the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 03 May 2018
| 03 May 2018
Advanced source apportionment of carbonaceous aerosols by coupling offline AMS and radiocarbon size-segregated measurements over a nearly 2-year period
Athanasia Vlachou
Kaspar R. Daellenbach
Carlo Bozzetti
Benjamin Chazeau
Gary A. Salazar
Soenke Szidat
Jean-Luc Jaffrezo
Christoph Hueglin
Urs Baltensperger
André S. H. Prévôt
Carbonaceous aerosols are related to adverse human health effects. Therefore, identification of their sources and analysis of their chemical composition is important. The offline AMS (aerosol mass spectrometer) technique offers quantitative separation of organic aerosol (OA) factors which can be related to major OA sources, either primary or secondary. While primary OA can be more clearly separated into sources, secondary (SOA) source apportionment is more challenging because different sources – anthropogenic or natural, fossil or non-fossil – can yield similar highly oxygenated mass spectra. Radiocarbon measurements provide unequivocal separation between fossil and non-fossil sources of carbon. Here we coupled these two offline methods and analysed the OA and organic carbon (OC) of different size fractions (particulate matter below 10 and 2.5 µm – PM10 and PM2.5, respectively) from the Alpine valley of Magadino (Switzerland) during the years 2013 and 2014 (219 samples). The combination of the techniques gave further insight into the characteristics of secondary OC (SOC) which was rather based on the type of SOC precursor and not on the volatility or the oxidation state of OC, as typically considered. Out of the primary sources separated in this study, biomass burning OC was the dominant one in winter, with average concentrations of 5.36 ± 2.64 µg m−3 for PM10 and 3.83 ± 1.81 µg m−3 for PM2.5, indicating that wood combustion particles were predominantly generated in the fine mode. The additional information from the size-segregated measurements revealed a primary sulfur-containing factor, mainly fossil, detected in the coarse size fraction and related to non-exhaust traffic emissions with a yearly average PM10 (PM2.5) concentration of 0.20 ± 0.24 µg m−3 (0.05 ± 0.04 µg m−3). A primary biological OC (PBOC) was also detected in the coarse mode peaking in spring and summer with a yearly average PM10 (PM2.5) concentration of 0.79 ± 0.31 µg m−3 (0.24 ± 0.20 µg m−3). The secondary OC was separated into two oxygenated, non-fossil OC factors which were identified based on their seasonal variability (i.e. summer and winter oxygenated organic carbon, OOC) and a third anthropogenic OOC factor which correlated with fossil OC mainly peaking in winter and spring, contributing on average 13 % ± 7 % (10 % ± 9 %) to the total OC in PM10 (PM2.5). The winter OOC was also connected to anthropogenic sources, contributing on average 13 % ± 13 % (6 % ± 6 %) to the total OC in PM10 (PM2.5). The summer OOC (SOOC), stemming from oxidation of biogenic emissions, was more pronounced in the fine mode, contributing on average 43 % ± 12 % (75 % ± 44 %) to the total OC in PM10 (PM2.5). In total the non-fossil OC significantly dominated the fossil OC throughout all seasons, by contributing on average 75 % ± 24 % to the total OC. The results also suggested that during the cold period the prevailing source was residential biomass burning while during the warm period primary biological sources and secondary organic aerosol from the oxidation of biogenic emissions became important. However, SOC was also formed by aged fossil fuel combustion emissions not only in summer but also during the rest of the year.
- Article
(3570 KB) - Full-text XML
-
Supplement
(1291 KB) - BibTeX
- EndNote





The field deployment of the time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Canagaratna et al., 2007) has advanced our understanding of aerosol chemistry and dynamics. The HR-ToF-AMS provides quantitative mass spectra of the non-refractory particle component, including, but not limited to, organic aerosol (OA), ammonium sulfate and nitrate, by combining the flash vaporization of particle species and the electron ionization of the resulting gases. The application of positive matrix factorization (PMF, Paatero, 1997) techniques has demonstrated that the collected OA mass spectra contain sufficient information to quantitatively distinguish aerosol sources. However, the cost and intensive maintenance requirements of this instrument significantly hinder its systematic, long-term deployment as part of a dense network and most applications are limited to few weeks of measurements (Jimenez et al., 2009; El Haddad et al., 2013; Crippa et al., 2013). This information is critical for model validation and policy directives. The Aerodyne aerosol chemical speciation monitors (ACSM, Ng et al., 2011; Fröhlich et al., 2013) were developed as a low-cost, low-maintenance alternative to the AMS; however, their reduced chemical resolution can limit the factor separation achievable by source apportionment.
The recent utilization of the AMS for the offline analysis of ambient filter samples (Daellenbach et al., 2016) has significantly broadened the spatial and temporal scales accessible to high-resolution AMS measurements (Daellenbach et al., 2017; Bozzetti et al., 2017a, b). In addition, the technique enables measurement of aerosol composition outside the normal size transmission window of the AMS; the standard AMS can measure up to only 1 µm, or ∼ 2.5 µm with a newly developed aerodynamic lens (Williams et al., 2013; Elser et al., 2016). This capability has been used to quantify the contributions of primary biological organic aerosol to OA in PM10 filters (Bozzetti et al., 2016). Finally, the offline AMS technique allows a retrospective reaction to critical air quality events. For example, one of the applications of this approach had been to examine a severe haze event in China which affected a total area of ∼ 1.3 million km2 and ∼ 800 million people (Huang et al., 2014).
A major limitation of the technique is the resolution of low water solubility fractions, as the recoveries of some of them are not accessible. Despite this, source apportionment results obtained using this technique are in good agreement with online AMS or ACSM measurements. PMF analysis of offline AMS data has yielded factors related with primary emissions from traffic, biomass burning and coal burning, and secondary organic aerosols (SOA) differentiated according to their different seasonal contributions. Nevertheless, the identification of SOA precursors using the AMS has proven challenging, due to the evolution of different precursors towards chemically similar species and the extensive fragmentation by the electron ionization used in the AMS.
The radiocarbon (14C) analysis of particulate matter has proven to be a powerful technique providing an unequivocal distinction between non-fossil (e.g. biomass burning and biogenic emissions) and fossil (e.g. traffic exhaust emissions and coal burning) sources (Lemire et al., 2002; Szidat et al., 2004, 2009). The measurement of the 14C content of total carbon (TC), which comprises the elemental carbon (EC) originating from combustion sources and the organic carbon (OC), had been the subject of many studies (Schichtel et al., 2008; Glasius et al., 2011; Genberg et al., 2011; Zhang et al., 2012, , 2016; Zotter et al., 2014b; Bonvalot et al., 2016). Results have shown that in European sites, especially in Alpine valleys, the non-fossil sources play an important role during winter due to biomass burning and in summer due to biogenic sources (Gelencsér et al., 2007; Zotter et al., 2014b). Moreover, at regional background sites close to urbanized areas in Europe (Dusek et al., 2017) as well as in megacities such as Los Angeles and Beijing, fossil OA may also exhibit significant contributions to the total OA (Zotter et al., 2014a; Zhang et al., 2017). However, the determination of the 14C content in EC and OC separately is challenging and therefore not often attempted for extended datasets.
The coupling of the offline AMS/PMF with radiocarbon analysis provides further insight into the sources of organic aerosols and in particular those related to SOA precursors. Such combination has been already attempted (Minguillón et al., 2011; Zotter et al., 2014a; Huang et al., 2014; Beekmann et al., 2015; Ulevicius et al., 2016); however, the focus has rather been on high OA concentration episodes, while little is known about the yearly cycle of the most important SOA precursors and the size resolution of the different fossil and non-fossil OA fractions.
Here, we present offline AMS measurements of a total of 219 samples, 154 of which are PM10 samples representative of the years 2013 and 2014 and 65 PM2.5 concurrent with PM10 samples for the year 2014 (January to September). 14C analysis was also performed on a subset of 33 PM10 samples, covering the year 2014. The size-segregated samples offered better insights into the mechanism by which the different fractions enter the atmosphere, while the coupling of offline AMS/PMF and 14C analysis provided a more profound understanding of the SOA fossil and non-fossil precursors on a yearly basis.
2.1 Site and sampling collection
Magadino is located in an Alpine valley in the Southern part of Switzerland, south of the Alps (Fig. S1 in the Supplement). The station (46∘9′37′′ N, 8∘56′2′′ E, 204 m a.s.l.) belongs to the Swiss National Air Pollution Monitoring Network (NABEL) and is classified as a rural background site. It is located relatively far from busy roads or residential areas and surrounded by agricultural fields and forests. It is ca. 1.4 km away from Cadenazzo train station, ca. 8 km from Lake Maggiore (Lago Maggiore) and ca. 7 km from the small Locarno Airport.
The filter samples under examination are 24 h integrated PM10 (from 4 January 2013 to 28 September 2014, with a 4-day interval) and PM2.5 (from 3 January to 28 September 2014, with a 4-day interval). PM was sampled and collected on 14 cm (exposed diameter) quartz fibre filters, using a high volume sampler (500 L min−1). After the sampling, filter samples and field blanks were wrapped in lint-free paper and stored at −20 ∘C.
2.2 Offline AMS method
The offline AMS method is thoroughly described by Daellenbach et al. (2016). Briefly, four punches of 16 mm diameter from each filter sample are extracted in 15 mL of ultrapure water (18.2 MΩ cm at 25 ∘C with total organic carbon, TOC, < 3 ppb), followed by insertion in an ultra-sonic bath for 20 min at 30 ∘C. The water-extracted samples are then filtered through a 0.45 µm nylon membrane syringe and inserted to an Apex Q nebulizer (Elemental Scientific Inc., Omaha, NE, USA) operating at 60 ∘C. The resulting aerosols generated in Ar (≥ 99.998 % vol., Carbagas, 3073, Gümligen, Switzerland) were dried by a Nafion dryer and subsequently injected and analysed by the HR-ToF-AMS.
To correct for the interference of NH4NO3 on the CO signal as described in Pieber et al. (2016), several dilutions of NH4NO3 in ultrapure water were measured regularly as well. The CO signal was then calculated as
where CO2,real represents the corrected CO signal, CO2,meas and NO3,meas are signals from the samples measured, and the correction factor was determined during the campaign by measuring aqueous NH4NO3.
2.3 14C analysis
Based on the instrumentation setup described in Agrios et al. (2015) and on the method described in Zotter et al. (2014b), radiocarbon analysis of TC and EC was conducted on a set of 33 filters. The 14C content of blank filters was measured for TC only, as there was no EC found on these filters. All the 14C results are given in fractions of modern carbon (fM) representing the 14C ∕ 12C ratios of each sample relative to the respective 14C ∕ 12C ratio of the reference year 1950 (Stuiver and Polach, 1977).
2.3.1 14C measurements of TC
For the determination of the 14C content of TC, a Sunset OC ∕ EC analyser (Model 4L, Sunset Laboratory, USA) equipped with a non-dispersive infrared (NDIR) detector was first used in order to combust each filter punch (1.5 cm2) under pure O2 (99.9995 %) at 760 ∘C for 400 s. The generated CO2 was then captured online by a zeolite trap within a gas inlet system (GIS) and then injected in the accelerator mass spectrometer (AMS*) mini carbon dating system (MICADAS) at the Laboratory for the Analysis of Radiocarbon with AMS* (LARA), University of Bern, Switzerland (Szidat et al., 2014) for 14C measurement. (Note that we used AMS* and AMS as abbreviations for the accelerator mass spectrometer and the aerosol mass spectrometer, respectively, to avoid confusion.)
The fM of TC underwent a blank correction following an isotopic mass balance approach:
where is the blank corrected fM; mCsample and mCb are the carbon mass in sample and blank, respectively; and fM,sample and fM,b are the fM measured for sample and blank, respectively. Error propagation was applied for the determination of the uncertainty. The fM,b was 0.61 ± 0.10 and the concentration of the blank 1.1 ± 0.2 µg C m−3.
2.3.2 14C measurements of EC
For the EC isolation of the samples, each filter punch (1.5 cm2) was analysed by the Sunset EC ∕ OC analyser with the use of the Swiss_4S protocol developed by Zhang et al. (2012). According to the protocol, the heating is conducted in four different steps under different gas conditions: step one under pure O2 at 375 ∘C for 150 s, step two under pure O2 at 475 ∘C for 180 s, step three under He (> 99.999 %) at 450 ∘C for 180 s followed by an increase in the temperature up to 650 ∘C for another 180 s, and step four under pure O2 at 760 ∘C for 150 s. Each filter sample was previously water extracted and dried, in order to minimize the positive artefact induced by the OC by removing the water-soluble OC (WSOC), which is known to produce charring (Piazzalunga et al., 2011; Zhang et al., 2012). By this method, the water-insoluble OC (WINSOC) was removed during the first three steps of the Swiss_4S protocol. In the fourth step, EC was combusted and then trapped in the GIS and measured by the AMS* MICADAS, as described above.
This protocol was preferred over the protocols commonly used in thermo-optical methods (EUSAAR 2 or NIOSH) because it optimises the separation of the two fractions OC and EC by minimizing (i) the positive artefact of charring produced by WSOC during the first three steps and (ii) the premature losses, during the removal of the WINSOC in the third step, of the less refractory part of EC which may preferentially originate from non-fossil sources such as biomass burning.
Following a similar principle to Zotter et al. (2014b), both charring and EC yield, which is the part of EC that remained on the filter after step three and before step four in the Swiss_4S protocol, were quantified and corrected for with the help of the laser mounted on the Sunset analyser. The laser transmittance is monitored continuously during the heating process. Charring in step three was quantified as
where ATN refers to the laser attenuation, is the maximum attenuation in step three, and and are the initial attenuations in step two and one, respectively.
The EC yield in step three was quantified as
The average charred OC was found to be 4 ± 2 % and the recovered EC for all samples was on average 71 ± 7 %.
As there is a linear relationship between the fraction of modern carbon for EC ( and the EC yield (Zhang et al., 2012), the slope can be used to extrapolate to 100 % EC yield. According to Zotter et al. (2014), a slope of 0.35 ± 0.11 was considered to correct all to 100 % of EC yield, such that
2.3.3 Calculation of 14C content of OC
The fraction of modern carbon of OC ( was calculated following a mass balance approach:
where TC and EC are the concentrations of total and elemental carbon, respectively, and and are the fractions of modern carbon of TC and EC, respectively. The uncertainty of was calculated by propagating the error of each component of Eq. (6).
