the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Nov 2018
| 08 Nov 2018
Is there an aerosol signature of chemical cloud processing?
Abdulmonam M. Aldhaif
Taylor Shingler
Ewan Crosbie
Luke Ziemba
Pedro Campuzano-Jost
Jose L. Jimenez
Armin Wisthaler
The formation of sulfate and secondary organic aerosol mass in the aqueous phase (aqSOA) of cloud and fog droplets can significantly contribute to ambient aerosol mass. While tracer compounds give evidence that aqueous-phase processing occurred, they do not reveal the extent to which particle properties have been modified in terms of mass, chemical composition, hygroscopicity, and oxidation state. We analyze data from several field experiments and model studies for six air mass types (urban, biogenic, marine, wild fire biomass burning, agricultural biomass burning, and background air) using aerosol size and composition measurements for particles 13–850 nm in diameter. We focus on the trends of changes in mass, hygroscopicity parameter κ, and oxygen-to-carbon (O ∕ C) ratio due to chemical cloud processing. We find that the modification of these parameters upon cloud processing is most evident in urban, marine, and biogenic air masses, i.e., air masses that are more polluted than very clean air (background air) but cleaner than heavily polluted plumes as encountered during biomass burning. Based on these trends, we suggest that the mass ratio (Rtot) of the potential aerosol sulfate and aqSOA mass to the initial aerosol mass can be used to predict whether chemical cloud processing will be detectable. Scenarios in which this ratio exceeds Rtot∼0.5 are the most likely ones in which clouds can significantly change aerosol parameters. It should be noted that the absolute value of Rtot depends on the considered size range of particles. Rtot is dominated by the addition of sulfate (Rsulf) in all scenarios due to the more efficient conversion of SO2 to sulfate compared to aqSOA formation from organic gases. As the formation processes of aqSOA are still poorly understood, the estimate of RaqSOA is likely associated with large uncertainties. Comparison to Rtot values as calculated for ambient data at different locations validates the applicability of the concept to predict a chemical cloud-processing signature in selected air masses.
- Article
(3917 KB) - Full-text XML
-
Supplement
(445 KB) - BibTeX
- EndNote





Clouds and, in particular, aerosol–cloud interactions represent one of the largest uncertainties in our current understanding of radiative forcing (IPCC, 2013). Thus, representing cloud chemistry in models is challenging, as the prediction of aerosol mass production in clouds is inherently impacted by the uncertainties in the description of cloud properties (e.g., liquid water content (LWC), drop size distribution, cloud lifetime, geographical location, altitude, and cloud density), in addition to uncertainties in the chemical mechanisms and precursors (Ervens, 2015). Airborne chemical measurements in clouds can be used to study cloud processing, but such measurements are relatively sparse and usually only represent snapshots of a few seconds of an aircraft transect. Several studies have been performed on mountaintops where hill-capped clouds cover the summit for extended period of times (Choularton et al., 1997; Herrmann et al., 2005; Li et al., 2017). While in “hill cloud” experiments more continuous datasets can be collected, they are limited in their geographical coverage and their interpretation is complicated by variable advection of various sources and air masses. Many studies show enhanced concentrations of sulfate, oxalate, and related organics in cloud-processed air compared to cloud-free air (Crahan et al., 2004; Sorooshian et al., 2006a; 2007a; Wonaschuetz et al., 2012). It has been recognized for several decades that globally a major fraction of sulfate is formed in clouds (Roelofs et al., 1998; Barth et al., 2000) and to a smaller extent also in deliquesced aerosol particles (Sievering et al., 1991; Alexander et al., 2005; Zheng et al., 2015). More recently, it has been shown that secondary organic aerosol (SOA) mass can also be formed by chemical reactions in cloud and aerosol water (aqSOA) (Surratt et al., 2010; Ervens et al., 2011; McNeill, 2015; Xu et al., 2015; Marais et al., 2016). However, SOA formation in clouds is not always observed. For example, Wagner et al. (2015) systematically analyzed vertical profiles in the SOA-dominated southeastern US and found that SOA formation in fair-weather cumulus clouds was statistically insignificant.
The formation processes, precursors, and conditions for aqSOA formation are more poorly quantified than for sulfate. Tracer compounds such as oxalic acid that are dominant products of aqueous-phase processes have been identified but usually only contribute a few percent to the total aerosol mass (Shen et al., 2012; Wang et al., 2012), and thus they do not reveal the general role of aqSOA formation to modify aerosol properties. In addition, oxalate might have additional, less dominant emission sources such as biomass burning (Narukawa et al., 1999; Falkovich et al., 2005; Zhang et al., 2017) and other sources (Huang and Yu, 2007). Bulk properties of OA have been shown to be modified differently by aqueous-phase processes than by surface or gas-phase reactions. These properties include the oxygen-to-carbon (O ∕ C) ratio, which is often higher in aqueous-phase-derived products (Ervens et al., 2011; Sorooshian et al., 2011; Waxman et al., 2013; Chakraborty et al., 2016) or hygroscopicity (Shingler et al., 2016). However, pathways that produce aerosol with high O ∕ C via gas-phase reactions are also possible (Chhabra et al., 2011; Ehn et al., 2014; Krechmer et al., 2015), which needs to be taken into account in the interpretation of case studies. Several laboratory studies have shown that organics that are formed in aqueous-phase reactions might absorb light (e.g., De Haan et al., 2010; Powelson et al., 2014; Laskin et al., 2015). However, these products only comprise a very small fraction of the total organic carbon and are likely formed in evaporating cloud droplets, i.e., on short timescales, and when solute concentrations become sufficiently high.
The addition of mass in clouds only occurs on activated particles and often leads to a distinct droplet mode that separates unactivated particles from activated ones (Hoppel et al., 1994). A similar effect of mode separation might be achieved by collision–coalescence within clouds (Feingold et al., 1996); however, these physical processes do not lead to a distinct change in chemical composition such as the production of aqueous-phase tracer compounds. While this size separation might be the most unequivocal microphysical tracer of chemical cloud processing, the change in bulk and/or size-resolved (physico)chemical properties of the aerosol population might also be used to identify cloud-processed aerosol. Not only total aerosol mass, but also its distribution throughout the particle population is important since particle size and composition determine particle atmospheric lifetime by dry and wet deposition (Maria et al., 2004) and the aerosol direct and indirect effects on climate (Lin et al., 2014). Thus, it is important to identify and quantify how cloud-derived products affect aerosol loading and properties.
In the current study, we apply a combination of model simulations and observations to explore a possible signature of chemical cloud processing in aerosol. Unlike other studies that focused on single parameters, such as modification of size distribution (Eck et al., 2012), aqSOA tracer compounds (Kawamura and Ikushima, 1993; Kawamura and Yasui, 2005; Agarwal et al., 2010), and the O ∕ C ratio (Chakraborty et al., 2016), we compare all of these properties in different air masses from clean (background air) to heavily polluted (biomass burning) as they were identified using highly instrumented aircraft data from SEAC4RS. Trends in model results are compared to those from other observational datasets in order to draw conclusions on a possible cloud-processing signature in different air mass types.
2.1 Datasets and air masses
2.1.1 SEAC4RS (Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys) aircraft field study results as input to model
A rich set of airborne data collected on the NASA DC-8 is used from SEAC4RS based in Houston, Texas, in August–September 2013 (Toon et al., 2016). SEAC4RS was a multi-platform field campaign addressing issues associated with atmospheric composition over North America. It included two test flights and 21 research flights with the DC-8 covering altitudes from the surface to above 10 km. Data from this campaign were used to initialize model simulations that will be discussed subsequently.
Owing to the broad range of conditions sampled, a variety of criteria used by Shingler et al. (2016) for the same dataset were applied to define the following air mass types:
-
biomass burning (BB)–wildfire: acetonitrile > 250 pptv or (when acetonitrile unavailable) CO > 250 ppbv in nonurban areas;
-
biomass burning–agricultural: same as BB–wildfire, with additional visual confirmation;
-
biogenic: isoprene + monoterpenes + methyl vinyl ketone (MVK) + methacrolein (MACR) > 2 ppbv and acetonitrile < 250 pptv;
-
marine: in planetary boundary layer (PBL), over ocean, and more than 40 km from the coast;
-
urban: in PBL; spatially over Houston (30.50∘ N, −94.60∘ W to 29.00∘ N, −96.10∘ W) or Los Angeles (34.17∘ N, −117.00∘ W to 33.44∘ N, −119.75∘ W); and
-
background–mixed: in PBL; did not fit into first five categories.
Relevant instruments used to obtain data to apply the criteria above included a proton-transfer-reaction mass spectrometer (PTR-MS) (de Gouw and Warneke, 2007) for selected species, including MACR, MVK, monoterpenes (sum of monoterpene isomers), isoprene, and acetonitrile. Isoprene levels in biomass burning plumes represented upper limits owing to interferences from other species such as furan. Furthermore, isoprene levels are higher in the biomass burning–agricultural category compared to the biomass burning–wildfire category owing to the aircraft having sampled the former much closer to its source compared to the more aged plumes of the latter. The MVK and MACR data are also vulnerable to an interference (ISOPOOH), which is most relevant in lower NO regions. As the corresponding NO and NO2 levels (measured by the NOAA NOyO3 instrument) in Table S1 in the Supplement are not very low, this potential interference is considered to be minor, except for the biogenic cases in which it is known to be substantial. Data for CO were obtained from a folded-path, differential absorption, mid-IR diode laser spectrometer (Sachse et al., 1987). Water vapor data were used from the diode laser hygrometer to identify the height of the PBL (Diskin et al., 2002). Table S1 also lists the concentrations of the following gases: HCHO (NASA In Situ Airborne Formaldehyde (ISAF) instrument), SO2 (Georgia Tech Chemical Ionization Mass Spectrometer; CIMS), H2O2 (Caltech CIMS), and O3 (NOAA NOyO3 instrument). It is further noted that the marine category is still impacted by anthropogenic pollution owing to transported continental pollution and ship exhaust and thus should not be regarded as representing pristine marine air masses.
An Aerodyne high-resolution time-of-flight aerosol mass spectrometer (HR-AMS) (DeCarlo et al., 2006; Canagaratna et al., 2007; Dunlea et al., 2009) was used for non-refractory composition of submicrometer particles, including the O ∕ C ratio of OA. Given that the HR-AMS, as operated and analyzed for SEAC4RS, did not quantify refractory and semi-refractory species, submicron sodium chloride and nitrate in the marine BL are not included in the AMS results.
Black carbon (BC) data were obtained with a humidified-dual single-particle soot photometer (HD-SP2) (Schwarz et al., 2015). Aerosol size distribution data were used from the Langley Aerosol Research Group Experiment (LARGE) instrument package from a scanning mobility particle sizer (SMPS; TSI, Inc., model 3080/3010; midpoint Dp between 11 and 316 nm) and an ultrahigh-sensitivity aerosol spectrometer (UHSAS; Droplet Measurement Technologies, Inc.; midpoint Dp between 63 and 891 nm). Sizing calibrations were performed frequently during the measurement period using polystyrene latex spheres and monodisperse ammonium sulfate particles for the SMPS and UHSAS, respectively. The two distributions were stitched together at the upper diameter bound of the SMPS, above which the UHSAS data were used (Sect. 3.2).
2.1.2 Datasets for identifying cloud processing
Data are analyzed from several other campaigns. More specifically, the following datasets are used:
- i.
water-soluble anions and cations from a particle-into-liquid sampler coupled to off-line ion chromatography (PILS-IC, Brechtel Mfg. Inc.; Sorooshian et al., 2006b) deployed on the Center for Interdisciplinary Remotely Piloted Aircraft Studies (CIRPAS) Twin Otter during the Gulf of Mexico Atmospheric Composition and Climate Study (GoMACCS) mission between August and September 2006 based in Houston, Texas;
- ii.
size-resolved aerosol composition from a micro-orifice uniform deposit impactor (MOUDI, MSP Corporation; Marple et al., 1991) at three ground sites in Arizona (Hayden, Tucson, Mount Lemmon; Sorooshian et al., 2012; Youn et al., 2015) and in Marina, California (Maudlin et al., 2015; Braun et al., 2017); and
- iii.
size-resolved aerosol hygroscopic growth factors g(RH) as measured by a humidified tandem differential mobility analyzer (HTDMA; Brechtel Manufacturing Inc., BMI model 3002; Wonaschütz et al., 2013) for samples collected at a ground site at Mount Lemmon in Arizona.
2.2 Model
2.2.1 Model description
A parcel model is used to simulate cloud processing in a transect of an air parcel along a prescribed trajectory through a cloud (Feingold and Kreidenweis, 2000; Ervens et al., 2004). Gas-phase chemistry occurs during the full simulation; the chemical scheme is based on the NCAR master mechanism (Kim et al., 2012). Gas-phase precursors for aqSOA include isoprene, toluene, xylene, and ethylene whose oxidation products (glyoxal and related compounds) are taken up into the aqueous phase and further oxidized (Ervens et al., 2004). These precursor compounds and SO2 are not replenished during the simulation to simulate emissions into a cloud away from emission sources. AqSOA formation from these compounds and sulfate formation by SO2 oxidation with H2O2 and O3 have been described previously (Ervens et al., 2014; McVay and Ervens, 2017). Aerosol mass formation outside clouds or on and/or in interstitial particles inside the clouds is not included to focus only on aerosol modification due to chemical processes in cloud droplets. We do not include non-oxidative aqSOA formation pathways in our model (e.g., IEPOX formation) as they have been shown to (i) occur on longer timescales and (ii) are most effective in wet aerosol compared to cloud droplets (Woo and McNeill, 2015). Thus, overall the predicted total aqSOA mass might represent an underestimate, while the formation rate might be overestimated. Particle growth is assumed to only occur via chemical mass addition; collision–coalescence processes are neglected.
It should be noted that we do not aim to reproduce observational results but rather seek trends in aerosol properties (O ∕ C ratio, hygroscopicity, mass) over a wide range of conditions in different air masses. The volume-weighted hygroscopicity parameter κ is calculated based on the equations given by Petters and Kreidenweis (2007), considering the individual aerosol fractions multiplied by κsulf=0.7, κaqSOA=0.5, , , and κorg=0.1. κorg refers to the initial organic aerosol fraction before cloud processing; this organic fraction is likely composed of both SOA and primary organic aerosol (POA). This estimate is based on measurements during SEAC4RS in which particles with high organic content exhibited κ∼0.1 (Shingler et al., 2016). The organics that are added by chemical reactions in the cloud water are referred to here as aqSOA. The hygroscopicity parameter for this added aqSOA mass (κaqSOA=0.5) is assumed to be the upper range of oxalate salts (Drozd et al., 2014) since oxalate (oxalic acid) is one of the major constituents of cloud aqSOA (Ervens et al., 2011). The initial κ values at t = 0 are the values that were determined from growth factor measurements during SEAC4RS (Shingler et al., 2016). The change in κ during the course of the simulation is calculated after each model time step (1 s), i.e., by taking into account the newly formed sulfate and aqSOA masses that are added to the initial mass.
