the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Aug 2019
| 13 Aug 2019
Biogenic and anthropogenic sources of aerosols at the High Arctic site Villum Research Station
Ingeborg E. Nielsen
Henrik Skov
Andreas Massling
Axel C. Eriksson
Manuel Dall'Osto
Heikki Junninen
Nina Sarnela
Robert Lange
Sonya Collier
Qi Zhang
Christopher D. Cappa
Jacob K. Nøjgaard
There are limited measurements of the chemical composition, abundance and sources of atmospheric particles in the High Arctic To address this, we report 93 d of soot particle aerosol mass spectrometer (SP-AMS) data collected from 20 February to 23 May 2015 at Villum Research Station (VRS) in northern Greenland (81∘36′ N). During this period, we observed the Arctic haze phenomenon with elevated PM1 concentrations ranging from an average of 2.3, 2.3 and 3.3 µg m−3 in February, March and April, respectively, to 1.2 µg m−3 in May. Particulate sulfate () accounted for 66 % of the non-refractory PM1 with the highest concentration until the end of April and decreasing in May. The second most abundant species was organic aerosol (OA) (24 %). Both OA and PM1, estimated from the sum of all collected species, showed a marked decrease throughout May in accordance with the polar front moving north, together with changes in aerosol removal processes. The highest refractory black carbon (rBC) concentrations were found in the first month of the campaign, averaging 0.2 µg m−3. In March and April, rBC averaged 0.1 µg m−3 while decreasing to 0.02 µg m−3 in May.
Positive matrix factorization (PMF) of the OA mass spectra yielded three factors: (1) a hydrocarbon-like organic aerosol (HOA) factor, which was dominated by primary aerosols and accounted for 12 % of OA mass, (2) an Arctic haze organic aerosol (AOA) factor and (3) a more oxygenated marine organic aerosol (MOA) factor. AOA dominated until mid-April (64 %–81 % of OA), while being nearly absent from the end of May and correlated significantly with , suggesting the main part of that factor is secondary OA. The MOA emerged late at the end of March, where it increased with solar radiation and reduced sea ice extent and dominated OA for the rest of the campaign until the end of May (24 %–74 % of OA), while AOA was nearly absent. The highest O∕C ratio (0.95) and S∕C ratio (0.011) was found for MOA. Our data support the current understanding that Arctic aerosols are highly influenced by secondary aerosol formation and receives an important contribution from marine emissions during Arctic spring in remote High Arctic areas. In view of a changing Arctic climate with changing sea-ice extent, biogenic processes and corresponding source strengths, highly time-resolved data are needed in order to elucidate the components dominating aerosol concentrations and enhance the understanding of the processes taking place.
- Article
(2053 KB) - Full-text XML
-
Supplement
(12749 KB) - BibTeX
- EndNote





Climate change driven by anthropogenic emission of greenhouse gases seriously impacts the Arctic, which has experienced average temperature increases of twice the global mean during the last 100 years (AMAP, 2015; IPCC, 2018). Warming has led to destabilization of permafrost (AMAP, 2017) and a longer melting season, resulting in a critical decrease in the sea-ice extent (Stroeve et al., 2007). The latter changes the Earth's albedo and results in positive sea-ice and snow-albedo feedbacks, causing further warming (Lenton, 2012). In addition to long-lived greenhouse gases such as CO2, atmospheric aerosols also have an impact on the radiation balance of the Earth. Aerosols affect the radiative balance in various ways. They can absorb and scatter solar radiation, causing either warming or cooling of the atmosphere, respectively. Aerosols can also impact the properties of clouds, for example affecting cloud reflectivity, by serving as cloud-condensation and ice nuclei (Twomey, 1977). Due to aerosols' climatic importance it is crucial to expand the knowledge regarding their chemical and physical properties in the Arctic to reduce the current uncertainty (IPCC, 2013) with respect to the overall effect of aerosols on Earth's energy budget.
It is well established that the aerosol concentration in the Arctic atmosphere is seasonally varying, resulting in higher loadings during winter and spring compared to summer and fall, often referred to as “Arctic haze” (Heidam et al., 1999, 2004; Tunved et al., 2013; Quinn et al., 2007; Barrie et al., 1981; Heidam, 1984). This is explained by a greater accessibility to the lower troposphere in the Arctic from anthropogenic source regions outside the Arctic, due to an expansion of the polar dome (AMAP, 2011) in winter and spring. In addition, during the Arctic winter strong temperature inversions create stable stratification where aerosol removal processes are strongly reduced prolonging their atmospheric lifetime (Stohl, 2006; Sodemann et al., 2011; AMAP, 2011). The air masses inside the wintertime dome are extremely dry, limiting aerosol wet deposition, while low turbulence caused by the stratification and slow vertical exchange reduces the dry deposition of aerosols (Sodemann et al., 2011; Stohl, 2006; Abbatt et al., 2019). The Arctic haze peaks in early spring (Heidam et al., 1999, 2004; Law and Stohl, 2007; Stohl, 2006; Abbatt et al., 2019). Arctic haze particles effectively scatter light (Andrews et al., 2011; Schmeisser et al., 2018) and act as cloud-condensation nuclei (CCN) (Earle et al., 2011; Komppula et al., 2005). Due to the expansion of the polar dome, a major part of the aerosol mass is long-range transported from source regions outside the Arctic where the primary source region has been identified as the northern part of Eurasia (Nguyen et al., 2013; Quinn et al., 2008; Heidam et al., 2004; Stohl et al., 2007; Christensen, 1997; Abbatt et al., 2019). Studies have shown that main constituents of Arctic aerosols are sulfate () and organics mixed with a minor fraction of nitrate (), ammonium (), black carbon (BC) and heavy metals (Quinn et al., 2007; Fenger et al., 2013; Nguyen et al., 2013; Frossard et al., 2011; Barrie et al., 1981). This is also the case at the High Arctic station, Villum Research Station (VRS) at Station Nord in northern Greenland, where this study was conducted. Rahn and Heidam (1981) have previously estimated the average chemical composition of Arctic sub-micrometer aerosols during winter and spring as 2 µg m−3 , 1 µg m−3 organic aerosol (OA), 0.3–0.5 µg m−3 BC and a few hundred ng m−3 of other compounds. Since then, and BC during winter and spring have declined at Alert, Mount Zeppelin, Utqiaġvik (formerly known as Barrow) and VRS (Heidam et al., 1999; Hirdman et al., 2010; AMAP, 2015). However, the total Arctic column burden may have increased (Sharma et al., 2013).
BC is the most important aerosol for absorbing solar radiation in the atmosphere. Of particular concern for the Arctic, BC deposited on snow and ice-covered surfaces changes the albedo, leading to increased absorption of solar radiation and direct heating of the surface (Bond et al., 2013). Consequently, melting accelerates, giving BC an important role especially in an Arctic context (Bond et al., 2013; Quinn et al., 2008; AMAP, 2011). Long-range transport of BC to the Arctic is very effective in midwinter, when removal processes are slowest. Transport reaches a minimum in late spring, where wet deposition becomes an important removal process (Abbatt et al., 2019; AMAP, 2015). Natural emissions from vegetation fires can be considerable in spring and early summer (Mahmood et al., 2016). Overall, the general seasonal cycle of BC in the Arctic is characterized by the highest concentrations being observed between January and April and the lowest concentrations being observed throughout the summer, with periodic spikes in concentration throughout the summer (Sharma et al., 2006). OA is also an important component of Arctic aerosols and is composed of many different molecules derived from either primary emissions or from secondary production. Consequently, there are often many distinct sources of OA. OA can typically contribute up to one-third of PM1 in the Arctic, though few studies have characterized this component in detail (Barrett et al., 2015; Brock et al., 2011; Frossard et al., 2011; Kawamura et al., 2010; Quinn et al., 2002; Shaw et al., 2010; Leaitch et al., 2018; Chang et al., 2011; Willis et al., 2018). Total OA is relatively constant or decreasing over time in late winter. However, during spring it increases, suggesting that there is photochemical production of OA (Willis et al., 2018). There is a need for more detailed measurements of OA composition in the Arctic to better understand the key sources and how these vary over time (Willis et al., 2018).
It is crucial to understand natural sources in addition to anthropogenic sources of Arctic aerosols. Marine and coastal marine locations constitute a large part of Arctic, and marine aerosols comprise both organic and inorganic constituents of primary and secondary origin. Production of primary marine aerosols is known to correlate with wind speed and possibly also other mechanisms (Willis et al., 2018). Primary marine organic aerosols in Arctic regions are believed to consist of water-soluble or surface-active organic compounds present in the surface water or water-insoluble microgels (Willis et al., 2018; Leck and Bigg, 2005; Orellana et al., 2011). Marine aerosols play an important role for the climate due to their optical properties and ability to alter cloud nucleation (Abbatt et al., 2019; Willis et al., 2018). Biogenic marine aerosols can scatter solar radiation, which will result in a negative radiative forcing. Biogenic marine aerosols can also coat soot particles, which may be transported from wildfires (AMAP, 2015), which could impact the CCN activity and absorption by the soot particles (Lange et al., 2018). Methanesulfonic acid (MSA), an oxidation product of dimethyl sulfide (DMS), is abundant in spring and summer (Abbatt et al., 2019) and is a key indicator of secondary marine aerosols. MSA levels have been associated with marginal sea ice moving north (Laing et al., 2013; Quinn et al., 2009; Sharma et al., 2012). A new satellite-based model suggests that DMS emissions in the Arctic have increased by 30 % per decade over the last 2 decades, due to both increased temperatures and decreased ice cover (Abbatt et al., 2019). A relationship between MSA and the frequency of new particle formation has also been inferred based on long-term observations (Dall'Osto et al., 2017), although MSA cannot be the nucleating part. This suggest that DMS is important for summertime particles. Another important natural source of Arctic aerosols is ammonia, which among other things is believed to originate from migrating sea bird colonies (Croft et al., 2016). Modeling studies have been shown to better capture particle burst and growth when an ammonia source from sea birds was included (Croft et al., 2016, 2019). Additionally, ammonia can also be transported from boreal wildfires from lower latitudes.
Many previous Arctic studies have been based on off-line analysis and filter measurements of ambient aerosols with a relatively low time resolution from hours up to a week (Heidam et al., 1999, 2004; Skov et al., 2006; Quinn et al., 2007, 2009; Massling et al., 2015; Leaitch et al., 2018; Sharma et al., 2012). Beside the low time resolution, a disadvantage of these types of measurements can be evaporate loss or adsorption of semi-volatile compounds (Lee et al., 2013; Dillner et al., 2009). Highly time-resolved in situ measurements can reduce these artifacts while also enabling the possibility of observing the variations in different chemical species on a much shorter timescale. In this way, it is possible to look into the processes behind the observed levels. In the last decade, aerosol mass spectrometry (AMS) (Canagaratna et al., 2007; DeCarlo et al., 2006; Jimenez et al., 2003; Drewnick et al., 2005; Jayne et al., 2000) has been widely used as an online method for quantitative analysis of chemical composition of atmospheric particles. With the addition of a laser vaporizer (Onasch et al., 2012), its application has been extended to include refractory aerosol components, including refractory black carbon (rBC).
In this study, the time-dependent concentrations of sub-micrometer particle composition including OA, , , , chloride (Cl−) and rBC are reported at the High Arctic site VRS. The measurements were conducted by application of a soot particle aerosol mass spectrometer (SP-AMS) and auxiliary measurements during the Arctic spring 2015, when concentrations are expected to peak. The objectives are to gain better insight into the processes influencing the chemical composition of High Arctic aerosols and to allocate potential sources and source types by use of positive matrix factorization (PMF).
2.1 Sampling site
The atmospheric measurements were carried out at VRS located at the Danish military station, Station Nord, in northern Greenland (Fig. S1, 81∘36′ N, 16∘40′ W, 24 m a.m.s.l.). VRS is situated in a region with a dry and cold climate where the annual precipitation is 188 mm and the annual mean temperature is −21 ∘C. The dominating wind direction is southwesterly with an average wind speed of 4 m s−1, as apparent from Fig. S1 (Rasch et al., 2016; Nguyen et al., 2013). The SP-AMS data were sampled in an atmospheric observatory containing two laboratories, whereas data from a multi-angle absorption photometer (MAAP) and a filter pack sampler were collected in a smaller co-located hut (Flyger's Hut) – both equipped with particle and gas inlets. The two measurement sites are located 2.5 km southeast of the military station and are only 300 m apart. Given the close proximity of the two laboratories and the lack of hyper-local sources, we expect both to sample largely the same air mass. A high-volume sampler (HVS) provided filter samples for off-line analysis. The HVS was located on the outskirts of the military station, 2.5 km from the main sampling site. More information concerning the supplementary instruments can be found in Supplement. All particulate measurements at the atmospheric observatory were conducted by drawing air through a slightly heated (absolute 5 ∘C) particle inlet custom-built by TROPOS (Leipzig, Germany). Sampling took place during a CRAICC (Cryosphere-Atmosphere Interactions in a Changing Arctic Climate) field campaign from 20 February to 23 May 2015.
