the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 15 Mar 2018
| 15 Mar 2018
Direct measurement of NO3 radical reactivity in a boreal forest
Jonathan Liebmann
Einar Karu
Nicolas Sobanski
Jan Schuladen
Mikael Ehn
Simon Schallhart
Lauriane Quéléver
Heidi Hellen
Hannele Hakola
Thorsten Hoffmann
Jonathan Williams
Horst Fischer
Jos Lelieveld
We present the first direct measurements of NO3 reactivity (or inverse lifetime, s−1) in the Finnish boreal forest. The data were obtained during the IBAIRN campaign (Influence of Biosphere-Atmosphere Interactions on the Reactive Nitrogen budget) which took place in Hyytiälä, Finland during the summer/autumn transition in September 2016. The NO3 reactivity was generally very high with a maximum value of 0.94 s−1 and displayed a strong diel variation with a campaign-averaged nighttime mean value of 0.11 s−1 compared to a daytime value of 0.04 s−1. The highest nighttime NO3 reactivity was accompanied by major depletion of canopy level ozone and was associated with strong temperature inversions and high levels of monoterpenes. The daytime reactivity was sufficiently large that reactions of NO3 with organic trace gases could compete with photolysis and reaction with NO. There was no significant reduction in the measured NO3 reactivity between the beginning and end of the campaign, indicating that any seasonal reduction in canopy emissions of reactive biogenic trace gases was offset by emissions from the forest floor. Observations of biogenic hydrocarbons (BVOCs) suggested a dominant role for monoterpenes in determining the NO3 reactivity. Reactivity not accounted for by in situ measurement of NO and BVOCs was variable across the diel cycle with, on average, ≈ 30 % “missing” during nighttime and ≈ 60 % missing during the day. Measurement of the NO3 reactivity at various heights (8.5 to 25 m) both above and below the canopy, revealed a strong nighttime, vertical gradient with maximum values closest to the ground. The gradient disappeared during the daytime due to efficient vertical mixing.
- Article
(2505 KB) - Full-text XML
-
Supplement
(901 KB) - BibTeX
- EndNote





Biogenic and anthropogenic volatile organic compounds (VOCs) have a significant impact on air quality and human health and knowledge of their tropospheric lifetimes, determined by the oxidizing capacity of the lowermost atmosphere, is a prerequisite to predicting future atmospheric composition and climate change (Lelieveld et al., 2008). Recent estimates (Guenther et al., 2012) suggest that about 1000 Tg of biogenic volatile organic compounds (BVOCs), are emitted annually by vegetation. The boreal forest covers an area of ≈ 15 million km2 worldwide, which is comparable to that covered by tropical rainforest (Eerdekens et al., 2009). Forests emit large amounts of unsaturated hydrocarbons in the form of the terpenoids such as, isoprene (2-methylbuta-1,3-diene, C5H8), monoterpenes (C10H16), and sesquiterpenes (C15H24) that have a significant impact on HOx (HO + HO2) and NOx (NO + NO2) budgets (Hakola et al., 2003; Tarvainen et al., 2005; Holzke et al., 2006; Lappalainen et al., 2009) and the formation of secondary organic particle (Hallquist et al., 2009).
Along with the reaction with O3, BVOCs are oxidized in the troposphere by reactions with OH and NO3 radicals. OH radical-induced oxidation mainly takes place during daytime with the NO3 radical (formed by reaction of O3 with NO2, Reaction R1) accounting for the major fraction of radical-induced loss of BVOC at nighttime (Wayne et al., 1991; Atkinson, 2000; Atkinson and Arey, 2003a, b; Brown and Stutz, 2012; Mogensen et al., 2015; Ng et al., 2017; Liebmann et al., 2017). The rapid photolysis of NO3 by sunlight (Reactions R5, R6) and reaction with NO (Reaction R2) typically reduces its lifetime to a few seconds during daytime. At nighttime, reaction of NO3 with NO2 results in thermal equilibrium between NO3 and N2O5 (Reactions R3, R4).
Reactions (R1) to (R6) do not represent a reduction of NOx as no reactive nitrogen species are removed from the gas phase. However, heterogeneous uptake of N2O5 to particles (R7) and the reaction of NO3 with BVOCs (forming either HNO3 or organic nitrates, see below) both result in the transfer of gas-phase NOx to particulate forms, thus reducing the rate of photochemical O3 formation from NO2 photolysis (Dentener and Crutzen, 1993).
In forested environments at low NOx the lifetime of NO3 with respect to chemical losses during the temperate months will generally be driven by the terpenoids (isoprene, monoterpenes, and sesquiterpenes), the reaction proceeding via addition to the carbon–carbon double bond to form nitrooxyalkyl peroxy radicals. The peroxy radicals react further (with HO2, NO, NO2, or NO3) to form multi-functional peroxides and organic nitrates, which can contribute to the generation and growth of secondary organic aerosols (Ehn et al., 2014; Fry et al., 2014; Ng et al., 2017; Liebmann et al., 2017) or be lost by deposition. The main processes outlining the role of NO3 in removing NOx from the atmosphere are summarized in Fig. 1. Clearly, the lifetime of NO3 with respect to reaction with BVOCs (the subject of this article) determines the relative rate of formation of inorganic nitrate via heterogeneous processes (HNO3) and organic nitrates, which have different lifetimes with respect to chemical and depositional loss and thus different efficiencies of NOx removal. It also indirectly determines the rate of generation of reactive chlorine (in the form of ClNO2) resulting from the heterogeneous reactions of N2O5 with chloride containing particles (Osthoff et al., 2008; Thornton et al., 2010; Phillips et al., 2012, 2016; Ammann et al., 2013).
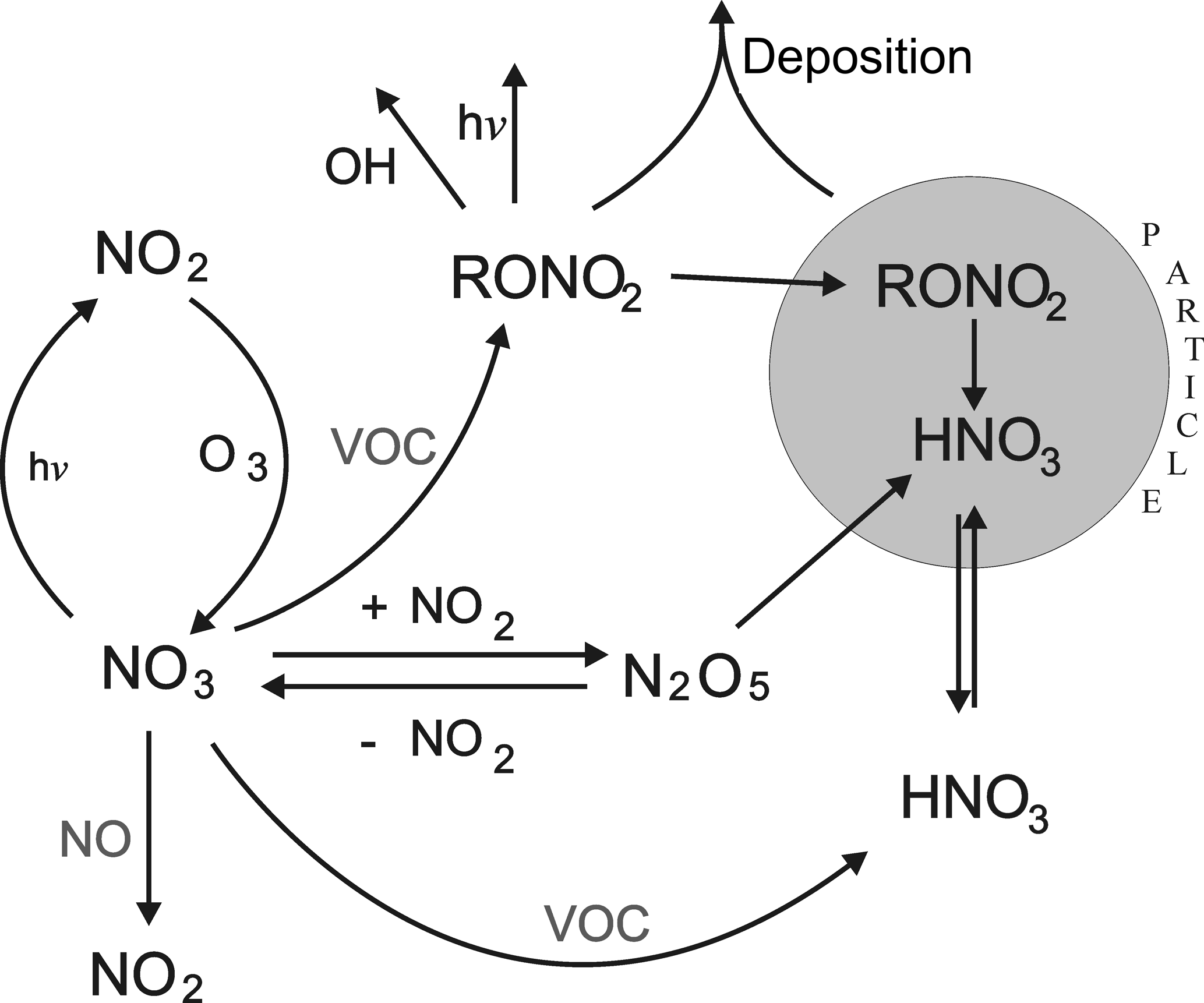
Figure 1Gas-phase formation and loss of tropospheric NO3 indicating processes which transfer reactive nitrogen to the particulate phase. RONO2 are alkyl nitrates. VOC is volatile organic compound.
Reactivity measurements have previously been applied to assess the overall loss rates of the OH radical in the boreal forest and to test for closure in its budget (Hens et al., 2014). In forested environments, the measured reactivity has generally been found to be significantly higher than that calculated from summing up reactivity due to individual reactive trace gases, (Sinha et al., 2010; Nölscher et al., 2012, 2016) resulting in an apparent “missing reactivity”. In a similar vein, O3 flux measurements in Californian pine forests required monoterpene emissions that were 10 times higher than measured in order to explain the O3 losses (Goldstein et al., 2004). These studies argue for the presence of monoterpenes/sesquiterpenes that are not detected by standard instruments used to measure BVOCs. Direct measurements of NO3 reactivity were not available until very recently (Liebmann et al., 2017); hence the reactivity of NO3 has traditionally been calculated from concentration measurements by assuming balanced production and loss terms (stationary state, see Sect. 3.4), or from measurements of the VOCs that contribute to its loss and the known rate constant for reaction of each VOC with NO3. The first method may break down when stationary state is not achieved (Brown et al., 2003). For example, Sobanski et al. (2016b) observed much lower stationary-state loss rates of NO3 compared to those calculated from measured VOC mixing ratios in a forested/urban location. It was therefore concluded that this was mainly the result of sampling from a low-lying residual layer with VOC emissions that were too close to the sampling point for NO3 concentrations to achieve stationary state. The second method relies on comprehensive measurement and accurate quantification of all VOCs that react with NO3, which, in a chemically complex environment such as a forest, may not always be possible.
In this paper we describe direct, point measurements of NO3 reactivity in ambient air in the boreal forest of southern Finland and analyse the results using ancillary measurements of NOx, NO3, O3, and biogenic hydrocarbons as well as meteorological parameters.
The IBAIRN campaign took place in September 2016 in the boreal forest at Hyytiälä, Finland. September marks the transition from late summer to autumn at Hyytiälä, with the number of daylight hours at the site changing from ≈ 14 to 11.5 from the beginning to the end of September, with the first widespread ground frost occurring close to the end of the campaign. The relative humidity within the canopy frequently reached 100 % at nighttime, though there was little rainfall during the study period. By the end of the campaign, the initially green leaves of deciduous trees had turned brown and fresh needle/leaf litter was accumulating on the forest floor. The daily profiles of temperature, relative humidity and the NO3 photolysis rate constant ( are displayed in Fig. 2.
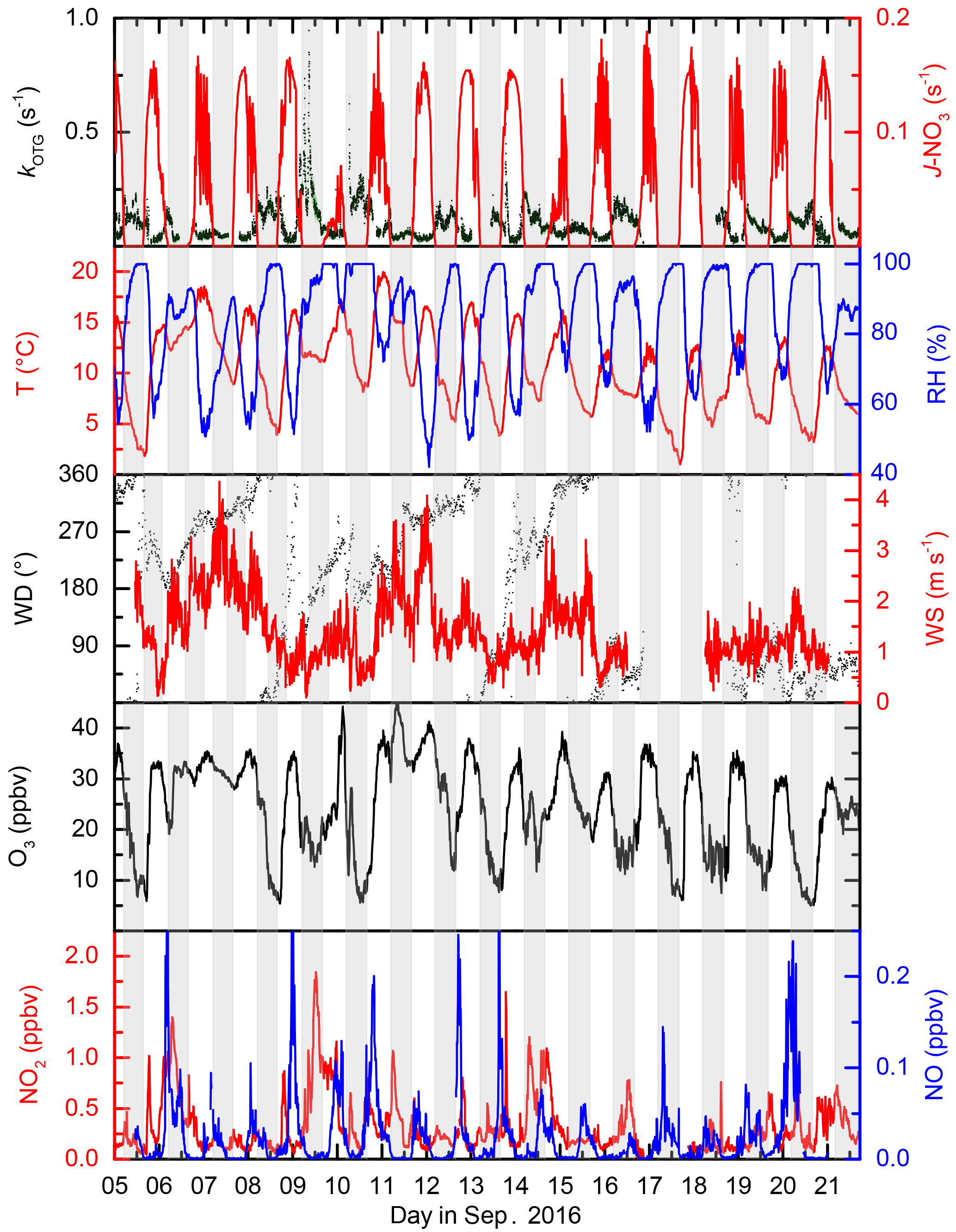
Figure 2Overview of measurements during IBAIRN. The grey shaded regions represent nighttime. The uncertainty in kOTG is given by the green shaded region. Measurements were obtained from the common inlet at a height of 8.5 m apart from the NO3 photolysis rate (taken from a height of 35 m on an adjacent tower), wind direction (WD) and wind speed (WS) (both at 16.5 m on the 128 m tower). A time series of kOTG is given in log scale in the Supplement (Fig. S1).