The O ∕ C ratio is molecular based and its evolution is calculated according to aqSOA products (O ∕ C(glyoxal) = 1, O ∕ C(glyoxylic acid) = 1.5, O ∕ C(oxalic acid) = 2, etc.) that are added to the initial organic fraction. While these compounds might be hydrated in cloud water and have O ∕ C = 2, in the dried aerosol phase, they are likely present in their dehydrated state. The bulk O ∕ C ratio is calculated by summing the total number of oxygen atoms across all organic species and dividing by the total number of carbon atoms. The evolution of particle sizes in 30 size classes (11 nm < aerodynamic diameter < 860 nm) is tracked.
2.2.2 Model simulations
The model simulations are initialized with air-mass-specific aerosol and gas-phase compositions for the six air masses. The cases differ in their initial concentrations of aerosol mass, mass fractions, particle number concentrations and initial κ, O ∕ C ratio (Table 1), and gas-phase mixing ratios (Table S1). It is assumed that the aerosol is internally mixed and particles of all sizes have the same composition, which is supported by the AMS size distributions for most cases, except some fresh biogenic plumes. For simplicity, we consider the same trajectory for all air masses in the parcel model in which an air parcel spends about 40 min of the 1 h long simulation time in the cloud (between ∼450 and 1100 s and 1800–3600 s, respectively). Cloud chemistry occurs only when a minimum total liquid water content, LWC > 0.01 g m−3, is exceeded. When RH drops below 100 % cloud droplets evaporate, together with some volatile organics. Even though organic acids (glyoxylic, oxalic, pyruvic) have relatively high vapor pressures, it is assumed that they stay in the particle phase as they contribute to aqSOA in the form of salts and complexes. The low pH value of aerosol water as observed during SEAC4RS might lead to evaporation of organic acids with low pKa values (Häkkinen et al., 2014; Nah et al., 2018). However, the fact that these acids are present in aerosol found during the campaign suggests a complex set of equilibria of acid partitioning between the gas and condensed phases as well as salt and the complex formation of partially dissolved carboxylates. Additional formation pathways may be missing from our model studies so that the production rate of organic acids (Lim et al., 2010) is, on the one hand, underestimated. On the other hand, loss reactions of organic acids might be underestimated, such as the photolysis of iron–dicarboxylato complexes (Weller et al., 2014).
All initial size distributions show some evidence of cloud processing (Sect. 3.2); however, our model exercise intends to show the extent to which such somewhat aged aerosol populations will be further altered due to aqueous-phase processing. In the following, we discuss both the modification of bulk and size-resolved parameters.
Table 1Initial aerosol properties for six air masses during SEAC4RS. These data are used as inputs to the box and parcel models in order to simulate aqueous-phase processing.
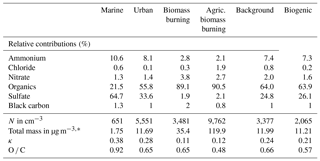
* Masses are given in standard m−3.
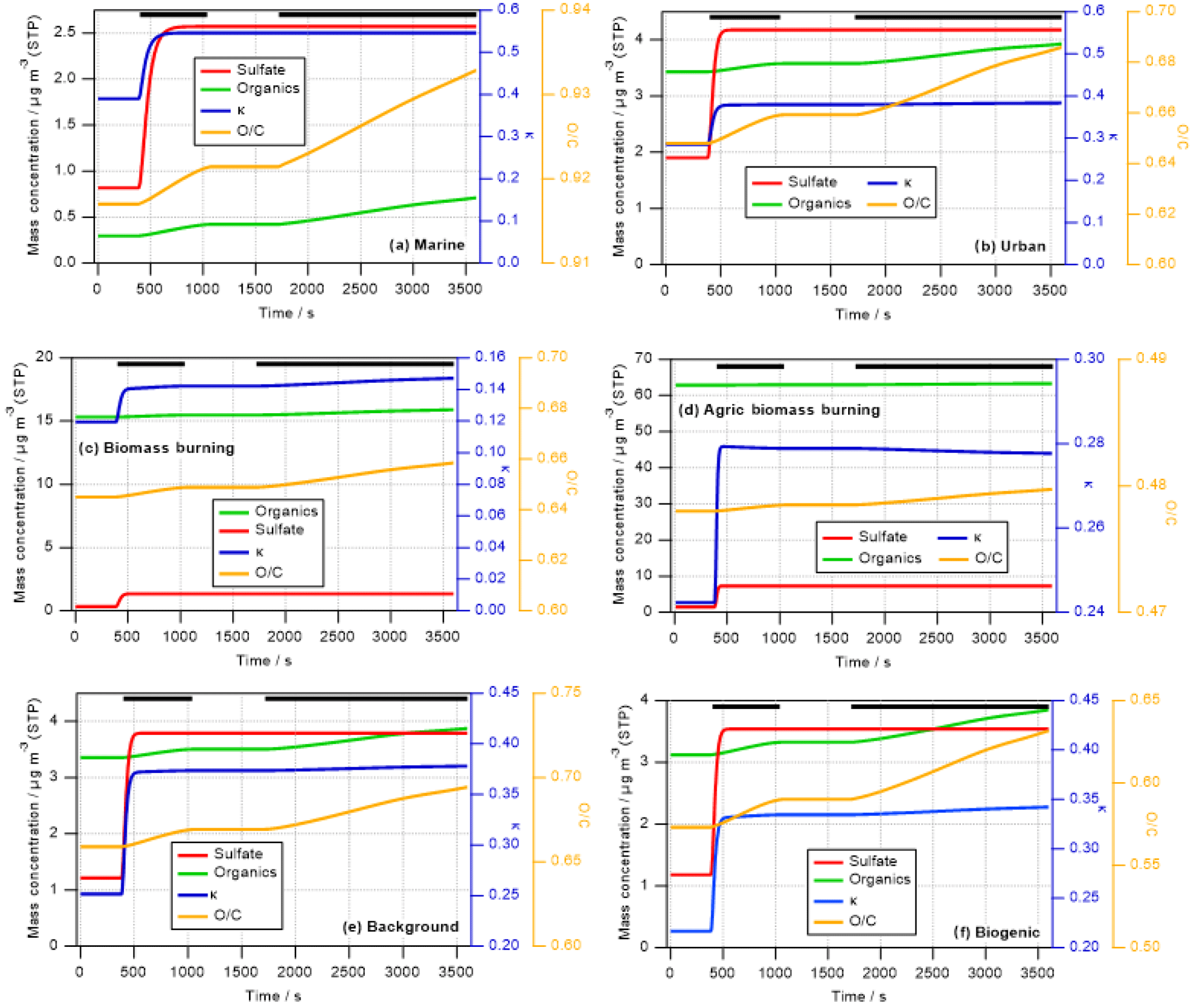
Figure 1Predicted change in aerosol properties due to cloud processing of six different air masses identified during SEAC4RS. Cloud processing simulations are performed for 1 h during which a cloud exists for ∼40 min. Green and red lines show predicted increases in organic and sulfate mass (left axis), respectively; blue and orange lines represent the change in hygroscopicity parameter κ (first right axis) and O ∕ C ratio (second right axis), respectively. The thick black lines near the top of the panels denote the in-cloud time.
3.1 Bulk parameters
3.1.1 Mass increase
Figure 1 shows the predicted increase in sulfate and aqSOA mass (left axis) over the course of the 1 h simulations, with the parcel spending about 40 min in the cloud. The in-cloud time is marked by the black lines at the top of each panel in Fig. 1.
In all cases, sulfate increases very rapidly and stays constant after all SO2 has been consumed. The increase in aqSOA is slower as it is formed in multiple oxidation steps from precursors (e.g., isoprene → glyoxal → glyoxylic acid → oxalic acid). In the time period between the two cloud passages (∼1100–1750 s), the lines are horizontal as no mass is added during that time and thus the aerosol mass and properties remain unchanged. These different timescales are in agreement with previous findings on the comparison of sulfate vs. OH-initiated aqSOA formation in clouds (Ervens et al., 2004), for which a similar chemical mechanism was applied, and in aqueous aerosol (El-Sayed et al., 2015). Rapid SO2 oxidation within clouds and fogs has been observed previously (Husain et al., 2000; Reilly et al., 2001). Our predicted sulfate formation rates (–10−5 M s−1 depending on the air mass) are in general agreement with those as found at Mount Tai, China, for moderately acidic and neutral cloud water (Shen et al., 2012).
The relative proportions of sulfate and aqSOA to total in-cloud mass addition depend on the air mass: while there is clearly more aqSOA than sulfate in the biomass burning and biogenic scenarios (Fig. 1c, d, and f), the masses formed in cloud in the urban and marine scenarios are more sulfate dominated, even though the total SO2 in these scenarios is less than the total VOC mixing ratios (Table S1). While SO2 is completely converted to sulfate (other sulfate precursors are not considered) on a short timescale, aqSOA mass yields from in-cloud VOC oxidation are much lower ( %), as in each oxidation step volatile compounds are formed (e.g., HCHO, CO2) that do not contribute to aqSOA (Ervens et al., 2008) but evaporate from the droplets.
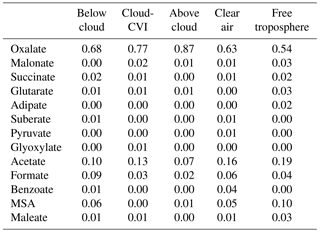
The absolute mass increase seems quite large (0.2–10 µg m−3 depending on air mass type). However, it should be remembered that our predictions might exaggerate real conditions as neither physical (deposition) nor chemical (oxidation of organics to volatile compounds in cloud water) sinks for aerosol mass are included in our model in order to tease out the clearest signature of aqueous-phase processing possible. The increase in sulfate in clouds has been predicted in many previous studies for a variety of air masses (e.g., Table 1 in Ervens, 2015). Cloud residues at Mount Tai, China, exhibited large fractions of sulfate and water-soluble organics (Li et al., 2011). In the latter study, both sulfate and aqSOA were found internally mixed in the droplet mode, which suggests that both were formed in clouds. Increasing organic mass with altitude in clouds (which can be considered analogous to processing time) has been observed in several previous field studies focused on marine and urban air masses (e.g., Sorooshian et al., 2007a; Wonaschuetz et al., 2012). Table 2 compares the distribution of individual organic acids to the total organic acid content in various air masses, as measured by the PILS-IC method on the CIRPAS Twin Otter during the 2006 GoMACCS campaign. The comparison of the oxalic acid contributions below, in, and above cloud suggests that these three air masses were connected and mass was transported vertically while it was processed in cloud, leading to an increasing oxalate fraction. In contrast to the GoMACCS and other measurements, no clear aqSOA signature was observed in an SOA-rich biogenic region (Wagner et al., 2015). Often, air above clouds might not be cloud-processed but transported horizontally, which may lead to erroneous interpretation of the role of cloud processing (Sect. 4). While the increase in oxalate clearly points to in-cloud mass formation due to aqueous-phase processes, in-cloud aerosol measurements are likely associated with some uncertainties, in particular due to the difficulties of sampling cloud droplets vs. interstitial particles, and issues associated with cloud droplet impacts on inlets (e.g., Murphy et al., 2004).
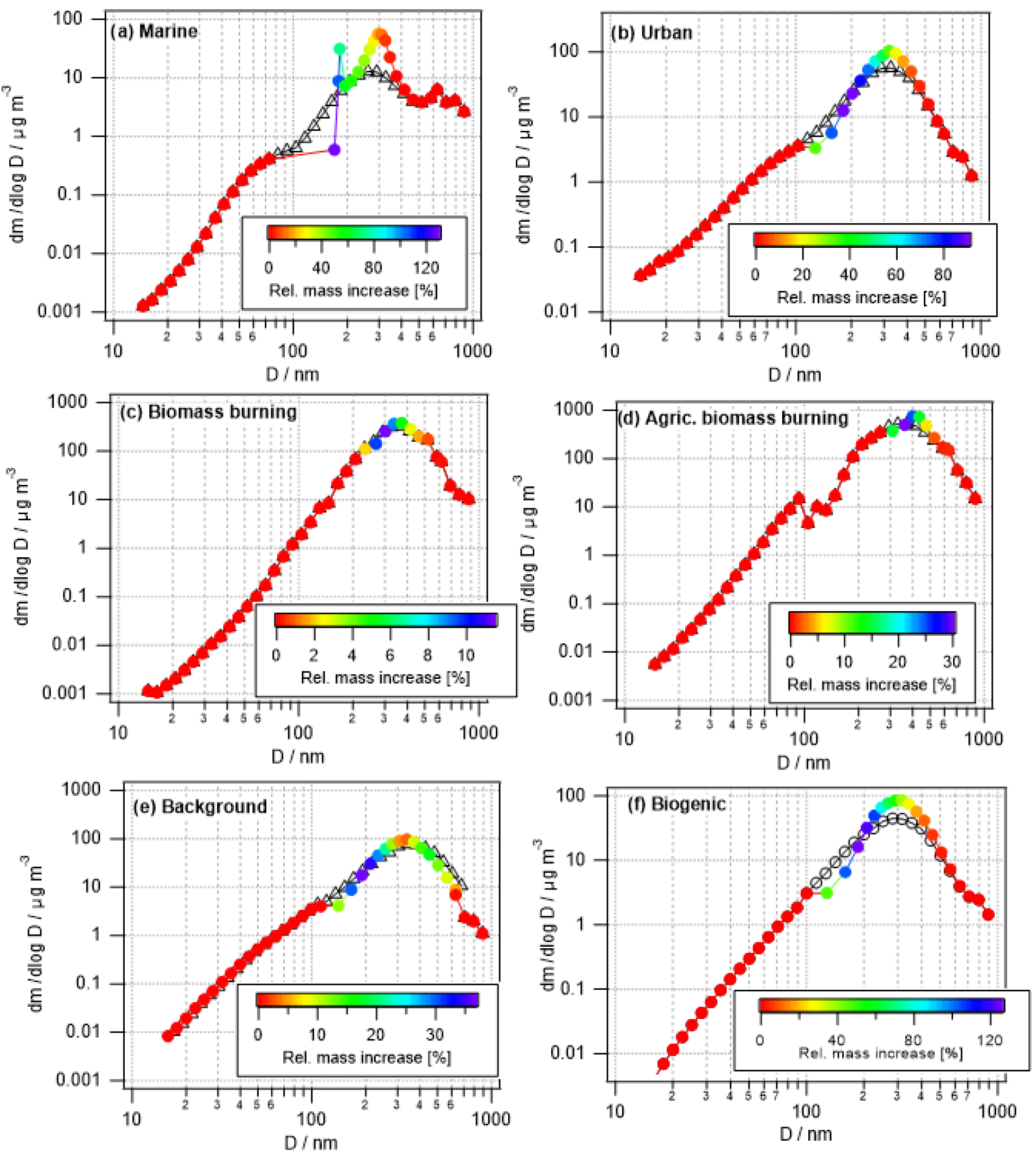
Figure 2Measured initial (black) and predicted cloud-processed (colored) mass distributions of aerosol particles in six air masses as identified during SEAC4RS. Color coding refers to the relative mass increase (Eq. 2).
