the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Aug 2019
| 27 Aug 2019
Halogen activation and radical cycling initiated by imidazole-2-carboxaldehyde photochemistry
Pablo Corral Arroyo
Raffael Aellig
Peter A. Alpert
Rainer Volkamer
Atmospheric aerosol particles can contain light-absorbing organic compounds, also referred to as brown carbon (BrC). The ocean surface and sea spray aerosol particles can also contain light-absorbing organic species referred to as chromophoric dissolved organic matter (CDOM). Many BrC and CDOM species can contain carbonyls, dicarbonyls or aromatic carbonyls such as imidazole-2-carboxaldehyde (IC), which may act as photosensitizers because they form triplet excited states upon UV–VIS light absorption. These triplet excited states are strong oxidants and may initiate catalytic radical reaction cycles within and at the surface of atmospheric aerosol particles, thereby increasing the production of condensed-phase reactive oxygen species (ROS). Triplet states or ROS can also react with halides, generating halogen radicals and molecular halogen compounds. In particular, molecular halogens can be released into the gas phase, which is one halogen activation pathway. In this work, we studied the influence of bromide and iodide on the photosensitized production and release of hydroperoxy radicals (HO2) upon UV irradiation of films in a coated wall flow tube (CWFT) containing IC in a matrix of citric acid (CA) irradiated with UV light. In addition, we measured the iodine release upon irradiation of IC ∕ CA films in the CWFT. We developed a kinetic model coupling photosensitized CA oxidation with condensed-phase halogen chemistry to support data analysis and assessment of atmospheric implications in terms of HO2 production and halogen release in sea spray particles. As indicated by the experimental results and confirmed by the model, significant recycling of halogen species occurred via scavenging reactions with HO2. These prevented the full and immediate release of the molecular halogen (bromine and iodine) produced. Recycling was stronger at low relative humidity, attributed to diffusion limitations. Our findings also show that the HO2 production from BrC or CDOM photosensitized reactions can increase due to the presence of halides, leading to high HO2 turnover, in spite of low release due to the scavenging reactions. We estimated the iodine production within sea salt aerosol particles due to iodide oxidation by ozone (O3) at M s−1 assuming O3 was in Henry's law equilibrium with the particle. However, using an O3 diffusion coefficient of cm2 s−1, iodine activation in an aged, organic-rich sea spray is estimated to be M s−1. The estimated iodine production from BrC photochemistry based on the results reported here amounts to M s−1 and indicates that BrC photochemistry can exceed O3 reactive uptake in controlling the rates of iodine activation from sea spray particles under dry or cold conditions where diffusion is slow within particles.
- Article
(1125 KB) - Full-text XML
-
Supplement
(415 KB) - BibTeX
- EndNote





Volatile halogen-containing species such as CH3X, CH2XY, HOX, XY and X2 (where X and Y can be Cl, Br and I) are known as activated halogen species (AHS). They are produced at the ocean surface, in snowpacks or in aerosol particles and emitted into the atmospheric gas phase. Their production is referred to as halogen activation. Halogen activation is driven by the oxidation of halides by ozone (Carpenter et al., 2013; Schmidt et al., 2016) and radicals (e.g., OH or NO3) (Sander and Crutzen, 1996), N2O5 (Behnke et al., 1997) or photochemical oxidation (Wang and Pratt, 2017; Wren et al., 2013). These volatile compounds can also be emitted to the atmosphere in the form of biogenic halogen-containing organic species (Org–X) (Hepach et al., 2016; Vogt et al., 1999) or by volcanos, among other processes (Simpson et al., 2015). AHS are precursors of reactive halogen species (RHS) such as the X atom or XO (Sherwen et al., 2016a), which affect oxidative processes in the gas phase (Saiz-Lopez et al., 2012). In the troposphere, for example, the presence of RHS shifts the HOx equilibrium (HO2↔OH) towards OH (Bloss et al., 2005; Chameides and Davis, 1980; von Glasow et al., 2004; Saiz-Lopez, 2012; Sommariva et al., 2012; Lary, 1996), especially for the case of IO (Schmidt et al., 2016; Stone et al., 2018; Saiz-Lopez et al., 2008; Bloss et al., 2005; Dix et al., 2013; Volkamer et al., 2015). RHS also influence the budgets of nitrogen oxides (NOx), organic compounds and organic peroxy radicals (Simpson et al., 2015). It has been observed that RHS of iodine produce ultrafine particles found in coastal areas (McFiggans et al., 2010; Mahajan et al., 2011). This new particle formation occurs via polymerization of I2O5 or HIO3 (Hoffmann et al., 2001; McFiggans et al., 2004; Saunders and Plane, 2006; Sherwen et al., 2016b; Sipila et al., 2016), which are produced by the (photo)oxidation of iodine precursor species such as I2 (Saiz-Lopez and Plane, 2004), HOI (Carpenter et al., 2013; Sherwen et al., 2016b) and Org–X (Carpenter, 2003). The production and cycling of AHS and RHS at the ocean surface or in sea spray particles are key processes to understand their release into the gas phase and the contributions to their emission fluxes (Pechtl et al., 2007; Carpenter et al., 2013; Herrmann et al., 2003).
Apart from O3, N2O5 and inorganic radicals, halogen activation can also be initiated by triplet excited states of light-absorbing organic compounds (Tinel et al., 2014; Jammoul et al., 2009). Typically referred to as brown carbon (BrC; Laskin et al., 2015), organic compounds absorbing in the UV-A–VIS range are ubiquitously present in atmospheric aerosols. Similar compounds also occur in marine or terrestrial water environments, where they are referred to as chromophoric dissolved organic matter (CDOM). The involvement of triplet-forming CDOM or BrC species, also termed photosensitizers, in radical chain oxidation and redox processes characterized by the interplay of organic radicals and reactive oxygen species (ROS) was first recognized in aquatic photochemistry (Canonica, 2000; McNeill and Canonica, 2016) and has since also been reported in atmospheric aerosol photochemistry (George et al., 2015).
Photosensitizers of atmospheric interest absorb above 300 nm and typically have carbonyl functions attached to an aromatic system (see absorption spectra in Fig. S1 in the Supplement; Canonica, 2000). Aromatic carbonyls may derive from the oxidation of aromatic (and phenolic) compounds in the atmosphere. They may also derive from multiphase chemistry of carbonyls in aqueous ammonium sulfate (AS) aerosol, as is the case for imidazole-2-carboxaldehyde (IC) derived from glyoxal, which is a globally important oxygenated volatile organic compound (OVOC) from biogenic VOC oxidation (Stavrakou et al., 2009). IC (absorption spectrum in Fig. S1) is an important photosensitizer (Aregahegn et al., 2013; Kampf et al., 2012; Yu et al., 2014; Corral-Arroyo et al., 2018; González Palacios et al., 2016) and is used as a proxy in the present study.
The concentration of photosensitizing BrC or CDOM species in marine and continental aerosol particles is high enough to represent a substantial source of triplets (O'Dowd and de Leeuw, 2007; Blanchard, 1964; Hoffman and Duce, 1976; Hunter and Liss, 1977; Cincinelli et al., 2001; Chen et al., 2016). When BrC is derived from biomass burning, its concentration is especially high (see review by Laskin et al., 2015). Halides are internally mixed with organics in continental aerosol particles originating from long-range transport or local sources and in marine environments at the ocean surface or in sea spray aerosol (Knopf et al., 2014). In the absence of direct measurements of excited triplet states in aerosols related to these environments, we may consider the steady-state concentration of triplet states in fog water of up to 10−13 M reported by Kaur and Anastasio (2018). Assuming that drying of such fog droplets generally leads to representative triplet concentrations, the upper limit of the concentration of triplet states in aerosol particles would be around 10−10 M due to the lower water activity. The concentration of iodide and bromide in sea spay aerosol particles may reach 10−6 M (Pechtl et al., 2007; Baker, 2004, 2005) and M (Herrmann et al., 2003), respectively. Using the abovementioned concentration and a rate coefficient of the reaction between a typical sensitizer triplet state and iodide of 5×109 M−1 s−1 (Tinel et al., 2014), we calculate that iodine activation may reach M s−1. In the absence of diffusion limitations, this leads to a short reactive lifetime of the order of seconds for iodide in the aqueous phase. De Laurentiis et al. (2012) suggested that excited triplet states could oxidize bromide faster than OH radicals in seawater. Some modeling studies of aerosol chemistry consider inorganic halogen chemistry to be important (Sherwen et al., 2016b, a); however, Pechtl et al. claimed that reactions with dissolved organic matter may be included as a relevant HOI deactivation pathway (Sarwar et al., 2016; Pechtl et al., 2007; Roveretto et al., 2019). Photosensitized halogen activation is less understood and has not been included in these models.