2.1 SMEAR II site
The measurements presented here were conducted at the “Station for Measuring Forest Ecosystem-Atmosphere Relations II” (SMEAR II) in Hyytiälä (61∘51′ N, 24∘17′ E) in southern Finland at 180 m above sea level (Hari and Kulmala, 2005). This site has been the focus of intensive research investigating BVOCs (Rinne et al., 2005; Holzke et al., 2006; Hakola et al., 2009; Lappalainen et al., 2009; Aaltonen et al., 2011) and their influence on O3 reactivity (Mogensen et al., 2015; Zhou et al., 2017), OH reactivity (Sinha et al., 2010; Nölscher et al., 2012) and simulations on NO3 lifetimes (Hakola et al., 2003; Peräkylä et al., 2014). SMEAR II is located 49 km north-east of Tampere (pop. ≈ 226 000; 430 inh. km−2) and 88 km south-west of Jyväskylä (pop. ≈ 137 000; 120 inh. km−2). Anthropogenic influence at the site is generally low, especially when the wind comes from the sparsely populated northern sector. Operations of a sawmill, a wood mill, and a pellet factory in Korkeakoski, 5 km southeast of Hyytiälä, can result in elevated levels of monoterpenes at SMEAR II (Eerdekens et al., 2009; Liao et al., 2011; Williams et al., 2011; Hakola et al., 2012). Furthermore, pollution from forest management as well as minor influences from nearby settlements with low population densities are possible.
Figure 2 shows the local wind speed and wind direction at a height of 16 m (close to the top of the canopy) during the campaign. The wind rose in Fig. 3a indicates that the prevailing wind was from the north-west and north-east sectors (≈ 60 % of the time) compared to 28 % from the southern sector, of which only ≈ 8 % came from the south-east. Nonetheless, two isolated plumes from Korkeakoski were evident as greatly increased values of the NO3 reactivity and BVOC levels, as discussed later. In general wind speeds at a height of 16 m were low, favouring a stable boundary layer during nighttime. Wind speed, wind direction, temperature, precipitation, and relative humidity were monitored at various heights on the 128 m SMEAR II tower. Details regarding these and other supporting measurements made at this site can be found elsewhere (Hari and Kulmala, 2005; Hari et al., 2013). The vegetation at the site consists mostly of Scots pine (Pinus sylvestris, > 60 %) with occasional Norway spruce (Picea abies), aspen (Populus sp.) and birch (Betula sp.). The most common vascular plants are lingonberry (Vaccinium vitis-idea L.), bilberry (Vaccinium myrtillus L.), wavy hair grass (Deschampsia flexuosa (L.) Trin.) and heather (Calluna vulgaris (L.) Hull.). The ground is covered with common mosses as such as Schreber's big red stem moss (Pleurozium schreberi (Brid.) Mitt.) and a dicranum moss (Dricanum Hedw. sp.). The canopy height is ≈ 20 m with an average tree density of 1370 stems (diameter at breast height > 5 cm) per hectare (Ilvesniemi et al., 2009).
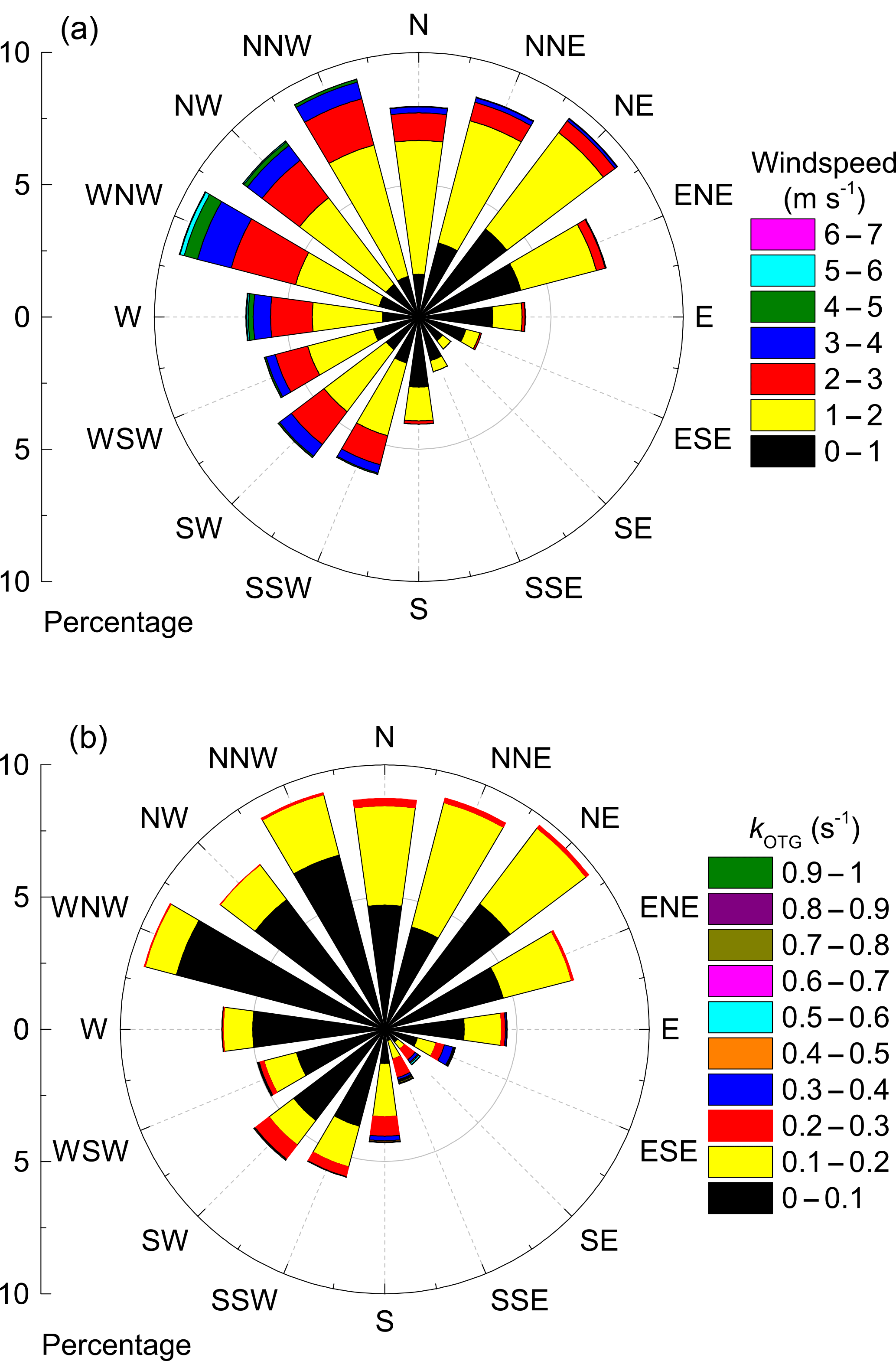
Figure 3(a) Wind rose coloured according to wind speed. (b) Wind rose coloured according to kOTG.
2.2 NO3 reactivity measurement
NO3 reactivity was measured using an instrument that was recently described in detail by Liebmann et al. (2017). Initially, 40 to 60 pptv of synthetically generated NO3 radicals (Reaction R1) were mixed with either zero air (ZA) or ambient air in a cylindrical flow-tube thermostatted to 21 ∘C. After a reaction time of 10.5 s, the remaining NO3 was detected by cavity-ring-down spectroscopy (CRDS) at 662 nm. The measurement cycle was typically 400 s for synthetic air and 1200 s for ambient air, with intermittent signal zeroing (every ≈ 100 s) by addition of NO.
The observed loss of NO3 in ambient air compared to ZA was converted to a reactivity via numerical simulation of a simple reaction scheme (Liebmann et al., 2017) using measured amounts of NO, NO2, and O3. The parameter obtained, kOTG, is a loss rate constant for NO3 from which contributions from NO and NO2 have been removed, and thus refers to reactive loss to organic trace gases (OTGs) only. Throughout the article, NO3 reactivity and kOTG are equivalent terms, with units of s−1. The dynamic range of the instrument was increased to 0.005–45 s−1 by automated, dynamic dilution of the air sample, the limit of detection being defined by the stability of the NO3 source. Online calibration of the reactivity using an NO standard was performed every 2 h for 10 min. The uncertainty of the measurement was between 0.005 and 0.158 s−1, depending mainly on dilution accuracy, NO levels, and the stability of the NO3 source (Liebmann et al., 2017).
The instrument was operated in a laboratory container located in a gravel-bedded clearing in the forest. Air samples were drawn at a flow rate of 2900 standard cubic centimetres per minute (sccm) through a 2 µm membrane filter (Pall Teflo) and 4 m of PFA tubing (6.35 mm OD) from the centre of a high-flow inlet (Ø = 15 cm, flow = 10 m3 min−1) which sampled at a height of 8 m, 3 m above the roof of the container and circa 8 m away from the forest edge. Several instruments sampled from the high-flow and we refer to this as the “common inlet”. Relative humidity and temperature were monitored in the common inlet using standard sensors (1st Innovative Sensor Technology, HYT939, ± 1.8 % RH). Vertical profiles of NO3 reactivity (8.5 to 25 m) were measured by attaching 30 m of PFA tubing directly to the flow tube and raising/lowering the open end (with membrane filter) using a rope hoist attached to a 30 m tower about 5 m from the container.
2.3 NO, NO2, O3, and NO3 measurements
NO was sampled from the common inlet using a modified commercial chemiluminescence detector (CLD 790 SR) based on the reaction between NO and O3 (ECO Physics, Dürnten, Switzerland). The detection limit for NO was 5 pptv for an integration period of 5 s, the total uncertainty (2 σ) was 20 % (Li et al., 2015). Ozone was measured by two instruments based on optical absorption, both sampling from the common inlet. These were a 2B-Technology, Model 202 and a Thermo Environmental Instruments Inc., Model 49 both with detection limits of ≈ 1 ppb. The two instruments had uncertainties (provided by manufacturer) of 5 and 2 % respectively. Agreement between the two O3 measurements was excellent (slope = 1.000 ± 0.001, offset of −0.21 ppbv, R2=0.98). Vertical profiles in O3 (up to 125 m) were made using a TEI 49 C analyser sampling from inlets at various heights on a tower located 130 m north–north-west of the measurement container. NO2 and NO3 were measured from the common inlet using a multi-channel, thermal dissociation-cavity ring down spectrometer (TD-CRDS) recently described in detail by (Sobanski et al., 2016a). NO3 radicals were detected at 662 nm with a detection limit of 1.3 pptv (1 min averaging) and an uncertainty of 25 %. NO2 was detected at 405 nm with an uncertainty of 6 % and a detection limit of 60 pptv (1 min averaging).
2.4 VOC measurements
Three different instruments were used to monitor VOCs, including (1) a gas chromatograph equipped with an atomic emission detector (GC-AED) which sampled from the common inlet; (2) a thermal desorption gas chromatograph with mass spectrometric detection (GC-MS) sampling about 1.5 m above the ground and ≈ 10 m away from the reactivity measurements; and (3) and a proton transfer reaction time of flight mass spectrometer (PTR-TOF-MS) located about 170 m away in dense forest and sampling at a height of ≈ 2.5 m above the ground.
2.4.1 GC-AED
The GC-AED consisted of a cryogenic pre-concentrator coupled to an Agilent 7890B GC and an atomic emission detector (JAS AEDIII, Moers, Germany). The GC-AED sampled air through a 15 m long, (1.27 cm) outer diameter PFA Teflon tube (flow rate = 20 L min−1, transmission time 3.3 s) which was heated to ≈ 10 ∘C above ambient. The instrument was calibrated in situ with an 84-component gravimetrically prepared gas-phase calibration reference standard with a stated accuracy of better than ±5 % (Apel-Riemer Environmental, Inc., Florida, USA). The average total uncertainty of the species measured from repeated calibration standard measurements combined with the flow measurements and calibration standard uncertainty was calculated to be 14 %. α-pinene, Δ-3-carene, β-pinene, camphene, and d-limonene were all calibrated individually. Detection limits for the monoterpene species were 1.0, 0.9, 0.4, 0.5, and 0.3 pptv respectively. As this is the first deployment of this instrument, more details are provided in the Supplement: a full description will be the subject of an upcoming publication.
2.4.2 GC-MS
The GC-MS was located in a container in a gravel-bedded clearing about 4 m from the edge of the forest and ≈ 30 m away from the common inlet. Air samples were taken every other hour (30 min sampling time) at a height of 1.5 m by drawing air at 1 L min−1 through a 1 m long fluorinated ethylene propylene (FEP) inlet (inner diameter 1/8 inch). Ozone was removed via a heated (120 ∘C) stainless steel tube (Hellén et al., 2012). VOCs were collected from a 40 mL min−1 subsample flow into the cold trap (Tenax TA/ Carbopack B) of the thermal desorption unit (TurboMatrix, 650, Perkin-Elmer) connected to a gas chromatograph (Clarus 680, Perkin-Elmer) with HP-5 column (60 m, inner diameter 0.25 mm, film thickness 1 µm) coupled to a mass spectrometer (Clarus SQ 8 T, Perkin-Elmer). The instrument was used for measurements of isoprene, monoterpenes, and aromatic hydrocarbons and was calibrated for all individual compounds using liquid standards in methanol solutions, which were injected into the Tenax TA/Carbopack B adsorbent tubes and analysed with the same method as the air samples. Detection limits for monoterpenes (α-pinene, camphene, β-pinene, 3Δ-carene, myrcene, p-cymene, limonene, 1,8-cineol, and terpinolene) were 0.2–1.2 pptv and for β-caryophyllene 0.8 pptv. The average total uncertainty (10 % for all monoterpenes and β-caryophyllene) was calculated from the reproducibility of the calibrations, uncertainty of the standard preparation and the uncertainty in the sampling flow.
