the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Oct 2018
| 19 Oct 2018
The oxidation regime and SOA composition in limonene ozonolysis: roles of different double bonds, radicals, and water
Yiwei Gong
Zhongming Chen
Huan Li
Volatile organic compounds play an important role in air quality and climate change, largely because they contribute to the formation of oxidizing compounds and secondary organic aerosol (SOA). In this study, a series of products, including peroxides and carbonyl compounds in both gaseous and particulate phases, were simultaneously detected to investigate the oxidation regime and SOA composition in limonene ozonolysis. The roles of different double bonds (DBs), radicals, and water were also examined. In our first investigation, we focused on representative oxidizing compounds produced in limonene ozonolysis, including stabilized Criegee intermediates (SCIs), OH radicals, and peroxides. The dependence of H2O2 and hydroxymethyl hydroperoxide (HMHP) formation on RH demonstrates that the reaction with water is an important reaction pathway for limonene SCIs, and the lower limit SCI yields of endocyclic and exocyclic DBs were estimated to be ∼0.24 and ∼0.43, respectively. The OH yield was determined by adding sufficient amounts of an OH scavenger, and the OH yields of endocyclic and exocyclic DBs were ∼0.65 and ∼0.24, respectively. These results indicate that in limonene ozonolysis the endocyclic DB is inclined to generate OH radicals through the hydroperoxide channel, while the exocyclic DB has a higher fraction of forming SCIs. Additionally, other gas-phase and particle-phase peroxides were also studied in this work. The formation of performic acid (PFA) and peracetic acid (PAA) was promoted significantly by increasing RH and the degree of oxidation, and the discrepancy between the experimental and model results suggested some missing formation pathways. Considerable generation of H2O2 from SOA in the aqueous phase was observed, especially at a high [O3] ∕ [limonene] ratio, which was mainly attributed to the hydration and decomposition of unstable peroxides in SOA such as peroxycarboxylic acids and peroxyhemiacetals. Different DBs and OH scavengers had a large impact on the particulate peroxides, and their stability indicated that the types of peroxides in SOA changed under different conditions. As for the contribution of peroxides to SOA, the results demonstrated that the mass fraction of particulate peroxides in limonene SOA was less than 0.2 at a low [O3] ∕ [limonene] ratio, while the mass fraction was 0.4–0.6 at a high [O3] ∕ [limonene] ratio. The partitioning behavior of peroxides showed that multi-generation oxidation helped produce more low-volatility peroxides, which partially explained the higher SOA yield. The partitioning behavior of carbonyls was also examined and the experimental partitioning coefficients (Kp) were found to be typically several orders of magnitude higher than the theoretical values. This study provided new insights into the oxidation regime and SOA composition in limonene ozonolysis, and limonene showed its specificity in many aspects when both endocyclic and exocyclic DBs were ozonated. We suggest that the atmospheric implications of terpenes containing more than one DB and the SOA composition, especially particulate peroxides, need further study.
- Article
(6263 KB) - Full-text XML
-
Supplement
(5761 KB) - BibTeX
- EndNote





As an important monoterpene, limonene has a high emission rate from both biogenic and anthropogenic sources that is second only to pinene (Atkinson and Arey, 2003; Clausen et al., 2001; Fellin and Otson, 1994; Griffin et al., 1999; Guenther et al., 1995; Lamb et al., 1993; Seifert et al., 1989; Sindelarova et al., 2014; Wolkoff et al., 2000). Total monoterpene emissions are estimated to be 50 Tg C yr−1, and limonene might comprise about 20 % of that (Stroud et al., 2005). In addition to massive emissions from vegetation, the extensive utilization of limonene in household and industrial processes makes it non-negligible in the atmosphere. Compared with its isomers α-pinene and β-pinene, an obvious structural feature of limonene is that it has two different double bonds (DBs), an endocyclic DB and an exocyclic DB, which complicates the behavior and the fate of limonene in the atmosphere. Ozonolysis is an important reaction pathway of limonene and it serves as a source of free radicals, intermediate products, and aerosol. The first step in alkene ozonolysis is the addition of O3 to the carbon–carbon double bond, forming an energy-rich primary ozonide (POZ). The POZ decomposes into two sets of carbonyls plus carbonyl oxides, which are called excited Criegee intermediates (ECIs) (Criegee, 1975; Fenske et al., 2000; Gutbrod et al., 1996, 1997; Kroll et al., 2001a, b). ECIs can isomerize through the hydroperoxide channel followed by OH production, rearrange to esters with subsequent decomposition, or undergo collisional stabilization, forming stabilized Criegee intermediates (SCIs) (Cremer et al., 1993; Presto and Donahue, 2004; Richard et al., 1999; Wegener et al., 2007).
In the last few years, the oxidizing compounds formed in alkene ozonolysis have received much attention because they are able to remove a series of trace gases and contribute to atmospheric oxidation capacity (Möller, 2009; Prinn, 2003; Taatjes et al., 2013). Representatives of these compounds include OH radicals, SCIs, and peroxides, etc. Investigations into the oxidation regime of alkene ozonolysis are essential to understanding of the formation of these critical products and evaluation of the actual impact of this reaction in the atmosphere. As an important source of OH radicals needed for OH-initiated reactions occurring in darkness (Kroll et al., 2001a, b), OH formation pathways in alkene ozonolysis have been extensively studied and the major pathway is considered to be the unimolecular decomposition of ECIs. Furthermore, some studies suggest that SCIs could also generate OH radicals through self-decomposition or reactions with other species (Anglada et al., 2002; Hasson et al., 2003; Kroll et al., 2001a, b; Tillmann et al., 2010; Zhang and Zhang, 2005). SCIs generated during the process have been shown to have a lifetime, usually on the order of sub-seconds (Kroll et al., 2001a, b; Long et al., 2018; Mauldin et al., 2012), that is sufficiently long to react with other trace species, such as H2O, SO2, NO2, carbonyls, alcohols, and carboxylic acids. (Sakamoto et al., 2017; Sipilä et al., 2014; Yao et al., 2014). A number of studies have focused on the reactions of SCIs containing 3 or fewer carbon atoms, and their unimolecular decomposition and reactions with water show strong structure dependence. The reactions with water monomers and water dimers are the main reaction pathways for CH2OO and anti-CH3CHOO, while the unimolecular decomposition would be more important for syn-CH3CHOO and (CH3)2COO (Huang et al., 2015; Lin et al., 2016; Welz et al., 2012, 2014; Taatjes et al., 2013). However, the reaction mechanisms for larger SCIs formed from biogenic alkenes ozonolysis are still unclear, since it is difficult to synthesize and observe them directly. Furthermore, reactions of alkenes with ozone can also generate a considerable amount of peroxides, which are considered to be important due to their oxidizability and role as a radicals' reservoir. H2O2 is the most crucial oxidant to oxidize S (IV), forming sulfuric acid and sulfate in the aqueous phase (Calvert et al., 1985; Penkett et al., 1979; Peña et al., 2001). And as a species accounting for 40 %–50 % of total global organic peroxides (Crounse et al., 2006; Khan et al., 2015), peroxycarboxylic acids (RC(O)OOH) play an important role in promoting atmospheric oxidation capacity and enhancing the acidity of the aqueous phase.
One important reason why limonene chemistry has attracted much attention is its high secondary organic aerosol (SOA) formation potential, which is higher than that of α-pinene since limonene is doubly unsaturated (Andersson-Sköld and Simpson 2001; Kroll and Seinfeld, 2008; Lane et al., 2008; Lee et al., 2006). Although progress has been made in simulating SOA formation with the theory of gas–particle partitioning, there are still large discrepancies between model and experimental results (Cocker et al., 2001; Griffin et al., 1999; Hoffmann et al., 1997; Odum et al., 1996; Pankow, 1994; Presto et al., 2005; Pye and Seinfeld, 2010). Laboratory studies of SOA formation in limonene ozonolysis mainly focused on the aerosol yields under different conditions and the identification of products in the particle phase (Calogirou et al., 1999; Glasius et al., 2000; Grosjean et al., 1992, 1993; Leungsakul et al., 2005; Ng et al., 2006; Pathak et al., 2012); however, the composition of limonene SOA still needs detailed study, especially the effect of different DBs. As a double-unsaturated terpene, the SOA formation process of limonene could be more complicated than that for single-unsaturated terpene, since multi-generation oxidation has a significant influence on SOA. In this study, two classes of species, peroxides and carbonyls, are studied for their behaviors in limonene SOA formation. Over the past few years, analyses of organic peroxides suggest that they are an important component in aerosol (Docherty et al., 2005; Heaton et al., 2007; Li et al., 2016; Mertes et al., 2012; Pathak et al., 2012), and particulate peroxides could cause negative health effects when they penetrate into lungs (Chen et al., 2011; Fischer and Maier, 2015; Gupta et al., 2014; Verma et al., 2009; Wragg et al., 2016). The reactive uptake and particle-phase reactions of carbonyls are believed to be responsible for fractions of aqueous SOA formation (Ervens et al., 2011; Mcneill et al., 2012), especially for the dicarbonyls glyoxal (GL) and methylglyoxal (MGL). Until now, there has been little research into the contribution of peroxides and carbonyls to limonene SOA and their behaviors in aerosol formation are only vaguely understood, to the best of our knowledge.
The focus of this study is to investigate the oxidation regime and SOA composition in limonene ozonolysis, especially for the roles of different DBs, radicals, and water. We consider the generation of SCIs, OH radicals, and peroxides in discussions of the oxidation regime in the reaction system. Furthermore, peroxides and carbonyls are taken as representatives to study their behaviors in SOA formation.
2.1 Chemicals
This study used the following: R-(+)-Limonene (Sigma-Aldrich, ≥99.0 %), 2-butanol (Sigma-Aldrich, 99.5 %), cyclohexane (Alfa Aesar, ≥99.9 %), potassium iodide (KI, Alfa Aesar, 99.9 %), hydrogen peroxide (H2O2, Alfa Aesar, 35 wt %), orthophosphoric acid (H3PO4, Fluka, 85 %–90 %), hemin (Sigma, ≥98.0 %), 4-hydroxyphenylacetic acid (Alfa Aesar, 99 %), formaldehyde (FA; Sigma-Aldrich, 37 wt %), acetaldehyde (AA; Amethyst Chemicals, 40 wt %), acetone (ACE; Fluka, ≥99.7 %), hydroxyacetone (HACE; Sigma-Aldrich, 90 %), GL (Sigma-Aldrich, 40 wt %), MGL (Sigma-Aldrich, 40 wt %), 2-butanone (Alfa Aesar, ≥99 %), acetonitrile (Alfa Aesar, ≥99.7 %), 2,4-dinitrophenylhydrazine (DNPH, TCI, 50 wt %), ammonia solution (NH3•H2O, Beijing Tongguang Fine Chemicals Company, 25.0 %–28.0 %), ammonium chloride (NH4Cl, Beijing Tongguang Fine Chemicals Company, ≥99.5 %), sulfuric acid (H2SO4, Xilong Chemical Company, 95.0 %–98.0 %), ultrapure water (18 MΩ, Millipore), N2 (≥99.999 %, Beijing Haikeyuanchuang Practical Gas Company Limited, Beijing, China), O2 (≥99.999 %, Beijing Haikeyuanchuang Practical Gas Company Limited, Beijing, China), polytetrafluoroethylene (PTFE) filter membrane (Whatman Inc., 47 mm in diameter), and quartz microfiber filters (Whatman Inc.).
2.2 Apparatus and procedures
A flow tube reactor (2 m length, 70 mm inner diameter, quartz wall), equipped with a water jacket for controlling the temperature, was used to investigate the ozonolysis of limonene. All the experiments were conducted at 298±0.5 K in darkness. O3 was generated by O2 photolysis in a 2 L quartz tube with a low-pressure Hg lamp, and the detailed quantification method of O3 was described in our previous study (Chen et al., 2008). H2O2 produced by UV irradiation of O2 and trace water was measured in control experiments and deducted from the results. Limonene gas was generated by passing N2 flow over liquid limonene in a diffusion tube at the selected temperature, and OH scavenger (2-butanol or cyclohexane) gas was generated with a bubbler. The concentrations of limonene and the OH scavenger were determined using a gas chromatograph with a flame ionization detector (GC-FID, Agilent 7890A, USA). Water vapor was generated by passing N2 through a water bubbler, which contained a carborundum disc submerged in ultrapure water (18 MΩ). The mixing gas, including limonene, OH scavenger, ozone, and dry or wet synthetic air (80 % N2 and 20 % O2), was successively introduced into the reactor. With a total flow rate of 2 standard L min−1, the residence time was estimated to be 240 s.
To explore the reaction mechanism of endocyclic and exocyclic DBs' ozonolysis and the effect of multi-generation oxidation in limonene ozonolysis, we conducted two sets of experiments with different ratios of ozone to limonene concentration. In the following, [O3] denotes the concentration of ozone, [limonene] denotes the concentration of limonene, and [O3] ∕ [limonene] denotes the concentration ratio of ozone to limonene. In the low [O3] ∕ [limonene] experiments, the initial concentrations of limonene and ozone were ∼280 and ∼500 ppbv, respectively, whereas in the high [O3] ∕ [limonene] experiments, the initial concentrations of limonene and ozone were ∼183 ppbv and ∼19 ppmv, respectively. In both sets of experiments, sufficient quantities of 2-butanol and cyclohexane were added to scavenge OH radicals in the RH range of 0 %–90 %. In the tables and figures, the low-ratio and high-ratio experiments are denoted by L and H, respectively, and the conditions in the absence of the scavenger, in the presence of 2-butanol, and in the presence of cyclohexane are denoted by No-sca, 2-But, and C-hex, respectively. The experimental conditions are listed in Table 1.