Figure 1 illustrates the catalytic cycle of a photosensitizer in an organic aerosol particle in the presence of halides. First, the photosensitizer (P) absorbs radiation. This is followed by singlet, P*(s), to triplet, P*(t), intersystem crossing. The triplet state is long lived and acts as an oxidant (Canonica, 2000) reacting with an electron donor, e.g., a halide ion (X−) or an organic H atom donor, producing a ketyl radical (PH• ∕ P•−). Oxygen competes with electron/H atom donors for the triplet and is able to produce singlet oxygen (1O2) from its reaction with the triplet. The ketyl radical passes on an electron or hydrogen atom to oxygen or another electron acceptor (e.g., NO2, Stemmler et al., 2006) producing HO2 and returning the photosensitizer to its ground state. The quantum yield in terms of oxidation of an electron donor and reduction of electron acceptor (e.g., formation of HO2) per absorbed photon is affected by competing processes, such as the deactivation of the singlet, deactivation of the triplet (phosphorescence, non-radiative decay and reaction with oxygen) and other radical reactions involving the reduced ketyl radical. The presence of organics that are highly reactive with triplet states increases the photosensitized HO2 radical production of imidazole-2-carboxaldehyde (IC) up to 20 M d−1 (Corral-Arroyo et al., 2018). The oxidation of the halide anion by the triplet state of IC leads to halide radicals (X• and ), and the ensuing halide radical–radical reactions produce molecular halogen compounds (Reactions R8–R11 and R14, Table 1). H2O2 is additionally produced by the self-reaction of HO2 and by the reaction between HO2 and . We do not consider further reactivity of H2O2 as it is not photolyzed at the wavelengths used in the present study. The oxidized species X2, and X• are likely recycled into X− by HO2 radicals (Reactions R5–R9, Table 1). However, a fraction of X2 may be released into the gas phase (Jammoul et al., 2009), and these recycling processes determine the effective efficiency in terms of halogen activated per photon absorbed by the photosensitizer.
Table 1Chemical reactions and the corresponding literature rate coefficients of halide and HO2 radical chemistry.
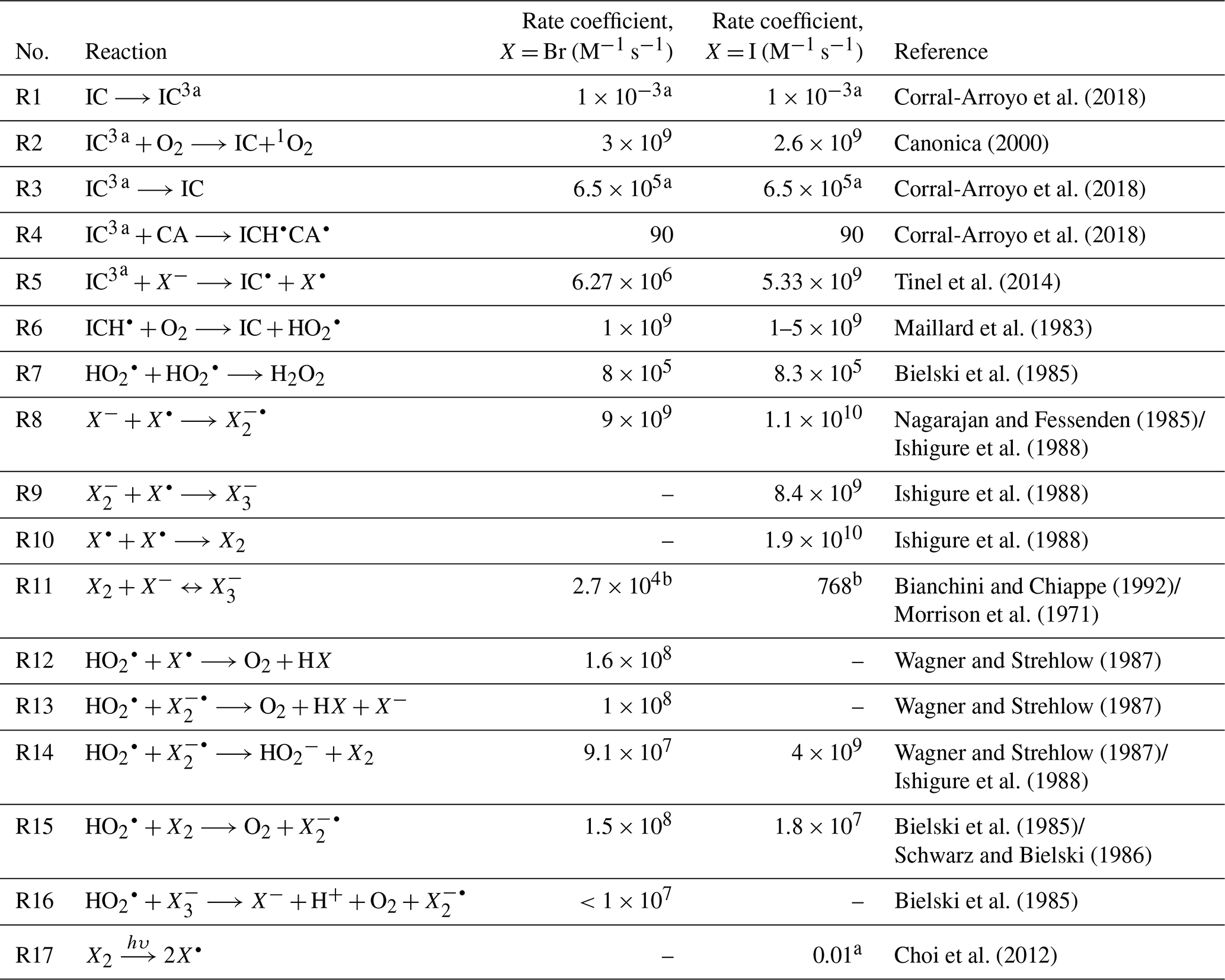
a First-order rate coefficient (s−1). b Equilibrium constant (M−1).
In this work, we quantify the effect of bromide and iodide on the HO2 production from IC photochemistry and evaluate the iodine activation resulting from the subsequent condensed-phase radical reactions by means of coated wall flow tube (CWFT) experiments. As a matrix, we use citric acid (CA) that serves as a proxy for non-absorbing highly oxidized and functionalized secondary organic compounds in the atmosphere, which are also ubiquitous in marine air (O'Dowd and de Leeuw, 2007). In solution, CA takes up or releases water gradually without phase change over the whole range of relative humidity (RH) values studied here (Lienhard et al., 2012; Zardini et al., 2008). This allowed us to carefully address the influence of the microphysical conditions on transport and chemical reactions. Finally, we discuss the relevance of our findings for atmospheric sea spray aerosol.
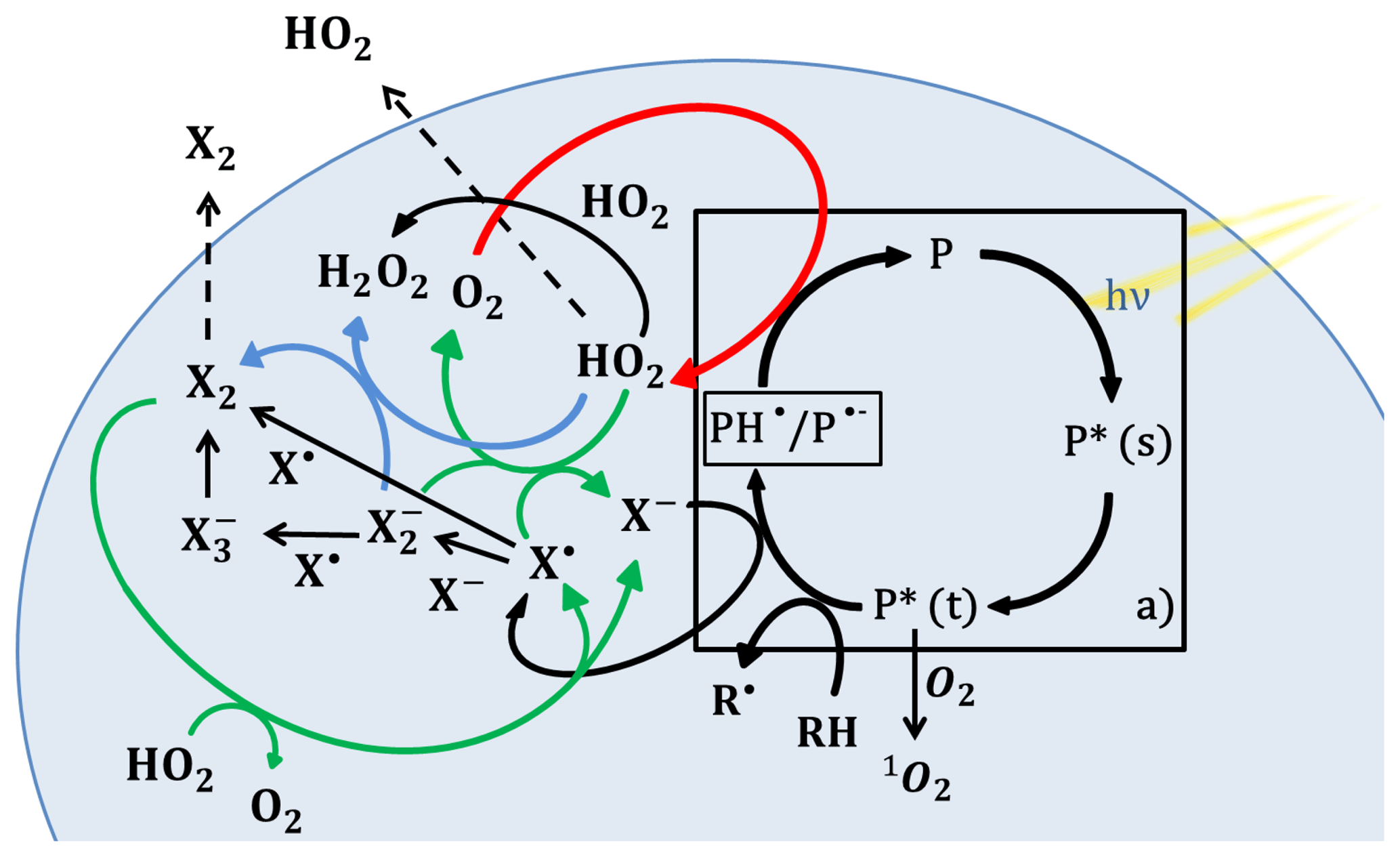
Figure 1Photochemical catalytic cycle of IC (box “a)”) and halide radical chemistry induced in a particle. IC is a photosensitizer (P) that first absorbs light, excites to its singlet state P*(s) and transitions to its triplet state P*(t), which reacts with an H atom/electron donor (RH and X−) to produce the reduced ketyl radical (PH•) and halide radicals (X•). The halide radicals can produce molecular halogen (X2) or by reacting with X−. PH• may transfer a H atom or electron to an acceptor, such as O2 producing HO2 radicals. HO2 can recycle the halide radicals previously produced into halides or further oxidize the to produce halogen molecules. HO2 radicals can be released into the gas phase or react within the particle with halide radicals or with itself. Solid lines refer to reactions, and dashed lines refer to transfer from the condensed to the gas phase. The red arrow indicates HO2 production, the green arrows indicate recycling of halides promoted by HO2 and the blue arrows indicate the reaction of with HO2 to form X2. Rate coefficients are provided in Table 1.
