the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Mar 2026
| 10 Mar 2026
Determining the key sources of uncertainty in dimethyl sulfide and methanethiol oxidation under tropical, temperate, and polar marine conditions
Benedict E. H. Harvey
Chiara Giorio
This study quantifies how uncertainties in the gas-phase rate constants used in the oxidation mechanisms of dimethyl sulfide (DMS) and methanethiol (CH3SH) (both major natural sources of sulfur to the atmosphere), affect products such as methanesulfonic acid and sulfuric acid, which influence cloud formation and climate.
We updated our previously reported DMS oxidation mechanism and extended it to include 9 halogen, 71 aqueous, and 4 CH3SH reactions. This updated mechanism was then run in box models covering temperate, tropical, and polar marine conditions based on field campaigns.
Constrained Monte Carlo sampling was employed to propagate the uncertainties in the mechanism. Uncertainties in the concentrations of the products were time-dependent and ranged from 10 %–200 % for most species, with methanesulfonic acid and sulfuric acid having the largest relative uncertainties.
Sensitivity analysis using the EASI RBD-FAST algorithm was performed to identify which reactions and processes were the largest sources of uncertainty for the modelled oxidation products. Individually, reactions involving the formation and loss of CH3SO2O2 were major contributors to the uncertainties in gas-phase methanesulfonic acid and sulfuric acid. Reactions of species with OH and rate constants based on structure-activity relationships were commonly found to significantly contribute to uncertainty in most of the DMS oxidation products studied. Large uncertainties associated with OCS were attributed to the photolysis of hydroperoxymethyl thioformate, which has not yet been studied experimentally or theoretically. We suggest that future work on DMS oxidation should prioritise these processes to reduce the uncertainty in the climate impact of marine sulfur species.
- Article
(3120 KB) - Full-text XML
-
Supplement
(7982 KB) - BibTeX
- EndNote




The oxidation of dimethyl sulfide (CH3SCH3, DMS), the largest natural source of sulfur in the atmosphere (Bates et al., 1992), and methanethiol (CH3SH, MeSH), a major source of sulfur in the marine atmosphere (Wohl et al., 2024), can result in the formation of sulfuric acid (H2SO4) and methanesulfonic acid (CH3SO3H, MSA). These oxidation products are known to contribute to cloud condensation nuclei (CCN) due to their low volatility and high hygroscopicity (Curry and Webster, 1999; Boy et al., 2005; Kulmala, 2003; Rosati et al., 2022). Additionally, gas-phase MSA and H2SO4 can contribute to new particle formation, which can affect the number density of CCN (Kerminen et al., 2018; Covert et al., 1992; Beck et al., 2021). As such, DMS and CH3SH oxidation products can affect cloud properties, and the representation of DMS chemistry in global models affects calculations of Earth's radiative forcing (Carslaw et al., 2013).
Although the emission and subsequent oxidation of DMS are important processes in climate models, they are currently not well represented. McCoy et al. (2020) identified that global models underestimated the number concentration of cloud droplets in the Southern Ocean and proposed that it could be due to the underestimation of DMS emissions, or the processes governing nucleation of new particles from DMS oxidation particles. Additionally, Fung et al. (2022) demonstrated that adjusting the DMS oxidation chemistry used in global models impacted the global sulfate burden in both the pre-industrial atmosphere and present day by 29 % and 8.8 %, respectively. These results demonstrate the importance of an accurate representation of DMS oxidation in Earth system models to improve their modelling of Earth's radiative balance, and subsequently, predictions of global average temperatures.
Following the theoretical work by Wu et al. (2015) that determined that hydroperoxymethyl thioformate (OCHSCH2OOH, HPMTF) was a plausible DMS oxidation product, further experimental and theoretical work studying the oxidation of DMS, and its products, has been conducted (Berndt et al., 2019; Ye et al., 2021, 2022; Goss and Kroll, 2024; Shen et al., 2022; Berndt et al., 2020, 2023; Jernigan et al., 2024; Chen et al., 2023; Vereecken et al., 2025; Jernigan et al., 2022; Li et al., 2021; Lily et al., 2023; Arathala and Musah, 2023; Rhyman et al., 2023). These studies have helped to improve our knowledge of DMS oxidation. However, significant uncertainties remain, and model simulations supported by the latest understanding of DMS chemistry still diverge in both the magnitudes of their predictions and the sensitivity to changes in oxidants and temperature (Cala et al., 2023; Jacob et al., 2024).
Numerical models are key tools for quantifying the impacts of uncertainties related to the chemistry of species in the atmosphere. Model sensitivity tests can be performed to identify which reactions are the largest contributors to the model outputs, such as the concentration of products (Tomlin, 2013). However, a reaction that the model is particularly sensitive to might already be well-characterised experimentally and further experimental/theoretical research may not yield further constraints. To identify which reactions should be explored further, uncertainty quantification can be used to demonstrate the uncertainty in output concentrations, along with determining which reactions contribute the most to that uncertainty. Hence, uncertainty quantification can guide where efforts to improve mechanisms will have the greatest impact (Tomlin, 2013; Dunker et al., 2020; Vasyunin et al., 2004).
In the context of DMS, there have been several previous studies that have performed uncertainty quantification related to its oxidation mechanism (Lucas and Prinn, 2005; Saltelli and Hjorth, 1995; Campolongo et al., 1999). Saltelli and Hjorth (1995) investigated the uncertainty in a 37-reaction OH-initiated DMS oxidation mechanism, considering polluted and non-polluted environments with constant oxidant concentrations (i.e. the diurnal cycle was not considered). They found that the reaction of CH3SOO contributed to SO2 uncertainty, while the decomposition of CH3SO3 did not seem important to H2SO4 formation. Campolongo et al. (1999) extended the work by Saltelli and Hjorth (1995), incorporating liquid-phase chemistry and temperature dependent uncertainties, indicating that both temperature dependence and aqueous-phase chemistry are needed to propagate uncertainties in DMS oxidation. Lucas and Prinn (2005) investigated the effect of uncertainties from a larger gas-phase mechanism (49 reactions), incorporating the oxidation from NO3 in addition to OH in a box model representing summertime in the Southern Ocean. In total they propagated the uncertainties from 58 parameters, including gas-phase chemistry, heterogeneous loss (from aerosol uptake and dry deposition), mixing, and DMS emissions. They demonstrated the importance of heterogeneous loss and found that reactions involving CH3SOO2 contribute to the uncertainty in MSA and H2SO4, however, they found the decomposition of CH3SO2 insignificant. Although a larger gas-phase mechanism was considered compared to Campolongo et al. (1999), the work by Lucas and Prinn (2005) lacked aqueous-phase reactions and temperature dependence, and only explored one case study. All these uncertainty quantification studies developed our understanding of the uncertainties in DMS oxidation chemistry, however they used limited descriptions of the chemistry (excluding halogen chemistry for example), lacked inclusion of the recently discovered HPMTF pathway, and most importantly none of the mechanisms were validated against comprehensive chamber studies. Thus, an uncertainty quantification study is needed that includes an extensive, updated and validated DMS oxidation mechanism, with both aqueous-phase chemistry and temperature dependence considered, to guide the work needed to reduce uncertainty in DMS chemistry in the future.
Selecting a suitable uncertainty quantification method to use for a DMS oxidation mechanism requires balancing scalability with the ability to capture nonlinear behaviour and interactions between reactions. Although local, one-at-a-time sampling methods have been used previously for uncertainty quantification in atmospheric chemistry due to being easier to interpret and less computationally expensive (Newsome and Evans, 2017), they are unable to capture interactions between different reactions (Saltelli and Annoni, 2010). The Sobol method is a popular and effective sensitivity analysis method that provides first and higher-order sensitivity indices (Sobol, 2001). However, it is computationally expensive and is not recommended for problems with over 70 sampled parameters (Stein et al., 2022). Another popular sensitivity analysis technique, the Morris method, is effective with large numbers of parameters and has been used for an analysis of the DMS oxidation mechanism (Campolongo et al., 2007), but it is a qualitative method and primarily provides a ranking of the parameters by importance (Goffart and Woloszyn, 2021). Although multiple linear regression provides simple and relatively fast quantitative analysis, it assumes linearity (Saltelli and Annoni, 2010). Alternatively, the Fourier amplitude sensitivity test (FAST) combined with random balanced design (RBD) provides estimations of first-order Sobol sensitivity indices (Tarantola et al., 2006; Tissot and Prieur, 2012). By combining this method with an algorithm named EASI (Effective Algorithm for computing global Sensitivity Indices) that also estimates first-order sensitivity indices (Plischke, 2010), the resultant EASI RBD-FAST method can be used on Latin hypercube sampling results (Goffart and Woloszyn, 2021), and performs well for large numbers of parameters (Stein et al., 2022). Due to the size and complexity of the DMS mechanism, EASI RBD-FAST is well suited to explore the uncertainties in the chemistry.
This study expands on our previous work (Jacob et al., 2024), where a comprehensive mechanism was developed and validated through chamber experiments. We apply a simple box model (Knote and Barre, 2022) to simulate the average conditions observed during field campaigns that have probed marine sulfur chemistry in different locations and under different conditions (Lee et al., 2010; Sommariva et al., 2004; Jones et al., 2008). The simplicity of the box model allows us to focus on the impacts of uncertainty in the chemical mechanisms. The uncertainties in the rate constants of the gas-phase DMS oxidation mechanism have been propagated to determine the uncertainty in the concentration of oxidation products. The three marine environments chosen to conduct this work, representing tropical, temperate, and polar conditions, enable us to explore the state-dependence of the chemical uncertainties. In addition to DMS, the oxidation of CH3SH has also been included owing to its importance as a source of reduced sulfur (Wohl et al., 2024) and its connection to the DMS oxidation mechanism via the CH3SH radical. Finally, halogen reactions and aqueous chemistry have been incorporated, to explore their effect on DMS and CH3SH oxidation. EASI RBD-FAST analysis has been performed on the results, identifying the reactions that contribute the most to the uncertainty in the concentration of each species. These reactions should be studied further, as they will have the largest impact on improving the representation of DMS and CH3SH oxidation in chemistry and climate models.
In light of work that was published after Jacob et al. (2024), the OH-initiated DMS oxidation mechanism has been updated and re-evaluated for this study, which is briefly discussed in the following paragraphs. The extension of this mechanism to include halogen and CH3SH chemistry is included in the supplement to this paper (Sect. S1 in the Supplement). In addition to gas-phase chemistry, aqueous chemistry has been included based on CAPRAM DMS module 1.0 (DM1.0) (Hoffmann et al., 2016), along with additional SO2, sulfate, and oxidant aqueous reactions from Wollesen de Jonge et al. (2021) (further details are also included in Sect. S1).
Our previous mechanism for the gas-phase OH-initiated oxidation of DMS has been updated in a manner consistent with the development methodology discussed in that work (Jacob et al., 2024). These updates include adjustments to CH3SOx chemistry due to work from Chen et al. (2023) and Berndt (2025), along with the removal of a dimethyl sulfone (CH3SO2CH3, DMSO2) formation pathway due to a study by Goss and Kroll (2024). These changes are discussed in more detail in Sect. S2.
An overview of the performance of the updated gas-phase mechanism against the Master Chemical Mechanism (MCM) v3.3.1 and mechanisms from Ye et al. (2022), Shen et al. (2022), and Jernigan et al. (2022), using the same five experiments used in Jacob et al. (2024) is given in Fig. 1. Compared to these mechanisms, the updated mechanism has an average fractional gross error within 0.05 of the best performing mechanism for 8 out of 14 products. Additional figures demonstrating the effect of these updates on the performance of the mechanism for each experiment can be found in Sect. S3.
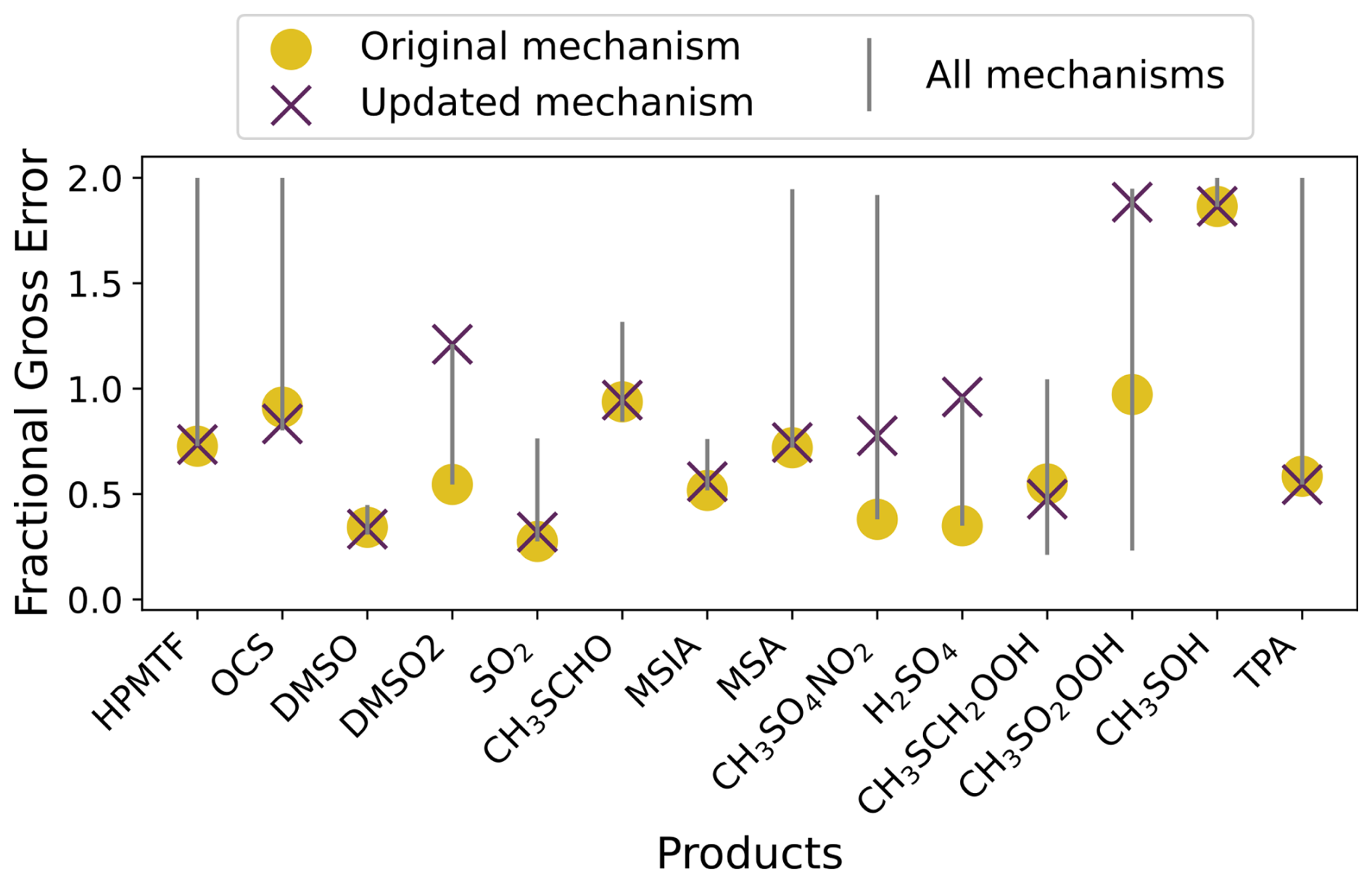
Figure 1The range in the average fractional gross error of four mechanisms (Jernigan, Shen, Ye and MCM, grey line) compared to the error in the updated mechanism (purple cross), and the original mechanism (yellow dot), for each product found in the experiments by Albu et al. (2008), Jernigan et al. (2022), Ye et al. (2022) and Shen et al. (2022).
We found that the updates did not affect the performance of our mechanism in the Ye et al. (2022) experiment 2a, or the Jernigan et al. (2022) experiment. In the Albu et al. (2008) experiment, the updated mechanism did not produce DMSO2, however, the formation of DMSO2 is likely due to elevated RO2 concentrations which are unlikely to occur in the marine atmosphere (Goss and Kroll, 2024). Additionally, although the updated mechanism forms less DMSO2 than measured in Ye et al. (2022) experiment 1, the formation of DMSO2 in that experiment is likely due to NOx reactions, which would not be prominent in the marine environment.
Our updated mechanism outperforms the MCM and mechanisms from Ye et al. (2022), Shen et al. (2022) and Jernigan et al. (2022) in the formation of SO2, H2SO4 and MSA in the Ye et al. (2022) experiment 1. The updated mechanism understandably performs less well than the original mechanism for these products, as the decomposition of CH3SO2 in the original mechanism was scaled to reproduce that experiment. The scaling was a temporary solution until further studies were performed. As such, the update, which includes the recent theoretical work by Chen et al. (2023) for the decomposition of CH3SO2 and reaction with oxygen, is in line with the methodology from Jacob et al. (2024). Finally, in the Shen et al. (2022) experiment, the updated mechanism overpredicts CH3SO2OOH and H2SO4 by factors of 40 and 4, respectively. The performance of the updated mechanism for these species demonstrates that further work is needed to understand CH3SO2O2 reactions (including reactions with HO2 and RO2). The updated mechanism was used in this study, with additional halogen and aqueous reactions discussed in Sect. S1. The full gas-phase mechanism used is given in Table S2 and a comparison among the original, updated and expanded mechanisms is demonstrated in Figs. S7–9.
Whilst the updated mechanism does not fully capture all species, as we have outlined above, the inclusion of the extra reactions allows a more rigorous quantification of uncertainties that are atmospherically relevant.
