the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Mar 2026
| 20 Mar 2026
Gas-phase products from nitrate radical oxidation of five monoterpenes: insights from free-jet flow-tube experiments
Yi Zhang
Hannu Koskenvaara
Jian Zhao
Formation of secondary organic aerosol (SOA), which affects climate and health, is largely driven by the gas-particle transfer of highly oxygenated organic molecules (HOMs). These HOMs form via autoxidation following reactions of volatile organic compounds (VOCs) with atmospheric oxidants. While the oxidation of monoterpenes, the most important biogenic VOCs for SOA formation, by ozone (O3) and the hydroxyl radical (OH) is well-studied, the role of the nitrate radical (NO3), a crucial nighttime oxidant, remains less understood. This study investigated NO3-initiated oxidation of five monoterpenes: α-pinene (AP), Δ-3-carene, limonene, β-pinene (BP), and β-myrcene. Using a newly built free-jet flow-tube system (8.8 s reaction time) and chemical ionization mass spectrometry (amine/nitrate ionization), we observed a wide range of peroxy radicals and closed-shell products. Product closure was reasonably achieved for AP, limonene, and myrcene (estimated to 50 %–70 %), but was lower for carene and BP (20 %–40 %). AP and limonene predominantly yielded C10H16O2 (molar yields >50 %), while a notably high signal for carene was the peroxy radical C10H16NO8, for myrcene the radical C10H16NO7, and for BP the accretion product C20H32N2O8. The distinct HOM yields further emphasize highly structure-dependent oxidation pathways: 6.5 % (myrcene), 6.1 % (carene), 1.8 % (BP), 1.1 % (limonene), and 0.8 % (AP). The HOM yields differ from those of ozonolysis, but overall HOM yields from NO3 oxidation are comparable in magnitude (0 %–10 %). This study provides comprehensive and quantitative distributions of NO3 oxidation products for the most common monoterpenes, providing important knowledge of their fast (aut)oxidation pathways.
- Article
(4545 KB) - Full-text XML
-
Supplement
(6126 KB) - BibTeX
- EndNote




Secondary organic aerosol (SOA) is a major component of atmospheric particulate matter (Hallquist et al., 2009; Jimenez et al., 2009), affecting climate through interactions with radiation and cloud formation (Boucher et al., 2013; Spracklen et al., 2011), and impacting human health via inhalation exposure (Kelly and Fussell, 2015). SOA formation is attributed to the gas-to-particle transfer of a wide range of oxidized products with low volatility (Donahue et al., 2012; Hallquist et al., 2009; Kroll and Seinfeld, 2008), which are generated from atmospheric degradation processes of volatile organic compounds (VOCs) (Atkinson and Arey, 2003). Recently, highly oxygenated organic molecules (HOMs) have been discovered to be a new and important group of VOC oxidation products, contributing significantly to SOA through condensation (Bianchi et al., 2019; Ehn et al., 2014) or even nucleation (Huang et al., 2024). Formed in the gas phase under atmospherically relevant conditions and generally containing more than five oxygen atoms (Bianchi et al., 2019), HOMs are produced through rapid peroxy radical (RO2) autoxidation (i.e., intramolecular H-shift or ring closures followed by O2 addition), following the initial oxidation of VOCs by atmospheric oxidants (Bianchi et al., 2019; Crounse et al., 2013; Ehn et al., 2014). Among all VOCs, monoterpenes (MTs), C10H16, represent one of the largest biogenic emissions, alone outnumbering the total anthropogenic VOC emissions, making them a crucial source of atmospheric condensation vapors (Guenther et al., 2012; McFiggans et al., 2019).
Although considerable progress has been made in understanding HOM formation from ozone (O3) and hydroxyl radical (OH) initiated oxidation of MTs (Berndt et al., 2016, 2018a; Berndt, 2021, 2022; Bianchi et al., 2019; Ehn et al., 2014; Iyer et al., 2021; Jokinen et al., 2015; Luo et al., 2024; Meder et al., 2025; Molteni et al., 2019; Shen et al., 2022; Zhao et al., 2015, 2024), the role of the nitrate radical (NO3), a key nighttime oxidant (Brown and Stutz, 2012), remains less well characterized. While NO3-initiated nocturnal oxidation can significantly contribute to total oxidized organic aerosol production in fall and winter (Liu et al., 2024), recent findings by Dewald et al. (2024) suggest that NO3-initiated organic nitrate formation can also be substantial during the daytime, underscoring the need for deeper insight into NO3-driven autoxidation processes. Moreover, gaseous organic nitrates from NO3 oxidation are potentially an important NOx reservoir, due to the possible rapid photolysis (Takeuchi et al., 2025; Wang et al., 2023). This further highlights the importance of an improved understanding of NO3 oxidation mechanisms.
Most studies over the last decades on NO3 oxidation used smog chambers to investigate SOA formation (Bates et al., 2022; Bell et al., 2022; Boyd et al., 2015; Claflin and Ziemann, 2018; Fry et al., 2014; Hallquist et al., 1999; Nah et al., 2016; Ng et al., 2017; Perraud et al., 2010; Spittler et al., 2006). Recently, HOMs were identified to play a vital role in SOA from the NO3 oxidation of MTs (Dam et al., 2022; Day et al., 2022; Draper et al., 2019; Graham et al., 2023; Guo et al., 2022; Harb et al., 2025; Shen et al., 2021). However, these studies either focused on one to two MTs or did not provide distributions covering products from less-oxygenated compounds to HOMs. Offering a controlled environment closely mimicking the atmosphere, smog chambers are good at studying complex multiphase chemistry and long-lived oxidation products, due to their typical residence times ranging from half an hour to several hours. In contrast, flow-tubes operate on timescales of seconds to minutes, providing valuable insights into fast autoxidation and short-lived radicals. The last decade witnessed a series of free-jet flow-tube experiments focusing on early-stage O3/OH oxidation products, including short-lifetime RO2 radicals, with multiple chemical ionization reagents applied (Berndt et al., 2015a, 2018a, b; Berndt, 2021, 2022). Specifically, the short reaction time of the flow-tube system (1.1–7.5 s; Berndt et al., 2015b) enables the direct comparison of the competing pathways of RO2 radicals (Bianchi et al., 2019): unimolecular termination or isomerization leading to autoxidation; and bimolecular reactions leading to the formation of alkoxy radicals (RO), closed-shell monomers (e.g., RC=O, ROH), or dimers (ROOR). The advantage of short-reaction-time flow-tube experiments for studying gas-phase oxidation mechanisms has only sparingly been applied to investigations of NO3-initiated oxidation processes.
In this study, we investigated NO3 oxidation mechanisms of five common MTs (α-pinene, Δ-3-carene, limonene, β-pinene, and β-myrcene) by directly comparing their early-stage gas-phase RO2 radicals and closed-shell products, using a newly built free-jet flow-tube system with short reaction times and a chemical ionization mass spectrometer. Avoiding the high level of NO3 typically produced by N2O5 thermal decomposition (Bates et al., 2022; Fry et al., 2014), we used a pre-reactor to generate the NO3 radical from the reaction NO2 + O3. The deployment of diethylamine (C4H11N) and isotopically labeled nitric acid (H15NO3) as reagent sources for the mass spectrometer allowed us to measure oxidation products with two or more oxygen atoms (Cai et al., 2024; Riva et al., 2019). By varying the concentrations of MTs, NO2, and O3, we identified the NO3 oxidation products, compared their concentrations, and explored their behaviors with increasing NO3 radicals. We also developed a simple 0-D box model, which describes the flow-tube experiments, to calculate the reacted MT concentrations. Finally, we estimated what fraction of all reaction products we could observe, as well as the HOM yields.
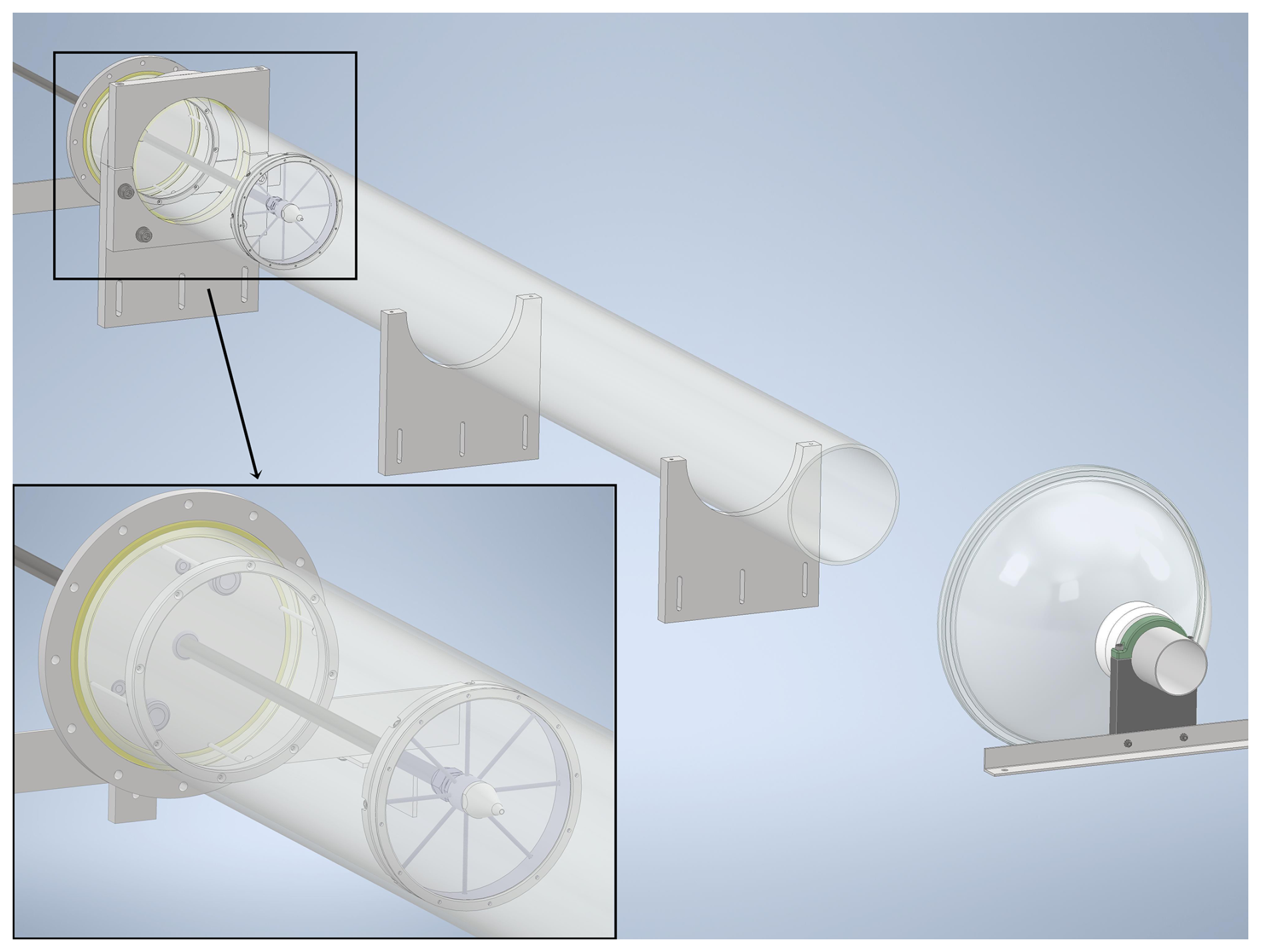
Figure 1Schematic of the free-jet flow-tube system. The gas-injecting side is shown in a zoomed-in view. An exhaust port with a film hood is placed downstream the flow-tube outlet. The reaction distance is defined as the distance from the nozzle to the sampling port, which is located at the center of the outlet (not shown).