2.2 The soot particle aerosol mass spectrometer
An SP-AMS (Aerodyne Research Inc.) was deployed at VRS for measuring mass concentration and chemical composition of sub-micrometer aerosols with a time resolution of 2 min. The SP-AMS is described in detail elsewhere (Onasch et al., 2012). In brief, the instrument samples aerosols into a vacuum chamber through an aerodynamic particle lens, which creates a narrow particle beam. In the vacuum chamber, the aerosols accelerate to a velocity depending on their vacuum aerodynamic diameter, enabling analysis of the aerosol size distribution. Subsequently, the aerosols undergo vaporization, ionization with 70 eV electron impact and detection with time-of-flight mass spectrometry. The vaporization of aerosols components in the SP-AMS can occur in two ways: (1) impaction on a tungsten surface at a temperature of 600 ∘C or (2) intersection with the beam of a continuous-wave 1064 nm intracavity Nd:YAG laser. The laser extends the application of the AMS to include refractory particulate matter (R-PM) since it enables vaporization of strongly infrared light absorbing particles, such as refractory BC (Onasch et al., 2012). In this study, high-resolution (HR) mass concentrations of , , , organics, Cl− and rBC are obtained from the SP-AMS.
The SP-AMS was operated for 2 min in laser-off mode and 2 min in laser-on mode in V mode and alternated between the mass spectrum mode and the particle time-of-flight (pToF) to obtain sub-micrometer particles (PM1). Non-refractory species are reported for time periods where the laser was off. The flow rate was controlled regularly with a Gilian Gilibrator (Sensidyne). During the first part of the campaign, ionization efficiency (IE) calibrations with ammonium nitrate particles were conducted on a weekly basis and during the last part every second week. To establish the detection limit and to enable adjustments of the fragmentation tables a high-efficiency particulate air (HEPA) filter was applied on a daily basis for a period of 30 to 60 min with a time resolution of 2 min. The lower detection limit of the different species was determined as 3 times the standard deviation of the mass concentration during the HEPA filter periods (Table 1). The data were analyzed with the standard AMS Igor Pro-based (version 6.35 Wavemetrics, Inc) software tools SQUIRREL (version 1.57G) and PIKA (version 1.16H), available at http://cires1.colorado.edu/jimenez-group/ToFAMSResources/ToFSoftware/index.html. The analysis followed the principles described in DeCarlo et al. (2006), Jimenez et al. (2003); Allan et al. (2004) and Onasch et al. (2012).
Table 1Detection limits. The detection limits for the SP-AMS are calculated from periods sampling through HEPA filters with a time resolution of 2 min (average from eight HEPA filter periods of 30 to 60 min over the entire campaign). The detection limit for the MAAP is from Massling et al. (2015).

The default relative ionization efficiency (RIE) values for OA, , and Cl− of 1.4, 1.2, 1.1 and 1.3, respectively, were applied, which are based on Canagaratna et al. (2007). A RIE of 3.5 was applied for . It should be noted that chloride reported in the current study is measured with laser off and is thus non-refractory chloride and largely excludes refractory species such as chloride in sea salt aerosols. Thus, reported Cl− in this study is most likely primarily a sum of organic Cl− and NH4Cl due to the acidic environment at VRS. However, the partitioning of chloride between different species has not been investigated further, since it is not within the scope of this study. A RIE for rBC of 0.46 was found from calibrations with Regal Black (a commercial carbon black). The appropriateness of this RIE for ambient Arctic rBC is discussed in Sect. 2.4. Calibrations with Regal Black and ammonium nitrate were done with the same frequency. Fragment ions from organic species can overlap with some of the marker ions for rBC. To minimize the organic contribution to the nominal rBC signal (at an organic contribution was especially evident), was used to quantify rBC. Thus, the signal was scaled with a factor of 1∕0.55 to match the fraction in the Regal Black mass spectra (Martinsson et al., 2015). The applied collection efficiency (CE) for non-refractory PM and rBC will be discussed in more detail in a subsequent section.
2.3 Auxiliary equipment
The aerosol light absorption was measured using a MAAP (Model 5012 Thermo Scientific) operated at a flow rate of 1 m3 h−1 with an inlet without a size cutoff. Aerosols were sampled on a filter in which the light absorption at 670 nm was measured by a photometer. Detailed information about the instrument can be found in Petzold and Schonlinner (2004) and previous MAAP measurements from VRS are published in Massling et al. (2015). The BC concentration is determined from the relationship between the aerosol light absorption coefficient and a specific aerosol absorption coefficient (Petzold and Schonlinner, 2004). The specific absorption coefficient describes BCs ability to absorb solar radiation at a specific wavelength, which depends on the age of the aerosol (Petzold et al., 1997; Sharma et al., 2002) and is often determined based on correlations with thermal–optical measurements of elemental carbon (EC) (Sharma et al., 2004). In this study, the MAAP's default value of 6.6 m2 g−1 has been applied based on Massling et al. (2015). Uncertainty in the conversion factor likely impacts the reported absolute concentrations and potentially the temporal variability. In addition, a scanning mobility particle sizer (SMPS) measured the particle number size distribution, which was used for validating the SP-AMS results. The SMPS is custom-built with a Vienna-type medium column and more information can be found in Lange et al. (2018). A description of the validation can be found in Supplement.
2.4 Comparison between instruments
A collection efficiency (CE) adjustment is normally applied to AMS data, which accounts for particle loss in the instrument caused by the inlet and the aerodynamic lens, beam divergence, and particle bounce effects (Canagaratna et al., 2007; Onasch et al., 2012). In this study, the parameterization developed by Middlebrook et al. (2012) has been used where a time-dependent CE is determined based on the aerosols chemical composition. Previous studies have shown an increasing CE with particle acidity, the content of nitrate and relative humidity (Quinn et al., 2006; Jayne et al., 2000; Matthew et al., 2008). The time-dependent CE varied with the majority (> 97 %) of values between 0.8 and 1 (Fig. S2). In this study, the high CE was due to acidic aerosols. This is also evident from Fig. S3a, showing that the theoretical predicted concentration necessary for neutralizing the mass concentration of inorganic anions is much larger than the actual concentration measured by the SP-AMS (slope =0.14). The acidity is explained by the high amount of sulfuric acid.
Applying the RIE for rBC of 0.46 determined from Regal Black calibrations, a good correlation between rBC and BCMAAP is found (Fig. S3b). While there is a strong linear relationship between the two (R2=0.83), the BCMAAP was about 3 times larger than the SP-AMS rBC (slope ). This indicates that the actual RIE for rBC was lower than the value of 0.46 determined during laboratory calibrations. A lower RIE can be explained by different particle size and a more complex morphology of the Arctic soot compared to the Regal Black used for calibration. An effective RIE is determined for rBC by forcing the SP-AMS measurements to match the MAAP measurements. For rBC an effective RIE of 0.15 () is hence applied in this study.
Comparison of the total PM1 mass concentration (sum of OA, , , , Cl− and rBC) with the calculated total volume from the SMPS assuming spherical particles was carried out to validate the SP-AMS results. The SMPS was operated to characterize particles having mobility diameters between 9 and 870 nm. This corresponds to a larger size range than sampled by the SP-AMS, which has 100 % transmission efficiency within aerodynamic diameters between 70 and 600 nm, and adjustment from aerodynamic diameter to mobility diameter further brings the SP-AMS into the SMPS range (DeCarlo et al., 2006; Allan et al., 2003). However, previous studies (Nguyen et al., 2016; Lange et al., 2018) have shown that the dominant particle size range at VRS during winter and spring months is within detection range of the SP-AMS. Thus, the number of particles from the SMPS exceeding the size range measured by the SP-AMS should be relatively small and thereby not influence the results, since particles in the lower end of the size distribution do not significantly contribute to volume. There was a generally reasonable temporal correspondence between the two measurements. Although there were some periods where they differed notably they were within the expected range given the accuracy of the two instruments. A more detailed discussion about the comparison between the two instruments is presented in Supplement (Fig. S5).
2.5 Positive matrix factorization
PMF analysis (Paatero, 1997; Paatero and Tapper, 1994; Lanz et al., 2007; Ulbrich et al., 2009) was conducted on the time-dependent organic mass spectra to determine OA factors and potential sources of OA. The analysis was carried out with the PMF Evaluation Tool Software (PET, v2.08D; available online at http://cires1.colorado.edu/jimenez-group/wiki/index.php/PMF-AMS_Analysis_Guide, last access: 8 August 2019) on mass spectra consisting of HR ions with m∕z values from 12 to 100. The detailed procedure is described elsewhere (Ulbrich et al., 2009; Zhang et al., 2011). The input HR mass spectra and error matrix with the appropriate ion fragments were generated in PIKA, where the error matrix was calculated as the sum of the quadrature of the electronic noise and Poisson counting for each ion (Allan et al., 2003). Isotopes were removed from both the data and error matrix since they would give additional weight to the parent ion in the PMF analysis.
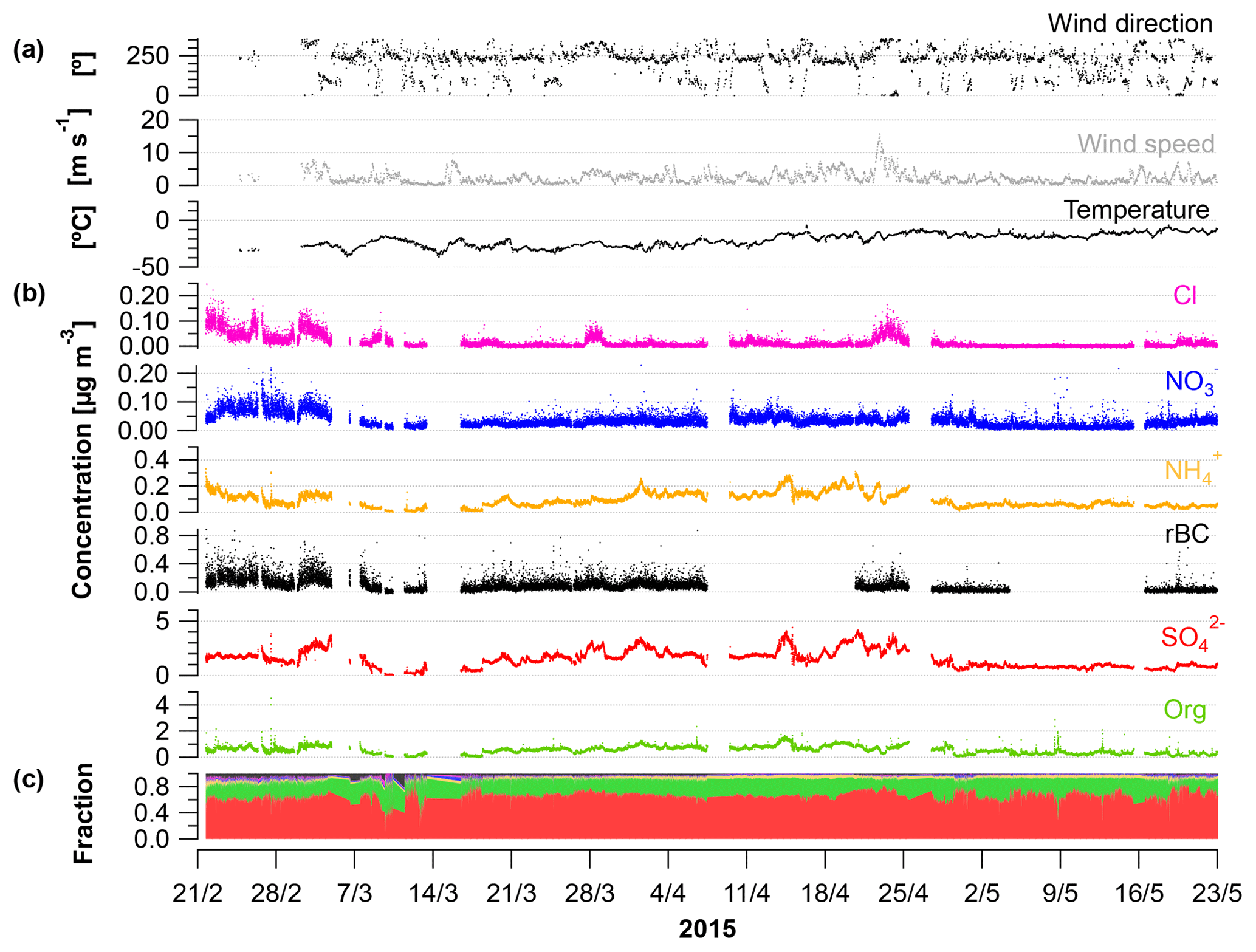
Figure 1Time series from 21 February to 23 May 2015 showing (a) wind direction (∘); mean wind speed (m s−1) and temperature (∘C); (b) concentrations of Cl, , , rBC, , and OA from the SP-AMS (µg m−3); and (c) fraction of the aerosol species to the total PM1.
As described in Ulbrich et al. (2009) “weak” ions with a signal-to-noise ratio (SNR) between 0.2 and 2 were down-weighted by a factor of 2, whereas “bad” ions with a SNR below 0.2 were removed from the data and error matrix. The PMF was executed in exploration mode with a range of factors (between 1 and 5). The robustness of the solutions was tested by setting different random starting points (SEED value of 0 to 10; steps =1) (Zhang et al., 2011). The detailed procedures for choosing the best solution were based on Zhang et al. (2011). A solution with three factors (Fig. 2) was identified after evaluating Q∕Qexp and residuals, interpreting the mass spectra and investigating the temporal correlation between the factor time series and potential tracer species (Ulbrich et al., 2009; Zhang et al., 2011). FPEAK and seed values were changed to test the stability of the three-factor solution and, based on the diagnostic plots, a three-factor solution was selected with a FPEAK and seed value of zero (Fig. S7). A four-factor solution was scientifically not meaningful with respect to the chemical composition and returned an O∕C ratio ≫1 for one of the factors. Hence, we do not observe a fourth “continental” factor, which has been previously observed during the ASCOS cruise track in the summer and fall seasons around Svalbard (Chang et al., 2011). If present, the continental factor is most likely of negligible abundance for which reason the PMF analysis cannot differentiate it from other oxygenated organic aerosol (OOA). Detailed information regarding the factor combination can be found in Supplement.
