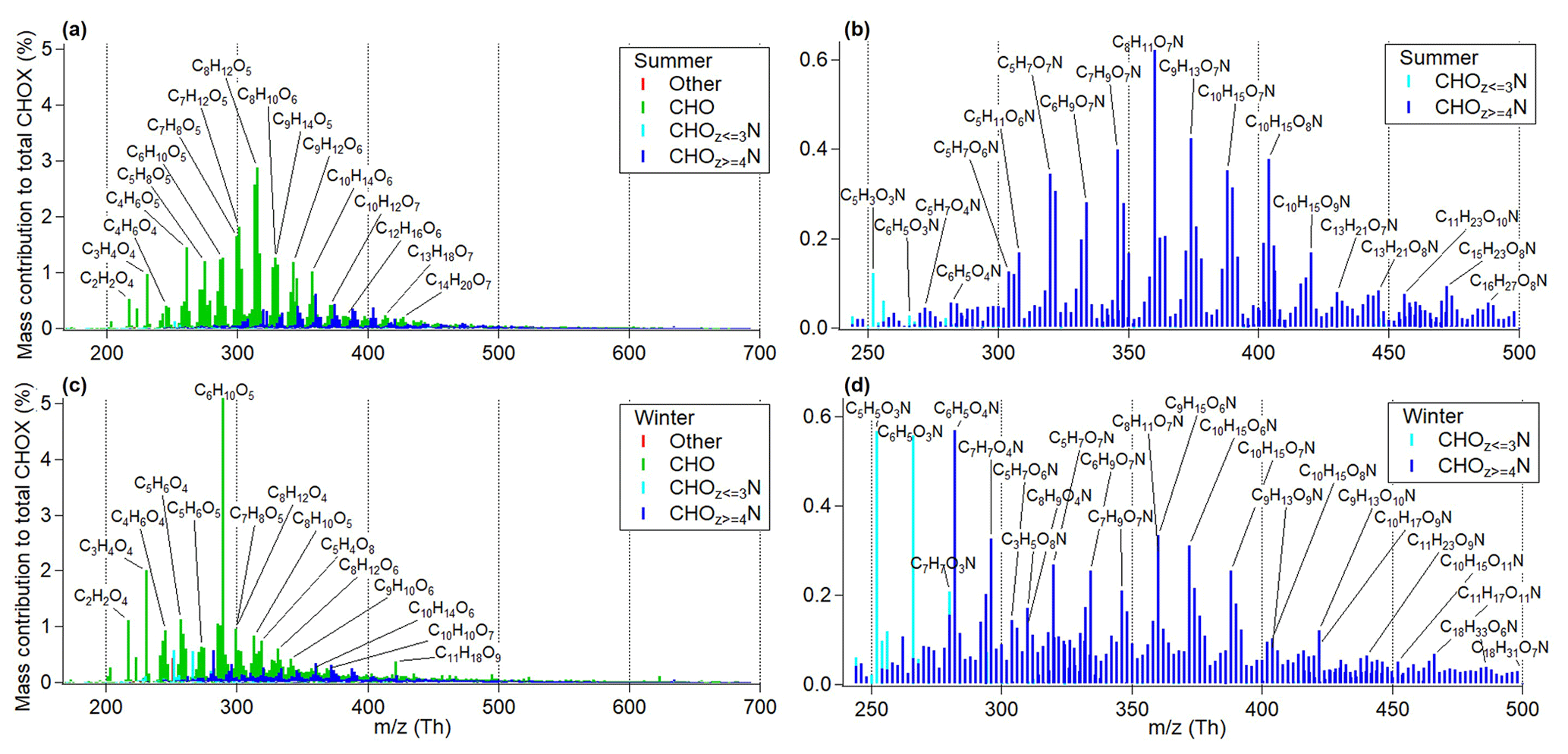
Figure 1CIMS mass spectra comparison of CHOX compounds (separated into CHO, CHON and other compounds) (a) and CHON compounds (b) in the summer period, as well as CHOX compounds (c) and CHON compounds (d) in the winter period as a function of m∕z (includes mass of I− ion; m∕z 126.9050 Th). Mass contributions of each compound were normalized to the sum of the mass of all detected CHOX compounds.
3.2 Molecular composition of OOA
Figure 1a and c show a comparison of CIMS mass spectral patterns of all CHOX compounds (Cx≥1Hy≥1Oz≥1X0−n detected as clustered with I−, with X being different atoms like N, S, Cl, or a combination thereof; 1808 out of a total of 2138 identified compounds and accounting for >96 % of total signals) and CHON compounds only for summer and winter (panels b, d). The mass spectra shown were normalized to the sum of the deposited mass of all detected CHOX compounds. Although the absolute CHOX mass concentrations are uncertain, the time series of the sum of the deposited mass of all detected CHOX compounds follows the trend of the OA concentrations measured by AMS quite well (Pearson's R: 0.95 for summer and 0.96 for winter). CHO compounds (compounds containing only C, H, and O atoms) are the dominating group and make up 79.4±3.3 % of total CHOX in summer and 74.6±2.2 % of total CHOX in winter, followed by CHON compounds with 20.1±3.4 % of total CHOX in summer and 24.6±2.4 % of total CHOX in winter. CHON compounds contribute relatively more mass in winter (also reflected in the organic-bound nitrate fraction – OrgNO3, i.e., organonitrates – from AMS data; summer: 0.3±0.2 µg m−3, winter: 1.7±1.1 µg m−3; determined assuming an ratio of OrgNO3 of 0.1; Farmer et al., 2010; Kiendler-Scharr et al., 2016; see Figs. S4 and S5), while CHO compounds contribute relatively more mass in summer. This is possibly due to the higher daytime O3 concentrations in summer and higher daytime NO2 concentrations in winter (Fig. S6) as well as different emission sources. The contributions of some biogenic marker compounds are higher in summer (Fig. 1a–b), particularly C8H12O5 (molecular formula corresponding to 2-hydroxyterpenylic acid identified in α-pinene SOA by Claeys et al., 2009; Kahnt et al., 2014) and C8H11O7N (identified in the laboratory as α-pinene oxidation product by Lee et al., 2016). We also observe good correlations (Pearson's R: 0.85; Fig. S7a) between our summer mass spectra and the summer daytime mass spectra acquired in 2016 near Karlsruhe (a city in southwest Germany, about 70 km northwest of Stuttgart; Huang et al., 2019), indicative of the regional nature of sources and/or chemistry in summer. We therefore conclude that the majority of the precursor VOCs for OOA presented here in summer are most likely of biogenic origin, despite the urban location of the measurement site.
