the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Jul 2018
| 10 Jul 2018
Efficient N2O5 uptake and NO3 oxidation in the outflow of urban Beijing
Haichao Wang
Zhijun Wu
Dongjie Shang
Zhaofeng Tan
Yujue Wang
Michael Le Breton
Shengrong Lou
Mingjin Tang
Yusheng Wu
Wenfei Zhu
Jing Zheng
Limin Zeng
Mattias Hallquist
Yuanhang Zhang
Nocturnal reactive nitrogen compounds play an important role in regional air pollution. Here we present the measurements of dinitrogen pentoxide (N2O5) associated with nitryl chloride (ClNO2) and particulate nitrate (p) at a suburban site of Beijing in the summer of 2016. High levels of N2O5 and ClNO2 were observed in the outflow of the urban Beijing air masses, with 1 min average maxima of 937 and 2900 pptv, respectively. The N2O5 uptake coefficients, γ, and ClNO2 yield, f, were experimentally determined from the observed parameters. The N2O5 uptake coefficient ranged from 0.012 to 0.055, with an average of 0.034 ± 0.018, which is in the upper range of previous field studies reported in North America and Europe but is a moderate value in the North China Plain (NCP), which reflects efficient N2O5 heterogeneous processes in Beijing. The ClNO2 yield exhibited high variability, with a range of 0.50 to unity and an average of 0.73 ± 0.25. The concentration of the nitrate radical (NO3) was calculated assuming that the thermal equilibrium between NO3 and N2O5 was maintained. In NOx-rich air masses, the oxidation of nocturnal biogenic volatile organic compounds (BVOCs) was dominated by NO3 rather than O3. The production rate of organic nitrate (ON) via NO3 + BVOCs was significant, with an average of 0.10 ± 0.07 ppbv h−1. We highlight the importance of NO3 oxidation of VOCs in the formation of ON and subsequent secondary organic aerosols in summer in Beijing.
- Article
(2889 KB) - Full-text XML
-
Supplement
(671 KB) - BibTeX
- EndNote





It has been well recognized that reactive nitrogen compounds, specifically the nitrate radical (NO3) and dinitrogen pentoxide (N2O5), play a key role in nighttime chemistry (Wayne et al., 1991; Brown and Stutz, 2012). NO3 is the most important oxidant in the nighttime and can be considered the nighttime analogue of the hydroxyl radical (OH) for certain volatile organic compounds (VOCs; Wayne et al., 1991; Benton et al., 2010). NO3 can initiate the removal of many kinds of anthropogenic and biogenic emissions after sunset. In NOx-rich plumes, NO3 is responsible for the vast majority of the oxidation of biogenic VOCs because of its rapid reactions with unsaturated hydrocarbons (Edwards et al., 2017). NO3 is predominantly formed by the reaction of NO2 with O3 (Reaction R1) and further reacts with NO2 to produce N2O5 (Reaction R2). N2O5 is rapidly decomposed back to NO3 (Reaction R3), NO3, and N2O5 are in dynamic equilibrium in the troposphere.
Photolysis of NO3 and the reaction of NO3 with NO are rapid, which leads to a daytime NO3 lifetime being shorter than 5 s with extremely low concentrations, whereas in low-NO air masses, the fate of NO3 is mainly controlled by the mixing ratios of various VOCs and N2O5 heterogeneous hydrolysis because the two terms are the dominating loss pathways of NO3 and N2O5. The VOC reaction is significant downwind of an urban area or a strongly urban-influenced forested area in summer. The NO3 oxidation of VOCs was responsible for more than 70 % of nocturnal NO3 loss in Houston (Stutz et al., 2010) and contributed approximately 50 % in a forest region in Germany (Geyer et al., 2001). The reactions of NO3 with several biogenic VOCs (BVOCs) produce considerable amounts of organic nitrates (ONs) with efficient yields, which act as important precursors of secondary organic aerosols (SOAs). The reaction of NO3 with isoprene has a SOA mass yield of 23.8 % (Ng et al., 2008). For the reaction with monoterpene, such as limonene, the SOA mass yield can reach 174 % at ambient temperatures (Boyd et al., 2017). The reactions of NO3 + BVOCs are critical to the studies of aerosols on regional and global scales (Fry et al., 2009; Rollins et al., 2009; Pye et al., 2010; Ng et al., 2017). For example, ON had extensive percentages of fine particulate nitrate (p) (34–44 %) in Europe (Kiendler-Scharr et al., 2016).
The heterogeneous hydrolysis of N2O5 produces soluble nitrate (HNO3 or ) and nitryl chloride (ClNO2) on chloride-containing aerosols (Reaction R4) (Finlayson-Pitts et al., 1989). This reaction is known to be an important intermediate in the NOx removal processes (Brown et al., 2006). The pseudo-first-order loss rate constant of N2O5 via heterogeneous uptake is given in Eq. (1) (Wahner et al., 1998).
Here c is the mean molecule speed of N2O5, Sa is the aerosol surface concentration, and γ (N2O5) is the N2O5 uptake coefficient. N2O5 heterogeneous hydrolysis is one of the major uncertainties of the NO3 budget since the N2O5 uptake coefficient can be highly variable and difficult to quantify (Brown and Stutz, 2012; Chang et al., 2011; Wang and Lu, 2016). Laboratory and field measurement studies have reported that the N2O5 uptake coefficient has large variability and ranges from < 0.001 to 0.1; the N2O5 uptake coefficient depends on relative humidity (RH), particle morphology, compositions (water content, nitrate, sulfate, and organic or mineral particles), and other factors (Wahner et al., 1998; Mentel et al., 1999; Hallquist et al., 2003; Thornton et al., 2003, 2005; Brown et al., 2006; Bertram and Thornton, 2009; Tang et al., 2012, 2014; Gaston et al., 2014; Gržinić et al., 2015; Tang et al., 2017). The coupled chemical mechanisms in ambient conditions are still not well understood. ClNO2 forms and accumulates with a negligible sink during the night and further photolyzes and liberates the chlorine radical (Cl) and NO2 after sunrise. Hundreds of parts per trillion by volume to parts per billion by volume of ClNO2 can lead to several parts per billion by volume of O3 enhancement and significant primary ROx production (Osthoff et al., 2008; Thornton et al., 2010; McLaren et al., 2010; Riedel et al., 2014; Sarwar et al., 2014; Tham et al., 2016).
Large amounts of NOx have been emitted for the past several decades in China, but comprehensive field studies of the nighttime chemical processes of reactive nitrogen oxides remain sparse. Previous studies have found high mixing ratios of NO3 associated with high NO3 reactivity in the megacities in China, including Shanghai, the Pearl River Delta (PRD), and Beijing (Li et al., 2012; S. S. Wang et al., 2013; D. Wang et al., 2015). The N2O5 concentration was elevated in Beijing (H. C. Wang et al., 2017a, b) but was moderate in other parts of the North China Plain (NCP), such as Wangdu, Jinan, and Mount Tai (Tham et al., 2016; X. F. Wang et al., 2017; Z. Wang et al., 2017). Recently, the N2O5 uptake coefficients were determined to be very high, even up to 0.1 in the NCP, but the reason is still not well studied (H. C. Wang et al., 2017b; X. F. Wang et al., 2017; Z. Wang et al., 2017). Reactive N2O5 chemistry was also reported in Hong Kong and showed the highest field-observed N2O5 concentration to date (Wang et al., 2016; Brown et al., 2016). Observations and model simulations revealed that fast heterogeneous uptake of N2O5 is an important pathway of p formation in China (H. C. Wang et al., 2017b; Z. Wang et al., 2017; Su et al., 2017); the reaction also contributed significantly to removal (Z. Wang et al., 2017; Brown et al., 2016). Moreover, chlorine activation from N2O5 uptake had a significant effect on daytime photolysis chemistry in China (Xue et al., 2015; Li et al., 2016; Tham et al., 2016; T. Wang et al., 2016).
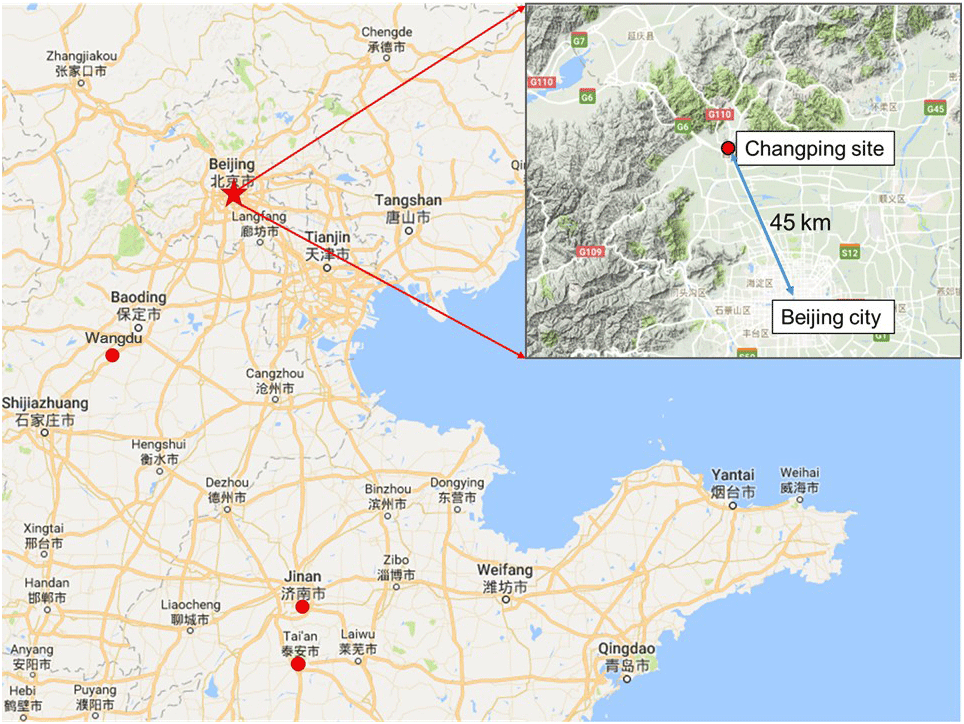
Figure 1Map of Beijing and the surrounding area. The red star shows the location of the Changping site, and red dots show other sites where previous N2O5 measurements were conducted in the North China Plain (NCP), including Wangdu, Jinan, and Mount Tai (Tai'an).
In this study, to quantify the contribution of NO3 and N2O5 chemistry to the atmospheric oxidation capacity and the NOx removal process in the outflow of urban Beijing, we report the measurement of N2O5, ClNO2, and related species in the surface layer of a suburban site in Beijing and determine the N2O5 heterogeneous uptake coefficients and ClNO2 yields. The nighttime NO3 oxidation of BVOCs and its impact on ON formation in a NOx-rich region were diagnosed. Finally, the nighttime NOx removal via NO3 and N2O5 chemistry was estimated and discussed.
2.1 The site
Within the framework of a Sino-Sweden Joint Research Programme, “Photochemical Smog in China”, a summer field campaign was conducted in Beijing to enhance our understanding of secondary chemistry via photochemical smog and heterogeneous reactions (Hallquist et al., 2016). The data presented here were collected at a regional site, PKU-CP (Peking University, Changping campus), from 23 May to 5 June 2016. The measurement site is located in the northern rural area of Beijing, approximately 45 km from the city center; the closest road is approximately 1 km to the south, and there is no major industry in the surrounding areas (Fig. 1). The site is surrounded to the north, east, and west by mountains. The general feature of this site is that it captures air masses with strong influences from both urban and biogenic emissions. Instruments were set up on the fifth floor of the main building of the campus with inlets approximately 12 m above the ground. Time is given in this paper as CNST (Chinese national standard time: UTC+8 h). During the campaign, sunrise was at 05:00 CNST and sunset was at 19:30 CNST.
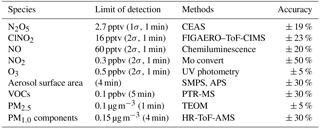
Table 1The observed gas and particle parameters used in this analysis during the campaign.
2.2 Instrument setup
A comprehensive suite of trace gas compounds and aerosol properties was measured in the field study, and the details are listed in Table 1. N2O5 was measured using a newly developed cavity-enhanced absorption spectrometer (CEAS; H. C. Wang et al., 2017a). In the CEAS, ambient N2O5 was thermally decomposed to NO3 in a perfluoroalkoxy alkane (PFA) tube (length: 35 cm, I.D.: 4.35 mm) heated to 120 ∘C and was then detected within a PFA resonator cavity; the cavity was heated to 80 ∘C to prevent NO3 reacting back to N2O5. Ambient gas was sampled with a 1.5 m sampling line (I.D.: 4.35 mm) with a flow rate of 2.0 L min−1. NO was injected for 20 s to destroy NO3 from N2O5 thermal decomposition in a 5 min cycle, and the corresponding measurements were then used as reference spectra. A Teflon polytetrafluoroethylene (PTFE) filter was used in front of the sampling module to remove ambient aerosol particles. The filter was replaced with a fresh one every hour to avoid the decrease in N2O5 transmission efficiency due to aerosol accumulation on the filter. The limit of detection (LOD) was 2.7 pptv (1σ), and the measurement uncertainty was 19 %.