3.1.2 Changes in bulk hygroscopicity
The initial κ is in all air masses lower than that of sulfate () and even lower than that assumed for aqSOA (κaqSOA=0.5) (Sect. 2.2.1 and Table 1). The predicted κ values (blue lines in Fig. 1, first right axis) increase immediately due to the rapid sulfate addition and then drop when aqSOA mass is added to the processed particles, corresponding to the changes in absolute masses and mass ratios (Sect. 3.1.1). It should be remembered that the simulations are set up such that they represent the decay of precursor gases without any replenishment during the simulation time. If mixing of additional gas-phase precursors occurred continuously into the cloud, the changes in κ might not be as temporally resolved as predicted in Fig. 1. In such a case, the distinct temporal changes in κ due to sulfate and aqSOA addition, respectively, might be more obscured, e.g., when other secondary organics are added simultaneously to the particles. The timescales of aqSOA formation might be different for other aqSOA formation processes, i.e., those that are not initiated by the OH radical, which would also change the slope of the mass and κ evolution in Fig. 1. In addition, in the ambient atmosphere the predicted trends in κ could be additionally obscured due to mixing with other air masses into the cloud. Overall, the changes in κ in the biomass burning cases (Fig. 2c and d) are overall very small (Δκ≤0.1) except during the sharp peak at the beginning when sulfate is added.
Similar trends in an increase in the hygroscopicity parameter κ have been observed previously; for example, Henning et al. (2014) demonstrated an increase of in a forest site in Thuringia (Germany). However, in a marine cloud no distinct difference in the hygroscopicity of particles due to cloud processing was detected (Swietlicki et al., 1999) since the preexisting particles consisted mostly of ammonium sulfate and the added mass was sulfate. In the Amazon during the wet season, the increase in particle size and hygroscopicity was small due to atmospheric processing but still more significant than in the dry season (Rissler et al., 2006). In that case, mostly organic material was added to the preexisting particles. These trends in the various scenarios are in qualitative agreement with the findings from our model studies in which the largest change in hygroscopicity is predicted to occur in biogenic areas.
Table 2Percent contributions of individual organic acids to total organic acid mass in aerosols in different regions of the lower troposphere during the 2006 GoMACCS campaign based in Houston, Texas (Sorooshian et al., 2007b). “Cloud-CVI” corresponds to droplet residual particle measurements, “clear air” is in the boundary layer in cloud-free conditions, and “free troposphere” is determined by meteorological sounding profiles.
3.1.3 Changes in bulk O ∕ C ratio
The oxygen-to-carbon (O ∕ C) ratio only reflects the composition of the organic portion of the aerosol (OA). The orange lines in Fig. 1 (second right axis) show the predicted bulk O ∕ C ratios as calculated based on predicted aqSOA formation (Sect. 2). In all cases, the O ∕ C ratio increases close to the beginning of the simulations, with the lines following the same trends as predicted for the aqSOA mass increase (Sect. 3.1.1). The increase in the O ∕ C ratio continues in the second passage of the parcel through the cloud. This increase is caused both by the oxidation of dissolved VOCs and the further oxidation of aqSOA products that have been formed in the cloud water (e.g., oxidation of glyoxylic to oxalic acid). Similar to the findings for the mass increase and κ, the largest increase in the O ∕ C ratio can be seen for the urban, marine, and biogenic cases with Δ(O ∕ C) ≤ 0.4. For the biomass burning cases, the changes are rather subtle with an increase of Δ(O ∕ C) ≤ 0.1. It should be noted that this change might even represent an overestimate as we neglect numerous physical and chemical processes that could lead to a weaker increase in Δ(O ∕ C) or even to its decrease. Such processes include non-oxidative reactions that lead to aqSOA, which will produce less-oxygenated aerosol. It was discussed that IEPOX might contribute significantly to aqSOA in wet aerosol (Budisulistiorini et al., 2017) or non-photochemical processes in fog (Sullivan et al., 2016). Wet deposition or further oxidation of oxygenated and highly soluble aqSOA constituents might lead to a removal of water-soluble organics and thus to an overall decrease in the bulk O ∕ C ratio.
There are not many studies that focus on modifications of the O ∕ C ratio in the aqueous phase. Gilardoni et al. (2016) found an increase in the O ∕ C ratio of Δ(O ∕ C) ∼ 0.2 upon fog processing in a biomass burning plume. The increase upon processing was similar to the predicted one with Δ(O ∕ C) ∼ 0.2 (Fig. 1c and d). During the Whistler Aerosol and Cloud Study (WACS 2010), Lee et al. (2012) found that in a biogenically influenced background site, the O ∕ C ratio was clearly enhanced upon cloud processing, similar to the O ∕ C ranges as shown in Fig. 1e and f. However, they pointed out uncertainties in translating the f44 signal from unit-mass-resolution AMS measurements into the O ∕ C ratio, as the relationships determined by Aiken et al. (2008) and Canagaratna et al. (2015) might not be generally valid for all species and ranges of O ∕ C ratios.
3.1.4 The ratio of potential added mass from precursor gases to initial aerosol mass
In order to significantly change the bulk properties of initial aerosol by additional sulfate and aqSOA mass, the newly formed mass has to comprise a substantial fraction of the total mass so that the volume-based κ and the molecular-based O ∕ C ratio are significantly changed. Based on this idea, we calculate an initial potential added mass due to aqueous processing of precursor-to-preexisting mass ratio for each air mass:
where [SO2] is the mass concentration of SO2 [µg m−3], the factor of 98∕64 accounts for the mass difference of H2SO4 vs. SO2, and [aqSOA prec] is the total mass concentration [µg m−3] of all VOCs that may act as precursors for aqSOA (Table S1). These precursors include isoprene, methyl vinyl ketone, methacrolein, toluene, xylene, and ethylene. The numerator is the potentially added mass, i.e., the mass that would be added to the initial mass m0 [µg m−3] due to cloud processing if the precursors were completely consumed. The VOC mixing ratio is multiplied with an approximate effective mass yield factor YaqSOA in order to account for the facts that (i) only part of the VOC mass will be converted into aqSOA and (ii) aqSOA species can be further oxidized to CO2; thus, unlike sulfate, it is not a preserved mass. An effective yield of 10 % is assumed in the remainder of this study based on model studies that have shown that the aqSOA yield from isoprene is at most 10 %, depending on cloud and NOx conditions (Ervens et al., 2008). This previous model study might not have included all aqSOA precursors and formation pathways, so the mass yields reported there might represent an underestimate. If more updated information on yields for specific precursors becomes available, the value of Y used in Eq. (1) can be updated accordingly. This parameter might be higher for aqSOA formation from oxygenated compounds but is not a conservative value over time due to the possibly efficient decrease in aqSOA products due to oxidation. Therefore, targeted experiments should be performed in order to refine this value for a variety of (oxygenated) VOCs.
Table 3Mass ratios of potential aerosol mass and initial aerosol mass m0 from SO2 (), aqSOA precursors (RaqSOA), and total mass ratios (Rtot) (Eq. 1).

In Table 3, the Rtot values for all six air masses are listed. The highest value (Rtot=0.97) is shown for the marine scenario, followed by the values for the biogenic (Rtot=0.31) and urban (Rtot=0.22) cases. The lowest values are shown for the biomass burning cases (Rtot=0.04 and Rtot=0.14 for wildfire and agricultural burning, respectively) with a similar value for the background case (R=0.07). While the VOC mixing ratios in the biomass burning air masses are relatively high, the inefficient conversion into aerosol mass (compared to sulfate) and the high preexisting aerosol concentrations lead to an overall low Rtot value. This is in agreement with previous studies that showed that in biomass burning plumes large fractions of organic material reside in the particle phase compared to the gas phase (Heald et al., 2008; Cubison et al., 2011). Even though the agricultural biomass burning air mass contains the highest SO2 mixing ratio among all six air masses, the added sulfate is not sufficient to alter the properties of the initial aerosol mass m0, which is also the highest among all cases (Table 1). All values are higher than the RaqSOA values for the same air mass. This trend suggests that generally the addition of sulfate to an initial aerosol population might more efficiently change the initial aerosol population than the addition of aqSOA. The contribution of aerosol processing by organics is highest in the biomass burning cases in which RaqSOA is 20 % and 6 % of Rtot, respectively. The trends in Table 3 give some guidance for which air masses a cloud-processing signature may be expected: the higher the ratio R, the more susceptible the preexisting aerosol mass is to be substantially enhanced by in-cloud mass formation.
Air masses in the southeastern US are usually categorized as biogenic, but only little evidence of cloud processing was observed, which was mostly ascribed to sulfate addition (Wagner et al., 2015). Applying the concept of the ratio R to these air masses, it can be shown that the initial mass of ∼10 µg m−3 was relatively high, whereas the precursor concentrations were comparably low ([SO2] ∼ 0.3 ppb, [isoprene] ∼ 1.5 ppb, [aromatics] < 1 ppb) (Hu et al., 2015; Wagner et al., 2015), resulting in , RaqSOA=0.02, and Rtot=0.1, respectively. In contrast, above Houston, cloud processing was observed (Wonaschuetz et al., 2012). The air masses there contained lower aerosol mass but higher precursors (m0∼5 µg m−3; [SO2] = 1.5 ppb; [aromatics] ∼ 8 ppb, [isoprene + MVK + MACR] ∼ 4 ppb), yielding , RaqSOA=0.8, and Rtot=1.6, respectively, even higher than the highest value in Table 3. In the experiments described by Wagner et al. (2015) and Wonaschuetz et al. (2012) similar particle size ranges (<50 up to 800 nm) were measured. If a narrower range of particle sizes were taken into account (e.g., only SMPS data up to D=316 nm; see Sect. 2.1.1), the denominator in Eq. (1) would be smaller and consequently the resulting R larger. Thus, by comparing R values from different experiments, it should be ensured that measurements of similar particle size ranges are considered.
3.2 Changes in size-resolved parameters
Bulk properties do not allow for any detailed conclusions about the effect of cloud processing on individual particles and thus on their resulting composition and size. While being more complex both in terms of measurements and model simulations, only size-resolved measurements and model studies permit such conclusions and are discussed subsequently. Chemical processes in cloud droplets might significantly change the properties of the droplet residuals. Depending on the activated fraction, this modification might change the bulk properties of the total aerosol population to different extents. It has been discussed by Ervens et al. (2014) that in small droplets, i.e., in particles with a relatively high surface-to-volume ratio, more efficient aqSOA formation can be expected as oxidation rates might be enhanced due to efficient oxidant and precursor uptake. The size of cloud droplets is not a strong function of the size and/or composition of the CCN but it is mostly determined by the growth history and competition for water vapor within the cloud. As size-resolved composition measurements from SEAC4RS are very noisy, any conclusions based on these data might be inconclusive.
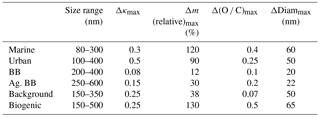
Table 4Summary of model results describing the aerosol properties upon cloud processing; Δκ, Δm, Δ(O ∕ C), and ΔDiam values denote the largest predicted change as shown in Figs. 3, 5, and S1, respectively.
3.2.1 Size-resolved mass increase
Many studies have discussed the formation of a droplet mode upon cloud processing, which is caused by mass addition to activated particles only. Such predicted evolution of the aerosol size distribution is shown for the six air masses in Fig. 2. Processed size distributions in Fig. 2 are overlaid on the initial size distributions (black symbols) and color coded by the relative mass increase, i.e.,
This relative mass increase is similar to the parameter R as defined in Eq. (1) in the sense that it shows the resulting mass increase upon complete processing of the precursors after cloud processing. In agreement with the trends as identified for the bulk masses (Sect. 3.1.1), the two biomass burning scenarios show the smallest relative mass increase with the largest values of ∼12 % and 30 %. In all other cases, the mass of some particles might double (relative mass increase ∼100 %). Table 4 summarizes the maximum relative mass increase for individual sizes, together with the particle size range that is mostly affected by cloud processing. In the two biomass burning scenarios, only particles with diameters > ∼250 nm show any processing. Due to the high particle number concentration in these cases, the maximum cloud supersaturation is suppressed because the numerous particles act as an efficient condensation sink for water vapor. Consequently, only a small fraction of the aerosol population is activated into cloud droplets. This small activated fraction explains the rather small changes in bulk κ and the O ∕ C ratio (Fig. 1). Cloud processing often leads to the separation of unactivated and activated particles within the aerosol size distribution due to the “Hoppel minimum”. In Fig. 2a, b, e, and f the particles around 100 nm are affected most strongly and also show some sign of this separation into a droplet mode. Thus, it is predicted that cloud processing leads to a shift to larger particle sizes and a narrowing of the size distribution (Feingold and Kreidenweis, 2000). In the biomass burning cases (Fig. 2c and d), the most affected particles are near the maximum of the main size mode and the shift to larger sizes is not as clear.
The absolute mass increase is in all cases several tens to hundreds of ng m−3 in the individual size classes that are separated by d(log bin width) ∼0.05 (Fig. S1 in the Supplement). Whereas this translates into doubling of particle mass in the cleaner air masses, the relative mass increase in the biomass burning cases is much smaller owing to the initial high particle loading, i.e., low R values (Eq. 1). Tracers of aqueous-phase processing have been identified, e.g., by Cook et al. (2017), in cloud samples that were affected by biomass burning plumes. However, such analyses do not reveal the extent to which the total aerosol population might have been altered in the cloud. Our results show that the modified particles only represent a small fraction of the total aerosol population.
Many previous studies have identified a droplet mode upon cloud processing. An overview article has been given by Eck et al. (2012). In Fig. 3, exemplary results of cloud processing in urban, marine, and remote air masses are shown. The relative mass increase in sulfate and oxalate is examined as a tracer for chemical cloud processing. For two sites, data were compared between a moist and dry period; more specifically, data were compared between monsoon months (July–September) and a dry period (June) for the urban area in Tucson, Arizona, and also between a monsoon period and a drier period in November for a remote site in Hayden, Arizona. Finally, data were compared between a fire period and a non-fire period in Marina, California, during the summer when there is persistent cloud coverage. A consistent feature for the two Arizona sites was that a peak in the relative mass increase (monsoon versus other periods) for sulfate and oxalate was between 0.32 and 0.56 µm, which is consistent with the droplet mode. While the fire and non-fire comparison does not contrast periods with varying moisture levels, it contrasts periods with varying amounts of precursors that still reveal the importance of aqueous processing in terms of greater mass production when precursors are more plentiful. The relative mass increase for the comparison of fire and non-fire conditions in the coastal–marine area with persistent cloud coverage was highest between 0.56 and 1 µm, in agreement with the larger critical diameter (i.e., smaller activated fraction) in Fig. 2. Analysis of fog-processed aerosol in Fresno, California, also revealed a clear signature in terms of size distribution and composition changes (Ge et al., 2012). In this latter study, both sulfate and aqSOA accumulated at particle sizes above ∼200 nm upon cloud processing. Similarly, tracers of aqSOA formation were detected in fog in the Po Valley (Gilardoni et al., 2014). Cloud-processed particles were observed in many targeted field experiments. For example, increased mass in large particles was detected during the HCCT experiment in which cloud-processed aerosol was analyzed (Henning et al., 2014).