Significantly higher contributions of C6H10O5 (molecular formula corresponding to levoglucosan, a tracer for biomass burning; Saarnio et al., 2010) are observed in winter compared to summer (Fig. 1c). Higher contributions of C6H5O3N, C7H7O3N, C6H5O4N, and C7H7O4N (molecular formulae corresponding to nitrated phenols, tracers for biomass burning identified by Mohr et al., 2013) are also observed in winter (Fig. 1d). Some of these compounds were also observed in the central European city of Zurich, Switzerland, in winter (Daellenbach et al., 2019). We cannot completely exclude the possibility that these compounds may have contributions from vehicular emissions (Tong et al., 2016). However, significantly higher contributions of levoglucosan and nitrated phenols indicate that biomass burning emissions are a dominant contributor to OOA in Stuttgart in winter. In addition to compounds from biomass burning, we also observe similar patterns of contributions of CHON compounds with m∕z between 300 and 400 Th (also dominated by C8H11O7N) and high contributions of C8H12O4 (molecular formula corresponding to terpenylic acid identified in α-pinene SOA by Claeys et al., 2009) and C8H12O6 (molecular formula corresponding to 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA) in α-pinene SOA identified by Szmigielski et al., 2007; Müller et al., 2012) in winter. After removing the five biomass burning tracer compounds (C6H10O5, C6H5O3N, C7H7O3N, C6H5O4N, and C7H7O4N), good correlations (Pearson's R: 0.70; Fig. S7b) are observed between summer mass spectra and winter mass spectra, indicating that biogenic emissions may also contribute significantly to the OOA particulate mass in winter. In addition, in both summer and winter, contributions of C7H8O5 (identified in the laboratory as toluene oxidation product by Hinks et al., 2018; Molteni et al., 2018) are observed, with higher contributions in summer (when the main wind direction was from the inner city of Stuttgart; see Fig. S3) than in winter, indicating anthropogenic influences related to traffic or industrial activities (EPA, 1994).

Figure 2Mass contribution of CHO and CHON compounds with different numbers of oxygen atoms as a function of the number of carbon atoms to total CHOX compounds for the summer (a, b) and winter (c, d) periods. The corresponding distribution for the other season is plotted as a gray dotted line.
In the following we will have a closer look at the bulk molecular composition for winter and summer daytime OOA measured by CIMS. Consistent with the O : C measured by AMS (winter: 0.61±0.12; summer: 0.55±0.10), higher O : C values are also observed by CIMS in winter (0.89±0.06) compared to summer (0.82±0.02), despite a lower ambient temperature and weaker global radiation in winter (see Fig. S2). The AMS O : C values are expected to be lower than those of the organic compounds measured by iodide CIMS, as the latter is selective towards oxygenated compounds (Lee et al., 2014). Mass contributions of CHO and CHON with different numbers of oxygen atoms per molecule to total CHOX compounds as a function of the number of carbon atoms are shown in Fig. 2. C8HO compounds exhibit the highest mass contributions in summer, while C6HO compounds surpass C8HO compounds in winter due to the large contributions of levoglucosan (C6H10O5; see Fig. 2a, c). The mass distribution of CHO compounds in winter also exhibits higher contributions from compounds with 1–6 carbon atoms and 4–5 (levoglucosan) or 7–9 oxygen atoms, while in summer higher contributions from compounds with 7–10 carbon atoms and 5–7 oxygen atoms are observed. This indicates that the slightly higher oxidation levels (or O : C) in winter are related to both shorter carbon chain lengths and a higher number of oxygen atoms of the OOA compounds compared to summer (see also Fig. S8). In addition, relatively higher contributions of compounds with a larger number of carbon atoms (C16–23) are also observed in winter (Fig. 2). A similar pattern can be found for CHON compounds (Fig. 2b, d). C9−10HON compounds exhibit the highest mass contributions in summer, similar to what we observed in 2016 in summer near the city of Karlsruhe, where these compounds were determined to originate from biogenic VOC emissions (Huang et al., 2019). The filters in Stuttgart were deposited during daytime, and therefore the chemistry involved in the formation of these CHON compounds is likely influenced by the reaction of organic peroxy radicals (RO2) with NOx; contributions of oxidation products formed via nighttime NO3 radical chemistry cannot be ruled out. In winter, C6HON relative contributions exceed those from C9−10HON compounds, similar to the pattern of CHO compounds and indicative of similar sources (biomass burning emissions). Furthermore, in summer CHON compounds are dominated by compounds with 6–9 oxygen atoms, while in winter significantly higher contributions from compounds with 5–7 carbon atoms and 3–4 oxygen atoms are observed, mostly due to nitrated phenols (; see also Fig. 1d).
The results imply the importance of nonfossil OA formation from biogenic and/or biomass burning influences in different seasons even in a city with high traffic emissions mainly based on fossil fuel combustion (LUBW, 2019). This is similar to previous studies in other European cities such as Barcelona, Spain (Mohr et al., 2012), and some megacities in China (Ni et al., 2019). In the next section, we investigate the volatility of OOA compounds measured by CIMS, which can influence their lifetime in the atmosphere and thus air quality.
3.3 Seasonal changes in the volatility of OOA
3.3.1 Volatility distribution
Effective saturation mass concentrations (Csat), a measure for the volatility of a compound, were parameterized for each CHO and CHON compound using the approach by Li et al. (2016),
and then corrected for the summer (24 ∘C) and winter (2 ∘C) periods (Stolzenburg et al., 2018; Donahue et al., 2011; Epstein et al., 2010):
We stress here that isomers cannot be differentiated with the Csat parameterization (Donahue et al., 2011) and that thermal fragmentation of organic compounds (Lopez-Hilfiker et al., 2015; Huang et al., 2018) during particle desorption with the FIGAERO can bias the Csat results towards higher volatilities. This will be discussed later. The CHO and CHON compounds were then grouped into a 25-bin volatility basis set (VBS; Donahue et al., 2006) based on their log10Csat values (Fig. 3). Organic compounds with Csat lower than 10−4.5 µg m−3, between 10−4.5 and 10−0.5 µg m−3, between 10−0.5 and 102.5 µg m−3, and between 102.5 and 106.5 µg m−3 are termed extremely low-volatility organic compounds (ELVOCs), low-volatility organic compounds (LVOCs), semi-volatile organic compounds (SVOCs), and intermediate-volatility organic compounds (IVOCs), respectively (Donahue et al., 2009). As shown in Fig. 3a, organic compounds with Csat between 102 and 103 µg m−3 make up the biggest mass contributions during daytime in both summer and winter (Fig. 3a). The dominating compounds in these volatility bins come from the group of C8−12HO compounds in summer and from the group of C1−7HO compounds with relatively higher O : C in winter (Fig. S9a, c). Dominant compounds are 2-hydroxyterpenylic acid (C8H12O5) and levoglucosan (C6H10O5) for summer and winter, respectively (compare also to Fig. 1a, c). Non-negligible contributions from C1−7HON compounds are also observed in these volatility bins in winter (Fig. S9b, d), mainly from nitrated phenols (; compare also to Fig. 1d). In winter we observe much lower contributions of IVOCs with Csat between 105 and 106 µg m−3 in the particle phase. However, we may also have contributions from thermal decomposition products of oligomers to these low-molecular-weight compounds (also reflected in the multimode thermograms for CHOX compounds with 1–5 carbon atoms; see Fig. S10). But for the larger compounds, such as dimers and trimers, contributions of thermal decomposition products become negligible (i.e., thermograms are unimodal; Huang et al., 2018; Wang and Ruiz, 2018). LVOCs and ELVOCs, which include compounds with larger molecular weight, exhibit higher mass contributions in winter (LVOC: 37.0±2.2 %; ELVOC: 15.9±3.5 %) than in summer (LVOC: 22.6±2.5 %; ELVOC: 4.8±1.2 %; see Fig. 3b–c). The average mass-weighted log10Csat value is 0.97±0.28 µg m−3 for summer and µg m−3 for winter.
