the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Jul 2018
| 19 Jul 2018
Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS
Michael Le Breton
Yujue Wang
Åsa M. Hallquist
Ravi Kant Pathak
Jing Zheng
Yudong Yang
Dongjie Shang
Marianne Glasius
Thomas J. Bannan
Qianyun Liu
Chak K. Chan
Carl J. Percival
Wenfei Zhu
Shengrong Lou
David Topping
Yuchen Wang
Jianzhen Yu
Keding Lu
Mattias Hallquist
A time-of-flight chemical ionization mass spectrometer (CIMS) utilizing the Filter Inlet for Gas and Aerosol (FIGAERO) was deployed at a regional site 40 km north-west of Beijing and successfully identified and measured 17 sulfur-containing organics (SCOs are organo/nitrooxy organosulfates and sulfonates) with biogenic and anthropogenic precursors. The SCOs were quantified using laboratory-synthesized standards of lactic acid sulfate and nitrophenol organosulfate (NP OS). The variation in field observations was confirmed by comparison to offline measurement techniques (orbitrap and high-performance liquid chromatography, HPLC) using daily averages. The mean total (of the 17 identified by CIMS) SCO particle mass concentration was 210 ± 110 ng m−3 and had a maximum of 540 ng m−3, although it contributed to only 2 ± 1 % of the organic aerosol (OA). The CIMS identified a persistent gas-phase presence of SCOs in the ambient air, which was further supported by separate vapour-pressure measurements of NP OS by a Knudsen Effusion Mass Spectrometer (KEMS). An increase in relative humidity (RH) promoted partitioning of SCO to the particle phase, whereas higher temperatures favoured higher gas-phase concentrations.
Biogenic emissions contributed to only 19 % of total SCOs measured in this study. Here, C10H16NSO7, a monoterpene-derived SCO, represented the highest fraction (10 %) followed by an isoprene-derived SCO. The anthropogenic SCOs with polycyclic aromatic hydrocarbon (PAH) and aromatic precursors dominated the SCO mass loading (51 %) with C11H11SO7, derived from methyl naphthalene oxidation, contributing to 40 ng m−3 and 0.3 % of the OA mass. Anthropogenic-related SCOs correlated well with benzene, although their abundance depended highly on the photochemical age of the air mass, tracked using the ratio between pinonic acid and its oxidation product, acting as a qualitative photochemical clock. In addition to typical anthropogenic and biogenic precursors the biomass-burning precursor nitrophenol (NP) provided a significant level of NP OS. It must be noted that the contribution analysis here is only representative of the detected SCOs. There are likely to be many more SCOs present which the CIMS has not identified.
Gas- and particle-phase measurements of glycolic acid suggest that partitioning towards the particle phase promotes glycolic acid sulfate production, contrary to the current formation mechanism suggested in the literature. Furthermore, the HSO4⋅ cluster measured by the CIMS was utilized as a qualitative marker for acidity and indicates that the production of total SCOs is efficient in highly acidic aerosols with high and organic content. This dependency becomes more complex when observing individual SCOs due to variability of specific VOC precursors.
- Article
(2913 KB) - Full-text XML
-
Supplement
(358 KB) - BibTeX
- EndNote





Atmospheric particulate matter (PM) is known to play a major role in affecting climate and air quality, leading to severe health issues, such as respiratory and cardiovascular degradation (Pope et al., 2002; Kim et al., 2015). Secondary organic aerosols (SOAs), formed through reactions of volatile organic compounds (VOCs) yielding semi-volatile products that partition into the aerosol phase, represent a significant fraction of PM (Hallquist et al., 2009). They remain the most poorly understood PM source due to the complexity of their chemical nature, resulting in discrepancies between observations and models (Heald et al., 2005). Annual average PM1 (particulate matter of diameter less than 1 µm) concentrations in Beijing reached 89.5 µg m−3 in 2013 and, although they recently dropped to 80.6 µg m−3, are still significantly above the Chinese National Ambient Air Quality Standard (CNAAQS, 35 µg m−3 annual average). The knowledge gap of PM primary emissions and secondary production limits scientifically based abatement strategies targeting effects of secondary pollution in highly polluted regions (Hallquist et al., 2016; Q. Zhang et al., 2012). Therefore, Beijing is an ideal case study region for intense measurement campaigns that increase our understanding of the sources and processes involved in atmospheric aerosol chemistry in megacities. A growing number of field studies in this region have been carried out in recent years that are specifically focused on the haze events investigating the composition of primary and secondary particle aerosols and their formation mechanisms (Guo et al., 2012, 2014, 2013; Huang et al., 2010; Hu et al., 2016, 2017; Li et al., 2017).
Organosulfates (OSs), here part of sulfur-containing organics (SCOs), are known important SOA components formed by reactions between reactive organic compounds and sulfate (Iinuma et al., 2007; Surratt et al., 2007, 2008), which is generated by the oxidation of SO2, primarily emitted by fossil fuel combustion (Wuebbles and Jain, 2001). OSs have previously been measured in ambient aerosols at a number of geographical locations, from remote regions to highly populated urban environments (Surratt et al., 2007, 2008; Kristensen and Glasius, 2011; Stone et al., 2012; H. Zhang et al., 2012; Worton et al., 2013; Shalamzari et al., 2013; Hansen et al., 2014), although their composition and contribution to organic mass can vary significantly (Huang et al., 2015). To date, several of their precursors are not known (e.g. Hansen et al., 2014). Mechanistic studies reveal multiple possible pathways for SCO formation, which depend on the availability of reactants in the atmosphere (Hettiyadura et al., 2015), increasing the complexity of understanding their formation and fate within models. Measurements of specific SCOs have been shown to individually contribute up to 1 % of the total organics (Olson et al., 2011; Liao et al., 2015).
Isoprene SCOs are hypothesized to be the most abundant precursor in the ambient atmosphere (Surratt et al., 2007; Liao et al., 2015) and are often used as markers of isoprene-derived SOA in field campaigns (H. Zhang et al., 2012). Aromatic SCOs considered to originate from anthropogenic sources have been recently observed in Lahore, Pakistan (Stone et al., 2012) and in urban sites in eastern Asia (Lin et al., 2012). Riva et al. (2015, 2016) have also previously probed the SCO formation potential from PAH and alkane oxidation in the presence of acidic sulfate aerosols. Glycolic acid sulfate (GAS) is considered another potentially important SCO due to its common abundance and possible sources (Olson et al., 2011; Liao et al., 2015). It is thought to form via a gas-phase precursor reaction with acidic aerosol sulfate or from the particle-phase reaction of methyl vinyl ketone with a sulfate particle, although both of these mechanisms are yet to be proven (Liao et al., 2015). GAS is also the only SCO to date which has been detected in the gas phase (Ehn et al., 2010), indicating the possible importance of gas in particle-phase partitioning of some SCOs.
SCOs are thought to be good tracers for heterogeneous aerosol phase chemistry and SOA formation since the known formation mechanisms involve reactive uptake of gas-phase organic species onto aerosol (Surratt et al., 2010). Due to their hydrophilic nature, polarity and relatively low volatility, they may significantly help nanoparticle growth and increase their potential to become cloud condensation nuclei (Smith et al., 2008). Therefore, it is imperative to improve our knowledge of SCO abundance, formation, distribution, precursors and fate to help to develop our understanding of SOA formation.
Mass spectrometry coupled with electrospray ionization is a common method used to detect SCOs (Iinuma et al., 2007; Reemtsma et al., 2006; Surratt et al., 2007; Gomez-Gonzalez et al., 2008). Liquid chromatography can efficiently separate aromatic and monoterpene-derived organic sulfates-containing aromatic rings or long alkyl chains and is used for speciation of SCOs (Stone et al., 2012). Furthermore, hydrophilic interaction liquid chromatography has been utilized as a very selective technique due to its ability to allow the SCO to retain a carboxyl group, enabling detection of a larger suite of compounds (Gao et al., 2006). The methods above often rely on sampling filters in the field and therefore provide a relatively low measurement frequency. This can limit the ability to evaluate production pathways when concentrations are often integrated over a period of hours or more. Further reactions on filters between the organics and sulfates have also been postulated to add a bias to the SCO concentration measured with respect to initial deposition onto the filter (Hettiyadura et al., 2017; Kristensen et al., 2016). Recently, a particle analysis laser mass spectrometer (PALMS) was utilized to measure a number of OSs over the USA, highlighting the ability of time-of-flight mass spectrometers to measure several SCOs at high time frequencies (Liao et al., 2015).
This study utilizes a Filter Inlet for Gas and Aerosol (FIGAERO) time-of-flight chemical ionization mass spectrometer (ToF-CIMS) for the measurement of ambient SCOs at a semi-rural site 40 km from Beijing, China. This instrument enables measurements of either the gas-phase components or thermally desorbed particles by a high-resolution mass spectrometer via a multi-port inlet, as described in detail by Lopez-Hilfiker et al. (2014). The soft and selective ionization technique and high time resolution coupled with the FIGAERO enables the simultaneous detection and measurement of SCOs in the gas and particle phase at ng m−3 concentrations. This work aims to identify dominant SCOs in Beijing and their precursors. The high time resolution measurements are utilized to probe their abundance under different chemical and environmental regimes, providing insight into their formation.
2.1 Site description
The data presented here were collected during the measurement campaign “Photochemical smog in China” with an initiative to enhance our understanding of SOA formation via photochemical smog in China (Hallquist et al., 2016). The campaign was coordinated by Peking University (PKU) and the University of Gothenburg with a focus on spring-/summertime pollution episodes in north-eastern China through gas and particle-phase measurements. The set-up was situated at a semi-rural site 40 km north-east of downtown Beijing close to Changping town (40.2207∘ N, 116.2312∘ E). All online instruments sampled from inlets on the fourth-floor laboratory (12 m above ground) at Peking University Changping Campus from 13 May to 23 June 2016, while filter measurements took place on the roof. The average temperature and relative humidity throughout the campaign were 23 ∘C and 44 % respectively. The wind speed averaged at 2 m s−1 from the south to the south-west. A total of four pollution episodes were observed during the campaign period, which are classified as sustained periods of high aerosol loading, reaching a maximum of 115 micrograms per cubic metre (µg m−3) for PM1. The episodes were dominated by organic and nitrate aerosols, although episode 3 contained high sulfate loading, equal to that of nitrate. The mass loading for the semi-rural site showed good correlation with the PKU campus measurement site (30 km south–south-west of the Changping site and 12 km north-west of downtown Beijing) throughout the campaign, allowing for extrapolation of the semi-rural site results to inner-city conditions. HYSPLIT back-trajectory results showed the pollution episodes often correlated with air masses coming from the direction of Beijing (south-south-east). Clean-air days mostly had north-westerly winds with clean air from the rural mountain regions north-west of Beijing and the measurement site.
A high-resolution time-of-flight aerosol mass spectrometer (ToF-AMS) was utilized to measure the mass concentrations and size distributions of non-refractory species in submicron aerosols, including organics, sulfate, ammonium and chloride (DeCarlo et al., 2006; Hu et al., 2013). The set-up of this instrument has been previously described by Hu et al. (2016). An IONICON Analytik high-sensitivity PTR-MS (proton transfer mass spectrometer) as described by de Gouw and Warneke et al. (2007) provided supporting precursor VOC measurements.
