the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Dec 2018
| 06 Dec 2018
Airborne and ground-based observations of ammonium-nitrate-dominated aerosols in a shallow boundary layer during intense winter pollution episodes in northern Utah
Alessandro Franchin
Dorothy L. Fibiger
Lexie Goldberger
Erin E. McDuffie
Alexander Moravek
Caroline C. Womack
Erik T. Crosman
Kenneth S. Docherty
William P. Dube
Sebastian W. Hoch
Ben H. Lee
Russell Long
Jennifer G. Murphy
Joel A. Thornton
Steven S. Brown
Munkhbayar Baasandorj
Ann M. Middlebrook
Airborne and ground-based measurements of aerosol concentrations, chemical composition, and gas-phase precursors were obtained in three valleys in northern Utah (USA). The measurements were part of the Utah Winter Fine Particulate Study (UWFPS) that took place in January–February 2017. Total aerosol mass concentrations of PM1 were measured from a Twin Otter aircraft, with an aerosol mass spectrometer (AMS). PM1 concentrations ranged from less than 2 µg m−3 during clean periods to over 100 µg m−3 during the most polluted episodes, consistent with PM2.5 total mass concentrations measured concurrently at ground sites. Across the entire region, increases in total aerosol mass above ∼2 µg m−3 were associated with increases in the ammonium nitrate mass fraction, clearly indicating that the highest aerosol mass loadings in the region were predominantly attributable to an increase in ammonium nitrate. The chemical composition was regionally homogenous for total aerosol mass concentrations above 17.5 µg m−3, with 74±5 % (average ± standard deviation) ammonium nitrate, 18±3 % organic material, 6±3 % ammonium sulfate, and 2±2 % ammonium chloride. Vertical profiles of aerosol mass and volume in the region showed variable concentrations with height in the polluted boundary layer. Higher average mass concentrations were observed within the first few hundred meters above ground level in all three valleys during pollution episodes. Gas-phase measurements of nitric acid (HNO3) and ammonia (NH3) during the pollution episodes revealed that in the Cache and Utah valleys, partitioning of inorganic semi-volatiles to the aerosol phase was usually limited by the amount of gas-phase nitric acid, with NH3 being in excess. The inorganic species were compared with the ISORROPIA thermodynamic model. Total inorganic aerosol mass concentrations were calculated for various decreases in total nitrate and total ammonium. For pollution episodes, our simulations of a 50 % decrease in total nitrate lead to a 46±3 % decrease in total PM1 mass. A simulated 50 % decrease in total ammonium leads to a 36±17 % µg m−3 decrease in total PM1 mass, over the entire area of the study. Despite some differences among locations, our results showed a higher sensitivity to decreasing nitric acid concentrations and the importance of ammonia at the lowest total nitrate conditions. In the Salt Lake Valley, both HNO3 and NH3 concentrations controlled aerosol formation.
- Article
(7318 KB) - Full-text XML
-
Supplement
(3477 KB) - BibTeX
- EndNote





Intense wintertime air pollution from particulate matter affects numerous locations in the United States (Chen et al., 2012; Lurmann et al., 2006) and around the world (Bessagnet et al., 2005; Gwaze et al., 2007; Petetin et al., 2016, Ricciardelli et al., 2017; Wang et al., 2014). In the US, pollution from fine particles (PM2.5) has been decreasing in the past decades. However, PM2.5 concentrations in Salt Lake City and surrounding valleys remain among the highest nationwide (EPA Air Quality, 2017). High PM2.5 episodes in these areas occur predominantly during winter, when persistent cold-air pools (PCAPs) form as a result of high-pressure ridges (Whiteman et al., 2014; Lareau et al., 2013). These events increase in frequency and severity during snow-covered periods (Green et al., 2015). Under PCAP conditions, emissions of pollutants and precursor gases are trapped in a shallow boundary layer, as vertical mixing is limited. Precursor gases react chemically, increasing the production of PM2.5 via secondary pathways. Almost every year, wintertime concentrations of PM2.5 exceed the limit of 35 µg m−3 in a 24 h average in the Logan, Ogden–Clearfield, Salt Lake City, and Provo–Orem Core-Based Statistical Areas (CBSAs, EPA Air Quality Statistics by City, 2016), leading to violations of the National Ambient Air Quality Standards (NAAQS) set by the Environmental Protection Agency (EPA). This air quality issue affects almost 80 % of the population of the state of Utah and has been associated with increased risk for stroke, heart disease, lung cancer, and both chronic and acute respiratory diseases (Beard et al., 2012; Pope et al., 2006; WHO, 2016).
Chemical composition measurements of PM and their precursor gases, together with a detailed understanding of the meteorology, are key to identifying sources and developing mitigation strategies. Previous studies showed that ammonium nitrate (NH4NO3) dominates the aerosol mass during pollution episodes (Hansen et al., 2010; Kelly et al., 2013; Kuprov et al., 2014; Long et al., 2003, 2005a, b; Mangelson et al., 1997; Silva et al., 2007). Analysis of ground-based data suggests that its formation, at least in the Salt Lake Valley and Cache Valley, is likely to be nitric acid limited (Kuprov et al., 2014; Mangelson et al., 1997). The Great Salt Lake is a potential source of chloride in the area, and chloride may also play a role during pollution episodes, although its role is much less clear. Hansen and co-workers found chloride (in the form of sodium chloride) to be a minor part of the PM2.5 mass fraction (1.3 % on average over the period of their study 22–31 January 2007 in Lindon, UT; Hansen et al., 2010). However, Kelly and co-workers, using two different factor analysis techniques, found that, after ammonium nitrate, ammonium chloride could also be a significant source of secondary wintertime PM2.5 (Kelly et al., 2013).
Formation of PM2.5 results in part from a complex interaction of chemical production mechanisms with boundary layer meteorology (Baasandorj et al., 2017a). Previous studies carried out in wintertime in San Joaquin Valley (CA) highlighted the critical role of nocturnal chemical production of nitrate aloft in the residual layer (i.e., that region decoupled from the stable nocturnal boundary layer influenced by previous-day surface emissions). Those studies suggested that nighttime nitrate formation within the residual layer was a major contributor to surface-level PM2.5 concentrations (Watson et al., 2002; Brown et al., 2006; Chow et al., 2006; Lurmann et al., 2006; Prabhakar et al., 2017). Studies in other regions of the US have reached similar conclusions regarding the role of nighttime processing aloft as a source of winter soluble nitrate (Stanier et al., 2012; Kim et al., 2014). Similarly, Baasandorj and co-workers found evidence in their ground-based observations in the Salt Lake City area for nitrate formation occurring at night and early morning photochemistry in the upper levels of a PCAP (Baasandorj et al., 2017a). Although the residual layer is inferred to be a critical region for understanding process-level atmospheric chemistry leading to winter ammonium nitrate in polluted regions, few air quality studies have directly probed residual layer chemical composition using an instrumented aircraft in a polluted winter boundary layer (Brown et al., 2013; McDuffie et al., 2018).
In this work, we present results of the Utah Winter Fine Particulate Study (UWFPS), an intensive measurement campaign combining airborne and surface-based measurements that took place in Salt Lake City and surrounding valleys in January and February 2017 (Figs. 1 and Supplement Fig. S1). In particular, we focus on the chemical and physical aerosol properties measured by an aerosol mass spectrometer (AMS) and an ultra-high-sensitivity aerosol spectrometer (UHSAS) aboard the National Oceanic and Atmospheric Administration (NOAA) Twin Otter research aircraft. The flights took place from 16 January to 12 February and were based out of the Salt Lake City International Airport, with flight plans extending up to Cache Valley, across the Great Salt Lake, and over the Salt Lake and Utah valleys. Flight plans included missed approaches at several local airports in the three main study regions to probe the vertical structure of the concentrations of aerosols and precursor gases. The NOAA Twin Otter aircraft was equipped with a suite of instruments that allowed comprehensive measurements of the aerosols and the gas-phase precursors that contribute to the pollution episodes in the area.
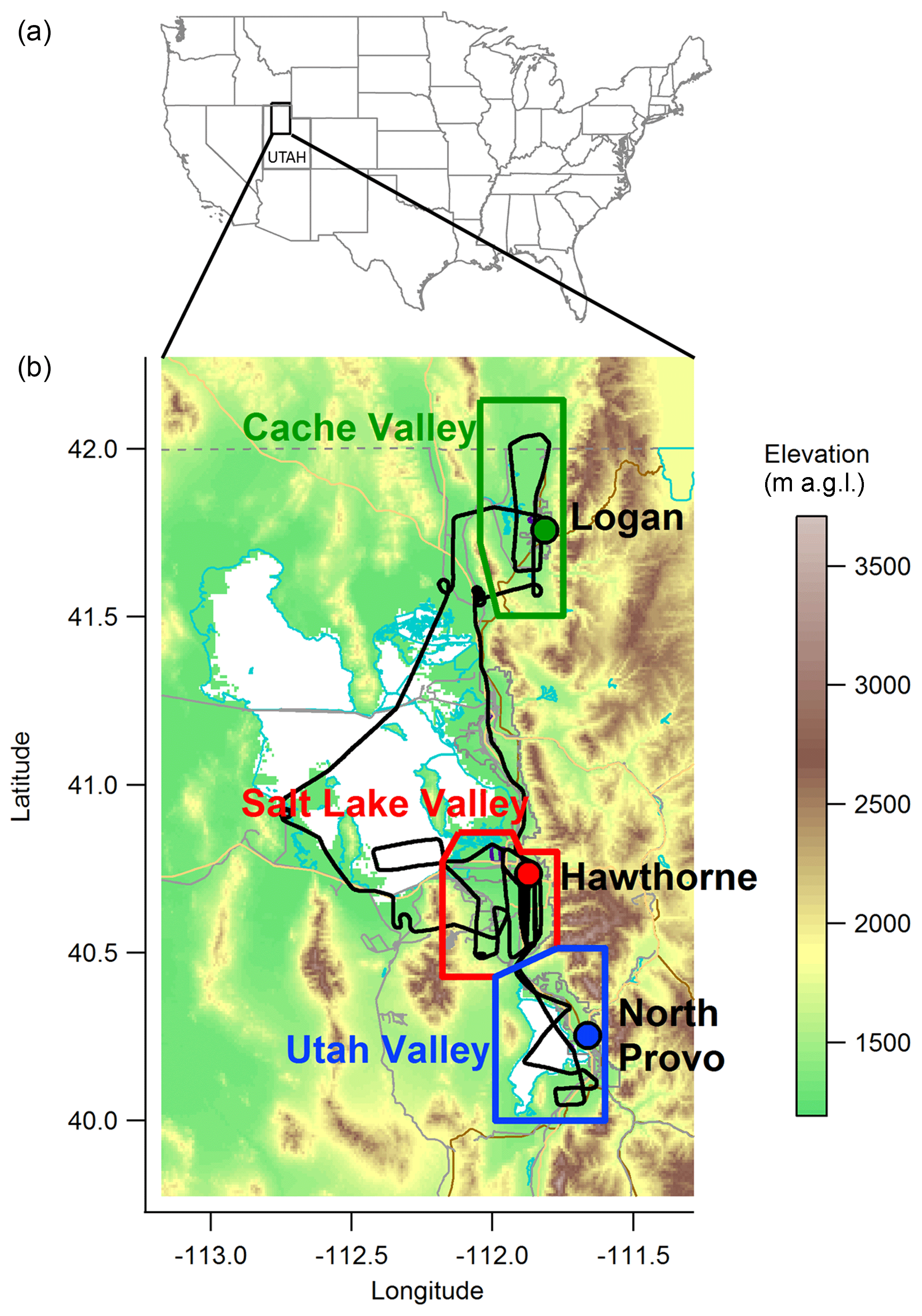
Figure 1Map showing the area studied during the UWFPS. (a) Map of the United States with the region of interest of this study highlighted by a black rectangle. (b) Detail of the area of interest colored by elevation as shown in the color bar. In black a typical flight track is shown. The Cache Valley study region is outlined in green, Salt Lake Valley in red, and Utah Valley in blue. The circles with the same color pattern and labeled in black represent the locations of the ground sites with data presented here.
The goal of this analysis is to achieve a better understanding of the processes that drive the conversion of precursor vapors into aerosol particles. We compared measured inorganic gas and particle composition to a thermodynamic model and used the model output to probe sensitivities to changes in that composition that are useful to developing effective PM2.5 control strategies.
The airborne measurements, carried out using the NOAA Twin Otter aircraft, were designed to characterize pollution episodes with a particular focus on ammonium nitrate aerosol formation. The aerosol measurements from the aircraft were compared with ground-site data of PM2.5, PM10, and non-refractory PM1 (NR-PM1) obtained by the Utah Department of Air Quality (UDAQ) and the U.S. Environmental Protection Agency (EPA; UWFPS team, 2017). The thermodynamic model ISORROPIA was used to compare predicted aerosol to vapor partitioning with the measurements.
2.1 Twin Otter aircraft instrumentation
2.1.1 Aerosol measurements
Aerosols were sampled using a perpendicular near-isokinetic inlet described and characterized previously for aircraft measurements (Perring et al., 2013). The inner diameter of the secondary diffuser of the inlet was modified from the original inlet (∅=2.5 mm in the modified version) to accommodate a 3 liter per minute (L min−1) total inlet flow at the lower cruise speed of the Twin Otter (∼250 km h−1). The total inlet flow was chosen in order to limit sampling line losses due to diffusion and evaporation. The sampled air downstream of the inlet probe was split into a 37 cm sampling line to the UHSAS and a 153 cm sampling line to the AMS with an excess flow of about 2.8 L min−1 that was discarded. The excess flow was monitored with a laminar flow element and a calibrated pressure transducer (Honeywell 480-5439-ND).
The AMS (Aerodyne Research Inc., Billerica, MA) measures the chemical composition of the non-refractory aerosol particles in the 70 to 850 nm range (NR-PM1) (Canagaratna et al., 2007; Drewnick et al., 2005; Jayne et al., 2000). The working principle of the AMS is to (1) focus ambient aerosols in a vacuum chamber with an aerodynamic lens, (2) evaporate the aerosol particles by impacting them onto a 600 ∘C surface, and (3) ionize the evaporated molecules with electron-impact ionization and analyze the resulting fragments with a mass spectrometer to obtain information on the chemical composition of aerosols. For the majority of sampling, the AMS collected bulk mass spectra (MS mode). Additionally, aerosol size distributions are obtained by operating the AMS in particle time-of-flight (pToF) mode wherein mass spectrometer signals are monitored as a function of the time required for particles of different sizes to traverse the length of the vacuum chamber. The NOAA AMS deployed on the Twin Otter aircraft (TO-AMS) was equipped with a pressure-controlled inlet to maintain a constant mass flow rate into the instrument as the aircraft changed altitude (Bahreini et al., 2008). The TO-AMS was also equipped with a light-scattering module (LS) that provided more accurate size information in the range of 200–1000 nm and on the collection efficiency (CE; Liao et al., 2017). A CE of 1 was used for this analysis based on the light scattering (see Supplement).