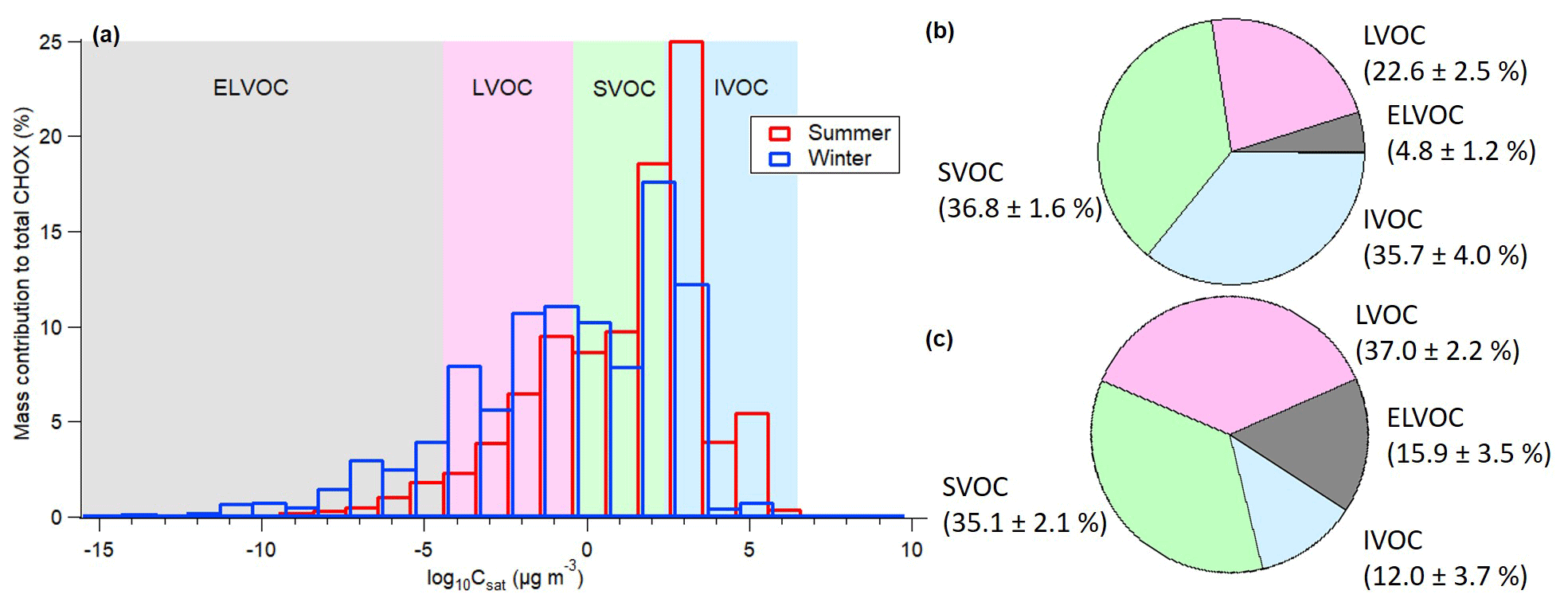
Figure 3(a) Volatility distribution for the summer (24 ∘C) and winter (2 ∘C) periods; resulting pie chart for the mass contributions of SVOCs, IVOCs, LVOCs, and ELVOCs in the summer (b) and winter (c) periods. Compounds more volatile than IVOCs with Csat higher than 106.5 µg m−3 (summer: 0.1±0.0 %; winter: 0.0±0.0 %) are not labeled in the pie chart.
The results indicate that even though the lower ambient temperatures in winter may lead to increased partitioning of SVOCs into the particle phase, the bulk winter OOA is less volatile. Similar results were also observed in Zurich, Switzerland, by Canonaco et al. (2015) based on AMS data. The lower volatility of OOA in Stuttgart in winter compared to summer can be partially explained by the higher contributions of compounds with shorter carbon chain lengths and a higher number of oxygen atoms in winter (i.e., higher O : C; see Fig. S8), as well as the relatively higher contribution of larger molecules (number of carbons atoms >16; see also Fig. 2). Differences in aging processes (functionalization, fragmentation, and oligomerization; Jimenez et al., 2009) between the seasons may also play a role, since Keller and Burtscher (2017) found that aging processes reduce the volatility of OA from biomass burning. Despite the lower ambient temperature during deposition in winter (2 ∘C) compared to the container temperature (25 ∘C), we assume that evaporation of highly volatile organic compounds during deposition in winter has a negligible influence on the main results of this study based on chamber experiments under similar temperature differences (e.g., Huang et al., 2018).