2.4.3 PTR-TOF-MS
The PTR-TOF-MS (PTR-TOF 8000, Ionicon Analytic GmbH) measures whole VOC spectra in real time (Jordan et al., 2009; Graus et al., 2010) with mass resolution of 4500 (full width at half maximum). The instrument was located in the main cottage, approximately 170 m away from the common inlet. Ambient air was sampled from 2.5 m above the ground, using a 3.5 m long (4 mm inner diameter) PTFE sampling air at 20 L min−1. A subsample flow of 1 L min−1 was passed via 10 cm of PTFE tubing (1.6 mm inner diameter), by way of a three-way valve and 15 cm of PEEK tubing (1 mm inner diameter) to the PTR-TOF-MS. The raw data was collected with 10 s resolution. The instrument measured total monoterpenes at and isoprene at , which were calibrated with a gas standard (Apel Riemer Environmental Inc., USA) containing isoprene and α-pinene. The calibration set up and routine are described in detail in Schallhart et al. (2016). The campaign average limit of detection (LOD, 3σ, 10 min time resolution) was 5.5 and 3.2 pptv for isoprene and monoterpenes respectively.
NOx mixing ratios were generally low during the campaign with NO2 between 0.1 to 1.84 ppbv with a campaign average of 0.32 ppbv showing little variation across the diel cycle. The mean daytime NO mixing ratio was 43 pptv while nighttime NO was close to or below the limit of detection (≈ 5 pptv) and its contribution to the loss of NO3 was generally insignificant (see below). Ozone mixing ratios showed large day/night differences with daily maxima between 30 and 40 ppbv, whilst nighttime values were as low as 5–10 ppbv. Possible reasons for the large changes in O3 across the diel cycle are addressed in Sect. 3.1.
3.1 NO3 reactivity and nighttime loss of O3
NO3 reactivity was measured from 5 September 12:00 UTC to 22 September 05:30 UTC; the 1 min averaged time series of kOTG is displayed in Fig. 2. The overall uncertainty in kOTG is given by the green, shaded region. NO3 photolysis and reaction with NO result in concentrations that are generally below the detection limit of modern instruments during daytime and steady-state calculations of NO3 reactivity are lower limit estimates. In contrast, our direct approach allows us to derive and analyse daytime values of kOTG as long as NOx measurements are available (see above). Figure 2 indicates that, in general, the NO3 reactivity was highest at nighttime; the maximum observed values in kOTG was 0.94 s−1, (at 21:00 UTC on 9 September) implying a lifetime of just 1 s and a very reactive air mass at this time. The mean nighttime value of kOTG was ≈ a factor ten lower at 0.11 s−1, the daytime mean even lower at 0.04 s−1. Broadly speaking, the nighttime NO3 lifetimes during IBAIRN were very short (≈ 10 s on average) compared to previous indirect, ground level measurements in other locations where several groups have reported lifetimes of hundreds to thousands of seconds (Heintz et al., 1996; Allan et al., 1999; Geyer et al., 2001; Aldener et al., 2006; Ambrose et al., 2007; Brown et al., 2009; Crowley et al., 2010, 2011; Sobanski et al., 2016b). Our short NO3 lifetimes are, however, compatible with the very low NO3 mixing ratios observed in forested regions with high rates of emission of biogenic trace gases (Gölz et al., 2001; Rinne et al., 2012; Ayres et al., 2015).
Figure 3b illustrates the dependence of kOTG on wind direction. Air masses from the northern sector were generally associated with lower reactivity (< 0.2 s−1) whereas all incidents of reactivity larger than 0.3 s−1 were associated with air masses from the SE sector. Enhanced reactivity from the south-east may be caused by emissions from the sawmill at Korkeakoski (Eerdekens et al., 2009), or a local woodshed storing freshly cut timber about 100 m away from the containers. This may have been compounded by the lower than average wind speeds associated with air masses from the south-east, which reduced the rate of exchange between the nocturnal boundary layer and above canopy air, effectively trapping ground-level emissions into a shallow boundary layer. Emissions from the sawmill reaching the site on the night of the 9–10 September provided a useful test of our method at high reactivity.
In order to examine the difference in daytime and nighttime NO3 reactivity and also explain the large nighttime variability in kOTG we categorize the nights into three broad types: (1) nights with strong temperature inversion where the NO3 reactivity was greatly increased compared to the previous or following day, (2) nights without temperature inversion with comparable (usually low) daytime and nighttime NO3 reactivity, and (3) events with unusually high NO3 reactivity. Figure 4 shows an expanded view of kOTG over a five day/night period (5–10 September) in which all three types are represented. It also plots the temperature at different heights as well as the RH and O3 measured in the common inlet at 8.5 m height.
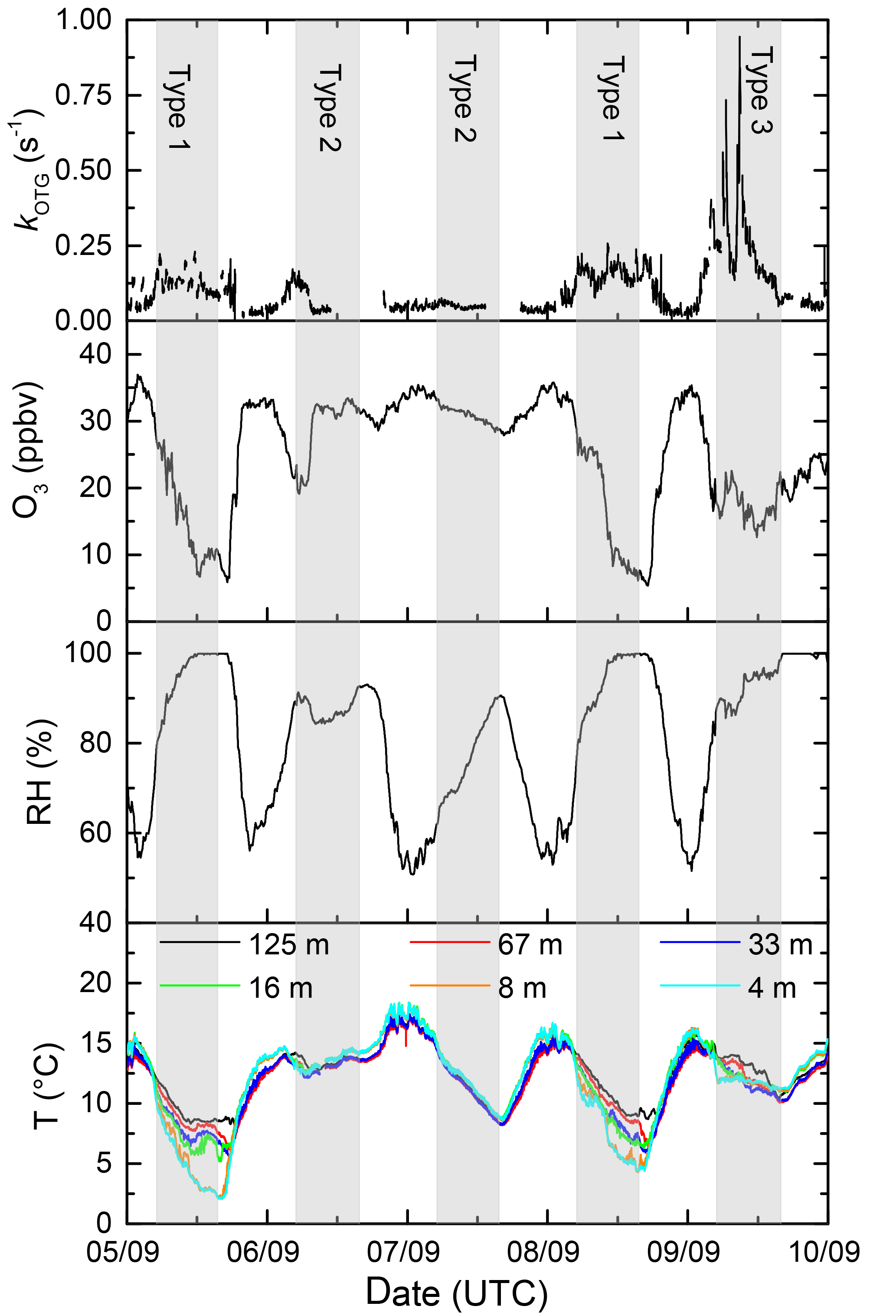
Figure 4Expanded view of five campaign days illustrating the three types (1–3) of night encountered. Type 1 has a strong vertical gradient in temperature (T) and significant O3 loss with relative humidity (RH) at 100 %. Type 2 (no temperature inversion), has little or no O3 loss. Type 3 is influenced by emissions from the Korkeakoski sawmill.
3.1.1 Type 1 and type 2 nights
Within this 5-day period, the nights on which the reactivity was high relative to the day (type 1) are the 5–6 and 8–9. These nights are characterized by large depletion in O3, a significant temperature inversion of 5–7 ∘C between heights of 8 and 128 m, and a relative humidity of 100 % directly after sunset. In contrast, two interspersed nights with comparable reactivity to daytime values (6–7, 7–8, type 2) display much weaker (if any) nighttime loss of O3 compared to levels during the previous day, no significant temperature inversion and a relative humidity less than 100 %. The observations within this period can be extended to all campaign days. Figure 5 presents the diel cycle of kOTG and O3 mixing ratios separated into nights of type 1 (with temperature inversion) and type 2 (no temperature inversion). The shaded regions represent the variability of the measured values. During 24 h periods in which the night was characterized by strong temperature inversion (panel (a) in Fig. 5), the O3 mixing ratios display a large diel variation, with a maximum of 35 ppbv at about 13:00 UTC dropping rapidly to a minimum of ≈ 13 ppb between midnight and sunrise at around 05:00 UTC. O3 depletion due to its slow reaction with NO2 (present at maximum 2 ppbv at night) does not contribute significantly to its loss even if all resultant NO3 reacts to form organic nitrates rather than to form N2O5 and re-release NOx.
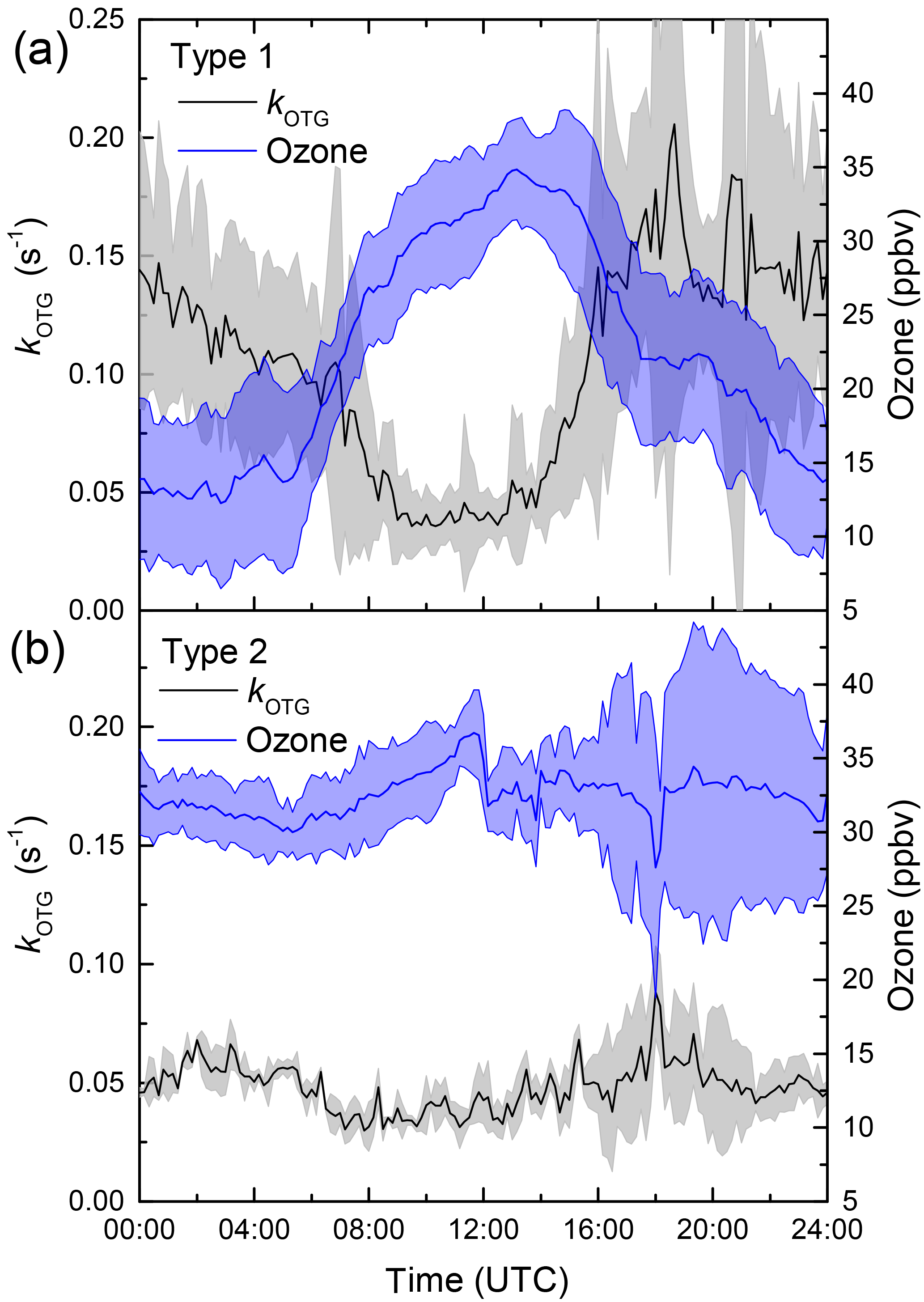
Figure 5Diel profiles of kOTG (black line) and O3 (blue line) on two different types of days/nights. (a) Type 1 (strong nighttime temperature inversion). (b) Type 2, (no temperature inversion). The shaded areas represent 2 σ uncertainty and indicate variability over the diel cycle.
The O3 mixing ratio shows an inverse diel profile to the NO3 reactivity raising the possibility that the rapid loss of ozone is linked to high NO3 reactivity; the large values of kOTG and rapid O3 depletion observed on nights with a significant temperature inversion are clear indicators that nighttime boundary layer dynamics plays a key role in controlling both the NO3 reactivity and O3 loss. A strong nocturnal temperature inversion will weaken the mixing within or ventilation of the lowermost boundary layer causing a build-up of reactive, biogenic emissions in the lower layer, and also prevent down-mixing of drier, O3-rich air leading to the apparent higher loss rate of O3 and higher relative humidity. The strong anti-correlation between kOTG and O3 may also provide a clue to the origin of the O3 loss. Whilst the generally high NO3 reactivity can, to a large extent, be explained by the presence of reactive trace gases (see Sect. 3.2), the precipitous loss of O3 on several nights when kOTG was high (see Figs. 2 and 4) may have components of both dry deposition and gas-phase reactions.
