the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 30 Jan 2018
| 30 Jan 2018
The effects of isoprene and NOx on secondary organic aerosols formed through reversible and irreversible uptake to aerosol water
Marwa M. H. El-Sayed
Diana L. Ortiz-Montalvo
Christopher J. Hennigan
Isoprene oxidation produces water-soluble organic gases capable of partitioning to aerosol liquid water. The formation of secondary organic aerosols through such aqueous pathways (aqSOA) can take place either reversibly or irreversibly; however, the split between these fractions in the atmosphere is highly uncertain. The aim of this study was to characterize the reversibility of aqSOA formed from isoprene at a location in the eastern United States under substantial influence from both anthropogenic and biogenic emissions. The reversible and irreversible uptake of water-soluble organic gases to aerosol water was characterized in Baltimore, Maryland, USA, using measurements of particulate water-soluble organic carbon (WSOCp) in alternating dry and ambient configurations. WSOCp evaporation with drying was observed systematically throughout the late spring and summer, indicating reversible aqSOA formation during these times. We show through time lag analyses that WSOCp concentrations, including the WSOCp that evaporates with drying, peak 6 to 11 h after isoprene concentrations, with maxima at a time lag of 9 h. The absolute reversible aqSOA concentrations, as well as the relative amount of reversible aqSOA, increased with decreasing NOx ∕ isoprene ratios, suggesting that isoprene epoxydiol (IEPOX) or other low-NOx oxidation products may be responsible for these effects. The observed relationships with NOx and isoprene suggest that this process occurs widely in the atmosphere, and is likely more important in other locations characterized by higher isoprene and/or lower NOx levels. This work underscores the importance of accounting for both reversible and irreversible uptake of isoprene oxidation products to aqueous particles.
- Article
(5706 KB) - Full-text XML
-
Supplement
(1032 KB) - BibTeX
- EndNote





Isoprene (2-methyl-1,3-butadiene, C5H8) is the most abundant non-methane organic compound emitted globally (Guenther et al., 2012). Isoprene oxidation stimulates tropospheric ozone production and contributes substantially to secondary organic aerosol (SOA) formation, thus impacting air quality and climate (Henze and Seinfeld, 2006; Pfister et al., 2008). In the southeastern United States, isoprene is likely the dominant SOA precursor during summer (Ying et al., 2015; Kim et al., 2015). The oxidation products of isoprene include compounds that partition to aerosol liquid water (ALW), such as isoprene epoxydiol (IEPOX), glyoxal, and methylglyoxal. These species do not partition to dry particles (Kroll et al., 2005; Nguyen et al., 2014), so their condensed-phase products are called aqueous SOA (aqSOA; Ervens et al., 2011). IEPOX uptake also depends on the inorganic composition and acidity of the seed particles (Surratt et al., 2010; Gaston et al., 2014; Budisulistiorini et al., 2017; Lin et al., 2012; Riedel et al., 2015). A body of work indicates that the uptake of water-soluble organic gases into atmospheric waters (clouds, fogs, and aerosol water) is an important pathway for SOA formation (Ervens et al., 2011). Isoprene oxidation products can also form SOA in the absence of aerosol water (Surratt et al., 2006; Nguyen et al., 2014), though the majority of regional-scale isoprene SOA is currently thought to form through aqueous pathways (Marais et al., 2016). Isoprene emissions show strong seasonal variations in most locations (Guenther et al., 2012), suggesting that aqSOA formation is similarly seasonal in nature. Indeed, SOA formed from IEPOX shows a pronounced seasonal signature in the southeastern USA that is consistent with isoprene emissions (Budisulistiorini et al., 2016; Xu et al., 2015).
Although substantial evidence from laboratory, modeling, and ambient studies indicates the importance of aqSOA formation, many uncertainties remain in understanding this pathway on a mechanistic level (McNeill, 2015). A significant uncertainty is the fate of aqSOA under conditions of water evaporation, such as in a cloud cycle or with diurnal changes in ambient relative humidity (RH). The formation of aqSOA is initiated by the equilibrium (and thus reversible) partitioning of water-soluble organic gases to liquid water (McNeill, 2015). In the aqueous phase, the dissolved organics can undergo reversible reactions such as hydration and oligomerization (De Haan et al., 2009) or irreversible reactions such as acid catalysis, reaction with inorganics, or radical reactions (e.g., Ervens et al., 2014; Ortiz-Montalvo et al., 2014; Lee et al., 2013). The former process implies that at least some of the dissolved organics will repartition back to the gas phase when water evaporates, while the latter process can form low-volatility products that remain in the particle phase even after the evaporation of water. Most clouds are non-precipitating (Pruppacher, 1986) and ALW changes throughout the day with changing RH (Nguyen et al., 2014; Khlystov et al., 2005). Thus, determining whether the uptake is reversible or irreversible is critical in understanding the fate of many oxidized organics in the atmosphere. While ambient studies provide evidence for both reversible and irreversible aqSOA formation (El-Sayed et al., 2016, 2015), the reasons underlying these differences are still unclear.
It is important to note that we define aqSOA as all organics present in the condensed phase through partitioning to liquid water, regardless of whether the uptake is reversible or irreversible. Although some definitions of aqSOA only include the organic material that is taken up into liquid water and remains in the particle phase after water evaporation (e.g., Ervens et al., 2011), we favor a more comprehensive definition since the organics contribute to aerosol effects on health and optical properties when they are in the condensed phase. Our definition is consistent with the treatment of other semi-volatile aerosol species such as ammonium nitrate. It is, however, important to distinguish reversible and irreversible aqSOA since the atmospheric lifetime of these compounds may differ significantly depending on their phase (Nguyen et al., 2015). Therefore, we define the low-volatility products that remain in the particle phase after the evaporation of liquid water as “irreversible aqSOA”, and the organic compounds taken up in liquid water that repartition back to the gas phase with water evaporation as “reversible aqSOA”.
Nitrogen oxides (NOx ≡ NO + NO2) may be one factor affecting the reversibility of isoprene aqSOA. NOx plays a critical role in the oxidation of volatile organic compounds (VOCs). This includes a major effect on the chemical pathway of isoprene oxidation, and on the resulting SOA yield (Kroll and Seinfeld, 2008; Ervens et al., 2008). NOx affects the volatility, oxidation state, and aging of isoprene-derived SOA (Xu et al., 2014). Recent modeling studies predict that isoprene oxidation in the eastern USA is split almost equally between high- and low-NOx pathways (Travis et al., 2016). Laboratory studies show significant evaporation of aqueous isoprene SOA particles when dried, indicating reversible aqSOA (Wong et al., 2015). This is consistent with the understanding of aqSOA formed from individual isoprene oxidation products thought to be predominantly IEPOX and glyoxal (Sareen et al., 2017). During the summer, model predictions suggest that glyoxal production from isoprene occurs almost equally through low- and high-NOx pathways in the eastern USA (Chan Miller et al., 2017). Glyoxal is taken up to ALW reversibly and irreversibly (Ortiz-Montalvo et al., 2012; Galloway et al., 2009). IEPOX is formed predominantly through the low-NOx pathway (Paulot et al., 2009; Surratt et al., 2010), and its uptake to ALW could be reversible or irreversible (Nguyen et al., 2014; Riedel et al., 2015). Therefore, potential differences in reversible aqSOA associated with NOx may be due to differences in IEPOX production under these chemical regimes. The aim of this study was to characterize the effects of isoprene and NOx on aqSOA formed reversibly and irreversibly at a site in the eastern USA heavily impacted by biogenic and anthropogenic emissions.
2.1 WSOC measurements
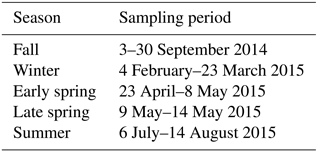
Ambient measurements were carried out across all four seasons in Baltimore, Maryland (Table 1). The experimental setup has been described in detail elsewhere (El-Sayed et al., 2016, 2015). Briefly, water-soluble organic carbon was measured in the gas phase (WSOCg) using a mist chamber (MC), and in the particle phase (WSOCp) using a particle into liquid sampler (PILS; Brechtel Manufacturing), both coupled to a total organic carbon (TOC) analyzer (model 900 Turbo, GE Analytical) operated in Turbo mode. The WSOCp measurement was alternated between an ambient channel (WSOCp) and a “dried” channel (WSOCp,dry) using an automated three-way valve (Brechtel Manufacturing). The WSOCp sample was at ambient RH while the WSOCp,dry sample passed through a silica gel diffusion dryer (Table S1 in the Supplement). Both the WSOCp and the WSOCp,dry samples pass through a parallel-plate carbon denuder (Sunset Laboratories) prior to sampling in the PILS. This reduces gas-phase interferences, which are minor in the PILS (Sullivan et al., 2004), and prevents the re-condensation of volatilized organic gases that evaporate in the dryer. Although some gas-phase organics may be lost to the silica gel (Faust et al., 2017), potentially perturbing the gas–particle equilibrium for the dry channel, sampling both channels through the carbon denuder should minimize such differences. Further, based upon the timescales of ambient organic aerosol (OA) equilibration (minutes to hours; Saha et al., 2017), it is highly unlikely that stripping gas-phase compounds would produce any appreciable OA evaporation with only the 7 s residence time encountered in our system. The diffusion dryer does not implement heating, so differences in the WSOCp concentrations between the two channels are due to WSOCp evaporation that results from ALW evaporation. Note that the WSOCp,dry channel has not been designed to dry particles completely to efflorescence (El-Sayed et al., 2016). WSOCp losses through the three-way valve and through the dried channel are less than 1 % (mass concentration basis; El-Sayed et al., 2016): no corrections to the data were applied. A ratio of OM ∕ OC = 2.1 was used to convert aerosol organic carbon (OC) into organic mass (OM), based upon characterizations of WSOCp in the eastern USA (Xu et al., 2017a).
WSOCp is operationally defined based upon the solubilities of the organics themselves and upon the level of dilution employed for the analysis (Psichoudaki and Pandis, 2013). In the eastern USA, the WSOCp measurement is often used as a surrogate for SOA, especially during summer (Weber et al., 2007). The measurement includes SOA formed through absorptive partitioning and through aqueous-mediated pathways (aqSOA). We consider any WSOCp that evaporates with drying to be reversible aqSOA, since this material exists in the condensed phase because of the aerosol water and partitions back to the gas phase when the water evaporates.
The WSOCp,dry measurement system employs a total drying time of ≈ 7 s. The residence time for equilibrium to take place in evaporating water/organic droplets is dependent on the specific organics as well as the aerosol inorganic chemical composition. Longer drying times may increase the amount of evaporated aqSOA in our system, indicating that our measurements provide a conservative (low) bound estimate on the concentration of reversible aqSOA and on the WSOCp,dry ∕ WSOCp ratio (El-Sayed et al., 2016).
The fully automated online system was housed in a temperature-controlled environmental enclosure (EKTO, Inc.) placed on the rooftop of the Engineering Building at the University of Maryland, Baltimore County (UMBC). The three samples: WSOCg, WSOCp, and WSOCp,dry were repeatedly measured in a 14 min cycle with sampling times of 4, 5, and 5 min, respectively. Dynamic blanks were measured regularly throughout each ambient sampling period. Factory calibrations of the TOC analyzer were regularly checked with sucrose solutions prepared to bracket the range of concentrations observed during ambient sampling.