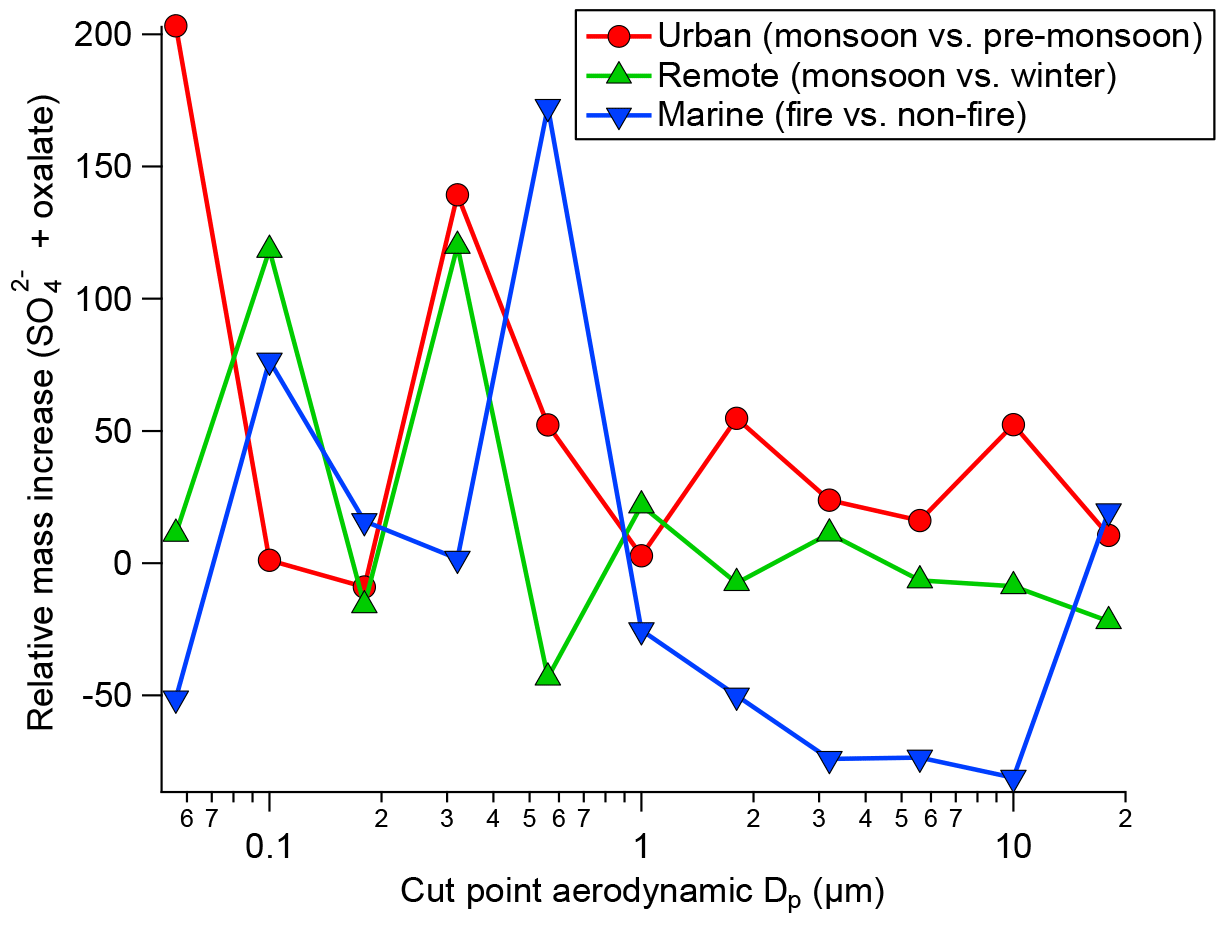
Figure 3Summary of the relative mass concentration increase for sulfate plus oxalate as a function of dry particle size for three scenarios: monsoon–pre-monsoon changes in an urban area (inner city Tucson, Arizona), monsoon–winter changes in a remote area in central Arizona (Hayden, Arizona), and fire–non-fire changes in a coastal–marine area with persistent cloud coverage in July–August (Marina, California).
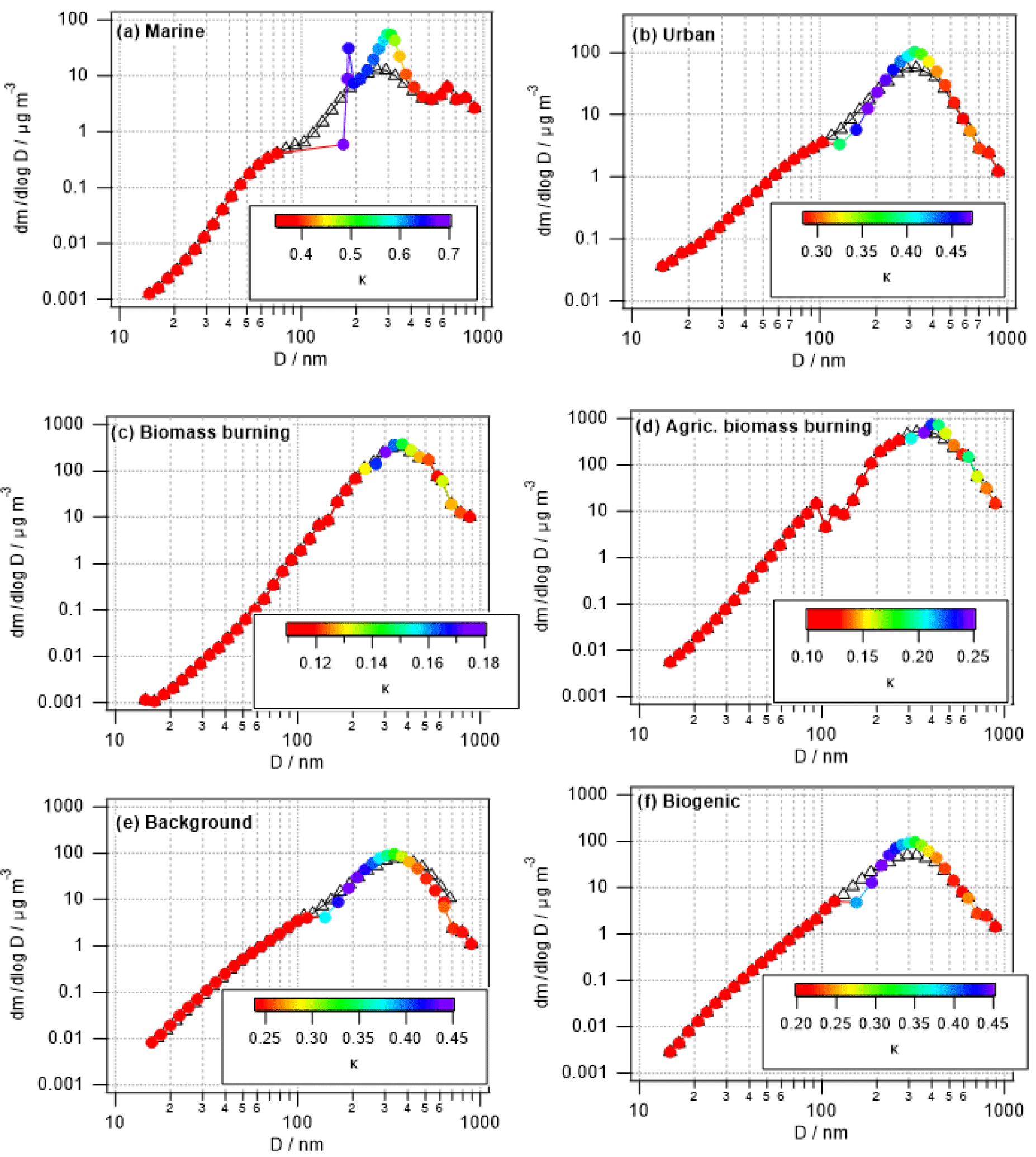
3.2.2 Changes in size-resolved hygroscopicity (κ)
Figure 4 shows the same parcel model results as in Fig. 3, but color coded by κ instead of the relative mass increase. Unlike Fig. 1 that shows the time evolution of κ, in Fig. 4 the model-predicted values after 1 h of processing are shown. Conclusions are similar to those in Sect. 3.1.2 as the smallest changes in κ are seen in the biomass burning cases in which only a small fraction of the aerosol population is processed (Table 4) and the high initial mass is not increased substantially by the addition of sulfate and aqSOA.
The hygroscopic growth factor g(RH) is a measure of particle hygroscopicity. HTDMA measurements of initial and cloud-processed aerosol can give evidence of cloud processing as more sulfate and aqSOA mass is added to larger particle sizes, enhancing the hygroscopicity of previously less hygroscopic particles. Figure 5 shows an example of a size distribution of aerosol hygroscopic growth (shown as κ) atop Mount Lemmon in Arizona in relation to chemical mass fractions of selected water-soluble species including inorganic and organic acid ions. The size ranges with the highest κ values exhibit the highest mass fractions of sulfate. The results show how it is difficult to isolate the impact of aqSOA. Although the contribution of oxalate to the total organic acid mass is highest for diameters in the range of 0.32–0.55 µm (41 %), that same stage exhibited the highest sulfate mass fraction (73 %) and inorganic mass fraction (88 %). This may have trumped the smaller effect of a change in the functionality of the organic fraction of the aerosol. In a separate study in the marine boundary layer off the California coast, Hersey et al. (2009) measured reduced size-resolved aerosol hygroscopic growth factors above the stratocumulus cloud top compared to the sub-cloud region as a result of enhanced bulk aerosol organic mass fractions above cloud. However, the air masses were different below and above cloud, with continental free tropospheric air enriched with organics residing above cloud top and more inorganic-rich aerosol below cloud bases. This observation demonstrates that comparisons of below- and above-cloud air should be performed carefully as differences in aerosol properties are not necessarily due to cloud processing. A recent study comparing inflow and outflow aerosol from deep convective storms revealed that although size-resolved κ values may not have exhibited a significant enhancement in the anvil outflows (and sometimes reduced values), the signature of aqueous processing could have been missed as a result of lateral entrainment and the mixing of less hygroscopic aerosol with the processed aerosol that entered at the storm cloud base (Sorooshian et al., 2017a); this might also be a consequence of different scavenging efficiencies of sulfate and organics, respectively (Yang et al., 2015). A case was profiled in which biomass burning aerosol with low hygroscopicity entrained into a storm and resulted in a lower mean κ value in the outflow compared to the inflow. An altitude-dependent entrainment model was applied to that analysis to show that the measured κ value exceeded that predicted for the outflow, revealing that a process, most likely aqueous processing, helped increase the hygroscopicity of the aerosol.
3.2.3 Changes in size-resolved O ∕ C ratio
The same figure as for mass increase and κ change is once more reproduced in Fig. S2 showing the change in the O ∕ C ratio throughout the aerosol distribution upon chemical cloud processing. In the marine case (Fig. S2a), the O ∕ C ratio is predicted to increase by about 0.5 units in the activated fraction. As the smallest activated particles are smaller than 100 nm and thus the activated fraction is substantial, this change in O ∕ C ratio is also reflected in the bulk O ∕ C ratio in Fig. 1 and translates into a high RaqSOA value (Eq. 1). Changes in the O ∕ C ratio are smaller in the urban case, as the strong increase in mass is mostly due to sulfate, which does not affect the O ∕ C ratio. Even though the organic content is higher in the biomass burning cases, the change in the O ∕ C ratio is smallest compared to the other cases due to the high initial mass. In biomass burning scenarios, cloud water might contain highly oxidized organics and thus a high O ∕ C ratio (Gilardoni et al., 2016; Cook et al., 2017). However, as the dissolved mass only comprises a small fraction of the total particle number, this oxidation might not affect bulk aerosol properties to a large extent.
Fog water analysis in the Indo-Gangetic plains revealed higher O ∕ C in small fog droplets with a difference of Δ(O ∕ C) ∼ 0.2 between small and large fog droplets (Chakraborty et al., 2016). Model studies explained this difference with the larger surface-to-volume ratio of smaller droplets, which allows for more efficient uptake of oxidants such as OH and aqSOA precursors from the gas phase (Ervens et al., 2014). As OH is assumed to be (one of) the most efficient oxidants of organics in cloud droplets, the resulting higher OH concentration leads to relatively more aqSOA and a higher O ∕ C ratio in small droplets. The fog study by Chakraborty et al. (2016) might not be directly comparable to the model results in Fig. S2, which contrasts activated and non-activated particles upon cloud processing. However, the fog studies show that drop size plays an important role for aqSOA formation. Given the high cloud drop number concentration in the biomass burning cases in the SEAC4RS biomass burning scenarios (a few thousand cm−3 as opposed to a few hundred cm−3 or less in the other cases), the smallest cloud droplets might be present in these cases. Thus, it can be expected that the aqSOA formation rates in such cloud droplets are highest (Ervens et al., 2014) due to the favorable total surface-to-volume ratio (McVay and Ervens, 2017). In fact, in both biomass burning scenarios, several tens of µg m−3 of organic mass are added (Fig. 1c and d) in agreement with observations of efficient aqSOA formation in cloud-processed biomass burning plumes (Gilardoni et al., 2016). However, this mass is not sufficient to change the properties of the preexisting aerosol mass (Sect. 3.1.4).