2.1 Experimental description
The setup to determine HO2 production in an irradiated laminar coated wall flow tube (CWFT) by scavenging HO2 with an excess of nitrogen monoxide (NO) has been described in detail in our previous work (González Palacios et al., 2016; Corral-Arroyo et al., 2018) and in the Supplement (Figs. S2 and S3). Tubes (1.2 cm inner diameter, 50 cm long, DURAN glass) coated with mixtures of IC ∕ CA ∕ NaI and IC ∕ CA ∕ NaBr on their inner surfaces were snugly fit into the temperature and relative humidity controlled CWFT as inserts surrounded by seven fluorescent lamps (UV-A range, Philips Cleo Effect 20 W: 300–420 nm, 41 cm, 2.6 cm o.d., see Fig. S1). The flows of N2 and O2 were set at 1 and 0.5 L min−1, respectively. The NO concentration (5–10 mL min−1 of a mix of N2 and NO at 100 ppm) was always high enough (1–2.5×1013 molecules cm−3) to efficiently scavenge ∼99 % of HO2 produced by the films within 20–50 ms, which was far less than our residence time of 2 s. NO was measured by a chemiluminescence detector (ECO PHYSICS CLD 77 AM). For experiments with bromide, we assumed that the concentration of bromide did not change over the timescale of our experiments and, therefore, the system was in steady-state under irradiation. Conversely, the concentration of iodide decreased rapidly (within tens of minutes), as the iodine is rapidly released into the gas phase. Therefore, we determined the NO loss from the first few minutes of irradiation for reporting HO2 production rates for experiments using iodide in films.
Iodine release into the gas phase was observed by converting all gas-phase iodine compounds to I2O5 following a procedure developed by Saunders and Plane (2006). Part of the flow from the reactor (0.1 L min−1 out of 1.5 L min−1) was mixed with 0.2 L min−1 of O2∕O3 (1 %), and this mixture was fed into a quartz reactor with 0.07 s residence time, which was irradiated with a Hg Pen-Ray lamp (184 nm). The O2∕O3 (1 %) mixture was produced by a discharge in pure O2 and quantified with a photometric ozone analyzer. In the quartz reactor, all iodine compounds were readily photolyzed and oxidized to I2O5, which polymerized and produced particles via homogeneous nucleation (Carpenter et al., 2013; Saunders and Plane, 2006). The resulting aerosol flow was led to a scanning mobility particle sizer (SMPS) through aerosol tubing with a residence time of around 20 s. The SMPS consisted of a homemade differential mobility analyzer (DMA, 93.5 cm long, 0.937 cm inner diameter and 1.961 outer diameter) and a condensation particle counter (CPC, Model 3775, TSI Inc.). The mass of the I2O5 particles was determined from their size distribution with the density assumed to be 2.3±0.3 g cm−3 following Saunders and Plane (2006). The particle mass was converted to an equivalent I2 release assuming the stoichiometry of I2O5. We were only able to measure particles ≥ 20 nm in diameter reliably (Fig. S4). This method does not distinguish between iodine and any other volatile iodine compound, which may also be oxidized to I2O5. HOI or IO might be produced in the films by the oxidation of halide radicals or molecular halogens, but they are likely not significant products in the absence of O3 in the CWFT. Hence, we rely on our proposed mechanism (Fig. 1) and assume that iodine activation is dominated by the production of I2.
Aqueous solutions containing halides (10−8, 10−5 and 0.01 M for iodide and 10−5 and 0.01 M for bromide) were prepared beforehand. For each experiment, 76.6 mg of CA and 4 mg of IC (2.5 mg of IC for the experiments measuring iodine release) were dissolved in different volumes of a halide solution in order to get different halide concentrations in the films. Once prepared, a solution was deposited in the glass tube while rolling and turning the tube in all directions at room temperature under a gentle flow of N2 humidified to the RH later used in experiments. This procedure was necessary to ensure homogeneous thin films checked by visual inspection and to prevent the film from drying out prior to the experiments. Freshly prepared solutions were always used to prepare the films. After final equilibration in the CWFT, concentrations in the film were 6 M for CA, 0.7 M for IC, between 10−8 and 0.01 M for iodide, and between 10−4 and 0.01 M for bromide (0.4 M of IC and 33 mM of iodide for iodine release measurements) at around 35 % RH at 20 ∘C. These were calculated assuming that the water content in the film was controlled by the hygroscopicity of CA only, as parameterized by Zardini et al. (2008). Films are expected to be liquid at 35 % RH and have a viscosity of 10–100 Pa s (Song et al., 2016). For iodide, only two measurements were made for each film, as iodide is consumed rapidly, while for bromide four to six consecutive measurements were made for each film. One measurement consisted of comparing the signals of NO before and after switching the UV lamps on or off. For the CWFT experiments, in which the release of I2 was measured, films were loaded with 2.5 mg of IC, 76.6 mg of CA (6.5 % in molar ratio) and 313 µg of NaI, corresponding to concentrations of 0.4 M, 6 M and 33 mM of IC, CA and iodide, respectively, and the iodine release into the gas phase at 34 % RH was followed uninterruptedly.
2.2 Chemicals
The chemicals used were imidazole-2-carboxaldehyde (> 99 %, Aldrich), citric acid (Fluka), sodium bromide (Sigma-Aldrich) and sodium iodide (Sigma-Aldrich).
3.1 HO2 production, scavenging and release
Figure 2 presents the HO2 radical release in the CWFT as a function of halide concentration from films loaded with IC ∕ CA ∕ NaBr and IC ∕ CA ∕ NaI. Error bars are the standard deviation of multiple measurements. The HO2 radical release exhibits first an increase starting from the baseline in the absence of halides and then a peak at about 8×1011 cm−2 min−1. The baseline HO2 release is due to HO2 production from the reaction of the ketyl radical PH• with O2 (blue solid line in Fig. 1), with the PH• being produced from the oxidation of CA by the triplet P*(t). The baseline was measured in this study and is consistent with our previous value (Corral-Arroyo et al., 2018; González Palacios et al., 2016). Increasing the halide content beyond the peak concentration observed in Fig. 2 resulted in a decrease of HO2 below the baseline. Our results can be explained by halides contributing to the reduction of P* due to their ability to donate an electron more efficiently than CA. This should have led to an increased production of PH• and, thus, increased production of HO2 (Fig. 1). The observed HO2 production and release is enhanced above the baseline from M for iodide and M for bromide. This implies that the rate coefficient for the reduction of the IC triplet (P*) by iodide is also 3 orders of magnitude faster than that for reduction by bromide, in line with the rate coefficients for R5 in Table 1 of 5.33×109 and 6.27×106 M−1 s−1, respectively, as measured by Tinel et al. (2014).