2.2 VOC and NOx measurements
Isoprene measurements from the Essex Photochemical Assessment Monitoring Stations (PAMS; AQS ID# 240053001) were provided by Maryland Department of the Environment (MDE). The Essex site represents the PAMS station closest to UMBC (≈ 20 km distance). Isoprene was measured by MDE every 6 days from September to May, and hourly during the summer (June, July, and August). The hourly isoprene measurements were automated following EPA method 142, using cryogenic preconcentration for sample collection followed by analysis via gas chromatography with flame ionization detection (GC-FID, Perkin Elmer Clarus 500). Hourly measurements of NOx were also carried out by MDE at the Essex site following method 74 (chemiluminescence). Data were acquired from the US Environmental Protection Agency (https://aqs.epa.gov/api).
A key assumption employed in this analysis is that the WSOC measurements made at UMBC are representative of conditions at Essex, the location of the NOx and isoprene measurements. Aerosol concentrations in the Baltimore–Washington, D.C. region are spatially uniform over tens of kilometers (Beyersdorf et al., 2016). Further, WSOCp concentrations in the eastern USA exhibit small spatial variations across urban-to-rural gradients during the summertime (Weber et al., 2007). These prior analyses showed that aerosol concentrations, and in particular WSOC, were not dependent on wind direction. Isoprene emissions in the eastern USA are regional in nature, due to the expansive coverage of broadleaf forests (Pye et al., 2013; Guenther et al., 2012). NOx emissions are spatially segregated from those of isoprene, and are far more localized. However, the isoprene–NOx chemical regime (high- or low-NOx) in the eastern USA is generally well represented with model resolution of 28 × 28 km, suggesting that the chemistry occurring on small scales, such as in individual power plant plumes, does not significantly affect the regional isoprene–NOx regime (Yu et al., 2016). NOx concentrations at Essex (20 km ENE of UMBC) and HU-Beltsville (35 km SSW of UMBC) are strongly correlated (R=0.89, Fig. S1 in the Supplement), likely due to the overwhelming contribution of mobile source emissions along the heavily traveled I-95 highway corridor to the region (Anderson et al., 2014). Together, this supports our analysis into the effects of isoprene and NOx on reversible aqSOA using the measurements described above.
An overview of the seasonal sampling periods is given in Table 1. Measurements were taken from 3 to 4 weeks on average during each of the four seasons. Note that the spring season has been divided into early (23 April to 8 May) and late (9 to 14 May) periods due to the differences in the WSOCp results observed during these times. The WSOCp measurements have been reported to be a good surrogate of the total SOA in the atmosphere (Weber et al., 2007; Kondo et al., 2007), which includes aqSOA as well as SOA formed through traditional gas-phase partitioning (Donahue et al., 2009). The formation of aqSOA was observed throughout the year, except for the early spring season. This observation was based on the relationship between the fraction of total WSOC in the particle phase, Fp (Fp = WSOCp ∕ (WSOCp + WSOCg)) as a function of RH in combination with seasonal ALW analyses (Hennigan et al., 2008). The individual results for the fall and summer have been previously reported (El-Sayed et al., 2016, 2015). A synthesis of aqSOA formation across all seasons is the subject of ongoing analysis.
3.1 Reversibility of aqSOA formation by season
Previous studies conducted by our group have provided evidence for both irreversible (El-Sayed et al., 2015) and reversible (El-Sayed et al., 2016) aqSOA formation during the fall and summer seasons, respectively. Figure 1 shows the WSOCp,dry ∕ WSOCp ratio across all of the seasons. A ratio of unity indicates that drying did not impact WSOCp while a ratio less than unity indicates that particle drying caused the evaporation of some WSOCp, and thus was considered reversible aqSOA (El-Sayed et al., 2016). Figure 1 shows that the WSOCp,dry ∕ WSOCp ratio was unity during the fall and winter, indicating that the WSOCp remained in the condensed phase upon drying. Therefore, the aqSOA formation that was observed occurred irreversibly (El-Sayed et al., 2015). In the early spring, the WSOCp,dry ∕ WSOCp ratio was also unity, but this was expected since no Fp-RH enhancement was observed and no significant aqSOA was observed during this period. Beginning in the late spring and continuing into the summer, the WSOCp,dry ∕ WSOCp ratio was systematically lower than unity. During both seasons, we observed systematic evaporation of some WSOCp as a result of the ALW evaporation. In the late spring, the WSOCp,dry ∕ WSOCp ratio was 0.92, on average, and decreased further during the summer when it reached an average of 0.87 (El-Sayed et al., 2016). This observation indicates that at least some of the aqSOA formation occurring in the late spring and summer seasons was reversible. WSOCp evaporation was higher during the night than during the day (Fig. S2), likely due to higher RH levels and higher ALW at night (Guo et al., 2015).
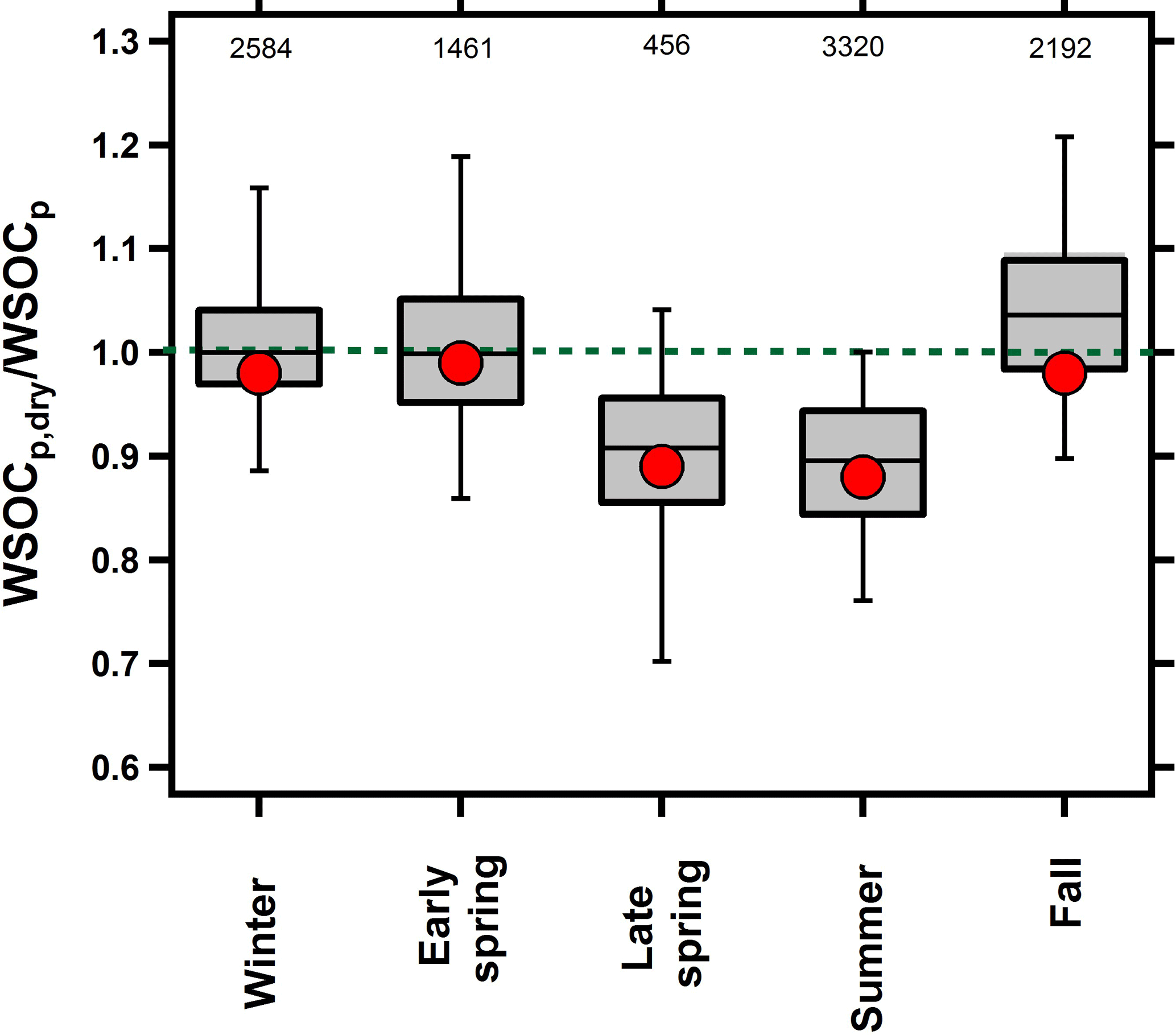
Figure 1Box plot of the overall seasonal WSOCp,dry ∕ WSOCp ratios. For each bin, mean (red marker), median (horizontal black line), 25th and 75th percentiles (lower and upper box values), and 5th and 95th percentiles (vertical lines) are shown. The dotted green line at unity is shown for visual reference. Numbers at the top represent the number of paired WSOCp,dry ∕ WSOCp measurements within each season.

Figure 2Annual climatology of isoprene concentrations in Essex, Maryland (2011 to 2015). Symbols represent average concentrations (in ppbC or nmol C mol−1) while error bars represent ±1σ.
We attribute the observed WSOCp evaporation during the late spring and summer seasons to aqSOA that partitions reversibly to ALW. The physical properties that affect SOA formed through absorptive partitioning (what Ervens et al., 2011, call gasSOA) and SOA formed through an aqueous mediated pathway (aqSOA) are fundamentally different (vapor pressure and gas solubility in water, respectively). Note that ALW can affect SOA formed through traditional absorptive partitioning by increasing the total concentration and decreasing the average molecular weight of the absorbing OM phase (Seinfeld and Pankow, 2003). Models predict that this phenomenon enhances SOA concentrations in the eastern USA (Pankow et al., 2015; Jathar et al., 2016) and that drying the particles will result in the evaporation of some semi-volatile SOA compounds in response to this perturbation (Pankow, 2010). However, the effect of ALW on gas–particle partitioning is more pronounced at low organic concentrations (1 to 2 µg m−3), and its sensitivity becomes less profound at higher OA levels (Pankow, 2010). Previous results from our group showed the opposite effect: evaporated WSOCp concentrations increased significantly with an increase in OA concentrations (El-Sayed et al., 2016). Further, the semi-volatile organic compounds most influenced by this water effect are predicted to be the less-oxidized, fresh SOA (Pankow, 2010). WSOCp is more strongly correlated with the LV-OOA (low-volatility oxygenated organic aerosol) factor identified by the Aerodyne aerosol mass spectrometer (AMS) compared to the SV-OOA (semi-volatile OOA) factor (Sun et al., 2011; Xu et al., 2017b; Kondo et al., 2007). This suggests that the evaporation of WSOCp was not due to the overall effects on OA partitioning (Jathar et al., 2016), but was due to the reversible partitioning of water-soluble organic gases to aerosol water. In the following sections, we characterize the reasons underlying the seasonal differences in WSOCp,dry ∕ WSOCp shown in Fig. 1.
3.2 Climatology of isoprene
Isoprene oxidation products are thought to be the most important precursors to aqSOA formation (Marais et al., 2016). Figure 2 shows the average annual climatology of isoprene in Baltimore, Maryland. These measurements were made at the MDE Essex site, a location ≈ 20 km from UMBC where the WSOC measurements were conducted. In the eastern USA, isoprene emissions are regional (Palmer et al., 2003); therefore, data from the Essex site will show consistent trends with those at UMBC. Isoprene concentrations in Baltimore tend to be very low in the winter and early spring seasons, with average monthly values of ≈ 0.2 ppbC (nmol mol−1), but they start to rise sharply at the beginning of May, and remain elevated (though variable) during the summer season. This is highly consistent with previously measured seasonal isoprene emissions in other parts of the eastern USA (Goldstein et al., 1998). Isoprene concentrations decrease dramatically in September (average decrease of 70 % from 1 to 30 September), and then remain low through the winter.