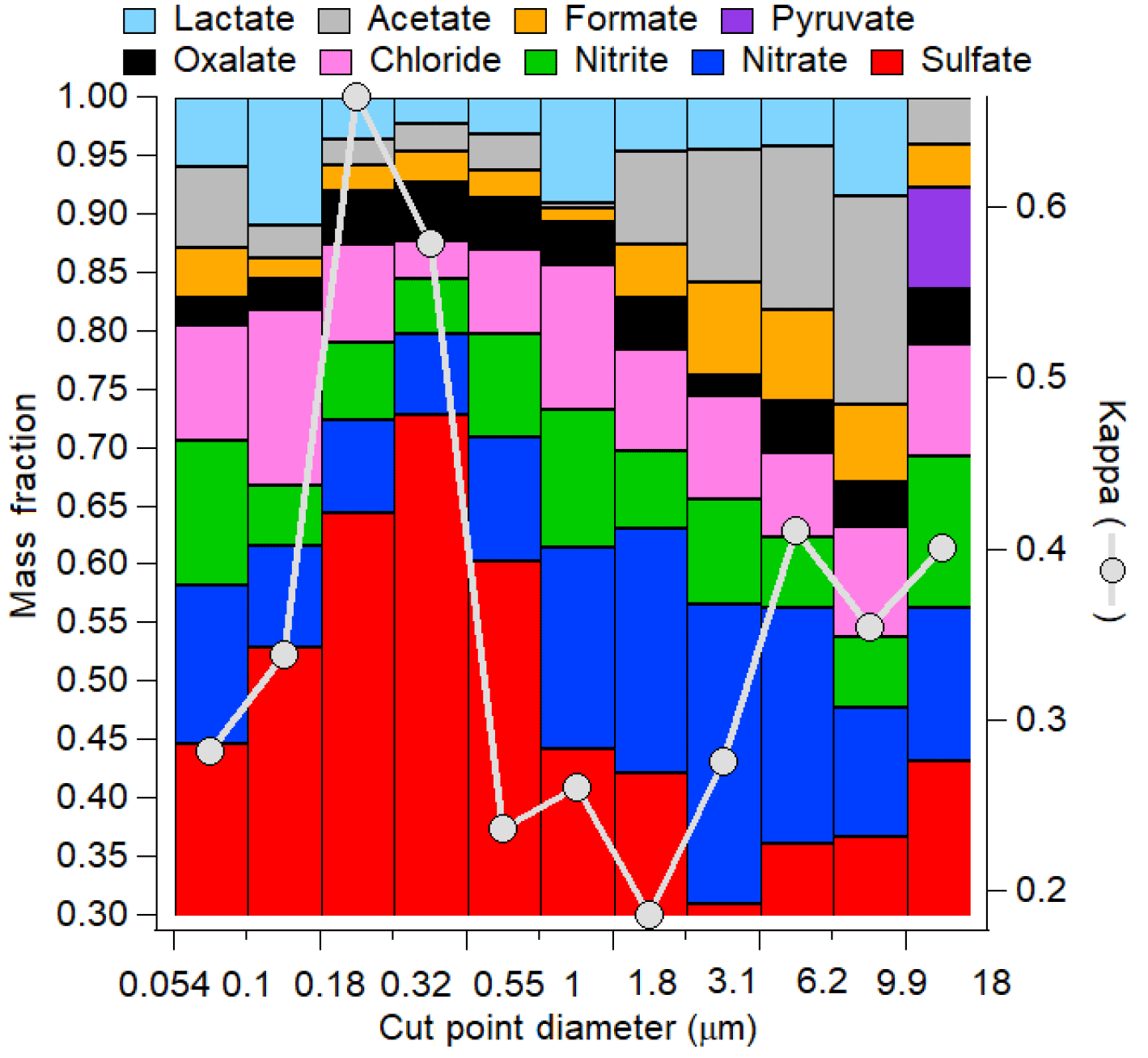
Figure 5Size-resolved aerosol hygroscopic growth (as κ) and chemical mass fractions as a function of dry particle diameter at a mountaintop site (Mount Lemmon, Arizona; February 2010).
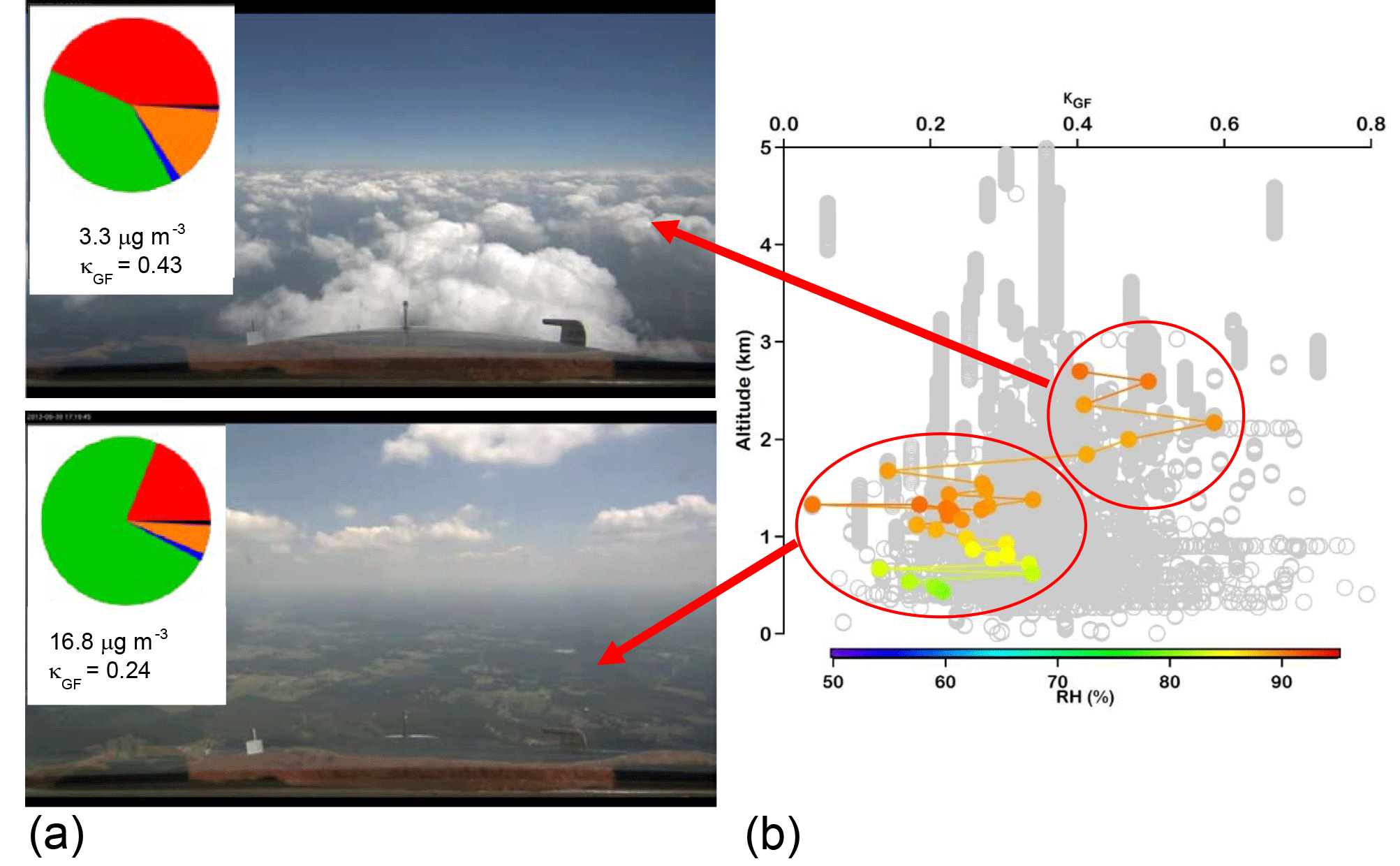
Figure 6(a) Photographs taken from the NASA DC-8 showing where the aircraft was relative to clouds during a SEAC4RS flight on 30 August 2013. The pie charts correspond to AMS chemical mass fractions for non-refractory aerosol (green: organic; red: sulfate; orange: ammonium; blue: nitrate) and for black carbon (in black) as measured by the HD-SP2 instrument. The average total submicrometer mass concentrations and growth-factor-derived κ values are shown below the pies for above-cloud-base and sub-cloud-base sampling. (b) Vertical profile of size-resolved GF-derived κ for particles with dry diameters between 180 and 400 nm with gray being all points during the flight and the colored points being for the specific measurements near the cloud field. While the RH values of the humidified channel of the DASH-SP are shown for each measurement by the cloud field, κ values are shown to allow for a fair comparison regardless of the humidified RH.
The previous model analysis suggests that detecting unambiguous evidence of a particle having undergone aqueous processing compared to clear-air processing is challenging in the ambient atmosphere. Evidence of cloud processing strongly depends on the air mass and its history. This is in sharp contrast to controlled laboratory experiments in which conditions can be optimized to detect an aqueous signature by an increase in O ∕ C ratio, κ, or the formation of tracer compounds (Lim et al., 2010; Lee et al., 2012). In the ambient atmosphere, several interferences might obscure the signature and/or lead to false conclusions.
- i.
Studies with vertically resolved measurements below, inside, and above cloud have an added complication that clouds can be decoupled from a significant portion of the sub-cloud layer (Wang et al., 2016), or there can be a very sharp temperature inversion immediately above the cloud top that leads to a different air mass above the tops associated with the free troposphere (Dadashazar et al., 2018). Thus, continuity in meteorological parameters, such as temperature and/or relative humidity, should be carefully taken into account before conclusions are drawn on cloud processing. Aircraft that fly even within 10 m above cloud top in the entrainment interface layer, such as in subtropical stratocumulus regions, still have influence from free tropospheric air masses (Dadashazar et al., 2018). Figure 6 demonstrates an example of an airborne experiment in which particles with higher κ were found above cloud than below and in cloud. However, this trend is coincidental as the air mass above did not originate from the cloud. In other studies, such an increase in κ was correctly attributed to sulfate addition due to chemical cloud processing (Shingler et al., 2016). Similar mixing of air masses from the free troposphere and the boundary layer was observed within the inter-cloud layer in the southeastern US (Wagner et al., 2015).
- ii.
While likely the majority of atmospheric oxalate is formed in clouds, it has been shown that oxalate might have additional sources, such as biomass burning (Narukawa et al., 1999; Falkovich et al., 2005; Zhang et al., 2017) or direct emissions (Huang and Yu, 2007). In addition, oxalate and other aqSOA compounds can become further oxidized in clouds, in particular in the presence of iron (Zuo and Hoigné, 1992, 1994; Furukawa and Takahashi, 2011; Kawamura et al., 2012; Sorooshian et al., 2013; Weller et al., 2013). Oxalic acid and other (weaker) organic acids might evaporate from acidic aerosols (Häkkinen et al., 2014; Nah et al., 2018). Thus, the lack of a clear oxalate increase is not necessarily indicative of processing in cloud-free air only. Finding correlations between aqueous organic tracer species and sulfate (Yu et al., 2005; Huang et al., 2006) does not necessarily demonstrate causality. Covariance alone (or the lack thereof) of tracer species (e.g., oxalate) with their aqueous precursors such as glyoxal or glyoxylate (e.g., Sorooshian et al., 2006a; Rinaldi et al., 2011) is not a sufficient indicator to conclude that chemical cloud processing is taking place.
- iii.
Aerosol composition can be altered during sampling due to possible fragmentation and volatilization of aqSOA products in counterflow virtual impactor (CVI) inlets used to isolate cloud droplet residual particles (Shingler et al., 2012; Prabhakar et al., 2014).
- iv.
Although still limited in their ability to provide direct proof, reports of size-resolved field measurements may miss out on the full story of an aqueous signature if only the submicrometer size range (e.g., droplet mode) is examined, as aqueous processing can influence the composition of aerosol in the coarse mode (e.g., Deshmukh et al., 2017).
- v.
Not only aqueous-phase processing in clouds but also in deliquesced aerosol particles can lead to aerosol mass (Volkamer et al., 2006; Perri et al., 2010; McNeill et al., 2012; Marais et al., 2016). While this is rather inefficient for sulfate, many laboratory and ambient studies suggested that aqSOA can be efficiently formed in cloud-free high-relative-humidity conditions. Characteristics of this aqSOA mass might be similar to cloud aqSOA (highly oxygenated and functionalized). Correlations of increased aqSOA mass with increasing relative humidity or cloud vs. non-cloud scenarios should be interpreted with caution (Youn et al., 2013). Relative humidity is usually higher in the morning, when pollution and the boundary layer thickness and mixing are different than in the afternoon. Similar to findings for nitrate (Lee et al., 2003), it can be expected that the temperature increase during the day might lead to a decrease in organic mass due to the volatilization of semivolatile SOA.
We have analyzed datasets from the SEAC4RS and other field experiments in order to identify various aerosol properties that might show evidence of aqueous-phase processing of aerosol particles within clouds. In total, three properties, namely mass increase, hygroscopicity (κ), and O ∕ C ratio, were explored by means of model studies for six different air masses (urban, marine, wildfire biomass burning, agricultural biomass burning, biogenic, and background). In order to quantify the susceptibility of an aerosol population to be significantly modified by clouds, we define a mass ratio Rtot, which is the ratio of possible precursor gases for aerosol mass formation (SO2, VOCs), i.e., the potential aerosol mass, and the initial aerosol mass.
Model results suggest that in moderately polluted air masses, such as in urban, marine, and biogenic scenarios, changes in particle mass and properties can be most easily identified. The biomass burning cases show the lowest values of Rtot (0.14 and 0.02, respectively), whereas the marine air mass is characterized by the highest value of Rtot=0.97. In general, Rtot is high in clean scenarios, whereas it is rather low in scenarios with high initial mass (e.g., biomass burning). The mass ratio (ratio of potential sulfate mass to initial aerosol mass) is larger than the values for RaqSOA (potential aqSOA mass versus initial aerosol mass). Thus, sulfate addition likely leads to more aerosol modification during cloud processing than aqSOA addition. Calculating this ratio for previous experiments in different air masses explains why in some cases (e.g., urban) cloud processing was observed, whereas it was not clearly detected in a clean biogenic scenario. It should be cautioned that only measurements of similar particle size ranges should be compared since this range will determine the initial aerosol mass that is used in the calculation of R.
Since the O ∕ C ratio only characterizes the organic aerosol fraction, this ratio might not change significantly due to chemical cloud processing. Other parameters that describe the total aerosol mass, such as mass increase or a change in the hygroscopicity parameter κ, might be more useful to detect a signature of chemical cloud processing.
Previously, the presence of tracer compounds of aqueous-phase processing, such as hydroxymethanesulfonate (Munger et al., 1986), oxalate (e.g., Huang et al., 2006; Sorooshian et al., 2010; Wonaschuetz et al., 2012), oligomers (Mazzoleni et al., 2010), or light-absorbing products (Laskin et al., 2015), has been used to detect the influence of aqueous processing. These compounds usually only comprise a small fraction of the total aerosol mass and thus give only limited quantitative information on the role of aqueous-phase processing in the modification of aerosol. These tracer compounds might have chemical sinks, such as the complexation of oxalate with iron or other trace metals and subsequent photolysis (e.g., Sorooshian et al., 2013) or the decay or oxidation of hydroxymethanesulfonate (Kok et al., 1986; Whiteaker and Prather, 2003). It is likely that water-soluble organic particle constituents (e.g., SOA from sources other than aqueous-phase processes) become oxidized to volatile compounds within cloud droplets and thus the total SOA mass might decrease, whereas aqSOA material is added. Evaporation of organic acids from aerosol particles at low pH might lead to a further decrease in aqSOA. The analysis and interpretation of datasets acquired near clouds should be performed with care, and it should be ensured that air masses in and above clouds are coupled.
Overall, it can be stated that there is no unambiguous answer to the initial question in the title of this study as to whether there is always a clear signature of chemical cloud processing on aerosol. The extent to which aerosol properties are modified by chemical processes in clouds depends primarily on the initial aerosol mass, particle number concentration, and sulfate and aqSOA precursor gases, as quantified by the mass ratio Rtot. In addition, parameters such oxidant levels, cloud water pH, and lifetime will also affect in-cloud mass formation rates. Size-resolved measurements can provide evidence of whether a droplet mode exists that is formed from the addition of cloud-derived mass. Our model results show that this droplet mode can be expected to be mostly comprised of sulfate, whereas a modification of aerosol properties due to aqSOA formation is likely to be small. Our findings are expected to provide guidance on future field and model studies targeting the role of cloud processing in aerosol properties and total ambient aerosol loading. The lack of a signature does not imply that no aqueous-phase processing occurs. In such cases the signature might have been masked by other processes, which include physical and chemical removal processes of aerosol mass.