ClNO2 and N2O5 were also detected using a time-of-flight chemical ionization mass spectrometer (ToF-CIMS) with the Filter Inlet for Gas and Aerosols (FIGAERO; Lopez-Hilfiker et al., 2014; Bannan et al., 2015). Briefly, the gas-phase species were measured via a 2 m long, 6 mm outer-diameter PFA inlet while the particles were simultaneously collected on a Teflon filter via a separate 2 m long, 10 mm outer-diameter copper tubing inlet; both had flow rates of 2 L min−1. The gas phase was measured for 25 min at 1 Hz, and the FIGAERO instrument was then switched to place the filter in front of the ion molecule region; it was then heated incrementally to 200 ∘C to desorb all the mass from the filter to be measured in the gas phase, which resulted in high-resolution thermograms. Formic acid calibrations were performed daily using a permeation source maintained at 40 ∘C. Post-campaign laboratory calibrations of N2O5 were first normalized to the campaign formic acid calibrations to account for any change in sensitivity (Le Breton et al., 2014). Then, ClNO2 measurements were quantified by passing the N2O5 over a wetted NaCl bed to produce ClNO2. The decrease in N2O5 from the reaction with NaCl was assumed to be equal to the concentration of ClNO2 produced (i.e., 100 % yield). The sensitivities of the CIMS to N2O5 and ClNO2 were found to be 9.5 and 1.2 ion counts per pptv Hz−1, respectively, with errors of 23 and 25 % for ClNO2 and N2O5, respectively. The LODs for ClNO2 and N2O5 were 16 and 8 pptv, respectively. An intercomparison of N2O5 measurements between the CEAS and FIGAERO–ToF-CIMS showed good agreement; another paper on chlorine photochemical activation during this campaign gives detailed intercomparison results of N2O5 measured with the two different techniques (Le Breton et al., 2018).
Submicron aerosol composition (PM1.0), including nitrate, sulfate, chloride, ammonium, and organic compounds, were measured using a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) (De Carlo et al., 2006; Zheng et al., 2017). Particle number and size distribution (PNSD) were measured with a scanning mobility particle sizer (SMPS, TSI 3936) and an aerosol particle sizer (APS, TSI 3321) (Yue et al., 2009). The SMPS measured the particles in the range between 3.5 and 523.3 nm in diameter, and the APS measured the particles with a diameter range from 597.6 nm to 10.0 µm. Sa was calculated based on the dry-state particle number and geometric diameter in each size bin (3.5 nm–2.5 µm). Dry-state Sa was corrected to wet-particle-state Sa for particle hygroscopicity by a growth factor. The growth factor, f(RH) = 1 + 8.77 × (RH/100)9.74, was derived from the measurement of aerosol extinction as a function of RH in autumn in Beijing and is valid for 30 % < RH < 90 % (Liu et al., 2013). The uncertainty of the wet aerosol surface areas was estimated to be ∼ 30 %, associated with the error from the dry PNSD measurement (∼ 20 %) and the growth factor (∼ 20 %). During this measurement, fine particles below 500 nm contributed to more than 90 % of the total Sa.
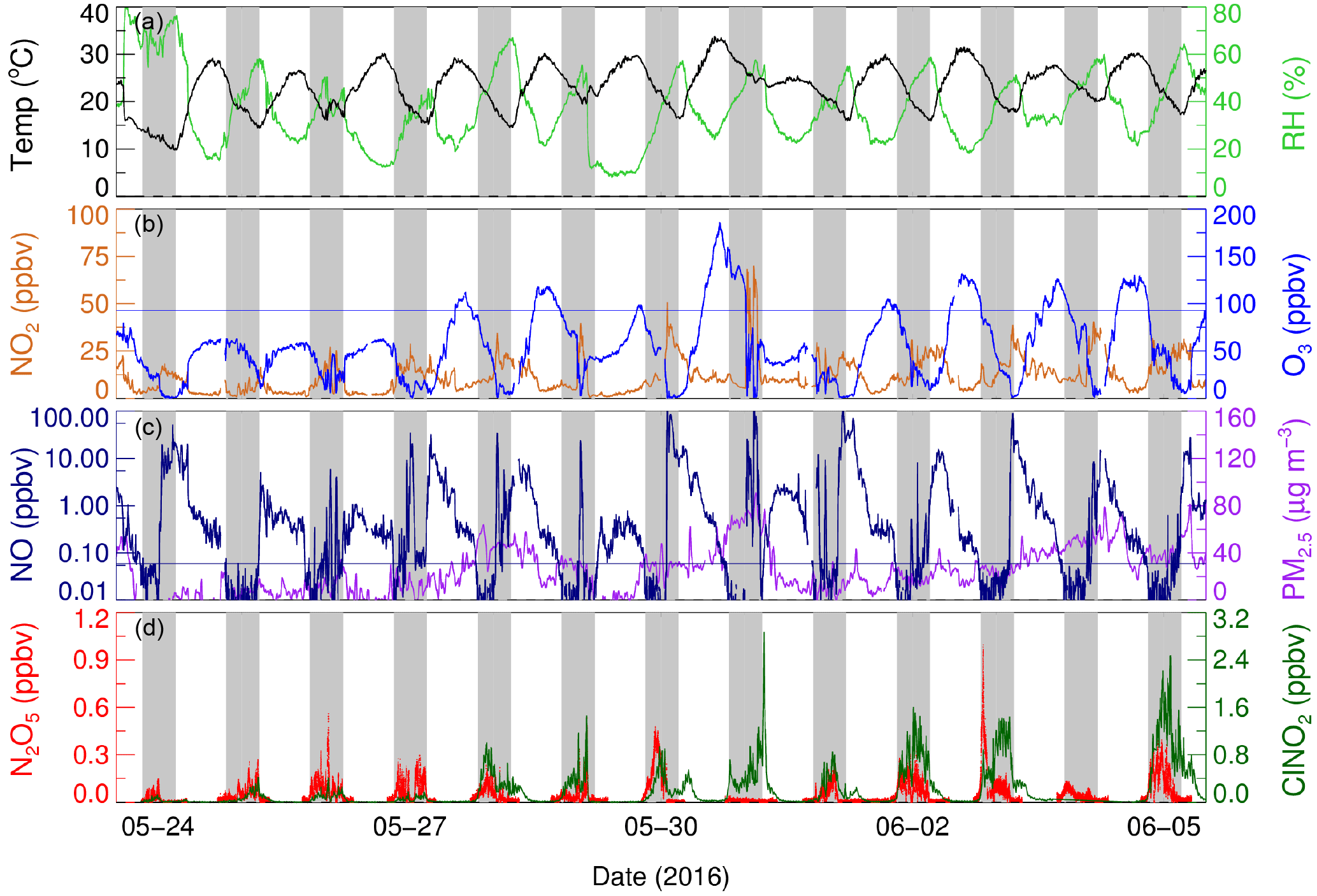
Figure 2Time series of N2O5, ClNO2, and other relevant parameters. The blue line in the O3 panel denotes the Chinese national air quality standard for O3 (ca. 93 ppbv for the surface conditions). The black line in the NO panel denotes 0.06 ppbv.
VOCs were measured by proton-transfer-reaction mass spectrometry (PTR-MS) with a time resolution of 5 min (de Gouw and Warneke, 2007; Wang et al., 2014). A commercial instrument (Thermo Fisher Scientific model 42i) equipped with a molybdenum catalytic converter was used to monitor NOx. The LODs were 60 pptv (1 min) for NO and 300 pptv (1 min) for NO2, with both at a 20 % precision (Tan et al., 2017). The molybdenum catalytic technique not only converts NO2 to NO but also converts ambient NOy such as peroxyacetyl nitrate (PAN) and HNO3. Therefore, the measured NO2 concentration corresponded to NO2 + NOy and was normally higher than the real concentration, especially in an aged air mass with high NOx conditions. In this study, we used a factor of 0.6 to correct the nighttime NO2 concentration (a detailed explanation is in the Supplement Sect. S1 and Fig. S1). The correction factor (0.6) is the average of the correction factors during nighttime. The standard deviation of the daytime correction factor for all the air masses experienced at the Changping site was determined to be 0.27 (1σ). If this uncertainty is extended to the nighttime correction factor, the resulting uncertainty of the nighttime correction is 45 %. The uncertainty of NO2 is 50 % when further including the associated measurement uncertainty from calibrations. O3 was measured by a commercial instrument using ultraviolet (UV) absorption (Thermo Fisher Scientific model 49i); the LOD was 0.5 ppbv, with an uncertainty of 5 %. The mass concentration of PM2.5 was measured using a standard tapered-element oscillating microbalance (TEOM, 1400A analyzer). Meteorological parameters included relative humidity, temperature, pressure, wind speed, and wind direction and were available during the campaign. Photolysis frequencies were calculated from the spectral actinic photon flux density measured with a spectroradiometer (Bohn et al., 2008).
3.1 Overview
During the campaign, the meteorological conditions of the site included high temperature and low RH; the temperature ranged from 10 to 34 ∘C and was 23 ± 5 ∘C on average, and RH ranged from 10 to 80 %, with an average of 37 ± 15 %. Because of the special terrain of the observation site, the local wind was measured by the in situ meteorological stations; the site has a typical mountain–valley breeze that cannot reflect the general air mass movement patterns at slightly higher altitudes. Figure S2 shows the calculated backward trajectories using the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model (Draxler and Rolph, 2003). These images show the 24 h backward particle dispersion trajectories for 12:00 local time (CNST) as the starting time during 23 May–5 July 2016. The arrivals of air masses were mainly from the northwest and the south. Therefore, we meteorologically separated the measurement period into two parts. The first 3 days show that the air masses came from the north or northwest; the air masses represent the background region (defined as background air mass, BAM). The air masses after 26 May originated from the polluted NCP and passed over urban Beijing; they were characterized by large NOx emissions and severe photochemical pollution (defined as urban air mass, UAM).
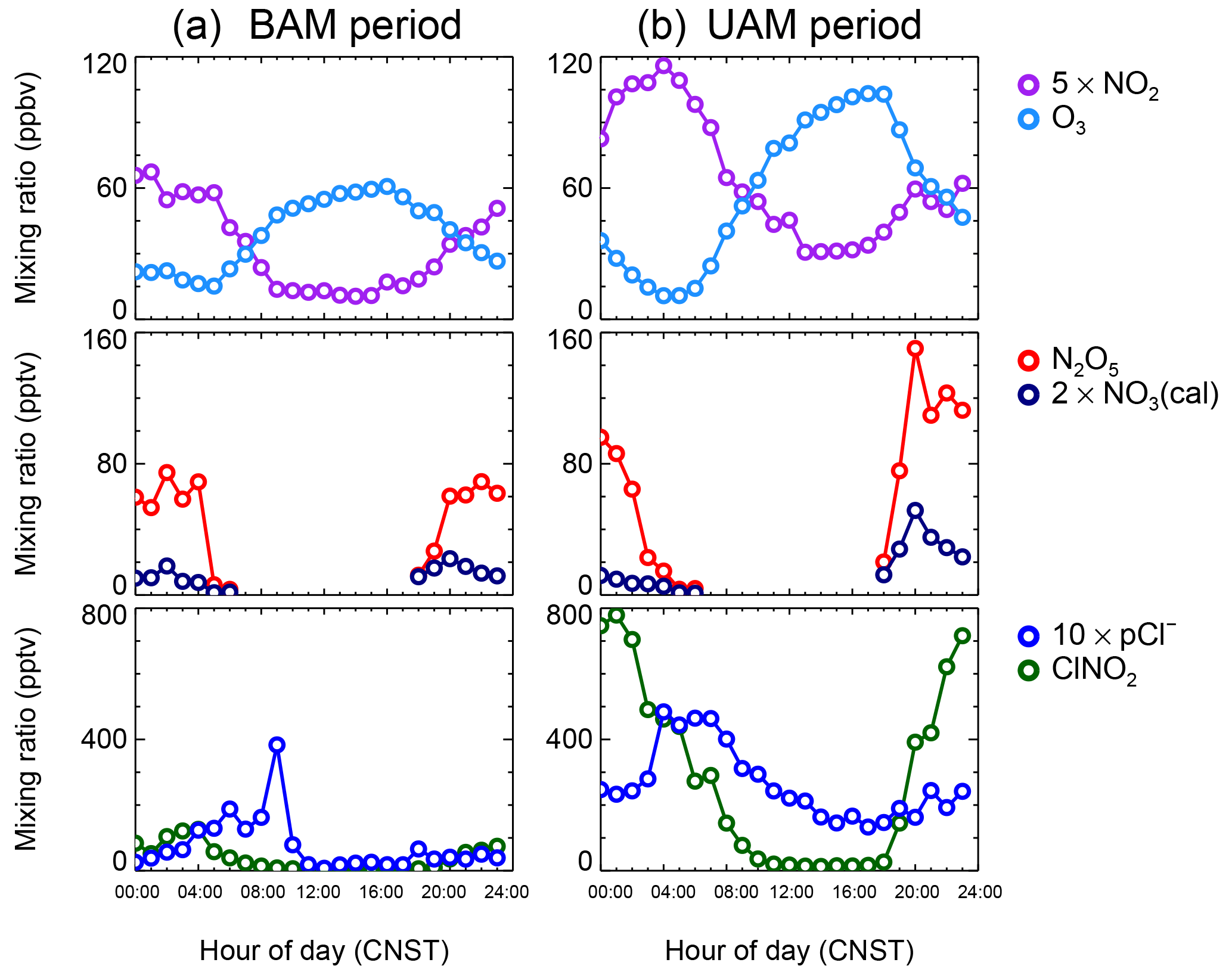
Figure 3Mean diurnal profiles of 5 × NO2, O3, N2O5, 2 × NO3 (calculated), ClNO2, and 10 × pCl−. Panel (a) depicts the background air mass (BAM) period and (b) depicts the urban air mass (UAM) period.