3.3 Effect of isoprene on reversible aqSOA
During the late spring, the onset of reversible aqSOA formation corresponds to the dramatic increase in isoprene concentrations (Fig. 2). Observations of the AMS IEPOX factor (Budisulistiorini et al., 2016) and chemical markers for isoprene SOA (Kleindienst et al., 2007) show similarly sharp transitions in the spring and fall in the southeastern USA. The highest reversible aqSOA levels were observed during the summer when isoprene emissions were at their maximum. Other VOCs, such as monoterpenes, also contribute to SOA in the eastern USA (Xu et al., 2015), but monoterpene and isoprene SOA tracers show distinctly different temporal patterns in the eastern USA. Isoprene SOA peaks during the summer, but monoterpene SOA tracers exhibit similar (or lower) concentrations in the summer compared to other seasons (Kleindienst et al., 2007; Ding et al., 2008). Further, monoterpene SOA is typically associated with semi-volatile and less-oxidized OA factors in the AMS analysis (Xu et al., 2015; Jimenez et al., 2009) but WSOCp is poorly correlated with these factors (Timonen et al., 2013; Xu et al., 2016). On the basis of these prior studies and the results in Figs. 1 and 2, we attribute the reversible aqSOA in Baltimore to isoprene.
Due to the magnitude of regional isoprene emissions and its predicted contribution to SOA, we would expect relationships between isoprene and both WSOCg and WSOCp concentrations. However, simple correlations between isoprene and WSOC are not expected due to dramatic differences in their atmospheric lifetimes. Under typical summertime conditions, the oxidation of isoprene to form WSOCg will take a few hours (Hodzic et al., 2014). These oxidation products can undergo further reactions to form lower-volatility compounds that partition to the aerosol phase contributing to WSOCp, a process that is expected to take several hours (Ng et al., 2006; Atkinson and Arey, 2003).
The relationship between isoprene and WSOC (both WSOCp and WSOCg) was characterized for the summer, when hourly isoprene data were available. To account for the differences in the expected time frame for transformation of isoprene into WSOCg and WSOCp, we analyzed the WSOC concentrations as a function of isoprene with a variable time lag. We investigated the relationship between the isoprene concentrations at time t and the WSOC concentrations at t+n, where n is the time lag, which was systematically varied from 0 to 13 h. For example, a 1 h time lag indicates that the isoprene concentrations at t are compared to the WSOC concentrations measured t+1 h after those of isoprene. An offset of zero indicates that the timing of the WSOC measurements is aligned with the timing of the isoprene measurements. During each hour, there were four to five WSOCp and WSOCg measurements corresponding to one isoprene sample; therefore, hourly averages of WSOC were calculated to provide a consistent basis for analysis.
First, the isoprene–WSOCg relationship was analyzed for time lags in the range of 0 to 6 h (Fig. 3). The WSOCg data were binned based on the corresponding isoprene concentrations; each marker represents the median of the WSOCg concentration within each isoprene concentration bin. At 0 to 2 h time lags, no relationship was observed between isoprene and WSOCg. This was anticipated because isoprene has a typical atmospheric lifetime of 1 to 2 h against oxidation by OH (Atkinson and Arey, 2003). However, with a time lag of 3 h, an increase in isoprene concentrations was coincident with an increase in WSOCg concentrations. This effect was observed for time lags up to 5 h, as illustrated by the solid blue lines in Fig. 3. Across the entire summer, a 5 ppbC (nmol mol−1) increase in isoprene concentrations was associated with a median increase of 2.0 µg C m−3 in WSOCg. When the time lag between isoprene and WSOCg was more than 5 h, there was no longer a relationship between isoprene and WSOCg concentrations. This observation highlights the effect of isoprene on the formation of water-soluble organic gases. Isoprene and WSOCg showed similar diurnal profiles during the summer, especially when the time lag was considered (Fig. S3). Overall, this suggests that fresh isoprene emissions take about 3 to 5 h to form WSOCg in an urban environment during typical summertime conditions. Note that the measurement of WSOCg only includes compounds with effective Henry's law constants above ≈ 103 M atm−1 (≈ 101 mol m−3 Pa−1) (Spaulding et al., 2002), so the MC does not efficiently sample many first-generation isoprene oxidation products, such as methacrolein ( M atm−1, or mol m−3 Pa−1) or methyl vinyl ketone ( M atm−1, or mol m−3 Pa−1; Sander, 2015).
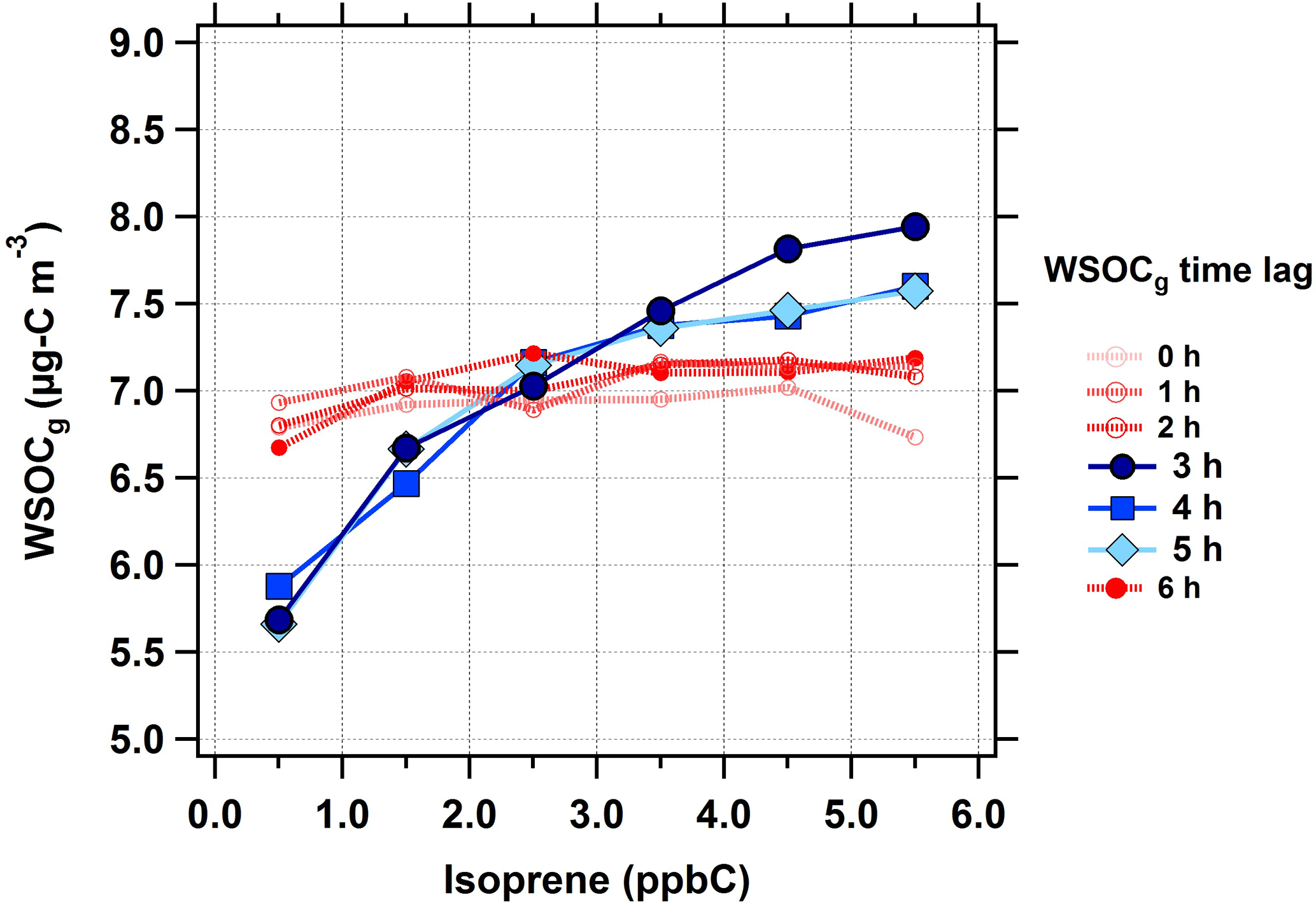
Figure 3Median WSOCg concentrations as a function of isoprene concentrations (in ppbC or nmol C mol−1) at different WSOCg time lags during the summer. The following isoprene concentrations bins were defined as follows: < 1 ppbC, 1 to 2 ppbC, 2 to 3 ppbC, 3 to 4 ppbC, 4 to 5 ppbC, and > 5 ppbC. Scatter and box plots showing individual data are presented in Fig. S6.
The relationship between isoprene and evaporated WSOCp (i.e., reversible aqSOA) was characterized using the same time lag analysis, extended from n = 0 to 13 h. At time lags less than 5 h, there was no relationship between isoprene and evaporated WSOCp concentrations (red dotted lines in Fig. 4). However, the amount of evaporated WSOCp increased with increasing isoprene concentrations when the evaporated WSOCp time lag was in the range of 6 to 11 h (green solid lines in Fig. 4). The highest response of evaporated WSOCp to isoprene was found for a time lag of 9 h. At this time lag, an increase of 5 ppbC (nmol C mol−1) in isoprene concentrations led to a median increase of 0.7 µg m−3 in evaporated WSOCp. Beyond the 11 h time lag, no relationship was observed between isoprene and evaporated WSOCp levels (blue dotted lines in Fig. 4). The average evaporated WSOCp concentrations showed a similar increase with increasing isoprene, but were even higher than the median levels (Fig. S4). For example, at a 9 h time lag, a 5 ppbC (nmol C mol−1) increase in isoprene corresponded to an average increase in evaporated WSOCp of 1.6 µg m−3. The 6 to 11 h time lag between isoprene and the evaporated WSOCp is consistent with the predicted kinetics of isoprene epoxydiol-derived secondary organic aerosol (IEPOX-SOA) formation in the eastern USA (Budisulistiorini et al., 2017). This observed 6 to 11 h time lag between isoprene and the evaporated WSOCp is likely due to multi-generational oxidation (Carlton et al., 2009; Hodzic et al., 2014; Paulot et al., 2009). Alternately, it could be that the isoprene oxidation products that partition reversibly to liquid water were formed relatively quickly (< 6 h), but responded to the diurnal cycle in ALW, which peaks in the eastern USA in the early morning hours (Guo et al., 2015). The observed delay time could also be the combination of these factors. Consistent with Figs. 3 and 4, there was also a strong relationship between the WSOCg concentration and the time-offset evaporated WSOCp concentration (Fig. S5). The above observations suggest that isoprene is strongly linked with the formation of reversible aqSOA in the eastern USA. Based on this relationship, we next consider the effect of NOx on reversible aqSOA formation since NOx is critical to isoprene oxidation chemistry (Kroll et al., 2006).