2.3.4 Nuclear bomb peak correction
The expected fM coming from fossil samples should be equal to zero due to the complete decay of 14C until now, whereas the fM from non-fossil samples is expected to be unity. However, due to the extensive nuclear bomb testing during the late 1950s and early 1960s, the radiocarbon amount in the atmosphere increased dramatically because of the high neutron flux during the explosions. Therefore the measured fM of non-fossil samples may exhibit values greater than one (Levin et al., 2010a). To correct for this effect, the fM is normalized to a reference non-fossil fraction (fNF,ref) which represents the amount of 14C currently in the atmosphere compared to 1950, before the nuclear bomb tests. As EC comes from either biomass burning or fossil sources, the non-fossil fraction of EC (fNF,EC) equals the fM coming from biomass burning (fM,bb). The latter was estimated by a tree growth model (Mohn et al., 2008) and was equal to 1.101. The non-fossil fraction of OC (fNF,OC) is calculated as
where fM,bio (= 1.023) is the fraction of modern carbon of biogenic sources and was estimated from 14CO2 measurements in Schauinsland (Levin et al., 2010a). The fractions of biogenic sources (pbio) and biomass burning (pbb) to the total non-fossil sources were set to 0.5 since both sources are important in Magadino during the year (biomass burning in winter, biogenic sources in summer).
2.4 Additional measurements
Organic and elemental carbon fractions were determined by a Sunset EC ∕ OC analyser with the use of the EUSAAR-2 thermal-optical transmittance protocol (Cavalli et al., 2010). Water-soluble organic carbon was measured by a total organic carbon analyser (Jaffrezo et al., 2005) with the use of catalytic oxidation of water-extracted filter samples and detection of the resulting CO2 with an NDIR. The concentrations of major ionic species (K+, Na+, Mg2+, Ca2+, NH, Cl−, NO and SO as well as methane sulfonic acid (MSA) were determined by ion chromatography (Jaffrezo et al., 1998). Anhydrous sugars (levoglucosan, mannosan, galactosan) were analysed by an ion chromatograph (Dionex ICS3000) using high-performance anion exchange chromatography (HPAEC) with pulsed amperometric detection. Cellulose was analysed by performing enzymatic conversion of cellulose to D-glucose (Kunit and Puxbaum, 1996) and D-glucose was determined by HPAEC.
3.1 Method
The obtained organic mass spectra from the offline AMS measurements were analysed by positive matrix factorization (Paatero and Tapper, 1994; Ulbrich et al., 2009). PMF attempts to solve the bilinear matrix equation,
by following the weighted least-squares approach. In the case of aerosol mass spectrometry, i represents the time index, j the fragment and k the factor number. If Xij is the matrix of the organic mass spectral data and si,j the corresponding error matrix, Gi,k the matrix of the factor time series, Fk,j the matrix of the factor profiles and Ei,j the model residual matrix, then PMF determines Gi,k and Fk,j such that the ratio of the Frobenius norm of Ei,j over si,j is minimized. The allowed Gi,k and Fk,j are always non-negative. The input error matrix si,j includes the measurement uncertainty (ion-counting statistics and ion-to-ion signal variability at the detector) (Allan et al., 2003) as well as the blank variability. Fragments with a signal-to-noise ratio (SNR) below 0.2 were removed and the ones with SNR lower than 2 were down-weighted by a factor of 3, as recommended by Paatero and Hopke (2003). Both input data and error matrices were scaled to the calculated water-soluble organic matter (WSOMi) concentration:
where is determined from the AMS measurements and WSOCi is the water-soluble OC measured by the TOC analyser.
The Source Finder toolkit (SoFi v.4.9, Canonaco et al., 2013) for IGOR Pro software package (Wavemetrics, Inc., Portland, OR, USA) was used to run the PMF algorithm. The PMF was solved by the multilinear engine 2 (ME-2, Paatero, 1999), which allows the constraining of the Fk,j elements to vary within a certain range defined by the scalar α (), such that the modelled equals
Here we constrained only the hydrocarbon-like factor (HOA) from high-resolution mass spectra analysed by Crippa et al. (2013).
3.2 Sensitivity analysis
To understand the variability of our dataset we explored 4–10 factor solutions and retained the 7-factor solution as the best representation of the data. The exploration of the PMF solutions is thoroughly described in Sect. S.1 in the Supplement.
We assessed the accuracy of PMF results by bootstrapping the input data (Davison and Hinkley, 1997). New input data and error matrices were created by randomly resampling the time series from the original input matrix (223 samples in total: 219 + 4 remeasurements from the PM10 samples), with replacement; i.e. any sample from the whole population can be resampled more than once. Each sample measurement included on average blocks of 12 mass spectral repetitions; therefore, resampling was performed on the blocks. Out of the 223 original samples, some of them were represented several times, while some others not at all. Overall, the resampled data made up on average 64 ± 2 % of the total original data per bootstrap run. We performed 180 bootstrap runs, with each of the generated matrices being perturbed by varying the Xij element within twice the corresponding error matrix si,j. Within the resampling operation, the α value used to set the HOA constraining strength was varied between 0 and 1 with an increment of 0.1 to assess the sensitivity of the results on the α value.
To select the physically plausible solutions we applied two criteria:
-
We accepted solutions where the average absolute concentrations of all factors in PM2.5 did not statistically significantly exceed their concentrations in PM10. For this we performed a paired t test with a significance level of 0.01 (Fig. S2 and Table S1 in the Supplement).
-
We excluded outlier solutions identified by examining the correlation of factor time series from bootstrap runs with their respective factor time series from the average of all bootstrap runs. The rejected solutions included factors that did not correlate with the corresponding average factor time series, meaning that one of the factors was not separated (Fig. S3 in the case of water-soluble primary biological organic carbon, PBOC).
In total 24 bootstrap runs were retained after the application of the aforementioned criteria.
3.3 Recoveries
In order to rescale the WSOC concentration of a factor k to its total concentration OCk, we used factor recoveries (Rk) as proposed by Daellenbach et al. (2016). First, the WSOMk was calculated as
where
and
is estimated from the input data matrix for the PMF.
The WSOMk was converted to WSOCk to fit the measured OC concentrations (determined by the Sunset EC ∕ OC analyser). The WSOCk was determined as
where is calculated from each factor profile.
Finally, the recoveries were applied following Eq. (15):
To assess the recoveries and their uncertainties, we evaluated the sum of OCi,k against the measured OC (OCi,measured) by fitting Eq. (16). The starting values for the Rk fitting were based on Bozzetti et al. (2016) (for RPBOA) and Daellenbach et al. (2016) except RSCOA, which was randomly varied between 0 and 1 (increment: 10−4). While RHOA and RSCOA were constrained, RPBOA, RBBOA, RWOOA, RAOOA and RSOOA were determined by a non-negative multilinear fit (see below in Sect. 4.3 for a description of these PMF factors from offline AMS results). The multilinear fit was chosen to be non-negative because a negative Rk would mean a negative concentration of WSOCk or OCk. The fit was performed 100 times for each of the retained bootstrap solutions.
Each fit was initiated by perturbing the OCi,k and the WSOCi,k concentrations within their uncertainties, assuming a normal distribution of errors, to assess the influence of measurement precision on Rk. Additionally, we introduced a constant 5 % accuracy bias corresponding to the OC and WSOC measurement accuracy.
To select the environmentally meaningful solutions we applied the following criteria:
-
To retain the recoveries that achieved the OC mass closure, we estimated the OC residuals and discarded solutions where OC residuals were statistically different from 0 within 1 standard deviation for each size fraction individually and for winter and summer individually.
-
We also examined the dependence between the WSOC residuals and each factor WSOCi,k (t test, α=0.001). Overall, 55 % of the solutions were retained.
-
The physically plausible range of the recoveries is [0, 1]. However, the mathematically possible range can exceed the upper limit. Rk larger than 1 would mean that WSOCk is larger than OCk and is, therefore, non-physical. For this reason, out of the accepted solutions that survived the previous two criteria, the retained Rk combinations were weighted according to their physical interpretability. More specifically, fitting results with Rk larger than 1 were down-weighted according to the measurement uncertainties of WSOC and OC (see Sect. S.2, Fig. S4).
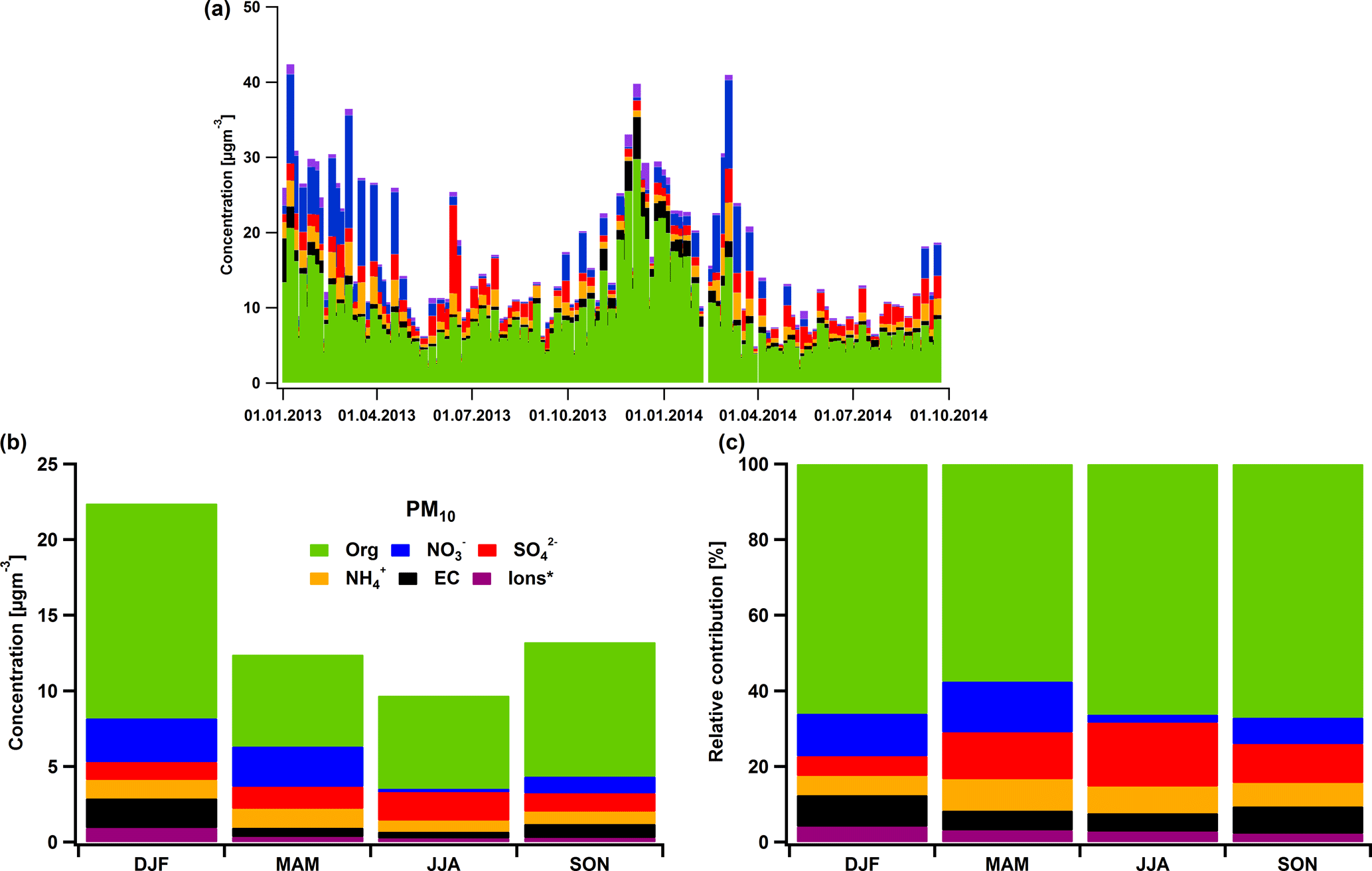
Figure 1Concentrations of OM, EC and major ionic species for the years 2013 and 2014 (a), their seasonal concentrations (b) and relative contributions to the total measured mass within the particulate matter (PM10) (c). The sum of the ions Na+, K+, Mg2+, Ca2+ and Cl− are included in the indication “Ions*”.
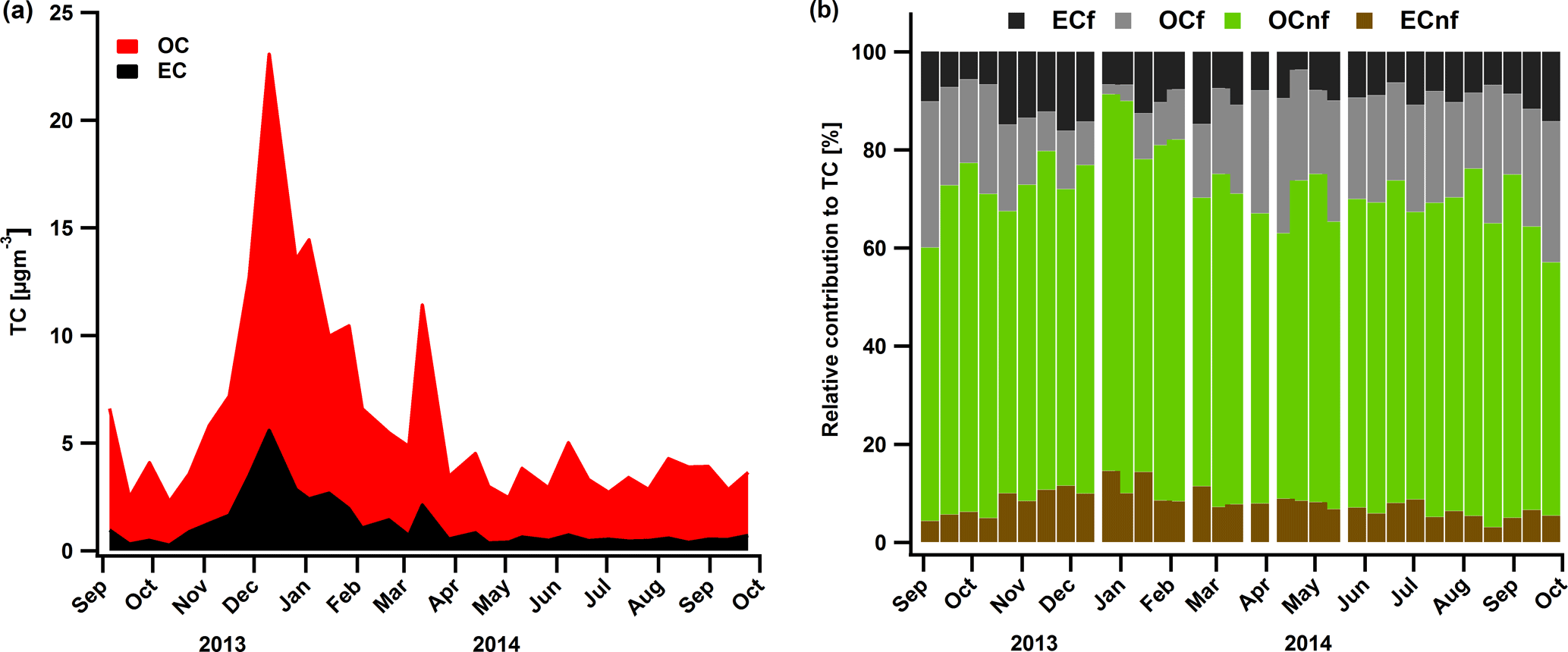
Figure 2Time series of OC and EC (a) concentrations in PM10. 14C analysis results with the relative contributions of EC fossil, OC fossil, OC non-fossil and EC non-fossil to the TC (b).