After the oxidation of the halide ion by the triplet state, it is expected that a cascade of fast reactions takes place leading to the production of and molecular halogens (X2). Most of these halogen species react rapidly with HO2 (Reactions R5–R9 in Table 1), which explains the drop of the HO2 release at high halide concentrations. Additionally, HO2 radicals also undergo self-reaction, meaning that this scavenging pathway will be more relevant at high halide concentrations, where more HO2 is produced (8×105 M−1s−1) (Bielski et al., 1985). The reaction of HO2 with , the main HO2 scavenging reaction (R14; Table 1) is faster for the iodine species than for the bromine species, which induces a suppression of the HO2 release at lower concentrations for iodide than for bromide. In this way, the majority of HO2 is scavenged before being released into the gas phase for films with concentrations of iodide above 10−3 M and of bromide of 10−2 M. The ratio of the rate coefficients of the triplet with iodide and bromide (Reaction R5) is higher than the ratio of the rate coefficients of HO2 with iodine and bromine species, which induce the recycling (Reactions R12–R16). We suspect that this is the reason why the HO2 release decreases more quickly with concentration for bromide than for iodide.
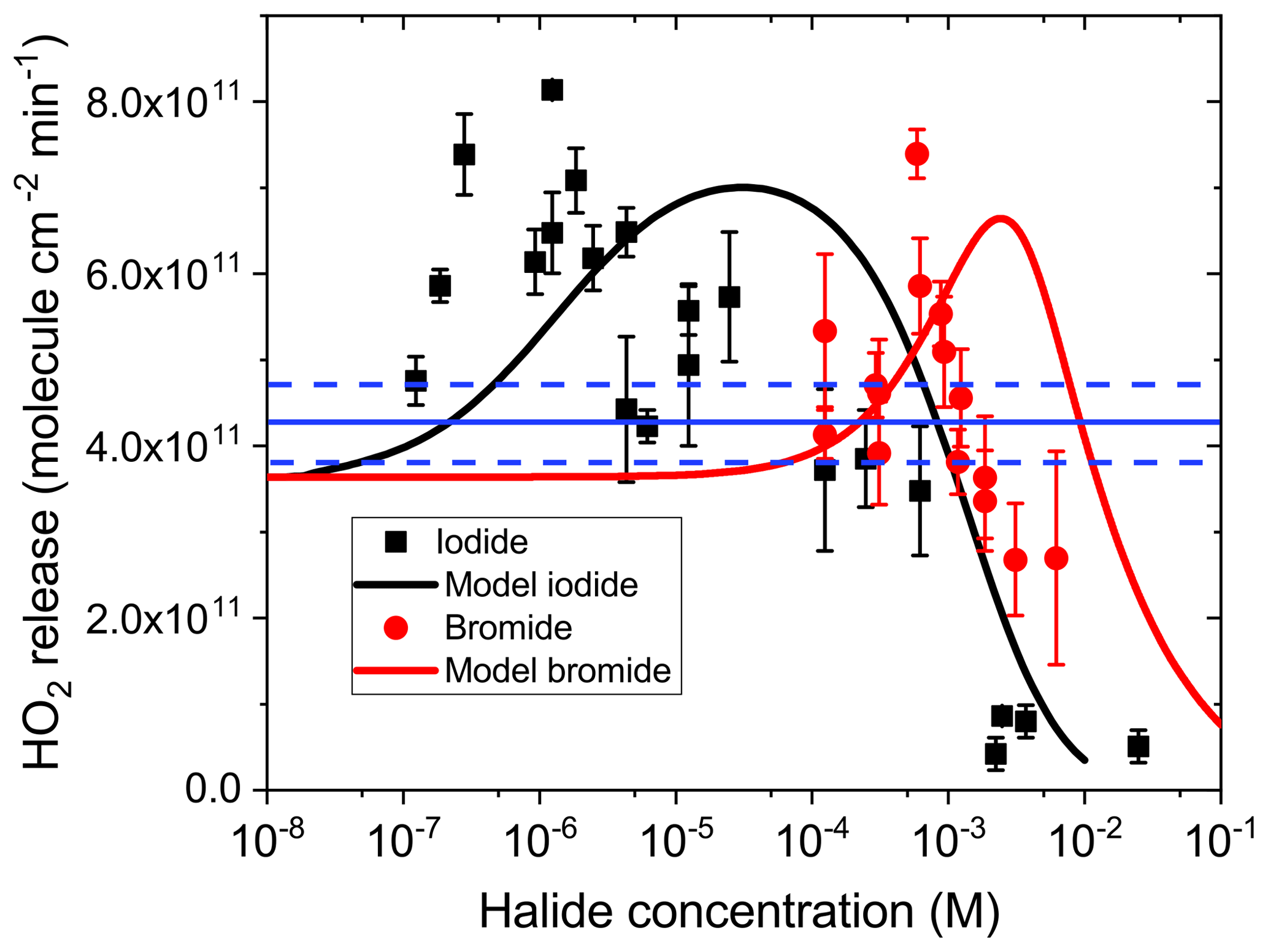
Figure 2HO2 release at 34 % RH from films with 4 mg of IC, 76.8 mg of CA, and various concentrations of bromide (red circles) and iodide (black squares). Error bars indicate the standard deviation of measurements in the same film. The blue line and the dashed blue lines indicate measured HO2 production and uncertainty, respectively, from films with the same IC and CA concentration but in the absence of halides. Solid black and red lines are fits using the model described in the text below.
In our recent work (Corral-Arroyo et al., 2018), a steady-state kinetic model was developed treating IC photochemistry and HO2 release from films of IC ∕ CA as a function of concentration of IC, relative humidity, film thickness or additional organic triplet scavengers. Here, we extended that model to include the scavenging of the triplet state of IC by halides. The inter-halogen conversion reactions (R8–R11) and a set of HO2 scavenging reactions (R12–R16) were added (see Table 1). We also added the photolysis of iodine by integrating the product of the irradiance spectrum of the lamps used (Fig. S1) and the absorption spectrum of iodine (Choi et al., 2012). Due to the fast equilibrium between I2 and , their concentration ratio remained fixed by the equilibrium constant (Table 1), effectively meaning that both have the same sources and sinks. Further details of the reactions and rate coefficients are given in the Supplement. Using the literature values for each reaction, the model captured the general trend of HO2 release with a maximum and a downward slope upon increasing concentrations of halides. However, the model overpredicted the HO2 release at middle and high concentrations of halides (10−5–10−1 M). Therefore, in the process of optimization to adjust the model output to observations, the inter-halogen conversion reactions (R8–R11) were kept at their literature values, whereas the HO2 scavenging reactions (R12–R16) were decreased as described in the Supplement. Such adjustments were justified because literature rate coefficients measured in dilute aqueous solution may not necessarily be the same at a high solute strength, which was the case for our films. There is evidence that hydrogen bonded transition states are involved in electron transfer (Ivkovic-Jensen and Kostic, 1997), proton coupled electron transfer, hydrogen abstraction reactions (Mitroka et al., 2010) and quenching reactions between triplets and salts (Kunze et al., 1997). Thus, reduced activity of reactants and water may act to reduce reaction rates. However, we refrained from adding more and ill-constrained processes and parameters to achieve a better apparent fit. As shown in Fig. 2, the maximum HO2 release rates are reproduced considering the scatter in the data. The position of the maximum is determined by the ratio between the scavenging of triplet states by halides and the HO2 scavenging reactions. The predicted maxima are shifted towards higher halide concentrations compared with our observations. This can be explained if CA derived radicals reacted with halogen radicals to produce halogen-containing organic compounds, as already observed in aquatic media (Roveretto et al., 2019), which could result in a partial scavenging of halogens. Another feature captured by our model is the fact that the downward slopes of observed HO2 production are greater for films containing bromide than for those containing iodide.