The time series of N2O5, ClNO2, and other relevant species are shown in Fig. 2, and nighttime statistical results are listed in Table S1 in the Supplement. The daily 8 h maximum of O3 concentration exceeded 93 ppbv (Chinese national air quality standard) for 8 of 12 days, and all the O3-polluted air masses came from the urban region. When the air masses were from the background region, the daily maximum of O3 was only approximately 60 ppbv, much lower than that from the urban region. The NO2 concentration was elevated, with a nocturnal average value over 10 ppbv during the UAM period. The nocturnal nitrate radical production rate, P(NO3), was large, with an average of 1.2 ± 0.9 ppbv h−1, which is comparable with rates previously reported in the NCP and Hong Kong (Tham et al., 2016; Brown et al., 2016; Z. Wang et al., 2017; X. F. Wang et al., 2017). The daily peaks of N2O5 were 100–500 pptv most nights; the maximum of 937 pptv in a 1 min average was observed near 20:00 CNST on the early night of 2 June, when the P(NO3) was up to 4 ppbv h−1. The average mixing ratio of N2O5 was 73 ± 90 pptv, which is much higher than recent measurements reported in northern China (Tham et al., 2016; X. F. Wang et al., 2017; Z. Wang et al., 2017), but much lower than that observed in the residual layer of the outflow from the PRD region, where N2O5 was up to 7.7 ppbv (Wang et al., 2016). With an elevated O3 mixing ratio in the first half of the night, the NO lifetime was only several minutes, and the mixing ratio of NO concentration was observed below the detection limit. During the second half of the night when the O3 concentration was low, high levels of NO could occasionally be observed, and N2O5 dropped to zero because of the fast titration by NO, as during the events that occurred in the second half of the nights of 24, 28, and 30 May. The PM2.5 mass concentration was moderate during the measurement period, with an average of 26 ± 21 µg m−3, and the average Sa was 560 ± 340 µm2 cm−3. Elevated ClNO2 was observed with a daily maximum of over 800 pptv (1 min average) during the UAM period. The maximum of ClNO2 was observed with 2900 pptv in the morning (05:30 CNST) of 31 May. The observed ClNO2 in Beijing was comparable with that reported in the NCP (Tham et al., 2016; X. F. Wang et al., 2017; Z. Wang et al., 2017), but slightly higher than that measured at coastal (e.g., Osthoff et al., 2008) and inland sites (e.g., Thornton et al., 2010). Overall, high ClNO2 observed at this site suggested that fast N2O5 heterogeneous hydrolysis and effective ClNO2 yields are common in Beijing.
3.2 Mean diurnal profiles
The mean diurnal profiles of the measured NO2, O3, N2O5, and ClNO2 and the particle chloride content are shown in Fig. 3, as well as the calculated NO3 based on the thermal equilibrium of NO2, NO3, and N2O5. Figure 3a shows the average results of the BAM period, and Fig. 3b shows those of the UAM period. NO2 and O3 from the UAM, as well as the mixing ratios of N2O5, NO3 and ClNO2, were much higher than those from the BAM, but the daily variation tendencies of those species in the two kinds of air masses were similar. N2O5 began to accumulate in the late afternoon and increased sharply after sunset. A peak occurred near 20:00 CNST and decreased below the instrument detection limit at sunrise. The time at which N2O5 maxima occurred is similar to our previous observation in urban Beijing (H. C. Wang et al., 2017b). However, the decrease rate of the observed N2O5 after the peak time was much slower than that in urban Beijing, where the N2O5 dropped to zero in 2–4 h, which suggests a relatively slow N2O5 loss rate in suburban Beijing. The daily average peaks of N2O5 during the BAM period and the UAM period were 75 and 150 pptv, respectively. The calculated NO3 diurnal profile was quite similar to N2O5, and the daily average peaks of NO3 during the BAM and UAM periods were approximately 11 and 27 pptv, respectively. The uncertainty of NO3 calculation was estimated to be 67 % according to Eq. (2), which is dominated by the uncertainty of NO2 measurement.
The observed ClNO2 concentrations showed a clear increase after sunset; ClNO2 reached a maximum before sunrise for the BAM period but around midnight for the UAM period. The diurnal peak of ClNO2 in the BAM period was 125 pptv, whereas the diurnal peak of ClNO2 was over 780 pptv in the UAM period, and 6 times as high as that in the UAM period. Particulate chloride (Cl−) is a key factor that affects the ClNO2 yield on aerosol surface. Higher particle chloride leads to higher ClNO2 yield and promotes the N2O5 conversion to ClNO2 (e.g., Finlayson-Pitts et al., 1989; Behnke et al., 1997), whereas the particle chloride content during the measurement was below 60 pptv and was much lower than the mixing ratio of ClNO2, suggesting a continuously nighttime Cl source replenished to support ClNO2 formation. HYSPLIT showed that the air masses mainly came from the continental, not coastal, regime, suggesting that large amounts of Cl− were not replenished by NaCl from marine sources, but they possibly replenished by gas-phase HCl through the acid displacement reaction (Ye et al., 2016). Cl− was found to be strongly correlated with CO and SO2, likely originating from an anthropogenic source, such as power plants or combustion sources (Le Breton et al., 2018). According to the mass balance, the gas-phase HCl for supporting the production of ClNO2 is several parts per billion by volume per night. The required HCl source indicated the ratio HCl ∕ pCl− is about 10–30, which was found to be consistent with the following observation in Beijing. Although the HCl measurement was not available in this study, note that up to 10 ppbv of HCl was observed in urban Beijing in September 2016; we propose that gas-phase HCl was sufficient to support ClNO2 formation.
After sunrise, ClNO2 was photolyzed and decreased with the increasing photolysis intensity; however, ClNO2 can still survive until noon with an averaged daily maximum of J(ClNO2) of 1.7 × 10−4 s−1. Similar to the studies reported in London, Texas, and Wangdu (Bannan et al., 2015; Faxon et al., 2015; Tham et al., 2016), we observed sustained elevated ClNO2 events after sunrise on 5 of 12 days. For example, on the morning of 30 May, ClNO2 increased fast after sunrise and up to 500 pptv at 08:00 CNST. Such a high ClNO2 increase was impossible to attribute to the local chemical formation since N2O5 dropped to almost zero and the required N2O5 uptake coefficients were unrealistically high. A previous study suggested that abundant ClNO2 produced in the residual layer at night and downward transport in the morning may help to explain this phenomenon (Tham et al., 2016).
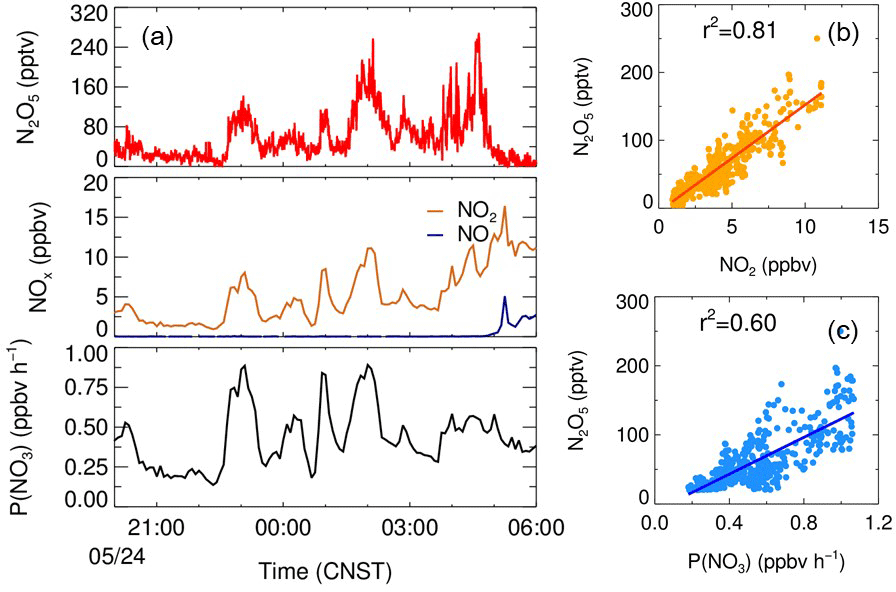
Figure 4The correlation of the mixing ratio of N2O5 and NO2 and the production rate of NO3 on the night of 24 May.
3.3 Variation in N2O5 in the background air masses
During the BAM period, the O3 concentration was well in excess of NO2. In the NO3 and N2O5 formation processes, the limited NO2 in the high-O3 region indicates that the variation in NO2 is more essential to the variation in the N2O5 concentration. As shown in Fig. 4, during the night of 24 May (20:00–04:00 CNST), the local emission of NO was negligible. The O3 concentration was larger than 25 ppbv, much higher than NO2 and free of the local NO emissions. The N2O5 concentration was highly correlated with NO2 (R2=0.81) and the NO3 production rate (R2=0.60), suggesting the N2O5 concentration was solely a response to the NO2 concentration in the BAM when enough O3 was present.
Table 2Summary of the field-observed ambient ClNO2 ∕ N2O5.

a Daily average results. b Power plant plume cases at Mt. Tai in Shandong, China. c Estimated according to Mielke et al. (2013).
3.4 Elevated ClNO2-to-N2O5 ratio
Large day-to-day variabilities in N2O5 and ClNO2 were observed during the measurement period. Following the work of Osthoff et al. (2008), Mielke et al. (2013), Phillips et al. (2012), and Bannan et al. (2015), we used the concentration ratio of ClNO2 to N2O5 to describe the conversion capacity of N2O5 to ClNO2. Note that the loss of N2O5 by dry deposition would drive up the ClNO2 : N2O5. The nighttime peak values and mean values of ClNO2 : N2O5 were used to calculate the daily ratios (Table S2); the calculation period is from 19:30 to 05:00 CNST the next day. The average nighttime ratio ranged from 0.7 to 42.0, with a mean of 7.7 and a median of 6.0. ClNO2 formation was effective, with ClNO2 : N2O5 ratios larger than 1 : 1 throughout the campaign, except for the night of 26 May, when the ratio was 0.7 : 1. Previous observations of the ClNO2 : N2O5 ratios are summarized in Table 2. Compared with the results conducted in similar continental regions in Europe and America (0.2–3.0), the ratios in this work were significantly higher and consistent with recent studies in the NCP (Tham et al., 2016; X. F. Wang et al., 2017; Z. Wang et al., 2017), which suggests that high ClNO2 : N2O5 ratios were ubiquitous in the NCP and implies that ClNO2 yield via N2O5 uptake is efficient.
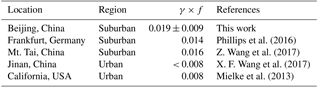
Table 3Summary of the average γ×f values derived in the field observations.
4.1 Determination of N2O5 uptake coefficients
A composite term, γ×f, was used to evaluate the production of ClNO2 from N2O5 heterogeneous hydrolysis (Mielke et al., 2013). γ×f was estimated by fitting the observed ClNO2 in a time period when the nighttime concentrations of ClNO2 increased continuously. The increased ClNO2 was assumed to be solely from the N2O5 uptake. The fitting was optimized by changing the input of γ×f associated with the measured N2O5 and Sa, until the ClNO2 increase was well reproduced (Eq. 3).