The campaign averaged, diel variation of ozone at different heights (4 to 125 m) as measured at the SMEAR II tower (Fig. S2 of the Supplement) indicate that the most rapid losses of ozone are at the lowest heights, around and below the canopy. Ozone is generally removed from the lower troposphere by both stomatal and non-stomatal deposition, the latter involving loss to surfaces and soil. The reactive, gas-phase loss mechanisms of O3 and NO3 are in some ways similar, as both react with NO to form NO2, or with unsaturated VOCs by addition to the double bond. We estimated the loss rate constant for O3 due to its reaction with terpenes using approximate ambient mixing ratios from 20:00 to 00:00 UTC on the 20 September for d-limonene (20 pptv), α-pinene (400 pptv), Δ-carene (100 pptv), and β-pinene (100 pptv) and using literature rate constants for the O3 + terpene reactions. The calculated O3 loss (only 2 % from 20:00 to 00:00 UTC) is clearly insufficient to explain the IBAIRN observations. We also note that the presence of high concentrations of terpenes when the site was impacted by the Korkeakoski sawmill resulted in the largest NO3 reactivity observed, but did not lead to large O3 losses (Fig. 4). As leaf stomata are closed during nighttime, the decrease in O3 can be attributed either to non-stomatal deposition or chemical sinks due to reaction with reactive biogenic trace gases (not the measured monoterpenes) and NO. Previous studies of O3 loss in forests have highlighted the potential role of unidentified, reactive organic compounds (Kurpius and Goldstein, 2003; Goldstein et al., 2004; Holzinger et al., 2006; Rannik et al., 2012). In contrast to monoterpenes, which react only slowly with O3 (rate constants are ≈ 10−16–10−17 cm3 molecule−1 s−1), sesquiterpenes can react rapidly; for example, for β-caryophyllene the rate coefficient is cm3 molecule−1 s−1 (IUPAC, 2017). The presence of sesquiterpenes would therefore provide an explanation for the observations of high NO3 reactivity and rapid O3 loss. We examine the potential role of sesquiterpenes in more detail in Sect. 3.2 where the contribution of measured terpenoids to NO3 reactivity is discussed. We also note that recent modelling studies using Hyytiälä data (Chen et al., 2018; Zhou et al., 2017) conform that O3 depletion events are associated with the formation of a shallow boundary layer and high relative humidity. Zhou et al. (2017) conclude that chemical reaction plays only a minor role in ozone loss processes during the night, which was suggested to be dominated by deposition to wet surfaces at relative humidity > 70 %, which is in accord with laboratory investigations (Sun et al., 2016).
3.1.2 Type 3 nights
The period between the evening and midnight on the 9 September is an example of a type 3 night, with extremely high NO3 reactivity, which was not accompanied by significant O3 depletion, temperature inversion or a RH of 100 %. The apparently anomalously high reactivity on this night can be traced back to a change in wind direction, which swept from easterly to southerly during this period, bringing air that was impacted by monoterpene emissions from the sawmill in Korkeakoski. High mixing ratios of terpenoids in air masses that have passed over the sawmill have been documented frequently (Eerdekens et al., 2009; Sinha et al., 2010; Liao et al., 2011; Hakola et al., 2012; Nölscher et al., 2012). Other occurrences of sawmill contaminated air during IBAIRN were on the 10 September from 18:40 to 19:00 UTC and on the 14 September from 06:30 to 08:00 UTC, HYSPLIT back-trajectories (GDAS global, 0.5∘), indicating that the air mass passed over Korkeakoski ≈ 0.5 h prior to reaching the SMEAR II site.
3.2 Comparison of kOTG with NO3 reactivity derived from VOC measurements
In this section we compare kOTG with NO3 reactivity calculated from ambient VOC mixing ratios. During IBAIRN, three instruments (GC-MS, GC-AED, and PTR-TOF) measuring VOCs were deployed (see Sect. 2.5 for details). As the PTR-TOF reports only a summed mixing ratio of all monoterpenes, ΣMT(PTR-TOF), we first generated an equivalent parameter for the two GCs, ΣMT(GC-MS) and ΣMT(GC-AED). For the GC-MS, α-pinene, β-pinene, Δ-carene, d-limonene, camphene, myrcene, and terpinolene were considered whereas for the GC-AED, α-pinene, β-pinene, Δ-carene, camphene, and d-limonene were taken into account. The ΣMT data are displayed as a time series in Fig. 6, which indicates large differences between the three measurements as highlighted in Fig. S3 of the Supplement). While the ΣMT(GC-AED) and ΣMT(PTR-TOF) data are in reasonable agreement, especially when mixing ratios were large, the values reported by the GC-MS are consistently and significantly lower (factor 2 to > 10) than those of the others instruments. The time dependent variability in the differences in ΣMT reported by the GC-MS, GC-AED, and PTR-TOF is a strong indication that the cause is most likely related to instrument location and inhomogeneity in terpene emissions within the forest. Whilst the GC-AED sampled from the common inlet at a height of 8.5 m, which was also used for the NO3 reactivity measurements, the inlet of the GC-MS was ≈ 10 m away and sampled 1.5 m above the gravel covered clearing, very close to the side of the container which housed the instrument. The PTR-TOF-MS was located roughly 170 m away in a wooden cottage directly surrounded by dense forest and sampled close to the forest floor at a height of ≈ 1.5 m. With very low within-canopy wind speeds, especially during nighttime, both horizontal as well as vertical mixing in the forest and in the clearing are weak so that each VOC measurement may, to some extent, reflect the mixture and total amount of BVOCs that are very locally emitted. This aspect was examined by comparing individual monoterpenes measured by the GC-MS and the GC-AED. The results, presented as correlation plots for 4 monoterpenes in Fig. S4 of the Supplement, show that the monoterpene ratios measured by the two instruments (GC-AED/GC-MS), were variable with values of 1.69 ± 0.06 for α-pinene, 2.51 ± 0.09 for β-pinene, 4.29 ± 0.21 for Δ-carene, and 0.45 ± 0.03 for d-limonene. A similar picture emerges for isoprene, for which the GC-AED measured mixing ratios that were a factor 2–5 larger than measured by the GC-MS. The variable relative concentrations of monoterpenes reported by each instrument is further evidence of the inhomogeneity of emissions within the forest and also the influence of different tree chemotypes within single tree-families in Hyytiälä, which can exhibit vastly different emission rates of various monoterpenes (Bäck et al., 2012; Yassaa et al., 2012).
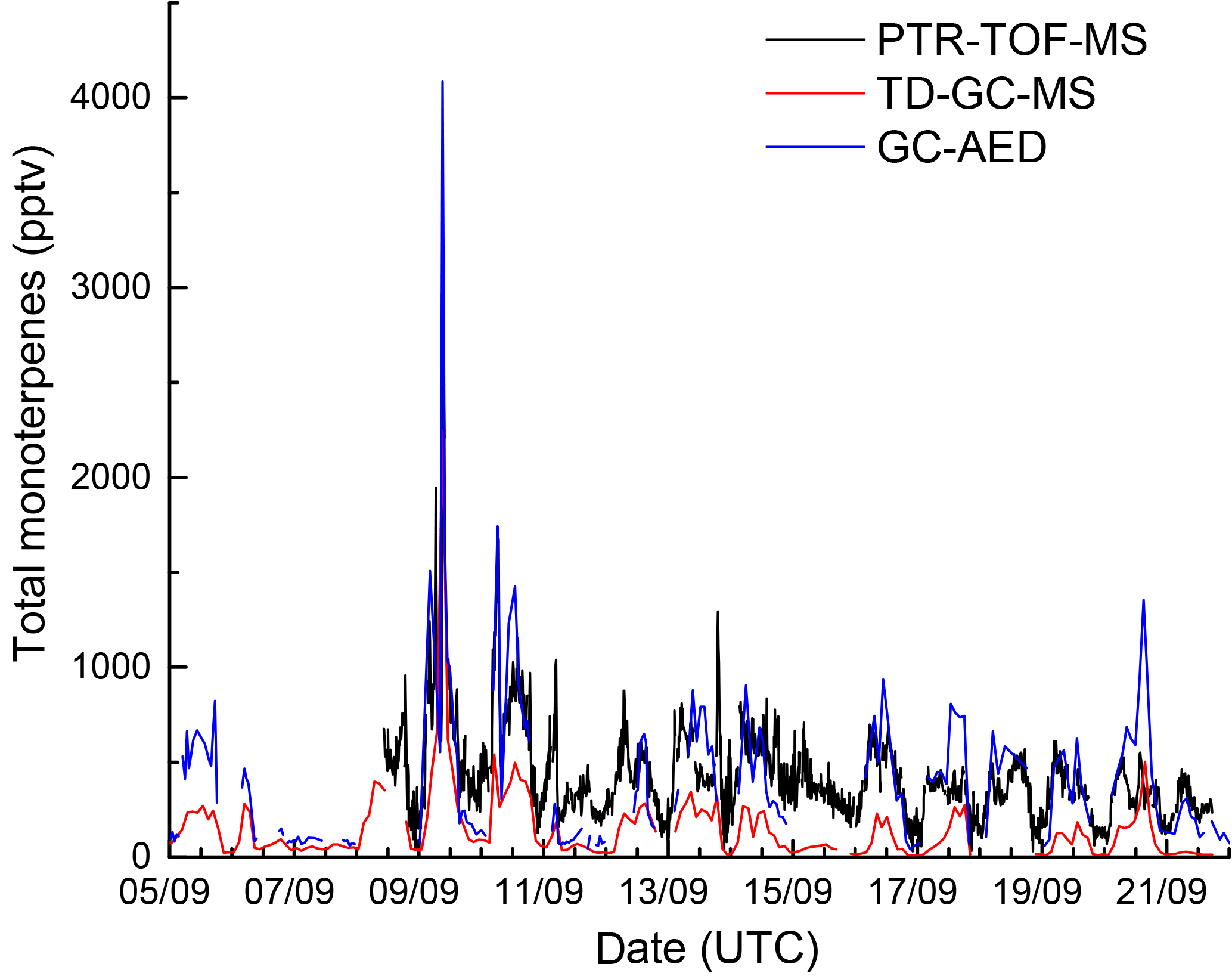
Figure 6Time series of total monoterpenes from GC-AED (black), GC-MS (red), and PTR-TOF-MS (blue). The data are reproduced as histograms in Fig. S3 of the Supplement.
For the purpose of comparing our point measurements of kOTG with NO3 reactivity calculated from BVOC measurements, we restricted our analysis to the data set obtained by the GC-AED, which sampled from the same inlet. Nonetheless, when comparing measured NO3 mixing ratios with those calculated from NO3 reactivity and its production term (see Sect. 3.4) we use both GC-based datasets.
The loss rate constant, kOTG, represents chemical reactions of [NO3] with all organic trace gases present, and can be compared to the loss rate constant (kGC-AED) obtained from the concentrations of VOCs in the same air mass as measured by the GC-AED, and the rate coefficient for reaction with NO3:
A difference in the values of kOTG and kGC-AED is defined as missing reactivity (s−1):
where [Ci] is the measured VOC concentration and ki the corresponding rate constant. The rate constants used in these calculations of kGC-AED were taken from the IUPAC evaluation (IUPAC, 2017). Figure 7 (lower panel) shows the concentrations of the monoterpenes as measured by the GC-AED. The dominant monoterpene was α-pinene followed by Δ-carene, β-pinene, d-limonene, and camphene. The GC-AED also detected myrcene and linalool and some other terpenes but the very low mixing ratios meant that none of them contributed significantly to NO3 loss.
In Fig. 7 (upper panel) we overlay the time series of kOTG and kGC-AED. For clarity of presentation we have omitted to plot the overall uncertainty of each measurement, which was calculated as described previously (Liebmann et al., 2017) and is plotted in Fig. S6 of the Supplement. The correlation between kOTG and kGC-AED is displayed as Fig. S7 of the Supplement and indicates, on average, that measured organics accounted for ≈ 70 % of the total NO3 reactivity.

Figure 7Upper plot: comparison of kGC-AED (red data points) and measured NO3 reactivity (kOTG, black data points). The fractional missing reactivity (blue data points) was calculated as (. Lower plot: mixing ratios of individual monoterpenes as measured by the GC-AED and their contribution to the NO3 reactivity. The grey shaded areas represent nighttime.
The uncertainty associated with kGC-AED was calculated by propagating uncertainty in the mixing ratios of the individual terpenes (14 %, mainly resulting from uncertainty in the calibration standard and the calibration reproducibility) and assuming 15 % uncertainty in the rate coefficients for reactions of NO3 with each terpene. The values of kOTG and kGC-AED do not agree within their combined uncertainties, indicating that the missing reactivity calculated in Eq. (3) is statistically significant. Figure 8 plots the time series of the fractional contribution to kGC-AED made by monoterpenes detected by the GC-AED. The GC-AED derived NO3 reactivity is dominated by α-pinene and Δ-carene and to a lesser extent d-limonene, with minor contributions from β-pinene, camphene, and isoprene.
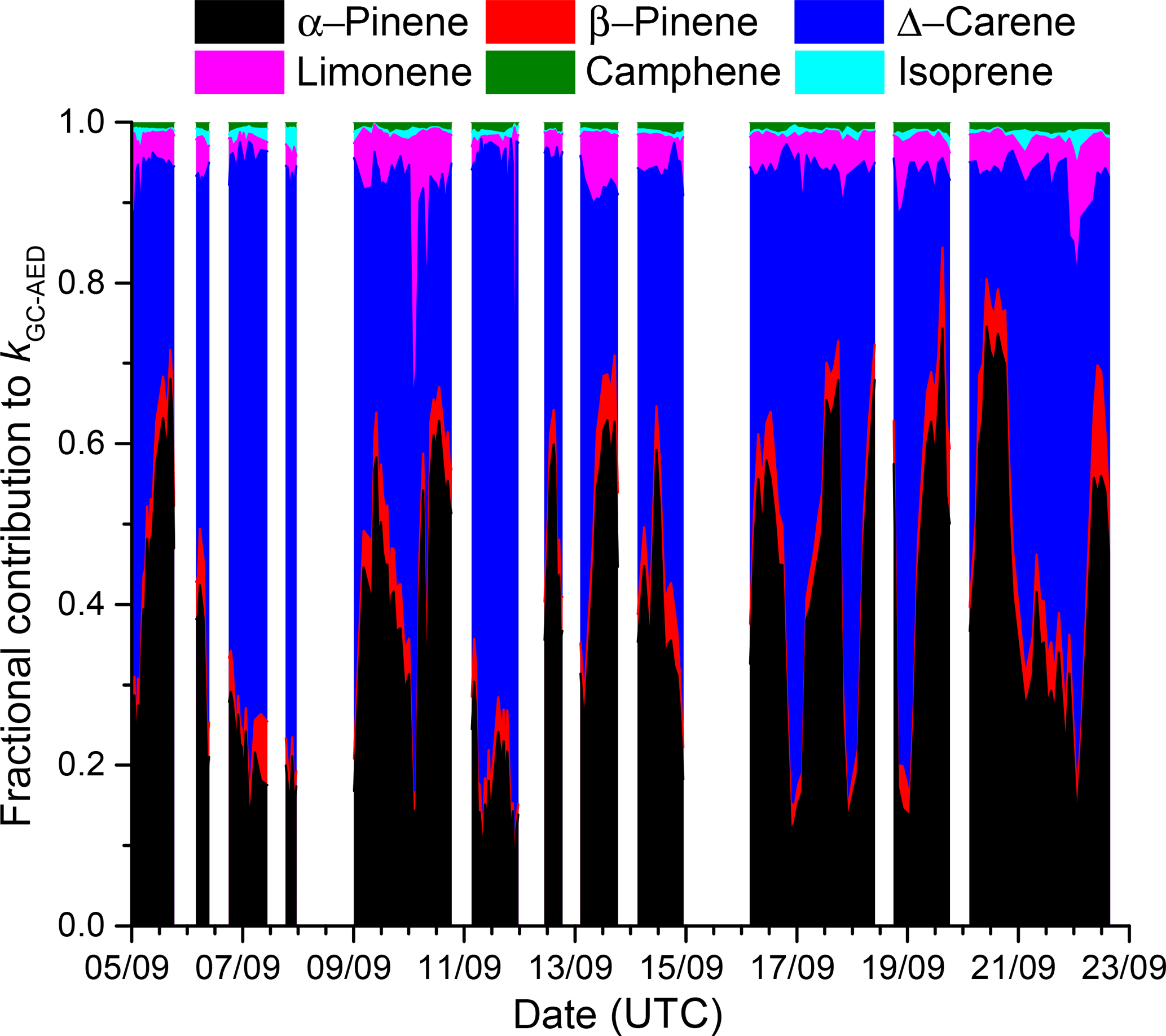
Figure 8Fractional contribution of individual monoterpenes (measured by the GC-AED) to kGC-AED indicating the dominant role of α-pinene and Δ-carene.