The results of the uncertainty analysis of the DMS mechanism depend on environmental conditions such as sunlight, temperature, deposition velocity, concentrations of oxidants, and boundary layer height. These environmental conditions differ based on the location and time of year. Ideally, the uncertainty quantification and sensitivity analysis would be conducted in as many conditions as possible to cover a large parameter space. However, thousands of box model simulations are needed to conduct each of these experiments, and the availability of data from field campaigns to constrain these environmental conditions is limited. As such, in this work, three marine boundary layer regimes representing tropical, temperate, and polar marine environments were chosen. Cape Verde, Cape Grim, and Halley Station cover these three environments and are locations where fieldwork has been conducted in the past (Lee et al., 2010; Sommariva et al., 2004; Jones et al., 2008). Although these field campaigns only represent a subset of the marine environment at specific points in time, they provide realistic, observation-based constraints for the models. Table 1 provides a summary of the average conditions (chemical and meteorological) used to constrain the box models, obtained from fieldwork or ERA5 data and described in Sects. S6 and S7.
Table 1A summary of the conditions in Cape Verde, Cape Grim, and Halley Station box model runs which, unless specified, are fieldwork daily averages over the chosen days (Natural Environment Research Council et al., 2006; Allan et al., 1987; Natural Environment Research Council et al., 2005). For the Cape Grim data, the gas-phase concentrations are daytime averages (11:00 a.m.–02:00 p.m. AEDT), and the aerosol concentrations are averages of measurements taken in February 1989 and 1990 from a different Cape Grim campaign (Andreae et al., 1999).
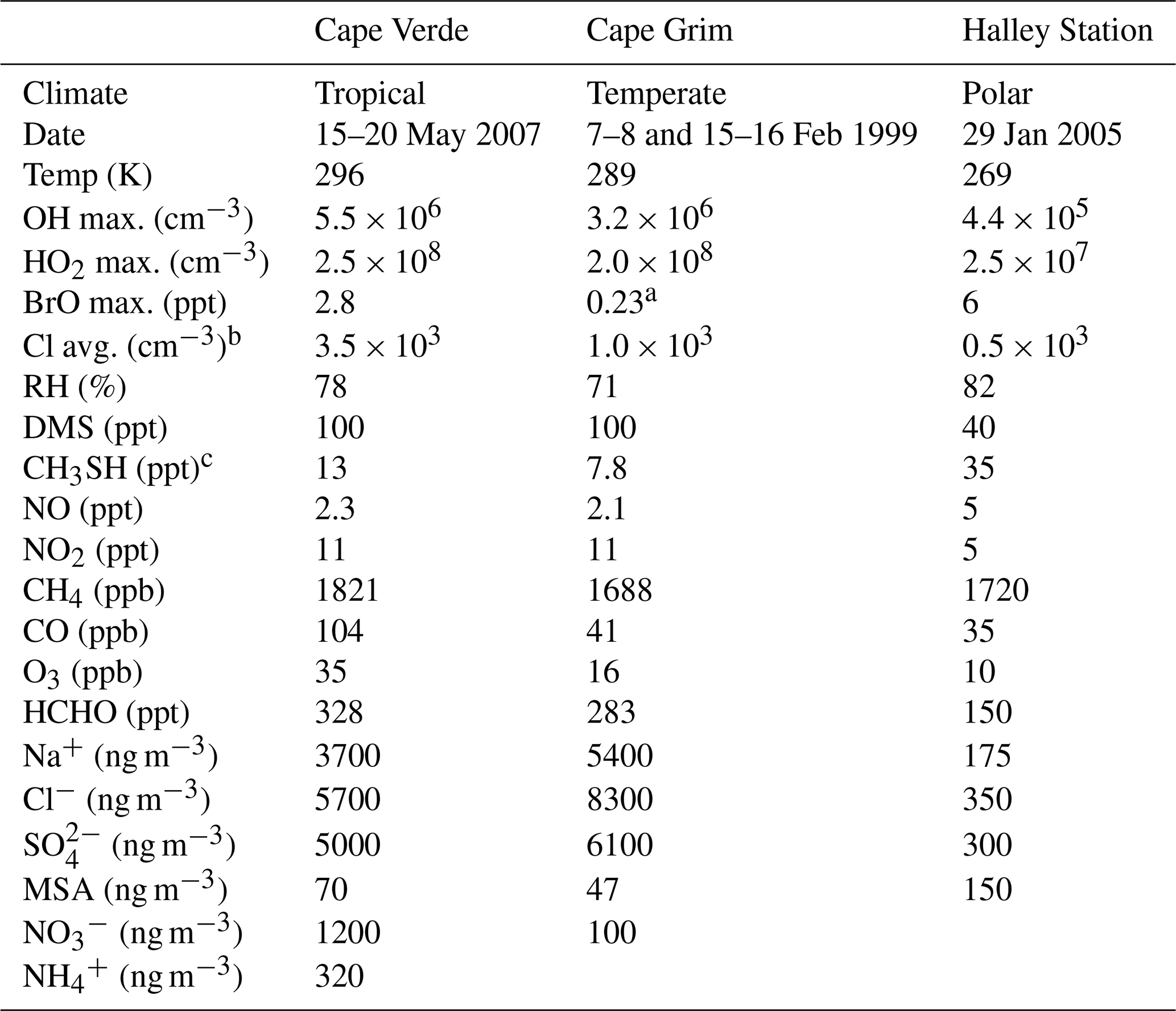
a Cape Grim BrO concentration is based on the average mixing ratio (0.1 ppt) from ATom measurements below 2 km in marine environments (Veres et al., 2020). b Gas-phase chlorine atom concentrations are annual mean surface concentrations from GEOS-Chem modelling (Wang et al., 2021). c These are modelled average CH3SH mixing ratios, which were obtained after applying the emissions from Wohl et al. (2024) (described in the text).
The field campaign data were averaged hourly to provide the input pressure, boundary layer height, temperature, and relative humidity.
Dilution of all species was based on boundary layer height; as the height increased, “background” air based on the initial concentrations of non-sulfur species was added to dilute the concentrations of species in the box. Photolysis rates were calculated using a similar approach to the MCM (Saunders et al., 2003), using a zenith angle based on the latitude, longitude, and date of the fieldwork. Clear sky conditions were assumed for photolysis. The aerosol liquid water content, aerosol pH and dry deposition velocities calculated from available data were used directly as parameters in the model (see Supplement for more details). Box model simulations for each location lasted for eight days, with the parameters of the box models (such as temperature and photolysis rate constants) repeated for each day, allowing the concentrations of the species to stabilise. The initial concentrations and constant emissions of DMS, NOx, CH4, CO, O3 and HCHO were adjusted in each box model until the average concentration given in Table 1 was obtained, or in the case of Cape Grim, the daytime averages (11:00 a.m.–02:00 p.m.). Additional loss factors for OH and HO2 radicals were included and adjusted until the maximum radical concentrations aligned with the measured values.
Although not measured in the campaigns, methanethiol (CH3SH) emissions were included in the box models based on the global emission fields from Wohl et al. (2024) which were developed using measured CH3SH seawater concentrations. The ratios of CH3SH flux to total volatile methylated sulfur flux (combining CH3SH and DMS) from Wohl et al. (2024) were 18 %, 15 %, and 23 % at the latitudes corresponding to Cape Verde, Cape Grim, and Halley Station, respectively, during the season when the fieldwork campaigns were conducted. These ratios correspond to a CH3SH flux that is 22 %, 18 % and 30 % of the DMS flux, respectively, and the corresponding ratio for each site was used to determine the CH3SH emissions in each marine box model.
As including a full halogen mechanism was outside the scope of this work, BrO and Cl concentrations were calculated offline. The concentration of BrO follows a diurnal cycle similar to a top-hat distribution (Saiz-Lopez et al., 2007; Read et al., 2008); this distribution was replicated in this work using the broad time-dependent photolysis rate of O3 forming O(3P) and scaled to the maximum concentration of BrO used for the marine runs. The diurnal cycle of Cl follows a narrow distribution (Chang et al., 2004), and as such, the photolysis rate of O3 forming O(1D) was used to provide the diurnal cycle. Chlorine atom concentrations in the box models were based on annual mean surface concentrations from GEOS-Chem calculations (Wang et al., 2021). Daily average concentrations of 3.5×103, 1.0×103 and 0.5×103 molec. cm−3 were used for Cape Verde, Cape Grim and Halley Station, respectively. The diurnal cycle applied to the average concentrations resulted in maximum Cl concentrations of 1.4×104, 0.4×104 and 0.1×104 molec. cm−3, which are within an order of magnitude of Cl atom concentrations measured in the marine environment (Saiz-Lopez and von Glasow, 2012).
The initial conditions, emissions, and OH and HO2 loss rates, along with time-dependent BrO mixing ratios, Cl concentrations, pressure, temperature, dry deposition and boundary layer height used in the three box models, can be found in Sect. S8. The zero-dimensional models were run using BOXMOX (Knote and Barre, 2022), a KPP wrapper (Sandu and Sander, 2006). The microphysics used in the model to incorporate monodisperse aerosols was based on the SPACCIM box model (Wolke et al., 2005) and described in further detail in Sect. S1.1. Although this work focuses on the gas-phase concentrations of sulfur species, the concentrations of several aqueous species for the three marine regimes are provided in Sect. S9.
Figure 2 shows the loss rate of DMS through different reaction pathways in the base run of the different box models on the last day of the model runs. These rates are affected both by temperature and oxidant concentration and, as such, differ between the different marine scenarios. In the tropical and polar regimes, the primary oxidation mechanism is by reaction with BrO (49 % and 96 %, respectively), followed by reaction with OH (29 % abstraction and 14 % addition in the tropical regime, 1.4 % abstraction and 2.7 % addition in the polar regime). In the temperate model, reaction with OH was the primary oxidation mechanism (49 % abstraction, 33 % addition), followed by reaction with BrO (12 %). The reaction with Cl atoms is minor (0.2 %–3 %), and loss from the aqueous reaction of DMS with O3 is negligible (< 1 %). NO3 oxidation contributes a maximum of 5%, in the tropical conditions. Although the fraction of oxidation through OH abstraction in the temperate and tropical box models is similar to modelled global oxidation (27 %–37 %), the oxidation through BrO addition is less representative of global oxidation (8 %–18 %) in the tropical and polar regimes (Khan et al., 2016; Fung et al., 2022; Chen et al., 2018; Tashmim et al., 2024). The measured BrO mixing ratio in the polar region is representative of elevated BrO formation in polar sea-ice regions (typically in spring), which is potentially due to the uplifting of brine-coated snow (Seo et al., 2020). Elevated bromine compounds in coastal sites (such as Cape Verde) can arise from increased concentrations of biological marine sources such as macroalgae and plankton (Quack and Wallace, 2003; Carpenter and Liss, 2000; Butler et al., 2007). Although column measurements of BrO indicate concentrations of 1–2 ppt throughout the free troposphere, BrO concentrations are typically lower in the marine boundary layer outside coastal regions (Wang et al., 2015; Veres et al., 2020), which could explain the difference between global BrO-initiated oxidation of DMS, and the tropical and polar box models used in this work.
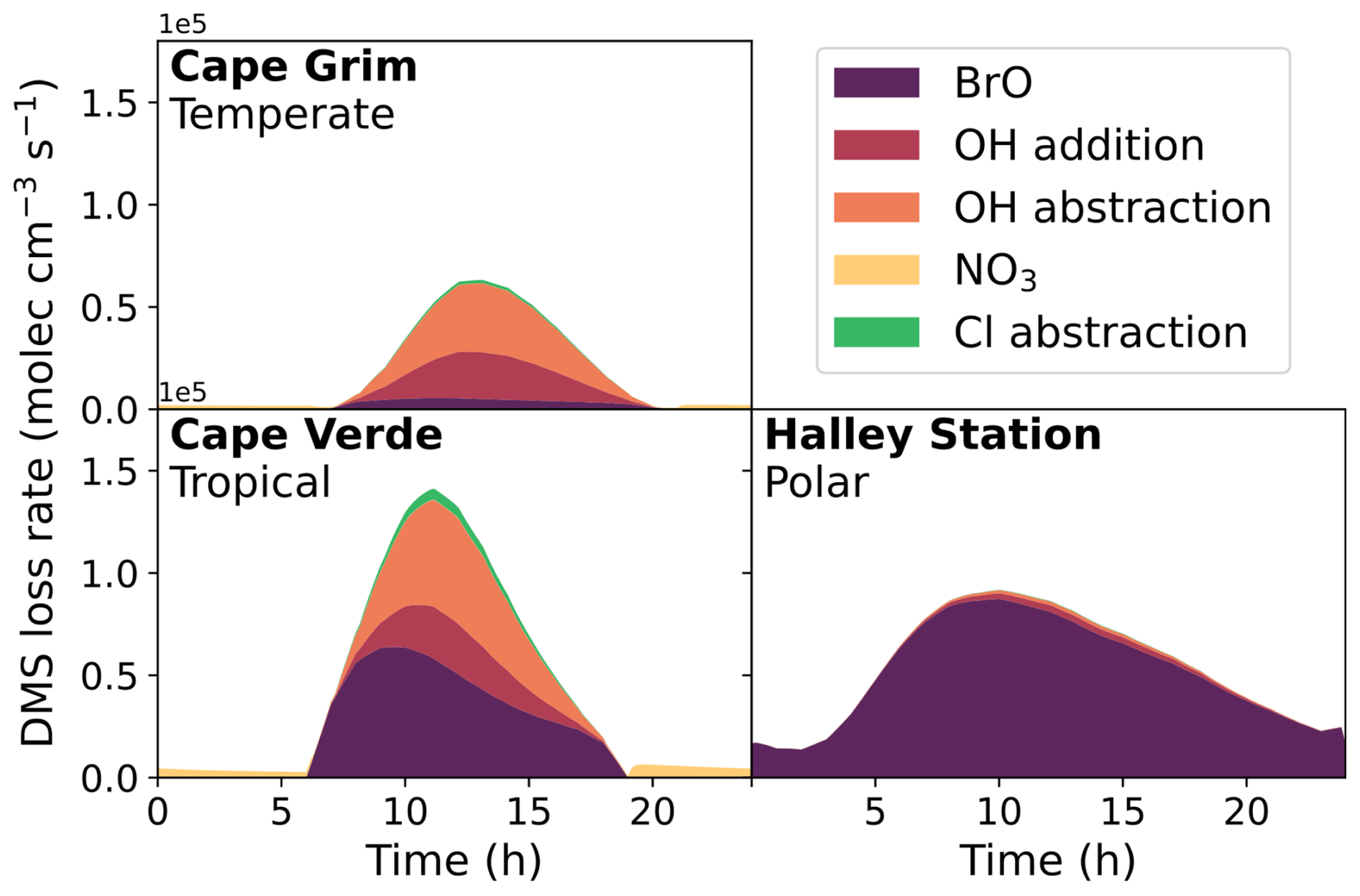
Figure 2The rate of loss of DMS from the major oxidation pathways, BrO, OH, NO3 and Cl, in the three marine regime scenarios on the last day of the eight-day box model run.
The combined RO2 and HO2 concentration was measured at Halley Station and Cape Grim. In both cases, the total mixing ratio was less than 15 ppt over the chosen days. Our model slightly over-predicts the RO2 + HO2 mixing ratio in Cape Grim (maximum of 26 ppt) and underestimates it in Halley Station (maximum of 5 ppt).
Hereafter, the Cape Verde, Cape Grim, and Halley Station box models will be referred to as the tropical, temperate, and polar box models, respectively.
4.1 Determining uncertainty in rate constants
In the literature, there is no consistent method of determining the uncertainties that should be attributed to rate constants (Atkinson et al., 2006; Burkholder et al., 2019; Dunker et al., 2020; Vasyunin et al., 2004). For reactions that have been evaluated by review panels, such as IUPAC or NASA panel reports (Atkinson et al., 2006; Burkholder et al., 2019), estimates of uncertainty factors are provided for rate constants. Neither of these panels use statistical methods to determine these factors, due to the limited data available, and instead rely on expert knowledge of the techniques and experiments, and consideration of systematic errors. Additionally, the uncertainty factors depend on the number of studies that measured a specific reaction, and the agreement between those studies.
In this work, we have developed a methodology to assign uncertainties to rate constants, which is roughly based on the methodology used by IUPAC (Atkinson et al., 2006) due to it being transparent and data-driven. Our framework is illustrated in Fig. 3 and described in the rest of this section, with the exceptions to our methodology outlined in Sect. S10.
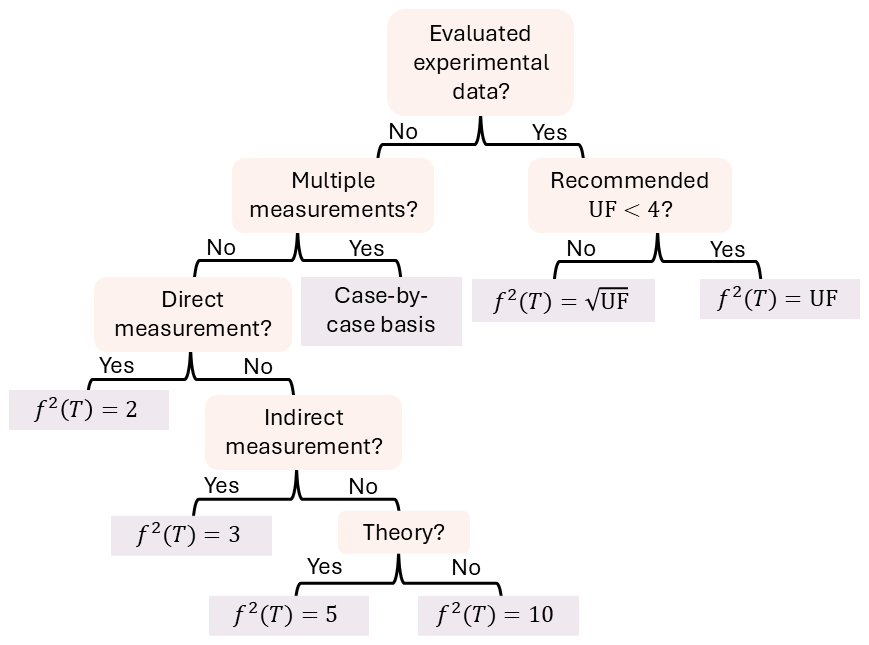
Figure 3The framework for assigning uncertainty factors to rate constants used in this work, where UF stands for uncertainty factor, and represents the 95 % confidence interval uncertainty factor recommended by the NASA panel report. In this flowchart, “evaluated experimental data” refers to reactions evaluated by the NASA panel report (Burkholder et al., 2019). Details and exceptions to this framework can be found in the text.