3.1 Time series
Time-dependent OA, , , , Cl− and rBC concentrations [µg m−3] measured by the SP-AMS are presented in Fig. 1 together with temperature [∘C], mean wind speed [m s−1] and wind direction [∘] for the time period 21 February to 23 May 2015. Weekly average concentrations can be found in Fig. S6. Figure 1c shows the time-dependent mass fraction of the different species. The total measured PM1 concentration during the field study may seem relatively high, averaging 2.3 µg m−3 – ranging from 2.3, 2.3 and 3.3 µg m−3 in February, March and April, respectively, to 1.2 µg m−3 in May. It should be emphasized that this average does not consider particulate water, NaCl and elements, such as K, Ca, Si, Al and Fe. These elements may additionally contribute 0.1–0.2 µg m−3 to PM1 (Nguyen et al., 2013; Heidam et al., 2004). The measurement period covers the Arctic late winter and spring where high aerosol loadings are expected due to the favorable conditions for long-range transport of aerosols from midlatitudes and slow particle removal rates. With regard to PM1 concentration, we hence observe the typical Arctic haze phenomenon. Generally, the area around VRS is dominated by winds from the southwest (Nguyen et al., 2013), which is also evident during this campaign (Fig. S1). As expected, no diurnal pattern is observed for any of the chemical species. These are mainly transported from long distances. For example, the source regions that contributed to ground-level SOx at VRS were assigned to Western Europe (7 %), Eastern Europe (9 %), Asia (2 %), North America (7 %) and Russia being the main emitter by far (75 %) (Heidam et al., 2004). During summer, the atmospheric circulation is confined within the Arctic region and is considered essentially local. Thus, marine biogenic sources that peak during spring and summer are expected to originate from within the region. Arctic sites show similar increases in key particulate pollutants in winter and early spring, where maximum sulfate concentrations may reach 3 µg m−3 as compared to average summer concentrations of 0.1 µg m−3 (Quinn et al., 2007). For example, typical PM1 concentrations were 0.1–0.2 µg m−3 in August to September during the ASCOS expedition (Chang et al., 2011). Sulfate is dominated by anthropogenic sources, accounting for 65 % at Alert (Norman et al., 1999) and 75 % Svalbard (Udisti et al., 2016) as annual averages. On the contrary, biogenic sources accounted for 63 % of sulfate in size fractions smaller than 490 nm at Alert during summer (Ghahremaninezhad et al., 2016).
During the entire campaign, is the dominant species that on average makes up almost 70 % of the PM1 mass concentration, with the highest concentration until the end of April and decreasing in May (Fig. 1b–c). This is in accordance with previous findings for at VRS based on measurements with lower time resolution (Nguyen et al., 2013; Fenger et al., 2013; Heidam et al., 2004). Atmospheric is mainly formed as secondary inorganic and only a minor fraction is from primary emissions (Massling et al., 2015). Secondary is formed by atmospheric oxidation of sulfur dioxide (SO2) and to some extent DMS (as the long-range transport occurs over sea ice) and is dependent on the oxidative capacity of the atmosphere, e.g., the concentration of hydroxyl radicals (OH). Secondary long-range transported depends on atmospheric oxidation of SO2 at the vicinity of the source regions, whereas local transformation (close to VRS) of SO2 leads to higher concentration of from March, where solar radiation is sufficient, with peak radiation exceeding 100 W m−2 (Fig. 3). This is consistent with results reported from other Arctic sites (Quinn et al., 2007; Gong et al., 2010; Heidam et al., 2004; Skov et al., 2017). Previous studies suggest that the main source of SO2 and at VRS is long-range transport of anthropogenic emissions mainly originating from Siberia (Heidam et al., 2004; Nguyen et al., 2013). In winter and early spring, direct emissions of sea salt sulfate and photooxidation of oceanic emissions of DMS were expected to play a minor role, since the ocean surrounding VRS is frozen at that time of year (Heidam et al., 2004). However, a recent study using both airplane measurements and modeling suggests that long-range transport of DMS is significant during spring (Ghahremaninezhad et al., 2017). From the beginning of April, the sea ice extent of the Northern Hemisphere is markedly reduced and, at the same time, solar radiation increases (Fig. 3). In this period, we observe MSA as an ion in the SP-AMS at m∕z 78.9854. MSA is formed by atmospheric oxidation of DMS, which results from bacterial breakdown of dimethylsulfoniopropionate produced by marine phytoplankton and microalgae (Carpenter et al., 2012). In this study, MSA emerges steadily and peaks the end of April (see Sect. 3.2). Oxidation of DMS may involve the hydroxyl radical, ozone, and halogen radicals such as Cl− and BrO (Barnes et al., 2006; Hoffmann et al., 2016).
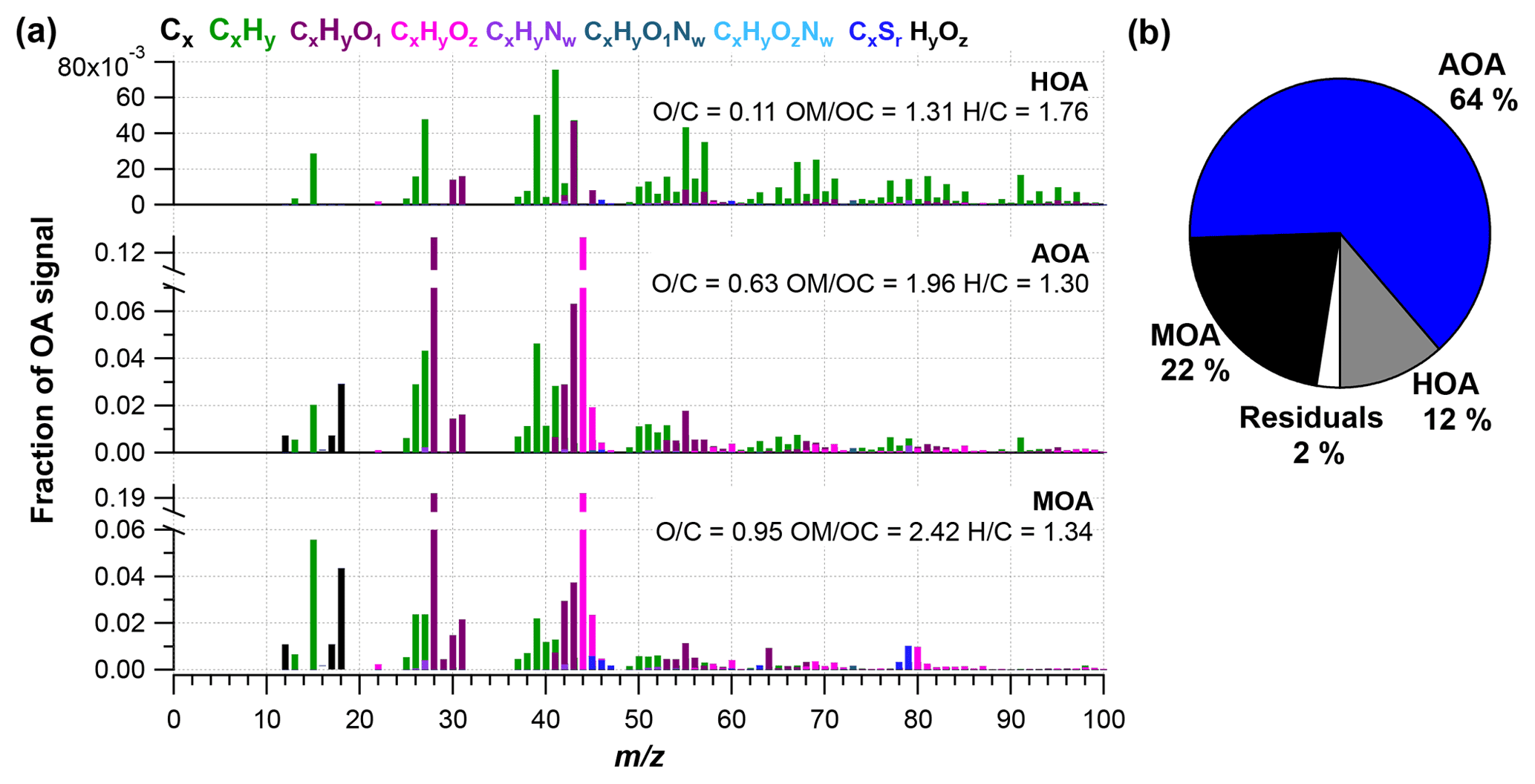
Figure 2(a) High-resolution mass spectra of PMF factors, including hydrocarbon-like organic aerosol (HOA), Arctic haze organic aerosol (AOA) and marine organic aerosol (MOA), and (b) the factor share of ambient mass concentration. O∕C, OM∕OC and H∕C ratios are presented for each factor.
In this study, the OA fraction is the second largest contributor to PM1 where weekly averages showed a clear decrease from mid-April relative to concentrations in February and March (Fig. 1). The OA time-dependent concentration shows relatively large peaks during shorter time periods, which in some cases can be attributed to a change in wind direction from southwesterly to northerly winds (around 10∘, Fig. S1). While these wind directions were registered on a few occasions, they potentially provided local pollution from the military station located three kilometers away from the measurement site. These peaks have not been discarded and the impacts of local pollution will be discussed further in Sect. 3.2.
Particulate is found in much lower concentrations compared to OA and but with the same transition pattern as the two other species. For the campaign, a significant correlation is found between and . However, it is known that and do not originate from the same sources. SO2, a key precursor to , originates from combustion of fossil fuel and is oxidized to in the atmosphere. In contrast, ammonia (NH3), which is the precursor of , derives largely in winter and spring from long-range transport of emissions from biomass burning and agriculture (Fisher et al., 2011), whereas in summertime NH3 emissions from seabird colonies can play a significant role (Croft et al., 2016). The strong correlation between and (R2=0.70) suggests that the acidity of the particles is reasonably constant over time. This is furthermore in agreement with the general assumption that is bound irreversibly to (e.g., Seinfeld and Pandis, 2006), in this case as ammonium bisulfate. Particle-bound has a much longer lifetime than NH3 (Baek and Aneja, 2004) and is therefore transported as even to the High Arctic.
The average concentrations of and Cl− are 0.03 and 0.02 µg m−3, respectively, which is close to the detection limits. These concentration levels are lower compared to what has previously been observed at VRS (Fenger et al., 2013; Heidam et al., 2004). However, the SP-AMS does not typically measure refractory chloride at normal vaporizer temperatures, such as NaCl (Canagaratna et al., 2007). However, Ovadnevaite et al. (2012) has demonstrated how the AMS could be calibrated to measure NaCl in high time resolution. Moreover, Fenger et al. (2013) found that the overall size distribution of chloride and differed from , with Cl− and mainly found in supermicrometer particles (> 1 µm) not detectable by SP-AMS. Based on the size of the particles and air mass back-trajectories, Fenger et al. (2013) suggested that the particles originate from local and regional sources (frost flowers and refreezing leads). Only during certain periods with specific wind directions were and Cl− found in accumulation mode particles, which were ascribed to long-range transported particles (Fenger et al., 2013). Current research has suggested that blowing snow might be a much more dominant source of sea salt aerosols compared to frost flowers (Huang and Jaeglé, 2017).
The highest rBC loadings are found in the first month of the campaign (February), averaging 0.2 µg m−3. In March and April, the average is 0.1 µg m−3 which then decreases to 0.02 µg m−3 in May. As with OA, some of the spikes in the rBC time series are related to a change in wind direction and likely the result of local pollution from the military station. All data are included here and missing time periods of rBC (during April and May) are due to technical problems with the SP-AMS laser. BC is primarily emitted from both anthropogenic and natural combustion sources (Bond et al., 2013). Upon emission, aerosols containing BC grow by condensation and coagulation into the accumulation mode. These accumulation mode BC-containing particles can be transported over longer distances during the Arctic haze period and may serve as cloud seeds in the late spring, when precipitation begins to be important in the Arctic (Bond et al., 2013; AMAP, 2011; Massling et al., 2015; Garrett et al., 2011). Further, condensational growth of the BC-containing particles may increase the absorption by these particles (Cappa et al., 2012; Liu et al., 2015). Previous studies have found a correlation between BC and at different Arctic stations (Massling et al., 2015; Eckhardt et al., 2015; Hirdman et al., 2010). These studies suggest that the two species are internally mixed and possibly undergo similar transport patterns. Furthermore, comparable correlation slopes were found for the different Arctic locations, which suggest that source regions of BC and could be similar throughout the Arctic. An even more recent study suggests that only a minor part of ambient aerosols contained rBC inclusions (Kodros et al., 2018). We find a significant correlation between the two species (students t test, level of significance 99.995), consistent with previous studies. However, we also find that the R2 value is relatively low (0.18). The reason for this is that there are periods with particularly high rBC concentrations, likely originating from local emission sources (e.g., the military base), which will be investigated further in the following section. Additionally, in April and May from DMS oxidation will make up a larger fraction of total and thereby reduce the ratio between rBC and , which is also evident from Fig. S4.
