the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Jul 2026
| 02 Jul 2026
Interface-dominated hydroxymethanesulfonate and its isomer formation provides key mechanisms for reconciling the atmospheric sulfur budget gap in polluted and cold environments
Yang Liu
Xiaohua Yang
Yuchen Zhang
Hydroxymethanesulfonate (HMS) is a critical source of particulate sulfur, formed by formaldehyde (HCHO) and sulfur dioxide (SO2) in droplets. Current models relying on bulk aqueous-phase HMS formation only explain ∼ one-third of unexplained sulfate concentrations, leaving gaps in atmospheric sulfur budget, especially in polluted and cold environments. Using Born–Oppenheimer molecular dynamics simulations, we explored HMS and its isomer hydroxymethyl sulfite (HMSi) formation mechanisms across aqueous phase and air–water/ice interfaces. Air–water interfaces enable nearly barrierless HMS formation (0.6 kcal mol−1) via unique stepwise water-mediated proton transfer, preferring HMS over HMSi (0.6 vs. 6.1 kcal mol−1), which contrasts sharply with the competitive pathways observed in the bulk aqueous phase (7.7 vs. 7.6 kcal mol−1). In contrast, protonation of formaldehyde under strongly acidic conditions reverses reaction selectivity, favoring HMSi formation over HMS. Importantly, these reaction mechanisms remain viable at air–ice interfaces in cold environments including polar areas and the upper troposphere, revealing ice surfaces as previously overlooked yet significant sites for atmospheric organosulfate formation. Our findings suggest that interfacial mechanisms may provide efficient pathways for HMS and HMSi formation in both polluted and cold environments, helping to reconcile model-observation discrepancies in the atmospheric sulfur budget.
- Article
(6487 KB) - Full-text XML
-
Supplement
(2501 KB) - BibTeX
- EndNote




Atmospheric fine particulate matter (PM) impacts human health, visibility, ecosystems, and global climate (Zhang et al., 2015). These fine particles can exist not only as liquid aerosol droplets but also transform into ice crystals under cold atmospheric conditions (Knopf et al., 2018). Among these constituents, sulfates represent critical species driving atmospheric particle formation (Sipila et al., 2010; Zhang, 2010; Zhong et al., 2019). Despite decades of research, a persistent discrepancy between modeled and observed sulfate concentrations remains, particularly in polluted, strongly acidic aerosol environments such as winter haze episodes in northern China and remote cold regions (Wang et al., 2014, 2016, 2025; Campbell et al., 2024). This gap reveals critical deficiencies in our understanding of atmospheric sulfur cycling, particularly regarding organosulfur intermediates that may contribute significantly to sulfate formation.
Hydroxymethane sulfonate (HMS), formed from the reaction of formaldehyde (HCHO) and sulfur dioxide (SO2) in cloud droplets (Reactions R1–R3), serves as an important tracer for cloud and fog processing (Wei et al., 2020). However, HMS has often been overlooked as a potential contributor to atmospheric sulfur budgets, primarily because common aerosol measurement techniques (e.g., commercial aerosol mass spectrometry) frequently misidentify HMS as sulfate, thereby compromising the accuracy of sulfate simulation models (Moch et al., 2018). With advances in analytical techniques, recent field studies have detected HMS in diverse environments worldwide, including urban areas such as Beijing, tropical regions such as Singapore, remote cold regions such as Alaska, and marine settings (Campbell et al., 2022; Moch et al., 2020; Song et al., 2019; Zhao et al., 2024). HMS impacts atmospheric chemistry not only through its widespread presence but also via its subsequent chemical transformations to sulfate (Lai et al., 2023, 2024). Multiple studies have demonstrated that HMS formation can substantially contribute to ambient sulfate concentrations (Huang et al., 2023; Zhang et al., 2024b). These findings highlight the potentially significant contribution of HMS to atmospheric sulfur chemistry. Song et al. (2019) estimated that HMS could account for approximately one-third of the discrepancy between model-simulated and observed sulfate in haze events.
Nevertheless, current studies on the HCHO and SO2 reaction mechanisms suffer from critical limitations that hinder the closure of the sulfur budget gap, especially in polluted and cold atmospheric environments. Chen and Zhao (2020) employed density functional theory calculations with implicit solvent models to study aqueous-phase reactions between HCHO and bisulfite (HOSO). Their study revealed that this reaction can produce hydroxymethyl sulfite (HMSi, Reaction R4), a structural isomer of HMS with distinct sulfur-oxygen binding, as a competitive product in aqueous-phase (Chen and Zhao, 2020). However, implicit solvent models, which treat the solvent as a structureless continuum, inherently lack the molecular-level detail necessary to capture subtle mechanistic variations and dynamic solvent-solute interactions (Zhang et al., 2017). In contrast, explicit solvent models allow reactive intermediates and transition states to interact directly with individual solvent molecules, fully capturing the dynamic influence of solvation on reaction pathways (Norjmaa et al., 2021). This gap in molecular-level understanding of HMS/HMSi aqueous formation mechanisms remains a nonnegligible barrier to accurately modeling atmospheric sulfur cycling.
In addition to bulk aqueous phase, air-water interfaces represent ubiquitous yet fundamentally distinct reactive environments in atmospheric systems, including cloud droplets, aerosols, and sea-salt particles. The molecular environment at these interfaces differs significantly from bulk aqueous phases, characterized by unique hydrogen bonding networks that can enhance the adsorption and condensation of atmospheric species onto aerosols (Li et al., 2021; Ruiz-Lopez et al., 2020). Notably, both HCHO and bisulfite (HOSO) demonstrate preferential accumulation at air-water interfaces (Martins-Costa et al., 2012; Yang et al., 2019), creating localized concentration enhancements and reactive microenvironments. These distinct interfacial properties may substantially enhance reaction kinetics or facilitate entirely novel reaction mechanisms compared to bulk solution (Ning et al., 2023, 2024). Despite their potential significance, a systematic understanding of how HMS and HMSi formation mechanisms differ between bulk and interfacial environments and whether reaction selectivity toward HMS versus HMSi varies at air-water interfaces, remains absent. This knowledge gap directly limits the accuracy of organosulfur formation representations in atmospheric models.
Additionally, the chemical complexity introduced by extreme aerosol acidity (pH) presents another critical knowledge gap. Based on global surface-layer aerosol pH estimated by Li et al. (2022) using annual-mean (2016) GEOS-Chem simulations coupled with E-AIM thermodynamic calculations, aerosol pH in the range of −1 to 1 occurs over at least ∼ 20 % of the global surface area (Li et al., 2022) where the model successfully converges (RH ≥ 60 % and T ≥ 263.15 K). These highly acidic regions span diverse environments including marine and continental systems (Angle et al., 2021; Ding et al., 2019; Jia et al., 2020; Kakavas et al., 2021; Pye et al., 2018; Zheng et al., 2020; Zhou et al., 2022). These highly acidic environments trigger fundamental changes in formaldehyde speciation, leading to the formation of protonated formaldehyde (HCHOH+). Experimental evidence (Jayne et al., 1996) has demonstrated that HCHOH+ can exist and accumulate at droplet surfaces under such acidic conditions, establishing its potential significance in atmospheric reaction. Despite this potential significance, the interfacial chemistry of protonated formaldehyde with sulfur species remains uncharacterized. Hence, the contributions of HMS and HMSi to atmospheric sulfur budgets in these acidic environments also remain poorly understood.
In addition to air-water interfaces, ice crystals exhibit high surface-to-volume ratios, providing extensive reactive interfaces that play crucial roles in atmospheric chemistry (George et al., 2015; Kerbrat et al., 2008; Zhong et al., 2020). Notably, HCHO and SO2, the precursors to HMS and HMSi, are ubiquitous in cold regions (Sumner and Shepson, 1999; Anderson et al., 2017; Höpfner et al., 2015; Joppe et al., 2024). However, it remains unclear whether air–ice interfaces favor HMS or HMSi formation. If ice surfaces do facilitate HMS or HMSi formation, current models, which overlook the organosulfur chemistry on ice surface, are likely to substantially underestimate their contributions to atmospheric sulfur budgets.
To address these critical knowledge gaps, we employed Born–Oppenheimer molecular dynamics (BOMD) simulations with metadynamics enhancement and high-level quantum chemical calculations to elucidate HMS and HMSi formation mechanisms in polluted and cold environments. Our specific objectives are: (i) to reveal aqueous-phase reaction mechanisms with explicit solvation, focusing on the competitive HMS/HMSi formation pathways; (ii) to clarify the formation mechanisms and selectivity rules of HMS/HMSi at the air-water interface; (iii) to determine how protonated formaldehyde alters reaction pathways and selectivity in strongly acidic environments; (iv) to assess the feasibility of HMS and HMSi formation at the air–ice interface at temperatures characteristic of polar regions and the upper troposphere; (v) to systematically characterize the interfacial dynamics of HMS and HMSi products, including their hydrogen bonding networks, orientational preferences, and implications for atmospheric oxidation. By integrating these investigations, we aim to reveal interfacial sulfur cycling mechanisms, reconcile model-observation discrepancies, and improve organosulfur chemistry representations in global atmospheric models.
2.1 Quantum Chemistry Calculations
Geometry optimizations of stationary points (reactants, products, and transition states) were performed at the B3LYP/6-311+G(d,p) level (Krishnan et al., 1980; McLean and Chandler, 1980; Stephens et al., 1994) with the DFT-D3 dispersion correction of Grimme et al. (2010) using Gaussian 16 package (Frisch et al., 2016). Harmonic frequency analyses were conducted at the same level. The transition states are verified to be connected with the corresponding reactants or products by means of intrinsic reaction coordinate (IRC) calculations. The solvation model based on density implicit solvent model (SMD) was applied to represent the aqueous phase. To obtain more accurate electronic energies, single-point energy calculations were subsequently performed at the DLPNO-CCSD(T) level of theory employing the aug-cc-pVTZ basis set (Guo et al., 2018; Riplinger and Neese, 2013; Riplinger et al., 2013). These calculations were carried out using the ORCA 5.0.4 quantum chemistry program package (Neese, 2011). Fukui function analysis evaluated the nucleophilic reactivity of atoms within the HOSO anion. Electron density difference maps were analyzed to reveal charge transfer between reactant fragments. All wave function analyses were performed by Multiwfn 3.8 (dev) code (Lu and Chen, 2012, 2023; Zhang and Lu, 2021).