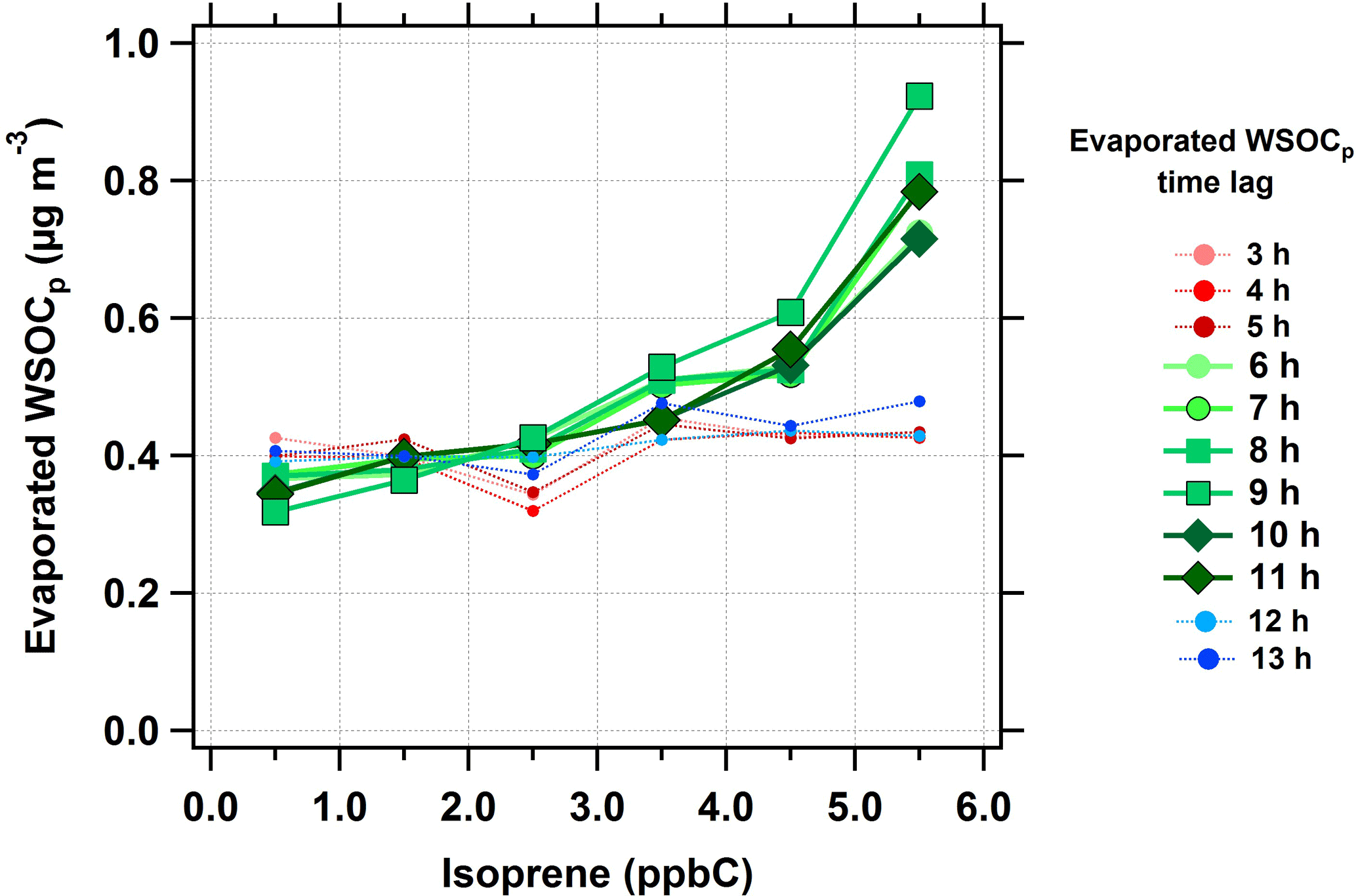
Figure 4Median evaporated WSOCp concentrations as a function of isoprene concentrations (ppbC or nmol C mol−1) at different evaporated WSOCp time lags during the summer. Scatter and box plots showing individual data are presented in Fig. S6.
Note that we assume that the WSOCp measurement is a surrogate for SOA (Weber et al., 2007). However, WSOCp is weakly correlated with lightly oxygenated components in OA, such as the SV-OOA factor often resolved by the AMS (Timonen et al., 2013). Thus, our analysis would likely be a poor method for some SOA systems, for example α-pinene ozonolysis (Jimenez et al., 2009). As discussed above, the WSOCg measurement does not efficiently sample compounds with low Henry's law constants, including some first-generation isoprene oxidation products (Hodzic et al., 2014). These measurement limitations contribute to the 6 to 11 h and 3 to 5 h time lags for the isoprene associations with evaporated WSOCp and WSOCg, respectively. For many compounds, multi-generation oxidation contributes significantly to SOA formation (Ng et al., 2006), and this is almost certainly the case for atmospheric SOA (Jimenez et al., 2009). However, shorter lag times may be observed with other instruments sensitive to early generation oxidation products.
3.4 Effect of NOx on reversible aqSOA
Figure 5 shows the relationship between evaporated WSOCp and the NOx ∕ isoprene ratio during the summer. For this analysis, hourly NOx ∕ isoprene ratios and the hourly evaporated WSOCp concentrations with a 9 h time lag were used, since this timing corresponded to the maximum evaporated WSOCp. Figure 5 shows that the amount of evaporated WSOCp decreased substantially with an increase in the NOx ∕ isoprene ratio. At low NOx ∕ isoprene ratios (less than 0.5 ppb ppbC−1, or 0.5 mol (mol C)−1), the amount of evaporated WSOCp was at its maximum (average of 1.4 µg m−3); however at NOx ∕ isoprene ratios more than 15 ppb ppbC−1 (mol (mol C)−1), the evaporated WSOCp was as low as 0.2 µg m−3. Generally, the evaporated WSOCp decreased with the increase in NOx ∕ isoprene ratios, but flattened out beyond NOx ∕ isoprene ratios of ≈ 5 ppb ppbC−1 (mol (mol C)−1).
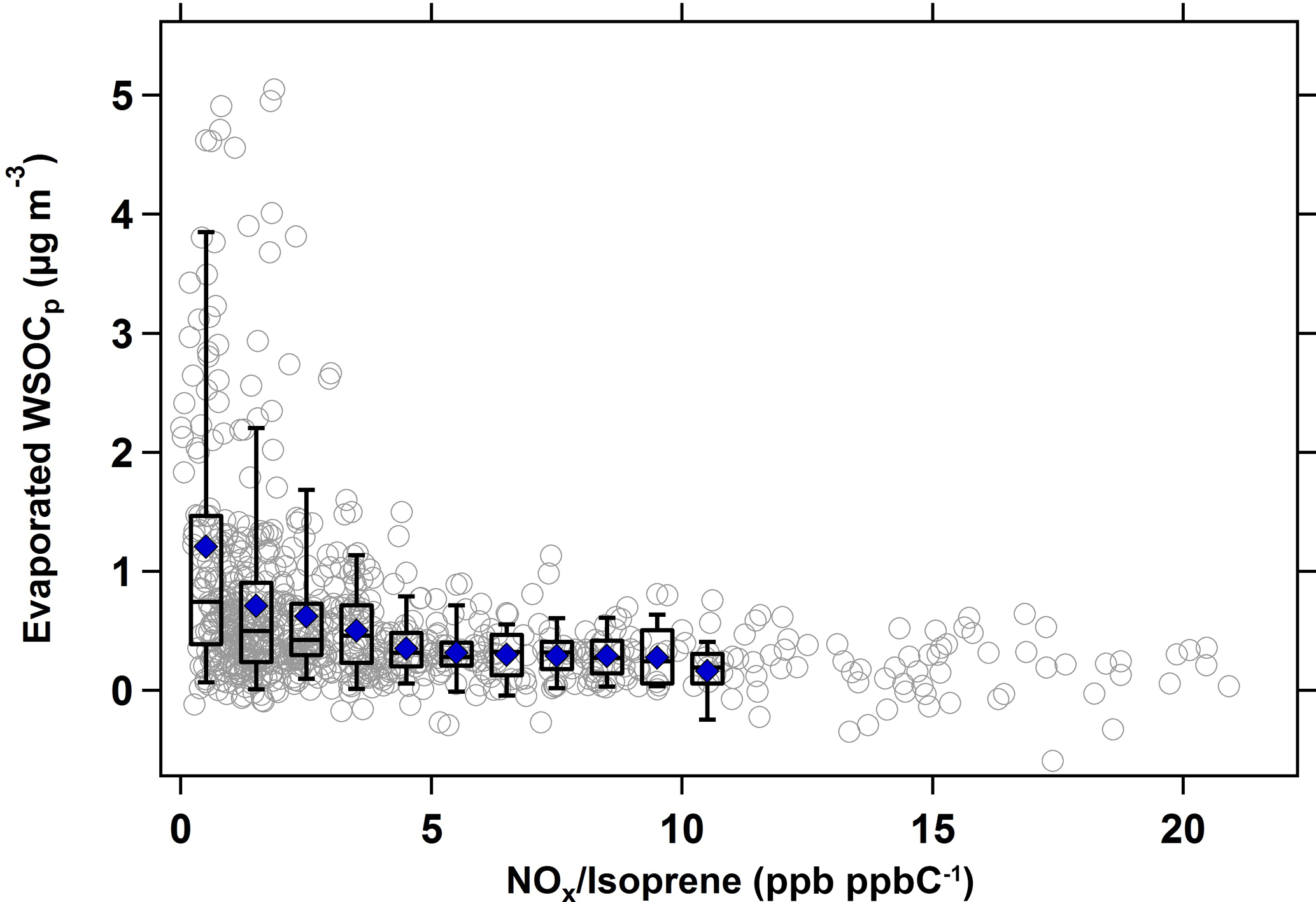
Figure 5Scatter and box plots of evaporated WSOCp (9 h time lag) as a function of NOx ∕ isoprene ratio (ppb ppbC−1 or mol (mol C)−1 in the summer. Bins were chosen to include at least 50 data points. For each bin, mean (blue marker), median (horizontal black line), 25th and 75th percentiles (lower and upper box values), and 5th and 95th percentiles (vertical lines) are shown.
Similarly, the effect of NOx ∕ isoprene ratios on WSOCp concentrations during the summer is shown in Fig. 6. As in Fig. 5, the hourly NOx ∕ isoprene ratios were compared against the hourly WSOCp concentrations at a time lag of 9 h. At NOx ∕ isoprene ratios of less than 0.5 ppb ppbC−1 (mol (mol C)−1), the average WSOCp concentration was ≈ 5 µg m−3, but it decreased substantially to ≈ 1.5 µg m−3 (almost summertime WSOCp background levels) at NOx ∕ isoprene ratios above 15 ppb ppbC−1 (mol (mol C)−1).