4.1 PM10 composition
PM10 in Magadino has been characterized by high carbonaceous concentrations during winter (Gianini et al., 2012a; Zotter et al., 2014b). This is clearly illustrated in Fig. 1 where an overview of the PM10 composition is presented in Fig. 1a with Fig. 1b and c summarizing the concentrations and relative contributions of each component to the total PM10 averaged per season. The peaks of OM and EC during winter (daily averages up to 26 and 5.9 µg m−3, respectively) are indications of the increased wood-burning activity. Other Alpine sites close to Magadino, such as Roveredo and San Vittore in Switzerland, have also exhibited high OM concentrations due to residential wood burning (Szidat et al., 2007, for PM10 in Roveredo, Lanz et al., 2010, for PM1 in Roveredo and Zotter et al., 2014b, for PM10 in San Vittore and Roveredo). The organic contribution dominated the inorganic fraction not only in winter, but also throughout both years (Fig. 1c). Note that the EC concentrations are much lower in spring compared to winter (Fig. 1b). The main inorganic aerosols contributing to the total PM are NO, SO and NH. NO represented the second major component of PM10, exhibiting a seasonal cycle with higher concentrations during winter (2.9 µg m−3). The notable discrepancy of NO concentrations between the first (2013) and second (2014) winter could be explained by the lower temperatures in January–February 2013 compared to 2014. Conversely, SO showed a rather stable yearly cycle with slightly higher concentrations in summer (1.9 µg m−3) compared to winter (1.3 µg m−3), despite a shallower boundary layer height in winter.
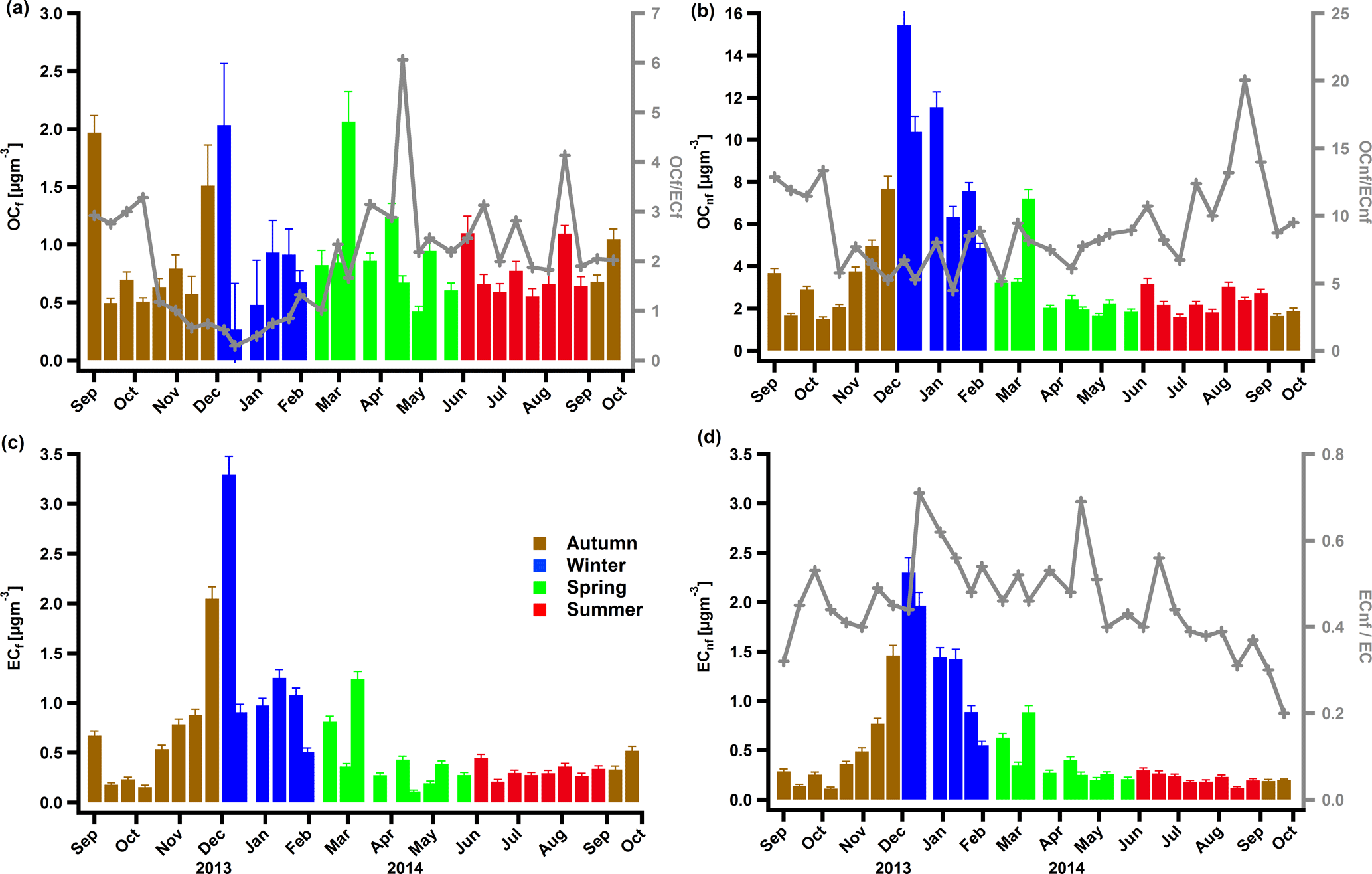
Figure 3Concentrations in PM10 of OCf (a), OCnf (b), ECf (c) and ECnf (d) colour-coded by seasons. The ratios OCf ∕ ECf, OCnf ∕ ECnf, and ECnf ∕ EC are also displayed in (a), (b) and (d), respectively.
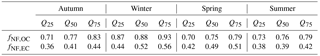
Table 1Median OC and EC non-fossil fractions per season in PM10 with interquartile range.
4.2 14C analysis results
So far radiocarbon results have been reported mostly for relatively short periods of time (Bonvalot et al., 2016), mainly describing high concentration events, and only a few studies report measurements on a yearly basis (Genberg et al., 2011; Gilardoni et al., 2011; Zotter et al., 2014b; Zhang et al., 2016, 2017; Dusek et al., 2017). Here, for a subset of 33 PM10 filters from the year 2014, we present yearly contributions of OCnf, OCf, ECnf and ECf.
Overall the total carbon concentrations followed a yearly pattern mainly caused by the shallow planetary boundary layer and the enhanced biomass burning activity during winter, with OC reaching on average (± 1 standard deviation) 9.4 ± 4.5 and EC 2.6 ± 1.5 µg m−3 (Fig. 2a). During the rest of the year, TC remained rather stable with much lower concentrations (OCavg=3.7 ± 1.9 and ECavg=0.8 ± 0.7 µg m−3). 14C results indicate that non-fossil sources prevail over the fossil ones in Magadino. More specifically, we found that in winter on average ± 0.1 and ± 0.1, which is in agreement with the reported fractions by Zotter et al., 2014b (fNF,OC = 0.8 ± 0.1 and ± 0.2). Table 1 summarizes the fNF per fraction season wise.
OCnf was the dominant part of TC throughout the year with contributions of up to 80 % in winter and 71 % in summer (Fig. 2b) and average concentrations of 8.5 ± 4.2 and 2.4 ± 0.6 µg m−3 in winter and summer, respectively (Fig. 3b). Such high contributions in winter strongly indicate that biomass burning (BB) from residential heating is the main source of carbonaceous aerosols in this region, similar to previous reports (Jaffrezo et al., 2005; Puxbaum et al., 2007; Sandradewi et al., 2008; Favez et al., 2010; Zotter et al., 2014b). The coefficient of determination R2 between OCnf and levoglucosan, a characteristic marker for BB, was 0.92 (Fig. S7a), and the slope (OCnf ∕ levoglucosan = 4.8 ± 0.3) lies within the reported range by Zotter et al. (2014b) for Magadino (which was 6.9 ± 2.6).
The concentration of ECnf was significantly higher in winter (average 1.3 ± 0.7 µg m−3) compared to the rest of the year (spring average: 0.4 ± 0.2 µg m−3, summer average: 0.21 ± 0.06 µg m−3, autumn average: 0.43 ± 0.41 µg m−3) (Fig. 3d). ECnf is considered to originate solely from BB, for instance from residential wood burning in winter. This assumption is supported by the very high correlation (R2=0.95) with levoglucosan (Fig. S7b) and the slope (ECnf ∕ levoglucosan = 0.82 ± 0.03) which is also in agreement with the literature (Zotter et al., 2014b; Herich et al., 2014).
The strong correlation between OCnf and ECnf, driven mainly by the winter data points, supports the fact that OCnf is mostly from biomass burning in winter (Fig. S6a). In late spring, summer and early autumn, the contribution of ECnf decreased significantly (on average to 0.23 ± 0.07 µg m−3). The low correlation of OCnf and ECnf during this period (Fig. S6a), in combination with the increase in the OCnf ∕ ECnf ratio in summer (Fig. 3b), suggests that a part of the secondary OCnf originates from non-combustion sources, e.g. biogenic/natural sources.
In total, the relative contribution of the fossil fraction to the TC was 27 %. Excluding winter, ECf exhibited slightly higher concentrations than ECnf (Fig. 3c and d). The average concentrations of ECf were 1.26 ± 0.93, 0.41 ± 0.35, 0.31 ± 0.07 and 0.63 ± 0.56 µg m−3 for winter, spring, summer and autumn, respectively (Fig. 3c). The increase in ECf witnessed in winter could be mainly attributed to the shallower planetary boundary layer (PBL) rather than to an increase in the emissions (Fig. S8a). The sources of ECf in the coarse (PM10–PM2.5) size fraction are typically related to resuspension of abrasion products of vehicle tires or brake wear (Bukowieki et al., 2010; Zhang et al, 2013). The fine part of ECf is due to fossil fuel burning, here mostly due to traffic exhaust emissions. It is significantly correlated with NOx (Fig. S8b) and the ECf ∕ NOx = 0.020 ratio lies within the reported slopes (Zotter et al., 2014b, and references therein).
The contribution of OCf to TC decreased during winter (8 %) but remained roughly stable throughout the rest of the year (22 % in spring, 21 % in summer and 19 % in autumn, Fig. 2b) with average concentrations 0.87 ± 0.30, 0.96 ± 0.12, 0.89 ± 0.14 and 0.76 ± 0.10 µg m−3 for winter, spring, summer and autumn, respectively (Fig. 3a). The low correlation overall observed between OCf and ECf (Fig. S6b) may indicate that a fraction of OCf is not directly emitted but formed as secondary OC (SOC) from fossil-fuel-related emissions (e.g. traffic). This is supported by low OCf ∕ ECf ratios in winter (on average 0.7 ± 0.3) and much higher values in spring and summer (on average 2.7 ± 1.1) (Fig. 3a). The low ratios are consistent with tunnel measurement studies (Li et al., 2016; Chirico et al., 2011; El Haddad et al., 2009) and the increase in OCf ∕ ECf in spring and summer above these values is an indication of anthropogenic SOA formation. We also note that fossil SOA may be formed by other sources besides traffic. A recent study revealed that fossil SOA is produced by the oxidation of volatile chemical products coming from petrochemical sources (McDonald et al., 2018).
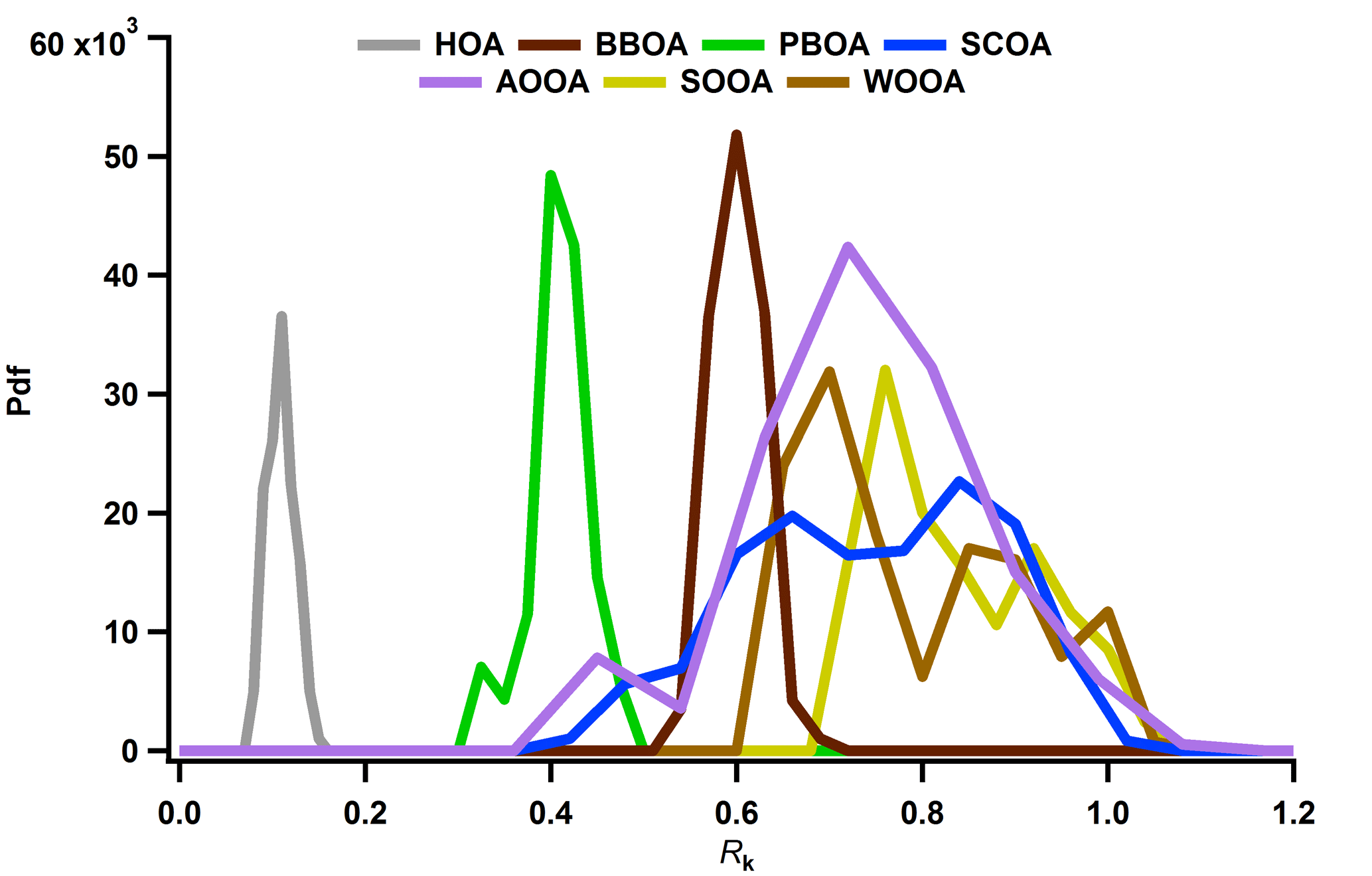
Figure 4Probability density functions of factor recoveries: hydrocarbon-like OA (HOA) in grey, biomass burning OA (BBOA) in dark brown, sulfur-containing OA (SCOA) in blue, primary biological OA (PBOA) in green, anthropogenic oxygenated OA (AOOA) in purple, summer oxygenated OA (SOOA) in yellow and winter oxygenated OA (WOOA) in light brown.