In Fig. 9 we plot the diel profiles of kOTG and kGC-AED (s−1) averaged for the whole campaign. To do this, we interpolated the values of kOTG, obtained with 60 s time resolution averaged to 900 s data onto the low-time resolution (≈ 60 min) GC-AED dataset. In the lower panels of Fig. 9 we plot two separate diel profiles, separating the data into nights with (middle panel) and without (lower panel) strong temperature inversion. The missing reactivity (in s−1) across the entire diel profile is between ≈ 0.02 and 0.07, the larger value encountered during nighttime. In contrast, the fraction of missing reactivity within the campaign averaged diel cycle was observed during daytime (≈ 60 %), with only 30 % missing at nighttime. The lowermost panel of Fig. 9 highlights the fact that kOTG was lower during campaign day/night periods with no temperature inversion and shows that it is roughly constant across the diel cycle. Likewise, the diel cycle in the reactivity attributed to the monoterpenes is also constant, with a missing reactivity of between 0.02 and 0.04 s−1. A different picture emerges for the diel cycle considering only the days/nights with strong temperature inversion. On average, we see a much higher nighttime reactivity, which is tracked in its diel profile by that calculated from the measured monoterpenes. In this case, the missing reactivity is generally higher and more variable, with values between 0 and 0.1 s−1.
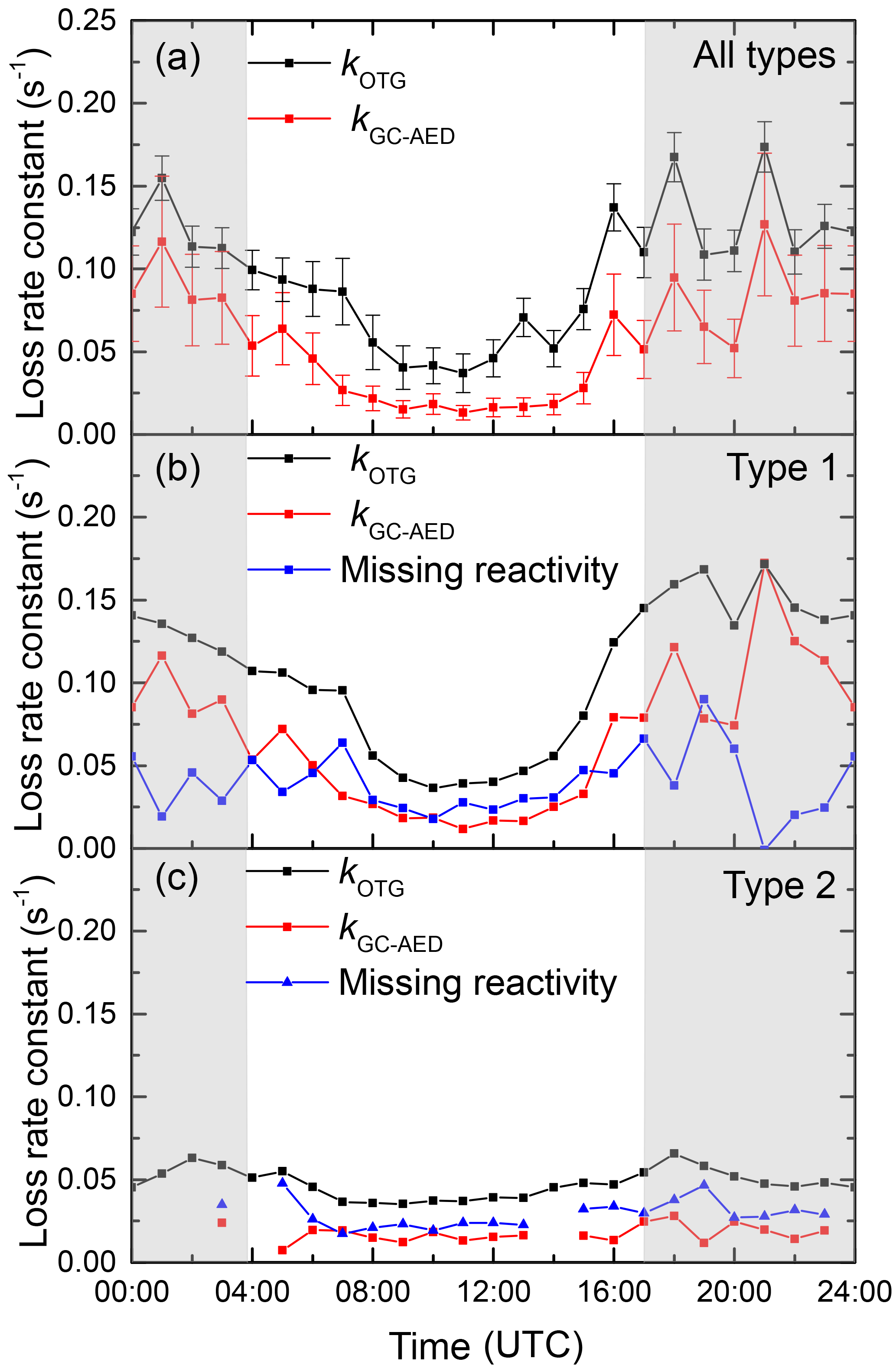
Figure 9(a) Campaign averaged diel cycle of NO3 reactivity (kOTG) and the reactivity calculated from the monoterpenes reported by the GC-AED. The error bars represent the overall uncertainty in each parameter and not variability. Panels (b) and (c) show data from type 1 nights (significant nocturnal temperature inversion) and type 2 nights (weak or no nocturnal temperature inversion) respectively.
Although statistically significant, the fraction of reactivity missing is much smaller than that reported for OH at this site (Nölscher et al., 2012) whereby up to 90 % of the observed reactivity was unaccounted for when the forest was under stress due to high temperatures. For OH, the fractional missing reactivity was also greatest when the overall reactivity was high, which is in contrast with the situation for NO3 where missing reactivity was highest when the overall reactivity was low (i.e. during daytime). The OH radical reacts with most hydrocarbons and many inorganic trace gases and may be considered unselective in its reactivity, whereas NO3 is a more specific oxidant of VOCs, its reactions in the forest dominated by addition to unsaturated VOCs or reaction with NO.
As the nighttime mixing ratios of NO were low (< 5 pptv apart from the night 20–21 September when a mixing ratio of ≈ 25 pptv was measured), its contribution to the overall nighttime loss of NO3 was insignificant. Figure S8 of the Supplement indicates that, averaged over the entire campaign, NO accounted for less than 2 % of the reactive loss of NO3 at night. A different picture emerges for daytime, for which the campaign averaged contribution of NO to the overall chemical reactivity of NO3 peaked at 40 % at about 10:00 UTC. However, even during daytime, the average missing reactivity of 0.025 s−1 (Fig. 9) would require an extra 40 pptv of NO to account for it, which is clearly not within the total uncertainty of the NO measurement.
A more plausible explanation for the missing NO3 reactivity is incomplete detection of all reactive BVOCs by the GC-AED, which does not report mixing ratios of some hydrocarbons such as 2-methyl-3-buten-2-ol, p-cymene, and 1,8 cineol, which the GC-MS showed to be present. The GC-MS mixing ratios of these species (which react slowly with NO3) were however too low for them to contribute significantly, even taking into account the potentially larger concentrations at the common inlet.
We also consider the potential role of sesquiterpenes. Mixing ratios of β-caryophyllene reported by the GC-MS were generally low, with a maximum value of 25 pptv. However, the rate coefficient reported (Shu and Atkinson, 1995) for the reaction of NO3 with β-caryophyllene is large ( cm3 molecule−1 s−1) and sesquiterpenes at levels of 10 s of pptv can contribute significantly to NO3 loss rates. Like monoterpenes, the emissions of sesquiterpenes are driven by temperature, with tree emissions most important during the hottest months (Duhl et al., 2008). Whilst previous studies at this site (Hakola et al., 2006) found no correlation between the β-caryophyllene and monoterpene emissions of an enclosed Scots pine branch, we find that β-caryophyllene mixing ratios (reported by the GC-MS) are correlated with those of several monoterpenes measured by the same instrument. This is illustrated in Fig. S5 of the Supplement which indicates β-caryophyllene/monoterpene ratios (α-pinene β-pinene and Δ-carene) of 0.061 ± 0.002 (R2 0.86), 0.294 ± 0.011 (R2 0.86), and 0.181 ± 0.007 (R2 0.84), respectively. As the monoterpenes and sesquiterpenes have very different lifetimes with respect to chemical loss, we have excluded the sawmill impacted data (red data point) as sesquiterpenes are unlikely to survive the ≈ 0.5 h. transport time from Korkeakoski due to their rapid reaction with O3. The high levels of β-caryophyllene measured may indicate that the source during IBAIRN is unlikely to be Scots pine, the emissions from which are strongly temperature dependent during the summer months but low and independent of temperature in September (Hakola et al., 2006).
A rough estimate of the β-caryophyllene mixing ratio at the common inlet may be obtained from the GC-AED measurement of α-pinene and the α-pinene/β-caryophyllene ratios measured by the GC-MS (see above). The resulting β-caryophyllene mixing ratios lie between 10 and 60 pptv, which, based on a rate constant of cm3 molecule−1 s−1, results in a contribution to NO3 reactivity of up to 0.03 s−1. As β-caryophyllene emissions from pine tree needles reveals a strong temperature dependence (Hakola et al., 2006) it seems unlikely that this is an important source of β-caryophyllene during the relatively cold September nights of the IBAIRN campaign and its emissions from other sources, especially those at ground level including soil may be more important (Insam and Seewald, 2010; Penuelas et al., 2014).
In summary, the BVOC measurements indicate that NO3 reactivity in this boreal environment is dominated by reaction with monoterpenes with, on average, 70 % of the reactivity during nighttime and 40 % of the reactivity during daytime explained by α- and β-pinene, Δ-carene, limonene, and camphene. Unidentified monoterpenes/sesquiterpenes are likely to account for a significant fraction of the VOC-derived missing reactivity.
3.3 NO3 levels: measurements versus calculations using production and loss terms
Previous estimates of NO3 reactivity (often reported as its inverse lifetime) have relied on NO3 concentration measurements and the assumption that the production and loss of NO3 are in stationary state. By combining kOTG and other loss processes such as photolysis and reaction with NO with the NO3 production term, we can also calculate the NO3 concentration:
where k1 is the rate constant (cm3 molecule−1 s−1) for reaction of NO2 with O3 and k2 is the rate constant (cm3 molecule−1 s−1) for the reaction of NO3 with NO and is its photolysis rate constant (s−1). was calculated from actinic flux measurements (spectral radiometer, Metcon GmbH; Meusel et al., 2016) and NO3 cross sections/quantum yields from an evaluation (Burkholder et al., 2016). This expression does not consider indirect loss of NO3 via heterogeneous loss processes of N2O5, which, given the high levels of BVOC (short NO3 lifetimes) and low aerosol surface area, cannot contribute significantly.
Figure 10 plots the time series of measured NO3 mixing ratios (1 min averages, blue lines) for the entire campaign, which indicates that NO3 was always below the detection limit of 1.3 pptv, which is defined by variation in the zero-signal rather than random noise (Sobanski et al., 2016a). The fact that the measured NO3 mixing ratios are slightly negative (by ≈ 0.2 pptv) is due to a few percent NO2 contamination of the NO sample used to zero the NO3 signal. We also plot the time series (black line) of the stationary state NO3 mixing ratios, [NO3]ss, calculated according to (Eq. 3). The low NOx levels and moderate O3 levels combine to result in a weak production rate for NO3 of less than 0.03 pptv s−1 for the entire campaign resulting in predicted levels of [NO3]ss of less than 0.2 pptv. On two nights, higher mixing ratios close to 1 pptv (nights of 6–7 and 10–11) are predicted, a result of elevated production rates due to higher NO2 levels.