2.2 Born–Oppenheimer Molecular Dynamics Simulations
Born–Oppenheimer molecular dynamics (BOMD) simulations were performed using the CP2K package (Kuhne et al., 2020). All BOMD simulations employed the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional (Perdew et al., 1996) with the DFT-D3 dispersion corrections of Grimme et al. (2010). This D3 correction has been shown to improve the description of water at the water–air interface at the GGA level (Dodia et al., 2019). A double-ζ Gaussian basis set (DZVP-MOLOPT-SR-GTH) was used for valence electrons in conjunction with Goedecker–Teter–Hutter (GTH) pseudopotentials for core electrons (Goedecker et al., 1996; VandeVondele and Hutter, 2007). The plane wave and Gaussian basis set cutoff energies were set to 300 and 40 Ry, respectively. The simulations were conducted in the canonical (NVT) ensemble using the canonical sampling through velocity rescaling (CSVR) thermostat (Bussi et al., 2007) with a time step of 1 fs. For aqueous-phase simulations, a cubic box of 1.7 nm × 1.7 nm × 1.7 nm containing 128 water molecules was employed. The air–water interface model was constructed by expanding the z-axis of this cubic box to 4.7 nm while keeping the xy-plane dimensions (1.7 nm × 1.7 nm) and water molecules (128) unchanged, which generated vacuum regions above and below the aqueous slab to establish two air–water interfaces. Free energy profiles were computed via well-tempered metadynamics (see Sect. S1 in the Supplement for details) as implemented in PLUMED (Bonomi et al., 2009). Collective variables for HMS and HMSi formation are defined in Fig. S1 in the Supplement. Convergence was assessed by monitoring Gaussian hill height evolution (Fig. S2), collective variable sampling behavior (Fig. S3), and the time evolution of free energy profiles (Fig. S4). Detailed configurations for air–ice interface simulations are provided in the Supplement (Fig. S5).
3.1 Bulk-Phase Mechanisms with Explicit Solvation
Bulk aqueous reactions represent an important pathway for HCHO–HOSO interactions in atmospheric chemistry. Although previous DFT studies employing implicit solvation models have provided valuable understanding of this reaction (Chen and Zhao, 2020), explicit solvation approaches offer complementary insights by capturing discrete water-reactant interactions and dynamic hydrogen bonding that may play important roles in determining reaction mechanisms and kinetics. To provide a more detailed molecular-level picture of bulk aqueous HMS formation, we employed BOMD simulations with metadynamics to investigate the HCHO–HOSO reaction with explicit water molecules.
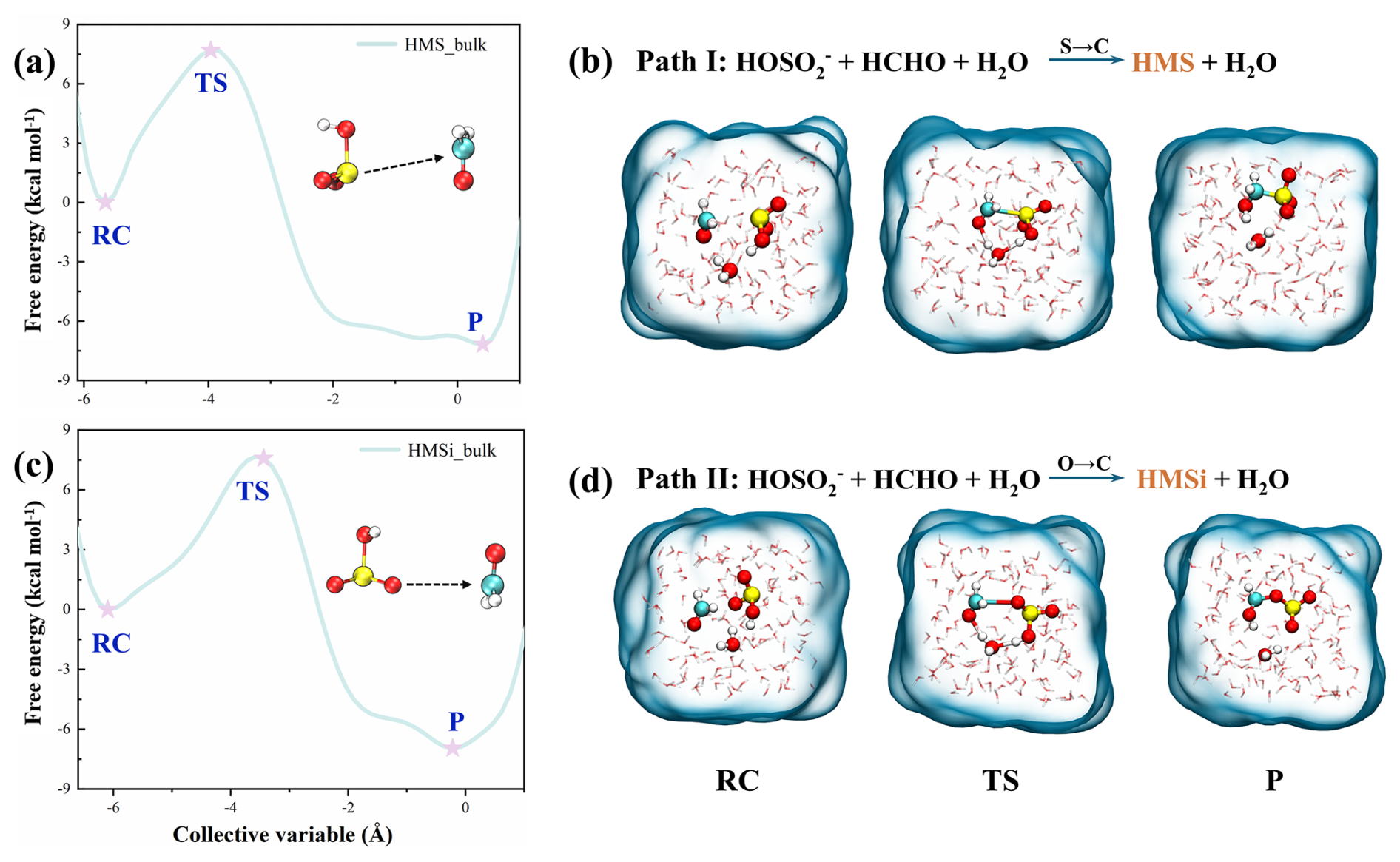
Figure 1Snapshot structures captured from the metadynamics-biased BOMD simulations of HCHO and HOSO reaction in the bulk phase. (a) Free energy profile for Path I. (b) Snapshot structures along Path I. (c) Free energy profile for Path II. (d) Snapshot structures along Path II. RC, TS, and P denote the reactant complexes, transition states, and products, respectively.
Given that HOSO contains multiple potential nucleophilic sites (sulfur, non-hydroxyl oxygens, and hydroxyl oxygen), we first assessed their relative reactivities through preliminary quantum chemical calculations (Fig. S6). These analyses confirmed that the hydroxyl oxygen O(H) in HOSO exhibits significantly lower nucleophilic reactivity compared to the sulfur atom and non-hydroxyl oxygen atoms. Furthermore, reactions involving the O(H) did not yield viable products. Based on these findings, reactions involving the O(H) were excluded from subsequent metadynamics simulations. As illustrated in Fig. 1, two distinct reaction pathways dominate the bulk-phase HCHO + HOSO reaction. In Path I (Fig. 1b), the sulfur atom of the HOSO attacks the carbon atom of formaldehyde, forming a seven-membered ring transition state with the participation of a water molecule. The calculated free energy barrier for the formation of HMS is 7.7 kcal mol−1. Path II (Fig. 1d) proceeds via nucleophilic attack of a non-hydroxyl oxygen atom of HOSO on the carbonyl carbon atom of HCHO. This pathway proceeds through a ring transition state mediated by water molecules (Fig. 1d). The free energy barrier for HMSi formation via this route is 7.6 kcal mol−1. These values are in close agreement with barriers derived from high-level quantum chemical calculations (Fig. S7), validating the reliability of the simulation results. The comparable barriers for HMS and HMSi formation in bulk water (7.7 vs. 7.6 kcal mol−1) indicate that both isomers form competitively in bulk water. Despite these moderate free energy barriers, bulk phase HMS formation alone is insufficient to reconcile the atmospheric sulfur budget gap. Atmospheric modeling studies incorporating bulk-phase HMS chemistry can account for only approximately one-third of the discrepancy between modeled and observed sulfate concentrations during severe haze episodes (Song et al., 2019). This shortfall may be partly attributed to the high surface-to-volume ratios of atmospheric aerosols and cloud droplets, where interfacial processes may play a more significant role than bulk-phase reactions.
3.2 Rapid and Selective HMS Formation at the Air-Water Interface
Given the limitations of bulk-phase chemistry identified above, understanding HCHO-HOSO reactivity at air–water interfaces becomes critical. Recent investigations reveal significant accumulation of both HCHO and HOSO at the air-water interface (Martins-Costa et al., 2012; Yang et al., 2019). This interfacial enrichment suggests potentially significant atmospheric reaction mechanisms previously overlooked. Herein, we employed BOMD simulations to investigate these interfacial heterogeneous reactions at the air-water interface.
Similar to bulk-phase reactions, interfacial processes proceed via two distinct pathways. Remarkably, HMS formation via Path I proceeds rapidly at the air–water interface. Starting from reactant complex configurations optimized at the B3LYP-D3/6-311+G(d,p) level, unbiased BOMD trajectories reveal that the reaction completes within ∼ 3 ps (Figs. 2a, S8), in contrast to the bulk-phase reaction. During the reaction, the distance between the C atom of HCHO and the S atom of HOSO gradually decreases to approximately 1.95 Å (Fig. 2d, e), with a corresponding Mayer bond order of 0.57, indicating that an S–C bonding interaction has been established at this intermediate stage. The resulting anionic intermediate structurally resembles HMS, differing only in proton location, which remains on the O atom of the HOSO. Electron density difference analysis demonstrates decreased electron density surrounding the S atom with concurrent increase at the formaldehyde O atom during intermediate formation (Fig. 2f). Mulliken population analysis quantifies this charge transfer: the charge on S atom in HOSO increases from approximately 0.6 to 0.81 e, while the charge on O atom in HCHO decreases from approximately −0.3 to −0.57 e (Fig. S9). These analyses can provide clear evidence for electron transfer from HOSO to HCHO in the initial nucleophilic addition step. Subsequently, at approximately 2.42 ps, a transition state structure forms wherein the O2 atom of proximal H2O approaches the H2 atom of the reaction intermediate, while the H1–O2 bond in H2O lengthens. Interfacial water molecules facilitate the final step of the reaction, completing HMS formation while shortening the S–C bond from approximately 1.95 to 1.81 Å (Fig. 2g). Unlike the bulk-phase reaction, where S–C bond formation and proton transfer occur synchronously (Fig. 1b), the interfacial HMS formation proceeds via a stepwise mechanism: the S–C bond forms initially, followed by water molecule-mediated proton transfer from HOSO.