2.2 ToF-CIMS set-up
Gas and particle-phase species were measured using an iodide ToF-CIMS coupled to the FIGAERO inlet (Lopez-Hilfiker et al., 2014). The ToF-CIMS can be operated in either negative or positive ionization modes, and a variety of reagent ion sources can be used. In this work the ToF-CIMS was operated in single-reflection mode. The negative iodide ion (I−) was used as the reagent in all experiments. Dry ultra-high-purity N2 was passed over a permeation tube containing liquid CH3I (Alfa Aesar, 99 %), and the flow was passing a Tofwerk X-ray ion source type P (operated at 9.5 kV and 150 µA) to produce the ionization ions. The ionized gas was then directed to the ion–molecule reaction (IMR) through an orifice (Ø = 1 µm). Reaction products (e.g. compound X) were identified by their corresponding cluster ions, XI− or the deprotonated ion, allowing for the collection of whole-molecule data. The nominal reagent and sample flow rates into the IMR chamber of the instrument were 3.5 and 2 litres per minute (L min−1) respectively. The IMR itself was temperature controlled at 40 ∘C and operated at a nominal pressure of 500 mbar. The ToF-CIMS was configured to measure singularly charged ions with a mass-to-charge ratio (m∕z) of 7–620 and a reduced mass range in order to compensate for the lower count rate emitted by the soft X-ray source with respect to the polonium-235 radioactive source as commonly deployed. The tuning was optimized to increase sensitivity, which resulted in a spectral resolution of 3500. In some instances during the campaign, the mass range was changed to a higher mass range (1000 m∕z) to ensure no major contributing peaks were being unaccounted for. Perfluoropentanoic acid was utilized as a mass calibrant up to m∕z 527 through its dimer and trimer. This range of mass calibration peaks also limited accurate peak identification above m∕z 620.
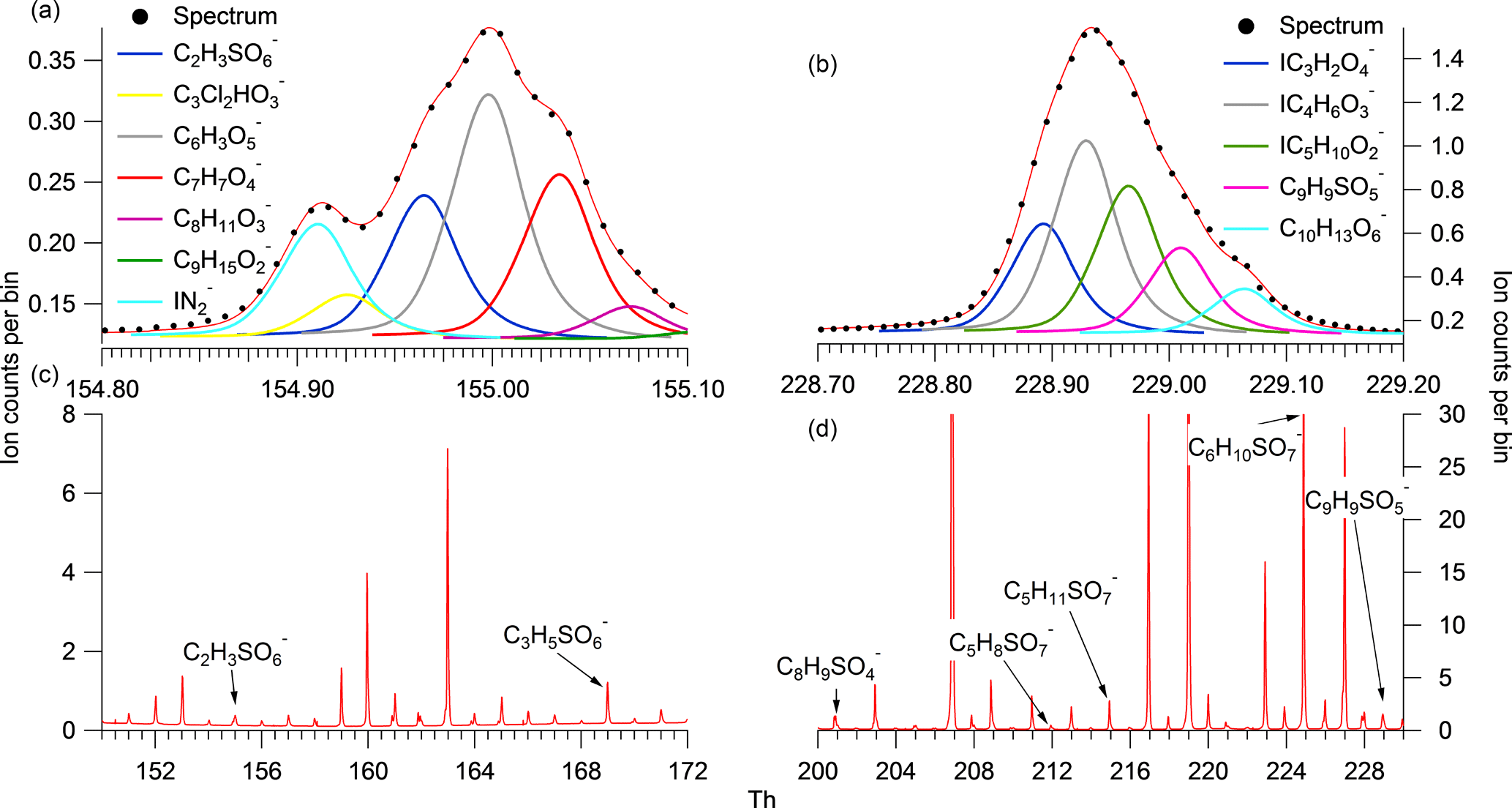
Figure 1Panels (c) and (d) display the average campaign mass spectrum for different regions on the spectra, which are further expanded to specific HR fittings for individual peaks in panels (a) and (b).
The FIGAERO inlet collected particles on a Zefluor® PTFE membrane filter. The aerosol sample line was composed of 12 mm copper tubing, while 12 mm Teflon tubing was used for the gas sample line. The FIGAERO was operated in a cyclic pattern, with 25 min of gas-phase sampling and simultaneous particle collection, followed by a 20 min period during which the filter was shifted into positioned over the IMR inlet and the collected particle mass was desorbed. Desorption was facilitated by a 2 LPM flow of heated UHP N2 over the filter. The temperature of the N2 was increased from 20 to 250 ∘C in 15 min (3.5 ∘C min−1), followed by a 5 min temperature soak time to ensure that all remaining mass that volatilizes at 250 ∘C was removed from the filter. The resulting desorption time series profiles allowed for a distinct separation of measured species as a function of their thermal properties.
Table 1SCOs identified at the Changping site with their masses, chemical names and potential precursors.
2.3 SCO measurement
2.3.1 Identification
Spectral analysis was performed using Towfware V2.5.11. The average peak shape for the tuning utilized for this campaign was used to calculate the mass resolution and optimization of the baseline fit. The mass spectrum was mass calibrated (allowing for accurate centroid peak position to be estimated, improving on a Gaussian assumption) using four ions up to mass 527 (the dimer of perfluoropentanoic acid) and applying a custom peak shape to achieve accurate peak identification below 5 ppm error across the mass range (0–620 AMU). A time series of the mass calibrant error for the entire campaign (Fig. S1 in the Supplement) illustrates how the error deviated by only ±1 throughout the measurement period. This provides confidence that variation in the signal and peak positioning did not result in large errors of identification and quantification of the analysed peaks. A priori unknown peaks were added to resolve overlapping peaks on the spectra until the residual was less than 5 %. Each unknown peak was assigned a chemical formula using the peaks exact mass maxima to five decimal places and also isotopic ratios of subsequent minor peaks. An accurate fitting was characterized by a ppm error of less than 5 and subsequent accurate fittings of isotopic peaks. An example of the spectra and peak fittings can be found in Fig. 1, highlighting the mass spectral fit for GAS and C9H9SO5. Although the structure cannot be determined with CIMS, it is assumed that no fragmentation of larger SCO species contribute to the SCO identified due to the soft ionization technique employed. The SCOs were identified in the spectra as negative ions assumed to be formed by hydrogen removal. Here, we present 17 SCOs that were identified in the mass spectra, which are displayed in Table 1 with their respective exact mass, formula, literature nomenclature and possible precursors. The peak fittings for all 17 SCOs are presented in the Supplement (Fig. S2). All 17 SCOs represented a significant signal in the average desorption spectra from the particle-phase analysis. It must be noted that gas-phase spectra at times contained other ions at a similar mass to the SCOs that contributed to higher counts than the SCO. This may result in a variable error to the measurement, although this should be at a minimum due to the use of a custom peak shape and low general mass calibration error of the spectra. The SCOs detected ions that ranged from 154.96 m∕z (GAS) to 294.06 m∕z (C10H11NSO7). The number of oxygen in the SCO ranged from O3 (C7H7SO3) to O7 (C5H8SO7). It is acknowledged that the CIMS may not detect all SCOs in the ambient air due to peak-fitting resolution limitations and limits of detection, therefore enabling the possibility for misrepresentation of the dominant SCO and an underestimation of total abundance. However, no physical features of the SCO (structure, O : C ratio, mass, etc.) should prevent the CIMS from identifying the major SCO in the Beijing ambient air. Consequently, we assumed that the measured SCOs represent a significant enough fraction to enable analysis of of the relationship between individual SCOs and the total SCO measurement.
2.3.2 Quantification of SCOs
The OS and nitrooxy organosulfates (NOS) calibrations are normalized to formic acid calibrations (as described in Le Breton et al., 2012, 2013) to account for any drift in sensitivity throughout the campaign. This relative sensitivity technique has been previously utilized for N2O5 and ClNO2 and has been verified with laboratory experiments (Le Breton et al., 2014). As a result of low mass range of the SCOs, common functionality, relatively small change in polarity and lack of available stable SCO standards, we calibrated for two SCOs (lactic acid sulfate (LAS) and NP OS) and applied an average sensitivity for all the SCOs detected in Beijing. The ToF-CIMS sensitivity utilizing iodide as a reagent ion is known to vary by up to 3 orders of magnitude; therefore, further work is necessary to develop SCO standards and assess possible variations in sensitivity. NP OS is available commercially from Sigma-Aldrich and was utilized to calibrate for the NOSs. L(+) lactic acid from Sigma-Aldrich (95 %) was utilized as the preliminary agent for lactic acid sulfate synthesis and was produced using the same technique as used by Olson et al. (2011). Briefly, a solution of 76.1 mg, 1.29 mmol, lactic acid in 2 mL di-methyl-formamide (DMF) was added dropwise to sulfur trioxide pyridine (0.96 g, 7.75 mmol) in 2 mL DMF at 0 ∘C. The solution is then stirred for 1 h at 0 ∘C and 40 min at room temperature and is re-cooled to 0 ∘C. Trimethylamine (0.23 mL, 1.66 mmol) was added for quenching and the mixture wad was stirred for a further 1 h. The solvent is then evaporated under a vacuum and NMR is directly utilized to calculate the purity which was found to be 8.2 %.
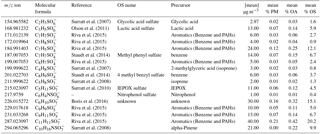
A known mass of the solid calibrant (NP OS and lactic acid sulfate) was added to three different volumes of milliQ water to produce different concentration standards. A known volume of each solution was then placed onto the FIGAERO filter and a desorption cycle was run. The total ion counts for the high-resolution (HR) SCO peak relates directly to the sensitivity of the system with respect to total ion counts per molecule reaching the detector. Figure 2 shows a 3-point calibration curve for NP OS and the corresponding thermogram, mass spectra and peak fit. The sensitivity of LAS and NP OS calibrations was calculated to be 2.0 and 1.6 ion counts per ppt Hz−1 respectively. All SCO were calibrated using the LAS sensitivity and all NOS using the NP OS sensitivity.
During desorption of both SCOs, fragmentation of the organic core and sulfate group was observed, resulting in a desorption profile at m∕z 97 (the bisulfate ion) and the deprotonated organic mass, i.e. C3H5O3 for lactic acid. A number of different temperature ramping rates were measured with the FIGAERO to further probe the fragmentation and it was found that an increase in ramp rate (∘C min−1) decreased the calculated sensitivity due to an increase in fragmentation. This not only serves to highlight how the calibration tests of a species must mimic the exact measurement conditions but also suggests potential interferences from fragmentation on the organic m∕z's. The relatively low concentration of the organic precursor with respect to the SCO results in little error in quantification, although this ratio may significantly change in different air masses and a number of products of organic oxidation may fragment, resulting in a significant error. This fragmentation can also be observed within the high-resolution thermograms of the FIGAERO as a double desorption and further highlights the necessity for detailed thermogram analysis to accurately deconvolve desorption relevant only to particle loss from the filter and not fragmentation or ion chemistry in the IMR. The fragmentation is considered to be constant throughout the campaign. The error for the SCO measurements may vary for each individual SCO, possibly due to structure, volatility and fragmentation. It is commonly accepted within the literature that a functional group sensitivity can be applied to compounds lacking calibration (e.g. Lee et al., 2016 for organic nitrates, ONs). Here we calculate an average error of 52 % for the SCOs, calculated using the standard deviation of the NP OS calibration time series data.