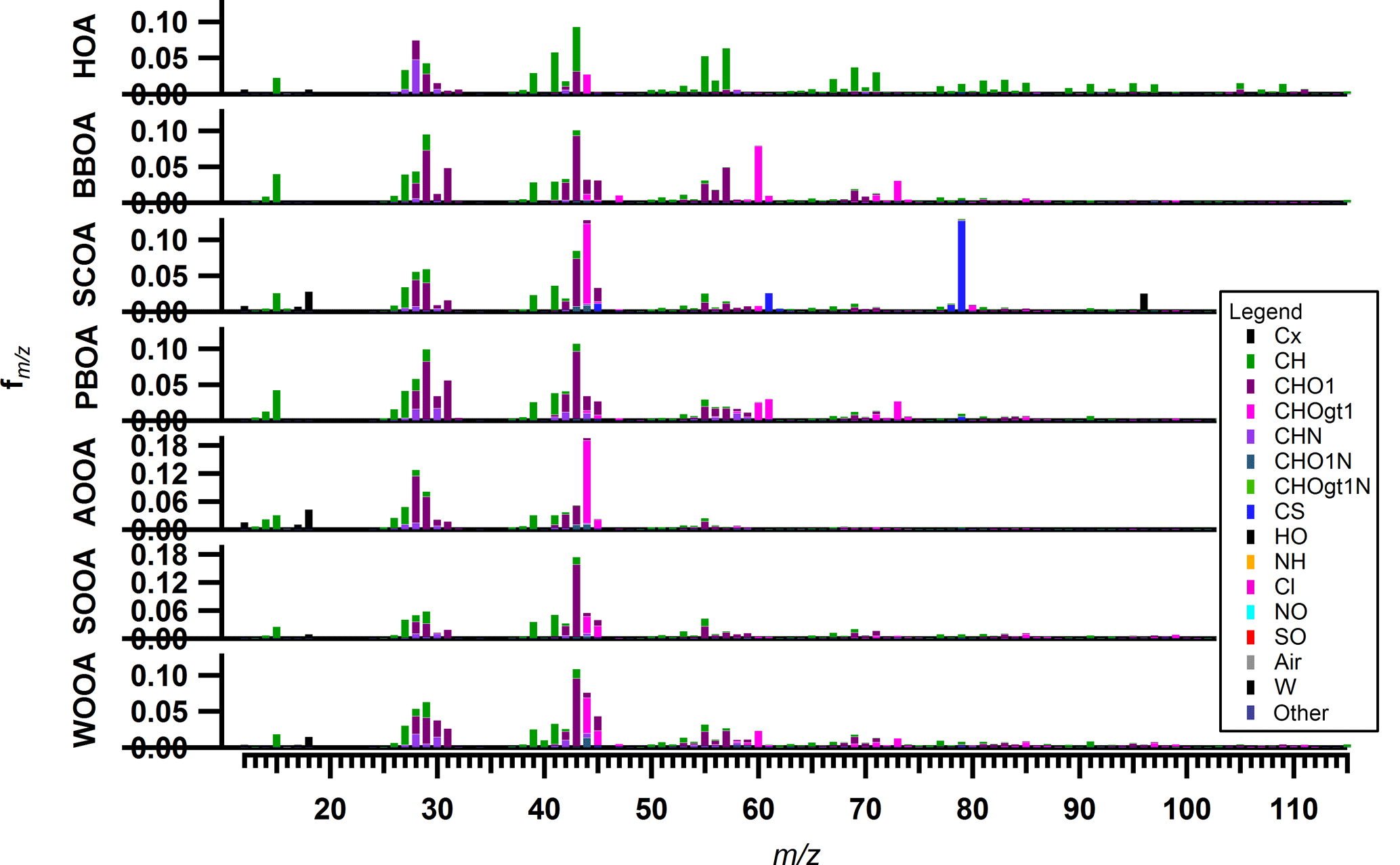
Figure 5Offline AMS/PMF (ME-2) factor profiles: hydrocarbon-like OA (HOA), biomass burning OA (BBOA), sulfur-containing OA (SCOA), primary biological OA (PBOA), anthropogenic oxygenated OA (AOOA), summer oxygenated OA (SOOA) and winter oxygenated OA (WOOA).
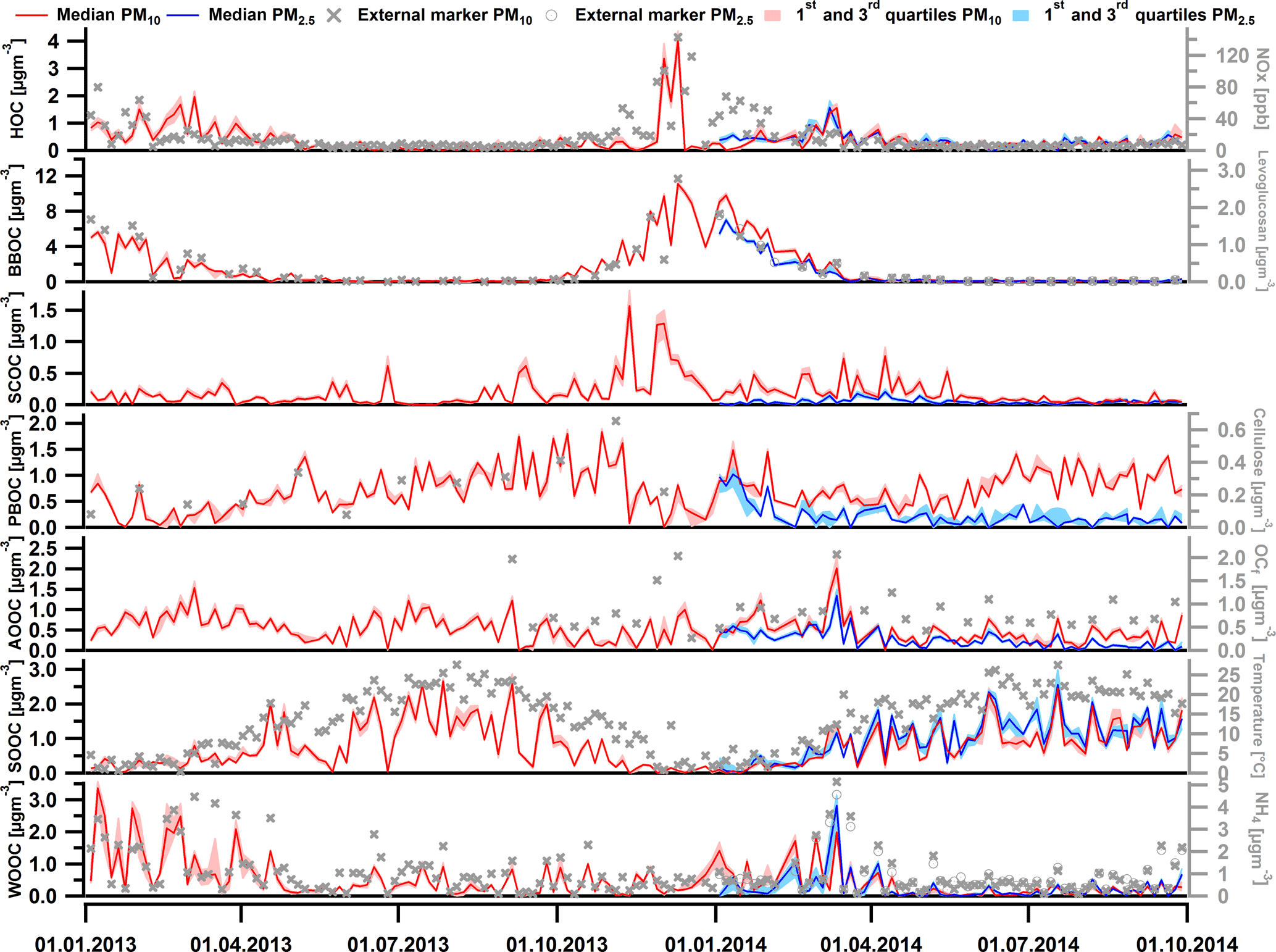
Figure 6Factor (in red for PM10 and blue for PM2.5) and external marker (in grey markers) time series for the two size fractions: HOC and NOx, BBOC and levoglucosan, SCOC, PBOC and cellulose, AOOC and OCf, SOOC and temperature, and WOOC and NH. Note that here, different from Fig. 5, the factors are quantified according to their carbon mass concentration, with HOC, BBOC, SCOC, PBOC, AOOC, SOOC, and WOOC referring to hydrocarbon-like organic carbon (OC), biomass burning OC, sulfur-containing OC, primary biological OC, anthropogenic oxygenated OC, summer oxygenated OC, and winter oxygenated OC, respectively.
4.3 Offline AMS analysis results: factor interpretation
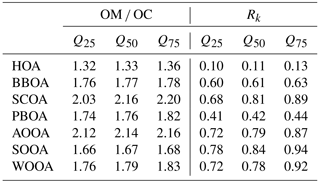
In this section, we will interpret the PMF outputs. The factor recoveries for all factors, Rk, determined as described in Sect. 3.3, are shown in Fig. 4. Factor mass spectra are displayed in Fig. 5. The contribution of the different factors to OA is presented in Fig. 6. In addition, for some cases we will discuss the factor contribution to OC to check the consistency of our results with previous literature reports. Recovery values determined and used in this study will also be compared for each factor to previous values. Median values of the recoveries as well as the OM ∕ OC ratios with their interquartile range are compiled in Table 2. The Rk values were in general consistent with previous reports (Daellenbach et al., 2016, 2017; Bozzetti et al., 2016). Here we report for the first time the recoveries of each SOA factor individually which were in agreement with the ones reported by Daellenbach et al. (2016). The consistency of the recovery results with not only previous offline AMS/PMF studies but also with online AMS measurements (Xu et al., 2017) points out that this method is rather robust and universal for different datasets.
Hydrocarbon-like OA (HOA), typically associated with traffic emissions, was constrained using the reference HOA high-resolution profile from Crippa et al. (2013). The resulting factor profile (Fig. 5) exhibited a low OM ∕ OC (Table 2) and the time series followed the one from NOx (Fig. 6). As the offline AMS technique requires water-extracted samples, it is expected that HOA, which mostly contains water-insoluble material, will be poorly represented. This is also shown by the low recovery RHOA,median which was estimated to be 0.11 (Q25=0.10 and Q75=0.13) as reported in Daellenbach et al. (2016) (Fig. 4). Therefore, the correlation between HOA and NOx was weak (Fig. S9). However, the HOA/NOx ratio was 0.017 for PM10 and 0.008 for PM2.5 and these values are consistent with already reported ones in the literature (Daellenbach et al., 2017; Lanz et al., 2007). In addition, the HOC time series followed a similar yearly cycle as ECf (Fig. S10a) and the HOC ∕ OCf ratio was 0.37 ± 0.12 (Fig. S10b), in agreement with Zotter et al. (2014a).
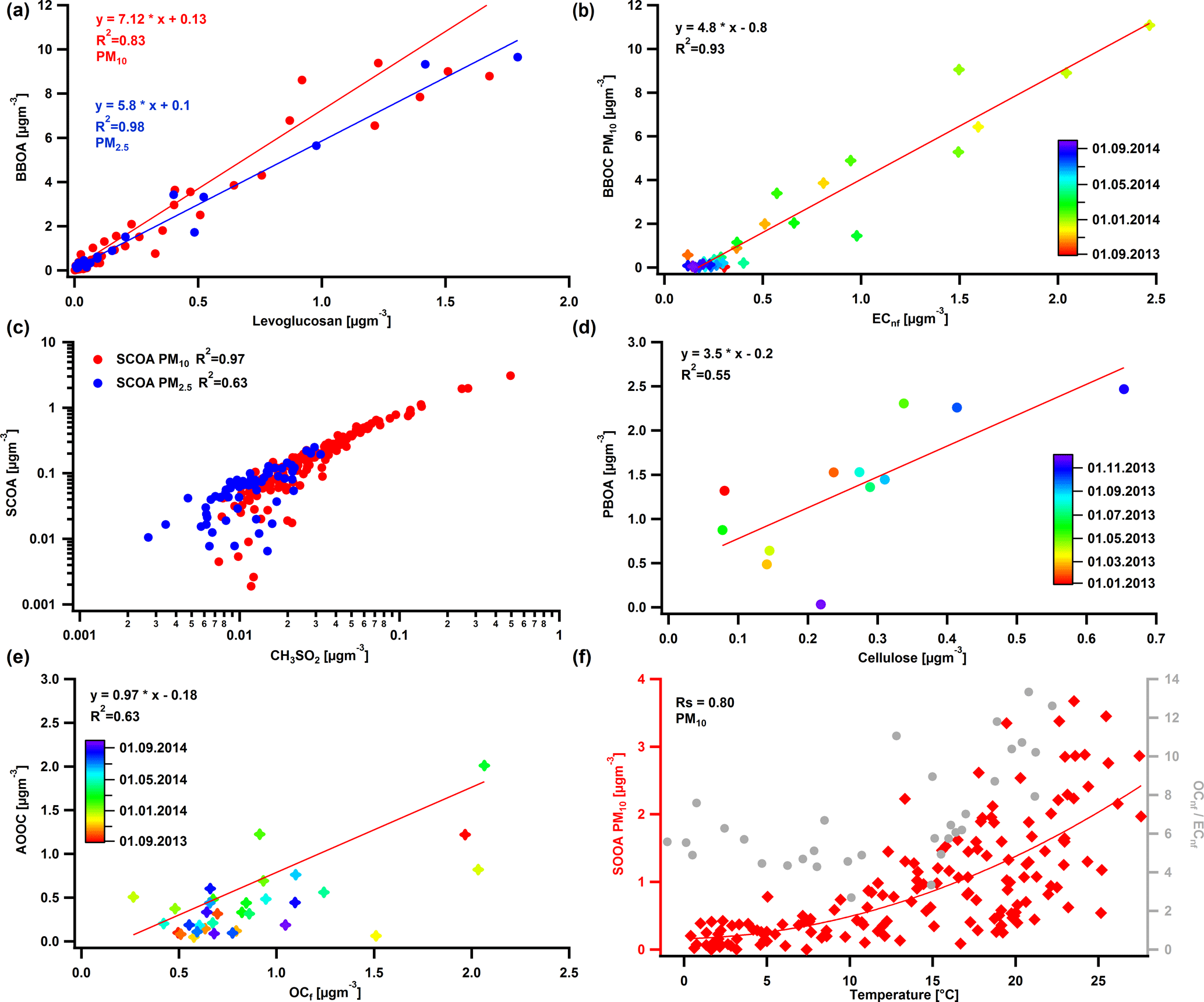
Figure 7Correlations between BBOA and levoglucosan for the two size fractions (a), BBOC and ECnf for PM10 (b), SCOA and CH3SO for the two size fractions (c) (the regression lines show a linear relationship), PBOA and cellulose for PM10 (d), AOOC and OCf (the regression fit was weighted by the standard deviation of AOOC) (e), and SOOA and daily averaged temperature as well as OCnf ∕ ECnf ratio and temperature for PM10 (f).