The majority of the kinetic data used in atmospheric chemistry models come from the NASA and IUPAC panel reports. It is important to stress that the uncertainty factors chosen by the NASA panel report and IUPAC differ, especially regarding single measurements. In their evaluation of kinetic data, the IUPAC panel assigns rate constants that are based on a single measurement an uncertainty factor of 2 or 3.2 (with a maximum of 5), depending on the reliability of the measurement (Atkinson et al., 2006). The NASA panel report uses larger bounds regarding high levels of uncertainty; for their 95 % confidence interval at 298 K (f2(298 K), equivalent to 2σ in normal distributions), their uncertainty factors range from 4 to 100 when reactions have only been measured once. One example of this difference is the reaction of CH3SOO with NO2, demonstrated in Fig. 4. Although both review panels recommend the same rate constant (2.2 cm3 molec.−1 s−1) for the reaction, IUPAC recommends a factor of 2 uncertainty, whereas the 95 % confidence interval recommended by the NASA panel report (JPL) is a factor of 4.
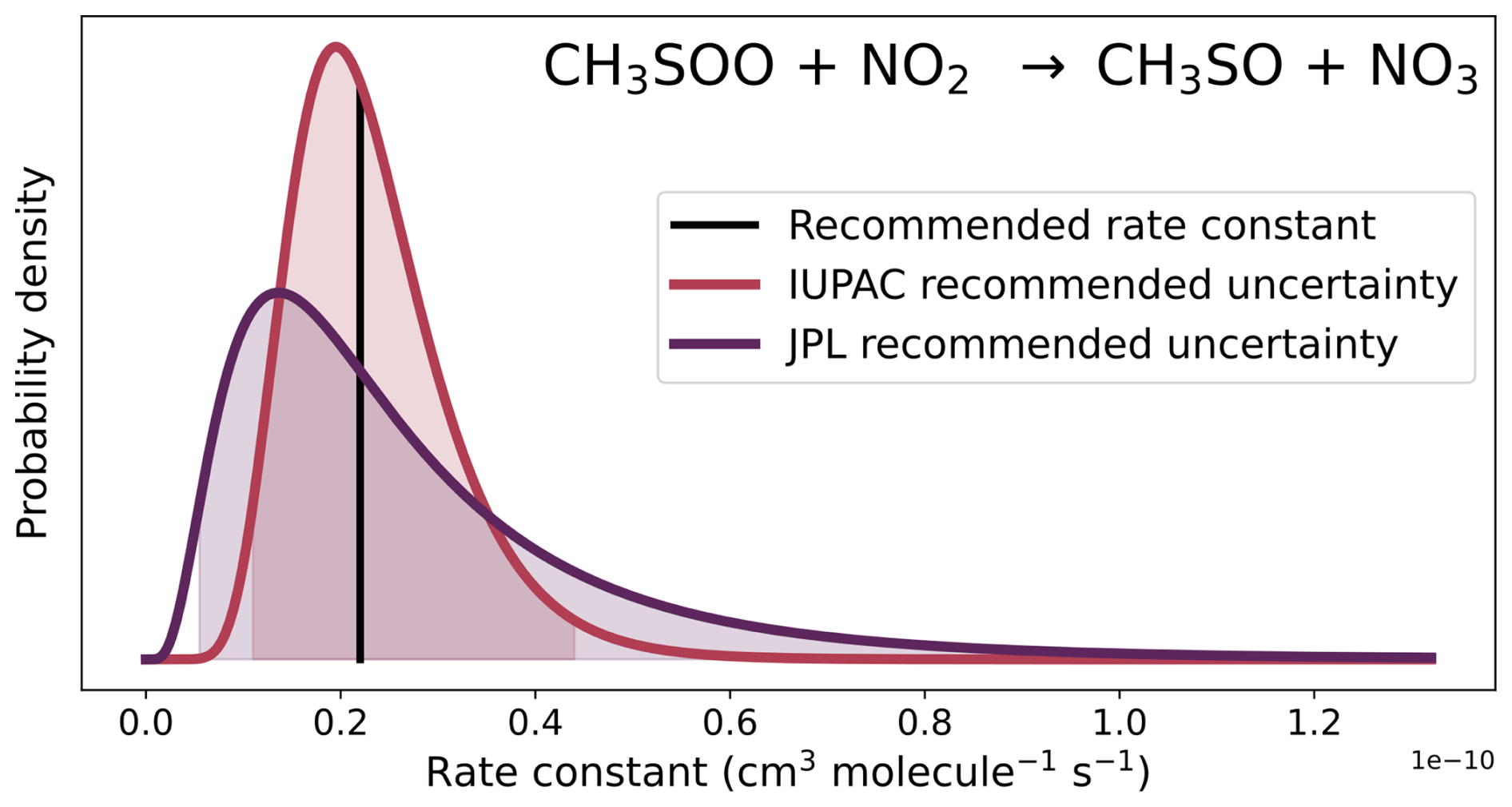
Figure 4The recommended rate constant for the reaction of CH3SOO with NO2 (black line), and the probability distributions for that rate constant at 298 K from the IUPAC (red line), and NASA panel report (JPL, purple line) recommendations. The shaded areas represent the 95 % confidence intervals in the rate constants.
Although the methodology of assigning uncertainties in this work was based on IUPAC, the evaluation of DMS oxidation chemistry in the NASA panel report is more up-to-date than the IUPAC evaluation. As such, the rate constants of our mechanism are based on the NASA panel report when available, and the uncertainties for those rate constants from that report (which had also been updated) were used in this work. If the uncertainty factor from the NASA panel report included temperature dependence (g), the average temperature of the marine regime (shown in Table 1) was used to calculate the uncertainty factor for that box model run. However, to have a consistent framework of attributing uncertainties, when the 95 % confidence interval recommended by the NASA panel report used an uncertainty factor of 4 or larger, the square root of that factor was used instead, bringing the factor closer to the uncertainty factors recommended by IUPAC, as described in the previous paragraph. In the case of the rate constant displayed in Fig. 4, a factor of 2 was used in this work. These larger uncertainty factors, which are all integers, are not based on a statistical analysis, but are estimates; the larger uncertainty factors recommended by the NASA panel report have been reduced to align with the IUPAC framework.
In addition to evaluated data from the NASA panel report, the mechanism used in this work includes (i) reactions from recent studies that have not yet been evaluated, (ii) theoretical calculations, and (iii) structure-activity relationships. When a rate constant has been measured once experimentally but not evaluated, it was given an uncertainty factor of 2 or 3, depending on the reliability of the measurement, following the guidelines from IUPAC (Atkinson et al., 2006). Indirect measurements are considered less reliable as they depend on other reactions and rate constants that may not be fully understood (Atkinson et al., 2006). As such, indirect measurements are given an uncertainty factor of 3, and direct measurements are given an uncertainty factor of 2. Theoretical calculations were given an uncertainty factor of 5, in accordance with the factor of 5 uncertainty attributed to rate constants from the theory calculations by Chen et al. (2023), and the factor of 3 uncertainty attributed to the calculations from Jernigan et al. (2022). Although theoretical calculations from Vereecken et al. (2025) and Lv et al. (2019) were used in this work and may have higher uncertainties, the model was not sensitive to these reactions within the conditions used in this work. However, applying an uncertainty factor of 5 to theoretical calculations may not apply to other chemical mechanisms, which could result in an underestimation of the uncertainty. Finally, uncertainty factors of 10 have been attributed to reactions that are based on structure-activity relationships or estimates. The structure-activity relationships (SARs) used in this work derive from the Master Chemical Mechanism (MCM) and are based on carbon-based chemistry (Saunders et al., 2003), whereas the estimates, mostly from Yin et al. (1990), consider sulfur chemistry, bond dissociation energies, and ab initio calculations. The source attributed to each of the rate constants used in this work can be found in Table S2.
Previous studies have tended to use much narrower uncertainty bounds than we apply in this work. In a study by Newsome and Evans (2017), the authors performed an uncertainty analysis of tropospheric atmospheric chemistry and used the 1σ uncertainty factors from the NASA panel report in their analysis. Dunker et al. (2020) also used 1σ factors from the NASA panel report, supplementing them with IUPAC recommendations and assigning uncertainty factors themselves when recommendations were not available. Their uncertainty factors ranged from 1.05–10. When Vasyunin et al. (2004) investigated the influence of rate constant uncertainties in astrochemical modelling results, they assumed an uncertainty factor of 2 when a structure-activity relationship was used or the uncertainty was not provided for a rate constant.
4.2 Sampling of uncertainty
The uncertainty factors of rate constants provide a range of probable values. However, the “true” value will most likely be close to the median, which corresponds to the measured, calculated or estimated rate constant. This probability can be reflected by a probability density function for the rate constants. Due to the skewed nature of the uncertainty factors and the requirement for rate constants to be positive, a log-normal distribution is typically used as the probability density function (Stewart and Thompson, 1996), as demonstrated in Fig. 4. The uncertainty factors recommended by the NASA panel report and IUPAC define upper and lower bounds which correspond approximately to the 95 % confidence interval of a log-normal distribution, and the recommended rate constant represents the median value in the distribution. As such, the log-normal distribution is well suited to represent the uncertainty of the rate constants in this work.
A purely random sampling of this distribution would be computationally expensive, as each value sampled for a rate constant would require a box model simulation. To approach this, a constrained Monte Carlo method, Latin hypercube sampling, has been chosen to reduce the number of simulations needed to represent the uncertainty distribution (Saltelli and Annoni, 2010). This method divides the probability distribution into bins of equally sized probabilities, and randomly samples from within those bins (each bin contains one sample). The number of bins corresponds to the total number of simulations run. This global sampling method is done simultaneously and independently for all reactions to cover the whole possible uncertainty space.
4.3 Calculating sensitivity indices
The Latin hypercube sampling described above yields uncertainty ranges for the modelled concentrations of species. However, further analysis is needed to understand the contribution of specific parameters (rate constants) to the modelled uncertainty. The EASI RBD-FAST method used in this work (through the SALib coding library in Python) provides first-order sensitivity indices, which are a measure of the contribution of one parameter (reaction/process) to the uncertainty of the output (concentration of a species) (Herman and Usher, 2017). For reaction i, the first-order sensitivity index, Si, ranges from zero to one. In a linear system, the sum of these first-order sensitivity indices for all n reactions/processes (where n=166 in this case) will equal one. However, in nonlinear systems, second-order sensitivity indices, which describe the contributions from interactions between reactions, become increasingly important. In these systems, the total of first-order contributions is less than one, due to higher-order sensitivity indices:
To determine how many simulations are required for reliable results from EASI RBD-FAST, the analysis should be repeated with different numbers of simulations to evaluate when the results converge (Stein et al., 2022). This analysis has been done for the tropical box model at midday and a sample size of 2000 simulations was chosen based on that work (discussed in detail in Sect. S11).
The results of the constrained Monte Carlo sampling for all three marine regimes over the last modelled day are given in Fig. 5, with the shaded regions corresponding to the 90 % confidence interval at each time step of gas-phase concentrations from the 2000 box model simulations. We focus on a subset of species simulated by the mechanism, with the species chosen being major oxidation products that were measured in laboratory chamber studies (Albu et al., 2008; Jernigan et al., 2022; Ye et al., 2022; Shen et al., 2022), along with the precursors CH3SH and DMS. The distributions of mixing ratios for these species across the simulations at midday are given in Sect. S12.
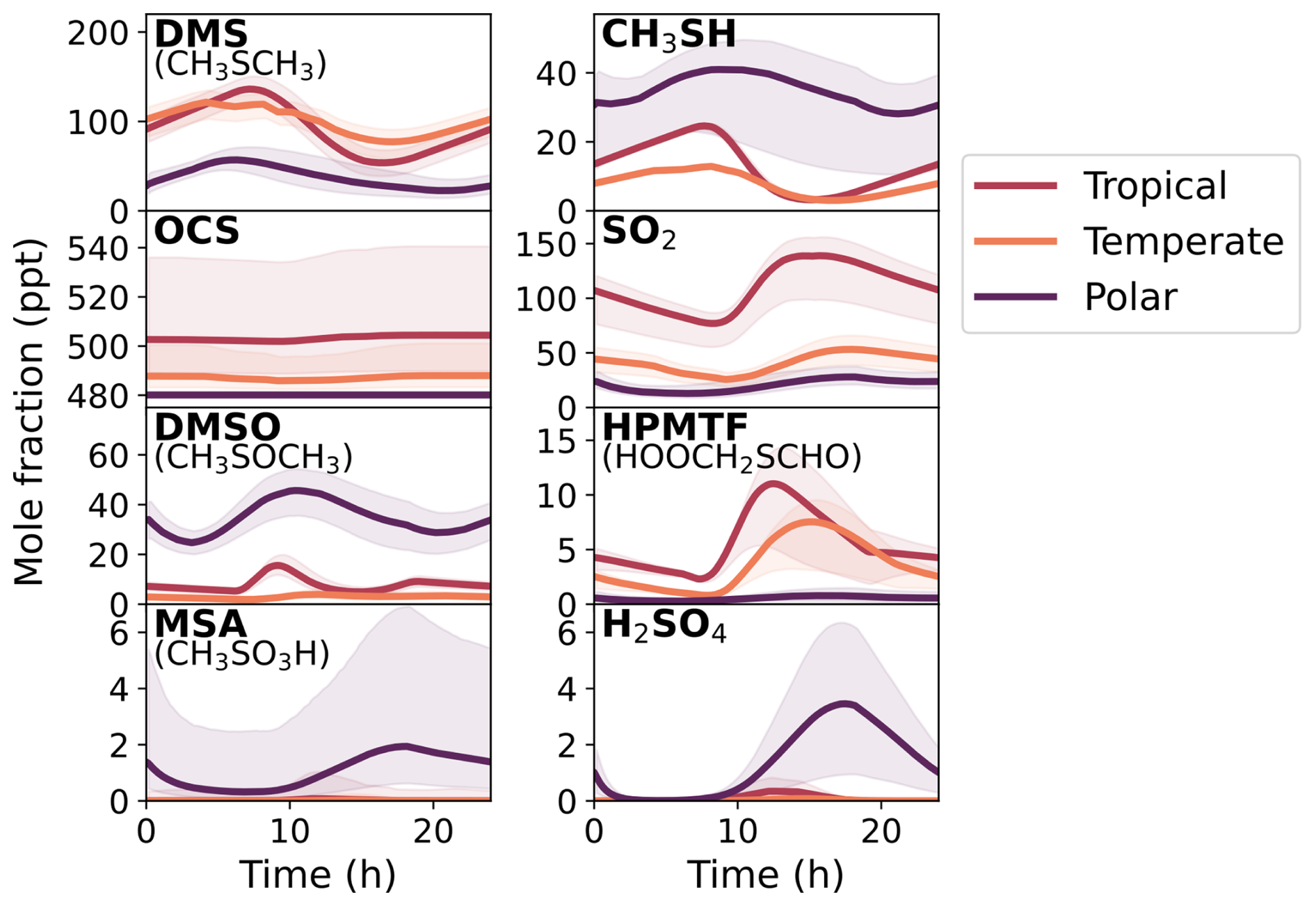
Figure 5The concentration of gas-phase DMS, SO2, OCS, DMSO, CH3SH, HPMTF, MSA, and H2SO4 in the base run (solid lines) on the last day of the eight-day box model run, along with the 90 % confidence interval based on the 2000 Monte Carlo simulations (shaded area). The Cape Verde, Cape Grim, and Halley Station represent the tropical, temperate, and polar marine regimes, respectively. For OCS, a mixing ratio of 480 ppt has been added to incorporate measured background mixing ratios.
These results (Fig. 5) provide an overview of the uncertainty in the modelled concentrations from the uncertainty in the chemical mechanism. The focus of this work is to explore the uncertainties in the gas-phase mechanism. Both the gas-phase rate constants along with aqueous partitioning coefficients have been perturbed, but none of the uncertainty from input parameters, such as liquid water content, emissions, or dry deposition velocities, has been included. Additionally, uncertainties from the aqueous chemistry or adsorption rate have not been included, which would provide additional uncertainties through the loss of gas-phase species. Nevertheless, aqueous chemistry was included in these box models as it improves the representation of the loss pathways of the gas-phase species through aerosol uptake. Finally, whilst our aim would be to have fully quantitative results, the results presented here must be viewed as semi-quantitative, as they are dependent on the uncertainty factors chosen for each rate constant, some of which are not based on statistical analysis, and do not account for the uncertainty of “missing chemistry” (reactions that are not in our mechanism but may be occurring in the atmosphere).
Figure 5 shows that the concentration of MSA had the largest relative uncertainty, with average upper uncertainties of 3000 %, 4000 % and 400 % for the tropical, temperate, and polar box models, respectively. The relative uncertainty was smaller during the daylight hours, when the diurnal concentration of MSA peaked (from around 12:00 p.m. to 05:00 p.m.), but larger at night – hinting at specific processes that dominate the uncertainty. H2SO4 and OCS were also highly uncertain, with average upper uncertainties ranging from 100 %–200 % (except for OCS in the polar regime, which had an upper uncertainty of 340 %). However, including a background mixing ratio of 480 ppt, due to the long lifetime of OCS (Cartwright et al., 2023), decreases the total uncertainty of OCS to within ±7 % for all regimes. Although the average uncertainty in the concentration of CH3SH was within ±12 % in the tropical and temperate regimes, an average upper uncertainty of 25 % and lower uncertainty of 55 % was found for CH3SH in the polar regime. The average uncertainties for HPMTF across the tropical, temperate, and polar regimes were within ±39 %, ±43 % and ±86 %, respectively. DMS and SO2 had average uncertainties within ±30 % in the tropical and temperate regimes, and within ±50 % in the polar regime. Finally, the average uncertainty of dimethyl sulfoxide (CH3SOCH3, DMSO) ranged from ±25 %–49 % across all regimes.