Figure 2The desorption profile of NP OS 3-step calibrations for 0.1, 0.2 and 0.3 µL 1000 ppm solution is displayed in panel (c). Its corresponding average stick spectra are shown in (a) and the sum of counts per molecule loading for each calibration is shown in (b).
The limitation of FIGAERO temperature ramps to 250 ∘C may result in further error as some SCOs may not be fully desorbed from the filter due to their low vapour pressures. To evaluate the mass left on a filter, several double desorption cycles were run in which mass is collected and desorbed, such as in standard use. This is run by reheating the same filter once cooled to attain a second thermogram of the same filter. The second thermogram exhibited an average of 90 % reduction of counts for the SCO, although the NOSs had an average decrease of 82 % counts. This indicates that most but not all mass is removed from the filters when desorbing. For the interpretation of the results of the field campaign, this effect will induce a small distortion in the time evolution of SCOs when compared to other parameters; e.g. 9 % of NOSs will remain on the filter and be subjected to the subsequent desorption cycle.
2.3.3 Offline and online measurement comparison of SCOs
Filter measurements for orbitrap and HPLC MS analysis were taken diurnally at the same sampling site, although from a different inlet and location in the building. The CIMS hourly desorption data were averaged over the corresponding collection period to attain daytime and night-time CIMS data points. The period between 23 May and 1 June was selected due to all instrument measurements being undisturbed during this period. It must be noted that CIMS data lack one data point daily, while the background filter measurements were taken. The CIMS, orbitrap and HPLC do not measure all of the same species. Here a comparison of a total of five ions is presented with the HPLC and two are from the orbitrap. A further extensive comparison is described in an accompanying paper. Figure S3 illustrates the time series of CIMS (hourly and diurnal) measurements of GAS and IEPOX sulfate alongside the HPLC measurements. The diurnal data agree well with an R value of 0.78 and 0.82 for GAS and IEPOX sulfate respectively. The sum of the time series (GAS; IEPOX sulfate, LAS, C4H7SO7, C5H11SO7 and C5H7SO7) for HPLC and C9H11SO5, and C9H8SO5 for orbitrap is displayed in the top panel. In general, the time series agree well and have a good correlation (R = 0.7 and 0.81 for HPLC and orbitrap respectively), illustrating the ability of CIMS to agree with the offline methods and measure the SCOs accurately at low and high time resolutions.
2.4 Knudsen Effusion Mass Spectrometer (KEMS)
The KEMS technique was utilized to measure the vapour pressure of SCOs observed in the gas-phase measurements by the CIMS. The KEMS technique is able to measure vapour pressures from 10−1 to 10−8 pascal (Pa), ranging from volatile organic compounds to extremely low-volatility organic compounds. A full description of the technique can be found in Booth et al. (2009, 2010) and the measurements of a series of compounds over a large vapour-pressure (VP) range in a recent intercomparison study from this instrument can be found in Krieger et al. (2018). Briefly, the instrument consists of a temperature-controlled Knudsen effusion cell, suitable for controlled generation of a molecular beam of the sample organic compounds in a vacuum chamber, coupled to a quadrupole mass spectrometer. The cell has a chamfered effusing orifice with a size ≤ 1∕10 the mean free path of the gas molecules in the cell. This ensures the orifice does not significantly disturb the thermodynamic equilibrium of the samples in the cell (Hilpert, 2001). The system is calibrated using the mass spectrometer signal from a sample of known vapour pressure, in this case malonic acid (vapour pressure at 298 K = 5.25 × 10−4 Pa; Booth et al., 2012). A load lock chamber allows the ionizer filament to be left on, then a new sample of unknown vapour pressure can be measured. Solid-state vapour pressures measured in the KEMS can then be converted to subcooled liquid vapour pressures using the melting point, enthalpy and entropy of fusion, which are obtained by using a differential scanning calorimeter (DSC) (TA instruments Q200).
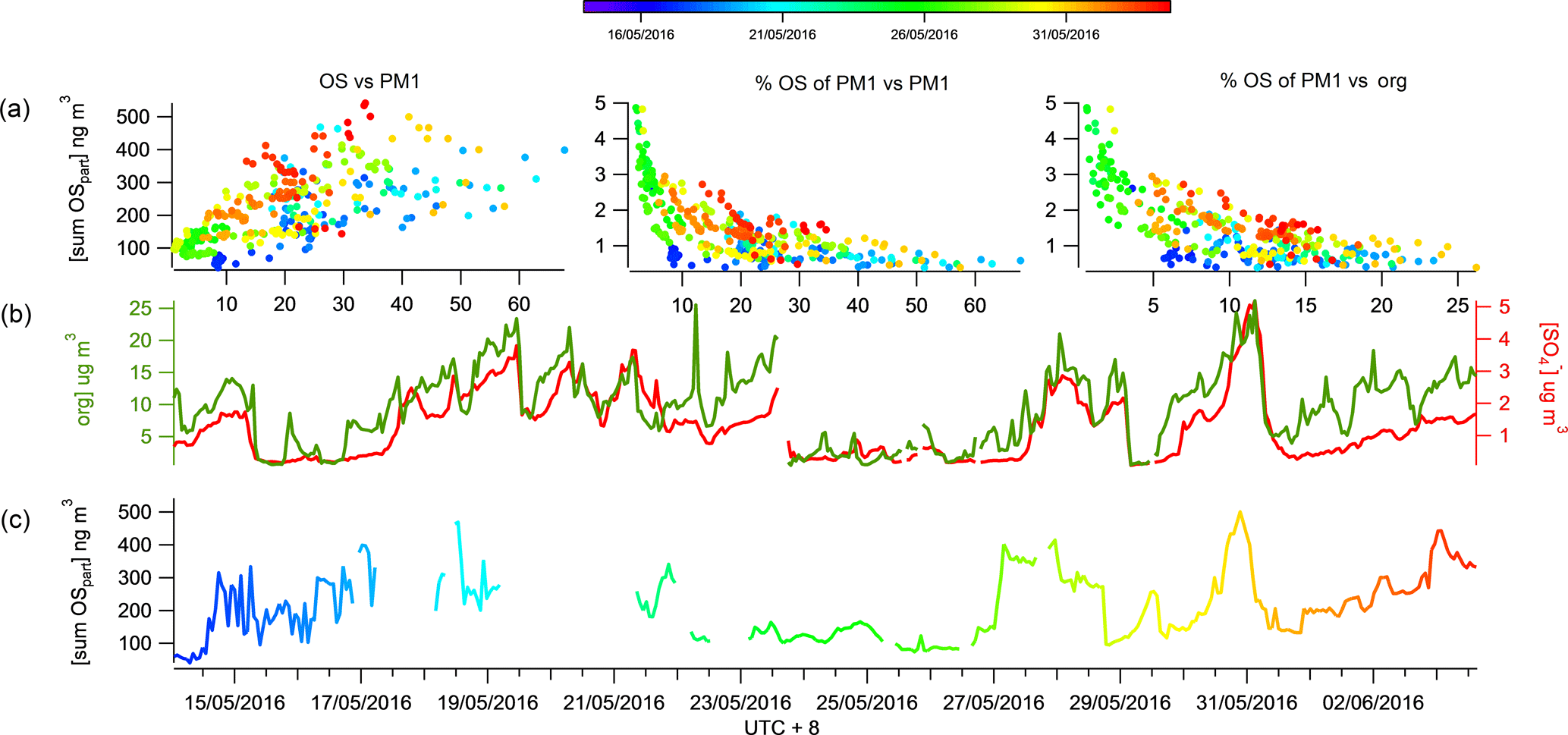
Figure 3The time series of total SCO (colour coded by time) is displayed in panel (c). The time series of the AMS organic (green) and sulfate (red) are displayed in panel (b) and the correlation of SCO to PM1, mass fraction of PM1 and organics are displayed in panel (a).
3.1 SCO contribution to PM1 at Changping
The SCOs measured at the Changping site had a mean campaign concentration of 210 ± 110 ng m−3 (Table 1). The highest concentration of total SCOs during the campaign was 540 ng m−3 and the lowest 40 ng m−3; thus they are omnipresent and have significant sources during most atmospheric conditions. These concentrations are consistent with Stone et al. (2012), reporting an average SCO concentration of 700 ng m−3 in a number of rural and urban sites in Asia. A mean SCO contribution to organic aerosol (OA) in the work presented here was calculated to be 2.0 ± 1 % (Table 1), within the range of values calculated by Stone et al. (2012) (0.8 to 4.5 %), further supporting evidence that the SCO contribution to PM1 mass is relatively low in Asia. The CIMS cannot claim to measure total SCO, rather than singularly identify and measure SCOs contributing to the total mass loading. Therefore, the SCO contribution reported in this work should be considered a lower limit. The Liao et al. (2015) study also supports the idea that the SCO contribution to PM1 mass in anthropogenically dominated regions is less significant than that from biogenically dominating air masses by observing a significantly higher contribution of IEPOX sulfate to PM1 mass on the eastern coast of the USA (1.4 %) than the western coast (0.2 %).
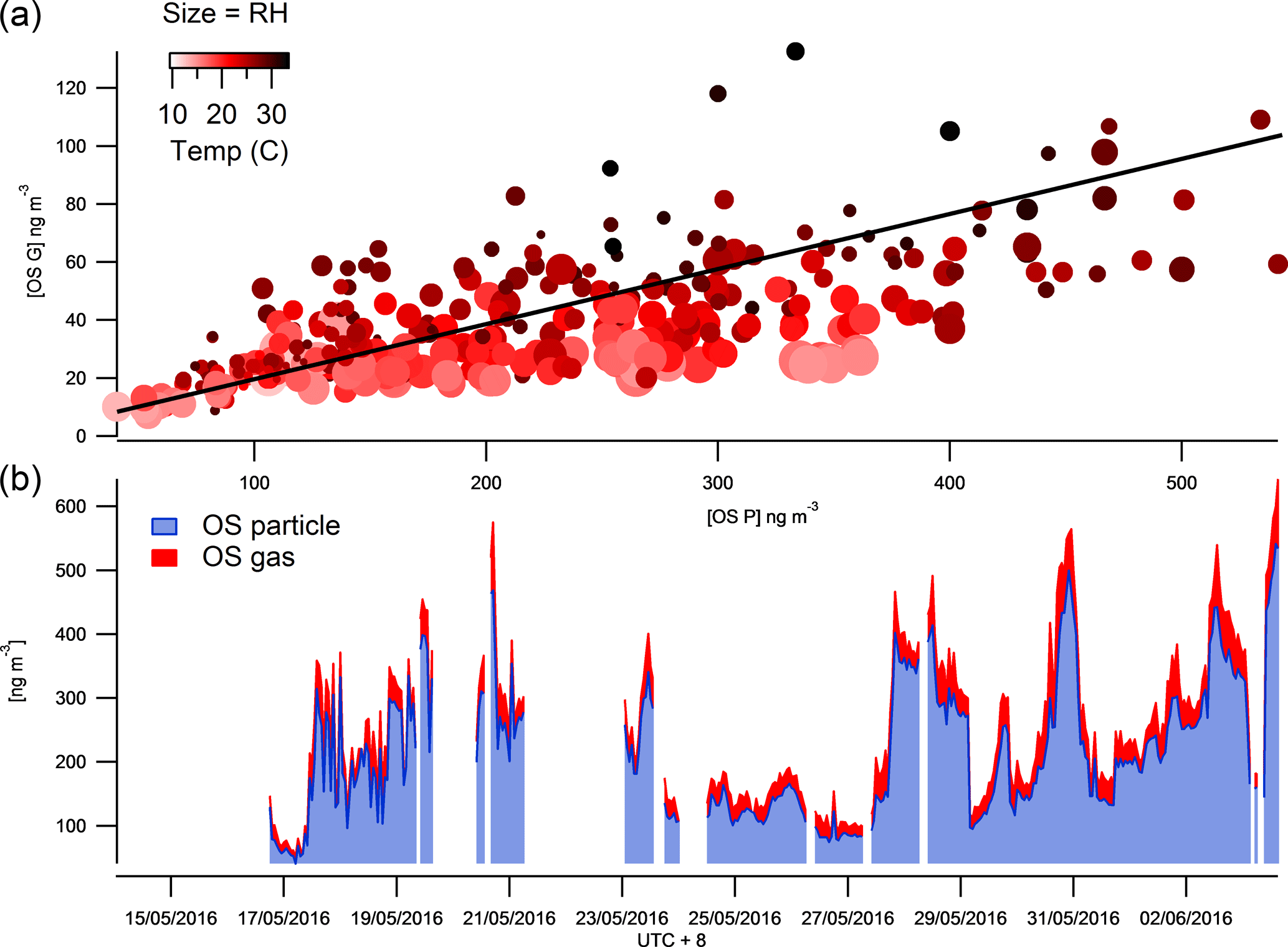
Figure 4The time series of total SCOs in the gas and particle phases (b) and their correlation colour coded by temperature and size binned by relative humidity (a).