Biomass burning OA (BBOA) was identified by its significant contributions of the oxygenated fragments C2H4O (at m∕z 60) and C3H5O (at m∕z 73), common markers for wood burning formed by fragmentation of anhydrous sugars (Alfarra et al., 2007) (Fig. 5). It was also identified by its distinct seasonal variation which exhibited exclusively high concentrations in winter, reaching up to 20.0 ± 0.7 µg m−3 for PM10 in December 2013 and 12.3 ± 0.5 µg m−3 for PM2.5 in January 2014 (Fig. 6). The median value for the OM ∕ OC ratio was 1.8 and the RBBOA was consistent with the low end of the reported one by Daellenbach et al. (2016) (Table 2). The identification of this factor as BBOA was further confirmed by its remarkable correlation with levoglucosan. Similar to levoglucosan, this factor did not exhibit a significant difference between PM2.5 and PM10 concentrations (Fig. S5a), suggesting that most of these particles are present in the fine mode, consistent with previous observations (Levin et al., 2010b). The BBOA/levoglucosan ratio was 7.1 for PM10 and 5.8 for PM2.5, which falls into the range reported by Daellenbach et al. (2017) and was also consistent with the ratio reported by Bozzetti et al. (2016). The difference of BBOA/levoglucosan for the two size fractions is due to four samples in BBOA PM10 with high concentrations. Lastly, BBOC showed a strong correlation with ECnf, with a slope of 4.9 (Fig. 7b) which fell within the range of the compiled ECnf/BBOC ratios in Ulevicius et al. (2016).
Table 3Season-wise average (± 1 standard deviation) concentrations (in µg m−3) of different OA factors per size fraction. Note that for the 2 different years the months per season can vary.

Sulfur-containing OA (SCOA) was identified by its spectral fingerprint which is described by a high contribution of the fragment CH3SO (at m∕z 79) (Fig. 5) and high OM ∕ OC ratio (Table 2). The RSCOA (Fig. 4, Table 2) showed a much broader distribution than the rest of the primary OC recoveries yet more limited towards the strongly water-soluble fractions compared to Daellenbach et al. (2017). SCOA concentrations were higher in the coarse fraction compared to PM2.5 (Figs. 6 and 7c, S5) and exhibited higher concentrations during autumn and winter compared to summer (Table 3). A similar profile had previously been linked to a marine origin by Crippa et al. (2013) in Paris; however, Daellenbach et al. (2017) found that SCOA in Switzerland was rather a primary locally emitted source with no marine origin due to its anti-correlation with methane sulfonic acid (MSA). Here we confirm that SCOA did not follow the MSA time series (Fig. S11) but rather the time series of NOx. These observations suggest that this factor is connected to a primary coarse particle episodic source related to traffic.
Primary biological OA (PBOA) exhibited significant contributions from the fragment C2H5O (part of m∕z 61) (Fig. 5) and was more enhanced in summer and spring (Fig. 6). The RPBOA (Fig. 4, Table 2) met the high end of RPBOA in Bozzetti et al. (2016). PBOA appeared mostly in the coarse mode (Table 3, Fig. S5). The mass spectral features, the seasonality and coarse contribution suggested the biological nature of this factor possibly including plant debris. Additional support of this interpretation is provided by the correlation of PBOA with cellulose (Fig. 7d), a polymer mostly found in the cell wall of plants. The correlation improved if only data from summer and spring were considered. The outliers here were the late autumn and winter points when BBOA was more important and PBOA could not as easily be separated by the PMF technique.
One out of the three oxygenated OAs (OOA) was identified as a highly oxidized factor, due to the significant contribution of the fragment CO (Fig. 5) and the high OM ∕ OC ratio (Table 2) which was consistent with the reported OM ∕ OC ratio by Turpin et al. (2001) for non-urban aerosols. This factor peaked mainly in winter and spring and the PM2.5 size fraction exhibited higher concentrations during this period compared to the coarse size fraction (Table 3, Fig. 6). The water solubility of this oxygenated factor was high (Fig. 4, Table 2), which is consistent with the literature values (Daellenbach et al., 2016, 2017) that refer to the sum of all oxygenated factors, as well as with reported water-soluble fractions for highly oxidized compounds (Xu et al., 2017). The yearly median concentration for PM10 was 0.97 µg m−3 (Q25 = 0.86 and Q75=1.09 µg m−3), which accounts for approximately 13 % of the total OA. Out of all the possible correlations with external markers, this factor correlated best with OCf (Fig. 7e); therefore, we chose to name it anthropogenic OOA (AOOA) (see also discussion in Sect. 4.4.2). Both AOOC and OCf followed very similar annual cycles (Fig. S12) with average AOOC ∕ OCf = 0.97 ± 2.49. This observation along with the increase in OCf ∕ ECf as already discussed in Sect. 4.2 could indicate that this factor is linked to secondary organic aerosol from traffic emissions or to transported air masses from industrialized areas. It may also be connected to the oxidation of volatile chemical products such as pesticides, coatings, printing inks or cleaning agents (McDonald et al., 2018). Further discussion about AOOC can be found in Sect. 4.4.
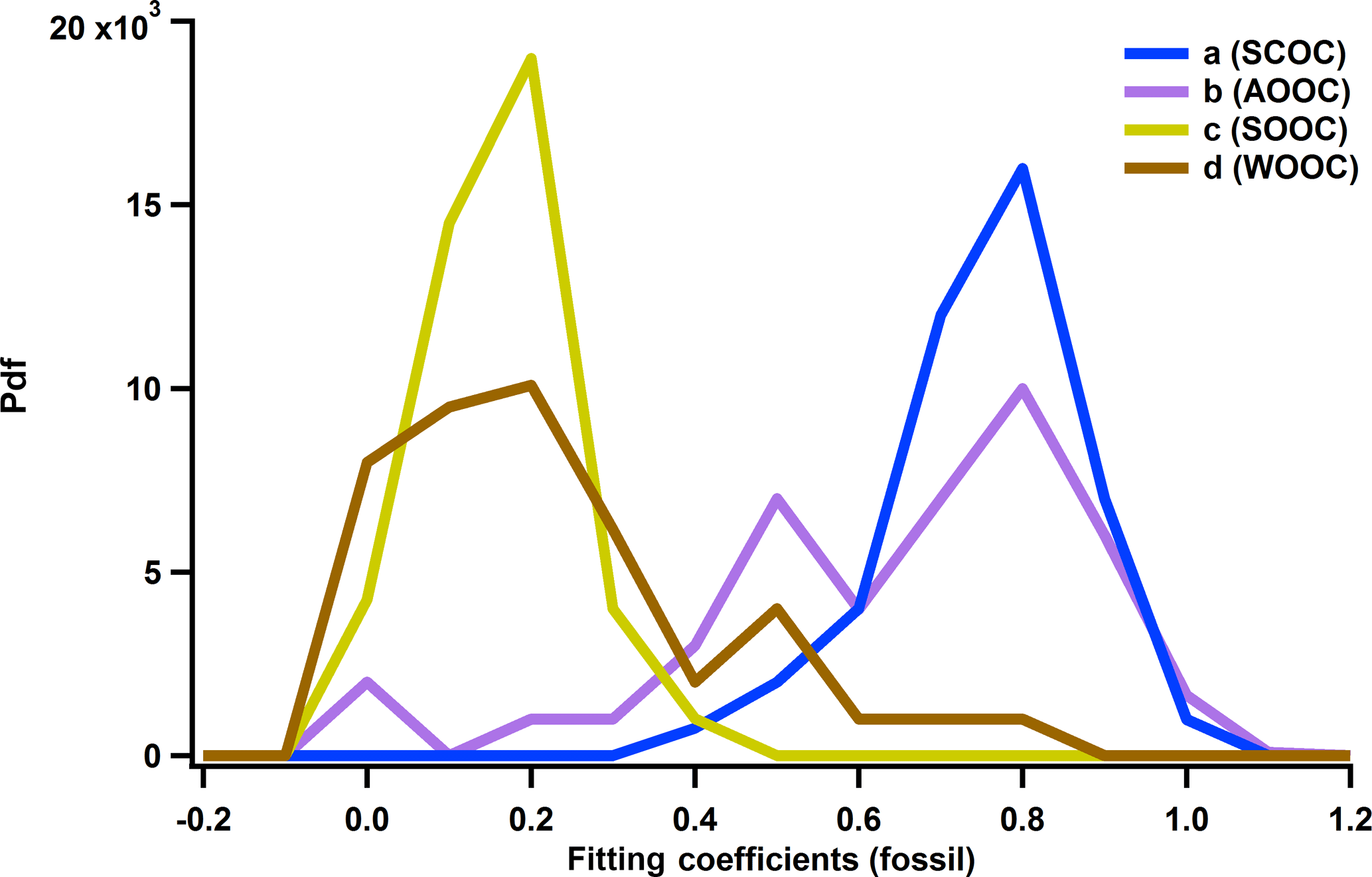
Figure 8Probability density functions of the fitting coefficients of the relative fossil contributions: SCOC in blue, AOOC in purple, SOOC in yellow and WOOC in light brown.
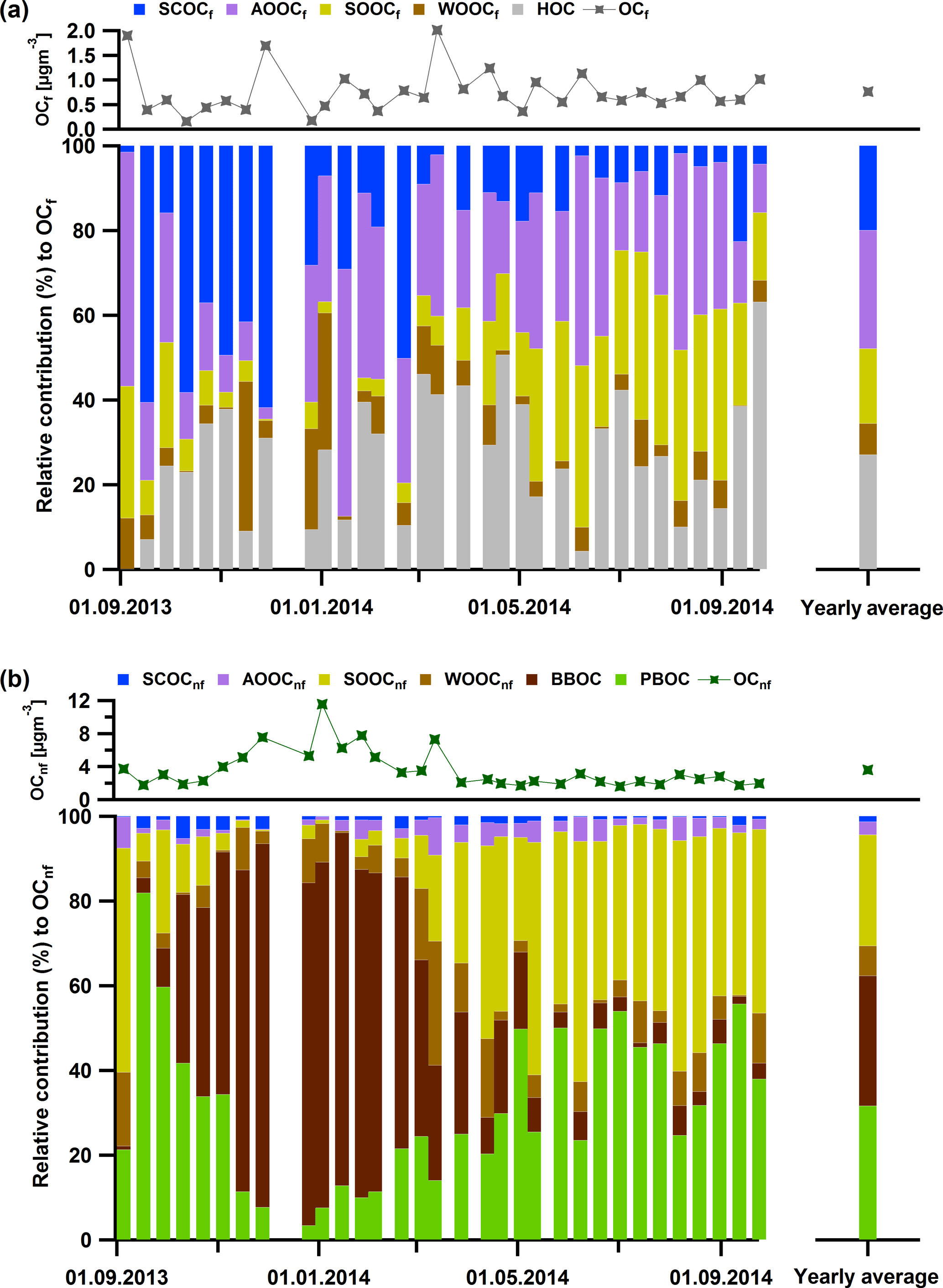
Figure 9Relative contributions to the fossil OC per factor (PM10) (a) and to the non-fossil OC per factor (PM10) (b): BBOC in dark brown, SCOCf and SCOCnf in blue, PBOC in green, AOOCf and AOOCnf in purple, SOOCf and SOOCnf in yellow, and WOOCf and WOOnf in light brown. Note that the total non-fossil concentrations (dark green markers) are on average 6 times higher compared to the fossil ones (dark grey markers).