5.1 Comparison with observations
Whilst our results demonstrate that many of the modelled species have narrow uncertainty bounds, not varying largely from their base run, several species exhibit large uncertainty in their simulated profiles. We can assess whether the uncertainty bounds of our simulations result in concentrations that are realistic by comparing the simulated mixing ratios of the species with larger uncertainty bounds to literature data.
OCS is a ubiquitous source of sulfur in the atmosphere. Its long tropospheric lifetime of around 2.5 years (Cartwright et al., 2023) makes it challenging to simulate in a box model. Rather than include the full OCS sources and sinks, our box model runs have focused on the contribution of DMS and CH3SH to OCS formation. Although the uncertainty for OCS is over 150 % in the tropical and temperate box models, the upper bound for OCS formed solely from DMS oxidation within the eight-day box model run was only 60 ppt. To rectify the lack of full sources and sinks of OCS, a mixing ratio of 480 ppt was added to OCS as a post-processing step based on the measured average OCS mixing ratio of 480 ppt (Davidson et al., 2021). The resultant OCS mixing ratios are shown in Fig. 5.
MSA and H2SO4 had upper uncertainty bounds up to 6.9 ppt (1.9 ×108 molec. cm−3) and 6.3 ppt (1.7 ×108 molec. cm−3) in the polar regime, respectively. In the tropical and temperate box models, the upper uncertainty bounds reached 1.0 ppt for MSA (2.6 ×107 molec. cm−3) and 0.8 ppt for H2SO4 (2.0 ×107 molec. cm−3). Measurements of gas-phase MSA and H2SO4 concentrations range from 105–107 molec. cm−3 in clean Southern Ocean air (Baccarini et al., 2021), and up to 2 ×107 and 1.1 ×108 molec. cm−3 for MSA and H2SO4, respectively, at Mace Head (Berresheim et al., 2002), a temperate marine environment. This comparison indicates that the upper uncertainty bounds for the polar regime may be unrealistic, however, the base concentrations are within observed concentrations.
Performing EASI RBD-FAST analysis on the results of the 2000 Latin hypercube sampling, shown in Fig. 5, provides first-order sensitivity indices which can be used to determine which reactions are contributing most to the uncertainties. A first-order sensitivity index describes the contribution of one reaction to the uncertainty in the concentration of a species at one time point.
As an example, the first-order sensitivity indices for HPMTF in the three regimes at midday are provided in Table 2. These results can be interpreted as follows: OH-initiated oxidation of HPMTF contributes 37 % to the uncertainty in the HPMTF concentration in the tropical regime, making it the largest contributor for that regime, followed by the Norrish Type I photolysis of HPMTF (resulting in the loss of the HCO radical, 24 %), which is the largest contributor to the uncertainty in the temperate regime (24 %). Sampling the model output at different times of the day leads to slightly different sensitivity indices and is explored in Sect. 6.1.
Table 2The estimated first-order sensitivity indices (as a percentage) for HPMTF in the three marine regimes at midday, along with the uncertainty factor associated with each reaction. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
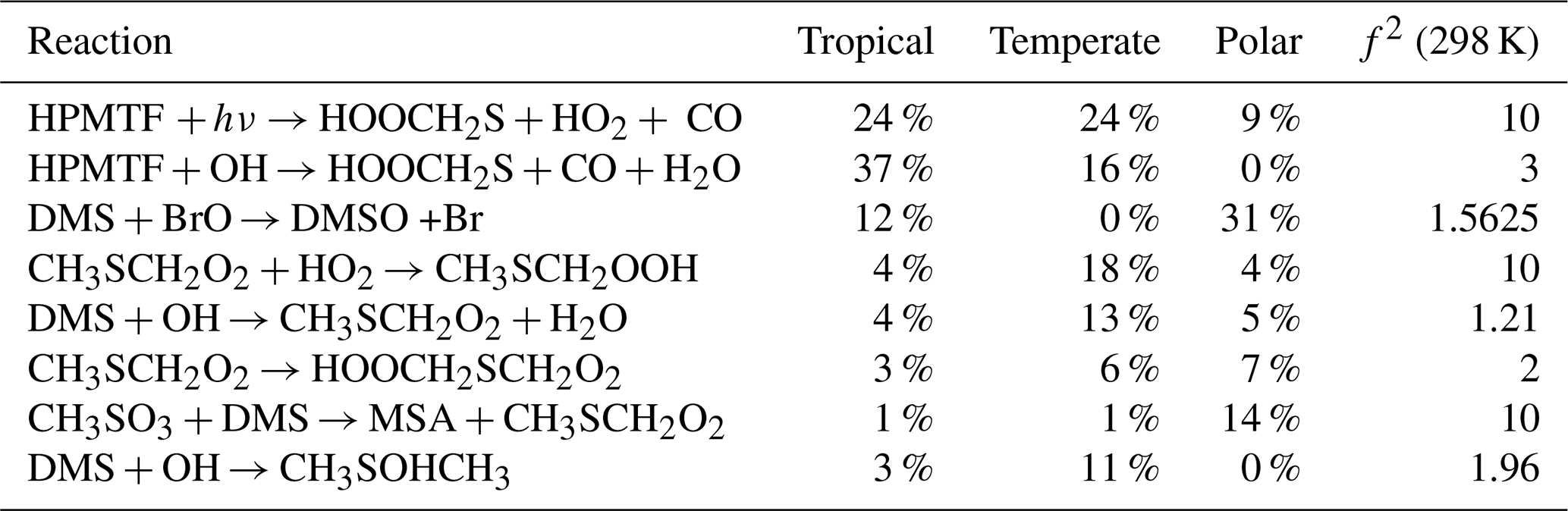
To visualise the role that specific reactions play, those that contribute at least 5 % to the uncertainty in one marine regime for MSA, OCS, HPMTF, and SO2 at midday are displayed on a simplified DMS oxidation scheme in Fig. 6. The mechanism is shown in black, with colours used to indicate reactions that contribute to uncertainty in different species. Tables including these reactions and their contributions, along with DMSO and H2SO4, can be found in Appendix A.
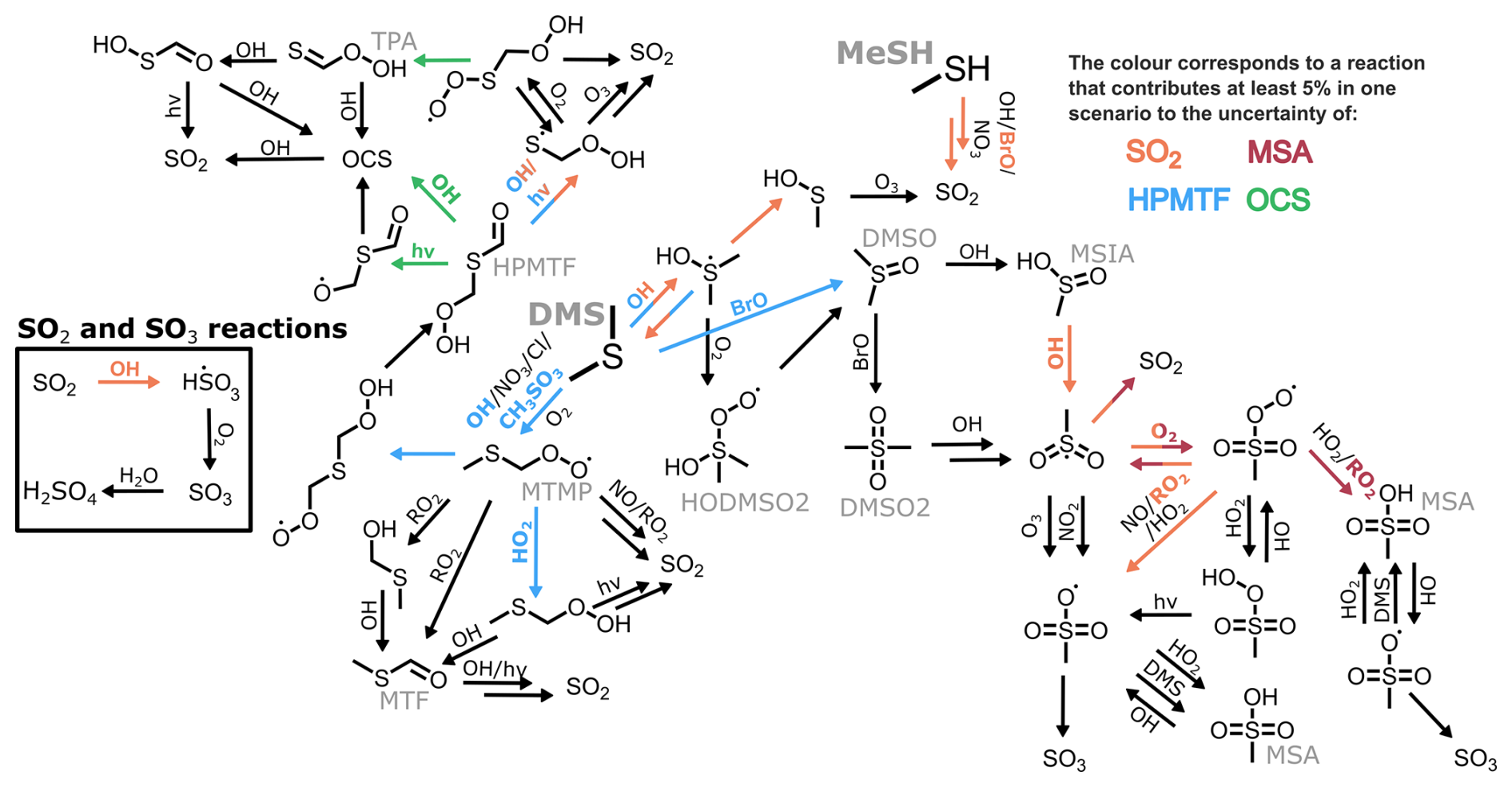
Figure 6A simplified DMS oxidation mechanism highlighting reactions that contribute at least 5 % to the uncertainty of SO2, MSA, HPMTF and OCS, through orange, maroon, blue, and green arrows (respectively).
The EASI RBD-FAST method does not provide estimates of second-order sensitivity indices, defined as the contributions to the uncertainty due to interactions between two reactions. For one species, if the sum of these first-order sensitivity indices is close to one, then the uncertainty is well represented by first-order interactions (independent contributions from individual reactions). To calculate this total, all values that contributed less than 0.75 % to the uncertainty were excluded, as with the chosen sample size of 2000, negative sensitivity indices of up to −0.75 % were found (these negative values are model approximation errors and are an indication of the noise in the sensitivity indices). The total first-order contributions (total SI) are provided in Table 3.
Table 3The total first-order sensitivity indices (Total SI) of key species in the three marine regimes, along with R2 values from a multiple linear regression model.
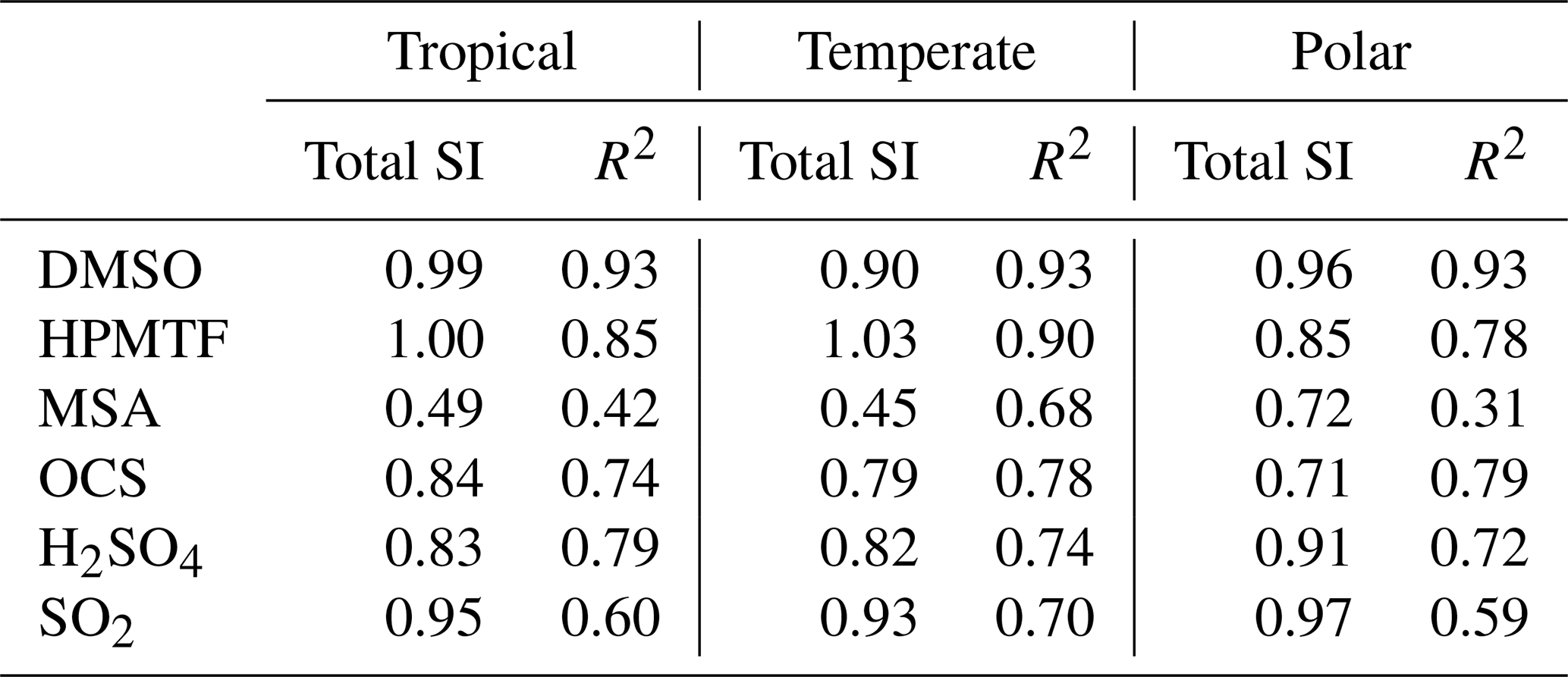
The total SI is above 80 % for DMSO, HPMTF, H2SO4, and SO2, however, it is as low as 45 % for MSA in the temperate regime; consequently, only 45 % of the uncertainty in MSA in that regime can be attributed to specific reactions from the DMS oxidation mechanism in this study. A low total SI is usually due to a nonlinear relationship, resulting in more contributions from the interactions between reactions. The nonlinearity was explored by analysing the simulation results with a multiple linear regression model which also measured the contribution of each reaction to the uncertainty in the concentration of the sulfur species at midday, based on Rodriguez and Dabdub (2003). The resultant R2 values are given in Table 3. In three cases (MSA and DMSO in the temperate regime, and OCS in the polar regime), the R2 value was higher than the total SI calculated. Otherwise, the total SI was larger than the R2 value, which demonstrates that the lower total sensitivity indices are due to nonlinearities. Second-order sensitivity indices would provide more information on the contribution of specific reactions to the uncertainty, however, a full Sobol sensitivity analysis, which would provide second-order sensitivity indices, is unfeasible, with over 160 parameters (reactions/processes) being explored (Stein et al., 2022).
6.1 Time dependence of the uncertainty
The sensitivity analysis thus far has focused on the contribution of reactions to the uncertainty in the concentration of different species at midday; in this section, the time dependence is explored for SO2.
The first column of Fig. 7 displays the rate of formation and loss of SO2 from different reactions and processes. The SO2 formation from the reaction of CH3SO with O3 and the isomerisation of CH3SOO into CH3SO2 (which decomposes into SO2 due to the internal energy following the isomerisation) are grouped together in purple. In the marine conditions used in this work, CH3SO and CH3SOO arise from CH3S reacting with O3 and O2, respectively. In these conditions, over 79 % of the CH3S formed in the base run is from the reaction of CH3SH with OH or BrO, with 2 %–12 % from the decomposition of CH3SCH2O, which stem from the reaction of CH3SCH2OO with RO2 or NO. This highlights the important role that CH3SH makes to oxidised sulfur species in the marine environment.
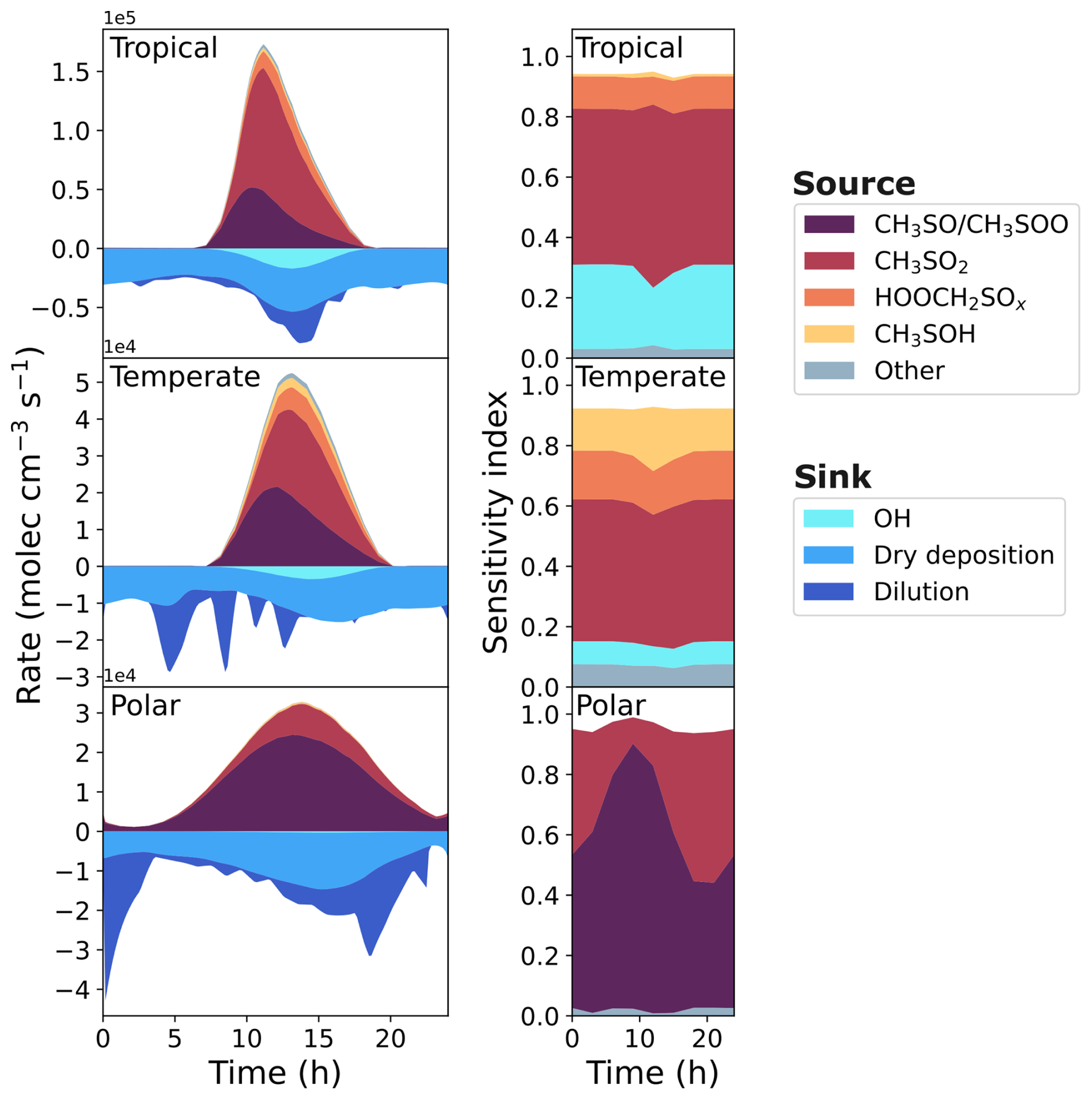
Figure 7The rate of production and loss of SO2 in the base run of the three marine regimes (first column), and the corresponding grouped sensitivity indices for the uncertainty in SO2 concentration (second column).