The observation of a higher relative contribution of SCOs to total organics in more remote regions compared to a densely populated urban area supports the idea that SCOs provide a higher contribution to PM in aged air due to their secondary production pathways. Similarly to Lahore (as studied by Stone et al., 2012), Beijing has many strong primary anthropogenic sources which will dominate the mass loading and therefore will initially contribute to a lower fraction of the total concentration from secondary production due to limited processing near the source. Throughout the campaign, a good correlation (R2=0.66) was observed between an increase in ΔSCO mass and PM1 mass, although the SCO contribution to PM1 decreased exponentially (Fig. 3), indicating that the pollution episodes contain a lower fraction of SCOs with respect to total PM1. This result suggests that SCOs do not play as large a role as expected even though their precursors (organics and sulfate) are abundant within the episodes, indicating that the conditions of their formation may be more vital than the absolute concentrations of precursors.
3.2 Gas- to particle-phase partitioning of SCOs
The FIGAERO ToF-CIMS data exhibited indications of SCOs in both the particle and gas phase. Previous studies have supported the existence of gas-phase GAS in ambient air (e.g. Ehn et al., 2010), although some works have attributed other measurement techniques on the detection of gas-phase SCO to result from measurement artefacts (Hettiyadura et al., 2017; Kristensen et al., 2016). Once all HR peaks have been identified, the batch fitting and HR time series for the whole data set is processed and then separated into gas-phase measurements and particle-phase desorption profile time series. The data are background corrected, i.e. subtraction of both the gas-phase background periods and blank filter desorption. Upon analysis of the resultant data, significant concentrations of gas-phase SCOs were observed. Figure 4 depicts the overall sum of SCO mass concentration time series in the gas and particle phase. The mean contribution from gas-phase SCO to total SCO was found to be 11.6 %, 23 ± 8 ng m−3. This suggests that a significant amount of SCO is always present in the gas phase, and factors that influence gas-to-particle partitioning influence the level of this contribution. These changes in contribution also reduce the possibilities for memory effect, e.g. one possibility is the deposition of SCOs onto the IMR walls during the temperature ramp of the desorption, which in time may degas and be observed in the gas phase. This would likely result in a constant ratio of particle- to gas-phase concentrations and would likely cause a hysteresis in the observed gas-phase measurements with respect to the particle phase, which was not observed.
The vapour pressure of NP OS was measured using the KEMS instrument in the laboratory to establish the existence of gas-phase SCOs. This technique has recently been employed to measure the vapour pressure of NP (Bannan et al., 2017). The KEMS experiments found the solid-state vapour pressure of NP OS to be 5.07 × 10−5 Pa at 298 K. Assuming an average subcooled liquid correction for all compounds measured in the Bannan et al. (2017) study, as no DSC data are available, the subcooled liquid vapour pressure of NP OS is 2.32 × 10−4 Pa. This vapour pressure lies within the semi-volatile organic compound range, therefore supporting the potential partitioning of SCOs to the gas phase under ambient conditions. To further validate the CIMS and KEMS findings, one can evaluate different compound VPs from the FIGAERO data utilizing the Tmax and compare them to literature values. The CIMS, using Tmax, estimated VPs of malonic, succinic and glutaric acid at 2 × 10−3, 1.85 × 10−3, 1 × 10−3 Pa, which compare well to values presented by Bilde et al. (2015) of 6.2 × 10−3, 1.3 × 10−3, 1 × 10−3 Pa. Using this agreement for well-known substances we notice the Tmax of SCO to be in the range in which it can provide significant gas-phase concentration. Still, the observed presence of gas-phase GAS and IEPOX-OS does not agree with previous studies of these compounds (Stone et al., 2012; Hettiyadura et al., 2017). Therefore, one needs to be cautious and deeper analysis into exact VPs and partitioning from the present work must be performed to assess whether their gas-phase presence could be fully confirmed. So far we note that fragmentation of organic species (oligomers) during desorption could lead to a potential artefact and a lower Tmax at a monomer peak (Stark et al., 2017 and Lopez-Hilfiker et al., 2016). However, here we identify and expect no dimers or oligomers that could fragment to form the identified SCOs. Furthermore, the higher mass organics are likely to have a much higher VP than the lower mass SCOs and provide a second Tmax which would produce a lower VP value due to the greater energy required to break the bonds. Analysis of Tmax throughout the campaign shows no double peak thermograms and an acceptable stability of Tmax (Fig. S4). Tmax varied by up to 14 ∘C and appeared to correlate well with particulate loading, similarly to that observed by Huang et al. (2018), who suggested that this is a result of diffusion limitations within the particle matrix. If the data are tentatively analysed to assess the mechanism regarding their partitioning, aerosol liquid water content would affect the partitioning of gas-phase compounds to aerosols (Zhang et al., 2007). Data point size coding of the correlation of the gas- and particle-phase SCO concentrations indicates partitioning towards the aerosol phase at lower relative humidities (Fig. 4). Conversely, as the temperature increases (as indicated by the red shading) the SCOs partition further towards the gas phase, as thermodynamically expected.
Further work is necessary to validate these findings and determine the mechanisms and importance of gas-phase SCO abundance in ambient air. For example, the high contribution in the gas phase could be perturbed if equilibrium between condensation to particle-phase and gas-phase formation has not been established. It must be noted that a correct calibration of Tmax with VP would be necessary to extract such information, but qualitatively the relative VP compared to NP OS could be utilized as a reliable scale due to its independent calibration by KEMS.
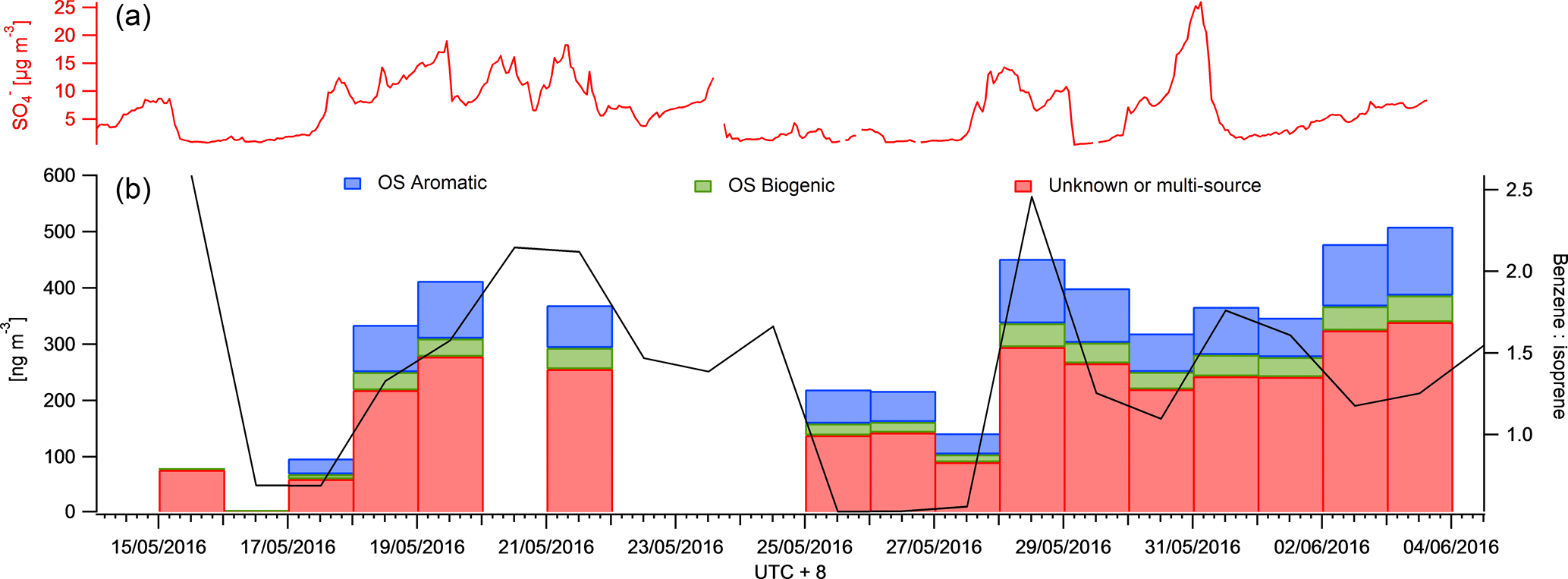
Figure 5A time series of the mean daily benzene to isoprene ratio as a marker for anthropogenic and biogenic influence (black) is displayed in panel (a). The CIMS data were also binned to provide mean daily SCO concentrations for aromatic (blue) and biogenic (green) precursor SCOs (b). The red bars represent SCOs with an unknown source or SCO produced via both biogenic and anthropogenic pathways. The AMS concentration is also presented to indicate the availability of sulfur in the particle phase.
4.1 SCO sources at the Changping site
SCOs are known to have biogenic and anthropogenic sources and some have multiple sources from both, e.g. GAS (Hettiyadura et al., 2017; Hansen et al., 2014). Burning events are known to emit high levels of organics and nitrates and potentially sulfur, depending on the type of fuel used. This enables biomass burning to be a potential anthropogenic and biogenic source of SCOs. The site at Changping was influenced by both regional anthropogenic pollution from the Beijing area and localized anthropogenic activity (industry, biomass burning and traffic) but also by emissions from biogenic sources, as it is situated in a semi-rural area with forest, vegetation and plantations. This was evident from the benzene and isoprene PTR-MS measurements, which have mean campaign concentrations of 0.55 ± 0.4 and 0.27 ± 0.19 ppb respectively with maxima of 5 and 1.5 ppb respectively. Thus, as shown in Fig. 5, mean daily concentrations of PTR-MS measurements were utilized to evaluate whether the ratio between benzene and isoprene can indicate a higher mass loading and contribution of aromatic and biogenic SCOs measured in this work. Data on days with incomplete time series have been removed to ensure the presented data represent a full mean of the day concentration. A good correlation between the benzene : isoprene ratio and sum of SCOs is observed. It suggests an increase in relative anthropogenic emissions promotes an increase in total SCO loading. It should be noted that C6H10SO7 has no known precursor in the literature, although it contributes significantly to the SCO mass loading in this work (16 %).
4.1.1 Biogenic and anthropogenic SCOs
Biogenic SCOs are known to be comprised of monoterpene-, sesquiterpene- and isoprene-derived SCOs which have been identified in rural, suburban and urban areas around the world and have been shown to be a major constituent of SOA (Surratt et al., 2008; Shalamzari et al., 2014; Liao et al., 2015). IEPOX sulfate is commonly found to be the most dominant SCO at many locations and was also identified at the Changping site. The IEPOX sulfate mean concentration represented 0.11 % of the OA mass, agreeing well with concentrations found in western USA (significant anthropogenic emissions) and lower than the eastern USA as expected due to higher biogenic and isoprene emissions (Liao et al., 2015). Although IEPOX sulfate is considered one of the most abundant individual organic molecules in aerosols (Chan et al., 2010), here our results show that it only contributed to 2 % of the SCO mass and was the eighth most abundant SCO in the particle phase. Additionally, two other isoprene-derived SCOs, C5H8SO7 and C4H8SO7, were measured by the CIMS with mean campaign concentrations of 2 and 3 ng m−3 respectively and a contribution of 0.02 % to OA mass. The highest-contributing biogenic SCO to the ambient air was a NOS, C10H16NSO7, a known NOS derived from alpha-Pinene oxidation. This NOS had a mean campaign concentration of 21 ng m−3 and a 0.2 % contribution to OA mass.