2.1 Flow-tube system
The experiments (temperature: 298 ± 2 K; pressure: 1 atm; dry conditions: relative humidity <1 %) were carried out in a newly constructed free-jet flow-tube system (Fig. 1), located in the Physicum building at the University of Helsinki. Similar to the design by Berndt et al. (2015b), our flow-tube system consists of a quartz tube (length: 2 m, inner diameter: 15 cm) and a movable stainless-steel injector (length: 1.8 m, inner diameter: 8 mm) with a nozzle (inner diameter: 3 mm). A total flow of 100 L min−1 is supplied by a 250 L min−1 zero-air generator (ZA-737-250, Tisch Environmental) and regulated by various mass flow controllers (MKS, Inc.). Oxidants such as O3 or NO3 are carried by 5 L min−1 of zero air to the central injector and then mixed into the 95 L min−1 main gas flow, which contains reactants such as various VOCs. The narrow nozzle at the end of the injector enables a high flow velocity, causing turbulent mixing at the beginning of the reaction. As the flow slows down, it gradually becomes laminar, as also observed visually using smoke (see Sect. S1 in the Supplement for details). The open outlet design and the exhaust port placed downstream (Fig. 1) help maintain laminar flow while sampling takes place at the center of the outlet. The experimental design allows investigations under nearly wall-free conditions (Berndt et al., 2015b). To enable the use of a box model for simulating reactions inside the free-jet flow-tube system, effective reaction times were experimentally determined via ozonolysis of 2,3-dimethyl-2-butene (TME, ≥ 99 %, Sigma-Aldrich) at varying reaction distances (Fig. S1 and Sect. S1) (Berndt et al., 2015b): 2.5 s (50 cm), 4.6 s (70 cm), 6.4 s (90 cm), and 8.3 s (110 cm).
2.2 Experiment design
Using the free-jet flow-tube system at an effective reaction time of 8.3 s, we investigated the formation of RO2 radicals and closed-shell products in the NO3 oxidation of five MTs, i.e., α-pinene (AP, 99 %, Sigma-Aldrich), Δ-3-carene (99 %, Sigma-Aldrich), limonene (96 %, Sigma-Aldrich), β-pinene (BP, 99 %, Sigma-Aldrich), and β-myrcene (90 %, Sigma-Aldrich). These precursors were injected into the main gas flow using a 1.5 L min−1 zero-air flow and a syringe pump system (Fusion F100T2, Chemyx, Inc.).
O3 and NO2, generated from an ozone generator (Dasibi 1008-PC) and a NO2 cylinder (0.1 % NO2 in N2, Linde Gas) respectively, were mixed in an external 0.7 L cylindrical glass reactor (Meder et al., 2025) to produce NO3 and N2O5. The total flow through the pre-reactor was fixed at 1 L min−1, resulting in a reaction time of 42 s and concentrations of O3 and NO2 ([O3]–1800 ppb and [NO2]–8600 ppb) approximately 100 times higher than those in the free-jet flow-tube after dilution. An additional 4 L min−1 of zero air was added to the pre-reactor outflow before it entered the injector. The NO3 concentration in the reaction region of the flow-tube was sustained at a few ppt (Fig. S2), which is comparable to atmospheric levels (Ayres et al., 2015; Bates et al., 2022). NO levels were negligible due to the lack of NO2 photolysis, consistent with the very low signals from our NOx analyzer and the lack of detectable products from NO termination.
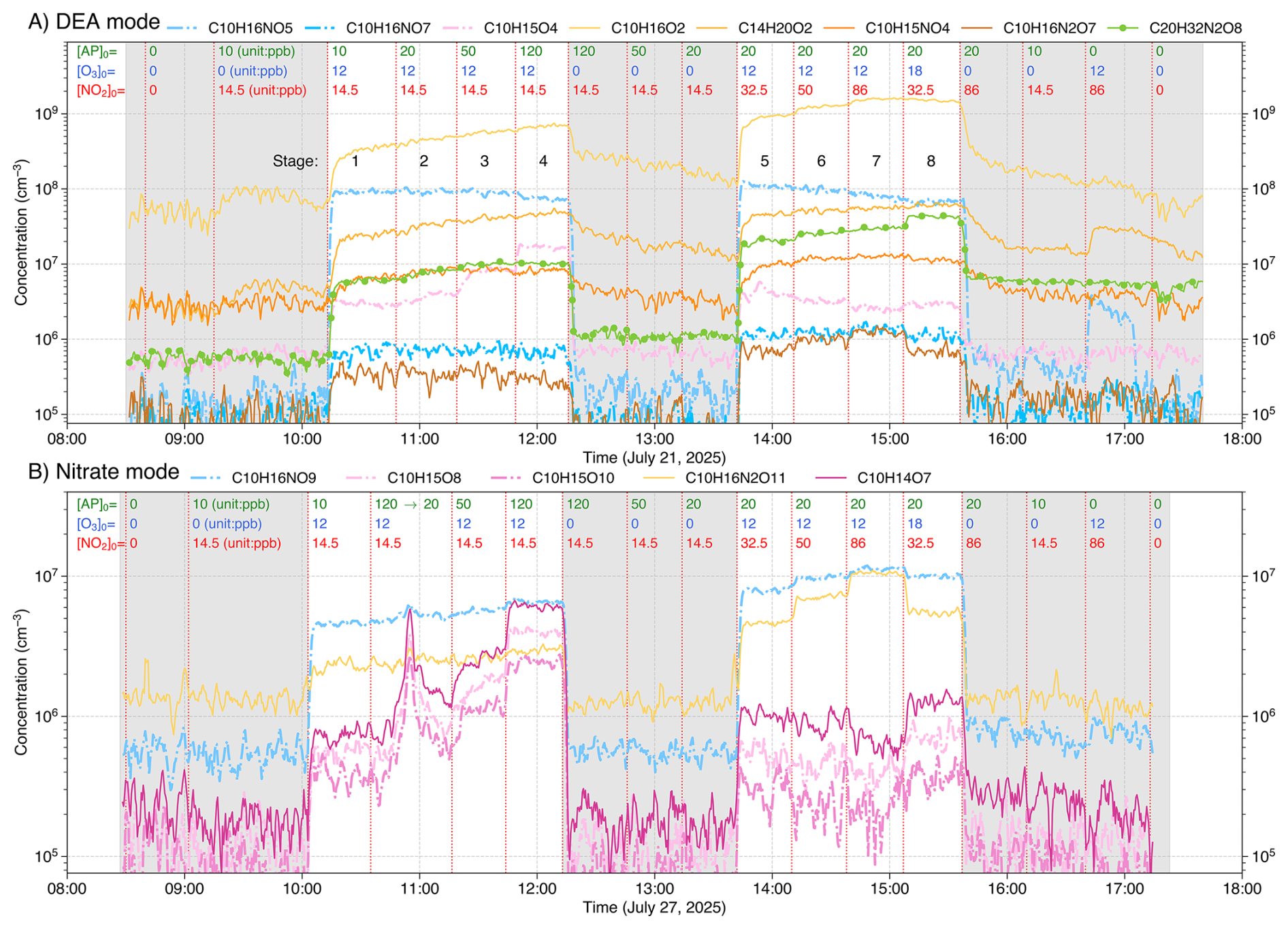
Figure 2Timeseries of major products from α-pinene (AP) + NO3 experiments, measured using both DEA (A) and nitrate (B) modes. Concentrations (in cm−3) of radicals, closed-shell monomers, and closed-shell dimers are shown as dash-dot lines, solid lines, and solid lines with markers, respectively. Products are color-coded by origin: expected NO3 oxidation products (radicals, closed-shell monomers, and dimers) are shown in blue, yellow-orange, and green colors, respectively, while expected O3/OH oxidation products are shown in pink. Grey shaded areas represent background stages. Initial precursor concentrations (in ppb) shown at the top of each subplot correspond to values present in the total flow of 100 L min−1, without any chemical conversion. The effective reaction time is 8.8 s. Note that the sharp spike in C10H15O8,10 and C10H14O7 at approximately 11:00 (B) resulted from an initial AP injection of 120 ppb, which was subsequently corrected to 20 ppb. While a mistake initially, this confirms that these three species originate from O3/OH oxidation.
The experiments were designed to first ramp up MT concentrations in four stages, followed by a ramp-up of NO2, with background stages included throughout (Fig. 2). This approach allowed us to distinguish products formed via NO3 oxidation from those formed by O3/OH oxidation (see Sect. 3.1). The initial concentrations of each stage shown at the top of each subplot in Fig. 2 refer to the values present in the total flow of 100 L min−1, without any chemical conversion. Since NO2 and O3 were reacted in the pre-reactor before entering the flow-tube, their actual concentrations at the start of the oxidation process were slightly lower than the initial values. No particles were present in the input gas, and particle formation did not occur due to low reactant conversion and the short reaction time (Sect. S1).
2.3 Instrumentation
Gas-phase RO2 radicals and closed-shell products containing two or more oxygen atoms were measured using a chemical ionization mass spectrometer (CIMS, Tofwerk AG/Aerodyne Research, Inc.), coupled with an Eisele-type inlet (Eisele and Tanner, 1993) and operated with either diethylamine (DEA, C4H11N, ≥ 99.5 %, Sigma-Aldrich) or isotopically labeled nitric acid (H15NO3, ∼10 N in H2O, 98 atom % 15N, Sigma-Aldrich) as the reagent ion source (referred to as DEA- or nitrate-CIMS). The DEA mode allows detection of compounds with two or more oxygen atoms, while the nitrate mode is highly selective for HOMs (Cai et al., 2024; Ehn et al., 2014; Riva et al., 2019). The 15N-labeled nitric acid was used in this study to unambiguously identify the organic nitrate products by distinguishing the N originating from unlabeled oxidant NO3 (formed via NO2 + O3) from the labeled reagent ions. The sampling flow rate was 10 L min−1, and additional operational details are provided in Sect. S2. The CIMS was equipped with a long time-of-flight mass spectrometer (LTOF), achieving a mass resolution of ∼ 7000 at mass-to-charge ratio () of 149 Th in DEA mode, and ∼ 8500 at 125 Th in nitrate mode. Sampling was conducted through a 90° bent 3/4-inch stainless-steel inlet tube (total length: 40 cm; 25 cm from the Eisele inlet to the bend, and 15 cm from the bend to the flow-tube outlet), positioned to sample from the center of the open outlet of the flow-tube. The 90° bend allows side sampling without disturbing the laminar outflow. The reaction time within the inlet is calculated to be 0.46 s, resulting in a total effective reaction time of roughly 8.8 s (at the reaction distance of 110 cm) for measurements taken by the CIMS. The same inlet tube was used in sulfuric acid calibration experiments for the CIMS (Kürten et al., 2012). Based on a two-dimensional (2-D) flow reactor calibration model (He et al., 2023), a calibration factor of 7.3×109 cm−3 (at least ± 50 %) was determined, and applied for both DEA and nitrate mode for product quantification (see Sect. S2).
A Vocus proton-transfer-reaction time-of-flight mass spectrometer (Vocus PTR-TOF, Tofwerk AG) was used to validate the syringe pump system (see Sect. S2 for details). Initial concentrations of O3 and NO2 were measured using gas monitors: a photometric O3 analyzer (model 400, Teledyne API) and an NO-NO2 analyzer (model T200UP, Teledyne API), respectively (see Sect. S2 for details). The O3 analyzer was also used to quantify O3 consumption during the reaction time characterization experiments involving the reaction TME + O3. Data from both the Vocus PTR-TOF and DEA-/nitrate-CIMS were analyzed using the Igor-based Tofware software (Tofware_3_3_0).
2.4 Box model
We constructed a simple 0-D box model to describe the chemical reactions occurring in the flow-tube system with a defined effective reaction time. Rather than simulating specific RO2 or closed-shell products, the model was primarily used to estimate the concentrations of reacted MTs (or equivalently, the total amount of products) resulting from NO3, O3, and OH oxidation, based on the dominant reaction pathways (Table S1 in the Supplement). Specifically, in the case of the flow-tube system, the model was used in batch mode, meaning no continuous input was applied and only the initial concentrations of precursors were set. The simulation spans the entire experimental setup, from the pre-reactor through the injector to the free-jet flow-tube, as illustrated in Fig. S2. The NO3 oxidation product molar yields (or fractions) were calculated via dividing the measured product concentrations by the modeled MT concentration ([MT]) reacted by the NO3 radical (Shen et al., 2021).