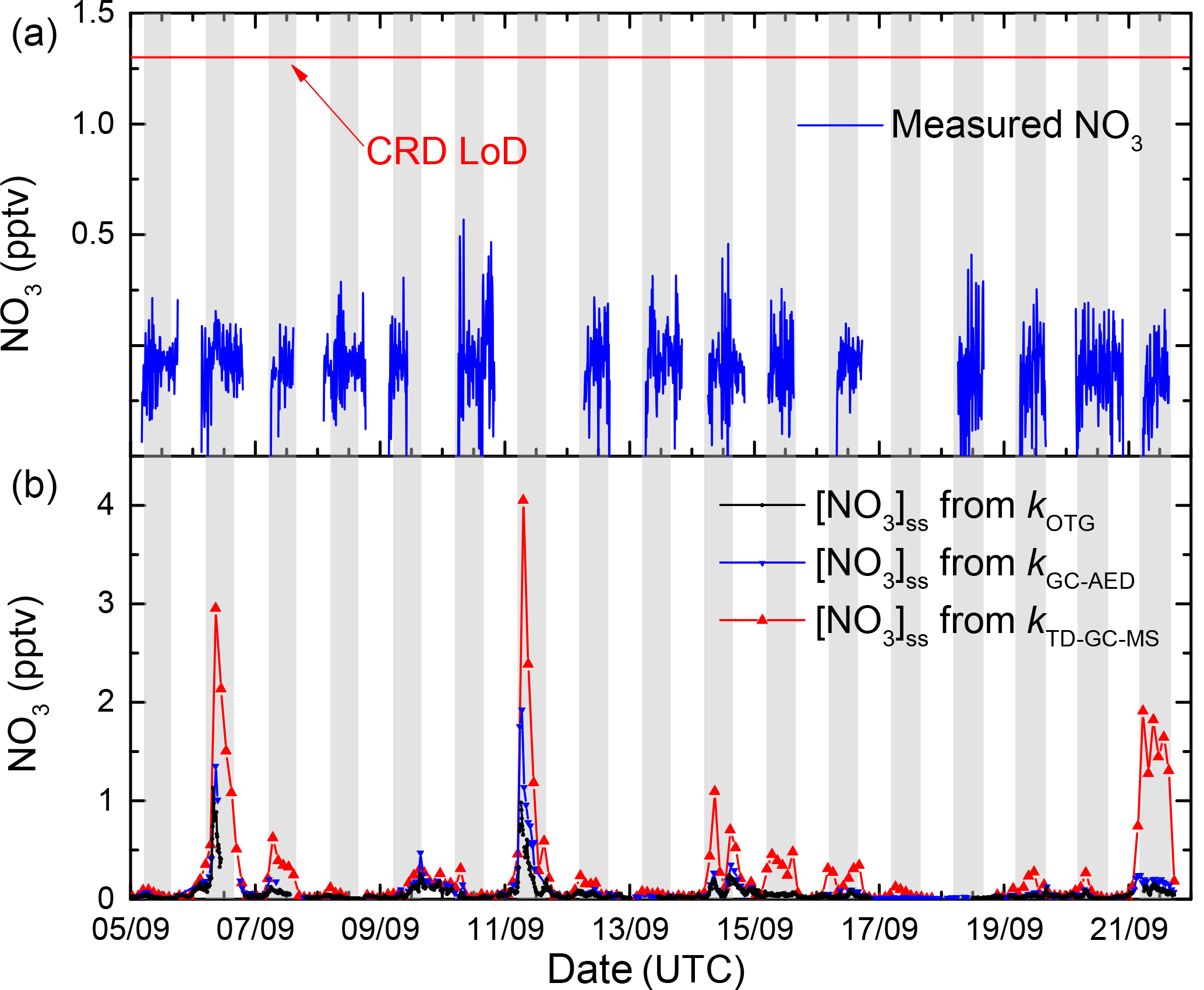
Figure 10Stationary state NO3 mixing ratios calculated from the production term (k1[NO2][O3]) and using either kOTG + k2[NO] + (b, black line), kGC-MS + k2[NO] + (b, red line), or kGC-MS + k2[NO] + (b, blue line) as loss terms. For comparison, the measured NO3 mixing ratios are also plotted (a, blue line) as well as the 1.3 pptv limit of detection (horizontal red line).
The advantages of directly measured kOTG rather than reactivity calculations based on measurements of reactive trace gases is illustrated by plotting the predicted NO3 levels based on the reactive hydrocarbons reported by the GC-AED and GC-MS, i.e. use of kGC-MS and kGC-AED rather than kOTG. Use of the GC-MS data, which reported the lowest levels of biogenic hydrocarbons, would lead to the prediction of measurable amounts (up to 4 pptv) of NO3 on several nights, contradicting our NO3 measurements and previous reports (Rinne et al., 2012) of very low NO3 levels at this site.
3.4 Vertical gradient in NO3 reactivity
Both column and point measurements of tropospheric NO3 indicate a strong vertical gradient in its mixing ratio with significantly elevated levels aloft (Aliwell and Jones, 1998; Allan et al., 2002; von Friedeburg et al., 2002; Stutz et al., 2004; Brown et al., 2007a, b; Brown and Stutz, 2012). The NO3 gradient is the result of lower production rates close to the ground, where O3 levels are depleted due to deposition and also lower loss rates aloft as the concentration of reactive traces gases from ground level emissions decreases with altitude. High resolution data (Brown et al., 2007b) indicate that the largest gradient in NO3 concentration is often found in the lowermost 50 m. Nighttime monoterpene mixing ratios in forested, boreal regions have been found to display a vertical gradient, with highest mixing ratios at lower levels (Holzinger et al., 2005; Rinne et al., 2005; Eerdekens et al., 2009). This is a result of direct emissions, such as monoterpenes from the trees at canopy level; and emissions of monoterpenes and sesquiterpenes from rotting leaf litter into a shallow, stratified boundary layer, suggesting that reactive species close to the ground will dominate in controlling the NO3 lifetime and thus mixing ratio (Aaltonen et al., 2011). We explored this by measuring kOTG at various heights above ground, including measurements from a few metres below the canopy, to a few metres above the tree tops. Altogether we recorded 14 vertical profiles on the 18 September 2016, five obtained during the daytime (10:15–05:15 UTC) and nine obtained at nighttime (16:00–24:00 UTC).
Figure 11 displays the averaged nighttime and daytime values of kOTG recorded at 8.5, 12.0, 17.0, 22.0, and 27.0 m. The total time to take a single profile was < 15 mins. During the day (black data points), we find no significant vertical gradient in NO3 reactivity, which was roughly constant at ≈ 0.03 s−1. In contrast, the average nighttime vertical profile (red data points) reveals a strong gradient in kOTG with the highest values slightly below canopy height (8.5 to 12.5 m) with a rapid decrease above. At 20 m and above, daytime and nighttime values of kOTG were comparable. These observations are qualitatively consistent with gradients in monoterpene mixing ratios in this forest (Rinne et al., 2005) and with the conclusion of Mogensen et al. (2015), who considered NO3 reactions with monoterpenes and sesquiterpenes emitted from Scots pines at canopy height for the exceptionally warm summer of 2010. The modelled, nighttime vertical gradient in NO3 described by Mogensen et al. (2015) displays a maximum at 12 m but differs from the measured gradient from IBAIRN in that lower reactivity was modelled at the lowest heights, which may be expected as the model considered only emissions of reactive BVOCs from trees and not from ground sources. In contrast to the vertical gradient measured during IBAIRN, the modelled NO3 reactivity showed highest values during daytime, coincident with the maximum NO mixing ratio (Mogensen et al., 2015) but was generally lower than our measured values. Mogensen et al. (2015) indicate that the model is likely to underestimate the NO3 reactivity due to compounds that cannot be measured by GC-MS as well as by the unknown products of their oxidation.
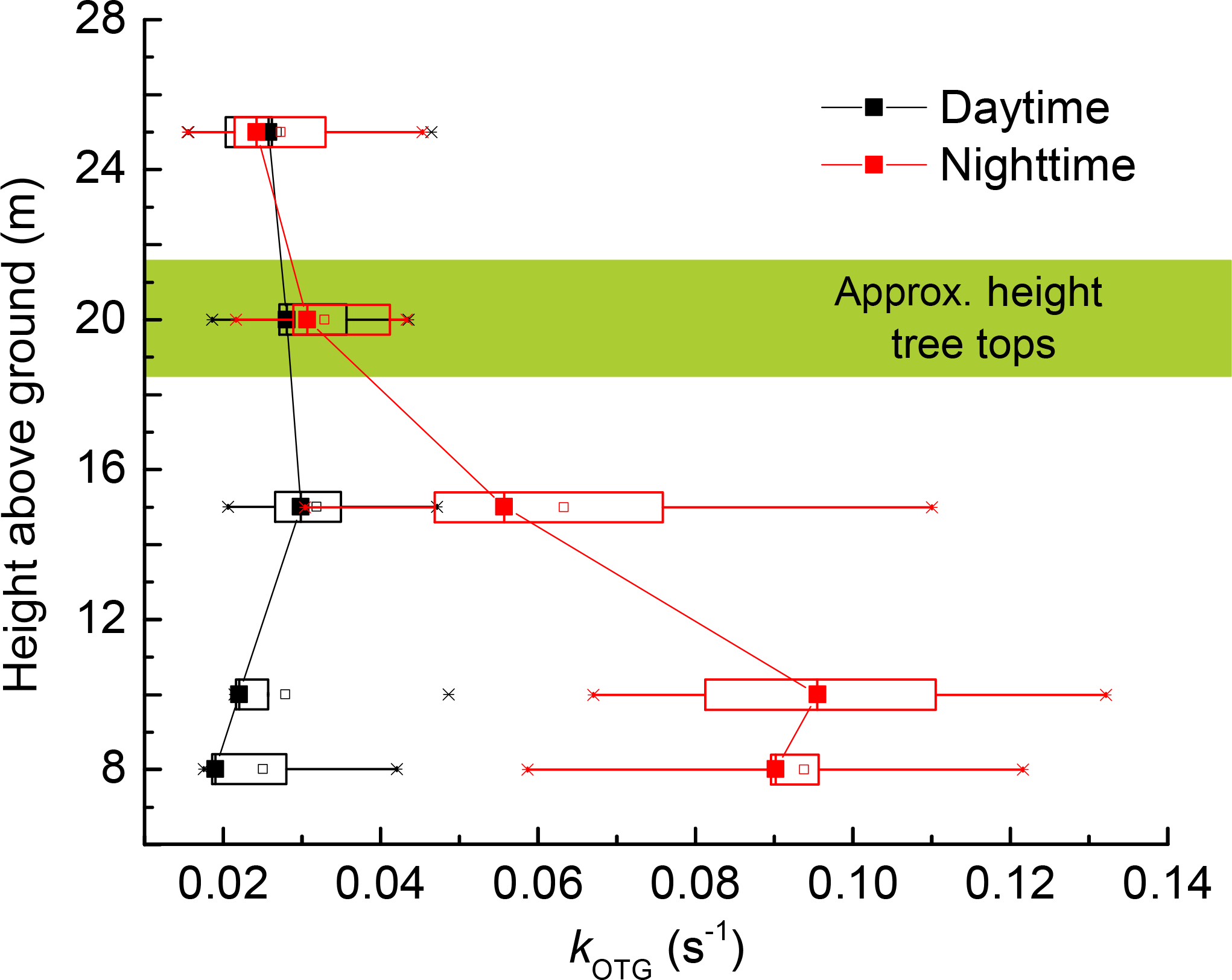
Figure 11Vertical profiles of NO3 reactivity (kOTG) on 17–18 September 2016. The data represent the average of five profiles during the day and nine profiles during the night.
The increase in kOTG below the canopy may be caused by ground level emissions of reactive trace gases from tree and plant debris or other flora (mosses, lichens) at forest-floor level. α-pinene and Δ3-carene, emissions from ground level may vary with litter quality and quantity, soil microbial activity and the physiological stages of plants (Warneke et al., 1999; Insam and Seewald, 2010; Aaltonen et al., 2011; Penuelas et al., 2014). Previous work in the tropical forest has indicated that sesquiterpenes concentrations can peak at ground level rather than within the canopy (Jardine et al., 2011), although the applicability of this result to the boreal forest is unclear.
We conclude that high rates of emission of reactive gases into the stratified nocturnal boundary layer along with ventilation and dilution above canopy height result in strong nocturnal gradients in NO3 reactivity. During the daytime, efficient turbulent mixing removes the gradient. We did not obtain a vertical profile of kOTG on a night when the temperature inversion was absent, but expect it would be significantly weaker, as is the gradient in O3 on such nights.
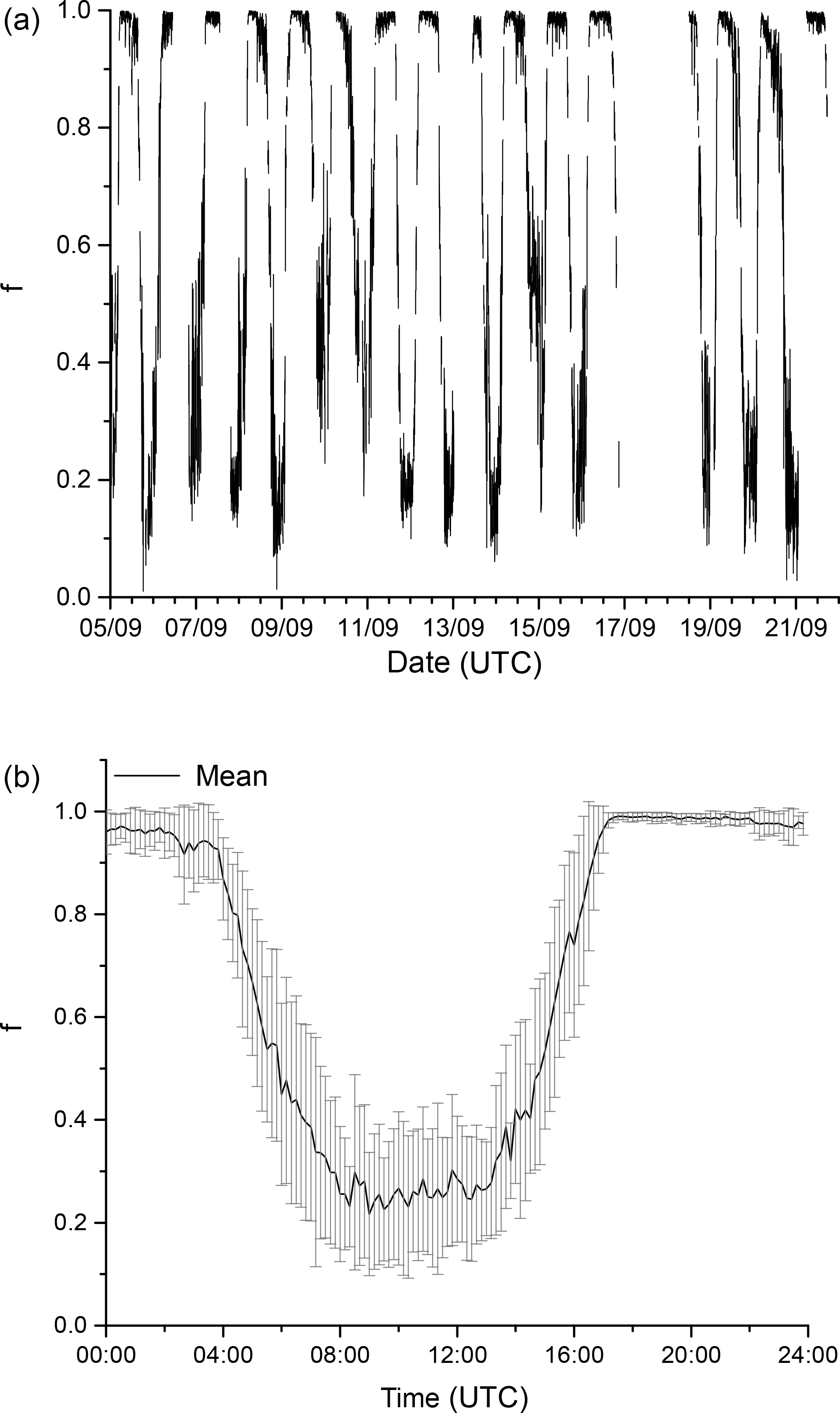
Figure 12The fraction, f, of the total NO3 loss with organic trace gases as a time series (a) and as a campaign averaged, diel cycle (b) where .
3.5 High NO3 reactivity and its contribution to NOx loss
The high reactivity of NO3 towards organic trace gases in the boreal environment means that other loss processes, including formation of N2O5 or reaction with NO are suppressed. To a first approximation we can assume that, at nighttime, in the absence of NO and sunlight, each NO3 radical formed in the reaction of NO2 with O3 will react with a biogenic hydrocarbon, resulting in formation of an organic nitrate at a yield of between 20 and 100 %, depending on the identity of the organic reactant (Ng et al., 2017). The large values for kOTG obtained during the day mean that a significant fraction of the NO3 formed can be converted to organic nitrates rather than result in re-formation of NO2 via reaction with NO or photolysis. The fraction, f, of NO3 that will react with organic trace gases is given by
Figure 12 illustrates the time series (upper plot) and the campaign averaged diel cycle (lower plot) for f which varies between ≈ 0.1 and 0.4 at the peak of the actinic flux, the variation is largely caused by day-to-day variability in insolation. As the spectral radiometer was located at a height of 35 m, will be slightly overestimated around midday as light levels within the canopy are lower. The overestimation will be magnified during the early morning and late afternoon when the forest is in shade at lower levels but the spectral radiometer is not. The daytime values for f are thus lower limits. With typical daytime NO levels of 50–100 pptv, the term [NO]k2 contributes ≈ 0.03–0.06 s−1 to NO3 loss, whereas has maxima of close to 0.1 s−1 each day. For comparison daytime values of kOTG of ≈ 0.05 s−1 were often observed (Fig. 2).
The diel cycle for f shows that even at the peak of the actinic flux, on average circa 20 % of the NO3 formed will react with an organic trace gas rather than be photolysed or react with NO in this environment. This implies that, in the summer–autumn boreal forest, NO3 reactions may represent a significant loss of NOx not only during the nighttime but over the full diel cycle, with a significant enhancement in the daytime production of alkyl nitrates, generally assumed to proceed only via reactions of organic peroxy radicals with NO.
The first direct measurements of NO3 reactivity to organic trace gases (kOTG) in the boreal forest indicate that NO3 is very short lived in this environment with lifetimes generally less than 10 s, mainly due to reaction with monoterpenes. The highest NO3 reactivities were encountered during nights with strong temperature inversions and a relative humidity of 100 %, and were accompanied by rapid O3 depletion, together highlighting the important role of nocturnal boundary layer dynamics in controlling canopy-level NO3 reactivity. The daytime reactivity was sufficiently large that reactions of NO3 with organic trace gases could compete with photolysis and the reaction with NO, so that NO3-induced losses of NOx and the formation of organic nitrates was significant. Measurements of the vertical profile in NO3 reactivity indicate a strong gradient during nighttime, with the highest reactivity observed below canopy height, highlighting a potential role for emissions of reactive trace gases from the forest floor. The hydrocarbons measured did not fully account for the observed NO3 reactivity, indicating the presence of unsaturated organic trace gases that were not identified, sesquiterpenes being potential candidates.
The IBAIRN data will be released at the end of 2018 when they can be obtained on request (via John Crowley) from the owners.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-3799-2018-supplement.
The authors declare that they have no conflict of interest.
We are grateful to ENVRIplus for partial financial support of the IBAIRN
campaign. We also thank Uwe Parchatka for provision of the NO
dataset.
The article processing charges for
this open-access
publication were covered by the Max Planck
Society.
Edited by: Dwayne
Heard
Reviewed by: two anonymous referees
Aaltonen, H., Pumpanen, J., Pihlatie, M., Hakola, H., Hellén, H., Kulmala, L., Vesala, T., and Bäck, J.: Boreal pine forest floor biogenic volatile organic compound emissions peak in early summer and autumn, Agr. Forest Meteorol., 151, 682–691, doi:https://doi.org/10.1016/j.agrformet.2010.12.010, 2011.
Aldener, M., Brown, S. S., Stark, H., Williams, E. J., Lerner, B. M., Kuster, W. C., Goldan, P. D., Quinn, P. K., Bates, T. S., Fehsenfeld, F. C., and Ravishankara, A. R.: Reactivity and loss mechanisms of NO3 and N2O5 in a polluted marine environment: Results from in situ measurements during New England Air Quality Study 2002, J. Geophys. Res.-Atmos., 111, D23S73, https://doi.org/10.1029/2006JD007252, 2006.
Aliwell, S. R. and Jones, R. L.: Measurements of tropospheric NO3 at midlatitude, J. Geophys. Res.-Atmos., 103, 5719–5727, 1998.
Allan, B. J., Carslaw, N., Coe, H., Burgess, R. A., and Plane, J. M. C.: Observations of the nitrate radical in the marine boundary layer, J. Atmos. Chem., 33, 129–154, 1999.
Allan, B. J., Plane, J. M. C., Coe, H., and Shillito, J.: Observations of NO3 concentration profiles in the troposphere, J. Geophys. Res.-Atmos., 107, 4588, https://doi.org/10.1029/2002jd002112, 2002.
Ambrose, J. L., Mao, H., Mayne, H. R., Stutz, J., Talbot, R., and Sive, B. C.: Nighttime nitrate radical chemistry at Appledore island, Maine during the 2004 international consortium for atmospheric research on transport and transformation, J. Geophys. Res.-Atmos., 112, D21302, doi:10.1.1029/2007JD008756, 2007.
Ammann, M., Cox, R. A., Crowley, J. N., Jenkin, M. E., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume VI – heterogeneous reactions with liquid substrates, Atmos. Chem. Phys., 13, 8045–8228, https://doi.org/10.5194/acp-13-8045-2013, 2013.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, 2003a.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003b.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bäck, J., Aalto, J., Henriksson, M., Hakola, H., He, Q., and Boy, M.: Chemodiversity of a Scots pine stand and implications for terpene air concentrations, Biogeosciences, 9, 689–702, https://doi.org/10.5194/bg-9-689-2012, 2012.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, 2012.
Brown, S. S., Stark, H., and Ravishankara, A. R.: Applicability of the steady state approximation to the interpretation of atmospheric observations of NO3 and N2O5, J. Geophys. Res.-Atmos., 108, 4539, https://doi.org/10.1029/2003JD003407, 2003.
Brown, S. S., Dube, W. P., Osthoff, H. D., Stutz, J., Ryerson, T. B., Wollny, A. G., Brock, C. A., Warneke, C., De Gouw, J. A., Atlas, E., Neuman, J. A., Holloway, J. S., Lerner, B. M., Williams, E. J., Kuster, W. C., Goldan, P. D., Angevine, W. M., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Vertical profiles in NO3 and N2O5 measured from an aircraft: Results from the NOAA P-3 and surface platforms during the New England Air Quality Study 2004, J. Geophys. Res.-Atmos., 112, D22304, https://doi.org/10.1029/2007jd008893, 2007a.
Brown, S. S., Dubé, W. P., Osthoff, H. D., Wolfe, D. E., Angevine, W. M., and Ravishankara, A. R.: High resolution vertical distributions of NO3 and N2O5 through the nocturnal boundary layer, Atmos. Chem. Phys., 7, 139–149, https://doi.org/10.5194/acp-7-139-2007, 2007b.
Brown, S. S., deGouw, J. A., Warneke, C., Ryerson, T. B., Dubé, W. P., Atlas, E., Weber, R. J., Peltier, R. E., Neuman, J. A., Roberts, J. M., Swanson, A., Flocke, F., McKeen, S. A., Brioude, J., Sommariva, R., Trainer, M., Fehsenfeld, F. C., and Ravishankara, A. R.: Nocturnal isoprene oxidation over the Northeast United States in summer and its impact on reactive nitrogen partitioning and secondary organic aerosol, Atmos. Chem. Phys., 9, 3027–3042, https://doi.org/10.5194/acp-9-3027-2009, 2009.
Burkholder, J. B., Sander, S. P., Abbatt, J., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15–10, Jet Propulsion Laboratory, Pasadena, available at: http://jpldataeval.jpl.nasa.gov, 2016.
Chen, X., Quéléver, L. L. J., Fung, P. L., Kesti, J., Rissanen, M. P., Bäck, J., Keronen, P., Junninen, H., Petäjä, T., Kerminen, V.-M., and Kulmala, M.: Observations of ozone depletion events in a Finnish boreal forest, Atmos. Chem. Phys., 18, 49–63, https://doi.org/10.5194/acp-18-49-2018, 2018.
Crowley, J. N., Schuster, G., Pouvesle, N., Parchatka, U., Fischer, H., Bonn, B., Bingemer, H., and Lelieveld, J.: Nocturnal nitrogen oxides at a rural mountain-site in south-western Germany, Atmos. Chem. Phys., 10, 2795–2812, https://doi.org/10.5194/acp-10-2795-2010, 2010.
Crowley, J. N., Thieser, J., Tang, M. J., Schuster, G., Bozem, H., Beygi, Z. H., Fischer, H., Diesch, J.-M., Drewnick, F., Borrmann, S., Song, W., Yassaa, N., Williams, J., Pöhler, D., Platt, U., and Lelieveld, J.: Variable lifetimes and loss mechanisms for NO3 and N2O5 during the DOMINO campaign: contrasts between marine, urban and continental air, Atmos. Chem. Phys., 11, 10853–10870, https://doi.org/10.5194/acp-11-10853-2011, 2011.
Dentener, F. J. and Crutzen, P. J.: Reaction of N2O5 on tropospheric aerosols – Impact on the global distributions of NOx, O3, and OH, J. Geophys. Res.-Atmos., 98, 7149–7163, 1993.
Duhl, T. R., Helmig, D., and Guenther, A.: Sesquiterpene emissions from vegetation: a review, Biogeosciences, 5, 761–777, https://doi.org/10.5194/bg-5-761-2008, 2008.
Eerdekens, G., Yassaa, N., Sinha, V., Aalto, P. P., Aufmhoff, H., Arnold, F., Fiedler, V., Kulmala, M., and Williams, J.: VOC measurements within a boreal forest during spring 2005: on the occurrence of elevated monoterpene concentrations during night time intense particle concentration events, Atmos. Chem. Phys., 9, 8331–8350, https://doi.org/10.5194/acp-9-8331-2009, 2009
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkle, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Geyer, A., Alicke, B., Konrad, S., Schmitz, T., Stutz, J., and Platt, U.: Chemistry and oxidation capacity of the nitrate radical in the continental boundary layer near Berlin, J. Geophys. Res.-Atmos., 106, 8013–8025, 2001.
Goldstein, A. H., McKay, M., Kurpius, M. R., Schade, G. W., Lee, A., Holzinger, R., and Rasmussen, R. A.: Forest thinning experiment confirms ozone deposition to forest canopy is dominated by reaction with biogenic VOCs, Geophys. Res. Lett., 31, L22106, https://doi.org/10.1029/2004gl021259, 2004.
Gölz, C., Senzig, J., and Platt, U.: NO3-initiated oxidation of biogenic hydrocarbons, Chemosphere – Global Change Science, 3, 339–352, https://doi.org/10.1016/S1465-9972(01)00015-0, 2001.
Graus, M., Muller, M., and Hansel, A.: High Resolution PTR-TOF: Quantification and Formula Confirmation of VOC in Real Time, J. Am. Soc. Mass Spectrom., 21, 1037–1044, 2010.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hakola, H., Tarvainen, V., Laurila, T., Hiltunen, V., Hellén, H., and Keronen, P.: Seasonal variation of VOC concentrations above a boreal coniferous forest, Atmos. Environ., 37, 1623–1634, doi:https://doi.org/10.1016/S1352-2310(03)00014-1, 2003.
Hakola, H., Tarvainen, V., Bäck, J., Ranta, H., Bonn, B., Rinne, J., and Kulmala, M.: Seasonal variation of mono- and sesquiterpene emission rates of Scots pine, Biogeosciences, 3, 93–101, https://doi.org/10.5194/bg-3-93-2006, 2006.
Hakola, H., Hellén, H., Tarvainen, V., Bäck, J., Patokoski, J., and Rinne, J.: Annual variations of atmospheric VOC concentrations in a boreal forest, Boreal Environ. Res., 14, 722–730, 2009.
Hakola, H., Hellén, H., Hemmilä, M., Rinne, J., and Kulmala, M.: In situ measurements of volatile organic compounds in a boreal forest, Atmos. Chem. Phys., 12, 11665–11678, https://doi.org/10.5194/acp-12-11665-2012, 2012.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hari, P. and Kulmala, M.: Station for Measuring Ecosystem–Atmosphere Relations (SMEAR II), Boreal Environ. Res., 10, 315–322, 2005.
Hari, P., Heliövaara, K., and Kulmala, L.: Physical and Physiological Forest Ecology: Physical and Physiological Forest Ecology, edited by: Hari, P., Heliövaara, K., and Kulmala, L., Springer, 2013.
Heintz, F., Platt, U., Flentje, H., and Dubois, R.: Long-term observation of nitrate radicals at the tor station, Kap Arkona (Rugen), J. Geophys. Res.-Atmos., 101, 22891–22910, 1996.
Hellén, H., Kuronen, P., and Hakola, H.: Heated stainless steel tube for ozone removal in the ambient air measurements of mono- and sesquiterpenes, Atmos. Environ., 57, 35–40, doi:https://doi.org/10.1016/j.atmosenv.2012.04.019, 2012.
Hens, K., Novelli, A., Martinez, M., Auld, J., Axinte, R., Bohn, B., Fischer, H., Keronen, P., Kubistin, D., Nölscher, A. C., Oswald, R., Paasonen, P., Petäjä, T., Regelin, E., Sander, R., Sinha, V., Sipilä, M., Taraborrelli, D., Tatum Ernest, C., Williams, J., Lelieveld, J., and Harder, H.: Observation and modelling of HOx radicals in a boreal forest, Atmos. Chem. Phys., 14, 8723–8747, https://doi.org/10.5194/acp-14-8723-2014, 2014.
Holzinger, R., Lee, A., Paw, K. T., and Goldstein, U. A. H.: Observations of oxidation products above a forest imply biogenic emissions of very reactive compounds, Atmos. Chem. Phys., 5, 67–75, https://doi.org/10.5194/acp-5-67-2005, 2005.
Holzinger, R., Lee, A., McKay, M., and Goldstein, A. H.: Seasonal variability of monoterpene emission factors for a ponderosa pine plantation in California, Atmos. Chem. Phys., 6, 1267–1274, https://doi.org/10.5194/acp-6-1267-2006, 2006.
Holzke, C., Hoffmann, T., Jaeger, L., Koppmann, R., and Zimmer, W.: Diurnal and seasonal variation of monoterpene and sesquiterpene emissions from Scots pine (Pinus sylvestris L.), Atmos. Environ., 40, 3174–3185, https://doi.org/10.1016/j.atmosenv.2006.01.039, 2006.
Ilvesniemi, H., Levula, J., Ojansuu, R., Kolari, P., Kulmala, L., Pumpanen, J., Launiainen, S., Vesala, T., and Nikinmaa, E.: Long-term measurements of the carbon balance of a boreal Scots pine dominated forest ecosystem, Boreal Environ. Res., 14, 731–753, 2009.
Insam, H. and Seewald, M. S. A.: Volatile organic compounds (VOCs) in soils, Biol. Fertil. Soils, 46, 199–213, https://doi.org/10.1007/s00374-010-0442-3, 2010.
IUPAC (Ammann, M., Cox, R. A., Crowley, J. N., Herrmann, H., Jenkin, M. E., McNeill, V. F., Mellouki, A., Rossi, M. J., Troe, J., and Wallington, T. J.): Task Group on Atmospheric Chemical Kinetic Data Evaluation, available at: http://iupac.pole-ether.fr/index.html (last access: October 2017), 2017.
Jardine, K., Serrano, A. Y., Arneth, A., Abrell, L., Jardine, A., van Haren, J., Artaxo, P., Rizzo, L. V., Ishida, F. Y., Karl, T., Kesselmeier, J., Saleska, S., and Huxman, T.: Within-canopy sesquiterpene ozonolysis in Amazonia, J. Geophys. Res.-Atmos., 116, D19301, https://doi.org/10.1029/2011jd016243, 2011.
Jordan, A., Haidacher, S., Hanel, G., Hartungen, E., Mark, L., Seehauser, H., Schottkowsky, R., Sulzer, P., and Mark, T. D.: A high resolution and high sensitivity proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF-MS), Int. J. Mass Spectrom., 286, 122–128, 2009.
Kurpius, M. R. and Goldstein, A. H.: Gas-phase chemistry dominates O3 loss to a forest, implying a source of aerosols and hydroxyl radicals to the atmosphere, Geophys. Res. Lett., 30, 1371, https://doi.org/10.1029/2002gl016785, 2003.
Lappalainen, H. K., Sevanto, S., Bäck, J., Ruuskanen, T. M., Kolari, P., Taipale, R., Rinne, J., Kulmala, M., and Hari, P.: Day-time concentrations of biogenic volatile organic compounds in a boreal forest canopy and their relation to environmental and biological factors, Atmos. Chem. Phys., 9, 5447–5459, https://doi.org/10.5194/acp-9-5447-2009, 2009.
Lelieveld, J., Butler, T. M., Crowley, J. N., Dillon, T. J., Fischer, H., Ganzeveld, L., Harder, H., Lawrence, M. G., Martinez, M., Taraborrelli, D., and Williams, J.: Atmospheric oxidation capacity sustained by a tropical forest, Nature, 452, 737–740, 2008.
Li, J., Reiffs, A., Parchatka, U., and Fischer, H.: In situ measurements of atmospheric CO and its correlation with NOx and O3 at a rural mountain site, Metrol. Meas. Sys., XXII, 25–38, 2015.
Liao, L., Dal Maso, M., Taipale, R., Rinne, J., Ehn, M., Junninen, H., Aijala, M., Nieminen, T., Alekseychik, P., Hulkkonen, M., Worsnop, D. R., Kerminen, V. M., and Kulmala, M.: Monoterpene pollution episodes in a forest environment: indication of anthropogenic origin and association with aerosol particles, Boreal Environ. Res., 16, 288–303, 2011.
Liebmann, J. M., Schuster, G., Schuladen, J. B., Sobanski, N., Lelieveld, J., and Crowley, J. N.: Measurement of ambient NO3 reactivity: design, characterization and first deployment of a new instrument, Atmos. Meas. Tech., 10, 1241–1258, https://doi.org/10.5194/amt-10-1241-2017, 2017.
Meusel, H., Kuhn, U., Reiffs, A., Mallik, C., Harder, H., Martinez, M., Schuladen, J., Bohn, B., Parchatka, U., Crowley, J. N., Fischer, H., Tomsche, L., Novelli, A., Hoffmann, T., Janssen, R. H. H., Hartogensis, O., Pikridas, M., Vrekoussis, M., Bourtsoukidis, E., Weber, B., Lelieveld, J., Williams, J., Pöschl, U., Cheng, Y., and Su, H.: Daytime formation of nitrous acid at a coastal remote site in Cyprus indicating a common ground source of atmospheric HONO and NO, Atmos. Chem. Phys., 16, 14475–14493, https://doi.org/10.5194/acp-16-14475-2016, 2016.
Mogensen, D., Gierens, R., Crowley, J. N., Keronen, P., Smolander, S., Sogachev, A., Nölscher, A. C., Zhou, L., Kulmala, M., Tang, M. J., Williams, J., and Boy, M.: Simulations of atmospheric OH, O3 and NO3 reactivities within and above the boreal forest, Atmos. Chem. Phys., 15, 3909–3932, https://doi.org/10.5194/acp-15-3909-2015, 2015.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Nölscher, A. C., Williams, J., Sinha, V., Custer, T., Song, W., Johnson, A. M., Axinte, R., Bozem, H., Fischer, H., Pouvesle, N., Phillips, G., Crowley, J. N., Rantala, P., Rinne, J., Kulmala, M., Gonzales, D., Valverde-Canossa, J., Vogel, A., Hoffmann, T., Ouwersloot, H. G., Vilà-Guerau de Arellano, J., and Lelieveld, J.: Summertime total OH reactivity measurements from boreal forest during HUMPPA-COPEC 2010, Atmos. Chem. Phys., 12, 8257–8270, https://doi.org/10.5194/acp-12-8257-2012, 2012.
Nölscher, A. C., Yanez-Serrano, A. M., Wolff, S., de Araujo, A. C., Lavric, J. V., Kesselmeier, J., and Williams, J.: Unexpected seasonality in quantity and composition of Amazon rainforest air reactivity, Nat. Commun., 7, 10383, https://doi.org/10.1038/ncomms10383, 2016.
Osthoff, H. D., Roberts, J. M., Ravishankara, A. R., Williams, E. J., Lerner, B. M., Sommariva, R., Bates, T. S., Coffman, D., Quinn, P. K., Dibb, J. E., Stark, H., Burkholder, J. B., Talukdar, R. K., Meagher, J., Fehsenfeld, F. C., and Brown, S. S.: High levels of nitryl chloride in the polluted subtropical marine boundary layer, Nat. Geosci., 1, 324–328, 2008.
Penuelas, J., Asensio, D., Tholl, D., Wenke, K., Rosenkranz, M., Piechulla, B., and Schnitzler, J. P.: Biogenic volatile emissions from the soil, Plant Cell Environ., 37, 1866–1891, https://doi.org/10.1111/pce.12340, 2014.
Peräkylä, O., Vogt, M., Tikkanen, O. P., Laurila, T., Kajos, M. K., Rantala, P. A., Patokoski, J., Aalto, J., Yli-Juuti, T., Ehn, M., Sipila, M., Paasonen, P., Rissanen, M., Nieminen, T., Taipale, R., Keronen, P., Lappalainen, H. K., Ruuskanen, T. M., Rinne, J., Kerminen, V. M., Kulmala, M., Back, J., and Petaja, T.: Monoterpenes' oxidation capacity and rate over a boreal forest: temporal variation and connection to growth of newly formed particles, Boreal Environ. Res., 19, 293–310, 2014.
Phillips, G. J., Tang, M. J., Thieser, J., Brickwedde, B., Schuster, G., Bohn, B., Lelieveld, J., and Crowley, J. N.: Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions, Geophys. Res. Lett., 39, L10811, https://doi.org/10.1029/2012GL051912, 2012.
Phillips, G. J., Thieser, J., Tang, M., Sobanski, N., Schuster, G., Fachinger, J., Drewnick, F., Borrmann, S., Bingemer, H., Lelieveld, J., and Crowley, J. N.: Estimating N2O5 uptake coefficients using ambient measurements of NO3, N2O5, ClNO2 and particle-phase nitrate, Atmos. Chem. Phys., 16, 13231–13249, https://doi.org/10.5194/acp-16-13231-2016, 2016.
Rannik, Ü., Altimir, N., Mammarella, I., Báck, J., Rinne, J., Ruuskanen, T. M., Hari, P., Vesala, T., and Kulmala, M.: Ozone deposition into a boreal forest over a decade of observations: evaluating deposition partitioning and driving variables, Atmos. Chem. Phys., 12, 12165–12182, https://doi.org/10.5194/acp-12-12165-2012, 2012.
Rinne, J., Ruuskanen, T. M., Reissell, A., Taipale, R., Hakola, H., and Kulmala, M.: On-line PTR-MS measurements of atmospheric concentrations of volatile organic compounds in a European boreal forest ecosystem, Boreal Environmental Research, 10, 425–436, 2005.
Rinne, J., Markkanen, T., Ruuskanen, T. M., Petäjä, T., Keronen, P., Tang, M. J., Crowley, J. N., Rannik, Ü., and Vesala, T.: Effect of chemical degradation on fluxes of reactive compounds – a study with a stochastic Lagrangian transport model, Atmos. Chem. Phys., 12, 4843–4854, https://doi.org/10.5194/acp-12-4843-2012, 2012.
Schallhart, S., Rantala, P., Nemitz, E., Taipale, D., Tillmann, R., Mentel, T. F., Loubet, B., Gerosa, G., Finco, A., Rinne, J., and Ruuskanen, T. M.: Characterization of total ecosystem-scale biogenic VOC exchange at a Mediterranean oak-hornbeam forest, Atmos. Chem. Phys., 16, 7171–7194, https://doi.org/10.5194/acp-16-7171-2016, 2016.
Shu, Y. H. and Atkinson, R.: Atmospheric lifetimes and fates of a series of sesquiterpenes, J. Geophys. Res.-Atmos., 100, 7275–7281, https://doi.org/10.1029/95jd00368, 1995.
Sinha, V., Williams, J., Lelieveld, J., Ruuskanen, T. M., Kajos, M. K., Patokoski, J., Hellen, H., Hakola, H., Mogensen, D., Boy, M., Rinne, J., and Kulmala, M.: OH Reactivity Measurements within a Boreal Forest: Evidence for Unknown Reactive Emissions, Environ. Sci. Technol., 44, 6614–6620, https://doi.org/10.1021/es101780b, 2010.
Sobanski, N., Schuladen, J., Schuster, G., Lelieveld, J., and Crowley, J. N.: A five-channel cavity ring-down spectrometer for the detection of NO2, NO3, N2O5, total peroxy nitrates and total alkyl nitrates, Atmos. Meas. Tech., 9, 5103–5118, https://doi.org/10.5194/amt-9-5103-2016, 2016a.
Sobanski, N., Tang, M. J., Thieser, J., Schuster, G., Pöhler, D., Fischer, H., Song, W., Sauvage, C., Williams, J., Fachinger, J., Berkes, F., Hoor, P., Platt, U., Lelieveld, J., and Crowley, J. N.: Chemical and meteorological influences on the lifetime of NO3 at a semi-rural mountain site during PARADE, Atmos. Chem. Phys., 16, 4867–4883, https://doi.org/10.5194/acp-16-4867-2016, 2016b.
Stutz, J., Alicke, B., Ackermann, R., Geyer, A., White, A., and Williams, E.: Vertical profiles of NO3, N2O5, O3, and NOX in the nocturnal boundary layer: 1. Observations during the Texas Air Quality Study 2000 J. Geophys. Res.-Atmos., 109, D12306, https://doi.org/10.1029/2003JD004209, 2004.
Sun, S., Moravek, A., Trebs, I., Kesselmeier, J., and Sorgel, M.: Investigation of the influence of liquid surface films on O3 and PAN deposition to plant leaves coated with organic/inorganic solution, J. Geophys. Res.-Atmos., 121, 14239–14256, 2016.
Tarvainen, V., Hakola, H., Hellén, H., Bäck, J., Hari, P., and Kulmala, M.: Temperature and light dependence of the VOC emissions of Scots pine, Atmos. Chem. Phys., 5, 989–998, https://doi.org/10.5194/acp-5-989-2005, 2005.
Thornton, J. A., Kercher, J. P., Riedel, T. P., Wagner, N. L., Cozic, J., Holloway, J. S., Dube, W. P., Wolfe, G. M., Quinn, P. K., Middlebrook, A. M., Alexander, B., and Brown, S. S.: A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 464, 271–274, https://doi.org/10.1038/nature08905, 2010.
von Friedeburg, C., Wagner, T., Geyer, A., Kaiser, N., Vogel, B., Vogel, H., and Platt, U.: Derivation of tropospheric NO3 profiles using off-axis differential optical absorption spectroscopy measurements during sunrise and comparison with simulations, J. Geophys. Res.-Atmos., 107, 4168, https://doi.org/10.1029/2001JD000481, 2002.
Warneke, C., Karl, T., Judmaier, H., Hansel, A., Jordan, A., Lindinger, W., and Crutzen, P. J.: Acetone, methanol and other partially oxidized volatile organic emissions from dead plant matter by abiological proceses: Significance for atmospheric HOx chemistry, Global. Biogeochem. Cy., 13, 9–17, 1999.
Wayne, R. P., Barnes, I., Biggs, P., Burrows, J. P., Canosa-Mas, C. E., Hjorth, J., Le Bras, G., Moortgat, G. K., Perner, D., Poulet, G., Restelli, G., and Sidebottom, H.: The nitrate radical: Physics, chemistry, and the atmosphere, Atmos. Environ. A-Gen., 25, 1–206, 1991.
Williams, J., Crowley, J., Fischer, H., Harder, H., Martinez, M., Petäjä, T., Rinne, J., Bäck, J., Boy, M., Dal Maso, M., Hakala, J., Kajos, M., Keronen, P., Rantala, P., Aalto, J., Aaltonen, H., Paatero, J., Vesala, T., Hakola, H., Levula, J., Pohja, T., Herrmann, F., Auld, J., Mesarchaki, E., Song, W., Yassaa, N., Nölscher, A., Johnson, A. M., Custer, T., Sinha, V., Thieser, J., Pouvesle, N., Taraborrelli, D., Tang, M. J., Bozem, H., Hosaynali-Beygi, Z., Axinte, R., Oswald, R., Novelli, A., Kubistin, D., Hens, K., Javed, U., Trawny, K., Breitenberger, C., Hidalgo, P. J., Ebben, C. J., Geiger, F. M., Corrigan, A. L., Russell, L. M., Ouwersloot, H. G., Vilà-Guerau de Arellano, J., Ganzeveld, L., Vogel, A., Beck, M., Bayerle, A., Kampf, C. J., Bertelmann, M., Köllner, F., Hoffmann, T., Valverde, J., González, D., Riekkola, M.-L., Kulmala, M., and Lelieveld, J.: The summertime Boreal forest field measurement intensive (HUMPPA-COPEC-2010): an overview of meteorological and chemical influences, Atmos. Chem. Phys., 11, 10599–10618, https://doi.org/10.5194/acp-11-10599-2011, 2011.
Yassaa, N., Song, W., Lelieveld, J., Vanhatalo, A., Bäck, J., and Williams, J.: Diel cycles of isoprenoids in the emissions of Norway spruce, four Scots pine chemotypes, and in Boreal forest ambient air during HUMPPA-COPEC-2010, Atmos. Chem. Phys., 12, 7215–7229, https://doi.org/10.5194/acp-12-7215-2012, 2012.
Zhou, P., Ganzeveld, L., Rannik, Ü., Zhou, L., Gierens, R., Taipale, D., Mammarella, I., and Boy, M.: Simulating ozone dry deposition at a boreal forest with a multi-layer canopy deposition model, Atmos. Chem. Phys., 17, 1361–1379, https://doi.org/10.5194/acp-17-1361-2017, 2017.