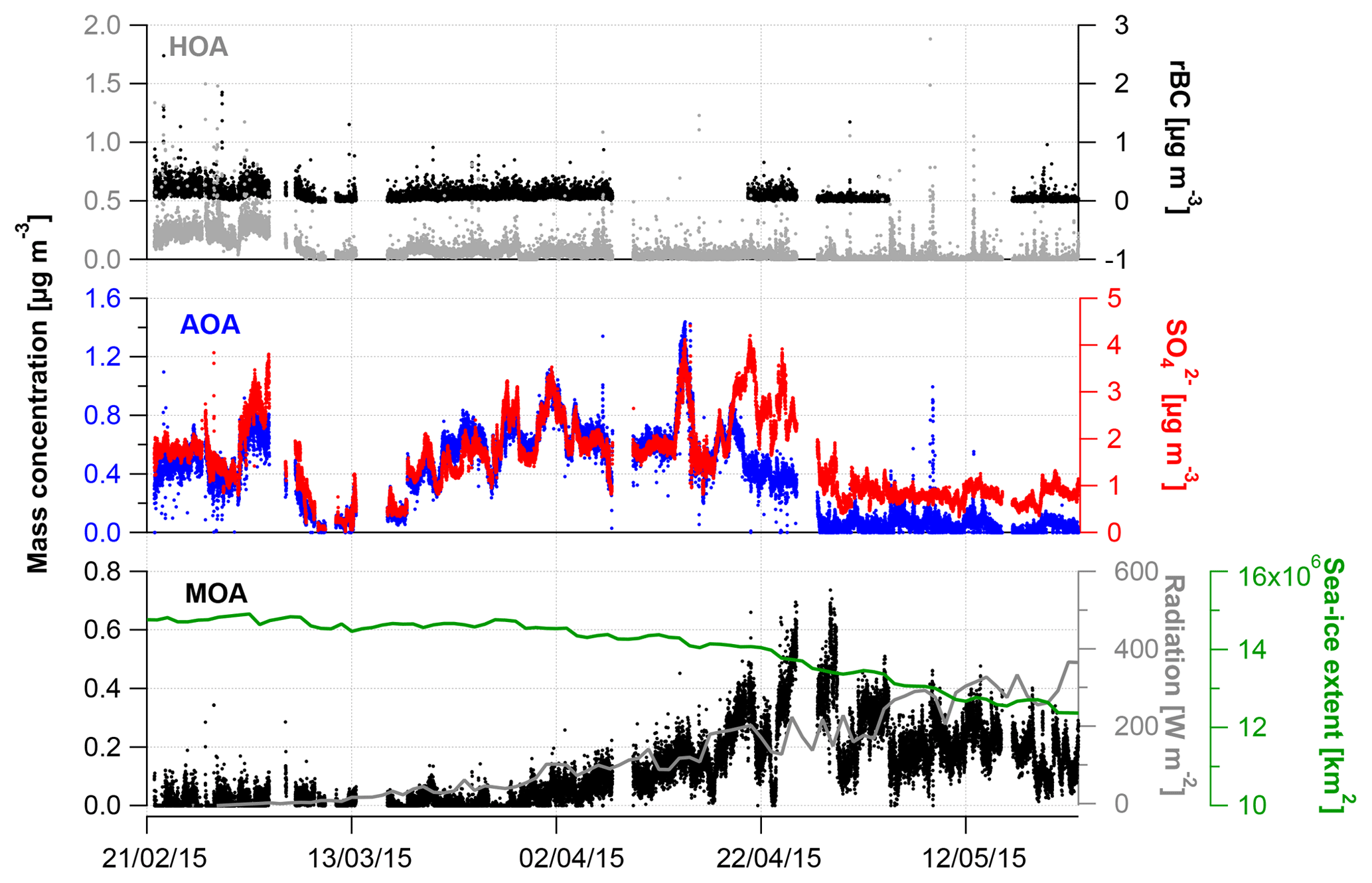
Figure 3Time series for hydrocarbon-like organic aerosol (HOA), Arctic haze organic aerosol (AOA), marine organic aerosol (MOA) and tracers (rBC, ). Sea ice extension in the Northern Hemisphere and shortwave radiation (daily average) are included in the time series for MOA (see text).
3.2 Source apportionment
The PMF analysis was conducted for the HR OA mass spectra with one to five PMF factors and a three-factor solution was chosen (more details can be found in Supplement). Figure 2 shows the mass spectral profiles of the three different factors for the entire campaign period. Figure 3 illustrates time series for the factors and Table 2 shows the correlation of each factor with tracer species. Figure 4 illustrates the average mass concentration (µg m−3) and the mass fraction of the factors in February, March, April and May. The PMF analysis yielded three factors: (1) a hydrocarbon-like organic aerosol factor (HOA), (2) an oxygenated Arctic haze organic aerosol factor (AOA) dominating winter and early spring, and (3) a more oxygenated marine organic aerosol factor (MOA), which builds up in late spring and becomes the dominating OA throughout late spring. The identification of these factors is discussed below.
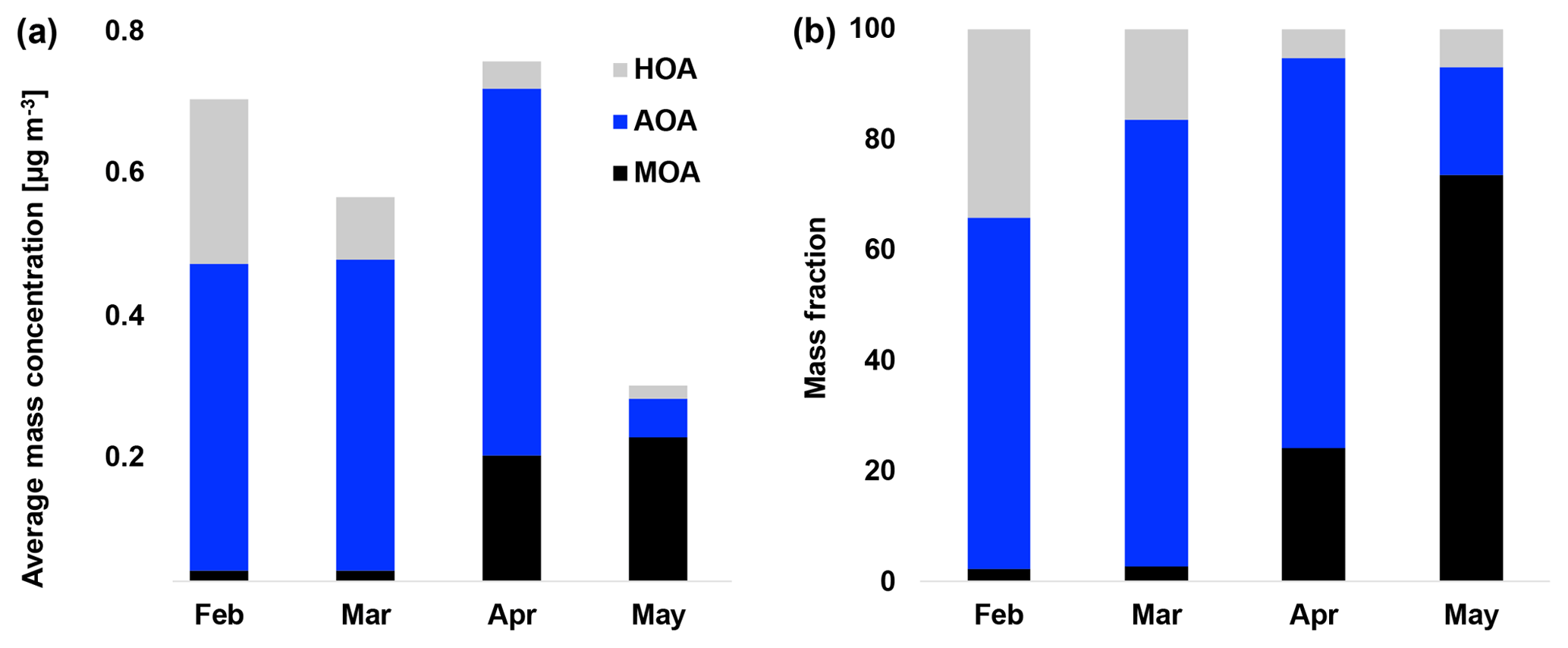
Figure 4(a) Average mass concentration (µg m−3) of hydrocarbon-like organic aerosol (HOA), Arctic haze organic aerosol (AOA) and marine organic aerosol (MOA) in February, March, April and May. (b) Mass fraction of HOA, AOA and MOA in February, March, April and May.
Table 2R2 correlations between PMF factors and tracers (rBC, MSA, and ).
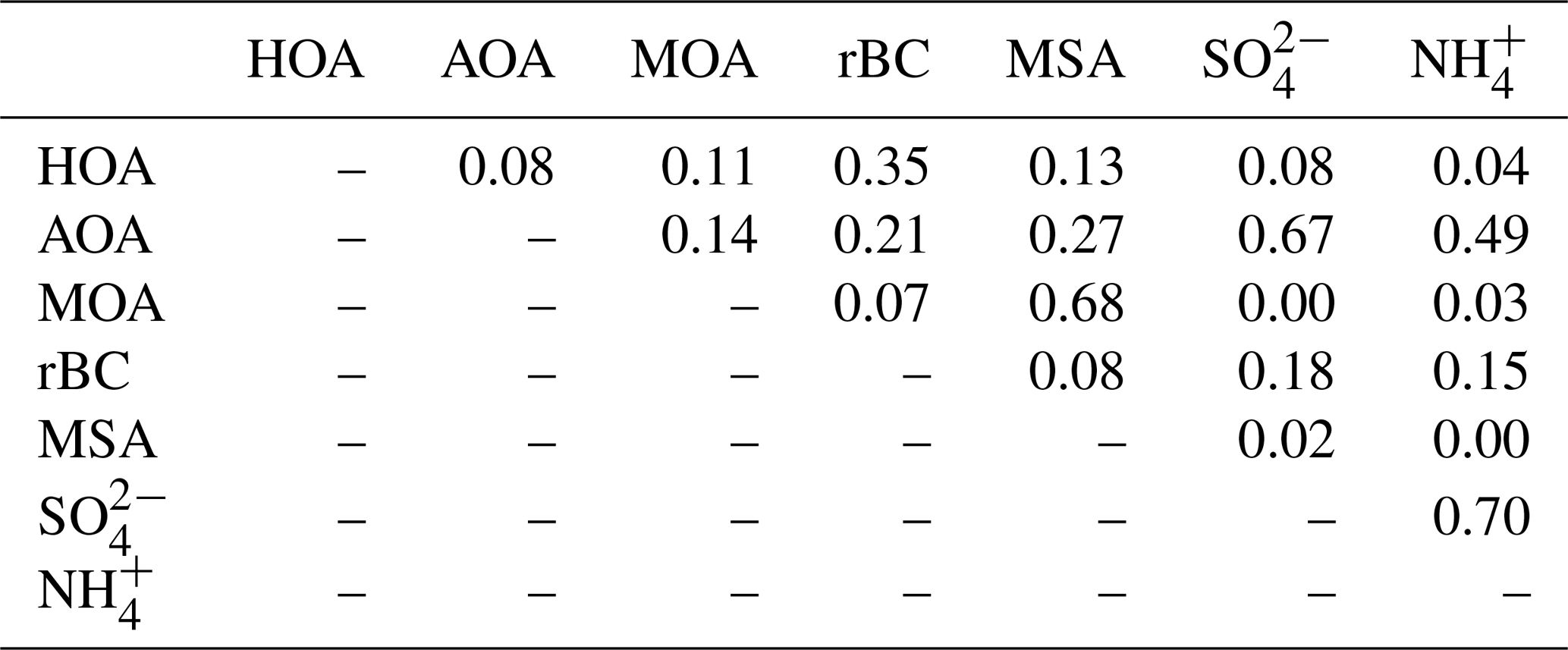
The HOA factor is characterized by hydrocarbon fragments, especially at m∕z 41, 43, 55, 57, 67, 69 and 71 (, , , , , and , respectively) from chemically reduced organic emissions. The O∕C ratio of 0.11, high signal at m∕z 57 and the absence of is a characteristic of primary combustion sources of fossil origin, which is similar to other HOA factors found in previous studies (Zhang et al., 2005; Aiken et al., 2009) and at other Arctic locations (Frossard et al., 2011). The very small contribution from the at and the very small abundances of typical biomass burning OA (BBOA) marker ions at m∕z 60 () and m∕z 73 () in the HOA factor spectrum suggests that the HOA factor is not mixed with BBOA. This finding is consistent with previous results that indicate BBOA levels are typically very low, based on measurements of levoglucosan in the Arctic, (Zangrando et al., 2013). The time series of HOA and rBC showed a moderate correlation (R2=0.35), which is consistent with the HOA factor being of primary origin. The relatively low R2 value (Table 2) can be partly explained by rBC being internally mixed with and transported with the AOA factor. The HOA time series is generally higher in concentration at the beginning of the measurement period (Fig. 4). The time series of HOA reveals a number of shorter periods with high mass loading, which could be caused by local pollution from the military station 2 km north of the measurement site, due to a change in wind direction or exhaust plumes from snow scooters and heavy-duty vehicles occasionally clearing the road nearby the measurement station for snow (see the wind rose in Fig. S1). It is not trivial to distinguish local events and, in this case, the possible local contamination was investigated by comparing high HOA peaks (> 0.45 µg m−3) with size distribution measurements from the SMPS (Lange et al., 2018). Periods which were attributed to local contamination accounted for less than 1 % of OA concentration. Therefore, essentially the entire HOA concentration is assigned to long-range transportation, possibly sources with different ratios of HOA and rBC, which would explain the moderate correlation between HOA and rBC.