3.1 Experiment overview and the determination of NO3 oxidation products and HOMs
A total of ten sets of NO3 oxidation experiments were conducted, two for each of the five MTs using both DEA- and nitrate-CIMS, with time series presented in Figs. 2 and S3–S6. The experimental procedure varied the initial concentrations of MT (i.e., [MT]0), NO2, and O3 across distinct stages (concentrations for each stage shown directly at the top of the plots in Figs. 2 and S3–S6). The sequence of these stages included: two initial background stages (with and without MTs); four MT-ramping stages (varying [MT]0 from 10 to 120 ppb); three background stages (ramping [MT]0 down to 20 ppb); three NO2-ramping stages (varying [NO2]0 from 32.5 to 86 ppb); a single O3 perturbation stage (with decreased [NO2]0 and a 50 % increase in [O3]0); and finally, three different background stages. The experimental stages, excluding background stages, are numbered from 1 to 8 (Fig. 2).
During the MT-ramping stages (Stages 1–4 in Fig. 2), products from O3 or OH (produced from MT + O3) oxidation are expected to increase substantially, as the chemical conversion ratio of both MT and O3 is small (Sect. S1). In contrast, NO3 oxidation products would show a much slower increase, limited by the NO3 radical concentration. This differential behavior is clearly illustrated in Fig. S7, where the ratio of reacted [MT] by NO3 to total reacted [MT] by all oxidants is shown to decrease significantly as [MT]0 is ramped from 10 to 120 ppb. Conversely, during the NO2-ramping stages (Stages 5–7), NO3 oxidation products would increase notably due to the rising NO3 radical concentration, while O3/OH oxidation products would decrease slightly due to enhanced O3 consumption by NO2. In the O3 perturbation stage (Stage 8), where [O3]0 was increased by 50 %, O3/OH oxidation products are expected to increase accordingly, while NO3 oxidation products are expected to stay almost unchanged or even decrease due to the lower [NO2]0. This systematic design allowed us to attribute product origins. For example, in the AP experiments (Fig. 2), compounds such as C10H15O and C10H14O7 were identified as ozonolysis products, while C10H16NO and C10H16O2 were identified as NO3 oxidation products. It should be noted that for the DEA mode (Figs. 2A, and S3A–S6A), the injection of MT without added oxidants increased the signal for certain products (e.g., C10H16O2), possibly due to MT impurity or surface reactions. Consequently, the background stages with MT injection were used for background subtractions.
The volatility of multifunctional organic molecules is a function of their carbon number and the type and quantity of functional groups they contain. While a high oxygen-to-carbon ratio (O : C) typically indicates low volatility, it has been suggested that the nitrate group (–ONO2) contributes to volatility reduction by an amount comparable to a single hydroxyl group (–OH) (Chuang and Donahue, 2016). Therefore, products with a nitrate group (e.g., those initiated by NO3 radicals) necessitate a higher total oxygen atom count than the common threshold (≥6 oxygen atoms) established for non-nitrate HOMs from O3/OH oxidation (Bianchi et al., 2019) to reach an equivalent Low Volatility Organic Compound (LVOC) or Extremely LVOC (ELVOC) regime.
O3, OH or NO3 can initiate oxidation by directly adding to a double bond, leading to a C-centered radical followed by fast O2 addition, which yields a primary RO2 (Atkinson and Arey, 2003) that can then undergo autoxidation (Ehn et al., 2014). The primary peroxy radicals will contain 2–5 O atoms, depending on oxidant and whether the cleavage of the double bond results in fragmentation or not. It is worth noting that a minor oxidation pathway of H-abstraction by OH radical was previously reported (Shen et al., 2022; Vereecken and Peeters, 2000), producing a sequence of radicals such as C10H15O. However, we did not see any noticeable amount of radical C10H15Ox that could be attributed to NO3 oxidation, meaning that its possible H-abstraction pathways were negligible in our flow-tube experiments.
The recommended definition for HOMs is that they have undergone autoxidation and contain 6 or more O-atoms. We take a slightly more restrictive approach here, since NO3 oxidation adds more O-atoms to the RO2 than the more often studied O3 and OH oxidation. When using the term “HOM”, we use the following criteria: RO2 radicals with 8 or more oxygen atoms (C10H16NOx, x≥8), closed-shell products with 7 or more oxygen atoms (C10H15,17NOx, x≥7), and peroxynitrates with 10 or more oxygen atoms (C10H16N2Ox, x≥10); HOM dimers are defined as species with 9 or more oxygen atoms (starting from C20H32N2O9, formed by C10H16NO5 + C10H16NO6). We also include “cross dimers” from NO3-initiated RO2 reacting with O3- or OH-initiated RO2, i.e. C20H31NOx/C20H33NOx with x≥8 or x≥7, respectively, as the initial RO2 from O3/OH oxidation has four or three oxygen atoms.
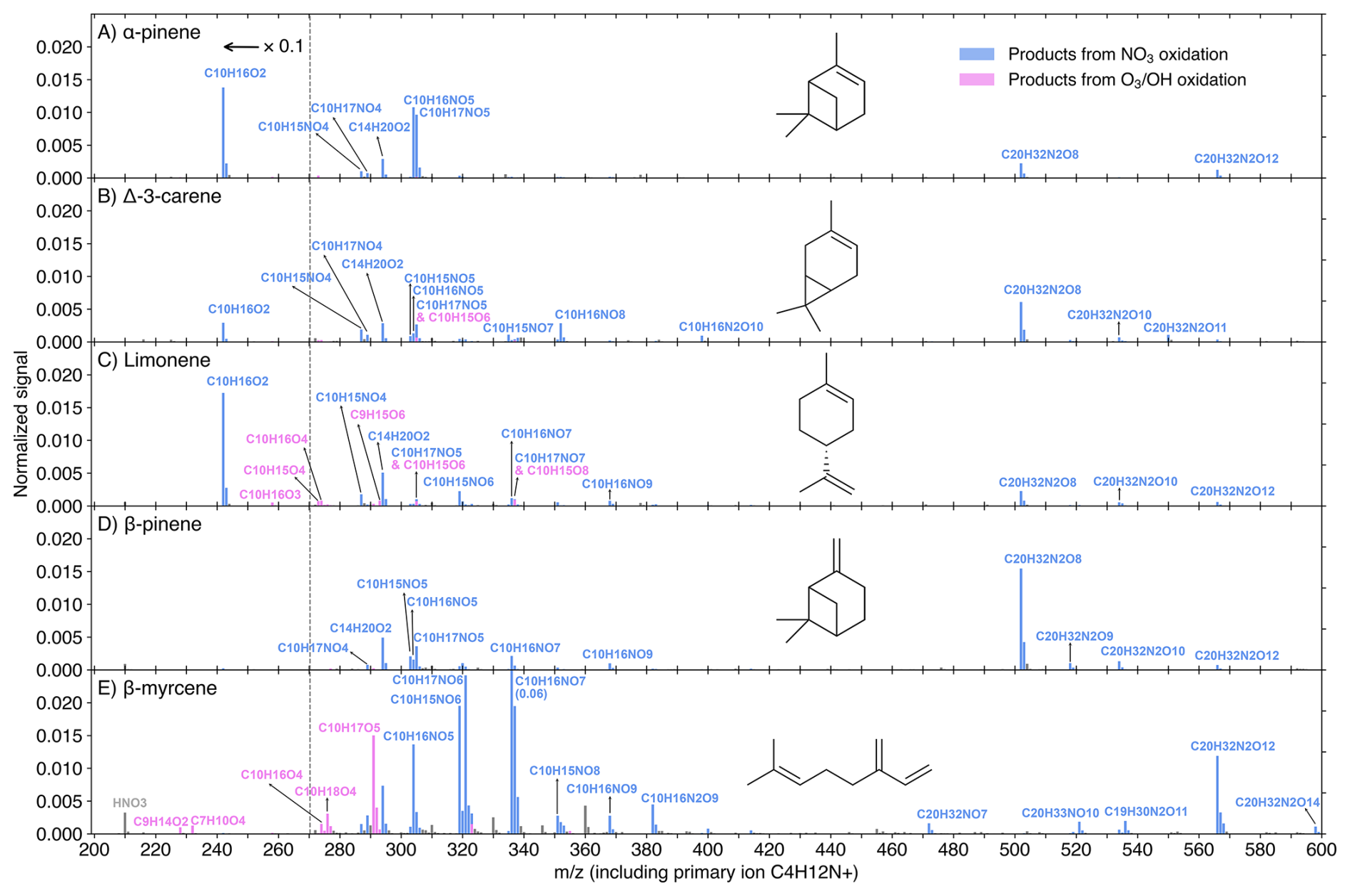
Figure 3DEA-CIMS spectra (10 min average) of MT + NO3 reactions at the experimental stage 6. Initial concentrations were [MT]0=20 ppb, [O3]0=12 ppb, and [NO2]0=50 ppb, with an effective reaction time of 8.8 s. The spectra display the normalized peak signals (see Eq. S4) and were corrected by subtracting background signals. Blue bars represent products (including both radicals and closed-shell species) from NO3 oxidation, pink bars show products from O3 or OH oxidation, and grey bars are for peaks not of interest. All compounds of interest were charged by clustering with the C4H12N+ reagent ion, which is omitted from the displayed molecular formulas for clarity. Peaks below 271 Th are divided by 10.
A comparison of concentrations for major monomer compounds (O ≥7), dimers (O ≥11), and peroxynitrates (O ≥10) between nitrate and DEA modes is presented in Fig. S8. For all MTs except carene, the first-autoxidation RO2 (C10H16NO7) was detected much more efficiently by the DEA mode. Conversely, the nitrate mode exhibited greater selectivity for products resulting from two or more autoxidation steps (i.e., RO2 with O ≥9, or O ≥8 if one RO2 to RO conversion occurred) for all MTs except myrcene. Closed-shell monomers corresponding to these two-autoxidation-step RO2 species (with O ≥8 or 7) showed comparable concentrations between the two modes, with differences being less than one order of magnitude only for BP and myrcene. Therefore, the monomer results are consistent with the fact that nitrate-CIMS is usually utilized for investigating HOMs due to its high selectivity (Bianchi et al., 2019; Riva et al., 2019). It is notable that some defined HOM dimers exhibited higher signals in the DEA mode (Fig. S8). Furthermore, the HOM dimers C20H32N2O9,10 were absent from nitrate spectra (Fig. 4), despite their non-negligible signals in the DEA spectrum of MTs such as BP (Fig. 3D).
3.2 Peroxy radicals and closed-shell products from DEA- and nitrate-CIMS
As the DEA and nitrate modes collectively cover a wide range of oxidation products (O ≥2), their representative spectra (Figs. 3 and 4) provide insightful molecular information on the NO3 oxidation products of the five MTs. Normalized signals are displayed for the experimental stage 6 ([MT]0=20 ppb, [O3]0=12 ppb, and [NO2]0=50 ppb), where the NO3 radical dominates the oxidation (Fig. S7), making these spectra highly representative of the NO3-initiated chemistry. It is worth noting that products in the DEA mode were charged by clustering with C4H12N+, while products in the nitrate mode were charged by clustering with either a primary ion monomer (NO or 15NO or a dimer (H15NO3⋅ 15NO or HNO3⋅ 15NO or HNO3⋅ NO) (see Fig. S9 for detailed charging scheme). For further investigation of the molar yields and trends of individual products as a function of reacted [MT] by NO3 radicals, Figs. 5 and S10 utilize data from five experimental stages (2 and 5–8) with [MT]0=20 ppb and the NO3 oxidation was the dominant oxidation process (Fig. S7).
The DEA-CIMS spectra (Fig. 3) show that the closed-shell species C10H16O2 was the dominant product for limonene, AP, and carene, with yields exceeding 50 % for the former two (Fig. 5A and C). This finding is consistent with earlier chamber studies which identified pinonaldehyde and endolim as the major NO3 oxidation products of AP (reported yields of 39 %–71 %) and limonene (reported yields of 25 %–72 %), respectively (Hallquist et al., 1999; Spittler et al., 2006; Wängberg et al., 1997). Although the yield of caronaldehyde from carene + NO3 was previously estimated to be minor (2 %–3 %; Hallquist et al., 1999), our results suggest that C10H16O2 represents the largest single molar yield for carene at approximately 10 % (Fig. 5B). The formation of C10H16O2 can be attributed to bimolecular reactions of the peroxy radical C10H16NO5 forming the nitrooxy alkoxy radical (RO) C10H16NO4. This RO radical then undergoes nitrooxy-side β-scission (i.e., breaking the C–C bond in ⋅ O–C–C–ONO2), a process followed by NO2 loss, resulting in the C10H16O2 product (Fig. S11) (Bates et al., 2022; Draper et al., 2019; Kurtén et al., 2017; Novelli et al., 2021). The comparative low C10H16O2 yield observed from carene + NO3 is consistent with its alkoxy radical C10H16NO4 tending toward alkyl-side β-scission (i.e., breaking of the RC–CO⋅ bond), which favors further autoxidation over C10H16O2 formation (Fig. S11) (Draper et al., 2019; Kurtén et al., 2017).