Here t0 and t denote the start time and end time, respectively; the calculation time duration was normally several hours. [ClNO2](t0) is the observed concentration at t0 and set as the fitting offset. Note that the transport leads to the bias of the N2O5 uptake coefficient and ClNO2 yield. But the small variation in the mixing ratio of CO (< 5 %) during each analysis time period suggested the transport process is not important to the increasing ClNO2. The derived γ×f was found to be constant with small uncertainties for optimization (see Table S3). The γ×f had moderate variability and ranged from 0.008 to 0.035 with an average of 0.019 ± 0.009. Table 3 summarizes the γ×f values derived in the previous field observations. The value in suburban Germany was between 0.001 and 0.09, with an average of 0.014 (Phillips et al., 2016), and the average value in Mt. Tai, China, was approximately 0.016 (Z. Wang et al., 2017). The average γ×f in this study was comparable with that of the two suburban sites, whereas at an urban site of Jinan, China (X. F. Wang et al., 2017), the value was lower than 0.008 and comparable with that in the CalNex-LA campaign. The three sets of γ×f values from suburban regions were about twice as large as those in urban regions, which implies that the ClNO2 formation efficiency in the aged air masses in suburban regions was higher than in the urban region. The difference of the overall yield between the two regions may be caused by particle properties or other factors (Riemer et al., 2009; Gaston et al., 2014; Gržinić et al., 2015; Bertram and Thornton, 2009).
According to reaction R4, soluble nitrate and ClNO2 were formed by N2O5 heterogeneous uptake, with yields of 2−f and f, respectively. Following the recent work of Phillips et al. (2016), we used the observed p and ClNO2 formation rates to derive individual γ and f. The calculations assumed that the relevant properties of the air mass are conserved and that the losses of produced species are negligible; additionally, the N2O5 uptake coefficients and the ClNO2 yield are independent of particle size. The nights characterized by the following two features were chosen for further analysis: (1) significant correlations between p and ClNO2 were present (R2>0.5), which suggested that, to a good approximation, both ClNO2 and p are produced only by N2O5 heterogeneous uptake. The reason for excluding other nights with low correction (R2<0.2) was that ClNO2 and p may be affected by the effective transport or other production pathways, and these contributions cannot be well quantified. Therefore the selection of a high correction of ClNO2 with p may lead to a bias as the contribution from other formation pathways and the transport were neglected. (2) During an increasing period of p, an equivalent or faster increase in ammonium to p was also observed, which means enough gas-phase ammonia was repartitioned to form ammonium nitrate and suppress the release of HNO3. The ammonia-rich conditions (22 ± 9 ppbv on average) in Beijing demonstrated that the degassing of HNO3 at night can be effectively buffered by the high concentrations of ammonia presented in the NCP (Liu et al., 2017). Both gas–particle repartitioning of HNO3 and nighttime-produced HNO3 will result in the overestimation of γ and the underestimation of f. The daytime-produced HNO3 will soon be in a new equilibrium rapidly on the timescale of total nitrate chemical production, and the nighttime formation of HNO3 is normally not important; thus the nocturnal HNO3 uptake impact is negligible. During this campaign, five nights were eligible for the following analysis. The observational data of N2O5, ClNO2, p, and Sa were averaged to 5 min for the following analysis. The formations of p and ClNO2 were integrated to reproduce the increasing p and ClNO2 by inputting an initial γ and f. The offset of p and ClNO2 is the measured p and ClNO2 concentration at the start time. γ and f were optimized based on the Levenberg–Marquardt algorithm until good agreement between the observed and predicted concentrations of p and ClNO2 was obtained (Phillips et al., 2016). Figure 5 depicts an example of the fitting results on 28 May. The predicted N2O5 uptake coefficient and ClNO2 yield were 0.017 and 1.0, respectively. The uncertainty on each individual fitting is varied from 55 to 100 % due to the variability in and measurement uncertainties of p and ClNO2. Five sets of values of γ and f obtained are listed in Table 4. N2O5 uptake coefficients ranged from 0.012 to 0.055, with an average of 0.034 ± 0.018, and the ClNO2 yield ranged from 0.50 to unity, with an average of 0.73 ± 0.25. The errors from each derivation were 55 % and came from the field measurements of Sa, N2O5, p, and ClNO2.
Table 4List of the N2O5 uptake coefficients and the yield of ClNO2 in this campaign.

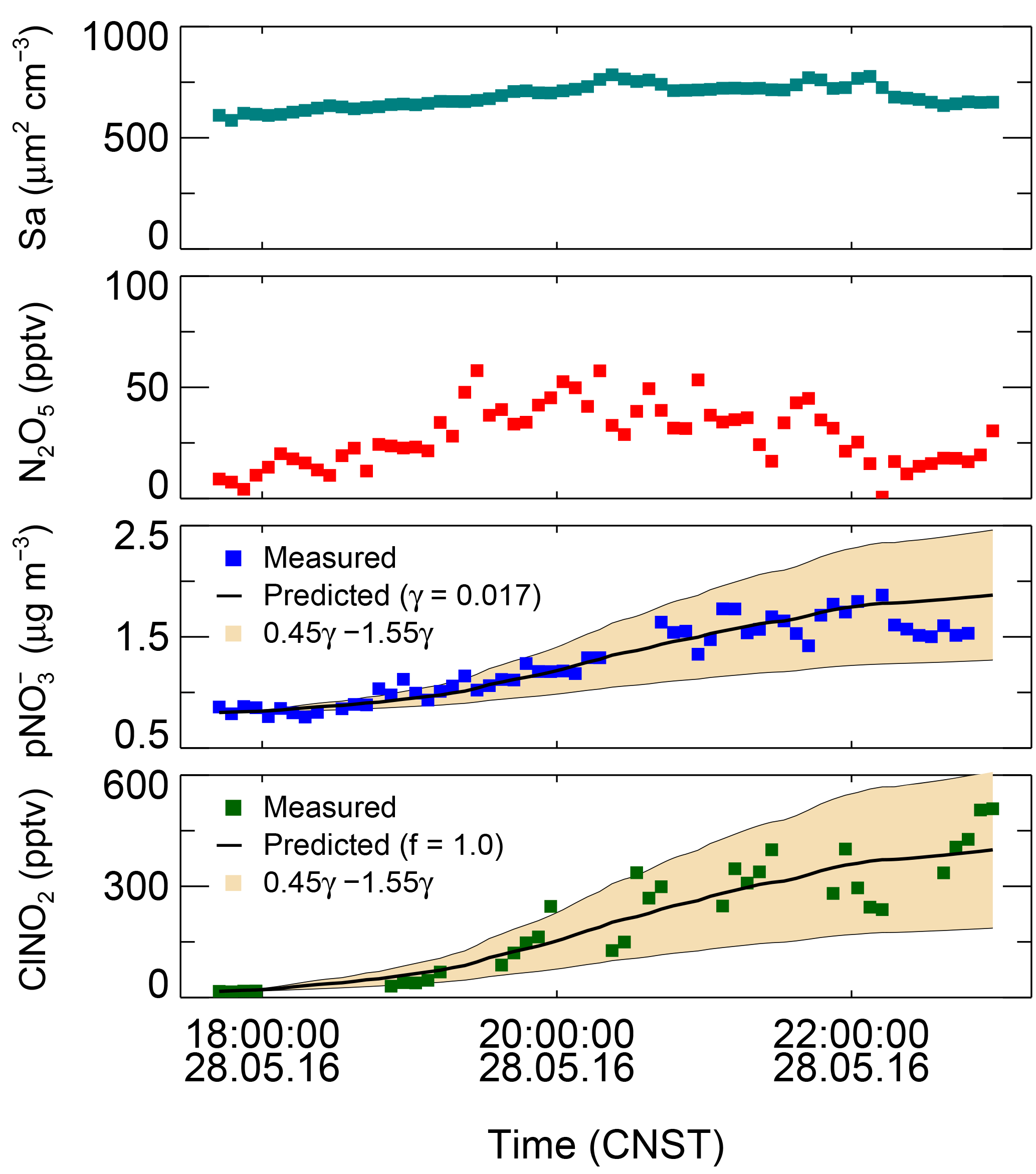
Figure 5The best fit of γ and f to reproduce the observed ClNO2 and p with an offset on 28 May. The black lines are the predicted results of the integrated and ClNO2 by using the observed Sa and N2O5.
The average γ value was consistent with the results determined by the same method at a rural site in Germany (Phillips et al., 2016) but was higher than those in the UK and North America where they used other derivation methods, including the steady-state lifetime method (Morgan et al., 2015; Brown et al., 2006, 2009), the iterated box model (Wagner et al., 2013), and direct measurement based on an aerosol flow reactor (Bertram et al., 2009; Riedel et al., 2012). The steady-state lifetime method is very sensitive to NO2 concentration, and since the NO2 measurement suffered with ambient NOy interference, we did not apply the steady-state lifetime method in this study (Brown et al., 2003). Nonetheless, the derived γ in Beijing showed good agreement with the recent results derived with the steady-state method in Jinan and Mt. Tai (X. F. Wang et al., 2017; Z. Wang et al., 2017). The consistency eliminates the discrepancy possibly brought about by the differences in analysis methods. Therefore, we suggest that fast N2O5 uptake was a ubiquitous feature that existed in the NCP. In this study, sulfate is the dominant component of PM1.0, accounting for more than 30 % of its mass concentration, which may be the reason for the elevated N2O5 uptake coefficient present in Beijing, like the results for high sulfate air mass over Ohio and western Pennsylvania (Brown et al., 2006). Previous studies have shown that the N2O5 uptake coefficient strongly depends on the liquid water content, p, and organic mass. Liquid water content promotes N2O5 uptake, whereas p and organic mass inhibit N2O5 uptake (e.g., Thornton et al., 2003; Wahner et al., 1998; Bertram and Thornton, 2009). Because of the limited data set of N2O5 uptake coefficients in this work, the function dependence studies on the determined N2O5 uptake coefficients with the parameters mentioned above were not convincing. More valid data are needed in the further studies of the N2O5 uptake mechanism. With respect to f, the values are comparable to those observed in Germany (Phillips et al., 2016) and are similar to those estimated in the power plant plume in Mt. Tai with high chloride content (Z. Wang et al., 2017).
4.2 N2O5 lifetime and reactivity
The lifetime of N2O5 was estimated using the steady-state method, assuming that the production and loss of N2O5 was in balance after a period following sunset. Equation (4) for the steady-state approximation has been frequently applied in analyzing the fate of N2O5 (Platt et al., 1980; Allan et al., 1999; Brown et al., 2003).
Here τss(N2O5) denotes the steady-state lifetime of N2O5 and Lss(N2O5) denotes the loss term of N2O5 corresponding to the steady-state lifetime. A numerical model was used to check the validity of the steady-state approximation (Brown et al., 2003); details are given in Fig. S3. The results show that the steady state can generally be achieved within 30 min. In this study, the steady-state lifetime was only calculated from 20:00 to the next day at 04:00 CNST. The time periods with a NO concentration larger than 0.06 ppbv (instrument LOD) were excluded as the steady state is easily disturbed. The overall N2O5 loss rate (k(N2O5)) can be calculated by accumulating each individual loss term in Eq. (5), including the N2O5 heterogeneous hydrolysis and the reaction of NO3 with VOCs.
The NO3 heterogeneous uptake and the loss of N2O5 via gas-phase reactions were assumed to be negligible (Brown and Stutz, 2012). denotes the reaction rate constants of the reaction of NO3 + VOCsi. Isoprene and monoterpene were used in this calculation.