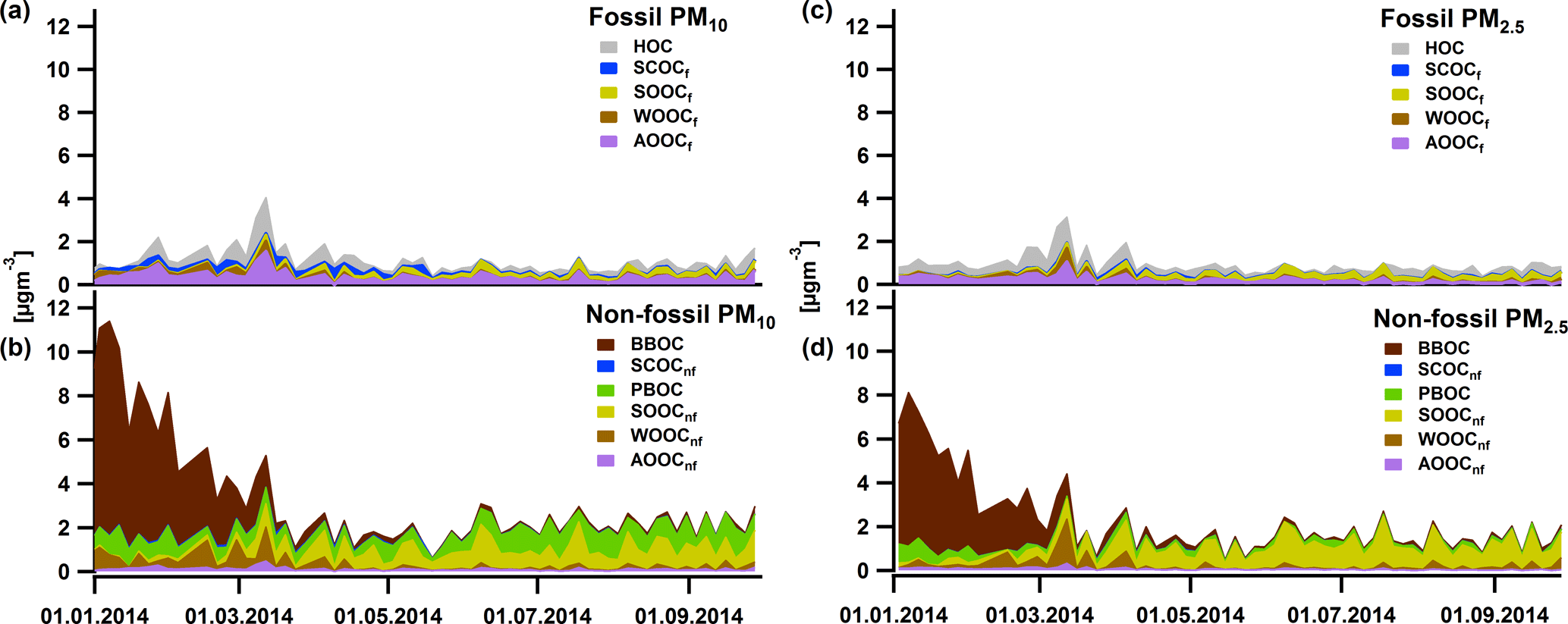
Figure 10Yearly cycles of fossil PM10 (a), non-fossil PM10 (b), fossil PM2.5 (c), and non-fossil PM2.5 (d) OC factors: BBOC in dark brown, SCOCf and SCOCnf in blue, PBOC in green, AOOCf and AOOCnf in purple, SOOCf and SOOCnf in yellow, and WOOCf and WOOnf in light brown. Note that the covered time periods in (a), (b) and (c), (d) are different.

Figure 11Averaged contributions of the fossil and non-fossil primary and secondary OC to the total OC season wise for PM10.
Summer oxygenated OA (SOOA) was mainly identified by the high contribution of the fragment C2H3O+ (m∕z 43) (Fig. 5) (fC2H3O) as well as its seasonal behaviour (Fig. 6). Like all the oxygenated OA factors, it was highly water soluble (Fig. 4, Table 2). The highest concentrations were witnessed in July with values of 4.4 µgm−3 for PM10 in 2013 and 4.3 µgm−3 for PM2.5 in 2014. The bulk contribution of this factor was present in the PM2.5 fraction (Table 3, Fig. S5). The seasonal variability of SOOA followed the daily temperature average (Fig. 6). In fact, SOOA exponentially increased with temperature (Fig. 7f). Such behaviour was also observed in Daellenbach et al. (2017), where they connected this factor to the oxidation of terpene emissions and therefore to biogenic SOA formation. The exponential dependence of SOOA with temperature was also similar to the temperature dependence of the biogenic SOA concentrations from a Canadian terpene-rich forest, reported by Leaitch et al. (2011). A similar factor was identified with an online instrument in Zurich during summer 2011, where the semi-volatile OOA was mainly formed by biogenic sources as the high temperatures favour the biogenic emissions compared to the rest (Canonaco et al., 2015). Finally, the O ∕ C ratio (0.37) fell into the range of the reported O ∕ C ratios measured by chamber-generated SOA (Aiken et al., 2008), which was similar to biogenic SOA produced in flow tubes (Heaton et al., 2007).
Named after its seasonal behaviour (Daellenbach et al., 2017), the third oxygenated factor, winter oxygenated OA (WOOA), exhibited the highest concentrations during winter. WOOA mass spectrum exhibited elevated contributions of the fragment C2H3O+ (Fig. 5), but lower compared to SOOA (for WOOA fC2H3O). It also exhibited a slightly enhanced contribution of the fragment C2H4O which can be an indication that this factor originated from aged biomass burning emissions. Moreover, a similar mass spectral pattern (peaks of fragments C3H3O+, C3H5O, C4H5O and C5H7O at m∕z 55, 73, 85 and 99, respectively) to the one coming from oxygenated products from a wood-burning experiment (Bruns et al., 2015) was found. The recovery of this factor manifested high values (Table 2) and consisted mainly of fine-mode particles (Fig. S5). WOOA also correlated with NH (Fig. S13), which is directly connected to the inorganic secondary ions NO and SO.
4.4 Coupling of offline AMS and 14C analyses
In this section of the paper we will show the combined results of AMS/PMF and radiocarbon analyses. The first part will elaborate on the technical aspect of the analysis by presenting the calculation of the contribution of each factor to the fossil OC. In the second part, a thorough description of each fossil and non-fossil major source will be given. The time series of each fossil and non-fossil fraction for the whole AMS dataset is illustrated in Fig. 10. Contributions of the primary and secondary OC to the total OC will be also discussed and shown in Fig. 11.
4.4.1 Calculation of fossil and non-fossil fraction per factor
To combine the AMS/PMF with the 14C results, the identified sources from AMS/PMF were divided into fossil and non-fossil fractions. HOC was fully assigned to fossil sources assuming that the percentage of biofuel content is negligible. BBOC and PBOC were considered totally non-fossil. To explore the fossil and non-fossil nature of the rest of the factors, we performed multilinear regression using Eq. (17):
where a, b, c and d are the fitting coefficients, weighted by the relative uncertainty of OC HOCi. To investigate the stability of the solution, we obtained distributions of the fitting coefficients by performing 100 bootstrap runs where input data were randomly selected (Fig. 8). The median values (and first and third quartiles) were as follows: a=0.81 (Q25=0.73, Q75=0.88), b=0.77 (Q25=0.54, Q75=0.85), c=0.21 (Q25=0.15, Q75=0.26) and d=0.23 (Q25=0.13, Q75=0.39).
We chose to apply the multilinear regression to the fossil fraction because for the non-fossil part, the errors related to fitting coefficients were very high and the dependences of the OCnf on the input factors were not statistically significant (p values > 0.1).
To calculate the non-fossil part of each factor k (kOCnf), we used the following equation:
This analysis suggests that the major fossil primary sources were HOC and SCOC (81 % ± 11 % fossil), while AOOC (77 % ± 23 % fossil) was the only major fossil secondary source. In terms of the non-fossil sources, the dominating primary sources included BBOC and PBOC, whereas the most important secondary sources were SOOC (79 % ± 11 % non-fossil) and WOOC (77 % ± 23 % non-fossil).
4.4.2 Contribution of fossil and non-fossil, primary and secondary OC to the total OC
The results point out that 81 % ± 11 % (average and 1 standard deviation) of SCOC was fossil (SCOCf). Taking into account the enhanced contribution of SCOC in the coarse size fraction, its sulfur content and its fossil nature, we assume that this factor is linked to primary anthropogenic sources related to traffic, such as tire wear, resuspension of road dust (Bukowiecki et al., 2010), resuspension from asphalt concrete (Gehrig et al., 2010) or asphalt mixture abrasion (in bituminous binder, Fullova et al., 2017). The contribution of SCOCf to the OCf was more important during autumn and winter (up to 62 %, Fig. 9a) in contrast to spring and summer (on average 9 % ± 5 %), while on average the contribution to the OCf was 20 % ± 19 %. The concentrations in winter and autumn were similar and on average for PM10 (PM2.5) 0.22 ± 0.21 µg m−3 (0.03 ± 0.03 µg m−3) (Fig. 10, Table S2), which accounted for 73 % of the total SCOC for this period. However, the contribution of SCOCf to the total OC for the coarse size fraction was not high (5 % ± 8 % on average).
The combined 14C ∕ AMS analysis supported the initial hypothesis that AOOC was mainly related to the oxidation of fossil fuel combustion emissions (e.g. traffic), as AOOC was 77 % ± 23 % fossil (AOOCf) on average. The average contribution of AOOCf to the OCf was 28 % ± 14 % (Fig. 9a), larger than SCOCf, while its contribution to the total OC was 10 % ± 5 % for the coarse OC and 7 % ± 7 % of the fine OC. The yearly cycle exhibited elevated contributions in winter and spring compared to summer and autumn with average values for PM10: 0.47 ± 0.22, 0.43 ± 0.30, 0.39 ± 0.23 and 0.29 ± 0.23 µg m−3, respectively (Fig. 10, Table S2). In winter and spring most of the mass concentration came from the PM2.5 size range in contrast to the other two seasons.
The fossil fractions of SOOC (SOOCf) and WOOC (WOOCf) were low (21 and 23 %, respectively) and could also be attributed to traffic emissions or less likely (due to low emissions) to aged aerosols from residential fossil fuel heating. SOOCf was important during summer with contributions up to 40 % to the OCf and WOOCf was more distinctively present during a few days in autumn and winter (up to 35 % to the OCf) in contrast to the rest of the year (Fig. 9a).
From the non-fossil sources, apart from non-fossil SCOC (SCOCnf) and non-fossil AOOC (AOOCnf), the rest of the factors exhibited a very distinct yearly cycle with BBOC contributing up to 86 % to the OCnf in late autumn and winter (Fig. 9b, yearly average 28 % ± 30 %) and with PBOC and SOOCnf becoming more important in late spring, summer and early autumn with contributions up to 82 and 57 %, respectively (Fig. 9b).
SOOC was 79 % non-fossil which supported the AMS/PMF results: the significance of non-fossil SOOC (SOOCnf) during summer can be attributed to SOA formation from biogenic emissions. The average contribution of SOOCnf to OCnf was 25 % ± 19 % (Fig. 9b). SOOCnf was more pronounced in PM2.5 (on average 1.12 ± 0.40 µg m−3 in summer and 0.75 ± 0.35 µg m−3 in spring, Fig. 10, Table S2). This factor along with PBOC was the main and almost equally important source of OC during spring and summer, with PBOC contributing to OC in the coarse mode (on average 35 % ± 16 % from April to August 2014) and SOOCnf in the fine mode (46 % ± 15 % from April to August 2014). PBOC made up 30 % ± 18 % of the OCnf and the average concentrations of PBOCcoarse for 2014 were 1.00 ± 0.23 µg m−3 in summer and 0.56 ± 0.21 µg m−3 in spring.
Non-fossil WOOC (WOOCnf) dominated over WOOCf (77 % over 23 %). The average yearly contribution to OCnf was low (6 % ± 6 %, Fig. 9b); however, WOOCnf,coarse was apparent during the cold period especially in 2013 with concentrations of 0.88 ± 0.74 µgm−3 on average for winter (0.28 ± 0.28 µg m−3 for autumn) (Fig. 10). In 2014 the concentrations dropped for winter (autumn) with 0.53 ± 0.43 µg m−3 (0.15 ± 0.13 µg m−3) for PM10 and 0.22 ± 0.19 µg m−3 (0.21 ± 0.21 µg m−3) for PM2.5. Based on its yearly cycle (Fig. 10b and d) WOOCnf could be linked to aged OA influenced by wintertime and early spring biomass burning emissions. Therefore, not only AOOCf but also WOOCnf can be related to anthropogenic activities. In other studies (Daellenbach et al., 2017; Bozzetti et al., 2016) this factor was more pronounced; however, in our case in winter most of the OCnf was related to primary biomass burning.
Overall for PM10 the non-fossil primary OC contributions were more important during autumn (57 %) and winter (75 %), whereas in spring and summer the non-fossil secondary OC contributions became more pronounced (32 and 40 %, respectively) (Fig. 11). The dominance of the SOC during the warm period is likely related to the stronger solar radiation which favours the photo-oxidation of biogenic volatile organic compounds and to the elevated biogenic volatile organic compounds emissions.
The coupling of offline AMS and 14C analyses allowed a detailed characterization of the carbonaceous aerosol in the Alpine valley of Magadino for the years 2013–2014. The seasonal variation along with the two size-segregated measurements (PM10 and PM2.5) gave insights into the source apportionment, by for example quantifying the resuspension of road dust or asphalt concrete and estimating its contribution to the OC or by identifying SOC based on SOC precursors. More specifically, seven sources including four primary and three secondary ones were identified. The non-fossil primary sources were dominating during autumn and winter, with BBOC exhibiting by far the highest concentrations. During spring and summer again two non-fossil sources, PBOC in the coarse fraction and SOOCnf in the fine mode, prevailed over the fossil ones. The size-segregated measurements and 14C analysis enabled a better understanding of the primary SCOC factor, which was enhanced in the coarse fraction and was mainly fossil, suggesting that it may originate from resuspension of road dust or tire – asphalt abrasion. The results also showed that SOC was formed mainly by biogenic sources during summer and anthropogenic sources during winter. However, SOC formed possibly by oxidation of traffic emissions or volatile chemical products was also apparent during summer (AOOCf). AOOCf was also important during winter along with SOC linked to transported non-fossil carbonaceous aerosols coming from anthropogenic activities such as biomass burning (WOOCnf).
The data are available upon request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-6187-2018-supplement.
The authors declare that they have no conflict of interest.
This work is funded by the Swiss Federal Office for the Environment (FOEN),
OSTLUFT and the cantons of Basel, Graubünden, Ticino, Thurgau and Valais.
The LABEX OSUG@2020 (ANR-10-LABX-56) funded analytical instruments at IGE.