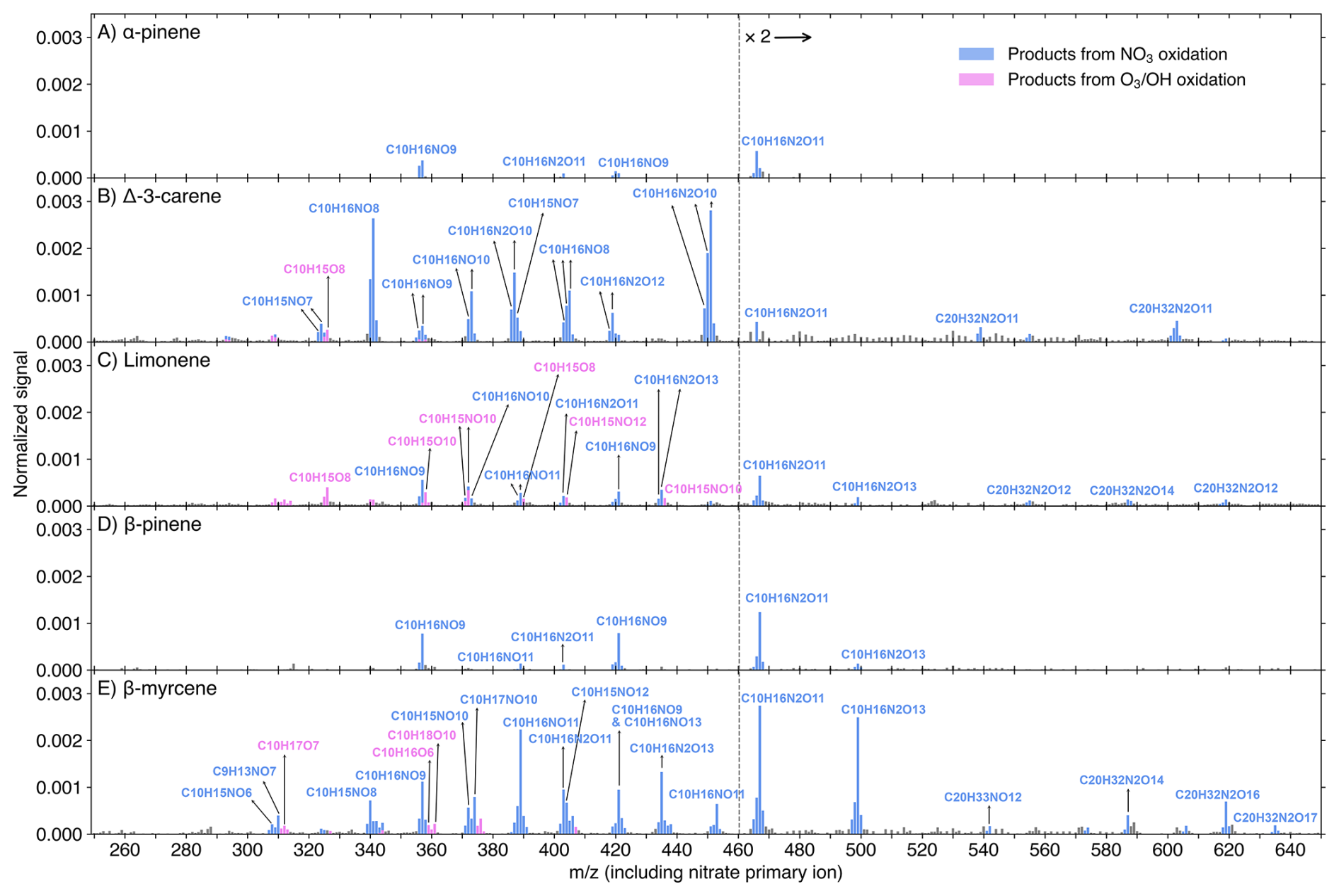
Figure 4Nitrate-CIMS spectra (10 min average) of MT + NO3 reactions at the experimental stage 6. Initial concentrations were [MT]0=20 ppb, [O3]0=12 ppb, and [NO2]0=50 ppb, with an effective reaction time of 8.8 s. The spectra display the normalized peak signals (see Eq. S3) and were corrected by subtracting background signals. Blue bars represent products (including both radicals and closed-shell species) from NO3 oxidation, pink bars show products from O3 or OH oxidation, and grey bars are for peaks not of interest. Compounds of interest were charged by clustering with either a primary ion monomer (e.g., 15NO or a dimer (e.g., H15NO3⋅ 15NO), which is omitted from the displayed molecular formulas for clarity. Note that, in some cases, multiple arrows point to a single formula from different because one molecule can cluster with both 15N-labeled primary ions and non-labeled ones, resulting in two or three adjacent mass-to-charge ratios corresponding to the same molecule. The detailed charging schemes are shown in Fig. S9. Peaks above 460 Th are multiplied by 2.
This favored alkyl-side β-scission for carene would initiate a sequence of RO2 radicals (C10H16NO), aligning with the high signals of C10H16NO8,10 observed (Figs. 3B–5B). The relative low concentration of C10H16NO6 suggests rapid autoxidation to the more oxygenated C10H16NO8. Conversely, the absence of C10H16NO8 and the low signals of C10H16NO6 for AP and limonene align with their high yields of C10H16O2 (Figs. 3–5), reinforcing that the alkyl-side β-scission is unfavored for these two MTs. For limonene, NO3 addition could also occur at the exocyclic double bond forming C10H16NO5, then leading to the formation of C10H16O2 via bimolecular reactions or initiation of autoxidation (Mayorga et al., 2022). The dominance of odd-oxygen-number RO2 (C10H16NO7,9) for AP and limonene (Figs. 3–5) indicates “direct” autoxidation steps from C10H16NO5 with minor RO2 to RO conversion. It is noteworthy that, for limonene, the second NO3 addition to the double bond of C10H16O2 could directly produce C10H16NO7, followed by autoxidation to C10H16NO9,11 (Guo et al., 2022). But this second NO3 addition was unfavored due to the short effective reaction time at 8.8 s. Overall, the comparatively low signals and yields of each HOM for AP and limonene (panels A and C in Figs. 4, 5, and S10), suggest that they do not possess ring-opening processes as effective as carene for fostering subsequent autoxidation, and the second NO3 addition for limonene is negligible.
In contrast, BP and myrcene, which do not have the same endocyclic double bond as the other three MTs, did not generate C10H16O2 in NO3 oxidation (Fig. 3). BP has the highest yields of the radical C10H16NO6 and closed-shell species C10H15,17NO5 (formed from bimolecular reactions of C10H16NO6) among the five MTs (Figs. 3 and 5), supporting the mechanism proposed by Claflin and Ziemann (2018), where the RO radical C10H16NO4 from bimolecular reactions of C10H16NO5, experiences a ring-opening process leading to C10H16NO6. Subsequent bimolecular reactions can convert C10H16NO6 to the RO radical C10H16NO5, which can consequently trigger another ring-opening to form C10H16NO7 after O2 addition. With less constraints by rigid cyclic structures (Kurtén et al., 2015), C10H16NO7 is expected to easily undergo autoxidation steps. The presence of considerable C10H16NO7,9 and the absence of C10H16NO8 (Figs. 3D and 4D) suggest the second ring-opening process and subsequent autoxidation. Although abundant C10H16NO8 was observed in an earlier chamber study focusing on BP + NO3 (Shen et al., 2021), it was not formed fast as C10H16NO9 at the beginning based on their timeseries, suggesting that C10H16NO8 likely resulted from C10H16NO7 undergoing RO-forming bimolecular reactions followed by one autoxidation step. Furthermore, due to the short reaction time of 8.8 s in our flow-tube experiments, we did not detect second-generation peroxy radicals (i.e., those resulting from two NO3 additions) with two nitrogen atoms, which were reported in Shen et al. (2021).
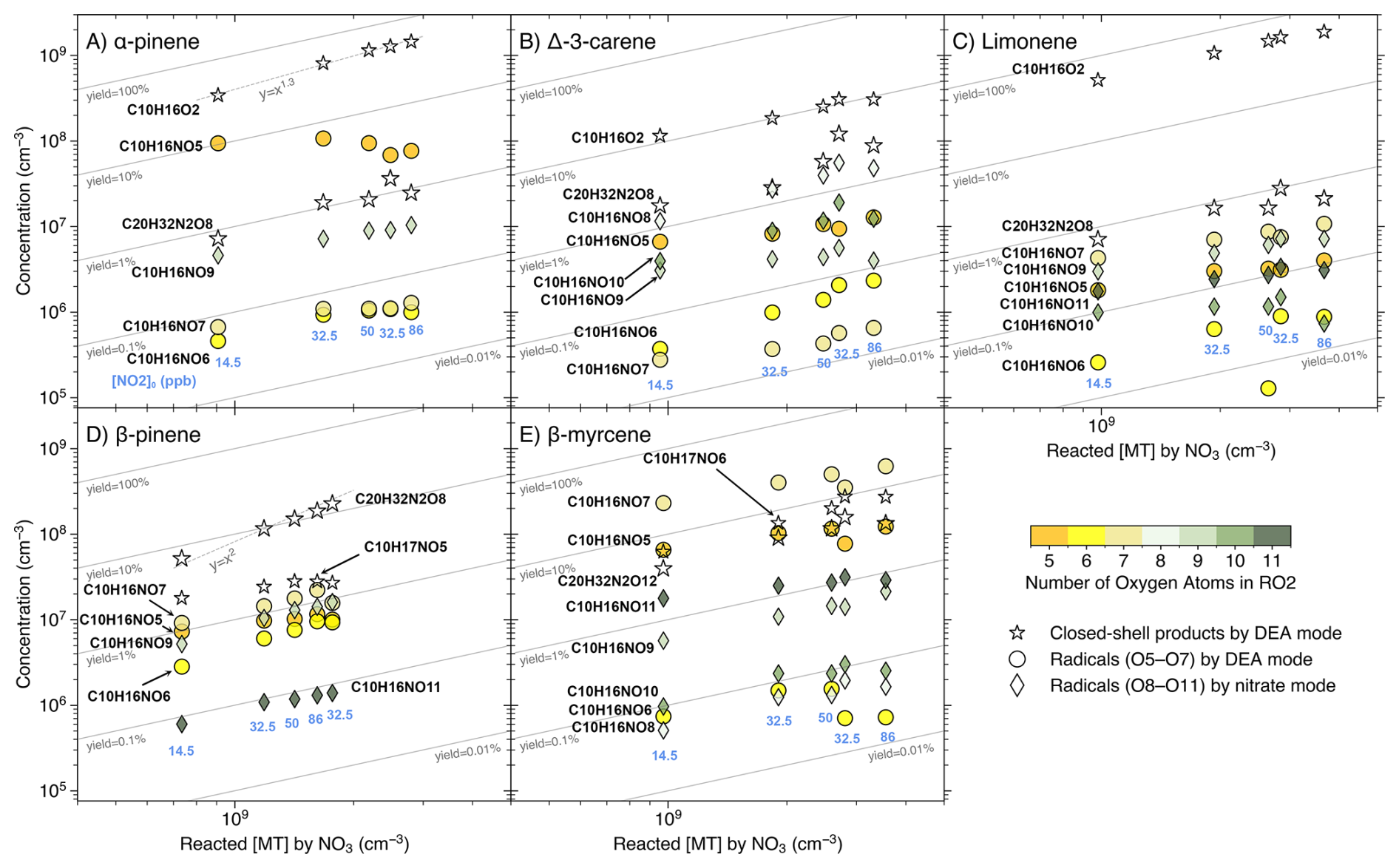
Figure 5Concentrations (in cm−3) of the dominant closed-shell monomer and dimer, and the RO2 radicals, from the MT + NO3 reactions, as a function of reacted [MT] by the NO3 radical. Data from experimental stages 2 and 5–8 are presented as 10 min averaged measurements taken at an effective reaction time of 8.8 s. The reacted [MT] by NO3 was varied primarily by increasing [NO2]0 (values in ppb shown beneath each column). The only exception is the transition from stage 7 to stage 8, where [O3]0 increased from 12 to 18 ppb and [NO2]0 decreased from 86 to 32.5 ppb (see Fig. 2). Radicals are grouped and colored by the number of oxygen atoms: circles represent radicals with O =5 to 7 measured by the DEA mode, and diamonds show radicals with O =8 to 11 measured by the nitrate mode. The dominant closed-shell monomer and dimer measured by the DEA mode are both shown by hollow stars, with the dimer distinguished by a larger marker size.
