the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 07 Aug 2025
| 07 Aug 2025
Atmospheric fate of organosulfates through gas-phase and aqueous-phase reactions with hydroxyl radicals: implications for inorganic sulfate formation
Narcisse Tsona Tchinda
Xiaofan Lv
Stanley Numbonui Tasheh
Julius Numbonui Ghogomu
Organosulfates are important tracers for aerosol particles, yet their influence on aerosol chemical composition remains poorly understood. This study uses quantum chemical calculations based on density functional theory to explore the reactions of some prevalent organosulfates, specifically methyl sulfate and glycolic acid sulfate, with hydroxyl radicals (HO•) in the gas phase and aqueous phase. Results indicate that all reactions initiate with hydrogen abstraction by HO• from CH3- in methyl sulfate and from -CH2- and -COOH in glycolic acid sulfate, followed by the further reaction of the resulting radicals through self-decomposition, interaction with O2 and, possibly, O3. We found that the hydrogen abstraction from the -COOH group in glycolic acid sulfate could lead to decarboxylation and eventually form similar products as methyl sulfate. The primary reaction products are inorganic sulfate, carbonyl compounds, and formic sulfuric anhydride. Rate constants of and cm3 molec.−1 s−1 at 298.15 K were determined for the gas-phase reactions of methyl sulfate and glycolic acid sulfate, respectively. The former value is consistent with a previous experimental report. Additionally, besides O2 as the primary oxidant in the fragmentation of organosulfates, this study unveils that O3 may be a complementary oxidant in this process, especially in environments enriched with ozone. Overall, this study elucidates mechanisms for HO•-initiated transformation of organosulfates and highlights the potential role of chemical substitution, thereby enhancing our understanding of their atmospheric chemistry and implications for inorganic sulfate formation, which are vital for evaluating their impact on aerosol properties and climate processes.
- Article
(5769 KB) - Full-text XML
-
Supplement
(1220 KB) - BibTeX
- EndNote




Atmospheric particulate matter is a complex mixture of inorganic and organic matter, with organic matter typically accounting for 20 %–90 % of the total mass (Hallquist et al., 2009; Stone et al., 2012). The concentration level of particulate matter and its evolution have important impacts on regional air quality, climate change and human health. In this regard, the study of organic aerosol concentration levels, sources, and secondary transformations is key to understanding the formation mechanism of compounds responsible for air pollution. Organosulfates, characterized by sulfate groups (R-OSO3H/R-OSO, where R is an alkyl or aryl group), constitute the most abundant component of organic aerosols. Organosulfates have been widely detected in aerosol particles from various environments in the Americas, Europe, Asia, and the Arctic over the past decades (Iinuma et al., 2007; Surratt et al., 2008; Nguyen et al., 2014; Kourtchev et al., 2016; Lin et al., 2014). Due to the presence of -OSO or -OSO3H functional groups in their structure, organosulfates are acidic and highly water-soluble, which enables them to enhance the hygroscopicity of aerosols, with potential climate impacts (Chan et al., 2011).
The formation pathways of organosulfates are complex and varied. They have been shown to be generated by non-homogeneous and multiphase reactions (Iinuma et al., 2007; Surratt et al., 2008). For example, Rudziński et al. (2009) and Worton et al. (2011) found that the photo-oxidation of a variety of biogenic volatile organic compounds, such as isoprene, α-pinene, and β-pinene, can lead to the formation of organosulfates. Nozière et al. (2010, 2015) suggested that the reaction products of sulfate anions with isoprene can yield organosulfates. Recent studies uncovered pathways by which organosulfates can be generated from the heterogeneous reaction of SO2 with unsaturated bonds in fatty acids (Shang et al., 2016; Passananti et al., 2016). Organosulfates derived from organic acids were recently identified and characterized in fine particulate matter samples collected in the southeastern US, with glycolic acid sulfate being the most abundant (Hettiyadura et al., 2017).
Despite extensive research on the concentration, composition, and formation mechanisms of organosulfates, the limited knowledge of molecular-level mechanisms of their transformations hinders further understanding of their atmospheric processes as well as their physico-chemical properties (Huang et al., 2015). Organosulfates primarily exist in the particulate phase due to their low volatility (Estillore et al., 2016; George and Abbatt, 2010), although a non-negligible fraction has been shown to always be present in the gas phase (Ehn et al., 2010; Le Breton et al., 2018) where they can react continuously with gas-phase oxidants (e.g., HO• radicals, O3, and NO3 radicals) at or near particle surfaces. The transformation of organosulfates generates not only new organic matter, but also inorganic sulfur species, such as HSO and SO. Consequently, the conversion of organosulfates can significantly alter the composition and physico-chemical properties of atmospheric particulate matter (Hettiyadura et al., 2017). However, little is known in this regard. A few recorded studies include the chemical transformation of methyl sulfate, ethyl sulfate, and an α-pinene-derived organosulfate by heterogeneous HO• oxidation (Kwong et al., 2018; Xu et al., 2022). Based on their observed reaction products, the authors suggested a mechanism proceeding through the formation of an alkoxy radical intermediate followed by fragmentation, yet the mechanism remains not fully elucidated. Moreover, organosulfates that can allow hydrogen abstraction at the β-position to the sulfate group could give rise to more complex mechanisms driven by the Russell mechanism (Russell, 1957) and the Bennett and Summers reaction (Bennett and Summers, 1974). While the presence of functional groups can exhibit specific features in the chemical transformation of organosulfates due to their complexity, their potential impact has not yet been thoroughly investigated.
Glycolic acid sulfate and methyl sulfate are two low molecular weight organic sulfates commonly found in the atmosphere, differing structurally by the α-substitution of a hydrogen atom by the carboxyl group. Both organosulfates have been detected at various locations around the world at concentrations in the ranges 1.08×106–5.01×107 molec. cm−3 for methyl sulfate (Hettiyadura et al., 2015; Peng et al., 2021) and 1.16×107–4.71×108 molec. cm−3 for glycolic acid sulfate (Huang et al., 2018; Hettiyadura et al., 2015; Wang et al., 2021; Hughes and Stone, 2019; Cai et al., 2020; Liao et al., 2015). This study employs quantum chemical calculations to explore the transformation pathways of these organosulfates in both gas-phase and aqueous-phase environments, emphasizing the effects of carboxyl substitution. Ultimately, this study assesses the potential for sulfate formation and the atmospheric implications of these reactions.
2.1 Electronic structure calculations and thermochemistry
Quantum chemical calculations were used to investigate the transformation reactions of two organosulfates (methyl sulfate and glycolic acid sulfate) in the gas phase and aqueous phase. Geometry optimizations of all reaction states on the energy surface were performed with density functional theory using the Gaussian 09 package (Frisch et al., 2013). Pre-optimizations were first carried out using the M06-2X functional (Zhao and Truhlar, 2008) and the 6-31+G(d,p) basis set, and the best structures were further refined by the M06-2X/6-311+g(2df,2pd) method to yield the final structures. Vibrational frequency analysis on the M06-2X/6-311+g(2df,2pd) optimized structures were performed using the same method under the rigid rotor-harmonic oscillator approximation at 298.15 K and 1 atm at the same level of theory to yield zero-point energies and the thermochemical parameters. The continuum solvation model based on the solute electron density was used to model the aqueous phase at the same level of theory as the gas phase. This model is particularly suitable for describing atmospheric processes and effectively resolving the energy barriers (Ostovari et al., 2018; Xu and Coote, 2019; Cheng et al., 2019). In the aqueous phase, the Gibbs free energies are calculated at a standard temperature of 298.15 K and by converting the standard pressure of 1 atm (in the gas phase) to the standard concentration of 1 M. Details are provided in Sect. S1 in the Supplement. Single-point energy calculations on the M06-2X/6-311+g(2df,2pd) structures were performed with the CCSD(T)-F12/cc-pVDZ-F12 method using Orca version 4.2.1 (Riplinger and Neese, 2013). The M06-2X/6-311+g(2df,2pd) method has successfully been used in a series of previous studies (Ding et al., 2023; Wang et al., 2024; Cheng et al., 2022) and has been proved to efficiently resolve the transition state configuration. Given the size of the system investigated here, this level of theory appears to be a good compromise between accuracy and computation time.