Oxygenated aerosols from numerous field campaigns in the Northern Hemisphere are deconvolved into HOA and OOA. OOA has been shown to account for a large fraction of OA and to be a good surrogate for secondary organic aerosols (SOA) in multiple studies (Ng et al., 2010; Zhang et al., 2007, 2011). Oxygen-containing functional groups produce m∕z 43 (C2H3O+) and m∕z 44 () fragments, which are prominent peaks in OOA mass spectra (Ng et al., 2010), including those of MOA and AOA found in this study. These factors are highly OOA factors with O∕C ratios of 0.63 and 0.95, respectively. According to Jimenez et al. (2009) these factors would be classified as low-volatility OOA (LV-OOA). There is strong evidence that OOA is secondary in nature and several studies of aging indicate that OA converges towards LV-OOA following numerous steps of atmospheric oxidation (Jimenez et al., 2009). The AOA is the most abundant factor from the beginning of the campaign through mid-April. AOA accounts for 64 % of OA mass for the entire field study but ranges from 64 %, 81 % and 71 % of OA in February, March and April to 20 % in May (Figs. 2b and 4). The dominating OA appears to originate from long-range transport into the region during winter and spring. At the end of April and onwards the factor was nearly absent, which is in agreement with increasing wet deposition in the spring and a contracting polar dome impairing long-range transport into northern Greenland (Abbatt et al., 2019). Generally, an OOA factor mainly consists of SOA but can also include oxygenated organic species from primary emissions (Zhang et al., 2005). In this case the AOA factor correlates significantly (level 99.995) with , which is mainly formed by atmospheric oxidation of SO2 suggesting the main part of the factor being SOA. The correlation is especially good until mid-April after which begins to correlate with MOA. The O∕C ratio of 0.63 also indicates a less oxidized and fresher SOA factor or an SOA formed from generally larger precursor volatile organic compounds (VOCs), similar to what has been found in previous studies (O∕C between 0.52 and 0.64; Aiken et al., 2008). The AOA mass spectrum also included mass spectral peaks at m∕z 60.021 () and 73.029 (). These fragments are often taken as being indicative of anhydrous sugar such as levoglucosan and thereby suggest that biomass burning makes some contribution to Arctic OA. However, SOA also contributed to the abundance of (Aiken et al., 2008, 2009; Cubison et al., 2011; Lee et al., 2010; Saarnio et al., 2013). Quantitatively, the expected abundance of from SOA did not exceed the measured concentration in this study. Biomass burning is generally assumed to play a significant role in the context of the composition of the Arctic aerosol (Stohl et al., 2013), where recent publications using isotopes of carbon reports that biomass burning or biofuel used to account for up to 57 % of EC at the Arctic station Zeppelin at Svalbard during high-pollution events in winter (Winiger et al., 2015). However, levoglucosan is prone to atmospheric oxidation by hydroxide radicals (OH) (Hennigan et al., 2010; Hoffmann et al., 2010), which could degrade the markers during transport to northern Greenland. This can explain the low abundance of levoglucosan markers measured in this study.
The MOA factor has a mass spectrum dominated by m∕z 28 and 44 (CO+ and ), of which the latter is probably a fragment from, e.g., organic acids and acid-derived species, such as esters (Duplissy et al., 2011). An O∕C of 0.95 reveals that the factor is highly oxidized and most likely photochemically aged. The MOA spectrum resembles a marine organic plume previously published from Mace Head, in the northeastern Atlantic Ocean, showing evidence of both primary and secondary organic aerosols of marine origin (Ovadnevaite et al., 2011). Most abundant peaks in this spectrum were oxygenated fragments at m∕z 28 and 44. Also prominent were m∕z 27, 39 and 41 from the CH family and m∕z 43 and 55 from the CHO family, which are also found in the MOA spectrum. The two spectra differ in terms of abundances of CH-like organic matter, but they are different from the marine organic aerosol factor published during the ASCOS expedition in the Central Arctic Ocean (Chang et al., 2011), which shows a closer resemblance with the mass spectrum of pure MSA, i.e., dominating peaks at m∕z 15, 48, 64 and 79. The distinct peak at m∕z 78.9854 is specific for MSA (Huang et al., 2017) and reveals that MOA has a secondary biogenic source (Becagli et al., 2013). The resemblance of MOA from this study with the mass spectrum from Mace Head and the high O∕C ratio of 0.95 indicate that MOA is composed of chemically aged aerosols from both oxidation of primary aerosols and secondary organic aerosols (Ovadnevaite et al., 2011; Fu et al., 2015). Aerosol growth has been correlated with the presence of MSA and other organic species (Willis et al., 2016).
Figures 3 and 4 illustrate HOA and AOA decreasing around mid-April, while MOA builds up from the end of March. In 2015, Arctic sunrise onset was on 28 February at VRS, where the sun became visible for a few minutes. Polar daytime initiates photochemistry and hence the production of OH radicals (Seinfeld and Pandis, 2006) and reactive halogen radicals (Hoffmann et al., 2016; Barnes et al., 2006). From mid-April, the sun is above the horizon all day until the beginning of September. Still, solar radiation varies over the day and hence the OH production. In contrast, the concentration of OH during buildup of Arctic haze is correspondingly low with ozone being the major oxidant during the dark winter. In Fig. 3, the daily averaged solar radiation (W m−2) and sea ice extent (km2) in the Northern Hemisphere are shown together with the time series of MOA. While MOA is less abundant during February and March, this factor greatly increases in April, when radiation exceeds approximately 100 W m−2. In April, the highest OA concentrations is observed where AOA accounts for around 70 % of OA (Fig. 4). In May, MOA becomes the dominating OA while AOA nearly disappears. At the same time, we observe the lowest concentration of OA (0.01 µg m−3) consisting of 75 % MOA (Fig. 4). This is significantly higher than observed at Alert by Narukawa et al. (2008), where marine organic matter contributed 45 % to aerosol total carbon in late spring (26 April–6 May 2000). However, direct comparison is difficult due to different methods and time periods (Narukawa et al., 2008). Until the beginning of April, the sea ice extent is constant at around 14.5 million square kilometers in the Northern Hemisphere (Fig. 3). Hereafter, about a month after the onset of polar daytime, the sea ice surface area starts to decline. After 6 weeks starting from a constant sea ice extent in mid-May, this is reduced by 2 million square kilometers corresponding to a 14 % loss of ice-covered surface area. Consequently, more open waters allow for higher DMS emissions (Abbatt et al., 2019) and atmospheric oxidation of DMS to MSA involving OH. This can be visualized from the strong coupling between DMS concentration and chlorophyll a from DMS-producing phytoplankton (Park et al., 2013). Moreover, Becagli et al. (2016) concluded that oceanic primary production was related to melting of sea ice and extension of marginal sea ice areas based on satellite-derived chlorophyll a and measurements of MSA (Becagli et al., 2016). Also, open leads and marginal ice zones provide primary marine aerosols (Willis et al., 2018). Indeed, previous findings suggest that biogenic productivity in open oceans and sea ice zones and the emission of DMS are responsible for increased new particle formation, as sea ice pack extent retreats (Dall'Osto et al., 2017). Quinn and co-workers reported increased concentrations of MSA at Utqiaġvik from 2000 to 2009 associated with the northward migration of the marginal ice zone (Quinn et al., 2009; Sharma et al., 2012; Laing et al., 2013). Of the four northernmost year-round manned observatories at Alert, Mount Zeppelin, VRS and Utqiaġvik, the highest MSA concentrations are measured at Mount Zeppelin, likely due to its proximity to open waters around Svalbard, which are a significant source of DMS from May to August (e.g., Lana et al., 2011). This contrasts with the situation around VRS, which is covered with ice for most of the year.
Considering the stronger oxidizing environment starting in April, we expect MOA to be abundant until fall (Chang et al., 2011). MOA constitutes 22 % of OA on average during our measurement period, ranging from 2 % to 3 % of OA in February and March to 24 % and 74 % of OA in April and May, respectively (Figs. 2b and 4). Thus, MOA is by far the most abundant OA from the end of April and onwards. MOA dominates the OA mass after polar sunrise and persists during polar daytime so the aerosol's optical impact might be substantial. At the same time, MOA dominates when the overall PM1 concentration is very low and particle numbers are low, and hence CCN concentrations can be low. The observed transition between AOA and MOA is in agreement with Narukawa et al. (2008), who observed a transition between fossil-fuel-influenced OA to marine OA. MOA may contain oxidation products of DMS and other VOCs of oceanic origin, as well as a variety of primary components including saccharides such as mannitol in addition to insoluble gels (Croft et al., 2019; Leck and Bigg, 2005; Orellana et al., 2011; Fu et al., 2013; Ovadnevaite et al., 2011). In line with our findings, modeling at several sites in the Canadian Arctic suggested that marine OA other than MSA may account for more than half of the summertime OA (Croft et al., 2019). These findings encourage further studies of optical properties and chemical composition and physicochemical parameters as CCN ability or hygroscopicity of aerosols prevailing during polar daytime.
In the transition from polar night to polar day we observed elevated PM1 concentration ranging from an average of 2.3, 2.3 and 3.3 µg m−3 in February, March and April, respectively, to 1.2 µg m−3 in May. We concluded to be the most abundant species in sub-micrometer aerosols with the highest concentration up to the end of April and decreasing in May. This is in accordance with previous findings from VRS but also at Alert (Norman et al., 1999) and Svalbard (Udisti et al., 2016), where has been apportioned to be 65 % and 75 % anthropogenic, respectively. While not previously quantified at VRS, OA was found to be the second largest contributor to PM1 (24 %). As for the other species, OA showed a decrease in concentration from mid-April relative to February and March. rBC concentrations were found to be highest in the first month and then decrease throughout the campaign with average concentrations of 0.2, 0.1, 0.1 and 0.02 µg m−3 in February, March, April and May, respectively.
Source apportionment analysis yielded three factors, identified as a hydrocarbon-like organic aerosol (HOA), arctic haze organic aerosol (AOA) and marine organic aerosol (MOA) with O∕C ratios of 0.11, 0.63 and 0.95, respectively. HOA, being the least oxidized factor, made up 12 % of OA, of which 1 % of OA was demonstrated to be contamination from the nearby military camp. AOA and MOA made up 86 % of OA averaged across the campaign, with AOA averaging 64 % and MOA 22 % (2 % residuals). AOA and MOA showed evidence of SOA. Furthermore, the resemblance of MOA to a previously published marine organic plume was indicative of MOA having a primary organic component. The sum of long-range-transported HOA and AOA make up the vast majority of OA during the Arctic haze period. AOA and MOA exhibit distinct temporal variability. The less oxidized AOA builds up during the Arctic haze period and dominates until early spring (64 %–81 % of OA), during which both the absolute and relative contribution to the OA burden decreases substantially. In contrast, MOA emerges only after early spring but is then by far the dominating OA from the end of April and onwards (24 %–74 % of OA). The fact that MOA emerges at a time where long-range transport is impaired by increased deposition and a contracting polar dome indicates that the sources of this factor are more regional in nature. This is supported by the confined atmospheric circulation within the Arctic region during summer (Heidam et al., 2004). This demonstrates the importance of biogenic sources in the Arctic, especially in the spring. In view of changing biogenic processes and corresponding source strengths of aerosol precursors in a changing Arctic climate with changing sea-ice extent, additional high-time-resolution measurements are urgently needed in order to elucidate the organic components dominating aerosol summer mass and number concentrations.
All data used in this publication are available to the community and can be accessed by request to the corresponding author, Ingeborg Elbæk Nielsen (ien@envs.au.dk), or Jacob Klenø Nøjgaard (jakn@envs.au.dk).
The supplement describes site information, supplementary instruments, collection efficiency, validation of SP-AMS data and key diagnostics for the PMF solution. The supplement related to this article is available online at: https://doi.org/10.5194/acp-19-10239-2019-supplement.
IEN and JKN carried out the field measurements, and IEN did the analysis of the SP-AMS data. JKN and IEN carried out the PMF analysis and took the lead in writing the manuscript. HS supervised the project and provided critical feedback, participated in the field campaign and helped shape the research and manuscript. HJ and NS helped monitor the SP-AMS during the field campaign and commented on the manuscript. SC, QZ and CDC helped interpret the SP-AMS data set and provided critical feedback on the manuscript. AM and RL participated in the field campaign, discussed the analysis and commented on the manuscript. ACE and MDO discussed the analysis and results and commented on the manuscript.
The authors declare that they have no conflict of interest.
The research was supported by the Arctic Centre of Research and iCLIMATE at Aarhus University, Cryosphere–Atmosphere Interaction in a Changing Arctic Climate (CRAICC) and WOOD combustion – detailed Monitoring related to Acute effects in Denmark (WOODMAD). Special thanks go to laboratory technician Bjarne Jensen and the staff at Station Nord for their great support during the field campaign. Sissel Bjørn Svendsen is gratefully acknowledged for her data control of the SMPS data, and the Villum Foundation is acknowledged for financing the new research station, Villum Research Station, at Station Nord.
This research has been financially supported by the Danish Environmental Protection Agency and Danish Energy Agency with means from the MIKA/DANCEA funds for environmental support to the Arctic Region (project nos. MST-113-0079, 2016-3025, MST-112-00298) and the Danish Council for Independent Research (project NUMEN, grant no. DFF-FTP-4005-00485B).
This paper was edited by Kostas Tsigaridis and reviewed by three anonymous referees.