All data from DC-3 and SEAC4RS are publicly available from the NASA Langley Research Center Atmospheric Science Data Center at https://www-air.larc.nasa.gov/missions/dc3-seac4rs/index.html (DC3, 2018) and https://www-air.larc.nasa.gov/missions/seac4rs/ (SEAC4RS, 2018), https://doi.org/10.5067/Aircraft/SEAC4RS/Aerosol-TraceGas-Cloud, respectively. CIRPAS Twin Otter data can be found elsewhere (Sorooshian et al., 2017b, 2018). Complete model results are available upon request from Barbara Ervens.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-16099-2018-supplement.
AS, AA, TS, EC, and LZ provided the particle physicochemical property data. PCJ and JJ provided the AMS data. AW provided the PTR-MS data. BE performed the model calculations. AS and BE wrote the paper.
The authors declare that they have no conflict of interest.
Armin Sorooshian was funded by Office of Naval Research grants N00014-10-1-0811,
N00014-11-1-0783, N00014-10-1-0200, N00014-04-1-0118, and N00014-16-1-2567
and NASA grants NNX12AC10G and NNX14AP75G. Taylor Shingler was supported with a NASA
Earth and Space Science Fellowship (NNX14AK79H). PTR-MS measurements during
SEAC4RS were supported by the Austrian Federal Ministry for Transport,
Innovation and Technology (BMVIT) through the Austrian Space Applications
Programme (ASAP) of the Austrian Research Promotion Agency (FFG). Tomas Mikoviny
is acknowledged for support with data collection and analysis. Pedro Campuzano-Jost
and Jose L. Jimenez were supported by NASA grants NNX12AC03G and NNX15AT96G. The authors
acknowledge Andreas Beyersdorf, Anne E. Perring, and Joshua P. Schwarz for
SEAC4RS data used, and they acknowledge several SEAC4RS participants
for providing gas-phase data (John Crounse, Alex Teng, NOyO3
team, Greg Huey, David Tanner, Xiaoxi Liu, and Thomas Hanisco).
Edited by: Manabu Shiraiwa
Reviewed by: two anonymous referees
Agarwal, S., Aggarwal, S. G., Okuzawa, K., and Kawamura, K.: Size distributions of dicarboxylic acids, ketoacids, α-dicarbonyls, sugars, WSOC, OC, EC and inorganic ions in atmospheric particles over Northern Japan: implication for long-range transport of Siberian biomass burning and East Asian polluted aerosols, Atmos. Chem. Phys., 10, 5839–5858, https://doi.org/10.5194/acp-10-5839-2010, 2010.
Aiken, A. C., Decarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: O ∕ C and OM ∕ OC ratios of primary, secondary and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Alexander, B., Park, R. J., Jacob, D. J., Li, Q. B., Yantosca, R. M., Savarino, J., Lee, C. C. W., and Thiemens, M. H.: Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes, J. Geophys. Res.-Atmos., 110, D10307, https://doi.org/10.1029/2004JD005659, 2005.
Barth, M. C., Rasch, P. J., Kiehl, J. T., Benkovitz, C. M., and Schwartz, S. E.: Sulfur chemistry in the National Center for Atmospheric Research Community Climate Model: Description, evaluation, features, and sensitivity to aqueous chemistry, J. Geophys. Res.-Atmos., 105, 1387–1415, https://doi.org/10.1029/1999jd900773, 2000.
Braun, R. A., Dadashazar, H., MacDonald, A. B., Aldhaif, A. M., Maudlin, L. C., Crosbie, E., Aghdam, M. A., Hossein Mardi, A., and Sorooshian, A.: Impact of Wildfire Emissions on Chloride and Bromide Depletion in Marine Aerosol Particles, Environ. Sci. Technol., 51, 9013–9021, https://doi.org/10.1021/acs.est.7b02039, 2017.
Budisulistiorini, S. H., Nenes, A., Carlton, A. G., Surratt, J. D., McNeill, V. F., and Pye, H. O. T.: Simulating Aqueous-Phase Isoprene-Epoxydiol (IEPOX) Secondary Organic Aerosol Production During the 2013 Southern Oxidant and Aerosol Study (SOAS), Environ. Sci. Technol., 51, 5026–5034, https://doi.org/10.1021/acs.est.6b05750, 2017.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the Aerodyne Aerosol Mass Spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Chakraborty, A., Ervens, B., Gupta, T., and Tripathi, S. N.: Characterization of organic residues of size-resolved fog droplets and their atmospheric implications, J. Geophys. Res.-Atmos., 121, 4317–4332, https://doi.org/10.1002/2015JD024508, 2016.
Chhabra, P. S., Ng, N. L., Canagaratna, M. R., Corrigan, A. L., Russell, L. M., Worsnop, D. R., Flagan, R. C., and Seinfeld, J. H.: Elemental composition and oxidation of chamber organic aerosol, Atmos. Chem. Phys., 11, 8827–8845, https://doi.org/10.5194/acp-11-8827-2011, 2011.
Choularton, T. W., Colvile, R. N., Bower, K. N., Gallagher, M. W., Wells, M., Beswick, K. M., Arends, B. G., Möls, J. J., Kos, G. P. A., Fuzzi, S., Lind, J. A., Orsi, G., Facchini, M. C., Laj, P., Gieray, R., Wieser, P., Engelhardt, T., Berner, A., Kruisz, C., Möller, D., Acker, K., Wieprecht, W., Lüttke, J., Levsen, K., Bizjak, M., Hansson, H. C., Cederfelt, S. I., Frank, G., Mentes, B., Martinsson, B., Orsini, D., Svenningsson, B., Swietlicki, E., Wiedensohler, A., Noone, K. J., Pahl, S., Winkler, P., Seyffer, E., Helas, G., Jaeschke, W., Georgii, H. W., Wobrock, W., Preiss, M., Maser, R., Schell, D., Dollard, G., Jones, B., Davies, T., Sedlak, D. L., David, M. M., Wendisch, M., Cape, J. N., Hargreaves, K. J., Sutton, M. A., Storeton-West, R. L., Fowler, D., Hallberg, A., Harrison, R. M., and Peak, J. D.: The great dun fell cloud experiment 1993: An overview, Atmos. Environ., 31, 2393–2405, https://doi.org/10.1016/S1352-2310(96)00316-0, 1997.
Cook, R. D., Lin, Y. H., Peng, Z., Boone, E., Chu, R. K., Dukett, J. E., Gunsch, M. J., Zhang, W., Tolic, N., Laskin, A., and Pratt, K. A.: Biogenic, urban, and wildfire influences on the molecular composition of dissolved organic compounds in cloud water, Atmos. Chem. Phys., 17, 15167–15180, https://doi.org/10.5194/acp-17-15167-2017, 2017.
Crahan, K. K., Hegg, D., Covert, D. S., and Jonsson, H.: An exploration of aqueous oxalic acid production in the coastal marine atmosphere, Atmos. Environ., 38, 3757–3764, https://doi.org/10.1016/j.atmosenv.2004.04.009, 2004.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Dadashazar, H., Braun, R. A., Crosbie, E., Chuang, P. Y., Woods, R. K., Jonsson, H. H., and Sorooshian, A.: Aerosol characteristics in the entrainment interface layer in relation to the marine boundary layer and free troposphere, Atmos. Chem. Phys., 18, 1495–1506, https://doi.org/10.5194/acp-18-1495-2018, 2018.
DC3: Deep Convective Clouds & Chemistry, available at: https://www-air.larc.nasa.gov/missions/dc3-seac4rs/index.html, last access: August 2018.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earth's atmosphere using proton-transfer-reaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, https://doi.org/10.1002/mas.20119, 2007.
De Haan, D. O., Hawkins, L. N., Kononenko, J. A., Turley, J. J., Corrigan, A. L., Tolbert, M. A., and Jimenez, J. L.: Formation of Nitrogen-Containing Oligomers by Methylglyoxal and Amines in Simulated Evaporating Cloud Droplets, Environ. Sci. Technol., 45, 984–991, https://doi.org/10.1021/es102933x, 2010.
Deshmukh, D. K., Kawamura, K., Deb, M. K., and Boreddy, S. K. R.: Sources and formation processes of water-soluble dicarboxylic acids, ω-oxocarboxylic acids, α-dicarbonyls, and major ions in summer aerosols from eastern central India, J. Geophys. Res.-Atmos., 122, 3630–3652, https://doi.org/10.1002/2016JD026246, 2017.
Diskin, G. S., Podolske, J. R., Sachse, G. W., and Slate, T. A.: Open-path airborne tunable diode laser hygrometer. Diode Lasers and Applications in Atmospheric Sensing, in: SPIE Proceedings, edited by: Fried, A., International Society for Optical Engineering, Seattle, WA, USA, 196–204, 2002.
Drozd, G., Woo, J., Häkkinen, S. A. K., Nenes, A., and McNeill, V. F.: Inorganic salts interact with oxalic acid in submicron particles to form material with low hygroscopicity and volatility, Atmos. Chem. Phys., 14, 5205–5215, https://doi.org/10.5194/acp-14-5205-2014, 2014.
Dunlea, E. J., DeCarlo, P. F., Aiken, A. C., Kimmel, J. R., Peltier, R. E., Weber, R. J., Tomlinson, J., Collins, D. R., Shinozuka, Y., McNaughton, C. S., Howell, S. G., Clarke, A. D., Emmons, L. K., Apel, E. C., Pfister, G. G., van Donkelaar, A., Martin, R. V., Millet, D. B., Heald, C. L., and Jimenez, J. L.: Evolution of Asian aerosols during transpacific transport in INTEX-B, Atmos. Chem. Phys., 9, 7257–7287, https://doi.org/10.5194/acp-9-7257-2009, 2009.
Eck, T. F., Holben, B. N., Reid, J. S., Giles, D. M., Rivas, M. A., Singh, R. P., Tripathi, S. N., Bruegge, C. J., Platnick, S., Arnold, G. T., Krotkov, N. A., Carn, S. A., Sinyuk, A., Dubovik, O., Arola, A., Schafer, J. S., Artaxo, P., Smirnov, A., Chen, H., and Goloub, P.: Fog- and cloud-induced aerosol modification observed by the Aerosol Robotic Network (AERONET), J. Geophys. Res.-Atmos., 117, D07206, https://doi.org/10.1029/2011jd016839, 2012.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
El-Sayed, M. M. H., Wang, Y., and Hennigan, C. J.: Direct atmospheric evidence for the irreversible formation of aqueous secondary organic aerosol, Geophys. Res. Lett., 42, 5577–5586, https://doi.org/10.1002/2015GL064556, 2015.
Ervens, B.: Modeling the Processing of Aerosol and Trace Gases in Clouds and Fogs, Chem. Rev., 115, 4157–4198, https://doi.org/10.1021/cr5005887, 2015.
Ervens, B., Feingold, G., Frost, G. J., and Kreidenweis, S. M.: A modeling study of aqueous production of dicarboxylic acids, 1. Chemical pathways and speciated organic mass production, J. Geophys. Res.-Atmos., 109, D15205, https://doi.org/10.1029/2003JD004387, 2004.
Ervens, B., Carlton, A. G., Turpin, B. J., Altieri, K. E., Kreidenweis, S. M., and Feingold, G.: Secondary organic aerosol yields from cloud-processing of isoprene oxidation products, Geophys. Res. Lett., 35, L02816, https://doi.org/10.1029/2007gl031828, 2008.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Ervens, B., Sorooshian, A., Lim, Y. B., and Turpin, B. J.: Key parameters controlling OH-initiated formation of secondary organic aerosol in the aqueous phase (aqSOA), J. Geophys. Res.-Atmos., 119, 3997–4016, https://doi.org/10.1002/2013JD021021, 2014.
Falkovich, A. H., Graber, E. R., Schkolnik, G., Rudich, Y., Maenhaut, W., and Artaxo, P.: Low molecular weight organic acids in aerosol particles from Rondonia, Brazil, during the biomass-burning, transition and wet periods, Atmos. Chem. Phys., 5, 781–797, https://doi.org/10.5194/acp-5-781-2005, 2005.
Feingold, G. and Kreidenweis, S.: Does cloud processing of aerosol enhance droplet concentrations?, J. Geophys. Res., 105, 24351–24361, https://doi.org/10.1029/2000JD900369, 2000.
Feingold, G., Kreidenweis, S. M., Stevens, B., and Cotton, W. R.: Numerical simulations of stratocumulus processing of cloud condensation nuclei through collision-coalescence, J. Geophys. Res., 101, 21391–21402, https://doi.org/10.1029/96JD01552, 1996.
Furukawa, T. and Takahashi, Y.: Oxalate metal complexes in aerosol particles: implications for the hygroscopicity of oxalate-containing particles, Atmos. Chem. Phys., 11, 4289–4301, https://doi.org/10.5194/acp-11-4289-2011, 2011.
Ge, X., Zhang, Q., Sun, Y., Ruehl, C. R., and Setyan, A.: Effect of aqueous-phase processing on aerosol chemistry and size distributions in Fresno, California, during wintertime, Environ. Chem., 9, 221–235, https://doi.org/10.1071/EN11168, 2012.
Gilardoni, S., Massoli, P., Giulianelli, L., Rinaldi, M., Paglione, M., Pollini, F., Lanconelli, C., Poluzzi, V., Carbone, S., Hillamo, R., Russell, L. M., Facchini, M. C., and Fuzzi, S.: Fog scavenging of organic and inorganic aerosol in the Po Valley, Atmos. Chem. Phys., 14, 6967–6981, https://doi.org/10.5194/acp-14-6967-2014, 2014.
Gilardoni, S., Massoli, P., Paglione, M., Giulianelli, L., Carbone, C., Rinaldi, M., Decesari, S., Sandrini, S., Costabile, F., Gobbi, G. P., Pietrogrande, M. C., Visentin, M., Scotto, F., Fuzzi, S., and Facchini, M. C.: Direct observation of aqueous secondary organic aerosol from biomass-burning emissions, P. Natl. Acad. Sci. USA, 113, 10013–10018, https://doi.org/10.1073/pnas.1602212113, 2016.
Häkkinen, S. A. K., McNeill, V. F., and Riipinen, I.: Effect of Inorganic Salts on the Volatility of Organic Acids, Environ. Sci. Technol., 48, 13718–13726, https://doi.org/10.1021/es5033103, 2014.
Heald, C. L., Goldstein, A. H., Allan, J. D., Aiken, A. C., Apel, E., Atlas, E. L., Baker, A. K., Bates, T. S., Beyersdorf, A. J., Blake, D. R., Campos, T., Coe, H., Crounse, J. D., DeCarlo, P. F., de Gouw, J. A., Dunlea, E. J., Flocke, F. M., Fried, A., Goldan, P., Griffin, R. J., Herndon, S. C., Holloway, J. S., Holzinger, R., Jimenez, J. L., Junkermann, W., Kuster, W. C., Lewis, A. C., Meinardi, S., Millet, D. B., Onasch, T., Polidori, A., Quinn, P. K., Riemer, D. D., Roberts, J. M., Salcedo, D., Sive, B., Swanson, A. L., Talbot, R., Warneke, C., Weber, R. J., Weibring, P., Wennberg, P. O., Worsnop, D. R., Wittig, A. E., Zhang, R., Zheng, J., and Zheng, W.: Total observed organic carbon (TOOC) in the atmosphere: a synthesis of North American observations, Atmos. Chem. Phys., 8, 2007–2025, https://doi.org/10.5194/acp-8-2007-2008, 2008.
Henning, S., Dieckmann, K., Ignatius, K., Schäfer, M., Zedler, P., Harris, E., Sinha, B., van Pinxteren, D., Mertes, S., Birmili, W., Merkel, M., Wu, Z., Wiedensohler, A., Wex, H., Herrmann, H., and Stratmann, F.: Influence of cloud processing on CCN activation behaviour in the Thuringian Forest, Germany during HCCT-2010, Atmos. Chem. Phys., 14, 7859–7868, https://doi.org/10.5194/acp-14-7859-2014, 2014.
Herrmann, H., Wolke, R., Müller, K., Brüggemann, E., Gnauk, T., Barzaghi, P., Mertes, S., Lehmann, K., Massling, A., Birmili, W., Wiedensohler, A., Wieprecht, W., Acker, K., Jaeschke, W., Kramberger, H., Svrcina, B., Bächmann, K., Collett Jr., J. L., Galgon, D., Schwirn, K., Nowak, A., van Pinxteren, D., Plewka, A., Chemnitzer, R., Rüd, C., Hofmann, D., Tilgner, A., Diehl, K., Heinold, B., Hinneburg, D., Knoth, O., Sehili, A. M., Simmel, M., Wurzler, S., Majdik, Z., Mauersberger, G., and Müller, F.: FEBUKO and MODMEP: Field measurements and modelling of aerosol and cloud multiphase processes, Atmos. Environ., 39, 4169–4183, https://doi.org/10.1016/j.atmosenv.2005.02.004, 2005.
Hersey, S. P., Sorooshian, A., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Aerosol hygroscopicity in the marine atmosphere: a closure study using high-time-resolution, multiple-RH DASH-SP and size-resolved C-ToF-AMS data, Atmos. Chem. Phys., 9, 2543–2554, https://doi.org/10.5194/acp-9-2543-2009, 2009.
Hoppel, W. A., Frick, G. M., Fitzgerald, J. W., and Larson, R. E.: Marine Boundary layer measurements of new particle formation and the effects nonprecipitating clouds have on aerosol size distribution, J. Geophys. Res., 99, 14443–14459, 1994.
Hu, L., Millet, D. B., Baasandorj, M., Griffis, T. J., Travis, K. R., Tessum, C. W., Marshall, J. D., Reinhart, W. F., Mikoviny, T., Müller, M., Wisthaler, A., Graus, M., Warneke, C., and de Gouw, J.: Emissions of C6–C8 aromatic compounds in the United States: Constraints from tall tower and aircraft measurements, J. Geophys. Res.-Atmos., 120, 826–842, https://doi.org/10.1002/2014JD022627, 2015.
Huang, X.-F. and Yu, J. Z.: Is vehicle exhaust a significant primary source of oxalic acid in ambient aerosols?, Geophys. Res. Lett., 34, L02808, https://doi.org/10.1029/2006gl028457, 2007.
Huang, X.-F., Yu, J. Z., He, L.-Y., and Yuan, Z.: Water-soluble organic carbon and oxalate in aerosols at a coastal urban site in China: Size distribution characteristics, sources, and formation mechanisms, J. Geophys. Res., 111, D22212, https://doi.org/10.1029/2006jd007408, 2006.
Husain, L., Rattigan, O. V., Dutkiewicz, V., Das, M., Judd, C. D., Khan, A. R., Richter, R., Balasubramanian, R., Swami, K., and Walcek, C. J.: Case studies of the SO2 + H2O2 reaction in clouds, J. Geophys. Res.-Atmos., 105, 9831–9841, 2000.
IPCC: Climate Change 2013: The Physical Science Basis, in: Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, edited by: Stocker, T. F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S. K., Boschung, J., Nauels, A., Xia, Y., Bex, V., and Midgley, P. M., Cambridge University Press, Cambridge, UK and New York, NY, USA, 1535 pp., 2013.
Kawamura, K, and Ikushima, K.: Seasonal Changes in the Distribution of Dicarboxylic Acids in the Urban Atmosphere, Environ. Sci. Technol., 27, 2227–2235, 1993.
Kawamura, K. and Yasui, O.: Diurnal changes in the distribution of dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban Tokyo atmosphere, Atmos. Environ., 39, 1945–1960, 2005.
Kawamura, K., Ono, K., Tachibana, E., Charriére, B., and Sempéré, R.: Distributions of low molecular weight dicarboxylic acids, ketoacids and α-dicarbonyls in the marine aerosols collected over the Arctic Ocean during late summer, Biogeosciences, 9, 4725–4737, https://doi.org/10.5194/bg-9-4725-2012, 2012.
Kim, S. W., Barth, M. C., and Trainer, M.: Influence of fair-weather cumulus clouds on isoprene chemistry, J. Geophys. Res.-Atmos., 117, D10302, https://doi.org/10.1029/2011jd017099, 2012.
Kok, G. L., Gitlen, S. N., and Lazrus, A. L.: Kinetics of the Formation and Decomposition of Hydroxymethanesulfonate, J. Geophys. Res. , 91, 2801–2804, 1986.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., Clair, J. M. S., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Laskin, A., Laskin, J., and Nizkorodov, S. A.: Chemistry of Atmospheric Brown Carbon, Chem. Rev., 115, 4335–4382, https://doi.org/10.1021/cr5006167, 2015.
Lee, A. K. Y., Hayden, K. L., Herckes, P., Leaitch, W. R., Liggio, J., Macdonald, A. M., and Abbatt, J. P. D.: Characterization of aerosol and cloud water at a mountain site during WACS 2010: secondary organic aerosol formation through oxidative cloud processing, Atmos. Chem. Phys., 12, 7103–7116, https://doi.org/10.5194/acp-12-7103-2012, 2012.
Lee, S., Murphy, D. M., Thomson, D. S., and Middlebrook, A. M.: Nitrate and oxidized organic ions in single particle mass spectra during the 1999 Atlanta Supersite Project, J. Geophys. Res., 108, 8417, https://doi.org/10.1029/2001JD001455, 2003.
Li, W., Li, P., Sun, G., Zhou, S., Yuan, Q., and Wang, W.: Cloud residues and interstitial aerosols from non-precipitating clouds over an industrial and urban area in northern China, Atmos. Environ., 45, 2488–2495, https://doi.org/10.1016/j.atmosenv.2011.02.044, 2011.
Li, Y., Barth, M. C., Patton, E. G., and Steiner, A. L.: Impact of In-Cloud Aqueous Processes on the Chemistry and Transport of Biogenic Volatile Organic Compounds, J. Geophys. Res.-Atmos., 122, 11131–11153, https://doi.org/10.1002/2017JD026688, 2017.
Lim, Y. B., Tan, Y., Perri, M. J., Seitzinger, S. P., and Turpin, B. J.: Aqueous chemistry and its role in secondary organic aerosol (SOA) formation, Atmos. Chem. Phys., 10, 10521–10539, https://doi.org/10.5194/acp-10-10521-2010, 2010.
Lin, G., Sillman, S., Penner, J. E., and Ito, A.: Global modeling of SOA: the use of different mechanisms for aqueous phase formation, Atmos. Chem. Phys., 14, 5451–5475, https://doi.org/10.5194/acp-14-5451-2014, 2014.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
Maria, S. F., L. M. Russell, M. K. Gilles, and Myeni, S. C. B.: Organic Aerosol Growth Mechanisms and Their Climate Forcing Implications, Science, 306, 1921–1924, 2004.
Marple, V. A., Rubow, K. L., and Behm, S. M.: A Microorifice Uniform Deposit Impactor (MOUDI): Description, Calibration, and Use, Aerosol Sci. Tech., 14, 434–446, https://doi.org/10.1080/02786829108959504, 1991.
Maudlin, L. C., Wang, Z., Jonsson, H. H., and Sorooshian, A.: Impact of wildfires on size-resolved aerosol composition at a coastal California site, Atmos. Environ., 119, 59–68, https://doi.org/10.1016/j.atmosenv.2015.08.039, 2015.
Mazzoleni, L. R., Ehrmann, B. M., Shen, X., Marshall, A. G., and Collett, J. L.: Water-soluble atmospheric organic matter in fog: Exact masses and chemical formula identification by ultrahigh-resolution Fourier transform ion cyclotron resonance mass Spectrometry, Environ. Sci. Technol., 44, 3690–3697, https://doi.org/10.1021/es903409k, 2010.
McNeill, V. F.: Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols, Environ. Sci. Technol., 49, 1237–1244, https://doi.org/10.1021/es5043707, 2015.
McNeill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-Phase Secondary Organic Aerosol and Organosulfate Formation in Atmospheric Aerosols: A Modeling Study, Environ. Sci. Technol., 46, 8075–8081, https://doi.org/10.1021/es3002986, 2012.
McVay, R. and Ervens, B.: A microphysical parameterization of aqSOA and sulfate formation in clouds, Geophys. Res. Lett., 44, 7500–7509, 2017.
Munger, J. W., Tiller, C., and Hoffmann, M. R.: Identification of Hydroxymethanesulfonate in Fog Water, Science, 231, 247–249, 1986.
Murphy, D. M., Cziczo, D. J., Hudson, P. K., Thomson, D. S., Wilson, J. C., Kojima, T., and Buseck, P. R.: Particle Generation and Resuspension in Aircraft Inlets when Flying in Clouds, Aerosol Sci. Tech., 38, 401–409, https://doi.org/10.1080/02786820490443094, 2004.
Nah, T., Guo, H., Sullivan, A. P., Chen, Y., Tanner, D. J., Nenes, A., Russell, A., Ng, N. L., Huey, L. G., and Weber, R. J.: Characterization of aerosol composition, aerosol acidity, and organic acid partitioning at an agriculturally intensive rural southeastern US site, Atmos. Chem. Phys., 18, 11471–11491, https://doi.org/10.5194/acp-18-11471-2018, 2018.
Narukawa, M., Kawamura, K., Takeuchi, N., and Nakajima, T.: Distribution of dicarboxylic acids and carbon isotopic compositions in aerosols from 1997 Indonesian forest fires, Geophys. Res. Lett., 26, 3101–3104, https://doi.org/10.1029/1999GL010810, 1999.
Perri, M. J., Lim, Y. B., Seitzinger, S. P., and Turpin, B. J.: Organosulfates from glycolaldehyde in aqueous aerosols and clouds: Laboratory studies, Atmos. Environ., 44, 2658–2664, 2010.
Petters, M. D. and Kreidenweis, S. M.: A single parameter representation of hygroscopic growth and cloud condensation nucleus activity, Atmos. Chem. Phys., 7, 1961–1971, https://doi.org/10.5194/acp-7-1961-2007, 2007.
Powelson, M. H., Espelien, B. M., Hawkins, L. N., Galloway, M. M., and De Haan, D. O.: Brown Carbon Formation by Aqueous-Phase Carbonyl Compound Reactions with Amines and Ammonium Sulfate, Environ. Sci. Technol., 48, 985–993, https://doi.org/10.1021/es4038325, 2014.
Prabhakar, G., Sorooshian, A., Toffol, E., Arellano, A. F., and Betterton, E. A.: Spatiotemporal distribution of airborne particulate metals and metalloids in a populated arid region, Atmos. Environ., 92, 339–347, https://doi.org/10.1016/j.atmosenv.2014.04.044, 2014.
Reilly, J. E., Rattigan, O. V., Moore, K. M., Judd, C., Sherman, D. E., Dutkiewicz, V. A., Kreidenweis, S. M., Husain, L., and Collett, J. L.: Drop size dependent S(IV) oxidation in chemically heterogenous radiation fogs, Atmos. Environ., 35, 5717–5728, 2001.
Rinaldi, M., Decesari, S., Carbone, C., Finessi, E., Fuzzi, S., Ceburnis, D., O'Dowd, C., Sciare, J., Burrows, J., Vrekoussis, M., Ervens, B., Tsigaridis, K., and Facchini, M. C.: Evidence of a natural marine source of oxalic acid and a possible link to glyoxal, J. Geophys. Res., 116, D16204, https://doi.org/10.1029/2011JD015659, 2011.
Rissler, J., Vestin, A., Swietlicki, E., Fisch, G., Zhou, J., Artaxo, P., and Andreae, M. O.: Size distribution and hygroscopic properties of aerosol particles from dry-season biomass burning in Amazonia, Atmos. Chem. Phys., 6, 471–491, https://doi.org/10.5194/acp-6-471-2006, 2006.
Roelofs, G., Lelieveld, J., and Ganzeveld, L.: Simulation of global sulfate distribution and the influence on effective cloud drop radii with a coupled photochemistry sulfur cycle model, Tellus B, 50, 224–242, https://doi.org/10.1034/j.1600-0889.1998.t01-2-00002.x, 1998.
Sachse, G. W., Hill, G. F., Wade, L. O., and Perry, M. G.: Fast-response, high-precision carbon monoxide sensor using a tunable diode laser absorption technique, J. Geophys. Res.-Atmos., 92, 2071–2081, https://doi.org/10.1029/JD092iD02p02071, 1987.
Schwarz, J. P., Perring, A. E., Markovic, M. Z., Gao, R. S., Ohata, S., Langridge, J., Law, D., McLaughlin, R., and Fahey, D. W.: Technique and theoretical approach for quantifying the hygroscopicity of black-carbon-containing aerosol using a single particle soot photometer, J. Aerosol Sci., 81, 110–126, https://doi.org/10.1016/j.jaerosci.2014.11.009, 2015.
SEAC4RS: Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys, available at: https://www-air.larc.nasa.gov/missions/seac4rs/, last access: August 2018.
Shen, X., Lee, T., Guo, J., Wang, X., Li, P., Xu, P., Wang, Y., Ren, Y., Wang, W., Wang, T., Li, Y., Carn, S. A., and Collett Jr., J. L.: Aqueous phase sulfate production in clouds in eastern China, Atmos. Environ., 62, 502–511, https://doi.org/10.1016/j.atmosenv.2012.07.079, 2012.
Shingler, T., Dey, S., Sorooshian, A., Brechtel, F. J., Wang, Z., Metcalf, A., Coggon, M., Mülmenstädt, J., Russell, L. M., Jonsson, H. H., and Seinfeld, J. H.: Characterisation and airborne deployment of a new counterflow virtual impactor inlet, Atmos. Meas. Tech., 5, 1259–1269, https://doi.org/10.5194/amt-5-1259-2012, 2012.
Shingler, T., Crosbie, E., Ortega, A., Shiraiwa, M., Zuend, A., Beyersdorf, A., Ziemba, L., Anderson, B., Thornhill, L., Perring, A. E., Schwarz, J. P., Campazano-Jost, P., Day, D. A., Jimenez, J. L., Hair, J. W., Mikoviny, T., Wisthaler, A., and Sorooshian, A.: Airborne characterization of subsaturated aerosol hygroscopicity and dry refractive index from the surface to 6.5 km during the SEAC4RS campaign, J. Geophys. Res:.-Atmos., 121, 4188–4210, https://doi.org/10.1002/2015JD024498, 2016.
Sievering, H., Boatman, J., Galloway, J., Keene, W., Kim, Y., Luria, M., and Ray, J.: Heterogeneous sulfur conversion in sea-salt aerosol particles: the role of aerosol water content and size distribution, Atmos. Environ. A, 25, 1479–1487, https://doi.org/10.1016/0960-1686(91)90007-T, 1991.
Sorooshian, A., Brechtel, F. J., Ervens, B., Feingold, G., Varutbangkul, V., Bahreini, R., Murphy, S., Holloway, J. S., Atlas, E. L., Anlauf, K., Buzorius, G., Jonsson, H., Flagan, R. C., and Seinfeld, J. H.: Oxalic acid in clear and cloudy atmospheres: Analysis of data from International Consortium for Atmospheric Research on Transport and Transformation 2004, J. Geophys. Res.-Atmos., 111, D23S45, https://doi.org/10.1029/2005JD006880, 2006a.
Sorooshian, A., Brechtel, F. J., Ma, Y., Weber, R. J., Corless, A., Flagan, R. C., and Seinfeld, J. H.: Modeling and Characterization of a Particle-into-Liquid Sampler (PILS), Aerosol Sci. Tech., 40, 396–409, https://doi.org/10.1080/02786820600632282, 2006b.
Sorooshian, A., Lu, M.-L., Brechtel, F. J., Jonsson, H., Feingold, G., Flagan, R. C., and Seinfeld, J. H.: On the source of organic acid aerosol layers above clouds, Environ. Sci. Technol., 41, 4647–4654, 2007a.
Sorooshian, A., Ng, N. L., Chan, A. W. H., Feingold, G., Flagan, R. C., and Seinfeld, J. H.: Particulate organic acids and overall water-soluble aerosol composition measurements from the 2006 Gulf of Mexico Atmospheric Composition and Climate Study (GoMACCS), J. Geophys. Res.-Atmos., 112, D13201, https://doi.org/10.1029/2007JD008537, 2007b.
Sorooshian, A., Murphy, S. M., Hersey, S., Bahreini, R., Jonsson, H., Flagan, R. C., and Seinfeld, J. H.: Constraining the contribution of organic acids and AMS m/z 44 to the organic aerosol budget: On the importance of meteorology, aerosol hygroscopicity, and region, Geophys. Res. Lett., 37, L21807, https://doi.org/10.1029/2010gl044951, 2010.
Sorooshian, A., Wonaschütz, A., Jarjour, E. G., Hashimoto, B. I., Schichtel, B. A., and Betterton, E. A.: An aerosol climatology for a rapidly growing arid region (southern Arizona): Major aerosol species and remotely sensed aerosol properties, J. Geophys. Res.-Atmos., 116, D19205, https://doi.org/10.1029/2011jd016197, 2011.
Sorooshian, A., Wang, Z., Coggon, M. M., Jonsson, H. H., and Ervens, B.: Observations of Sharp Oxalate Reductions in Stratocumulus Clouds at Variable Altitudes: Organic Acid and Metal Measurements During the 2011 E-PEACE Campaign, Environ. Sci. Technol., 47, 7747–7756, https://doi.org/10.1021/es4012383, 2013.
Sorooshian, A., Csavina, J., Shingler, T., Dey, S., Brechtel, F. J., Sáez, A. E., and Betterton, E. A.: Hygroscopic and Chemical Properties of Aerosols Collected near a Copper Smelter: Implications for Public and Environmental Health, Environ. Sci. Technol., 46, 9473–9480, https://doi.org/10.1021/es302275k, 2012.
Sorooshian, A., Shingler, T., Crosbie, E., Barth, M. C., Homeyer, C. R., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Thornhill, K. L., Ziemba, L. D., Blake, D. R., and Fried, A.: Contrasting aerosol refractive index and hygroscopicity in the inflow and outflow of deep convective storms: Analysis of airborne data from DC3, J. Geophys. Res.-Atmos., 122, 4565–4577, https://doi.org/10.1002/2017JD026638, 2017a.
Sorooshian, A., MacDonald, A. B., Dadashazar, H., Bates, K. H., Coggon, M. M., Craven, J. S., Crosbie, E., Hersey, S. P., Hodas, N., Lin, J. J., Marty, A. N., Maudlin, L. C., Metcalf, A. R., Murphy, S. M., Padro, L. T., Prabhakar, G., Rissman, T. A., Shingler, T., Varutbangkul, V., Wang, Z., Woods, R. K., Chuang, P. Y., Nenes, A., Jonsson, H. H., Flagan, R. C., and Seinfeld, J. H.: A multi-year data set on aerosol–cloud–precipitation–meteorology interactions for marine stratocumulus clouds, Figshare, https://doi.org/10.6084/m9.figshare.5099983.v3, 2017b.
Sorooshian, A., MacDonald, A. B., Dadashazar, H., Bates, K. H., Coggon, M. M., Craven, J. S., Crosbie, E., Hersey, S. P., Hodas, N., Lin, J. J., Marty, A. N., Maudlin, L. C., Metcalf, A. R., Murphy, S. M., Padro, L. T., Prabhakar, G., Rissman, T. A., Shingler, T., Varutbangkul, V., Wang, Z., Woods, R. K., Chuang, P. Y., Nenes, A., Jonsson, H. H., Flagan, R. C., and Seinfeld, J. H.: A multi-year data set on aerosol–cloud–precipitation–meteorology interactions for marine stratocumulus clouds, Sci. Data, 5, 180026, https://doi.org/10.1038/sdata.2018.26, 2018.
Sullivan, A. P., Hodas, N., Turpin, B. J., Skog, K., Keutsch, F. N., Gilardoni, S., Paglione, M., Rinaldi, M., Decesari, S., Facchini, M. C., Poulain, L., Herrmann, H., Wiedensohler, A., Nemitz, E., Twigg, M. M., and Collett Jr., J. L.: Evidence for ambient dark aqueous SOA formation in the Po Valley, Italy, Atmos. Chem. Phys., 16, 8095–8108, https://doi.org/10.5194/acp-16-8095-2016, 2016.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Swietlicki, E., Berg, O. H., Zhou, J., and Aalto, P.: The effect of cloud processing on the hygroscopic properties of aerosol particle in the marine boundary layer: Preliminary results from a hill cap cloud experiment during the ACE-2 Experiment, Trans. Ecol. Environm., 28, 475–479, 1999.
Toon, O. B., Maring, H., Dibb, J., Ferrare, R., Jacob, D. J., Jensen, E. J., Luo, Z. J., Mace, G. G., Pan, L. L., Pfister, L., Rosenlof, K. H., Redemann, J., Reid, J. S., Singh, H. B., Thompson, A. M., Yokelson, R., Minnis, P., Chen, G., Jucks, K. W., and Pszenny, A.: Planning, implementation, and scientific goals of the Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys (SEAC4RS) field mission, J. Geophys. Res.-Atmos., 121, 4967–5009, https://doi.org/10.1002/2015JD024297, 2016.
Volkamer, R., Jimenez, J. L., SanMartini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006.
Wagner, N. L., Brock, C. A., Angevine, W. M., Beyersdorf, A., Campuzano-Jost, P., Day, D., de Gouw, J. A., Diskin, G. S., Gordon, T. D., Graus, M. G., Holloway, J. S., Huey, G., Jimenez, J. L., Lack, D. A., Liao, J., Liu, X., Markovic, M. Z., Middlebrook, A. M., Mikoviny, T., Peischl, J., Perring, A. E., Richardson, M. S., Ryerson, T. B., Schwarz, J. P., Warneke, C., Welti, A., Wisthaler, A., Ziemba, L. D., and Murphy, D. M.: In situ vertical profiles of aerosol extinction, mass, and composition over the southeast United States during SENEX and SEAC4RS: observations of a modest aerosol enhancement aloft, Atmos. Chem. Phys., 15, 7085–7102, https://doi.org/10.5194/acp-15-7085-2015, 2015.
Wang, Z., Wang, T., Guo, J., Gao, R., Xue, L., Zhang, J., Zhou, Y., Zhou, X., Zhang, Q., and Wang, W.: Formation of secondary organic carbon and cloud impact on carbonaceous aerosols at Mount Tai, North China, Atmos. Environ., 46, 516–527, https://doi.org/10.1016/j.atmosenv.2011.08.019, 2012.
Wang, Z., Ramirez, M. M., Dadashazar, H., MacDonald, A. B., Crosbie, E., Bates, K. H., Coggon, M. M., Craven, J. S., Lynch, P., Campbell, J. R., Aghdam, M. A., Woods, R. K., Jonsson, H., Flagan, R. C., Seinfeld, J. H., and Sorooshian, A.: Contrasting cloud composition between coupled and decoupled marine boundary layer clouds, J. Geophys. Res.-Atmos., 121, 11679–11691, https://doi.org/10.1002/2016JD025695, 2016.
Waxman, E. M., Dzepina, K., Ervens, B., Lee-Taylor, J., Aumont, B., Jimenez, J. L., Madronich, S., and Volkamer, R.: Secondary organic aerosol formation from semi- and intermediate-volatility organic compounds and glyoxal: Relevance of O ∕ C as a tracer for aqueous multiphase chemistry, Geophys. Res. Lett., 40, 1–5, https://doi.org/10.1002/grl.50203, 2013.
Weller, C., Horn, S., and Herrmann, H.: Effects of Fe(III)-concentration, speciation, excitation-wavelength and light intensity on the quantum yield of iron(III)-oxalato complex photolysis, J. Photoch. Photobiol. A, 255, 41–49, https://doi.org/10.1016/j.jphotochem.2013.01.014, 2013.
Weller, C., Tilgner, A., Bräuer, P., and Herrmann, H.: Modelling the impact of iron-carboxylate photochemistry on radical budget and carboxylate degradation in cloud droplets and particles, Environ. Sci. Technol., 48, 5652–5659, https://doi.org/10.1021/es4056643, 2014.
Whiteaker, J.,and Prather, K. A.: Hydroxymethanesulfonate as a tracer for fog processing of individual aerosol particles, Atmos. Environ., 37, 1033–1043, 2003.
Wonaschuetz, A., Sorooshian, A., Ervens, B., Chuang, P. Y., Feingold, G., Murphy, S. M., de Gouw, J., Warneke, C., and Jonsson, H. H.: Aerosol and gas re-distribution by shallow cumulus clouds: An investigation using airborne measurements, J. Geophys. Res.-Atmos., 117, D17202, https://doi.org/10.1029/2012jd018089, 2012.
Wonaschütz, A., Coggon, M., Sorooshian, A., Modini, R., Frossard, A. A., Ahlm, L., Mülmenstädt, J., Roberts, G. C., Russell, L. M., Dey, S., Brechtel, F. J., and Seinfeld, J. H.: Hygroscopic properties of smoke-generated organic aerosol particles emitted in the marine atmosphere, Atmos. Chem. Phys., 13, 9819–9835, https://doi.org/10.5194/acp-13-9819-2013, 2013.
Woo, J. L. and McNeill, V. F.: simpleGAMMA v1.0 – a reduced model of secondary organic aerosol formation in the aqueous aerosol phase (aaSOA), Geosci. Model Dev., 8, 1821–1829, https://doi.org/10.5194/gmd-8-1821-2015, 2015.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Yang, Q., Easter, R. C., Campuzano-Jost, P., Jimenez, J. L., Fast, J. D., Ghan, S. J., Wang, H., Berg, L. K., Barth, M. C., Liu, Y., Shrivastava, M. B., Singh, B., Morrison, H., Fan, J., Ziegler, C. L., Bela, M., Apel, E., Diskin, G. S., Mikoviny, T., and Wisthaler, A.: Aerosol transport and wet scavenging in deep convective clouds: A case study and model evaluation using a multiple passive tracer analysis approach, J. Geophys. Res.-Atmos., 120, 8448–8468, https://doi.org/10.1002/2015JD023647, 2015.
Youn, J.-S., Wang, Z., Wonaschütz, A., Arellano, A., Betterton, E. A., and Sorooshian, A.: Evidence of aqueous secondary organic aerosol formation from biogenic emissions in the North American Sonoran Desert, Geophys. Res. Lett., 40, 3468–3472, https://doi.org/10.1002/grl.50644, 2013.
Youn, J. S., Crosbie, E., Maudlin, L. C., Wang, Z., and Sorooshian, A.: Dimethylamine as a major alkyl amine species in particles and cloud water: Observations in semi-arid and coastal regions, Atmos. Environ., 122, 250–258, https://doi.org/10.1016/j.atmosenv.2015.09.061, 2015.
Yu, J. Z., Huang, X.-F., Xu, J., and Hu, M.: When Aerosol sulfate goes up, so does Oxalate: Implication for the Formation Mechanisms of Oxalate, Environ. Sci. Technol., 39, 128–133, 2005.
Zhang, J., Liu, L., Wang, Y., Ren, Y., Wang, X., Shi, Z., Zhang, D., Che, H., Zhao, H., Liu, Y., Niu, H., Chen, J., Zhang, X., Lingaswamy, A. P., Wang, Z., and Li, W.: Chemical composition, source, and process of urban aerosols during winter haze formation in Northeast China, Environ. Pollut., 231, 357–366, https://doi.org/10.1016/j.envpol.2017.07.102, 2017.
Zheng, B., Zhang, Q., Zhang, Y., He, K. B., Wang, K., Zheng, G. J., Duan, F. K., Ma, Y. L., and Kimoto, T.: Heterogeneous chemistry: a mechanism missing in current models to explain secondary inorganic aerosol formation during the January 2013 haze episode in North China, Atmos. Chem. Phys., 15, 2031–2049, https://doi.org/10.5194/acp-15-2031-2015, 2015.
Zuo, Y. and Hoigné, J.: Formation of Hydrogen Peroxide and Depletion of Oxalic Acid in Atmospheric Water by Photolysis of Iron(III)-Oxalato-Complexes, Environ. Sci. Technol., 26, 1014–1022, 1992.
Zuo, Y. and Hoigné, J.: Photochemical Decomposition of Oxalic, Glyoxalic and Pyruvic Acid Catalysed by Iron in Atmospheric Waters, Atmos. Environ., 28, 1231–1239, 1994.