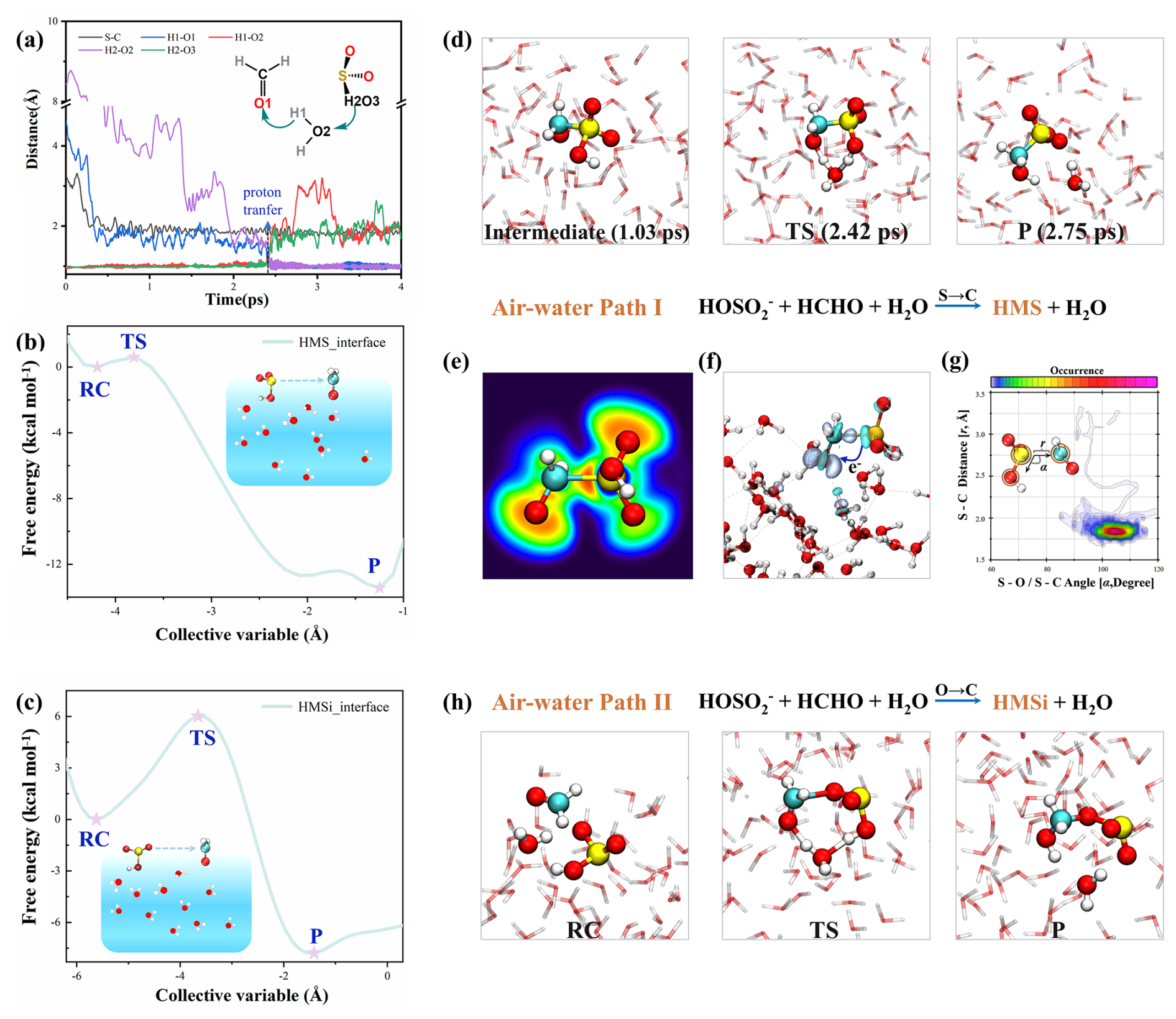
Figure 2Mechanistic investigation of HCHO and HOSO at the air-water interface. (a) Time evolution of key bond distances during HMS formation, with corresponding structural snapshots shown in (d). (b) Free energy profile for the HMS formation via Path I. (c) Free energy profile for the HMSi formation via Path II, with representative structural snapshots presented in (h). (e) Color-mapped electron localization function analysis illustrating bond reorganization in key intermediates during HMS formation. (f) Electron density difference (Δρ) of the reaction intermediates (gray region: Δρ>0; cyan region: Δρ<0). (g) Combined distribution functions involving the bond distance (r, Å) and angular distribution function (α, degree) for HCHO and HOSO.
To obtain quantitative free energy profiles and systematically explore the interfacial reaction pathways, we performed computationally expensive metadynamics-biased BOMD simulations. Metadynamics simulations reveal an exceptionally low free energy barrier of 0.6 kcal mol−1 for interfacial HMS formation (Fig. 2b), corroborating the fast reaction observed in unbiased trajectories. We validated our BOMD findings using high-level quantum chemical calculations using a ten-water-molecule cluster model (W10) at the DLPNO-CCSD(T)/aug-cc-pVTZ//B3LYP-D3/6-311+G(d,p) level. These high-level calculations further validated the near-barrierless HMS formation, providing complementary evidence to our metadynamics simulations (Fig. S10).
Our results differ from Li et al. (2025)'s recent report of a ∼ 7.6 kcal mol−1 free energy barrier for this process. This discrepancy stems from their use of a single distance collective variable d(C⋯S) in metadynamics simulations, which overlooked the critical water-mediated cyclic proton transfer mechanism. In contrast to the rapid formation of HMS, no fast formation of HMSi was observed across multiple simulation trajectories. Metadynamics simulations revealed a higher free energy barrier of 6.1 kcal mol−1 for HMSi formation at the interface (Fig. 2c), with the reaction mechanism paralleling that observed in the bulk phase. Although this barrier represents a 1.5 kcal mol−1 reduction from the bulk-phase value (7.6 kcal mol−1), it remains substantially higher than the negligible barrier for HMS formation.
To elucidate the potential contributor to the enhanced interfacial reactivity, we analyzed the solvation environments of HCHO and HOSO at the interface versus bulk water (Fig. S11). The partial solvation characteristic of the air-water interface results in reduced water coordination around both reactants relative to their bulk-phase counterparts, potentially facilitating reactant approach. Furthermore, the HOMO–LUMO gap of the reactive complex is smaller at the interface than in bulk water, suggesting that the partially solvated environment may contribute to the reaction acceleration (Fig. S12) (Ishiyama et al., 2022; Kusaka et al., 2021). It is important to note that the interfacial acceleration mechanism is multifaceted. Beyond partial solvation effects, other factors including the interfacial electric field and synergistic enthalpy-entropy effects play crucial roles in promoting HMS formation at the air-water interface (Li et al., 2025). These computational findings demonstrate a clear mechanistic preference for HMS formation over HMSi at the air-water interface, with a markedly lower free energy barrier (ΔG=0.6 kcal mol−1 for HMS versus 6.1 kcal mol−1 for HMSi). The exceptionally low barrier indicates that HMS formation proceeds very rapidly, especially in the polluted regions with abundant HCHO and SO2 in the atmosphere, which can provide an important theoretical foundation for more accurate simulation of atmospheric sulfur budgets.
3.3 Product Selectivity Reversal in Strongly Acidic Environments
The strongly acidic nature of many atmospheric environments, as discussed earlier, leads to protonation of HCHO, forming HCHOH+. Experimental studies (Jayne et al., 1996) have demonstrated that when pH drops below 2, formaldehyde undergoes efficient uptake by acidic droplets, leading to its protonation and formation of HCHOH+. However, the detailed mechanistic consequences of this protonation, especially as they manifest in heterogeneous environments, remain poorly understood. To capture the complex reactivity landscape of HCHOH+ with HOSO under strongly acidic conditions, we conducted 25 independent BOMD simulations (detailed configurations in Fig. S13). Figure 3a displays the temporal evolution of product formation across all simulations, with each horizontal line representing an individual BOMD trajectory. The colored markers indicate specific chemical transformations. Hydroxymethanesulfonic acid (HMSA) and hydroxymethyl hydrogen sulfite (HMHSi) formed within 2 ps in most trajectories. The subsequent deprotonation processes led to the formation of HMS and HMSi within approximately 10 ps. The mechanistic diversity enabled by formaldehyde protonation under strongly acidic conditions represents a fundamental departure from reactions involving neutral formaldehyde molecules. Figure 3b illustrates the three distinct mechanistic pathways identified through our simulations. Path A proceeds via nucleophilic attack by sulfur atom of HOSO at the carbonyl carbon of HCHOH+, leading to HMSA formation before transforming to HMS. Path B involves nucleophilic attack by either O1 or O2 of HOSO on the carbonyl carbon, forming HMSi.