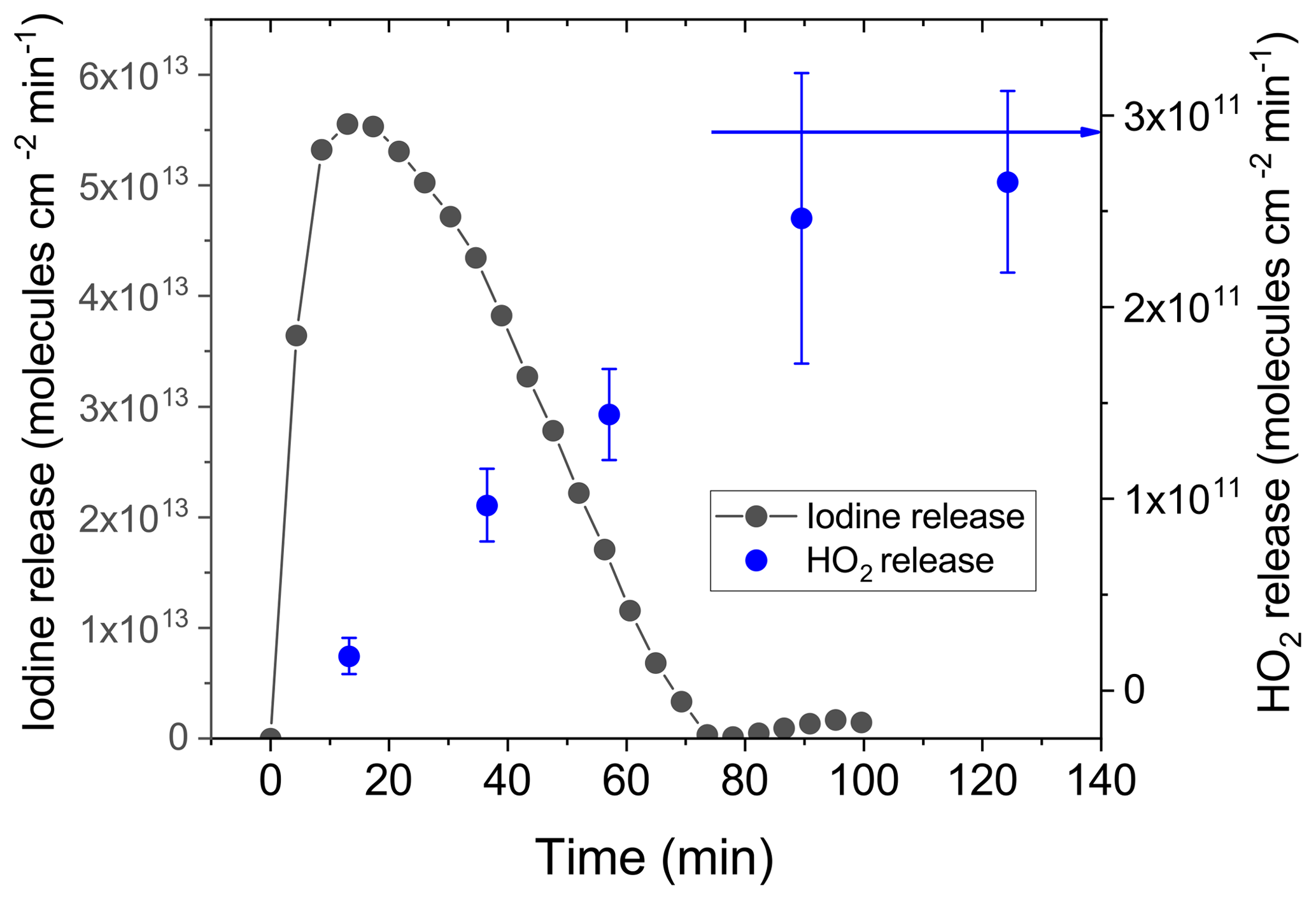
Figure 3Iodine release calculated from the measured mass size distribution of iodine oxide particles produced by the oxidation of iodine species released from the CWFT. The left y axis is expressed as equivalent I2 release (gray circles), and the right y axis is the corresponding HO2 release (blue circles) into the gas phase versus time while irradiating a film in the CWFT loaded with 2.5 mg of IC, 76.8 mg of CA and 313 µg of NaI and equilibrated at 34 % RH (33 mM I−). The blue arrow indicates the HO2 release expected for the film in the absence of iodide.
3.2 Iodine activation
Figure 3 shows the release of iodine and the corresponding HO2 release from a single film continually irradiated for 140 min. Iodine release strongly increased with irradiation, peaking after only several minutes of irradiation and falling off over the following 60 min. The maximum of the iodine release was 5.5×1013 molecules min−1 cm−2. When normalized to the initial amount of iodide present in the film, this corresponds to a iodide lifetime of around 8400 s – thus a little over 2 h. The steady-state model prediction is 4.9×1015 molecules min−1 cm−2 at the initial concentration of iodide, which cannot be directly compared to the measurement as the measurement with the SMPS could not resolve a sharp initial release. Furthermore, it should be noted that the model is not following the system over time. The corresponding HO2 release versus time was measured with a separate film under the same conditions and within the same range of time. Initially, HO2 is entirely depleted as expected for the high iodide concentration of 33 mM used (see Fig. 2). Then, HO2 release increases linearly until 90 min when a steady state is obtained at 3×1011 molecules min−1 cm−2, which is the same as that measured in the absence of iodide (Corral-Arroyo et al., 2018) (blue arrow in Fig. 3 and blue line in Fig. 2). When comparing to Fig. 2, the evolution of the HO2 release with time indicates that a drop in the iodide concentration from 33 mM to below 10−4 M occurred. The total integrated I2O5 mass measured over the whole observation period corresponds to 70(±10) % of the iodide added to the film. As indicated in the Supplement, we could not measure the mass from particles with a diameter smaller than 20 nm, so the mass calculated is a lower limit of the real mass released from the film. In combination with the synchronized behavior of both releases (HO2 and I2), this indicates that iodide is nearly completely depleted in our films after 100 min of irradiation and presumably most of the iodide is converted into molecular iodine, consistent with the lifetime estimate based on the observed maximum release rate. Alternatively, sinks of halides in the films could be the reaction of halide radicals (I• or I) and of HOI or HOBr with organics producing Org–X (Abrahamsson et al., 2018 ;Gilbert et al., 1988; Roveretto et al., 2019) or further oxidation of iodine to iodate, which was beyond the scope of our study.
The efficiency of the iodine activation depends on the different competing processes occurring in the P catalytic cycle and the processes involving halogen radical chemistry (Fig. 1). Oxygen, CA and halides compete for the triplet. Once the triplet oxidizes the halide, the radicals produced can be recycled back to halide or produce the molecular X2 compounds bromine and iodine. X2 can be recycled back to (recycling B) or escape to the gas phase. In spite of the inability of the steady-state model to follow the iodide depletion over time, we can use the model to assess these recycling pathways. For iodine, the model predicts that around 50 % of halogen atoms produced are released to the gas phase as molecular halogen (40 % RH, cm2 s−1 and cm2 s−1) indicating that the fate of around half of iodide radicals is recycling and the other half is leaving the condensed phase as iodine. We argue that HO2, X• and X− compete for , and when X2 is finally produced, it can diffuse out or react with HO2 to produce . According to our model, halogen atom recycling does not change significantly with RH; however, this is not the case for molecular halogens. The predicted efficiency in the release of molecular iodine was about 85 %–95 % at a RH of 40 % and decreased to 45 %–65 % when the diffusion coefficient was decreased by 1 order of magnitude. For molecular bromine, the efficiency of ∼99 % dropped by 2.5 % when was decreased by the same amount. Increasing the diffusion coefficient by 1 order of magnitude increased the efficiency in the release of molecular iodine or bromine to 97 %–99.5 % and almost 100 %, respectively. Thus, changing diffusivity due to a change in RH may have a strong impact on the cycling and, thus, the fate of X2, but not for X radicals.
We investigated the influence of halides on the photochemistry of imidazole-2-carboxaldehyde and its oxidative capacity. The addition of both iodide and bromide increased the HO2 radical production in the system IC ∕ CA. This can be explained by the oxidation of halide ions by the IC triplet being several orders of magnitude faster than the corresponding oxidation of CA (when M then ) (Tinel et al., 2014; Corral-Arroyo et al., 2018). The halogen radical species resulting from the reaction with the triplet scavenge away the HO2 produced, preventing it from leaving the film and thereby maintaining its capacity to participate in redox cycles with the halide species.
Typical concentrations of iodide and bromide in sea spray particles are 10−6 M (Baker, 2004, 2005; Pechtl et al., 2007) and M (Herrmann et al., 2003), respectively. At the sea surface many kinds of chromophoric organic compounds are present, including biomolecules, carbonylic and carboxylic compounds (CDOM) (Chen et al., 2016; Quinn et al., 2015), which are uplifted along with sea spray particles (Hunter and Liss, 1977; Cincinelli et al., 2001). Based on our results, halides are concentrated enough in atmospheric aerosol particles to contribute to the radical production. Assessment of chlorine activation via IC as a chromophore and sensitizer reacting with chloride, which is present in higher concentrations in sea salt aerosol particles (∼ 5.4 M) (Herrmann et al., 2003), was beyond the scope of this study. While the ratio of chloride to bromide or iodide is higher than the inverse ratio of the corresponding rate coefficients (Tinel et al., 2014), the complex radical chemistry and kinetics require detailed attention in order to understand the impacts on chlorine activation and photosensitized HO2 production. Halogen activation depends on the kinetics of the triplet states with halide ions and on the recycling reaction that control the halogen and HO2 yields, so that different relative yields of the two may be expected for different photosensitizing BrC or CDOM species. Furthermore, interactions among the halogens, i.e., bromine with iodine, or either of them with chlorine, have not yet been considered here. An additional aspect is that primary organics present in nascent sea spray particles or on the ocean surface may themselves scavenge triplet states with rates of the same order of magnitude as iodide (Canonica, 2000), thus diminishing the capacity for halogen activation. Although, we suspect that the complex secondary radicals, e.g., alkoxy radicals, would still propagate the triplet-induced capacity to oxidize halides.