Table 1Experimental conditions.
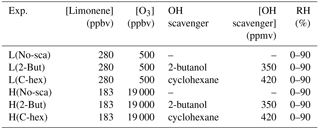
Note: L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca,no scavenger; 2-But, 2-butanol; C-hex, cyclohexane; RH, relative humidity.
According to previous studies, the rate constant of the endocyclic DB reaction with ozone in limonene ozonolysis is cm3 molecule−1 s−1 (Atkinson, 1990; Shu and Atkinson, 1994), while the exocyclic DB reaction with ozone is about 30 times slower than the endocyclic DB reaction (Zhang et al., 2006). Based on those rate constants, we estimate that at a low [O3] ∕ [limonene] ratio, less than 1 % exocyclic DB is ozonated, so this situation mainly represents first-generation oxidation. In this circumstance, because the ozone concentration is low, the OH reaction would impact the amount of limonene consumed by O3. In the presence of the OH scavenger, ∼42 % endocyclic DB reacted with O3, while in the absence of the scavenger, ∼38 % endocyclic DB reacted with O3. At a high [O3] ∕ [limonene] ratio, more than 99 % endocyclic DB and about 51 % exocyclic DB reacted with ozone, and since the ozone concentration in this situation was high, the OH effect on the ozonolysis was presumed to be unimportant. The latter condition, which contained multi-generation oxidation processes, was more likely to occur since the ratio of [O3] to [limonene] was similar to the ratio in the real atmosphere.
It should be noted that one advantage of the flow tube reactor is that the wall would be in equilibrium with the gas phase after a stationary period. According to our observations, this process usually requires about 2 h. Thus, to stabilize the system and diminish the wall effect to the greatest extent possible, we ran the reactor for 2 h prior to taking measurements. And after the experiments, the reactor was rinsed with ultrapure water and blown dry with N2.
2.3 Products' analysis
To better investigate the gas–particle partitioning of products formed in limonene ozonolysis, we analyzed the gas-phase and particle-phase products simultaneously. We measured total peroxides and a series of low-molecule-weight (LMW) peroxides. In the following discussion we regard those peroxides that could be detected by high performance liquid chromatography (HPLC) to be LMW peroxides, while we take the peroxides undetermined by HPLC to be high-molecule-weight (HMW) peroxides. For particle-phase peroxides' detection, a PTFE filter was used for the SOA collection, and the mass of SOA was measured by a semi-micro balance (Sartorius, Germany). Since the control experiment showed that a long-duration collection led to the loss of some peroxides in particles, the collection time was controlled to be 3 h for each filter, and the accuracy of the particulate products' analysis was discussed in the Supplement. Each loaded PTFE filter was extracted with 20 mL H3PO4 solution (pH 3.5) using a shaker (Shanghai Zhicheng ZWY 103D, China) at 180 rpm and 4 ∘C for 15 min, and the extraction efficiency was confirmed in our previous work (Li et al., 2016). For gas-phase peroxides' detection, the air samples through the filter were collected in a glass coil collector at a temperature of 4 ∘C with H3PO4 solution (pH 3.5) serving as the rinsing solution. The coil collector is around 30 cm long and its effective length is about 100 cm. The coil is similar with that used in earlier studies (Grossmann et al., 2003; Sauer et al., 1996, 1997), and the diagram of the thermostatic coil collector is provided in the Supplement.
The method for detecting the peroxides was reported in our previous studies (Hua et al., 2008; Li et al., 2016) and is only briefly described here. LMW peroxides were analyzed using HPLC (Agilent 1100, USA) coupled with post-column derivatization and fluorescence detection online. Peroxides separated by column chromatography reacted with p-hydroxyphenylacetic acid (POPHA) to form POPHA dimers under the catalysis of hemin, and then the dimers were quantified using a fluorescence detector. The standard solution of peroxides was prepared and used for calibration in each measurement. The detection limit for the gas-phase LMW peroxides was about 22 pptv, and in the particle phase the detection limit was around 0.068 ng µg−1 for H2O2. The accuracy of this method was estimated to be around 7 %, with the precision of the measurement results usually within 20 %. The concentration of total peroxides (H2O2, ROOH, and ROOR′) was determined using the iodometric spectrophotometric method, which is based on the reaction of peroxides and iodide ions (Docherty et al., 2005; Mutzel et al., 2013). Briefly, excessive KI solution was added to samples purged of O2. After remaining in darkness 12–24 h for the derivatization, the produced I ions were quantified at 420 nm using a UV–VIS spectrophotometer (SHIMADZU UV-1800, Japan). The efficiency of the total peroxides' measurement was discussed in our previous work (Li et al., 2016), and the detection limit of the total peroxides was about 0.92 ppbv in the gas phase and 0.025 µg µg−1 in the particle phase. The accuracy of this method was estimated to be 10 %, with the precision of the measurement results usually within 20 %.
To measure the particle-phase carbonyls, SOA was collected onto a quartz microfiber filter for 3 h. After collection, the filter was placed upside down in a conical flask, and then 5 mL acetonitrile, 1 mL DNPH saturated solution, and 50 µL H2SO4 solution (0.25 M) were added into the flask in sequence. The flask was shaken at 180 rpm and 4 ∘C for 3 h and kept in darkness for 12–24 h prior to detection. For the gas-phase carbonyls' measurement, gas through the filter was directly introduced into a Horibe tube, which was placed in a cold trap (Beijing Tiandijingyi TH-95-15-G, China) at about −98 ∘C to freeze the products in the tube. The Horibe tube was composed of an inlet tube (25 cm length, 4 cm O.D.), a coil (7 laps, 1 cm O.D.), and an outlet tube with a carborundum disc. The diagram of the Horibe tube is shown in the Supplement. After collection, 10 mL acetonitrile was added to rinse the inside of the Horibe tube to dissolve carbonyl compounds, and then the solution was mixed with DNPH saturated solution and H2SO4 solution to derivatize for 12–24 h in darkness. The derived samples were analyzed using HPLC with UV detection (Agilent 1100, USA). Details of this process are provided in our previous report (Wang et al., 2009). The detection limit of carbonyl compounds was about 0.3 ppbv in the gas phase, and in the particle phase it was 0.15 ng µg−1 in terms of FA. The accuracy of this method was estimated to be 8 % and the precision of the measurement results was within 30 %. Since the yields of both ACE and MGL were low, especially under some conditions, their uncertainty might be high and the precision was within 80 %.
2.4 Wall loss experiments
To increase the accuracy of the results, we designed and conducted a series of control experiments to quantify the wall loss effect, including gaseous products and aerosol wall loss evaluation. Gas-containing peroxide constituents were generated by passing N2 through a diffusion tube, at a controlled temperature of 4 ∘C, which contained a certain peroxide solution. The synthetic method of multiple organic peroxides was described in our previous work (Huang et al., 2013). Gas-containing carbonyl constituents were prepared by injecting a liquid substance into an evacuated steel canister (15 L, Entech Instruments). Then N2 was added constantly until the pressure in the canister reached 30 psi. The outlet of the canister was linked with a mass flow controller that regulated the gas flow rate. The gas-containing peroxides or carbonyls were mixed with synthetic air at different RH levels and introduced into the reactor at a rate of 2 standard L min−1, and the concentrations of peroxides and carbonyls were controlled to be similar to the concentrations of products observed in limonene ozonolysis. After the gas mixture was introduced into the flow tube, a period of approximately 2 h was needed for the gas and wall to become balanced, and then the measurements began. The aerosol wall loss experiment used two-stage reaction equipment that contained two flow tube reactors. In order to explore the wall loss effect on pure SOA, the first reactor was used to generate aerosol, so that limonene and O3 had sufficient time to react completely. The gas-containing aerosol from the first reactor was mixed with synthetic air at different RH levels and introduced into the second reactor at a rate of 2 standard L min−1. The particles at the inlet and outlet of the second reactor were collected on PTFE filters and measured by a balance to calculate the SOA concentration. The wall loss fraction of the gas-phase constituent or SOA was determined as the difference between the inlet and outlet concentrations divided by the inlet concentration as Eq. (S1) in the Supplement. The wall loss experiments were conducted in the RH range of 0 %–90 %, and the profiles of loss fractions as a function of RH could be used to correct products and SOA yields to diminish the wall loss effect.
3.1 Wall loss correction
The wall loss fractions of LMW peroxides increased with increasing RH, and this tendency was especially obvious for performic acid (PFA) and peracetic acid (PAA), whose loss fractions increased successively with RH. For H2O2 and hydroxymethyl hydroperoxide (HMHP), their wall loss fractions went up quickly above 50 % RH, yet they did not have a large change below 50 % RH. Generally speaking, HMHP had the highest wall loss fraction, reaching ∼0.25 at 90 % RH, while PFA had the lowest wall loss fraction. The average of these LMW peroxides loss fractions was used to correct the wall loss effect for gas-phase HMW peroxides.
As for carbonyl compounds, the variation of their wall loss fractions with RH was not very obvious. The loss curves of FA, AA, and ACE were somewhat irregular. The wall loss fractions of HACE, GL, and MGL were lowest at 10 % RH, and then they gradually arose with increasing water vapor concentration. The higher loss fractions of GL and MGL compared with other carbonyls could be attributed to their inclination towards hydration. FA had the lowest wall loss fraction around 0.03, while MGL had the highest loss fraction around 0.12.
The wall loss effect on SOA was also investigated from 0 % to 90 % RH. The SOA wall loss fraction was ∼0.06 in dry conditions, then it increased slightly with water vapor concentration. The SOA loss fraction was ∼0.11 and ∼0.17 at 50 % and 90 % RH, respectively. The dependencies of the peroxides, carbonyls, and SOA wall loss fractions on RH are provided in the Supplement.
3.2 SCIs generation
The reaction between SCIs and water, which produces α-hydroxyalkyl hydroperoxides (HAHPs) decomposing to H2O2, carbonyls, and carboxylic acids, is thought to be an essential source of these compounds and serves as a principle source of H2O2 formation without light (Becker et al., 1990, 1993; Gäb et al., 1985; Sauer et al., 1999). Figure 1 shows the dependence of H2O2 yield and HMHP yield on RH, and the six profiles in each subgraph represent conditions at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger (2-butanol or cyclohexane). The molar yield used here is defined as the ratio of the product's number of moles to the number of moles of limonene consumed, as given by Eq. (S2). It was obvious that, even in different cases, the variations of H2O2 yield and HMHP yield with RH had similar tendencies, both of which increased significantly from 0 % RH to 70 % RH, and then they approached the limiting values. There was little effect of the OH scavenger. At a low [O3] ∕ [limonene] ratio, the maximum H2O2 yield was ∼24.00 % without the OH scavenger, ∼24.60 % with 2-butanol, and ∼22.95 % with cyclohexane. At a high [O3] ∕ [limonene] ratio, the maximum H2O2 yield reached ∼41.20 % without the OH scavenger, ∼41.80 % with 2-butanol, and ∼40.50 % with cyclohexane. As for HMHP, its yield was much higher at a high [O3] ∕ [limonene] ratio (∼5.43 %) than at a low [O3] ∕ [limonene] ratio (∼0.62 %), with the specific information found in Fig. 1. It is usually believed that H2O2 has two generation pathways: HO2 self-reaction and HAHPs' decomposition. The former pathway is considered as a main contributor to H2O2 during daytime, while the latter is regarded to occur without photochemistry. In the reaction system discussed here, when we applied a box model coupled with the limonene reaction mechanism extracted from Master Chemical Mechanism (MCM) v3.3 (http://mcm.leeds.ac.uk/MCMv3.3.1, last access: 15 March 2018) to simulate the reaction, the yield of H2O2 formed from the HO2 self-reaction was estimated to be less than 0.1 % under both dry and wet conditions. When the 2-butanol or cyclohexane reaction mechanism was taken into consideration, the contribution of this pathway to H2O2 formation was still very limited; hence, it was assumed that the HO2 self-reaction was not important for H2O2 generation in this reaction system. Under dry conditions, a small amount of water vapor might desorb from the flow tube wall and participate in reactions, resulting in a small amount of H2O2 formation. Thus, the high H2O2 yield observed here might be mainly attributed to SCIs' reaction with water.