Although the reactions of HOOCH2SO and HOOCH2SOO have not been studied experimentally or computationally, their rate constants in this work use structure-activity relationships based on the reactions of CH3SO and CH3SOO, respectively. The HOOCH2SO reaction with O3 and the isomerisation of HOOCH2SOO (and subsequent decomposition) forming SO2 have been grouped as HOOCH3SOx. HOOCH2SO and HOOCH2SOO both arise from reactions of HOOCH2S, which forms from the photolysis of HPMTF, and the reaction of OH with HPMTF.
Another major source of SO2 is the decomposition of CH3SO2, which arises from the reaction of methanesulfinic acid (CH3SO2H, MSIA) with OH. In our mechanism, MSIA is the sole product of the reaction of DMSO with OH, with DMSO originating from an addition reaction of BrO, Cl, or OH with DMS.
Finally, SO2 can form from the reaction of CH3SOH with O3. CH3SOH is a minor product from the addition reaction of OH with DMS, as the addition reaction primarily forms DMSO (after subsequent reaction with O2).
Loss of SO2 through dilution occurs when the boundary layer height increases in the model, while dry deposition depends on wind speed and frictional velocity (see Supplement for details). As the effect of clouds has not been included in these box models, loss of SO2 through aqueous reactions is negligible.
The second column of Fig. 7 shows the contribution of grouped reactions to the uncertainty in the SO2 concentration. Any reactions that directly form or react with a species that forms SO2 (e.g. CH3SO2) have been grouped with that species. In the case of CH3SO2, that includes the uncertainty from the reaction of MSIA and OH (forming CH3SO2), and the reaction of CH3SO2 with O2. Additionally, reactions that indirectly contribute to the formation of species are also grouped with that species; the reaction of CH3SH with BrO forming CH3S is included in the CH3SO/CH3SOO grouping, as over 99 % of the CH3S produced in the base run reacts with O3 and O2 to form CH3SO and CH3SOO, respectively. Finally, reactions that indirectly contribute to the loss of a species are grouped with that species; since the reaction of CH3SO2 with O2 forming CH3SO2O2 is reversible, reactions of CH3SO2O2 with RO2 have been grouped with CH3SO2, as they reduce the concentration of CH3SO2. The groupings of reactions that contribute at least 5 % to the uncertainty of SO2 in one marine regime are given in Table A1.
In the temperate and tropical marine regimes, the contributions of different reactions to the uncertainty in SO2 concentration are not very time-dependent, with some variation observed in the contributions at midday. However, the sensitivity indices in the polar regime do show time dependence; the contribution of CH3SO/CH3SOO reactions range from 40 %–88 % depending on the time of day, with a peak contribution at around 09:00 a.m. In this work, sensitivity indices are a measure of the effect of the variance of one rate constant on the variance in the concentration at one time point. The sensitivity indices indicate that the concentration of SO2 in the polar box model is primarily dependent on the reaction between CH3SH and BrO (f2(T)=10) in the morning, and becomes increasingly dependent on the decomposition of CH3SO2 (f2(T)=5) in the evening. As shown in Fig. 2, 96 % of the oxidation of DMS occurs through the addition of BrO, forming DMSO. However, the broader diurnal distribution of BrO compared to OH radicals, the major chemical sink of DMSO, results in a build-up of DMSO until 10:00 a.m. (Fig. 5), with the rate of loss of DMSO through OH oxidation reaching a maximum around 1 pm. The MSIA formed from DMSO oxidation must also react with OH radicals to form CH3SO2, which is the source of SO2. Conversely, SO2 formed from the BrO-initiated oxidation of CH3SH is independent of OH concentration. Additionally, variance in the rate constant of the reaction of BrO with CH3SH results in a large variance in the concentration of CH3SH (Fig. 5); in the lower uncertainty bound (when the rate constant is largest), the maximum concentration, 23.0 ppt around 06:00 a.m., is a factor of 2.2 higher than the minimum concentration, 10.5 ppt around 08:00 p.m. Although the rate constant is larger in this case, the lower CH3SH concentrations in the afternoon and evening would result in less SO2 produced through the oxidation of CH3SH at those times.
The combination of the time dependence of CH3SH concentration, and the reactions of OH with DMSO becoming more important in the afternoon (after a buildup of DMSO in the morning), results in the time dependence in sensitivity indices of SO2 concentration. Although it is important to consider the time dependence of the sensitivity indices, the following results focus on midday concentrations, at which time the rates of DMS oxidation are generally higher, due to the higher concentrations of oxidants (see Fig. 2). However, summary figures similar to Figs. 8 and 9 showing sensitivity indices using concentrations at midnight, 06:00 a.m. and 06:00 p.m. are included in Sect. S13.
6.2 Contributions of reaction types
To provide an overview of the sensitivity results at midday, the contributions of reactions to the total uncertainty have been grouped into reaction types, displayed in Fig. 8. These groupings are based on the reactant involved (i.e. BrO or O2). In the case of unimolecular reactions, isomerisation and decomposition have been grouped into the unimolecular category, with photolysis reactions separated. Reactions where CH3SO3 radicals abstract hydrogen from H-atom donors such as DMS, HO2 and HCHO are grouped as “H-donor” reactions. The remaining HO2 reactions are grouped with RO2. Additionally, the contribution from the uncertainty in Henry's law constants has been categorised as “aqueous partitioning”. Finally, minor reaction types not described here were lumped together in the “other” category. In Fig. 8, all reactions have been grouped into a category, and as such, the total contribution is the total of the first-order sensitivity indices.
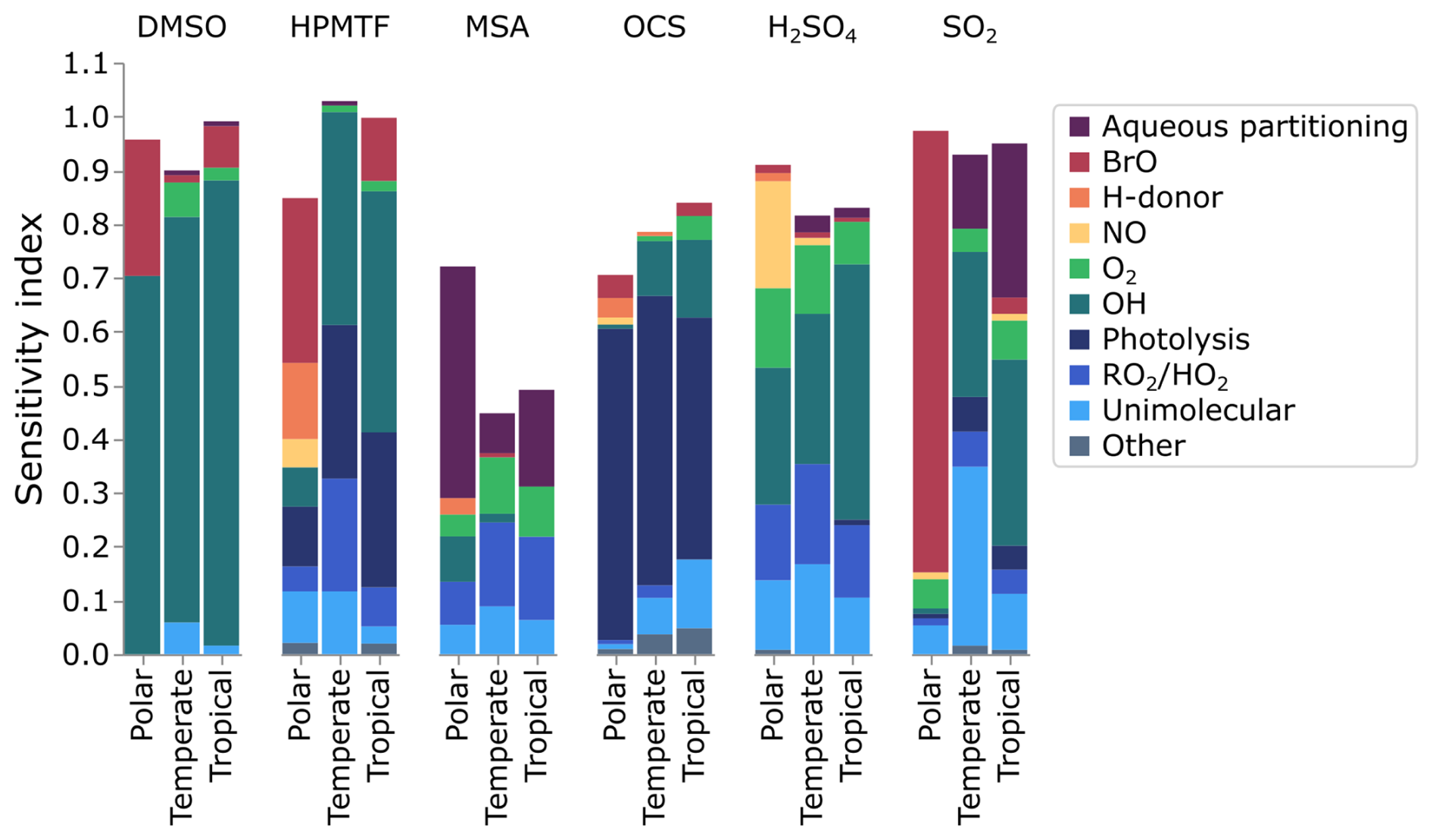
Figure 8The contribution of different processes (reactions and phase transfers) to the uncertainty in gas-phase DMSO, HPMTF, MSA, OCS, H2SO4, and SO2.
Photolysis reactions contribute 45 %–58 % of the OCS uncertainty and contribute 11 %–29 % of the uncertainty in HPMTF. In both cases, this is due to the photolysis reactions of HPMTF, which have never been studied and were attributed an uncertainty factor of 10. These photolysis reactions, the Norrish Type 1 reaction resulting in the loss of HCO, and the photolysis of the hydroperoxyl group, are based on the photolysis of C3H7CHO and CH3OOH, respectively.
OH reactions contribute over 10 % of the uncertainty of DMSO, HPMTF, OCS, H2SO4 and SO2 in the temperate and tropical marine regimes, due to the reactions of OH with SO2, MSIA, DMSO, and HPMTF. The uncertainty in these reactions is generally low, as they have been explored experimentally, with a maximum uncertainty factor of 3.23 (for the reaction of SO2 with OH at 269 K). However, their contribution to the total uncertainty demonstrates their importance.
The reactions of BrO primarily affect DMSO and HPMTF in the tropical and polar regimes due to the reaction of DMS and BrO (contributing 8 %–31 %, f2(T)= 1.58–1.81). BrO-initiated oxidation of DMS forms DMSO (CH3SOCH3), from which HPMTF (HOOCH2SCHO) cannot be produced. As such, the dependence on the reaction to HPMTF formation is inversely related; a faster rate of reaction of DMS with BrO results in a decrease in HPMTF production. The reaction of BrO with CH3SH is the largest contributor to the uncertainty in SO2 in the polar regime (82 %, f2(T)=10), as described in the previous section.
The reactions with HO2 and RO2 primarily affect the concentration of MSA (8 %–16 %) and H2SO4 (13 %–19 %), with reactions with NO also contributing to the uncertainty in the concentration of H2SO4; the importance of these reactions arises from the reactions of CH3SO2O2 and CH3SO3. CH3SO2O2 can react with RO2 and HO2 to form MSA or CH3SO3, while the reaction with NO solely forms CH3SO3. Although CH3SO3 can react with a H-donor (such as HO2 or DMS) to form MSA, these reactions contribute less than 3 % to the uncertainty in MSA; the rate constant of the reaction of CH3SO3 with HO2 recommended by the Master Chemical Mechanism (Jenkin et al., 1997) has been reduced by a factor of 50 in our updated mechanism based on an analysis of measurements by Berndt (2025) (see Sect. S2 for more information). The fate of CH3SO3 in this work is primarily decomposition into SO3, which forms H2SO4.
The importance of unimolecular reactions is predominantly due to two decomposition reactions (CH3SO2 and CH3SOHCH3) and two isomerisation reactions (CH3SCH2OO and HOOCH2SOO). The decomposition of CH3SO2 forms SO2 (based on Chen et al. (2023) theory, f2(T)=5), and is in competition with the reversible addition of O2, forming CH3SO2O2. Additionally, the decomposition of CH3SOHCH3 to form CH3SOH (f2(T)=10), which is considered a minor pathway, contributes to the uncertainty in SO2 as CH3SOH is converted to SO2 through reaction with O3. The isomerisation reaction of CH3SCH2OO has been measured directly by Assaf et al. (2023) (f2(T)=2), while the isomerisation of HOOCH2SOO forming thioperformic acid (S = CHOOH, TPA) and HO2 is based on theory from Jernigan et al. (2022), and assigned an uncertainty factor of 5.
6.3 Contributions of source types
In addition to grouping the reactions by the type of reaction, they have been grouped by the source of their rate constants used in the mechanism. Figure 9 shows the contribution to uncertainty for different species and regimes, grouped into the sources of the rate constants. In general, the uncertainties in rate constants were based on recommendations from the NASA panel report (defined as “evaluated experiments” in this work); when these recommendations were not available, the methodology outlined in Sect. 4.1 was used. This methodology is based on the reliability of the source producing the rate constant, for example, direct measurements are generally more reliable than indirect measurements. However, if a direct measurement has not been performed at atmospheric pressures (such as the reaction of CH3SH and BrO), it may have a larger uncertainty than indirect measurements. In this work, all theory-based reactions have been attributed an uncertainty factor of 5, and structure-activity relationships (SARs) and estimates a factor of 10 (see Table S2 for more details).
Figure 9 demonstrates that the sources of uncertainty differ between species and marine environments, and can be used to identify how those uncertainties can be reduced. We can use this figure to make some recommendations and general conclusions. We suggest that species whose uncertainties are primarily due to structure-activity relationships or estimates should be focused on by theoreticians, as their work would decrease those uncertainties. When the uncertainty is due to theory, we encourage experimentalists to study those reactions, while rate constants based on a single measurement should also undergo further experimentation to either corroborate or improve the measurement precision (preferably through direct measurements). Finally, large uncertainty factors in the evaluated data could also be reduced through further experiments. As such, Fig. 9 provides a summary of how the uncertainties in the concentrations of DMS oxidation products can be reduced.