The acyclic structure, featuring three double bonds, makes the reaction of myrcene with the NO3 radical mechanistically distinct from the other four cyclic MTs. The dominance of odd-oxygen-number peroxy radicals C10H16NO suggest the fast autoxidation steps of myrcene comparing to RO-forming bimolecular reactions (Figs. 3E–5E). Moreover, the ring closures between RO2 and double bonds are normally fast, leading to the formation of C–O–O–C peroxide groups (Vereecken et al., 2021). The proposed ring-closure processes are supported by the relative detection efficiency of myrcene + NO3 products by DEA and nitrate modes (Fig. S8E): only when RO2 radicals reached C10H16NO10,11 and dimers reached C20H32N2O18, were they detected more efficiently by the nitrate mode. Because after two ring-closure steps (which consume the remaining two double bonds following NO3 addition), those species would just begin to possess –OH/–OOH groups (via H-shift), which enhance the detection efficiency by the nitrate mode as hydrogen bond donors (Hyttinen et al., 2015). Considering bimolecular reactions RO2 + R'O2 forming ROH and R'C=O (Bianchi et al., 2019), the substantial amount of closed-shell species C10H15,17NO potentially indicates that these RO2 bimolecular reactions were fast for myrcene (Figs. 3E, 4E, and S10E).
It's notable that for AP, the concentration of the closed-shell monomer C10H17NO5 is almost 100 times higher than that of the RO2 radical C10H16NO6, without the presence of the closed-shell C10H15NO5 (Figs. 3A, 5A, and S10A). This suggests that the formation of C10H17NO5 is likely due to the reaction C10H16NO5 + HO2, which was reported as an important pathway with a high molar yield (Bates et al., 2022). The potential involvement of HO2 in other MTs could also be indicated by the relative abundance of some closed-shell monomers. For instance, the C10H17NO5 was more abundant than C10H15NO5 for both carene and BP (Fig. 3).
An unexpected observation is the considerable amount of C14H20O2 across all MTs (Fig. 3). Since this species always increased when both MTs and NO3 were available and followed trends similar to other NO3 products (Figs. 2 and S3–S6), it is attributed to NO3 oxidation. Its presence even for BP and myrcene (Figs. S5 and S6), which did not have C10H16O2, rules out the initial speculation that it was formed by C10H16O2 clustering with a C4H4 (which is possibly a fragment of the primary ion and had constant high concentration for all MTs; see Fig. S5A for example), and suggests the involvement of some unknown fragmentation processes.
The RO2 bimolecular reactions also form dimers (ROOR') besides RO radicals and closed-shell monomers (Bianchi et al., 2019). The dimer C20H32N2O8 (from two C10H16NO5) was the dominant dimer for all MTs except myrcene, where the most abundant dimer was C20H32N2O12, likely formed from two C10H16NO7 (Fig. 3). It's notable that among all MTs, BP has the highest yield of C20H32N2O8, which is approximately an order of magnitude higher than its corresponding peroxy radical C10H16NO5 (Figs. 3 and 5). Claflin and Ziemann (2018) proposed that in BP + NO3, C20H32N2O8 was formed in the particle phase by two closed-shell monomers, however, our observation suggests that the C20H32N2O8 could be formed rapidly in the gas phase. It remains unclear why BP in particular has such a high yield of this dimer. While we cannot rule out that this signal is impacted by some undesired chemical or instrumental effects, it is puzzling why such an effect would be so dramatic only for BP.
The dimer patterns overall match the RO2 radical distributions. AP, limonene, and myrcene, having major RO2 radicals with odd oxygen number, produced series of even-oxygen-number dimers C20H32N2Ox (Figs. 3–4). On the contrary, with considerable even-oxygen-number RO2 radicals present alongside the odd ones, carene and BP also have noticeable amounts of odd-oxygen-number dimers C20H32N2O11 (via C10H16NO5 + C10H16NO8) and C20H32N2O9 (via C10H16NO5 + C10H16NO6), respectively. In the DEA spectrum of myrcene (Fig. 3E), the formation of dimers C20H32NO7 and C19H30N2O11 was likely due to the fragmentation occurring during bimolecular reactions of two NO3-initiated RO2 (namely NO3-RO2), while C20H33NO10 could be from a NO3-RO2 (C10H16NO7) reacting with an OH-RO2 (C10H17O5).
From both DEA and nitrate spectra, we observed abundant peroxynitrates (RO2NO2) from NO3-RO2 + NO2, such as C10H16N2O10 for carene (Figs. 3B and 4B) and C10H16N2O for myrcene (Figs. 3E and 4E), and the yields of these RO2NO2 expectedly increased with higher NO2 concentrations (Fig. S10). Earlier studies using NO2 + O3 as NO3 source also reported noticeable amounts of RO2NO2 (Dam et al., 2022; Draper et al., 2019; Guo et al., 2022; Mayorga et al., 2022). These RO2NO2 were especially significant among the HOMs in nitrate spectra (Fig. 4) and tended to be charged by clustering with dimer primary ions (e.g., H15NO3⋅ 15NO (Fig. S9). Furthermore, we also observed a noticeable amount of C10H15NO10,12 in NO3 + limonene (Figs. 4C and S4B), as previously reported by Guo et al. (2022). However, instead of attributing these species directly to NO3 oxidation products, we regard them as RO2NO2 formed from C10H15O8,10 (O3-RO2) + NO2, since their behavior in the time series mirrored that of C10H15O8,10 during stages 1–4 (Fig. S4). Generally, more oxygenated RO2 radicals exhibited higher conversion to RO2NO2 (Fig. S12). This is likely due to the formation of more stable RO2NO2 structures, such as peroxyacetyl nitrates (PANs), which feature longer atmospheric lifetimes (Atkinson, 2000; Russell et al., 2025).
Generally, the concentrations of most NO3 products increased monotonically with the rising reacted [MT] by the NO3 radicals (Figs. 5 and S10). The most notable exception is that for AP, the peroxy radical C10H16NO5 and its closed-shell monomer C10H17NO5 significantly decreased with increasing reacted [MT], beyond expectation (Figs. 5A and S10A). As all stages in Figures 5 and S10 have the same [MT]0, the reacted [MT] by NO3 is directly proportional to the amount of NO3 in the system. The faster-than-linear increase of C10H16O2 (which approximately follows a 1:1.3 line, as shown in Fig. 5A) suggests that with increasing [NO3], the peroxy radical C10H16NO5 was converted to C10H16O2 more efficiently. Also, AP has the highest yield of the radical C10H16NO5 among all five MTs studied (Fig. 5), proposing that both autoxidation and bimolecular reactions are comparatively slow. The findings appear to be consistent with the previous studies that estimated the SOA yield of AP + NO3 to be near 0 % using N2O5 as NO3 source (Fry et al., 2014), as high [NO3] could significantly enhance the role of the RO2 + NO3 reaction in the experiments, leading to the formation of the comparatively volatile C10H16O2 (Bates et al., 2022). We observed that only the dimer C20H32N2O8 from BP + NO3 follows a near-quadratic trend with the reacted [MT] (Fig. 5D). This indicates that the reaction coefficient of the dimer-forming bimolecular reaction is the fastest compared to other competing pathways, which is consistent with C20H32N2O8 being the main product (Figs. 3D, 5D, and S10D). It is noteworthy that the data points corresponding to the column with [NO2]0=32.5 ppb but the higher reacted [MT] (stage 8), often deviated from the general trend (Figs. 5 and S10). This deviation occurred because the increased [O3]0 in stage 8 enhanced the production of O3/OH oxidation products, such as O3/OH-RO2 radicals, which subsequently affected the concentrations of NO3 oxidation products.
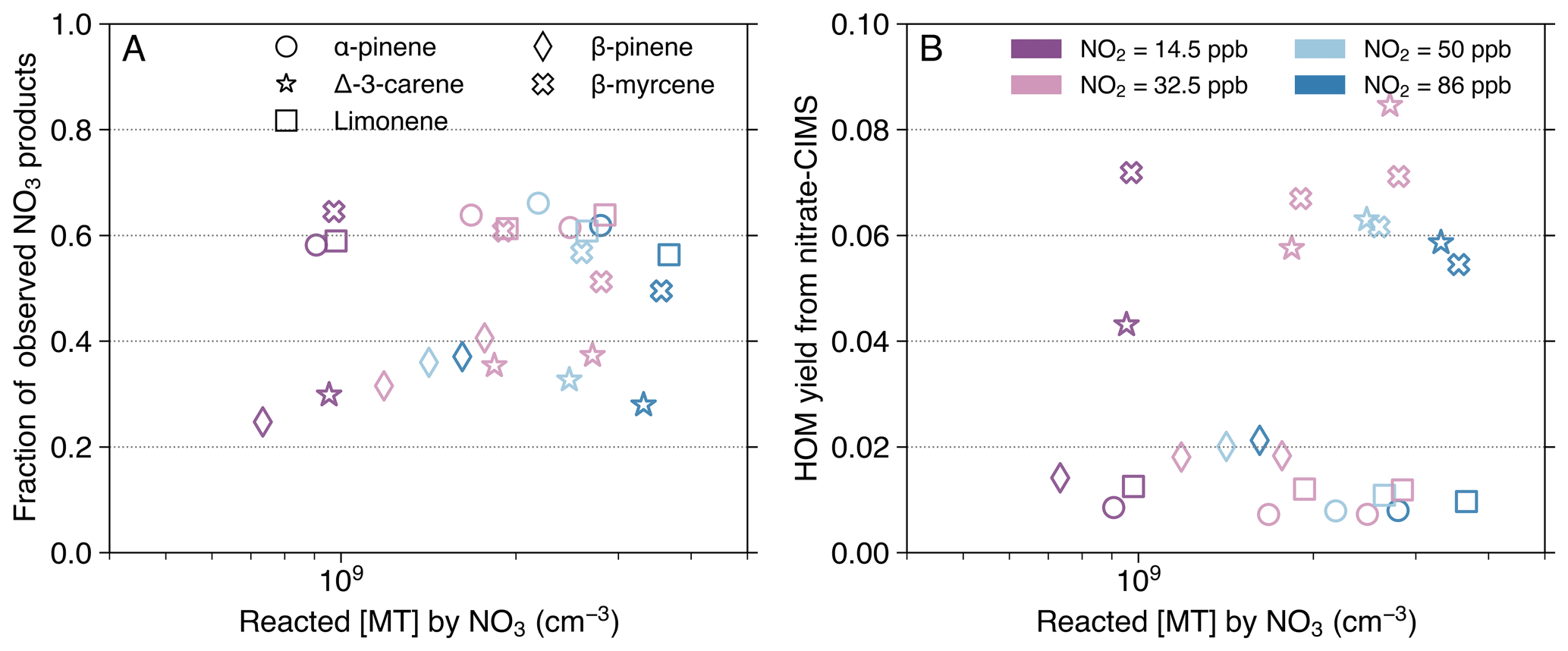
Figure 6Fraction of observed NO3-initiated products out of the total NO3-oxidized MT (A) and HOM yield (B), as a function of reacted [MT] by the NO3 radical. Data from experimental stages 2 and 5–8 are presented as 10 min averaged measurements taken at an effective reaction time of 8.8 s. The HOM yield (B) is solely based on nitrate-CIMS measurement. The calculation for the fraction of observed NO3-initiated products (A) combines HOM concentrations from the nitrate mode with less oxygenated products from the DEA mode. Exceptions were made for some HOM dimers: C20H32N2O9,10 (for all MTs) and C20H32N2O12 (for myrcene) were taken from the DEA mode due to their absence in the nitrate spectra (Fig. 4E). Dimer concentrations were multiplied by a factor of 2 to represent their monomer-equivalent contribution. Different MTs are distinguished by marker shape: circle (AP), star (carene), square (limonene), diamond (BP), and cross (myrcene). Marker colors denote the distinct [NO2]0 used in the experiments. Note that stages 5 and 8 have the same [NO2]0=32.5 ppb, but stage 8 has a higher O3 level and thus a higher reacted [MT] by NO3.