In Sect. 1 we estimated the maximum extent of iodine activation in a solution containing 10−6 M based on a steady-state triplet concentration of 10−10 M to be M s−1, corresponding to a rather short lifetime of iodide of only a few seconds. Based on the results obtained, we can refine this number. We note that the experiment in Fig. 3 cannot be directly extrapolated to atmospheric conditions due to the high 33 mM iodide concentration used, which suppresses the triplet concentration to 10−12 M. In addition, the viscous films were 3.4 µm thick; thus, they were beyond atmospheric particle size ranges. Therefore, we run model calculations with 10−6 M iodide, a film thickness of 0.5 µm and with the IC concentration adjusted such that the triplet concentration at steady state reached 10−10 M. Under these conditions, the iodine release is estimated to be M s−1 at 35 % RH and roughly a factor of 2 larger when the diffusion coefficients are set to the cm2 s−1 range for a low-viscosity liquid. Next, we compare these rates to the estimated oxidation rate by O3, first, in a reaction–diffusion limited regime. To accomplish this, we calculate the uptake coefficient, γ, of O3 under the assumption that the reaction proceeds in the reacto-diffusive kinetic regime; thus, for a viscous particle with low diffusivity
where R is the universal gas constant and the mean thermal velocity of ozone, , is 318 m s−1 at T=25 ∘C. We calculate that using a Henry's law constant, , of 0.14 M atm−1 (Berkemeier et al., 2016); a diffusion coefficient of ozone, , of cm2 s−1 (Berkemeier et al., 2016), which corresponds to an aqueous CA particle at ∼40 % RH at room temperature or to a CA particle at ∼70 % RH at −20 ∘C (Lienhard et al., 2014); a bulk reaction rate coefficient, , of 4.2×109 M−1 s−1 (Magi et al., 1997); and the same particle phase iodide concentration, [I]b, of 10−6 M, as above. Particle size effects or contributions from a surface reaction (Moreno et al., 2018) were neglected for this simple comparison. The rate of O3 uptake, U (in molecules s−1 particle−1) and, thus, of iodide oxidation (as an upper limit to iodine activation) can be calculated by
where is the concentration of ozone in the gas phase (in molecule cm−3) and r is the radius of the particle. For a gas-phase mixing ratio of 100 ppb (2.5×1012 molecule cm−3) and a particle diameter of 500 nm, normalization to the particle volume yields an iodide turnover of M s−1, which is an order of magnitude below that estimated for the photosensitized oxidation under comparable conditions.
Conversely, we can consider a more dilute aqueous particle or an aqueous particle that is dominated by inorganic ions only, where the liquid-phase diffusion coefficient is high and the solubility of O3 is lower, M atm−1 as in pure water (Sander, 2015). O3 remains well-mixed throughout the particle due to the low iodide content. For the same iodide content and O3 mixing ratio as above, the iodine activation would become M s−1; thus, it would be about a factor of 5 higher than the estimate for the photosensitized oxidation under conditions with high RH and high diffusivity. We note that at 0∘ zenith angle, the solar actinic flux is about 3 times greater than the UV lamps we used in the experiment; therefore, the excitation rates of IC may be 3 times faster than what was used here. We conclude that photosensitized iodine production is relevant for sea spray aerosol particles containing chromophores under lower RH conditions or lower temperature when the reactive uptake of ozone is slow. Under humid conditions and with less organics present the activation via reaction with ozone may dominate, although still with a significant contribution from photosensitized chemistry.
We noted the existence of a cycling in halide radical chemistry that shuts down the HOx chemistry and, simultaneously, prevent the release of molecular halogens to the gas phase. Furthermore, this cycling strongly depends on the diffusion properties of the matrix, reaching a greater cycling efficiency when diffusion is slow and a lower efficiency when diffusion is fast. Even so, the release is not entirely reduced under a wide range of diffusion regimes and a large fraction of the iodine produced (50 %–100 %) will be released. Based on the model predictions, we suspect that bromine activation behaves in a similar way to iodine activation, as the impacts on HO2 release were similar.
The data underlying Figs. 2 and 3 and the MATLAB codes of the steady-state model calculations are available in the Supplement.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-10817-2019-supplement.
PCA, MA, RV and PAA designed the study. PCA and RA performed the experiments and analyzed the data. PCA wrote the paper, with contributions from all co-authors.
The authors declare that they have no conflict of interest.
We would like to thank Mario Birrer for technical support. Peter A. Alpert appreciates support by a PSI CROSS project and the “PSI-FELLOW” program, which is cofunded by the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant no. 701647. Rainer Volkamer is thankful for financial support from the National Science Foundation – Atmospheric and Geospace Sciences (US).
This research has been supported by the Swiss National Science Foundation (grant no. 163074), the European Union's Horizon 2020 (grant no. 701647), and the National Science Foundation – Atmospheric and Geospace Sciences (US) (grant no. 1620530).
This paper was edited by Dwayne Heard and reviewed by two anonymous referees.
Abrahamsson, K., Granfors, A., Ahnoff, M., Cuevas, C. A., and Saiz-Lopez, A.: Organic bromine compounds produced in sea ice in Antarctic winter, Nature Comm., 9, 5291, https://doi.org/10.1038/s41467-018-07062-8, 2018.
Aregahegn, K. Z., Nozière, B., and George, C.: Organic aerosol formation photo-enhanced by the formation of secondary photosensitizers in aerosols, Faraday Discuss., 165, 123–134, https://doi.org/10.1039/c3fd00044c, 2013.
Baker, A. R.: Inorganic iodine speciation in tropical Atlantic aerosol, Geophys. Res. Lett., 31, L23S02, https://doi.org/10.1029/2004gl020144, 2004.
Baker, A. R.: Marine aerosol iodine chemistry: The importance of soluble organic iodine, Environ. Chem., 2, 295–298, https://doi.org/10.1071/en05070, 2005.
Behnke, W., George, C., Scheer, V., and Zetzsch, C.: Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments, J. Geophys. Res., 102, 3795–3804, https://doi.org/10.1029/96jd03057, 1997.
Berkemeier, T., Steimer, S. S., Krieger, U. K., Peter, T., Poschl, U., Ammann, M., and Shiraiwa, M.: Ozone uptake on glassy, semi-solid and liquid organic matter and the role of reactive oxygen intermediates in atmospheric aerosol chemistry, Phys. Chem. Chem. Phys., 18, 12662–12674, https://doi.org/10.1039/c6cp00634e, 2016.
Bianchini, R. and Chiappe, C.: Stereoselectivity and reversibility of electrophylic bromine addition to stilbenes in chloroform – Influence of bromide tribromide pentabromide equilibrium in the counteranion of the ionic intermediates, J. Org. Chem., 57, 6474–6478, https://doi.org/10.1021/jo00050a021, 1992.
Bielski, B. H. J., Cabelli, D. E., Arudi, R. L., and Ross, A. B.: Reactivity of Radicals in Aqueous Solution, J. Phys. Chem. Ref. Data, 14, 1041–1100, https://doi.org/10.1063/1.555739, 1985.
Blanchard, D. C.: Sea-to-air transport of surface active material, Science, 146, 396–397, https://doi.org/10.1126/science.146.3642.396, 1964.
Bloss, W. J., Lee, J. D., Johnson, G. P., Sommariva, R., Heard, D. E., Saiz-Lopez, A., Plane, J. M. C., McFiggans, G., Coe, H., Flynn, M., Williams, P., Rickard, A. R., and Fleming, Z. L.: Impact of halogen monoxide chemistry upon boundary layer OH and HO2 concentrations at a coastal site, Geophys. Res. Lett., 32, L06814, https://doi.org/10.1029/2004gl022084, 2005.
Canonica, S., Hellrung, B., and Wirz, J.: Oxidation of phenols by triplet aromatic ketones in aqueous solution, J. Phys. Chem. A, 104, 1226–1232, https://doi.org/10.1021/jp9930550, 2000.
Carpenter, L. J.: Iodine in the marine boundary layer, Chem. Rev., 103, 4953–4962, https://doi.org/10.1021/cr0206465, 2003.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nature Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Chameides, W. L. and Davis, D. D.: Iodine – Its possible role in tropospheric photochemistry, J. Geophys. Res., 85, 7383–7398, https://doi.org/10.1029/JC085iC12p07383, 1980.
Chen, Q., Miyazaki, Y., Kawamura, K., Matsumoto, K., Coburn, S., Volkamer, R., Iwamoto, Y., Kagami, S., Deng, Y., Ogawa, S., Ramasamy, S., Kato, S., Ida, A., Kajii, Y., and Mochida, M.: Characterization of Chromophoric Water-Soluble Organic Matter in Urban, Forest, and Marine Aerosols by HR-ToF-AMS Analysis and Excitation–Emission Matrix Spectroscopy, Environ. Sci. Technol., 50, 10351–10360, https://doi.org/10.1021/acs.est.6b01643, 2016.
Choi, S., Baik, S., Park, S., Park, N., and Kim, D.: Implementation of Differential Absorption LIDAR (DIAL) for Molecular Iodine Measurements Using Injection-Seeded Laser, J. Opt. Soc. Korea, 16, 325–330, 2012.
Cincinelli, A., Desideri, P. G., Lepri, L., Checchini, L., Del Bubba, M., and Udisti, R.: Marine contribution to the chemical composition of coastal and inland Antarctic snow, Int. J. Environ. Anal. Chem., 79, 283–299, https://doi.org/10.1080/03067310108044390, 2001.
Corral-Arroyo, P., Bartels-Rausch, T., Alpert, P. A., Dumas, S., Perrier, S., George, C., and Ammann, M.: Particle phase photosensitized radical production and aerosol aging, Environ. Sci. Technol., 52, 7680–7688, https://doi.org/10.1021/acs.est.8b00329, 2018.