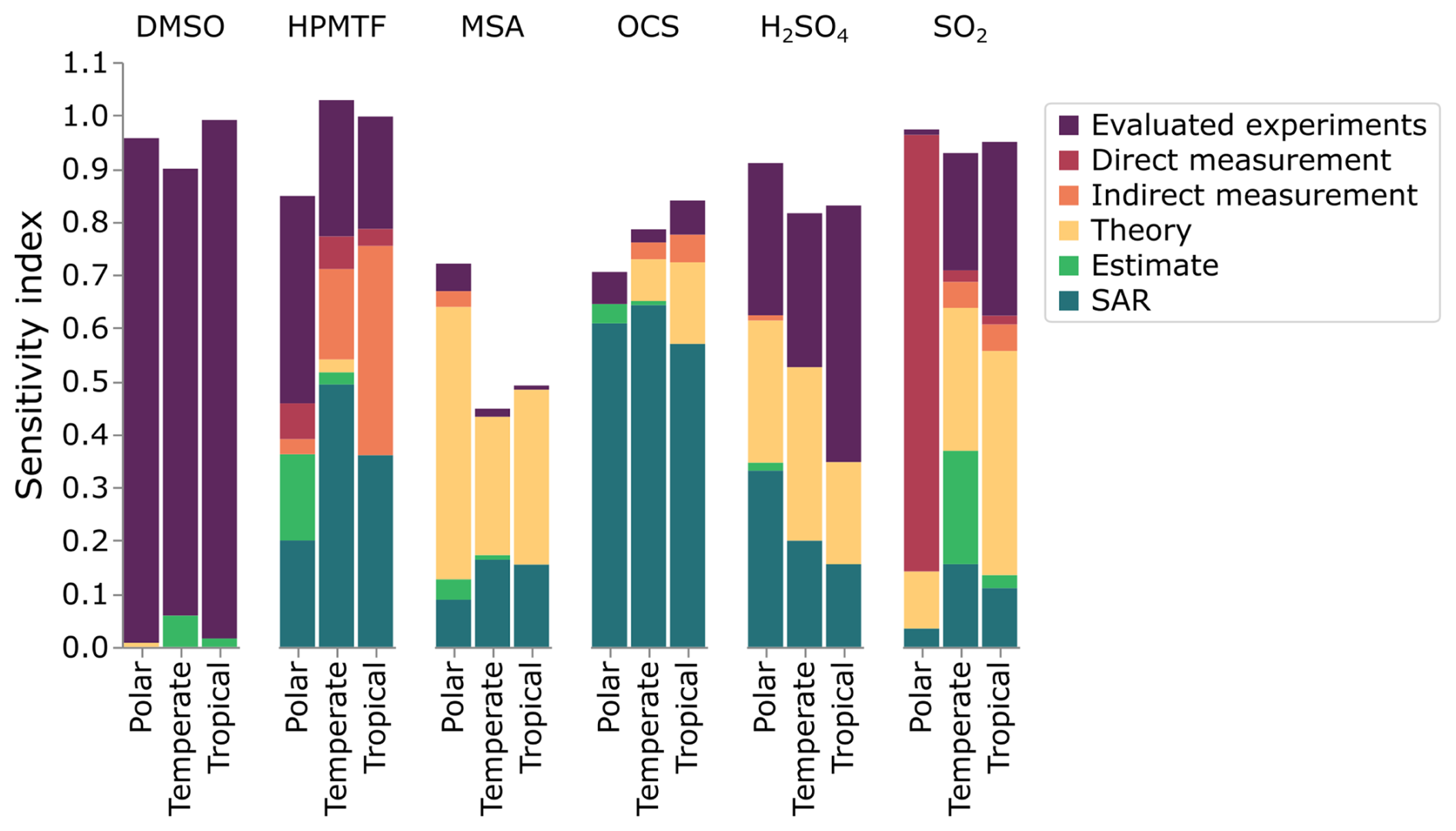
Figure 9The contribution of different sources of rate constants to the uncertainty in gas-phase DMSO, HPMTF, MSA, OCS, H2SO4 and SO2. In this work, “evaluated experiments” refers to reactions evaluated by the NASA panel report (Burkholder et al., 2019).
The uncertainty of DMSO concentration, which was an average of ±25 %–49 % in the regimes, is primarily due to reactions that have been evaluated (84 %–98 %). Three reactions that have been evaluated by the Burkholder et al. (2019) NASA panel report contribute the most to the uncertainty in DMSO concentration: the reaction of DMSO with OH radicals (contributing 40 %–82 %, f2(T)= 1.47–2.07), the reaction of DMS with BrO (contributing 8 %–25 % in the tropical and polar regimes, f2(T)= 1.58–1.81), and the addition reaction of OH with DMS (contributing 35 % in the temperate regime, f2(T)=1.96).
The contribution to uncertainty from direct measurements is smaller than the contribution from indirect experiments, with the exception of HPMTF and SO2 in the polar regime. In the polar regime, the primary contribution from an indirect experiment is the uncertainty from the reaction of CH3SH and BrO for SO2 (contributing 82 %, f2(T)=10), and the isomerisation of CH3SCH2O2 for HPMTF (contributing 7 %, f2(T)=2).
Rate constants arising from theory contribute the most to the uncertainty of MSA (26 %–51 %), and SO2 in the temperate and tropical regimes (27 % and 42 %, respectively). For both species, this is due to the decomposition of CH3SO2 forming SO2 (f2(T)=5), and the reversible addition of oxygen to CH3SO2 (f2(T)=5), along with the uncertainty in Henry's law constant for MSIA (f2(T)=55). Note that the larger uncertainty factors for Henry's law constants reflect the assigned uncertainties in the NASA panel report (see Sect. S10 and Table S1 for more information). Additionally, the Henry's law constant for MSA contributes 7 %–17 % of the uncertainty in the concentration of MSA (f2(T)=55).
Finally, structure-activity relationships (SARs), which have an uncertainty factor of 10, contribute over 57 % of the uncertainty in OCS concentration and over 15 % in HPMTF and H2SO4. SAR-based rate constants are the largest contributor to the uncertainty in OCS concentration due to the photolysis of the hydroperoxyl group from HPMTF, which has never been studied, and is based on the photolysis of CH3OOH. Additionally, the reactions of CH3SO2O2 with RO2 and NO, based on structure-activity relationships, contribute to the uncertainties in SO2, MSA, and H2SO4.
This work builds on previous efforts (Lucas and Prinn, 2005; Saltelli and Hjorth, 1995; Campolongo et al., 1999) to quantify the uncertainty in the DMS oxidation mechanism and identify key reactions that drive the sensitivity of the species involved. We have developed an expanded and up-to-date mechanism, evaluated against a wide range of experimental conditions, that accounts for not only the OH-initiated oxidation of DMS, but also the halogen reactions and aqueous uptake of DMS and its oxidation species, and for the first time coupled in CH3SH, given its recently recognised importance in the marine sulfur cycle (Wohl et al., 2024). Our work identifies the key reactions, reaction types, and rate constant sources that contribute the most to the uncertainty in the gas-phase concentrations of DMS, CH3SH, SO2, OCS, DMSO, HPMTF, MSA and H2SO4 in tropical, temperate, and polar marine environments. The gas-phase reactions that had the largest overall contributions (determined through a sum of the sensitivity indices over all three marine regimes) are included in Table 4.
Table 4Gas-phase reactions identified as the largest overall contributors to the uncertainty of MSA, H2SO4, SO2, OCS, and HPMTF. The uncertainty factors at 298 K (f2), the sources of the rate constants, and the species that are affected by the reactions are also included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
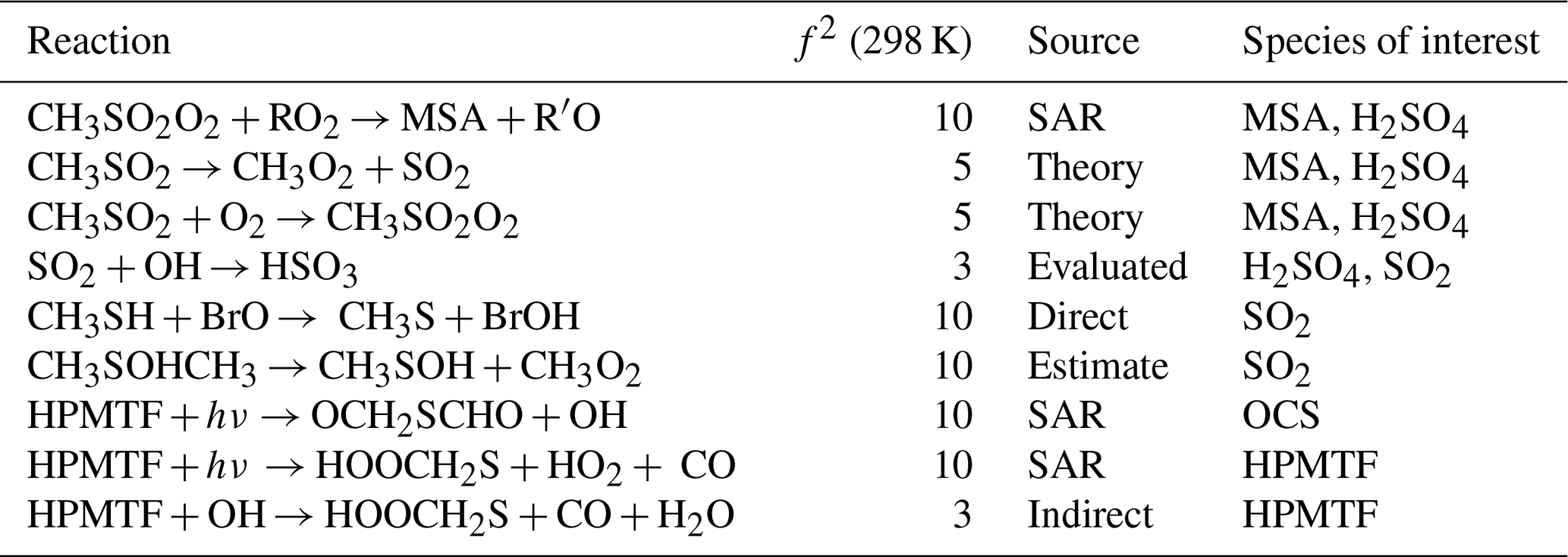
Our work identified that uncertainties in the kinetics of DMS oxidation are particularly important for OCS. Over the eight-day model simulations we found that OCS produced in the model varied by up to 150 %, mostly arising from the photolysis of HPMTF, which has never been studied experimentally or theoretically. Future studies on these photolysis reactions are needed. Uncertainties of up to ±55 % were found for CH3SH in the polar region, where CH3SH emissions are generally highest (Wohl et al., 2024). The uncertainty in CH3SH stems from its reaction with BrO, which has only been measured at pressures up to 3 Torr; further experiments at atmospheric pressures are required. Finally, uncertainties in gas-phase MSA and H2SO4 are based on a range of reactions, however further study into reactions of CH3SO2O2 with RO2 and NO, reactions of CH3SOx with oxygen, the decomposition of CH3SO2, and Henry's law constant for MSA would reduce their uncertainties. This analysis does not include uncertainty from reactions that are not in our mechanism; more chamber studies and fieldwork are needed to identify new products, and “missing” chemistry. Additionally, the results depend on the uncertainty factors chosen for each reaction; for reactions that have not been extensively studied, the uncertainty factors may be underestimated.
Our work has demonstrated the usefulness of the EASI RBD-FAST method in atmospheric chemistry; with the nonlinearity observed in some species (such as MSA with an R2 value as low as 0.31), multiple linear regression is unsuitable. However, as uncertainty analysis relies on attributing uncertainties to rate constants, uncertainty propagation becomes difficult with mechanisms that include hundreds or thousands of reactions. To improve the feasibility of this work, we recommend that databases of chemical mechanisms, such as the Master Chemical Mechanism (MCM), should include uncertainty factors for each reaction, and crucially details on how those uncertainty factors were derived.
This analysis relied on fieldwork to constrain chemical and physical parameters, however, concentrations of gas-phase DMS oxidation products, along with CH3SH and halogen concentrations (with the exception of BrO in two campaigns), were not available. Additional fieldwork exploring DMS oxidation in different marine regimes would allow a more thorough evaluation of how realistic the uncertainties are and explore whether the observations are captured within the uncertainties. Marine field campaigns conducted in the remote ocean should be prioritised, as they would allow an investigation of marine conditions with a reduced influence of bromine and anthropogenic emissions observed in coastal regions.
As we've shown in Table 4, in some cases, the largest contributors to uncertainty have been underexplored. However, some reactions will require significant advancements to lower the uncertainty in their kinetics, as these sit at the bounds of what is possible with current technology (such as the reaction of SO2 with OH). A balance is needed to tackle both types of reactions, and we urge modellers to engage with experimentalists to explore similar systems (e.g. the oxidation of other compounds important for air quality and climate). Our results will enable further studies that combine these uncertainties with observational constraints to produce improved mechanisms of the oxidation of DMS and CH3SH.
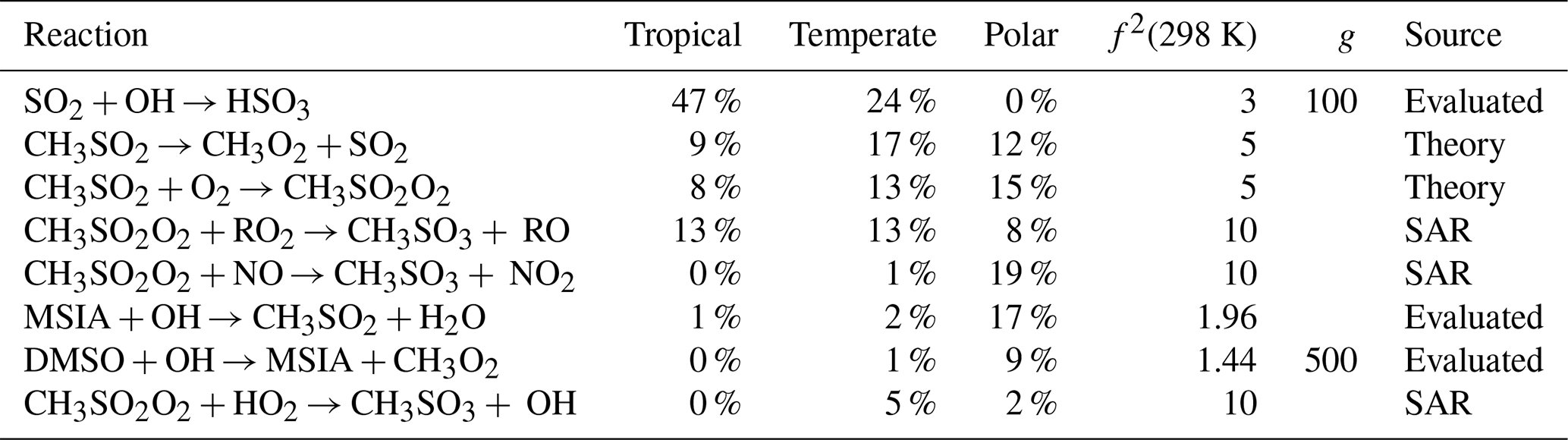
Table A1The estimated first-order sensitivity indices for SO2 in the three marine regimes at midday, along with the uncertainty factor at 298 K associated with each reaction (f2 (298 K)), the temperature dependence of the uncertainty if included (g), and the source of the rate constant. Additionally, the chosen groupings of the reactions, relevant for Fig. 7, are given. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
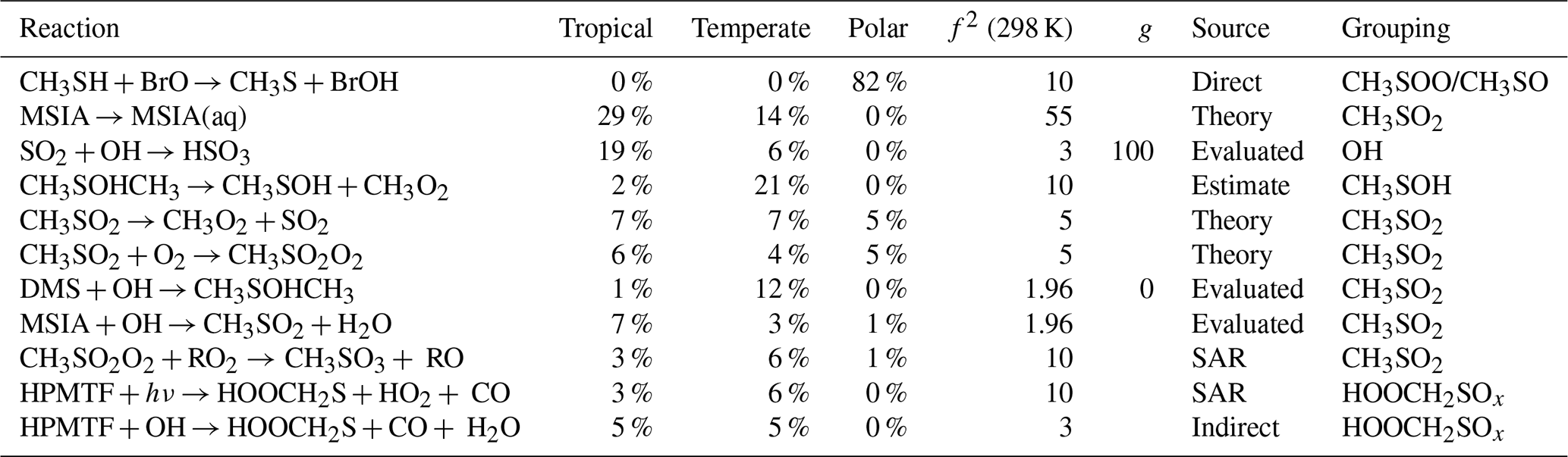
Table A2The estimated first-order sensitivity indices for DMSO (CH3SOCH3) in the three marine regimes at midday, along with the uncertainty factor at 298 K associated with each reaction (f2 (298 K)), the temperature dependence of the uncertainty if included (g), and the source of the rate constant. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
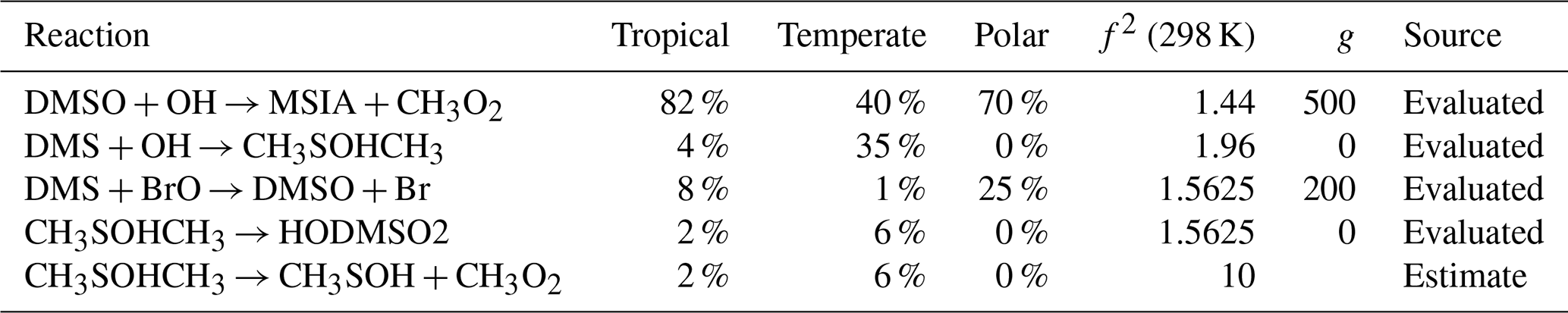
Table A3The estimated first-order sensitivity indices for MSA (CH3SO3H) in the three marine regimes at midday, along with the uncertainty factor at 298 K associated with each reaction (f2 (298 K)), the temperature dependence of the uncertainty if included (g), and the source of the rate constant. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
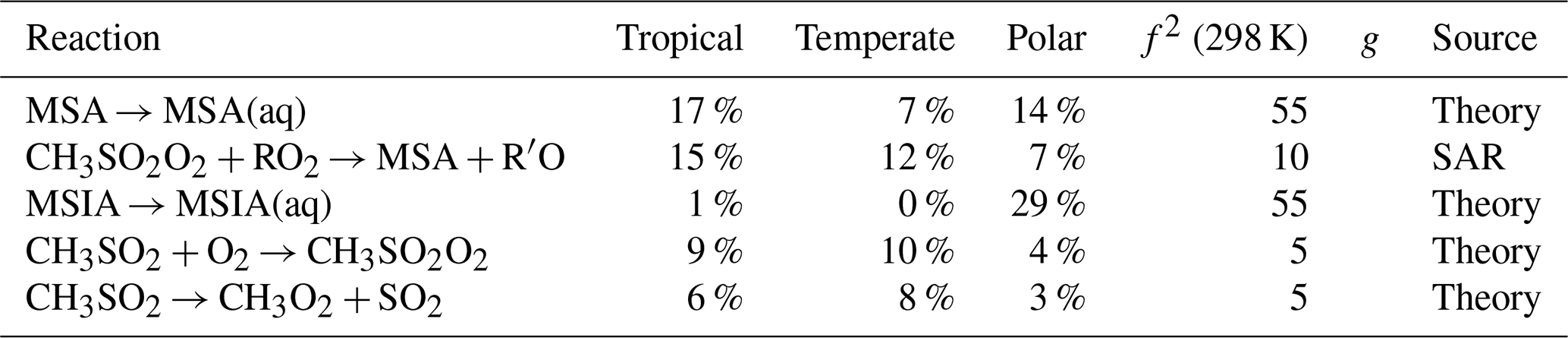
Table A4The estimated first-order sensitivity indices for OCS in the three marine regimes at midday, along with the uncertainty factor at 298 K associated with each reaction (f2 (298 K)), the temperature dependence of the uncertainty if included (g), and the source of the rate constant. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
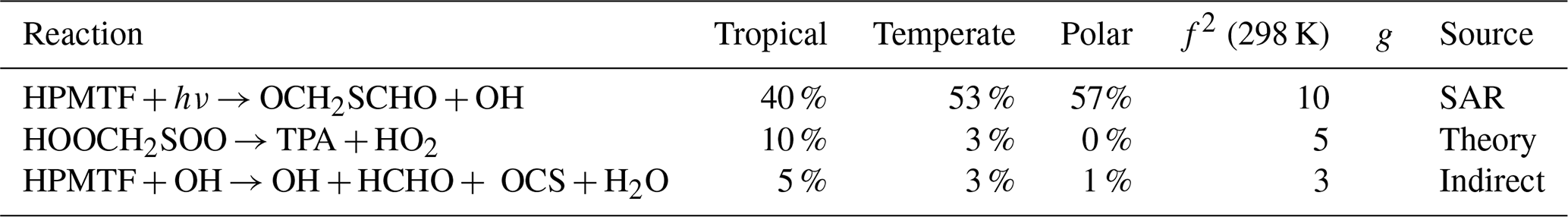
Table A5The estimated first-order sensitivity indices for H2SO4 in the three marine regimes at midday, along with the uncertainty factor at 298 K associated with each reaction (f2 (298 K)), the temperature dependence of the uncertainty if included (g), and the source of the rate constant. Only reactions that contribute at least 5 % to the uncertainty in one regime have been included. In these reactions O2 has not been conserved, and has been added to radicals when their reaction with O2 is fast.