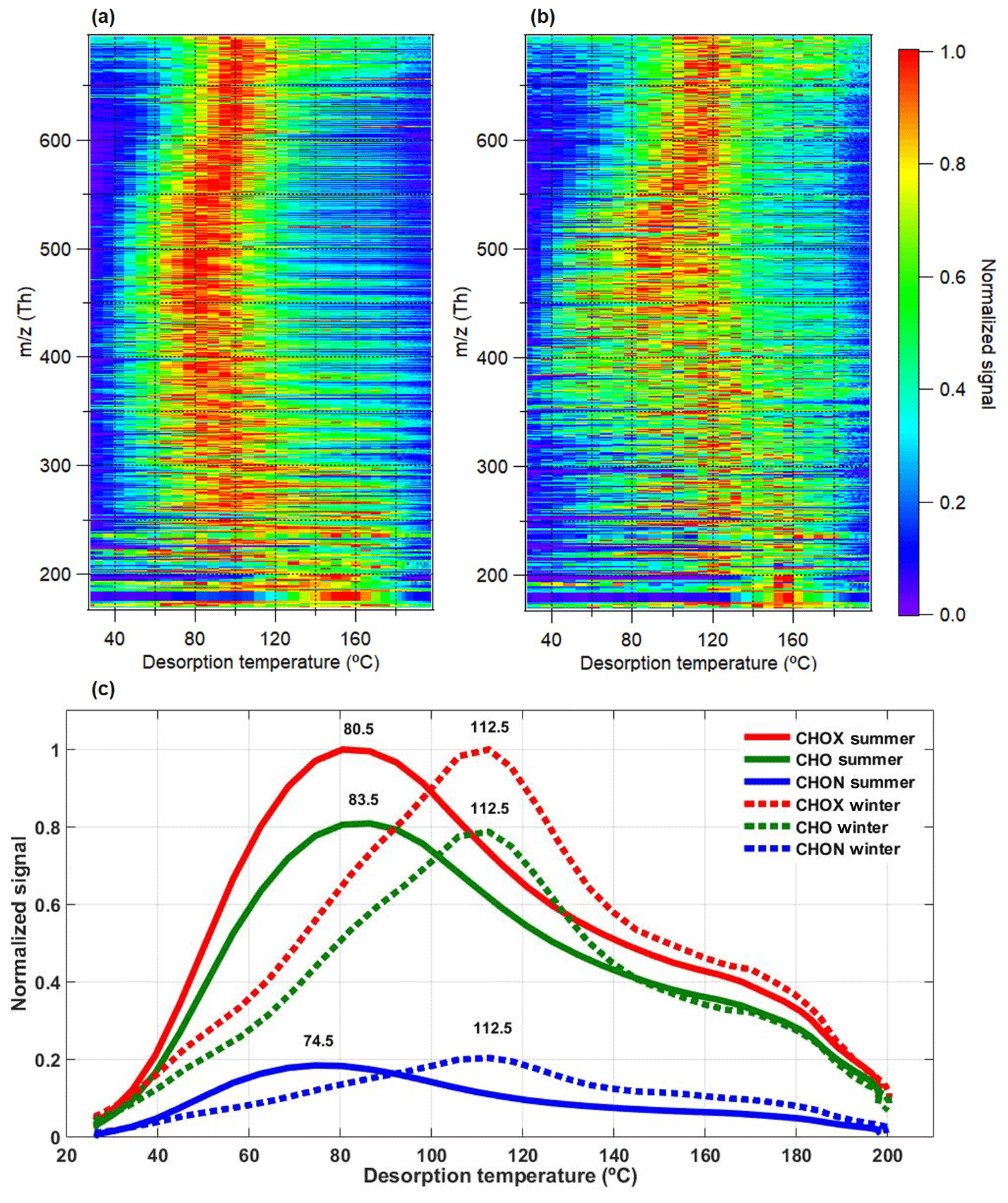
Figure 4Comparison of campaign average high-resolution two-dimensional (2-D) thermograms of CHOX compounds for the summer (a) and winter (b) periods vs. m∕z (includes mass of I− ion; m∕z 126.9050 Th) and the sum thermograms of CHOX, CHO, and CHON compounds (c). The 2-D thermograms and sum thermograms were normalized to their maximum values.
3.3.2 Variation of the maximum desorption temperatures (Tmax)
Thermograms resulting from the thermal desorption of the filter samples were analyzed. Tmax, the maximum desorption temperatures at which the signals of a compound peak, were compared for summer and winter. Since the CIMS instrument configuration and setup were kept exactly the same during these two measurements and the temperature is kept constant in our laboratory throughout the year, we do not expect significant shifts in Tmax due to the experimental setup. Figure 4a–b show the campaign average high-resolution two-dimensional (2-D) thermogram of CHOX compounds, a framework recently developed to investigate the OOA thermal desorption behavior over the entire m∕z and Tmax range (Wang and Ruiz, 2018). Each thermogram of each individual compound in the 2-D space was normalized to its maximum signal. Due to the CHOX compounds containing at least one carbon atom, one hydrogen atom, and one oxygen atom and being detected as clustered with I− (m∕z 126.9050 Th), the smallest m∕z in the 2-D thermogram is 168 Th. As shown in Fig. 4a–b, the majority of the OOA compounds exhibit higher Tmax with a wider spread across different CHOX compounds in winter (114.4±17.1 ∘C, average ±1 standard deviation) compared to summer (96.8±18.2 ∘C; see also Fig. S11). For the summer period, Tmax decreases from 160 to 60 ∘C for m∕z 168–280 Th, stays relatively constant at 60–110 ∘C for m∕z 280–550 Th, and increases from 60 to 120 ∘C for m∕z >550 Th; for the winter period, Tmax decreases from 160 to 80 ∘C for m∕z 168–280 Th, stays relatively constant at 2 Tmax regions (one region at 80–100 ∘C and the other one at 110–130 ∘C) for m∕z 280–550 Th, and increases from 80 to 130 ∘C for m∕z >550 Th (see also Fig. S11). The high Tmax values for m∕z <280 Th (SVOC range) result from multimode thermograms with thermal decomposition of larger molecules (Lopez-Hilfiker et al., 2015). A similar picture can be seen in the campaign average thermograms for the sum of the signals of all CHOX, CHO, and CHON compounds detected (normalized to the maximum; Fig. 4c). The pattern of the summer 2-D thermogram in Stuttgart, particularly the zigzag-like behavior of, e.g., m∕z 280–380 Th (see Fig. S12a), is comparable to that from SOA derived from chlorine-initiated oxidation of n-alkanes at high RH (67 %) and high NOx conditions observed by Wang and Ruiz (2018) and was explained by increased hydroxyl functionalization over ketone functionalization. The winter 2-D thermogram in Stuttgart also has a zigzag-like pattern but less pronounced and at higher Tmax (see Fig. S12b).
The results indicate a generally lower apparent volatility (i.e., higher Tmax) of bulk OOA in winter, in agreement with the results based on the Csat parametrization (see also Fig. 3). Recent studies show that the Tmax of a compound can be influenced by isomers (Thompson et al., 2017), thermal fragmentation of larger molecules during the heating of the filter (Lopez-Hilfiker et al., 2015), variations in filter mass loading (Huang et al., 2018; Wang and Ruiz, 2018), and/or differences in particle viscosity (Huang et al., 2018). Since deposited organic mass loadings on the filter samples were similar for summer and winter, we can exclude a mass loading effect on the Tmax results presented here. However, we also visually observed that the filter samples in winter were more blackish, possibly due to the higher black carbon (BC) concentrations during daytime (10:00–16:00) in winter (1247±112 ng m−3) compared to summer (1032±311 ng m−3). If (and how) the higher BC concentrations can affect the desorption behaviors of organic compounds (i.e., interactions between organic compounds and BC) is still unknown and requires further laboratory studies. Higher O : C (Buchholz et al., 2019) and/or higher oligomer mass fractions (Huang et al., 2018; compare to LVOC and ELVOC mass contributions in Fig. 3b–c) have been shown to be correlated with higher Tmax, which is in agreement with our mass spectral observations in winter. Higher inorganic sulfate concentrations in winter (see Fig. S1) might also play a role in the formation of low-volatility but thermally unstable organic compounds, which can only be detected as their decomposition products with CIMS (Gaston et al., 2016; Riva et al., 2019). If assuming that the winter thermograms are more influenced by the thermal decomposition of oligomers than the summer thermograms, which artificially shifts the molecular-formula-based volatility distribution towards higher volatility, the winter OOA is expected to be even less volatile.