2.2 Reaction kinetics
Reactions were modeled by assuming the pseudo-steady state approximation of the reactant complex formed from the interaction between initial reactants (organosulfate (OS = methyl sulfate or glycolic acid sulfate) and hydroxyl radical (HO•)). Based on this approximation, initial reactants are in equilibrium with the reactant complex that further rearranges through a transition state (TS) configuration to form the product complex according to the following reaction:
The reaction kinetics analysis was conducted by applying transition state theory (Truhlar et al., 1996). Based on this theory applied to Reaction (R1), the biomolecular rate constant (kbim) is given as
where Keq is the equilibrium constant of formation of the reactant complex, and kuni is the unimolecular rate constant of the transformation of the reactant complex to the product complex, respectively expressed by the following equations:
In the above equations, ΔGeq is the Gibbs free energy of the formation of the reactant complex from initial reactants, h is Planck's constant, kB is Boltzmann's constant, ΔG# is the Gibbs free energy of the barrier separating the reactant complex from the product complex, R is the molar gas constant, T is the absolute temperature, c0 is the standard concentration (with values of 1 M and 2.46×1019 molec. cm−3 in the aqueous phase and in the gas phase, respectively, at 1 atm and 298.15 K), and κ is the Eckart tunneling coefficient (calculated by solving the Schrodinger equation for an asymmetrical one-dimensional Eckart potential (Eckart, 1930)).
While Eq. (1) is valid for calculating the biomolecular rate constants of gas-phase reactions, for aqueous-phase reactions, it is corrected by taking into account the contribution of the molecular diffusion described by Collins–Kimball theory (Collins and Kimball, 1949). This leads to the overall rate constant for Reaction (R1) in the aqueous phase as follows:
kD is the steady-state Smoluchowski rate constant calculated as (Smoluchowski, 1917)
In Eq. (5), ROS,OH stands for the reaction distance between the organosulfate and hydroxyl radical, defined as the sum of their respective radii, ROS and ROH. NA is the Avogadro number and D is the sum of the diffusion coefficients of reactants (Truhlar, 1985). For a reactant i in water, the diffusion coefficient Di is given by the Stokes-Einstein approach (Einstein, 1905) as follows:
The radii of reactants (organosulfate and hydroxyl radical), assumed to be spherical, were calculated by Eq. (6) using the Multiwfn software (Lu and Chen, 2012). All numerical values for radii, diffusion coefficients of reactants and the steady-state Smoluchowski rate constants are provided in Tables S1 and S2 in the Supplement.
The reactions were explored both in the gas phase and the aqueous phase. A previous study indicated that, in the gas phase, methyl sulfate and glycolic acid sulfate are likely to be hydrated under relevant atmospheric temperature and humidity (Tsona and Du, 2019a). Hence, in this study the reactions with HO• in the gas phase have been explored by considering methyl sulfate hydrates (CH3-O-SO3H•••(H2O)n=0–2) and glycolic acid sulfate hydrates (HOOC-CH2-O-SO3H•••(H2O)n=0–2).
3.1 Mechanism of methyl sulfate reaction with HO• radicals
3.1.1 CH3-O-SO3H•••(H2O) HO• reaction in the gas phase
Our calculations indicate that the CH3-O-SO3H•••(H2O)n=0–2 reaction with HO• proceeds through the formation of an intermediate reactant complex, •OH•••CH3-O-SO3H•••(H2O)n=0–2, in which the interaction between HO• and CH3-O-SO3H•••(H2O)n=0–2 is mainly established through the hydrogen atom of HO• and one oxygen atom of CH3-O-SO3H. This intermediate reactant complex readily reacts through a hydrogen abstraction from the methyl group of CH3-O-SO3H by HO• to form the H2C•-O-SO3H•••(H2O)n+1 product complex. A distinct feature was observed for n= 2, where the sulfate group was deprotonated, forming an ion pair with H3O+ (i.e, H2C•-O-SO,H3O+•••(H2O)2). This is likely to be because for this configuration the minimum amount of water (2 molecules) necessary to achieve the stability of H3O+ in the gas phase has been reached (Markovitch and Agmon, 2007; Heindel et al., 2018). Although the presence of water has a weak effect on the formation of the intermediate reactant complex, it substantially stabilizes the transition state towards the formation of the product complex by reducing the Gibbs free energy barrier from 6.45 kcal mol−1 for the CH3-O-SO3H + HO• reaction to 3.52 kcal mol−1 for the CH3-O-SO3H•••(H2O)2+ HO• reaction. Energetics and structural details of this reaction are given in Table 1 and Fig. 1. The observed catalytic effect of water has been generally observed in some previous H-abstraction reactions (Wang et al., 2019; Wang et al., 2017; Zhang et al., 2024; Wang et al., 2024).