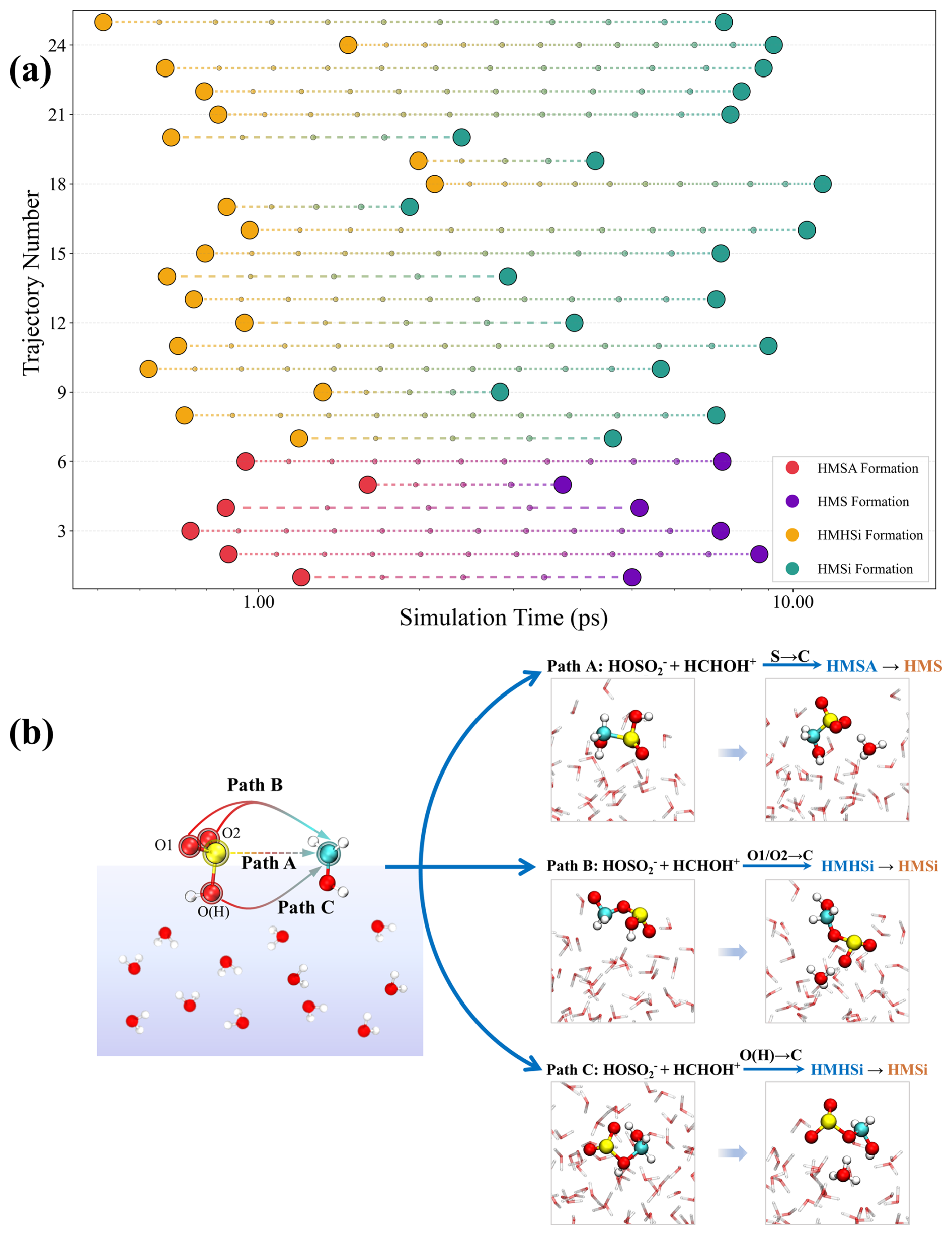
Figure 3Mechanistic investigation of the reaction between HCHOH+ and HOSO in acidic environment. (a) Time evolution of product formation across 25 independent BOMD simulations. Each horizontal line represents a single trajectory, with the starting point indicating the formation time of intermediate species (HMSA or HMHSi) and the end point indicating the formation time of final products (HMS or HMSi) after deprotonation. Colored markers represent different reaction products: HMSA (red), HMS (purple), HMHSi (yellow), and HMSi (green). Simulation time is shown in picoseconds (ps). (b) The three distinct mechanistic pathways (Paths A, B, and C).
Our trajectory analysis shows no significant preference between O1 and O2 attack, suggesting that both oxygen atoms experience similar activation in the interfacial acidic environment. The subsequent deprotonation of HMHSi to HMSi proceeds through water-mediated proton transfer, with interfacial water molecules playing a crucial catalytic role. Perhaps most remarkably, Path C demonstrates that the typically unreactive hydroxyl oxygen O(H) of HOSO becomes a viable nucleophile under acidic conditions.
Although the O(H) site of HOSO is generally weakly nucleophilic (Fig. S6), protonation fundamentally alters the electronic structure of HCHO by introducing a new δ-hole on the oxygen atom while substantially intensifying the δ-hole at the carbonyl carbon (Fig. S14). These multiple electron-deficient sites work synergistically to attract even weak nucleophiles such as O(H), thereby enabling direct HMSi formation. This reactivity highlights the unique ability of highly acidic interfaces to modulate electronic structure, thereby activating HCHOH+ toward unconventional nucleophilic pathways. Statistical analysis of the product distribution reveals a notable preference for HMSi formation under acidic conditions: among the 25 trajectories, 6 proceeded through Path A to form HMS, while 19 yielded HMSi via either Path B or Path C, establishing an HMS : HMSi ratio of approximately 1 : 3. This product distribution contrasts sharply with the preferential HMS formation observed at neutral pH interfaces, highlighting the profound influence of acidity on reaction selectivity. The pH-dependent reaction selectivity becomes even more pronounced under extreme acidic conditions. Under extremely acidic conditions (pH ≈ 0), the speciation of sulfurous acids at interfaces undergoes significant shifts. As observed by Buttersack et al. (2024) HSO becomes the predominant species at the air-water interface under extremely acidic conditions. With the sulfur atom protonated and therefore unavailable for nucleophilic attack, S–C bond formation leading to HMS becomes inhibited, yielding only HMSi (Fig. S15). These results establish HMSi as a potentially significant contributor to atmospheric sulfur budgets in strongly acidic environments. From an analytical perspective, HMS and HMSi differ in charge distribution, molecular geometry, and bonding arrangements at the sulfur center, which may in principle enable their separation by targeted ion chromatography and their differentiation by high-resolution tandem mass spectrometry. Both techniques have been successfully applied to HMS detection (Wei et al., 2020; Campbell et al., 2022), although dedicated method development and experimental verification will be required to confirm the resolution of these two isomers in ambient samples.
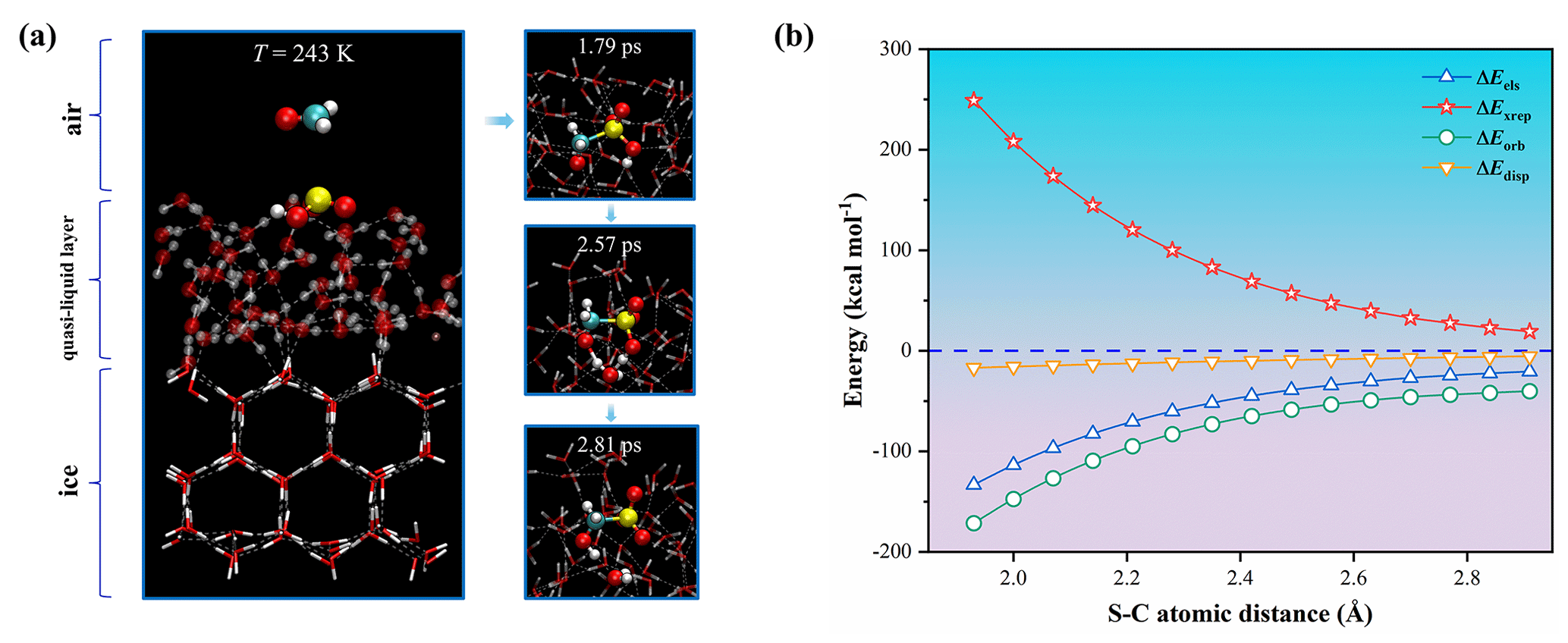
Figure 4Interfacial reaction of HCHO + HOSO on the ice surface. (a) Snapshot structures of BOMD simulation for the air–quasi-liquid-layer (QLL) interface at T=243 K. (b) Energy decomposition analysis (sobEDAw) plotted against S–C atomic distance (Å) during the reaction between HCHO and HOSO fragments, where ΔEels (blue triangles) represents the electrostatic interaction energy, ΔExrep (red stars) denotes the exchange repulsion term, ΔEorb (green circles) indicates the orbital interaction energy and ΔEdisp (yellow triangles) shows the dispersion term. Additional energy decomposition details provided in Sect. S2.