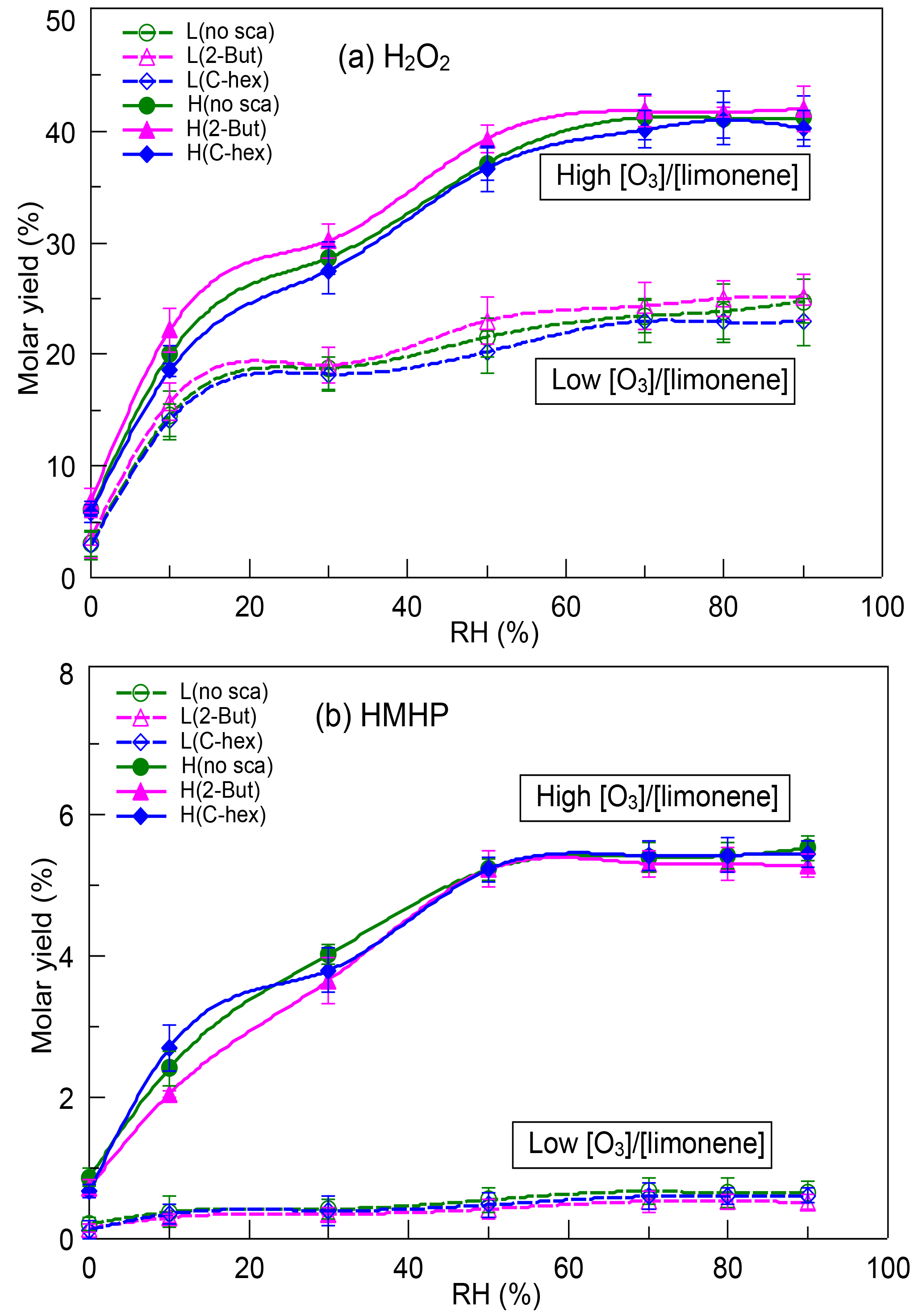
Figure 1Dependence of (a) H2O2 yield and (b) HMHP yield on relative humidity (RH) at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger (2-butanol or cyclohexane). H2O2, hydrogen peroxide; HMHP, hydroxymethyl hydroperoxide; L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
From 0 % to 70 % RH, both the H2O2 yield and HMHP yield were promoted significantly by increasing RH, indicating that the reaction with water gradually became dominant for limonene SCIs. Jiang et al. (2013) suggested that the formation of HAHPs was the most favorable pathway for limonene SCIs' reaction with H2O, and the subsequent decomposition of HAHPs was thought to be prior to generation of aldehyde and H2O2 (Chen, 2016; Kumar, 2014). Although theoretical calculation results indicate that HAHPs' decomposition is slow, some studies proved that water and acid molecules could greatly promote the decomposition process (Anglada et al., 2002, 2011; Aplincourt and Anglada, 2003; Crehuet et al., 2001), and H2O2 formation from HAHPs' decomposition occurs very rapidly (Chen et al., 2016; Winterhalter et al., 2000). The fact that few HAHPs larger than HMHP were identified in alkene ozonolysis also provided evidence for the rapid decomposition of large HAHPs. Above 70 % RH, the appearance of the limiting values of H2O2 yield and HMHP yield suggested that the water vapor concentration was high enough that the bimolecular reaction of SCIs and H2O suppressed the unimolecular decomposition of SCIs and the reactions of SCIs with other products. In previous studies, the unimolecular decomposition of SCIs and the reactions of SCIs with water showed strong structure dependence. For CH2OO and anti-CH3CHOO, the atmospheric lifetimes of their bimolecular reactions with H2O and (H2O)2 were less than 1 ms, while their lifetimes of unimolecular reaction were much longer (Lin et al., 2016; Sheps et al., 2014; Taatjes et al., 2013; Welz et al., 2012). For syn-CH3CHOO and (CH3)2COO, the atmospheric lifetimes of their bimolecular reactions with H2O and (H2O)2 were more than 100 ms, while their lifetimes of unimolecular reaction were just a few milliseconds at 298 K (Drozd et al., 2017; Huang et al., 2015; Long et al., 2018; Sheps et al., 2014; Taatjes et al., 2013; Welz et al., 2014). For larger SCIs their atmospheric lifetimes of unimolecular reaction and bimolecular reactions with H2O and (H2O)2 were not quantified. Tillmann et al. (2010) inferred that about 46 % of ECIs formed from α-pinene ozonolysis would stabilize and at 44 % RH about 65 % of SCIs formed H2O2 after reacting with water and 28 % of SCIs decomposed. Yao et al. (2014) showed that a large fraction of ECIs formed from α-cedrene were stabilized and the unimolecular decomposition of SCIs was suppressed by their bimolecular reactions. Although we could not estimate the rates of SCIs' decomposition and their reaction with water, the results here prove that the reaction with water was an essential route for limonene SCIs, and the rapid decomposition of HAHPs made an important contribution to H2O2 formation.
According to the experimental conditions elaborated in Sect. 2.2, we tried to calculate the contribution of endocyclic DB and exocyclic DB ozonolysis to H2O2 and HMHP formation, and furthermore, infer the SCIs generated in the ozonolysis of different DBs. The predicted SCI yield was derived by combining the limiting yields of H2O2 and HMHP together, as in Eq. (S3), based on the assumption that the limiting yield of H2O2 was equal to the large SCI yield (Hasson, 2001a, b). Considering that a portion of SCIs might undergo unimolecular decomposition, a lower limit yield of SCIs formed in limonene ozonolysis was estimated here. Additionally, as described in Sect. 3.3, the results demonstrated that the OH yield was not obviously affected by RH, and we speculated that the fraction of SCIs that decomposed and formed OH radicals was small. It was observed that the OH scavenger did not have a large impact on the SCIs' measurement results, while a big difference existed between the ozonolysis of two DBs. The SCI yield of the endocyclic DB ozonolysis was around 24.45 %, yet the exocyclic DB ozonolysis had a larger stabilization fraction of ECIs, which was about 42.90 %. It should be noted that, since our experiments just isolated the ozonolysis of different DBs according to their discrepancy in reaction rates, the SCI yield of the exocyclic DB ozonolysis estimated here was not for a specific product formed from endocyclic DB ozonolysis but was the sum of first-generation products with a remaining double bond, as shown in Fig. 2. The results meant that, even though exocyclic DB ozonolysis is much slower than endocyclic DB, it plays a non-negligible role in generating SCIs in limonene ozonolysis.

Figure 2The reaction scheme of endocyclic and exocyclic double bonds in limonene ozonolysis.
3.3 OH radicals generation
In this study, OH yield was determined by adding sufficient 2-butanol as a scavenger and measuring how much 2-butanone was generated. Aschmann et al. (2002) suggested that using 2-butanol to measure OH formation would be more accurate than using cyclohexane. The yield of 2-butanone formed from the reaction of 2-butanol and OH was detected previously (Aschmann et al., 2002; Baxley and Wells, 1998; Chew and Atkinson, 1996), and we used 0.66, which is the average of the reported values (Forester and Wells, 2011), to calculate OH radicals' yield. The results showed that the yield of OH radicals produced from endocyclic DB ozonolysis was 0.65±0.21, while for exocyclic DB, the OH yield was 0.24±0.13. The OH generation did not show a clear dependence on water vapor concentration. The OH yield of endocyclic DB ozonolysis was within the range of published values, while the OH yield of exocyclic DB ozonolysis was determined to be higher than that reported by Herrmann et al. (2010). The effect of water on OH formation in alkene ozonolysis is still under debate as there are discrepancies among the existing publications. Anglada et al. (2002) used quantum mechanical calculations to indicate that water could increase OH production, and Tillmann et al. (2010) reported higher OH yield under humid conditions. Nevertheless, more studies showed that OH formation was independent of water vapor concentration (Aschmann et al., 2002; Atkinson and Aschmann, 1993; Atkinson et al., 1992; Berndt et al., 2003; Forester and Wells, 2011; Hasson et al., 2003). The fact that the OH yields of both unsaturated bonds were not obviously affected by RH suggests that the major pathway of OH generation in limonene ozonolysis is the decomposition of ECIs through hydroperoxide channel. The possibility that other OH formation pathways also exist cannot be totally excluded, but we speculate that any other pathways are not significant. Hence, it could be concluded that in limonene ozonolysis, the endocyclic DB is inclined to generate OH radicals through ECIs' decomposition, while the exocyclic DB has a higher fraction of stabilization-forming SCIs.
3.4 Peroxycarboxylic acids generation
In our experiments, two kinds of peroxycarboxylic acids, PFA (CH(O)OOH) and PAA (CH3C(O)OOH), were observed, and the yields of PFA and PAA both showed large discrepancies between the low-ratio and high-ratio experiments (Fig. 3). The yields of PFA and PAA at a high [O3] ∕ [limonene] ratio were about 3 times as much as those at a low [O3] ∕ [limonene] ratio. When the exocyclic DB was ozonated, the generation of PFA and PAA was enhanced to a large extent compared with only endocyclic DB ozonolysis, indicating that exocyclic DB ozonolysis had an important impact on PFA and PAA. As far as we know, this was the first time that such high yields of PFA and PAA in alkene ozonolysis have been reported. For both PFA and PAA, the highest molar yields were observed when no OH scavenger was used, demonstrating that the OH reaction contributed to part of their formation. When OH radicals were scavenged, we speculated that PFA and PAA were mainly formed through ECIs' isomerization and decomposition following the generation of HC(O)OO and CH3C(O)OO radicals. These radicals could further react with HO2 and form peroxycarboxylic acids, which provided a plausible explanation for the higher yields of PFA and PAA observed in experiments with 2-butanol instead of cyclohexane.

Figure 3Dependence of (a) PFA yield and (b) PAA yield on relative humidity (RH) at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger (2-butanol or cyclohexane). PFA, performic acid; PAA, peracetic acid; L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
The yields of PFA and PAA had a positive relationship with water vapor concentration in all the cases, reaching the highest level at 90 % RH. Model simulation results significantly underestimated the PAA formation and ignored the RH effect, indicating some missing pathways of PAA generation related to water. Because of the deficiency of PFA mechanism in MCM, we did not simulate the PFA formation. However, the similar variation tendency of PFA and PAA yields could provide some evidence for the assumption that they might have similar formation mechanisms. In previous studies, the dominant formation pathway of PAA was considered to be the reaction of CH3C(O)OO radicals with HO2 radicals (Groß et al., 2014; Lightfoot et al., 1992; Winiberg et al., 2016), while the formation pathway of PFA had not been identified yet and was speculated to be the reaction of HC(O)OO radicals with HO2 radicals (Liang et al., 2015). The rate constant of the RO2 reaction with HO2 was investigated in a series of studies and was thought to be unaffected by water (Atkinson et al., 1999; Lightfoot et al., 1992; Tyndall et al., 2001; Wallington et al., 1992). Therefore, the increase of PFA and PAA yields with RH might be attributed to the promoting effect of water on HC(O)OO and CH3C(O)OO radical generation. Because the highly reactive peroxides were easy to lose on the wall, it might be hard for chamber studies, which usually last for hours, to observe these peroxycarboxylic acids. In field observations, PAA is widely reported in remote and urban areas (Lee et al., 2000; Liang et al., 2013; Zhang et al., 2010), yet there are few reports on PFA existence in the atmosphere (Liang et al., 2015). The results here demonstrate that limonene ozonolysis could contribute to PFA and PAA formation, and although its high instability and reactivity make PFA difficult to observe, it may have a short duration in the atmosphere.
Table 2H2O2 generation per particle mass (ng ug−1) in SOA formed with different OH scavengers in the relative humidity (RH) range of 0 %–90 % at a low [O3] ∕ [limonene] ratio.

No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
3.5 Particulate peroxides and H2O2 generation
3.5.1 H2O2 evolution at a low [O3] ∕ [limonene] ratio
H2O2 generation from SOA in the aqueous phase is thought to be a possible way of producing H2O2 in cloud water or releasing H2O2 continuously after inhalation. Here, we quantitatively measured H2O2 generation from SOA produced in limonene ozonolysis in different cases. Considering that some unstable constituents would decay rapidly when SOA extract solution was kept at room temperature (298 K), we chose to keep the SOA solution at 4 ∘C to maintain the stability of the samples and prolong their storage time. In the low-ratio experiments, the concentration of total peroxides in solution was nearly stable in 48 h. The H2O2 concentration went through a brief period of increase and then became stable. After the aqueous H2O2 concentration reached a plateau, the amount of particulate H2O2 per particle mass formed in different cases, from 0 % to 90 % RH, was calculated and shown in Table 2. Regardless of whether the OH scavenger was used or not, SOA produced at higher RH was inclined to have a higher capacity of producing H2O2. When no OH scavenger was used, the lowest H2O2 content per particle mass was ∼1.13 ng µg−1 in dry conditions, and the highest value was ∼2.45 ng µg−1 at 90 % RH. When 2-butanol was used the changing trend of H2O2 generation resembled the condition without the scavenger, and the minimum was ∼1.33 ng µg−1 at 0 % RH, while the maximum was ∼2.89 ng µg−1 at 80 % RH, which was a little higher than the value at 90 % RH. It was interesting to note that, in the presence of cyclohexane, the trend of H2O2 generation in SOA solution differed greatly from both of the above. Even under dry conditions, the H2O2 content per particle mass could reach ∼3.22 ng µg−1, and the maximum was ∼4.63 ng µg−1 at 90 % RH.