The model input and output files for the three marine regimes, including the updated mechanism used, can be found through Apollo (https://doi.org/10.17863/CAM.124752, Jacob et al., 2026).
The supplement related to this article is available online at https://doi.org/10.5194/acp-26-3567-2026-supplement.
BEHH wrote the initial uncertainty propagation code while supervised by LSDJ. LSDJ extended this code, constructed the box models, attributed the uncertainty factors, extended the chemical mechanism, performed the analysis, and wrote the paper under the supervision of ATA and CG. All authors were involved in helpful discussions and contributed to the paper.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. The authors bear the ultimate responsibility for providing appropriate place names. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
Lorrie S. D. Jacob acknowledges the Cambridge Australia Scholarships and the Cambridge Trust for stipend and tuition support.
This research has been supported by the Natural Environment Research Council (grant no. NE/W009412/1).
This paper was edited by Kelvin Bates and reviewed by two anonymous referees.
Albu, M., Barnes, I., Becker, K. H., Patroescu-Klotz, I., Benter, T., and Mocanu, R.: FT-IR Product Study on the OH Radical Initiated Oxidation of Dimethyl Sulfide: Temperature and O2 Partial Pressure Dependence, in: Simulation and Assessment of Chemical Processes in a Multiphase Environment, edited by: Barnes, I. and Kharytonov, M. M., Springer Science, Dortdrecht, 501–513, https://doi.org/10.1007/978-1-4020-8846-9_41, 2008. a, b, c
Allan, B., Ayers, G., Baker, J., Brough, N., Carpenter, L., Creasey, D., Fraser, P., Galbally, I., Gillett, R., Heard, D., Kivlighon, L., Kochhar, M., Krummel, P., Lee, J., Lewis, A., Meyer, M., Mills, G., Monks, P., Pilling, M., Salisbury, G., Steele, P., Sturrock, G., and Weeks, I.: Southern Ocean Atmospheric Photochemistry Experiment 2 (SOAPEX-2): atmospheric constituents concentration measurements from Cape Grim, Tasmania, NCAS British Atmospheric Data Centre [data set], http://catalogue.ceda.ac.uk/uuid/a266f328ead407624dde4bb5c9e2e6a2/ (last access: 27 February 2026), 1987. a
Andreae, M. O., Elbert, W., Cai, Y., Andrea, T. W., and Gras, J.: Non-sea-salt sulfate, methanesulfonate, and nitrate aerosol concentrations and size distributions at Cape Grim, Tasmania, J. Geophys. Res.-Atmos., 104, 21695–21706, https://doi.org/10.1029/1999JD900283, 1999. a
Arathala, P. and Musah, R. A.: Atmospheric degradation of dimethyl sulfone mediated by OH, Cl and NO3, and the C-centered dimethyl sulfone radical + 3O2 reaction: a kinetics and mechanistic study, Atmos. Environ., 315, 119990, https://doi.org/10.1016/j.atmosenv.2023.119990, 2023. a
Assaf, E., Finewax, Z., Marshall, P., Veres, P. R., Neuman, J. A., and Burkholder, J. B.: Measurement of the intramolecular hydrogen-shift rate coefficient for the CH3SCH2OO radical between 314 and 433 K, J. Phys. Chem. A, 127, 2336–2350, https://doi.org/10.1021/acs.jpca.2c09095, 2023. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a, b, c, d, e, f
Baccarini, A., Dommen, J., Lehtipalo, K., Henning, S., Modini, R. L., Gysel-Beer, M., Baltensperger, U., and Schmale, J.: Low-volatility vapors and new particle formation over the Southern Ocean during the Antarctic Circumnavigation Expedition, J. Geophys. Res.-Atmos., 126, e2021JD035126, https://doi.org/10.1029/2021JD035126, 2021. a
Bates, T. S., Lamb, B. K., Guenther, A., Dignon, J., and Stoiber, R. E.: Sulfur emissions to the atmosphere from natural sources, J. Atmos. Chem., 14, 315–337, https://doi.org/10.1007/BF00115242, 1992. a
Beck, L. J., Sarnela, N., Junninen, H., Hoppe, C. J. M., Garmash, O., Bianchi, F., Riva, M., Rose, C., Peräkylä, O., Wimmer, D., Kausiala, O., Jokinen, T., Ahonen, L., Mikkilä, J., Hakala, J., He, X.-C., Kontkanen, J., Wolf, K. K. E., Cappelletti, D., Mazzola, M., Traversi, R., Petroselli, C., Viola, A. P., Vitale, V., Lange, R., Massling, A., Nøjgaard, J. K., Krejci, R., Karlsson, L., Zieger, P., Jang, S., Lee, K., Vakkari, V., Lampilahti, J., Thakur, R. C., Leino, K., Kangasluoma, J., Duplissy, E.-M., Siivola, E., Marbouti, M., Tham, Y. J., Saiz-Lopez, A., Petäjä, T., Ehn, M., Worsnop, D. R., Skov, H., Kulmala, M., Kerminen, V.-M., and Sipilä, M.: Differing mechanisms of new particle formation at two Arctic sites, Geophys. Res. Lett., 48, e2020GL091334, https://doi.org/10.1029/2020GL091334, 2021. a
Berndt, T.: Methanesulfonic acid (MSA) and SO3 formation from the addition channel of atmospheric dimethyl sulfide oxidation, Chem. Commun., 61, 1443–1446, https://doi.org/10.1039/D4CC05913A, 2025. a, b
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast peroxy radical isomerization and OH recycling in the reaction of OH radicals with dimethyl sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019. a
Berndt, T., Chen, J., Møller, K. H., Hyttinen, N., Prisle, N. L., Tilgner, A., Hoffmann, E. H., Herrmann, H., and Kjaergaard, H. G.: SO2 formation and peroxy radical isomerization in the atmospheric reaction of OH radicals with dimethyl disulfide, Chem. Commun., 56, 13634–13637, https://doi.org/10.1039/D0CC05783E, 2020. a
Berndt, T., Hoffmann, E. H., Tilgner, A., Stratmann, F., and Herrmann, H.: Direct sulfuric acid formation from the gas-phase oxidation of reduced-sulfur compounds, Nat. Commun., 14, 4849, https://doi.org/10.1038/s41467-023-40586-2, 2023. a
Berresheim, H., Elste, T., Tremmel, H. G., Allen, A. G., Hansson, H.-C., Rosman, K., Dal Maso, M., Mäkelä, J. M., Kulmala, M., and O'Dowd, C. D.: Gas-aerosol relationships of H2SO4, MSA, and OH: observations in the coastal marine boundary layer at Mace Head, Ireland, J. Geophys. Res.-Atmos., 107, D198100, https://doi.org/10.1029/2000JD000229, 2002. a
Boy, M., Kulmala, M., Ruuskanen, T. M., Pihlatie, M., Reissell, A., Aalto, P. P., Keronen, P., Dal Maso, M., Hellen, H., Hakola, H., Jansson, R., Hanke, M., and Arnold, F.: Sulphuric acid closure and contribution to nucleation mode particle growth, Atmos. Chem. Phys., 5, 863–878, https://doi.org/10.5194/acp-5-863-2005, 2005. a
Burkholder, J. B., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 19, Tech. rep., JPL Publication 19-5, Jet Propulsion Laboratory, Pasadena, http://jpldataeval.jpl.nasa.gov/ (last access: 27 February 2026), 2019. a, b, c, d, e
Butler, J. H., King, D. B., Lobert, J. M., Montzka, S. A., Yvon-Lewis, S. A., Hall, B. D., Warwick, N. J., Mondeel, D. J., Aydin, M., and Elkins, J. W.: Oceanic distributions and emissions of short-lived halocarbons, Global Biogeochem. Cy., 21, GB1023, https://doi.org/10.1029/2006GB002732, 2007. a
Cala, B. A., Archer-Nicholls, S., Weber, J., Abraham, N. L., Griffiths, P. T., Jacob, L., Shin, Y. M., Revell, L. E., Woodhouse, M., and Archibald, A. T.: Development, intercomparison, and evaluation of an improved mechanism for the oxidation of dimethyl sulfide in the UKCA model, Atmos. Chem. Phys., 23, 14735–14760, https://doi.org/10.5194/acp-23-14735-2023, 2023. a
Campolongo, F., Saltelli, A., Jensen, N. R., Wilson, J., and Hjorth, J.: The role of multiphase chemistry in the oxidation of dimethylsulphide (DMS). A latitude dependent analysis, J. Atmos. Chem., 32, 327–356, https://doi.org/10.1023/A:1006154618511, 1999. a, b, c, d
Campolongo, F., Cariboni, J., and Saltelli, A.: An effective screening design for sensitivity analysis of large models, Environ. Model. Softw., 22, 1509–1518, https://doi.org/10.1016/j.envsoft.2006.10.004, 2007. a
Carpenter, L. J. and Liss, P. S.: On temperate sources of bromoform and other reactive organic bromine gases, J. Geophys. Res.-Atmos., 105, 20539–20547, https://doi.org/10.1029/2000JD900242, 2000. a
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013. a
Cartwright, M. P., Pope, R. J., Harrison, J. J., Chipperfield, M. P., Wilson, C., Feng, W., Moore, D. P., and Suntharalingam, P.: Constraining the budget of atmospheric carbonyl sulfide using a 3-D chemical transport model, Atmos. Chem. Phys., 23, 10035–10056, https://doi.org/10.5194/acp-23-10035-2023, 2023. a, b
Chang, C.-T., Liu, T.-H., and Jeng, F.-T.: Atmospheric concentrations of the Cl atom, CIO radical, and HO radical in the coastal marine boundary layer, Environ. Res., 94, 67–74, https://doi.org/10.1016/j.envres.2003.07.008, 2004. a
Chen, J., Lane, J. R., Bates, K. H., and Kjaergaard, H. G.: Atmospheric gas-phase formation of methanesulfonic acid, Environ. Sci. Technol., 57, 21168–21177, https://doi.org/10.1021/acs.est.3c07120, 2023. a, b, c, d, e
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018. a
Covert, D. S., Kapustin, V. N., Quinn, P. K., and Bates, T. S.: New particle formation in the marine boundary layer, J. Geophys. Res.-Atmos., 97, 20581–20589, https://doi.org/10.1029/92JD02074, 1992. a
Curry, J. A. and Webster, P. J.: Nucleation and diffusional growth, Chap. 5, in: Thermodynamics of Atmospheres and Oceans, edited by: Holton, J. R., vol. 65, Academic Press, Boulder, 129–158, https://doi.org/10.1016/S0074-6142(99)80027-8, 1999. a
Davidson, C., Amrani, A., and Angert, A.: Tropospheric carbonyl sulfide mass balance based on direct measurements of sulfur isotopes, P. Natl. Acad. Sci. USA, 118, e2020060118, https://doi.org/10.1073/pnas.2020060118, 2021. a
Dunker, A. M., Wilson, G., Bates, J. T., and Yarwood, G.: Chemical sensitivity analysis and uncertainty analysis of ozone production in the comprehensive air quality model with extensions applied to eastern Texas, Environ. Sci. Technol., 54, 5391–5399, https://doi.org/10.1021/acs.est.9b07543, 2020. a, b, c
Fung, K. M., Heald, C. L., Kroll, J. H., Wang, S., Jo, D. S., Gettelman, A., Lu, Z., Liu, X., Zaveri, R. A., Apel, E. C., Blake, D. R., Jimenez, J.-L., Campuzano-Jost, P., Veres, P. R., Bates, T. S., Shilling, J. E., and Zawadowicz, M.: Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing, Atmos. Chem. Phys., 22, 1549–1573, https://doi.org/10.5194/acp-22-1549-2022, 2022. a, b
Goffart, J. and Woloszyn, M.: EASI RBD-FAST: an efficient method of global sensitivity analysis for present and future challenges in building performance simulation, J. Build. Eng., 43, 103129, https://doi.org/10.1016/j.jobe.2021.103129, 2021. a, b
Goss, M. B. and Kroll, J. H.: Chamber studies of OH + dimethyl sulfoxide and dimethyl disulfide: insights into the dimethyl sulfide oxidation mechanism, Atmos. Chem. Phys., 24, 1299–1314, https://doi.org/10.5194/acp-24-1299-2024, 2024. a, b, c
Herman, J. and Usher, W.: SALib: an open-source Python library for Sensitivity Analysis, J. Open Source Softw., 2, 97, https://doi.org/10.21105/joss.00097, 2017. a
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016. a
Jacob, L. S. D., Giorio, C., and Archibald, A. T.: Extension, development, and evaluation of the representation of the OH-initiated dimethyl sulfide (DMS) oxidation mechanism in the Master Chemical Mechanism (MCM) v3.3.1 framework, Atmos. Chem. Phys., 24, 3329–3347, https://doi.org/10.5194/acp-24-3329-2024, 2024. a, b, c, d, e, f
Jacob, L. S. D., Harvey, B. E. H., Giorio, C., and Archibald, A. T.: Input and output files supporting Determining the key sources of uncertainty in dimethyl sulfide and methanethiol oxidation under tropical, temperate, and polar marine conditions, Apollo [code, data set], https://doi.org/10.17863/CAM.124752, 2026. a
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jernigan, C. M., Fite, C. H., Vereecken, L., Berkelhammer, M. B., Rollins, A. W., Rickly, P. S., Novelli, A., Taraborrelli, D., Holmes, C. D., and Bertram, T. H.: Efficient production of carbonyl sulfide in the low-NOx oxidation of dimethyl sulfide, Geophys. Res. Lett., 49, e2021GL096838, https://doi.org/10.1029/2021GL096838, 2022. a, b, c, d, e, f, g, h
Jernigan, C. M., Rivard, M. J., Berkelhammer, M. B., and Bertram, T. H.: Sulfate and carbonyl sulfide production in aqueous reactions of hydroperoxymethyl thioformate, ACS ES&T Air, 1, 397–404, https://doi.org/10.1021/acsestair.3c00098, 2024. a
Jones, A. E., Wolff, E. W., Salmon, R. A., Bauguitte, S. J.-B., Roscoe, H. K., Anderson, P. S., Ames, D., Clemitshaw, K. C., Fleming, Z. L., Bloss, W. J., Heard, D. E., Lee, J. D., Read, K. A., Hamer, P., Shallcross, D. E., Jackson, A. V., Walker, S. L., Lewis, A. C., Mills, G. P., Plane, J. M. C., Saiz-Lopez, A., Sturges, W. T., and Worton, D. R.: Chemistry of the Antarctic Boundary Layer and the Interface with Snow: an overview of the CHABLIS campaign, Atmos. Chem. Phys., 8, 3789–3803, https://doi.org/10.5194/acp-8-3789-2008, 2008. a, b
Kerminen, V.-M., Chen, X., Vakkari, V., Petäjä, T., Kulmala, M., and Bianchi, F.: Atmospheric new particle formation and growth: review of field observations, Environ. Res. Lett., 13, 103003, https://doi.org/10.1088/1748-9326/aadf3c, 2018. a
Khan, M., Gillespie, S., Razis, B., Xiao, P., Davies-Coleman, M., Percival, C., Derwent, R., Dyke, J., Ghosh, M., Lee, E., and Shallcross, D.: A modelling study of the atmospheric chemistry of DMS using the global model, STOCHEM-CRI, Atmos. Environ., 127, 69–79, https://doi.org/10.1016/j.atmosenv.2015.12.028, 2016. a
Knote, C. and Barre, J.: BOXMOX extension to KPP, https://mbees.med.uni-augsburg.de/boxmodeling/ (last access: 27 February 2026), 2022. a, b
Kulmala, M.: How particles nucleate and grow, Science, 302, 1000–1001, https://doi.org/10.1126/science.1090848, 2003. a