The TO-AMS was operated in MS mode (6 s, with 4 s open to sample and 2 s closed for background correction) and pToF mode (4 s) in 27 sub-cycles of 10 s in total. We measured in LS mode for 30 s every 4.5 min. The aircraft AMS measurements are all reported in units of micrograms per standard cubic meter of air, for which standard conditions are 273 K and 1 atm. Details of the AMS calibration procedures for the UWFPS are presented in the Supplement. The 10 s average limit of detection for nitrate (), ammonium (), organic species, sulfate (), chloride (Cl−), and the total aerosol mass were 0.04, 0.09, 0.33, 0.03, 0.07, and 0.38 µg m−3, respectively. The uncertainty in the total AMS mass concentrations is estimated to be 20 % (Bahreini et al., 2009). It is important to note that during the campaign refractory species such as sea and/or lake salt (mostly sodium chloride), road salt (mostly magnesium chloride), dust (mostly alkali salts and silicon oxides), and black carbon (from diesel exhaust or wood combustion) were not measured.
The UHSAS (Droplet Measurement Technologies, Longmont, CO) was used to measure the dry size distributions of aerosol particles in the range of 70 to 1000 nm. The working principle of the UHSAS is based on optical scattering. The aerosol particles enter the instrument and scatter the light produced by a solid-state laser (1054 nm, 1 kW cm−2). Mirrors capable of collecting light over a large solid angle (22–158∘) direct the scattered light to two different photodiodes: an avalanche photodiode for detecting the smallest particles and a low-gain PIN photodiode for detecting particles in the upper size range. The signals generated by the scattered light are used for particle sizing since the amount of light scattered correlates strongly with particle size (Cai et al., 2008). UHSAS data during the UWFPS were recorded every 3 s and are reported here per standard cubic centimeter (273 K and 1 atm). The relative humidity in the sampling line was calculated to be less than 30 %. The UHSAS inlet flow was monitored with a linear flow element and a calibrated pressure transducer (Honeywell 480-5439-ND) in order to correct for variations in the inlet flow (Brock et al., 2011). The uncertainty during UWFPS was 2 % on the sizing, 14 % on the number concentration, 20 % on the total surface, and 35 % on the total volume.
2.1.2 Gas-phase measurements
On the Twin Otter aircraft, we obtained measurements of several gas-phase species, including those relevant for inorganic gas–aerosol partitioning in the region: nitric acid (HNO3), hydrochloric acid (HCl), and ammonia (NH3). We used these aircraft gas-phase species, as well as data from aerosol-phase nitrate, chloride, and ammonium, to calculate the ratios of gas to the total ammonium or total nitrate for estimating which species is the limiting reagent in ammonium nitrate formation and for the input to thermodynamic modeling with ISORROPIA (see Sect. 2.3).
The University of Washington high-resolution time-of-flight iodide adduct ionization mass spectrometer, hereafter referred to as HRToF-CIMS, was used to measure a suite of reactive inorganic gaseous halogen (HCl, Cl2, Br2, ClNO2, etc.) and nitrogen oxide (HNO3, N2O5, HONO, etc.) species. The major components of the CIMS were described previously (Lee et al., 2014) with upgrades implemented prior to UWFPS to improve in-flight calibration and sampling protocol (Lee et al., 2018). Ambient air was drawn into the CIMS at a rate of ∼22 L min−1 through a 39 cm long 1.6 cm inner diameter (ID) polytetrafluoroethylene (PTFE) inlet, resulting in a mean residence time of about ∼0.21 s. The CIMS instrument subsamples 2 standard L min−1 from the centerline of the main flow traveling through the 1.6 cm ID inlet with the remainder of the flow being pulled radially through a circular slot around and downstream of the ion molecule reaction (IMR) region entrance, to minimize sampling air that has been in contact with the inlet surface. Instrument background levels were determined every 60 s for a duration of 5 s by overflowing the IMR with ultra-high-purity nitrogen (UHP N2). The UHP N2 was introduced at the entrance of the IMR region, which represents the dominant source (> 80 %) of the sum of the background levels originating from the inlet and IMR region under field conditions. The molecular formulae of the compounds listed above are readily identified given its mass resolving power (4500–5500 Th Th−1; Junninen et al., 2010), with no known spectrally interfering species. For nitric acid (HNO3) and hydrogen chloride (HCl), the calibration uncertainty for both is about 30 %, while their limits of detection (LODs), determined by accounting for their calibration coefficients and variabilities in their background levels, were 60 and 160 ppt, respectively.
A continuous-wave quantum cascade tunable infrared laser differential absorption spectrometer (QC-TILDAS, Aerodyne Research Inc., Billerica, MA; Tevlin et al., 2017) was used to measure ammonia (NH3) concentrations from the Twin Otter aircraft. Prior to installation, the instrument was optimized for a lower total weight, allowing its operation on the Twin Otter aircraft. Furthermore, it was equipped with a pressure control system to account for changing ambient pressure with altitude. Ambient air was aspirated at a flow rate of 4 L min−1 through the multi-pass absorption cell (0.5 L, 76 m effective path length) and NH3 absorption was detected at 965.3 cm−1. In order to minimize adsorption of ammonia on the inlet walls, an additional bypass flow rate of 16 L min−1 was introduced to purge the 50 cm long inlet tubing. In addition, the inlet was heated to approximately 40 ∘C to further reduce tubing wall effects. Evaporation of ammonia from NH4NO3 aerosols was minimized by removing the particulate matter using a PFA (Teflon) virtual impactor upstream of the QC-TILDAS absorption cell. The uncertainty of the NH3 measurement during UWFPS was 150 ppt (1σ at 1 Hz sample frequency). To account for potential systematic errors, caused by changes in cabin temperature, for example, zero measurements were performed regularly during flights. The LOD (3σ) was 450 ppt at the 1 Hz sample frequency and 90 ppt for a 30 s averaging interval.
Nitrogen oxides (NO, NO2), total reactive nitrogen (NOy), and ozone (O3) were measured at 1 s time resolution using the NOAA custom-built nitrogen oxide cavity ring-down spectrometer (NOxCaRD). The instrument directly measures NO2 by optical absorption at 405 nm and quantitatively converts NO and O3 into NO2 by reaction with excess O3 or NO, respectively, in two separate channels (Fuchs et al., 2009; Washenfelder et al., 2011). A fourth channel converts NOy to NO and NO2 by thermal dissociation in a quartz inlet at 650 ∘C and subsequently converts any NO to NO2 in excess O3 (Wild et al., 2014). The measurement accuracy was 5 % for O3, NOx, and NO2 and 12 % for NOy. Periodic zeros were measured for 30 s every 5–7 min. Measurements were less accurate during periods of rapid altitude change due to a minor pressure dependence in the background zeros in the NO channel that could not be fully corrected during post-processing.
2.1.3 Meteorological parameters
A commercial met probe (Avantech) measured meteorological parameters (ambient temperature, pressure, relative humidity with respect to liquid water, wind speed, and wind direction), the global positioning satellite (GPS) location including altitude above sea level, and aircraft parameters (heading, pitch, and roll). Wind data were compromised for some flights, making only partial coverage available. The aircraft GPS altitude above sea level was converted into altitude above ground level using USGS data (https://gis.utah.gov/data/elevation-and-terrain/, last access: 30 November 2018).
2.2 PM2.5 ground-based measurements
As part of the Utah air monitoring network, UDAQ continually monitors PM2.5, PM10, nitrogen oxides or NOx (NO and NO2), ozone (O3), and meteorological parameters according to EPA guidelines and operates 24 h Federal Reference Method (FRM) samplers for PM2.5 for NAAQS compliance. Here, we used PM2.5 data from two UDAQ sites: Hawthorne, the main air monitoring station for Salt Lake City (40.734397∘ N, 111.8721∘ W), situated on the Salt Lake Valley floor at ∼1306 m above sea level (a.s.l.), and North Provo (lat 40.253826, long −111.663311), situated in a central location of the Utah Valley at ∼1402 m a.s.l. We also used PM2.5 and NR-PM1 data obtained by the EPA at a temporary sampling station located in Logan (lat 41.758875, long −111.815122), on the Utah State University campus in the Cache Valley at ∼1405 m a.s.l. These three sites are located in three sub-basins in proximity of the Great Salt Lake, in northern Utah.
2.3 Thermodynamic calculations using ISORROPIA
Cold temperatures and the presence of both nitric acid and ammonia are conducive to forming ammonium nitrate in the aerosol phase. To examine phase partitioning of these compounds for the flight data, we used the thermodynamic model ISORROPIA (v2.1) (Fountoukis and Nenes, 2007; Nenes et al., 1998). We run ISORROPIA in “forward” mode, in which the total (gas + particulate) measured concentrations of various inorganic species are input into the model along with meteorological data (temperature and relative humidity). From these input parameters, concentrations for each phase are calculated using the van 't Hoff equation assuming thermodynamic equilibrium. The Kelvin effect is neglected in the model calculations and all the aerosol particles are assumed to have the same composition.
The inorganic species used for input to ISORROPIA were simplified here to the available flight data for total nitrate (), total ammonium (), total chloride (), and sulfate (). The composition of the particle phase (, , and ) was from the TO-AMS, therefore NR-PM1. As the AMS does not quantitatively measure refractory sodium or its salts (e.g., sodium chloride, nitrate, or sulfate) under normal operating conditions, the model input for particulate sodium was set to zero with the assumption that this does not affect the partitioning of the acidic gases with their corresponding ammonium salts. In other words, the equilibrium vapor pressures of the inorganic acids examined here are constrained only by their ammonium salts and the vapor pressures of the acidic gases over their sodium salts are considered to be negligible. Because nitric acid is known to displace chloride from sodium, potassium, calcium, and magnesium salts (Bondy et al., 2017; Metzger et al., 2006; Sudheer and Rengarajan, 2015; Trebs et al., 2005), aerosols containing these refractory species could act as a sink for HNO3 and source of HCl in this environment. It is also important to note that the term “total nitrate” in this paper refers to gas-phase plus NR-PM1 nitrate measured with an AMS. While we estimate that particles smaller than 70 nm (lower end of the transmission efficiency of the AMS) contributed less than 0.5 % to the total mass, with our definition we may exclude a non-negligible part of nitrate from the coarse mode.
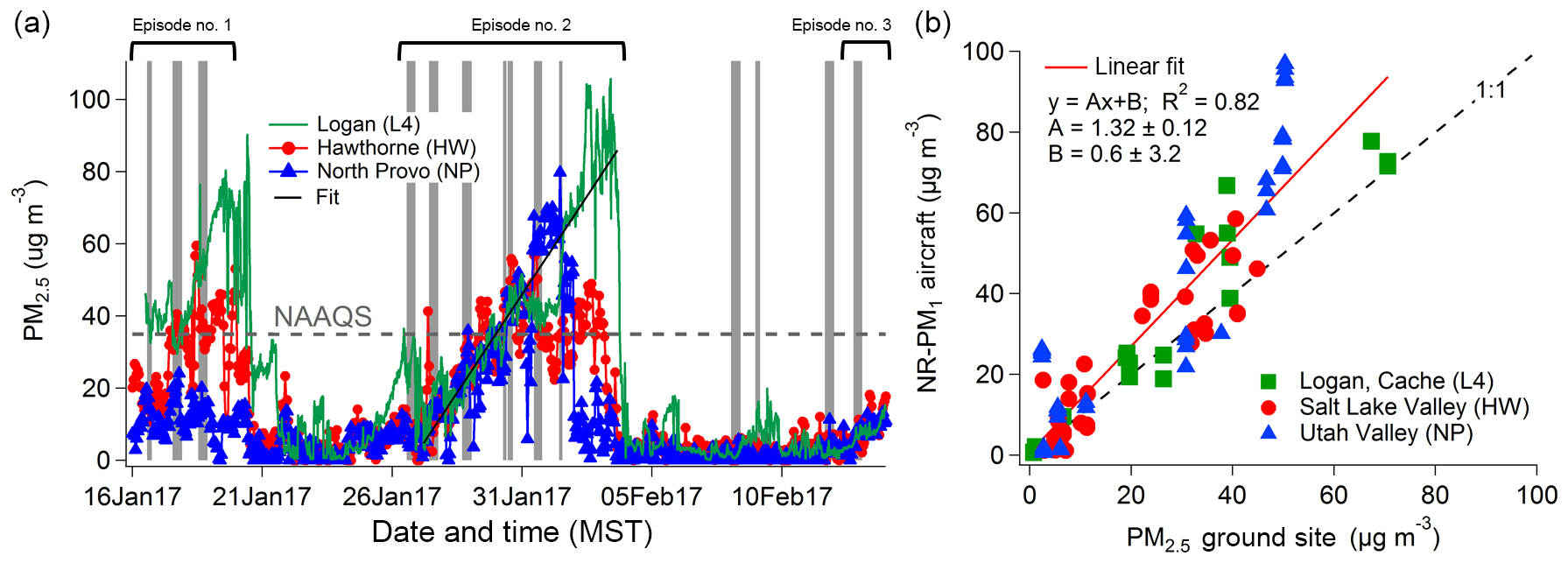
Figure 2PM2.5 measurements from three ground sites during UWFPS. (a) Time series of PM2.5 measured in Logan (L4), Cache Valley, in green; at Hawthorne Elementary School (HW), Salt Lake Valley, in red; and in North Provo (NP), Utah Valley, in blue. The grey shaded areas correspond to the times of Twin Otter flights; the horizontal dashed line indicates the National Ambient Air Quality Standard (NAAQS) of 35 µg m−3 for 24 h averaged PM2.5. The black solid line is a fit over all the data of the increase in PM2.5 during Episode 2. The slope is 10.89±0.08 µg m−3 day−1 (slope ± 95 % CI). (b) Comparison of the non-refractory (NR) PM1 measured with the AMS from the Twin Otter for altitudes lower than 50 m a.g.l. during the missed approaches at the airports closest to the ground site measurements. The dashed line is the 1:1 line and the red solid line is a linear fit; correlation coefficient, fit coefficients, and their 95 % CI are reported in the legend.