Table 1Electronic energy changes (ΔE) and Gibbs free energy changes (ΔG at 298.15 K and 1 atm) for all intermediate species in the reaction of methyl sulfate with HO• radicals. Energy units are kcal mol−1. “RC” stands for intermediate reactant complex, “TS” stands for transition state, “PD” stands for product, and “nw” stands for the number of added water molecules to the reaction of methyl sulfate with HO• radicals.
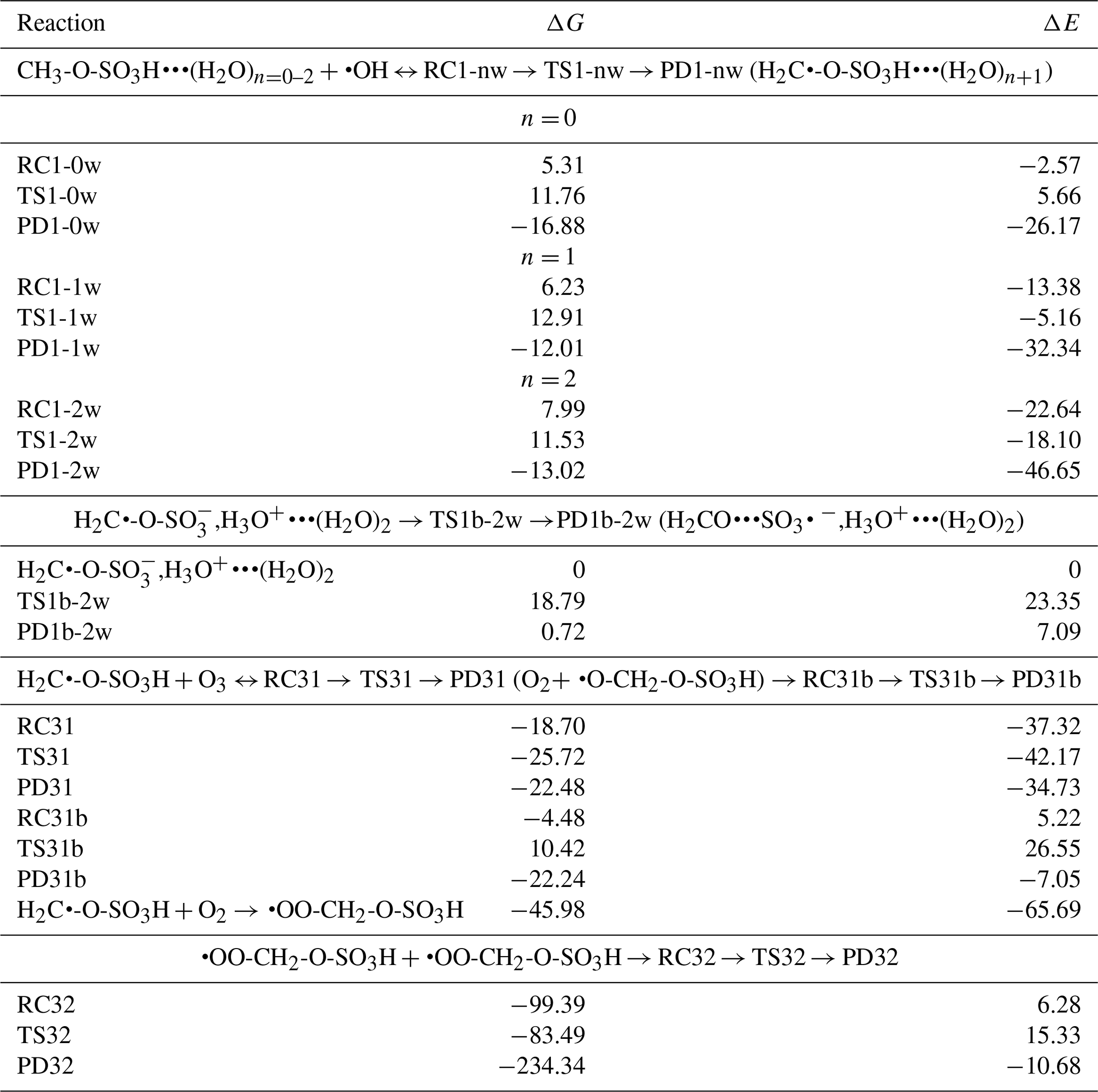
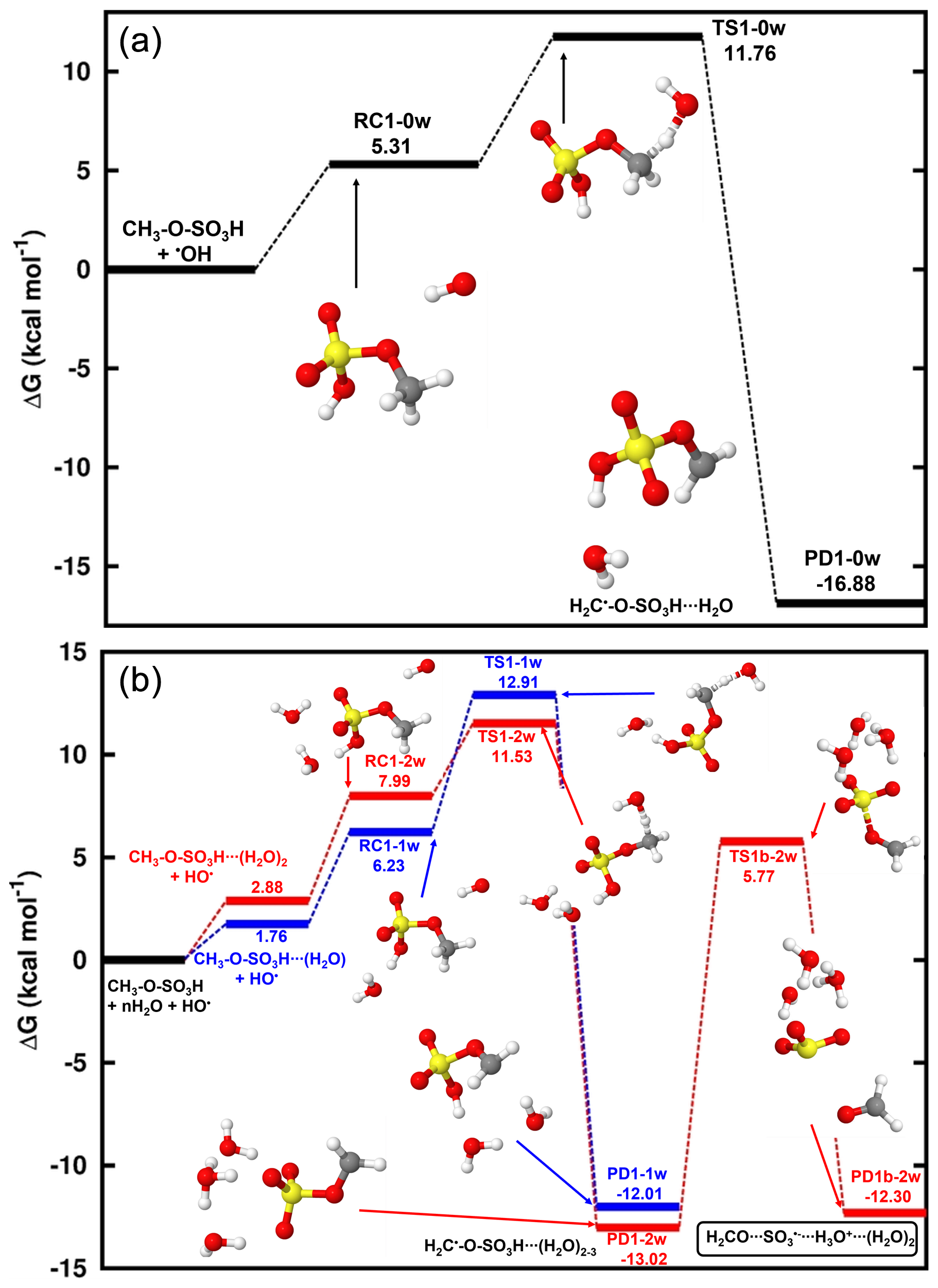
Figure 1Gibbs free energy changes (in kcal mol−1) and optimized structures for all intermediates in the reaction of CH3-O-SO3H•••(H2O)n=0–2 with HO• radicals. The top panel is the reaction in the absence of water and the bottom panel is the reaction in the presence of water, where the blue indicates the monohydrated reaction and the red line indicates the dihydrated reaction. The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
Unimolecular rate constants for the transformation of •OH•••CH3-O-SO3H•••(H2O)n to H2C•-O-SO3H•••(H2O)n+1 at 298 K were determined to be 1.15×108, 7.89×107 and 1.59×1010 s−1, corresponding to atmospheric lifetimes of 9 ns, 13 ns, and 63 ps, for n= 0, 1, and 2, respectively, for the intermediate reactant complex. The lifetimes in the picosecond and nanosecond regimes indicate that •OH•••CH3-O-SO3H•••(H2O)n=0–2 complexes are indeed highly reactive and would most likely react fast before they could experience collisions with other abundant atmospheric oxidants (Bork et al., 2012). Hence, the immediate fate of •OH•••CH3-O-SO3H•••(H2O)n=0–2 is undoubtedly hydrogen abstraction by HO• to form H2C•-O-SO3H•••(H2O)n+1. The bimolecular rate constant for this reaction is determined to be cm3 molec.−1 s−1 at 298.15 K. As a radical, H2C•-O-SO3H•••(H2O)n+1 would eventually undergo further decomposition depending on its stability, atmospheric lifetime, and surrounding atmospheric conditions.
The stability of H2C•-O-SO3H•••(H2O)n+1, examined relative to decomposition through the reverse reaction back to the •OH•••CH3-O-SO3H•••(H2O)n complex, reveals barrier heights of 28.64, 24.92, and 24.55 kcal mol−1 for n= 0, 1, and 2, corresponding to unimolecular rate constants of , , and s−1, respectively. These low-rate constants indicate that, once formed, H2C•-O-SO3H•••(H2O)n+1 would not react back to form initial reactants before possible collision with atmospheric oxidants have occurred. Beside hypothesizing self-decomposition to be a likely outcome of H2C•-O-SO3H, observed atmospheric lifetimes indicate that its fate would depend upon collisions with most atmospheric oxidants, including O2, O3, and NO2. Considering that water is known to evaporate fast from atmospheric species, H2C•-O-SO3H would readily react in both its unhydrated and hydrated forms.
Based on these observations, the fate of H2C•-O-SO3H was examined relative to the following decomposition processes:
Our attempts to optimize the self-decomposition of H2C•-O-SO3H•••(H2O)n failed except for n= 3 (Reaction R2). This was as expected since the gas-phase stability of H3O+ can only be achieved by solvation with at least two water molecules (Markovitch and Agmon, 2007; Heindel et al., 2018).
Reaction (R2) proceeds by one S-O bond breaking and a hydrogen transfer from -SO3H to H2O to release the H2CO•••SO3•−,H3O+•••(H2O)2 complex. The transition state in this process was located at 18.79 kcal mol−1 above H2C•-O-SO,H3O+•••(H2O)2. To confirm the location of the free electron on the different states of H2C•-O-SO3H•••(H2O)3 decomposition, the analysis of the charge distribution was performed. As shown in Fig. 2a, the electronic charge initially located on the CH2- fragment progressively migrates along the H2C•-O-SO3H core to ultimately rest on SO3, leaving CH2O electronically neutral. Although SO3•− and H2CO are expected products of this decomposition, our calculations show that the outcome of this reaction can only be moderate due to the relatively high energy barrier. Several experimental and theoretical studies showed that SO would quickly hydrate in the gas phase to form a SO(H2O)n cluster wherein the oxidation to sulfuric acid occurs in a mechanism stabilized and catalyzed by a free electron (Bork et al., 2013; Tsona and Du, 2019b; Fehsenfeld and Ferguson, 1974; Svensmark et al., 2007; Enghoff and Svensmark, 2008). Considering this outcome for SO in the gas phase, H2CO and inorganic sulfate are expected products of the gas-phase reaction of methyl sulfate with HO• at ambient conditions. The calculated unimolecular rate constant of this decomposition at 298.15 K, s−1, indicates that this process can account for the fate of H2C•-O-SO3H only under the conditions of low oxidants. The products predicted by our calculations were observed in a previous experimental study by Kwong et al. (2018) for the same reaction, and the mechanisms from Russell (1957) and Bennett and Summers (1974) were speculated by the authors to explain this formation. Besides these mechanisms, Reaction (R2) in this study might be a complementary mechanism to the formation of inorganic sulfate and formaldehyde from the reaction of methyl sulfate with HO•, at least at a certain degree of hydration or humidity.