Abbatt, J. P. D., Leaitch, W. R., Aliabadi, A. A., Bertram, A. K., Blanchet, J.-P., Boivin-Rioux, A., Bozem, H., Burkart, J., Chang, R. Y. W., Charette, J., Chaubey, J. P., Christensen, R. J., Cirisan, A., Collins, D. B., Croft, B., Dionne, J., Evans, G. J., Fletcher, C. G., Galí, M., Ghahremaninezhad, R., Girard, E., Gong, W., Gosselin, M., Gourdal, M., Hanna, S. J., Hayashida, H., Herber, A. B., Hesaraki, S., Hoor, P., Huang, L., Hussherr, R., Irish, V. E., Keita, S. A., Kodros, J. K., Köllner, F., Kolonjari, F., Kunkel, D., Ladino, L. A., Law, K., Levasseur, M., Libois, Q., Liggio, J., Lizotte, M., Macdonald, K. M., Mahmood, R., Martin, R. V., Mason, R. H., Miller, L. A., Moravek, A., Mortenson, E., Mungall, E. L., Murphy, J. G., Namazi, M., Norman, A.-L., O'Neill, N. T., Pierce, J. R., Russell, L. M., Schneider, J., Schulz, H., Sharma, S., Si, M., Staebler, R. M., Steiner, N. S., Thomas, J. L., von Salzen, K., Wentzell, J. J. B., Willis, M. D., Wentworth, G. R., Xu, J.-W., and Yakobi-Hancock, J. D.: Overview paper: New insights into aerosol and climate in the Arctic, Atmos. Chem. Phys., 19, 2527–2560, https://doi.org/10.5194/acp-19-2527-2019, 2019.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O∕C and OM∕OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Aiken, A. C., Salcedo, D., Cubison, M. J., Huffman, J. A., DeCarlo, P. F., Ulbrich, I. M., Docherty, K. S., Sueper, D., Kimmel, J. R., Worsnop, D. R., Trimborn, A., Northway, M., Stone, E. A., Schauer, J. J., Volkamer, R. M., Fortner, E., de Foy, B., Wang, J., Laskin, A., Shutthanandan, V., Zheng, J., Zhang, R., Gaffney, J., Marley, N. A., Paredes-Miranda, G., Arnott, W. P., Molina, L. T., Sosa, G., and Jimenez, J. L.: Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0) – Part 1: Fine particle composition and organic source apportionment, Atmos. Chem. Phys., 9, 6633–6653, https://doi.org/10.5194/acp-9-6633-2009, 2009.
Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R.: Quantitative sampling using an Aerodyne aerosol mass spectrometer: 1. Techniques of data interpretation and error analysis, J. Geophys. Res.-Atmos., 108, 4283, https://doi.org/10.1029/2003jd001607, 2003.
Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., Canagaratna, M. R., Jayne, J. T., and Worsnop, D. R.: A generalised method for the extraction of chemically resolved mass spectra from Aerodyne aerosol mass spectrometer data, J. Aerosol Sci., 35, 909–922, https://doi.org/10.1016/j.jaerosci.2004.02.007, 2004.
AMAP: The Impact of Black Carbon on Arctic Climate (2011), edited by: Quinn, P. K., Stohl, A., Arneth, A., Berntsen, T., Burkhart, J. F., Christensen, J., Flanner, M., Kupiainen, K., Lihavainen, H., Shepherd, M., Shevchenko, V., Skov, H., and Vestreng, V., AMAP Technical Report No. 4 (2011), Arctic Monitoring and Assessment Programme (AMAP), Oslo, 72 pp., 2011.
AMAP: AMAP Assessment 2015: Black carbon and ozone as Arctic climate forcers, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, vii + 166 pp., 2015.
AMAP: Snow, Water, Ice and Permafrost in the Arctic (SWIPA) 2017, Arctic Monitoring and Assessment Programme (AMAP), Oslo, Norway, xiv + 269 pp., 2017.
Andrews, E., Ogren, J. A., Bonasoni, P., Marinoni, A., Cuevas, E., Rodriguez, S., Sun, J. Y., Jaffe, D. A., Fischer, E. V., Baltensperger, U., Weingartner, E., Coen, M. C., Sharma, S., Macdonald, A. M., Leaitch, W. R., Lin, N. H., Laj, P., Arsov, T., Kalapov, I., Jefferson, A., and Sheridan, P.: Climatology of aerosol radiative properties in the free troposphere, Atmos. Res., 102, 365–393, https://doi.org/10.1016/j.atmosres.2011.08.017, 2011.
Baek, B. H. and Aneja, V. P.: Measurement and analysis of the relationship between ammonia, acid gases, and fine particles in eastern North Carolina, J. Air Waste Manage. Assoc., 54, 623–633, https://doi.org/10.1080/10473289.2004.10470933, 2004.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl sulfide and dimethyl sulfoxide and their oxidation in the atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Barrett, T. E., Robinson, E. M., Usenko, S., and Sheesley, R. J.: Source Contributions to Wintertime Elemental and Organic Carbon in the Western Arctic Based on Radiocarbon and Tracer Apportionment, Environ. Sci. Technol., 49, 13733–13733, https://doi.org/10.1021/acs.est.5b05128, 2015.
Barrie, L. A., Hoff, R. M., and Daggupaty, S. M.: The Influence of Mid-Latitudinal Pollution Sources on Haze in the Canadian Arctic, Atmos. Environ., 15, 1407–1419, https://doi.org/10.1016/0004-6981(81)90347-4, 1981.
Becagli, S., Lazzara, L., Fani, F., Marchese, C., Traversi, R., Severi, M., di Sarra, A., Sferlazzo, D., Piacentino, S., Bommarito, C., Dayan, U., and Udisti, R.: Relationship between methanesulfonate (MS-) in atmospheric particulate and remotely sensed phytoplankton activity in oligo-mesotrophic central Mediterranean Sea, Atmos. Environ., 79, 681–688, https://doi.org/10.1016/j.atmosenv.2013.07.032, 2013.
Becagli, S., Lazzara, L., Marchese, C., Dayan, U., Ascanius, S. E., Cacciani, M., Caiazzo, L., Di Biagio, C., Di Iorio, T., di Sarra, A., Eriksen, P., Fani, F., Giardi, F., Meloni, D., Muscari, G., Pace, G., Severi, M., Traversi, R., and Udisti, R.: Relationships linking primary production, sea ice melting, and biogenic aerosol in the Arctic, Atmos. Environ., 136, 1–15, https://doi.org/10.1016/j.atmosenv.2016.04.002, 2016.
Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T., DeAngelo, B. J., Flanner, M. G., Ghan, S., Karcher, B., Koch, D., Kinne, S., Kondo, Y., Quinn, P. K., Sarofim, M. C., Schultz, M. G., Schulz, M., Venkataraman, C., Zhang, H., Zhang, S., Bellouin, N., Guttikunda, S. K., Hopke, P. K., Jacobson, M. Z., Kaiser, J. W., Klimont, Z., Lohmann, U., Schwarz, J. P., Shindell, D., Storelvmo, T., Warren, S. G., and Zender, C. S.: Bounding the role of black carbon in the climate system: A scientific assessment, J. Geophys. Res.-Atmos., 118, 5380–5552, https://doi.org/10.1002/jgrd.50171, 2013.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Cappa, C. D., Onasch, T. B., Massoli, P., Worsnop, D. R., Bates, T. S., Cross, E. S., Davidovits, P., Hakala, J., Hayden, K. L., Jobson, B. T., Kolesar, K. R., Lack, D. A., Lerner, B. M., Li, S.-M., Mellon, D., Nuaaman, I., Olfert, J. S., Petäjä, T., Quinn, P. K., Song, C., Subramanian, R., Williams, E. J., and Zaveri, R. A.: Radiative absorption enhancements due to the mixing state of atmospheric black carbon, Science, 337, 1078–1081, https://doi.org/10.1126/science.1223447, 2012.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/c2cs35121h, 2012.
Chang, R. Y.-W., Leck, C., Graus, M., Müller, M., Paatero, J., Burkhart, J. F., Stohl, A., Orr, L. H., Hayden, K., Li, S.-M., Hansel, A., Tjernström, M., Leaitch, W. R., and Abbatt, J. P. D.: Aerosol composition and sources in the central Arctic Ocean during ASCOS, Atmos. Chem. Phys., 11, 10619–10636, https://doi.org/10.5194/acp-11-10619-2011, 2011.
Christensen, J. H.: The Danish eulerian hemispheric model – a three-dimensional air pollution model used for the arctic, Atmos. Environ., 31, 4169–4191, https://doi.org/10.1016/s1352-2310(97)00264-1, 1997.
Croft, B., Wentworth, G. R., Martin, R. V., Leaitch, W. R., Murphy, J. G., Murphy, B. N., Kodros, J. K., Abbatt, J. P. D., and Pierce, J. R.: Contribution of Arctic seabird-colony ammonia to atmospheric particles and cloud-albedo radiative effect, Nat. Commun., 7, 13444, https://doi.org/10.1038/ncomms13444, 2016.
Croft, B., Martin, R. V., Leaitch, W. R., Burkart, J., Chang, R. Y.-W., Collins, D. B., Hayes, P. L., Hodshire, A. L., Huang, L., Kodros, J. K., Moravek, A., Mungall, E. L., Murphy, J. G., Sharma, S., Tremblay, S., Wentworth, G. R., Willis, M. D., Abbatt, J. P. D., and Pierce, J. R.: Arctic marine secondary organic aerosol contributes significantly to summertime particle size distributions in the Canadian Arctic Archipelago, Atmos. Chem. Phys., 19, 2787–2812, https://doi.org/10.5194/acp-19-2787-2019, 2019.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Dall'Osto, M., Beddows, D. C. S., Tunved, P., Krejci, R., Strom, J., Hansson, H. C., Yoon, Y. J., Park, K. T., Becagli, S., Udisti, R., Onasch, T., O'Dowd, C. D., Simo, R., and Harrison, R. M.: Arctic sea ice melt leads to atmospheric new particle formation, Sci. Rep.-UK, 7, 3318, https://doi.org/10.1038/s41598-017-03328-1, 2017.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Dillner, A. M., Phuah, C. H., and Turner, J. R.: Effects of post-sampling conditions on ambient carbon aerosol filter measurements, Atmos. Environ., 43, 5937–5943, https://doi.org/10.1016/j.atmosenv.2009.08.009, 2009.
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new time-of-flight aerosol mass spectrometer (TOF-AMS) – Instrument description and first field deployment, Aerosol Sci. Technol., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005.
Duplissy, J., DeCarlo, P. F., Dommen, J., Alfarra, M. R., Metzger, A., Barmpadimos, I., Prevot, A. S. H., Weingartner, E., Tritscher, T., Gysel, M., Aiken, A. C., Jimenez, J. L., Canagaratna, M. R., Worsnop, D. R., Collins, D. R., Tomlinson, J., and Baltensperger, U.: Relating hygroscopicity and composition of organic aerosol particulate matter, Atmos. Chem. Phys., 11, 1155–1165, https://doi.org/10.5194/acp-11-1155-2011, 2011.
Earle, M. E., Liu, P. S. K., Strapp, J. W., Zelenyuk, A., Imre, D., McFarquhar, G. M., Shantz, N. C., and Leaitch, W. R.: Factors influencing the microphysics and radiative properties of liquid-dominated Arctic clouds: Insight from observations of aerosol and clouds during ISDAC, J. Geophys. Res.-Atmos., 116, D00t09, https://doi.org/10.1029/2011jd015887, 2011.
Eckhardt, S., Quennehen, B., Olivié, D. J. L., Berntsen, T. K., Cherian, R., Christensen, J. H., Collins, W., Crepinsek, S., Daskalakis, N., Flanner, M., Herber, A., Heyes, C., Hodnebrog, Ø., Huang, L., Kanakidou, M., Klimont, Z., Langner, J., Law, K. S., Lund, M. T., Mahmood, R., Massling, A., Myriokefalitakis, S., Nielsen, I. E., Nøjgaard, J. K., Quaas, J., Quinn, P. K., Raut, J.-C., Rumbold, S. T., Schulz, M., Sharma, S., Skeie, R. B., Skov, H., Uttal, T., von Salzen, K., and Stohl, A.: Current model capabilities for simulating black carbon and sulfate concentrations in the Arctic atmosphere: a multi-model evaluation using a comprehensive measurement data set, Atmos. Chem. Phys., 15, 9413–9433, https://doi.org/10.5194/acp-15-9413-2015, 2015.
Fenger, M., Sørensen, L. L., Kristensen, K., Jensen, B., Nguyen, Q. T., Nøjgaard, J. K., Massling, A., Skov, H., Becker, T., and Glasius, M.: Sources of anions in aerosols in northeast Greenland during late winter, Atmos. Chem. Phys., 13, 1569–1578, https://doi.org/10.5194/acp-13-1569-2013, 2013.
Fisher, J. A., Jacob, D. J., Wang, Q. Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z. F., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate-ammonium aerosol in the Arctic in winter-spring, Atmos. Environ., 45, 7301–7318, https://doi.org/10.1016/j.atmosenv.2011.08.030, 2011.
Frossard, A. A., Shaw, P. M., Russell, L. M., Kroll, J. H., Canagaratna, M. R., Worsnop, D. R., Quinn, P. K., and Bates, T. S.: Springtime Arctic haze contributions of submicron organic particles from European and Asian combustion sources, J. Geophys. Res.-Atmos., 116, D05205, https://doi.org/10.1029/2010jd015178, 2011.