3.5.2 H2O2 evolution at a high [O3] ∕ [limonene] ratio
In the experiments with a high [O3] ∕ [limonene] ratio, SOA produced with different scavengers was found to have different rates of H2O2 generation in solution at 4 ∘C, according to which we determined appropriate detection frequency and total duration for the three kinds of SOA. SOA produced without the OH scavenger had the lowest rate of H2O2 generation, so an 8-day measurement result was reported here. For SOA produced in the presence of 2-butanol, H2O2 generation was a little faster than the former and reached the limiting value within 6 days. However, SOA produced with cyclohexane had a much faster rate than both of the above, and the H2O2 concentration in solution became stable within 3 days. For these three types of SOA, the total peroxides' concentration decreased slightly in the analysis duration, which is clarified in the Supplement.
It is obvious that SOA produced in the high-ratio experiments has a greater ability of generating H2O2, and Fig. 4 shows the time profiles of H2O2 evolution of different kinds of SOA. When no OH scavenger was used, H2O2 concentration in solution rose constantly for 6 days then became stable. The limiting value of the H2O2 generation was influenced by RH, which was ∼5.28 ng µg−1 at 0 % RH, then increased gradually with increasing RH until 80 % RH (∼14.45 ng µg−1). In the experiments with 2-butanol, the H2O2 concentration kept rising in the first 4 days, then it became stable. Under dry conditions, the limiting value of the H2O2 content was ∼7.60 ng µg−1, then it increased until 50 % RH. SOA produced above 50 % RH did not show any obvious difference, and the highest H2O2 content was ∼15.50 ng µg−1. When cyclohexane was added, the H2O2 concentration in solution increased rapidly on the first day then tended to become stable. The limiting value of H2O2 generation was also affected by water vapor concentration, yet the promoting effect was not very significant, and no large difference was observed above 30 % RH. The limiting H2O2 content per particle mass was ∼16.64 ng µg−1 in dry conditions and was ∼30.00 ng µg−1 above 30 % RH.
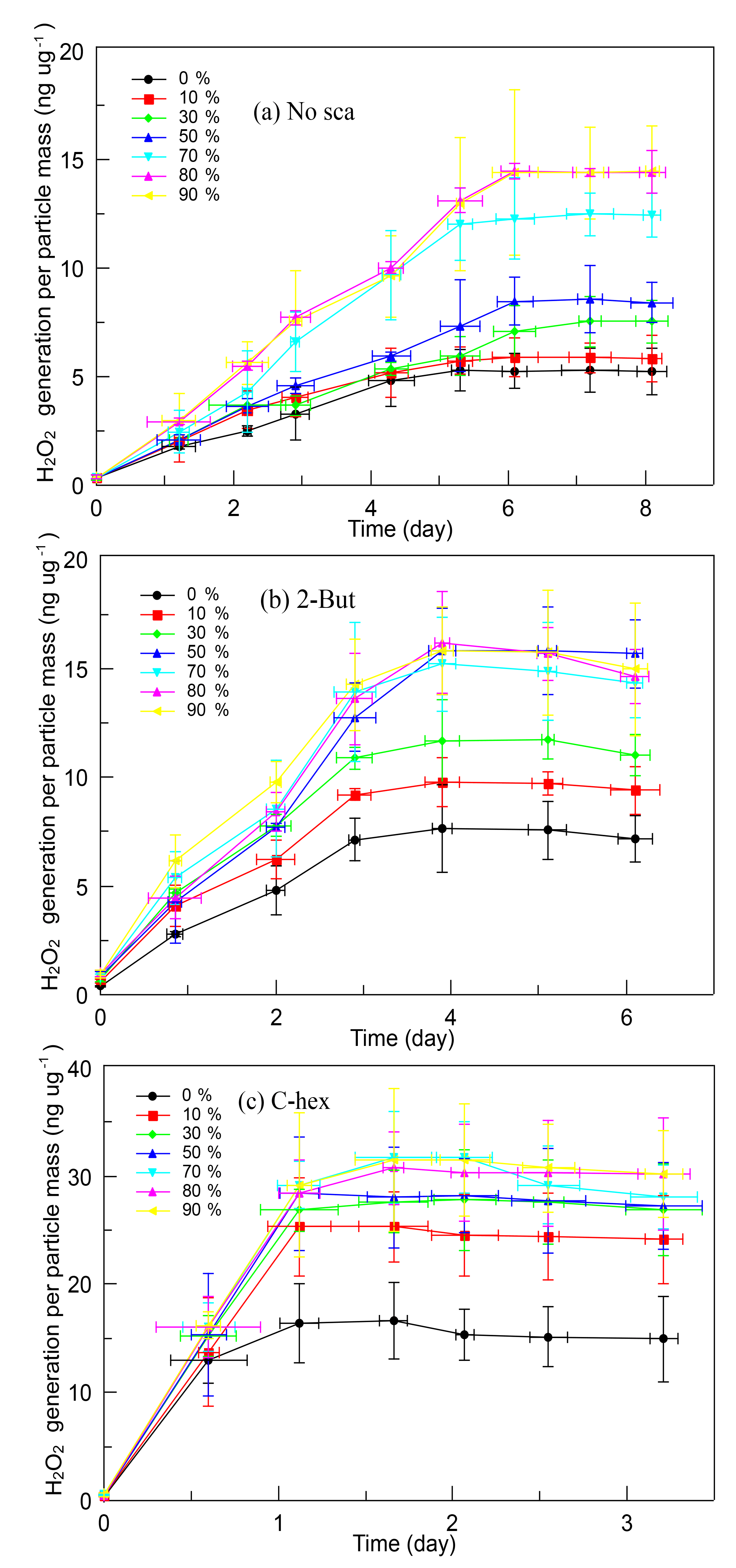
Figure 4Time profiles of H2O2 evolution per particle mass of different SOA formed (a) without the OH scavenger, (b) with 2-butanol, and (c) with cyclohexane in the relative humidity (RH) range of 0 %–90 % at a high [O3] ∕ [limonene] ratio. H2O2, hydrogen peroxide; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
3.5.3 Different stabilities of particulate peroxides
According to the measurement results of the total peroxides and H2O2 in the SOA solution, the particulate peroxides could be roughly divided into two categories: one is unstable and could decompose or hydrate to generate H2O2, the other one is stable and could nearly remain unchanged in several days. Supposing that all the peroxides contained one peroxy bond, the molar fractions of stable or unstable peroxides could be calculated as follows:
where φi is the molar fraction of stable or unstable peroxides in particles, Ni (mol) is the number of moles of stable or unstable peroxides in particles, and Ntotal (mol) is the number of moles of total peroxides in particles. At a low [O3] ∕ [limonene] ratio, the molar fraction of unstable peroxides in SOA was around 0.11–0.13 in the case of adding no scavenger, which was similar to the case of adding 2-butanol. In the presence of cyclohexane, the molar fraction of unstable peroxides would increase to 0.20–0.32. At a high [O3] ∕ [limonene] ratio, the molar fractions of unstable peroxides in the case of adding no scavenger and adding 2-butanol were also similar, with both of them in the range 0.13–0.25, yet this value would reach ∼0.50 when cyclohexane was used. Detailed information is provided in the Supplement. Model results showed that most peroxides produced in the reaction were ROOH and R(O)OOH; in addition, some studies proposed that peroxyhemiacetals also made an important contribution to aerosol (Tobias and Ziemann, 2000; Tobias et al., 2000). The acetal reaction producing peroxyhemiacetals is reversible, so part of the peroxyhemiacetals in SOA may have hydrolyzed to form some peroxides. To investigate the stabilities of ROOH and R(O)OOH, we synthesized methyl hydroperoxide (MHP) and ethyl hydroperoxide (EHP) to represent ROOH. PFA and PAA were used to represent R(O)OOH. All the synthesis solutions were stored at 4 ∘C, which was the same as the experimental condition. PFA and PAA were found to decompose and generate H2O2 within several days, yet MHP and EHP maintained stable. Hence, we speculate that peroxycarboxylic acids and peroxyhemiacetals might be the main components of unstable peroxides in particles, and they contribute to H2O2 generation in the aqueous phase.
The amount of H2O2 generation at a low [O3] ∕ [limonene] ratio measured here was comparable with the published value of H2O2 produced from α-pinene SOA in solution (Li et al., 2016; Wang et al., 2011). However, at a high [O3] ∕ [limonene] ratio, the H2O2 generation level increased significantly, which proved that the multi-generation oxidation improved the formation of peroxycarboxylic acids and peroxyhemiacetals in particles. These results demonstrated that SOA produced in limonene ozonolysis could behave as an important source of H2O2 in the aqueous phase, and they also showed the difference between the SOA formed from single-unsaturated monoterpene ozonolysis and double-unsaturated monoterpene ozonolysis.
3.5.4 The effect of the OH scavenger
Prior research has provided evidence for OH radicals' formation in ozonolysis experiments, and the OH scavenger is often used to avoid the disturbance of the OH reaction. Both 2-butanol and cyclohexane are commonly used OH scavengers, and Chew and Atkinson (1996) showed that there was no difference in their abilities to scavenge OH radicals. However, it should be noted that the OH scavenger could convert OH radicals into a mixture of hydroperoxy (HO2) and alkylperoxy (RO2) radicals, and a higher [HO2] / [RO2] ratio is observed when 2-butanol is used (Docherty and Ziemann, 2003; Jonsson et al., 2008a). As shown in the Sect. 3.6.1, the fact that both at a low and high [O3] ∕ [limonene] ratio, the SOA yield was higher with 2-butanol than cyclohexane suggested that the increase in HO2 concentration promoted SOA formation. This result is consistent with that suggested by Keywood et al. (2004) who observed the higher [HO2] / [RO2] ratio, resulting in a higher SOA yield in cyclohexene ozonolysis, while Docherty and Ziemann (2003) showed that an increased [HO2] / [RO2] ratio inhibited aerosol formation in β-pinene ozonolysis.
Some studies demonstrated that acid and peroxide products were sensitive to OH scavengers (Ahmad et al., 2017; Henry and Donahue, 2011; Ma et al., 2008). Here, through investigating the stabilities of particulate peroxides formed in different conditions, we showed that the choice of the OH scavenger would influence the types of particulate peroxides produced in the reaction. In Sect. 3.5.3, we suggest that peroxycarboxylic acids and peroxyhemiacetals be the main components of unstable peroxides in particles. The peroxyhemiacetals formed by heterogeneous reactions of peroxides and aldehydes could dissociate into these species, yet the formation of peroxyhemiacetals was not considered to be affected by the OH scavenger since both the peroxides and carbonyl compounds observed in the gas phase did not show large differences when the OH scavenger changed. We speculate that the impact of the OH scavenger on particulate peroxides could be mainly attributed to the formation of peroxycarboxylic acids under a different [HO2] / [RO2] ratio. Keywood et al. (2004) indicated that the OH scavenger impacted the HO2 reaction with acyl peroxy radicals forming acid and peracid products, which were considered to have low volatility. With higher HO2 concentration, the SOA yield in the presence of 2-butanol was higher than that in the presence of cyclohexane, since the HO2–acyl peroxy reactions contributed to some low-volatility products. However, the RO2 radicals formed from cyclohexane may also participate in reactions and help produce more unstable peroxycarboxylic acids which could partition into the particle phase.
Table 3The SOA yield and mass fraction of particulate peroxides at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger from 0 % to 90 % relative humidity (RH).
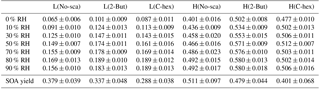
Note: L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
3.6 Contribution of peroxides to SOA
3.6.1 SOA formation
The yield of SOA produced from limonene ozonolysis in different cases ranging from 0 % to 90 % RH was measured. The SOA yield is defined as the ratio of aerosol mass concentration to the mass concentration of limonene consumed, as given by Eq. (S4). In the six different cases, SOA yield was determined to be unaffected by water, yet it showed strong dependence on the reactants' ratio and the use of the OH scavenger, as shown in Table 3. Whether at a low or high [O3] ∕ [limonene] ratio, the case of no OH scavenger had the highest SOA yield, which suggested the effect of OH reaction on aerosol formation, and the order of SOA yield was no scavenger > 2-butanol > cyclohexane. The case with the highest SOA yield was at a high [O3] ∕ [limonene] ratio without the OH scavenger (0.511±0.097), while the lowest SOA yield was detected at a low [O3] ∕ [limonene] ratio with cyclohexane (0.288±0.038).
3.6.2 Peroxides' mass fraction
In this study, we used the iodometric method to analyze the particle-phase total peroxides' content in limonene ozonolysis. The iodometric method is commonly regarded as a standard method for detecting total peroxides and it could almost quantify all kinds of peroxides (Bonn et al., 2004; Jenkin, 2004). The mass fraction used here is defined as the ratio of particulate peroxides' mass to SOA mass, as given in Eq. (S5). The average molecular weight of peroxides in particles is assumed to be 300 g mol−1, which is estimated to be slightly less than the molecular weight of peroxyhemiacetals, and this value has been used to calculate the mass of particulate peroxides in some studies (Docherty et al., 2005; Nguyen et al., 2010; Surratt et al., 2006). The mass fractions of peroxides in the six cases from 0 % to 90 % RH are summarized in Table 3. In each case, the peroxides' mass fraction in SOA increased slightly with RH, and there was a significant difference between the low-ratio and high-ratio experiments, while there was little effect of the OH scavenger. The mass fraction of peroxides at a low [O3] ∕ [limonene] ratio was usually below 0.2; however, at a high [O3] ∕ [limonene] ratio the peroxides' mass fraction increased to 0.4–0.6. Docherty et al. (2005) reported that the peroxides' mass fraction in α-pinene SOA was ∼0.47, while for β-pinene SOA the fraction was ∼0.85. Li et al. (2016) observed that peroxides could account for ∼0.21 in α-pinene SOA. We are the first to report the mass fraction of peroxides in SOA derived from limonene ozonolysis, highlighting the important role of organic peroxides in SOA composition, especially when multi-generation oxidation happens.