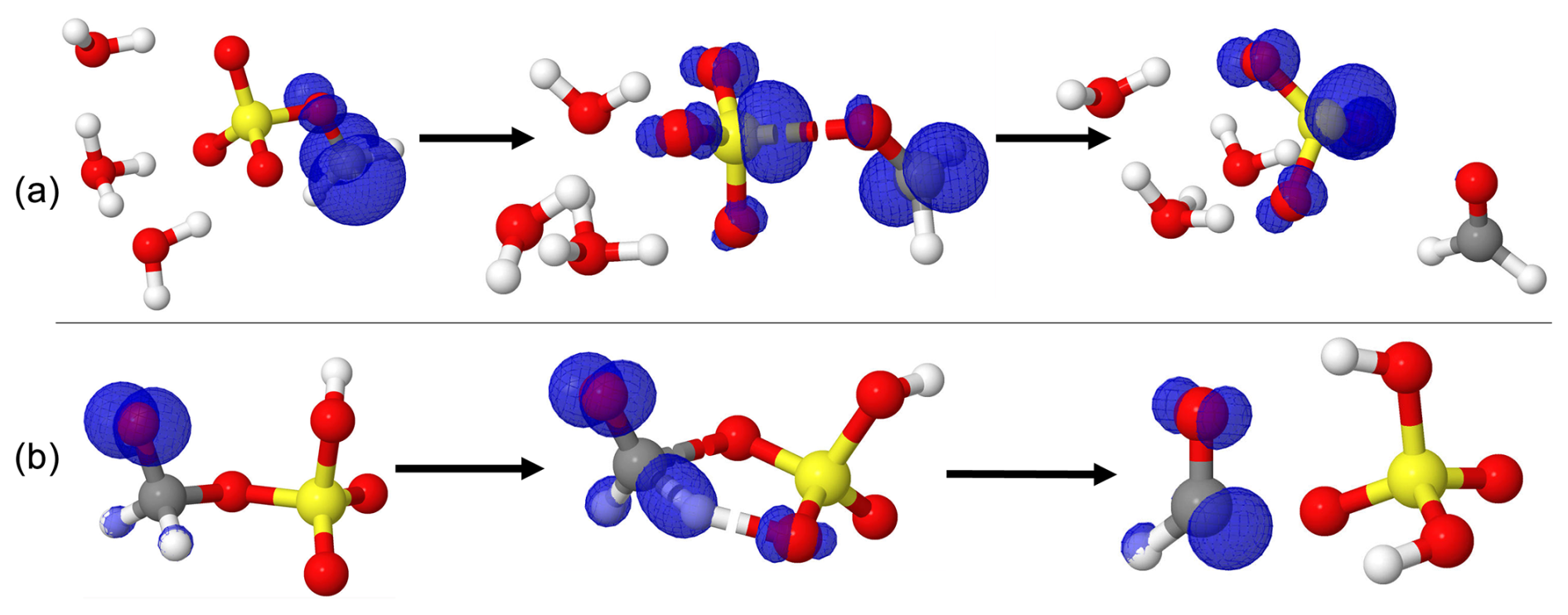
Figure 2Representation of the spin density (blue color) on the electronic states of the decomposition of (a) H2C•-O-SO3H•••(H2O)3, and (b) •O-H2C-O-SO3H. From left to right are the reactant, transition state, and product complex, respectively. The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
Besides the self-decomposition of H2C•-O-SO3H, we further examined its reactions with NO2 (Reaction R3), O3 (Reaction R4), and O2 (Reaction R5). We found that for Reaction (R3), SO H2CO + NO formation would be prevented by a high Gibbs free energy barrier (∼74 kcal mol−1), regardless of the number of water molecules involved. Energetics of this reaction are given in Table S3 and Fig. S1 in the Supplement. This high energy barrier is likely related to the difficulty in breaking the ON-O and C-O bonds to release the products. We conclude that Reaction (R3) is likely without atmospheric significance to the chemistry of CH3-O-SO3H under relevant atmospheric conditions.
The reaction of H2C•-O-SO3H•••(H2O)n with O3 was examined thereafter. Regardless of the presence or absence of water, this reaction proceeds through a submerged energy barrier, with the general mechanism following two main steps: the interaction of H2C•-O-SO3H with O3 and the unimolecular decomposition of the resulting intermediate product. The interaction of H2C•-O-SO3H•••(H2O)n with O3 led to exergonic formation of the O3•••H2C•-O-SO3H•••(H2O)n intermediate reactant complex. The formed reactant complexes are further transformed through an oxygen transfer from O3 to H2C•-O-SO3H•••(H2O)n via transition state configurations to form alkoxyl radicals •O-CH2-O-SO3H•••(H2O)n. The energy barrier in this transformation in the absence of water is located 7.02 kcal mol−1 below the corresponding reactant complex and is weakly altered by the presence of water. The exergonic formation of O3•••H2C•-O-SO3H•••(H2O)n and the submerged barrier in its transformation are indicative of instantaneous formation of •O-CH2-O-SO3H•••(H2O)n from H2C•-O-SO3H•••(H2O)n with O3.
Figure 3Gibbs free energy changes (in kcal mol−1) and optimized structures for all intermediates in the reaction of H2C•-O-SO3H with O3 (a) and O2 (b). The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
Optimized structures and energetics of all reaction states in these reactions are given in Figs. 3, S2, Tables 1 and S4. The direct product of this transformation is a sulfonate alkoxy radical, •O-H2C-O-SO3H, which further decomposes through the C-O bond cleavage and hydrogen transfer from •O-CH2- to -SO3H to form the H2SO4•••HCO• product complex with substantial energy gain. The electronic charge distribution on •O-H2C-O-SO3H in the reactant and on HCO• in the product complex was confirmed by our electronic charge analysis as shown in Figs. 2b and S3. The barrier height to this decomposition is significantly lowered from 14.90 kcal mol−1 (for H2C•-O-SO3H) to 6.59 kcal mol−1 (for H2C•-O-SO3H•••(H2O)3). These correspond, respectively, to unimolecular rate constants of 7.38×101 and 9.06×107 s−1 at 298.15 K. The particular stability of the transition state in H2C•-O-SO3H•••(H2O)3 decomposition can be attributed to the mediation of the additional water molecule in the hydrogen transfer from •O-CH2- to -SO3H. This is in line with the demonstrated increasing catalytic role of water with increasing number of water molecules in SO3 hydrolysis to sulfuric acid (Larson et al., 2000; Hofmann and Schleyer, 1994; Hofmann-Sievert and Castleman, 1984; Morokuma and Muguruma, 1994; Loerting and Liedl, 2000). The currently presented mechanism for H2SO4•••HCO• formation from C-O bond cleavage was suggested by Huang et al. (2018) to explain bisulfate formation from the fragmentation of organosulfates. Compared to H2C•-O-SO3H self-decomposition and reaction with NO2, the reaction with O3 is the most energetically and kinetically favorable process. Nonetheless, considering all the processes by Reactions (R2)–(R4), it is obvious that the main products of the gas-phase reaction of methyl sulfate with HO• are formaldehyde (H2CO) and sulfuric acid (H2SO4). This agrees with the experimental observation by Kwong et al. (2018).
