the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 09 Jul 2026
| 09 Jul 2026
The impact of CO on secondary organic aerosols formed from the mixture of α-pinene and n-dodecane
Guangzhao Xie
Aristeidis Voliotis
Thomas J. Bannan
Yunqi Shao
Huihui Wu
Dawei Hu
Gordon McFiggans
Secondary organic aerosol (SOA) formation is strongly influenced by atmospheric conditions. Achieving atmospheric relevance in chamber experiments is essential for understanding and predicting the impacts of SOA on air quality and climate. However, many chamber studies are conducted under simplified conditions or with a single SOA precursor. Here, we investigated the impact of CO on SOA particle mass yields and chemical composition from α-pinene (a biogenic volatile organic compound, VOC), n-dodecane (an anthropogenic intermediate-volatility organic compound, IVOC), and their mixture in the presence of nitrogen oxides () in the Manchester Aerosol Chamber (MAC) using online measurements. The results show that the influence of CO differed between single- and mixed-precursor systems. In the single-precursor systems, CO significantly suppressed SOA particle mass yields, whereas no such suppression was observed in the mixture. Moreover, compared with the single-precursor systems, CO exerted a diminished impact on the organic peroxy (RO2) radical reaction pathways in the mixture, with the extent of this change differing between α-pinene and n-dodecane. These findings highlight the importance of accounting for atmospheric complexity in laboratory studies.
- Article
(2863 KB) - Full-text XML
-
Supplement
(1377 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA) constitutes a substantial fraction of ambient aerosol and has significant impacts on air quality, climate and human health. It is formed through the oxidation of gas-phase organic compounds followed by gas-particle partitioning (Atkinson and Arey, 2003; Hallquist et al., 2009; Jimenez et al., 2009; Ramanathan et al., 2001; Robinson et al., 2007). These processes are complex and strongly influenced by atmospheric conditions (Hallquist et al., 2009; Kroll and Seinfeld, 2008; Xu et al., 2015). Despite extensive research, achieving a comprehensive understanding and accurate prediction of SOA formation remain challenging (Kenagy et al., 2024; Shrivastava et al., 2017).
Laboratory studies and atmospheric modelling are two key approaches for investigating atmospheric SOA (Burkholder et al., 2017). Model parameterisations are largely derived from laboratory studies, and the accuracy of model predictions strongly depends on the atmospheric relevance of experimental conditions employed (Burkholder et al., 2017; Kanakidou et al., 2005; Kenagy et al., 2024). The ambient atmosphere comprises a complex mixture of biogenic and anthropogenic emissions, including a wide range of gas-phase organic compounds and inorganic trace gases (Gu et al., 2021; Guenther et al., 1995). Field measurements have provided evidence that anthropogenic emissions can modulate SOA formed from biogenic precursors (Budisulistiorini et al., 2015; Shilling et al., 2013; Xu et al., 2015). However, many laboratory experiments are conducted under simplified conditions or with a single SOA precursor, which may introduce uncertainties when extrapolating these results to atmospheric models (Kenagy et al., 2024; Shrivastava et al., 2017; Tsigaridis et al., 2014).
Organic peroxy radicals (RO2) play a central role in SOA formation (Kroll and Seinfeld, 2008; Ziemann and Atkinson, 2012). They can undergo bimolecular termination reactions with hydroperoxyl radicals (HO2), other RO2 radicals, or nitrogen oxides (), as well as unimolecular termination (Atkinson, 2000; Goldman et al., 2021; Molteni et al., 2019; Ziemann and Atkinson, 2012). Recent studies have focused on the autoxidation pathways of RO2 radicals that produce highly oxygenated molecules (HOMs), which are considered potentially important contributors to SOA formation owing to their extremely low volatility (Bianchi et al., 2019; Ehn et al., 2014; Pospisilova et al., 2020). In real atmospheric environments, the coexistence of multiple SOA precursors and various inorganic trace gases introduces additional chemical complexity into the system (McFiggans et al., 2019; Xu et al., 2015). Such complexity can substantially modify RO2 reaction pathways, thereby influencing product distributions and yields.
An increasing number of studies have focused on mixtures of multiple precursors. McFiggans et al. (2019) demonstrated that mixing α-pinene with isoprene substantially suppresses SOA formation from α-pinene, reducing SOA mass formation by about 60 % and SOA yield by 40 %. This suppression was attributed to two main mechanisms. First, isoprene, which exhibits a relatively low yield, efficiently competes with α-pinene for available OH, thereby suppressing the formation of α-pinene-derived RO2 radicals. Second, isoprene-derived RO2 radicals can scavenge HOM-RO2 derived from α-pinene, leading to the formation of products with higher volatility. More broadly, mixing effects on SOA particle mass yields have also been observed for other precursor combinations. For example, in multi-precursor systems consisting of two monoterpenes (α-pinene and limonene), SOA formation from α-pinene was enhanced by approximately 50 %, while that from limonene was reduced by about 20 % (Takeuchi et al., 2022). More recent studies have extended such investigations to ternary mixtures comprising biogenic (α-pinene and isoprene) and anthropogenic (o-cresol) precursors, and have also shown that the overall SOA particle mass yields in the mixture deviate from those predicted by additive calculations (Voliotis et al., 2022a). These findings suggest that simple linear addition of SOA particle mass yields from individual components may lead to inaccurate estimates of total SOA formation in mixed-precursor systems.
Atmospheric inorganic trace gases, such as CO and NOx, can alter oxidant levels and RO2 reaction pathways (Atkinson, 2000; Baker et al., 2024; Chen et al., 2022; Kang et al., 2025; Kroll and Seinfeld, 2008; Lane et al., 2008; Pullinen et al., 2020; Pye et al., 2019; Sarrafzadeh et al., 2016). In laboratory experiments, SOA precursor concentrations are often higher than those typically observed in the ambient atmosphere for practical reasons (Ziemann and Atkinson, 2012). This can lead to relatively low HO2RO2 ratios compared with atmospheric conditions, favouring RO2+RO2 reactions over RO2+HO2 reactions (Ziemann and Atkinson, 2012). The former forms accretion products, which may have extremely low volatility and are expected to contribute to SOA formation, potentially leading to an overestimation of SOA particle mass yields (Kenagy et al., 2024; Peräkylä et al., 2023; Ziemann and Atkinson, 2012). The presence of CO can directly consume OH and produce HO2 radicals, thereby shifting the HO2RO2 ratio and increasing the importance of the RO2 termination via HO2 (Lu and Khalil, 1993). Previous studies have quantified the effect of CO on SOA production. McFiggans et al. (2019) showed that CO suppressed α-pinene dimer (containing 17–20 carbon atoms) formation by a factor of two, while the amounts of HOMs were suppressed by factors of 4–5. Baker et al. (2024) further demonstrated that, under constant OH conditions, the addition of CO increased the HO2RO2 ratio from approximately to about , leading to a ∼60 % reduction in the abundance of HOM-accretion products and a ∼30 % decrease in the SOA formation potential of HOMs. However, these studies were conducted under NOx-free conditions. In the ambient atmosphere, high concentrations of CO are often co-emitted with other anthropogenic pollutants, such as NOx. NOx can react with ROx radicals (), thereby influencing ROx cycling and, consequently, the formation of SOA and O3 (Chen et al., 2022; Clapp and Jenkin, 2001; Pusede et al., 2015). RO2 radicals react rapidly with NO to form alkoxy radicals (RO) or organic nitrates (Atkinson, 2000; Chen et al., 2022; Kang et al., 2025; Ziemann and Atkinson, 2012). RO2 can also react with NO2 to form peroxynitrates; however, these species are generally thermally unstable, except at very low temperatures or when derived from acylperoxy radicals (Atkinson, 2000; Goldman et al., 2021; Ziemann and Atkinson, 2012). The effects of NOx on SOA particle mass yields have been extensively studied. Sarrafzadeh et al. (2016) reported that, in β-pinene photooxidation experiments under low-NOx conditions, SOA particle mass yields increased with rising NOx concentrations, which they attributed to enhanced OH concentrations. However, after removing the effect of OH, the yields decreased with increasing NOx. Pullinen et al. (2020) revealed that higher NOx concentrations reduced the formation of gas-phase α-pinene HOM-accretion products, leading to a lower SOA particle mass yield. When CO and NOx coexist, oxidant levels and RO2 reaction pathways are influenced by multiple interacting processes. These interactions contribute to the complexity of the ambient atmosphere. It is therefore important to investigate SOA formation in systems containing multiple trace gases.
In this study, we employed a photochemical system incorporating mixtures of biogenic and anthropogenic SOA precursors together with multiple inorganic trace gases commonly associated with anthropogenic emissions. Within this framework, we investigated the impact of CO on SOA particle mass yields and chemical composition from α-pinene, n-dodecane, and their mixture in the presence of NOx. Based on changes in chemical composition, we inferred shifts in RO2 reaction pathways and their potential influence on yields. α-Pinene (C10H16) is the most abundant monoterpene in the troposphere and contributes significantly to the global SOA budget (Andreae and Crutzen, 1997; Lee et al., 2006). n-Dodecane (C12H26) serves as a proxy for anthropogenic intermediate-volatility organic compounds (IVOCs), being widely present in fuels and emitted primarily as a non-combusted hydrocarbon (Zhao et al., 2015). Experiments were conducted in the Manchester Aerosol Chamber (MAC), using online instruments to characterise particle- and gas-phase compounds.
Table 1Summary of experimental conditions.
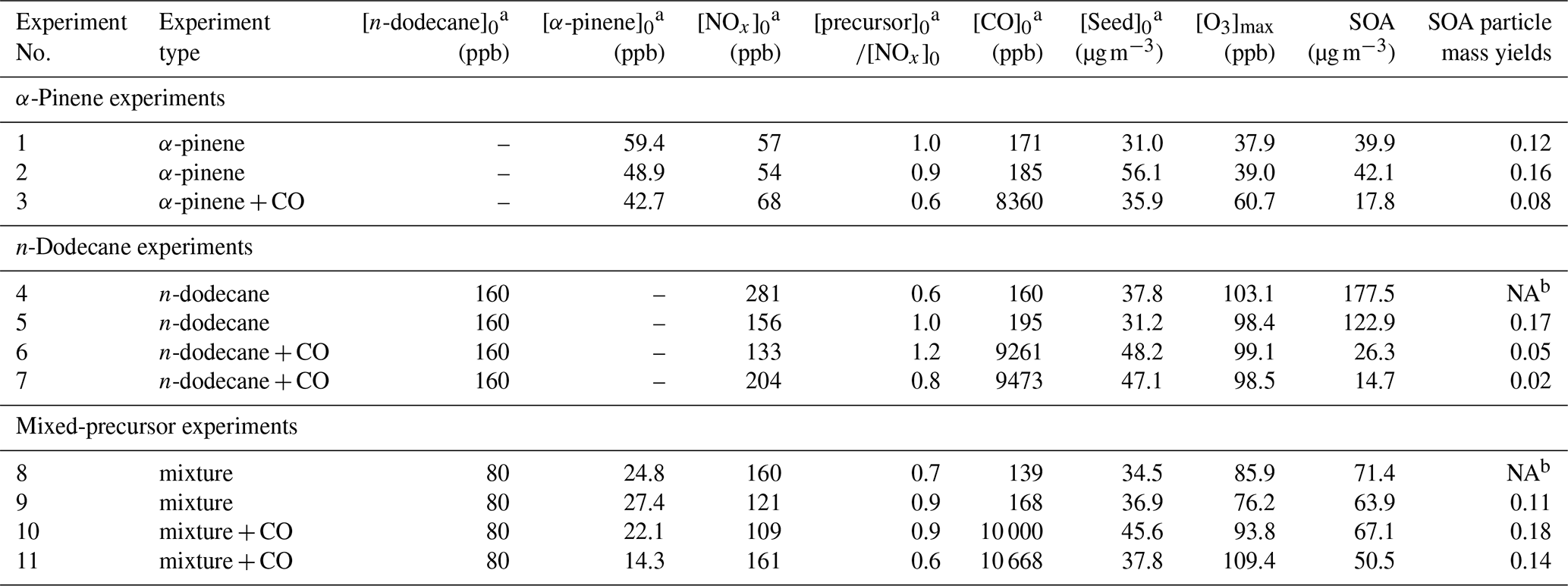
a The subscript “0” indicates the initial concentration.
b NA: no available data.