3.6.3 Peroxides' partitioning
The molar yields of HMW peroxides in both the gas phase and the particle phase were determined in different cases. There was little effect of RH and the OH scavenger, while at a high [O3] ∕ [limonene] ratio the HMW peroxides' yield increased by ∼10 % in contrast with the yield at a low [O3] ∕ [limonene] ratio. Furthermore, the gas-phase HMW peroxides and particle-phase HMW peroxides were discussed separately, which is shown in Fig. 5. For gas-phase HMW peroxides, the molar yield showed a decreasing tendency with increasing RH, which was obvious at a high [O3] ∕ [limonene] ratio. For particle-phase HMW peroxides, the molar yield showed a slight increasing dependence on RH and their yield was promoted significantly by the degree of oxidation. A possible explanation for the RH effect was that the water content in aerosol would increase when RH increased, and this promoted the uptake of some compounds into particles. However, some research proved that the effect of water on the partitioning of organic products seemed to be small, especially when no inorganic seed particles were used (Jonsson et al., 2006, 2008a, b). Thus, we propose that the promoting effect of water on particulate HMW peroxides can be mainly attributed to the effect of water participating in some gas-phase and particle-phase reactions, resulting in the formation of more low-volatility peroxides. The contribution of multi-generation oxidation products to SOA formation in limonene ozonolysis has been stated by some studies (Hoffmann et al., 1997; Ng et al., 2006), and the results here indicate that organic peroxides might account for a considerable proportion of these products. Multi-generation oxidation helps produce more low-volatility peroxides in the gas phase that could partition into particles, and moreover, it accelerates the occurrence of some heterogeneous reactions by providing more reactive species.
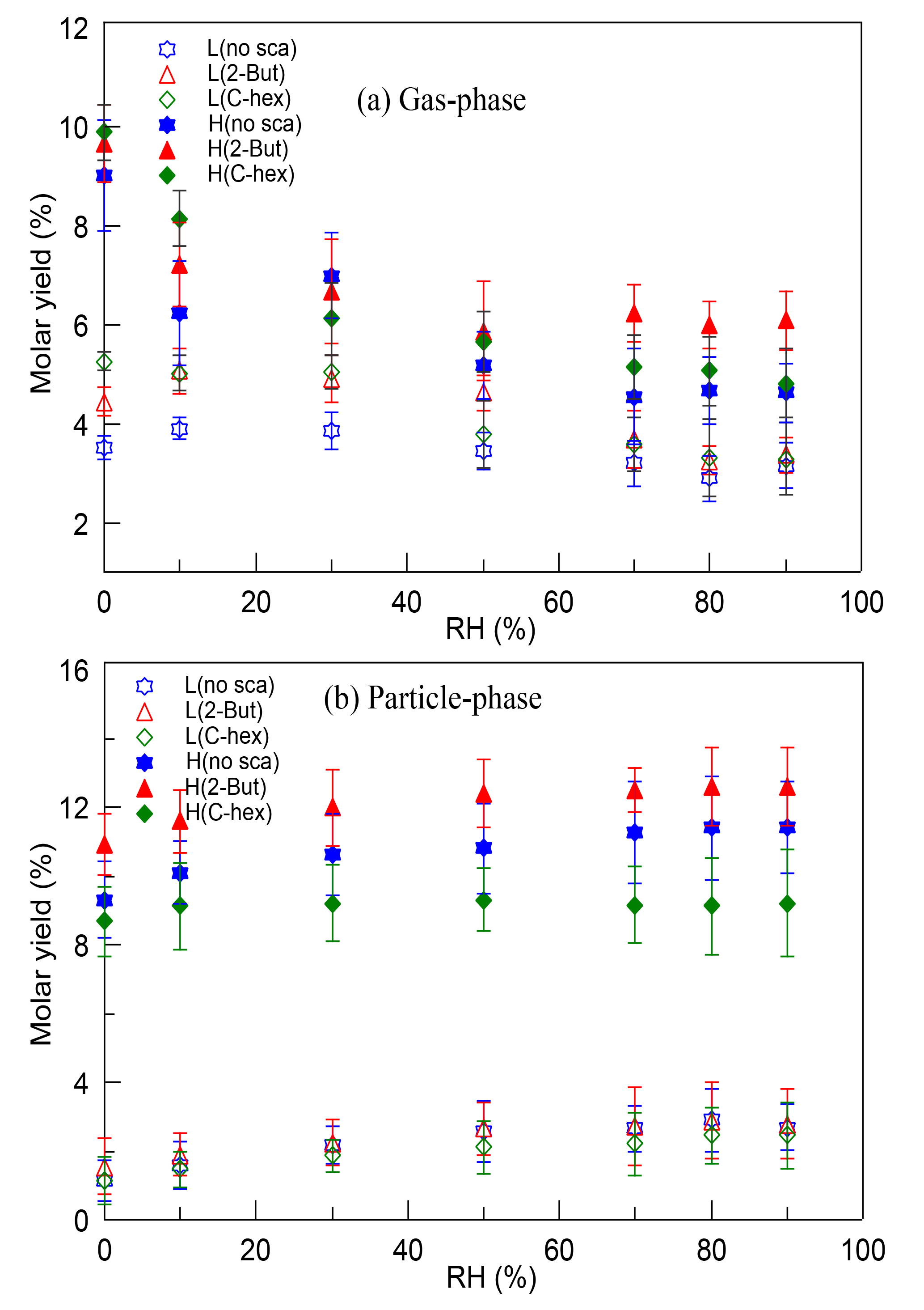
Figure 5The variation of (a) gas-phase and (b) particle-phase high-molecular-weight peroxides' molar yields with relative humidity (RH) at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger (2-butanol or cyclohexane). L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane.
Table 4Yields (%) of carbonyls at a low or high [O3] ∕ [limonene] ratio in the presence or absence of the OH scavenger.

Note: –, below detection limit; L, low [O3] ∕ [limonene] ratio; H, high [O3] ∕ [limonene] ratio; No-sca, no scavenger; 2-But, 2-butanol; C-hex, cyclohexane; HACE, hydroxyacetone; FA, formaldehyde; AA, acetaldehyde; ACE, acetone; GL, glyoxal; MGL, methylglyoxal.
3.7 Contribution of carbonyls to SOA
3.7.1 Carbonyls' formation
Carbonyl compounds including HACE, FA, AA, ACE, GL, and MGL were detected in the reaction. Unlike peroxides, whose generation often showed obvious increasing dependence on RH, only FA yield would increase with RH and the production of other carbonyls was not significantly affected by water. The yields of HACE, AA, ACE, GL, and MGL in different cases are summarized in Table 4. The fact that HACE formed without the OH scavenger but did not form when cyclohexane was used indicated that HACE might be generated from OH reactions. As for AA and ACE, both of their yields in the presence of cyclohexane were found to be lower than the case without the scavenger, especially at a low [O3] ∕ [limonene] ratio. This suggested that the OH reaction contributed to a portion of their formation and the endocyclic DB ozonolysis did not tend to generate AA and ACE. It was speculated that these three kinds of carbonyls could also be generated from the reaction of 2-butanol or that HO2 radicals promoted their formation, so the presence of 2-butanol increased their yields. As regards to GL and MGL, RH and the OH scavenger did not have a large influence, and both of their yields at a high [O3] ∕ [limonene] ratio were higher than the conditions of a low [O3] ∕ [limonene] ratio. The generation of FA had a positive dependence on RH, which was illustrated in the Supplement and the total variation range could be found in Table 4.
3.7.2 Experimental and theoretical partitioning coefficients
A parameter that has been used widely to describe the partitioning feature of a compound is described as follows (Pankow, 1994; Pankow and Bidleman, 1992):
where Kp, i (m3 µg−1) is the partitioning coefficient of compound i, TSP (µg m−3) is the concentration of total suspended particulate matter, and Fi (µg m−3) and Ai (µg m−3) are the particulate and gaseous concentrations of compound i, respectively. The measured partitioning coefficients of FA, AA, and ACE were on the magnitude of 10−5 m3 µg−1, and the partitioning coefficients of HACE, GL, and MGL were on the magnitude of 10−4 m3 µg−1.
Theoretical gas–particle partitioning coefficients of these compounds were calculated using the absorption equilibrium equation defined by Pankow (1994), which has been used widely to estimate the ability of a substance to partition into the particulate phase and to predict SOA yield in models (Griffin et al., 1999; Hohaus et al., 2015; Odum et al., 1996; Yu et al., 1999):
where R is the ideal gas constant ( m3 atm mol−1 K−1), T is the temperature (K), fOM is the mass fraction of TSP that is the absorbing organic matter (OM) and is equal to 1 here, MWOM (g mol−1) is the mean molecular weight of the OM phase, which is estimated to be 130 g mol−1 in this study, ζi is the activity coefficient of compound i in the OM phase, which is assumed to be unity, and (Torr) is the vapor pressure of compound i, which is predicted using the method of Moller et al. (2008). The calculated gas–particle partitioning coefficients of HACE, FA, AA, ACE, GL, and MGL were m3 µ g−1, m3 µg−1, m3 µg−1, m3 µg−1, m3 µg−1, and m3 µg−1, respectively, at 298 K. The experimental Kp value of HACE was about 10 000 times larger than the theoretical value, and for other carbonyls, the experimental Kp value was about 100 000 times larger than the theoretical value. The gap between the experimental Kp and predicted Kp of GL and MGL we estimated was comparable with the results of Healy et al. (2008, 2009), but it was higher than that of Ortiz et al. (2013). The fact that the gas–particle partitioning coefficients of carbonyls observed were much higher than the theoretical values indicated that carbonyl compounds made a more important contribution to SOA formation than estimated. Although the partitioning coefficients measured in experiments showed a huge difference with the calculated values, a relationship between the measured Kp and the vapor pressure of carbonyl compounds was observed. Figure 6 shows the dependence of the measured lg(Kp) and predicted lg(Kp) on lg(p0), and their linear fitting curves. The slope of the linear fitting equation of the predicted lg(Kp) versus lg(p0) was −0.964, and R2 was 0.998. The slope of the linear fitting equation of measured lg(Kp) versus lg(p0) was −0.484, and R2 was 0.750, which indicated that the lg(Kp) of carbonyls observed in laboratory also had a negative correlation with lg(p0). A plausible explanation for the large difference between the measured and predicted Kp was that carbonyl compounds were easy to polymerize and react with other species on particles, resulting in these carbonyls existing in the form of hydrates and oligomers in SOA (Corrigan et al., 2008; Hastings et al., 2005; Kroll et al., 2005; Volkamer et al., 2007). The hydrates and oligomers of carbonyls have much lower vapor pressures than their precursors, and they could reversibly return to their carbonyl monomers during analysis (Healy et al., 2008; Ortiz et al., 2013; Toda et al., 2014).

Figure 6The relationship of measured and predicted partitioning coefficients (m3 µg−1) versus vapor pressure (atm) of carbonyls produced in limonene ozonolysis. Kp, partitioning coefficients; p0, vapor pressure, HACE, hydroxyacetone; FA, formaldehyde; AA, acetaldehyde; ACE, acetone; GL, glyoxal; MGL, methylglyoxal.
This work reports on an experimental study of the oxidation regime and SOA composition in limonene ozonolysis with respect to the roles of different DBs, radicals, and water. A series of oxidizing products including SCIs, OH radicals, and peroxides in both gaseous and particulate phases were detected. Based on the variation of H2O2 and HMHP generation on RH, the importance of the reaction of limonene SCIs with water was confirmed and the yield of SCIs could be estimated to be ∼0.24 for endocyclic DB and ∼0.43 for exocyclic DB. OH yields of endocyclic and exocyclic DBs were indirectly determined to be ∼0.65 and ∼0.24, demonstrating the different reaction mechanisms of different DBs in limonene ozonolysis. The formation of two peroxycarboxylic acids, PFA and PAA, was observed, and their high yields were first reported in alkene ozonolysis. The yields of PFA and PAA increased with RH and the degree of oxidation, showing the effect of water and the exocyclic DB oxidation on their formation. The H2O2 generation from SOA in solution provided evidence for the ability of SOA to contribute oxidants in the aqueous phase. And through investigating the stabilities of particulate peroxides formed under different conditions, we showed that the choice of the OH scavenger would influence the types of particulate peroxides produced in the reaction. The partitioning behaviors of peroxides and carbonyls were discussed in this paper, and the results showed their importance to SOA formation. Particulate peroxides could account for less than 0.2 in limonene SOA at a low [O3] ∕ [limonene] ratio and 0.4–0.6 at a high [O3] ∕ [limonene] ratio, which proved the important role of peroxides in SOA composition, especially when multi-generation oxidation happened. Due to their ability to polymerize and react with other species on particles, the carbonyl compounds often exist in SOA in the form of hydrates and oligomers, whose vapor pressures are much lower than that of their precursors. The partitioning coefficients of carbonyls observed in the laboratory were always several orders of magnitude higher than the theoretical values, indicating that their contribution to SOA was higher than estimated. Limonene is unique in many aspects because of its different DBs, suggesting that terpenes with multiple double bonds may have more complex effects on the atmosphere than previously thought. The results demonstrated the effect of the degree of oxidation, radical chemistry, and water on limonene SOA composition, while some particulate products, especially peroxides, should be studied further. Limonene chemistry in the atmosphere is characterized by the multi-generation oxidation that happens to different DBs of limonene. More laboratory evidence is needed to investigate whether the phenomena could be observed in other terpenes' oxidation that contain more than one DB.
The data are accessible by contacting the corresponding author (zmchen@pku.edu.cn).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-15105-2018-supplement.
YG and ZC designed the study. YG conducted the experiments, analyzed data, and wrote the paper. ZC helped to interpret the results and modify the manuscript. HL contributed to some methods of analysis and helped to modify the paper.
The authors declare that they have no conflict of interest.
We gratefully acknowledge the National Natural Science Foundation of China
(grant 21477002) and the National Key Research and Development Program of
China (grant 2016YFC0202704) for financial support.
Edited by: Markus Ammann
Reviewed by: two anonymous referees
Ahmad, W., Coeur, C., Cuisset, A., Coddeville, P., and Tomas, A.: Effects of scavengers of Criegee intermediates and OH radicals on the formation of secondary organic aerosol in the ozonolysis of limonene, J. Aerosol. Sci., 110, 70–83, https://doi.org/10.1016/j.jaerosci.2017.05.010, 2017.
Andersson-Sköld, Y. and Simpson, D.: Secondary organic aerosol formation in northern Europe: A model study, J. Geophys. Res., 106, 7357–7374, https://doi.org/10.1029/2000JD900656, 2001.
Anglada, J. M., Aplincourt, P., Bofill, J. M., and Cremer, D.: Atmospheric formation of OH radicals and H2O2 from alkene ozonolysis under humid conditions, Chem. Phys. Chem., 3, 215–221, https://doi.org/10.1002/1439-7641(20020215)3:2<215::Aid-Cphc215>3.3.Co;2-V, 2002.
Anglada, J. M., González, J., and Torrent-Sucarrat, M.: Effects of the substituents on the reactivity of carbonyl oxides. A theoretical study on the reaction of substituted carbonyl oxides with water, Phys. Chem. Chem. Phys., 13, 13034–13045, https://doi.org/10.1039/C1CP20872A, 2011.
Aplincourt, P. and Anglada, J. M.: Theoretical studies of the isoprene ozonolysis under tropospheric conditions. 2. Unimolecular and water-assisted decomposition of the α-hydroxy hydroperoxides, J. Phys. Chem. A, 107, 5812–5820, https://doi.org/10.1021/jp034203w, 2003.
Aschmann, S. M., Arey, J., and Atkinson, R.: OH radical formation from the gas-phase reactions of O3 with a series of terpenes, Atmos. Environ., 36, 4347–4355, https://doi.org/10.1016/S1352-2310(02)00355-2, 2002.
Atkinson, R.: Gas-phase tropospheric chemistry of organic compounds: A review, Atmos. Environ. Part A, 24, 1–41, https://doi.org/10.1016/0960-1686(90)90438-S, 1990.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Atkinson, R. and Aschmann, S. M.: OH radical production from the gas-phase reactions of O3 with a series of alkenes under atmospheric conditions, Environ. Sci. Technol., 27, 1357–1363, 1993.
Atkinson, R., Aschmann, S. M., Arey, J., and Shorees, B.: Formation of OH radicals in the gas phase reactions of O3 with a series of terpenes, J. Geophys. Res., 97, 6065–6073, https://doi.org/10.1029/92JD00062, 1992.
Atkinson, R., Baulch, D. L., Cox, R. A., Hampson, R. F., Kerr, J. A., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry, organic species: Supplement VII, J. Phys. Chem. Ref. Data, 28, 191–393, https://doi.org/10.1063/1.556048, 1999.
Baxley, J. S. and Wells, J. R.: The hydroxyl radical reaction rate constant and atmospheric transformation products of 2-butanol and 2-pentanol, Int. J. Chem. Kinet., 30, 745–752, https://doi.org/10.1002/(Sici)1097-4601(1998)30:10<745::Aid-Kin7>3.0.Co;2-V, 1998.
Becker, K. H., Brockmann, K. J., and Bechara, J.: Production of hydrogen peroxide in forest air by reaction of ozone with terpenes, Nature, 346, 256–268, https://doi.org/10.1038/346256a0, 1990.
Becker, K. H., Bechara, J., and Brockmann, K. J.: Studies on the formation of H2O2 in the ozonolysis of alkenes, Atmos. Environ. Part A, 27, 57–61, https://doi.org/10.1016/0960-1686(93)90070-F, 1993.
Berndt, T., Böge, O., and Stratmann, F.: Gas-phase ozonolysis of α-pinene: gaseous products and particle formation, Atmos. Environ., 37, 3933–3945, https://doi.org/10.1016/S1352-2310(03)00501-6, 2003.
Bonn, B., Kuhlmann, R. V., and Lawrence, M. G.: High contribution of biogenic hydroperoxides to secondary organic aerosol formation, Geophys. Res. Lett., 31, L10108, https://doi.org/10.1029/2003GL019172, 2004.
Calogirou, A., Larsen, B. R., and Kotzias, D.: Gas-phase terpene oxidation products: a review, Atmos. Environ., 33, 1423–1439, https://doi.org/10.1016/S1352-2310(98)00277-5, 1999.
Calvert, J. G., Lazrus, A., Kok, G. L., Heikes, B. G., Walega, J. G., Lind, J., and Cantrell, C. A.: Chemical mechanisms of acid generation in the troposphere, Nature, 317, 27–35, https://doi.org/10.1038/317027a0, 1985.
Chen, L., Wang, W. L., Wang, W. N., Liu, Y. L., Liu, F. Y., Liu, N., and Wang, B. Z.: Water-catalyzed decomposition of the simplest Criegee intermediate CH2OO, Theor. Chem. Acc., 135, 131, https://doi.org/10.1007/s00214-016-1894-9, 2016.
Chen, X., Hopke, P. K., and Carter, W. P. L.: Secondary organic aerosol from ozonolysis of biogenic volatile organic compounds: chamber studies of particle and reactive oxygen species formation, Environ. Sci. Technol., 45, 276–282, https://doi.org/10.1021/es102166c, 2011.
Chen, Z. M., Wang, H. L., Zhu, L. H., Wang, C. X., Jie, C. Y., and Hua, W.: Aqueous-phase ozonolysis of methacrolein and methyl vinyl ketone: a potentially important source of atmospheric aqueous oxidants, Atmos. Chem. Phys., 8, 2255–2265, https://doi.org/10.5194/acp-8-2255-2008, 2008.
Chew, A. A. and Atkinson, R.: OH radical formation yields from the gas-phase reactions of O3 with alkenes and monoterpenes, J. Geophys. Res., 101, 28649–28653, https://doi.org/10.1029/96JD02722, 1996.
Clausen, P. A., Wilkins, C. K., Wolkoff, P., and Nielsen, G. D.: Chemical and biological evaluation of a reaction mixture of R-(+)-limonene/ozone: Formation of strong airway irritants, Environ. Int., 26, 511–522, https://doi.org/10.1016/S0160-4120(01)00035-6, 2001.
Cocker, D. R., Flagan, R. C., and Seinfeld, J. H.: State-of-the-art chamber facility for studying atmospheric aerosol chemistry, Environ. Sci. Technol., 35, 2594–2601, https://doi.org/10.1021/es0019169, 2001.
Corrigan, A. L., Hanley, S. W., and Hann, D. O. D.: Uptake of glyoxal by organic and inorganic aerosol, Environ. Sci. Technol., 42, 4428–4433, https://doi.org/10.1021/es7032394, 2008.
Crehuet, R., Anglada, J. M., and Bofill, J. M.: Tropospheric formation of hydroxymethyl hydroperoxide, formic acid, H2O2, and OH from carbonyl oxide in the presence of water vapor: A theoretical study of the reaction mechanism, Chem. Eur. J., 7, 2227–2235, https://doi.org/10.1002/1521-3765(20010518)7:10<2227::AID-CHEM2227>3.0.CO;2-O, 2001.
Cremer, D., Gauss, J., Kraka, E., Stanton, J. F., and Bartlett, R. J.: A CCSD (T) investigation of carbonyl oxide and dioxirane. Equilibrium geometries, dipole moments, infrared spectra, heats of formation and isomerization energies, Chem. Phys. Lett., 209, 547–556, https://doi.org/10.1016/0009-2614(93)80131-8, 1993.
Criegee, R.: Mechanism of ozonolysis, Angew. Chem. Int. Edit., 14, 745–752, https://doi.org/10.1002/anie.197507451, 1975.
Crounse, J. D., Mckinney, K. A., Kwan, A. J., and Wennberg, P. O.: Measurement of gas-phase hydroperoxides by chemical ionization mass spectrometry, Anal. Chem., 78, 6726–6732, https://doi.org/10.1021/ac0604235, 2006.
Docherty, K. S. and Ziemann, P. J.: Effects of stabilized Criegee intermediate and OH radical scavengers on aerosol formation from reactions of β-pinene with O3, Aerosol Sci. Technol., 37, 877–891, https://doi.org/10.1080/02786820300930, 2003.
Docherty, K. S., Wu, W., Lim, Y. B., and Ziemann, P. J.: Contributions of organic peroxides to secondary aerosol formed from reactions of monoterpenes with O3, Environ. Sci. Technol., 39, 4049–4059, https://doi.org/10.1021/es050228s, 2005.
Drozd, G. T., Kurén, T., Donahue, N. M., and Lester, M. I.: Unimolecular decay of the dimethyl-substituted Criegee intermediate in alkene ozonolysis: decay time scales and the importance of tunneling, J. Phys. Chem. A, 121, 6036–6045, https://doi.org/10.1021/acs.jpca.7b05495, 2017.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Fellin, P. and Otson, R.: Assessment of the influence of climatic factors on concentration levels of volatile organic compounds (VOCs) in canadian homes, Atmos. Environ., 28, 3581–3586, https://doi.org/10.1016/1352-2310(94)00204-X, 1994.
Fenske, J. D., Hasson, A. S., Ho, A. W., and Paulson, S. E.: Measurement of absolute unimolecular and bimolecular rate constants for CH3CHOO generated by the trans-2-butene reaction with ozone in the gas phase, J. Phys. Chem. A, 104, 9921–9932, https://doi.org/10.1021/jp0016636, 2000.
Fischer, R. and Maier, O.: Interrelation of oxidative stress and inflammation in neurodegenerative disease: role of TNF, Oxid. Med. Cell Longev., 2015, 610813, https://doi.org/10.1155/2015/610813, 2015.
Forester, C. D. and Wells, J. R.: Hydroxyl radical yields from reactions of terpene mixtures with ozone, Indoor Air, 21, 400–409, https://doi.org/10.1111/j.1600-0668.2011.00718.x, 2011.
Gäb, S., Hellpointner, E., Turner, W. V., and Koŕte, F.: Hydroxymethyl hydroperoxide and bis(hydroxymethyl) peroxide from gas-phase ozonolysis of naturally occurring alkenes, Nature, 316, 535–536, https://doi.org/10.1038/316535a0, 1985.
Glasius, M., Lahaniati, M., Calogirou, A., Di Bella, D., Jensen, N. R., Hjorth, J., Kotzias, D., and Larsen, B. R.: Carboxylic acids in secondary aerosols from oxidation of cyclic monoterpenes by ozone, Environ. Sci. Technol., 34, 1001–1010, https://doi.org/10.1021/es990445r, 2000.
Griffin, R. J., Cocker, D. R., Seinfeld, J. H., and Dabdub, D.: Estimate of global atmospheric organic aerosol from oxidation of biogenic hydrocarbons, Geophys. Res. Lett., 26, 2721–2724, https://doi.org/10.1029/1999GL900476, 1999.
Grosjean, D., Williams, E. L., and Seinfeld, J. H.: Atmospheric oxidation of selected terpenes and related carbonyls: gas-phase carbonyl products, Environ. Sci. Technol., 26, 1526–1533, https://doi.org/10.1021/es00032a005, 1992.
Grosjean, D., Williams, E. L., Grosjean, E., Andino, J. M., and Seinfeld, J. H.: Atmospheric oxidation of biogenic hydrocarbons: reaction of ozone with β-pinene, d-limonene, and trans-caryophyllene, Environ. Sci. Technol., 27, 2754–2758, https://doi.org/10.1021/es00049a014, 1993.
Grossmann, D., Moortgat, G. K., Kibler, M., Schlomski, S., Bachmann, K., Alicke, B., Geyer, A., Platt, U., Hammer, M. U., Vogel, B., Mihelcic, D., Hofzumahaus, A., Holland, F., and Volz-Thomas, A.: Hydrogen peroxide, organic peroxides, carbonyl compounds, and organic acids measured at Pabstthum during BERLIOZ, J. Geophys. Res., 108, 8250, https://doi.org/10.1029/2001JD001096, 2003.
Groß, C. B. M., Dillon, T. J., Schuster, G., Lelieveld, J., and Crowley, J. N.: Direct kinetic study of OH and O3 formation in the reaction CH3C(O)O2 with HO2, J. Phys. Chem. A, 118, 974–985, https://doi.org/10.1021/jp412380z, 2014.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Gupta, R. K., Patel, A. K., Shah, N., Choudhary, A. K., Jha, U. K., Yadav, U. C., Gupta, P. K., and Pakuwal, U.: Oxidative stress and antioxidants in disease and Cancer: a review, Asian Pac. J. Cancer P., 15, 4405–4409, https://doi.org/10.7314/APJCP.2014.15.11.4405, 2014.
Gutbrod, R., Schindler, R. N., Kraka, E., and Cremer, D.: Formation of OH radicals in the gas phase ozonolysis of alkenes: the unexpected role of carbonyl oxides, Chem. Phys. Lett., 252, 221–229, https://doi.org/10.1016/0009-2614(96)00126-1, 1996.
Gutbrod, R., Kraka, E., Schindler, R. N., and Cremer, D.: Kinetic and theoretical investigation of the gas-phase ozonolysis of isoprene: Carbonyl oxides as an important source for OH radicals in the atmosphere, J. Am. Chem. Soc., 119, 7330–7342, https://doi.org/10.1021/ja970050c, 1997.
Hasson, A. S., Orzechowska, G., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes: 1. Ethene, trans-2-butene, and 2,3-dimethyl-2-butene, J. Geophys. Res., 106, 34131–34142, https://doi.org/10.1029/2001JD000597, 2001a.
Hasson, A. S., Ho, A. W., Kuwata, K. T., and Paulson, S. E.: Production of stabilized Criegee intermediates and peroxides in the gas phase ozonolysis of alkenes: 2. Asymmetric and biogenic alkenes, J. Geophys. Res., 106, 34143–34153, https://doi.org/10.1029/2001JD000598, 2001b.
Hasson, A. S., Chung, M. Y., Kuwata, K. T., Converse, A. D., Krohn, D., and Paulson, S. E.: Reaction of Criegee intermediates with water vapor – an additional source of OH radicals in alkene ozonolysis, J. Phys. Chem. A, 107, 6176–6182, https://doi.org/10.1021/jp0346007, 2003.
Hastings, W. P., Koehler, C. A., Bailey, E. L., and Haan, D. O. D.: Secondary organic aerosol formation by glyoxal hydration and oligomer formation: Humidity effects and equilibrium shifts during analysis, Environ. Sci. Technol., 39, 8728–8735, https://doi.org/10.1021/es050446l, 2005.
Healy, R. M., Wenger, J. C., Metzger, A., Duplissy, J., Kalberer, M., and Dommen, J.: Gas/particle partitioning of carbonyls in the photooxidation of isoprene and 1,3,5-trimethylbenzene, Atmos. Chem. Phys., 8, 3215–3230, https://doi.org/10.5194/acp-8-3215-2008, 2008.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of relative humidity on gas/particle partitioning and aerosol mass yield in the photooxidation of p-xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Heaton, K. J., Dreyfus, M. A., Wang, S. Y., and Johnston, M. V.: Oligomers in the early stage of biogenic secondary organic aerosol formation and growth, Environ. Sci. Technol., 41, 6129–6136, https://doi.org/10.1021/es070314n, 2007.
Henry, K. M. and Donahue, N. M.: Effect of the OH radical scavenger hydrogen peroxide on secondary organic aerosol formation from α-pinene ozonolysis, Aerosol Sci. Technol., 45, 696–700, https://doi.org/10.1080/02786826.2011.552926, 2011.
Herrmann, F., Winterhalter, R., Moortgat, G. K., and Williams, J.: Hydroxyl radical (OH) yields from the ozonolysis of both double bonds for five monoterpenes, Atmos. Environ., 44, 3458–3464, https://doi.org/10.1016/j.atmosenv.2010.05.011, 2010.
Hoffmann, T., Odum, J. R., Bowman, F., Collins, D., Klockow, D., Flagan, R. C., and Seinfeld, J. H.: Formation of organic aerosols from the oxidation of biogenic hydrocarbons, J. Atmos. Chem., 26, 189–222, https://doi.org/10.1023/A:1005734301837, 1997.
Hohaus, T., Gensch, I., Kimmel, J., Worsnop, D. R., and Kiendler-Scharr, A.: Experimental determination of the partitioning coefficient of β-pinene oxidation products in SOAs, Phys. Chem. Chem. Phys., 17, 14796–14804, https://doi.org/10.1039/c5cp01608h, 2015.
Hua, W., Chen, Z. M., Jie, C. Y., Kondo, Y., Hofzumahaus, A., Takegawa, N., Chang, C. C., Lu, K. D., Miyazaki, Y., Kita, K., Wang, H. L., Zhang, Y. H., and Hu, M.: Atmospheric hydrogen peroxide and organic hydroperoxides during PRIDE-PRD'06, China: their concentration, formation mechanism and contribution to secondary aerosols, Atmos. Chem. Phys., 8, 6755–6773, https://doi.org/10.5194/acp-8-6755-2008, 2008.
Huang, D., Chen, Z. M., Zhao, Y., and Liang, H.: Newly observed peroxides and the water effect on the formation and removal of hydroxyalkyl hydroperoxides in the ozonolysis of isoprene, Atmos. Chem. Phys., 13, 5671–5683, https://doi.org/10.5194/acp-13-5671-2013, 2013.
Huang, H. L., Chao, W., and Lin, J. J. M.: Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2, P. Natl. Acad. Sci., 112, 10857–10862, https://doi.org/10.1073/pnas.1513149112, 2015.
Jenkin, M. E.: Modelling the formation and composition of secondary organic aerosol from α- and β-pinene ozonolysis using MCM v3, Atmos. Chem. Phys., 4, 1741–1757, https://doi.org/10.5194/acp-4-1741-2004, 2004.
Jiang, L., Lan, R., Xu, Y. S., Zhang, W. J., and Yang, W.: Reaction of stabilized Criegee intermediates from ozonolysis of limonene with water: Ab Initio and DFT study, Int. J. Mol. Sci., 14, 5784–5805, https://doi.org/10.3390/ijms14035784, 2013.
Jonsson, A. M., Hallquist, M., and Ljungström, E.: Impact of humidity on the ozone initiated oxidation of limonene, 3-carene, and α-pinene, Environ. Sci. Technol., 40, 188–194, https://doi.org/10.1021/es051163w, 2006.
Jonsson, A. M., Hallquist, M., and Ljungström, E.: Influence of OH scavenger on the water effect on secondary organic aerosol formation from ozonolysis of limonene, 3-carene, and α-pinene, Environ. Sci. Technol., 42, 5938–5944, https://doi.org/10.1021/es702508y, 2008a.
Jonsson, Å. M., Hallquist, M., and Ljungström, E.: The effect of temperature and water on secondary organic aerosol formation from ozonolysis of limonene, δ3-carene and α-pinene, Atmos. Chem. Phys., 8, 6541–6549, https://doi.org/10.5194/acp-8-6541-2008, 2008b.
Keywood, M. D., Kroll, J. H., Varutbangkul, V., Bahreini, R., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from cyclohexene ozonolysis: Effect of OH scavenger and the role of radical chemistry, Environ. Sci. Technol., 38, 3343–3350, https://doi.org/10.1021/es049725j, 2004.
Khan, M. A. H., Cooke, M. C., Utembe, S. R., Xiao, P., Morris, W. C., Derwent, R. G., Archibald, A. T., Jenkin, M. E., Percival, C. J., and Shallcross, D. E.: The global budgets of organic hydroperoxides for present and pre-industrial scenarios, Atmos. Environ., 110, 65–74, https://doi.org/10.1016/j.atmosenv.2015.03.045, 2015.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Clarke, J. S., Donahue, N. M., Anderson, J. G., and Demerjian, K. L.: Mechanism of HOx formation in the gas-phase ozone-alkene reaction. 1. Direct, pressure-dependent measurements of prompt OH yields, J. Phys. Chem. A, 105, 1554–1560, https://doi.org/10.1021/jp002121r, 2001a.
Kroll, J. H., Sahay, S. R., Anderson, J. G., Demerjian, K. L., and Donahue, N. M.: Mechanism of HOx formation in the gas-phase ozone-alkene reaction. 2. Prompt versus thermal dissociation of carbonyl oxides to form OH, J. Phys. Chem. A, 105, 4446–4457, https://doi.org/10.1021/jp004136v, 2001b.
Kroll, J. H., Ng, N. L., Murphy, S. M., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds, J. Geophys. Res., 110, D23207, https://doi.org/10.1029/2005JD006004, 2005.
Kumar, M., Busch, D. H., Subramaniam, B., and Thompson, W. H.: Role of tunable acid catalysis in decomposition of α-hydroxyalkyl hydroperoxides and mechanistic implications for tropospheric chemistry, J. Phys. Chem. A, 118, 9701–9711, https://doi.org/10.1021/jp505100x, 2014.
Lamb, B., Gay, D., Westberg, H., and Pierce, T.: A biogenic hydrocarbon emission inventory for the U.S.A. using a simple forest canopy model, Atmos. Environ. Part A, 27, 1673–1690, https://doi.org/10.1016/0960-1686(93)90230-V, 1993.
Lane, T. E., Donahue, N. M., and Pandis, S. N.: Simulating secondary organic aerosol formation using the volatility basis-set approach in a chemical transport model, Atmos. Environ., 42, 7439–7451, https://doi.org/10.1016/j.atmosenv.2008.06.026, 2008.
Lee, A., Goldstein, A. H., Keywood, M. D., Gao, S., Varutbangkul, V., Bahreini, R., Ng, N. L., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes, J. Geophys. Res., 111, D07302, https://doi.org/10.1029/2005JD006437, 2006.
Lee, M. H., Heikes, B. G., and O'Sullivan, D. W.: Hydrogen peroxide and organic hydroperoxide in the troposphere: a review, Atmos. Environ., 34, 3475–3494, https://doi.org/10.1016/S1352-2310(99)00432-X, 2000.
Leungsakul, S., Jaoui, M., and Kamens, R. M.: Kinetic mechanism for predicting secondary organic aerosol formation from the reaction of d-limonene with ozone, Environ. Sci. Technol., 39, 9583–9594, https://doi.org/10.1021/es0492687, 2005.
Li, H., Chen, Z., Huang, L., and Huang, D.: Organic peroxides' gas-particle partitioning and rapid heterogeneous decomposition on secondary organic aerosol, Atmos. Chem. Phys., 16, 1837–1848, https://doi.org/10.5194/acp-16-1837-2016, 2016.
Liang, H., Chen, Z. M., Huang, D., Zhao, Y., and Li, Z. Y.: Impacts of aerosols on the chemistry of atmospheric trace gases: a case study of peroxides and HO2 radicals, Atmos. Chem. Phys., 13, 11259–11276, https://doi.org/10.5194/acp-13-11259-2013, 2013.
Liang, H., Chen, Z. M., Huang, D., Wu, Q. Q., and Huang, L. B.: Understanding atmospheric peroxyformic acid chemistry: observation, modeling and implication, Atmos. Chem. Phys. Discuss., 15, 2055–2084, https://doi.org/10.5194/acpd-15-2055-2015, 2015.
Lightfoot, P. D., Cox, R. A., Crowley, J. N., Destriau, M., Hayman, G. D., Jenkin, M. E., Moortgat, G. K., and Zabel, F.: Organic peroxy radicals: Kinetics, spectroscopy and tropospheric chemistry, Atmos. Environ. Part A, 26, 1805–1961, https://doi.org/10.1016/0960-1686(92)90423-I, 1992.
Lin, L. C., Chang, H. T., Chang, C. H., Chao, W., Smith, M. C., Chang, C. H., Lin, J. J. M., and Takahashi, K.: Competition between H2O and (H2O)2 reactions with CH2OO ∕ CH3CHOO, Phys. Chem. Chem. Phys., 18, 4557–4568, https://doi.org/10.1039/c5cp06446e, 2016.
Long, B., Bao, J. L., and Truhlar, D. G.: Unimolecular reaction of acetone oxide and its reaction with water in the atmosphere, P. Natl. Acad. Sci. USA, 115, 6135–6140, https://doi.org/10.1073/pnas.1804453115, 2018.
Ma, Y., Russell, A. T., and Marston, G.: Mechanisms for the formation of secondary organic aerosol components from the gas-phase ozonolysis of α-pinene, Phys. Chem. Chem. Phys., 10, 4294–4312, https://doi.org/10.1039/b803283a, 2008.
Mauldin, R. L., Berndt, T., Sipilä, M., Paasonen, P., Petäjä, T., Kim, S., Kurtén, T., Stratmann, F., Kerminen, V. M., and Kulmala, M.: A new atmospherically relevant oxidant of sulphur dioxide, Nature, 488, 193–196, https://doi.org/10.1038/nature11278, 2012.
Mcneill, V. F., Woo, J. L., Kim, D. D., Schwier, A. N., Wannell, N. J., Sumner, A. J., and Barakat, J. M.: Aqueous-phase secondary organic aerosol and organosulfate formation in atmospheric aerosols: A modeling study, Environ. Sci. Technol., 46, 8075–8081, https://doi.org/10.1021/es3002986, 2012.
Mertes, P., Pfaffenberger, L., Dommen, J., Kalberer, M., and Baltensperger, U.: Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP), Atmos. Meas. Tech., 5, 2339–2348, https://doi.org/10.5194/amt-5-2339-2012, 2012.
Möller, D.: Atmospheric hydrogen peroxide: Evidence for aqueous-phase formation from a historic perspective and a one-year measurement campaign, Atmos. Environ., 43, 5923–5936, https://doi.org/10.1016/j.atmosenv.2009.08.013, 2009.
Moller, B., Rarey, J., and Ramjugernath, D.: Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions, J. Mol. Liq., 143, 52–63, https://doi.org/10.1016/j.molliq.2008.04.020, 2008.
Mutzel, A., Rodigast, M., Iinuma, Y., Böge, O., and Herrmann, H.: An improved method for the quantification of SOA bound peroxides, Atmos. Environ., 67, 365–369, https://doi.org/10.1016/j.atmosenv.2012.11.012, 2013.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of first- versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Nguyen, T. B., Bateman, A. P., Bones, D. L., Nizkorodov, S. A., Laskin, J., and Laskin, A.: High-resolution mass spectrometry analysis of secondary organic aerosol generated by ozonolysis of isoprene, Atmos. Environ., 44, 1032–1042, https://doi.org/10.1016/j.atmosenv.2009.12.019, 2010.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/particle partitioning and secondary organic aerosol yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Ortiz, R., Shimada, S., Sekiguchi, K., Wang, Q., and Sakamoto, K.: Measurements of changes in the atmospheric partitioning of bifunctional carbonyls near a road in a suburban area, Atmos. Environ., 81, 554–560, https://doi.org/10.1016/j.atmosenv.2013.09.045, 2013.
Pankow, J. F.: An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 28, 189–193, https://doi.org/10.1016/1352-2310(94)90094-9, 1994.
Pankow, J. F. and Bidleman, T. F.: Interdependence of the slopes and intercepts from log-log correlations of measured gas-particle partitioning and vapor pressure 1. theory and analysis of available data, Atmos. Environ. Part A, 26, 1071–1080, https://doi.org/10.1016/0960-1686(92)90039-N, 1992.
Pathak, R. K., Salo, K., Emanuelsson, E. U., Cai, C., Lutz, A., Hallquist, A. M., and Hallquist, M.: Influence of ozone and radical chemistry on limonene organic aerosol production and thermal characteristics, Environ. Sci. Technol., 46, 11660–11669, https://doi.org/10.1021/es301750r, 2012.
Peña, R. M., García, S., Herrero, C., and Lucas, T.: Measurements and analysis of hydrogen peroxide rainwater levels in a Northwest region of Spain, Atmos. Environ., 35, 209–219, https://doi.org/10.1016/S1352-2310(00)00246-6, 2001.
Penkett, S. A., Jones, B. M. R., Brich, K. A., and Eggleton, A. E. J.: The importance of atmospheric ozone and hydrogen peroxide in oxidising sulphur dioxide in cloud and rainwater, Atmos. Environ., 13, 123–137, https://doi.org/10.1016/0004-6981(79)90251-8, 1979.
Presto, A. A. and Donahue, N. M.: Ozonolysis fragment quenching by nitrate formation: The pressure dependence of prompt OH radical formation, J. Phys. Chem. A, 108, 9096–9104, https://doi.org/10.1021/jp047162s, 2004.
Presto, A. A., Hartz, K. E. H., and Donahue, N. M.: Secondary organic aerosol production from terpene ozonolysis. 1. Effect of UV radiation, Environ. Sci. Technol., 39, 7036–7045, https://doi.org/10.1021/es050174m, 2005.
Prinn, R. G.: The cleansing capacity of the atmosphere, Annu. Rev. Environ. Resour., 28, 29–57, https://doi.org/10.1146/annurev.energy.28.011503.163425, 2003.
Pye, H. O. T. and Seinfeld, J. H.: A global perspective on aerosol from low-volatility organic compounds, Atmos. Chem. Phys., 10, 4377–4401, https://doi.org/10.5194/acp-10-4377-2010, 2010.
Sakamoto, Y., Yajima, R., Inomata, S., and Hirokawa, J.: Water vapour effects on secondary organic aerosol formation in isoprene ozonolysis, Phys. Chem. Chem. Phys., 19, 3165–3175, https://doi.org/10.1039/c6cp04521a, 2017.
Sauer, F., Schuster, G., Schäfer, C., and Moortgat, G. K.: Determination of H2O2 and organic peroxides in cloud- and rain-water on the Kleiner Feldberg during FELDEX, Geophys. Res. Lett., 23, 2605–2608, https://doi.org/10.1029/96GL02475, 1996.
Sauer, F., Limbach, S., and Moortgat, G. K.: Measurements of hydrogen peroxide and individual organic peroxides in the marine troposphere, Atmos. Environ., 31, 1173–1184, https://doi.org/10.1016/S1352-2310(96)00289-0, 1997.
Sauer, F., Schäfer, C., Neeb, P., Horie, O., and Moortgat, G. K.: Formation of hydrogen peroxide in the ozonolysis of isoprene and simple alkenes under humid conditions, Atmos. Environ., 33, 229–241, https://doi.org/10.1016/S1352-2310(98)00152-6, 1999.
Seifert, B., Mailahn, W., Schulz, C., and Ullrich, D.: Seasonal variation of concentrations of volatile organic compounds in selected German homes, Environ. Int., 15, 397–408, https://doi.org/10.1016/0160-4120(89)90054-8, 1989.
Sheps, L., Scully, A. M., and Au, K.: UV absorption probing of the conformer-dependent reactivity of a Criegee intermediate CH3CHOO, Phys. Chem. Chem. Phys., 16, 26701–26706, https://doi.org/10.1039/c4cp04408h, 2014.
Shu, Y. H. and Atkinson, R.: Rate constants for the gas-phase reactions of O3 with a series of terpenes and OH radical formation from the O3 reactions with sesquiterpenes at 296±2 K, Int. J. Chem. Kinet., 26, 1193–1205, https://doi.org/10.1002/kin.550261207, 1994.
Sindelarova, K., Granier, C., Bouarar, I., Guenther, A., Tilmes, S., Stavrakou, T., Müller, J.-F., Kuhn, U., Stefani, P., and Knorr, W.: Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years, Atmos. Chem. Phys., 14, 9317–9341, https://doi.org/10.5194/acp-14-9317-2014, 2014.
Sipilä, M., Jokinen, T., Berndt, T., Richters, S., Makkonen, R., Donahue, N. M., Mauldin III, R. L., Kurtén, T., Paasonen, P., Sarnela, N., Ehn, M., Junninen, H., Rissanen, M. P., Thornton, J., Stratmann, F., Herrmann, H., Worsnop, D. R., Kulmala, M., Kerminen, V.-M., and Petäjä, T.: Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids, Atmos. Chem. Phys., 14, 12143–12153, https://doi.org/10.5194/acp-14-12143-2014, 2014.
Stroud, C., Makar, P., Karl, T., Guenther, A., Geron, C., Turnipseed, A., Nemitz, E., Baker, B., Potosnak, M., and Fuentes, J. D.: Role of canopy-scale photochemistry in modifying biogenic-atmosphere exchange of reactive terpene species: Results from the CELTIC field study, J. Geophys. Res., 110, D17303, https://doi.org/10.1029/2005JD005775, 2005.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical composition of secondary organic aerosol formed from the photooxidation of isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Taatjes, C. A., Welz, O., Eskola, A. J., Savee, J. D., Scheer, A. M., Shallcross, D. E., Rotavera, B., Lee, E. P. F., Dyke, J. M., Mok, D. K. W., Osborn, D. L., and Percival, C. J.: Direct measurements of conformer-dependent reactivity of the Criegee intermediate CH3CHOO, Science, 340, 177–180, https://doi.org/10.1126/science.1234689, 2013.
Tillmann, R., Hallquist, M., Jonsson, Å. M., Kiendler-Scharr, A., Saathoff, H., Iinuma, Y., and Mentel, Th. F.: Influence of relative humidity and temperature on the production of pinonaldehyde and OH radicals from the ozonolysis of α-pinene, Atmos. Chem. Phys., 10, 7057–7072, https://doi.org/10.5194/acp-10-7057-2010, 2010.
Tobias, H. J. and Ziemann, P. J.: Thermal desorption mass spectrometric analysis of organic aerosol formed from reactions of aerosol formed from reactions of 1-tetradecene and O3 in the presence of alcohols and carboxylic acids, Environ. Sci. Technol., 34, 2105–2115, https://doi.org/10.1021/es9907156, 2000.
Tobias, H. J., Docherty, K. S., Beving, D. E., and Ziemann, P. J.: Effect of relative humidity on the chemical composition of secondary organic aerosol formed from reactions of 1-tetradecene and O3, Environ. Sci. Technol., 34, 2116–2125, https://doi.org/10.1021/es991057s, 2000.
Toda, K., Yunoki, S., Yanaga, A., Takeuchi, M., Ohira, S., Dasgupta, P. K., Formaldehyde content of atmospheric aerosol, Environ. Sci. Technol., 48, 6636–6643, https://doi.org/10.1021/es500590e, 2014.
Tyndall, G. S., Cox, R. A., Granier, C., Lesclaux, R., Moortgat, G. K., Pilling, M. J., Ravishankara, A. R., and Wallington, T. J.: Atmospheric chemistry of small organic peroxy radicals, J. Geophys. Res., 106, 12157–12182, https://doi.org/10.1029/2000JD900746, 2001.
Verma, V., Ning, Z., Cho, A. K., Schauer, J. J., Shafer, M. M., and Sioutas, C.: Redox activity of urban quasi-ultrafine particles from primary and secondary sources, Atmos. Environ., 43, 6360–6368, https://doi.org/10.1016/j.atmosenv.2009.09.019, 2009.
Volkamer, R., Martini, F. S., Molina, L. T., Salcedo, D., Jimenez, J. L., and Molina, M. J.: A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol, Geophys. Res. Lett., 34, L19807, https://doi.org/10.1029/2007GL030752, 2007.
Wallington, T. J., Dagaut, P., and Kurylo, M. J.: UV absorption cross section kinetics and mechanisms for peroxy radicals in the gas phase, Chem. Rev., 92, 667–710, https://doi.org/10.1021/cr00012a008, 1992.
Wang, H. L., Zhang, X., and Chen, Z. M.: Development of DNPH/HPLC method for the measurement of carbonyl compounds in the aqueous phase: applications to laboratory simulation and field measurement, Environ. Chem., 6, 389–397, https://doi.org/10.1071/EN09057, 2009.
Wang, Y., Kim, H., and Paulson, S. E.: Hydrogen peroxide generation from α- and β-pinene and toluene secondary organic aerosols, Atmos. Environ., 45, 3149–3156, https://doi.org/10.1016/j.atmosenv.2011.02.060, 2011.
Wegener, R., Brauers, T., Koppmann, R., Bares, S. R., Rohrer, F., Tillmann, R., Wahner, A., Hansel, A., and Wisthaler, A.: Simulation chamber investigation of the reactions of ozone with short-chained alkenes, J. Geophys. Res., 112, D13301, https://doi.org/10.1029/2006JD007531, 2007.
Welz, O., Savee, J. D., Osborn, D. L., Vasu, S. S., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Direct kinetic measurements of Criegee intermediate (CH2OO) formed by reaction of CH2I with O2, Science, 335, 204–207, https://doi.org/10.1126/science.1213229, 2012.
Welz, O., Eskola, A. J., Sheps, L., Rotavera, B., Savee, J. D., Scheer, A. M., Osborn, D. L., Lowe, D., Booth, A. M., Xiao, P., Khan, M. A. H., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: Rate coefficients of C1 and C2 Criegee intermediate reactions with formic and acetic acid near the collision limit: direct kinetics measurements and atmospheric implications, Angew. Chem. Int. Ed., 53, 4547–4550, https://doi.org/10.1002/anie.201400964, 2014.
Winiberg, F. A. F., Dillon, T. J., Orr, S. C., Groß, C. B. M., Bejan, I., Brumby, C. A., Evans, M. J., Smith, S. C., Heard, D. E., and Seakins, P. W.: Direct measurements of OH and other product yields from the HO2 + ?CH3C(O)O2 reaction, Atmos. Chem. Phys., 16, 4023–4042, https://doi.org/10.5194/acp-16-4023-2016, 2016.
Winterhalter, R., Neeb, P., Grossmann, D., Kolloff, A., Horie, O., and Moortgat, G.: Products and mechanism of the gas phase reaction of ozone with β-pinene, J. Atmos. Chem., 35, 165–197, https://doi.org/10.1023/A:1006257800929, 2000.
Wolkoff, P., Clausen, P. A., Wilkins, C. K., and Nielsen, G. D.: Formation of strong airway irritants in terpene/ozone mixtures, Indoor Air, 10, 82–91, https://doi.org/10.1034/j.1600-0668.2000.010002082.x, 2000.
Wragg, F. P. H., Fuller, S. J., Freshwater, R., Green, D. C., Kelly, F. J., and Kalberer, M.: An automated online instrument to quantify aerosol-bound reactive oxygen species (ROS) for ambient measurement and health-relevant aerosol studies, Atmos. Meas. Tech., 9, 4891–4900, https://doi.org/10.5194/amt-9-4891-2016, 2016.
Yao, L., Ma, Y., Wang, L., Zheng, J., Khalizov, A., Chen, M. D., Zhou, Y. Y., Qi, L., and Cui, F. P.: Role of stabilized Criegee Intermediate in secondary organic aerosol formation from the ozonolysis of α-cedrene, Atmos. Environ., 94, 448–457, https://doi.org/10.1016/j.atmosenv.2014.05.063, 2014.
Yu, J. Z., Cocker, D. R., Griffin, R. J., Flagan, R. C., and Seinfeld, J. H.: Gas-phase ozone oxidation of monoterpenes: gaseous and particulate products, J. Atmos. Chem., 34, 207–258, https://doi.org/10.1023/A:1006254930583, 1999.
Zhang, D. and Zhang, R. Y.: Ozonolysis of α-pinene and β-pinene: kinetics and mechanism, J. Chem. Phys., 122, 114308, https://doi.org/10.1063/1.1862616, 2005.
Zhang, J. Y., Hartz, K. E. H., Pandis, S. N., and Donahue, N. M.: Secondary organic aerosol formation from limonene ozonolysis: homogeneous and heterogeneous influences as a function of NOx, J. Phys. Chem. A, 110, 11053–11063, https://doi.org/10.1021/jp062836f, 2006.
Zhang, X., Chen, Z. M., He, S. Z., Hua, W., Zhao, Y., and Li, J. L.: Peroxyacetic acid in urban and rural atmosphere: concentration, feedback on PAN-NOx cycle and implication on radical chemistry, Atmos. Chem. Phys., 10, 737–748, https://doi.org/10.5194/acp-10-737-2010, 2010.