Contrary to the self-decomposition of H2C•-O-SO3H and reactions with NO2 and O3 that are favorable with H2C•-O-SO3H hydrates, we were unable to fully optimize the O2 reaction with H2C•-O-SO3H hydrates but with the unhydrated system, instead. This led to exergonic formation of the •OO-CH2-O-SO3H peroxyl radical with 45.98 kcal mol−1 Gibbs free energy gain. The chemistry of peroxyl radicals has been the subject of several experimental and theoretical studies. It is widely accepted that the peroxyl radical would predominantly decompose to form alkoxyl radicals or alcohols along with carbonyl compounds through tetroxide intermediates (Russell, 1957). However, although the end products from this decomposition have been verified experimentally, the mechanisms have often been deemed unlikely due to inconsistency between thermodynamic experiments and computational studies (Nangia and Benson, 1980; Zhang et al., 2012; Liang et al., 2011). Moreover, the effort to elucidate alcohols and carbonyl compounds formation from the decomposition of the tetroxide in a previous study could not be achieved due to the impossibility of determining the corresponding transition states (Ghigo et al., 2003), while alkoxyl radical formation was observed to simply correspond to the dissociation of the tetroxide. A more recent theoretical study specifically focusing on the decomposition pathways of the tetroxide intermediate indicated that although substantial uncertainties may exist in their computed energetics, alkoxyl radicals are likely primary products from atmospherically relevant peroxyl radicals (Salo et al., 2022). Following the above reasoning, our calculations indicate that two molecules of •OO-CH2-O-SO3H recombine to form a tetroxide that can further decompose to generate two alkoxyl radicals •O-CH2-O-SO3H clustered to molecular oxygen. Then, the two •O-CH2-O-SO3H radicals quickly interact with O2 to form H2O2 and formic sulfuric anhydride (HC(O)-O-SO3H) (Reaction R5). The latter product has been identified to enhance new particle formation (An, 2024). Energetics and structures of all intermediates in this reaction are given in Fig. 3.
Considering the recombination of two H2C•-O-SO3H molecules as another likely fate of H2C•-O-SO3H in the absence of other reaction partners such as O2, O3, or NO2 besides self-decay, our preliminary calculations indicate that this recombination would occur fast and would proceed through C-C bond formation via a barrierless process. However, the relevance of the resulting product is not clearly established and its fate may mostly depend on reactions initiated by a hydrogen abstraction from the -CH2- group.
The studied reaction of methyl sulfate with HO• radicals in the gas phase shows an example of the main processes through which organosulfates may be converted into inorganic sulfates. Humidity is seen to play a non-negligible role in the effective reaction of methyl sulfate while from the kinetics point of view, O3 is a key oxidant besides O2 in the intermediate steps.
3.1.2 CH3-O-SO HO• reaction in the aqueous phase
In the aqueous phase, methyl sulfate (CH3-O-SO undergoes similar steps as its electronically neutral counterpart to form the product complex H2C•-O-SOH2O. Figure 4 shows the Gibbs free energy surface for this reaction at 298.15 K and 1 M concentration, along with the structures of all stationary states. Further energetics details for this reaction are given in Table 2. We determined a Gibbs free energy barrier of 3.54 kcal mol−1 for this reaction (4.69 kcal mol−1 lower than in the CH3-O-SO3H + HO• reaction), and a bimolecular rate constant of 7.87×106 M−1 s−1. This value is about 13 times lower than a previous experimental value (Gweme and Styler, 2024), and the difference can be attributed both to computational errors and environmental factors such as pH, temperature, and ionic strength. The formation of H2C•-O-SOH2O occurred with substantial Gibbs free energy gain, −14.28 kcal mol−1, stable enough toward decomposition to initial reactants for which the energy barrier 23.89 kcal mol−1 is found (see Fig. 4). Based on this energy barrier height, we determined an atmospheric lifetime of 0.61 days for H2C•-O-SOH2O. This indicates that besides self-decomposition as an alternative chemical fate, H2C•-O-SOH2O will live long enough to experience collisions with abundant atmospheric oxidants. While NO2 dissolves effectively in water and is known to react with molecular water to produce nitric acid which is highly soluble in water (Ford and Miranda, 2020; Tan and Piri, 2013; Lee and Schwartz, 1981; England and Corcoran, 1974), the stability of O3 solubility in water is readily affected by various factors including ozone concentration, pH, and ultraviolet light (Lovato et al., 2009). Depending on environmental conditions, ozone can react via a direct reaction pathway involving molecular ozone or by an indirect route involving reactive intermediates that arise from its decomposition (Buehler et al., 1984; Buhler et al., 1984; Staehelin et al., 1984; Staehelin and Hoigne, 1982), whereas the reaction with NO2 would be equivalent to explicitly assessing the reaction with NO. Moreover, O3 is known to be 13 times more soluble in water than O2 (Seinfeld and Pandis, 1998). Hence, besides the self-decay reaction, the fate of H2C•-O-SO was examined via reactions with O2 and O3:
While inspecting the vibrational modes of H2C•-O-SO, it is obvious that dissociation along the H2C•O-SO3 bond to form H2CO + SO3•− (Reaction R6) would be a possible chemical fate. The analysis of electronic charge distribution on H2C•-O-SO confirms that the unpaired electron initially on CH2- gradually migrates to completely rest on SO3 in the products, leaving CH2O uncharged (see Fig. S4). This process was examined and a Gibbs free energy barrier height of 18.90 kcal mol−1 was found (see Fig. 4), which corresponds to a unimolecular rate constant of s−1 at 298.15 K. This is nearly equal to the rate constant of the similar step ( s−1) in the reaction of CH3-O-SO3H under humid conditions. The predicted relatively low-rate constant of H2C•-O-SO decomposition to H2CO and SO3•− can only account for the CH3-O-SO fate under the conditions of low oxidants concentrations.
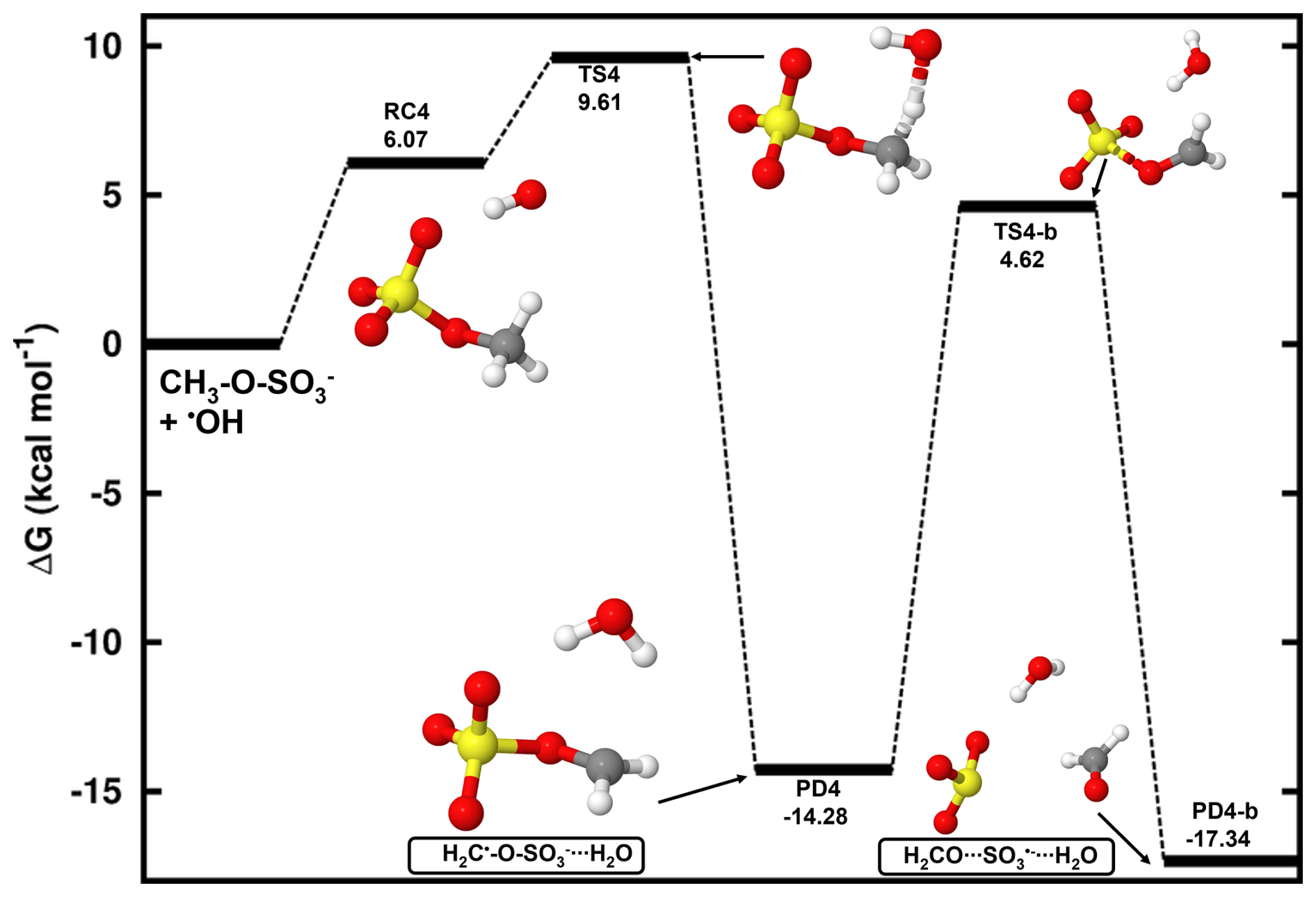
Figure 4Gibbs free energy changes (in kcal mol−1) and optimized structures for all intermediates in the reaction of CH3-O-SO with HO• radicals. The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
Table 2Gibbs free energy changes (ΔG) for all intermediate species in the reaction of deprotonated methyl sulfate with HO• radicals at 298.15 K and 1 M. Energy units are kcal mol−1. “RC” stands for intermediate reactant complex, “TS” stands for transition state, and “PD” stands for product.