The ISORROPIA output was compared directly with UWFPS measurements. We then performed several simulations designed to test the model response to decreasing the total nitrate and/or ammonium in order to simulate how much of a reduction in total nitrate and/or total ammonium would affect the total aerosol mass. When we compared the total NR-PM1 mass at different conditions, we used only the inorganic mass, as we did not have enough information on the organic constituents to include them in the model.
3.1 Comparison of ground sites and airborne observations
The UWFPS field-intensive measurement period encompassed two complete major pollution episodes (Episodes 1 and 2) and the beginning of a third minor pollution episode (Episode 3, Fig. 2a). All three ground sites, Logan in Cache Valley, Hawthorne in Salt Lake Valley, and North Provo in Utah Valley, showed similar increases in PM2.5 mass concentrations as a function of time. However, we observed some slight differences that are probably related to the availability of precursor gases and oxidants at the three sites as well as the various basin topographical and meteorological features (Baasandorj et al., 2017a).
The 2016/17 winter had several multiday pollution episodes and the PM2.5 temporal evolution of each individual episode during the UWFPS study period was similar to that from past years (Fig. S2). PM2.5 levels in the Salt Lake Valley (in red) often reach a plateau after an initial increase regionally, whereas PM2.5 concentrations continue to increase in the Utah and Cache valleys as long as the PCAP persists. Often a PCAP erodes first in Utah Valley and persists the longest in Cache Valley, which is surrounded by mountains and sheltered more effectively from mixing induced by large-scale winds. Similar changes in aerosol mass and volume as a function of time were also observed with the aircraft instruments over the study region. TO-AMS NR-PM1 measurements near the ground sites (within 8 km horizontally and altitudes less than 50 m above ground level, a.g.l.) during the study period correlated well (R2=0.82) with the PM2.5 ground site data measured at the same time and agreed within the measurement uncertainties (Fig. 2b). Hence, aerosol measurements from the aircraft were representative of and consistent with the ground observations. During the first pollution episode, both the aircraft and ground-based aerosol mass concentrations were the highest in Cache Valley (∼70 and ∼90 µg m−3, respectively) and the lowest in Utah Valley (∼10 and ∼25 µg m−3, respectively). During the second pollution episode, the highest mass concentrations observed at the ground sites in Cache Valley were up to 100 µg m−3, in Utah Valley were ∼70 µg m−3, and in Salt Lake Valley were up to 60 µg m−3. These variations among valleys in peak PM2.5 concentrations are characteristic and are due to variations in sources and meteorological processes (Baasandorj et al., 2018) (Fig. S2). During UWFPS, PM2.5 was on average around 70 % of PM10 in the three valleys (Fig. S3).
3.2 Aerosol chemical composition during polluted and clean conditions
For the purposes of this analysis, we define clean conditions, when the total aerosol mass (NR-PM1) is ≤2 µg m−3 and polluted conditions when the total aerosol mass is > 17.5 µg m−3. Our definition of polluted conditions follows the classification used by Whitemann et al. (2014) that links a total aerosol mass > 17.5 µg m−3 to PCAP conditions. NR-PM1 composition differed substantially between polluted and clean conditions (Fig. 3). During polluted conditions (44 % of the Twin Otter data) the dominant species was ammonium nitrate (74±5 %, mean ± standard deviation), while organic species, sulfate (mostly ammonium sulfate), and chloride (in the form of ammonium chloride) constituted a minor fraction of the mass (18±3 %, 6±3 %, and 2±2 %, respectively). The largest aerosol mass concentrations of ammonium nitrate occurred at temperatures below the ice frost point. During clean conditions (56 % of the data), the organic fraction dominated the mass of the aerosols (50±13 %), ammonium nitrate was the second most abundant species (31±9 %) followed by ammonium sulfate and chloride (13±7 % and 6±6 %, respectively). During pollution Episode 2, we observed at ground level an average increase in PM2.5 over time of about 11 µg m−3 day−1. The increase due to aerosol-phase ammonium nitrate was 8.2±0.4 µg m−3 day−1, and the remaining contribution of 2.8±0.1 µg m−3 day−1 was due to the other chemical species according to the mass fractions indicated above. Furthermore, the change in chemical composition was uniform across the three different valleys (Fig. 4) and across all other areas in the studied region (Fig. S4) for both airplane and ground-based measurements (Fig. S5), although detailed ground-based composition measurements were limited to the site in the Cache Valley. We can therefore say that aerosol mass concentrations were predominantly due to growth in ammonium nitrate across the entire region. This uniformity occurs despite heterogeneity in emissions, with relatively large urban emissions from Salt Lake City and relatively large agricultural emissions in the Cache Valley.
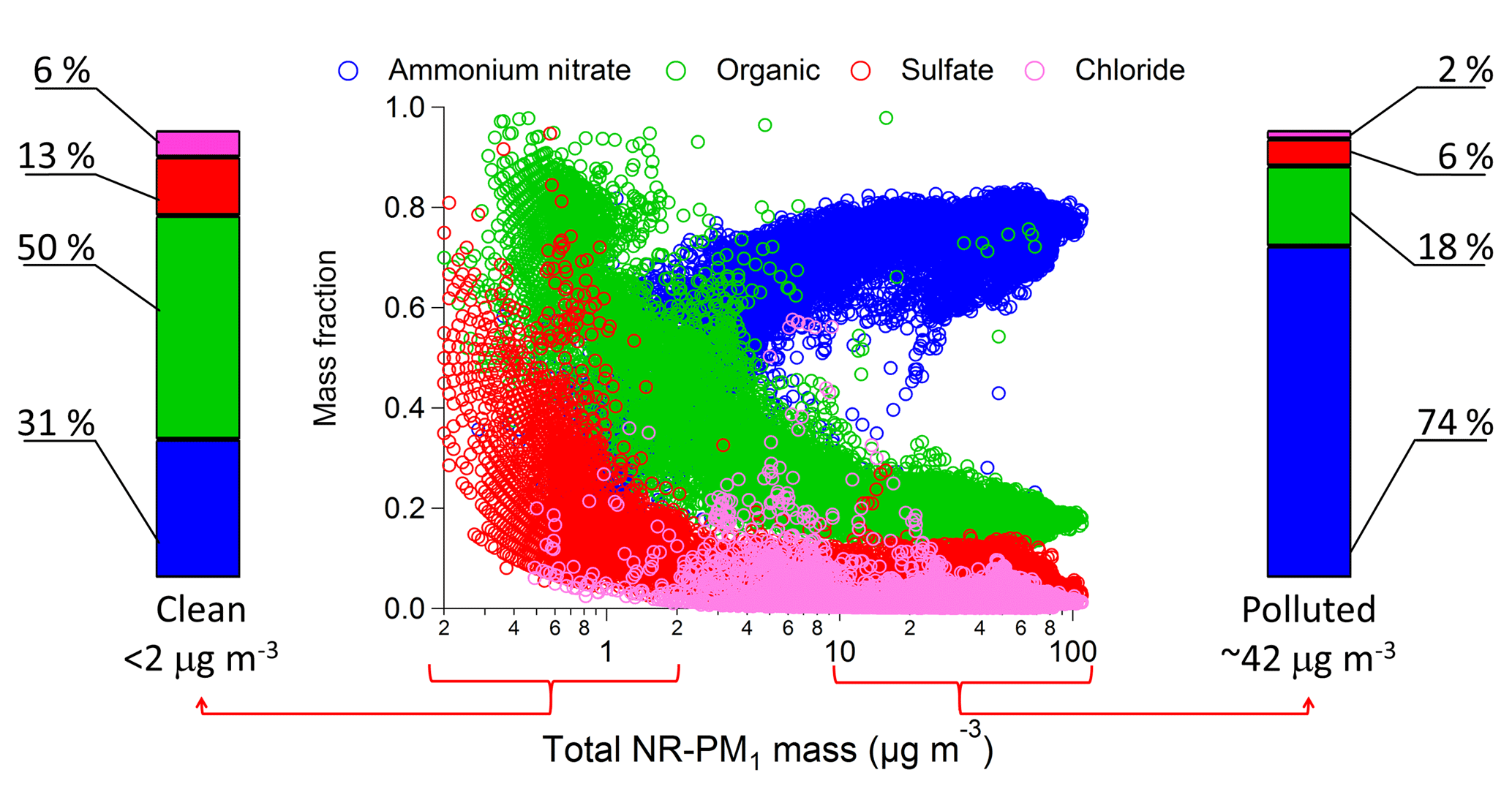
Figure 3The center shows aerosol mass fraction as a function of the total mass of NR-PM1 measured from the Twin Otter. Ammonium nitrate is in blue, organic species in green, and sulfate and chloride in red and pink, respectively. The bar chart on the left corresponds to the average of the mass fractions when the total aerosol mass is < 2 µg m−3 (clean conditions). The bar chart to the right corresponds to the average of the mass fractions when the total aerosol mass is > 17.5 µg m−3 (polluted conditions) and altitude is < 900 m a.g.l. (below the boundary layer).
A comparison of the total aerosol mass measured with the AMS with the dry aerosol volume measured with the UHSAS is shown in Fig. S6. The scatter plot gives an average slope of 1.8 g cm−3, which is comparable to the density of ammonium nitrate (1.72 g cm−3), within the combined measurement uncertainty. The aerosol-phase ion balance (Fig. 5) shows that the aerosols were completely neutralized, indicating that the measured nitrate, sulfate, and chloride are predominantly in the form of their corresponding ammonium salts (ammonium nitrate, ammonium sulfate, and ammonium chloride). The data points in orange have a low ammonium nitrate mass fraction and are below the 1:1 line (more acidic). Those correspond to measurements in the plume of the US magnesium plant (Fig. S1) where we observed a high organic (74.0±1.2 %), high chloride (6±0.6 %), and low ammonium mass fraction (3±0.6 %). It is important to note that those AMS measurements of the US magnesium plume reported in the current dataset carry a larger uncertainty than the rest of the dataset. The organic component of the aerosol mass was high (up to 50 µg m−3) and it is likely to be underestimated as CE = 1 was used (CE is < 1 for aerosols that are not ammonium nitrate and usually assumed = 0.5 for organic aerosol). In the plume, the reported nitrate mass is overestimated as the peak at 30, which is normally attributed to the NO fragment, showed a contribution from organic fragments. The plume presented a high aerosol chloride mass, in the form of ammonium chloride at concentrations up to 3.9 µg m−3.
3.3 Contribution of residential wood combustion to the aerosol mass and other organic aerosol sources to the total aerosol mass
Figures 3 and 4 show that the mass fraction of organic material in the particulate phase on a regional basis was generally less than 20 % during polluted conditions. There are very few data points in these aircraft observations containing higher organic mass fractions at the highest aerosol mass concentrations, which is consistent with sources of wood combustion organic aerosol varying widely (Shrivastava et al., 2007; Kleindienst et al., 2010; Woody et al., 2016). Hence, this organic aerosol fraction of roughly 20 % places an upper limit on the average direct contribution (i.e., from primary aerosol emissions or formation of secondary organic aerosol from wood combustion volatile organic compounds) of wood combustion to the regional total aerosol mass.
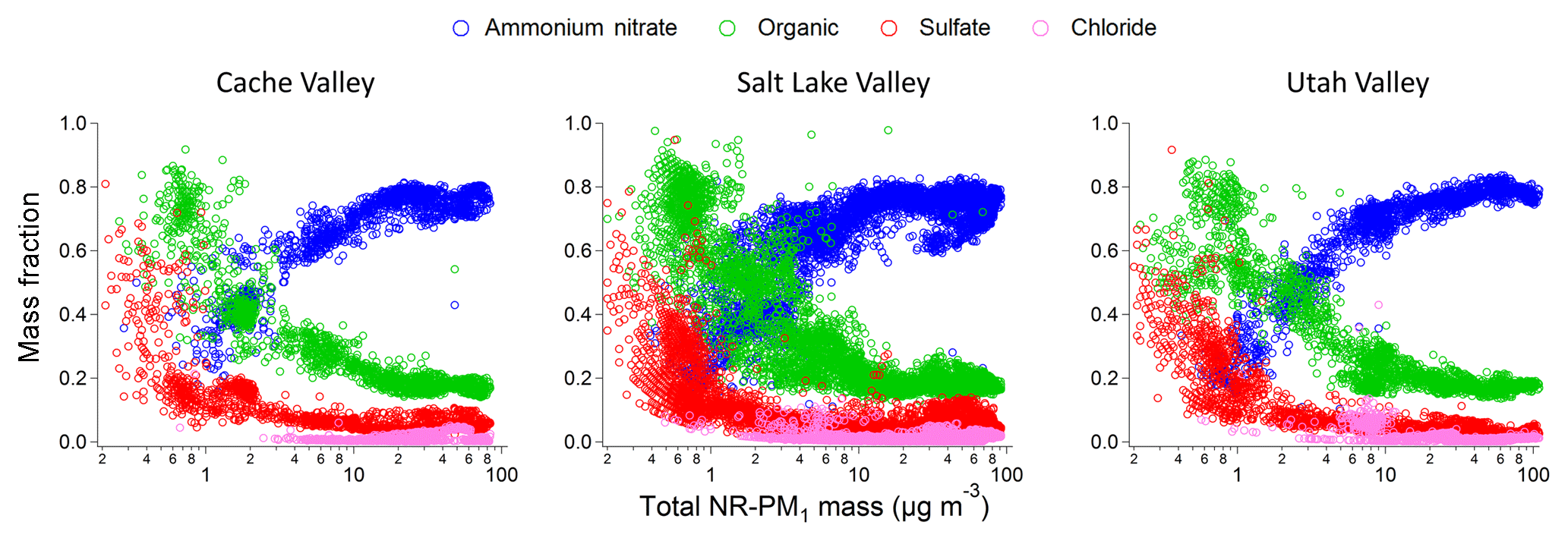
Figure 4Aerosol mass fraction as a function of total NR-PM1 mass for each of the three main study areas measured from the Twin Otter. Color code same as Fig. 3.