Anthropogenic SCOs, including PAH-derived SCOs have received more attention in recent studies due to their identification (Nozière et al., 2010; Hansen et al., 2015). Aromatic SCOs and sulfonates have only recently been identified as atmospherically abundant SCOs (Riva et al., 2015). In this work we find that the PAH-derived SCO C11H11SO7 is the most dominant SCO in Beijing with a mean concentration of 40 ng m−3, contributing to 20 % of the total SCO mass and 0.4 % of the OA mass. This SCO has been identified in laboratory studies as an SCO forming from the photo-oxidation of 2-methyl naphthalene, one of the most abundant gas-phase PAHs and is thought to represent a missing source of urban SOA (Riva et al., 2015). This work presents the possible significance of PAH SCOs in Beijing and further evidence that photo-oxidation of PAHs represents a greater SOA potential than currently recognized. A further eight anthropogenic aromatic-derived SCOs were identified as common components of the PM1, representing more than half of the total SCOs with C7H5SO4 contributing to 24 ng m−3 and 0.23 % OA mass. The total anthropogenic-related SCOs had a mean mass of 120 ng m−3 and contributed to 1.2 % of the OA mass.
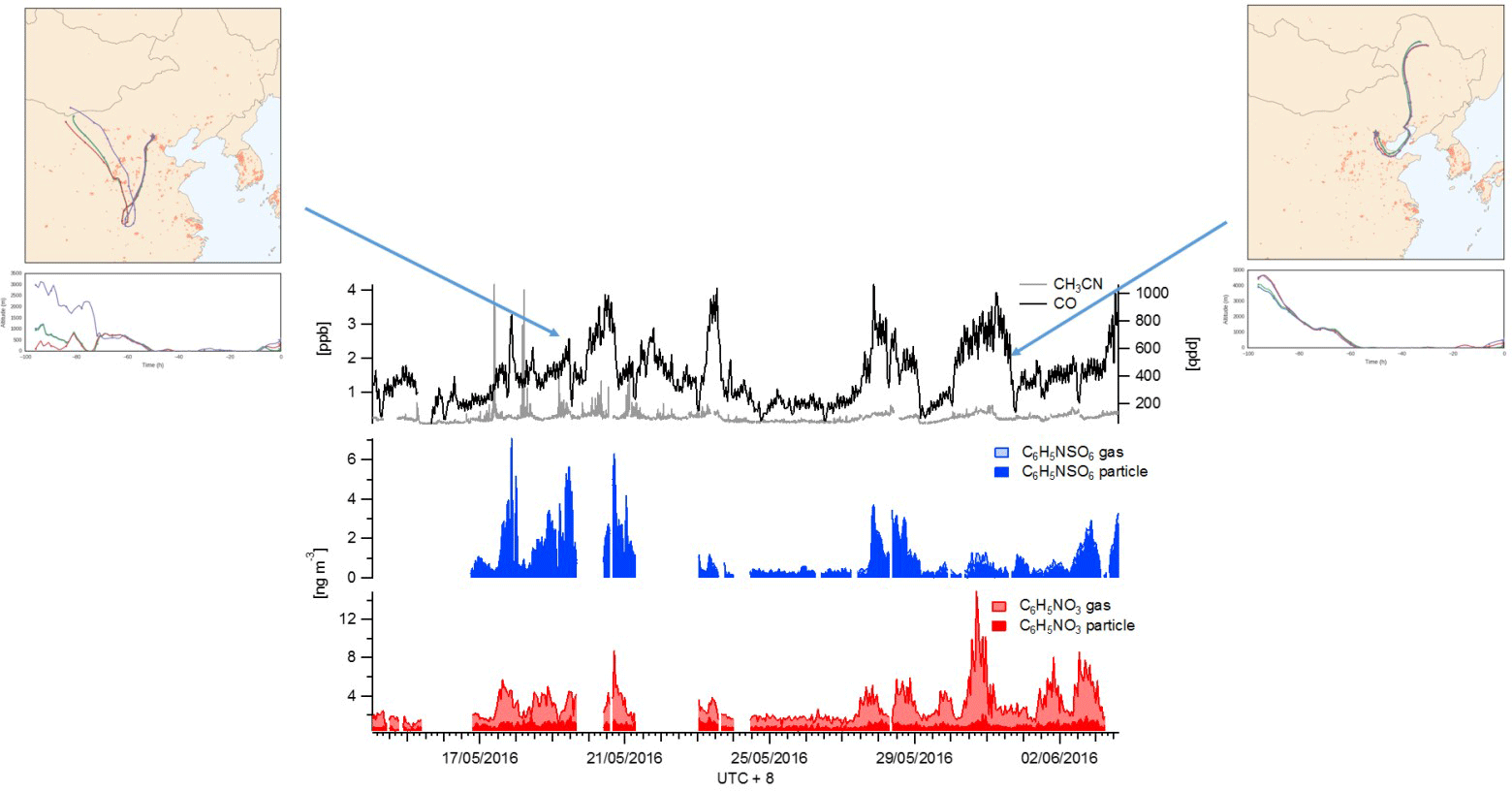
Figure 6The time series of gas- and particle-phase NP (red), NP OS (blue) and gas-phase acetonitrile (grey) and CO (black) between 16 and 3 June.
4.1.2 Biomass-burning source of SCOs
NP (a product of benzene oxidation and nitration) has previously been detected in the gas and aerosol phase (Harrison et al., 2005) and is an important component of brown carbon (Mohr et al., 2013). NP has primary sources, such as vehicle exhausts and biomass burning (Inomata et al., 2013 and Mohr et al., 2013), and secondary sources via the photo-oxidation of aromatic hydrocarbons in the atmosphere (Harrison et al., 2005). High levels on anthropogenic activity, biomass burning and strong photochemistry in Beijing therefore enable this region to be a strong potential source of NP. Both NP and NP OS diurnal profiles exhibit an increase in the morning (06:00 onwards), although NP OS appears to increase more rapidly in concentration. The early-morning biomass burning and anthropogenic activities are likely to contribute to the production of both species, although the higher sulfate content of the biomass-burning emissions may promote a faster production of NP OS and conversion of NP to NP OS. Both compounds continue to increase with a photochemical profile with one peak at midday but also a peak around 16:00, likely to be a second source of the day from anthropogenic activity. The NP OS continues to increase until sunset, which could result from further photochemical production from the NP emitted throughout the day, whereas NP falls off after the 16:00 peak. The campaign time series for NP and NP OS can be seen in Fig. 6. Unlike its precursor and most other pollutant markers measured in this work, including all other SCOs, NP OS exhibits higher concentrations between 17 and 22 May compared to 28 May–3 June. The only compound with a similar campaign profile is acetonitrile (a marker for biomass burning), which has significantly enhanced concentrations between 06:00 and 08:00 from 17 to 22 May. Back trajectories of these two time periods show that the air mass during the first period comes from the west, a more rural region of China that is known to be influenced heavily by biomass burning, whereas the second time period has wind directions mainly bringing in air masses that have gone through the Tianjin and Beijing areas. It is therefore hypothesized that the NP OS, which peaks later in the day than the NP and acetonitrile, is a secondary product formed from the biomass burning and has aged after being emitted from air masses further away. Here NP can have more local sources of biomass burning and traffic, which can then contribute to NP OS production, but at a slower timescale. In this data set it appears to have a lower production due to the limited oxidation of local air masses.
4.2 SCO production mechanisms
4.2.1 Precursor analysis
The availability of the organic precursors of SCOs is a limiting factor for the SCO production rate. The measurement of the precursors in the gas and particle phase by CIMS enables a more descriptive mechanism to be outlined as the partitioning of the precursor will vary the distribution between gas and particle production pathways and therefore the rate of corresponding SCO production. Glycolic acid (GA) has, on average, 75 % of its mass in the gas phase for the measurement period, whereas GAS is dominantly in the particle phase (Fig. 7). The GAS particle-phase concentration is observed to increase as the mass loading increases and the GA gas and particle concentrations increase, although the partitioning of the GA towards the gas phase restricts the SCO production. This can be seen in Fig. 7, as for a given concentration, the data with warm colours (red), representing a high fraction of precursor GA in the particle phase, generally provide a higher concentration of particulate GAS.
The main formation mechanism of GAS is thought to be via the reaction of GA in the gas phase with an acidic aerosol sulfate (Liao et al., 2015), contrary to what is observed here. Although an increase in GAS is observed to correlate with the GA, it appears that a partitioning towards the particle phase promotes GAS production. An R2 correlation of 0.68 is observed between GAp and GASp, whereas an R2 of 0.4 is observed between GAg and GASp.
The sum of benzene SCOs exhibits a good correlation to the gas-phase benzene time series (Fig. 8), although their abundance should also rely on the availability of sulfur in the particle phase and the age of the air mass if it is assumed that they are formed via secondary reactions of primary pollutants. In order to assess how the SCO production rates may vary due to these factors, two distinct high-benzene SCO events with similar benzene concentrations were scrutinized, i.e. 29 May and 1 June. The first period has lower concentrations, higher H2SO4 levels and a higher total benzene SCO concentration. The exact age (or time for oxidation) of compounds in an air mass are without an extensive modelling study complicated to derive. However, as proxies with which to attain an approximation about oxidation state one may use some trace compounds. Monoterpene oxidation by the hydroxyl radical (OH) or O3 results in the formation of multifunctional organic acids such as pinonic acid which can then be further oxidized by OH to form 3-methyl-1,2,3-butane-tricarboxylic acid (MBTCA), both of which are measured by CIMS. Therefore, in an air mass containing monoterpene emissions, as known here through the identification of their products such as C10H16NO7S, we can utilize the ratio of pinonic acid and MBTCA; as tracers of monoterpene SOA processing as detected in ambient aerosols in Europe, USA and the Amazon (Gao et al., 2006) as a relative photochemical clock. During the high-benzene event on 1 June, according to the pinonic acid: MBTCA ratio, the air mass is less oxidized relative to the air mass on 29 May (Fig. 8). This would allow less time for secondary production and explain the relatively lower concentration of SCOs, irrespective of higher concentrations and similar benzene concentrations.

Figure 7Campaign time series of glycolic acid (red) and GAS (blue) in the particle and gas phase. The top panel illustrates the correlation between GAS in the particle phase and .