Considering the H2C•-O-SO interaction with O3 (Reaction R7), this reaction is completely downhill, and it follows two main steps: formation of an alkoxy radical and decomposition of the latter into HSO and HCO• radicals (see Fig. 5). The first step is highly exergonic, with the reactant complex (O3•••H2C•-O-SO) being formed with −52.05 kcal mol−1 Gibbs free energy change. The decomposition of O3•••H2C•-O-SO into the O2•••O•-H2C-O-SO product complex is almost instantaneous, with the Gibbs free energy barrier located 5.98 kcal mol−1 below the reactant. As O2 evaporates from the product complex, the resulting alkoxy radical, •O-H2C-O-SO, is rapidly decomposed to HSO and HCO• by overcoming a relatively low energy barrier (7.92 kcal mol−1). The charge analysis confirms that during •O-H2C-O-SO decomposition, the unpaired electron effectively delocalizes from the oxygen atom of the alkoxy functional group to concentrate on the carbon atom of HCO (see Fig. S4), leaving HSO as one of the main products. These products were also identified as primary products in the CH3-O-SO reaction with •OH, although a different formation mechanism was proposed (Kwong et al., 2018). This study demonstrates that the reaction with O3 is distinctly thermodynamically and kinetically favorable, therefore highlighting the presented mechanism to be a determinant step in the oxidation of methyl sulfate by •OH radicals. While the role of HSO in the atmosphere is clearly established, for example in aerosol formation, HCO• has never been observed in aqueous media. Its presence has only been revealed through indirect observations and it has been suggested that it reacts fast with surrounding water to form formaldehyde (Jensen et al., 2010).
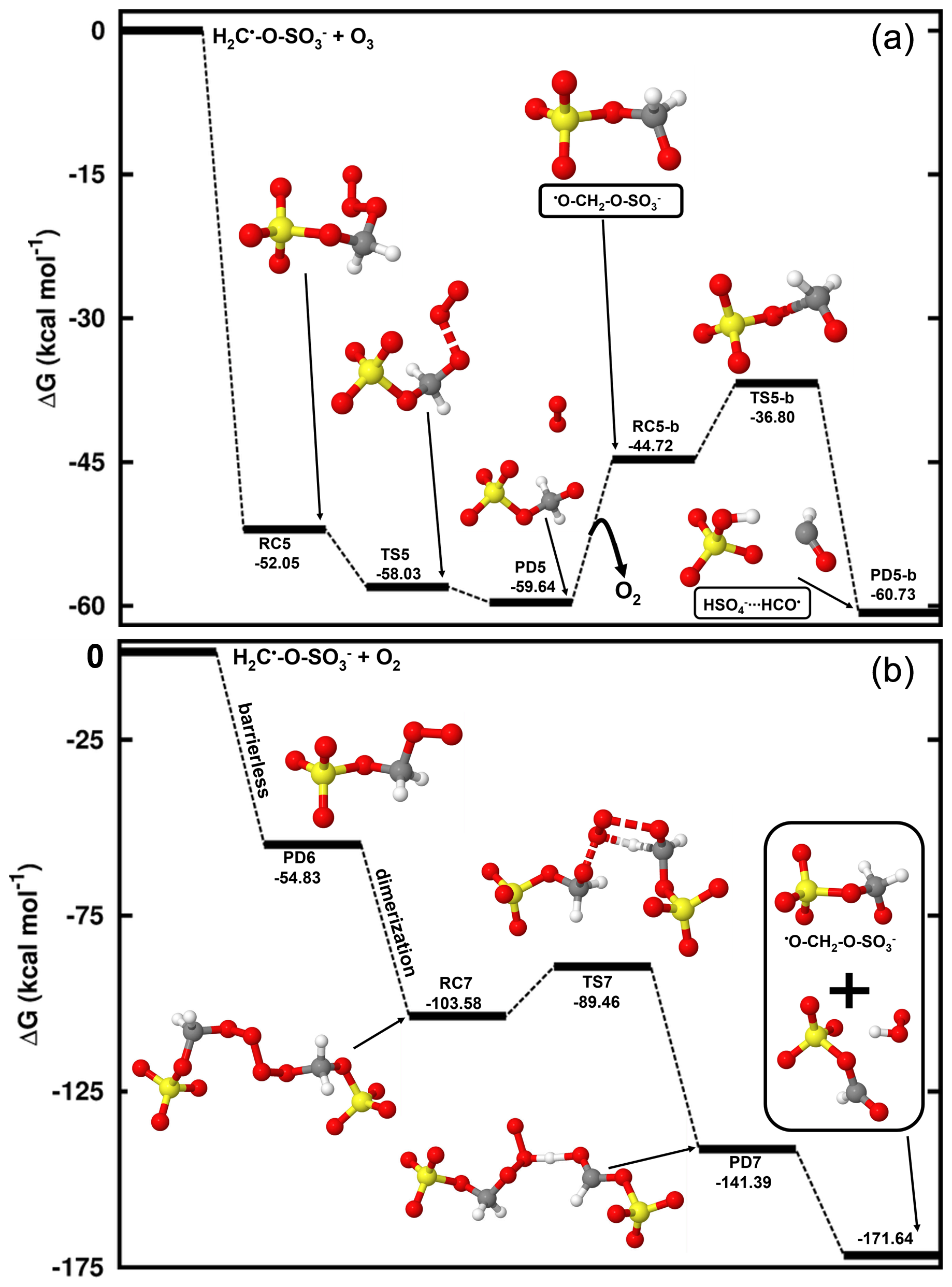
Figure 5Gibbs free energy changes (in kcal mol−1) and optimized structures for all intermediates in the reaction of CH2•-O-SO with O3 (a) and O2 (b). The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
H2C•-O-SO can also react fast with O2 (Reaction R8) to form a peroxyl radical (•OO-CH2-O-SO) through a barrierless process with the release of 54.83 kcal mol−1 Gibbs free energy. Based on the above on the chemistry of peroxyl radicals, two molecules of •OO-CH2-O-SO can combine to form a tetroxide, which can further decompose to generate •O-CH2-O-SO by overcoming a Gibbs free energy barrier of 14.12 kcal mol−1 (see Fig. 5). The alkoxy radical •O-CH2-O-SO can readily decompose to form HSO and HCO• by overcoming a barrier of 7.92 kcal mol−1, and as explained above and exemplified in Fig. 5, its formation from H2C•-O-SO reaction with O2 is less thermodynamically favorable than with O3, despite the overall rate of the former should be higher than that of the latter due to the high atmospheric concentration of O2. It follows from the above mechanisms that in most atmospherically relevant conditions, the pathway for •O-CH2-O-SO formation from the H2C•-O-SO reaction with O3 can readily complement that from Bennett and Summers that involves reaction with O2 (Bennett and Summers, 1974).
In addition to the mechanisms speculated by experimental studies (Kwong et al., 2018), the combination of mechanisms both in the gas phase and the aqueous phase presented in this study provides additional pathways for inorganic sulfate formation from the reaction of methyl sulfate with HO• radicals, namely the decomposition of H2C•-O-SO or H2C•-O-SO3H and their reactions with O3. These processes can drive significant changes in the chemical composition of aerosol, especially in terms of sulfate mass loadings.