De Laurentiis, E., Minella, M., Maurino, V., Minero, C., Mailhot, G., Sarakha, M., Brigante, M., and Vione, D.: Assessing the occurrence of the dibromide radical (Br) in natural waters: Measures of triplet-sensitised formation, reactivity, and modelling, Sci. Total Environ., 439, 299–306, https://doi.org/10.1016/j.scitotenv.2012.09.037, 2012.
Dix, B., Baidar, S., Bresch, J. F., Hall, S. R., Schmidt, K. S., Wang, S., and Volkamer, R.: Detection of iodine monoxide in the tropical free troposphere, P. Natl. Acad. Sci. USA, 110, 2035–2040, https://doi.org/10.1073/pnas.1212386110, 2013.
George, C., Ammann, M., D'Anna, B., Donaldson, D. J., and Nizkorodov, S. A.: Heterogeneous Photochemistry in the Atmosphere, Chem. Rev., 115, 4218–4258, https://doi.org/10.1021/cr500648z, 2015.
Gilbert, B. C., Stell, J. K., Peet, W. J., and Radford, K. J.: Generation and reactions of the chlorine atom in aqueous solution, J. Chem. Soc., Faraday Trans., 84, 3319–3330, https://doi.org/10.1039/f19888403319, 1988.
González Palacios, L., Corral Arroyo, P., Aregahegn, K. Z., Steimer, S. S., Bartels-Rausch, T., Nozière, B., George, C., Ammann, M., and Volkamer, R.: Heterogeneous photochemistry of imidazole-2-carboxaldehyde: HO2 radical formation and aerosol growth, Atmos. Chem. Phys., 16, 11823–11836, https://doi.org/10.5194/acp-16-11823-2016, 2016.
Hepach, H., Quack, B., Tegtmeier, S., Engel, A., Bracher, A., Fuhlbrügge, S., Galgani, L., Atlas, E. L., Lampel, J., Frieß, U., and Krüger, K.: Biogenic halocarbons from the Peruvian upwelling region as tropospheric halogen source, Atmos. Chem. Phys., 16, 12219–12237, https://doi.org/10.5194/acp-16-12219-2016, 2016.
Herrmann, H., Majdik, Z., Ervens, B., and Weise, D.: Halogen production from aqueous tropospheric particles, Chemosphere, 52, 485–502, https://doi.org/10.1016/s0045-6535(03)00202-9, 2003.
Hoffman, E. J. and Duce, R. A.: Factors influencing organic-carbon content of marine aerosols – Laboratory study, J. Geophys. Res., 81, 3667–3670, https://doi.org/10.1029/JC081i021p03667, 1976.
Hoffmann, T., O'Dowd, C. D., and Seinfeld, J. H.: Iodine oxide homogeneous nucleation: An explanation for coastal new particle production, Geophys. Res. Lett., 28, 1949–1952, https://doi.org/10.1029/2000gl012399, 2001.
Hunter, K. A. and Liss, P. S.: Input of organic material to oceans – Air-sea interactions and organic chemical composition of sea-surface, Marine Chem., 5, 361–379, https://doi.org/10.1016/0304-4203(77)90029-9, 1977.
Ishigure, K., Shiraishi, H., and Okuda, H.: Radiation-chemistry of aqueous iodine systems under nuclear-reactor accident conditions, Rad. Phys. Chem., 32, 593–597, 1988.
Ivkovic-Jensen, M. M. and Kostic, N. M.: Effects of viscosity and temperature on the kinetics of the electron-transfer reaction between the triplet state of zinc cytochrome c and cupriplastocyanin, Biochem., 36, 8135–8144, https://doi.org/10.1021/bi970327l, 1997.
Jammoul, A., Dumas, S., D'Anna, B., and George, C.: Photoinduced oxidation of sea salt halides by aromatic ketones: a source of halogenated radicals, Atmos. Chem. Phys., 9, 4229–4237, https://doi.org/10.5194/acp-9-4229-2009, 2009.
Kampf, C. J., Jakob, R., and Hoffmann, T.: Identification and characterization of aging products in the glyoxal/ammonium sulfate system – implications for light-absorbing material in atmospheric aerosols, Atmos. Chem. Phys., 12, 6323–6333, https://doi.org/10.5194/acp-12-6323-2012, 2012.
Kaur, R. and Anastasio, C.: First Measurements of Organic Triplet Excited States in Atmospheric Waters, Environ. Sci. Technol., 52, 5218–5226, https://doi.org/10.1021/acs.est.7b06699, 2018.
Knopf, D. A., Alpert, P. A., Wang, B., O'Brien, R. E., Kelly, S. T., Laskin, A., Gilles, M. K., and Moffet, R. C.: Microspectroscopic imaging and characterization of individually identified ice nucleating particles from a case field study, J. Geophys. Res., 119, 10365–10381, https://doi.org/10.1002/2014JD021866, 2014.
Kunze, A., Muller, U., Tittes, K., Fouassier, J. P., and Morlet-Savary, F.: Triplet quenching by onium salts in polar and nonpolar solvents, J. Photoch. Photobio. A, 110, 115–122, https://doi.org/10.1016/s1010-6030(97)00178-0, 1997.
Lary, D. J.: Gas phase atmospheric bromine photochemistry, J. Geophys. Res., 101, 1505–1516, https://doi.org/10.1029/95jd02463, 1996.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of atmospheric brown carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lienhard, D. M., Bones, D. L., Zuend, A., Krieger, U. K., Reid, J. P., and Peter, T.: Measurements of Thermodynamic and Optical Properties of Selected Aqueous Organic and Organic-Inorganic Mixtures of Atmospheric Relevance, J. Phys. Chem. A, 116, 9954–9968, https://doi.org/10.1021/jp3055872, 2012.
Lienhard, D. M., Huisman, A. J., Bones, D. L., Te, Y. F., Luo, B. P., Krieger, U. K., and Reid, J. P.: Retrieving the translational diffusion coefficient of water from experiments on single levitated aerosol droplets, Phys. Chem. Chem. Phys., 16, 16677–16683, https://doi.org/10.1039/c4cp01939c, 2014.
Lignell, H., Hinks, M. L., and Nizkorodov, S. A.: Exploring matrix effects on photochemistry of organic aerosols, P. Natl. Acad. Sci. USA, 111, 13780–13785, https://doi.org/10.1073/pnas.1322106111, 2014.
Magi, L., Schweitzer, F., Pallares, C., Cherif, S., Mirabel, P., and George, C.: Investigation of the uptake rate of ozone and methyl hydroperoxide by water surfaces, J. Phys. Chem. A, 101, 4943–4949, https://doi.org/10.1021/jp970646m, 1997.
Mahajan, A. S., Sorribas, M., Gómez Martín, J. C., MacDonald, S. M., Gil, M., Plane, J. M. C., and Saiz-Lopez, A.: Concurrent observations of atomic iodine, molecular iodine and ultrafine particles in a coastal environment, Atmos. Chem. Phys., 11, 2545–2555, https://doi.org/10.5194/acp-11-2545-2011, 2011.
Maillard, B., Ingold, K. U., and Scaiano, J. C.: Rate constants for the reactions of free-radicals with oxygen in solution, J. Am. Chem. Soc., 105, 5095–5099, https://doi.org/10.1021/ja00353a039, 1983.
McFiggans, G., Coe, H., Burgess, R., Allan, J., Cubison, M., Alfarra, M. R., Saunders, R., Saiz-Lopez, A., Plane, J. M. C., Wevill, D., Carpenter, L., Rickard, A. R., and Monks, P. S.: Direct evidence for coastal iodine particles from Laminaria macroalgae – linkage to emissions of molecular iodine, Atmos. Chem. Phys., 4, 701–713, https://doi.org/10.5194/acp-4-701-2004, 2004.
McFiggans, G., Bale, C. S. E., Ball, S. M., Beames, J. M., Bloss, W. J., Carpenter, L. J., Dorsey, J., Dunk, R., Flynn, M. J., Furneaux, K. L., Gallagher, M. W., Heard, D. E., Hollingsworth, A. M., Hornsby, K., Ingham, T., Jones, C. E., Jones, R. L., Kramer, L. J., Langridge, J. M., Leblanc, C., LeCrane, J.-P., Lee, J. D., Leigh, R. J., Longley, I., Mahajan, A. S., Monks, P. S., Oetjen, H., Orr-Ewing, A. J., Plane, J. M. C., Potin, P., Shillings, A. J. L., Thomas, F., von Glasow, R., Wada, R., Whalley, L. K., and Whitehead, J. D.: Iodine-mediated coastal particle formation: an overview of the Reactive Halogens in the Marine Boundary Layer (RHaMBLe) Roscoff coastal study, Atmos. Chem. Phys., 10, 2975–2999, https://doi.org/10.5194/acp-10-2975-2010, 2010.
McNeill, K. and Canonica, S.: Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci. Proc. Imp., 18, 1381–1399, https://doi.org/10.1039/c6em00408c, 2016.
Mitroka, S., Zimmeck, S., Troya, D., and Tanko, J. M.: How Solvent Modulates Hydroxyl Radical Reactivity in Hydrogen Atom Abstractions, J. Am. Chem. Soc., 132, 2907–2913, https://doi.org/10.1021/ja903856t, 2010.