2.1 Generic peroxy radical chemistry
The analysis has been informed by the prevailing generic peroxy radical chemistry. RO2 radicals can undergo bimolecular termination reactions with HO2 radicals, other RO2 radicals, or NOx, leading to the formation of closed-shell products (Atkinson, 2000; Ziemann and Atkinson, 2012).
Hydroperoxides:
Carbonyls and alcohols:
Organic nitrates:
Peroxynitrates:
Accretion products:
RO2 radicals can also undergo unimolecular reactions that lead to the formation of carbonyls (Goldman et al., 2021; Molteni et al., 2019).
QOOH is a key oxidation intermediate formed via intramolecular hydrogen abstraction by RO2 radicals.
Besides closed-shell products, RO2 radicals can also form RO radicals (Orlando et al., 2003).
HOMs are formed via autoxidation pathways of RO2 radicals (Bianchi et al., 2019; Goldman et al., 2021).
These reaction pathways compete with one another, thereby influencing the distribution of products.
α-Pinene photooxidation is expected to produce C10H15Ox and C10H17Ox as major RO2 families. The C10H17Ox family is initiated via OH addition to α-pinene (Berndt et al., 2016; Jenkin et al., 1997; Kang et al., 2025; Vereecken and Peeters, 2004). RO2+HO2 termination (Reaction R1) of C10H17Ox forms C10H18On hydroperoxides, and RO2+RO2 termination (Reaction R2) yields C10H16On carbonyls and C10H18On alcohols. Unimolecular termination (Reaction R6) of C10H17Ox generates C10H16On carbonyls. The C10H15Ox family is formed via hydrogen abstraction from α-pinene or from first-generation oxidation products (e.g., pinonaldehyde), as well as directly from ozonolysis through the vinyl hydroperoxide pathway (Jenkin et al., 1997; Johnson and Marston, 2008; Kang et al., 2025). RO2+HO2 termination (Reaction R1) of C10H15Ox forms C10H16On hydroperoxides, whereas RO2+RO2 termination (Reaction R2) produces C10H14On carbonyls and C10H16On alcohols. Unimolecular termination (Reaction R6) of C10H15Ox generates C10H14On carbonyls. RO2+RO2 reactions (Reaction R2) between C10H15Ox and C10H17Ox radicals lead to the formation of C10H14On carbonyls and C10H18On alcohols, and/or C10H16On carbonyls and alcohols.
The main RO2 radicals expected from n-dodecane photooxidation are C12H25Ox family (Zhang et al., 2014). RO2+HO2 termination (Reaction R1) yields C12H26On hydroperoxides, while RO2+RO2 termination (Reaction R2) produces C12H24On carbonyls and C12H26On alcohols. Unimolecular termination (Reaction R6) of C12H25Ox generates C12H24On carbonyls.
In the mixture, RO2 radicals originating from different precursors can undergo cross-reactions. Reactions (Reaction R2) between C10H15Ox and C12H25Ox yield C10H14On carbonyls and C12H26On alcohols or C12H24On carbonyls and C10H16On alcohols. Similarly, Reactions (Reaction R2) between C10H17Ox and C12H25Ox lead to the formation of C10H16On carbonyls and C12H26On alcohols, or C12H24On carbonyls and C10H18On alcohols.
RO radicals can undergo unimolecular decomposition, isomerisation, or react with O2 (Orlando et al., 2003). Reaction of RO radicals with O2 leads to the formation of carbonyl compounds and HO2 radicals:
RO radicals derived from C10H15Ox can form C10H14On carbonyls via this pathway, and those derived from C12H25Ox yield C12H24On carbonyls.
Theoretically, C10H14On and C12H24On carbonyls can be formed via multiple pathways, including RO2+RO2 reactions (Reaction R2), unimolecular termination of RO2 radicals (Reaction R6), and reaction of RO radicals with O2 (Reaction R11). However, previous studies have demonstrated that, under ambient-temperature conditions and in the presence of NOx, unimolecular termination pathways are not expected to be dominant in RO2 chemistry (Goldman et al., 2021; Goss et al., 2025). In addition, RO radicals derived from α-pinene generally favour fragmentation owing to the low energy barrier for C–C bond scission (Dibble, 2001). For linear RO radicals formed from long-chain alkanes, isomerisation dominates over reactions with O2 (Atkinson, 2007; Ziemann and Atkinson, 2012). On this basis, both unimolecular termination and RO+O2 reactions are expected to make only minor contributions and are therefore not explicitly considered in this study.
Therefore, C10H14On and C12H24On carbonyls are expected to be formed predominantly via RO2+RO2 reactions. In contrast, C10H16On, C10H18On, and C12H26On species can be produced not only through RO2+RO2 reactions but also via RO2+HO2 pathways. Accordingly, changes in the relative abundances of C10H14On and C12H24On compounds are used as indicators to assess the influence of CO on RO2 chemistry. In general, the presence of CO is expected to reduce the relative contribution of RO2+RO2 termination, which would be reflected in decreased relative abundances of C10H14On and C12H24On species.
2.2 Experimental setup and procedure
The experiments were conducted in the 18 m3 MAC at the University of Manchester. The chamber comprises a fluorinated ethylene propylene (FEP) Teflon bag supported by three rectangular frames. Further details of the chamber are provided in Shao et al. (2022). The irradiation source, consisting of two xenon arc lamps (XBO 6000W/HSLA OFR, Osram) and a series of halogen lamps (50W/4700K MR16, Solux), is mounted inside the chamber and generates irradiation over the wavelength range of 290–800 nm to mimic the atmospheric radiation spectrum. The corresponding actinic flux spectrum is presented in Shao et al. (2022). The photolysis rate of NO2 () was . To promote OH radical production, an additional UVC lamp (TUV 130W XPT SE UNP/20, Philips) was installed, with more than 90 % of its length masked to prevent excessive irradiation. The liquid precursors (α-pinene, analytical standard, Sigma-Aldrich; n-dodecane, anhydrous, ≥99.0 %, Sigma-Aldrich) were initially injected via syringe into a heated glass bulb to facilitate vaporisation, after which the vapours were carried into the chamber by electronic capture device-grade nitrogen (ECD N2). NOx was introduced from a custom-made cylinder using ECD N2 as the carrier gas. NO2 served as the source of O3, and the subsequent O3 photolysis generated OH radicals, thereby initiating photochemical oxidation. The initial precursorNOx ratios were controlled within the range of 0.6–1.2, while the initial NO2/NO ratios ranged between 1.5 and 2.5. Seed particles with a mass concentration of were generated by nebulising aqueous ammonium sulfate solutions ((NH4)2SO4, ACS reagent, ≥99.0 %, Sigma-Aldrich) using an aerosol generator (ATM 230, Topas). During seed injection, the carrier air was passed through the humidifier, ensuring the deliquescence of the seeds as they were generated. These particles provided a condensation surface for the oxidation products, thereby reducing wall losses and suppressing nucleation (Nah et al., 2017).
The initial experimental conditions are summarised in Table 1. Each experiment typically consisted of four steps:
- (i)
Pre-experiment: Repeated flush-fill cycles were conducted to achieve a low-background condition. During these cycles, the chamber was flushed for approximately 7 min and then refilled with clean air at the same flow rate, with this procedure repeated for about 1.5 h. Subsequently, SOA precursors, NOx, CO, and seed aerosols were introduced into the chamber. The temperature and relative humidity were adjusted to approximately 25 °C and 50±5 %, respectively.
- (ii)
Stabilisation: The chamber was kept in the dark for 20–30 min to stabilise initial conditions prior to illumination.
- (iii)
Experiment: When the lights were turned on, photooxidation and subsequent SOA formation were initiated. Each “experiment” phase lasted for approximately 5 h.
- (iv)
Post-experiment: After the lights were turned off, the chamber underwent repeated flush-fill cycles for approximately 1 h. It was then filled with O3 at a high concentration (≥1 ppm) and left to soak overnight to oxidise and remove residual O3-reactive organic species.
2.3 Iso-reactivity conditions
OH radicals served as the primary oxidant in our experiments. All experiments were initiated under iso-reactivity conditions with respect to OH (Voliotis et al., 2022b; Voliotis et al., 2021). Specifically, the total OH reactivity was kept constant between single- and mixed-precursor systems. In the mixed-precursor system, SOA precursor concentrations were set such that each contributed equally to the total OH reactivity. Under these conditions, each precursor had an equal initial probability of reacting with OH and producing first-generation oxidation products (Voliotis et al., 2022b; Voliotis et al., 2021). The initial reactivity was calculated using the following equation:
where Cprecursor,i is the concentration of precursor i (molecule cm−3), and kOH,i is the reaction rate coefficient of precursor i with OH (). The reaction rate coefficients for α-pinene and n-dodecane with OH are and , respectively (Atkinson, 2003; Dash et al., 2014). As α-pinene exhibits greater reactivity towards OH than n-dodecane, a higher initial concentration of n-dodecane was used to achieve iso-reactivity in the experiments. The target mixing ratios of α-pinene were 40 ppb in the single-precursor system and 20 ppb in the mixed-precursor system, while those of n-dodecane were 160 and 80 ppb, respectively. The ratio of α-pinene to n-dodecane falls within the range observed in urban and roadside environments (Okada et al., 2012). The initial CO concentration was also determined according to the principle of iso-reactivity. The reaction rate coefficient of CO with OH is (Amedro et al., 2012).
2.4 Instrumentation
Near-real-time gas- and particle-phase composition was measured using a Filter Inlet for Gases and Aerosols coupled to a Chemical Ionisation Time-of-Flight Mass Spectrometer (FIGAERO-CIMS, Aerodyne Research Inc.). SOA precursors were measured in real time using a Vocus Proton-Transfer Reaction Time-of-Flight Mass Spectrometer (Vocus PTR-ToF-MS, Tofwerk). The non-refractory submicron aerosol particle composition, including sulfate, nitrate, ammonium, chloride, and organics, was measured in real time using a Compact Time-of-Flight Aerosol Mass Spectrometer (C-ToF-AMS, Aerodyne Research Inc.). NO and NO2 were measured using a chemiluminescence NO–NO2–NOx analyser (Model 42i, Thermo Fisher Scientific Inc.). O3 and CO were measured using a UV absorption O3 analyser (Model 49C, Thermo Fisher Scientific Inc.) and a CO analyser (Model 48i, Thermo Fisher Scientific Inc.), respectively. The mass concentration of seed aerosols in the 20–500 nm size range was measured using a Differential Mobility Particle Sizer (DMPS), consisting of a Vienna-design differential mobility analyser (DMA) coupled to a Condensation Particle Counter (CPC, model 3775, TSI Inc.) (Alfarra et al., 2012). The availability of instruments for each experiment is listed in Table S1 in the Supplement.
2.4.1 FIGAERO-CIMS
The FIGAERO system enables simultaneous characterisation of gas- and particle-phase species by sampling gases through one inlet while collecting particulate matter on a filter via a separate sampling port (Bannan et al., 2019; Lopez-Hilfiker et al., 2014). The instrument was operated in negative-ion mode using I− as the reagent ion, generated by passing CH3I and N2 over a 210Po radioactive source. It was run in a cyclic mode consisting of the following procedure:
- (i)
30 min of gas-phase sampling and simultaneous particle collection onto a PTFE filter (2.0 µm pore size, Zefluor; filters were pre-heated to 200 °C to remove potential contaminants) both at 1 L min−1. During this step, the instrument was flushed with N2 for 0.5 min every 4.5 min to obtain the gas-phase instrument background signal.
- (ii)
25 min of temperature-programmed thermal desorption of the collected particles, with the temperature ramped from ambient to 200 °C.
- (iii)
15 min of isothermal soaking at 200 °C.
- (iv)
20 min of cooling from 200 °C to ambient temperature.
- (v)
2 min of N2 flushing to clean the instrument.
Each cycle spanned approximately 1.5 h, and each experiment comprised four such cycles. In the final cycle, the photochemical reaction was terminated after procedure (i), corresponding to the completion of particle sampling (Fig. S1 in the Supplement).
To account for background species in the chamber, background measurements were conducted weekly. During these measurements, all components (SOA precursors, seed particles, CO, and NOx) were injected under the same conditions as in the regular experiments, while the chamber was kept in the dark. Data obtained during these background measurements were subtracted from the corresponding gas- and particle-phase data acquired during the “experiment” phase.