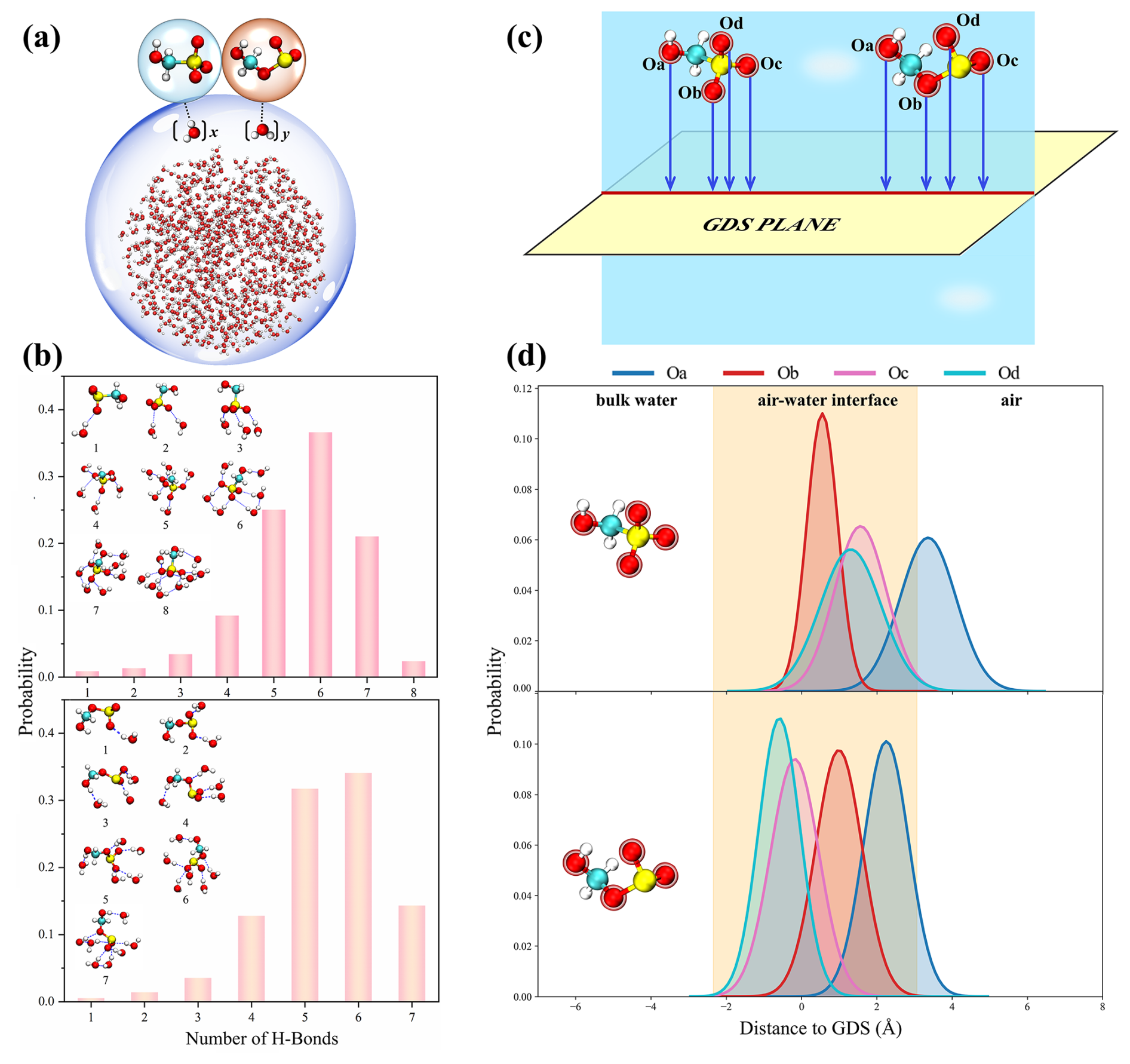
Figure 5(a) Schematics showing on the hydration behavior of HMS/HMSi on the water surface, where x and y represent the number of interfacial water molecules bound by HMS and HMSi, respectively, via hydrogen bonding. (b) Histograms of probabilities of HMS (upper panel) and HMSi (lower panel) conformations combining different numbers of water molecules. (c) Schematic illustration of the spatial orientation of oxygen atoms (Oa, Ob, Oc, and Od) in HMS and HMSi molecules with respect to the Gibbs dividing surface (GDS), defined as the plane where water density equals half its bulk value. (d) Probability distributions of distinct oxygen atoms across the interface for HMS (upper panel) and HMSi (lower panel), with the yellow zone indicating the interfacial region determined by GDS and water density profiles. Additional methodological details are provided in Supplement.
3.4 Previously Unrecognized Reactions at the Air-Ice Interface
Although air–water interfaces have been widely recognized as critical sites for heterogeneous reactions, the cold conditions prevalent in the upper troposphere and polar regions give rise to an additional interfacial environment in the form of atmospheric ice. These ice surfaces provide a distinct medium for heterogeneous chemistry beyond conventional water vapor and aqueous surfaces, particularly relevant given that HCHO and SO2 are ubiquitous throughout the atmosphere, including regions where temperatures favor ice formation (Anderson et al., 2017; Bai et al., 2023; Höpfner et al., 2015; Joppe et al., 2024; Sumner and Shepson, 1999). Despite the atmospheric abundance of ice crystals and the widespread distribution of these reactive species, the heterogeneous chemistry occurring at ice surfaces remains poorly characterized compared to reactions in other atmospheric environments. Considering the potential significance of ice-surface heterogeneous reactions for atmospheric sulfur cycling, we investigated the reaction between HCHO and HOSO at the air-ice interface to elucidate the associated reaction pathway. Given that the freezing process can lead to ion redistribution, we employed classical molecular dynamics (MD) simulations to track HOSO spatial distribution during ice formation (see Sect. S3 for details). The results showed that HOSO ions preferentially accumulate at the ice-air interface (Fig. S16), which is likely attributed to their exclusion from the growing ice crystal during crystallization (Ning et al., 2024; Tsironi et al., 2020). Next, we explored the reaction between HCHO and HOSO at the air-ice interface, with a quasi-liquid layer (QLL) on the ice crystal (Nagata et al., 2019) at T=243 K as shown in Fig. 4a. This temperature represents a typical polar temperature (Zhang et al., 2024a).
Energy decomposition analysis (Lu and Chen, 2023) (Fig. 4b; see Sect. S2 for methodology) revealed that the interaction between HCHO and HOSO fragments is driven by electrostatic (ΔEels) and orbital interaction (ΔEorb) components, with modest contributions from dispersion forces (ΔEdisp). Electronic structure analysis (Fig. S17) confirmed HOSO as the active electron donor throughout the reaction pathway. These results show that strong electrostatic attraction and orbital interactions jointly facilitate the approach of the two reactants and promote S–C bond formation on the ice surface. Subsequent water-mediated proton transfer yielded HMS. In addition, we investigated HMS formation at temperatures characteristic of the upper troposphere and lower stratosphere. The reaction remains feasible at 223 K, which represents a typical temperature of the high-altitude free troposphere (Li et al., 2024), and HMS formation proceeded rapidly in unbiased BOMD trajectories, completing within ∼ 3 ps through similar mechanistic pathways (Fig. S18). In contrast, HMSi formation was not observed in unbiased trajectories. Metadynamics simulations revealed free energy barriers of 5.4 kcal mol−1 at 243 K and 5.7 kcal mol−1 at 223 K for HMSi formation (Figs. S19 and S20). Additionally, the protonated formaldehyde pathway remained viable on ice surfaces (Figs. S21 and S22). Our simulations revealed three distinct reaction pathways on ice surfaces (Fig. S21): nucleophilic attack at the sulfur atom (Path A, forming HMS), at oxygen atoms O1/O2 (Path B, forming HMSi), and at the hydroxyl group O(H) (Path C, also forming HMSi). Furthermore, when HSO serves as the sulfur species, the reaction exclusively yields HMSi (Fig. S22).
Our mechanistic findings provide a potential explanation for field observations reporting enhanced HMS concentrations during cold atmospheric periods (Song et al., 2019; Campbell et al., 2024). While HMS formation involves multiple complex atmospheric processes, our results demonstrate that ice-mediated heterogeneous chemistry may significantly enhance this process through some key contributing factors. First, the substantially increased Henry law constants for both HCHO and SO2 at reduced temperatures facilitate enhanced partitioning of these precursors to condensed phases (Campbell et al., 2022). Second, efficient HMS formation on ice surfaces establishes a previously unrecognized heterogeneous pathway operating across cold atmospheric environments from polar regions to the upper troposphere. This ice-mediated chemistry represents an important yet underappreciated contributor to global organosulfur compound formation and atmospheric sulfur cycling. It should be noted that real atmospheric aerosol and cloud-droplet interfaces are chemically more complex than the simplified system examined here, and they may contain dissolved oxidants, organic species, transition metal ions, and other S(IV) components that could influence interfacial reactivity by competing for reactive sites or modifying the local environment. In future work, we intend to confirm the impacts of other atmospheric components.
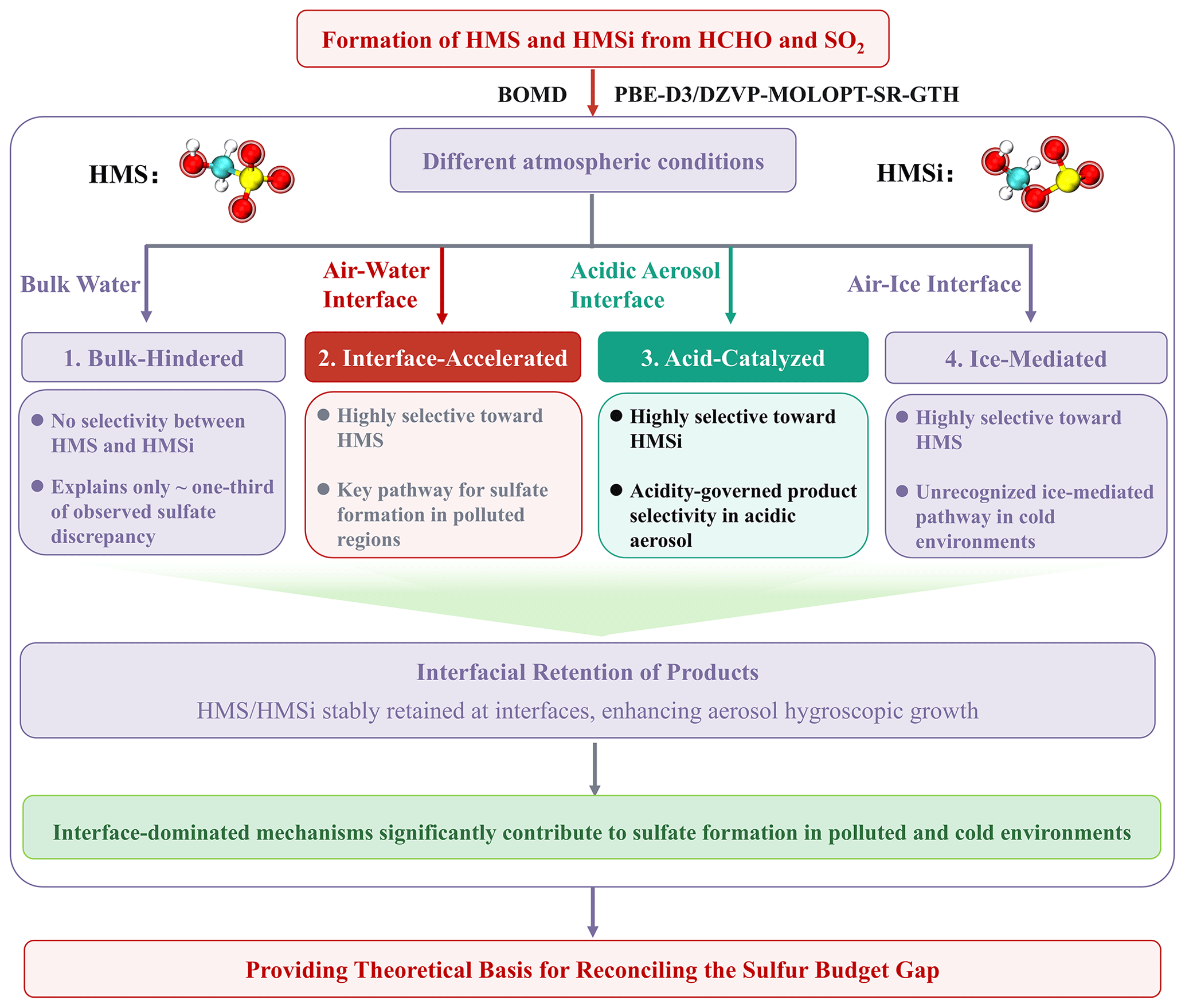
Scheme 1Conceptual overview of four atmospheric scenarios for HMS and HMSi formation (bulk aqueous phase, air–water interface, strongly acidic aerosol, and air–ice interface) and their respective contributions toward reconciling the atmospheric sulfur budget gap. The four labeled panels represent distinct reaction regimes: (1) Bulk-Hindered, (2) Interface-Accelerated, (3) Acid-Catalyzed, and (4) Ice-Mediated.