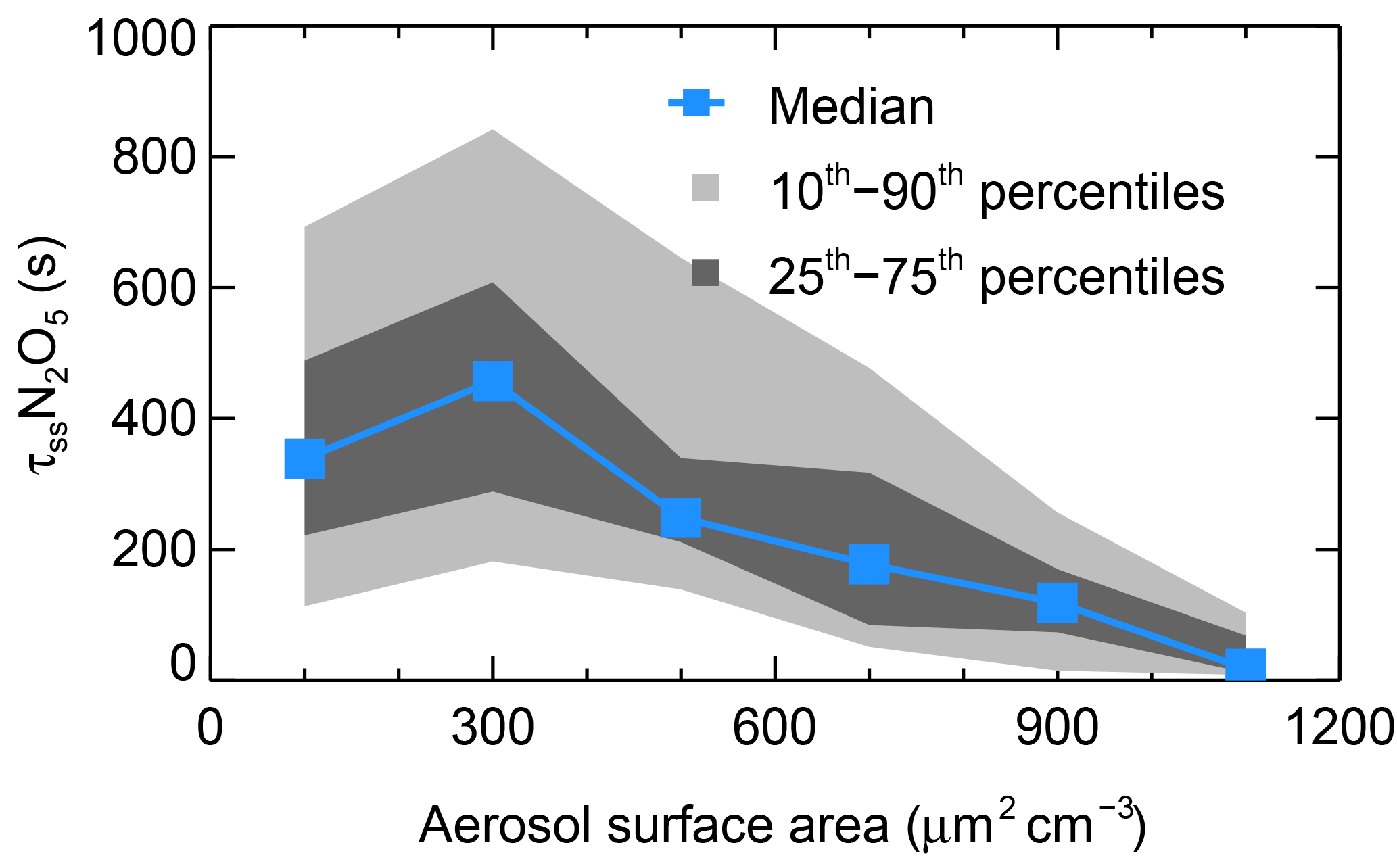
Figure 6The dependence of N2O5 lifetime on aerosol surface area. Data were selected from 20:00 to 04:00 CNST and are shown as medians, 25–75th percentile ranges, and 10–90th percentile ranges, as shown in the legend.
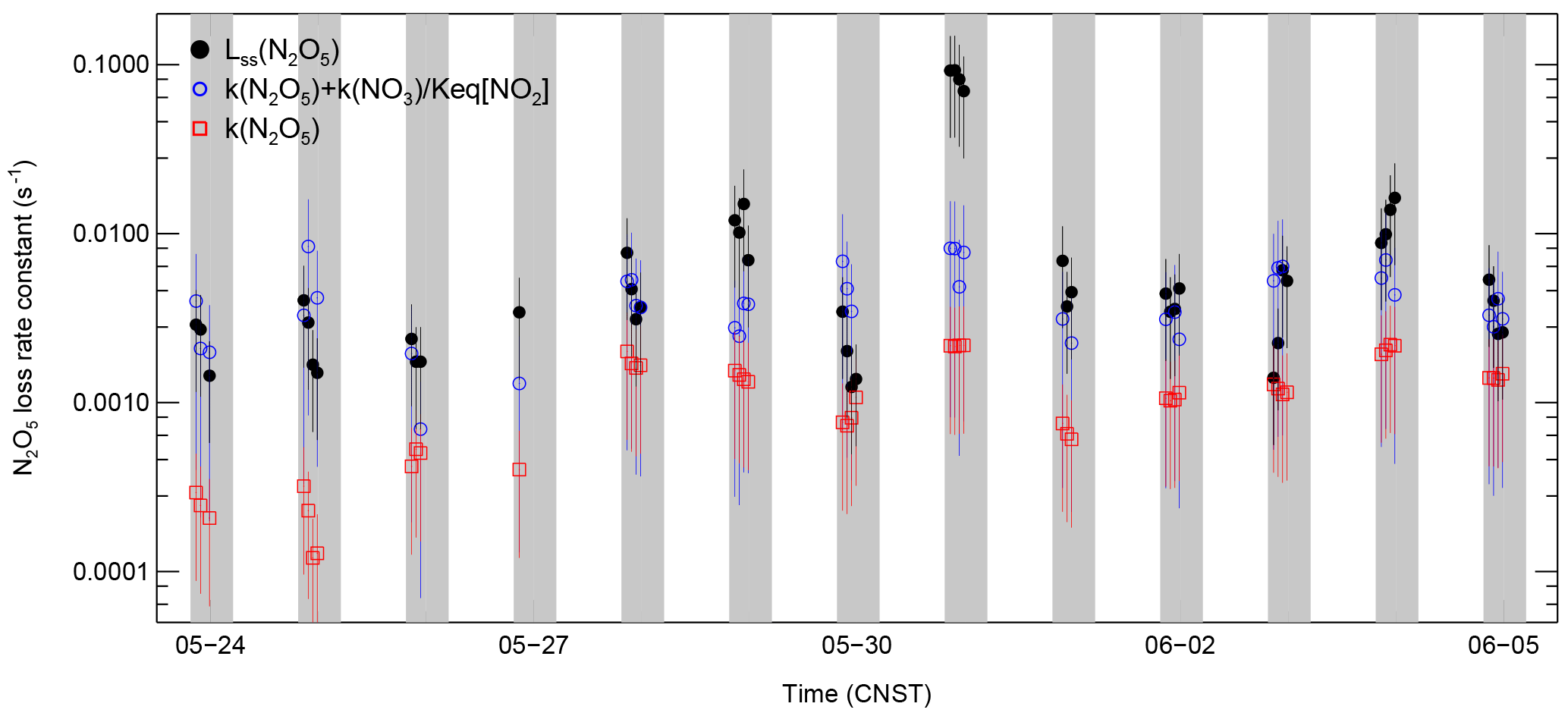
Figure 7Time series of the individual N2O5 loss terms and the loss rate constant of N2O5 in steady state (Lss(N2O5)).
The N2O5 loss rate coefficient by heterogeneous hydrolysis was calculated by using an average γ of 0.034. The time series of the steady-state lifetime of N2O5 is shown in Fig. S4. The N2O5 steady-state lifetime ranged from < 5 to 1260 s, with an average of 270 ± 240 s, and large variability was shown during the campaign. The N2O5 lifetimes during the BAM period were higher than those during the UAM period, which is predictable since the clean air mass has lower N2O5 reactivity because of much lower aerosol loading. Two extremely short N2O5 lifetime cases were captured on the nights of 30 May and 3 June, with peak values below 200 s throughout those nights. Figure 6 shows that the N2O5 lifetime had a very clear negative dependence on the ambient Sa when larger than 300 µm2 cm−3, which indicates that the N2O5 heterogeneous uptake plays an important role in the regulation of N2O5 lifetime. The study conducted in the residual layer of Hong Kong showed a similar tendency despite the overall N2O5 lifetime being shorter at this site (Brown et al., 2016). Additionally, a negative dependence of N2O5 lifetime on RH was reported in Hong Kong but was not observed in this study (Fig. S5).
Figure 7 shows the time series of the overall N2O5 loss rate constant as well as the N2O5 steady-state loss rate constant. The overall N2O5 loss rate constant was calculated from the individual terms (Eq. 3). The uncertainties of the N2O5 steady-state loss rate constant and the overall k(N2O5) are estimated to be 67 and 95 %, respectively (Eqs. 6 and 7). The largest error sources were from the corrected NO2 measurements.
On the night of 29 May, the steady-state loss rate constant was much lower than the overall k(N2O5); on the nights of 28 May and 3 June, the Lss(N2O5) values calculated with the steady-state method were much higher than the overall k(N2O5), but these discrepancies were in the range of the uncertainties. The steady-state loss rate constant in the case of 30 May was approximately 10 times larger than the overall loss rate constant, and this difference was outside of the range of uncertainty. The reason for the larger difference on this night is not understood from the available measurements. In general, the overall N2O5 loss rate constant and the steady-state N2O5 loss rate constant were comparable, taking into consideration the uncertainties. The average N2O5 loss rate constant contributed by the N2O5 heterogeneous hydrolysis was s−1. The average NO3 loss rate constant by the reaction of NO3 with VOCs was 0.015 ± 0.007 s−1, which is comparable with the previous results in suburban Beijing in 2006 (H. C. Wang et al., 2017b), in which the contribution to the N2O5 reactivity was ± s−1 on average. Compared with N2O5 loss via direct heterogeneous hydrolysis, the indirect loss via NO3 + VOCs was dominant, accounting for approximately 67 %. Because only a subset of the suite of organic species at the site was measured, the calculated loss rate constant via NO3 + VOCs represents a lower limit. Therefore, the N2O5 loss via NO3 + VOCs may occupy a larger proportion. The overall loss rate constant from NO3 + VOCs and N2O5 uptake was ± s−1 on average, which was reasonably lower than the steady-state N2O5 loss rate constant of ± s−1 on average. The gap may be explained by the unmeasured reactive VOCs or the unaccounted for NO that was near the instrumental limit of detection.
4.3 NO3-induced nocturnal oxidation of VOCs
Recent studies have suggested that the fate of BVOCs after sunset is dominated by NOx or O3, with variation in the ratio of NOx to BVOCs and that the nighttime oxidation is located in the transition region between NOx domination and O3 domination in the United States (Edwards et al., 2017). During this campaign, the nocturnal average concentrations of isoprene and monoterpene were 156 ± 88 and 86 ± 42 pptv, respectively. We used isoprene and monoterpene to represent a lower limit mixing ratio of total BVOCs; the average ratio of NOx ∕ BVOCs was larger than 10 and exhibited small variation during the BAM and UAM periods. The value was much higher than the critical value (NOx ∕ BVOC = 0.5) of the transition regime proposed by Edwards et al. (2017), which suggests that the oxidation of BVOCs in Beijing was NOx dominated and the nighttime fate of BVOCs was controlled by NO3. Since the reaction of NO3 with BVOCs has a high mass yield, the nocturnal ON production may be important in the high NOx ∕ BVOC region.
The first-order loss rate of VOCs initialized by oxidants, k(VOCsi), is defined as VOC reactivity and expressed as Eq. (8). Here, we only consider the reaction of VOCs with O3 and NO3. denotes the reaction rate constants of VOCsi with O3.
During this campaign, VOC reactivity could be determined with the measured O3 and calculated NO3. Figure 8 depicts four kinds of VOC reactivity distribution during nighttime, including the isoprene (ISO), monoterpene (MNT), double bond at the end or terminal position of the molecule (OLT), and alkenes with the double bond elsewhere in the molecule (OLI). The reaction rates were cited from the Regional Atmospheric Chemistry Mechanism version 2 (RACM2; Goliff et al., 2013). Previous measurement indicated the main detectable monoterpenes were α-pinene and β-pinene in summer in Beijing (Ying Liu, personal communication, 2018). Here we assumed α-pinene and β-pinene occupy half and half in the monoterpene with an uncertainty of 50 %. The rate coefficients of α-pinene and β-pinene with NO3 were referred to in Atkinson and Arey (2003). The uncertainty of the calculated mixing ratio of NO3 is 67 %, and the overall uncertainty of monoterpene reactivity was calculated to be 85 % with Gaussian propagation. The uncertainties of other kinds of VOCs was calculated to be 75 % by assuming the uncertainty of rate coefficient was 30 %. The VOC reactivity was dominated by NO3 oxidation and contributed up to 90 % in total; less than 10 % of VOCs were oxidized by O3 during the nighttime. Even though the NO3 concentration is in the lower range, NO3 is still responsible for more than 70 % nocturnal BVOC oxidation, and the results further confirmed that the oxidation of BVOCs is controlled by NO3 rather than O3 in summer in Beijing.