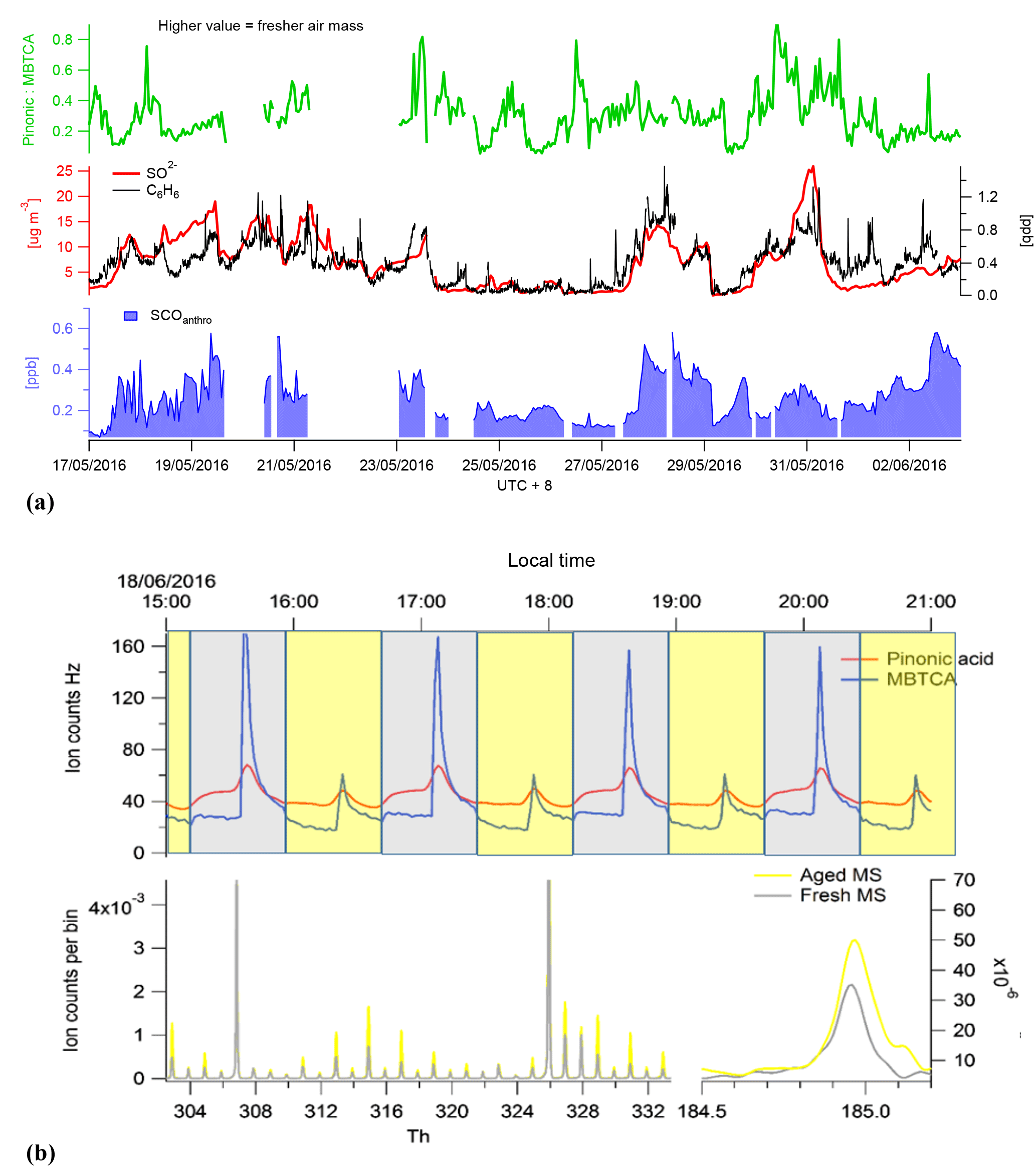
Figure 8(a) Total benzene-/PAH-derived SCO (SCOanthro) time series and respective and benzene concentrations. The indicator of photochemical aging (pinonic acid: MBTCA) is plotted in green. Panel (b) illustrates the mass spectral difference between fresh and aged air masses through Go:PAM and respective time series for pinonic acid and MBTCA.
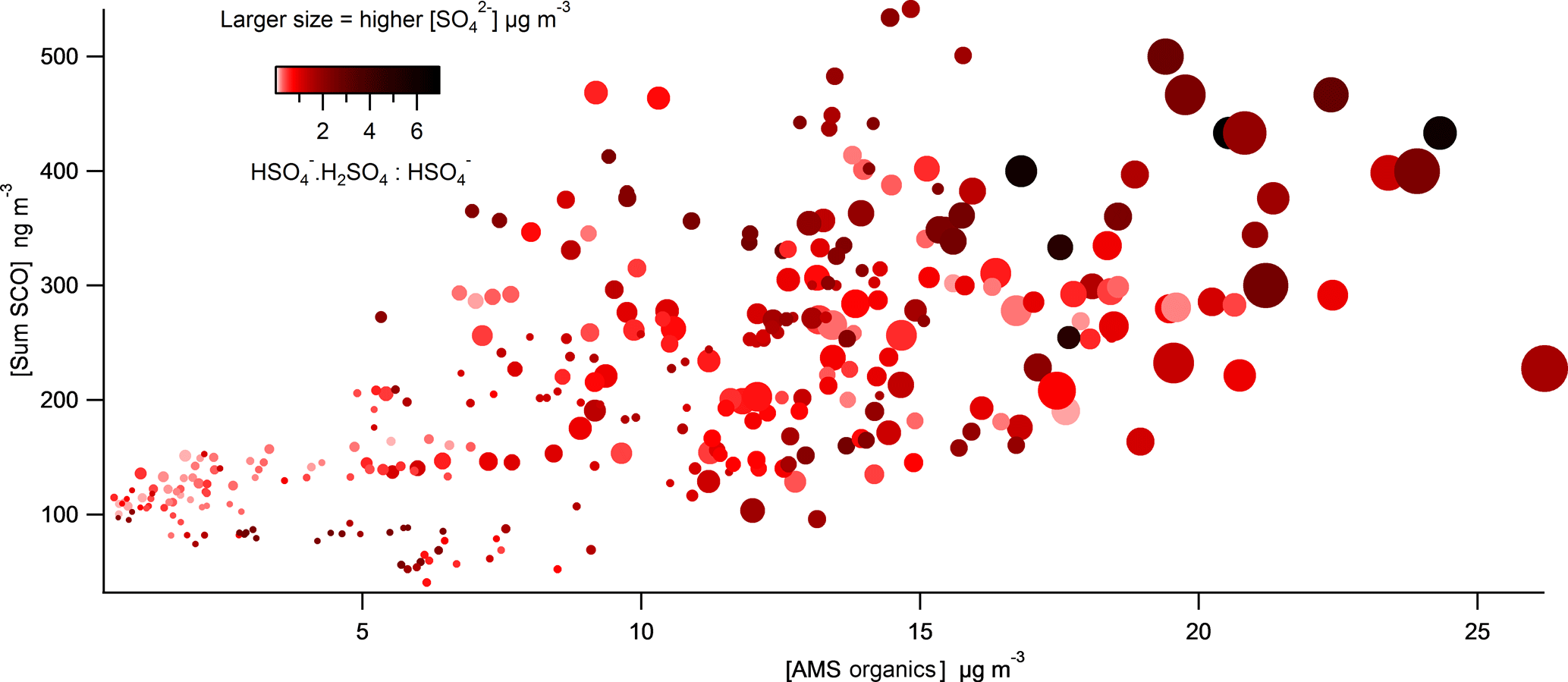
Figure 9Correlation plot of total SCOs vs. total particle-phase organics as a function of acidity (⋅H2SO4 : HSO4 are colour coded) and (data point size spanning concentrations from 0.2 to 16 µg m−3).
To further elaborate the Gothenburg Potential Aerosol Mass (Go:PAM) reactor (Watne et al, 2018) was tested and utilized to simulate aging of the air mass during periods of the campaign. Here the ratio between pinonic acid and MBTCA was observed to increase by an average of 3 during aging within the Go:PAM, which has been calculated to be the OH exposure equivalent of 2 days in the ambient atmosphere. As this ratio increased with aging, the SCO concentration also increased exponentially, further supporting the secondary production of SCO in photochemically aged air mass. Although limited data are available here for simultaneous Go:PAM and CIMS measurements, the results indicate the potential utilization of the chamber to probe secondary production processes.
4.2.2 Aerosol acidity
The molecular ion was identified in the mass spectra throughout the campaign, which has previously been detected by Liao et al. (2015) using a particle analysis laser mass spectrometer to measure SCOs. They attribute this mass to a cluster of with sulfuric acid (H2SO4). Particles in the presence of H2SO4, and therefore high acidity, form this cluster, whereas neutralized ions are likely to favour the unclustered form. Therefore, the ratio between the cluster and the bisulfate ion increases with increasing aerosol acidity (Murphy et al., 2007; Carn et al., 2011). Liao et al. (2015) validate the appropriateness of this cluster as a marker for aerosol acidity thorough comparisons with a thermodynamic model with inputs of gas and aerosol phase measurements. Acidity was also calculated utilizing the gas- and particle-phase H2SO4 and liquid H+ ion concentration analysed using an offline technique, as described by Guo et al. (2010), from diurnal samples taken at the site. This method showed good agreement with the integrated diurnal counts of the ion. Therefore, we employ the ⋅ cluster in this work as a qualitative scale for particle acidity utilizing similar assumptions. Figure 9 shows how total SCO mass concentration generally increased as total organic mass from the AMS increased. The correlation indicates that higher acidity (darker colours) tends to promote formation of SCOs when in the presence of high levels of organics and (larger symbol sizes), supporting the growing consensus that aerosol acidity plays an important role in ambient SCO formation. This importance of acidity agrees well with both the acid-catalysed epoxydiol ring opening formation mechanism (Surratt et al., 2010) and the sulfate radical initiated SCO formation because efficient formation of sulfate radicals also requires acidity (Schindelka et al., 2013).
The FIGAERO ToF-CIMS was successfully utilized for the ambient detection of 17 SCOs in Beijing in the gas and aerosol phase with limits of detection in the ng m3 range. Good agreement with offline filter measurements further supports the robustness of this method for high and low time resolution measurements of SCOs. Further calibrations and comparisons to total SCO measurements are required to evaluate its performance limitation with regard to sensitivity application and peak identification. The SCOs measured by CIMS contributed to 2 % of the OA at the semi-rural site, highlighting the relatively low contribution of SCOs in Beijing, an anthropogenically dominated environment. This calculation from CIMS may only be valid to infer each individual SCO contribution to total SOC mass as limitations in SCO identification and quantification limit the CIMS ability for total SCO measurements. The significance of their secondary production pathway prevailed, although they are still present in relatively fresh air masses. Contributions of SCO to total organics (2.0 ± 1 %), sulfate (15 ± 19 %) and PM (1.0 ± 1.4 %) indicate the concentrations observed in Beijing result from highly processed ambient air masses.
Gas-phase SCOs were identified for all the SCOs measured at the site, contributing to on average 12 % of the total SCO mass. The possibility of gas-phase SCOs in ambient air was supported by KEMS vapour-pressure measurements of NP OS and derived Tmax values, which suggest a vapour pressure in the semi-volatile range. The partitioning towards the gas phase was more efficient at high atmospheric temperatures, while lower relative humidities promoted partitioning in the particle phase.
Biogenic SCOs contributed a small fraction of the total SCO mass at Changping and were dominated by an α-Pinene-derived OS with 0.2 % contribution to the OA mass. IEPOX sulfate was only the eighth most abundant SCO measured, contrary to common reports that it is one of the most abundant SCOs. Anthropogenic precursors contributed to more than half of the SCO mass loading with a PAH-derived SCO contributing to as much as 1.2 % of the OA mass. Benzene-derived SCOs correlated well with gas-phase benzene levels and were heavily influenced by photochemical aging. The contribution of each benzene-derived SCO to total benzene-derived SCO mass varied daily and throughout the campaign, highlighting the complexity of the atmospheric processing and composition of SCO. Significant contributions from aromatic SCOs highlight the importance of anthropogenically emitted organics in the Beijing region and their contribution to the Beijing outflow and subsequent photochemistry. NP OS was attributed to biomass-burning emissions due to a co-occurrence with high levels of acetonitrile. This highlights the importance of anthropogenic emissions and their contribution to SOA from the urban Beijing outflow.
A qualitative CIMS marker for aerosol acidity highlighted the increase in SCO production rate in acidic aerosols in the presence of high and organics. The correlation of SCO production and RH becomes more complex for individual SCOs, which cannot be resolved within the framework of this study.
The data are to be uploaded to the following server https://snd.gu.se/sv (last access: 15 July 2018).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-18-10355-2018-supplement.
MaH, ÅMH, RKP, MiH, and SG were the project leaders for this measurement campaign. MLB and YujW operated the CIMS. QL and CKC led PAM operation and analysis. TJB, DT, and CJP led the KEMS analysis. MG, YucW, and JY supported organosulfate analysis. DS, WZ, and SL supported with AMS analysis. KL supported with general data analysis and project management.
The authors declare that they have no conflict of interest.
The work was done under the framework research programme on “Photochemical
smog in China” financed by the Swedish Research Council (639-2013-6917).
The National Natural Science Foundation of China (21677002) and the National
Key Research and Development Programme of China (2016YFC0202003) also helped
to fund this work.