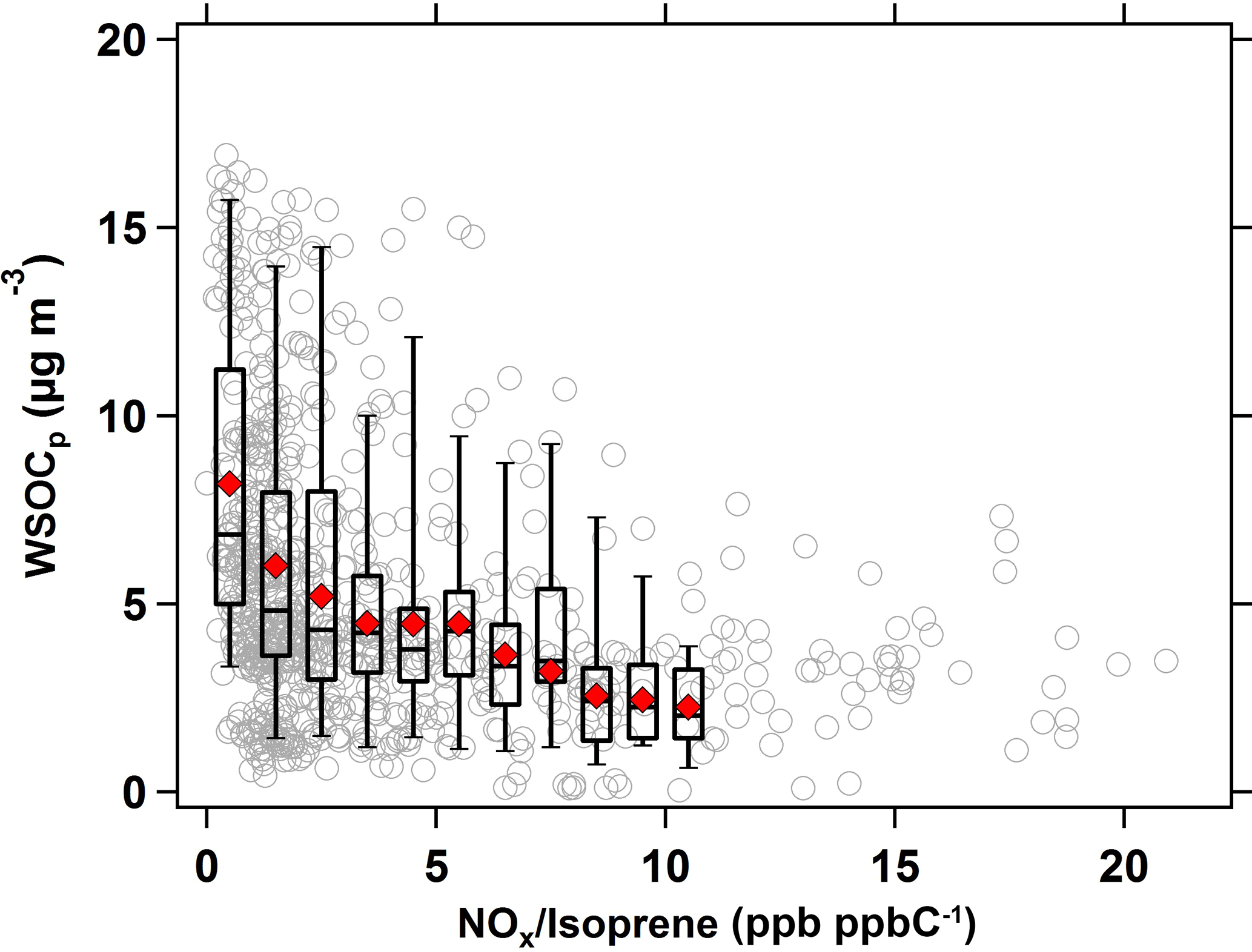
Figure 6Scatter plot of WSOCp as a function of NOx ∕ isoprene ratio (ppb ppbC−1 or mol (mol C)−1) in the summer. Symbols and bins are consistent with those defined in Fig. 5.
If isoprene is indeed associated with the evaporated WSOCp that we observed during the late spring and summer, then a logical question is why we did not observe this phenomenon during measurements throughout September (Fig. 1, El-Sayed et al., 2015). Although isoprene emissions decrease dramatically during September, there are still periods with elevated concentrations. Here, we analyze the effects of NOx and isoprene on the reversibility of isoprene aqSOA by considering the average daily NOx ∕ isoprene ratios during the late spring, summer, and fall. For this analysis, daily averages were used due to the lack of hourly isoprene measurements during the late spring and fall. The relationship between the WSOCp,dry ∕ WSOCp and NOx ∕ isoprene ratios across all three seasons is shown in Fig. 7. Figures 5 and 6 show that the relationships of the NOx ∕ isoprene ratio with WSOCp and evaporated WSOCp are qualitatively similar. However, it is clear from Fig. 7 that WSOCp and the evaporated WSOCp are affected differently by NOx ∕ isoprene. The days in which average NOx ∕ isoprene ratios were higher than 5 ppb ppbC−1 (mol (mol C)−1) were characterized by WSOCp,dry ∕ WSOCp ratios very close to unity, indicating irreversible aqSOA. On the other hand, the days in which NOx ∕ isoprene ratios were lower than 5 ppb ppbC−1 (mol (mol C)−1) were all characterized by WSOCp,dry ∕ WSOCp ratios lower than unity, indicating some reversible aqSOA on these days. Further, the WSOCp,dry ∕ WSOCp ratio decreased with decreasing NOx ∕ isoprene ratios under these conditions. IEPOX is produced under low-NOx conditions with very limited formation in NOx rich environments (Zhang et al., 2017), whereas glyoxal can be produced from both low-and high-NOx pathways with higher yields at high-NOx conditions (Chan Miller et al., 2017). Based on our observations, this suggests that IEPOX was more abundant during the late spring and summer and was responsible for the reversible aqSOA formed under the lower NOx ∕ isoprene conditions. These results provide an explanation for the variability in the seasonal occurrence of reversible aqSOA in the eastern USA.
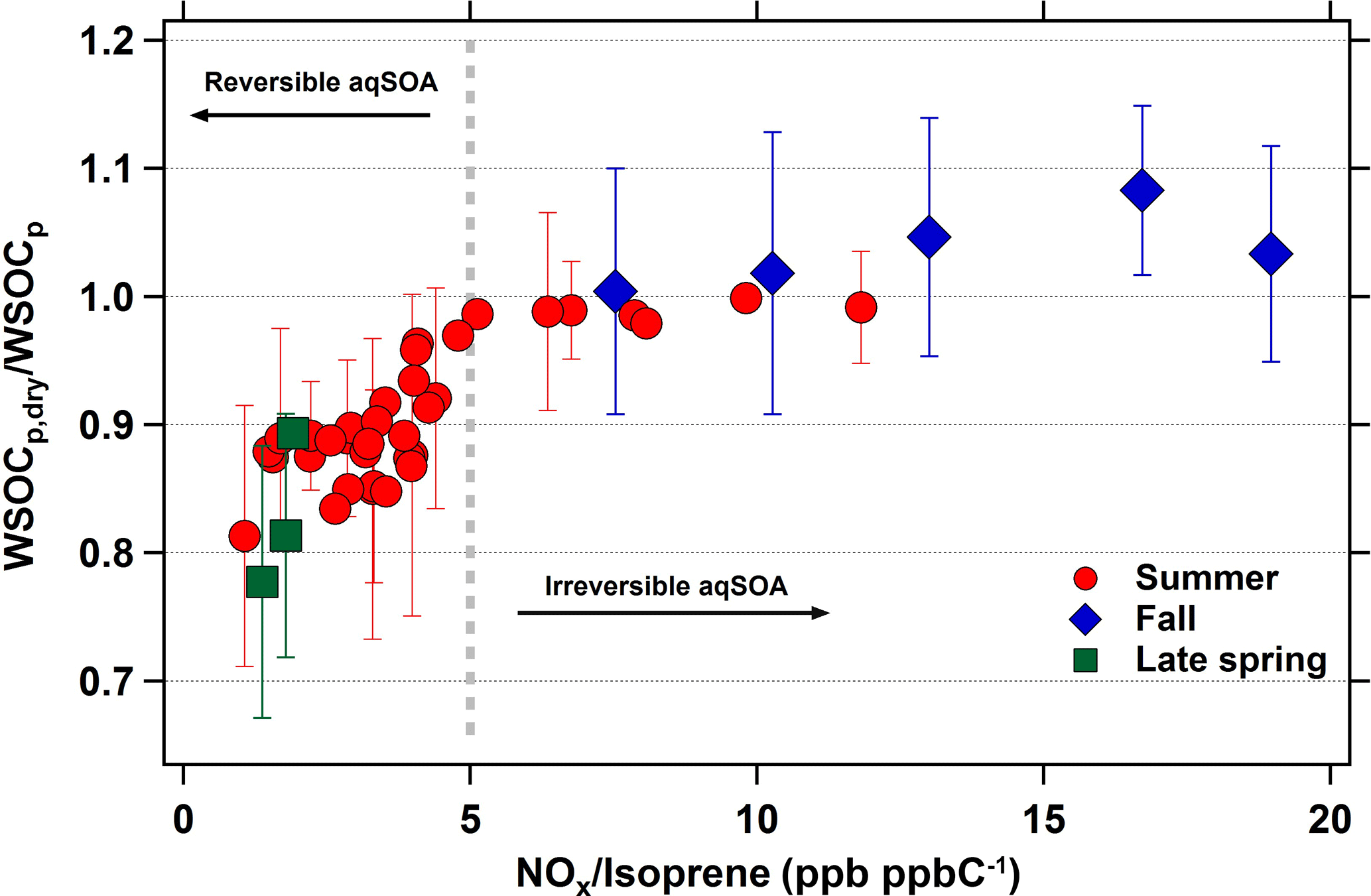
Figure 7Daily average WSOCp,dry ∕ WSOCp ratios as a function of daily average NOx ∕ isoprene ratios (ppb ppbC−1 or mol (mol C)−1). Gray dotted line is representative of the transition zone from reversible to irreversible aqSOA conditions. Error bars represent ±1σ, and are shown for one-third of the summertime data for clarity.
There is uncertainty in the absolute NOx ∕ isoprene ratio that represents the transition to reversible aqSOA. Although NOx concentrations at Essex are strongly correlated with those at a site 50 km away (HU-Beltsville), the absolute NOx concentrations are approximately 2 times higher at Essex (Fig. S1), due to its closer proximity to downtown Baltimore. Therefore, although Fig. 7 suggests that reversible aqSOA formation occurs at NOx ∕ isoprene ratios below 5 ppb ppbC−1 (mol (mol C)−1), transitions at lower ratios may be observed in other areas.
These results represent the first observations to characterize the seasonal occurrence of reversible aqSOA formation. The results suggest an important effect on aerosol measurements that implement drying, which may not measure (or may incompletely measure) reversible aqSOA. Our results suggest that this is especially relevant in areas with high isoprene emissions. For example, Zhang et al. (2012) observed substantial loss of WSOCp (≈ 30 % on average) from federal reference method (FRM) filters in the southeastern USA. It is likely that reversible aqSOA contributed to this measurement artifact, although direct comparisons to our WSOCp,dry measurement would be needed to test this hypothesis. These compounds are important, since they contribute to aerosol effects – visibility, aerosol optical depth (AOD), health, climate – when they are in the condensed phase.
We hypothesize that the evaporation of WSOCp observed with drying during the late spring and summer is due to the reversible partitioning of IEPOX to aerosol water, or to other low-NOx isoprene oxidation products such as multifunctional hydroperoxides (Y. Liu et al., 2016; Krechmer et al., 2015; Riva et al., 2016; J. Liu et al., 2016). This is supported by strong associations between the evaporated WSOCp and isoprene concentrations using the time lag analysis. It is further supported by the decreasing WSOCp,dry ∕ WSOCp ratios with decreasing NOx ∕ isoprene ratios. Note that Sareen et al. (2017) predict very low dissolved IEPOX in the eastern USA during summer (< 0.01 µg m−3), suggesting reversibly formed reaction products are the dominant contributors to reversible aqSOA.