Moreno, C. G., Gálvez, O., López-Arza Moreno, V., Espildora-García, E. M., and Baeza-Romero, M. T.: A revisit of the interaction of gaseous ozone with aqueous iodide. Estimating the contributions of the surface and bulk reactions, Phys. Chem. Chem. Phys., 20, 27571–27584, https://doi.org/10.1039/C8CP04394A, 2018.
Morrison, M., Bayse, G. S., and Michaels, A. W.: Determination of spectral properties of aqueous I2 and and equilibrium constant, Anal. Biochem., 42, 195–201, https://doi.org/10.1016/0003-2697(71)90026-1, 1971.
Nagarajan, V. and Fessenden, R. W.: Flash-photolysis of transient radicals, 1. Cl, Br, I and SCN2, J. Phys. Chem., 89, 2330–2335, https://doi.org/10.1021/j100257a037, 1985.
O'Dowd, C. D. and de Leeuw, G.: Marine aerosol production: a review of the current knowledge, Philos. Tr. R. Soc. S.-A, 365, 1753–1774, https://doi.org/10.1098/rsta.2007.2043, 2007.
Pechtl, S., Schmitz, G., and von Glasow, R.: Modelling iodide – iodate speciation in atmospheric aerosol: Contributions of inorganic and organic iodine chemistry, Atmos. Chem. Phys., 7, 1381–1393, https://doi.org/10.5194/acp-7-1381-2007, 2007.
Quinn, P. K., Collins, D. B., Grassian, V. H., Prather, K. A., and Bates, T. S.: Chemistry and Related Properties of Freshly Emitted Sea Spray Aerosol, Chem. Rev., 115, 4383–4399, https://doi.org/10.1021/cr5007139, 2015.
Roveretto, M., Li, M. C., Hayeck, N., Bruggemann, M., Emmelin, C., Perrier, S., and George, C.: Real-Time Detection of Gas-Phase Organohalogens from Aqueous Photochemistry Using Orbitrap Mass Spectrometry, ACS Earth Space Chem., 3, 329–334, https://doi.org/10.1021/acsearthspacechem.8b00209, 2019.
Saiz-Lopez, A. and Plane, J. M. C.: Novel iodine chemistry in the marine boundary layer, Geophys. Res. Lett., 31, L04112, https://doi.org/10.1029/2003gl019215, 2004.
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/c2cs35208g, 2012.
Saiz-Lopez, A., Plane, J. M. C., Mahajan, A. S., Anderson, P. S., Bauguitte, S. J.-B., Jones, A. E., Roscoe, H. K., Salmon, R. A., Bloss, W. J., Lee, J. D., and Heard, D. E.: On the vertical distribution of boundary layer halogens over coastal Antarctica: implications for O3, HOx, NOx and the Hg lifetime, Atmos. Chem. Phys., 8, 887–900, https://doi.org/10.5194/acp-8-887-2008, 2008.
Saiz-Lopez, A., Plane, J. M., Baker, A. R., Carpenter, L. J., von Glasow, R., Martin, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric chemistry of iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012.
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015.
Sander, R. and Crutzen, P. J.: Model study indicating halogen activation and ozone destruction in polluted air masses transported to the sea, J. Geophys. Res., 101, 9121–9138, https://doi.org/10.1029/95jd03793, 1996.
Sarwar, G., Kang, D., Foley, K., Schwede, D., Gantt, B., and Mathur, R.: Technical note: Examining ozone deposition over seawater, Atmos. Environ., 141, 255–262, https://doi.org/10.1016/j.atmosenv.2016.06.072, 2016.
Saunders, R. W. and Plane, J. M. C.: Fractal growth modelling of nanoparticles, J. Aerosol Sci., 37, 1737–1749, https://doi.org/10.1016/j.jaerosci.2006.08.007, 2006.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res., 121, 11819–11835, https://doi.org/10.1002/2015jd024229, 2016.
Schwarz, H. A. and Bielski, B. H. J.: Reactions of HO2 and O with iodine and bromine and the I and i atom reduction potentials, J. Phys. Chem., 90, 1445–1448, https://doi.org/10.1021/j100398a045, 1986.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016a.
Sherwen, T., Evans, M. J., Spracklen, D. V., Carpenter, L. J., Chance, R., Baker, A. R., Schmidt, J. A., and Breider, T. J.: Global modeling of tropospheric iodine aerosol, Geophys. Res. Lett., 43, 10012–10019, https://doi.org/10.1002/2016gl070062, 2016b.
Simpson, W. R., Brown, S. S., Saiz-Lopez, A., Thornton, J. A., and Glasow, R.: Tropospheric halogen chemistry: sources, cycling, and impacts, Chem. Rev., 115, 4035–4062, https://doi.org/10.1021/cr5006638, 2015.
Sipila, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Perakyla, O., Rissanen, M. P., Ehn, M., Vehkamaki, H., Kurten, T., Berndt, T., Petaja, T., Worsnop, D., Ceburnis, D., Kerminen, V. M., Kulmala, M., and O'Dowd, C.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Sommariva, R., Bloss, W. J., and von Glasow, R.: Uncertainties in gas-phase atmospheric iodine chemistry, Atmos. Environ., 57, 219–232, https://doi.org/10.1016/j.atmosenv.2012.04.032, 2012.
Song, Y. C., Haddrell, A. E., Bzdek, B. R., Reid, J. P., Bannan, T., Topping, D. O., Percival, C., and Cai, C.: Measurements and Predictions of Binary Component Aerosol Particle Viscosity, J. Phys. Chem. A, 120, 8123–8137, https://doi.org/10.1021/acs.jpca.6b07835, 2016.
Stavrakou, T., Müller, J.-F., De Smedt, I., Van Roozendael, M., Kanakidou, M., Vrekoussis, M., Wittrock, F., Richter, A., and Burrows, J. P.: The continental source of glyoxal estimated by the synergistic use of spaceborne measurements and inverse modelling, Atmos. Chem. Phys., 9, 8431–8446, https://doi.org/10.5194/acp-9-8431-2009, 2009.
Stemmler, K., Ammann, M., Donders, C., Kleffmann, J., and George, C.: Photosensitized reduction of nitrogen dioxide on humic acid as a source of nitrous acid, Nature, 440, 195–198, https://doi.org/10.1038/nature04603, 2006.
Stone, D., Sherwen, T., Evans, M. J., Vaughan, S., Ingham, T., Whalley, L. K., Edwards, P. M., Read, K. A., Lee, J. D., Moller, S. J., Carpenter, L. J., Lewis, A. C., and Heard, D. E.: Impacts of bromine and iodine chemistry on tropospheric OH and HO2: comparing observations with box and global model perspectives, Atmos. Chem. Phys., 18, 3541–3561, https://doi.org/10.5194/acp-18-3541-2018, 2018.
Tinel, L., Dumas, S., and George, C.: A time-resolved study of the multiphase chemistry of excited carbonyls: Imidazole-2-carboxaldehyde and halides, C. R. Chimie, 17, 801–807, https://doi.org/10.1016/j.crci.2014.03.008, 2014.
Vogt, R., Sander, R., Von Glasow, R., and Crutzen, P. J.: Iodine chemistry and its role in halogen activation and ozone loss in the marine boundary layer: A model study, J. Atmos. Chem., 32, 375–395, https://doi.org/10.1023/a:1006179901037, 1999.
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2–O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015.
von Glasow, R., von Kuhlmann, R., Lawrence, M. G., Platt, U., and Crutzen, P. J.: Impact of reactive bromine chemistry in the troposphere, Atmos. Chem. Phys., 4, 2481–2497, https://doi.org/10.5194/acp-4-2481-2004, 2004.
Wagner, I. and Strehlow, H.: On the flash-photolysis of bromide ions in aqueous solutions, Ber. Bunsen Phys. Chem., 91, 1317–1321, 1987.
Wang, S. Y. and Pratt, K. A.: Molecular Halogens Above the Arctic Snowpack: Emissions, Diurnal Variations, and Recycling Mechanisms, J. Geophys. Res.-Atmos., 122, 11991–12007, https://doi.org/10.1002/2017jd027175, 2017.
Wren, S. N., Donaldson, D. J., and Abbatt, J. P. D.: Photochemical chlorine and bromine activation from artificial saline snow, Atmos. Chem. Phys., 13, 9789–9800, https://doi.org/10.5194/acp-13-9789-2013, 2013.
Yu, L., Smith, J., Laskin, A., Anastasio, C., Laskin, J., and Zhang, Q.: Chemical characterization of SOA formed from aqueous-phase reactions of phenols with the triplet excited state of carbonyl and hydroxyl radical, Atmos. Chem. Phys., 14, 13801–13816, https://doi.org/10.5194/acp-14-13801-2014, 2014.
Zardini, A. A., Sjogren, S., Marcolli, C., Krieger, U. K., Gysel, M., Weingartner, E., Baltensperger, U., and Peter, T.: A combined particle trap/HTDMA hygroscopicity study of mixed inorganic/organic aerosol particles, Atmos. Chem. Phys., 8, 5589–5601, https://doi.org/10.5194/acp-8-5589-2008, 2008.