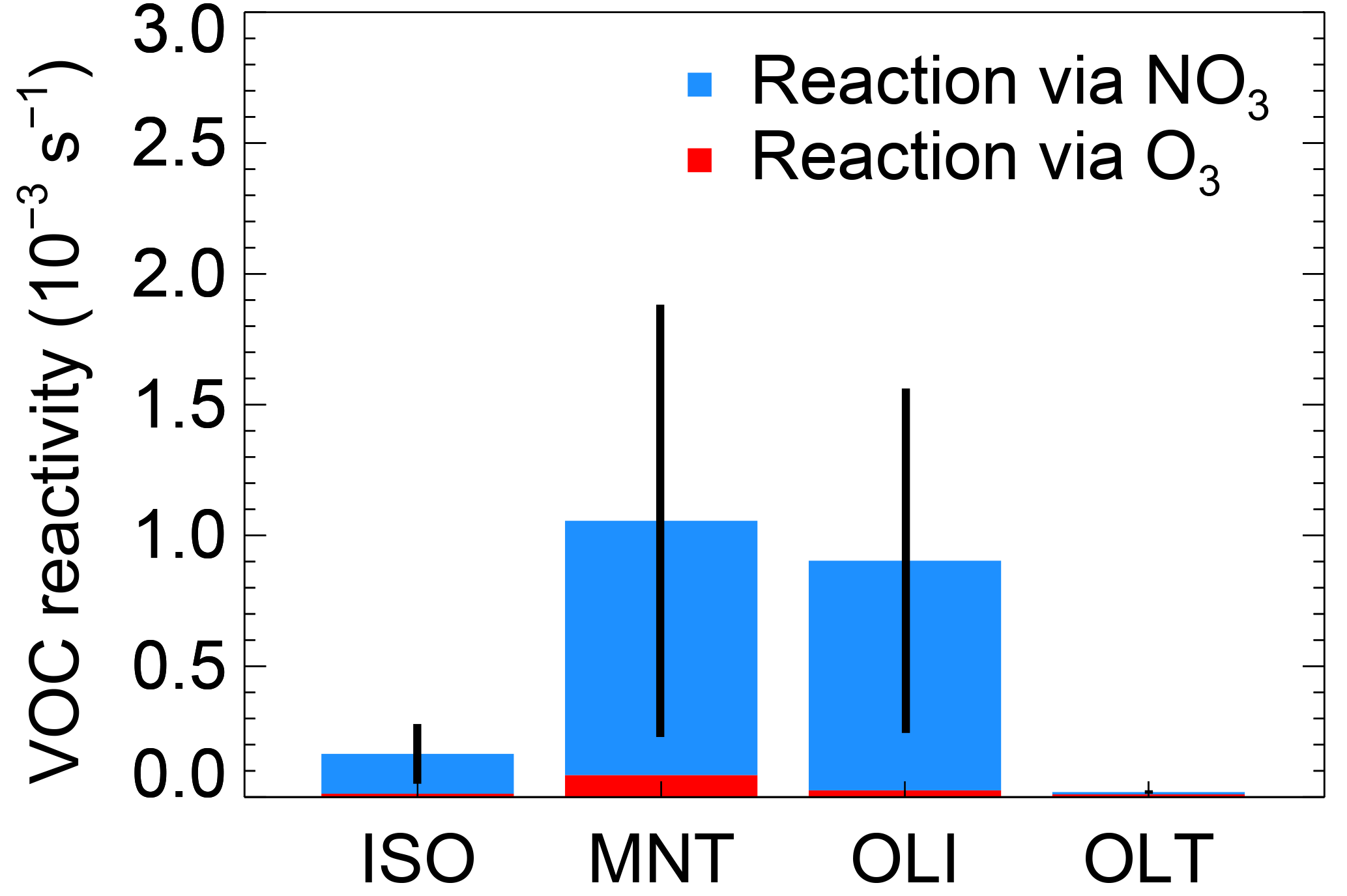
Figure 8The nighttime VOC reactivity of NO3 and O3 (defined as the first-order loss rate of VOCs initialized by oxidants, including NO3 and O3); the VOCs are classified as isoprene (ISO), monoterpene (MNT), terminal alkenes (OLT), and internal alkenes (OLI). The data were selected from 20:00 to the next day at 04:00 CNST.
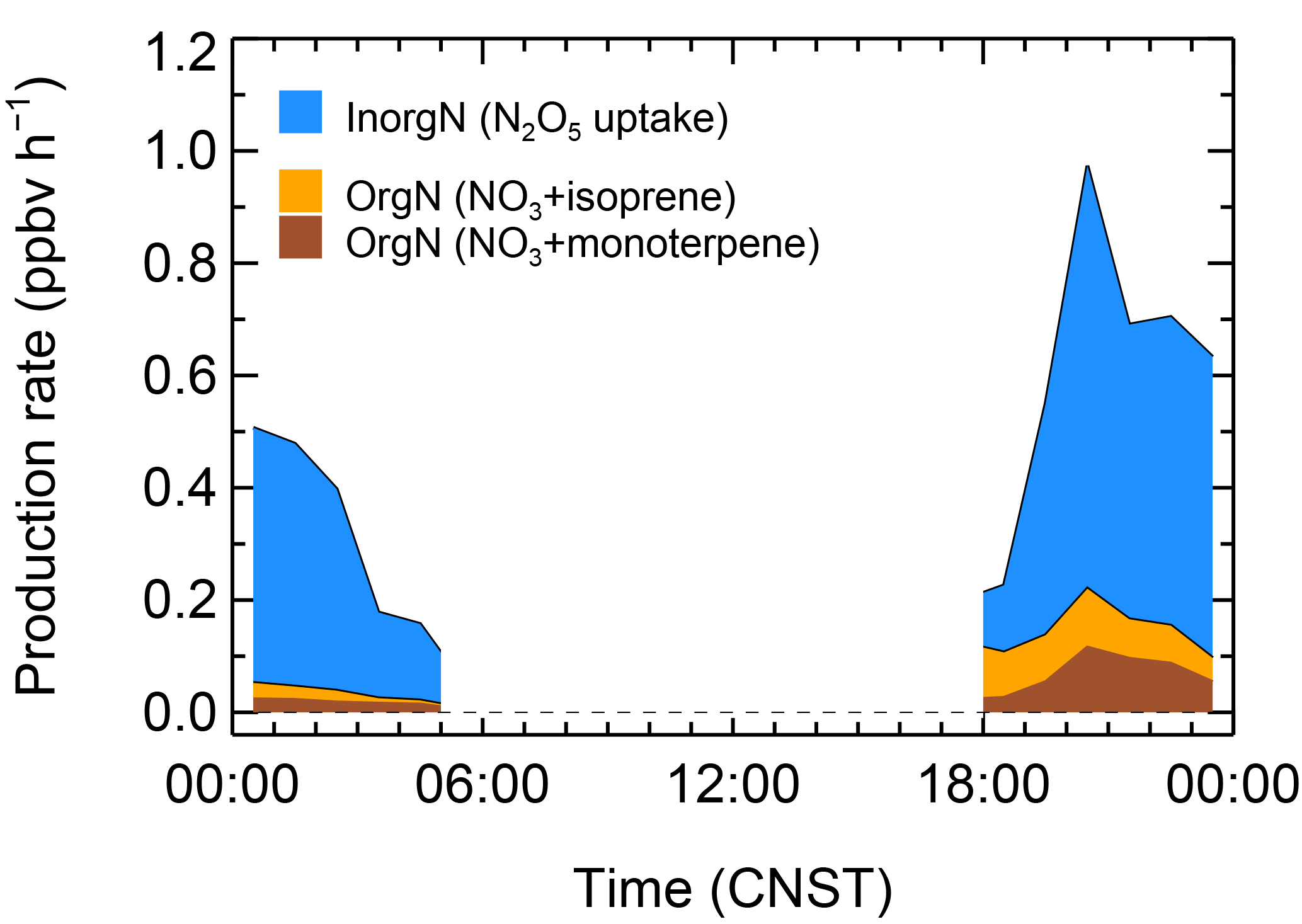
Figure 9The nighttime production rate of organic and inorganic nitrates; the inorganic nitrates were calculated from N2O5 heterogeneous hydrolysis, and ONs were calculated with the NO3 reacted with isoprene and monoterpene.
For calculating nocturnal ON production from NO3 oxidation of isoprene and monoterpene, as well as inorganic nitrate production via N2O5 heterogeneous uptake over the same period, the ClNO2 yield was set to the determined average value of 0.55. The organic nitrate yield of the reaction of NO3 with isoprene was set to 0.7, from Rollins et al. (2009). The yield from the reaction of NO3 with monoterpene was represented by NO3 + α-pinene and was set to 0.16, following Spittler et al. (2006). As α-pinene and β-pinene have very different ON yields, the yield set in the study was an upper limit for α-pinene-initialized ON, but has a relatively low yield for the β-pinene-initialized ON (e.g., Hallquist et al., 1999). Although the yield from the NO3 oxidation of isoprene is much higher than that of monoterpene, the total ON production was dominated by the oxidation of NO3 with monoterpene because the reaction of NO3 with monoterpene is much faster than that with isoprene. Because of the lack of measurement of alkenes and other VOCs that can react with NO3 and form ON, the calculated nighttime ON production rate analyzed here served as a lower estimation.
Figure 9 depicts the mean diurnal profiles of the nocturnal formation rates of inorganic nitrates and ON. The average production rate of ON was up to 0.10 ± 0.07 ppbv h−1, which was higher than that predicted at a suburban site in Beijing in 2006, with an average value of 0.06 ppbv h−1 (H. C. Wang et al., 2017c). In the high NOx ∕ BVOC air masses, the inorganic nitrate formation was proposed to increase with the increase in sunset NOx ∕ BVOC (Edwards et al., 2017). The formation rate of inorganic nitrate via N2O5 uptake was significant, with an average of 0.43 ± 0.12 ppbv h−1, and was much larger than the ON formation. NOx was mainly removed as the inorganic nitrate format by nocturnal NO3–N2O5 chemistry in Beijing. Overall, the NO3–N2O5 chemistry led to significant NOx removal, with 0.54 ppbv h−1 accounted for by the organic and inorganic nitrates, and the integral NOx removal was approximately 5 ppbv per night. Since ONs are important precursors of the SOAs, NO3 oxidation was very important from the perspective of organic aerosol formation and regional particulate matter (e.g., Ng et al., 2008).
We reported an intensive field study of NO3–N2O5 chemistry at a downwind suburban site in Beijing during the summer of 2016. High levels of ClNO2 and N2O5 were observed, with maxima of 2.9 and 937 pptv (1 min), respectively. The N2O5 uptake coefficient was estimated to be in the range of 0.010–0.055, with an average value of 0.034 ± 0.018, and the corresponding ClNO2 yield was derived to be in the range of 0.5–1.0, with an average value of 0.73 ± 0.25. The elevated ClNO2 levels and ClNO2 ∕ N2O5 ratios are comparable with those in chloride-rich regions in the NCP. The results highlight fast N2O5 heterogeneous hydrolysis and efficient ClNO2 formation in the outflow of urban Beijing.
Since the NO3–N2O5 chemical equilibrium favors NO3 in summer with high temperature, the elevated NO3 dominated the nocturnal degradation of BVOCs and could lead to efficient ON formation. Because the air masses in Beijing featured high NOx ∕ BVOC ratios (> 10), our results suggest that the nocturnal NO3 oxidation of BVOCs was NOx dominated. Because of the extremely high NOx emissions, the formation of ON may not be sensitive to the reduction of NOx but rather to the change of unsaturated VOCs (e.g., BVOCs), which is similar to the daytime photochemical O3 pollution (e.g., Lu et al., 2010) diagnosed for this area. This suggests that control of the unsaturated VOCs would moderate O3 pollution and ON particulate matter in parallel. Moreover, reduction of NOx would also be helpful in reducing the p formation via N2O5 heterogeneous hydrolysis under such high NOx ∕ BVOC ratios (Edwards et al., 2017).
The observational data and meteorological parameters used in this study are available from the corresponding authors upon request (k.lu@pku.edu.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-9705-2018-supplement.
SG, MiH, and MaH organized the field campaign. KL and HW designed the experiments on N2O5 chemistry. HW and KL analyzed the data. HW wrote the manuscript with input from KL. All authors contributed to measurements, discussing results, and commenting on the manuscript.
The authors declare that they have no conflict of interest.
This work was supported by the National Natural Science Foundation of
China (grant nos. 91544225, 41375124, 21522701, 41421064, 91744204), the
National Science and Technology Support Program of China (no. 2014BAC21B01),
the Strategic Priority Research Program of the Chinese Academy of Sciences
(grant no. XDB05010500), and the program on “Photochemical smog in China”
financed by the Swedish Research Council (639-2013-6917). The authors
gratefully acknowledge the Peking University and Gothenburg University
science team for their technical support and discussions during the
Changping campaign.
Edited by: Steven Brown
Reviewed by: three anonymous referees
Allan, B. J., Carslaw, N., Coe, H., Burgess, R. A., and Plane, J. M. C.: Observations of the nitrate radical in the marine boundary layer, J. Atmos. Chem., 33, 129–154, https://doi.org/10.1023/A:1005917203307, 1999.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Bannan, T. J., Booth, A. M., Bacak, A., Muller, J. B. A., Leather, K. E., Le Breton, M., Jones, B., Young, D., Coe, H., Allan, J., Visser, S., Slowik, J. G., Furger, M., Prevot, A. S. H., Lee, J., Dunmore, R. E., Hopkins, J. R., Hamilton, J. F., Lewis, A. C., Whalley, L. K., Sharp, T., Stone, D., Heard, D. E., Fleming, Z. L., Leigh, R., Shallcross, D. E., and Percival, C. J.: The first UK measurements of nitryl chloride using a chemical ionization mass spectrometer in central London in the summer of 2012, and an investigation of the role of Cl atom oxidation, J. Geophys. Res.-Atmos., 120, 5638–5657, https://doi.org/10.1002/2014jd022629, 2015.