3.2 Reaction mechanism of glycolic acid sulfate with HO• radicals
Hydrogen abstraction from glycolic acid sulfate could occur both from -CH2- and -COOH groups according to the following reactions:
The mechanism of Reaction (R9) is similar to that of the hydrogen abstraction from methyl sulfate that forms an alkyl radical. Through this process, glycolic acid sulfate readily undergoes a hydrogen abstraction from the -CH2- group by HO•, resulting in HOOC-CH•-O-SO3H formation. The reactant complex in this process lies at 4.65 kcal mol−1 at 298.15 K and 1 atm, and the transition state for its conversion is located 2.13 kcal mol−1 above the reactant complex. We determined a bimolecular rate constant of the overall reaction to be cm3 molec.−1 s−1 for this reaction at 298.15 K. This shows that hydrogen abstraction by •OH from glycolic acid sulfate is more favorable than from methyl sulfate, hereby highlighting the enhancing effect of the carboxyl substituent. The further chemistry of HOOC-CH•-O-SO3H is examined through reactions with O3 and O2. Our calculations show that contrary to the reaction with H2C•-O-SO3H, O3 hardly reacts with HOOC-CH•-O-SO3H as the O3•••HOOC-CH•-O-SO3H formation is highly endergonic at standard conditions. However, the reaction with O2 is seen to be fast, proceeding through formation of the reactant complex that is readily converted to HOOC-CH(OO)•-O-SO3H. The observed negative Gibbs free energy barrier (−9.02 kcal mol−1 below the reactant complex) in this conversion indicates that the formation of HOOC-CH(OO)•-O-SO3H is almost instantaneous at standard conditions. Two molecules of HOOC-CH(OO)•-O-SO3H develop into a tetroxide that then decomposes to HC(O)-O-SO3H and HOOC-CH(O)•-O-SO3H. The energetics of this reaction are provided in Fig. 6 and Table S5.
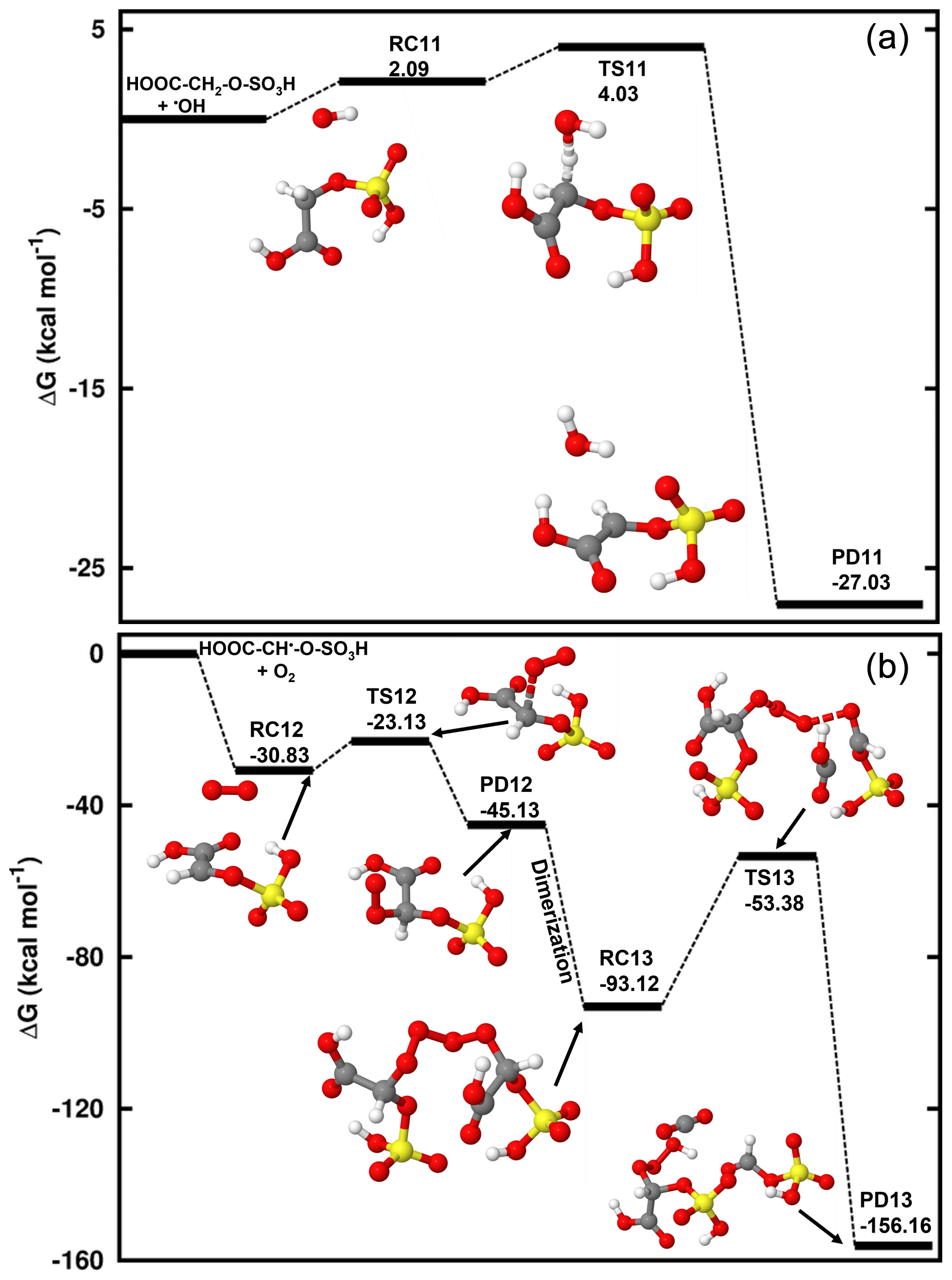
Figure 6Gibbs free energy changes (in kcal mol−1) and optimized structures for all intermediates in the reaction of HOOC-CH2-O-SO3H reaction with HO• radicals proceeding through hydrogen abstraction from the -CH2- group (a) and in the HOOC-CH•-O-SO3H reaction with O2 (b). The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
The abstraction from -COOH led to •OC(O)-CH2-O-SO3H (Reaction R10) that further decomposes to methylene sulfate radical (H2C•-O-SO3H) and CO2. The structures and energetics of all intermediate states of this reaction are given in Fig. S5 and Table S6. At the same level of theory, we determined a biomolecular rate constant of cm3 molec.−1 s−1, two orders of magnitude lower than the hydrogen abstraction from the -CH2- group. This indicates that hydrogen abstraction from glycolic acid would follow two competitive pathways although the pathway leading the alkyl radical would be somewhat preferred. It can be inferred that for organosulfates that have a -COOH substituent at the β-position relative to the sulfate group, decarboxylation would be a possible outcome of their decomposition. The chemistry of H2C•-O-SO3H was assessed in Sect. 3.1 above.
We further investigated the HO•-initiated reaction of glycolic acid sulfate in the aqueous phase, where the deprotonated state, HOOC-CH2-O-SO, is predominant. The preliminary interaction in this reaction is similar to that in the reaction of CH3-O-SO, with HO• abstracting the hydrogen atom from the -CH2- group to form the product complex, HOOC-CH•-O-SOH2O. The energies and structures of all different stationary states of this reaction are given in Fig. 7, while further energetics details are provided in Table 2. A bimolecular rate constant of 7.29×108 M−1 s−1 is determined for this reaction, in good agreement with the experimental report by Buxton et al. (1988). HOOC-CH•-O-SOH2O formation is highly exergonic, with −18.69 kcal mol−1 Gibbs free energy at 298.15 K and 1 M. Its decomposition back to the initial reactants is prevented by a substantially high energy barrier of 29.60 kcal mol−1. Based on this backward process, an atmospheric lifetime of 6.30×108 s is predicted for HOOC-CH•-O-SOH2O under relevant atmospheric conditions, long enough for this product complex to be subject to collisions with nearly all relevant atmospheric oxidants. H2O further dissociates from the product complex, leaving bare HOOC-CH•-O-SO to undergo the following decomposition processes:
Following Reaction (R11), HOOC-CH•-O-SO can undergo O-SO3 bond cleavage and form the HOOC-CHO•••SO3•− product complex at a unimolecular rate constant of s−1. The electronic charge analysis (shown in Fig. S6a) confirms the distribution of the unpaired electron on SO3 while HOOC-CHO is electrically neutral. Knowing that SO3•− has no other atmospheric chemical fate than inorganic sulfate, it follows that glycolic acid sulfate transformation by a HO•-initiated reaction would produce glyoxylic acid and sulfate at a nearly equal rate constant as CH2•-O-SO for a similar mechanism. The significance of this reaction will, however, depend on the rates of HOOC-CH•-O-SO reactions via other pathways.