Fu, P. Q., Kawamura, K., Chen, J., Charrière, B., and Sempéré, R.: Organic molecular composition of marine aerosols over the Arctic Ocean in summer: contributions of primary emission and secondary aerosol formation, Biogeosciences, 10, 653–667, https://doi.org/10.5194/bg-10-653-2013, 2013.
Fu, P. Q., Kawamura, K., Chen, J., Qin, M. Y., Ren, L. J., Sun, Y. L., Wang, Z. F., Barrie, L. A., Tachibana, E., Ding, A. J., and Yamashita, Y.: Fluorescent water-soluble organic aerosols in the High Arctic atmosphere, Sci. Rep.-UK, 5, 9845, https://doi.org/10.1038/srep09845, 2015.
Garrett, T. J., Brattstrom, S., Sharma, S., Worthy, D. E. J., and Novelli, P.: The role of scavenging in the seasonal transport of black carbon and sulfate to the Arctic, Geophys. Res. Lett., 38, L16805, https://doi.org/10.1029/2011GL048221, 2011.
Ghahremaninezhad, R., Norman, A.-L., Abbatt, J. P. D., Levasseur, M., and Thomas, J. L.: Biogenic, anthropogenic and sea salt sulfate size-segregated aerosols in the Arctic summer, Atmos. Chem. Phys., 16, 5191–5202, https://doi.org/10.5194/acp-16-5191-2016, 2016.
Gong, S. L., Zhao, T. L., Sharma, S., Toom-Sauntry, D., Lavoue, D., Zhang, X. B., Leaitch, W. R., and Barrie, L. A.: Identification of trends and interannual variability of sulfate and black carbon in the Canadian High Arctic: 1981–2007, J. Geophys. Res.-Atmos., 115, D07305, https://doi.org/10.1029/2009jd012943, 2010.
Heidam, N. Z.: The Components of the Arctic Aerosol, Atmos. Environ., 18, 329–343, https://doi.org/10.1016/0004-6981(84)90107-0, 1984.
Heidam, N. Z., Wahlin, P., and Christensen, J. H.: Tropospheric gases and aerosols in Northeast Greenland, J. Atmos. Sci., 56, 261–278, https://doi.org/10.1175/1520-0469(1999)056<0261:tgaain>2.0.co;2, 1999.
Heidam, N. Z., Christensen, J., Wåhlin, P., and Skov, H.: Arctic atmospheric contaminants in NE Greenland: levels, variations, origins, transport, transformations and trends 1990–2001, Sci. Total Environ., 331, 5–28, https://doi.org/10.1016/j.scitotenv.2004.03.033, 2004.
Hennigan, C. J., Sullivan, A. P., Collett, J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010gl043088, 2010.
Hirdman, D., Burkhart, J. F., Sodemann, H., Eckhardt, S., Jefferson, A., Quinn, P. K., Sharma, S., Ström, J., and Stohl, A.: Long-term trends of black carbon and sulphate aerosol in the Arctic: changes in atmospheric transport and source region emissions, Atmos. Chem. Phys., 10, 9351–9368, https://doi.org/10.5194/acp-10-9351-2010, 2010.
Hoffmann, D., Tilgner, A., Iinuma, Y., and Herrmann, H.: Atmospheric Stability of Levoglucosan: A Detailed Laboratory and Modeling Study, Environ. Sci. Technol., 44, 694–699, https://doi.org/10.1021/es902476f, 2010.
Hoffmann, E. H., Tilgner, A., Schrodner, R., Brauera, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Huang, J. and Jaeglé, L.: Wintertime enhancements of sea salt aerosol in polar regions consistent with a sea ice source from blowing snow, Atmos. Chem. Phys., 17, 3699–3712, https://doi.org/10.5194/acp-17-3699-2017, 2017.
Huang, S., Poulain, L., van Pinxteren, D., van Pinxteren, M., Wu, Z. J., Herrmann, H., and Wiedensohler, A.: Latitudinal and Seasonal Distribution of Particulate MSA over the Atlantic using a Validated Quantification Method with HR-ToF-AMS, Environ. Sci. Technol., 51, 418–426, https://doi.org/10.1021/acs.est.6b03186, 2017.
IPCC: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 1535 pp., 2013.
IPCC: Summary for Policymakers, in: Global Warming of 1.5 ∘C. An IPCC Special Report on the impacts of global warming of 1.5 ∘C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty, edited by: Masson-Delmotte, V., Zhai, P., Pörtner, H.-O., Roberts, D., Skea, J., Shukla, P. R., Pirani, A., Moufouma-Okia, W., Péan, C., Pidcock, R., Connors, S., Matthews, J. B. R., Chen, Y., Zhou, X., Gomis, M. I., Lonnoy, E., Maycock,T., Tignor, M., and Waterfield, T., World Meteorological Organization, Geneva, Switzerland, 32 pp., 2018.
Jayne, J. T., Leard, D. C., Zhang, X., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an Aerosol Mass Spectrometer for Size and Composition Analysis of Submicron Particles, Aerosol Sci. Technol., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Jimenez, J. L., Jayne, J. T., Shi, Q., Kolb, C. E., Worsnop, D. R., Yourshaw, I., Seinfeld, J. H., Flagan, R. C., Zhang, X. F., Smith, K. A., Morris, J. W., and Davidovits, P.: Ambient aerosol sampling using the Aerodyne Aerosol Mass Spectrometer, J. Geophys. Res.-Atmos., 108, 8425, https://doi.org/10.1029/2001jd001213, 2003.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kawamura, K., Kasukabe, H., and Barrie, L. A.: Secondary formation of water-soluble organic acids and alpha-dicarbonyls and their contributions to total carbon and water-soluble organic carbon: Photochemical aging of organic aerosols in the Arctic spring, J. Geophys. Res.-Atmos., 115, D21306, https://doi.org/10.1029/2010jd014299, 2010.
Kodros, J. K., Hanna, S. J., Bertram, A. K., Leaitch, W. R., Schulz, H., Herber, A. B., Zanatta, M., Burkart, J., Willis, M. D., Abbatt, J. P. D., and Pierce, J. R.: Size-resolved mixing state of black carbon in the Canadian high Arctic and implications for simulated direct radiative effect, Atmos. Chem. Phys., 18, 11345–11361, https://doi.org/10.5194/acp-18-11345-2018, 2018.
Komppula, M., Lihavainen, H., Kerminen, V. M., Kulmala, M., and Viisanen, Y.: Measurements of cloud droplet activation of aerosol particles at a clean subarctic background site, J. Geophys. Res.-Atmos., 110, D06204, https://doi.org/10.1029/2004jd005200, 2005.
Laing, J. R., Hopke, P. K., Hopke, E. F., Husain, L., Dutkiewicz, V. A., Paatero, J., and Viisanen, Y.: Long-term trends of biogenic sulfur aerosol and its relationship with sea surface temperature in Arctic Finland, J. Geophys. Res.-Atmos., 118, 776, 11770–11776, https://doi.org/10.1002/2013jd020384, 2013.
Lana, A., Bell, T. G., Simo, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, Gb1004, https://doi.org/10.1029/2010gb003850, 2011.
Lange, R., Dall'Osto, M., Skov, H., Nojgaard, J. K., Nielsen, I. E., Beddowse, D. C. S., Simob, R., Harrison, R. M., and Massling, A.: Characterization of distinct Arctic aerosol accumulation modes and their sources, Atmos. Environ., 183, 1–10, https://doi.org/10.1016/j.atmosenv.2018.03.060, 2018.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Law, K. S. and Stohl, A.: Arctic Air Pollution: Origins and Impacts, Science, 315, 1537–1540, https://doi.org/10.1126/science.1137695, 2007.
Leaitch, W. R., Russell, L. M., Liu, J., Kolonjari, F., Toom, D., Huang, L., Sharma, S., Chivulescu, A., Veber, D., and Zhang, W.: Organic functional groups in the submicron aerosol at 82.5∘ N, 62.5∘ W from 2012 to 2014, Atmos. Chem. Phys., 18, 3269–3287, https://doi.org/10.5194/acp-18-3269-2018, 2018.
Leck, C. and Bigg, E. K.: Source and evolution of the marine aerosol – A new perspective, Geophys. Res. Lett., 32, L19803, https://doi.org/10.1029/2005gl023651, 2005.
Lee, B. P., Li, Y. J., Yu, J. Z., Louie, P. K. K., and Chan, C. K.: Physical and chemical characterization of ambient aerosol by HR-ToF-AMS at a suburban site in Hong Kong during springtime 2011, J. Geophys. Res.-Atmos., 118, 8625–8639, https://doi.org/10.1002/jgrd.50658, 2013.
Lee, T., Sullivan, A. P., Mack, L., Jimenez, J. L., Kreidenweis, S. M., Onasch, T. B., Worsnop, D. R., Malm, W., Wold, C. E., Hao, W. M., and Collett, J. L.: Chemical Smoke Marker Emissions During Flaming and Smoldering Phases of Laboratory Open Burning of Wildland Fuels, Aerosol Sci. Technol., 44, I–V, https://doi.org/10.1080/02786826.2010.499884, 2010.
Lenton, T. M.: Arctic climate tipping points, A Journal of the Human Environment, 41, 10–22, https://doi.org/10.1007/s13280-011-0221-x, 2012.
Liu, S., Aiken, A. C., Gorkowski, K., Dubey, M. K., Cappa, C. D., Williams, L. R., Herndon, S. C., Massoli, P., Fortner, E. C., Chhabra, P. S., Brooks, W. A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., China, S., Sharma, N., Mazzoleni, C., Xu, L., Ng, N. L., Liu, D., Allan, J. D., Lee, J. D., Fleming, Z. L., Mohr, C., Zotter, P., Szidat, S., and Prevot, A. S. H.: Enhanced light absorption by mixed source black and brown carbon particles in UK winter, Nat. Commun., 6, 8435, https://doi.org/10.1038/ncomms9435, 2015.
Mahmood, R., von Salzen, K., Flanner, M., Sand, M., Langner, J., Wang, H. L., and Huang, L.: Seasonality of global and Arctic black carbon processes in the Arctic Monitoring and Assessment Programme models, J. Geophys. Res.-Atmos., 121, 7100–7116, https://doi.org/10.1002/2016jd024849, 2016.
Martinsson, J., Eriksson, A. C., Nielsen, I. E., Malmborg, V. B., Ahlberg, E., Andersen, C., Lindgren, R., Nystrom, R., Nordin, E. Z., Brune, W. H., Svenningsson, B., Swietlicki, E., Boman, C., and Pagels, J. H.: Impacts of Combustion Conditions and Photochemical Processing on the Light Absorption of Biomass Combustion Aerosol, Environ. Sci. Technol., 49, 14663–14671, https://doi.org/10.1021/acs.est.5b03205, 2015.
Massling, A., Nielsen, I. E., Kristensen, D., Christensen, J. H., Sørensen, L. L., Jensen, B., Nguyen, Q. T., Nøjgaard, J. K., Glasius, M., and Skov, H.: Atmospheric black carbon and sulfate concentrations in Northeast Greenland, Atmos. Chem. Phys., 15, 9681–9692, https://doi.org/10.5194/acp-15-9681-2015, 2015.
Matthew, B. M., Middlebrook, A. M., and Onasch, T. B.: Collection efficiencies in an Aerodyne Aerosol Mass Spectrometer as a function of particle phase for laboratory generated aerosols, Aerosol Sci. Technol., 42, 884–898, https://doi.org/10.1080/02786820802356797, 2008.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of Composition-Dependent Collection Efficiencies for the Aerodyne Aerosol Mass Spectrometer using Field Data, Aerosol Sci. Technol., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Narukawa, M., Kawamura, K., Li, S. M., and Bottenheim, J. W.: Stable carbon isotopic ratios and ionic composition of the high-Arctic aerosols: An increase in δ13C values from winter to spring, J. Geophys. Res.-Atmos., 113, D02312, https://doi.org/10.1029/2007jd008755, 2008.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Nguyen, Q. T., Skov, H., Sørensen, L. L., Jensen, B. J., Grube, A. G., Massling, A., Glasius, M., and Nøjgaard, J. K.: Source apportionment of particles at Station Nord, North East Greenland during 2008–2010 using COPREM and PMF analysis, Atmos. Chem. Phys., 13, 35–49, https://doi.org/10.5194/acp-13-35-2013, 2013.
Nguyen, Q. T., Glasius, M., Sørensen, L. L., Jensen, B., Skov, H., Birmili, W., Wiedensohler, A., Kristensson, A., Nøjgaard, J. K., and Massling, A.: Seasonal variation of atmospheric particle number concentrations, new particle formation and atmospheric oxidation capacity at the high Arctic site Villum Research Station, Station Nord, Atmos. Chem. Phys., 16, 11319–11336, https://doi.org/10.5194/acp-16-11319-2016, 2016.
Norman, A. L., Barrie, L. A., Toom-Sauntry, D., Sirois, A., Krouse, H. R., Li, S. M., and Sharma, S.: Sources of aerosol sulphate at Alert: Apportionment using stable isotopes, J. Geophys. Res.-Atmos., 104, 11619–11631, https://doi.org/10.1029/1999jd900078, 1999.
Onasch, T. B., Trimborn, A., Fortner, E. C., Jayne, J. T., Kok, G. L., Williams, L. R., Davidovits, P., and Worsnop, D. R.: Soot Particle Aerosol Mass Spectrometer: Development, Validation, and Initial Application, Aerosol Sci. Technol., 46, 804–817, https://doi.org/10.1080/02786826.2012.663948, 2012.