Edited by: Willy Maenhaut
Reviewed by: three anonymous referees
Bannan, T. J., Booth, A. M., Jones, B. T., O'Meara, S., Barley, M. H., Riipinen, I., Percival, C. J., and Topping, D.: Measured saturation vapor pressures of phenolic and nitro-aromatic compounds, Environ. Sci. Technol., 51, 3922–3928, 2017.
Bilde, M., Barsanti, K., Booth, M., Cappa, C. D., Donahue, N. M., Emanuelsson, E. U., McFiggans, G., Krieger, U. K., Marcolli, C., Topping, D., Ziemann, P., Barley, M., Clegg, S., Dennis-Smither, B., Hallquist, M., Hallquist, A. M., Khlystov, A., Kulmala, M., Mogensen, D., Percival, C. J., Pope, F., Reid, J. P., Ribeiro da Silva, M. A. V., Rosenoern, T., Salo, K., Soonsin, V., Yli-Juuti, T., Prisle, N. L., Pagels, J., Rarey, J., Zardini, A. A., and Riipinen, I.: Saturation vapor pressures and transition enthalpies of low-volatility organic molecules of atmospheric relevance: from dicarboxylic acids to complex mixtures, Chem. Rev., 115, 4115–4156, 2015.
Booth, A. M., Markus, T., McFiggans, G., Percival, C. J., Mcgillen, M. R., and Topping, D. O.: Design and construction of a simple Knudsen Effusion Mass Spectrometer (KEMS) system for vapour pressure measurements of low volatility organics, Atmos. Meas. Tech., 2, 355–361, https://doi.org/10.5194/amt-2-355-2009, 2009.
Booth, A. M., Barley, M. H., Topping, D. O., McFiggans, G., Garforth, A., and Percival, C. J.: Solid state and sub-cooled liquid vapour pressures of substituted dicarboxylic acids using Knudsen Effusion Mass Spectrometry (KEMS) and Differential Scanning Calorimetry, Atmos. Chem. Phys., 10, 4879–4892, https://doi.org/10.5194/acp-10-4879-2010, 2010.
Booth, A. M., Bannan, T., Barley, M. H., Topping, D. O., McFiggans, G., and Percival, C. J.: The role of ortho, meta, para isomerism in measured solid state and derived sub-cooled liquid vapour pressures of substituted benzoic acids, Roy. Soc. Chem., 2, 4430–4443, 2012.
Boris, A. J., Lee, T., Park, T., Choi, J., Seo, S. J., and Collett Jr., J. L.: Fog composition at Baengnyeong Island in the eastern Yellow Sea: detecting markers of aqueous atmospheric oxidations, Atmos. Chem. Phys., 16, 437–453, https://doi.org/10.5194/acp-16-437-2016, 2016.
Carn, S. A, Froyd, K. D., Anderson, B. E., Wennberg,P., Crounse, J., Spencer, K., Dibb, J. E., Krotkov, N. A., Browell, E. V., Hair, J. W., Diskin, G., Sachse, G., and Vay, S.: In situ measurements of tropospheric volcanic plumes in Ecuador and Colombia during TC4, J. Geophys. Res., 116, D00J24, https://doi.org/10.1029/2010JD014718, 2011.
Chan, M. N., Surratt, J. D., Claeys, M., Edgerton, E. S., Tanner, R. L., Shaw, S. L., Zheng, M., Knipping, E. M., Eddingsaas, N. C., Wennberg, P. O., and Seinfeld, J. H.: Characterization and quantification of isoprene-derived epoxydiols in ambient aerosol in the Southeastern United States, Environ. Sci. Technol., 44,12, 4590–4596, 2010.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., and Docherty, K. S.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, 2006.
de Gouw, J. and Warneke, C.: Measurements of volatile organic compounds in the earths atmosphere using proton-transferreaction mass spectrometry, Mass Spectrom. Rev., 26, 223–257, 2007.
Ehn, M., Junninen, H., Petäjä, T., Kurtén, T., Kerminen, V.-M., Schobesberger, S., Manninen, H. E., Ortega, I. K., Vehkamäki, H., Kulmala, M., and Worsnop, D. R.: Composition and temporal behavior of ambient ions in the boreal forest, Atmos. Chem. Phys., 10, 8513–8530, https://doi.org/10.5194/acp-10-8513-2010, 2010.
Gao, S., Surratt, J. D., Knipping, E. M., Edgerton, E. S., Shahgholi, M., and Seinfeld, J. H.: Characterization of polar organic components in fine aerosols in the southeastern United States: Identity, origin, and evolution, J. Geophys. Res.-Atmos., 111, D07302, https://doi.org/10.1029/2005JD006601, 2006.
Gómez-González, Y., Surratt, J. D., Cuyckens, F., Szmigielski, R., Vermeylen, R., Jaoui, M., Lewandowski, M., Offenberg, J. H., Kleindienst, T. E., Edney, E. O., Blockhuys, F., Van Alsenoy, C., Maenhaut, W., and Claeys, M.: Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(-) electrospray ionization mass spectrometry, J. Mass Spectrom., 43, 371–382, 2008.
Guo, S., Hu, M., Wang, Z. B., Slanina, J., and Zhao, Y. L.: Size-resolved aerosol water-soluble ionic compositions in the summer of Beijing: implication of regional secondary formation, Atmos. Chem. Phys., 10, 947–959, https://doi.org/10.5194/acp-10-947-2010, 2010.
Guo, S., Hu, M, Guo, Q. F., Zhang, X., Zheng, M., Zheng, J., Chang, C. C., Schauer, J. J., and Zhang, R. Y.: Primary sources and secondary formation of organic aerosols in Beijing, China, Environ. Sci. Technol., 46, 9846–9853, 2012.
Guo, S., Hu, M., Guo, Q., Zhang, X., Schauer, J. J., and Zhang, R.: Quantitative evaluation of emission controls on primary and secondary organic aerosol sources during Beijing 2008 Olympics, Atmos. Chem. Phys., 13, 8303–8314, https://doi.org/10.5194/acp-13-8303-2013, 2013.
Guo, S., Hu, M., Zamora, M, Peng, J. F., Shang, D. J., Zheng, J., Du Z. F., Wu, Z. J., Shao, M., Zeng, L. M., Molina, M., and Zhang, R.: Elucidating severe urban haze formation in China, P. Natl. Acad. Sci. USA, 111, 17373–17378, 2014.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Hallquist, M., Munthe, J., Hu, M., Mellqvist, J., Wang, T., Chan, C. K., Gao, J., Boman, J., Guo, S., Hallquist, Å. M., Moldanova, J., Pathak, R. K., Pettersson, J. B. C., Pleijel, H., Simpson, D., and Thynell, M.: Photochemical smog in china: Scientific challenges and implications for air quality policies, Nat. Sci. Rev., 3, 401–403, 2016.
Hansen, A. M. K., Kristensen, K., Nguyen, Q. T., Zare, A., Cozzi, F., Nøjgaard, J. K., Skov, H., Brandt, J., Christensen, J. H., Ström, J., Tunved, P., Krejci, R., and Glasius, M.: Organosulfates and organic acids in Arctic aerosols: speciation, annual variation and concentration levels, Atmos. Chem. Phys., 14, 7807–7823, https://doi.org/10.5194/acp-14-7807-2014, 2014.
Hansen, A. M. K., Hong, J., Raatikainen, T., Kristensen, K., Ylisirniö, A., Virtanen, A., Petäjä, T., Glasius, M., and Prisle, N. L.: Hygroscopic properties and cloud condensation nuclei activation of limonene-derived organosulfates and their mixtures with ammonium sulfate, Atmos. Chem. Phys., 15, 14071–14089, https://doi.org/10.5194/acp-15-14071-2015, 2015.
Harrison, M. A. J., Barra, S., Borghesi, D., Vione, D., Arsene, C., and Iulian Olariu, R.: Nitrated phenols in the atmosphere: a review, Atmos. Environ., 39, 231–248, 2005.
Heald, C. L., Jacob, D. J., Park, R. J., Russell, L. M., Huebert, B. J., Seinfeld, J. H., Liao, H., and Weber, R. J.: A large organic aerosol source in the free troposphere missing from current models, Geophys. Res. Lett., 32, L18809, https://doi.org/10.1029/2005GL023831, 2005.
Hettiyadura, A. P. S., Stone, E. A., Kundu, S., Baker, Z., Geddes, E., Richards, K., and Humphry, T.: Determination of atmospheric organosulfates using HILIC chromatography with MS detection, Atmos. Meas. Tech., 8, 2347–2358, https://doi.org/10.5194/amt-8-2347-2015, 2015.
Hettiyadura, A. P. S., Jayarathne, T., Baumann, K., Goldstein, A. H., de Gouw, J. A., Koss, A., Keutsch, F. N., Skog, K., and Stone, E. A.: Qualitative and quantitative analysis of atmospheric organosulfates in Centreville, Alabama, Atmos. Chem. Phys., 17, 1343–1359, https://doi.org/10.5194/acp-17-1343-2017, 2017.
Hilpert, K.: Potential of mass spectrometry for the analysis of inorganic high temperature vapors, Fresen. J. Anal. Chem., 370, 471–478, 2001.
Hu, W. W., Hu, M., Yuan, B., Jimenez, J. L., Tang, Q., Peng, J. F., Hu, W., Shao, M., Wang, M., Zeng, L. M., Wu, Y. S., Gong, Z. H., Huang, X. F., and He, L. Y.: Insights on organic aerosol aging and the influence of coal combustion at a regional receptor site of central eastern China, Atmos. Chem. Phys., 13, 10095–10112, https://doi.org/10.5194/acp-13-10095-2013, 2013.
Hu, W. W., Hu, M., Hu, W., Jimenez, J. L., Yuan, B., Chen, W., Wang, M., Wu, Y., Chen, C., Wang, Z., Peng, J., Yang, K., Zeng, L., and Shao, M.: Chemical composition, sources and aging process of sub-micron aerosols in Beijing: contrast between summer and winter, J. Geophys. Res.-Atmos., 121, 1955–1977, 2016.
Hu, W. W., Hu, M., Hu, W.-W., Zheng, J., Chen, C., Wu, Y., and Guo, S.: Seasonal variations in high time-resolved chemical compositions, sources, and evolution of atmospheric submicron aerosols in the megacity Beijing, Atmos. Chem. Phys., 17, 9979–10000, https://doi.org/10.5194/acp-17-9979-2017, 2017.
Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K.: Analysis of Organic Sulfur Compounds in Atmospheric Aerosols at the HKUST Supersite in Hong Kong Using HR-ToF-AMS, Environ. Sci. Technol., 49, 3672–3679, 2015.
Huang, W., Saathoff, H., Pajunoja, A., Shen, X., Naumann, K.-H., Wagner, R., Virtanen, A., Leisner, T., and Mohr, C.: α-Pinene secondary organic aerosol at low temperature: chemical composition and implications for particle viscosity, Atmos. Chem. Phys., 18, 2883–2898, https://doi.org/10.5194/acp-18-2883-2018, 2018.
Huang, X.-F., He, L.-Y., Hu, M., Canagaratna, M. R., Sun, Y., Zhang, Q., Zhu, T., Xue, L., Zeng, L.-W., Liu, X.-G., Zhang, Y.-H., Jayne, J. T., Ng, N. L., and Worsnop, D. R.: Highly time-resolved chemical characterization of atmospheric submicron particles during 2008 Beijing Olympic Games using an Aerodyne High-Resolution Aerosol Mass Spectrometer, Atmos. Chem. Phys., 10, 8933–8945, https://doi.org/10.5194/acp-10-8933-2010, 2010.
Iinuma, Y., Muller, C., Berndt, T., Boge, O., Claeys, M., and Herrmann, H.: Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol, Environ. Sci. Technol., 41, 6678–6683, 2007.
Inomata, S., Tanimoto, H., Fujitani, Y., Sekimoto, K., Sato, K., Fushimi, A., Yamada, H., Hori, S., Kumazawa, Y., Shimono, A., and Hikida, T.: On-line measurements of gaseous nitro-organic compounds in diesel vehicle exhaust by proton-transfer-reaction mass spectrometry, Atmos. Environ., 73, 195–203, 2013.
Kim, J. Y., Lee, E. Y., Choi, I., Kim, J., and Cho, K. H.: Effects of the particulate matter 2.5 (PM1) on lipoprotein metabolism, uptake and degradation, and embryo toxicity, Mol. Cells., 38, 1096–1104, 2015.
Krieger, U. K., Siegrist, F., Marcolli, C., Emanuelsson, E. U., Gøbel, F. M., Bilde, M., Marsh, A., Reid, J. P., Huisman, A. J., Riipinen, I., Hyttinen, N., Myllys, N., Kurtén, T., Bannan, T., Percival, C. J., and Topping, D.: A reference data set for validating vapor pressure measurement techniques: homologous series of polyethylene glycols, Atmos. Meas. Tech., 11, 49–63, https://doi.org/10.5194/amt-11-49-2018, 2018.
Kristensen, K. and Glasius, M.: Organosulfates and oxidation products from biogenic hydrocarbons in fine aerosols from a forest in North West Europe during spring, Atmos. Environ., 45, 4546–4556, 2011.
Kristensen, K., Bilde, M., Aalto, P. P., Petäjä, T., and Glasius, M.: Denuder/filter sampling of organic acids and organosulfates at urban and boreal forest sites: gas/particle distribution and possible sampling artifacts. Atmos. Environ., 130, 36–53, 2016.
Le Breton, M., McGillen, M. R., Muller, J. B. A., Bacak, A., Shallcross, D. E., Xiao, P., Huey, L. G., Tanner, D., Coe, H., and Percival, C. J.: Airborne observations of formic acid using a chemical ionization mass spectrometer, Atmos. Meas. Tech., 5, 3029–3039, https://doi.org/10.5194/amt-5-3029-2012, 2012.
Le Breton, M., Bacak, A., Muller, J. B. A., O'Shea, S. J., Xiao, P., Ashfold, M. N. R., Cooke, M. C., Batt, R., Shallcross, D. E., Oram, D. E., Forster, G., Bauguitte, S. J.-B., Palmer, P. I., Parrington, M., Lewis, A. C., Lee, J. D., and Percival, C. J.: Airborne hydrogen cyanide measurements using a chemical ionisation mass spectrometer for the plume identification of biomass burning forest fires, Atmos. Chem. Phys., 13, 9217–9232, https://doi.org/10.5194/acp-13-9217-2013, 2013.
Le Breton, M., Bacak, A., Muller, J. B. A., Bannan, T. J., Kennedy, O., Ouyang, B., Xiao, P., Bauguitte, S. J. B., Shallcross, D. E., Jones, R. L., Daniels, M. J. S., Ball, S. M., and Percival, C. J.: The first airborne comparison of N2O5 measurements over the UK using a CIMS and BBCEAS during the RONOCO campaign, Anal. Methods-UK, 6, 9731–9743, 2014.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521 https://doi.org/10.1073/pnas.1508108113, 2016.
Li, Y. J., Sun, Y., Zhang, Q., Li, X., Li, M., Zhou, Z., and Chan, C.: Real-time chemical characterization of atmospheric particulate matter in China: A review, Atmos. Environ., 158, 270–304, 2017.
Liao, J., Froyd, K. D., Murphy, D. M., Keutsch, F. N., Yu, G., Wennberg, P. O., St. Clair, J. M., Crounse, J. D., Wisthaler, A., Mikoviny, T., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Ryerson, T. B., Pollack, I. B., Peischl, J., Anderson, B. E., Ziemba, L. D., Blake, D. R., Meinardi, S., and Diskin, G.: Airborne measurements of organosulfates over the continental US, J. Geophys. Res.-Atmos., 120, 2990–3005, 2015.
Lin, P., Yu, J. Z., Engling, G., and Kalberer, M.: Organosulfates in humic-like substance fraction isolated from aerosols at seven locations in East Asia: A study by ultra-high-resolution mass spectrometry, Environ. Sci. Technol., 46, 13118–13127, 2012.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lopez-Hilfiker, F. D., Mohr, C., D'Ambro, E. L., Lutz, A., Riedel, T. P., Gaston, C. J., Lyer, S., Zhang, Z., Gold, A., Surratt, J. D., Lee, B. H., Kurten, T., Hu, W. W., Jiminez, J., Hallquist, M., and Thornton, J. A: Molecular composition and volatility of organic aerosol in the southeastern U.S.: Implications for IEPOX derived SOA, Environ. Sci. Technol., 50, 2200–2209, https://doi.org/10.1021/acs.est.5b04769, 2016.
Mohr, C., Lopez-Hilfiker, F. D., Zotter, P., Prévôt, A. S. H., Xu, L., Ng, N. L., Herndon, S. C., Williams, L. R., Franklin, J. P., Zahniser, M. S., Worsnop, D. R., Knighton, W. B., Aiken, A. C., Gorkowski, K. J., Dubey, M. K., Allan, J. D., and Thornton, J. A.: Contribution of nitrated phenols to wood burning brown carbon light absorption in Detling, United Kingdom during winter time, Environ. Sci. Technol., 47, 6316–6324, 2013.
Murphy, D. M., Cziczo, D. J., Hudson, P. K., and Thomson, D. S.: Carbonaceous material in aerosol particles in the lower stratosphere and tropopause region, J. Geophys. Res., 112, D04203, https://doi.org/10.1029/2006JD007297, 2007.
Nozière, B., Ekstrom, S., Alsberg, T., and Holmstrom, S.: Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 37, L05806, https://doi.org/10.1029/2009GL041683, 2010.
Olson, C. N., Galloway, M. M., Yu, G., Hedman, C. J., Lockett, M. R., Yoon, T., Stone, E. A., Smith, L. M., and Keutsch, F. N.: Hydroxycarboxylic acid-derived organosulfates: Synthesis, stability, and quantification in ambient aerosol, Environ. Sci. Technol., 45, 15, 6468–6474, 2011.
Pope, C. A., Burnett, R. T., Thun, M. J., Calle, E. E., Krewski, D., Ito, K., and Thurston, G. D.: Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution, J. Am. Med. Assoc., 287, 1132–1141, 2002.
Reemtsma, T., Weiss, S., Mueller, J., Petrovic, M., Gonzalez, S., Barcelo, D., Ventura, F., and Knepper, T. P.: Polar pollutants entry into the water cycle by municipal wastewater: A European perspective, Environ. Sci. Technol., 40, 5451–5458, https://doi.org/10.1021/es060908a, 2006.
Riva, M., Tomaz, S., Cui, T., Lin, Y-H, Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an unrecognized secondary anthropogenic source of organosulfates and sulfonates: gas-phase oxidation of polycyclic aromatic hydrocarbons in the presence of sulfate aerosol, Environ. Sci. Technol., 49, 6654–6664, 2015.
Riva, M., Da Silva Barbosa, T., Lin, Y.-H., Stone, E. A., Gold, A., and Surratt, J. D.: Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes, Atmos. Chem. Phys., 16, 11001–11018, https://doi.org/10.5194/acp-16-11001-2016, 2016.
Schindelka, J., Iinuma, Y., Hoffmann, D., and Herrmann, H.: Sulfate radical-initiated formation of isoprene-derived organosulfates in atmospheric aerosols, Faraday Discuss., 165, 237–259, 2013.
Shalamzari, M. S., Ryabtsova, O., Kahnt, A., Vermeylen, R., Hérent, M.-F., Quetin-Leclercq, J., Van Der Veken, P., Maenhaut, W., and Claeys, M.: Mass spectrometric characterization of organosulfates related to secondary organic aerosol from isoprene, Rapid Commun. Mass Sp., 27, 784–794, 2013.
Shalamzari, M. S., Kahnt, A., Vermeylen, R., Kleindienst, T. E., Lewandowski, M., Cuyckens, F., Maenhaut, W., and Claeys, M.: Characterization of polar organosulfates in secondary organic aerosol from the green leaf volatile 3-Z-hexenal, Environ. Sci. Technol., 48, 12671–12678, 2014.
Smith, J. N., Dunn, M. J., VanReken, T. M., Iida, K., Stolzenburg, M. R, McMurry, P. H., and Huey, L. G.: Chemical composition of atmospheric nanoparticles formed from nucleation in Tecamac, Mexico: Evidence for an important role for organic species in nanoparticle growth, Geophys. Res. Lett., 35, L04808, https://doi.org/10.1029/2007GL032523, 2008.
Stark, H., Yatavelli, R. L. N., Thompson, S. L., Kang, H., Krechmer, J. E., Kimmel, J. R., Palm, B. B., Hu, W., Hayes, P. L., Day, D. A., Campuzano-Jost, P., Canagaratna, M. R., Jayne, J. T., Worsnop, D. R., and Jimenez, J. L.: Impact of Thermal Decomposition on Thermal Desorption Instruments: Advantage of Thermogram Analysis for Quantifying Volatility Distributions of Organic Species, Environ. Sci. Technol., 51, 8491–8500, https://doi.org/10.1021/acs.est.7b00160, 2017.
Staudt, S., Kundu, S., He, X., Lehmler, H. J., Lin, Y. H., Cui, T. Q., Kristensen, K., Glasius, M., Zhang, X., Weber, R., Surratt, J. D., and Stone, E. A.: Aromatic organosulfates in atmospheric aerosols: Synthesis, characterization, and abundance, Atmos. Environ., 94, 366–373, 2014.
Stone, E. A., Yang, L., Yu, L. E., and Rupakheti, M.: Characterization of organosulfates in atmospheric aerosols at Four Asian locations, Atmos. Environ., 47, 323–329, 2012.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, 2007.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organic sulfate formation in biogenic secondary organic aerosol, J. Phys. Chem., 112, 8345–8378, 2008.
Surratt, J. D., Chan, A. W. H., Eddingsaas, N. C., Chan, M. N., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 15, 6640–6645, https://doi.org/10.1073/pnas.0911114107, 2010.
Watne, Å. K., Pschichoudaki, M., Ljungström, E., Le Breton, M., Hallquist, M., Jerksjö, M., Fallgren, H., Jutterström, S., and Hallquist, Å. M.: Fresh and oxidized emissions from in-use transit buses running on diesel, biodiesel and CNG, Environ. Sci. Technol. Lett., 52, 7720–7728, https://doi.org/10.1021/acs.est.8b01394, 2018.
Worton, D. R., Surratt, J. D., Lafranchi, B. W., Chan, A. W., Zhao, Y., Weber, R. J., Park, J. H., Gilman, J. B., de Gouw, J., Park, C., Schade, G., Beaver, M., Clair, J. M., Crounse, J., Wennberg, P., Wolfe, G. M., Harrold, S., Thornton, J. A., Farmer, D. K., Docherty, K. S., Cubison, M. J., Jimenez, J. L., Frossard, A. A., Russell, L. M., Kristensen, K., Glasius, M., Mao, J., Ren, X., Brune, W., Browne, E. C., Pusede, S. E., Cohen, R. C., Seinfeld, J. H., and Goldstein, A. H.: Observational insights into aerosol formation from isoprene, Environ. Sci. Technol., 47, 11403–11413, 2013.
Wuebbles, D. J. and Jain, A. K.: Concerns about climate change and the role of fossil fuel use, Fuel. Process. Technol., 71, 99–119, 2001.
Zhang, H., Worton, D. R., Lewandowski, M., Ortega, J., Rubitschun, C. L., Park, J.-H., Kristensen, K., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Gilman, J., Kuster, W. C., de Gouw, J., Park, C., Schade, G. W., Frossard, A. A., Russell, L., Kaser, L., Jud, W., Hansel, A., Cappellin, L., Karl, T., Glasius, M., Guenther, A., Goldstein, A. H., Seinfeld, J. H., Gold, A., Kamens, R. M., and Surratt, J. D.: Organosulfates as tracers for secondary organic aerosol (SOA) formation from 2-methyl-3-buten-2-ol (MBO) in the atmosphere, Environ. Sci. Technol., 46, 9437–9446, 2012.
Zhang, Q., Jimenez, J. L., Worsnop, D. R., and Canagaratna, M.: A case study of urban particle acidity and its influence on secondary organic aerosol, Environ. Sci. Technol., 41, 3213–3219, https://doi.org/10.1021/es061812j, 2007.
Zhang, Q., He, K., and Huo, H.: Policy: Cleaning China's air, Nature, 484, 161–162, 2012.