The FIGAERO-CIMS data were analysed using the Tofware package (v4.0.0) in Igor Pro 7.0.8 (WaveMetrics ©). I−, H2OI−, CH2O2I−, and were used for mass-to-charge calibration (calibration error≤3 ppm). High-resolution peak identification and fitting were performed in the range of 200–550 (iodide adducts), which contained the vast majority of the total signal. Owing to the lack of available calibration standards and potential variability in instrument sensitivity across different oxygenated organic compounds, quantitative analysis using I−-CIMS remains challenging (Lee et al., 2014). As a result, a uniform instrument sensitivity was assumed for all detected products. Additional uncertainties arise from the thermal decomposition in the FIGAERO. As shown in Fig. S2 in the Supplement, several compounds with relatively low carbon numbers exhibited comparatively high average carbon oxidation state () values and elevated maximum desorption temperature (Tmax). However, these species together accounted for less than 10 % of the total signal, indicating that the impact of thermal decomposition on the chemical composition was limited.
2.4.2 Vocus PTR-ToF-MS
The Vocus PTR-ToF-MS provides high-sensitivity and fast-response measurements of organic compounds without the need for pre-concentration or chromatographic separation. Compared to traditional PTR-MS, the Vocus employs a focusing ion-molecule reactor (IMR) consisting of a glass tube that is mounted inside a radio frequency (RF) quadrupole, with an axial electric field applied along the tube. This design enhances ion transmission efficiency and suppresses the clustering of ions with water molecules, thereby improving sensitivity and lowering the limit of detection (Jensen et al., 2023; Krechmer et al., 2018; Yuan et al., 2017).
In our experiments, the ion source was supplied with a 20 sccm flow of water vapor. The IMR was operated at 60 °C and 2.0 mbar, with an axial voltage of approximately 568 V and an RF amplitude of 450 V at 1.3 MHz. The reduced electric field strength () was 141 Td. Measurements were conducted on a 5 min cycle, consisting of 4 min of sampling followed by 1 min of instrumental background measurement. Instrument calibration was conducted daily. The calibration curve for α-pinene is presented in Fig. S3 in the Supplement. Owing to the absence of an n-dodecane calibration standard, direct quantification was not feasible. Moreover, n-dodecane undergoes extensive fragmentation during ionisation, and its protonated molecular ion signal is subject to interference from overlapping species. Therefore, alternative approaches were adopted for its quantification: (i) the initial mixing ratios were taken as the target values (160 ppb in the single-precursor system and 80 ppb in the mixed-precursor system), and (ii) the relative consumption of n-dodecane was inferred from the temporal evolution of the fragment ion (Fig. S4 in the Supplement). However, interference from other oxidation products or fragments cannot be fully excluded and may have led to an overestimation of SOA particle mass yields. Nevertheless, this uncertainty is unlikely to affect the overall trends or relative differences in yields.
2.4.3 C-ToF-AMS
A detailed description of the C-ToF-AMS can be found in Drewnick et al. (2009). Ionization efficiency (IE) and relative ionization efficiency (RIE) calibrations were carried out using size-selected NH4NO3 and (NH4)2SO4 particles. The average IE of NH4NO3 was determined to be , while the RIE for and were 4.71±0.24 and 1.13±0.01, respectively. These values are comparable to those reported in the literature (Canagaratna et al., 2007; Lannuque et al., 2023).
In this study, the organic aerosol (OA)/sulfate correction method was applied to correct for chamber wall losses in the SOA particle mass concentrations measured by AMS (Wang et al., 2018). This method assumes that the loss rate constants of OA and seed aerosols are identical, and that seed concentrations are affected solely by wall loss. The corrected particle mass concentration is given by:
where represents the SOA-to-sulfate ratio derived from AMS measurements, and Cseed(0) denotes the sulfate concentration at the beginning of the experiment.
SOA particle mass yields (YSOA) for each system were derived from SOA particle mass concentrations measured by AMS and precursor concentrations measured by PTR. It is defined as the mass of SOA particles formed per unit of precursor consumed (Gao et al., 2022):
For the single-precursor systems, Δprecursor (µg m−3) denotes the consumption of α-pinene or n-dodecane, whereas in the mixed-precursor system it refers to the total consumption of α-pinene and n-dodecane. In this study, the SOA particle mass yield refers to the overall yield and is calculated as the ratio of the total SOA particle mass formed to the total precursor consumed at the end of the experiment.
Figure 1 presents the temporal evolution of O3, precursor decay, and SOA particle mass concentrations during the photochemical reactions. Solid and dashed lines represent experiments conducted in the absence and presence of CO, respectively. The corresponding time series of NO, NO2, and CO are shown in Fig. S5 in the Supplement. These observations form the basis for evaluating the influence of CO on SOA particle formation and mass yields across different systems. Detailed results from the α-pinene, n-dodecane, and mixed-precursor experiments are presented in the following Sections.
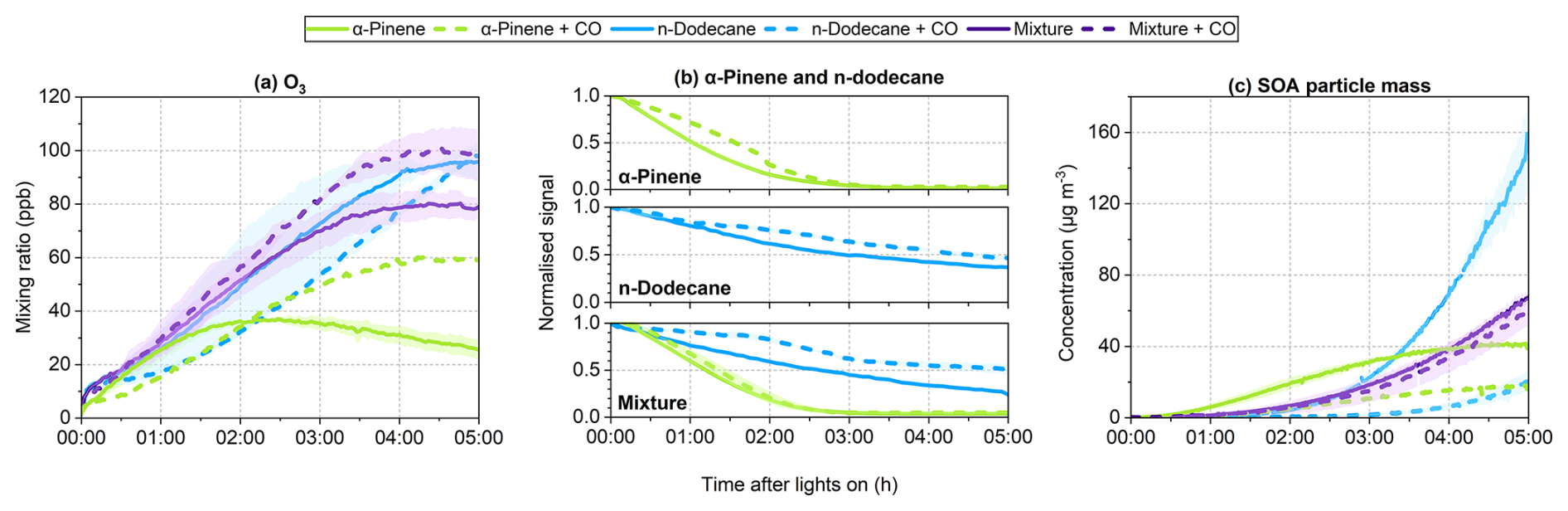
Figure 1Time series of (a) O3, (b) normalised SOA precursor signals, and (c) SOA particle mass concentrations during the photochemical reaction of α-pinene, n-dodecane and their mixture. Time 0 corresponds to the start of step (iii) (Sect. 2.2), when the chamber lights were turned on. Solid and dashed lines denote experiments conducted without and with CO, respectively. Where duplicate experiments were available, the lines represent the mean values, and the shaded area indicates the range between replicates (Table 1).
3.1 α-Pinene
3.1.1 SOA particle mass yields
The initial O3 concentration in the chamber was negligible. Upon illumination, O3 gradually accumulated, peaking at 38.5 ppb approximately two hours after lights on in the absence of CO, and then declined over time (Fig. 1a). In the presence of CO, the peak O3 concentration (60.7 ppb) was observed near the end of the experiment.
The initial α-pinene/NOx ratio in the α-pinene experiments was approximately 0.8 (Table 1). In the absence of CO, NOx concentrations declined during the first two hours of the reaction and subsequently stabilised. In contrast, in the presence of CO, NOx declined continuously throughout the experiment (Fig. S5). α-Pinene was almost entirely consumed within three hours under both conditions (Fig. 1b). Notably, the initial consumption rate was lower in the presence of CO. After approximately two hours, however, the decay rate increased and eventually converged with that observed in the absence of CO.
Compared to the experiment without CO, SOA particle mass increased more slowly in the presence of CO, resulting in substantially lower SOA particle mass concentrations (Fig. 1c). In both cases, the concentrations stabilised during the final hour of the reaction. By the end of the experiment, SOA particle mass concentrations reached 41.0 µg m−3 in the absence of CO and 17.8 µg m−3 in its presence. Correspondingly, the α-pinene SOA particle mass yield decreased from 0.14–0.08.
3.1.2 SOA particle chemical composition
Owing to the absence of data from the final two FIGAERO cycles in the α-pinene experiment with CO, the analysis of SOA particle composition was based on the second cycle, corresponding to two hours of reaction, by which time substantial SOA mass had already formed.
Figure 2a presents the high-resolution mass spectra of particle-phase compounds from α-pinene experiments conducted with and without CO, together with their differences. The products were mainly distributed within the molecular mass range of 150–280 Da. Under both conditions, C8H10O5 was the most abundant compound. Based on elemental composition, the compounds were classified into CHO and CHON groups. CHON species accounted for 21 % of the total signal in the absence of CO and 28 % in its presence.
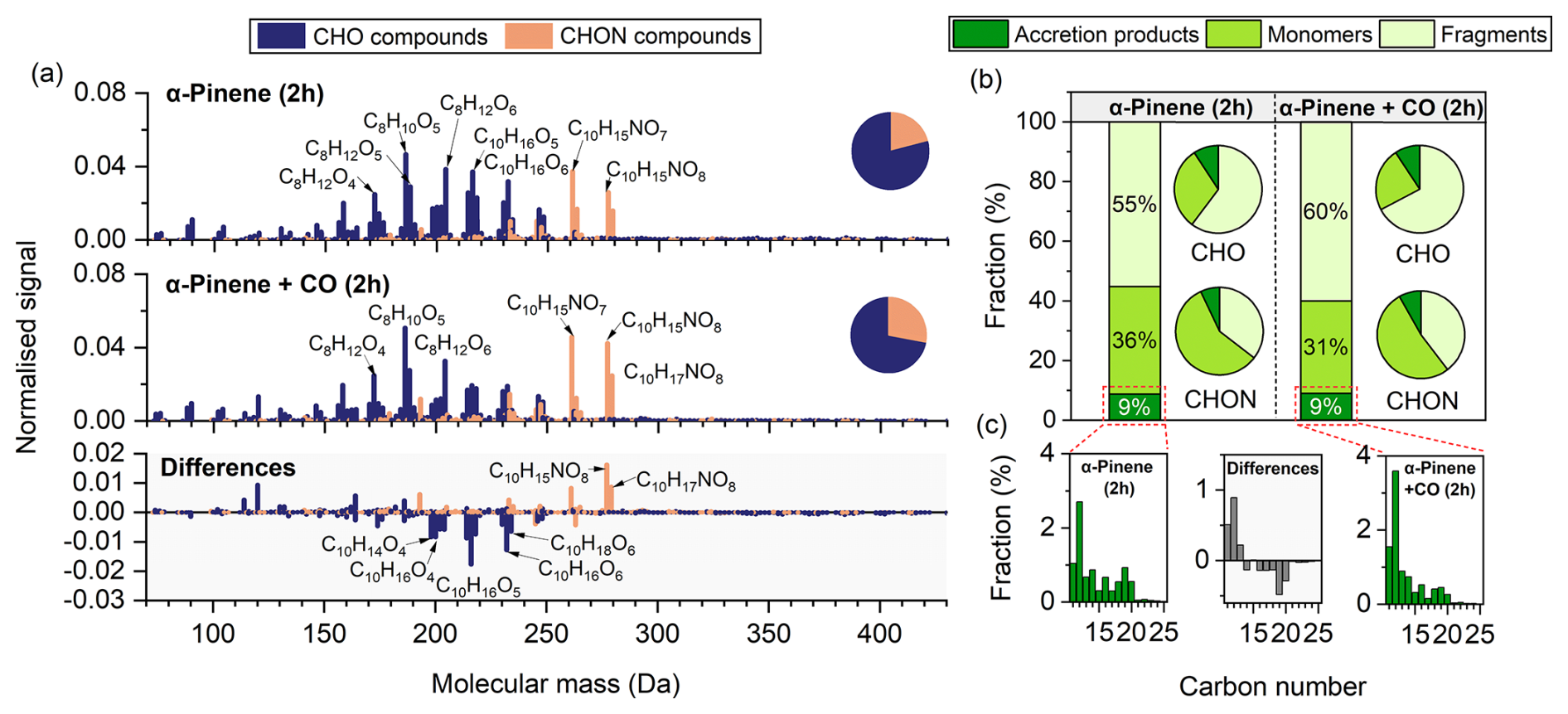
Figure 2(a) High-resolution mass spectra of particle-phase compounds measured by FIGAERO-CIMS in α-pinene experiments conducted with and without CO, and the corresponding difference spectra (with CO minus without CO). Prominent peaks are labelled with their corresponding molecular formulas. All signal intensities are normalised to 1. Pie charts display the proportions of CHO and CHON groups. (b) Fractions of α-pinene-derived fragments (C<10), monomers (C=10), and accretion products (C>10) in the absence and presence of CO. Bar charts represent their relative contributions to the total signal, while pie charts show their distribution within the CHO and CHON groups. (c) Carbon number distributions of accretion products in the absence (left) and presence (right) of CO. The middle panel shows the differences between the two conditions (with CO minus without CO).