Orellana, M. V., Matrai, P. A., Leck, C., Rauschenberg, C. D., Lee, A. M., and Coz, E.: Marine microgels as a source of cloud condensation nuclei in the high Arctic, P. Natl. Acad. Sci. USA, 108, 13612–13617, https://doi.org/10.1073/pnas.1102457108, 2011.
Ovadnevaite, J., O'Dowd, C., Dall'Osto, M., Ceburnis, D., Worsnop, D. R., and Berresheim, H.: Detecting high contributions of primary organic matter to marine aerosol: A case study, Geophys. Res. Lett., 38, L02807, https://doi.org/10.1029/2010gl046083, 2011.
Ovadnevaite, J., Ceburnis, D., Canagaratna, M., Berresheim, H., Bialek, J., Martucci, G., Worsnop, D. R., and O'Dowd, C.: On the effect of wind speed on submicron sea salt mass concentrations and source fluxes, J. Geophys. Res.-Atmos., 117, D16201, https://doi.org/10.1029/2011jd017379, 2012.
Paatero, P. and Tapper, U.: Positive Matrix Factorization – a Nonnegative Factor Model with Optimal Utilization of Error-Estimates of Data Values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203, 1994.
Paatero, P.: Least squares formulation of robust non-negative factor analysis, Chemom. Intell. Lab. Syst., 37, 23–35, https://doi.org/10.1016/s0169-7439(96)00044-5, 1997.
Park, K. T., Lee, K., Yoon, Y. J., Lee, H. W., Kim, H. C., Lee, B. Y., Hermansen, O., Kim, T. W., and Holmen, K.: Linking atmospheric dimethyl sulfide and the Arctic Ocean spring bloom, Geophys. Res. Lett., 40, 155–160, https://doi.org/10.1029/2012gl054560, 2013.
Petzold, A., Kopp, C., and Niessner, R.: The dependence of the specific attenuation cross-section on black carbon mass fraction and particle size, Atmos. Environ., 31, 661–672, https://doi.org/10.1016/s1352-2310(96)00245-2, 1997.
Petzold, A. and Schonlinner, M.: Multi-angle absorption photometry – a new method for the measurement of aerosol light absorption and atmospheric black carbon, J. Aerosol Sci., 35, 421–441, https://doi.org/10.1016/j.jaerosci.2003.09.005, 2004.
Quinn, P. K., Miller, T. L., Bates, T. S., Ogren, J. A., Andrews, E., and Shaw, G. E.: A 3-year record of simultaneously measured aerosol chemical and optical properties at Barrow, Alaska, J. Geophys. Res.-Atmos., 107, 4130, https://doi.org/10.1029/2001jd001248, 2002.
Quinn, P. K., Bates, T. S., Coffman, D., Onasch, T. B., Worsnop, D., Baynard, T., de Gouw, J. A., Goldan, P. D., Kuster, W. C., Williams, E., Roberts, J. M., Lerner, B., Stohl, A., Pettersson, A., and Lovejoy, E. R.: Impacts of sources and aging on submicrometer aerosol properties in the marine boundary layer across the Gulf of Maine, J. Geophys. Res.-Atmos., 111, D23s36, https://doi.org/10.1029/2006jd007582, 2006.
Quinn, P. K., Shaw, G., Andrews, E., Dutton, E. G., Ruoho-Airola, T., and Gong, S. L.: Arctic haze: current trends and knowledge gaps, Tellus B, 59, 99–114, https://doi.org/10.1111/j.1600-0889.2006.00238.x, 2007.
Quinn, P. K., Bates, T. S., Baum, E., Doubleday, N., Fiore, A. M., Flanner, M., Fridlind, A., Garrett, T. J., Koch, D., Menon, S., Shindell, D., Stohl, A., and Warren, S. G.: Short-lived pollutants in the Arctic: their climate impact and possible mitigation strategies, Atmos. Chem. Phys., 8, 1723–1735, https://doi.org/10.5194/acp-8-1723-2008, 2008.
Quinn, P. K., Bates, T. S., Schulz, K., and Shaw, G. E.: Decadal trends in aerosol chemical composition at Barrow, Alaska: 1976–2008, Atmos. Chem. Phys., 9, 8883–8888, https://doi.org/10.5194/acp-9-8883-2009, 2009.
Rahn, K. A. and Heidam, N. Z.: Progress in Arctic Air Chemistry, 1977–1980 – a Comparison of the 1st and 2nd Symposia, Atmos. Environ., 15, 1345–1348, https://doi.org/10.1016/0004-6981(81)90339-5, 1981.
Rasch, M., Frandsen, E. R., Skov, H., and Hansen, J. L.: Site Manual – Villum Research Station Station Nord Greenland, Aarhus University, 2016.
Saarnio, K., Teinilä, K., Saarikoski, S., Carbone, S., Gilardoni, S., Timonen, H., Aurela, M., and Hillamo, R.: Online determination of levoglucosan in ambient aerosols with particle-into-liquid sampler – high-performance anion-exchange chromatography – mass spectrometry (PILS–HPAEC–MS), Atmos. Meas. Tech., 6, 2839–2849, https://doi.org/10.5194/amt-6-2839-2013, 2013.
Schmeisser, L., Backman, J., Ogren, J. A., Andrews, E., Asmi, E., Starkweather, S., Uttal, T., Fiebig, M., Sharma, S., Eleftheriadis, K., Vratolis, S., Bergin, M., Tunved, P., and Jefferson, A.: Seasonality of aerosol optical properties in the Arctic, Atmos. Chem. Phys., 18, 11599–11622, https://doi.org/10.5194/acp-18-11599-2018, 2018.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric chemistry and physics: from air pollution to climate change, 2nd edition, Wiley, New York, ISBN 978-0-471-72017-1, 2006.
Sharma, S., Brook, J. R., Cachier, H., Chow, J., Gaudenzi, A., and Lu, G.: Light absorption and thermal measurements of black carbon in different regions of Canada, J. Geophys. Res.-Atmos., 107, 4771, https://doi.org/10.1029/2002JD002496, 2002.
Sharma, S., Lavoue, D., Cachier, H., Barrie, L. A., and Gong, S. L.: Long-term trends of the black carbon concentrations in the Canadian Arctic, J. Geophys. Res.-Atmos., 109, D15203, https://doi.org/10.1029/2003JD004331, 2004.
Sharma, S., Andrews, E., Barrie, L. A., Ogren, J. A., and Lavoue, D.: Variations and sources of the equivalent black carbon in the high Arctic revealed by long-term observations at Alert and Barrow: 1989–2003, J. Geophys. Res.-Atmos., 111, D14208, https://doi.org/10.1029/2005JD006581, 2006.
Sharma, S., Chan, E., Ishizawa, M., Toom-Sauntry, D., Gong, S. L., Li, S. M., Tarasick, D. W., Leaitch, W. R., Norman, A., Quinn, P. K., Bates, T. S., Levasseur, M., Barrie, L. A., and Maenhaut, W.: Influence of transport and ocean ice extent on biogenic aerosol sulfur in the Arctic atmosphere, J. Geophys. Res.-Atmos., 117, D12209, https://doi.org/10.1029/2011jd017074, 2012.
Sharma, S., Ishizawa, M., Chan, D., Lavoue, D., Andrews, E., Eleftheriadis, K., and Maksyutov, S.: 16-year simulation of Arctic black carbon: Transport, source contribution, and sensitivity analysis on deposition, J. Geophys. Res.-Atmos., 118, 943–964, https://doi.org/10.1029/2012JD017774, 2013.
Shaw, P. M., Russell, L. M., Jefferson, A., and Quinn, P. K.: Arctic organic aerosol measurements show particles from mixed combustion in spring haze and from frost flowers in winter, Geophys. Res. Lett., 37, L10803, https://doi.org/10.1029/2010gl042831, 2010.
Skov, H., Wahlin, P., Christensen, J., Heidam, N. Z., and Petersen, D.: Measurements of elements, sulphate and SO2 in Nuuk Greenland, Atmos. Environ., 40, 4775–4781, https://doi.org/10.1016/j.atmosenv.2006.03.057, 2006.
Skov, H., Massling, A., Nielsen, I. E., Nordstrøm, C., Bossi, R., Vorkamp, K., Christensen, J., Larsen, M. M., MHansen, K. M., Liisberg, J. B., and Poulsen, M. B.: AMAP core – atmospheric part: from 1990 to 2015, Results from Villum Research Station, Technical report no. 101, Aarhus University, DCE – Danish Centre for Environment and Energy, 2017.
Sodemann, H., Pommier, M., Arnold, S. R., Monks, S. A., Stebel, K., Burkhart, J. F., Hair, J. W., Diskin, G. S., Clerbaux, C., Coheur, P.-F., Hurtmans, D., Schlager, H., Blechschmidt, A.-M., Kristjánsson, J. E., and Stohl, A.: Episodes of cross-polar transport in the Arctic troposphere during July 2008 as seen from models, satellite, and aircraft observations, Atmos. Chem. Phys., 11, 3631–3651, https://doi.org/10.5194/acp-11-3631-2011, 2011.
Stohl, A.: Characteristics of atmospheric transport into the Arctic troposphere, J. Geophys. Res.-Atmos., 111, D11306, https://doi.org/10.1029/2005jd006888, 2006.
Stohl, A., Berg, T., Burkhart, J. F., Fjæraa, A. M., Forster, C., Herber, A., Hov, Ø., Lunder, C., McMillan, W. W., Oltmans, S., Shiobara, M., Simpson, D., Solberg, S., Stebel, K., Ström, J., Tørseth, K., Treffeisen, R., Virkkunen, K., and Yttri, K. E.: Arctic smoke – record high air pollution levels in the European Arctic due to agricultural fires in Eastern Europe in spring 2006, Atmos. Chem. Phys., 7, 511–534, https://doi.org/10.5194/acp-7-511-2007, 2007.
Stohl, A., Klimont, Z., Eckhardt, S., Kupiainen, K., Shevchenko, V. P., Kopeikin, V. M., and Novigatsky, A. N.: Black carbon in the Arctic: the underestimated role of gas flaring and residential combustion emissions, Atmos. Chem. Phys., 13, 8833–8855, https://doi.org/10.5194/acp-13-8833-2013, 2013.
Stroeve, J., Holland, M. M., Meier, W., Scambos, T., and Serreze, M.: Arctic sea ice decline: Faster than forecast, Geophys. Res. Lett., 34, L09501, https://doi.org/10.1029/2007gl029703, 2007.
Tunved, P., Ström, J., and Krejci, R.: Arctic aerosol life cycle: linking aerosol size distributions observed between 2000 and 2010 with air mass transport and precipitation at Zeppelin station, Ny-Ålesund, Svalbard, Atmos. Chem. Phys., 13, 3643–3660, https://doi.org/10.5194/acp-13-3643-2013, 2013.
Twomey, S.: The influence of pollution on the shortwave albedo of clouds, J. Atmos. Sci., 34, 1149–1152, https://doi.org/10.1175/1520-0469(1977)034<1149:TIOPOT>2.0.CO;2, 1977.
Udisti, R., Bazzano, A., Becagli, S., Bolzacchini, E., Caiazzo, L., Cappelletti, D., Ferrero, L., Frosini, D., Giardi, F., Grotti, M., Lupi, A., Malandrino, M., Mazzola, M., Moroni, B., Severi, M., Traversi, R., Viola, A., and Vitale, V.: Sulfate source apportionment in the Ny-Alesund (Svalbard Islands) Arctic aerosol, Rend Lincei-Sci Fis, 27, 85–94, https://doi.org/10.1007/s12210-016-0517-7, 2016.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Willis, M. D., Burkart, J., Thomas, J. L., Köllner, F., Schneider, J., Bozem, H., Hoor, P. M., Aliabadi, A. A., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Growth of nucleation mode particles in the summertime Arctic: a case study, Atmos. Chem. Phys., 16, 7663–7679, https://doi.org/10.5194/acp-16-7663-2016, 2016.
Willis, M. D., Leaitch, W. R., and Abbatt, J. P. D.: Processes Controlling the Composition and Abundance of Arctic Aerosol, Rev. Geophys., 56, 621–671, https://doi.org/10.1029/2018RG000602, 2018.
Winiger, P., Andersson, A., Yttri, K. E., Tunved, P., and Gustafsson, O.: Isotope-Based Source Apportionment of EC Aerosol Particles during Winter High-Pollution Events at the Zeppelin Observatory, Svalbard, Environ. Sci. Technol., 49, 11959–11966, https://doi.org/10.1021/acs.est.5b02644, 2015.
Zangrando, R., Barbaro, E., Zennaro, P., Rossi, S., Kehrwald, N. M., Gabrieli, J., Barbante, C., and Gambaro, A.: Molecular Markers of Biomass Burning in Arctic Aerosols, Environ. Sci. Technol., 47, 8565–8574, https://doi.org/10.1021/es400125r, 2013.
Zhang, Q., Alfarra, M. R., Worsnop, D. R., Allan, J. D., Coe, H., Canagaratna, M. R., and Jimenez, J. L.: Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry, Environ. Sci. Technol., 39, 4938–4952, https://doi.org/10.1021/es048568l, 2005.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K., DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A., Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., Zhang, Y. M., and Worsnop, D. R.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007gl029979, 2007.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Ulbrich, I. M., Ng, N. L., Worsnop, D. R., and Sun, Y.: Understanding atmospheric organic aerosols via factor analysis of aerosol mass spectrometry: a review, Anal. Bioanal. Chem., 401, 3045–3067, https://doi.org/10.1007/s00216-011-5355-y, 2011.