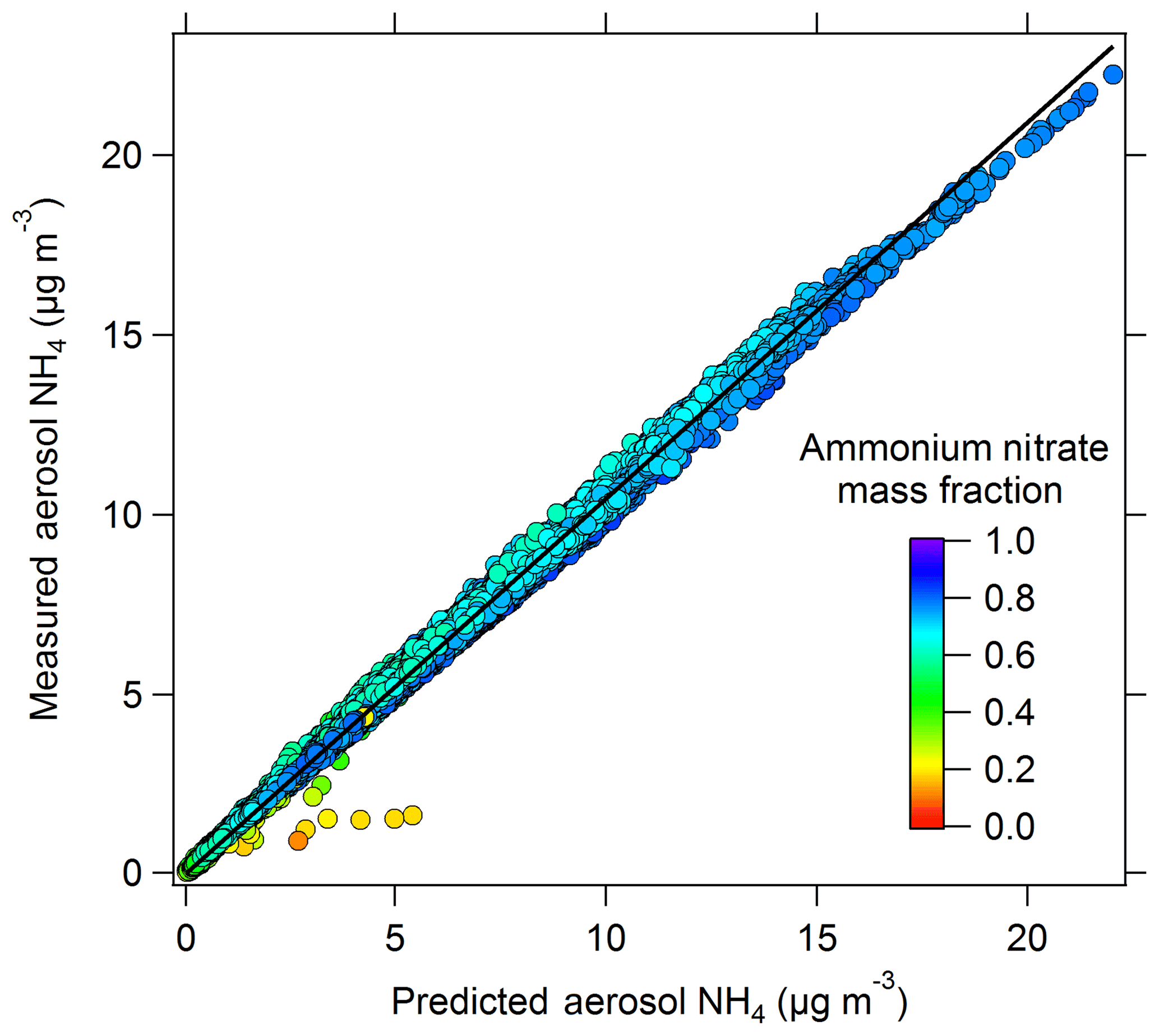
Figure 5Aerosol ion balance from the AMS measurements. On the x axis is the calculated mass of ammonium for fully neutralized aerosols ( = 18 × ( Cl−∕35.45)). On the y axis is the measured ammonium () in the aerosol particles. The black line is the 1:1 line, for which points on the line correspond to neutralized aerosols, and below the line are acidic aerosols. Data are color-coded by the ammonium nitrate mass fraction.
The organic signals in the mass spectra were examined to further estimate the contribution from wood combustion. Prior studies using an AMS instrument indicate that a potential marker of wood combustion is signalled at the mass-to-charge ratio (m∕z) of 60, which is a major ion fragment present in spectra from the known biomass burning marker species, levoglucosan (Alfarra et al., 2007). The fraction of the organic mass spectrum that is at m∕z 60 (termed f60) is about 8 % in spectra from levoglucosan (Alfarra et al., 2007) and is often significantly larger than the background value of 0.3 % (or 0.003) for air masses that are influenced by biomass burning (Cubison et al., 2011). During wintertime evenings, wood combustion is known to be a major contributor to the measured aerosol mass in several European suburban/urban sites, where f60 was measured on average to be 0.028 in Roveredo, Switzerland (Alfarra et al., 2007), 0.02 in Grenoble, France (Favez et al., 2010), and slightly less than 0.02 in Augsburg, Germany (Elsasser et al., 2013). Here we measured a median value (with the 25th and 75th percentiles) for f60 of 0.0035 (0.0031–0.0042) for the entire study during all conditions, indicating that the contribution of wood combustion to the organic mass fraction was generally fairly low compared to the European sites. Figure 6b shows the distribution of f60 for this study (light red) compared with previous studies in which the influence of biomass burning was negligible (green, blue, and dark red lines). Most of the spectra from our study had f60 close to the background value of 0.003; however the distribution shows a somewhat higher tail for our f60 values (17 % of the data). By comparing with the average fractions from the European wintertime studies, we estimate that the fraction of organic aerosol from wood combustion in these higher f60 cases was about 30 % of the total organic mass, i.e., about 6 % of the total NR-PM1.
Low values of f60 could be due to the initial wood combustion aerosol emissions having low f60 (Heringa et al., 2012; Schneider et al., 2006) or aging in the atmosphere causing f60 to decrease (Corbin et al., 2015; Ortega et al., 2013) before the aerosol was measured on the aircraft. Hence, relatively higher signals from aged aerosols could potentially indicate conversion of the fresh, primary biomass burning aerosols to lower f60 due to aging. The common marker for aged, oxidized organic aerosols is the peak at m∕z 44, which corresponds to the fragment (Allan et al., 2004). m∕z 44 correlates with the elemental oxygen ∕ carbon ratio (O : C) (Aiken et al., 2007, 2008; Canagaratna et al., 2015) and indicates organic aerosol aging (Ng et al., 2010; Cubison et al., 2011). A plot of f44 (i.e., the ratio of m∕z 44 to the total organic aerosol mass) versus f60 for our study is shown along with Cubison and co-workers' previous data in Fig. 6a. The data of freshly emitted biomass burning aerosols shown in Cubison et al. (2011) tended to have high f60 (0.02 to 0.04) with relatively low f44 (about 0.05) that changed to lower f60 (< 0.02) and higher f44 (0.15 to 0.23) as the aerosols aged. For the UWFPS study, however, f60 was generally low and f44 varied considerably.
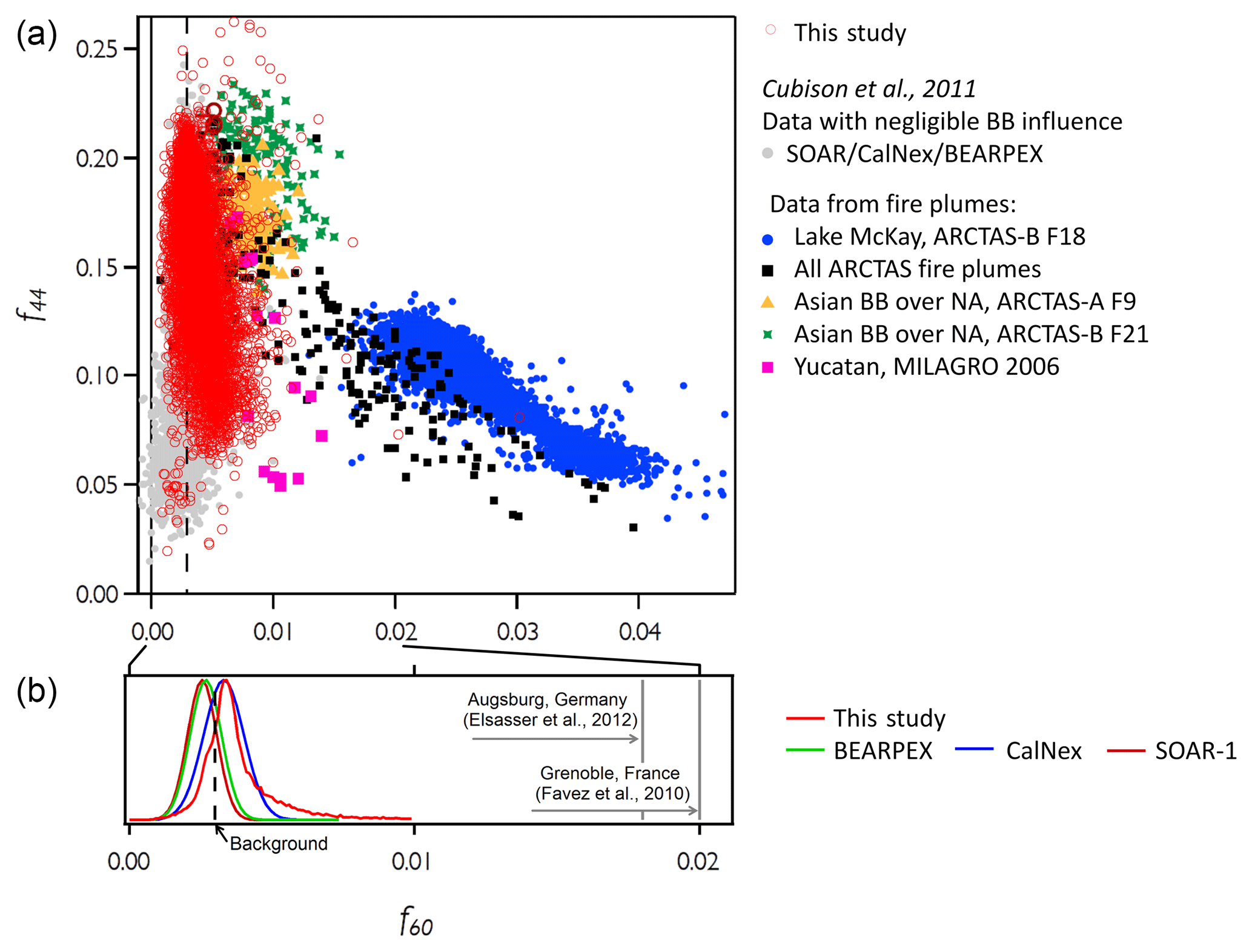
Figure 6(a) Fraction of the organic fragment at divided by the total organic mass (f44) as a function of the fraction of the organic fragment at m∕z = 60 divided by the total organic mass (f60). f44 and f60 are commonly used as markers for oxidized organic aerosol and for freshly emitted biomass burning aerosol, respectively The red open circles are the data measured during the entire UWFPS. The other symbols show data from previous studies (see Cubison el al., 2011, and references therein), with colored (non-red) points for the biomass burning (BB) plumes and grey points for data with negligible BB influence. The dashed black line corresponds to 0.003, the AMS background value for f60. (b) Frequency distribution of f60 values for this study (light red) and for previous studies with negligible BB influence (green, blue, and dark red lines). The vertical grey solid lines show f60 values for locations where the influence of wood combustion on organic aerosol was strong.
Since f44 was not consistently high, the organic aerosol was not always highly oxidized, indicating that peaks other than m∕z 60 were possibly important in the organic aerosol for the UWFPS dataset. Another marker to consider is f57 (ratio of m∕z 57 to the total organic signal in the AMS), which corresponds to a characteristic fragment from hydrocarbon-like organic aerosol (HOA) from lubricating oil and/or diesel fuel and related to traffic emissions (Canagaratna et al., 2004; Zhang et al., 2005). We observed f57 median values of 0.009, 0.012, and 0.010 for the Cache, Salt Lake, and Utah valleys, respectively. The reported f57 for the New York City diesel bus exhaust in Canagaratna et al. (2004) and for the Pittsburgh HOA in Zhang et al. (2005) was 0.081 and 0.075, respectively. This indicates that some of the organic fraction of NR-PM1 from the UWFPS study could have been from traffic emissions and was highest in the Salt Lake Valley as expected based on the higher urban density there. Note that this marker peak at m∕z 57 could also be present in railroad and aircraft emissions.
Previous ground-based observations in this region indicate significant spatial and temporal variability in the contribution of residential wood combustion (Cropper et al., 2018; Mouteva et al., 2017; Baasandorj et al., 2017b). The data reported here indicate that organic material from wood combustion is not the dominant mass fraction on a regional scale. However, smoke emissions could contribute to most of the aerosol mass at smaller scales or in near-source regions (Kelly et al., 2013).
Wood combustion may also have an indirect effect on the formation of inorganic aerosol via secondary pathways involving oxidation of volatile organic compounds emitted from this source. Other tracers of various organic aerosol emissions and secondary formation (e.g., black carbon, carbon monoxide) were not obtained from the aircraft during this project, making it difficult to determine the most important organic aerosol sources for the region.
3.4 Vertical profiles
Vertical profiles show that the aerosol volume and mass concentration was variable as a function of height. Within 1 km a.g.l. we observed layers of aerosols at different heights in each location at different times of the day at different evolutionary stages of the pollution episodes in several individual vertical profiles (Fig. S7). These changes in aerosol concentrations as a function of height were measured by both the UHSAS and the AMS (Fig. 7). The possible causes for these layers could be related to a difference in sources, losses, or to the many transport and mixing mechanisms that occur in complex terrain, which are difficult to constrain with our limited vertical measurements during the study (Baasandorj et al., 2018).

Figure 7Comparison among selected vertical profiles. On the left the aerosol volume, measured with the UHSAS (in red), and NR-PM1 mass measured with the AMS (in grey) are shown as a function of elevation in meters above ground level. On the right vertical profiles of ozone concentration (light blue) and potential temperature (orange) for the same three missed approaches are shown. The missed approach in Cache Valley took place on 28 January at 18:38 MST at the Logan-Cache Airport (LGU), the one in Salt Lake Valley on 28 January at 20:22 MST at Salt Lake City International (SLC), and the one in Utah Valley on 30 January at 13:24 MST at the Provo Municipal Airport (PVU).
Figure 8 shows the average vertical profiles of NR-PM1 and ammonium nitrate mass fraction for Cache Valley, Salt Lake Valley, and Utah Valley during Episode 2. On average, the Cache and Salt Lake valleys showed either a constant or a slight increase in aerosol mass with altitude, up to about 200 m a.g.l. in Cache and up to about 400 m a.g.l. in Salt Lake Valley. Utah Valley shows a general decrease in aerosol mass with altitude, shallower in the first 300 m a.g.l. and steeper between 300 and 450 m a.g.l. The Cache and Salt Lake valleys also show a decrease in aerosol mass with altitude from 200 to 450 m a.g.l. and from 400 to 700 m a.g.l., respectively. The average vertical profiles of all three valleys show a very clear transition where the aerosol mass is very low above 500 m a.g.l. in Cache Valley, 700 m a.g.l. in Salt Lake Valley, and 650 m a.g.l. in Utah Valley. The average vertical profiles of the ammonium mass fraction show for all three valleys that 75 % or more of the aerosol mass is ammonium nitrate and that this is true for the first 300 m a.g.l. in Cache Valley, 500 m a.g.l. in Salt Lake Valley, and 300 m a.g.l. in Utah Valley. Above those heights, the variability in the chemical composition of the aerosol particles increases and the ammonium nitrate fraction decreases. The marked difference aloft, where the aerosol mass is very low, is also characterized by a fraction of ammonium nitrate of 50 % or less, similar in composition to what we observed at lower altitudes in clean conditions.