3.5 Interfacial Dynamics of Products
To further analyze the atmospheric implications of these products, the dynamic interfacial behavior of HMS and HMSi was characterized through quantitative analysis of hydrogen-bonded water molecules at the air-water interface. As shown in Fig. 5, the reaction products are stabilized at the interface through hydrogen bonds (HBs) with surrounding water molecules. Specifically, HMS and HMSi exhibit the highest probability of forming hydrogen bonds with 4–7 interfacial water molecules, exceeding 80 % probability. These products remain bound to the interface through multiple hydrogen-bonding interactions with water molecules, which consequently impedes their transfer into the gas phase. This interfacial retention reflects their strong hydrophilicity, as evidenced by the extensive hydration distribution of HMS and HMSi under different relative humidities (RH = 25 %–100 %) and temperatures (T = 240–298 K; Fig. S23), which further suggests their potential to enhance aerosol hygroscopic growth.
To further elucidate the interfacial behavior of HMS and HMSi, we investigated the positional correlations between their active sites (Oa, Ob, Oc, and Od) and the air–water interface. Figure 5c depicts the distances between these active sites and the Gibbs dividing surface (GDS) to determine their positions at the interface, while Fig. 5d quantifies the probability distributions of each oxygen atom (Oa, Ob, Oc, and Od) across the air-water interface. For HMS, the Ob, Oc, and Od atoms predominantly reside within the interfacial region (yellow shaded area), while the hydroxyl oxygen, designated as Oa, exhibits maximum probability at positive distances from the GDS plane. This indicates preferential orientation of the hydroxyl group toward the gas phase. For HMSi, while the distribution pattern is comparable, the probability distribution curve for Oa shows a significant reduction in extension toward the gas phase compared to HMS. Notably, the spatial distributions of HMS and HMSi at the air-ice interface exhibit similar orientational preferences to those observed at the air-water interface (Fig. S24). This differential orientation of the hydroxyl oxygen may have atmospheric implications, as the greater gas-phase exposure of the hydroxyl group in HMS compared to HMSi suggests a potential enhancement of interfacial oxidation due to orientation preferences, which could facilitate its interaction with atmospheric oxidants such as hydroxyl radicals (OH) and oxygen (O2). These molecular-level insights into the interfacial behavior of HMS and HMSi provide mechanistic understanding of their atmospheric processing and environmental fate, enabling a more accurate assessment of the contributions of organosulfur compounds to aerosol formation.
The overall framework of this study is summarized in Scheme 1. This study clarifies the formation mechanisms of HMS and its isomer HMSi via Born–Oppenheimer molecular dynamics simulations combined with high-level quantum chemical calculations. Our findings provide critical theoretical insights for resolving the persistent atmospheric sulfur budget gap in polluted winter haze episodes and cold environments including polar regions and the upper troposphere.
In the bulk aqueous phase, HMS and HMSi form competitively with near-identical free energy barriers of 7.7 and 7.6 kcal mol−1, respectively. However, bulk aqueous phase contributions to atmospheric sulfur cycling remain limited because aerosols and ice crystals exhibit high surface-to-volume ratios that favor interfacial chemistry. At air–water interfaces in polluted aerosols, HMS formation proceeds with a substantially reduced barrier of 0.6 kcal mol−1 through stepwise water-mediated proton transfer, while HMSi formation faces a higher barrier of 6.1 kcal mol−1, suggesting that interfacial chemistry may substantially enhance HMS formation relative to the bulk aqueous phase. This interfacial selectivity reverses under strongly acidic conditions where pH drops below 2. In these acidic environments, protonated formaldehyde activates unconventional nucleophilic sites on bisulfite, shifting product distribution to favor HMSi with an HMS to HMSi ratio of approximately 1 to 3. At extreme acidity approaching pH 0, HMSi forms exclusively. Based on global surface-layer aerosol pH estimated using annual-mean GEOS-Chem simulations coupled with E-AIM thermodynamic calculations, aerosol pH in the range of −1 to 1 occurs over at least ∼ 20 % of the global surface area where the model successfully converges (Li et al., 2022).
HMS and HMSi are stably retained at air-water interfaces through four to seven hydrogen bonds with surrounding water molecules, occurring with greater than 80 % probability. Their strong hydrophilicity prevents gas-phase evaporation while enhancing aerosol hygroscopic growth. Despite comparable interfacial positioning of oxygen atoms, HMS exhibits a crucial structural distinction: its hydroxyl group orients preferentially toward the gas phase relative to HMSi at both air-water and air-ice interfaces. This enhanced gas-phase exposure may render HMS more accessible to atmospheric oxidants including hydroxyl radicals, potentially facilitating its interfacial oxidation due to orientation preferences. These molecular insights provide a critical basis for assessing organosulfur contributions to aerosol formation and their atmospheric fate.
Collectively, these mechanistic insights provide essential molecular-level understanding for reconciling the persistent discrepancies between modeled and observed sulfate levels. This work establishes a foundation for improving atmospheric sulfur budget models across polluted and cold regions, ultimately enhancing our ability to predict air quality and assess climate impacts. Extending these mechanistic findings to atmospheric models requires further effort, including additional simulations across a wider temperature range and improved characterization of aerosol acidity.
Data available on request from the authors.
Details on computational methods. The supplement related to this article is available online at https://doi.org/10.5194/acp-26-9357-2026-supplement.
XZ designed and supervised the research. YL and AN performed the quantum chemical calculations and the BOMD simulations. YL, AN, XY, LL, YZ, and XZ analyzed data. YL wrote the paper. XY, LL, and XZ reviewed the paper. All authors commented on the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We acknowledge financial support from the National Science Fund for Distinguished Young Scholars and the National Natural Science Foundation of China.
This work was supported by the National Science Fund for Distinguished Young Scholars (grant no. 22225607) and the National Natural Science Foundation of China (grant nos. 22376013 and 22306011). L. Liu thanks the Beijing Institute of Technology Research Fund Program for Young Scholars.
This paper was edited by Hinrich Grothe and reviewed by two anonymous referees.
Anderson, D. C., Nicely, J. M., Wolfe, G. M., Hanisco, T. F., Salawitch, R. J., Canty, T. P., Dickerson, R. R., Apel, E. C., Baidar, S., Bannan, T. J., Blake, N. J., Chen, D., Dix, B., Fernandez, R. P., Hall, S. R., Hornbrook, R. S., Gregory Huey, L., Josse, B., Jöckel, P., Kinnison, D. E., Koenig, T. K., Le Breton, M., Marécal, V., Morgenstern, O., Oman, L. D., Pan, L. L., Percival, C., Plummer, D., Revell, L. E., Rozanov, E., Saiz-Lopez, A., Stenke, A., Sudo, K., Tilmes, S., Ullmann, K., Volkamer, R., Weinheimer, A. J., and Zeng, G.: Formaldehyde in the Tropical Western Pacific: Chemical sources and sinks, convective transport, and representation in CAM-Chem and the CCMI models, J. Geophys. Res.-Atmos., 122, 11201–11226, https://doi.org/10.1002/2016JD026121, 2017.
Angle, K. J., Crocker, D. R., Simpson, R. M. C., Mayer, K. J., Garofalo, L. A., Moore, A. N., Mora Garcia, S. L., Or, V. W., Srinivasan, S., Farhan, M., Sauer, J. S., Lee, C., Pothier, M. A., Farmer, D. K., Martz, T. R., Bertram, T. H., Cappa, C. D., Prather, K. A., and Grassian, V. H.: Acidity across the interface from the ocean surface to sea spray aerosol, P. Natl. Acad. Sci. USA, 118, https://doi.org/10.1073/pnas.2018397118, 2021.
Bai, F.-Y., Chi, T.-X., Liu, X.-H., Meng, T.-T., Ni, S., and Zhao, Z.: Metal-free catalysis on the reactions of nitric acid with aliphatic aldehydes: A new potential source of organic nitrates, Atmos. Environ., 299, 119673, https://doi.org/10.1016/j.atmosenv.2023.119673, 2023.
Bonomi, M., Branduardi, D., Bussi, G., Camilloni, C., Provasi, D., Raiteri, P., Donadio, D., Marinelli, F., Pietrucci, F., Broglia, R. A., and Parrinello, M.: PLUMED: A portable plugin for free-energy calculations with molecular dynamics, Comput. Phys. Commun., 180, 1961–1972, https://doi.org/10.1016/j.cpc.2009.05.011, 2009.
Bussi, G., Donadio, D., and Parrinello, M.: Canonical sampling through velocity rescaling, J. Chem. Phys., 126, 014101, https://doi.org/10.1063/1.2408420, 2007.
Buttersack, T., Gladich, I., Gholami, S., Richter, C., Dupuy, R., Nicolas, C., Trinter, F., Trunschke, A., Delgado, D., Corral Arroyo, P., Parmentier, E. A., Winter, B., Iezzi, L., Roose, A., Boucly, A., Artiglia, L., Ammann, M., Signorell, R., and Bluhm, H.: Direct observation of the complex S(IV) equilibria at the liquid-vapor interface, Nat. Commun., 15, 8987, https://doi.org/10.1038/s41467-024-53186-5, 2024.
Campbell, J. R., Battaglia, M., Jr., Dingilian, K., Cesler-Maloney, M., St Clair, J. M., Hanisco, T. F., Robinson, E., DeCarlo, P., Simpson, W., Nenes, A., Weber, R. J., and Mao, J.: Source and Chemistry of Hydroxymethanesulfonate (HMS) in Fairbanks, Alaska, Environ. Sci. Technol., 56, 7657–7667, https://doi.org/10.1021/acs.est.2c00410, 2022.
Campbell, J. R., Michael, B., Jr., Dingilian, K. K., Cesler-Maloney, M., Simpson, W. R., Robinson, E. S., DeCarlo, P. F., Temime-Roussel, B., D'Anna, B., Holen, A. L., Wu, J., Pratt, K. A., Dibb, J. E., Nenes, A., Weber, R. J., and Mao, J.: Enhanced aqueous formation and neutralization of fine atmospheric particles driven by extreme cold, Sci. Adv., 10, eado4373, https://doi.org/10.1126/sciadv.ado4373, 2024.
Chen, K. and Zhao, J.: Theoretical investigation of a potentially important formation pathway of organosulfate in atmospheric aqueous aerosols, Sci. Rep., 10, 6299, https://doi.org/10.1038/s41598-020-61968-2, 2020.