Laboratory studies have found reversible and irreversible uptake of IEPOX to aqueous particles (Nguyen et al., 2014; Riedel et al., 2015). However, ambient studies generally suggest that IEPOX-SOA has very low volatility (Lopez-Hilfiker et al., 2016; Hu et al., 2016). This could be due to challenges measuring the reversible aqSOA by the methods used to derive volatilities. For example, it is unclear how the instruments employed by Lopez-Hilfiker et al. (2016) and Hu et al. (2016) respond to reversible IEPOX reaction products present in the aqueous phase. It could also be that the evaporated WSOCp we observe is contributed by other low-NOx isoprene oxidation products. Approximately 30 % of isoprene-derived SOA generated under NOx-free conditions partitioned reversibly to aerosol water, but the molecular identities of the reversible aqSOA were not determined (Wong et al., 2015). Although the experiments of Wong et al. (2015) were performed in a chemical regime where IEPOX formation is favored, it did not contribute to the SOA in their experiments due to high OH levels. Given the absence of IEPOX-SOA in the experiments of Wong et al. (2015), the atmospheric relevance of their results needs further review. The uptake of other, non-IEPOX, low-NOx oxidation products may explain such observations (J. Liu et al., 2016; Y. Liu et al., 2016; Riva et al., 2016; Krechmer et al., 2015). Overall, identifying the molecular composition of the reversible aqSOA that is associated with low-NOx isoprene oxidation will require targeted field measurements.
The effect of water evaporation on WSOCp also has important implications for the representation of SOA formation in models. The results in Fig. 1 show that ≈ 10 to 15 % of the total WSOCp evaporates with drying during the late spring and summer, on average. This suggests that the fraction of aqSOA that is formed reversibly is much higher than 15 %, since the measurement of WSOCp includes aqSOA and compounds formed through traditional SOA partitioning (e.g., Donahue et al., 2009). Further, the fraction of WSOCp that evaporates with drying is variable, with values of up to 60 % for individual measurements (El-Sayed et al., 2016). Models that include aqSOA and aerosol multi-phase chemistry can improve predictions of OA (e.g., Carlton et al., 2008; Marais et al., 2016). A complication of model evaluations is that comparisons of modeled OA concentrations to ambient measurements may be problematic if the measurements themselves are subject to the bias discussed above. For this reason, accounting for both reversible and irreversible uptake of water-soluble organic gases to liquid water is critical (McNeill, 2015). Our observations, supported by laboratory studies (Faust et al., 2017), suggest that treatment of aqSOA as an irreversible uptake process is not consistent with actual phenomena occurring in the atmosphere, especially in the eastern USA. Although likely due to a different mechanism, Y. Liu et al. (2016) and Riva et al. (2017) also showed that isoprene oxidation forms semi-volatile compounds that re-partition back to the gas phase after forming SOA.
The lifetime of organic compounds in the atmosphere is strongly dependent on their phase (Pye et al., 2017). Oxygenated organic compounds in the gas-phase often have much shorter lifetimes than particle-phase organics due to significantly higher dry deposition velocities (Nguyen et al., 2015) and photolysis rates (Fu et al., 2008). The reversible uptake of WSOCg to aerosol water may effectively shield these species from such loss processes, resulting in enhanced transport. Thus, accounting for the reversible partitioning of water-soluble organic gases to aerosol water would likely improve model predictions of these compounds.
NOx plays a critical role in the oxidation of VOCs, including effects on the composition and quantity of SOA produced. Herein, we show that NOx strongly affects the amount and nature of SOA produced in an urban area that is under substantial influence from biogenic emissions. Higher concentrations of WSOCp were associated with decreasing NOx ∕ isoprene ratios. The fraction of WSOCp that evaporated with drying was also inversely related to NOx ∕ isoprene. In the future, isoprene concentrations are predicted to increase in response to changes in temperature and land use associated with climate change (Heald et al., 2008; Sanderson et al., 2003). The eastern USA is currently undergoing a transition from high- to low-NOx chemical regimes (Travis et al., 2016; Edwards et al., 2017), and NOx levels are likely to continue decreasing (He et al., 2013). This suggests future NOx ∕ isoprene ratios will generally decrease across the eastern USA, as well, resulting in increased production of reversible aqSOA. The current results are from the greater Baltimore metropolitan area; although we observe a range of NOx concentrations and NOx ∕ isoprene ratios, these measurements are representative of an urban environment. Thus, we may expect WSOCp,dry ∕ WSOCp ratios to be even lower in more rural environments impacted by isoprene emissions.
In addition to NOx, sulfate also strongly affects SOA formation from isoprene through its separate contributions to ALW, particle acidity, and aqueous chemistry (Xu et al., 2015; Nguyen et al., 2014; Surratt et al., 2010). Laboratory studies have not yet elucidated the role of each factor in the reversibility of isoprene SOA, and we do not have sufficient sulfate data to characterize such effects with our analysis. However, it is worth noting that particle acidity is not likely a factor in the relative split between reversible and irreversible aqSOA formed from isoprene. Studies predict that particles in the eastern USA are highly acidic throughout the year (Weber et al., 2016; Battaglia et al., 2017; Guo et al., 2016, 2015), and acidity is not a limiting factor in isoprene SOA formation during the summer (Budisulistiorini et al., 2016; Xu et al., 2015). The implication from our observations is that reversible aqSOA from isoprene forms even in the presence of such persistently acidic particles. This further questions the treatment of isoprene SOA as an irreversible uptake process in models.
The eastern USA is undergoing a transition from a high- to low-NOx chemical regime, which has broad implications for nighttime chemistry, ozone production, and SOA formation (Travis et al., 2016; Marais et al., 2016; Edwards et al., 2017). Using a time lag analysis, we show that NOx ∕ isoprene strongly affects concentrations of SOA in the eastern USA, including SOA formed through the reversible uptake of water-soluble organic gases to aqueous particles. Lower NOx leads to a higher fraction of aqueous SOA formed reversibly. Our measurements from an urban area suggest that this process is even more important in other, more rural environments.
Predictions of future NOx and isoprene emissions in response to regulations, technology, and climate change also suggest that this process may increase in importance going forward. Such an inference is complicated by concurrent reductions in SO2 emissions in the USA and other developed nations. The consequent decreases in sulfate may offset the effects of NOx reductions on isoprene SOA (de Sá et al., 2017). However, we stress that prior studies into the NOx–sulfate–isoprene system have not systematically determined how these species affect the reversibility of isoprene SOA. Therefore, while we hypothesize that future decreases in NOx and increases in isoprene will increase reversible isoprene SOA (or at least the reversible fraction), the role of changing sulfate will also need to be considered. Future laboratory and modeling studies will be needed to address this question directly.
We hypothesize that IEPOX uptake to aqueous particles is responsible for the reversible aqSOA, but other low-NOx isoprene oxidation products are possible as well (Wong et al., 2015). We quantify reversible aqSOA through observations of WSOCp evaporation that results from drying. Ultimately, molecular composition measurements made concurrently with our WSOC system are required to identify the chemical species responsible for this phenomenon. The evaporation of WSOCp with drying occurred systematically during the late spring and summer, and was linked to isoprene and NOx. This has importance for a wide range of aerosol measurements that implement drying. It also has importance for modeling multi-phase SOA formation, as simplified treatment of irreversible uptake does not represent actual atmospheric processes.
The NOx and isoprene data are available from the EPA (https://aqs.epa.gov/api). The WSOC data are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-1171-2018-supplement.
The authors declare that they have no conflict of interest.
Certain commercial equipment, instruments or materials are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology.
This work was
supported by the National Science Foundation through award
CHE-1454763.
Edited by: Jason
Surratt
Reviewed by: three anonymous referees
Anderson, D. C., Loughner, C. P., Diskin, G., Weinheimer, A., Canty, T. P., Salawitch, R. J., Worden, H. M., Fried, A., Mikoviny, T., and Wisthaler, A.: Measured and modeled CO and NOy in DISCOVER-AQ: An evaluation of emissions and chemistry over the eastern US, Atmos. Environ., 96, 78–87, 2014.
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Battaglia Jr., M. A., Douglas, S., and Hennigan, C. J.: Effect of the Urban Heat Island on Aerosol pH, Environ. Sci. Technol., 51, 13095, https://doi.org/10.1021/acs.est.7b02786, 2017.
Beyersdorf, A. J., Ziemba, L. D., Chen, G., Corr, C. A., Crawford, J. H., Diskin, G. S., Moore, R. H., Thornhill, K. L., Winstead, E. L., and Anderson, B. E.: The impacts of aerosol loading, composition, and water uptake on aerosol extinction variability in the Baltimore–Washington, D.C. region, Atmos. Chem. Phys., 16, 1003–1015, https://doi.org/10.5194/acp-16-1003-2016, 2016.
Budisulistiorini, S. H., Baumann, K., Edgerton, E. S., Bairai, S. T., Mueller, S., Shaw, S. L., Knipping, E. M., Gold, A., and Surratt, J. D.: Seasonal characterization of submicron aerosol chemical composition and organic aerosol sources in the southeastern United States: Atlanta, Georgia,and Look Rock, Tennessee, Atmos. Chem. Phys., 16, 5171–5189, https://doi.org/10.5194/acp-16-5171-2016, 2016.
Budisulistiorini, S. H., Nenes, A., Carlton, A. G., Surratt, J. D., McNeill, V. F., and Pye, H. O. T.: Simulating Aqueous-Phase Isoprene-Epoxydiol (IEPOX) Secondary Organic Aerosol Production During the 2013 Southern Oxidant and Aerosol Study (SOAS), Environ. Sci. Technol., 51, 5026–5034, https://doi.org/10.1021/acs.est.6b05750, 2017.
Carlton, A. G., Turpin, B. J., Altieri, K. E., Seitzinger, S. P., Mathur, R., Roselle, S. J., and Weber, R. J.: CMAQ Model Performance Enhanced When In-Cloud Secondary Organic Aerosol is Included: Comparisons of Organic Carbon Predictions with Measurements, Environ. Sci. Technol., 42, 8798–8802, https://doi.org/10.1021/es801192n, 2008.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Chan Miller, C., Jacob, D. J., Marais, E. A., Yu, K., Travis, K. R., Kim, P. S., Fisher, J. A., Zhu, L., Wolfe, G. M., Hanisco, T. F., Keutsch, F. N., Kaiser, J., Min, K.-E., Brown, S. S., Washenfelder, R. A., González Abad, G., and Chance, K.: Glyoxal yield from isoprene oxidation and relation to formaldehyde: chemical mechanism, constraints from SENEX aircraft observations, and interpretation of OMI satellite data, Atmos. Chem. Phys., 17, 8725–8738, https://doi.org/10.5194/acp-17-8725-2017, 2017.
De Haan, D. O., Corrigan, A. L., Tolbert, M. A., Jimenez, J. L., Wood, S. E., and Turley, J. J.: Secondary Organic Aerosol Formation by Self-Reactions of Methylglyoxal and Glyoxal in Evaporating Droplets, Environ. Sci. Technol., 43, 8184–8190, https://doi.org/10.1021/es902152t, 2009.
de Sá, S. S., Palm, B. B., Campuzano-Jost, P., Day, D. A., Newburn, M. K., Hu, W., Isaacman-VanWertz, G., Yee, L. D., Thalman, R., Brito, J., Carbone, S., Artaxo, P., Goldstein, A. H., Manzi, A. O., Souza, R. A. F., Mei, F., Shilling, J. E., Springston, S. R., Wang, J., Surratt, J. D., Alexander, M. L., Jimenez, J. L., and Martin, S. T.: Influence of urban pollution on the production of organic particulate matter from isoprene epoxydiols in central Amazonia, Atmos. Chem. Phys., 17, 6611–6629, https://doi.org/10.5194/acp-17-6611-2017, 2017.
Ding, X., Zheng, M., Yu, L., Zhang, X., Weber, R. J., Yan, B., Russell, A. G., Edgerton, E. S., and Wang, X.: Spatial and seasonal trends in biogenic secondary organic aerosol tracers and water-soluble organic carbon in the southeastern United States, Environ. Sci. Technol., 42, 5171–5176, 2008.
Donahue, N. M., Robinson, A. L., and Pandis, S. N.: Atmospheric organic particulate matter: From smoke to secondary organic aerosol, Atmos. Environ., 43, 94–106, 2009.