Edited by: Eleanor Browne
Reviewed by: two anonymous referees
Agrios, K., Salazar, G. A., Zhang, Y. L., Uglietti, C., Battaglia, M., Luginbühl, M., Ciobanu, V. G., Vonwiller, M., and Szidat, S.: Online coupling of pure O2 thermo-optical methods – 14C AMS for source apportionment of carbonaceous aerosols study, Nucl. Instrum. Meth. B., 361, 288–293, https://doi.org/10.1016/j.nimb.2015.06.008, 2015.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, Z. B., Alfarra, M. R., Prevot, A. S., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O ∕ C and OM ∕ OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Alfarra, M. R., Prévôt, A. S. H., Szidat, S., Sandradewi, J.,Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U.: Identification of the mass spectral signature of organic aerosols from wood burning emissions, Environ. Sci. Technol., 41, 5770–5777, https://doi.org/10.1021/es062289b, 2007.
Allan, J. D., Jimenez, J. L., Williams, P. I., Alfarra, M. R., Bower, K. N., Jayne, J. T., Coe, H., and Worsnop, D. R.: Quantitative sampling using an Aerodyne aerosol mass spectrometer – 1. Techniques of data interpretation and error analysis, J. Geophys. Res.-Atmos., 108, 4090, https://doi.org/10.1029/2002JD002358, 2003.
Beekmann, M., Prévôt, A. S. H., Drewnick, F., Sciare, J., Pandis, S. N., Denier van der Gon, H. A. C., Crippa, M., Freutel, F., Poulain, L., Ghersi, V., Rodriguez, E., Beirle, S., Zotter, P., von der Weiden-Reinmüller, S. L., Bressi, M., Fountoukis, C., Petetin, H., Szidat, S., Schneider, J., Rosso, A., El Haddad, I., Megaritis, A., Zhang, Q. J., Michoud, V., Slowik, J. G., Moukhtar, S., Kolmonen, P., Stohl, A., Eckhardt, S., Borbon, A., Gros, V., Marchand, N., Jaffrezo, J. L., Schwarzenboeck, A., Colomb, A., Wiedensohler, A., Borrmann, S., Lawrence, M., Baklanov, A., and Baltensperger, U.: In situ, satellite measurement and model evidence on the dominant regional contribution to fine particulate matter levels in the Paris megacity, Atmos. Chem. Phys., 15, 9577–9591, https://doi.org/10.5194/acp-15-9577-2015, 2015.
Bonvalot, L., Tuna, T., Fagault, Y., Jaffrezo, J. L., Jacob, V., Chevrier, F., and Bard, E.: Estimating contributions from biomass burning and fossil fuel combustion by means of radiocarbon analysis of carbonaceous aerosols: application to the Valley of Chamonix, Atmos. Chem. Phys., 16, 13753–13772, https://doi.org/10.5194/acp-16-13753-2016, 2016.
Bozzetti, C., Daellenbach, K., R., Hueglin, C., Fermo, P., Sciare, J., Kasper-Giebl, A., Mazar, Y., Abbaszade, G., El Kazzi, M., Gonzalez, R., Shuster Meiseles, T., Flasch, M., Wolf, R., Křepelová, A., Canonaco, F., Schnelle-Kreis, J., Slowik, J. G., Zimmermann, R., Rudich, Y., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: Size-resolved identification, characterization, and quantification of primary biological organic aerosol at a European rural site, Environ. Sci. Technol., 50, 3425–3434, https://doi.org/10.1021/acs.est.5b05960, 2016.
Bozzetti, C., Sosedova, Y., Xiao, M., Daellenbach, K. R., Ulevicius, V., Dudoitis, V., Mordas, G., Byčenkienė, S., Plauškaitė, K., Vlachou, A., Golly, B., Chazeau, B., Besombes, J.-L., Baltensperger, U., Jaffrezo, J.-L., Slowik, J. G., El Haddad, I., and Prévôt, A. S. H.: Argon offline-AMS source apportionment of organic aerosol over yearly cycles for an urban, rural, and marine site in northern Europe, Atmos. Chem. Phys., 17, 117–141, https://doi.org/10.5194/acp-17-117-2017, 2017a.
Bozzetti, C., El Haddad, I., Salameh, D., Daellenbach, K. R., Fermo, P., Gonzalez, R., Minguillón, M. C., Iinuma, Y., Poulain, L., Elser, M., Müller, E., Slowik, J. G., Jaffrezo, J.-L., Baltensperger, U., Marchand, N., and Prévôt, A. S. H.: Organic aerosol source apportionment by offline-AMS over a full year in Marseille, Atmos. Chem. Phys., 17, 8247–8268, https://doi.org/10.5194/acp-17-8247-2017, 2017b.
Bruns E., Krapf, M., Orasche, J., Huang, Y., Zimmermann, R., Drinovec, L., Močnik, G., El-Haddad, I., Slowik, J. G., Dommen, J., Baltensperger, U., and Prévôt, A. S. H.: Characterization of primary and secondary wood combustion products generated under different burner loads, Atmos., Chem., Phys., 15, 2825–2841, https://doi.org/10.5194/acp-15-2825-2015, 2015.
Bukowiecki, N., Lienemann, P., Hill, M., Furger, M., Richard, A., Amato, F., Prevot, A. S. H., Baltensperger, U., Buchmann, B., and Gehrig, R.: PM10 emission factors for non-exhaust particles generated by road traffic in an urban street canyon and along a freeway in Switzerland, Atmos. Environ., 44, 2330–2340, https://doi.org/10.1016/j.atmosenv.2010.03.039, 2010.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the Aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Canonaco, F., Crippa, M., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: SoFi, an IGOR-based interface for the efficient use of the generalized multilinear engine (ME-2) for the source apportionment: ME-2 application to aerosol mass spectrometer data, Atmos. Meas. Tech., 6, 3649–3661, https://doi.org/10.5194/amt-6-3649-2013, 2013.
Canonaco, F., Slowik, J. G., Baltensperger, U., and Prévôt, A. S. H.: Seasonal differences in oxygenated organic aerosol composition: implications for emissions sources and factor analysis. Atmos. Chem. Phys. 15, 6993–7002, https://doi.org/10.5194/acp-15-6993-2015, 2015.
Cavalli, F., Viana, M., Yttri, K. E., Genberg, J., and Putaud, J.-P.: Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol, Atmos. Meas. Tech., 3, 79–89, https://doi.org/10.5194/amt-3-79-2010, 2010.
Chirico, R., Prevot A. S. H., DeCarlo P. F., Heringa M. F., Richter R., Weingartner E., and Baltensperger U.: Aerosol and trace gas vehicle emission factors measured in a tunnel using an aerosol mass spectrometer and other on-line instrumentation, Atmos. Environ., 45, 2182–2192, https://doi.org/10.1016/j.atmosenv.2011.01.069, 2011.
Crippa, M., El Haddad, I., Slowik, J. G., DeCarlo, P.F., Mohr, C., Heringa, M. F., Chirico, R., Marchand, N., L., Sciare, J., Baltensperger, U., and Prévôt, A. S. H.: Identification of marine and continental aerosol sources in Paris using high resolution aerosol mass spectrometry, J. Geophys. Res., 118, 1950–1963, https://doi.org/10.1002/jgrd.50151, 2013.
Daellenbach, K. R., Bozzetti, C., Krepelova, A., Canonaco, F., Huang, R.-J., Wolf, R., Zotter, P., Crippa, M., Slowik, J., Zhang, Y., Szidat, S., Baltensperger, U., Prévôt, A. S. H., and El Haddad, I.: Characterization and source apportionment of organic aerosol using offline aerosol mass spectrometry, Atmos. Meas. Tech., 9, 23–39, https://doi.org/10.5194/amt-9-23-2016, 2016.
Daellenbach K. R., Stefenelli G., Bozzetti C., Vlachou A., Fermo P., Gonzalez R., Piazzalunga A., Colombi C., Canonaco F., Kasper-Giebl A., Jaffrezo J.-L., Bianchi F., Slowik J. G., Baltensperger U., El-Haddad I., and Prévôt A. S. H.: Long-term chemical analysis and organic aerosol source apportionment at 9 sites in Central Europe: Source identification and uncertainty assessment, Atmos. Chem. Phys., 17, 13265–13282, https://doi.org/10.5194/acp-2017-124, 2017.
Davison, A. C. and Hinkley, D. V.: Bootstrap Methods and Their Application, Cambridge University Press, Cambridge, UK, 582 pp., 1997.
Dusek, U., Hitzenberger, R., Kasper-Giebl, A., Kistler, M., Meijer, H. A. J., Szidat, S., Wacker, L., Holzinger, R., and Röckmann, T.: Sources and formation mechanisms of carbonaceous aerosol at a regional background site in the Netherlands: insights from a year-long radiocarbon study, Atmos. Chem. Phys., 17, 3233–3251, https://doi.org/10.5194/acp-17-3233-2017, 2017.
El Haddad, I., Marchand, N., Dron, J., Temime-Roussel, B., Quivet, E., Wortham, H., Jaffrezo, J-L., Baduel, C., Voisin, D., Besombes, J. L., and Gille, G.: Comprehensive primary particulate organic characterization of vehicular exhaust emissions in France, Atmos. Environ., 43, 6190–6198, https://doi.org/10.1016/j.atmosenv.2009.09.001, 2009.
El Haddad, I., D'Anna, B., Temime-Roussel, B., Nicolas, M., Boreave, A., Favez, O., Voisin, D., Sciare, J., George, C., Jaffrezo, J. L., Wortham, H., and Marchand, N.: Towards a better understanding of the origins, chemical composition and aging of oxygenated organic aerosols: case study of a Mediterranean industrialized environment, Marseille, Atmos. Chem. Phys., 13, 7875–7894, https://doi.org/10.5194/acp-13-7875-2013, 2013.
Elser, M., Huang, R.-J., Wolf, R., Slowik, J. G., Wang, Q., Canonaco, F., Li, G., Bozzetti, C., Daellenbach, K. R., Huang, Y., Zhang, R., Li, Z., Cao, J., Baltensperger, U., El-Haddad, I., Prévôt, A. S. H., and André, S. H.: New insights into PM2.5 chemical composition and sources in two major cities in China during extreme haze events using aerosol mass spectrometry, Atmos. Chem. Phys., 16, 3207–3225, https://doi.org/10.5194/acp-16-3207-2016, 2016.
Favez, O., El Haddad, I., Piot, C., Boréave, A., Abidi, E., Marchand, N., Jaffrezo, J.-L., Besombes, J.-L., Personnaz, M.-B., Sciare, J., Wortham, H., George, C., and D'Anna, B.: Inter-comparison of source apportionment models for the estimation of wood burning aerosols during wintertime in an Alpine city (Grenoble, France), Atmos. Chem. Phys., 10, 5295–5314, https://doi.org/10.5194/acp-10-5295-2010, 2010.
Fröhlich, R., Cubison, M. J., Slowik, J. G., Bukowiecki, N., Prevot, A. S. H., Baltensperger, U., Schneider, J., Kimmel, J. R., Gonin, M., Rohner, U., Worsnop, D. R., and Jayne J. T.: The ToF-ACSM: a portable aerosol chemical speciation monitor with TOFMS detection, Atmos. Meas. Tech., 6, 3225–3241, https://doi.org/10.5194/amt-6-3225-2013, 2013.
Fullova, D., Durcanska D., and Hegrova, J.: Particulate matter mass concentrations produced from pavement surface abrasion, MATEC Web of Conferences, 117, 00048, https://doi.org/10.1051/matecconf/201711700048, 2017.
Gehrig, R., Zeyer, K., Bukowiecki, N., Lienemann, P., Poulikakos, L. D., Furger, M., and Buchmann, B.: Mobile load simulators – A tool to distinguish between the emissions due to abrasion and resuspension of PM10 from road surfaces, Atmos. Environ., 44, 4937–4943, https://doi.org/10.1016/j.atmosenv.2010.08.020, 2010.
Gelencsér, A., May, B., Simpson, D., Sánchez-Ochoa, A., Kasper-Giebl, A., Puxbaum, H., Caseiro, A., Pio, C., and Legrand, M.: Source apportionment of PM2.5 organic aerosol over Europe: Primary/secondary, natural/anthropogenic, and fossil/biogenic origin, J. Geophys. Res.-Atmos., 112, D23S04, https://doi.org/10.1029/2006JD008094, 2007.
Genberg, J., Hyder, M., Stenström, K., Bergström, R., Simpson, D., Fors, E. O., Jönsson, J. Å., and Swietlicki, E.: Source apportionment of carbonaceous aerosol in southern Sweden, Atmos. Chem. Phys., 11, 11387–11400, https://doi.org/10.5194/acp-11-11387-2011, 2011.
Gianini, M. F. D., Gehrig, R., Fischer, A., Ulrich, A., Wichser, A., and Hueglin, C.: Chemical composition of PM10 in Switzerland: An analysis for 2008/2009 and changes since 1998/1999, Atmos. Environ., 54, 97–106, https://doi.org/10.1016/j.atmosenv.2012.02.037, 2012.
Gilardoni, S., Vignati, E., Cavalli, F., Putaud, J. P., Larsen, B. R., Karl, M., Stenström, K., Genberg, J., Henne, S., and Dentener, F.: Better constraints on sources of carbonaceous aerosols using a combined 14C – macro tracer analysis in a European rural background site, Atmos. Chem. Phys., 11, 5685–5700, https://doi.org/10.5194/acp-11-5685-2011, 2011.
Glasius, M., la Cour, A., and Lohse, C.: Fossil and nonfossil carbon in fine particulate matter: A study of five European cities, J. Geophys. Res., 116, D11302, https://doi.org/10.1029/2011JD015646, 2011.
Heaton, K. J., Dreyfus, M. A., Wang, S., and Johnston, M. V.: Oligomers in the early stage of biogenic secondary organic aerosol formation and growth, Environ. Sci. Technol., 41, 6129–6136, https://doi.org/10.1021/es070314n, 2007.
Herich, H., Gianini, M. F. D., Piot, C., Močnik, G., Jaffrezo, J. L., Besombes, J. L., Prévôt, A. S. H., and Hueglin, C.: Overview of the impact of wood burning emissions on carbonaceous aerosols and PM in large parts of the Alpine region, Atmos. Environ., 89, 64–75, https://doi.org/10.1016/j.atmosenv.2014.02.008, 2014.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J., Han, Y., Dällenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Jaffrezo, J.-L., Calas, T., and Bouchet, M.: Carboxylic acids measurements with ionic chromatography, Atmos. Environ., 32, 2705–2708, https://doi.org/10.1016/S1352-2310(98)00026-0, 1998.