In this paper, the chemical composition and volatility of OA particles were investigated during July–August 2017 and February–March 2018 in the city of Stuttgart, one of the most polluted cities in Germany. The average organic mass loadings measured by AMS are lower in summer (5.1±3.2 µg m−3) than in winter (8.4±5.6 µg m−3), but the relative contributions of OA to total non-refractory PM2.5 mass measured by AMS are higher in summer (68.8±13.4 %) compared to winter (34.8±9.5 %). This can be explained by the differences in emission sources between the seasons, boundary layer height dynamics, and/or meteorological conditions (Canonaco et al., 2015; Daellenbach et al., 2019; Baumbach and Vogt, 2003). CIMS mass spectra from filter samples collected at the measurement site during daytime (10:00–16:00) show OOA compounds in summer have O : C of 0.82±0.02 and are more influenced by biogenic emissions (as shown by, e.g., tracers of α-pinene oxidation products), while OOA compounds in winter have slightly higher O : C (0.89±0.06) and are more influenced by biomass burning emissions (as shown by, e.g., signals of levoglucosan and nitrated phenols).
The apparent volatility of the OOA compounds varies between the two seasons. OOA in winter is found to be less volatile, which is reflected in the higher contributions of LVOCs and ELVOCs in the VBS, as well as in the higher maximum desorption temperatures (Tmax) of the organic compounds desorbing from the particles deposited on the filter samples. A potential reason for the lower apparent volatility of winter OOA is the increased residence time of air masses over Stuttgart due to stronger surface inversion and thus a longer atmospheric aging time of the OOA compounds, leading to a reduction in volatility (Keller and Burtscher, 2017; D'Ambro et al., 2018; Jimenez et al., 2009). This is also consistent with the higher O : C and the higher mass contributions of larger-molecular-weight LVOCs and ELVOCs in winter. Since the OOA observed in the winter period also shows an influence from biogenic emissions, the sources for the LVOCs and ELVOCs may be partly biogenic. In addition, interactions between different species and/or particles (particle matrix; Huang et al., 2018) due to higher BC, OA, and inorganic concentrations, such as the intermolecular interactions between biomass burning compounds and biogenic or anthropogenic organic compounds and/or the interactions between organic compounds, inorganic compounds, and BC, might also play a role in the reduction of the volatility of aerosol particles in winter. Overall, the lower apparent volatility of the winter OOA compounds could be caused by higher O : C but may also be related to the higher particle viscosity due to the higher mass contributions of larger-molecular-weight LVOCs and ELVOCs, interactions between different species and/or particles deposited on the filter (particle matrix; Huang et al., 2018), and/or thermal decomposition of large molecules.
The results suggest that whereas lower temperatures in winter may lead to increased partitioning of SVOCs into the particle phase, this does not result in a higher overall volatility of OOA in winter and that the difference in sources and/or chemistry between the seasons plays a more important role. Our study provides insights into the seasonal variation of the molecular composition and volatility of ambient OA particles during daytime and into their potential sources, which is important for air pollution mitigation in urban locations. Our study shows the important contributions of nonfossil OA from biogenic and biomass burning even in an urban area with high traffic emissions mainly based on fossil fuel combustion (LUBW, 2019). As a consequence, in addition to mitigating traffic emissions, reducing emissions of anthropogenic OOA precursors from, e.g., industry and biomass burning may contribute to reducing the environmental and health effects of air pollution.
Data are available upon request to the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-11687-2019-supplement.
WH operated AMS and took the filter samples during the two field campaigns, analyzed the filters by CIMS in the laboratory, did the CIMS and AMS data analysis, produced all figures, and wrote and edited the paper; HS organized the campaign, did the trace gas, CPC, and black carbon data analysis, and provided suggestions for the data interpretation and discussion; XS operated AMS and took the filter samples during the field campaigns; RR took the filter samples during the summer campaign; TL gave general advice and comments for this paper; CM provided suggestions for the data analysis, interpretation, and discussion and edited the paper. All authors contributed to the final text.
The authors declare that they have no conflict of interest.
Technical support by the staff at IMK-AAF, as well as financial support by the China Scholarship Council (CSC) for Wei Huang and Xiaoli Shen, is gratefully acknowledged. Support by the Deutsche Bahn AG, the University of Stuttgart, and the partners of the project “Three-Dimensional Observation of Atmospheric Processes in Cities (3DO)” (http://uc2-3do.org/, last access: 22 August 2019) is gratefully acknowledged.
The article processing charges for this open-access publication were covered by a Research Centre of the Helmholtz Association.
This paper was edited by Eleanor Browne and reviewed by two anonymous referees.
Baklanov, A., Molina, L. T., and Gauss, M.: Megacities, air quality and climate, Atmos. Environ., 126, 235–249, https://doi.org/10.1016/j.atmosenv.2015.11.059, 2016.
Bari, M. A., Baumbach, G., Kuch, B., and Scheffknecht, G.: Air pollution in residential areas from wood-fired heating, Aerosol. Air. Qual. Res., 11, 749–757, https://doi.org/10.4209/aaqr.2010.09.0079, 2011.
Baumbach, G. and Vogt, U.: Influence of inversion layers on the distribution of air pollutants in urban areas, Water Air Soil Poll., 3, 67–78, https://doi.org/10.1023/A:1026098305581, 2003.
Bhattu, D.: Primary Organic Aerosols, in: Air pollution and Control, edited by: Sharma, N., Agarwal, A., Eastwood, P., Gupta, T., and Singh, A., Energy, Environment, and Sustainability, Springer, Singapore, 109–117, 2018.
Buchholz, A., Lambe, A. T., Ylisirniö, A., Li, Z., Tikkanen, O.-P., Faiola, C., Kari, E., Hao, L., Luoma, O., Huang, W., Mohr, C., Worsnop, D. R., Nizkorodov, S. A., Yli-Juuti, T., Schobesberger, S., and Virtanen, A.: Insights into the O : C-dependent mechanisms controlling the evaporation of α-pinene secondary organic aerosol particles, Atmos. Chem. Phys., 19, 4061–4073, https://doi.org/10.5194/acp-19-4061-2019, 2019.
Canonaco, F., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: Seasonal differences in oxygenated organic aerosol composition: implications for emissions sources and factor analysis, Atmos. Chem. Phys., 15, 6993–7002, https://doi.org/10.5194/acp-15-6993-2015, 2015.