Lee, J. D., McFiggans, G., Allan, J. D., Baker, A. R., Ball, S. M., Benton, A. K., Carpenter, L. J., Commane, R., Finley, B. D., Evans, M., Fuentes, E., Furneaux, K., Goddard, A., Good, N., Hamilton, J. F., Heard, D. E., Herrmann, H., Hollingsworth, A., Hopkins, J. R., Ingham, T., Irwin, M., Jones, C. E., Jones, R. L., Keene, W. C., Lawler, M. J., Lehmann, S., Lewis, A. C., Long, M. S., Mahajan, A., Methven, J., Moller, S. J., Müller, K., Müller, T., Niedermeier, N., O'Doherty, S., Oetjen, H., Plane, J. M. C., Pszenny, A. A. P., Read, K. A., Saiz-Lopez, A., Saltzman, E. S., Sander, R., von Glasow, R., Whalley, L., Wiedensohler, A., and Young, D.: Reactive Halogens in the Marine Boundary Layer (RHaMBLe): the tropical North Atlantic experiments, Atmos. Chem. Phys., 10, 1031–1055, https://doi.org/10.5194/acp-10-1031-2010, 2010. a, b
Li, J., Tsona, N. T., Tang, S., Zhang, X., and Du, L.: Influence of water on the gas-phase reaction of dimethyl sulfide with BrO in the marine boundary layer, ACS Omega, 6, 2410–2419, https://doi.org/10.1021/acsomega.0c05945, 2021. a
Lily, M., Hynniewta, S., Lv, X., Chandra, A. K., Tchinda, N. T., and Du, L.: Assessing the isomerization of a primary intermediate ( radical) in dimethyl sulfide degradation in the marine boundary layer, ACS Earth Space Chem., 7, 2129–2138, https://doi.org/10.1021/acsearthspacechem.3c00209, 2023. a
Lucas, D. D. and Prinn, R. G.: Parametric sensitivity and uncertainty analysis of dimethylsulfide oxidation in the clear-sky remote marine boundary layer, Atmos. Chem. Phys., 5, 1505–1525, https://doi.org/10.5194/acp-5-1505-2005, 2005. a, b, c, d
Lv, G., Zhang, C., and Sun, X.: Understanding the oxidation mechanism of methanesulfinic acid by ozone in the atmosphere, Sci. Rep., 9, 2045–2322, https://doi.org/10.1038/s41598-018-36405-0, 2019. a
McCoy, I. L., McCoy, D. T., Wood, R., Regayre, L., Watson-Parris, D., Grosvenor, D. P., Mulcahy, J. P., Hu, Y., Bender, F. A.-M., Field, P. R., Carslaw, K. S., and Gordon, H.: The hemispheric contrast in cloud microphysical properties constrains aerosol forcing, P. Natl. Acad. Sci. USA, 117, 18998–19006, https://doi.org/10.1073/pnas.1922502117, 2020. a
Natural Environment Research Council, Bauguitte, S., Bloss, W., Clemitshaw, K., Fleming, Z., Heard, D., Jackson, A., Jones, A., Lee, J., Lewis, A., Mills, G., Rankin, A., Read, K., Roscoe, H., Salmon, R., Walker, S., and Wolff, E.: Chemistry of the Antarctic Boundary Layer and the Interface with Snow (CHABLIS): meteorological and atmospheric chemistry field measurements, NCAS British Atmospheric Data Centre [data set], http://catalogue.ceda.ac.uk/uuid/6a5cf2c6e142975e71ff340e0c41777d/ (last access: 27 February 2026), 2005. a
Natural Environment Research Council, Whalley, L., Pszenny, A., and Keene, W.: Reactive halogens in the marine boundary layer (RHaMBLe) campaign at Cape Verde (2007), NCAS British Atmospheric Data Centre [data set], http://catalogue.ceda.ac.uk/uuid/a2d86deca5264e38bce22b8c96f01d99/ (last access: 27 February 2026), 2006. a
Newsome, B. and Evans, M.: Impact of uncertainties in inorganic chemical rate constants on tropospheric composition and ozone radiative forcing, Atmos. Chem. Phys., 17, 14333–14352, https://doi.org/10.5194/acp-17-14333-2017, 2017. a, b
Plischke, E.: An effective algorithm for computing global sensitivity indices (EASI), Reliab. Eng. Syst. Saf., 95, 354–360, https://doi.org/10.1016/j.ress.2009.11.005, 2010. a
Quack, B. and Wallace, D. W. R.: Air-sea flux of bromoform: controls, rates, and implications, Global Biogeochem. Cy., 17, GB1023, https://doi.org/10.1029/2002GB001890, 2003. a
Read, K. A., Mahajan, A. S., Carpenter, L. J., Evans, M. J., Faria, B. V. E., Heard, D. E., Hopkins, J. R., Lee, J. D., Moller, S. J., Lewis, A. C., Mendes, L., McQuaid, J. B., Oetjen, H., Saiz-Lopez, A., Pilling, M. J., and Plane, J. M. C.: Extensive halogen-mediated ozone destruction over the tropical Atlantic Ocean, Nature, 453, 1232–1235, https://doi.org/10.1038/nature07035, 2008. a
Rhyman, L., Lee, E. P. F., Ramasami, P., and Dyke, J. M.: A study of the thermodynamics and mechanisms of the atmospherically relevant reaction dimethyl sulphide (DMS) with atomic chlorine (Cl) in the absence and presence of water using electronic structure methods, Phys. Chem. Chem. Phys., 25, 4780–4793, https://doi.org/10.1039/D2CP05814F, 2023. a
Rodriguez, M. A. and Dabdub, D.: Monte Carlo uncertainty and sensitivity analysis of the CACM chemical mechanism, J. Geophys. Res.-Atmos., 108, D154443, https://doi.org/10.1029/2002JD003281, 2003. a
Rosati, B., Isokääntä, S., Christiansen, S., Jensen, M. M., Moosakutty, S. P., Wollesen de Jonge, R., Massling, A., Glasius, M., Elm, J., Virtanen, A., and Bilde, M.: Hygroscopicity and CCN potential of DMS-derived aerosol particles, Atmos. Chem. Phys., 22, 13449–13466, https://doi.org/10.5194/acp-22-13449-2022, 2022. a
Saiz-Lopez, A. and von Glasow, R.: Reactive halogen chemistry in the troposphere, Chem. Soc. Rev., 41, 6448–6472, https://doi.org/10.1039/C2CS35208G, 2012. a
Saiz-Lopez, A., Mahajan, A. S., Salmon, R. A., Bauguitte, S. J.-B., Jones, A. E., Roscoe, H. K., and Plane, J. M. C.: Boundary layer halogens in coastal Antarctica, Science, 317, 348–351, https://doi.org/10.1126/science.1141408, 2007. a
Saltelli, A. and Annoni, P.: How to avoid a perfunctory sensitivity analysis, Environ. Model. Softw., 25, 1508–1517, https://doi.org/10.1016/j.envsoft.2010.04.012, 2010. a, b, c
Saltelli, A. and Hjorth, J.: Uncertainty and sensitivity analyses of OH-initiated dimethyl sulphide (DMS) oxidation kinetics, J. Atmos. Chem., 21, 187–221, https://doi.org/10.1007/BF00696755, 1995. a, b, c, d
Sandu, A. and Sander, R.: Technical note: Simulating chemical systems in Fortran90 and Matlab with the Kinetic PreProcessor KPP-2.1, Atmos. Chem. Phys., 6, 187–195, https://doi.org/10.5194/acp-6-187-2006, 2006. a
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003. a, b
Seo, S., Richter, A., Blechschmidt, A.-M., Bougoudis, I., and Burrows, J. P.: Spatial distribution of enhanced BrO and its relation to meteorological parameters in Arctic and Antarctic sea ice regions, Atmos. Chem. Phys., 20, 12285–12312, https://doi.org/10.5194/acp-20-12285-2020, 2020. a
Shen, J., Scholz, W., He, X.-C., Zhou, P., Marie, G., Wang, M., Marten, R., Surdu, M., Rörup, B., Baalbaki, R., Amorim, A., Ataei, F., Bell, D. M., Bertozzi, B., Brasseur, Z., Caudillo, L., Chen, D., Chu, B., Dada, L., Duplissy, J., Finkenzeller, H., Granzin, M., Guida, R., Heinritzi, M., Hofbauer, V., Iyer, S., Kemppainen, D., Kong, W., Krechmer, J. E., Kürten, A., Lamkaddam, H., Lee, C. P., Lopez, B., Mahfouz, N. G. A., Manninen, H. E., Massabò, D., Mauldin, R. L., Mentler, B., Müller, T., Pfeifer, J., Philippov, M., Piedehierro, A. A., Roldin, P., Schobesberger, S., Simon, M., Stolzenburg, D., Tham, Y. J., Tomé, A., Umo, N. S., Wang, D., Wang, Y., Weber, S. K., Welti, A., Wollesen de Jonge, R., Wu, Y., Zauner-Wieczorek, M., Zust, F., Baltensperger, U., Curtius, J., Flagan, R. C., Hansel, A., Möhler, O., Petäjä, T., Volkamer, R., Kulmala, M., Lehtipalo, K., Rissanen, M., Kirkby, J., El-Haddad, I., Bianchi, F., Sipilä, M., Donahue, N. M., and Worsnop, D. R.: High gas-phase methanesulfonic acid production in the OH-initiated oxidation of dimethyl sulfide at low temperatures, Environ. Sci. Technol., 56, 13931–13944, https://doi.org/10.1021/acs.est.2c05154, 2022. a, b, c, d, e, f
Sobol, I.: Global sensitivity indices for nonlinear mathematical models and their Monte Carlo estimates, Math. Comput. Simul., 55, 271–280, https://doi.org/10.1016/S0378-4754(00)00270-6, 2001. a
Sommariva, R., Haggerstone, A.-L., Carpenter, L. J., Carslaw, N., Creasey, D. J., Heard, D. E., Lee, J. D., Lewis, A. C., Pilling, M. J., and Zádor, J.: OH and HO2 chemistry in clean marine air during SOAPEX-2, Atmos. Chem. Phys., 4, 839–856, https://doi.org/10.5194/acp-4-839-2004, 2004. a, b
Stein, B. V., Raponi, E., Sadeghi, Z., Bouman, N., Van Ham, R. C. H. J., and Bäck, T.: A comparison of global sensitivity analysis methods for explainable AI with an application in genomic prediction, IEEE Access, 10, 103364–103381, https://doi.org/10.1109/ACCESS.2022.3210175, 2022. a, b, c, d
Stewart, R. W. and Thompson, A. M.: Kinetic data imprecisions in photochemical rate calculations: means, medians, and temperature dependence, J. Geophys. Res.-Atmos., 101, 20953–20964, https://doi.org/10.1029/96JD01708, 1996. a
Tarantola, S., Gatelli, D., and Mara, T.: Random balance designs for the estimation of first order global sensitivity indices, Reliab. Eng. Syst. Saf., 91, 717–727, https://doi.org/10.1016/j.ress.2005.06.003, 2006. a
Tashmim, L., Porter, W. C., Chen, Q., Alexander, B., Fite, C. H., Holmes, C. D., Pierce, J. R., Croft, B., and Ishino, S.: Contribution of expanded marine sulfur chemistry to the seasonal variability of dimethyl sulfide oxidation products and size-resolved sulfate aerosol, Atmos. Chem. Phys., 24, 3379–3403, https://doi.org/10.5194/acp-24-3379-2024, 2024. a
Tissot, J.-Y. and Prieur, C.: Bias correction for the estimation of sensitivity indices based on random balance designs, Reliab. Eng. Syst. Saf., 107, 205–213, https://doi.org/10.1016/j.ress.2012.06.010, 2012. a
Tomlin, A. S.: The role of sensitivity and uncertainty analysis in combustion modelling, Proc. Combust. Inst., 34, 159–176, https://doi.org/10.1016/j.proci.2012.07.043, 2013. a, b
Vasyunin, A. I., Sobolev, A. M., Wiebe, D. S., and Semenov, D. A.: Influence of uncertainties in the rate constants of chemical reactions on astrochemical modeling results, Astron. Lett., 30, 566–576, https://doi.org/10.1134/1.1784498, 2004. a, b, c
Vereecken, L., Novelli, A., Taraborrelli, D., and Wahner, A.: Perhemiacetal formation and Cl/NO3-initiated chemistry of hydroperoxymethylthioformate (HPMTF) in atmospheric DMS oxidation, Environ. Sci.: Atmos., 5, 181–190, https://doi.org/10.1039/D4EA00134F, 2025. a, b
Veres, P. R., Andrew Neuman, J., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J. F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Paul Bui, T., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020. a, b
Wang, S., Schmidt, J. A., Baidar, S., Coburn, S., Dix, B., Koenig, T. K., Apel, E., Bowdalo, D., Campos, T. L., Eloranta, E., Evans, M. J., DiGangi, J. P., Zondlo, M. A., Gao, R.-S., Haggerty, J. A., Hall, S. R., Hornbrook, R. S., Jacob, D., Morley, B., Pierce, B., Reeves, M., Romashkin, P., ter Schure, A., and Volkamer, R.: Active and widespread halogen chemistry in the tropical and subtropical free troposphere, P. Natl. Acad. Sci. USA, 112, 9281–9286, https://doi.org/10.1073/pnas.1505142112, 2015. a
Wang, X., Jacob, D. J., Downs, W., Zhai, S., Zhu, L., Shah, V., Holmes, C. D., Sherwen, T., Alexander, B., Evans, M. J., Eastham, S. D., Neuman, J. A., Veres, P. R., Koenig, T. K., Volkamer, R., Huey, L. G., Bannan, T. J., Percival, C. J., Lee, B. H., and Thornton, J. A.: Global tropospheric halogen (Cl, Br, I) chemistry and its impact on oxidants, Atmos. Chem. Phys., 21, 13973–13996, https://doi.org/10.5194/acp-21-13973-2021, 2021. a, b
Wohl, C., Villamayor, J., Galí, M., Mahajan, A. S., Fernández, R. P., Cuevas, C. A., Bossolasco, A., Li, Q., Kettle, A. J., Williams, T., Sarda-Esteve, R., Gros, V., Simó, R., and Saiz-Lopez, A.: Marine emissions of methanethiol increase aerosol cooling in the Southern Ocean, Sci. Adv., 10, eadq2465, https://doi.org/10.1126/sciadv.adq2465, 2024. a, b, c, d, e, f, g
Wolke, R., Sehili, A., Simmel, M., Knoth, O., Tilgner, A., and Herrmann, H.: SPACCIM: a parcel model with detailed microphysics and complex multiphase chemistry, Atmos. Environ., 39, 4375–4388, https://doi.org/10.1016/j.atmosenv.2005.02.038, 2005. a
Wollesen de Jonge, R., Elm, J., Rosati, B., Christiansen, S., Hyttinen, N., Lüdemann, D., Bilde, M., and Roldin, P.: Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 21, 9955–9976, https://doi.org/10.5194/acp-21-9955-2021, 2021. a
Wu, R., Wang, S., and Wang, L.: New mechanism for the atmospheric oxidation of dimethyl sulfide. The importance of intramolecular hydrogen shift in a CH3SCH2OO radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015. a
Ye, Q., Goss, M. B., Isaacman-VanWertz, G., Zaytsev, A., Massoli, P., Lim, C., Croteau, P., Canagaratna, M., Knopf, D. A., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Organic sulfur products and peroxy radical isomerization in the OH oxidation of dimethyl sulfide, ACS Earth Space Chem., 5, 2013–2020, https://doi.org/10.1021/acsearthspacechem.1c00108, 2021. a
Ye, Q., Goss, M. B., Krechmer, J. E., Majluf, F., Zaytsev, A., Li, Y., Roscioli, J. R., Canagaratna, M., Keutsch, F. N., Heald, C. L., and Kroll, J. H.: Product distribution, kinetics, and aerosol formation from the OH oxidation of dimethyl sulfide under different RO2 regimes, Atmos. Chem. Phys., 22, 16003–16015, https://doi.org/10.5194/acp-22-16003-2022, 2022. a, b, c, d, e, f, g, h
Yin, F., Grosjean, D., and Seinfeld, J. H.: Photooxidation of dimethyl sulfide and dimethyl disulfide part I: mechanism development, J. Atmos. Chem., 11, 309–364, https://doi.org/10.1007/BF00053780, 1990. a
- Abstract
- Introduction
- Aqueous and gas-phase dimethyl sulfide and methanethiol oxidation mechanism
- Marine regimes modelled
- Uncertainty quantification
- Uncertainty analysis
- Sensitivity analysis
- Conclusions
- Appendix A: Sensitivity indices tables
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Aqueous and gas-phase dimethyl sulfide and methanethiol oxidation mechanism
- Marine regimes modelled
- Uncertainty quantification
- Uncertainty analysis
- Sensitivity analysis
- Conclusions
- Appendix A: Sensitivity indices tables
- Code and data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement