the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 22 Aug 2018
| 22 Aug 2018
Average versus high surface ozone levels over the continental USA: model bias, background influences, and interannual variability
Jean J. Guo
Arlene M. Fiore
Lee T. Murray
Daniel A. Jaffe
Jordan L. Schnell
Charles T. Moore
George P. Milly
US background ozone (O3) includes O3 produced from anthropogenic O3 precursors emitted outside of the USA, from global methane, and from any natural sources. Using a suite of sensitivity simulations in the GEOS-Chem global chemistry transport model, we estimate the influence from individual background sources versus US anthropogenic sources on total surface O3 over 10 continental US regions from 2004 to 2012. Evaluation with observations reveals model biases of +0–19 ppb in seasonal mean maximum daily 8 h average (MDA8) O3, highest in summer over the eastern USA. Simulated high-O3 events cluster too late in the season. We link these model biases to excessive regional O3 production (e.g., US anthropogenic, biogenic volatile organic compounds (BVOCs), and soil NOx, emissions), or coincident missing sinks. On the 10 highest observed O3 days during summer (O3_top10obs_JJA), US anthropogenic emissions enhance O3 by 5–11 ppb and by less than 2 ppb in the eastern versus western USA. The O3 enhancement from BVOC emissions during summer is 1–7 ppb higher on O3_top10obs_JJA days than on average days, while intercontinental pollution is up to 2 ppb higher on average versus on O3_top10obs_JJA days. During the summers of 2004–2012, monthly regional mean US background O3 MDA8 levels vary by up to 15 ppb from year to year. Observed and simulated summertime total surface O3 levels on O3_top10obs_JJA days decline by 3 ppb (averaged over all regions) from 2004–2006 to 2010–2012, reflecting rising US background (+2 ppb) and declining US anthropogenic O3 emissions (−6 ppb) in the model. The model attributes interannual variability in US background O3 on O3_top10obs days to natural sources, not international pollution transport. We find that a 3-year averaging period is not long enough to eliminate interannual variability in background O3 on the highest observed O3 days.
- Article
(2293 KB) - Full-text XML
-
Supplement
(9731 KB) - BibTeX
- EndNote





In the USA, ozone (O3) is regulated as a criteria pollutant under the National Ambient Air Quality Standard (NAAQS). The current NAAQS for ground-level O3, set in October 2015, states that the fourth highest daily maximum 8 h average (MDA8) O3, averaged across three consecutive years, cannot be 71 ppb or higher (U.S. Environmental Protection Agency, 2015). The 3-year average is nominally intended to smooth out fluctuations in O3 levels resulting from natural variability in meteorology within the timing constraints of the federal Clean Air Act for air quality planning. As even 1 ppb of excess O3 may be enough to push a county out of NAAQS attainment, it is relevant to understand which sources influence the severity and timing of the highest O3 events. Since measured O3 does not retain a signature of the source from which it was produced, estimates of background O3 rely on models, ideally evaluated closely with observations, to build confidence in the model capability for source attribution. Here we apply a global chemistry transport model alongside O3 observations to examine which sources are influencing average versus high-O3 events, and the extent to which they vary from year to year.
As US anthropogenic emissions of O3 precursors decline, the relative importance of US background O3 to total surface O3 rises. US background O3 is defined here as the O3 levels that would exist in the absence of US anthropogenic emissions of O3 precursors, nitrogen oxide (NOx) and non-methane volatile organic compounds (NMVOCs). US background O3 thus includes naturally occurring O3 as well as O3 produced from global methane (including US anthropogenic emissions) and from O3 precursor emissions outside of the USA. Jaffe et al. (2018) review the current understanding on US background O3 from models and observations and its relevance to air quality standard setting and implementation. Previous studies estimating background O3 over the USA found that background sources of O3, including stratospheric O3 intrusions (Lin et al., 2012, 2015a), increasing Asian anthropogenic emissions (Lin et al., 2015b), and more frequent wildfires in summer (Abatzoglou and Williams, 2016; Jaffe, 2011; Yang et al., 2015), may present challenges to obtaining the O3 standard, especially since regional emission controls may be offset by a warming climate (Fiore et al., 2015). At high-altitude sites in the western USA (WUS) in spring, the influence from stratospheric intrusions and foreign transport, combined with relatively deep planetary boundary layers, can lead to high background O3 events (Fiore et al., 2002; Zhang et al., 2011). Lin et al. (2017) investigated surface O3 trends over the USA from 1980 to 2014 with the GFDL AM3 model and found that emissions controls decreased the 95th percentile summer O3 values in the eastern USA (EUS) by 5–10 ppb over 1988–2014, but rising Asian emissions increased this O3 metric by 2–8 ppb at individual sites in the WUS over the period (Lin et al., 2017).
Earlier work in the GEOS-Chem model analyzing background O3 during a single meteorological year noted a tendency for the model to underestimate springtime O3 at high-altitude WUS sites but overestimate summertime O3 over the EUS (e.g., Fiore et al., 2002, 2003; Wang et al., 2009; Zhang et al., 2011, 2014). Identifying the extent to which these biases reflect poor representation of US anthropogenic versus background O3 sources is relevant for assessing uncertainties in estimates of background O3 on days when the O3 NAAQS is exceeded. We build upon prior studies by analyzing MDA8 O3 measurements and 9-year model simulations spanning 2004–2012 from the GEOS-Chem 3-D global chemistry transport model (CTM). A suite of sensitivity simulations in which different emissions of O3 precursors are perturbed allows us to identify which sources are contributing the most to observed high-O3 days and on the days with the highest model bias. We assess here whether biases in the model reflect problems in the modeled transported background O3 versus O3 produced within the US from both background and anthropogenic sources. In addition, the availability of these simulations for 2004–2012 allows us to investigate the year-to-year variability in background sources and the extent to which this variability is relevant for observed high events, and therefore, potentially to attaining the O3 NAAQS. Though coarse-resolution global models such as GEOS-Chem will mix emissions into the same grid cell that may remain separate in the real atmosphere, a global model is necessary to quantify background O3 transported intercontinentally, including that produced via oxidation of methane. We estimate the influence from various individual background sources on O3 concentrations and the interannual variability in background O3 levels with a focus on the highest 10 events in each of the 10 U.S. EPA regions during each summer (JJA) or year. We aim to answer the following questions: (1) which sources exert the strongest influence on O3 on the 10 days with the highest model biases against observations? (2) Which background sources influence total O3 the most on average versus the 10 highest O3 days? (3) Which sources influence the interannual variability of O3 in each region on average versus the 10 highest O3 days?
2.1 Observations
We use observed 2004–2012 MDA8 O3 data from the EPA Air Quality System (AQS) network of urban, suburban, and rural monitoring sites, the Clean Air Status and Trends Network (CASTNet), and the Mount Bachelor Observatory (https://digital.lib.washington.edu/researchworks/browse?type=subject&value=Mt.+Bachelor+Observatory, last access: 22 January 2018) in Oregon. MDA8 O3 values for the AQS sites were downloaded from https://aqs.epa.gov/aqsweb/airdata/download_files.html#Daily (2004–2012 data last updated 28 June 2013). This dataset includes 1644 total sites from the contiguous USA from 2004 to 2012, with 1207 to 1333 sites collecting data each year (U.S. Environmental Protection Agency, 2014) (Table S1 in the Supplement).
The CASTNet (ftp://ftp.epa.gov/castnet/data) O3 monitoring sites are located in rural areas away from emission sources and densely populated regions. CASTNet sites are designed to capture background O3 levels and characterize broad spatial and temporal O3 trends. We calculate the MDA8 O3 concentration from hourly values at 108 CASTNet sites with data between 2004 and 2012, requiring at least 18 h of data per day for each MDA8 O3 calculation.
The Mount Bachelor Observatory, established in 2004 by the University of Washington Jaffe Research Group, is located 2.7 km above sea level on the summit of Mount Bachelor, an extinct volcano in the Cascade Mountains of central Oregon. It provides an estimate of baseline O3 levels over the west coast of the USA (Baylon et al., 2016). Baseline O3 is defined as the O3 concentration at sites with negligible influence from local emissions (National Research Council, 2010). Baseline O3 is a measurable quantity and differs from background O3 in that it contains some influence from US anthropogenic emissions that were not recently emitted but contributed to the global background. This station is analyzed as a standalone site in Sect. 3.2, given the relevance of high-altitude measurements for downwind surface O3 (Stauffer et al., 2017). We take all hourly O3 concentrations from Mount Bachelor and calculate the MDA8 O3 concentrations for 2004–2012. Daily averages are included only if at least 18 h of data are available and monthly averages require at least 20 days with valid 24 h mean or MDA8 data. For our comparison to monthly average O3 at Mount Bachelor Observatory, we sample the model both at the level closest to 2.7 km and at the surface.
We use temperature data from a resolution gridded dataset developed by Fan and van den Dool (2008) from the Global Historical Climatology Network (GHCN) and the Climate Anomaly Monitoring System (CAMS). GHCN Gridded V2 data were provided by the NOAA/OAR/ESRL PSD, Boulder, Colorado, USA (https://www.esrl.noaa.gov/psd/). Each observational site is matched to the model grid cell it falls in and the average monthly temperature is computed by averaging across all the sites in each region.
In order to evaluate the GEOS-Chem model O3 simulation (described below in Sect. 2.3) at a spatial scale comparable to the coarse horizontal resolution global grid (), we use a grid of surface MDA8 O3 measurements, interpolated from the AQS, CASTNet, and Canadian NAPS networks (Schnell and Prather, 2017). We degrade this dataset to to match the horizontal resolution of the GEOS-Chem simulations. As we did not archive 3-D high-frequency data, all MDA8 O3 values from the model are sampled at the lowest surface layer for comparison to observational sites.
2.2 Analysis regions
Each observational site in the EPA AQS and CASTNet datasets is linked to 1 of the 10 U.S. EPA air quality regions (Fig. S1 in the Supplement) based on which state the site is in. The Mount Bachelor data were included with Region 10 (Pacific Northwest) sites even though this site is not a regulatory monitor. Following Reidmiller et al. (2009), we select two regions, the Southeast (Region 4) and Mountains and Plains (Region 8), as representative regions for the EUS and WUS for illustration purposes in the main text. Figures for the other eight regions are included in the Supplement.
To find the daily mean O3 concentration within each region, we first match each observational site to the model grid within which it falls. We then average across all sites in each region to obtain a regional mean MDA8 O3 value in the observations and in the model. From the regionally averaged observed MDA8 O3, we find (1) the 10 days with the highest observed O3 during each year (hereafter, O3_top10obs days), similar to the definition for extreme events used in Schnell et al. (2014), (2) the 10 days with the highest O3 observations during each season (hereafter, O3_top10obs_MAM, O3_top10obs_JJA, and O3_top10obs_SON), and (3) the fourth highest MDA8 O3 within each year. In addition, we sample the model to find the 10 days each year with the highest positive biases. There is at most a 2–6-day overlap between the top 10 O3_Base days and the top 10 most biased days in 2004–2012 across all regions, but during most years, the overlap is around 0–2 days. We restrict our analysis to examining the top 10 observed O3 days as these days are most relevant from a policy perspective. We use O3_top10obs as our primary metric, however, instead of the policy-relevant fourth highest O3 because the model bias is typically lower on O3_top10obs days (Fig. S2 versus Fig. S3). On the days when the fourth highest values occur, the model bias is generally more strongly negative in the West and South Central regions and more strongly positive in the Midwest than on O3_top10obs days (Figs. S2, S3). In addition, while the model rarely captures the exact day when the observed fourth highest MDA8 O3 event occurs, there is a 3–4-day overlap on average between the O3_top10obs days and the highest 10 MDA8 O3 days in the model. This overlap is similar to the 3- and 6-day overlap Jaffe et al. (2018) found in their regional models for 1 May to 29 September 2011.
2.3 GEOS-Chem model simulations
We use the GEOS-Chem v9_02 global 3-D chemical transport model (CTM) (http://www.geos-chem.org) simulations driven by Modern-Era Retrospective analysis for Research and Applications (MERRA) reanalysis meteorology from the NASA Global Modeling and Assimilation Office for 2004–2012 (Rienecker et al., 2011). The MERRA reanalysis is available at horizontal resolution, which we degrade here to horizontal resolution. MERRA meteorology captures summer mean surface temperatures to within 1–2 K across US regions and precipitation to within 0.5 mm d−1 except for over the Northern Great Plains where a positive bias exceeds 1 mm d−1, but the variance in summer mean precipitation is lower than observed in some regions (Bosilovich, 2013). While interannual variability in cloudiness observed at weather stations is largely captured by MERRA, the reanalysis generally underestimates cloud cover and thus overestimates observed downward surface shortwave fluxes (Free et al., 2016). Methane surface concentrations are prescribed each month using spatially interpolated surface distributions from NOAA Global Monitoring Division flash data. We use the standard v9_02 chemical mechanism which includes recycling of isoprene nitrates (Mao et al., 2013) in contrast to the mechanisms used in earlier versions of GEOS-Chem (e.g., Zhang et al., 2014 as discussed in Fiore et al., 2014). Anthropogenic base emissions are from the Emission Database for Global Atmospheric Research (EDGAR) version 3.2 FT2000 inventory (Olivier et al., 2005) for inorganic compounds and the REanalysis of the TROpospheric chemical composition (RETRO) inventory (Hu et al., 2015; Schultz, 2007) for organic compounds. Inorganic emissions are overwritten by regional inventories for the US (EPA National Emissions Inventory 2005), Canada (Criteria Air Contaminants), Mexico (Big Bend Regional Aerosol and Visibility Observational study; Kuhns and Green, 2003), Europe (European Monitoring and Evaluation Programme; Auvray and Bey, 2005), and South and East Asia (Streets et al., 2006). Separate global inventories are used for ammonia (Bouwman et al., 1997), black carbon (Bond et al., 2007; Leibensperger et al., 2012), and ethane (Xiao et al., 2008). Anthropogenic surface emissions have diurnal and monthly variability, some with additional weekly cycles, and are scaled each year on the basis of economic data and estimates provided by individual countries, where available (van Donkelaar et al., 2008). The model does not include daily variations in US anthropogenic emissions associated with higher electricity demand on hotter days (e.g., Abel et al., 2017). Aircraft emissions are from the Aviation Emissions Inventory Code (AEIC) inventory (Stettler et al., 2011) and shipping emissions are from the International Comprehensive Ocean-Atmosphere Data Set (ICOADS; Lee et al., 2011; Wang et al., 2008). Biomass burning emissions follow the interannually varying monthly Global Fire Emissions Database version 3 (GFED3) inventory driven by satellite observations of fire activity (Giglio et al., 2010; van der Werf et al., 2010). Biofuel emissions are constant (Yevich and Logan, 2003). Biogenic VOC (volatile organic compound) emissions from terrestrial plants follow the Model of Emissions of Gases and Aerosols from Nature (MEGAN) scheme version 2.1 (Guenther et al., 2012) and vary with meteorology (Barkley et al., 2011). Global and US emissions are 29.5 and 5.2 Tg N yr−1, respectively, for anthropogenic NOx emissions (including biofuels), 4.2 and 0.1 Tg N yr−1 for biomass burning, 8.7 and 0.9 Tg N yr−1 for soil NOx, 6.7 and 1.0 Tg N yr−1 for lightning NOx, and 466.1 and 20.6 Tg C yr−1 for isoprene emissions. Emissions for NOx sources and isoprene are provided globally and within the USA for each year in Table S3.
We first perform a base simulation (O3_Base) with all emissions turned on for 2003–2012. We conduct a parallel suite of sensitivity simulations, in which selected sources are removed. In all simulations, we discard 2003 from our analysis as initialization. Our first set of sensitivity simulations estimates three different “background” definitions: (1) “North American background” (denoted O3_NAB) in which anthropogenic emissions within Canada, Mexico, and the USA are set to zero, but methane surface abundances are kept at present-day values; (2) “US background” (O3_USB), which is similar to O3_NAB except only US anthropogenic emissions are set to zero; and (3) “Natural background” (O3_NAT), in which all anthropogenic emissions have been removed globally and methane is prescribed at preindustrial levels. We estimate Canadian and Mexican influence (O3_CA+MX) on US O3 by subtracting O3_NAB from O3_USB; the influence from intercontinental pollution transport plus global methane (O3_ICT+CH4) is estimated by subtracting O3_NAT from O3_NAB. A second set of sensitivity simulations enables us to estimate the contribution of individual background sources to total simulated surface O3 by subtracting a simulation with that source shut off from the O3_Base simulation: (1) O3_NALNOx by turning off North American lightning NOx, (2) O3_SNOx by zeroing out global soil NOx, (3) O3_BVOC by zeroing out terrestrial biogenic VOC emissions (we also examine this “O3_noBVOC” simulation in Sect. 3.3), and (4) O3_BB by zeroing out biomass burning emissions, as summarized in Table 1. Due to nonlinearities in atmospheric photochemistry, these “zero out” estimates of source contributions depend on the presence of all other precursor emissions at present-day levels (e.g., the impact of BVOC emissions is sensitive to the amount of anthropogenic NOx emissions in the Base simulation). This set of model simulations does not directly isolate stratospheric O3 or Asian influences. Previous work has shown that stratospheric O3 can increase springtime O3 levels by 17–40 ppb in the WUS when MDA8 O3 levels are 70–85 ppb, and Asian emissions can contribute 8–15 ppb to MDA8 O3 on days above 60 ppb (Lin et al., 2012, 2015a). Stratospheric and Asian influences are included in O3_USB, Asian influences are included in O3_ICT+CH4, and O3_NAT includes stratospheric O3, biogenic emissions of O3 precursors, wildfires, and lightning NOx. As O3_BVOC includes O3 produced from biogenic VOC reacting with both natural and anthropogenic NOx, O3_USA and O3_BVOC are not additive. O3_BVOC thus contributes to both O3_USA and O3_USB.
Table 1Sensitivity simulations with the GEOS-Chem model and their application to estimate sources of ground-level O3.

3.1 MDA8 O3 distributions
To evaluate the ability of our coarse-resolution model to capture observed high-O3 events, we compare the MDA8 O3 averaged over each of the 10 EPA regions simulated by GEOS-Chem to the observations in two ways. In the first method, we use the Schnell and Prather (2017) gridded dataset degraded to the model resolution and sample the model directly at each of the degraded Schnell grid cells prior to calculating the regional average. In the second method, we sample the model grid cell containing each individual observational site (EPA AQS, CASTNet, and Mount Bachelor Observatory) prior to calculating the regional average. The model is biased positively with either method (Fig. 1a, b), but the shape of the model distribution constructed with the latter approach (Fig. 1b) better matches the observed distribution than that of the former (Fig. 1a). Matching individual sites to the nearest model grid (Fig. 1b) yields a better estimate of high-O3 days; the model overestimates the percentage of days above 70 ppb by about 3 times when we match them to individual measurement sites (3.14 % of days are above 70 ppb in the observations versus 9.92 % in model) but by about 10 times in comparison to the re-gridded Schnell et al. (2014) dataset (0.37 % of days are above 70 ppb in the observations versus 3.91 % in the re-gridded dataset).

Figure 1Frequency distribution of regionally averaged US MDA8 O3 values from 2004 to 2012 in the (a) Schnell and Prather (2017) dataset interpolated to and (b) at individual observational sites prior to averaging over each of the 10 EPA regions (total number of points is 9 years × 365 or 366 days × 10 regions) in the observations (blue) and the GEOS-Chem model (orange). (c) As in panel (b) but for the 10 most biased days in each region (total number of points is 9 years × 10 days × 10 regions). The line drawn at 70 ppb in panels (a, b) is the current O3 NAAQS level.
Simulated seasonal mean MDA8 averaged over the full 2004–2012 period is higher than observed by 5–30 ppb (Fig. 2a, b, c), with the largest biases typically occurring in the Northeast and Midwest. The model bias is highest in summer (JJA) (15–30 ppb at most sites), followed by fall (SON) (10–20 ppb) (Fig. 2a, b, c). Recent work in a newer version of GEOS-Chem attributes some of the positive model bias in the EUS to excessive NOx emissions in the 2011 National Emission Inventory (NEI) (Travis et al., 2016), an inability of the model to resolve vertical mixing in the boundary layer, and a weak response to cloud cover (Travis et al., 2017). Travis et al. (2016) find that the 3.5 Tg N yr−1 NEI 2011 estimate for US fuel NOx emissions is too high and contributes to excessive surface O3. Our simulations include even higher US fuel NOx emissions of 4.4 Tg N yr−1 during 2010–2012 (Table S3), implying that some portion of the model O3 bias reflects excessively high anthropogenic NOx emissions (Travis et al., 2016). The low bias in cloud cover in the MERRA meteorology and associated overestimate in downward shortwave surface radiation (Free et al., 2016) may also contribute to excessive O3 production in the model. The model is closest to the observations in spring, with a positive bias usually <10 ppb over the eastern states and generally within ±5 ppb over most western sites (Fig. 2a, b, c).
3.2 Baseline O3 at Mount Bachelor
Mount Bachelor Observatory (MBO) regularly samples free tropospheric O3 and is rarely influenced by local anthropogenic emissions (Reidmiller et al., 2009). It is, therefore, a valuable site for examining baseline O3. In Fig. S4, we compare the observed 24 h and MDA8 O3 concentrations at MBO for 2004–2012. The observed O3 concentrations vary from year to year, and by definition, MDA8 O3 is a few ppb higher than the 24 h mean mixing ratios. However, the seasonal pattern is similar across both metrics, with a springtime peak, a maximum in April, and a secondary summertime peak in July.
Figure 3 compares modeled and observed monthly mean 24 h O3 concentrations at the grid box that contains Mount Bachelor. For the model, we examine O3_Base and O3_USB 24 h average concentrations at 2.7 km and the height of the Mount Bachelor Observatory, as well as at the surface. It is important to note that the diurnal variations on the mountain may not be well captured by the CTM, due to upslope (daytime) ∕ downslope (nighttime) flow. We focus on the 24 h average because we only archived hourly O3 fields from the model at the surface and, thus, do not have the MDA8 O3 metric available at 2.7 km. The year-to-year variability is smaller in the model than observed (narrower shaded range). In all months, the O3_Base and O3_USB values are higher, by 9–14 and 11–21 ppb, respectively, at 2.7 km than at the surface. The model captures the magnitude of the observed springtime peak at 2.7 km, but summertime values are too high, with an overall peak in August. O3_USB contributes a greater fraction to O3_Base at 2.7 km (92–94 %) than at the surface (72–94 %). The simulated seasonal cycle differs at the surface, peaking in spring (March–April) and in September. In 2012, the observations show equivalent springtime and summertime peaks, more similar to the modeled seasonal cycle. While the observations generally decline from spring into summer, the model indicates an increase, leading to a substantial model overestimate during summer in most years. This model bias occurs across much of the USA as we show below.
Our sensitivity simulations enable us to interpret the sources contributing to the simulated seasonal distribution. The model indicates that at MBO, O3_USB is the major component of O3_Base, including during the summertime overestimate. In turn, the model indicates that the seasonality of O3_USB is largely driven by O3_NAT, which includes the influence from biogenic VOCs and NOx and lightning NOx, as well as stratospheric O3. O3_ICT+CH4 contributes around 15 ppb at 2.7 km and 5–10 ppb at the surface (Fig. 3). The model does suggest a springtime peak influence from O3_ICT+CH4 in the WUS, consistent with earlier work (e.g., Dentener et al., 2010). Even at this baseline site, the model indicates that O3_USA enhances monthly mean O3 by at least a few ppb at 2.7 km; at the surface, the model simulates a seasonal cycle for O3_USA that is typical of photochemical production from regional precursor emissions. O3_CA+MX is less than a few ppb at MBO whether the model is sampled at 2.7 km or the surface (not shown).
Table 2Summary information for each region. Columns show the model bias and O3 abundances in the O3_Base, O3_USB, and O3_USA simulations, and in the observations as well as for daily average temperature (1) on the O3_top10obs days in each season (average of 2004–2012), (2) across all days in each season (average of 2004–2012), and (3) the difference between these values, rounded to the nearest whole number.
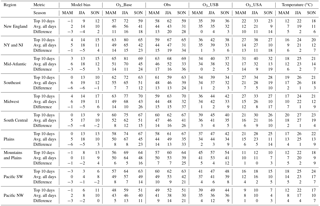
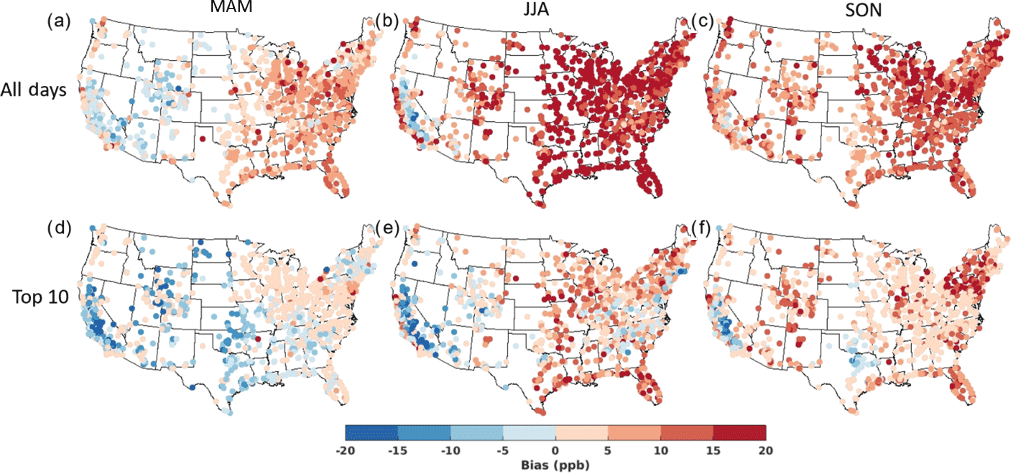
Figure 2Average MDA8 O3 model bias (O3_Base – observed) on all days in (a) MAM, (b) JJA, and (c) SON versus on the (d) O3_top10obs_MAM, (e) O3_top10obs_JJA, and (f) O3_top10obs_SON days at each observational site averaged across 2004–2012.
3.3 Magnitude and timing of high-O3 events
On O3_top10obs days, the model biases are typically lower than on average days (Fig. 2, Table 2; see also year-by-year maps in Fig. S2). At some WUS sites, the model underestimates O3 levels during the highest events by 10–20 ppb. The model systematically underestimates O3 in the Central Valley of California in all three seasons, which we attribute to the inability of the coarse model resolution to resolve topographical gradients and valley circulations (or stagnation) in this region which experiences some of the highest observed O3 in the nation.
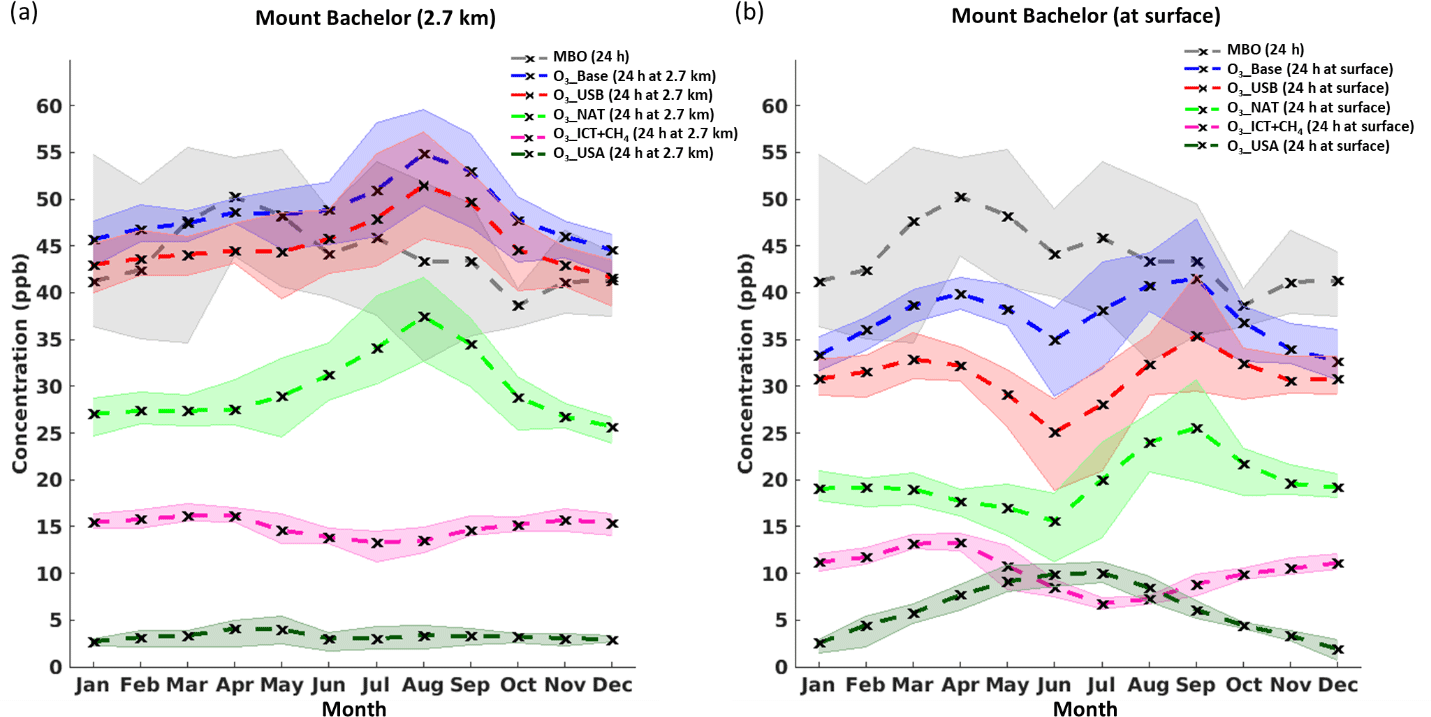
Figure 3Monthly 2004–2012 average 24 h O3 concentrations at Mount Bachelor Observatory. Observations (grey) are the same in both panels. Simulations from the GEOS-Chem model are sampled in the grid cell containing Mount Bachelor at (a) 2.7 km (the height of the Mount Bachelor Observatory) and at (b) the surface: O3_Base (blue), O3_USB (red), O3_NAT (light green), O3_ICT+CH4 (pink), and O3_USA (dark green). The shaded ranges span the highest and lowest years.
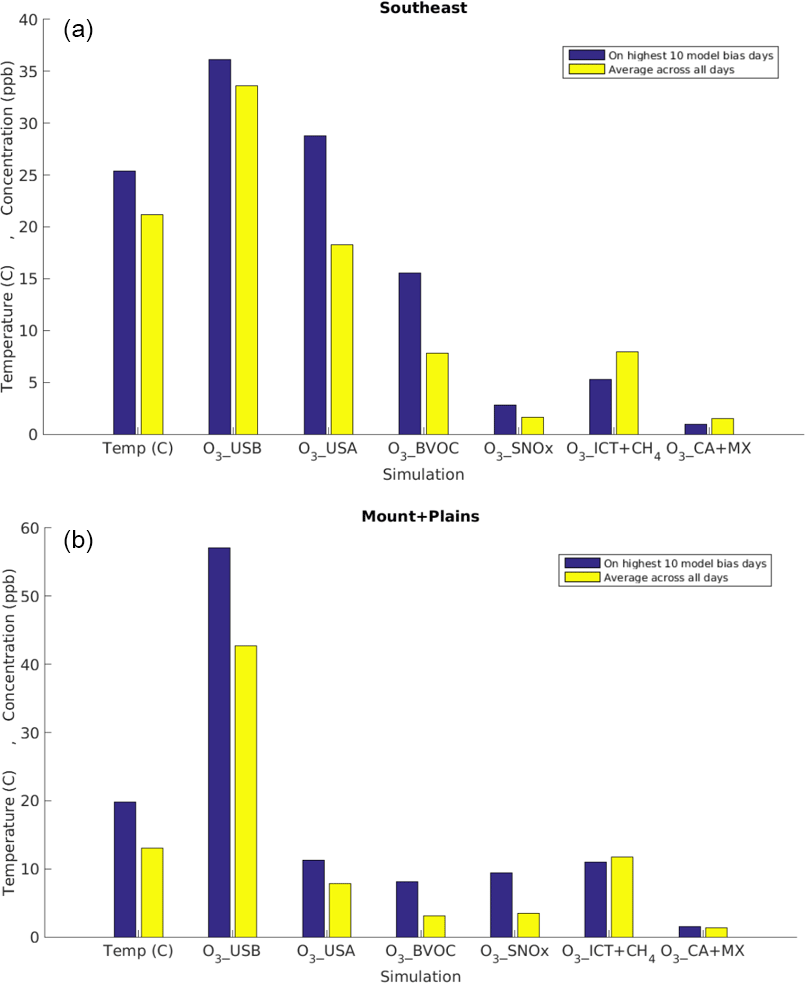
Figure 4Multi-year (2004–2012) March–October average temperature and MDA8 O3 source contributions estimated with the GEOS-Chem model in the (a) Southeast and (b) Mountain and Plains regions on the 10 most biased days (blue) versus averaged across all days (yellow). Note that the two regions are on different scales.
We compare the MDA8 O3 distributions in the observations versus the model (O3_Base) during the 10 most biased days in each of the 10 regions across the 9 years (900 total events). These “most-biased” days in the model tend to fall around the observed median (Fig. 1c) during the warm season (June–October), with almost 40 % of the days falling in August alone (Fig. S5), and are 9–45 ppb higher than the observations (circles in Fig. S6). We analyze the perturbation simulations (Table 1) to identify which sources influence simulated O3 most strongly on the most-biased days versus on average (i.e., all 365 or 366 days), which we assume are also likely the main drivers of the bias. In all regions, the largest sources on the most-biased model days are O3_USA (3–30 ppb higher MDA8 O3 than on average with the exception of the Pacific Southwest (SW) where O3_USA is smaller than on average days), O3_BVOC (by 1–15 ppb), and O3_SNOx (by 1–10 ppb; Figs. 4, S6). By contrast, O3_ICT+CH4 is up to a few ppb higher on average days than on the most-biased model days.
To explore possible drivers of model biases across the different seasons, we evaluate the timing of the highest 10 events across each year in the O3_Base, O3_USB, and O3_noBVOC (BVOCs shut off) simulations for each region (900 events). We bin these 900 events by month and calculate the percentage of the total events that fall within each month. Note that all the top 10 days fall between March and October. The standard model (O3_Base) underestimates the occurrence of high events early in the O3 season (March–June) and overestimates them later in the season (July–September) (Fig. S7). While the model indicates that most top 10 O3 days fall between July and August (35 % each), the observations show that the months of May through August each contain around 15–25 %, with the maximum in June at 25 %. Both O3_noBVOC and O3_USB shift the relative timing of the 10 highest O3 events towards April and May compared to O3_Base, but the shortage of high springtime O3 events remains (Fig. S7). The lack of high events in spring may reflect in part poor representation of stratospheric O3 intrusions at the coarse resolution of the CTM (Lin et al., 2012; Zhang et al., 2014), in addition to the role of US anthropogenic and BVOC emissions in the temporal mismatch as indicated by the improvements to the timing that occur in the O3_USB (US anthropogenic emissions shut off) and O3_noBVOC simulations. In addition to contributions from these sources, poor representation of O3 sinks may contribute to the model biases. For example, Makar et al. (2017) suggest that failing to represent canopy turbulence and shading effects on photolysis can lead to high-O3 biases in models.
3.4 Interannual variability
Figure S8 shows the Pearson correlations coefficients (r) between monthly average observed and O3_Base values from 2004 to 2012. In May, correlations are generally strong (r≥0.9) in the Mid-Atlantic and Southeast regions, but much lower (r=0.2) in the New England region. This pattern may reflect shortcomings in representing the onset of BVOC emissions. In July, the regions flip, with lower correlations in the Southeast and higher correlations in New England. At some sites in the WUS, lower correlations occur during summer months, which may be tied to excessive influence from lightning NOx advected from Mexico (see also Zhang et al., 2011, 2014) or anomalous events such as wildfires that are not well captured by the model.
In general, correlations only average about r=0.2 in the winter and early spring over much of the USA (Fig. S8); the drivers for these weak correlations may be connected to the model tendency to underestimate the occurrence of springtime high-O3 events. From May to September, however, the months during which high-O3 events are most likely to occur, the correlation between 2004 and 2012 observed and simulated O3 monthly averages over much of the contiguous USA exceeds r=0.7 (Fig. S8). We conclude that the model broadly captures monthly variations from year to year during the warm season and can thus be applied to interpret the role of background sources in contributing to interannual variations during most of the high-O3 season. We note that Clifton et al. (2017) found that the GEOS-Chem model does not capture interannual variability in deposition velocities observed at Harvard Forest, MA, but it is unclear to what extent this process would amplify or dampen interannual variability associated with changes in emissions.
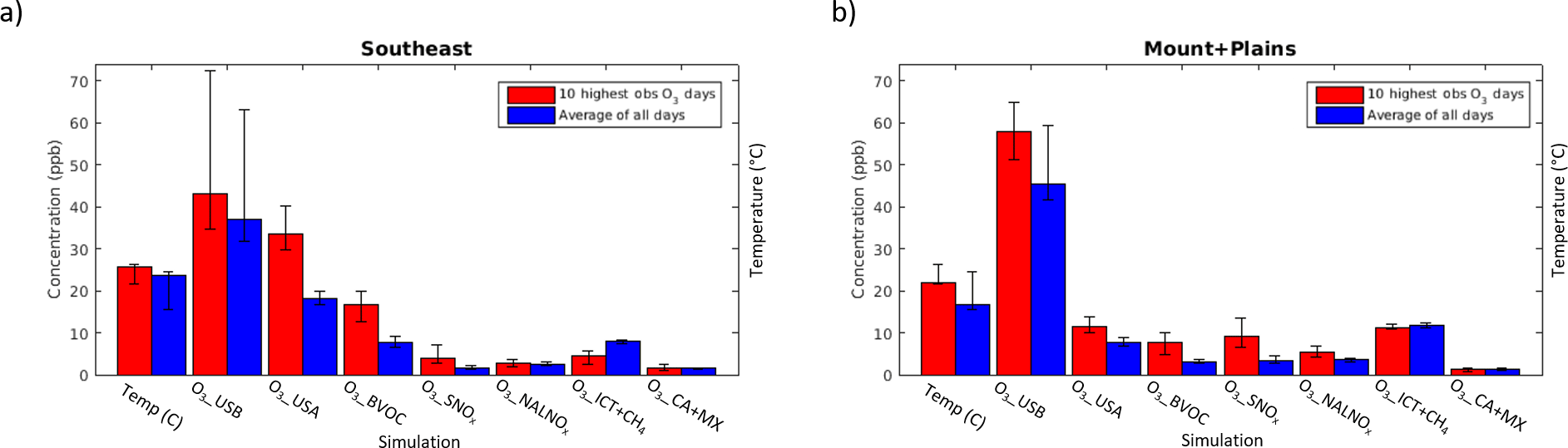
Figure 5Average 2004–2012 influence of each sensitivity simulation on O3_Base in the (a) Southeast and (b) Mountains and Plains regions on MDA8 O3_top10obs_JJA days (red) versus averaged across all days (blue). Error bars show the concentration on the lowest versus highest year for each sensitivity simulation in each region. Daily 24 h average temperature is also shown.
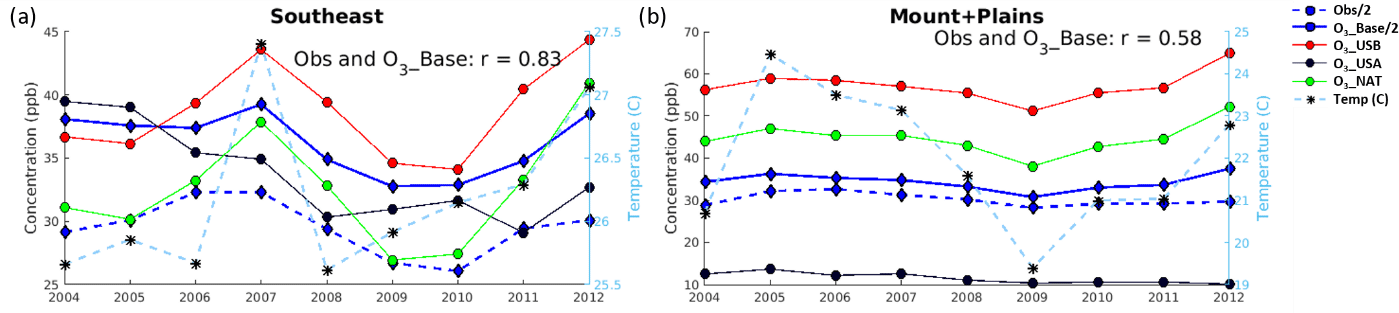
Figure 6Average yearly MDA8 O3_top10obs_JJA concentrations for observations (divided by 2 to fit on the same axes; blue dashed line), O3_Base (divided by 2; blue solid line), O3_USB (red), O3_USA (black), O3_NAT (green), and daily average temperature (in ∘C; light blue) in the (a) Southeast and (b) Mountains and Plains regions on the O3_top10obs_JJA days.
In Tables 2 and 3, we report the influence of the O3 sources defined in Table 1 on average versus O3_top10obs days separately for spring (MAM), summer (JJA), and fall (SON) (10 days from each of the 9 simulation years for 900 events for each region and season). We also report the difference in source influences between average and O3_top10obs days, which we interpret as the enhancement from that source relative to average conditions.
We first consider the average ranges in MDA8 O3 contributed by the various sources. Both O3_USA and O3_USB tend to follow the seasonal cycle of O3_Base, with highest abundances in summer. The model indicates that O3_USB is 30–50 ppb (range over regions) during summer and highest over the WUS. O3_USA is generally 20–30 ppb over the EUS in summer, but only 10–20 ppb over the WUS (Table 2). O3_ICT+CH4 averages 2–13 ppb over all regions and is highest in spring (8–13 ppb compared to 2–11 ppb in summer and 6–12 ppb in fall) (Table 3, Figs. 5, S9). O3_NALNOx has a relatively minor influence (at most 1.5 ppb) in all regions and seasons. The influence from O3_CA+MX is generally less than a couple of ppb except in New York (NY) and New Jersey (NJ), and in New England, where it can be as much as 4–7 ppb (Table 3, Fig. S9).
We interpret the “difference” lines in Tables 2 and 3 as the enhancements from each source on high days in each season (O3_top10obs_MAM, O3_top10obs_JJA, O3_top10obs_SON) relative to average conditions. Over all regions, O3_BVOC and O3_SNOx influence O3_Base more on O3_top10obs days (for all seasons) than on average, whereas O3_ICT+CH4 is typically lower by up to 3 ppb on O3_top10obs days (for all seasons) than on average days (Tables 2, 3, Figs. 5, S9). O3_USA is 8–11 ppb higher on O3_top10obs_JJA days versus the average over the New England, NY and NJ, Mid-Atlantic, Midwest, and South Central regions, but only up to 5 ppb higher over other regions (Table 2, Figs. 5, S9). The model indicates an even stronger anthropogenic enhancement (up to 19 ppb) on O3_top10obs_SON days in some EUS regions (Table 2). O3_USB is enhanced on O3_top10obs_JJA days by 2–12 ppb relative to the average, with the smallest enhancements occurring in the Mid-Atlantic, Southeast, and Midwest regions, and the largest enhancements occurring in the Pacific Northwest (NW). In contrast to all the other regions, O3_USB is the dominant source enhancing O3_top10days_JJA over the Mountains and Plains, Pacific NW, and Pacific SW regions (4–12 ppb for O3_USB but <5 ppb from either O3_USA or O3_BVOC). In line with earlier work reviewed by Jaffe et al. (2018), enhanced O3_USA dominates O3_top10obs_JJA days over much of the USA, whereas in the WUS, O3_USB enhancements exceed O3_USA enhancements on O3_top10days_JJA. O3_BVOC enhances O3_top10obs days (for all seasons) by up to 9 ppb, with the influence often largest in fall (when O3 formation is more sensitive to VOC; e.g., Jacob et al., 1995). We re-emphasize that BVOCs contribute both to O3_USA when reacting with anthropogenic NOx and to O3_USB when reacting with all other NOx sources. In contrast to the sources discussed above, O3_ICT+CH4 influences average days by up to a few ppb more than on O3_top10obs days (for all seasons), with the largest differences between average and high days occurring in EUS regions (1–3 ppb lower on O3_top10obs days (for all seasons) in New England, NY and NJ, and Mid-Atlantic; Table 3, Figs. 5, S9). O3_NALNOx is at most 2 ppb higher than average on O3_top10obs days. The O3_CA+MX influence is roughly equivalent (generally to within a ppb) on average versus O3_top10obs days during all seasons.
Table 3Summary information for each region. Each column shows the concentration for each background O3 source influence (1) on the O3_top10obs days in each season (average of 2004–2012), (2) across all days in each season (average of 2004–2012), and (3) the difference between these values, rounded to the nearest whole number.
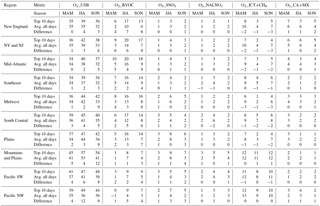
Table 4Change in MDA8 O3 concentrations from 2004–2006 to 2010–2012 in the observations, O3_Base, O3_USB, O3_USA, and temperature on O3_top10obs_JJA days.

Despite its high mean bias and seasonal phase shift, the model does capture some of the observed interannual variability in observed O3_top10obs_JJA MDA8 O3 concentrations (Figs. 6, S10; r=0.5 to ≥0.9). Comparing the 2004–2006 period with 2010–2012, both observed and simulated MDA8 O3 concentrations on O3_top10obs_JJA days hold steady or decrease across all regions. This change reflects opposing influences in the model: rising O3_USB (by 2 ppb averaged over all regions) and declining O3_USA concentrations (by 6 ppb averaged over all regions) (Figs. 6, S10, Table 4). We note that over the Pacific NW there is a 4 ppb decrease in O3_USB from 2004–2006 to 2010–2012. Over this period, temperatures generally warm over the EUS, but slightly cool in the WUS. Within the 10 regions, the model captures the sign of the changes in MDA8 O3 over this period but not the magnitude (Table 4). The model monthly mean temperatures in the model (from the MERRA reanalysis) closely match the observed GHCN+CAMS dataset (Table S4). Table 4 shows that regions with O3_USB increases generally experienced rising temperatures over this period, as the 2010–2012 period includes two of the warmest years on record. Figure 6 shows that O3_NAT tracks with O3_USB and temperature. Dips in MDA8 O3 occur during years with cooler temperatures (2008–2009) and increases occur in years with warmer temperatures (2011–2012), indicating that year-to-year variability in O3_USB on O3_top10obs_JJA days is primarily driven in the model by natural sources sensitive to meteorology rather than international O3 transport (Figs. 6, S10). Although 2012 was the hottest year on average between 2004 and 2012 (except in the Pacific NW where 2004 was warmer by about a degree), it was not the hottest summer in all regions.
We find that O3_USB drives the interannual variability on O3_top10obs_JJA days in the WUS ( for O3_USB versus O3_Base, whereas for O3_USA versus O3_Base; Table S5). In NY and NJ, the Southeast, Midwest, South Central, and Plains regions, O3_USB and O3_USA both contribute to the interannual variability on O3_top10obs_JJA days ( for both O3_USB and O3_USA versus O3_Base), while in New England and the Mid-Atlantic regions, O3_USA drives the interannual variability more than O3_USB (r=0.64 and 0.72 for O3_USA versus O3_Base but only 0.28 and 0.54, respectively, for O3_USB versus O3_Base; Table S5).
Year-to-year variations in monthly average O3_USB are relatively large, with 10–15 ppb differences between the highest and lowest O3_USB years during the warmest months (Figs. 7, S11). Seasonal variations also differ by region, especially during summer. For example, the western US regions have a smooth seasonal cycle, with O3_USB concentrations rising from January to a peak in July and August, and then declining again. Interannual and seasonal variability in O3_USB are generally greater in the Eastern U.S. regions than in the Mountains and Plains region and the Plains region (Figs. 7, S11). Year-to-year variability in O3_BVOC is smaller than O3_USB, with a maximum range of about 10 ppb between the highest and lowest years during August (Figs. 7, S12). O3_SNOx ranges by a few ppb throughout the summer in the Southeast and by up to 6 ppb over the Mountains and Plains in August (Figs. 7, S13).
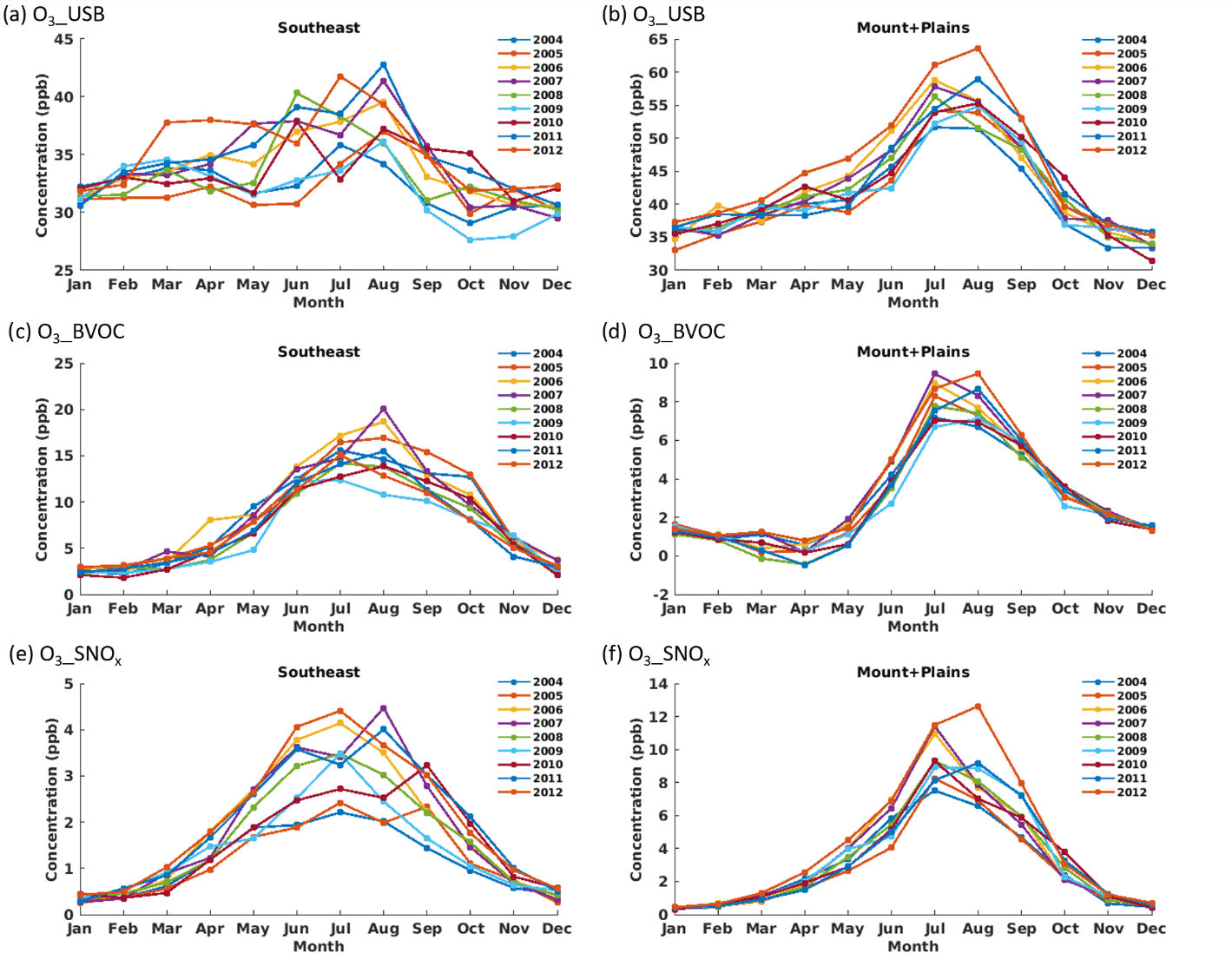
Figure 7Monthly average MDA8 O3_USB (a, b), O3_BVOC (c, d), and O3_SNOx (e, f) concentrations in the Southeast (a, c, e) and Mountains and Plains (b, d, f) regions. Colored lines denote individual years.

Figure 8Anomaly on the MDA8 O3_top10obs_JJA days relative to the 2004–2012 average in the Southeast (a, c) and in the Mountains and Plains (b, d) regions. Panels (a, b) show the observations, O3_Base, O3_USB, O3_USA, and daily average temperature (in ∘C) on O3_top10obs_JJA days. Panels (c, d) show O3_BVOC, O3_SNOx, O3_NALNOx, O3_BB, O3_ICT+CH4, and O3_CA+MX.
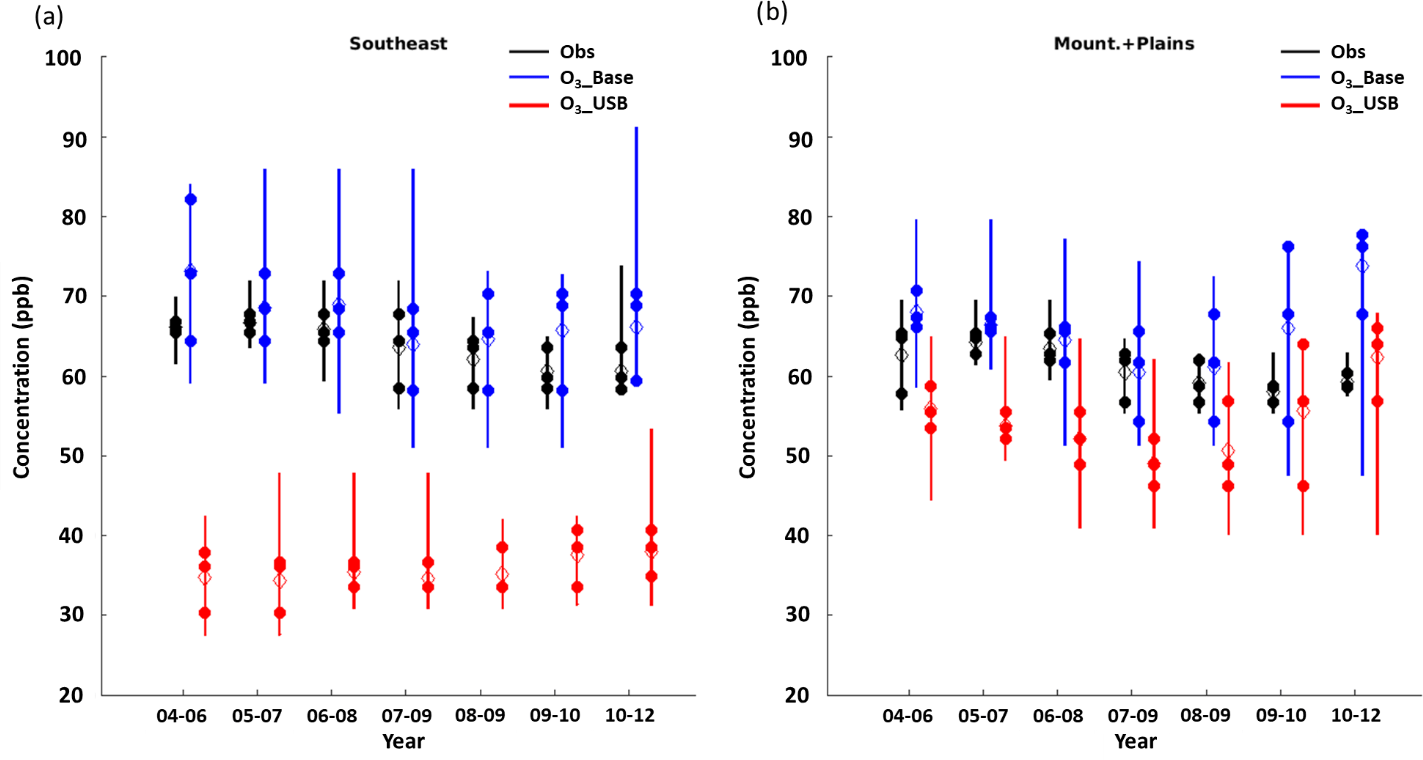
Figure 9The three fourth highest values (solid dots) used to calculate the 3-year average of the fourth highest MDA8 O3 day (hollow diamond). Vertical bars show the range between the highest and lowest O3_top10obs days across each 3-year span (i.e, across 30 total points) occurring between March and October in the (a) Southeast and (b) Mountains and Plains regions in the observations (black), and the O3_Base (blue) and O3_USB (red) simulations sampled on the same days as the top 10 observed values.
O3_USA anomalies relative to the 2004–2012 average illustrate declining influence in all regions, with negative anomalies after 2007 on both O3_top10obs and average days (Figs. 8, S14). This finding is well established by earlier work demonstrating decreases in high-O3 concentrations as a result of regional NOx emissions reductions over the past few decades (Cooper et al., 2012, 2014; Jaffe et al., 2018; Young et al., 2017). O3_BVOC is the main driver of the high and low O3 anomalies (up to ±5 ppb on O3_top10obs_JJA days) from year to year (Figs. 8, S15).
Specific events can affect O3 in any given year. For example, in 2008, there were extensive fires across much of California in May, June, and July. In 2008, the Pacific SW region, which includes California, Nevada, and Arizona, shows a positive anomaly in O3_BB (>1 ppb) on the O3_top10obs days, stronger than during any other year in that region (Fig. S15). If we restrict our analysis solely to Reno, NV, the anomaly for O3_BB was 7 ppb in July 2008 relative to the 2004–2012 July average (not shown). We emphasize that a single location can be more strongly influenced by a specific source than the regional averages on which we have focused.
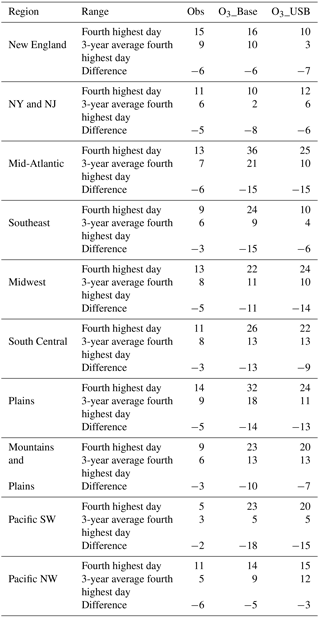
Currently, the U.S. EPA uses a 3-year averaging period of the fourth highest MDA8 O3 to assess compliance with the O3 NAAQS. We evaluate the extent to which this 3-year averaging period removes interannual variability in meteorology (the grounds for the averaging) (Figs. 9, S16). The observed range is generally much smaller than the model estimate. We find that the 3-year average of the fourth highest day decreases the range by 2–6 and 5–18 ppb in the observations and O3_Base, respectively, when compared to taking the fourth highest day in any given year when we look across all regions (Table 5). However, the 3-year average of the fourth highest day still ranges from 3 to 9 and 2 to 11 ppb in the observations and O3_Base, respectively, across all regions (compared to 5–15 and 10–36 ppb in the observations and O3_Base on the fourth highest day in each individual year). Thus, while averaging across the years decreases the spread, variability remains. In keeping with our previous analysis of the O3_top10obs days, we compare the spread of the fourth highest O3 day in each of the 3 years to the range of the O3_top10obs days across each 3-year span; the fourth highest days can range almost as widely as the O3_top10obs days in some years, but in other years, are clustered closer together (Fig. 9). Figure 9 shows that the range in O3_top10obs days for O3_Base generally correlates with O3_USB in the WUS, suggesting that O3_USB is the dominant influence on the high days there, but there is little correlation in the EUS. We conclude that a 3-year smoothing period is not long enough to eliminate the interannual variability in MDA8 O3 levels entirely, and in the WUS, this interannual variability tends to reflect variations in O3_USB.
Table 5Summary information for each region. The first row next to each region reports the range across 2004–2012 of the fourth highest MDA8 O3 values from each of the 9 individual years for the observations, O3_Base, and O3_USB. The second row reports the range across 2004–2012 of each of the 3-year averages of the fourth highest values (7 values) in each region for the observations, O3_Base, and O3_USB.
As air quality controls decrease US anthropogenic precursor emissions to O3, the relative importance of the background influence on total surface O3 increases. We use O3 MDA8 concentrations spanning 2004–2012 from the EPA AQS, CASTNet, and Mount Bachelor Observatory sites, and sensitivity simulations from the global GEOS-Chem 3-D chemistry transport model to estimate the influence from various individual background sources on O3 in each of the 10 EPA regions in the continental USA. The global scale of the GEOS-Chem model allows us to quantify intercontinental transport (including global methane) in addition to regional natural and anthropogenic sources of O3. The sensitivity simulations span 9 years, allowing us to examine the role of these sources in contributing to interannual variability. Our analysis contrasts average- and high-O3 days.
Correlations between monthly averages across 2004–2012 show that the model captures monthly variations from year to year, especially during summer (JJA). The model shows substantial variability in simulated monthly US background O3 concentrations from year to year, on the order of 10–15 ppb between 2004 and 2012 in summer (Fig. 7). We find that the extent to which the current 3-year averaging period for assessing compliance with the National Ambient Air Quality Standard for O3 succeeds in smoothing out interannual variability depends on the range in consecutive years, and thus varies by region and time period, but is generally not long enough to completely eliminate the interannual variability in background O3 (Fig. 9).
We find substantial biases in the severity (+0–19 ppb in maximum daily 8 h average (MDA8) O3) and timing of high-O3 events in the model. The model underestimates the frequency of high events in spring, possibly associated with stratospheric intrusions (Fiore et al., 2014; Zhang et al., 2011, 2014). Future efforts would benefit from quantifying the stratospheric (as well as Asian) influence alongside the other background sources we consider. We find a stronger influence of US anthropogenic emissions on regionally averaged MDA8 O3 (up to 30 ppb), and from BVOCs (up to 15 ppb) and soil NOx (up to 10 ppb) on the 10 most biased days as compared to average days. We conclude that regional production of O3 is driving the pervasive high positive model bias in summer, as opposed to transported background O3, although our sensitivity simulations do not allow us to rule out the possibility of a coincident missing sink.
Our finding that BVOC emissions contribute to the summertime surface O3 biases could reflect poor representation of the emissions (and subsequent oxidation chemistry). Earlier work has noted that MEGAN BVOC emissions are too high over California (Bash et al., 2016), southeast Texas (Kota et al., 2015), the Ozarks in southern Missouri (Carlton and Baker, 2011), and across much of the USA (Wang et al., 2017). One recent model study uniformly reduced MEGAN isoprene emissions by 20 % (Li et al., 2018), but we did not apply any such scaling here. In regions that are highly NOx-sensitive, additional isoprene should not strongly influence O3, as found over southeast Texas (Kota et al., 2015). While not eliminated entirely, the summertime model bias does lessen in the simulation with BVOC emissions set to zero, suggesting that the O3 bias is indeed exacerbated if BVOC emissions are overestimated in the model.
On the 10 days with the highest observed MDA8 O3 values (O3_top10obs) in each season, the model indicates that US anthropogenic and biogenic VOC emissions are the most important drivers, relative to average days, over most regions (Tables 2, 3). O3_top10obs_MAM and O3_top10obs_SON days (i.e., the 10 highest spring and fall MDA8 O3 days) are up to 9 ∘C warmer than average, but O3_top10obs_JJA days (i.e., the 10 highest summer MDA8 O3 days) are only 1–2 ∘C warmer than average (Table 2). US anthropogenic emissions enhance O3_top10obs_JJA days by 5–11 ppb above average in the eastern US regions, but by less than 2 ppb over the three western regions. Over these westernmost regions, US background O3 is 4–12 ppb higher on O3_top10obs_JJA days than on average (Table 2). Across the continental USA, biogenic VOC emissions enhance O3 by 1–7 ppb above average on O3_top10obs_JJA days, while intercontinental pollution is either similar or up to 2 ppb higher on average days (Table 3). Analysis of our simulations thus indicates that the highest O3 events are associated with regional O3 production rather than transported background.
From 2004–2006 to 2010–2012, MDA8 O3 concentrations on O3_top10obs_JJA days vary from year to year, but show little overall trend, decreasing by 3 ppb in both the observations and the model averaged over all regions (Fig. 6, Table 4). With our sensitivity simulations, we interpret this lack of an overall trend as a balance between rising US background O3 (by 2 ppb for O3_USB from 2004–2006 to 2010–2012 averaged over all regions) and declining US anthropogenic emissions (by 6 ppb for O3_USA from 2004–2006 to 2010–2012 averaged over all regions). The declining influence of US anthropogenic emissions on O3_top10obs_JJA days is consistent with earlier work showing high-O3 concentrations decreasing in response to regional precursor emissions controls since the late 1990s (e.g., Cooper et al., 2012, 2014; Frost et al., 2006; Simon et al., 2016).
In contrast to previous work, including with the GEOS-Chem model (e.g., Fiore et al., 2014 and references therein), we find that US background O3 tends to be higher in summer than in spring in most regions. This likely reflects differences in the isoprene chemistry, specifically the isoprene nitrates, between our version of GEOS-Chem (Mao et al., 2013) and older versions that treat isoprene nitrates as greater sinks for NOx and thereby suppress O3 production. The coarse resolution of our model will excessively mix isoprene and soil NOx sources (e.g., Yu et al., 2016), and thus may exaggerate the relative importance of enhanced background O3 resulting from soil NOx and isoprene. Nevertheless, the model skill at capturing the observed year-to-year variability in the regionally averaged 10 highest days lends some confidence to its attribution of this variability to natural sources (e.g., Fig. 6). Future work with high-resolution models (e.g., at the regional scale, ideally with boundary conditions that include source attributions from a global model) is needed, along with observational evidence, to quantify the extent to which biogenic VOC and NOx contribute to the highest observed O3 levels in the warm season. The importance of temperature-sensitive sources like biogenic VOC and NOx emissions to background O3 implies that in a warmer climate, these background influences on O3 will play an even more important role in driving up O3 levels.
Data behind the figures are provided online in the Columbia University Academic Commons (https://doi.org/10.7916/D8Z90W3X).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-12123-2018-supplement.
JG and AF contributed to the development of the concepts and ideas presented in this paper and wrote the manuscript, along with input and comments from all co-authors. LM performed the model simulation, DJ produced the hourly Mount Bachelor data, JS provided the gridded observational dataset, and GM contributed to creating the MDA8 O3 model dataset, helped process the CASTNet data, and calculated emissions values. CM contributed to the development of ideas related to the EPA 3-year averaging section as well as ideas related to the policy relevance of the paper.
The authors declare that they have no conflict of interest.
This article is part of the special issue “Global and regional assessment of intercontinental transport of air pollution: results from HTAP, AQMEII and MICS”. It is not associated with a conference.
We acknowledge insightful discussions with Gail Tonnesen and Pat Dolwick, and
comments on an earlier version from Serena Chung (all at U.S. EPA), and Mike
Previdi (Lamont-Doherty Earth Observatory) and Vaishali Naik (Geophysical
Fluid Dynamics Laboratory). We gratefully acknowledge support from NASA AQAST
(NNX12AF15G) and NASA HAQAST (NNX16AQ20G). This project has been funded in
part by the United States Environmental Protection Agency under assistance
agreement RD83587801 to AMF. The contents of this document do not necessarily
reflect the views and policies of the Environmental Protection Agency, nor
does the EPA endorse trade names or recommend the use of commercial products
mentioned in this document. This is Lamont contribution no. 8243. Data behind
the figures are provided online in the Columbia University Academic
Commons.
Edited by: Frank Dentener
Reviewed by: three anonymous referees
Abatzoglou, J. T. and Williams, A. P.: Impact of anthropogenic climate change on wildfire across western US forests, P. Natl. Acad. Sci. USA, 113, 11770–11775, https://doi.org/10.1073/pnas.1607171113, 2016.
Abel, D., Holloway, T., Kladar, R. M., Meier, P., Ahl, D., Harkey, M., and Patz, J.: Response of Power Plant Emissions to Ambient Temperature in the Eastern United States, Environ. Sci. Technol., 51, 5838–5846, https://doi.org/10.1021/acs.est.6b06201, 2017.
Auvray, M. and Bey, I.: Long-range transport to Europe: Seasonal variations and implications for the European ozone budget, J. Geophys. Res., 110, D11303, https://doi.org/10.1029/2004JD005503, 2005.
Barkley, M. P., Palmer, P. I., Ganzeveld, L., Arneth, A., Hagberg, D., Karl, T., Guenther, A., Paulot, F., Wennberg, P. O., Mao, J., Kurosu, T. P., Chance, K., Müller, J. F., De Smedt, I., Van Roozendael, M., Chen, D., Wang, Y., and Yantosca, R. M.: Can a “state of the art” chemistry transport model simulate Amazonian tropospheric chemistry?, J. Geophys. Res., 116, D16302, https://doi.org/10.1029/2011JD015893, 2011.
Bash, J. O., Baker, K. R., and Beaver, M. R.: Evaluation of improved land use and canopy representation in BEIS v3.61 with biogenic VOC measurements in California, Geosci. Model Dev., 9, 2191–2207, https://doi.org/10.5194/gmd-9-2191-2016, 2016.
Baylon, P. M., Jaffe, D. A., Pierce, R. B., and Gustin, M. S.: Interannual Variability in Baseline Ozone and Its Relationship to Surface Ozone in the Western U.S., Environ. Sci. Technol., 50, 2994–3001, https://doi.org/10.1021/acs.est.6b00219, 2016.
Bond, T. C., Bhardwaj, E., Dong, R., Jogani, R., Jung, S., Roden, C., Streets, D. G., and Trautmann, N. M.: Historical emissions of black and organic carbon aerosol from energy-related combustion, 1850–2000, Global Biogeochem. Cy., 21, GB2018, https://doi.org/10.1029/2006GB002840, 2007.
Bosilovich, M. G.: Regional climate and variability of NASA MERRA and recent reanalyses: U.S. summertime precipitation and temperature, J. Appl. Meteorol. Clim., 52, 1939–1951, https://doi.org/10.1175/JAMC-D-12-0291.1, 2013.
Bouwman, A. F., Lee, D. S., Asman, W. A. H., Dentener, F. J., Van Der Hoek, K. W., and Olivier, J. G. J.: A global high-resolution emission inventory for ammonia, Global Biogeochem. Cy., 11, 561–587, https://doi.org/10.1029/97GB02266, 1997.
Carlton, A. G. and Baker, K. R.: Photochemical modeling of the ozark isoprene volcano: MEGAN, BEIS, and their impacts on air quality predictions, Environ. Sci. Technol., 45, 4438–4445, https://doi.org/10.1021/es200050x, 2011.
Clifton, O. E., Fiore, A. M., Munger, J. W., Malyshev, S., Horowitz, L. W., Shevliakova, E., Paulot, F., Murray, L. T., and Griffin, K. L.: Interannual variability in ozone removal by a temperate deciduous forest, Geophys. Res. Lett., 44, 542–552, https://doi.org/10.1002/2016GL070923, 2017.
Cooper, O. R., Gao, R.-S., Tarasick, D., Leblanc, T., and Sweeney, C.: Long-term ozone trends at rural ozone monitoring sites across the United States, 1990–2010, J. Geophys. Res., 117, D22307, https://doi.org/10.1029/2012JD018261, 2012.
Cooper, O. R., Parrish, D. D., Ziemke, J., Balashov, N. V., Cupeiro, M., Galbally, I. E., Gilge, S., Horowitz, L., Jensen, N. R., Lamarque, J.-F., Naik, V., Oltmans, S. J., Schwab, J., Shindell, D. T., Thompson, A. M., Thouret, V., Wang, Y., and Zbinden, R. M.: Global distribution and trends of tropospheric ozone: An observation-based review, Elem. Sci. Anth., 2, 000029, https://doi.org/10.12952/journal.elementa.000029, 2014.
Dentener, F., Keating, T., and Akimoto, H.: Hemispheric Transport of Air Pollution 2010: Part A – Ozone and Particulate Matter, Air Pollut. Stud., 17, 305, https://doi.org/10.18356/2c908168-en, 2010.
Fan, Y. and van den Dool, H.: A global monthly land surface air temperature analysis for 1948-present, J. Geophys. Res., 113, D01103, https://doi.org/10.1029/2007JD008470, 2008.
Fiore, A. M., Jacob, D. J., Field, B. D., Streets, D. G., Fernandes, S. D., and Jang, C.: Linking ozone pollution and climate change: The case for controlling methane, Geophys. Res. Lett., 29, 1919, https://doi.org/10.1029/2002GL015601, 2002.
Fiore, A. M., Jacob, D. J., Liu, H., Yantosca, R. M., Fairlie, T. D., and Li, Q.: Variability in surface ozone background over the United States: Implications for air quality policy, J. Geophys. Res., 108, 4787, https://doi.org/10.1029/2003jd003855, 2003.
Fiore, A. M., Oberman, J. T., Lin, M., Zhang, L., Clifton, O. E., Jacob, D. J., Naik, V., Horowitz, L. W., Pinto, J. P., and Milly, G. P.: Estimating North American background ozone in U.S. surface air with two independent global models: Variability, uncertainties, and recommendations, Atmos. Environ., 96, 284–300, https://doi.org/10.1016/j.atmosenv.2014.07.045, 2014.
Fiore, A. M., Vaishali, N., and Leibensperger, E. M.: Air Quality and Climate Connections, J. Air Waste Manage., 65, 645–685, 2015.
Free, M., Sun, B., and Yoo, H. L.: Comparison between total cloud cover in four reanalysis products and cloud measured by visual observations at U.S. weather stations, J. Climate, 29, 2015–2021, https://doi.org/10.1175/JCLI-D-15-0637.1, 2016.
Frost, G. J., McKeen, S. A., Trainer, M., Ryerson, T. B., Neuman, J. A., Roberts, J. M., Swanson, A., Holloway, J. S., Sueper, D. T., Fortin, T., Parrish, D. D., Fehsenfeld, F. C., Flocke, F., Peckham, S. E., Grell, G. A., Kowal, D., Cartwright, J., Auerbach, N., and Habermann, T.: Effects of changing power plant NOx emissions on ozone in the eastern United States: Proof of concept, J. Geophys. Res., 111, D12306, https://doi.org/10.1029/2005JD006354, 2006.
Giglio, L., Randerson, J. T., van der Werf, G. R., Kasibhatla, P. S., Collatz, G. J., Morton, D. C., and DeFries, R. S.: Assessing variability and long-term trends in burned area by merging multiple satellite fire products, Biogeosciences, 7, 1171–1186, https://doi.org/10.5194/bg-7-1171-2010, 2010.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Hu, L., Millet, D. B., Baasandorj, M., Griffis, T. J., Travis, K. R., Tessum, C. W., Marshall, J. D., Reinhart, W. F., Mikoviny, T., Müller, M., Wisthaler, A., Graus, M., Warneke, C., and de Gouw, J.: Emissions of C6-C8 aromatic compounds in the United States: Constraints from tall tower and aircraft measurements, J. Geophys. Res.-Atmos., 120, 826–842, https://doi.org/10.1002/2014JD022627, 2015.
Jacob, D. J., Horowitz, L. W., Munger, J. W., Heikes, B. G., Dickerson, R. R., Artz, R. S. and Keene, W. C.: Seasonal transition from NOx- to hydrocarbon-limited conditions for ozone production over the eastern United States in September, J. Geophys. Res., 100, 9315–9315, https://doi.org/10.1029/94JD03125, 1995.
Jaffe, D. A.: Relationship between surface and free tropospheric ozone in the Western U.S., Environ. Sci. Technol., 45, 432–438, https://doi.org/10.1021/es1028102, 2011.
Jaffe, D. A., Cooper, O. R., Fiore, A. M., Henderson, B. H., Gail, S., Russell, A. G., Henze, D. K., Langford, A. O., Lin, M. and Moore, T.: Scientific assessment of background ozone over the U.S.: implications for air quality management, Elem. Sci. Anthr., 6, 56, https://doi.org/10.1525/elementa.309, 2018.
Kota, S. H., Schade, G., Estes, M., Boyer, D., and Ying, Q.: Evaluation of MEGAN predicted biogenic isoprene emissions at urban locations in Southeast Texas, Atmos. Environ., 110, 54–64, https://doi.org/10.1016/j.atmosenv.2015.03.027, 2015.
Kuhns, H. and Green, M.: Big Bend Regional Aerosol and Visibility Observational (BRAVO) Study Emissions Inventory, Desert Research Institute, NV, USA, available at: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.462.8648&rep=rep1&type=pdf (last access: 29 January 2018), 2003.
Lee, C., Martin, R. V., van Donkelaar, A., Lee, H., Dickerson, R. R., Hains, J. C., Krotkov, N., Richter, A., Vinnikov, K., and Schwab, J. J.: SO2 emissions and lifetimes: Estimates from inverse modeling using in situ and global, space-based (SCIAMACHY and OMI) observations, J. Geophys. Res., 116, D06304, https://doi.org/10.1029/2010JD014758, 2011.
Leibensperger, E. M., Mickley, L. J., Jacob, D. J., Chen, W.-T., Seinfeld, J. H., Nenes, A., Adams, P. J., Streets, D. G., Kumar, N., and Rind, D.: Climatic effects of 1950–2050 changes in US anthropogenic aerosols – Part 2: Climate response, Atmos. Chem. Phys., 12, 3349–3362, https://doi.org/10.5194/acp-12-3349-2012, 2012.
Li, J., Mao, J., Fiore, A. M., Cohen, R. C., Crounse, J. D., Teng, A. P., Wennberg, P. O., Lee, B. H., Lopez-Hilfiker, F. D., Thornton, J. A., Peischl, J., Pollack, I. B., Ryerson, T. B., Veres, P., Roberts, J. M., Neuman, J. A., Nowak, J. B., Wolfe, G. M., Hanisco, T. F., Fried, A., Singh, H. B., Dibb, J., Paulot, F., and Horowitz, L. W.: Decadal changes in summertime reactive oxidized nitrogen and surface ozone over the Southeast United States, Atmos. Chem. Phys., 18, 2341–2361, https://doi.org/10.5194/acp-18-2341-2018, 2018.
Lin, M., Fiore, A. M., Cooper, O. R., Horowitz, L. W., Langford, A. O., Levy, H., Johnson, B. J., Naik, V., Oltmans, S. J., and Senff, C. J.: Springtime high surface ozone events over the western United States: Quantifying the role of stratospheric intrusions, J. Geophys. Res., 117, D00V22, https://doi.org/10.1029/2012JD018151, 2012.
Lin, M., Fiore, A. M., Horowitz, L. W., Langford, A. O., Oltmans, S. J., Tarasick, D., and Rieder, H. E.: Climate variability modulates western US ozone air quality in spring via deep stratospheric intrusions, Nat. Commun., 6, 7105, https://doi.org/10.1038/ncomms8105, 2015a.
Lin, M., Horowitz, L. W., Cooper, O. R., Tarasick, D., Conley, S., Iraci, L. T., Johnson, B. J., Leblanc, T., Petropavlovskikh, I., and Yates, E. L.: Revisiting the evidence of increasing springtime ozone mixing ratios in the free troposphere over western North America, Geophys. Res. Lett., 42, 8719–8728, https://doi.org/10.1002/2015GL065311, 2015b.
Lin, M., Horowitz, L. W., Payton, R., Fiore, A. M., and Tonnesen, G.: US surface ozone trends and extremes from 1980 to 2014: quantifying the roles of rising Asian emissions, domestic controls, wildfires, and climate, Atmos. Chem. Phys., 17, 2943–2970, https://doi.org/10.5194/acp-17-2943-2017, 2017.
Makar, P. A., Staebler, R. M., Akingunola, A., Zhang, J., McLinden, C., Kharol, S. K., Pabla, B., Cheung, P., and Zheng, Q.: The effects of forest canopy shading and turbulence on boundary layer ozone, Nat. Commun., 8, 15243, https://doi.org/10.1038/ncomms15243, 2017.
Mao, J., Horowitz, L. W., Naik, V., Fan, S., Liu, J., and Fiore, A. M.: Sensitivity of tropospheric oxidants to biomass burning emissions: implications for radiative forcing, Geophys. Res. Lett., 40, 1241–1246, https://doi.org/10.1002/grl.50210, 2013.
National Research Council: Global Sources of Local Pollution: An Assessment of Long-Range Transport of Key Air Pollutants to and from the United States, The National Academies Press, Washington, DC, https://doi.org/10.17226/12743, 2010.
Olivier, J. G. J., Van Aardenne, J. A., Dentener, F. J., Pagliari, V., Ganzeveld, L. N., and Peters, J. A. H. W.: Recent trends in global greenhouse gas emissions:regional trends 1970–2000 and spatial distributionof key sources in 2000, Environmental Sciences, 2, 81–99, https://doi.org/10.1080/15693430500400345, 2005.
Reidmiller, D. R., Fiore, A. M., Jaffe, D. A., Bergmann, D., Cuvelier, C., Dentener, F. J., Duncan, B. N., Folberth, G., Gauss, M., Gong, S., Hess, P., Jonson, J. E., Keating, T., Lupu, A., Marmer, E., Park, R., Schultz, M. G., Shindell, D. T., Szopa, S., Vivanco, M. G., Wild, O., and Zuber, A.: The influence of foreign vs. North American emissions on surface ozone in the US, Atmos. Chem. Phys., 9, 5027–5042, https://doi.org/10.5194/acp-9-5027-2009, 2009.
Rienecker, M. M., Suarez, M. J., Gelaro, R., Todling, R., Bacmeister, J., Liu, E., Bosilovich, M. G., Schubert, S. D., Takacs, L., Kim, G. K., Bloom, S., Chen, J., Collins, D., Conaty, A., Da Silva, A., Gu, W., Joiner, J., Koster, R. D., Lucchesi, R., Molod, A., Owens, T., Pawson, S., Pegion, P., Redder, C. R., Reichle, R., Robertson, F. R., Ruddick, A. G., Sienkiewicz, M., and Woollen, J.: MERRA: NASA's modern-era retrospective analysis for research and applications, J. Climate, 24, 3624–3648, https://doi.org/10.1175/JCLI-D-11-00015.1, 2011.
Schnell, J. L. and Prather, M. J.: Co-occurrence of extremes in surface ozone, particulate matter, and temperature over eastern North America, P. Natl. Acad. Sci. USA, 114, 2854–2859, https://doi.org/10.1073/pnas.1614453114, 2017.
Schnell, J. L., Holmes, C. D., Jangam, A., and Prather, M. J.: Skill in forecasting extreme ozone pollution episodes with a global atmospheric chemistry model, Atmos. Chem. Phys., 14, 7721–7739, https://doi.org/10.5194/acp-14-7721-2014, 2014.
Schultz, M., Consortium, R., and Mota, B.: REanalysis of the TROpospheric chemical composition over the past 40 years (RETRO), Eur. Geosci. Union, 1179, 2003.
Simon, H., Wells, B., Baker, K. R., and Hubbell, B.: Assessing temporal and spatial patterns of observed and predicted ozone in multiple urban areas, Environ. Health Persp., 124, 1443–1452, https://doi.org/10.1289/EHP190, 2016.
Stauffer, R. M., Thompson, A. M., Oltmans, S. J., and Johnson, B. J.: Tropospheric ozonesonde profiles at long-term U.S. monitoring sites: 2. Links between Trinidad Head, CA, profile clusters and inland surface ozone measurements, J. Geophys. Res.-Atmos., 122, 1261–1280, https://doi.org/10.1002/2016JD025254, 2017.
Stettler, M. E. J., Eastham, S., and Barrett, S. R. H.: Air quality and public health impacts of UK airports. Part I: Emissions, Atmos. Environ., 45, 5415–5424, https://doi.org/10.1016/j.atmosenv.2011.07.012, 2011.
Streets, D. G., Zhang, Q., Wang, L., He, K., Hao, J., Wu, Y., Tang, Y., and Carmichael, G. R.: Revisiting China's CO emissions after the Transport and Chemical Evolution over the Pacific (TRACE-P) mission: Synthesis of inventories, atmospheric modeling, and observations, J. Geophys. Res., 111, D14306, https://doi.org/10.1029/2006JD007118, 2006.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Travis, K. R., Jacob, D. J., Keller, C. A., Kuang, S., Lin, J., Newchurch, M. J., and Thompson, A. M.: Resolving ozone vertical gradients in air quality models, Atmos. Chem. Phys. Discuss., https://doi.org/10.5194/acp-2017-596, 2017.
U.S. Environmental Protection Agency: Map of EPA Regions, available at: https://cfpub.epa.gov/roe/images/locator_maps/map-1.png (last access: 2 December 2015), 2012.
U.S. Environmental Protection Agency: AirData – Download Data, available from: https://aqs.epa.gov/aqsweb/airdata/download_files.html#Daily (last access: 19 February 2016), 2014.
U.S. Environmental Protection Agnecy: Implementation of the 2015 Primary Ozone NAAQS: Issues Associated with Background Ozone White Paper for Discussion, 2015.
van der Werf, G. R., Randerson, J. T., Giglio, L., Collatz, G. J., Mu, M., Kasibhatla, P. S., Morton, D. C., DeFries, R. S., Jin, Y., and van Leeuwen, T. T.: Global fire emissions and the contribution of deforestation, savanna, forest, agricultural, and peat fires (1997–2009), Atmos. Chem. Phys., 10, 11707–11735, https://doi.org/10.5194/acp-10-11707-2010, 2010.
van Donkelaar, A., Martin, R. V., Leaitch, W. R., Macdonald, A. M., Walker, T. W., Streets, D. G., Zhang, Q., Dunlea, E. J., Jimenez, J. L., Dibb, J. E., Huey, L. G., Weber, R., and Andreae, M. O.: Analysis of aircraft and satellite measurements from the Intercontinental Chemical Transport Experiment (INTEX-B) to quantify long-range transport of East Asian sulfur to Canada, Atmos. Chem. Phys., 8, 2999–3014, https://doi.org/10.5194/acp-8-2999-2008, 2008.
Wang, C., Corbett, J. J., and Firestone, J.: Improving spatial representation of global ship emissions inventories, Environ. Sci. Technol., 42, 193–199, https://doi.org/10.1021/es0700799, 2008.
Wang, H., Jacob, D. J., Le Sager, P., Streets, D. G., Park, R. J., Gilliland, A. B., and van Donkelaar, A.: Surface ozone background in the United States: Canadian and Mexican pollution influences, Atmos. Environ., 43, 1310–1319, https://doi.org/10.1016/j.atmosenv.2008.11.036, 2009.
Wang, P., Schade, G., Estes, M., and Ying, Q.: Improved MEGAN predictions of biogenic isoprene in the contiguous United States, Atmos. Environ., 148, 337–351, https://doi.org/10.1016/j.atmosenv.2016.11.006, 2017.
Xiao, Y., Logan, J. A., Jacob, D. J., Hudman, R. C., Yantosca, R., and Blake, D. R.: Global budget of ethane and regional constraints on U.S. sources, J. Geophys. Res., 113, D21306, https://doi.org/10.1029/2007JD009415, 2008.
Yang, J., Tian, H., Tao, B., Ren, W., Pan, S., Liu, Y., and Wang, Y.: A growing importance of large fires in conterminous United States during 1984–2012, J. Geophys. Res.-Biogeo., 120, 2625–2640, https://doi.org/10.1002/2015JG002965, 2015.
Yevich, R. and Logan, J. A.: An assessment of biofuel use and burning of agricultural waste in the developing world, Global Biogeochem. Cy., 17, 1095, https://doi.org/10.1029/2002GB001952, 2003.
Young, P. J., Naik, V., Fiore, A. M., Gaudel, A., Guo, J., Lin, M. Y., Neu, J., Parrish, D. D., Rieder, H. E., Schnell, J. L., Tilmes, S., Wild, O., Zhang, L., Brandt, J., Delcloo, A., Doherty, R. M., Geels, C., Hegglin, M. I., Hu, L., Im, U., Kumar, R., Luhar, A., Murray, L. T., Plummer, D., Rodriguez, J., Saiz-Lopez, A., Schultz, M. G., Woodhouse, M., Zeng, G., and Ziemke, J.: Tropospheric Ozone Assessment Report (TOAR): Assessment of global-scale model performance for global and regional ozone distributions, variability, and trends, Elem. Sci. Anth., 84 pp., available at: http://eprints.lancs.ac.uk/88836/1/TOAR_Model_Performance_07062017.pdf (last access: 12 December 2017), 2017.
Yu, K., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Miller, C. C., Travis, K. R., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Cohen, R. C., Dibb, J. E., Fried, A., Mikoviny, T., Ryerson, T. B., Wennberg, P. O., and Wisthaler, A.: Sensitivity to grid resolution in the ability of a chemical transport model to simulate observed oxidant chemistry under high-isoprene conditions, Atmos. Chem. Phys., 16, 4369–4378, https://doi.org/10.5194/acp-16-4369-2016, 2016.
Zhang, L., Jacob, D. J., Downey, N. V., Wood, D. A., Blewitt, D., Carouge, C. C., van Donkelaar, A., Jones, D. B. A., Murray, L. T., and Wang, Y.: Improved estimate of the policy-relevant background ozone in the United States using the GEOS-Chem global model with horizontal resolution over North America, Atmos. Environ., 45, 6769–6776, https://doi.org/10.1016/j.atmosenv.2011.07.054, 2011.
Zhang, L., Jacob, D. J., Yue, X., Downey, N. V., Wood, D. A., and Blewitt, D.: Sources contributing to background surface ozone in the US Intermountain West, Atmos. Chem. Phys., 14, 5295–5309, https://doi.org/10.5194/acp-14-5295-2014, 2014.
- Abstract
- Introduction
- Observations and model simulations
- Model evaluation
- Influence of individual sources on average versus high-O3 days
- Interannual variability in the sources influencing high versus average ground-level O3
- Discussion and conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- References
- Supplement
- Abstract
- Introduction
- Observations and model simulations
- Model evaluation
- Influence of individual sources on average versus high-O3 days
- Interannual variability in the sources influencing high versus average ground-level O3
- Discussion and conclusions
- Data availability
- Author contributions
- Competing interests
- Special issue statement
- Acknowledgements
- References
- Supplement