Ding, J., Zhao, P., Su, J., Dong, Q., Du, X., and Zhang, Y.: Aerosol pH and its driving factors in Beijing, Atmos. Chem. Phys., 19, 7939–7954, https://doi.org/10.5194/acp-19-7939-2019, 2019.
Dodia, M., Ohto, T., Imoto, S., and Nagata, Y.: Structure and Dynamics of Water at the Water–Air Interface Using First-Principles Molecular Dynamics Simulations. II. NonLocal vs Empirical van der Waals Corrections, J. Chem. Theory Comput., 15, 3836–3843, https://doi.org/10.1021/acs.jctc.9b00253, 2019.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams-Young, D., Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 16, Revision A.03, Gaussian Inc., Wallingford, CT, USA, https://gaussian.com/gaussian16/ (last access: 20 November 2023), 2016.
George, C., Ammann, M., D'Anna, B., Donaldson, D. J., and Nizkorodov, S. A.: Heterogeneous photochemistry in the atmosphere, Chem. Rev., 115, 4218–4258, https://doi.org/10.1021/cr500648z, 2015.
Goedecker, S., Teter, M., and Hutter, J.: Separable dual-space Gaussian pseudopotentials, Phys. Rev. B. , 54, 1703–1710, https://doi.org/10.1103/physrevb.54.1703, 1996.
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 132, 154104, https://doi.org/10.1063/1.3382344, 2010.
Guo, Y., Riplinger, C., Becker, U., Liakos, D. G., Minenkov, Y., Cavallo, L., and Neese, F.: Communication: An improved linear scaling perturbative triples correction for the domain based local pair-natural orbital based singles and doubles coupled cluster method [DLPNO-CCSD(T)], J. Chem. Phys., 148, 011101, https://doi.org/10.1063/1.5011798, 2018.
Höpfner, M., Boone, C. D., Funke, B., Glatthor, N., Grabowski, U., Günther, A., Kellmann, S., Kiefer, M., Linden, A., Lossow, S., Pumphrey, H. C., Read, W. G., Roiger, A., Stiller, G., Schlager, H., von Clarmann, T., and Wissmüller, K.: Sulfur dioxide (SO2) from MIPAS in the upper troposphere and lower stratosphere 2002–2012, Atmos. Chem. Phys., 15, 7017–7037, https://doi.org/10.5194/acp-15-7017-2015, 2015.
Huang, X., Liu, Z., Ge, Y., Li, Q., Wang, X., Fu, H., Zhu, J., Zhou, B., Wang, L., George, C., Wang, Y., Wang, X., Su, J., Xue, L., Yu, S., Mellouki, A., and Chen, J.: Aerosol high water contents favor sulfate and secondary organic aerosol formation from fossil fuel combustion emissions, npj Clim. Atmos. Sci., 6, https://doi.org/10.1038/s41612-023-00504-1, 2023.
Ishiyama, T., Tahara, T., and Morita, A.: Why the Photochemical Reaction of Phenol Becomes Ultrafast at the Air-Water Interface: The Effect of Surface Hydration, J. Am. Chem. Soc., 144, 6321–6325, https://doi.org/10.1021/jacs.1c13336, 2022.
Jayne, J. T., Worsnop, D. R., Kolb, C. E., Swartz, E., and Davidovits, P.: Uptake of Gas-Phase Formaldehyde by Aqueous Acid Surfaces, J. Phys. Chem., 100, 8015–8022, https://doi.org/10.1021/jp953196b, 1996.
Jia, S., Chen, W., Zhang, Q., Krishnan, P., Mao, J., Zhong, B., Huang, M., Fan, Q., Zhang, J., Chang, M., Yang, L., and Wang, X.: A quantitative analysis of the driving factors affecting seasonal variation of aerosol pH in Guangzhou, China, Sci. Total. Environ., 725, 138228, https://doi.org/10.1016/j.scitotenv.2020.138228, 2020.
Joppe, P., Schneider, J., Kaiser, K., Fischer, H., Hoor, P., Kunkel, D., Lachnitt, H.-C., Marsing, A., Röder, L., Schlager, H., Tomsche, L., Voigt, C., Zahn, A., and Borrmann, S.: The influence of extratropical cross-tropopause mixing on the correlation between ozone and sulfate aerosol in the lowermost stratosphere, Atmos. Chem. Phys., 24, 7499–7522, https://doi.org/10.5194/acp-24-7499-2024, 2024.
Kakavas, S., Patoulias, D., Zakoura, M., Nenes, A., and Pandis, S. N.: Size-resolved aerosol pH over Europe during summer, Atmos. Chem. Phys., 21, 799–811, https://doi.org/10.5194/acp-21-799-2021, 2021.
Kerbrat, M., Pinzer, B., Huthwelker, T., Gäggeler, H. W., Ammann, M., and Schneebeli, M.: Measuring the specific surface area of snow with X-ray tomography and gas adsorption: comparison and implications for surface smoothness, Atmos. Chem. Phys., 8, 1261–1275, https://doi.org/10.5194/acp-8-1261-2008, 2008.
Knopf, D. A., Alpert, P. A., and Wang, B.: The Role of Organic Aerosol in Atmospheric Ice Nucleation: A Review, ACS Earth. Space. Chem., 2, 168–202, https://doi.org/10.1021/acsearthspacechem.7b00120, 2018.
Krishnan, R., Binkley, J. S., Seeger, R., and Pople, J. A.: Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions, J. Chem. Phys., 72, 650–654, https://doi.org/10.1063/1.438955, 1980.
Kuhne, T. D., Iannuzzi, M., Del Ben, M., Rybkin, V. V., Seewald, P., Stein, F., Laino, T., Khaliullin, R. Z., Schutt, O., Schiffmann, F., Golze, D., Wilhelm, J., Chulkov, S., Bani-Hashemian, M. H., Weber, V., Borstnik, U., Taillefumier, M., Jakobovits, A. S., Lazzaro, A., Pabst, H., Muller, T., Schade, R., Guidon, M., Andermatt, S., Holmberg, N., Schenter, G. K., Hehn, A., Bussy, A., Belleflamme, F., Tabacchi, G., Gloss, A., Lass, M., Bethune, I., Mundy, C. J., Plessl, C., Watkins, M., VandeVondele, J., Krack, M., and Hutter, J.: CP2K: An electronic structure and molecular dynamics software package – Quickstep: Efficient and accurate electronic structure calculations, J. Chem. Phys., 152, 194103, https://doi.org/10.1063/5.0007045, 2020.
Kusaka, R., Nihonyanagi, S., and Tahara, T.: The photochemical reaction of phenol becomes ultrafast at the air-water interface, Nat. Chem., 13, 306–311, https://doi.org/10.1038/s41557-020-00619-5, 2021.
Lai, D., Wong, Y. K., Xu, R., Xing, S., Ng, S. I. M., Kong, L., Yu, J. Z., Huang, D. D., and Chan, M. N.: Significant Conversion of Organic Sulfur from Hydroxymethanesulfonate to Inorganic Sulfate and Peroxydisulfate Ions upon Heterogeneous OH Oxidation, Environ. Sci. Tech. Let., 10, 773–778, https://doi.org/10.1021/acs.estlett.3c00472, 2023.
Lai, D., Schaefer, T., Zhang, Y., Li, Y. J., Xing, S., Herrmann, H., and Chan, M. N.: Deactivating Effect of Hydroxyl Radicals Reactivity by Sulfate and Sulfite Functional Groups in Aqueous Phase-Atmospheric Implications for Small Organosulfur Compounds, ACS ES&T Air, 1, 678–689, https://doi.org/10.1021/acsestair.4c00033, 2024.
Li, H., Wang, X., Zhong, J., Chu, B., Ma, Q., Zeng, X. C., Francisco, J. S., and He, H.: Mechanistic Study of the Aqueous Reaction of Organic Peroxides with HSO on the Surface of a Water Droplet, Angew. Chem. Int. Ed. Engl., 60, 20200–20203, https://doi.org/10.1002/anie.202105416, 2021.
Li, J., Ning, A., Liu, L., and Zhang, X.: Atmospheric Bases-Enhanced Iodic Acid Nucleation: Altitude-Dependent Characteristics and Molecular Mechanisms, Environ. Sci. Technol., https://doi.org/10.1021/acs.est.4c06053, 2024.
Li, J., Tang, W., Zhu, J., Yang, J., and He, X.: Hydroxymethanesulfonate formation accelerated at the air-water interface by synergistic enthalpy-entropy effects, Nat. Commun., 16, 5187, https://doi.org/10.1038/s41467-025-59712-3, 2025.
Li, M., Su, H., Zheng, G., Kuhn, U., Kim, N., Li, G., Ma, N., Poschl, U., and Cheng, Y.: Aerosol pH and Ion Activities of HSO and SO in Supersaturated Single Droplets, Environ. Sci. Technol., 56, 12863–12872, https://doi.org/10.1021/acs.est.2c01378, 2022.
Lu, T. and Chen, F.: Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 33, 580–592, https://doi.org/10.1002/jcc.22885, 2012.
Lu, T. and Chen, Q.: Simple, Efficient, and Universal Energy Decomposition Analysis Method Based on Dispersion-Corrected Density Functional Theory, J. Phys. Chem. A, 127, 7023–7035, https://doi.org/10.1021/acs.jpca.3c04374, 2023.
Martins-Costa, M. T., Anglada, J. M., Francisco, J. S., and Ruiz-Lopez, M. F.: Reactivity of volatile organic compounds at the surface of a water droplet, J. Am. Chem. Soc., 134, 11821–11827, https://doi.org/10.1021/ja304971e, 2012.
McLean, A. D. and Chandler, G. S.: Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18, J. Chem. Phys., 72, 5639–5648, https://doi.org/10.1063/1.438980, 1980.
Moch, J. M., Dovrou, E., Mickley, L. J., Keutsch, F. N., Cheng, Y., Jacob, D. J., Jiang, J., Li, M., Munger, J. W., Qiao, X., and Zhang, Q.: Contribution of Hydroxymethane Sulfonate to Ambient Particulate Matter: A Potential Explanation for High Particulate Sulfur During Severe Winter Haze in Beijing, Geophys. Res. Lett., 45, 11969–11979, https://doi.org/10.1029/2018gl079309, 2018.
Moch, J. M., Dovrou, E., Mickley, L. J., Keutsch, F. N., Liu, Z., Wang, Y., Dombek, T. L., Kuwata, M., Budisulistiorini, S. H., Yang, L., Decesari, S., Paglione, M., Alexander, B., Shao, J., Munger, J. W., and Jacob, D. J.: Global Importance of Hydroxymethanesulfonate in Ambient Particulate Matter: Implications for Air Quality, J. Geophys. Res.-Atmos., 125, e2020JD032706, https://doi.org/10.1029/2020JD032706, 2020.