Edwards, P. M., Aikin, K. C., Dube, W. P., Fry, J. L., Gilman, J. B., de Gouw, J. A., Graus, M. G., Hanisco, T. F., Holloway, J., Hubler, G., Kaiser, J., Keutsch, F. N., Lerner, B. M., Neuman, J. A., Parrish, D. D., Peischl, J., Pollack, I. B., Ravishankara, A. R., Roberts, J. M., Ryerson, T. B., Trainer, M., Veres, P. R., Wolfe, G. M., Warneke, C., and Brown, S. S.: Transition from high- to low-NOx control of night-time oxidation in the southeastern US, Nat. Geosci., 10, 490–495, https://doi.org/10.1038/ngeo2976, 2017.
El-Sayed, M. M. H., Wang, Y. Q., and Hennigan, C. J.: Direct atmospheric evidence for the irreversible formation of aqueous secondary organic aerosol, Geophys. Res. Lett., 42, 5577–5586, https://doi.org/10.1002/2015gl064556, 2015.
El-Sayed, M. M. H., Amenumey, D., and Hennigan, C. J.: Drying-Induced Evaporation of Secondary Organic Aerosol during Summer, Environ. Sci. Technol., 50, 3626–3633, https://doi.org/10.1021/acs.est.5b06002, 2016.
Ervens, B., Carlton, A. G., Turpin, B. J., Altieri, K. E., Kreidenweis, S. M., and Feingold, G.: Secondary organic aerosol yields from cloud-processing of isoprene oxidation products, Geophys. Res. Lett., 35, L02816, https://doi.org/10.1029/2007gl031828, 2008.
Ervens, B., Turpin, B. J., and Weber, R. J.: Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field and model studies, Atmos. Chem. Phys., 11, 11069–11102, https://doi.org/10.5194/acp-11-11069-2011, 2011.
Ervens, B., Sorooshian, A., Lim, Y. B., and Turpin, B. J.: Key parameters controlling OH-initiated formation of secondary organic aerosol in the aqueous phase (aqSOA), J. Geophys. Res.-Atmos., 119, 3997–4016, https://doi.org/10.1002/2013JD021021, 2014.
Faust, J. A., Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Role of Aerosol Liquid Water in Secondary Organic Aerosol Formation from Volatile Organic Compounds, Environ. Sci. Technol., 51, 1405–1413, https://doi.org/10.1021/acs.est.6b04700, 2017.
Fu, T. M., Jacob, D. J., Wittrock, F., Burrows, J. P., Vrekoussis, M., and Henze, D. K.: Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.-Atmos., 113, D15303, https://doi.org/10.1029/2007jd009505, 2008.
Galloway, M. M., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Flagan, R. C., Seinfeld, J. H., and Keutsch, F. N.: Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions, Atmos. Chem. Phys., 9, 3331–3345, https://doi.org/10.5194/acp-9-3331-2009, 2009.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive Uptake of an Isoprene-Derived Epoxydiol to Submicron Aerosol Particles, Environ. Sci. Technol., 48, 11178–11186, https://doi.org/10.1021/es5034266, 2014.
Goldstein, A. H., Goulden, M. L., Munger, J. W., Wofsy, S. C., and Geron, C. D.: Seasonal course of isoprene emissions from a midlatitude deciduous forest, J. Geophys. Res.-Atmos., 103, 31045–31056, https://doi.org/10.1029/98jd02708, 1998.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Guo, H., Xu, L., Bougiatioti, A., Cerully, K. M., Capps, S. L., Hite Jr., J. R., Carlton, A. G., Lee, S.-H., Bergin, M. H., Ng, N. L., Nenes, A., and Weber, R. J.: Fine-particle water and pH in the southeastern United States, Atmos. Chem. Phys., 15, 5211–5228, https://doi.org/10.5194/acp-15-5211-2015, 2015.
Guo, H., Sullivan, A. P., Campuzano-Jost, P., Schroder, J. C., Lopez-Hilfiker, F. D., Dibb, J. E., Jimenez, J. L., Thornton, J. A., Brown, S. S., and Nenes, A.: Fine particle pH and the partitioning of nitric acid during winter in the northeastern United States, J. Geophys. Res.-Atmos., 121, 10355–10376, https://doi.org/10.1002/2016JD025311, 2016.
He, H., Stehr, J. W., Hains, J. C., Krask, D. J., Doddridge, B. G., Vinnikov, K. Y., Canty, T. P., Hosley, K. M., Salawitch, R. J., Worden, H. M., and Dickerson, R. R.: Trends in emissions and concentrations of air pollutants in the lower troposphere in the Baltimore/Washington airshed from 1997 to 2011, Atmos. Chem. Phys., 13, 7859–7874, https://doi.org/10.5194/acp-13-7859-2013, 2013.
Heald, C. L., Henze, D. K., Horowitz, L. W., Feddema, J., Lamarque, J. F., Guenther, A., Hess, P. G., Vitt, F., Seinfeld, J. H., Goldstein, A. H., and Fung, I.: Predicted change in global secondary organic aerosol concentrations in response to future climate, emissions, and land use change, J. Geophys. Res.-Atmos., 113, D05211, https://doi.org/10.1029/2007jd009092, 2008.
Hennigan, C. J., Bergin, M. H., Dibb, J. E., and Weber, R. J.: Enhanced secondary organic aerosol formation due to water uptake by fine particles, Geophys. Res. Lett., 35, L18801, https://doi.org/10.1029/2008gl035046, 2008.
Henze, D. K. and Seinfeld, J. H.: Global secondary organic aerosol from isoprene oxidation, Geophys. Res. Lett., 33, L09812, https://doi.org/10.1029/2006gl025976, 2006.
Hodzic, A., Aumont, B., Knote, C., Lee-Taylor, J., Madronich, S., and Tyndall, G.: Volatility dependence of Henry's law constants of condensable organics: Application to estimate depositional loss of secondary organic aerosols, Geophys. Res. Lett., 41, 4795–4804, https://doi.org/10.1002/2014gl060649, 2014.
Hu, W., Palm, B. B., Day, D. A., Campuzano-Jost, P., Krechmer, J. E., Peng, Z., de Sá, S. S., Martin, S. T., Alexander, M. L., Baumann, K., Hacker, L., Kiendler-Scharr, A., Koss, A. R., de Gouw, J. A., Goldstein, A. H., Seco, R., Sjostedt, S. J., Park, J.-H., Guenther, A. B., Kim, S., Canonaco, F., Prévôt, A. S. H., Brune, W. H., and Jimenez, J. L.: Volatility and lifetime against OH heterogeneous reaction of ambient isoprene-epoxydiols-derived secondary organic aerosol (IEPOX-SOA), Atmos. Chem. Phys., 16, 11563–11580, https://doi.org/10.5194/acp-16-11563-2016, 2016.
Jathar, S. H., Cappa, C. D., Wexler, A. S., Seinfeld, J. H., and Kleeman, M. J.: Simulating secondary organic aerosol in a regional air quality model using the statistical oxidation model – Part 1: Assessing the influence of constrained multi-generational ageing, Atmos. Chem. Phys., 16, 2309–2322, https://doi.org/10.5194/acp-16-2309-2016, 2016.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525, https://doi.org/10.1126/science.1180353, 2009.
Khlystov, A., Stanier, C. O., Takahama, S., and Pandis, S. N.: Water content of ambient aerosol during the Pittsburgh air quality study, J. Geophys. Res.-Atmos., 110, D07S10, https://doi.org/10.1029/2004jd004651, 2005.
Kim, P. S., Jacob, D. J., Fisher, J. A., Travis, K., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Jimenez, J. L., Campuzano-Jost, P., Froyd, K. D., Liao, J., Hair, J. W., Fenn, M. A., Butler, C. F., Wagner, N. L., Gordon, T. D., Welti, A., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Teng, A. P., Millet, D. B., Schwarz, J. P., Markovic, M. Z., and Perring, A. E.: Sources, seasonality, and trends of southeast US aerosol: an integrated analysis of surface, aircraft, and satellite observations with the GEOS-Chem chemical transport model, Atmos. Chem. Phys., 15, 10411–10433, https://doi.org/10.5194/acp-15-10411-2015, 2015.
Kleindienst, T. E., Jaoui, M., Lewandowski, M., Offenberg, J. H., Lewis, C. W., Bhave, P. V., and Edney, E. O.: Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 41, 8288–8300, https://doi.org/10.1016/j.atmosenv.2007.06.045, 2007.
Kondo, Y., Miyazaki, Y., Takegawa, N., Miyakawa, T., Weber, R. J., Jimenez, J. L., Zhang, Q., and Worsnop, D. R.: Oxygenated and water-soluble organic aerosols in Tokyo, J. Geophys. Res.-Atmos., 112, D01203, https://doi.org/10.1029/2006jd007056, 2007.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., and Rivera-Rios, J. C.: Formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-NO oxidation, Environ. Sci. Technol., 49, 10330–10339, 2015.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation under high-NOx conditions, Geophys. Res. Lett., 32, L18808, https://doi.org/10.1029/2005gl023637, 2005.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol Formation from Isoprene Photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Lee, A. K. Y., Zhao, R., Li, R., Liggio, J., Li, S.-M., and Abbatt, J. P. D.: Formation of Light Absorbing Organo-Nitrogen Species from Evaporation of Droplets Containing Glyoxal and Ammonium Sulfate, Environ. Sci. Technol., 47, 12819–12826, https://doi.org/10.1021/es402687w, 2013.
Lin, P., Yu, J. Z., Engling, G., and Kalberer, M.: Organosulfates in humic-like substance fraction isolated from aerosols at seven locations in East Asia: A study by ultra-high-resolution mass spectrometry, Environ. Sci. Technol., 46, 13118–13127, 2012.
Liu, J., D'Ambro, E. L., Lee, B. H., Lopez-Hilfiker, F. D., Zaveri, R. A., Rivera-Rios, J. C., Keutsch, F. N., Iyer, S., Kurten, T., Zhang, Z., Gold, A., Surratt, J. D., Shilling J. E., and Thornton, J. A.: Efficient isoprene secondary organic aerosol formation from a non-IEPOX pathway, Environ. Sci. Technol., 50, 9872–9880, 2016.
Liu, Y., Kuwata, M., McKinney, K. A., and Martin, S. T.: Uptake and release of gaseous species accompanying the reactions of isoprene photo-oxidation products with sulfate particles, Phys. Chem. Chem. Phys., 18, 1595–1600, https://doi.org/10.1039/C5CP04551G, 2016.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Iyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jimenez, J., Hallquist, M., and Thornton, J. A.: Molecular Composition and Volatility of Organic Aerosol in the Southeastern U.S.: Implications for IEPOX Derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
McNeill, V. F.: Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols, Environ. Sci. Technol., 49, 1237–1244, https://doi.org/10.1021/es5043707, 2015.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of First- versus Second-Generation Products to Secondary Organic Aerosols Formed in the Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015.
Ortiz-Montalvo, D. L., Lim, Y. B., Perri, M. J., Seitzinger, S. P., and Turpin, B. J.: Volatility and Yield of Glycolaldehyde SOA Formed through Aqueous Photochemistry and Droplet Evaporation, Aerosol Sci. Technol., 46, 1002–1014, https://doi.org/10.1080/02786826.2012.686676, 2012.