Cappa, C. D. and Jimenez, J. L.: Quantitative estimates of the volatility of ambient organic aerosol, Atmos. Chem. Phys., 10, 5409–5424, https://doi.org/10.5194/acp-10-5409-2010, 2010.
Cappa, C. D. and Wilson, K. R.: Evolution of organic aerosol mass spectra upon heating: implications for OA phase and partitioning behavior, Atmos. Chem. Phys., 11, 1895–1911, https://doi.org/10.5194/acp-11-1895-2011, 2011.
Chan, C. K. and Yao, X.: Air pollution in mega cities in China, Atmos. Environ., 42, 1–42, https://doi.org/10.1016/j.atmosenv.2007.09.003, 2008.
Claeys, M., Iinuma, Y., Szmigielski, R., Surratt, J. D., Blockhuys, F., Van Alsenoy, C., Böge, O., Sierau, B., Gómez-González, Y., Vermeylen, R., Van der Veken, P., Shahgholi, M., Chan, A. W. H., Herrmann, H., Seinfeld, J. H., and Maenhaut, W.: Terpenylic acid and related compounds from the oxidation of α-pinene: Implications for new particle formation and growth above forests, Environ. Sci. Technol., 43, 6976–6982, https://doi.org/10.1021/es9007596, 2009.
Crippa, M., Canonaco, F., Lanz, V. A., Äijälä, M., Allan, J. D., Carbone, S., Capes, G., Ceburnis, D., Dall'Osto, M., Day, D. A., DeCarlo, P. F., Ehn, M., Eriksson, A., Freney, E., Hildebrandt Ruiz, L., Hillamo, R., Jimenez, J. L., Junninen, H., Kiendler-Scharr, A., Kortelainen, A.-M., Kulmala, M., Laaksonen, A., Mensah, A. A., Mohr, C., Nemitz, E., O'Dowd, C., Ovadnevaite, J., Pandis, S. N., Petäjä, T., Poulain, L., Saarikoski, S., Sellegri, K., Swietlicki, E., Tiitta, P., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Organic aerosol components derived from 25 AMS data sets across Europe using a consistent ME-2 based source apportionment approach, Atmos. Chem. Phys., 14, 6159–6176, https://doi.org/10.5194/acp-14-6159-2014, 2014.
D'Amato, G., Bergmann, K. C., Cecchi, L., Annesi-Maesano, I., Sanduzzi, A., Liccardi, G., Vitale, C., Stanziola, A., and D'Amato, M.: Climate change and air pollution: Effects on pollen allergy and other allergic respiratory diseases, Allergo J. Int., 23, 17–23, https://doi.org/10.1007/s40629-014-0003-7, 2014.
D'Ambro, E. L., Schobesberger, S., Zaveri, R. A., Shilling, J. E., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., and Thornton, J. A.: Isothermal evaporation of α-pinene ozonolysis SOA: volatility, phase state, and oligomeric composition, ACS Earth Space Chem., 2, 1058–1067, https://doi.org/10.1021/acsearthspacechem.8b00084, 2018.
Daellenbach, K. R., Kourtchev, I., Vogel, A. L., Bruns, E. A., Jiang, J., Petäjä, T., Jaffrezo, J.-L., Aksoyoglu, S., Kalberer, M., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: Impact of anthropogenic and biogenic sources on the seasonal variation in the molecular composition of urban organic aerosols: a field and laboratory study using ultra-high-resolution mass spectrometry, Atmos. Chem. Phys., 19, 5973–5991, https://doi.org/10.5194/acp-19-5973-2019, 2019.
de Gouw, J. A., Middlebrook, A. M., Warneke, C., Goldan, P. D., Kuster, W. C., Roberts, J. M., Fehsenfeld, F. C., Worsnop, D. R., Canagaratna, M. R., Pszenny, A. A. P., Keene, W. C., Marchewka, M., Bertman, S. B., and Bates, T. S.: Budget of organic carbon in a polluted atmosphere: Results from the New England Air Quality Study in 2002, J. Geophys. Res.-Atmos., 110, D16305, https://doi.org/10.1029/2004JD005623, 2005.
Donahue, N. M., Robinson, A. L., Stanier, C. O., and Pandis, S. N.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, https://doi.org/10.1021/es052297c, 2006.
Donahue, N. M., Robinson, A. L., and Pandis, S. N.: Atmospheric organic particulate matter: From smoke to secondary organic aerosol, Atmos. Environ., 43, 94–106, https://doi.org/10.1016/j.atmosenv.2008.09.055, 2009.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
El Haddad, I., D'Anna, B., Temime-Roussel, B., Nicolas, M., Boreave, A., Favez, O., Voisin, D., Sciare, J., George, C., Jaffrezo, J.-L., Wortham, H., and Marchand, N.: Towards a better understanding of the origins, chemical composition and aging of oxygenated organic aerosols: case study of a Mediterranean industrialized environment, Marseille, Atmos. Chem. Phys., 13, 7875–7894, https://doi.org/10.5194/acp-13-7875-2013, 2013.
EPA: Locating and estimating air emissions from sources of toluene, EPA-454/R-93-047, in: Research Triangle Park, North Carolina, United States, 3, 3–4, 1994.
Epstein, S. A., Riipinen, I., and Donahue, N. M.: A semiempirical correlation between enthalpy of vaporization and saturationconcentration for organic aerosol, Environ. Sci. Technol., 44, 743–748, https://doi.org/10.1021/es902497z, 2010.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Fuzzi, S., Baltensperger, U., Carslaw, K., Decesari, S., Denier van der Gon, H., Facchini, M. C., Fowler, D., Koren, I., Langford, B., Lohmann, U., Nemitz, E., Pandis, S., Riipinen, I., Rudich, Y., Schaap, M., Slowik, J. G., Spracklen, D. V., Vignati, E., Wild, M., Williams, M., and Gilardoni, S.: Particulate matter, air quality and climate: lessons learned and future needs, Atmos. Chem. Phys., 15, 8217–8299, https://doi.org/10.5194/acp-15-8217-2015, 2015.
Gaston, C. J., Lopez-Hilfiker, F. D., Whybrew, L. E., Hadley, O., McNair, F., Gao, H. L., Jaffe, D. A., and Thornton, J. A.: Online molecular characterization of fine particulate matter in Port Angeles, WA: Evidence for a major impact from residential wood smoke, Atmos. Environ., 138, 99–107, https://doi.org/10.1016/j.atmosenv.2016.05.013, 2016.