Nagata, Y., Hama, T., Backus, E. H. G., Mezger, M., Bonn, D., Bonn, M., and Sazaki, G.: The Surface of Ice under Equilibrium and Nonequilibrium Conditions, Acc. Chem. Res., 52, 1006–1015, https://doi.org/10.1021/acs.accounts.8b00615, 2019.
Neese, F.: The ORCA program system, WIRes Comput. Mol. Sci., 2, 73–78, https://doi.org/10.1002/wcms.81, 2011.
Ning, A., Zhong, J., Li, L., Li, H., Liu, J., Liu, L., Liang, Y., Li, J., Zhang, X., Francisco, J. S., and He, H.: Chemical Implications of Rapid Reactive Absorption of I2O4 at the Air-Water Interface, J. Am. Chem. Soc., 145, 10817–10825, https://doi.org/10.1021/jacs.3c01862, 2023.
Ning, A., Li, J., Du, L., Yang, X., Liu, J., Yang, Z., Zhong, J., Saiz-Lopez, A., Liu, L., Francisco, J. S., and Zhang, X.: Heterogenous Chemistry of I2O3 as a Critical Step in Iodine Cycling, J. Am. Chem. Soc., 146, 33229–33238, https://doi.org/10.1021/jacs.4c13060, 2024.
Norjmaa, G., Ujaque, G., and Lledós, A.: Beyond Continuum Solvent Models in Computational Homogeneous Catalysis, Top. Catal., 65, 118–140, https://doi.org/10.1007/s11244-021-01520-2, 2021.
Perdew, J. P., Burke, K., and Ernzerhof, M.: Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 77, 3865–3868, https://doi.org/10.1103/PhysRevLett.77.3865, 1996.
Pye, H. O. T., Zuend, A., Fry, J. L., Isaacman-VanWertz, G., Capps, S. L., Appel, K. W., Foroutan, H., Xu, L., Ng, N. L., and Goldstein, A. H.: Coupling of organic and inorganic aerosol systems and the effect on gas–particle partitioning in the southeastern US, Atmos. Chem. Phys., 18, 357–370, https://doi.org/10.5194/acp-18-357-2018, 2018.
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 034106, https://doi.org/10.1063/1.4773581, 2013.
Riplinger, C., Sandhoefer, B., Hansen, A., and Neese, F.: Natural triple excitations in local coupled cluster calculations with pair natural orbitals, J. Chem. Phys., 139, 134101, https://doi.org/10.1063/1.4821834, 2013.
Ruiz-Lopez, M. F., Francisco, J. S., Martins-Costa, M. T. C., and Anglada, J. M.: Molecular reactions at aqueous interfaces, Nat. Rev. Chem., 4, 459–475, https://doi.org/10.1038/s41570-020-0203-2, 2020.
Sipila, M., Berndt, T., Petaja, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin 3rd, R. L., Hyvarinen, A. P., Lihavainen, H., and Kulmala, M.: The role of sulfuric acid in atmospheric nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010.
Song, S., Gao, M., Xu, W., Sun, Y., Worsnop, D. R., Jayne, J. T., Zhang, Y., Zhu, L., Li, M., Zhou, Z., Cheng, C., Lv, Y., Wang, Y., Peng, W., Xu, X., Lin, N., Wang, Y., Wang, S., Munger, J. W., Jacob, D. J., and McElroy, M. B.: Possible heterogeneous chemistry of hydroxymethanesulfonate (HMS) in northern China winter haze, Atmos. Chem. Phys., 19, 1357–1371, https://doi.org/10.5194/acp-19-1357-2019, 2019.
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J.: Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields, J. Phys. Chem., 98, 11623–11627, https://doi.org/10.1021/j100096a001, 1994.
Sumner, A. L. and Shepson, P. B.: Snowpack production of formaldehyde and its effect on the Arctic troposphere, Nature, 398, 230–233, https://doi.org/10.1038/18423, 1999.
Tsironi, I., Schlesinger, D., Spah, A., Eriksson, L., Segad, M., and Perakis, F.: Brine rejection and hydrate formation upon freezing of NaCl aqueous solutions, Phys. Chem. Chem. Phys., 22, 7625–7632, https://doi.org/10.1039/c9cp05436g, 2020.
VandeVondele, J. and Hutter, J.: Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 127, 114105, https://doi.org/10.1063/1.2770708, 2007.
Wang, G., Zhang, R., Gomez, M. E., Yang, L., Levy Zamora, M., Hu, M., Lin, Y., Peng, J., Guo, S., Meng, J., Li, J., Cheng, C., Hu, T., Ren, Y., Wang, Y., Gao, J., Cao, J., An, Z., Zhou, W., Li, G., Wang, J., Tian, P., Marrero-Ortiz, W., Secrest, J., Du, Z., Zheng, J., Shang, D., Zeng, L., Shao, M., Wang, W., Huang, Y., Wang, Y., Zhu, Y., Li, Y., Hu, J., Pan, B., Cai, L., Cheng, Y., Ji, Y., Zhang, F., Rosenfeld, D., Liss, P. S., Duce, R. A., Kolb, C. E., and Molina, M. J.: Persistent sulfate formation from London Fog to Chinese haze, P. Natl. Acad. Sci. USA, 113, 13630–13635, https://doi.org/10.1073/pnas.1616540113, 2016.
Wang, G., Zhang, S., Wu, C., Zhu, T., Xu, X., Ge, S., Sun, H., Sun, Z., Wang, J., Ji, Y., Gao, J., Ren, Y., Li, H., Zhang, F., Wang, Y., and Seinfeld, J. H.: Atmospheric sulfate aerosol formation enhanced by interfacial anions, PNAS Nexus, 4, pgaf058, https://doi.org/10.1093/pnasnexus/pgaf058, 2025.
Wang, Y., Zhang, Q., Jiang, J., Zhou, W., Wang, B., He, K., Duan, F., Zhang, Q., Philip, S., and Xie, Y.: Enhanced sulfate formation during China's severe winter haze episode in January 2013 missing from current models, J. Geophys. Res.-Atmos., 119, https://doi.org/10.1002/2013jd021426, 2014.
Wei, L., Fu, P., Chen, X., An, N., Yue, S., Ren, H., Zhao, W., Xie, Q., Sun, Y., Zhu, Q.-F., Wang, Z., and Feng, Y.-Q.: Quantitative Determination of Hydroxymethanesulfonate (HMS) Using Ion Chromatography and UHPLC-LTQ-Orbitrap Mass Spectrometry: A Missing Source of Sulfur during Haze Episodes in Beijing, Environ. Sci. Tech. Let., 7, 701–707, https://doi.org/10.1021/acs.estlett.0c00528, 2020.
Yang, J., Li, L., Wang, S., Li, H., Francisco, J. S., Zeng, X. C., and Gao, Y.: Unraveling a New Chemical Mechanism of Missing Sulfate Formation in Aerosol Haze: Gaseous NO2 with Aqueous HSO3-/SO32-, J. Am. Chem. Soc., 141, 19312–19320, https://doi.org/10.1021/jacs.9b08503, 2019.
Zhang, J. and Lu, T.: Efficient evaluation of electrostatic potential with computerized optimized code, Phys. Chem. Chem. Phys., 23, 20323–20328, https://doi.org/10.1039/d1cp02805g, 2021.
Zhang, J., Zhang, H., Wu, T., Wang, Q., and van der Spoel, D.: Comparison of Implicit and Explicit Solvent Models for the Calculation of Solvation Free Energy in Organic Solvents, J. Chem. Theory. Comput., 13, 1034–1043, https://doi.org/10.1021/acs.jctc.7b00169, 2017.
Zhang, R.: Getting to the critical nucleus of aerosol formation, Science, 328, 1366–1367, https://doi.org/10.1126/science.1189732, 2010.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of Urban Fine Particulate Matter, Chem. Rev., 115, 3803–3855, https://doi.org/10.1021/acs.chemrev.5b00067, 2015.
Zhang, W., Zheng, D., Han, H., Wan, Z., Zhong, J., Ji, Y., Li, G., Francisco, J. S., and An, T.: Promoting Cl2O Generation from the HOCl + HOCl Reaction on Aqueous/Frozen Air-Water Interfaces, J. Am. Chem. Soc., 146, 31935–31944, https://doi.org/10.1021/jacs.4c11337, 2024a.
Zhang, Y., Han, R., Sun, X., Sun, C., Griffith, S. M., Wu, G., Li, L., Li, W., Zhao, Y., Li, M., Zhou, Z., Wang, W., Sheng, L., Yu, J. Z., and Zhou, Y.: Sulfate Formation Driven by Wintertime Fog Processing and a Hydroxymethanesulfonate Complex With Iron: Observations From Single-Particle Measurements in Hong Kong, J. Geophys. Res.-Atmos., 129, https://doi.org/10.1029/2023jd040512, 2024b.
Zhao, M., Shen, H., Zhang, J., Liu, Y., Sun, Y., Wang, X., Dong, C., Zhu, Y., Li, H., Shan, Y., Mu, J., Zhong, X., Tang, J., Guo, M., Wang, W., and Xue, L.: Carbonyl Compounds Regulate Atmospheric Oxidation Capacity and Particulate Sulfur Chemistry in the Coastal Atmosphere, Environ. Sci. Technol., 58, 17334–17343, https://doi.org/10.1021/acs.est.4c03947, 2024.
Zheng, G., Su, H., Wang, S., Andreae, M. O., Poschl, U., and Cheng, Y.: Multiphase buffer theory explains contrasts in atmospheric aerosol acidity, Science, 369, 1374–1377, https://doi.org/10.1126/science.aba3719, 2020.
Zhong, J., Li, H., Kumar, M., Liu, J., Liu, L., Zhang, X., Zeng, X. C., and Francisco, J. S.: Mechanistic Insight into the Reaction of Organic Acids with SO(3) at the Air-Water Interface, Angew. Chem. Int. Ed. Engl., 58, 8351–8355, https://doi.org/10.1002/anie.201900534, 2019.
Zhong, J., Wang, C., Zeng, X. C., and Francisco, J. S.: Heterogeneous Reactions of SO3 on Ice: An Overlooked Sink for SO3 Depletion, J. Am. Chem. Soc., 142, 2150–2154, https://doi.org/10.1021/jacs.9b11723, 2020.
Zhou, M., Zheng, G., Wang, H., Qiao, L., Zhu, S., Huang, D., An, J., Lou, S., Tao, S., Wang, Q., Yan, R., Ma, Y., Chen, C., Cheng, Y., Su, H., and Huang, C.: Long-term trends and drivers of aerosol pH in eastern China, Atmos. Chem. Phys., 22, 13833–13844, https://doi.org/10.5194/acp-22-13833-2022, 2022.