Ortiz-Montalvo, D. L., Häkkinen, S. A. K., Schwier, A. N., Lim, Y. B., McNeill, V. F., and Turpin, B. J.: Ammonium Addition (and Aerosol pH) Has a Dramatic Impact on the Volatility and Yield of Glyoxal Secondary Organic Aerosol, Environ. Sci. Technol., 48, 255–262, https://doi.org/10.1021/es4035667, 2014.
Palmer, P. I., Jacob, D. J., Fiore, A. M., Martin, R. V., Chance, K., and Kurosu, T. P.: Mapping isoprene emissions over North America using formaldehyde column observations from space, J. Geophys. Res.-Atmos., 108, 4180, https://doi.org/10.1029/2002jd002153, 2003.
Pankow, J. F.: Organic particulate material levels in the atmosphere: Conditions favoring sensitivity to varying relative humidity and temperature, P. Natl. Acad. Sci. USA, 107, 6682–6686, https://doi.org/10.1073/pnas.1001043107, 2010.
Pankow, J. F., Marks, M. C., Barsanti, K. C., Mahmud, A., Asher, W. E., Li, J., Ying, Q., Jathar, S. H., and Kleeman, M. J.: Molecular view modeling of atmospheric organic particulate matter: Incorporating molecular structure and co-condensation of water, Atmos. Environ., 122, 400–408, 2015.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Pfister, G. G., Emmons, L. K., Hess, P. G., Lamarque, J. F., Orlando, J. J., Walters, S., Guenther, A., Palmer, P. I., and Lawrence, P. J.: Contribution of isoprene to chemical budgets: A model tracer study with the NCAR CTM MOZART-4, J. Geophys. Res.-Atmos., 113, D05308, https://doi.org/10.1029/2007JD008948, 2008.
Pruppacher, H. R.: The Role of Cloudphysics in Atmospheric Multiphase Systems: Ten Basic Statements, in: Chemistry of Multiphase Atmospheric Systems, edited by: Jaeschke, W., Springer Berlin Heidelberg, Berlin, Heidelberg, 133–190, 1986.
Psichoudaki, M. and Pandis, S. N.: Atmospheric Aerosol Water-Soluble Organic Carbon Measurement: A Theoretical Analysis, Environ. Sci. Technol., 47, 9791–9798, https://doi.org/10.1021/es402270y, 2013.
Pye, H. O. T., Pinder, R. W., Piletic, I. R., Xie, Y., Capps, S. L., Lin, Y.-H., Surratt, J. D., Zhang, Z., Gold, A., Luecken, D. J., Hutzell, W. T., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Epoxide Pathways Improve Model Predictions of Isoprene Markers and Reveal Key Role of Acidity in Aerosol Formation, Environ. Sci. Technol., 47, 11056–11064, https://doi.org/10.1021/es402106h, 2013.
Pye, H. O. T., Murphy, B. N., Xu, L., Ng, N. L., Carlton, A. G., Guo, H., Weber, R., Vasilakos, P., Appel, K. W., Budisulistiorini, S. H., Surratt, J. D., Nenes, A., Hu, W., Jimenez, J. L., Isaacman-VanWertz, G., Misztal, P. K., and Goldstein, A. H.: On the implications of aerosol liquid water and phase separation for organic aerosol mass, Atmos. Chem. Phys., 17, 343–369, https://doi.org/10.5194/acp-17-343-2017, 2017.
Riedel, T. P., Lin, Y.-H., Budisulistiorini, S. H., Gaston, C. J., Thornton, J. A., Zhang, Z., Vizuete, W., Gold, A., and Surratt, J. D.: Heterogeneous Reactions of Isoprene-Derived Epoxides: Reaction Probabilities and Molar Secondary Organic Aerosol Yield Estimates, Environ. Sci. Technol. Lett., 2, 38–42, https://doi.org/10.1021/ez500406f, 2015.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., Canagaratna, M. R., and Surratt, J. D.: Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016.
Riva, M., Budisulistiorini, S. H., Zhang, Z., Gold, A., Thornton, J. A., Turpin, B. J., and Surratt, J. D.: Multiphase reactivity of gaseous hydroperoxide oligomers produced from isoprene ozonolysis in the presence of acidified aerosols, Atmos. Environ., 152, 314–322, 2017.
Saha, P. K., Khlystov, A., Yahya, K., Zhang, Y., Xu, L., Ng, N. L., and Grieshop, A. P.: Quantifying the volatility of organic aerosol in the southeastern US, Atmos. Chem. Phys., 17, 501–520, https://doi.org/10.5194/acp-17-501-2017, 2017.
Sander, R.: Compilation of Henry's law constants (version 4.0) for water as solvent, Atmos. Chem. Phys., 15, 4399–4981, https://doi.org/10.5194/acp-15-4399-2015, 2015.
Sanderson, M. G., Jones, C. D., Collins, W. J., Johnson, C. E., and Derwent, R. G.: Effect of climate change on isoprene emissions and surface ozone levels, Geophys. Res. Lett., 30, 1936, https://doi.org/10.1029/2003gl017642, 2003.
Sareen, N., Waxman, E. M., Turpin, B. J., Volkamer, R., and Carlton, A. G.: Potential of Aerosol Liquid Water to Facilitate Organic Aerosol Formation: Assessing Knowledge Gaps about Precursors and Partitioning, Environ. Sci. Technol., 51, 3327–3335, https://doi.org/10.1021/acs.est.6b04540, 2017.
Seinfeld, J. H. and Pankow, J. F.: Organic atmospheric particulate material, Annu. Rev. Phys. Chem., 54, 121–140, 2003.
Spaulding, R. S., Talbot, R. W., and Charles, M. J.: Optimization of a Mist Chamber (Cofer Scrubber) for Sampling Water-Soluble Organics in Air, Environ. Sci. Technol., 36, 1798–1808, https://doi.org/10.1021/es011189x, 2002.
Sullivan, A. P., Weber, R. J., Clements, A. L., Turner, J. R., Bae, M. S., and Schauer, J. J.: A method for on-line measurement of water-soluble organic carbon in ambient aerosol particles: Results from an urban site, Geophys. Res. Lett., 31, L13105, https://doi.org/10.1029/2004gl019681, 2004.
Sun, Y.-L., Zhang, Q., Schwab, J. J., Demerjian, K. L., Chen, W.-N., Bae, M.-S., Hung, H.-M., Hogrefe, O., Frank, B., Rattigan, O. V., and Lin, Y.-C.: Characterization of the sources and processes of organic and inorganic aerosols in New York city with a high-resolution time-of-flight aerosol mass apectrometer, Atmos. Chem. Phys., 11, 1581–1602, https://doi.org/10.5194/acp-11-1581-2011, 2011.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl Acad. Sci. USA, 107, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Timonen, H., Carbone, S., Aurela, M., Saarnio, K., Saarikoski, S., Ng, N. L., Canagaratna, M. R., Kulmala, M., Kerminen, V.-M., Worsnop, D. R., and Hillamo, R.: Characteristics, sources and water-solubility of ambient submicron organic aerosol in springtime in Helsinki, Finland, J. Aerosol Sci., 56, 61–77, https://doi.org/10.1016/j.jaerosci.2012.06.005, 2013.
Travis, K. R., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Zhu, L., Yu, K., Miller, C. C., Yantosca, R. M., Sulprizio, M. P., Thompson, A. M., Wennberg, P. O., Crounse, J. D., St. Clair, J. M., Cohen, R. C., Laughner, J. L., Dibb, J. E., Hall, S. R., Ullmann, K., Wolfe, G. M., Pollack, I. B., Peischl, J., Neuman, J. A., and Zhou, X.: Why do models overestimate surface ozone in the Southeast United States?, Atmos. Chem. Phys., 16, 13561–13577, https://doi.org/10.5194/acp-16-13561-2016, 2016.
Weber, R. J., Sullivan, A. P., Peltier, R. E., Russell, A., Yan, B., Zheng, M., de Gouw, J., Warneke, C., Brock, C., Holloway, J. S., Atlas, E. L., and Edgerton, E.: A study of secondary organic aerosol formation in the anthropogenic-influenced southeastern United States, J. Geophys. Res.-Atmos., 112, D13302, https://doi.org/10.1029/2007jd008408, 2007.
Weber, R. J., Guo, H., Russell, A. G., and Nenes, A.: High aerosol acidity despite declining atmospheric sulfate concentrations over the past 15 years, Nat. Geosci., 9, 282–285, 2016.
Wong, J. P. S., Lee, A. K. Y., and Abbatt, J. P. D.: Impacts of Sulfate Seed Acidity and Water Content on Isoprene Secondary Organic Aerosol Formation, Environ. Sci. Technol., 49, 13215–13221, https://doi.org/10.1021/acs.est.5b02686, 2015.
Xu, L., Kollman, M. S., Song, C., Shilling, J. E., and Ng, N. L.: Effects of NOx on the Volatility of Secondary Organic Aerosol from Isoprene Photooxidation, Environ. Sci. Technol., 48, 2253–2262, https://doi.org/10.1021/es404842g, 2014.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Xu, L., Guo, H., Weber, R. J., and Ng, N. L.: Chemical characterization of water-soluble organic aerosol in contrasting rural and urban environments in the southeastern United States, Environ. Sci. Technol., 51, 78–88, 2016.
Xu, L., Guo, H., Weber, R. J., and Ng, N. L.: Chemical Characterization of Water-Soluble Organic Aerosol in Contrasting Rural and Urban Environments in the Southeastern United States, Environ. Sci. Technol., 51, 78–88, https://doi.org/10.1021/acs.est.6b05002, 2017a.
Xu, W., Han, T., Du, W., Wang, Q., Chen, C., Zhao, J., Zhang, Y., Li, J., Fu, P., Wang, Z., Worsnop, D. R., and Sun, Y.: Effects of Aqueous-Phase and Photochemical Processing on Secondary Organic Aerosol Formation and Evolution in Beijing, China, Environ. Sci. Technol., 51, 762–770, https://doi.org/10.1021/acs.est.6b04498, 2017b.
Ying, Q., Li, J., and Kota, S. H.: Significant Contributions of Isoprene to Summertime Secondary Organic Aerosol in Eastern United States, Environ. Sci. Technol., 49, 7834–7842, https://doi.org/10.1021/acs.est.5b02514, 2015.
Yu, K., Jacob, D. J., Fisher, J. A., Kim, P. S., Marais, E. A., Miller, C. C., Travis, K. R., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Cohen, R. C., Dibb, J. E., Fried, A., Mikoviny, T., Ryerson, T. B., Wennberg, P. O., and Wisthaler, A.: Sensitivity to grid resolution in the ability of a chemical transport model to simulate observed oxidant chemistry under high-isoprene conditions, Atmos. Chem. Phys., 16, 4369–4378, https://doi.org/10.5194/acp-16-4369-2016, 2016.
Zhang, X., Liu, Z., Hecobian, A., Zheng, M., Frank, N. H., Edgerton, E. S., and Weber, R. J.: Spatial and seasonal variations of fine particle water-soluble organic carbon (WSOC) over the southeastern United States: implications for secondary organic aerosol formation, Atmos. Chem. Phys., 12, 6593–6607, https://doi.org/10.5194/acp-12-6593-2012, 2012.
Zhang, Y. J., Tang, L. L., Sun, Y. L., Favez, O., Canonaco, F., Albinet, A., Couvidat, F., Liu, D. T., Jayne, J. T., Wang, Z., Croteau, P. L., Canagaratna, M. R., Zhou, H. C., Prevot, A. S. H., and Worsnop, D. R.: Limited formation of isoprene epoxydiols-derived secondary organic aerosol under NOx-rich environments in Eastern China, Geophys. Res. Lett., 44, 2035–2043, https://doi.org/10.1002/2016gl072368, 2017.