3.3 Product closure and HOM yields
Product closure was reasonably reached for AP, limonene, and myrcene, given that the fractions of observed NO3-initiated products out of the total NO3-oxidized MT were calculated to be 50 %–70 % (Fig. 6A). Considering the inherent uncertainties stemming mainly from the calibration factor applied to CIMS quantification (Sect. S2) and the estimated coefficients of NO3 and N2O5 wall losses within the box model (Table S1), it is possible that we were able to observe almost all the products. On the contrary, carene and BP exhibited lower fractions, at 20 %–40 % (Fig. 6A). This reduced closure could be attributed to the unaccounted formation of products containing one O-atom, as these cannot be effectively detected by the DEA mode (Riva et al., 2019). An earlier flow-tube study reported that the yield of nopinone (C9H14O) from BP + NO3 varied substantially (20 %–80 %) across different experimental pressures and NO concentrations (Berndt et al., 1999). To our knowledge, no previous studies have explicitly identified and quantified any major products containing only one O-atom from carene + NO3, proposing either a new reaction pathway or a severely underestimated calibration factor for some carene products.
To maintain consistency with previous studies, which exclusively used nitrate-CIMS for HOM investigations (Dam et al., 2022; Day et al., 2022; Draper et al., 2019; Ehn et al., 2014; Guo et al., 2022; Harb et al., 2025; Li et al., 2024; Luo et al., 2022; Shen et al., 2021; Zhang et al., 2024a, b; Zhao et al., 2024), HOM yields were calculated solely using data from the nitrate mode in this study. However, we note that the HOM yields may be underestimated because of the low selectivity of nitrate-CIMS toward some HOM dimers with relatively low oxygen numbers (discussed in Sect. 3.1). One example is that the proposed formation of C–O–O–C groups inhibited the effective detection by the nitrate mode of some HOMs formed from myrcene + NO3 (such as C10H16NO9). Despite this underestimation, the acyclic myrcene exhibits the highest average HOM yield (∼6.5 %; Fig. 6B), as could be expected, since its open structure imposes fewer constraints on autoxidation compared to the cyclic MTs. Among the four cyclic MTs, NO3 oxidation of carene produced the highest average HOM yield at approximately 6.1 %, closely approaching that of myrcene. This is consistent with the finding that carene undergoes a ring-opening process (i.e., the alkyl-side β-scission of the nitrooxy alkoxy radical) that facilitates autoxidation (Sect. 3.2; Figs. 3–5, and S11) (Draper et al., 2019; Kurtén et al., 2017). The HOM yield of BP + NO3 (at ∼1.8 %) exceeds that of AP (∼0.8 %) and limonene (∼1.1 %). Directly compared to our results, Guo et al. (2022) reported a similar HOM yield (1.5 %) for limonene + NO3, while Shen et al. (2021) estimated a much higher HOM yield (4.8 %) for BP. Due to the difference of experimental setups and conditions (e.g., reaction time and NO3 source), direct quantitative comparison of HOM yields across various studies is often challenging. Nevertheless, if combining findings from earlier studies that have measured HOM yields from more than one MT using the same sampling system, we can conclude that HOM yields from ozonolysis can be listed in the order (from highest to lowest): limonene > carene > AP > myrcene > BP (Jokinen et al., 2015; Zhao et al., 2024). This order contrasts with the NO3 oxidation results in this study. The relatively low HOM yield of myrcene + O3 is likely due to decomposition into smaller fragments following the breaking of the primary ozonide (Atkinson and Arey, 2003). Conversely, Jokinen et al. (2015) reported myrcene having the highest HOM yield in OH oxidation (∼1 %), followed by limonene, BP, and AP.
We can compare the HOM yields from NO3 oxidation (Fig. 6B) to their corresponding SOA formation reported in earlier studies. These studies consistently found that AP has the lowest SOA yield (DeVault et al., 2022; Hallquist et al., 1999), even approaching zero (Fry et al., 2014). This is possibly due to excessive [NO3] that favors volatile product formation, as discussed in Sect. 3.2. However, the relative SOA yields for cyclic MTs vary across different studies (DeVault et al., 2022; Fry et al., 2014; Hallquist et al., 1999), thereby making comparisons to our HOM yields inconclusive. The discrepancies may relate to different conditions during the experiments and suggest that simultaneous measurements of HOMs might have shed some light on the relevant reaction pathways taking place in the experiments.
3.4 Implications, challenges, and limitations
Our findings validate the previous observations that small structural differences among MTs strongly influence the product distributions during oxidation, and demonstrate that the HOM yield of an individual MT from NO3 oxidation can differ significantly from that by O3/OH oxidation. Still, the overall HOM yields are comparable between oxidants, generally falling in the range 0 %–10 %, although yields from OH oxidation are typically the lowest (Bianchi et al., 2019; Jokinen et al., 2015; Zhao et al., 2024). Since HOMs are a large source of SOA (Ehn et al., 2014), the SOA formation is expected to vary between day and night, depending on the VOC distribution and the dominant oxidant (e.g., OH vs NO3). For instance, if limonene is the key precursor driving SOA formation during the day, its role would diminish at night because the rapid reaction limonene + NO3 yields far fewer HOMs and more of the volatile compound C10H16O2. The correlation between HOM formation and SOA mass is most evident at low OA mass concentrations, where extremely low-volatile HOM species dominate new particle formation and early growth. In more polluted environments with higher pre-existing OA mass, however, low-volatile and semi-volatile (i.e., non-HOM) oxidation products can contribute substantially to SOA mass. Therefore, HOM and SOA yields are not always closely related, and their relationship can vary significantly across different chemical regimes and environments (with differences in VOC composition, oxidant conditions, and background OA loadings). The difference between the relative magnitudes of HOM and SOA yields of each MT (DeVault et al., 2022; Fry et al., 2014; Hallquist et al., 1999) can partly be explained by the potentially underestimated HOM yields and contribution of non-HOM products to SOA formation, but also by differing reaction conditions between studies. In our flow-tube experiments, we have much shorter residence time compared to traditional chamber SOA yield experiments (seconds vs. hours), during which multigenerational products can form and contribute to SOA formation, for instance, NO3 addition to the first-generation product C10H16O2 from limonene + NO3 reactions. Moreover, particle-phase reactions occurring after condensation introduce additional uncertainties when directly comparing our HOM yields with previously reported SOA yield results. Overall, our results revealed that the NO3 oxidation mechanisms are highly structure-dependent, and their contribution to atmospheric particles varies significantly among different MTs.
A major challenge in studying NO3 chemistry is replicating atmospherically relevant RO2 radical fates. Since we used NO2 + O3 to supply NO3, the excessive NO2 relative to NO3 (Fig. S2) resulted in the formation of a noticeable amount of RO2NO2 (Figs. 3, 4, S9, S10, and S12). This competing pathway (RO2 + NO2) may obscure the importance of other reactions involving RO2 radicals. While using the thermal decomposition of N2O5 could avoid the RO2NO2-forming issue, it often provides too high [NO3], causing the bimolecular reaction RO2 + NO3 to dominate (Bates et al., 2022). For instance, this excessive [NO3] pushes AP + NO3 to form more volatile C10H16O2, underestimating the importance of AP in SOA formation. This highlights the inherent difficulty in studying NO3 + MTs mechanisms in laboratory settings.
One major limitation of our study is the incomplete product closure observed for carene and BP, possibly due to the DEA mode failing to detect products containing one O-atom. The formation of these unseen products can be investigated by future studies using instruments with better selectivity. Another limitation is the inability to perform dilution experiments without a well-designed dilution unit attached to the CIMS inlet, preventing us from ruling out potential ion-induced reactions taking place in the chemical ionization inlet (Berndt et al., 2016; Peräkylä et al., 2023). However, as the levels of reacted [MT] in this study are comparable to earlier flow-tube studies where ion-induced dimer formation was found not to be significant (Berndt et al., 2018a; Peräkylä et al., 2023), we do not expect such processes to be dominant in our experiments. The design of the dilution unit is crucial, as our unit caused too much turbulence that decreased all signals far beyond the expected dilution factor, warranting future study. It is worth noting that the reagents which are selective to low-oxygenated compounds (O <6), such as the DEA used in this study, can be very sensitive to contaminants. For example, nitric acid (HNO3) from the NO2 cylinder initially depleted our primary ion (C4H12N+), affecting quantification, but the addition of a HEPA filter (Whatman plc) after the gas cylinder removed most, though not all, of the HNO3. Moreover, the lack of more specific standard calibration compounds than sulfuric acid (Sect. S2) limits our CIMS quantification for the larger organic compounds.
It is notable that the box model used for estimating concentrations of reacted MTs does not capture turbulent mixing, and thus an instantaneous dilution of the injector flow into the main flow was assumed. However, the experimentally determined reaction times theoretically account for turbulent mixing. Nevertheless, some uncertainties may arise from different relative concentrations of oxidants and reactants. We did not explore the low levels of MTs where bimolecular reactions are negligible and RO2 concentrations increase linearly with reacted [MT]. Because this study does not focus on kinetics, and since bimolecular reactions commonly occur in the atmosphere, we believe our experimental conditions provide an atmospherically relevant distribution of radicals and closed-shell products.
This study utilized a newly built free-jet flow-tube system with a moveable injector that allows changing the reaction time to measure products from monoterpene (AP, carene, limonene, BP, myrcene) oxidation by NO3. Switching a CIMS between DEA and nitrate modes enabled us to investigate a wide range of gas-phase radicals and closed-shell products, at an effective reaction time of 8.8 s (8.3 s in the flow-tube at 110 cm reaction distance and 0.46 s in the CIMS sampling inlet). The nearly wall-free experimental conditions, direct comparison of the five MTs, and the wide coverage of oxidation products provide valuable insights into early-stage NO3 oxidation pathways including autoxidation. The design of our NO3 experiments allowed us to distinguish NO3-initiated products from O3/OH-initiated ones.
Consistent with previous studies (Draper et al., 2019; Hallquist et al., 1999; Kurtén et al., 2017; Spittler et al., 2006), NO3 oxidation of AP and limonene produced substantial C10H16O2 with yields exceeding 50 %, while carene produced much less C10H16O2 and instead underwent more efficient autoxidation. Several surprising features were also observed for the different MTs: i) for AP, with rising [NO3], the concentration of the primary RO2 radical C10H16NO5 did not increase as expected, but instead showed an increased yield of C10H16O2, ii) while our results agree with the previously proposed ring-opening process of BP following reaction with NO3, a dominant dimer C20H32N2O8 (with a yield of ∼10 % and a quadratic trend with reacted [BP]) was observed, suggesting very rapid accretion product formation in the gas phase, while an earlier study hypothesized that this dimer forms solely in the particle phase (Claflin and Ziemann, 2018), iii) the considerable amount of RO2 species C10H16NO from the acyclic myrcene suggests efficient autoxidation, but instead of H-shifts to form –OOH groups, we speculate that the RO2 undergo ring-closure processes with double bonds to form C–O–O–C groups, based both on expectations from its structure and on the relative detection efficiency of the products by DEA and nitrate modes.
The fractions of observed NO3-initiated products indicate that product closure was reasonably reached for AP, limonene, and myrcene, considering the uncertainties of CIMS quantification and modeled reacted [MT]. In contrast, the incomplete closure of carene and BP is likely due to the low sensitivity of the DEA mode toward products with only one O-atom. While the formation of nopinone, C9H14O, in BP + NO3 was previously reported, there may be unknown processes for carene to produce large amounts of one-oxygen products. The average HOM yields from NO3 oxidation follow the order: myrcene (6.5 %), carene (6.1 %), BP (1.8 %), limonene (1.1 %), and AP (0.8 %). This result confirms that NO3 oxidation yields significant amounts of HOMs, and the overall range is comparable to O3 oxidation (0 %–10 %).