The compounds can be categorised into three classes based on carbon number: monomers, fragments, and accretion products (Fig. 2b). Monomers derived from α-pinene consisted of C10 products, whereas fragment compounds contained fewer than 10 carbon atoms and accretion products contained more than 10. Fragments dominated under both conditions, accounting for 55 % and 60 % of the total signal in the absence and presence of CO, respectively. Within the CHO group, fragments contributed more than 60 %, with a substantial proportion distributed in the C7–C9 range (Fig. S11 in the Supplement). Monomers accounted for 36 % and 31 % in the absence and presence of CO, respectively, and were the dominant class within the CHON group, accounting for more than 50 %. The presence of CO led to a lower proportion of C10 CHO compounds (e.g., C10H16O4−6) and a higher proportion of C10 CHON compounds (e.g., C10H15NO7−8) (Fig. 2a). The overall fraction of accretion products remained constant at 9 % under both conditions. However, the proportion of C16–C24 accretion products was lower in the presence of CO (Fig. 2c).
The major RO2 radicals derived from α-pinene react via the Reactions (R1) and (R2) pathways to form the C10H14On, C10H16On, and C10H18On families. As shown in Fig. 3, in the absence of CO these species accounted for 11.0 %, 13.6 %, and 4.0 % of the CHO group, respectively, and decreased to 9.0 %, 9.5 %, and 2.4 % in its presence.
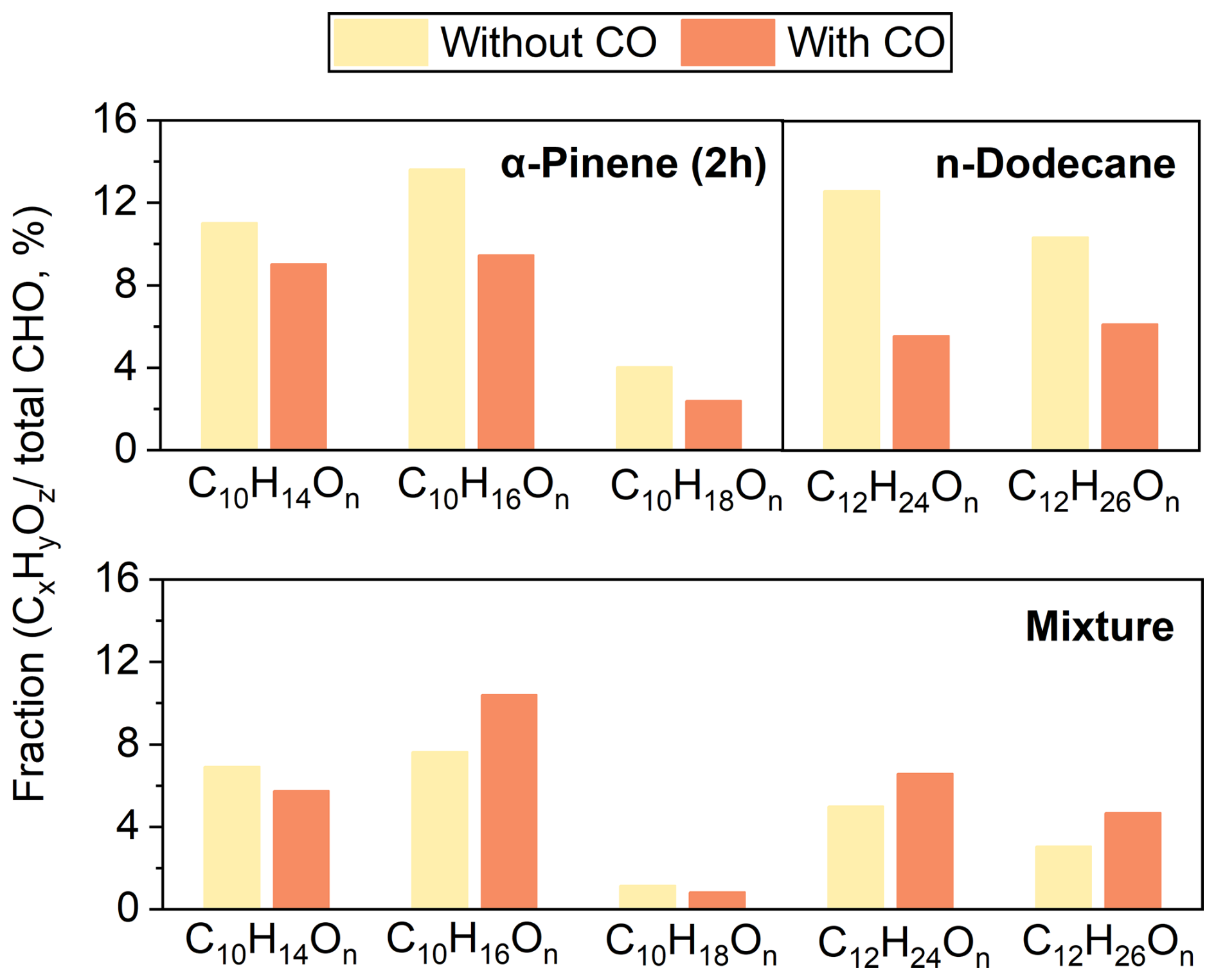
Figure 3Relative contributions of C10H14On, C10H16On, C10H18On, C12H24On, and C12H26On to the CHO group in the α-pinene, n-dodecane, and mixture systems in the absence and presence of CO.
3.2 n-Dodecane
3.2.1 SOA particle mass yields
In the n-dodecane experiments, O3 concentrations were generally higher in the absence of CO than in its presence (Fig. 1a). The temporal evolution of O3 differed markedly between the two conditions. In the absence of CO, O3 had nearly reached its peak by the end of the experiment, whereas in the presence of CO it continued to increase throughout the experiment. Despite these differences in formation rates and peak timing, the final O3 concentrations in both systems converged to similar levels, approaching 100 ppb.
The initial n-dodecane/NOx ratio was approximately 0.9 (Table 1). NOx concentrations declined steadily throughout the experiment under both conditions (Fig. S5). In the presence of CO, the decay rate of n-dodecane was lower (Fig. 1b). By the end of the experiment, 37 % of the initial n-dodecane remained unreacted in the absence of CO, whereas 47 % remained when CO was present.
In the absence of CO, the final SOA particle mass concentration reached 122.9 µg m−3, corresponding to a mass yield of 0.17 (Exp. 5). In the presence of CO, it reached 20.5 µg m−3, with a yield of 0.04.
3.2.2 SOA particle chemical composition
Compared to α-pinene, particle-phase products derived from n-dodecane exhibited generally higher molecular mass distributions, primarily within the range of 210–310 Da (Fig. 4a). In the absence of CO, the most abundant species were C12H25NO5, C12H24O5, and C12H26O3, whereas in the presence of CO, C12H25NO4, C12H23NO7, and C12H25NO6 dominated. CHON compounds accounted for 37 % and 43 % of the total signal in the absence and presence of CO, respectively.
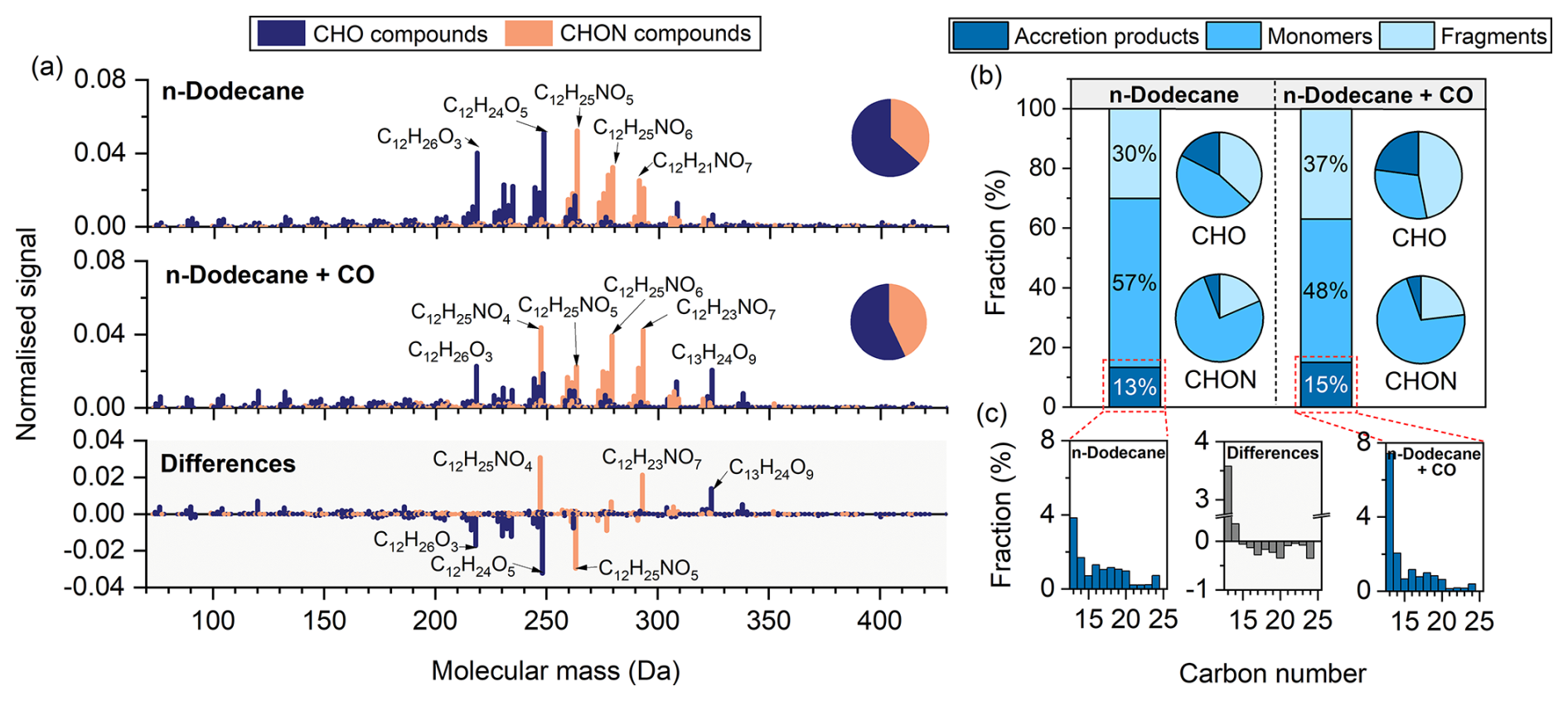
Figure 4(a) High-resolution mass spectra of particle-phase compounds measured by FIGAERO-CIMS in n-dodecane experiments conducted with and without CO, and the corresponding difference spectra (with CO minus without CO). Prominent peaks are labelled with their corresponding molecular formulas. All signal intensities are normalised to 1. Pie charts display the proportions of CHO and CHON groups. (b) Fractions of n-dodecane-derived fragments (C<12), monomers (C=12), and accretion products (C>12) in the absence and presence of CO. Bar charts represent their relative contributions to the total signal, while pie charts show their distribution within the CHO and CHON groups. (c) Carbon number distributions of accretion products in the absence (left) and presence (right) of CO. The middle panel shows the differences between the two conditions (with CO minus without CO).
In the n-dodecane systems, compounds containing 12 carbon atoms were classified as monomers, those with fewer than 12 as fragments, and those with more than 12 as accretion products (Fig. 4b). Monomers dominated under both conditions, accounting for 57 % of the total signal in the absence of CO and 48 % in its presence. Within the CHON group, monomers accounted for more than 70 %. The presence of CO led to a lower proportion of C12 CHO compounds (e.g., C12H24O5 and C12H26O3) and a higher proportion of C12 CHON compounds (e.g., C12H25NO4 and C12H23NO7) (Fig. 4a). However, a few exceptions were observed. For example, a series of highly oxygenated C13 CHO compounds, such as C13H24O9 and C13H22O10, accounted for a higher fraction in the presence of CO, whereas C12H25NO5 accounted for a higher fraction in the absence of CO. Fragments accounted for 30 % and 37 % in the absence and presence of CO, respectively. While the overall fraction of accretion products was comparable under both conditions, the presence of CO reduced the fraction of C16–C24 accretion products (Fig. 4c).
The major RO2 radicals derived from n-dodecane react via the Reactions (R1) and (R2) pathways to form the C12H24On and C12H26On families. As shown in Fig. 3, in the absence of CO these species accounted for 12.6 % and 10.4 % of the CHO group, respectively, and decreased to 5.6 % and 6.1 % in its presence.
3.3 Mixture
3.3.1 SOA particle mass yields
During the first hour of the reaction, O3 concentrations were comparable in the absence and presence of CO (Fig. 1a). Thereafter, O3 levels became higher in the presence of CO. In both cases, O3 concentrations peaked during the final hour, reaching 81.1 ppb without CO and 101.6 ppb with CO.
The initial precursorNOx ratio was approximately 0.8 (Table 1). NOx concentrations declined steadily throughout the reaction under both conditions (Fig. S5). In the mixture, the presence of CO led to lower decay rates for both α-pinene and n-dodecane compared to the experiment without CO (Fig. 1b). Nevertheless, α-pinene was fully consumed within three hours in both cases. By the end of the experiment, 25 % of the initial n-dodecane remained unreacted without CO, whereas 51 % remained with CO.
In the absence of CO, the final SOA particle mass concentration reached 63.9 µg m−3, corresponding to a mass yield of 0.11 (Exp. 9). In the presence of CO, it reached 58.8 µg m−3, with a yield of 0.16.
3.3.2 SOA particle chemical composition
Compared to single-precursor systems, the mixed-precursor system exhibited a broader molecular mass distribution, primarily ranging from 150–330 Da (Fig. 5a). In the absence of CO, C8H10O5, C8H12O6, and C12H24O5 showed the highest signal intensities, whereas in the presence of CO, C12H24O5, C8H10O5, and C12H25NO6 were most abundant. CHON compounds accounted for 30 % and 29 % of the total signal in the absence and presence of CO, respectively.
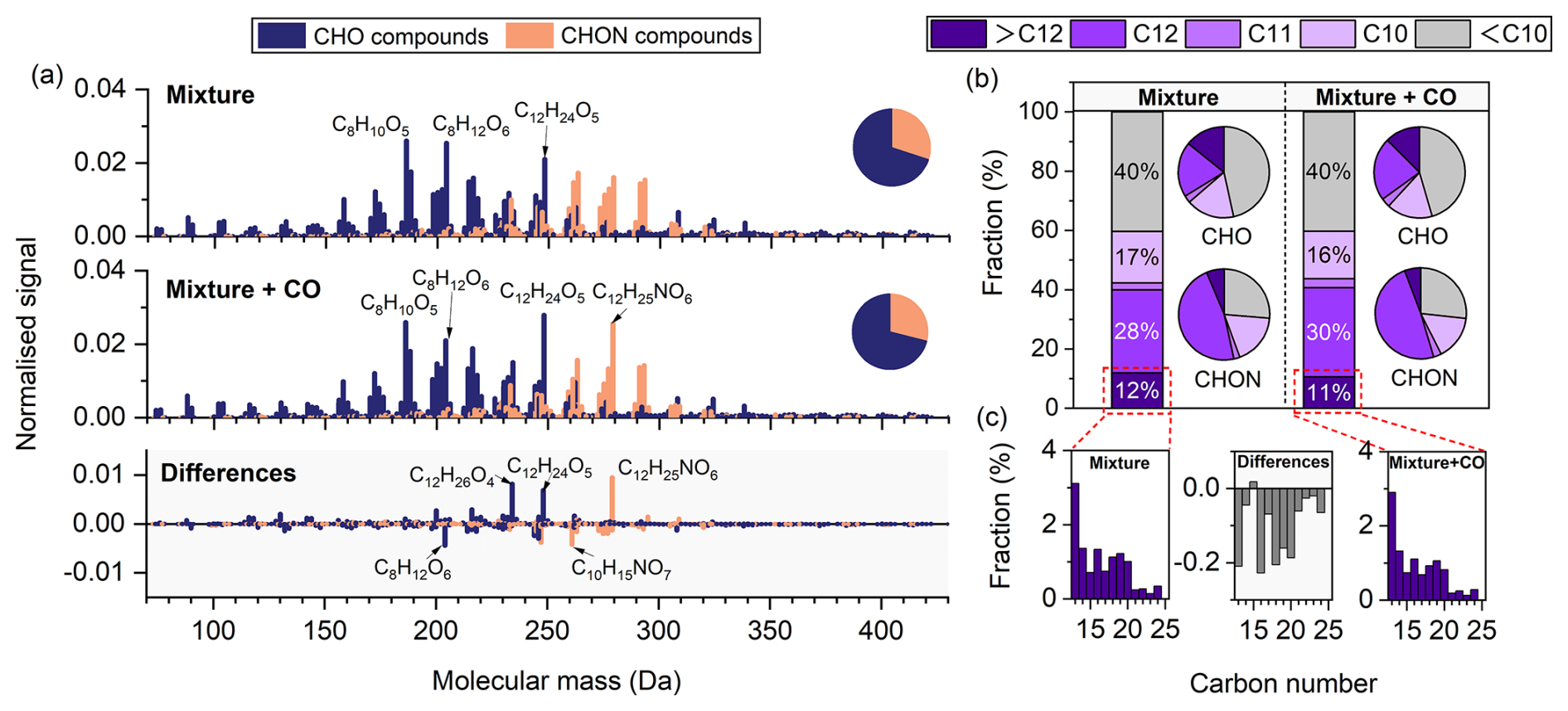
Figure 5(a) High-resolution mass spectra of particle-phase compounds measured by FIGAERO-CIMS in mixture experiments conducted with and without CO, and the corresponding difference spectra (with CO minus without CO). Prominent peaks are labelled with their corresponding molecular formulas. All signal intensities are normalised to 1. Pie charts display the proportions of CHO and CHON groups. (b) Fractions of particle-phase products with different carbon numbers in the absence and presence of CO. Bar charts represent their relative contributions to the total signal, while pie charts show their distribution within the CHO and CHON groups. (c) Carbon number distributions of products with more than 12 carbon atoms in the absence (left) and presence (right) of CO. The middle panel shows the differences between the two conditions (with CO minus without CO).
In the mixture, compounds with fewer than 10 carbon atoms were classified as fragments, while those containing more than 12 carbon atoms were considered accretion products. Fragments dominated under both conditions and accounted for 40 % of the total signal in each case. Accretion products accounted for 12 % and 11 % in the absence and presence of CO, respectively. Except for C15 species, the fractions of C13–C24 products decreased slightly in the presence of CO. In addition, the presence of CO resulted in an increased proportion of C12 CHO compounds (e.g., C12H26O4 and C12H24O5), and a reduced proportion of C10 CHON compounds (e.g., C10H15NO7) (Fig. 5a). Overall, changes in carbon number distribution were less pronounced in the mixed-precursor system than in the single-precursor systems (Fig. S11).
The bottom panel of Fig. 3 shows the relative contributions of C10H14On, C10H16On, C10H18On, C12H24On, and C12H26On to the CHO products in the mixture. In the presence of CO, the fractions of C10H14On and C10H18On decreased from 6.9 % and 1.2 % to 5.5 % and 0.8 %, respectively, whereas those of C10H16On, C12H24On, and C12H26On increased from 7.6 %, 5.0 %, and 3.1 % to 10.4 %, 6.6 %, and 4.7 %, respectively.
4.1 Photochemistry
The photochemical reactions in this study involved the simultaneous presence of NOx and CO, multiple oxidants (OH and O3), and multiple precursor species. The interactions among these factors substantially increase the complexity of the system, making it challenging to establish comparable experimental conditions across different precursor systems. In this study, two key approaches were adopted: (i) ensuring initial iso-reactivity towards OH radicals, and (ii) setting comparable initial precursorNOx ratios across systems. Additionally, an oxidant closure approach was employed to characterise the photochemical conditions. As OH radicals could not be directly measured in this study, their concentrations were estimated from the temporal evolution of O3 and the consumption of precursors, or alternatively from the depletion of CO (see details in the Supplement). This approach enabled a quantitative evaluation of the relative contributions of different oxidants to precursor oxidation.
Under idealised iso-reactivity conditions, all systems would exhibit comparable initial OH reactivity, and in the mixture each precursor molecule would initially have an equal probability of reacting with OH. In practice, however, O3 also contributed to precursor oxidation, and the differing reactivities of individual precursors towards O3 can modify the precursor decay and secondary oxidant formation, thereby influencing the reactivity. n-Dodecane was oxidised exclusively by OH radicals. For α-pinene, although OH remained the dominant photochemical sink in this study, the contribution of O3 to its decay was not negligible. As shown in Fig. S12 in the Supplement, the relative contributions of these oxidants evolved over time, with the role of O3 generally becoming more important as the reaction proceeded. In the α-pinene single-precursor system, on average approximately 80 % of α-pinene decay was attributable to OH oxidation, while the remaining ∼20 % was driven by ozonolysis. By comparison, the contribution of ozonolysis was slightly higher in the mixed-precursor system. Thus, fully comparable reactivity across different systems was difficult to maintain throughout the reaction when multiple oxidants were present. This reflects an inherent limitation of defining iso-reactivity with respect to a single oxidant in multi-oxidant systems.
The precursorNOx ratio is important for determining the chemical regime of O3 and SOA formation (Chen et al., 2022). However, when multiple precursors are involved, maintaining similar initial precursorNOx ratios may not be sufficient to establish comparable chemical regimes across systems. In this study, the temporal profiles of O3 and NOx differed substantially between the single- and mixed-precursor systems (Figs. 1a and S5). In the α-pinene system, O3 concentrations peaked after approximately two hours of reaction and subsequently declined, while NOx levels stabilised. By this point, over 80 % of α-pinene had been consumed, and the SOA particle formation rate began to decline (Fig. 1b and c). These trends may indicate a reduction in RO2+NO reactions, which would slow the conversion of NO to NO2 and thereby limit photochemical O3 production. In contrast, in the n-dodecane and mixture systems, over 50 % of n-dodecane was still unreacted after two hours, and the SOA particle formation rate continued to increase (Fig. 1b and c), indicating that RO2+NO reactions remained active. This sustained reactivity enabled continuous conversion of NO to NO2 and enhanced photochemical O3 production.
These results raise an important consideration for studies involving multiple precursors and oxidants. Even when initial OH reactivity and precursorNOx ratios are controlled, achieving fully comparable experimental conditions across such systems remains challenging. Given that the coexistence of multiple precursors and oxidants is a common feature of the ambient atmosphere, future laboratory studies should explore a broader range of precursorNOx ratios and systematically assess the effects of varying oxidants to improve our understanding of SOA formation under atmospherically relevant conditions.
The addition of CO further perturbed the photochemical processes, altering both oxidant levels and precursor decay rates. CO can consume OH radicals, preventing their reaction with SOA precursors (McFiggans et al., 2019). Based on the estimated OH concentrations, evidence for this oxidant scavenging effect was observed. During the initial stage of the reaction, CO reduced the OH concentrations by approximately 50 % to around (Fig. S6 in the Supplement). However, OH levels gradually recovered as the reaction progressed and eventually reached values comparable to those observed in the absence of CO (except for n-dodecane system). In the presence of CO, the reaction of CO with OH led to enhanced HO2 formation. Subsequent HO2+ NO reactions regenerated OH, thereby increasing radical propagation efficiency. In contrast, in the absence of CO, although O3 photolysis provided a primary source of OH, OH regeneration in the n-dodecane system was likely less efficient, consistent with the decline in OH concentrations. In both the α-pinene and mixture systems, however, OH concentrations continued to increase even without CO, indicating the presence of additional OH regeneration processes, such as OH formation during α-pinene ozonolysis. In addition to its impact on OH concentrations, the presence of CO also modified O3 levels. In the presence of CO, both the α-pinene and mixture systems exhibited higher peak O3 concentrations, whereas the n-dodecane system showed generally lower O3 levels. Variations in oxidant concentrations contributed to changes in SOA precursor decay rates (Fig. 1b). In the absence of CO, α-pinene was almost completely consumed within 3 h. In the presence of CO, its decay was initially suppressed; however, after approximately 2 h the decay rate increased, likely due to secondary OH production and elevated O3 concentrations. Such that α-pinene was nevertheless nearly fully consumed within 3 h. As a result, CO did not significantly affect the overall extent of α-pinene consumption. In contrast, for n-dodecane, the presence of CO not only slowed the oxidation rate but also reduced the overall extent of consumption, leaving a substantial fraction unreacted by the end of the experiment.
4.2 Effect of CO on SOA particle chemical composition
4.2.1 Single-precursor systems
The presence of CO led to several consistent changes in the chemical composition of SOA particles in both the α-pinene and n-dodecane systems, including an increased relative contribution of the CHON group and fragment species and a reduced fraction of C16–C24 accretion products (Figs. 2 and 4). In addition, the relative contributions of representative RO2+RO2 termination products (C10H14On and C12H24On) within the CHO group decreased (Fig. 3). These observations provide evidence for a similar shift in RO2 fate in the presence of CO in both systems. However, owing to the limitations of I−-CIMS measurements, the absolute contributions in individual reaction pathways cannot be fully constrained. The following discussion is therefore based partly on relative changes.
Organic nitrate concentrations were estimated from AMS measurements using the method described by Kiendler-Scharr et al. (2016). The results show that, in the single-precursor systems, the presence of CO led to a pronounced reduction in organic nitrate concentrations (Fig. S13 in the Supplement). This reduction can be attributed to two main factors. First, CO competes with SOA precursors for available OH (Figs. 1b and S6). Second, CO enhances HO2 formation, increasing the importance of the RO2+HO2 pathway and thereby altering RO2 reaction branching. In addition, lower NO concentrations were observed in the presence of CO (Fig. S5), consistent with enhanced conversion of NO to NO2 via the HO2+NO reaction. The increase in HO2 and decrease in NO reduced the likelihood of RO2 reacting with NO. Despite this absolute reduction, FIGAERO-CIMS results showed that the relative contributions of the CHON group and fragment products increased in the presence of CO (Figs. 2 and 4). CHON products are primarily formed through the pathway, and fragment species originate from the fragmentation of RO radicals (Atkinson, 2000; Ziemann and Atkinson, 2012). Owing to the rapid reaction of RO2 with NO and the high branching towards RO formation, reactions of RO2 with NO represent an important source of RO radicals under NOx conditions (Orlando et al., 2003; Ziemann and Atkinson, 2012). These observations therefore indicate that, in the presence of CO, the contribution of RO2+NO reactions decreased, but to a lesser extent than competing RO2 termination pathways.
AMS measurements showed a decrease in SOA particle mass concentrations in the presence of CO (Fig. 1c). In addition to OH scavenging, another important factor is that CO enhances competition between RO2+RO2 and RO2+HO2 reactions, thereby reducing the formation of accretion products (Baker et al., 2024; McFiggans et al., 2019; Peräkylä et al., 2023). Despite this reduction, CO did not significantly alter the overall fraction of accretion products. However, the relative contribution of C16–C24 species decreased (Figs. 2c and 4c), accompanied by an increase in C11–C15 species in the α-pinene system and C13–C14 species in the n-dodecane system. Accretion products with lower carbon numbers are expected to form via pathways that involve fragmentation of RO radicals (Kang et al., 2025), and their increased relative contribution is consistent with the elevated fraction of fragment products discussed above. In contrast, longer-chain accretion products are more likely to originate from RO2+RO2 reactions involving non-fragmented C10C12 RO2 radicals, including reactions between non-fragmented RO2 radicals and fragmented RO2 radicals (<C10), or between two non-fragmented RO2 radicals, yielding C20 and C24 accretion products in the α-pinene and n-dodecane systems, respectively. Combined with the reduced fractions of C10H14On and C12H24On families (Fig. 3), these observations indicate that CO preferentially suppressed RO2+RO2 chemistry, particularly pathways forming longer-chain accretion products.
Overall, in the single-precursor systems, CO reduced the contributions of both RO2+RO2 and RO2+NO reactions. However, reactions of RO2 with NO decreased to a lesser extent than competing RO2 termination pathways, and the reduction in RO2+RO2 termination was more pronounced for longer-chain accretion products than for shorter-chain ones.
4.2.2 Mixed-precursor system
Compared with the single-precursor systems, the influence of CO on SOA chemical composition differed in the mixed-precursor system. Specifically, (i) the presence of CO did not significantly alter the relative contributions of the CHON group and fragment species (Fig. 5a and b); (ii) the fractions of C13–C24 accretion products (excluding C15) slightly decreased (Fig. 5c); and (iii) within the CHO group, the fraction of the C10H14On family decreased, whereas that of the C12H24On family increased (Fig. 3).
In the mixed-precursor system, organic nitrate concentrations exhibited little variation in the presence of CO (Fig. S13), consistent with the largely unchanged relative contribution of the CHON group and fragment species. This suggests that the contribution of RO2+NO reactions was not substantially reduced under CO conditions.
SOA particle mass concentrations and the fraction of accretion products both decreased slightly in the presence of CO (Figs. 1c and 5), suggesting a slight reduction in the contribution of RO2+RO2 termination.
Moreover, CO led to a lower fraction of the C10H14On family in the mixture, consistent with the trend observed in the α-pinene single-precursor system. In contrast to the n-dodecane single-precursor system, however, the relative contribution of the C12H24On family increased in the presence of CO in the mixture. Together with the increase in the fraction of C12 species and decrease in that of C10 species (Fig. S11), these observations may indicate that CO affected RO2+RO2 termination involving α-pinene-derived RO2 more strongly than that involving n-dodecane-derived RO2.
Overall, in the mixed-precursor system, the influence of CO on RO2 termination pathways was less pronounced than in the single-precursor systems and may have affected n-dodecane- and α-pinene-derived RO2 to different extents.
Although the underlying mechanism cannot be fully resolved in this study, the observed changes in product distributions provide important evidence for shifts in RO2 reaction pathways in the mixed-precursor system under different conditions. As α-pinene and n-dodecane were used as representative precursors, these findings may be specific to the present system. Future chamber studies covering a broader range of precursor combinations are therefore needed to assess the generality of the observed behaviour.
4.3 Effect of CO on SOA particle mass yields
Figure 6 presents the SOA particle growth curves for each system. The slope of the curve represents the incremental SOA particle mass yield at a given stage of precursor consumption, while the final position of the curve reflects the overall yield achieved by the end of the experiment. The induction period is defined as the amount of SOA precursor consumed before SOA particle formation begins (Zhou et al., 2019). Compared with the α-pinene system, the n-dodecane system exhibited a longer induction period, while that of the mixed-precursor system lay in between. In the presence of CO, the induction period was extended in the n-dodecane system but remained largely unchanged in the α-pinene system. Notably, the induction period in the mixture system was shortened in the presence of CO. These behaviours suggest a distinct influence of CO on the SOA particle mass yields across different systems.
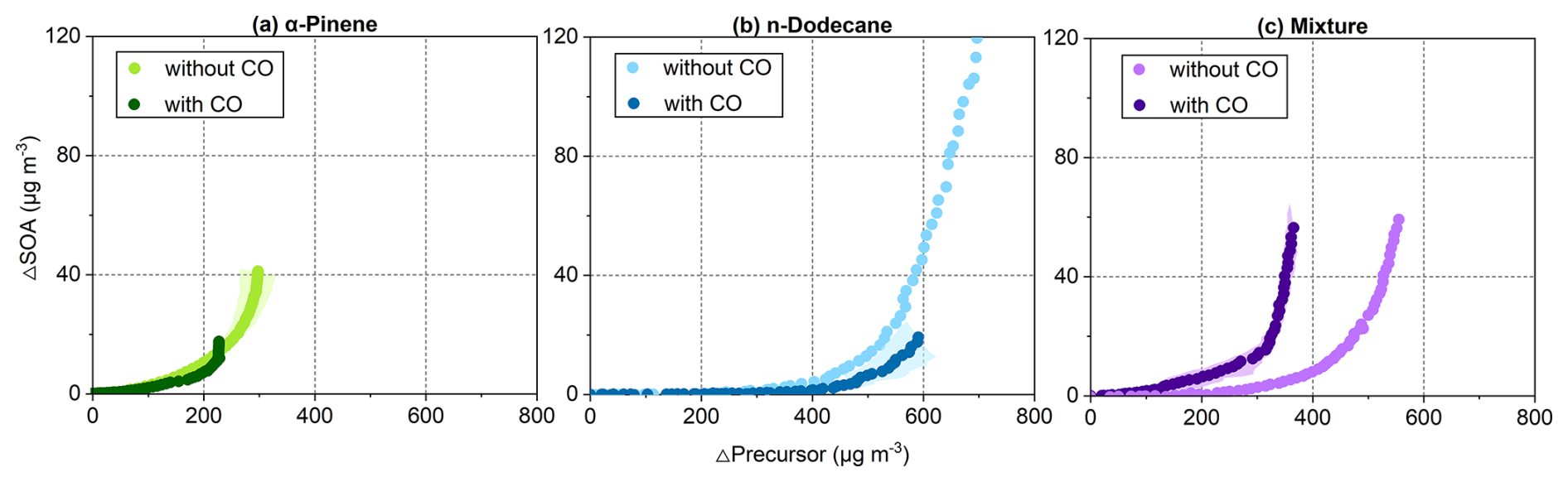
Figure 6Growth curves of SOA particles for (a) α-pinene, (b) n-dodecane, and (c) mixture experiments, defined as the ratio of SOA particle mass concentration to consumed precursor mass. Shaded areas represent the range between replicate experiments.
In the single-precursor systems, CO substantially reduced SOA formation, with a stronger effect for n-dodecane than for α-pinene. In the presence of CO, SOA particle mass concentrations and overall yields decreased by 83 % and 79 %, respectively, for n-dodecane, and by 57 % and 43 % for α-pinene. In contrast, the mixed-precursor system exhibited only an 8 % decrease in SOA mass concentration, and the overall yield increased slightly.
Chemical composition analysis indicates that, in the single-precursor systems, the contributions of accretion products derived from RO2+RO2 termination, particularly those with longer carbon chains, decreased in the presence of CO. These accretion products are expected to exhibit extremely low volatility and contribute efficiently to SOA formation (Peräkylä et al., 2023). At the same time, although the absolute concentration of organic nitrates decreased, the fractions of CHON and fragment products increased in the presence of CO. This suggests that RO2+NO reactions were also reduced, but less markedly than the competing RO2 termination pathways. Products formed via RO2+NO reactions are generally expected to exhibit higher volatility than those formed through RO2+HO2 and RO2+RO2 termination (Presto et al., 2005; Zhao et al., 2018). All these changes are therefore expected to shift the product distribution towards more volatile species, consistent with the observed decrease in SOA particle mass yields.
Compared with the single-precursor systems, changes in RO2 reaction pathways in the mixture appeared to exert a weaker influence on the formation of lower-volatility products. Consequently, SOA particle mass concentrations and yields behaved differently in the mixed-precursor system.
Competition between CO and SOA precursors for available OH was also a factor influencing the yields (McFiggans et al., 2019). However, the impact of differences in OH concentrations on SOA particle mass yields and chemical composition cannot be fully assessed in this study. Future work may need to re-adjust OH concentrations so that the systems can be maintained at comparable oxidation stages, thereby enabling more direct comparisons (Baker et al., 2024; McFiggans et al., 2019).
We established a photochemical system in the MAC that incorporated both biogenic and anthropogenic SOA precursors in the presence of CO and NOx. The results show that the influence of CO on SOA particle mass yields and chemical composition differed markedly between single- and mixed-precursor systems.
In the single-precursor systems, the presence of CO led to a notable reduction in SOA particle mass yields, with a stronger effect for n-dodecane than for α-pinene. By contrast, no such suppression was observed in the mixture. Chemical composition analysis indicated that, in the single-precursor systems, CO reduced the contributions of both RO2+RO2 and RO2+NO reactions. In the mixed-precursor system, however, RO2+NO reactions showed no evident reduction, while the decrease in RO2+RO2 termination was comparatively small. In addition, CO affected the two precursors to different extents in the mixture.
Although biogenic precursors contribute more substantially to SOA formation on a global scale, anthropogenic precursors can play a significant role in urban and suburban environments (Srivastava et al., 2022; Stone et al., 2010; Volkamer et al., 2006). Such regions are often characterised by elevated levels of co-emitted pollutants, such as CO and NOx, which can modify oxidant budgets and shift radical reaction pathways. Consequently, model parameterisations derived under single-precursor or idealised conditions may misrepresent SOA formation in non-pristine environments. Future laboratory studies should better capture the chemical complexity of the real atmosphere to improve the accuracy and applicability of SOA model parameterisations.
However, establishing experimental conditions that account for atmospheric chemical complexity while remaining comparable across different systems remains challenging. The nonlinear interactions among multiple precursors, inorganic trace gases, and oxidants substantially increase the complexity of the system. In this study, even when the initial OH reactivity and precursorNOx ratios were controlled, fully comparable conditions across such systems could not be achieved. This highlights the need for future work to systematically investigate SOA formation under controlled variations in oxidant levels and precursorNOx ratios to enhance the reliability and comparability of results.
All the data in the figures of this study are available upon request to the corresponding authors (g.mcfiggans@manchester.ac.uk and aristeidis.voliotis@manchester.ac.uk).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-9679-2026-supplement.
GX, AV, and GM conceived the study. GX and AV conducted the experiments. AV, TJB, YS, HW, DH provided assistance in instrument operation and data analysis. GX conducted the data analysis and wrote the manuscript with inputs from all the co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We thank colleagues from the Jülich and Gothenburg teams for valuable discussions, especially Thomas F. Mentel, Mattias Hallquist, and Sören R. Zorn. We acknowledge the use of ChatGPT (https://chatgpt.com/, last access: 9 March 2026) for assistance in language refinement of this manuscript.
This research has been supported by the China Scholarship Council (grant no. 202208330060), the Secondary Organic Aerosol Prediction in Realistic Atmospheres (SOAPRA) project (grant no. NE/V012665/1), and the Natural Environment Research Council (NERC) through the UK National Centre for Atmospheric Science (NCAS).
This paper was edited by Frank Keutsch and reviewed by two anonymous referees.
Alfarra, M. R., Hamilton, J. F., Wyche, K. P., Good, N., Ward, M. W., Carr, T., Barley, M. H., Monks, P. S., Jenkin, M. E., Lewis, A. C., and McFiggans, G. B.: The effect of photochemical ageing and initial precursor concentration on the composition and hygroscopic properties of β-caryophyllene secondary organic aerosol, Atmos. Chem. Phys., 12, 6417–6436, https://doi.org/10.5194/acp-12-6417-2012, 2012.
Amedro, D., Miyazaki, K., Parker, A., Schoernaecker, C., and Fittschen, C.: Atmospheric and kinetic studies of OH and HO2 by the FAGE technique, J. Environ. Sci., 24, 78–86, https://doi.org/10.1016/s1001-0742(11)60723-7, 2012.
Andreae, M. O. and Crutzen, P. J.: Atmospheric aerosols: biogeochemical sources and role in atmospheric chemistry, Science, 276, 1052–1058, https://doi.org/10.1126/science.276.5315.1052, 1997.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, https://doi.org/10.1016/s1352-2310(99)00460-4, 2000.
Atkinson, R.: Kinetics of the gas-phase reactions of OH radicals with alkanes and cycloalkanes, Atmos. Chem. Phys., 3, 2233–2307, https://doi.org/10.5194/acp-3-2233-2003, 2003.
Atkinson, R.: Rate constants for the atmospheric reactions of alkoxy radicals: An updated estimation method, Atmos. Environ., 41, 8468–8485, https://doi.org/10.1016/j.atmosenv.2007.07.002, 2007.
Atkinson, R. and Arey, J.: Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 103, 4605-4638, https://doi.org/10.1021/cr0206420, 2003.
Baker, Y., Kang, S., Wang, H., Wu, R., Xu, J., Zanders, A., He, Q., Hohaus, T., Ziehm, T., Geretti, V., Bannan, T. J., O'Meara, S. P., Voliotis, A., Hallquist, M., McFiggans, G., Zorn, S. R., Wahner, A., and Mentel, T. F.: Impact of ratio on highly oxygenated α-pinene photooxidation products and secondary organic aerosol formation potential, Atmos. Chem. Phys., 24, 4789–4807, https://doi.org/10.5194/acp-24-4789-2024, 2024.
Bannan, T. J., Le Breton, M., Priestley, M., Worrall, S. D., Bacak, A., Marsden, N. A., Mehra, A., Hammes, J., Hallquist, M., Alfarra, M. R., Krieger, U. K., Reid, J. P., Jayne, J., Robinson, W., McFiggans, G., Coe, H., Percival, C. J., and Topping, D.: A method for extracting calibrated volatility information from the FIGAERO-HR-ToF-CIMS and its experimental application, Atmos. Meas. Tech., 12, 1429–1439, https://doi.org/10.5194/amt-12-1429-2019, 2019.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Bianchi, F., Kurten, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Burkholder, J. B., Abbatt, J. P. D., Barnes, I., Roberts, J. M., Melamed, M. L., Ammann, M., Bertram, A. K., Cappa, C. D., Carlton, A. G., Carpenter, L. J., Crowley, J. N., Dubowski, Y., Georges, C., Heard, D. E., Herrmann, H., Keutsch, F. N., Kroll, J. H., McNeill, V. F., Ng, N. L., Nizkorodov, S. A., Orlando, J. J., Percival, C. J., Picquet-Varrault, B., Rudich, Y., Seakins, P. W., Surratt, J. D., Tanimoto, H., Thornton, J. A., Tong, Z., Tyndall, G. S., Wahner, A., Weschler, C. J., Wilson, K. R., and Ziemann, P. J.: The Essential Role for Laboratory Studies in Atmospheric Chemistry, Environ. Sci. Technol., 51, 2519–2528, https://doi.org/10.1021/acs.est.6b04947, 2017.
Canagaratna, M. R., Jayne, J. T., Jimenez, J. L., Allan, J. D., Alfarra, M. R., Zhang, Q., Onasch, T. B., Drewnick, F., Coe, H., Middlebrook, A., Delia, A., Williams, L. R., Trimborn, A. M., Northway, M. J., DeCarlo, P. F., Kolb, C. E., Davidovits, P., and Worsnop, D. R.: Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 26, 185–222, https://doi.org/10.1002/mas.20115, 2007.
Chen, T. Z., Zhang, P., Ma, Q. X., Chu, B. W., Liu, J., Ge, Y. L., and He, H.: Smog Chamber Study on the Role of NOx in SOA and O3 Formation from Aromatic Hydrocarbons, Environ. Sci. Technol., https://doi.org/10.1021/acs.est.2c04022, 2022.
Clapp, L. J. and Jenkin, M. E.: Analysis of the relationship between ambient levels Of O3, NO2 and NO as a function of NOx in the UK, Atmos. Environ., 35, 6391–6405, https://doi.org/10.1016/s1352-2310(01)00378-8, 2001.
Dash, M. R., Balaganesh, M., and Rajakumar, B.: Rate coefficients for the gas-phase reaction of OH radical with α-pinene: an experimental and computational study, Mol. Phys., 112, 1495–1511, https://doi.org/10.1080/00268976.2013.840395, 2014.
Dibble, T. S.: Reactions of the Alkoxy Radicals Formed Following OH-Addition to α-Pinene and β-Pinene. C–C Bond Scission Reactions, J. Am. Chem. Soc., 123, 4228–4234, https://doi.org/10.1021/ja003553i, 2001.
Drewnick, F., Hings, S. S., Alfarra, M. R., Prevot, A. S. H., and Borrmann, S.: Aerosol quantification with the Aerodyne Aerosol Mass Spectrometer: detection limits and ionizer background effects, Atmos. Meas. Tech., 2, 33–46, https://doi.org/10.5194/amt-2-33-2009, 2009.
Ehn, M., Thornton, J. A., Kleist, E., Sipila, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurten, T., Nielsen, L. B., Jorgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petaja, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Gao, L., Song, J., Mohr, C., Huang, W., Vallon, M., Jiang, F., Leisner, T., and Saathoff, H.: Kinetics, SOA yields, and chemical composition of secondary organic aerosol from β-caryophyllene ozonolysis with and without nitrogen oxides between 213 and 313 K, Atmos. Chem. Phys., 22, 6001–6020, https://doi.org/10.5194/acp-22-6001-2022, 2022.
Goldman, M. J., Green, W. H., and Kroll, J. H.: Chemistry of Simple Organic Peroxy Radicals under Atmospheric through Combustion Conditions: Role of Temperature, Pressure, and NOx Level, J. Phys. Chem. A, 125, 10303–10314, https://doi.org/10.1021/acs.jpca.1c07203, 2021.
Goss, M. B., Kenagy, H. S., Heald, C. L., and Kroll, J. H.: Re-Examining Chemical Conditions of Past Chamber Studies of Secondary Organic Aerosol Formation, ACS ES&T Air, 2, 2117–2130, https://doi.org/10.1021/acsestair.5c00112, 2025.
Gu, S., Guenther, A., and Faiola, C.: Effects of Anthropogenic and Biogenic Volatile Organic Compounds on Los Angeles Air Quality, Environ. Sci. Technol., 55, 12191–12201, https://doi.org/10.1021/acs.est.1c01481, 2021.
Guenther, A., Hewitt, C. N., Erickson, D., Fall, R., Geron, C., Graedel, T., Harley, P., Klinger, L., Lerdau, M., Mckay, W. A., Pierce, T., Scholes, B., Steinbrecher, R., Tallamraju, R., Taylor, J., and Zimmerman, P.: A global model of natural volatile organic compound emissions, J. Geophys. Res., 100, 8873–8892, https://doi.org/10.1029/94JD02950, 1995.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Jensen, A. R., Koss, A. R., Hales, R. B., and de Gouw, J. A.: Measurements of volatile organic compounds in ambient air by gas-chromatography and real-time Vocus PTR-TOF-MS: calibrations, instrument background corrections, and introducing a PTR Data Toolkit, Atmos. Meas. Tech., 16, 5261–5285, https://doi.org/10.5194/amt-16-5261-2023, 2023.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, E. J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Johnson, D. and Marston, G.: The gas-phase ozonolysis of unsaturated volatile organic compounds in the troposphere, Chem. Soc. Rev., 37, 699–716, https://doi.org/10.1039/B704260B, 2008.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kang, S., Wildt, J., Pullinen, I., Vereecken, L., Wu, C., Wahner, A., Zorn, S. R., and Mentel, T. F.: Formation of highly oxygenated organic molecules from α-pinene photooxidation: evidence for the importance of highly oxygenated alkoxy radicals, Atmos. Chem. Phys., 25, 15715–15740, https://doi.org/10.5194/acp-25-15715-2025, 2025.
Kenagy, H. S., Heald, C. L., Tahsini, N., Goss, M. B., and Kroll, J. H.: Can we achieve atmospheric chemical environments in the laboratory? An integrated model-measurement approach to chamber SOA studies, Sci. Adv., 10, eado1482, https://doi.org/10.1126/sciadv.ado1482, 2024.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Lane, T. E., Donahue, N. M., and Pandis, S. N.: Effect of NOx on Secondary Organic Aerosol Concentrations, Environ. Sci. Technol., 42, 6022–6027, https://doi.org/10.1021/es703225a, 2008.
Lannuque, V., D'Anna, B., Kostenidou, E., Couvidat, F., Martinez-Valiente, A., Eichler, P., Wisthaler, A., Müller, M., Temime-Roussel, B., Valorso, R., and Sartelet, K.: Gas–particle partitioning of toluene oxidation products: an experimental and modeling study, Atmos. Chem. Phys., 23, 15537–15560, https://doi.org/10.5194/acp-23-15537-2023, 2023.
Lee, A., Goldstein, A. H., Kroll, J. H., Ng, N. L., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Gas-phase products and secondary aerosol yields from the photooxidation of 16 different terpenes, J. Geophys. Res. Atmos., 111, https://doi.org/10.1029/2006jd007050, 2006.
Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Kurtén, T., Worsnop, D. R., and Thornton, J. A.: An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 48, 6309–6317, https://doi.org/10.1021/es500362a, 2014.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, Th. F., Lutz, A., Hallquist, M., Worsnop, D., and Thornton, J. A.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lu, Y. and Khalil, M. A. K.: Methane and carbon-monoxide in OH chemistry - The effects of feedbacks and reservoirs generated by the reactive products, Chemosphere, 26, 641–655, https://doi.org/10.1016/0045-6535(93)90450-j, 1993.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D. F., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, A. M., Simpson, D., Bergstrom, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Nah, T., McVay, R. C., Pierce, J. R., Seinfeld, J. H., and Ng, N. L.: Constraining uncertainties in particle-wall deposition correction during SOA formation in chamber experiments, Atmos. Chem. Phys., 17, 2297–2310, https://doi.org/10.5194/acp-17-2297-2017, 2017.
Okada, Y., Nakagoshi, A., Tsurukawa, M., Matsumura, C., Eiho, J., and Nakano, T.: Environmental risk assessment and concentration trend of atmospheric volatile organic compounds in Hyogo Prefecture, Japan, Environ. Sci. Pollut. Res., 19, 201–213, https://doi.org/10.1007/s11356-011-0550-0, 2012.
Orlando, J. J., Tyndall, G. S., and Wallington, T. J.: The Atmospheric Chemistry of Alkoxy Radicals, Chem. Rev., 103, 4657–4690, https://doi.org/10.1021/cr020527p, 2003.
Peräkylä, O., Berndt, T., Franzon, L., Hasan, G., Meder, M., Valiev, R. R., Daub, C. D., Varelas, J. G., Geiger, F. M., Thomson, R. J., Rissanen, M., Kurtén, T., and Ehn, M.: Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 145, 7780–7790, https://doi.org/10.1021/jacs.2c10398, 2023.
Pospisilova, V., Lopez-Hilfiker, F. D., Bell, D. M., El Haddad, I., Mohr, C., Huang, W., Heikkinen, L., Xiao, M., Dommen, J., Prevot, A. S. H., Baltensperger, U., and Slowik, J. G.: On the fate of oxygenated organic molecules in atmospheric aerosol particles, Sci. Adv., 6, eaax8922, https://doi.org/10.1126/sciadv.aax8922, 2020.
Presto, A. A., Huff Hartz, K. E., and Donahue, N. M.: Secondary Organic Aerosol Production from Terpene Ozonolysis. 1. Effect of UV Radiation, Environ. Sci. Technol., 39, 7036–7045, https://doi.org/10.1021/es050174m, 2005.
Pullinen, I., Schmitt, S., Kang, S., Sarrafzadeh, M., Schlag, P., Andres, S., Kleist, E., Mentel, T. F., Rohrer, F., Springer, M., Tillmann, R., Wildt, J., Wu, C., Zhao, D., Wahner, A., and Kiendler-Scharr, A.: Impact of NOx on secondary organic aerosol (SOA) formation from α-pinene and β-pinene photooxidation: the role of highly oxygenated organic nitrates, Atmos. Chem. Phys., 20, 10125–10147, https://doi.org/10.5194/acp-20-10125-2020, 2020.
Pusede, S. E., Steiner, A. L., and Cohen, R. C.: Temperature and Recent Trends in the Chemistry of Continental Surface Ozone, Chem. Rev., 115, 3898–3918, https://doi.org/10.1021/cr5006815, 2015.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Ramanathan, V., Crutzen, P. J., Kiehl, J. T., and Rosenfeld, D.: Atmosphere – Aerosols, climate, and the hydrological cycle, Science, 294, 2119–2124, https://doi.org/10.1126/science.1064034, 2001.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: Semivolatile emissions and photochemical aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Sarrafzadeh, M., Wildt, J., Pullinen, I., Springer, M., Kleist, E., Tillmann, R., Schmitt, S. H., Wu, C., Mentel, T. F., Zhao, D., Hastie, D. R., and Kiendler-Scharr, A.: Impact of NOx and OH on secondary organic aerosol formation from β-pinene photooxidation, Atmos. Chem. Phys., 16, 11237–11248, https://doi.org/10.5194/acp-16-11237-2016, 2016.
Shao, Y., Wang, Y., Du, M., Voliotis, A., Alfarra, M. R., O'Meara, S. P., Turner, S. F., and McFiggans, G.: Characterisation of the Manchester Aerosol Chamber facility, Atmos. Meas. Tech., 15, 539–559, https://doi.org/10.5194/amt-15-539-2022, 2022.
Shilling, J. E., Zaveri, R. A., Fast, J. D., Kleinman, L., Alexander, M. L., Canagaratna, M. R., Fortner, E., Hubbe, J. M., Jayne, J. T., Sedlacek, A., Setyan, A., Springston, S., Worsnop, D. R., and Zhang, Q.: Enhanced SOA formation from mixed anthropogenic and biogenic emissions during the CARES campaign, Atmos. Chem. Phys., 13, 2091–2113, https://doi.org/10.5194/acp-13-2091-2013, 2013.
Shrivastava, M., Cappa, C. D., Fan, J. W., Goldstein, A. H., Guenther, A. B., Jimenez, J. L., Kuang, C., Laskin, A., Martin, S. T., Ng, N. L., Petaja, T., Pierce, J. R., Rasch, P. J., Roldin, P., Seinfeld, J. H., Shilling, J., Smith, J. N., Thornton, J. A., Volkamer, R., Wang, J., Worsnop, D. R., Zaveri, R. A., Zelenyuk, A., and Zhang, Q.: Recent advances in understanding secondary organic aerosol: Implications for global climate forcing, Rev. Geophys., 55, 509–559, https://doi.org/10.1002/2016rg000540, 2017.
Srivastava, D., Vu, T. V., Tong, S., Shi, Z., and Harrison, R. M.: Formation of secondary organic aerosols from anthropogenic precursors in laboratory studies, npj Clim. Atmos. Sci., 5, 22, https://doi.org/10.1038/s41612-022-00238-6, 2022.
Stone, E. A., Hedman, C. J., Zhou, J., Mieritz, M., and Schauer, J. J.: Insights into the nature of secondary organic aerosol in Mexico City during the MILAGRO experiment 2006, Atmos. Environ., 44, 312–319, https://doi.org/10.1016/j.atmosenv.2009.10.036, 2010.
Takeuchi, M., Berkemeier, T., Eris, G., and Ng, N. L.: Non-linear effects of secondary organic aerosol formation and properties in multi-precursor systems, Nat. Commun., 13, https://doi.org/10.1038/s41467-022-35546-1, 2022.
Tsigaridis, K., Daskalakis, N., Kanakidou, M., Adams, P. J., Artaxo, P., Bahadur, R., Balkanski, Y., Bauer, S. E., Bellouin, N., Benedetti, A., Bergman, T., Berntsen, T. K., Beukes, J. P., Bian, H., Carslaw, K. S., Chin, M., Curci, G., Diehl, T., Easter, R. C., Ghan, S. J., Gong, S. L., Hodzic, A., Hoyle, C. R., Iversen, T., Jathar, S., Jimenez, J. L., Kaiser, J. W., Kirkevåg, A., Koch, D., Kokkola, H., Lee, Y. H., Lin, G., Liu, X., Luo, G., Ma, X., Mann, G. W., Mihalopoulos, N., Morcrette, J.-J., Müller, J.-F., Myhre, G., Myriokefalitakis, S., Ng, N. L., O'Donnell, D., Penner, J. E., Pozzoli, L., Pringle, K. J., Russell, L. M., Schulz, M., Sciare, J., Seland, Ø., Shindell, D. T., Sillman, S., Skeie, R. B., Spracklen, D., Stavrakou, T., Steenrod, S. D., Takemura, T., Tiitta, P., Tilmes, S., Tost, H., van Noije, T., van Zyl, P. G., von Salzen, K., Yu, F., Wang, Z., Wang, Z., Zaveri, R. A., Zhang, H., Zhang, K., Zhang, Q., and Zhang, X.: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 14, 10845–10895, https://doi.org/10.5194/acp-14-10845-2014, 2014.
Vereecken, L. and Peeters, J.: Nontraditional (Per)oxy Ring-Closure Paths in the Atmospheric Oxidation of Isoprene and Monoterpenes, J. Phys. Chem. A, 108, 5197–5204, https://doi.org/10.1021/jp049219g, 2004.
Voliotis, A., Wang, Y., Shao, Y., Du, M., Bannan, T. J., Percival, C. J., Pandis, S. N., Alfarra, M. R., and McFiggans, G.: Exploring the composition and volatility of secondary organic aerosols in mixed anthropogenic and biogenic precursor systems, Atmos. Chem. Phys., 21, 14251–14273, https://doi.org/10.5194/acp-21-14251-2021, 2021.
Voliotis, A., Du, M., Wang, Y., Shao, Y., Alfarra, M. R., Bannan, T. J., Hu, D., Pereira, K. L., Hamilton, J. F., Hallquist, M., Mentel, T. F., and McFiggans, G.: Chamber investigation of the formation and transformation of secondary organic aerosol in mixtures of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 22, 14147–14175, https://doi.org/10.5194/acp-22-14147-2022, 2022a.
Voliotis, A., Du, M., Wang, Y., Shao, Y., Bannan, T. J., Flynn, M., Pandis, S. N., Percival, C. J., Alfarra, M. R., and McFiggans, G.: The influence of the addition of isoprene on the volatility of particles formed from the photo-oxidation of anthropogenic–biogenic mixtures, Atmos. Chem. Phys., 22, 13677–13693, https://doi.org/10.5194/acp-22-13677-2022, 2022b.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, https://doi.org/10.1029/2006GL026899, 2006.
Wang, N., Jorga, S. D., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Particle wall-loss correction methods in smog chamber experiments, Atmos. Meas. Tech., 11, 6577–6588, https://doi.org/10.5194/amt-11-6577-2018, 2018.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017.
Zhang, X., Schwantes, R. H., Coggon, M. M., Loza, C. L., Schilling, K. A., Flagan, R. C., and Seinfeld, J. H.: Role of ozone in SOA formation from alkane photooxidation, Atmos. Chem. Phys., 14, 1733–1753, https://doi.org/10.5194/acp-14-1733-2014, 2014.
Zhao, D., Schmitt, S. H., Wang, M., Acir, I.-H., Tillmann, R., Tan, Z., Novelli, A., Fuchs, H., Pullinen, I., Wegener, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Effects of NOx and SO2 on the secondary organic aerosol formation from photooxidation of α-pinene and limonene, Atmos. Chem. Phys., 18, 1611–1628, https://doi.org/10.5194/acp-18-1611-2018, 2018.
Zhao, Y. L., Nguyen, N. T., Presto, A. A., Hennigan, C. J., May, A. A., and Robinson, A. L.: Intermediate Volatility Organic Compound Emissions from On-Road Diesel Vehicles: Chemical Composition, Emission Factors, and Estimated Secondary Organic Aerosol Production, Environ. Sci. Technol., 49, 11516–11526, https://doi.org/10.1021/acs.est.5b02841, 2015.
Zhou, C., Jang, M., and Yu, Z.: Simulation of SOA formation from the photooxidation of monoalkylbenzenes in the presence of aqueous aerosols containing electrolytes under various NOx levels, Atmos. Chem. Phys., 19, 5719–5735, https://doi.org/10.5194/acp-19-5719-2019, 2019.
Ziemann, P. J. and Atkinson, R.: Kinetics, products, and mechanisms of secondary organic aerosol formation, Chem. Soc. Rev., 41, 6582–6605, https://doi.org/10.1039/c2cs35122f, 2012.