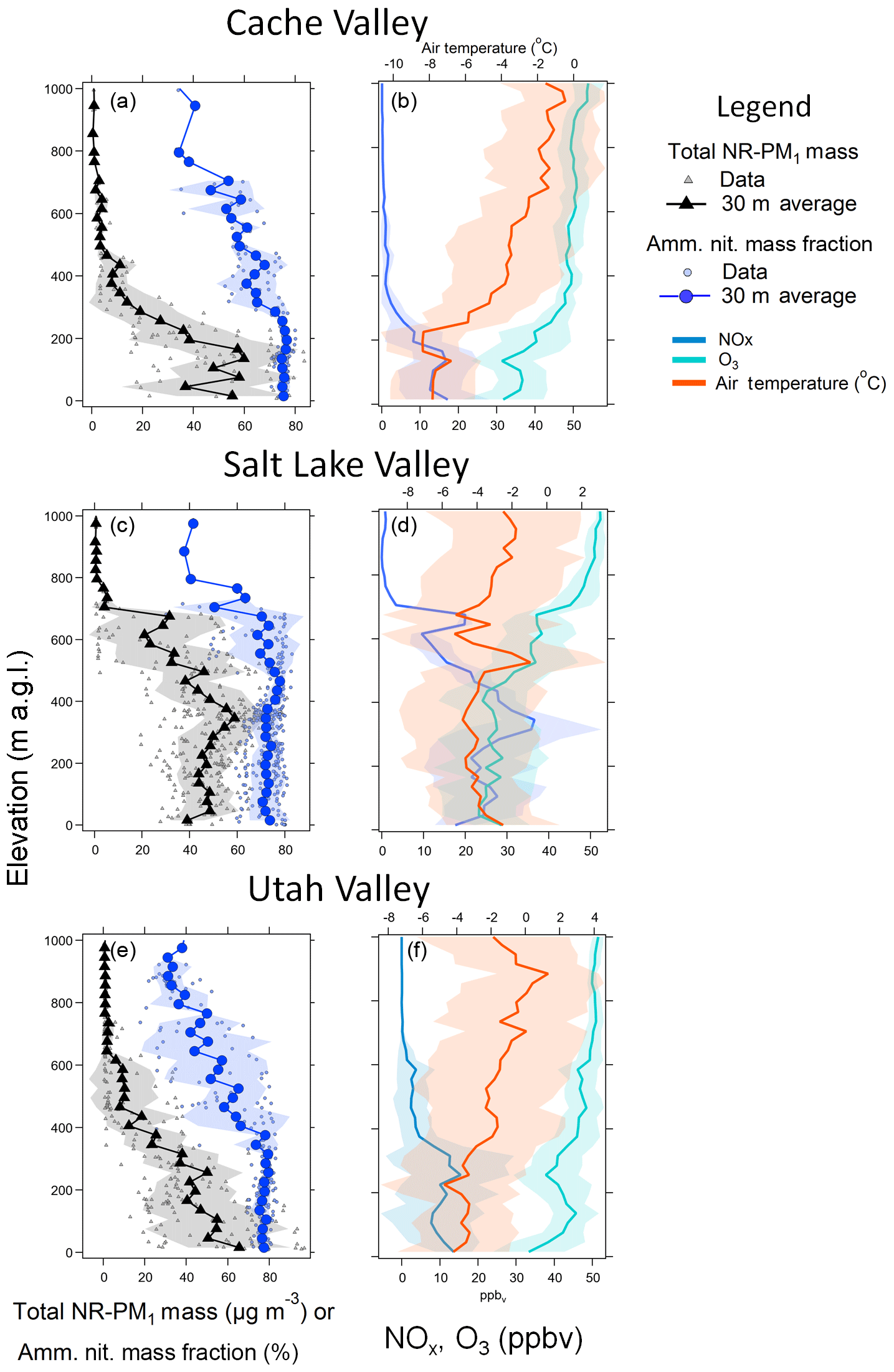
Figure 8On the left are the average vertical profiles of NR-PM1 mass (black triangles) and ammonium nitrate mass fraction (blue circles) measured during Episode 2. The triangular small grey markers and the small light blue circles are data used in the averages for all the missed approaches at the same airports as in Fig. 7. The black triangles and dark blue circles are averages over 30 m with their associated standard deviations. The right panels show corresponding vertical profiles of NOx (blue lines), O3 (aqua lines), and potential temperature (orange lines).
Interestingly, in some vertical profiles (Fig. S7), especially at intermediate mass loadings (from 2 to 17 µg m−3), higher mass loadings correspond to larger ammonium nitrate mass fractions (Fig. S7c, e, n, o). However, in other vertical profiles, changes in mass loadings do not correspond to a change in chemical composition (Fig. S7 d, j, l, m). The detection of larger fractions of ammonium nitrate at higher altitudes (Fig. S7f, n, o) might indicate formation of nitrate aloft. A clear change in chemical composition is always detectable between 500 and 1000 m a.g.l. for all locations. This change is related to the transition from the boundary layer, affected by regional sources, to the free troposphere affected by long-range transport and consists of a clear change from ammonium-nitrate-dominated aerosol to sulfate- and organic-dominated aerosol, concomitant with a decrease in aerosol concentration.
3.5 Limiting reagent of ammonium nitrate formation
A straightforward method of determining which gas-phase precursor (ammonia or nitric acid) is the limiting reagent in ammonium nitrate aerosol formation for the region is to compare the ratios of each species in the gas phase to the total: e.g., ammonia divided by total ammonium NH3 ∕ () and nitric acid divided by total nitrate HNO3 ∕ (). Figure 9 shows these ratios for the Cache, Salt Lake, and Utah valleys and Fig. S8 for the other areas of the study. For all three valleys, the nitric acid gas-phase fraction is nearly always smaller than the gas-phase fraction of ammonia. Because a stoichiometric 1:1 ratio of nitric acid to ammonia is needed to form ammonium nitrate, nitric acid is clearly limiting the formation of ammonium nitrate. The fraction of gas-phase nitric acid also tends to decrease sharply with increasing total mass concentrations, indicating that the available nitric acid partitions efficiently to the aerosol phase at relatively moderate aerosol mass concentrations. In contrast, the fraction of gas-phase ammonia is larger, irrespective of particle mass concentration, and decreases less steeply with increasing mass loadings (Fig. 9).
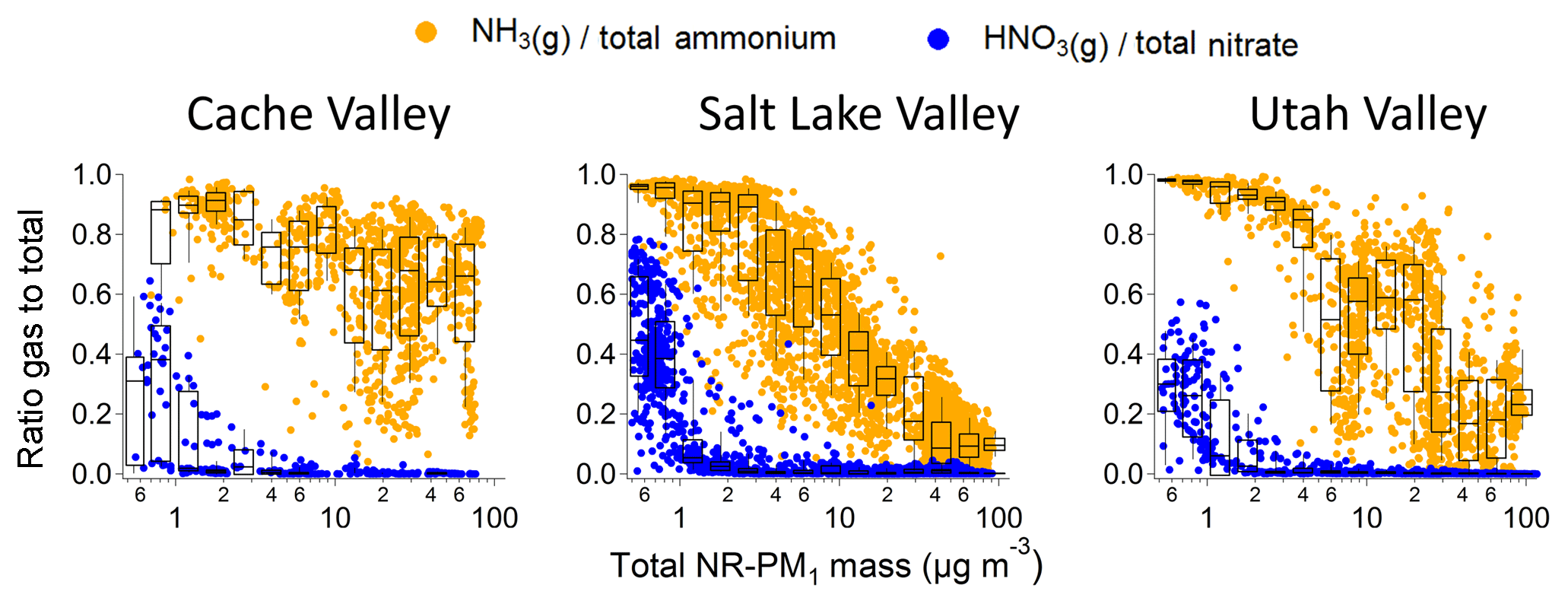
Figure 9Fraction of gas-phase species to total mass (gas + aerosol) plotted as a function of total observed aerosol mass. In orange is the ratio between gas-phase ammonia NH3 and total ammonium NH3(g) + . In blue is the ratio between gas-phase nitric acid HNO3 and total nitrate HNO3 + . Overlaid onto the data points are box-and-whisker plots for logarithmically spaced bins. The boundaries of the boxes are the 25th and 75th percentiles; the line in the box is the 50th percentile and the whiskers are 10th and 90th percentiles.
The relative importance of nitric acid in controlling the total aerosol mass varies among the three valleys. For the Cache Valley, the gas-phase fraction of nitric acid is always near zero due to relatively large amounts of total ammonium. The gas-phase fraction of nitric acid in the Utah Valley is frequently near zero, indicating that slightly more nitric acid (or less ammonia) is available there compared to the Cache Valley. In contrast, the gas-phase fraction of ammonia for the Salt Lake Valley is more frequently smaller than in the Cache and Utah valleys, especially at the largest total aerosol mass concentrations. Thus, during the most polluted conditions in the Salt Lake Valley, gas-phase ammonia is relatively less abundant, indicating a possible transition from nitric-acid-limited towards ammonia-limited ammonium nitrate formation as pollution episodes evolve.
A comparison between the measurements and the ISORROPIA model was carried out to investigate how effectively the measured partitioning between the aerosol and gas phases is represented by the thermodynamic model. We found that gas-phase ammonia, gas-phase hydrochloric acid, aerosol ammonium, and aerosol nitrate agree well with the model predictions (r2=0.984, 0.962, 0.984, and 0.997, respectively) while gas-phase nitric acid and aerosol chloride (as ammonium chloride) are not well represented by the model (r2=0.555 and 0.161, respectively; Fig. S9, Table S1 in the Supplement). It is important to note, however, that these two species have relatively lower mass concentrations and therefore have larger uncertainties associated with them. For example, the median HNO3 concentration for polluted events was 0.6 µg m3, comparable to 3 times the detection limit of the HRToF-CIMS. Furthermore, the detection limit is typically determined in low particulate nitrate conditions. Therefore, in “polluted conditions” often less than 2 % of particulate nitrate evaporating in the inlet system could explain the amount of gas-phase HNO3 detected by the HRToF-CIMS. The modeled gas-phase nitric acid was neither consistently higher nor lower than measured, with no clear dependency on temperature nor relative humidity. The aerosol chloride concentrations predicted by ISORROPIA were generally larger than measured. This overestimate of particle chloride by ISORROPIA has also been observed with other datasets (Haskins et al., 2018) and may affect the scenarios described below, especially for the effect of reducing total nitrate concentrations.
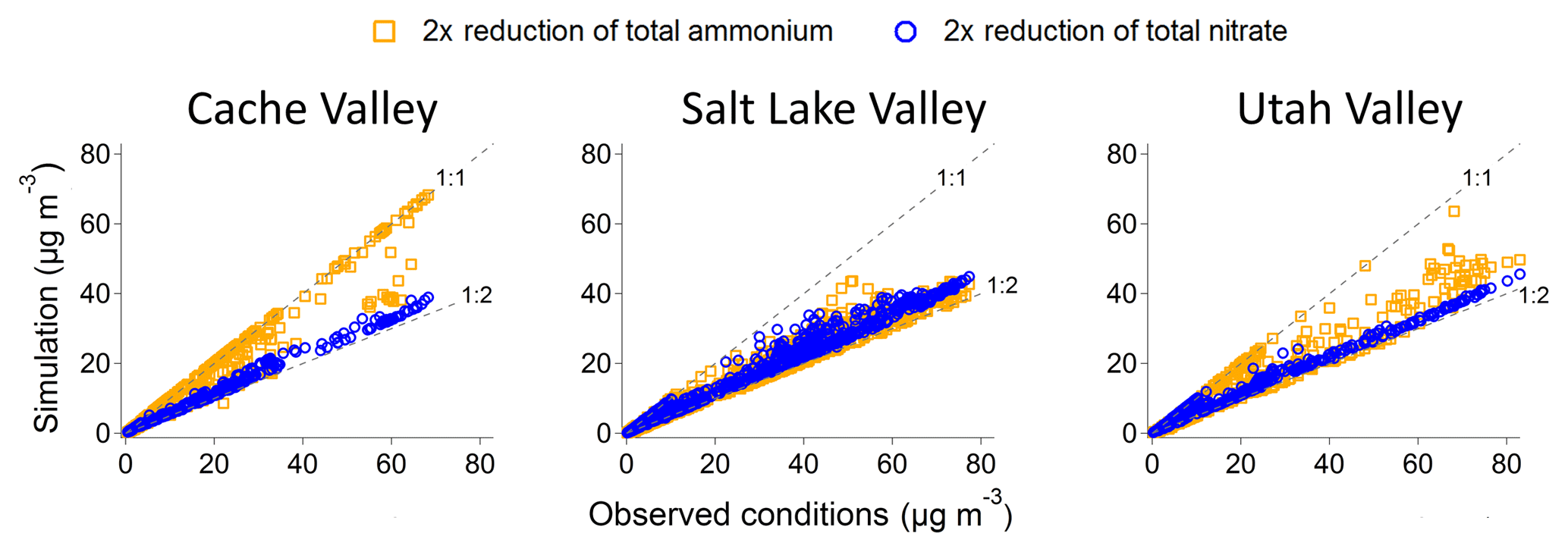
Figure 10Comparison of the simulated total inorganic aerosol mass (y axis) to the observed total inorganic aerosol mass with separate reductions in precursor species using ISORROPIA (x axis). Two different scenarios are shown: a 2-fold reduction of total ammonium (orange) and a 2-fold reduction in total nitrate (blue). The dashed grey lines in each plot represent the 1:1 line (i.e., no change in the simulated total inorganic mass in the aerosol phase) and the 1:2 line (i.e., a 2-fold reduction in the simulated total inorganic mass in the aerosol phase).
4.1 Simulated response to decreasing precursors
We carried out simulations of different scenarios using ISORROPIA (Figs. 10, S10, S11, and S12). Figure 10 shows the simulated decrease in total aerosol mass for two cases: one in which the total nitrate was reduced by a factor of 2 while keeping the total ammonium constant (blue) and one in which the total ammonium was reduced by a factor of 2 while keeping the total nitrate constant (orange). Reduction in total nitrate led to an approximately proportional decrease in total inorganic aerosol concentrations during polluted conditions and across all locations. Conversely, a decrease in ammonium gave a more variable response. In some regions, a 50 % decrease in total ammonium gave a decrease in aerosol mass of 50 % or close to it, while in other regions there was no change. Cache Valley did not respond to a decrease in total ammonium. In fact, most of the orange points for the simulated mass lay on the 1:1 line, indicating no decrease in total aerosol mass compared to the mass at current conditions. On the contrary, there was a proportional decrease in aerosol mass when the total nitrate was decreased (blue points along to 1:2 line). These results confirm that in this valley, there was an excess of gas-phase ammonia and the NH4NO3 formation chemistry is nitrate limited. The Salt Lake Valley responded to a reduction of either total nitrate or total ammonium (both blue and orange points are closer to the 1:2 line than the 1:1 line), suggesting that limiting either species could reduce aerosol formation. Despite the mass balance analysis showing excess reduced nitrogen, the thermodynamic model shows the total aerosol mass to be nearly proportionally sensitive to reductions in either reagent. The Utah Valley, similarly to the Salt Lake Valley, showed a response for the reduction of total nitrate and total ammonium, except for mass loadings lower than 20 µg m−3 for which a cluster of data points shows no response to the reduction of total ammonium. The area above the Great Salt Lake and Cache Valley was the least sensitive to the reduction of total ammonium while all other locations show varying responses for aerosol nitrate and chloride to the decrease in total ammonium. Interestingly, the aerosol chloride in the Salt Lake Valley and over the Great Salt Lake showed a slight increase for the decrease in total nitrate, pointing to a change in aerosol composition from ammonium nitrate to ammonium chloride. It is important to note that ISORROPIA generally overpredicts aerosol chloride (see Fig. S9), and decreases in total nitrate would potentially decrease the available total chloride from acid displacement reactions on the alkaline salts. Hence, the simulated total reduction of nitrate carries a larger uncertainty.
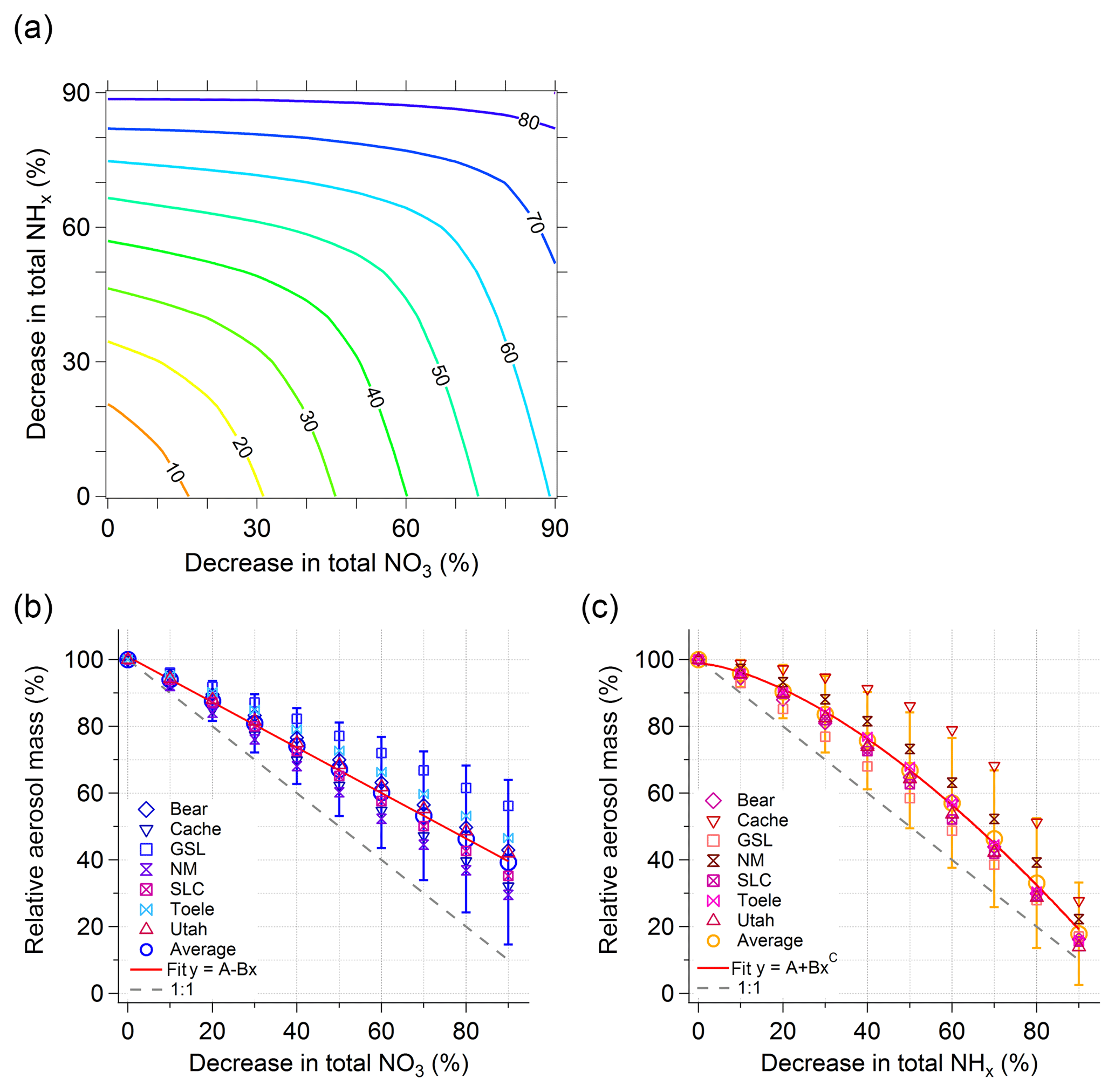
Figure 11(a) Contour plot showing the simulated decrease in total inorganic aerosol mass (contour lines, numbers shown in %) as a function of decrease in both total nitrate (x axis) and total ammonium (y axis). The data inputs used here are from the entire region. The decreases are relative to the observed conditions and expressed as percentages. Panels (b) and (c) show one-dimensional views of the surface plot shown in panel (a). Panels (b) and (c) are projections along the x and y axes, respectively. Panel (b) shows the simulated decrease in total inorganic NR-PM1 as a function of the decrease in total nitrate. In these simulations, the concentrations of total ammonium were kept constant and equal to the observed conditions. Panel (c) shows the simulated decrease in total inorganic NR-PM1 as a function of the decrease in total ammonium. In these simulations, the concentrations of total nitrate were kept constant and equal to the observed conditions. The open circles are averages over the entire region (same as panel a). The error bars are the standard deviations associated with the averages. The other markers are data relative to particular regions: Bear Valley, Cache Valley, the Great Salt Lake, North Metro, Salt Lake Valley, Tooele Valley, and Utah Valley. The dashed grey line is the 1:1 line and the red solid lines are a fit through the average points to guide the eye (linear fit in panel b and power law in panel c).
A comprehensive view of the sensitivity of total aerosol mass to reductions in total ammonium and nitrate is shown in Fig. 11a. This sensitivity study is based on 100 ISORROPIA simulations in which data for the entire region (the whole dataset) were used as the input; the total nitrate and/or the total ammonium were decreased at steps of 10 % and for each step the average total aerosol mass was calculated from the model output. For example, the 20 % contour line (yellow) shows the interpolation of the model output that leads to a 20 % reduction in total mass. This plot suggests that while reductions in both total ammonium and total nitrate would be effective overall, the most effective strategy to reduce the total aerosol mass in the region is through reduction of the total nitrate. The orange 10 % contour line is reached with total nitrate reductions of about 15 % without total ammonium reductions (bottom axis), whereas the total ammonium reduction would have to be more than 20 % to reach this contour line without total nitrate reductions (left axis). However, both reagents must decrease in order to achieve a reduction of total aerosol mass larger than 60 % relative to observed conditions. The importance of ammonium, once the total aerosol mass has been decreased, is shown as one-dimensional plots in Fig. 11b and c where the simulated decrease in total aerosol mass by decreasing only total nitrate or only total ammonium is shown.
Note that these predictions are somewhat simplified since the model includes total chloride () for which the aerosol chloride is ammonium chloride and changes in the amount of either total ammonium or total nitrate can cause additional effects for ammonium chloride partitioning. Furthermore, the model tends to overpredict the amount of aerosol ammonium chloride compared to observations. In the real environment, decreases in total nitrate would likely cause decreases in available total chloride because less displacement of chloride by nitric acid should occur from refractory salts such as sodium, calcium, or magnesium chlorides.
During the Utah Winter Fine Particulate Study (UWFPS), we measured gas precursors and aerosols from a NOAA Twin Otter aircraft over Salt Lake City and the surrounding valleys from 16 January to 12 February 2017. The NR-PM1 mass loadings for the region varied from below the AMS detection limit of 0.38 to 72.4 µg m−3 (second and 98th percentiles). The most severe pollution episodes occurred during persistent cold-air pool (PCAP) periods, when emissions were trapped near the surface in the three valleys studied (Salt Lake, Cache, and Utah). Aircraft measurements provide spatial and temporal evolution of the non-refractory aerosol chemical composition, as well as the aerosol spatial and temporal evolution.
The particle chemical composition during polluted conditions (aerosol mass > 17.5 µg m−3) was markedly different compared to clean conditions (aerosol mass ≤2 µg m−3). The aerosols during polluted conditions were characterized by large fractions of ammonium nitrate (74±5 %) while the fractions of organic species, sulfate (mostly ammonium sulfate), and chloride (in the form of ammonium chloride) were smaller (18±3 %, 6±3 %, and 2±2 %, respectively). This composition was uniform across all three valleys. The relatively low fraction of organic material indicated that wood combustion played a minor role in the direct formation of secondary aerosols during polluted conditions, as the amounts of the traditional biomass burning marker for the AMS spectra (f60) were similar to the ones measured in locations where biomass burning is negligible. However, we cannot completely exclude the role of wood combustion in the regional aerosol mass concentrations as aging in the atmosphere can cause the AMS markers to appear similar to background levels, and measurements of other biomass burning tracers such as acetonitrile and black carbon were not available to constrain the AMS observations. During clean conditions, the organic fraction dominated the mass of the aerosols (50±13 %), ammonium nitrate still remained the second most abundant species (31±9 %) followed by sulfate and chloride (13±7 % and 6±6 %, respectively).
Vertical profiles of the polluted region often showed varying layers of aerosol concentrations alternating. These variations did not always correspond to marked changes in chemical composition, although sometimes we observed a larger ammonium nitrate aerosol mass fraction below the top of the cold-air pool. Whether this observation is due to meteorology or chemistry remains unclear. Above the cold-air pool, the aerosol mass concentrations dropped considerably and the composition was characteristic of free tropospheric aerosols from long-range transport with relatively more sulfate and organic material and less ammonium nitrate.
Using gas-phase measurements of nitric acid and ammonia, we calculated the fraction of each species in the gas phase relative to the total (gas + aerosol). For all three valleys, the fraction of nitric acid in the gas phase was smaller than the gas-phase fraction of ammonia, indicating that the formation of ammonium nitrate in the aerosol phase was limited by the amount of nitric acid in the gas phase. The relative importance of nitric acid in controlling aerosol mass was largest for the Cache Valley, intermediate for the Utah Valley, and less important for the Salt Lake Valley. The gas-phase fraction of ammonia in the Salt Lake Valley is often less than 1 at the largest total aerosol mass concentrations, indicating that gas-phase nitric acid potentially becomes less limiting as pollution episodes evolve under PCAP conditions.
The largest aerosol mass concentrations of ammonium nitrate occurred at temperatures below the ice frost point. The comparison between measurements and the output of ISORROPIA showed agreement for ammonia, hydrochloric acid, aerosol ammonium, and aerosol nitrate. Measured nitric acid and the observed total chloride partitioning is not well represented by the model, with aerosol ammonium chloride consistently larger in the model. However, for these species the uncertainties in the model are larger than the other chemical species because their measured ambient concentrations were much smaller.
When we simulated a 50 % decrease in total (gas + aerosol) ammonium using ISORROPIA we found that Cache Valley did not show a decrease in total aerosol mass compared to measured total ammonium levels, while the Salt Lake and Utah valleys showed a decrease of 44 % and 37 %, respectively. When a 50 % decrease in total nitrate was simulated, a 44 % decrease in total aerosol mass was observed in all the three valleys, indicating that in all the locations NH4NO3 formation is controlled by nitric acid availability. However, with the evolution of the pollution episode under PCAP conditions over time, ammonium nitrate formation in the system appeared to become less nitric acid limited for Salt Lake Valley and Utah Valley, while Cache Valley always had excess ammonia.
The data used in this paper are accessible here: https://esrl.noaa.gov/csd/groups/csd7/measurements/2017uwfps/data.html (UWFPS team, 2017).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-17259-2018-supplement.
AF obtained and analyzed the UHSAS data, carried out the data analysis and wrote most of the paper. AF and AMM obtained and analyzed the aircraft based AMS data. DLF, EEM, CCW obtained and analyzed the NOxCARD data. LG, BHL, and JAT obtained and analyzed the I-CIMS data. AM and JGM obtained and analyzed the NH3 data. WPB provided technical support to the aircraft instruments. ETC and SWH provided input on the meteorology of the area. KSD obtained and analyzed the ground-based AMS data. MB and RL provided ground-based PM2.5 data. SBB and MB planned and supervised the measurement intensive period. All authors discussed the results and contributed to the final manuscript
The authors declare that they have no conflict of interest.
The authors would like to acknowledge the Utah Division of Air Quality for their
support and collaboration during the study. We thank the NOAA Aircraft
Operations Center for their dedication and professionalism, particularly the
pilots Robert Mitchel and Jason Clark for skillful navigation in complex
terrain and challenging meteorology. We thank Frank Erdesz for helping with
the installation of the UHSAS, Kyle Zarzana for helping with the calibration
and installation of the NOxCaRD, and Patrick Veres for helping with the
calibration of the HRToF-CIMS. We thank Tim Morphy and Teledyne for the loan
of the PM2.5 monitor at the Logan site. We thank Jon Abbatt at the
University of Toronto and the Canadian Aerosol Research Network, funded
by the Canada Foundation for Innovation, for lending the UHSAS.
Edited by: Joachim Curtius
Reviewed by: two anonymous referees
Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L.: Elemental analysis of organic species with electron ionization high-resolution mass spectrometry, Anal. Chem., 79, 8350–8358, https://doi.org/10.1021/ac071150w, 2007.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O/C and OM/OC ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Alfarra, M. R., Prevot, A. S. H., Szidat, S., Sandradewi, J., Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U.: Identification of the mass spectral signature of organic aerosols from wood burning emissions, Environ. Sci. Technol., 41, 5770–5777, https://doi.org/10.1021/es062289b, 2007.
Allan, J. D., Delia, A. E., Coe, H., Bower, K. N., Alfarra, M. R., Jimenez, J. L., Middlebrook, A. M., Drewnick, F., Onasch, T. B., Canagaratna, M. R., Jayne, J. T., and Worsnop, D. R.: A generalised method for the extraction of chemically resolved mass spectra from aerodyne aerosol mass spectrometer data, J. Aerosol. Sci., 35, 909–922, https://doi.org/10.1016/j.jaerosci.2004.02.007, 2004.
Baasandorj, M., Hoch, S. W., Bares, R., Lin, J. C., Brown, S. S., Millet, D. B., Martin, R., Kelly, K., Zarzana, K. J., Whiteman, C. D., Dube, W. P., Tonnesen, G., Jaramillo, I. C., and Sohl, J.: Coupling between Chemical and Meteorological Processes under Persistent Cold-Air Pool Conditions: Evolution of Wintertime PM2.5 Pollution Events and N2O5 Observations in Utah's Salt Lake Valley, Environ. Sci. Technol., 51, 5941–5950, https://doi.org/10.1021/acs.est.6b06603, 2017a.
Baasandorj, M., Eatough, D. J., Cropper, P. M., Overson, D. K., Hansen, J. C., Kuprov, R., and Lin, J. H.: West Valley City High Time Resolution Air Toxics Study Report, available at: https://documents.deq.utah.gov/air-quality/planning/technical-analysis/research/air-toxics/WVC/DAQ-2017-017121.pdf (last access: 30 November 2018), 2017b.
Baasandorj, M., Brown, S. S., Hoch, S., Crosman, E., Long R., Silva, P. Mitchell L., Hammond I., Martin, R., Bares R., Lin, J., Sohl J.,Page, J., McKeen, S., Pennell, C., Franchin, A., Middlebrook, A., Petersen, R., Hallar, G., Fibiger, D., Womack, C., McDuffie, E., Moravek, A., Murphy, J., Hrdina, A., Thornton, J., Goldberger, L., Lee, B., Riedel, T., Whitehill, A., Kelly, K., Hansen, J., and Eatough, D.: Utah Winter Fine Particulate Study Final Report, available at: https://www.esrl.noaa.gov/csd/groups/csd7/measurements/2017uwfps/finalreport.pdf, last access: 30 November 2018.
Bahreini, R., Dunlea, E. J., Matthew, B. M., Simons, C., Docherty, K. S., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., and Middlebrook, A. M.: Design and operation of a pressure-controlled inlet for airborne sampling with an aerodynamic aerosol lens, Aerosol. Sci. Tech., 42, 465–471, https://doi.org/10.1080/02786820802178514, 2008.
Bahreini, R., Ervens, B., Middlebrook, A. M., Warneke, C., de Gouw, J. A., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., Neuman, J. A., Ryerson, T. B., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J. S., Peischl, J., Richter, D., Walega, J., Weibring, P., Wollny, A. G., and Fehsenfeld, F. C.: Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas, J. Geophys. Res.-Atmos., 114, D00f16, https://doi.org/10.1029/2008jd011493, 2009.
Beard, J. D., Beck, C., Graham, R., Packham, S. C., Traphagan, M., Giles, R. T., and Morgan, J. G.: Winter Temperature Inversions and Emergency Department Visits for Asthma in Salt Lake County, Utah, 2003–2008, Environ. Health Persp., 120, 1385–1390, https://doi.org/10.1289/ehp.1104349, 2012.
Bessagnet, B., Hodzic, A., Blanchard, O., Lattuati, M., Le Bihan, O., Marfaing, H., and Rouil, L.: Origin of particulate matter pollution episodes in wintertime over the Paris Basin, Atmos. Environ., 39, 6159–6174, https://doi.org/10.1016/j.atmosenv.2005.06.053, 2005.
Bondy, A. L., Wang, B. B., Laskin, A., Craig, R. L., Nhliziyo, M. V., Bertman, S. B., Pratt, K. A., Shepson, P. B., and Ault, A. P.: Inland Sea Spray Aerosol Transport and Incomplete Chloride Depletion: Varying Degrees of Reactive Processing Observed during SOAS, Environ. Sci. Technol., 51, 9533–9542, https://doi.org/10.1021/acs.est.7b02085, 2017.
Brock, C. A., Cozic, J., Bahreini, R., Froyd, K. D., Middlebrook, A. M., McComiskey, A., Brioude, J., Cooper, O. R., Stohl, A., Aikin, K. C., de Gouw, J. A., Fahey, D. W., Ferrare, R. A., Gao, R.-S., Gore, W., Holloway, J. S., Hübler, G., Jefferson, A., Lack, D. A., Lance, S., Moore, R. H., Murphy, D. M., Nenes, A., Novelli, P. C., Nowak, J. B., Ogren, J. A., Peischl, J., Pierce, R. B., Pilewskie, P., Quinn, P. K., Ryerson, T. B., Schmidt, K. S., Schwarz, J. P., Sodemann, H., Spackman, J. R., Stark, H., Thomson, D. S., Thornberry, T., Veres, P., Watts, L. A., Warneke, C., and Wollny, A. G.: Characteristics, sources, and transport of aerosols measured in spring 2008 during the aerosol, radiation, and cloud processes affecting Arctic Climate (ARCPAC) Project, Atmos. Chem. Phys., 11, 2423–2453, https://doi.org/10.5194/acp-11-2423-2011, 2011.
Brown, S. G., Roberts, P. T., McCarthy, M. C., Lurmann, F. W., and Hyslop, N. P.: Wintertime vertical variations in particulate matter (PM) and precursor concentrations in the San Joaquin Valley during the California Regional coarse PM/fine PM Air Quality Study, J. Air Waste Manage., 56, 1267–1277, https://doi.org/10.1080/10473289.2006.10464583, 2006.
Brown, S. S., Thornton, J. A., Keene, W. C., Pszenny, A. A. P., Sive, B. C., Dube, W. P., Wagner, N. L., Young, C. J., Riedel, T. P., Roberts, J. M., VandenBoer, T. C., Bahreini, R., Ozturk, F., Middlebrook, A. M., Kim, S., Hubler, G., and Wolfe, D. E.: Nitrogen, Aerosol Composition, and Halogens on a Tall Tower (NACHTT): Overview of a wintertime air chemistry field study in the front range urban corridor of Colorado, J. Geophys. Res.-Atmos., 118, 8067–8085, https://doi.org/10.1002/jgrd.50537, 2013.
Cai, Y., Montague, D. C., Mooiweer-Bryan, W., and Deshler, T.: Performance characteristics of the ultra high sensitivity aerosol spectrometer for particles between 55 and 800 nm: Laboratory and field studies, J. Aerosol Sci., 39, 759–769, https://doi.org/10.1016/j.jaerosci.2008.04.007, 2008.
Canagaratna, M. R., Jayne, J. T., Ghertner, D. A., Herndon, S., Shi, Q., Jimenez, J. L., Silva, P. J., Williams, P., Lanni, T., Drewnick, F., Demerjian, K. L., Kolb, C. E., and Worsnop, D. R.: Chase studies of particulate emissions from in-use New York City vehicles, Aerosol Sci. Tech., 38, 555–573, https://doi.org/10.1080/02786820490465504, 2004.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Chen, L.-W. A., Watson, J. G., Chow, J. C., Green, M. C., Inouye, D., and Dick, K.: Wintertime particulate pollution episodes in an urban valley of the Western US: a case study, Atmos. Chem. Phys., 12, 10051–10064, https://doi.org/10.5194/acp-12-10051-2012, 2012.
Chow, J. C., Chen, L. W. A., Watson, J. G., Lowenthal, D. H., Magliano, K. A., Turkiewicz, K., and Lehrman, D. E.: PM2.5 chemical composition and spatiotemporal variability during the California Regional PM10/PM2.5 Air Quality Study (CRPAQS), J. Geophys. Res.-Atmos., 111, D10s04, https://doi.org/10.1029/2005jd006457, 2006.
Corbin, J. C., Keller, A., Lohmann, U., Burtscher, H., Sierau, B., and Mensah, A. A.: Organic Emissions from a Wood Stove and a Pellet Stove Before and After Simulated Atmospheric Aging, Aerosol Sci. Tech., 49, 1037–1050, https://doi.org/10.1080/02786826.2015.1079586, 2015.
Cropper, P. M., Eatough, D. J., Overson, D. K., Hansen, J. C., Caka, F., and Cary, R. A.: Use of a gas chromatography-mass spectrometry organic aerosol monitor for in-field detection of fine particulate organic compounds in source apportionment, J. Air Waste Manage., 68, 390–402, https://doi.org/10.1080/10962247.2017.1363095, 2018.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Drewnick, F., Hings, S. S., DeCarlo, P., Jayne, J. T., Gonin, M., Fuhrer, K., Weimer, S., Jimenez, J. L., Demerjian, K. L., Borrmann, S., and Worsnop, D. R.: A new time-of-flight aerosol mass spectrometer (TOF-AMS) – Instrument description and first field deployment, Aerosol Sci. Tech., 39, 637–658, https://doi.org/10.1080/02786820500182040, 2005.
Elsasser, M., Busch, C., Orasche, J., Schön, C., Hartmann, H., Schnelle-Kreis, J., and Zimmermann, R.: Dynamic changes of the aerosol composition and concentration during different burning phases of wood combustion, Energ. Fuel., 27, 4959–4968, 2013.
EPA Air Quality: available at: https://www.epa.gov/air-trends/air-quality-cities-and-counties (last access: 30 November 2018), 2017.
EPA Air Quality Statistics by City, available at: https://www.epa.gov/sites/production/files/2018-07/cbsafactbook2017.xlsx (last access: 30 November 2018), 2016.
Favez, O., El Haddad, I., Piot, C., Boréave, A., Abidi, E., Marchand, N., Jaffrezo, J.-L., Besombes, J.-L., Personnaz, M.-B., Sciare, J., Wortham, H., George, C., and D'Anna, B.: Inter-comparison of source apportionment models for the estimation of wood burning aerosols during wintertime in an Alpine city (Grenoble, France), Atmos. Chem. Phys., 10, 5295–5314, https://doi.org/10.5194/acp-10-5295-2010, 2010.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+-Ca2+-Mg2+--Na+---Cl−-H2O aerosols, Atmos. Chem. Phys., 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Fuchs, H., Dube, W. P., Lerner, B. M., Wagner, N. L., Williams, E. J., and Brown, S. S.: A Sensitive and Versatile Detector for Atmospheric NO2 and NOx Based on Blue Diode Laser Cavity Ring-Down Spectroscopy, Environ. Sci. Technol., 43, 7831–7836, https://doi.org/10.1021/es902067h, 2009.
Green, M. C., Chow, J. C., Watson, J. G., Dick, K., and Inouye, D.: Effects of Snow Cover and Atmospheric Stability on Winter PM2.5 Concentrations in Western U.S. Valleys, J. Appl. Meteorol. Clim., 54, 1191–1201, https://doi.org/10.1175/Jamc-D-14-0191.1, 2015.
Gwaze, P., Helas, G., Annegarn, H. J., Huth, J., and Piketh, S. J.: Physical, chemical and optical properties of aerosol particles collected over Cape Town during winter haze episodes, S. Afr. J. Sci., 103, 35–43, 2007.
Hansen, J. C., Woolwine, W. R., Bates, B. L., Clark, J. M., Kuprov, R. Y., Mukherjee, P., Murray, J. A., Simmons, M. A., Waite, M. F., Eatough, N. L., Eatough, D. J., Long, R., and Grover, B. D.: Semicontinuous PM2.5 and PM10 Mass and Composition Measurements in Lindon, Utah, during Winter 2007, J. Air Waste Manage., 60, 346–355, https://doi.org/10.3155/1047-3289.60.3.346, 2010.
Haskins, J. D., Jaeglé, L., Shah, V., Lee, B. H., Lopez-Hilfiker, F. D., Campuzano-Jost, P., Schroder, J. C., Day, D. A., Guo, H., Sullivan, A. P., Weber, R., Dibb, J., Campos, T., Jimenez, J. L., Brown, S. S., and Thornton, J. A.: Wintertimegas-particle partitioning and speciation of inorganic chlorine in the lower troposphere over the Northeast UnitedStates and Coastal Ocean, J. Geophys. Res.-Atmos., 123, https://doi.org/10.1029/2018JD028786, 2018.
Heringa, M. F., DeCarlo, P. F., Chirico, R., Lauber, A., Doberer, A., Good, J., Nussbaumer, T., Keller, A., Burtscher, H., Richard, A., Miljevic, B., Prevot, A. S. H., and Baltensperger, U.: Time-Resolved Characterization of Primary Emissions from Residential Wood Combustion Appliances, Environ. Sci. Technol., 46, 11418–11425, https://doi.org/10.1021/es301654w, 2012.
Jayne, J. T., Leard, D. C., Zhang, X. F., Davidovits, P., Smith, K. A., Kolb, C. E., and Worsnop, D. R.: Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Tech., 33, 49–70, https://doi.org/10.1080/027868200410840, 2000.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Kelly, K. E., Kotchenruther, R., Kuprov, R., and Silcox, G. D.: Receptor model source attributions for Utah's Salt Lake City airshed and the impacts of wintertime secondary ammonium nitrate and ammonium chloride aerosol, J. Air Waste Manage., 63, 575–590, https://doi.org/10.1080/10962247.2013.774819, 2013.
Kim, Y. J., Spak, S. N., Carmichael, G. R., Riemer, N., and Stanier, C. O.: Modeled aerosol nitrate formation pathways during wintertime in the Great Lakes region of North America, J. Geophys. Res.-Atmos., 119, 12420–12445, https://doi.org/10.1002/2014JD022320, 2014.
Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., Edney, E. O., Jaoui, M., Zheng, M., Ding, X. A., and Edgerton, E. S.: Contribution of Primary and Secondary Sources to Organic Aerosol and PM2.5 at SEARCH Network Sites, J. Air Waste Manage., 60, 1388–1399, https://doi.org/10.3155/1047-3289.60.11.1388, 2010.
Kuprov, R., Eatough, D. J., Cruickshank, T., Olson, N., Cropper, P. M., and Hansen, J. C.: Composition and secondary formation of fine particulate matter in the Salt Lake Valley: Winter 2009, J. Air Waste Manage., 64, 957–969, https://doi.org/10.1080/10962247.2014.903878, 2014.
Lareau, N. P., Crosman, E., Whiteman, C. D., Horel, J. D., Hoch, S. W., Brown, W. O. J., and Horst, T. W.: The Persistent Cold-Air Pool Study, B. Am. Meteorol. Soc., 94, 51–63, https://doi.org/10.1175/Bams-D-11-00255.1, 2013.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurten, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lee, B. H., Lopez-Hilfiker, F. D., Veres, P. R., McDuffie, E. E., Fibiger, D. L., Sparks, T. L., Ebben, C. J., Green, J. R., Schroder, J. C., Campuzano-Jost, P., Iyer, S., D'Ambro, E. L., Schobesberger, S., Brown, S. S., Wooldridge, P. J., Cohen, R. C., Fiddler, M. N., Bililign, S., Jimenez, J. L., Kurten, T., Weinheimer, A. J., Jaegle, L., and Thornton, J. A.: Flight Deployment of a High-Resolution Time-of-Flight Chemical Ionization Mass Spectrometer: Observations of Reactive Halogen and Nitrogen Oxide Species, J. Geophys. Res.-Atmos., 123, 7670–7686, https://doi.org/10.1029/2017JD028082, 2018.
Liao, J., Brock, C. A., Murphy, D. M., Sueper, D. T., Welti, A., and Middlebrook, A. M.: Single-particle measurements of bouncing particles and in situ collection efficiency from an airborne aerosol mass spectrometer (AMS) with light-scattering detection, Atmos. Meas. Tech., 10, 3801–3820, https://doi.org/10.5194/amt-10-3801-2017, 2017.
Long, R. W., Eatough, N. L., Mangelson, N. F., Thompson, W., Fiet, K., Smith, S., Smith, R., Eatough, D. J., Pope, C. A., and Wilson, W. E.: The measurement of PM2.5, including semi-volatile components, in the EMPACT program: results from the Salt Lake City Study, Atmos. Environ., 37, 4407–4417, https://doi.org/10.1016/S1352-2310(03)00585-5, 2003.
Long, R. W., Eatough, N. L., Eatough, D. J., Meyer, M. B., and Wilson, W. E.: Continuous Determination of Fine Particulate Matter Mass in the Salt Lake City Environmental Monitoring Project: A Comparison of Real-Time and Conventional TEOM Monitor Results, J. Air Waste Manage., 55, 1782–1796, https://doi.org/10.1080/10473289.2005.10464776, 2005a.
Long, R. W., Modey, W. K., Smith, P. S., Smith, R., Merrill, C., Pratt, J., Stubbs, A., Eatough, N. L., Eatough, D. J., Malm, W. C., and Wilson, W. E.: One- and Three-Hour PM2.5 Characterization, Speciation, and Source Apportionment Using Continuous and Integrated Samplers, Aerosol Sci. Tech., 39, 238–248, https://doi.org/10.1080/027868290925633, 2005b.
Lurmann, F. W., Brown, S. G., McCarthy, M. C., and Roberts, P. T.: Processes influencing secondary aerosol formation in the San Joaquin Valley during winter, J. Air Waste Manage., 56, 1679–1693, https://doi.org/10.1080/10473289.2006.10464573, 2006.
Mangelson, N. F., Lewis, L., Joseph, J. M., Cui, W. X., Machir, J., Williams, N. W., Eatough, D. J., Rees, L. B., Wilkerson, T., and Jensen, D. T.: The contribution of sulfate and nitrate to atmospheric fine particles during winter inversion fogs in Cache Valley, Utah, J. Air Waste Manage., 47, 167–175, https://doi.org/10.1080/10473289.1997.10464429, 1997.
McDuffie, E. E., Fibiger, D. L., Dube, W. P., Lopez-Hilfiker, F., Lee, B. H., Thornton, J. A., Shah, V., Jaegle, L., Guo, H. Y., Weber, R. J., Reeves, J. M., Weinheimer, A. J., Schroder, J. C., Campuzano-Jost, P., Jimenez, J. L., Dibb, J. E., Veres, P., Ebben, C., Sparks, T. L., Wooldridge, P. J., Cohen, R. C., Hornbrook, R. S., Apel, E. C., Campos, T., Hall, S. R., Ullmann, K., and Brown, S. S.: Heterogeneous N2O5 Uptake During Winter: Aircraft Measurements During the 2015 WINTER Campaign and Critical Evaluation of Current Parameterizations, J. Geophys. Res.-Atmos., 123, 4345–4372, https://doi.org/10.1002/2018JD028336, 2018.
Metzger, S., Mihalopoulos, N., and Lelieveld, J.: Importance of mineral cations and organics in gas-aerosol partitioning of reactive nitrogen compounds: case study based on MINOS results, Atmos. Chem. Phys., 6, 2549–2567, https://doi.org/10.5194/acp-6-2549-2006, 2006.
Mouteva, G. O., Randerson, J. T., Fahrni, S. M., Bush, S. E., Ehleringer, J. R., Xu, X. M., Santos, G. M., Kuprov, R., Schichtel, B. A., and Czimczik, C. I.: Using radiocarbon to constrain black and organic carbon aerosol sources in Salt Lake City, J. Geophys. Res.-Atmos., 122, 9843–9857, https://doi.org/10.1002/2017JD026519, 2017.
Nenes, A., Pandis, S. N., and Pilinis, C.: ISORROPIA: A new thermodynamic equilibrium model for multiphase multicomponent inorganic aerosols, Aquat. Geochem., 4, 123–152, https://doi.org/10.1023/A:1009604003981, 1998.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Perring, A. E., Schwarz, J. P., Gao, R. S., Heymsfield, A. J., Schmitt, C. G., Schnaiter, M., and Fahey, D. W.: Evaluation of a Perpendicular Inlet for Airborne Sampling of Interstitial Submicron Black-Carbon Aerosol, Aerosol Sci. Tech., 47, 1066–1072, https://doi.org/10.1080/02786826.2013.821196, 2013.
Petetin, H., Sciare, J., Bressi, M., Gros, V., Rosso, A., Sanchez, O., Sarda-Estève, R., Petit, J.-E., and Beekmann, M.: Assessing the ammonium nitrate formation regime in the Paris megacity and its representation in the CHIMERE model, Atmos. Chem. Phys., 16, 10419–10440, https://doi.org/10.5194/acp-16-10419-2016, 2016.
Pope, C. A., Muhlestein, J. B., May, H. T., Renlund, D. G., Anderson, J. L., and Horne, B. D.: Ischemic heart disease events triggered by short-term exposure to fine particulate air pollution, Circulation, 114, 2443–2448, https://doi.org/10.1161/Circulationaha.106.636977, 2006.
Prabhakar, G., Parworth, C. L., Zhang, X., Kim, H., Young, D. E., Beyersdorf, A. J., Ziemba, L. D., Nowak, J. B., Bertram, T. H., Faloona, I. C., Zhang, Q., and Cappa, C. D.: Observational assessment of the role of nocturnal residual-layer chemistry in determining daytime surface particulate nitrate concentrations, Atmos. Chem. Phys., 17, 14747–14770, https://doi.org/10.5194/acp-17-14747-2017, 2017.
Ricciardelli, I., Bacco, D., Rinaldi, M., Bonafe, G., Scotto, F., Trentini, A., Bertacci, G., Ugolini, P., Zigola, C., Rovere, F., Maccone, C., Pironi, C., and Poluzzi, V.: A three-year investigation of daily PM2.5 main chemical components in four sites: the routine measurement program of the Supersito Project (Po Valley, Italy), Atmos. Environ., 152, 418–430, https://doi.org/10.1016/j.atmosenv.2016.12.052, 2017.
Schneider, J., Weimer, S., Drewnick, F., Borrmann, S., Helas, G., Gwaze, P., Schmid, O., Andreae, M. O., and Kirchner, U.: Mass spectrometric analysis and aerodynamic properties of various types of combustion-related aerosol particles, Int. J. Mass Spectrom., 258, 37–49, https://doi.org/10.1016/j.ijms.2006.07.008, 2006.
Shrivastava, M. K., Subramanian, R., Rogge, W. F., and Robinson, A. L.: Sources of organic aerosol: Positive matrix factorization of molecular marker data and comparison of results from different source apportionment models, Atmos. Environ., 41, 9353–9369, https://doi.org/10.1016/j.atmosenv.2007.09.016, 2007.
Silva, P. J., Vawdrey, E. L., Corbett, M., and Erupe, M.: Fine particle concentrations and composition during wintertime inversions in Logan, Utah, USA, Atmos. Environ., 41, 5410–5422, https://doi.org/10.1016/j.atmosenv.2007.02.016, 2007.
Stanier, C., Singh, A., Adamski, W., Baek, J., Caughey, M., Carmichael, G., Edgerton, E., Kenski, D., Koerber, M., Oleson, J., Rohlf, T., Lee, S. R., Riemer, N., Shaw, S., Sousan, S., and Spak, S. N.: Overview of the LADCO winter nitrate study: hourly ammonia, nitric acid and PM2.5 composition at an urban and rural site pair during PM2.5 episodes in the US Great Lakes region, Atmos. Chem. Phys., 12, 11037–11056, https://doi.org/10.5194/acp-12-11037-2012, 2012.
Sudheer, A. K. and Rengarajan, R.: Time-resolved inorganic chemical composition of fine aerosol and associated precursor gases over an urban environment in western India: Gas-aerosol equilibrium characteristics, Atmos. Environ., 109, 217–227, https://doi.org/10.1016/j.atmosenv.2015.03.028, 2015.
Tevlin, A. G., Li, Y., Collett, J. L., McDuffie, E. E., Fischer, E. V., and Murphy, J. G.: Tall Tower Vertical Profiles and Diurnal Trends of Ammonia in the Colorado Front Range, J. Geophys. Res.-Atmos., 122, 12468–12487, https://doi.org/10.1002/2017JD026534, 2017.
Trebs, I., Metzger, S., Meixner, F. X., Helas, G. N., Hoffer, A., Rudich, Y., Falkovich, A. H., Moura, M. A. L., da Silva, R. S., Artaxo, P., Slanina, J., and Andreae, M. O.: The --Cl−--H2O aerosol system and its gas phase precursors at a pasture site in the Amazon Basin: How relevant are mineral cations and soluble organic acids?, J. Geophys. Res.-Atmos., 110, D07303, https://doi.org/10.1029/2004jd005478, 2005.
UWFPS team: UWFPS data, available at: https://esrl.noaa.gov/csd/groups/csd7/measurements/2017uwfps/data.html (last access: 30 November 2018), 2017.
Wang, D. X., Hu, J. L., Xu, Y., Lv, D., Xie, X. Y., Kleeman, M., Xing, J., Zhang, H. L., and Ying, Q.: Source contributions to primary and secondary inorganic particulate matter during a severe wintertime PM2.5 pollution episode in Xi'an, China, Atmos. Environ., 97, 182–194, https://doi.org/10.1016/j.atmosenv.2014.08.020, 2014.
Washenfelder, R. A., Wagner, N. L., Dube, W. P., and Brown, S. S.: Measurement of Atmospheric Ozone by Cavity Ring-down Spectroscopy, Environ. Sci. Technol., 45, 2938–2944, https://doi.org/10.1021/es103340u, 2011.
Watson, J. G. and Chow, J. C.: A wintertime PM2.5 episode at the fresno, CA, supersite, Atmos. Environ., 36, 465–475, https://doi.org/10.1016/S1352-2310(01)00309-0, 2002.
Whiteman, C. D., Hoch, S. W., Horel, J. D., and Charland, A.: Relationship between particulate air pollution and meteorological variables in Utah's Salt Lake Valley, Atmos. Environ., 94, 742–753, https://doi.org/10.1016/j.atmosenv.2014.06.012, 2014.
WHO: Ambient (outdoor) air quality and health, available at: http://www.who.int/mediacentre/factsheets/fs313/en/ (last access: 30 November 2018), 2016.
Wild, R. J., Edwards, P. M., Dube, W. P., Baumann, K., Edgerton, E. S., Quinn, P. K., Roberts, J. M., Rollins, A. W., Veres, P. R., Warneke, C., Williams, E. J., Yuan, B., and Brown, S. S.: A Measurement of Total Reactive Nitrogen, NOy, together with NO2, NO, and O3 via Cavity Ring-down Spectroscopy, Environ. Sci. Technol., 48, 9609–9615, https://doi.org/10.1021/es501896w, 2014.
Woody, M. C., Baker, K. R., Hayes, P. L., Jimenez, J. L., Koo, B., and Pye, H. O. T.: Understanding sources of organic aerosol during CalNex-2010 using the CMAQ-VBS, Atmos. Chem. Phys., 16, 4081–4100, https://doi.org/10.5194/acp-16-4081-2016, 2016.
Zhang, Q., Alfarra, M. R., Worsnop, D. R., Allan, J. D., Coe, H., Canagaratna, M. R., and Jimenez, J. L.: Deconvolution and quantification of hydrocarbon-like and oxygenated organic aerosols based on aerosol mass spectrometry, Environ. Sci. Technol., 39, 4938–4952, https://doi.org/10.1021/es048568l, 2005.