Overall, our results highlight that the product formation from MT + NO3 reactions is highly structure-dependent and behaves differently from O3 and OH oxidation. Our study provides individual product yields for the most abundant compounds, radical distributions, and HOM yields, which are all essential for correctly implementing MT + NO3 oxidation mechanisms into models. Future studies using instruments with better selectivity toward low-oxygenated compounds can validate whether our missing closure for carene and BP was indeed due to lack of sensitivity toward such compounds. Moreover, a comprehensive study on SOA yields of these different MTs together with HOM measurements will clarify to what extent HOMs are able to explain SOA formation from NO3 oxidation of monoterpenes.
Data are available upon request by contacting the corresponding authors. Data presented in the paper are archived in Zenodo (https://doi.org/10.5281/zenodo.17250833; Zhang et al., 2025).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-3933-2026-supplement.
ME and JYZ designed the study. JYZ and YZ conducted the experiments. JYZ analyzed the data with the help of ME and JZ. ME, JYZ, JZ, and HK designed the flow-tube system, with HK further developing the concept into a CAD model. JYZ prepared the manuscript with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
We thank Torsten Berndt for sharing information of his free-jet flow system. We thank Melissa Meder for her assistance in improving the experiment setup and conducting laminarity tests, and Otso Peräkylä for his ideas and help with the laminarity tests. We thank Yuanyuan Luo for the assistance on Vocus PTR-TOF. We thank Pekka Rantala, Petri Keronen, Pasi Aalto, Antti Lahti, Jarkko Mäntylä, and Tommy Chan for technical support and indispensable assistance during the experiments.
This work was supported by the Jane and Aatos Erkko Foundation (grant no. 220043). Jian Zhao thanks the Research Council of Finland (grant no. 1363283) and University of Helsinki funding (grant no. 70996217) for financial support.
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Sergey A. Nizkorodov and reviewed by two anonymous referees.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Atkinson, R. and Arey, J.: Atmospheric degradation of volatile organic compounds, Chem. Rev., 103, 4605–4638, https://doi.org/10.1021/cr0206420, 2003.
Ayres, B. R., Allen, H. M., Draper, D. C., Brown, S. S., Wild, R. J., Jimenez, J. L., Day, D. A., Campuzano-Jost, P., Hu, W., de Gouw, J., Koss, A., Cohen, R. C., Duffey, K. C., Romer, P., Baumann, K., Edgerton, E., Takahama, S., Thornton, J. A., Lee, B. H., Lopez-Hilfiker, F. D., Mohr, C., Wennberg, P. O., Nguyen, T. B., Teng, A., Goldstein, A. H., Olson, K., and Fry, J. L.: Organic nitrate aerosol formation via NO3 + biogenic volatile organic compounds in the southeastern United States, Atmos. Chem. Phys., 15, 13377–13392, https://doi.org/10.5194/acp-15-13377-2015, 2015.
Bates, K. H., Burke, G. J. P., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022.
Bell, D. M., Wu, C., Bertrand, A., Graham, E., Schoonbaert, J., Giannoukos, S., Baltensperger, U., Prevot, A. S. H., Riipinen, I., El Haddad, I., and Mohr, C.: Particle-phase processing of α-pinene NO3 secondary organic aerosol in the dark, Atmos. Chem. Phys., 22, 13167–13182, https://doi.org/10.5194/acp-22-13167-2022, 2022.
Berndt, T.: Peroxy Radical Processes and Product Formation in the OH Radical-Initiated Oxidation of α-Pinene for Near-Atmospheric Conditions, J. Phys. Chem. A, 125, 9151–9160, https://doi.org/10.1021/acs.jpca.1c05576, 2021.
Berndt, T.: Peroxy Radical and Product Formation in the Gas-Phase Ozonolysis of α-Pinene under Near-Atmospheric Conditions: Occurrence of an Additional Series of Peroxy Radicals O,O–C10H15O(O2)yO2 with y = 1–3, J. Phys. Chem. A, 126, 6526–6537, https://doi.org/10.1021/acs.jpca.2c05094, 2022.
Berndt, T., Boge, O., and Hermann, M.: Gas-Phase Reaction of NO3 Radicals with B-Pinene, in: Transactions on Ecology and the Environmen, 28, WIT Press, 79–83, 1999.
Berndt, T., Richters, S., Kaethner, R., Voigtländer, J., Stratmann, F., Sipilä, M., Kulmala, M., and Herrmann, H.: Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals, J. Phys. Chem. A, 119, 10336–10348, https://doi.org/10.1021/acs.jpca.5b07295, 2015a.
Berndt, T., Kaethner, R., Voigtländer, J., Stratmann, F., Pfeifle, M., Reichle, P., Sipilä, M., Kulmala, M., and Olzmann, M.: Kinetics of the unimolecular reaction of CH2OO and the bimolecular reactions with the water monomer, acetaldehyde and acetone under atmospheric conditions, Phys. Chem. Chem. Phys., 17, 19862–19873, https://doi.org/10.1039/C5CP02224J, 2015b.
Berndt, T., Richters, S., Jokinen, T., Hyttinen, N., Kurtén, T., Otkjær, R. V., Kjaergaard, H. G., Stratmann, F., Herrmann, H., Sipilä, M., Kulmala, M., and Ehn, M.: Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 7, 13677, https://doi.org/10.1038/ncomms13677, 2016.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chem. Int. Edit., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G., Forster, P., Kerminen, V.-M., Kondo, Y., Liao, H., Lohmann, U., Rasch, P., Satheesh, S. K., Sherwood, S., Stevens, B., and Zhang, X. Y.: Clouds and aerosols, in: Climate change 2013: The physical science basis, Contribution of working group I to the fifth assessment report of the intergovernmental panel on climate change, Cambridge University Press, 571–657, https://doi.org/10.1017/CBO9781107415324.016, 2013.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene+NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Brown, S. S. and Stutz, J.: Nighttime radical observations and chemistry, Chem. Soc. Rev., 41, 6405–6447, https://doi.org/10.1039/C2CS35181A, 2012.
Cai, R., Mikkilä, J., Bengs, A., Koirala, M., Mikkilä, J., Holm, S., Juuti, P., Meder, M., Partovi, F., Shcherbinin, A., Worsnop, D., Ehn, M., and Kangasluoma, J.: Extending the Range of Detectable Trace Species with the Fast Polarity Switching of Chemical Ionization Orbitrap Mass Spectrometry, Anal. Chem., 96, 8604–8612, https://doi.org/10.1021/acs.analchem.4c00650, 2024.
Chuang, W. K. and Donahue, N. M.: A two-dimensional volatility basis set – Part 3: Prognostic modeling and NOx dependence, Atmos. Chem. Phys., 16, 123–134, https://doi.org/10.5194/acp-16-123-2016, 2016.
Claflin, M. S. and Ziemann, P. J.: Identification and Quantitation of Aerosol Products of the Reaction of β-Pinene with NO3 Radicals and Implications for Gas- and Particle-Phase Reaction Mechanisms, J. Phys. Chem. A, 122, 3640–3652, https://doi.org/10.1021/acs.jpca.8b00692, 2018.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Dam, M., Draper, D. C., Marsavin, A., Fry, J. L., and Smith, J. N.: Observations of gas-phase products from the nitrate-radical-initiated oxidation of four monoterpenes, Atmos. Chem. Phys., 22, 9017–9031, https://doi.org/10.5194/acp-22-9017-2022, 2022.
Day, D. A., Fry, J. L., Kang, H. G., Krechmer, J. E., Ayres, B. R., Keehan, N. I., Thompson, S. L., Hu, W., Campuzano-Jost, P., Schroder, J. C., Stark, H., DeVault, M. P., Ziemann, P. J., Zarzana, K. J., Wild, R. J., Dubè, W. P., Brown, S. S., and Jimenez, J. L.: Secondary Organic Aerosol Mass Yields from NO3 Oxidation of α-Pinene and Δ-Carene: Effect of RO2 Radical Fate, J. Phys. Chem. A, 126, 7309–7330, https://doi.org/10.1021/acs.jpca.2c04419, 2022.
DeVault, M. P., Ziola, A. C., and Ziemann, P. J.: Products and Mechanisms of Secondary Organic Aerosol Formation from the NO3 Radical-Initiated Oxidation of Cyclic and Acyclic Monoterpenes, ACS Earth Space Chem., 6, 2076–2092, https://doi.org/10.1021/acsearthspacechem.2c00130, 2022.
Dewald, P., Seubert, T., Andersen, S. T., Türk, G. N. T. E., Schuladen, J., McGillen, M. R., Denjean, C., Etienne, J.-C., Garrouste, O., Jamar, M., Harb, S., Cirtog, M., Michoud, V., Cazaunau, M., Bergé, A., Cantrell, C., Dusanter, S., Picquet-Varrault, B., Kukui, A., Xue, C., Mellouki, A., Lelieveld, J., and Crowley, J. N.: NO3 reactivity during a summer period in a temperate forest below and above the canopy, Atmos. Chem. Phys., 24, 8983–8997, https://doi.org/10.5194/acp-24-8983-2024, 2024.
Donahue, N. M., Kroll, J. H., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set – Part 2: Diagnostics of organic-aerosol evolution, Atmos. Chem. Phys., 12, 615–634, https://doi.org/10.5194/acp-12-615-2012, 2012.
Draper, D. C., Myllys, N., Hyttinen, N., Møller, K. H., Kjaergaard, H. G., Fry, J. L., Smith, J. N., and Kurtén, T.: Formation of Highly Oxidized Molecules from NO3 Radical Initiated Oxidation of Δ-3-Carene: A Mechanistic Study, ACS Earth Space Chem., 3, 1460–1470, https://doi.org/10.1021/acsearthspacechem.9b00143, 2019.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res., 98, 9001–9010, https://doi.org/10.1029/93JD00031, 1993.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary Organic Aerosol Formation and Organic Nitrate Yield from NO3 Oxidation of Biogenic Hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Graham, E. L., Wu, C., Bell, D. M., Bertrand, A., Haslett, S. L., Baltensperger, U., El Haddad, I., Krejci, R., Riipinen, I., and Mohr, C.: Volatility of aerosol particles from NO3 oxidation of various biogenic organic precursors, Atmos. Chem. Phys., 23, 7347–7362, https://doi.org/10.5194/acp-23-7347-2023, 2023.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Guo, Y., Shen, H., Pullinen, I., Luo, H., Kang, S., Vereecken, L., Fuchs, H., Hallquist, M., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., Zhao, D., and Mentel, T. F.: Identification of highly oxygenated organic molecules and their role in aerosol formation in the reaction of limonene with nitrate radical, Atmos. Chem. Phys., 22, 11323–11346, https://doi.org/10.5194/acp-22-11323-2022, 2022.
Hallquist, M., Wängberg, I., Ljungström, E., Barnes, I., and Becker, K.-H.: Aerosol and Product Yields from NO3 Radical-Initiated Oxidation of Selected Monoterpenes, Environ. Sci. Technol., 33, 553–559, https://doi.org/10.1021/es980292s, 1999.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Harb, S., Cirtog, M., Alage, S., Cantrell, C., Cazaunau, M., Michoud, V., Pangui, E., Bergé, A., Giorio, C., Battaglia, F., and Picquet-Varrault, B.: Highly oxygenated molecules (HOMs) and secondary organic aerosol (SOA) formation from the oxidation of α- and β-phellandrenes by NO3 radicals, Atmos. Chem. Phys., 25, 11003–11024, https://doi.org/10.5194/acp-25-11003-2025, 2025.
He, X.-C., Shen, J., Iyer, S., Juuti, P., Zhang, J., Koirala, M., Kytökari, M. M., Worsnop, D. R., Rissanen, M., Kulmala, M., Maier, N. M., Mikkilä, J., Sipilä, M., and Kangasluoma, J.: Characterisation of gaseous iodine species detection using the multi-scheme chemical ionisation inlet 2 with bromide and nitrate chemical ionisation methods, Atmos. Meas. Tech., 16, 4461–4487, https://doi.org/10.5194/amt-16-4461-2023, 2023.
Huang, W., Junninen, H., Garmash, O., Lehtipalo, K., Stolzenburg, D., Lampilahti, J. L. P., Ezhova, E., Schallhart, S., Rantala, P., Aliaga, D., Ahonen, L., Sulo, J., Quéléver, L. L. J., Cai, R., Alekseychik, P., Mazon, S. B., Yao, L., Blichner, S. M., Zha, Q., Mammarella, I., Kirkby, J., Kerminen, V.-M., Worsnop, D. R., Kulmala, M., and Bianchi, F.: Potential pre-industrial–like new particle formation induced by pure biogenic organic vapors in Finnish peatland, Sci. Adv., 10, eadm9191, https://doi.org/10.1126/sciadv.adm9191, 2024.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015.