Behnke, W., George, C., Scheer, V., and Zetzsch, C.: Production and decay of ClNO2, from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments, J. Geophys. Res.-Atmos., 102, 3795–3804, https://doi.org/10.1029/96jd03057, 1997.
Benton, A. K., Langridge, J. M., Ball, S. M., Bloss, W. J., Dall'Osto, M., Nemitz, E., Harrison, R. M., and Jones, R. L.: Night-time chemistry above London: measurements of NO3 and N2O5 from the BT Tower, Atmos. Chem. Phys., 10, 9781–9795, https://doi.org/10.5194/acp-10-9781-2010, 2010.
Bertram, T. H. and Thornton, J. A.: Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride, Atmos. Chem. Phys., 9, 8351–8363, https://doi.org/10.5194/acp-9-8351-2009, 2009.
Bertram, T. H., Thornton, J. A., Riedel, T. P., Middlebrook, A. M., Bahreini, R., Bates, T. S., Quinn, P. K., and Coffman, D. J.: Direct observations of N2O5 reactivity on ambient aerosol particles, Geophys. Res. Lett., 36, L19803, https://doi.org/10.1029/2009gl040248, 2009.
Bohn, B., Corlett, G. K., Gillmann, M., Sanghavi, S., Stange, G., Tensing, E., Vrekoussis, M., Bloss, W. J., Clapp, L. J., Kortner, M., Dorn, H.-P., Monks, P. S., Platt, U., Plass-Dülmer, C., Mihalopoulos, N., Heard, D. E., Clemitshaw, K. C., Meixner, F. X., Prevot, A. S. H., and Schmitt, R.: Photolysis frequency measurement techniques: results of a comparison within the ACCENT project, Atmos. Chem. Phys., 8, 5373–5391, https://doi.org/10.5194/acp-8-5373-2008, 2008.
Boyd, C. M., Nah, T., Xu, L., Berkemeier, T., and Ng, N. L.: Secondary Organic Aerosol (SOA) from Nitrate Radical Oxidation of Monoterpenes: Effects of Temperature, Dilution, and Humidity on Aerosol Formation, Mixing, and Evaporation, Environ. Sci. Technol., 51, 7831–7841, 2017.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2cs35181a, 2012.
Brown, S. S., Stark, H., and Ravishankara, A. R.: Applicability of the steady state approximation to the interpretation of atmospheric observations of NO3 and N2O5, J. Geophys. Res.-Atmos., 108, 4539, https://doi.org/10.1029/2003jd003407, 2003.
Brown, S. S., Ryerson, T. B., Wollny, A. G., Brock, C. A., Peltier, R., Sullivan, A. P., Weber, R. J., Dube, W. P., Trainer, M., Meagher, J. F., Fehsenfeld, F. C., and Ravishankara, A. R.: Variability in nocturnal nitrogen oxide processing and its role in regional air quality, Science, 311, 67–70, https://doi.org/10.1126/science.1120120, 2006.
Brown, S. S., Dube, W. P., Fuchs, H., Ryerson, T. B., Wollny, A. G., Brock, C. A., Bahreini, R., Middlebrook, A. M., Neuman, J. A., Atlas, E., Roberts, J. M., Osthoff, H. D., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: Comparison to current model parameterizations, J. Geophys. Res.-Atmos., 114, D00f10, https://doi.org/10.1029/2008jd011679, 2009.
Brown, S. S., Dube, W. P., Tham, Y. J., Zha, Q. Z., Xue, L. K., Poon, S., Wang, Z., Blake, D. R., Tsui, W., Parrish, D. D., and Wang, T.: Nighttime chemistry at a high altitude site above Hong Kong, J. Geophys. Res.-Atmos., 121, 2457–2475, https://doi.org/10.1002/2015jd024566, 2016.
Chang, W. L., Bhave, P. V., Brown, S. S., Riemer, N., Stutz, J., and Dabdub, D.: Heterogeneous Atmospheric Chemistry, Ambient Measurements, and Model Calculations of N2O5: A Review, Aerosol Sci. Tech., 45, 665–695, 2011.
DeCarlo, P. F., Kimmel, J., Trimborn, A., Northway, M., Jayne, J. T., Aiken, A., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K., Worsnop, D. R., and Jimenez, J. L.: Field-deployable, high-resolution, time-of-flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, 2006.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transferreaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, 2007.
Draxler, R. R. and Rolph, G. D.: HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Tracker) Model access via NOAA ARL Ready Website, available at: http://www.arl.noaa.gov/ready/hysplit4.html (last access: 17 March 2017), NOAA Air Resources Laboratory, Silver Spring, MD, 2003.
Edwards, P. M., Aikin, K. C., Dube, W. P., Fry, J. L., Gilman, J. B., de Gouw, J. A., Graus, M. G., Hanisco, T. F., Holloway, J., Huber, G., Kaiser, J., Keutsch, F. N., Lerner, B. M., Neuman, J. A., Parrish, D. D., Peischl, J., Pollack, I. B., Ravishankara, A. R., Roberts, J. M., Ryerson, T. B., Trainer, M., Veres, P. R., Wolfe, G. M., Warneke, C., and Brown, S. S.: Transition from high- to low-NOx control of night-time oxidation in the southeastern US, Nat. Geosci., 10, 490, https://doi.org/10.1038/Ngeo2976, 2017.
Faxon, C. B., Bean, J. K., and Ruiz, L. H.: Inland Concentrations of Cl2 and ClNO2 in Southeast Texas suggest chlorine chemistry significantly contributes to atmospheric reactivity, Atmosphere, 6, 1487–1506, 2015.
Finlayson-Pitts, B. J., Ezell, M. J., and Pitts, J. N.: Formation of Chemically Active Chlorine Compounds by Reactions of Atmospheric Nacl Particles with Gaseous N2O5 and ClONO2, Nature, 337, 241–244, https://doi.org/10.1038/337241a0, 1989.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., dal Maso, M., Tillmann, R., Dorn, H.-P., Brauers, T., and Cohen, R. C.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Gaston, C. J., Thornton, J. A., and Ng, N. L.: Reactive uptake of N2O5 to internally mixed inorganic and organic particles: the role of organic carbon oxidation state and inferred organic phase separations, Atmos. Chem. Phys., 14, 5693–5707, https://doi.org/10.5194/acp-14-5693-2014, 2014.
Geyer, A., Alicke, B., Konrad, S., Schmitz, T., Stutz, J., and Platt, U.: Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.-Atmos., 106, 8013–8025, https://doi.org/10.1029/2000jd900681, 2001.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, 2013.
Gržinić, G., Bartels-Rausch, T., Berkemeier, T., Türler, A., and Ammann, M.: Viscosity controls humidity dependence of N2O5 uptake to citric acid aerosol, Atmos. Chem. Phys., 15, 13615–13625, https://doi.org/10.5194/acp-15-13615-2015, 2015.
Hallquist, M., Wangberg, I., Ljungstrom, E., Barnes, I., and Becker, K. H.: Aerosol and product yields from NO3 radical-initiated oxidation of selected monoterpenes, Environ. Sci. Technol., 33, 553–559, https://doi.org/10.1021/Es980292s, 1999.
Hallquist, M., Stewart, D. J., Stephenson, S. K., and Cox, R. A.: Hydrolysis of N2O5 on sub-micron sulfate aerosols, Phys. Chem. Chem. Phys., 5, 3453–3463, https://doi.org/10.1039/B301827j, 2003.
Hallquist, M., Munthe, J., Hu, M., Wang, T., Chan, C. K., Gao, J., Boman, J., Guo, S., Hallquist, A. M., Mellqvist, J., Moldanova, J., Pathak, R. K., Pettersson, J. B. C., Pleijel, H., Simpson, D., and Thynell, M.: Photochemical smog in China: scientific challenges and implications for air-quality policies, Natl. Sci. Rev., 3, 401–403, https://doi.org/10.1093/nsr/nww080, 2016.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Aijala, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, 2016.
Le Breton, M., Bacak, A., Muller, J. B. A., Bannan, T. J., Kennedy, O., Ouyang, B., Xiao, P., Bauguitte, S. J. B., Shallcross, D. E., Jones, R. L., Daniels, M. J. S., Ball, S. M., and Percival, C. J.: The first airborne comparison of N2O5 measurements over the UK using a CIMS and BBCEAS during the RONOCO campaign, Anal. Methods-UK, 6, 9731–9743, https://doi.org/10.1039/c4ay02273d, 2014.
Le Breton, M., Hallquist, Å. M., Pathak, R. K., Simpson, D., Wang, Y., Johansson, J., Zheng, J., Yang, Y., Shang, D., Wang, H., Liu, Q., Chan, C., Wang, T., Bannan, T. J., Priestley, M., Percival, C. J., Shallcross, D. E., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Chlorine oxidation of VOCs at a semi-rural site in Beijing: Significant chlorine liberation from ClNO2 and subsequent gas and particle phase Cl-VOC production, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2018-9, in review, 2018.
Li, Q., Zhang, L., Wang, T., Tham, Y. J., Ahmadov, R., Xue, L., Zhang, Q., and Zheng, J.: Impacts of heterogeneous uptake of dinitrogen pentoxide and chlorine activation on ozone and reactive nitrogen partitioning: improvement and application of the WRF-Chem model in southern China, Atmos. Chem. Phys., 16, 14875–14890, https://doi.org/10.5194/acp-16-14875-2016, 2016.
Li, S. W., Liu, W. Q., Xie, P. H., Qin, M., and Yang, Y. J.: Observation of Nitrate Radical in the Nocturnal Boundary Layer During a Summer Field Campaign in Pearl River Delta, China, Terr. Atmos. Ocean Sci., 23, 39–48, https://doi.org/10.3319/Tao.2011.07.26.01(a), 2012.
Liu, M. X., Song, Y., Zhou, T., Xu, Z. Y., Yan, C. Q., Zheng, M., Wu, Z. J., Hu, M., Wu, Y. S., and Zhu, T.: Fine particle pH during severe haze episodes in northern China, Geophys. Res. Lett., 44, 5213–5221, https://doi.org/10.1002/2017gl073210, 2017.
Liu, X. G., Gu, J. W., Li, Y. P., Cheng, Y. F., Qu, Y., Han, T. T., Wang, J. L., Tian, H. Z., Chen, J., and Zhang, Y. H.: Increase of aerosol scattering by hygroscopic growth: Observation, modeling, and implications on visibility, Atmos. Res., 132, 91–101, https://doi.org/10.1016/j.atmosres.2013.04.007, 2013.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lu, K. D., Zhang, Y. H., Su, H., Brauers, T., Chou, C. C., Hofzumahaus, A., Liu, S. C., Kita, K., Kondo, Y., Shao, M., Wahner, A., Wang, J. L., Wang, X. S., and Zhu, T.: Oxidant (O3 + NO2) production processes and formation regimes in Beijing, J. Geophys. Res.-Atmos., 115, D07303, https://doi.org/10.1029/2009JD012714, 2010.
McLaren, R., Wojtal, P., Majonis, D., McCourt, J., Halla, J. D., and Brook, J.: NO3 radical measurements in a polluted marine environment: links to ozone formation, Atmos. Chem. Phys., 10, 4187–4206, https://doi.org/10.5194/acp-10-4187-2010, 2010.
Mentel, T. F., Sohn, M., and Wahner, A.: Nitrate effect in the heterogeneous hydrolysis of dinitrogen pentoxide on aqueous aerosols, Phys. Chem. Chem. Phys., 1, 5451–5457, https://doi.org/10.1039/A905338g, 1999.
Mielke, L. H., Stutz, J., Tsai, C., Hurlock, S. C., Roberts, J. M., Veres, P. R., Froyd, K. D., Hayes, P. L., Cubison, M. J., Jimenez, J. L., Washenfelder, R. A., Young, C. J., Gilman, J. B., de Gouw, J. A., Flynn, J. H., Grossberg, N., Lefer, B. L., Liu, J., Weber, R. J., and Osthoff, H. D.: Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010, J. Geophys. Res.-Atmos., 118, 10638–10652, https://doi.org/10.1002/Jgrd.50783, 2013.