Guttikunda, S. K., Goel, R., and Pant, P.: Nature of air pollution, emission sources, and management in the Indian cities, Atmos. Environ., 95, 501–510, https://doi.org/10.1016/j.atmosenv.2014.07.006, 2014.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heald, C. L., Henze, D. K., Horowitz, L. W., Feddema, J., Lamarque, J.-F., Guenther, A., Hess, P. G., Vitt, F., Seinfeld, J. H., Goldstein, A. H., and Fung, I.: Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res.-Atmos., 113, D05211, https://doi.org/10.1029/2007JD009092, 2008.
Hebbert, M. and Webb, B.: Towards a Liveable Urban Climate: Lessons from Stuttgart, edited by: Gossop, C. and Nan, S., Liveable Cities: Urbanising World (ISOCARP 07), Routledge, London, 120–137, 2012.
Hinks, M. L., Montoya-Aguilera, J., Ellison, L., Lin, P., Laskin, A., Laskin, J., Shiraiwa, M., Dabdub, D., and Nizkorodov, S. A.: Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 18, 1643–1652, https://doi.org/10.5194/acp-18-1643-2018, 2018.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Huang, W., Saathoff, H., Pajunoja, A., Shen, X., Naumann, K.-H., Wagner, R., Virtanen, A., Leisner, T., and Mohr, C.: α-Pinene secondary organic aerosol at low temperature: chemical composition and implications for particle viscosity, Atmos. Chem. Phys., 18, 2883–2898, https://doi.org/10.5194/acp-18-2883-2018, 2018.
Huang, W., Saathoff, H., Shen, X., Ramisetty, R., Leisner, T., and Mohr, C.: Chemical characterization of highly functionalized organonitrates contributing to night-time organic aerosol mass loadings and particle growth, Environ. Sci. Technol., 53, 1165–1174, https://doi.org/10.1021/acs.est.8b05826, 2019.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353 2009.
Kahnt, A., Iinuma, Y., Mutzel, A., Böge, O., Claeys, M., and Herrmann, H.: Campholenic aldehyde ozonolysis: a mechanism leading to specific biogenic secondary organic aerosol constituents, Atmos. Chem. Phys., 14, 719–736, https://doi.org/10.5194/acp-14-719-2014, 2014.
Keller, A. and Burtscher, H.: Characterizing particulate emissions from wood burning appliances including secondary organic aerosol formation potential, J. Aerosol. Sci., 114, 21–30, https://doi.org/10.1016/j.jaerosci.2017.08.014, 2017.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kinney, P. L.: Interactions of climate change, air pollution, and human health, Curr. Environ Health Rep., 5, 179–186, https://doi.org/10.1007/s40572-018-0188-x, 2018.
Kleinman, L. I., Springston, S. R., Daum, P. H., Lee, Y.-N., Nunnermacker, L. J., Senum, G. I., Wang, J., Weinstein-Lloyd, J., Alexander, M. L., Hubbe, J., Ortega, J., Canagaratna, M. R., and Jayne, J.: The time evolution of aerosol composition over the Mexico City plateau, Atmos. Chem. Phys., 8, 1559–1575, https://doi.org/10.5194/acp-8-1559-2008, 2008.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W. W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H. Y., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J. M., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Li, Y., Pöschl, U., and Shiraiwa, M.: Molecular corridors and parameterizations of volatility in the chemical evolution of organic aerosols, Atmos. Chem. Phys., 16, 3327–3344, https://doi.org/10.5194/acp-16-3327-2016, 2016.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Carrasquillo, A. J., Daumit, K. E., Hunter, J. F., Kroll, J. H., Worsnop, D. R., and Thornton, J. A.: Phase partitioning and volatility of secondary organic aerosol components formed from α-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds, Atmos. Chem. Phys., 15, 7765–7776, https://doi.org/10.5194/acp-15-7765-2015, 2015.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular composition and volatility of organic aerosol in the Southeastern U.S.: Implications for IEPOX derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
LUBW: Luftreinhaltepläne für Baden-Württemberg (Grundlagenband 2017), in: LUBW State Institute for the Environment Baden-Württemberg, Karlsruhe, Germany, 2019.
Majewski, G., Czechowski, P. O., Badyda, A., and Brandyk, A.: Effect of air pollution on visibility in urban conditions. Warsaw case study, Environ. Prot. Eng., 40, 47–64, https://doi.org/10.5277/epe140204, 2014.
Marlier, M. E., Jina, A. S., Kinney, P. L., and DeFries, R. S.: Extreme air pollution in global megacities, Curr. Clim. Change Rep., 2, 15–27, https://doi.org/10.1007/s40641-016-0032-z, 2016.
Mayer, H.: Air pollution in cities, Atmos. Environ., 33, 4029–4037, https://doi.org/10.1016/S1352-2310(99)00144-2, 1999.
Mohr, C., DeCarlo, P. F., Heringa, M. F., Chirico, R., Slowik, J. G., Richter, R., Reche, C., Alastuey, A., Querol, X., Seco, R., Peñuelas, J., Jiménez, J. L., Crippa, M., Zimmermann, R., Baltensperger, U., and Prévôt, A. S. H.: Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data, Atmos. Chem. Phys., 12, 1649–1665, https://doi.org/10.5194/acp-12-1649-2012, 2012.
Mohr, C., Lopez-Hilfiker, F. D., Zotter, P., Prévôt, A. S. H., Xu, L., Ng, N. L., Herndon, S. C., Williams, L. R., Franklin, J. P., Zahniser, M. S., Worsnop, D. R., Knighton, W. B., Aiken, A. C., Gorkowski, K. J., Dubey, M. K., Allan, J. D., and Thornton, J. A.: Contribution of nitrated phenols to wood burning brown carbon light absorption in Detling, United Kingdom during winter time, Environ. Sci. Technol., 47, 6316–6324, https://doi.org/10.1021/es400683v, 2013.
Mohr, C., Lopez-Hilfiker, F. D., Yli-Juuti, T., Heitto, A., Lutz, A., Hallquist, M., D'Ambro, E. L., Rissanen, M. P., Hao, L. Q., Schobesberger, S., Kulmala, M., Mauldin III, R. L., Makkonen, U., Sipilä, M., Petäjä, T., and Thornton, J. A.: Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth, Geophys. Res. Lett., 44, 2958–2966, https://doi.org/10.1002/2017gl072718, 2017.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Ni, H., Huang, R.-J., Cao, J., Dai, W., Zhou, J., Deng, H., Aerts-Bijma, A., Meijer, H. A. J., and Dusek, U.: High contributions of fossil sources to more volatile organic aerosol, Atmos. Chem. Phys., 19, 10405–10422, https://doi.org/10.5194/acp-19-10405-2019, 2019.
Office for Environmental Protection: Luft: Erfolgreiche Maßnahmen zur Luftreinhaltung in Stuttgart: available at: https://www.stuttgart.de/item/show/15638 (last access: 2 September 2019), 2016.
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019.
Riva, M., Heikkinen, L., Bell, D. M., Peräkylä, O., Zha, Q., Schallhart, S., Rissanen, M. P., Imre, D., Petäjä, T., Thornton, J. A., Zelenyuk, A., and Ehn, M.: Chemical transformations in monoterpene-derived organic aerosol enhanced by inorganic composition, npj Clim. Atmos. Sci., 2, 1–9, https://doi.org/10.1038/s41612-018-0058-0, 2019.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: Semivolatile emissions and photochemical aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Roldin, P., Eriksson, A. C., Nordin, E. Z., Hermansson, E., Mogensen, D., Rusanen, A., Boy, M., Swietlicki, E., Svenningsson, B., Zelenyuk, A., and Pagels, J.: Modelling non-equilibrium secondary organic aerosol formation and evaporation with the aerosol dynamics, gas- and particle-phase chemistry kinetic multilayer model ADCHAM, Atmos. Chem. Phys., 14, 7953–7993, https://doi.org/10.5194/acp-14-7953-2014, 2014.
Saarnio, K., Aurela, M., Timonen, H., Saarikoski, S., Teinilä, K., Mäkelä, T., Sofiev, M., Koskinen, J., Aalto, P. P., Kulmala, M., Kukkonen, J., and Hillamo, R.: Chemical composition of fine particles in fresh smoke plumes from boreal wild-land fires in Europe, Sci. Total. Environ., 408, 2527–2542, https://doi.org/10.1016/j.scitotenv.2010.03.010, 2010.
Schwartz, J., Spix, C., Wichmann, H. E., and Malin, E.: Air pollution and acute respiratory illness in five German communities, Environ. Res., 56, 1–14, https://doi.org/10.1016/S0013-9351(05)80104-5, 1991.
Seinfeld, J. H., and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 3. ed., John Wiley & Sons, Inc., Hoboken, New Jersey, 2016.
Shen, X., Ramisetty, R., Mohr, C., Huang, W., Leisner, T., and Saathoff, H.: Laser ablation aerosol particle time-of-flight mass spectrometer (LAAPTOF): performance, reference spectra and classification of atmospheric samples, Atmos. Meas. Tech., 11, 2325–2343, https://doi.org/10.5194/amt-11-2325-2018, 2018.
Shrivastava, M., Easter, R. C., Liu, X., Zelenyuk, A., Singh, B., Zhang, K., Ma, P.-L., Chand, D., Ghan, S., Jimenez, J. L., Zhang, Q., Fast, J., Rasch, P. J., and Tiitta, P.: Global transformation and fate of SOA: Implications of low-volatility SOA and gas-phase fragmentation reactions, J. Geophys. Res.-Atmos., 120, 4169–4195, https://doi.org/10.1002/2014JD022563, 2015.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Mazon, S. B., Chen, D. X., Dias, A., Draper, D. C., Duplissy, J., Haddad, I., Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Süddeutsche Zeitung: Stuttgart löst als erste Stadt in Deutschland den Feinstaubalarm aus: available at: http://www.sueddeutsche.de/panorama/luftverschmutzung-stuttgart-loest-als-erste-stadt-in-deutschland-feinstaubalarm-aus-1.2822775 (last access: 18 January 2016), 2016.
Szmigielski, R., Surratt, J. D., Gómez-González, Y., Van der Veken, P., Kourtchev, I., Vermeylen, R., Blockhuys, F., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Seinfeld, J. H., Maenhaut, W., and Claeys, M.: 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol, Geophys. Res. Lett., 34, L24811, https://doi.org/10.1029/2007GL031338, 2007.
Thompson, S. L., Yatavelli, R. L. N., Stark, H., Kimmel, J. R., Krechmer, J. E., Day, D. A., Hu, W. W., Isaacman-VanWertz, G., Yee, L., Goldstein, A. H., Khan, M. A. H., Holzinger, R., Kreisberg, N., Lopez-Hilfiker, F. D., Mohr, C., Thornton, J. A., Jayne, J. T., Canagaratna, M., Worsnop, D. R., and Jimenez, J. L.: Field intercomparison of the gas/particle partitioning of oxygenated organics during the Southern Oxidant and Aerosol Study (SOAS) in 2013, Aerosol. Sci. Tech., 51, 30–56, https://doi.org/10.1080/02786826.2016.1254719, 2017.
Tong, H., Kourtchev, I., Pant, P., Keyte, I. J., O'Connor, I. P., Wenger, J. C., Pope, F. D., Harrison, R. M., and Kalberer, M.: Molecular composition of organic aerosols at urban background and road tunnel sites using ultra-high resolution mass spectrometry, Faraday Discuss., 189, 51–68, https://doi.org/10.1039/c5fd00206k, 2016.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Vaden, T. D., Imre, D., Beránek, J., Shrivastava, M., and Zelenyuk, A.: Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol, P. Natl. Acad. Sci. USA, 108, 2190–2195, https://doi.org/10.1073/pnas.1013391108, 2011.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006.
Wang, D. S. and Hildebrandt Ruiz, L.: Chlorine-initiated oxidation of n-alkanes under high-NOx conditions: insights into secondary organic aerosol composition and volatility using a FIGAERO–CIMS, Atmos. Chem. Phys., 18, 15535–15553, https://doi.org/10.5194/acp-18-15535-2018, 2018.
Williams, L. R., Gonzalez, L. A., Peck, J., Trimborn, D., McInnis, J., Farrar, M. R., Moore, K. D., Jayne, J. T., Robinson, W. A., Lewis, D. K., Onasch, T. B., Canagaratna, M. R., Trimborn, A., Timko, M. T., Magoon, G., Deng, R., Tang, D., de la Rosa Blanco, E., Prévôt, A. S. H., Smith, K. A., and Worsnop, D. R.: Characterization of an aerodynamic lens for transmitting particles greater than 1 micrometer in diameter into the Aerodyne aerosol mass spectrometer, Atmos. Meas. Tech., 6, 3271–3280, https://doi.org/10.5194/amt-6-3271-2013, 2013.
Yli-Juuti, T., Pajunoja, A., Tikkanen, O.-P., Buchholz, A., Faiola, C., Väisänen, O., Hao, L., Kari, E., Peräkylä, O., Garmash, O., Shiraiwa, M., Ehn, M., Lehtinen, K., and Virtanen, A.: Factors controlling the evaporation of secondary organic aerosol from α-pinene ozonolysis, Geophys. Res. Lett., 44, 2562–2570, https://doi.org/10.1002/2016GL072364, 2017.
Zhang, Q., Worsnop, D. R., Canagaratna, M. R., and Jimenez, J. L.: Hydrocarbon-like and oxygenated organic aerosols in Pittsburgh: insights into sources and processes of organic aerosols, Atmos. Chem. Phys., 5, 3289–3311, https://doi.org/10.5194/acp-5-3289-2005, 2005.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.