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015.
Kelly, F. J. and Fussell, J. C.: Air pollution and public health: emerging hazards and improved understanding of risk, Environ. Geochem. Hlth., 37, 631–649, https://doi.org/10.1007/s10653-015-9720-1, 2015.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Kurtén, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jørgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in α-Pinene Ozonolysis Products, J. Phys. Chem. A, 119, 11366–11375, https://doi.org/10.1021/acs.jpca.5b08948, 2015.
Kurtén, T., Møller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From α-Pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, https://doi.org/10.1021/acs.jpclett.7b01038, 2017.
Li, D., Huang, W., Wang, D., Wang, M., Thornton, J. A., Caudillo, L., Rörup, B., Marten, R., Scholz, W., Finkenzeller, H., Marie, G., Baltensperger, U., Bell, D. M., Brasseur, Z., Curtius, J., Dada, L., Duplissy, J., Gong, X., Hansel, A., He, X.-C., Hofbauer, V., Junninen, H., Krechmer, J. E., Kürten, A., Lamkaddam, H., Lehtipalo, K., Lopez, B., Ma, Y., Mahfouz, N. G. A., Manninen, H. E., Mentler, B., Perrier, S., Petäjä, T., Pfeifer, J., Philippov, M., Schervish, M., Schobesberger, S., Shen, J., Surdu, M., Tomaz, S., Volkamer, R., Wang, X., Weber, S. K., Welti, A., Worsnop, D. R., Wu, Y., Yan, C., Zauner-Wieczorek, M., Kulmala, M., Kirkby, J., Donahue, N. M., George, C., El-Haddad, I., Bianchi, F., and Riva, M.: Nitrate Radicals Suppress Biogenic New Particle Formation from Monoterpene Oxidation, Environ. Sci. Technol., 58, 1601–1614, https://doi.org/10.1021/acs.est.3c07958, 2024.
Liu, L., Hohaus, T., Franke, P., Lange, A. C., Tillmann, R., Fuchs, H., Tan, Z., Rohrer, F., Karydis, V., He, Q., Vardhan, V., Andres, S., Bohn, B., Holland, F., Winter, B., Wedel, S., Novelli, A., Hofzumahaus, A., Wahner, A., and Kiendler-Scharr, A.: Observational evidence reveals the significance of nocturnal chemistry in seasonal secondary organic aerosol formation, npj Clim. Atmos. Sci., 7, 207, https://doi.org/10.1038/s41612-024-00747-6, 2024.
Luo, Y., Garmash, O., Li, H., Graeffe, F., Praplan, A. P., Liikanen, A., Zhang, Y., Meder, M., Peräkylä, O., Peñuelas, J., Yáñez-Serrano, A. M., and Ehn, M.: Oxidation product characterization from ozonolysis of the diterpene ent-kaurene, Atmos. Chem. Phys., 22, 5619–5637, https://doi.org/10.5194/acp-22-5619-2022, 2022.
Luo, Y., Thomsen, D., Iversen, E. M., Roldin, P., Skønager, J. T., Li, L., Priestley, M., Pedersen, H. B., Hallquist, M., Bilde, M., Glasius, M., and Ehn, M.: Formation and temperature dependence of highly oxygenated organic molecules (HOMs) from Δ3-carene ozonolysis, Atmos. Chem. Phys., 24, 9459–9473, https://doi.org/10.5194/acp-24-9459-2024, 2024.
Mayorga, R., Xia, Y., Zhao, Z., Long, B., and Zhang, H.: Peroxy Radical Autoxidation and Sequential Oxidation in Organic Nitrate Formation during Limonene Nighttime Oxidation, Environ. Sci. Technol., 56, 15337–15346, https://doi.org/10.1021/acs.est.2c04030, 2022.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Meder, M., Graeffe, F., Luo, Y., Luo, J., Iyer, S., Valiev, R., Cai, R., Rissanen, M., Kurtén, T., Varelas, J. G., Geiger, F. M., Thomson, R. J., and Ehn, M.: Selective Deuteration Reveals the Importance of Multiple Branching Pathways in α-Pinene Autoxidation, J. Am. Chem. Soc., 147, 14131–14138, https://doi.org/10.1021/jacs.4c14462, 2025.
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Novelli, A., Cho, C., Fuchs, H., Hofzumahaus, A., Rohrer, F., Tillmann, R., Kiendler-Scharr, A., Wahner, A., and Vereecken, L.: Experimental and theoretical study on the impact of a nitrate group on the chemistry of alkoxy radicals, Phys. Chem. Chem. Phys., 23, 5474–5495, https://doi.org/10.1039/D0CP05555G, 2021.
Peräkylä, O., Berndt, T., Franzon, L., Hasan, G., Meder, M., Valiev, R. R., Daub, C. D., Varelas, J. G., Geiger, F. M., Thomson, R. J., Rissanen, M., Kurtén, T., and Ehn, M.: Large Gas-Phase Source of Esters and Other Accretion Products in the Atmosphere, J. Am. Chem. Soc., 145, 7780–7790, https://doi.org/10.1021/jacs.2c10398, 2023.
Perraud, V., Bruns, E. A., Ezell, M. J., Johnson, S. N., Greaves, J., and Finlayson-Pitts, B. J.: Identification of Organic Nitrates in the NO3 Radical Initiated Oxidation of α-Pinene by Atmospheric Pressure Chemical Ionization Mass Spectrometry, Environ. Sci. Technol., 44, 5887–5893, https://doi.org/10.1021/es1005658, 2010.
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019.
Russell, D. M., Kunkler, F., Shen, J., Kohl, M., DeVivo, J., Bhattacharyya, N., Xenofontos, C., Klebach, H., Caudillo-Plath, L., Simon, M., Ahongshangbam, E., Almeida, J., Amorim, A., Beckmann, H., Busato, M., Canagaratna, M. R., Chassaing, A., Cruz-Simbron, R., Dada, L., Holzbeck, P., Judmaier, B., Kaniyodical Sebastian, M., Koemets, P., Krüger, T., Liu, L., Martinez, M., Mentler, B., Morawiec, A., Onnela, A., Petäjä, T., Rato, P., Reza, M., Ruhl, S., Scholz, W., Sommer, E., Tomé, A., Tong, Y., Top, J., Umo, N. S., Unfer, G. R., Ward, R. X., Weissbacher, J., Yang, B., Yu, W., Zauner-Wieczorek, M., Zgheib, I., Zhang, J., Zheng, Z., El Haddad, I., Flagan, R. C., Hansel, A., Junninen, H., Kulmala, M., Lehtipalo, K., Lelieveld, J., Möhler, O., Schobesberger, S., Volkamer, R., Winkler, P. M., Worsnop, D. R., Christoudias, T., Pozzer, A., Donahue, N. M., Harder, H., Kirkby, J., He, X.-C., and Curtius, J.: Isoprene chemistry under upper-tropospheric conditions, Nat. Commun., 16, 8555, https://doi.org/10.1038/s41467-025-64229-w, 2025.
Shen, H., Zhao, D., Pullinen, I., Kang, S., Vereecken, L., Fuchs, H., Acir, I.-H., Tillmann, R., Rohrer, F., Wildt, J., Kiendler-Scharr, A., Wahner, A., and Mentel, T. F.: Highly Oxygenated Organic Nitrates Formed from NO3 Radical-Initiated Oxidation of β-Pinene, Environ. Sci. Technol., 55, 15658–15671, https://doi.org/10.1021/acs.est.1c03978, 2021.
Shen, H., Vereecken, L., Kang, S., Pullinen, I., Fuchs, H., Zhao, D., and Mentel, T. F.: Unexpected significance of a minor reaction pathway in daytime formation of biogenic highly oxygenated organic compounds, Sci. Adv., https://doi.org/10.1126/sciadv.abp8702, 2022.
Spittler, M., Barnes, I., Bejan, I., Brockmann, K. J., Benter, Th., and Wirtz, K.: Reactions of NO3 radicals with limonene and α-pinene: Product and SOA formation, Atmos. Environ., 40, 116–127, https://doi.org/10.1016/j.atmosenv.2005.09.093, 2006.
Spracklen, D. V., Jimenez, J. L., Carslaw, K. S., Worsnop, D. R., Evans, M. J., Mann, G. W., Zhang, Q., Canagaratna, M. R., Allan, J., Coe, H., McFiggans, G., Rap, A., and Forster, P.: Aerosol mass spectrometer constraint on the global secondary organic aerosol budget, Atmos. Chem. Phys., 11, 12109–12136, https://doi.org/10.5194/acp-11-12109-2011, 2011.
Takeuchi, M., Wang, Y., and Ng, N. L.: Rapid Photolysis of Gaseous Organic Nitrates Formed from Hydroxyl and Nitrate Radical Oxidation of α-Pinene and β-Pinene, ACS EST Air, https://doi.org/10.1021/acsestair.5c00183, 2025.
Vereecken, L. and Peeters, J.: Theoretical Study of the Formation of Acetone in the OH-Initiated Atmospheric Oxidation of α-Pinene, J. Phys. Chem. A, 104, 11140–11146, https://doi.org/10.1021/jp0025173, 2000.
Vereecken, L., Vu, G., Wahner, A., Kiendler-Scharr, A., and T. Nguyen, H. M.: A structure activity relationship for ring closure reactions in unsaturated alkylperoxy radicals, Phys. Chem. Chem. Phys., 23, 16564–16576, https://doi.org/10.1039/D1CP02758A, 2021.
Wang, Y., Takeuchi, M., Wang, S., Nizkorodov, S. A., France, S., Eris, G., and Ng, N. L.: Photolysis of Gas-Phase Atmospherically Relevant Monoterpene-Derived Organic Nitrates, J. Phys. Chem. A, 127, 987–999, https://doi.org/10.1021/acs.jpca.2c04307, 2023.
Wängberg, I., Barnes, I., and Becker, K. H.: Product and Mechanistic Study of the Reaction of NO3 Radicals with α-Pinene, Environ. Sci. Technol., 31, 2130–2135, https://doi.org/10.1021/es960958n, 1997.
Zhang, J., Zhao, J., Wollesen de Jonge, R., Sarnela, N., Roldin, P., and Ehn, M.: Evaluating the Applicability of a Real-Time Highly Oxygenated Organic Molecule (HOM)-Based Indicator for Ozone Formation Sensitivity at a Boreal Forest Station, Environ. Sci. Technol. Lett., 11, 1227–1232, https://doi.org/10.1021/acs.estlett.4c00733, 2024a.
Zhang, J., Zhao, J., Luo, Y., Mickwitz, V., Worsnop, D., and Ehn, M.: On the potential use of highly oxygenated organic molecules (HOMs) as indicators for ozone formation sensitivity, Atmos. Chem. Phys., 24, 2885–2911, https://doi.org/10.5194/acp-24-2885-2024, 2024b.
Zhang, J., Zhang, Y., Koskenvaara, H., Zhao, J., and Ehn, M.: Dataset for article “Gas-phase products from nitrate radical oxidation of five monoterpenes: insights from free-jet flow-tube experiments”, Zenodo [data set], https://doi.org/10.5281/zenodo.17250833, 2025.
Zhao, D. F., Kaminski, M., Schlag, P., Fuchs, H., Acir, I.-H., Bohn, B., Häseler, R., Kiendler-Scharr, A., Rohrer, F., Tillmann, R., Wang, M. J., Wegener, R., Wildt, J., Wahner, A., and Mentel, Th. F.: Secondary organic aerosol formation from hydroxyl radical oxidation and ozonolysis of monoterpenes, Atmos. Chem. Phys., 15, 991–1012, https://doi.org/10.5194/acp-15-991-2015, 2015.
Zhao, J., Mickwitz, V., Zhang, J., Alton, M., Canagaratna, M., Graeffe, F., Schobesberger, S., Worsnop, D., and Ehn, M.: Comparison of Gaseous and Particulate Highly Oxygenated Organic Molecules from the Ozonolysis of Terpenes, ACS EST Air, 1, 1294–1303, https://doi.org/10.1021/acsestair.4c00121, 2024.