Jaffrezo, J.-L., Aymoz, G., Delaval, C., and Cozic, J.: Seasonal variations of the water soluble organic carbon mass fraction of aerosol in two valleys of the French Alps, Atmos. Chem. Phys., 5, 2809–2821, https://doi.org/10.5194/acp-5-2809-2005, 2005.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prévôt, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Kunit, M. and Puxbaum, H.: Enzymatic determination of the cellulose content of atmospheric aerosols, Atmos. Environ., 30, 1233–1236, https://doi.org/10.1016/1352-2310(95)00429-7, 1996.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Lanz, V. A., Prévôt, A. S. H., Alfarra, M. R., Weimer, S., Mohr, C., DeCarlo, P. F., Gianini, M. F. D., Hueglin, C., Schneider, J., Favez, O., D'Anna, B., George, C., and Baltensperger, U.: Characterization of aerosol chemical composition with aerosol mass spectrometry in Central Europe: an overview, Atmos. Chem. Phys., 10, 10453–10471, https://doi.org/10.5194/acp-10-10453-2010, 2010.
Leaitch, W. R. Macdonald, A. M., Brickell, P. C., Liggio, J., Sjostedt, S. J., Vlasenko, A., Bottenheim, J. W., Huang, L., Li, S.-M., Liu, P. S. K., Toom-Sauntry, D., Hayden, K. A., Sharma, S., Shantz, N. C., Wiebe H. A., Zhang, W., Abbatt, J. P. D., Slowik, J. G., Chang, Rachel, Y.-W., Russell, L. M., Schwartz, R. E., Takahama, S., Jayne, J. T., and Ng, N. L.: Temperature response of the submicron organic aerosol from temperate forests, Atmos. Environ., 45, 6696–6704, https://doi.org/10.1016/j.atmosenv.2011.08.047, 2011.
Lemire, R. K., Allen, T. D., Klouda A. G., and Lewis W. C.: Fine particulate matter source attribution for Southeast Texas using 14C ∕ 13C ratios, J. Geophys. Res., 107, 4631, https://doi.org/10.1029/2002JD002339, 2002.
Levin, I., Naegler, T., Kromer, B., Diehl, M., Francey, R. J., Gomez-Pelaez, A. J., Steele, L. P., Wagenbach, D., Weller, R., and Worthy, D. E.: Observations and modelling of the global distribution and long-term trend of atmospheric 14CO2, Tellus B, 62, 26–46, https://doi.org/10.1111/j.1600-0889.2009.00446.x, 2010a.
Levin, E. J. T., McMeeking, G. R., Carrico, C. M., Mack, L. E., Kreidenweis, S. M., Wold, C. E., Moosmüller, H., Arnott, W. P., Hao, W. M., Collett, J. L., and Malm, W. C.: Biomass burning smoke aerosol properties measured during Fire Laboratory at Missoula Experiments (FLAME), J. Geophys. Res.-Atmos., 115, D18210, https://doi.org/10.1029/2009jd013601, 2010b.
Li, X. Dallmann, T. R., May, A. A., Tkacik, D. S., Lambe, A. T., Jayne, J. T., Croteau, P. L., and Presto, A. A.: Gas-particle partitioning of vehicle emitted primary organic aerosol measured in a traffic tunnel, Environ. Sci. Technol., 50, 12146–12155, https://doi.org/10.1021/acs.est.6b01666, 2016.
McDonald, B. C., de Gouw, J. A., Gilman, J. B., Jathar, S. H., Akherati, A., Cappa, C. D., Jimenez, J. L.,Lee-Taylor, J.,Hayes, P. L.,McKeen, S. A., Cui, Y. Y., Kim, S.-W.,Gentner, Drew R., Isaacman-VanWertz, G., Goldstein, A. H.,Harley, R. A., Frost, G. J., Roberts, J. M., Ryerson, T. B., and Trainer, M.: Volatile chemical products emerging as largest petrochemical source of urban organic emissions, Science, 359, 760–764, https://doi.org/10.1126/science.aaq0524, 2018.
Minguillón, M. C., Perron, N., Querol, X., Szidat, S., Fahrni, S. M., Alastuey, A., Jimenez, J. L., Mohr, C., Ortega, A. M., Day, D. A., Lanz, V. A., Wacker, L., Reche, C., Cusack, M., Amato, F., Kiss, G., Hoffer, A., Decesari, S., Moretti, F., Hillamo, R., Teinilä, K., Seco, R., Peñuelas, J., Metzger, A., Schallhart, S., Müller, M., Hansel, A., Burkhart, J. F., Baltensperger, U., and Prévôt, A. S. H.: Fossil versus contemporary sources of fine elemental and organic carbonaceous particulate matter during the DAURE campaign in Northeast Spain, Atmos. Chem. Phys., 11, 12067–12084, https://doi.org/10.5194/acp-11-12067-2011, 2011.
Mohn, J., Szidat, S., Fellner, J., Rechberger, H., Quartier, R., Buchmann, B., and Emmenegger, L.: Determination of biogenic and fossil CO2 emitted by waste incineration based on 14CO2 and mass balances, Bioresource Technol., 99, 6471–6479, https://doi.org/10.1016/j.biortech.2007.11.042, 2008.
Ng, N. L., Herndon, S. C., Trimborn, A., Canagaratna, M. R. Croteau, P. L., Onasch, T. B. Sueper, D., Worsnop, D. R., Zhang, Q., Sun, Y. L., and Jayne, J. T.: An Aerosol Chemical Speciation Monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol, Aerosol Sci. Tech., 45, 770–784, https://doi.org/10.1080/02786826.2011.560211, 2011.
Paatero, P.: Least squares formulation of robust non-negative factor analysis, Chemom. Intell. Lab. Syst., 37, 23–35, https://doi.org/10.1016/S0169-7439(96)00044-5, 1997.
Paatero, P.: The multilinear engine – A table-driven, least squares program for solving multilinear problems, including the n-way parallel factor analysis model, J. Comput. Graph. Stat., 8, 854–888, 1999.
Paatero, P. and Hopke, P. K.: Discarding or downweighting high-noise variables in factor analytic models, Anal. Chim. Acta, 490, 277–289, https://doi.org/10.1016/s0003-2670(02)01643-4, 2003.
Paatero, P. and Tapper U.: Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203,1994.
Piazzalunga, A., Bernardoni, V., Fermo, P., Valli, G., and Vecchi, R.: Technical Note: On the effect of water-soluble compounds removal on EC quantification by TOT analysis in urban aerosol samples, Atmos. Chem. Phys., 11, 10193–10203, https://doi.org/10.5194/acp-11-10193-2011, 2011.
Pieber, S. M., El Haddad, I., Slowik, J. G., Canagaratna, M. R., Jayne, J. T., Platt, S. M., Bozzetti, C., Daellenbach, K. R., Fröhlich, R., Vlachou, A., Klein, F., Dommen, J., Miljevic, B., Jimenez, J. L., Worsnop, D. R., Baltensperger, U., and Prévôt, A. S. H.: Inorganic salt interference on CO in aerodyne AMS and ACSM organic aerosol composition studies, Environ. Sci. Technol., 50, 10494–10503, https://doi.org/10.1021/acs.est.6b01035, 2016.
Puxbaum, H., Caseiro, A., Sànchez-Ochoa, A., Kasper-Giebl, A., Claeys, M., Gelencser, A, Legrand, M., Preunkert S., and Pio, C.: Levoglucosan levels at background sites in Europe for assessing the impact of biomass combustion on the European aerosol background, J. Geophys. Res., 112, D23S05, https://doi.org/10.1029/2006JD008114, 2007.
Sandradewi, J., Prévôt, A. S. H., Alfarra, M. R., Szidat, S.,Wehrli, M. N., Ruff, M., Weimer, S., Lanz, V. A., Weingartner, E., Perron, N., Caseiro, A., Kasper-Giebl, A., Puxbaum, H., Wacker, L., and Baltensperger, U.: Comparison of several wood smoke markers and source apportionment methods for wood burning particulate mass, Atmos. Chem. Phys. Discuss., 8, 8091–8118, https://doi.org/10.5194/acpd-8-8091-2008, 2008.
Schichtel, B. A., Malm, W. C., Bench, G., Fallon, S., McDade, C. E., Chow, J. C., and Watson, J. G.: Fossil and contemporary fine particulate carbon fractions at 12 rural and urban sites in the United States, J. Geophys. Res., 113, D02311, https://doi.org/10.1029/2007JD008605, 2008.
Stuiver, M. and Polach, H. A.: Reporting of 14C data, Radiocarbon, 19, 355–363, https://doi.org/10.1017/S0033822200003672, 1977.
Szidat, S., Jenk, T. M., Gäggeler, H. W., Synal, H. A., Fisseha, R., Baltensperger, U., Kalberer, M., Samburova, V., Reimann, S., Kasper-Giebl, A., and Hajdas, I.: Radiocarbon (14C)-deduced biogenic and anthropogenic contributions to organic carbon (OC) of urban aerosols from Zürich, Switzerland, Atmos. Environ., 38, 4035–4044, https://doi.org/10.1016/j.atmosenv.2004.03.066, 2004.
Szidat, S., Prévôt, A. S. H., Sandradewi, J., Alfarra, M. R., Synal, H.-A., Wacker, L., and Baltensperger, U.: Dominant impact of residential wood burning on particulate matter in Alpine valleys during winter, Geophys. Res. Lett., 34, L5820, https://doi.org/10.1029/2006gl028325, 2007.
Szidat, S., Ruff, M., Perron, N., Wacker, L. Synal, H. A., Hallquist, M., Shannigrahi, A. S., Yttri, K. E., Dye, C., and Simpson, D.: Fossil and non-fossil sources of organic carbon (OC) and elemental carbon (EC) in Göteborg, Sweden, Atmos. Chem. Phys, 9, 1521–1535, https://doi.org/10.5194/acp-9-1521-2009, 2009.
Szidat, S., Salazar, G. A., Vogel, E., Battaglia, M., Wacker, L., Synal, H.-A., and Türler, A.: 14C analysis and sample preparation at the new Bern Laboratory for the Analysis of Radiocarbon with AMS (LARA), Radiocarbon, 56, 561–566, https://doi.org/10.2458/56.17457, 2014.
Turpin, B. J. and Lim, H-J.: Species contributions to PM2.5 mass concentrations: revisiting common assumptions for estimating organic mass, Aerosol Sci. Technol., 35, 602–610, https://doi.org/10.1080/02786820119445, 2001.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Ulevicius, V., Byčenkienė, S., Bozzetti, C., Vlachou, A., Plauškaitė, K., Mordas, G., Dudoitis, V., Abbaszade, G.,Remeikis, V., Garbaras, A., Masalaite, A., Blees, J., Fröhlich, R., Dällenbach, K. R., Canonaco, F., Slowik, J. G., Dommen, J., Zimmermann, R., Schnelle-Kreis, J., Salazar, G. A., Agrios, K., Szidat, S., El Haddad, I., and Prévôt, A. S. H.: Fossil and non-fossil source contributions to atmospheric carbonaceous aerosols during extreme spring grassland fires in Eastern Europe, Atmos. Chem. Phys, 16, 5513–5529, https://doi.org/10.5194/acp-16-5513-2016, 2016.
Williams, L. R., Gonzalez, L. A., Peck, J., Trimborn, D., McInnis, J., Farrar, M. R., Moore, K. D., Jayne, J. T., Robinson, W. A., Lewis, D. K., Onasch, T. B., Canagaratna, M. R., Trimborn, A., Timko, M. T., Magoon, G., Deng, R., Tang, D., de la Rosa Blanco, E., Prévôt, A. S. H., Smith, K. A., and Worsnop, D. R.: Characterization of an aerodynamic lens for transmitting particles greater than 1 micrometer in diameter into the Aerodyne aerosol mass spectrometer, Atmos. Meas. Tech., 6, 3271–3280, https://doi.org/10.5194/amt-6-3271-2013, 2013.
Xu, L., Guo, H., Weber, R. J., and Ng, N. L.: Chemical characterization of water-soluble organic aerosol in contrasting rural and urban environments in the Southeastern United States, Environ. Sci. Technol., 51, 78–88, https://doi.org/10.1021/acs.est.6b05002, 2017.
Zhang, Y. L., Perron, N., Ciobanu, V. G., Zotter, P., Minguillón, M. C., Wacker, L., Prévôt, A. S. H., Baltensperger, U., and Szidat, S.: On the isolation of OC and EC and the optimal strategy of radiocarbon-based source apportionment of carbonaceous aerosols, Atmos. Chem. Phys., 12, 10841–10856, https://doi.org/10.5194/acp-12-10841-2012, 2012.
Zhang, Y. L., Zotter P., Perron N., Prévôt, A. S. H, Wacker L., and Szidat, S.: Fossil and non-fossil sources of different carbonaceous fractions in fine and coarse particles by radiocarbon measurement, Radiocarbon, 55, 1510–1520, https://doi.org/10.2458/azu_js_rc.55.16278, 2013.
Zhang, Y. L., Kawamura, K., Agrios, K., Lee, M., Salazar G., and Szidat, S.: Fossil and nonfossil sources of organic and elemental carbon aerosols in the outflow from Northeast China, Environ. Sci. Technol. 50, 6284–6292, https://doi.org/10.1021/acs.est.6b00351, 2016.
Zhang, Y. L., Ren, H., Sun, Y., Cao, F., Chang, Y., Liu, S., Lee, X., Agrios, K., Kawamura, K., Liu, D., Ren, L., Du, W., Wang, Z., Prevot, A. S. H., Szidat, S., and Fu, P. Q.: High contribution of non-fossil sources to submicrometer organic aerosols in Beijing, China, Environ. Sci. Technol. 51, 7842–7852, https://doi.org/10.1021/acs.est.7b01517, 2017.
Zotter, P., El-Haddad, I., Zhang, Y. L., Hayes, P. L., Zhang, X., Lin, Y.-H., Wacker, L., Schnelle-Kreis, J., Abbaszade, G., Zimmermann, R., Surratt, J. D., Weber, R., Jimenez, J. L., Szidat, S., Baltensperger, U., and Prévôt, A. S. H.: Diurnal cycle of fossil and nonfossil carbon using radiocarbon analyses during CalNex, J. Geophys. Res.-Atmos., 119, 6818–6835, https://doi.org/10.1002/2013jd021114, 2014a.
Zotter, P., Ciobanu, V. G., Zhang, Y. L., El-Haddad, I., Macchia, M., Daellenbach, K. R., Salazar, G. A., Huang, R.-J., Wacker, L., Hueglin, C., Piazzalunga, A., Fermo, P., Schwikowski, M., Baltensperger, U., Szidat, S., and Prévôt, A. S. H.: Radiocarbon analysis of elemental and organic carbon in Switzerland during winter-smog episodes from 2008 to 2012 – Part 1: Source apportionment and spatial variability, Atmos. Chem. Phys., 14, 13551–13570, https://doi.org/10.5194/acp-14-13551-2014, 2014b.