Morgan, W. T., Ouyang, B., Allan, J. D., Aruffo, E., Di Carlo, P., Kennedy, O. J., Lowe, D., Flynn, M. J., Rosenberg, P. D., Williams, P. I., Jones, R., McFiggans, G. B., and Coe, H.: Influence of aerosol chemical composition on N2O5 uptake: airborne regional measurements in northwestern Europe, Atmos. Chem. Phys., 15, 973–990, https://doi.org/10.5194/acp-15-973-2015, 2015.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, https://doi.org/10.1038/Ngeo177, 2008.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012gl051912, 2012.
Phillips, G. J., Thieser, J., Tang, M., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Platt, U., Perner, D., Winer, A. M., Harris, G. W., and Pitts, J. N.: Detection of NO3 in the Polluted Troposphere by Differential Optical-Absorption, Geophys. Res. Lett., 7, 89–92, https://doi.org/10.1029/Gl007i001p00089, 1980.
Pye, H. O. T., Chan, A. W. H., Barkley, M. P., and Seinfeld, J. H.: Global modeling of organic aerosol: the importance of reactive nitrogen (NOx and NO3), Atmos. Chem. Phys., 10, 11261–11276, https://doi.org/10.5194/acp-10-11261-2010, 2010.
Riedel, T. P., Bertram, T. H., Ryder, O. S., Liu, S., Day, D. A., Russell, L. M., Gaston, C. J., Prather, K. A., and Thornton, J. A.: Direct N2O5 reactivity measurements at a polluted coastal site, Atmos. Chem. Phys., 12, 2959–2968, https://doi.org/10.5194/acp-12-2959-2012, 2012.
Riedel, T. P., Wolfe, G. M., Danas, K. T., Gilman, J. B., Kuster, W. C., Bon, D. M., Vlasenko, A., Li, S.-M., Williams, E. J., Lerner, B. M., Veres, P. R., Roberts, J. M., Holloway, J. S., Lefer, B., Brown, S. S., and Thornton, J. A.: An MCM modeling study of nitryl chloride (ClNO2) impacts on oxidation, ozone production and nitrogen oxide partitioning in polluted continental outflow, Atmos. Chem. Phys., 14, 3789–3800, https://doi.org/10.5194/acp-14-3789-2014, 2014.
Riemer, N., Vogel, H., Vogel, B., Anttila, T., Kiendler-Scharr, A., and Mentel, T. F.: Relative importance of organic coatings for the heterogeneous hydrolysis of N2O5 during summer in Europe, J. Geophys. Res.-Atmos., 114, D17307, https://doi.org/10.1029/2008JD011369, 2009.
Rollins, A. W., Kiendler-Scharr, A., Fry, J. L., Brauers, T., Brown, S. S., Dorn, H.-P., Dubé, W. P., Fuchs, H., Mensah, A., Mentel, T. F., Rohrer, F., Tillmann, R., Wegener, R., Wooldridge, P. J., and Cohen, R. C.: Isoprene oxidation by nitrate radical: alkyl nitrate and secondary organic aerosol yields, Atmos. Chem. Phys., 9, 6685–6703, https://doi.org/10.5194/acp-9-6685-2009, 2009.
Sarwar, G., Simon, H., Xing, J., and Mathur, R.: Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere, Geophys. Res. Lett., 41, 4050–4058, https://doi.org/10.1002/2014gl059962, 2014.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, T., and Wirtz, K.: Reactions of NO3 radicals with limonene and alpha-pinene: Product and SOA formation, Atmos. Environ., 40, S116–S127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Stutz, J., Wong, K. W., Lawrence, L., Ziemba, L., Flynn, J. H., Rappengluck, B., and Lefer, B.: Nocturnal NO3 radical chemistry in Houston, TX, Atmos. Environ., 44, 4099–4106, https://doi.org/10.1016/j.atmosenv.2009.03.004, 2010.
Su, X., Tie, X. X., Li, G. H., Cao, J. J., Huang, R. J., Feng, T., Long, X., and Xu, R. G.: Effect of hydrolysis of N2O5 on nitrate and ammonium formation in Beijing China: WRF-Chem model simulation, Sci. Total. Environ., 579, 221–229, https://doi.org/10.1016/j.scitotenv.2016.11.125, 2017.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tang, M., Huang, X., Lu, K., Ge, M., Li, Y., Cheng, P., Zhu, T., Ding, A., Zhang, Y., Gligorovski, S., Song, W., Ding, X., Bi, X., and Wang, X.: Heterogeneous reactions of mineral dust aerosol: implications for tropospheric oxidation capacity, Atmos. Chem. Phys., 17, 11727–11777, https://doi.org/10.5194/acp-17-11727-2017, 2017.
Tang, M. J., Thieser, J., Schuster, G., and Crowley, J. N.: Kinetics and mechanism of the heterogeneous reaction of N2O5 with mineral dust particles, Phys. Chem. Chem. Phys., 14, 8551–8561, 2012.
Tang, M. J., Schuster, G., and Crowley, J. N.: Heterogeneous reaction of N2O5 with illite and Arizona test dust particles, Atmos. Chem. Phys., 14, 245–254, https://doi.org/10.5194/acp-14-245-2014, 2014.
Tham, Y. J., Wang, Z., Li, Q., Yun, H., Wang, W., Wang, X., Xue, L., Lu, K., Ma, N., Bohn, B., Li, X., Kecorius, S., Größ, J., Shao, M., Wiedensohler, A., Zhang, Y., and Wang, T.: Significant concentrations of nitryl chloride sustained in the morning: investigations of the causes and impacts on ozone production in a polluted region of northern China, Atmos. Chem. Phys., 16, 14959–14977, https://doi.org/10.5194/acp-16-14959-2016, 2016.
Thornton, J. A. and Abbatt, J. P. D.: N2O5 reaction on submicron sea salt aerosol: Kinetics, products, and the effect of surface active organics, J. Phys. Chem. A, 109, 10004–10012, https://doi.org/10.1021/Jp054183t, 2005.
Thornton, J. A., Braban, C. F., and Abbatt, J. P. D.: N2O5 hydrolysis on sub-micron organic aerosols: the effect of relative humidity, particle phase, and particle size, Phys. Chem. Chem. Phys., 5, 4593–4603, https://doi.org/10.1039/B307498f, 2003.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/Nature08905, 2010.
Wagner, N. L., Riedel, T. P., Young, C. J., Bahreini, R., Brock, C. A., Dube, W. P., Kim, S., Middlebrook, A. M., Ozturk, F., Roberts, J. M., Russo, R., Sive, B., Swarthout, R., Thornton, J. A., VandenBoer, T. C., Zhou, Y., and Brown, S. S.: N2O5 uptake coefficients and nocturnal NO2 removal rates determined from ambient wintertime measurements, J. Geophys. Res.-Atmos., 118, 9331–9350, https://doi.org/10.1002/Jgrd.50653, 2013.
Wahner, A., Mentel, T. F., and Sohn, M.: Gas-phase reaction of N2O5 with water vapor: Importance of heterogeneous hydrolysis of N2O5 and surface desorption of HNO3 in a large teflon chamber, Geophys. Res. Lett., 25, 2169–2172, https://doi.org/10.1029/98gl51596, 1998.
Wang, D., Hu, R. Z., Xie, P. H., Liu, J. G., Liu, W. Q., Qin, M., Ling, L. Y., Zeng, Y., Chen, H., Xing, X. B., Zhu, G. L., Wu, J., Duan, J., Lu, X., and Shen, L. L.: Diode laser cavity ring-down spectroscopy for in situ measurement of NO3 radical in ambient air, J. Quant. Spectrosc. Ra., 166, 23–29, https://doi.org/10.1016/j.jqsrt.2015.07.005, 2015.
Wang, H. C. and Lu, K. D.: Determination and Parameterization of the Heterogeneous Uptake Coefficient of Dinitrogen Pentoxide (N2O5), Prog. Chem., 28, 917–933, https://doi.org/10.7536/Pc151225, 2016.
Wang, H. C., Chen, J., and Lu, K.: Development of a portable cavity-enhanced absorption spectrometer for the measurement of ambient NO3 and N2O5: experimental setup, lab characterizations, and field applications in a polluted urban environment, Atmos. Meas. Tech., 10, 1465–1479, https://doi.org/10.5194/amt-10-1465-2017, 2017a.
Wang, H. C., Lu, K. D., Chen, X. R., Zhu, Q. D., Chen, Q., Guo, S., Jiang, M. Q., Li, X., Shang, D. J., and Tan, Z. F.: High N2O5 concentrations observed in urban Beijing: Implications of a large nitrate formation pathway, Environ. Sci. Technol. Lett., 10, 416–420, https://doi.org/10.1021/acs.estlett.7b00341, 2017b.
Wang, H. C., Lu, K. D., Tan, Z. F., Sun, K., Li, X., Hu, M., Shao, M., Zeng, L. M., Zhu, T., and Zhang, Y. H.: Model simulation of NO3, N2O5 and ClNO2 at a rural site in Beijing during CAREBeijing-2006, Atmos. Res., 196, 97–107, https://doi.org/10.1016/j.atmosres.2017.06.013, 2017c.
Wang, M., Shao, M., Chen, W., Yuan, B., Lu, S., Zhang, Q., Zeng, L., and Wang, Q.: A temporally and spatially resolved validation of emission inventories by measurements of ambient volatile organic compounds in Beijing, China, Atmos. Chem. Phys., 14, 5871–5891, https://doi.org/10.5194/acp-14-5871-2014, 2014.
Wang, S. S., Shi, C. Z., Zhou, B., Zhao, H., Wang, Z. R., Yang, S. N., and Chen, L. M.: Observation of NO3 radicals over Shanghai, China, Atmos. Environ., 70, 401–409, https://doi.org/10.1016/j.atmosenv.2013.01.022, 2013.
Wang, T., Tham, Y. J., Xue, L. K., Li, Q. Y., Zha, Q. Z., Wang, Z., Poon, S. C. N., Dube, W. P., Blake, D. R., Louie, P. K. K., Luk, C. W. Y., Tsui, W., and Brown, S. S.: Observations of nitryl chloride and modeling its source and effect on ozone in the planetary boundary layer of southern China, J. Geophys. Res.-Atmos., 121, 2476–2489, https://doi.org/10.1002/2015jd024556, 2016.
Wang, X. F., Wang, H., Xue, L. K., Wang, T., Wang, L. W., Gu, R. R., Wang, W. H., Tham, Y. J., Wang, Z., Yang, L. X., Chen, J. M., and Wang, W. X.: Observations of N2O5 and ClNO2 at a polluted urban surface site in North China: High N2O5 uptake coefficients and low ClNO2 product yields, Atmos. Environ., 156, 125–134, https://doi.org/10.1016/j.atmosenv.2017.02.035, 2017.
Wang, Z., Wang, W., Tham, Y. J., Li, Q., Wang, H., Wen, L., Wang, X., and Wang, T.: Fast heterogeneous N2O5 uptake and ClNO2 production in power plant and industrial plumes observed in the nocturnal residual layer over the North China Plain, Atmos. Chem. Phys., 17, 12361–12378, https://doi.org/10.5194/acp-17-12361-2017, 2017.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosamas, C. E., Hjorth, J., Lebras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The Nitrate Radical – Physics, Chemistry, and the Atmosphere, Atmos. Environ., 25, 1–203, https://doi.org/10.1016/0960-1686(91)90192-A, 1991.
Xue, L. K., Saunders, S. M., Wang, T., Gao, R., Wang, X. F., Zhang, Q. Z., and Wang, W. X.: Development of a chlorine chemistry module for the Master Chemical Mechanism, Geosci. Model Dev., 8, 3151–3162, https://doi.org/10.5194/gmd-8-3151-2015, 2015.
Ye, N. N. L., Dong, K. D., Wu, H. B., Zeng, Y. S., Zeng, L. M., and Zhang, Y. H.: A study of the Water-Soluble Inorgic Salts and Their Gases Precursors at Wangdu Site in the Summer Time, Acta Scientiarum Naturalium Universitatis, 52, 1109–1117, https://doi.org/10.13209/j.0479-8023.2016.116, 2016.
Yue, D. L., Hu, M., Wu, Z. J., Wang, Z. B., Guo, S., Wehner, B., Nowak, A., Achtert, P., Wiedensohler, A., Jung, J., Kim, Y. J., and Liu, S.: Characteristics of aerosol size distributions and new particle formation in the summer in Beijing, J. Geophys. Res.-Atmos., 114, D00g12, https://doi.org/10.1029/2008jd010894, 2009.
Zheng, J., Hu, M., Du, Z., Shang, D., Gong, Z., Qin, Y., Fang, J., Gu, F., Li, M., Peng, J., Li, J., Zhang, Y., Huang, X., He, L., Wu, Y., and Guo, S.: Influence of biomass burning from South Asia at a high-altitude mountain receptor site in China, Atmos. Chem. Phys., 17, 6853–6864, https://doi.org/10.5194/acp-17-6853-2017, 2017.