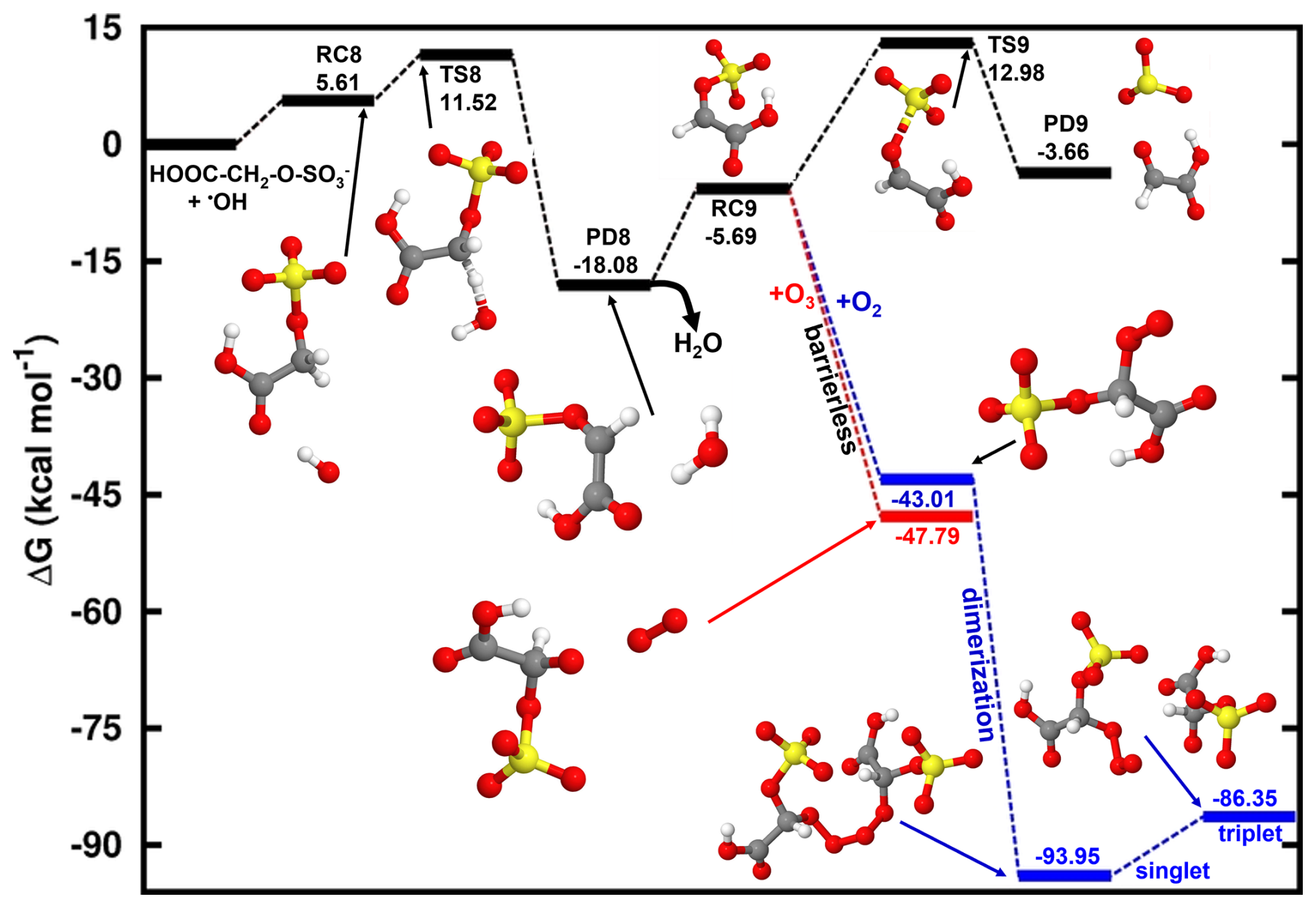
Figure 7Gibbs free energy changes (in kcal mol−1) and optimized structures for all stationary states in the reaction of HOOC-CH2-O-SO with HO• radicals, and subsequent reaction of the intermediate reactant complex with O3 (red line) and O2 (blue line). The sulfur atom is yellow, the oxygen atom is red, the carbon atom is gray, and the hydrogen atom is white.
Similar to H2C•-O-SO, the HOOC-CH•-O-SO reaction with O3 is completely downhill and directly undergoes an oxygen atom transfer for O3 to the -CH- group, forming the alkoxy radical, HOOC-CH(O)•-O-SO (Reaction R12). HOOC-CH(O)•-O-SO is susceptible to further decompose to CO2+ HCO• + HSO. However, we were unable to locate the appropriate transition state, which is seemingly associated with the mesomeric effect induced by the presence of an unpaired electron and entertained by the -COOH function. This situation was not observed in the decomposition of •O-CH2-O-SO.
Another reaction pathway for HOOC-CH•-O-SO reaction is by O2 addition (Reaction R13) in a barrierless process to form a peroxyl radical (HOOC-CH(OO•)-O-SO with the release of 43.01 kcal mol−1 Gibbs free energy. HOOC-CH(OO•)-O-SO can recombine with each other to form a tetroxide (OS-O-CH(COOH)-OOOO-CH(COOH)-O-SO) as shown in Fig. 7. However, we found that contrary to the case of •OO-CH2-O-SO, where the tetroxide could readily decompose to form the •O-CH2-O-SO radical in the singlet state, the fragmentation of OS-O-CH(COOH)-OOOO-CH(COOH)-O-SO to form the alkoxyl radical HOOC-CH(O)•-O-SO occurred on the triplet state instead. The triplet electronic state was shown from a recent study to be favorable to the decomposition of tetroxides to alkoxyl radicals from some atmospherically relevant peroxyl radicals (Salo et al., 2022). Figure S6b clearly shows the antibonding orbitals in the triplet state prior to the formation of HOOC-CH(O)•-O-SO upon which rests the unpaired electron.
3.3 Atmospheric implications
Organosulfates are important organic tracers for aerosols in the atmosphere. Although sufficient information on their sources and abundance has been gathered from previous studies, the understanding of the mechanisms of their transformation in the atmosphere remains incomplete. By investigating the decomposition mechanisms of two small atmospheric organosulfates (methyl sulfate and glycolic acid sulfate) by reaction with HO• radicals in this study, it was shown that the reaction of glycolic acid sulfate in the gas phase is more kinetically favorable than that of methyl sulfate, which can be attributed to the effect of -COOH substitution that stabilizes the intermediate reactant complex from glycolic acid. The chemical transformation of both organosulfates was seen to be more effective in the aqueous phase, with the reaction of methyl sulfate being more extensive than that of glycolic acid sulfate. The main products of these transformations are carbonyl compounds and inorganic sulfate, for which detailed mechanisms are provided. Not only does this study clarify the effect of substituents on the fragmentation of organosulfates, but it also complements previous experimental observations on methyl sulfate oxidation by HO• radicals (Kwong et al., 2018). For this reaction, we obtained a rate constant of cm3 molec.−1 s−1 in the gas phase, in agreement with the previously reported experimental value cm3 molec.−1 s−1) (Kwong et al., 2018). We also report a rate bimolecular rate constant of cm3 molec.−1 s−1 for the gas-phase reaction of HO• reaction with glycolic acid at 298.15 K. In the aqueous phase, we determined rate constants of 7.87×106 and 7.29×108 M−1 s−1 for the reaction of methyl sulfate and glycolic acid sulfate, respectively.
Among the three processes investigated (self-decomposition, reaction with O3, and reaction with O2), alkoxyl radicals can be formed both from alkyl radicals reactions with O2 and with O3. From the discussion above on reaction mechanisms and energetics, we clarify that besides the O2 reaction, the reaction with O3 could be a complementary intermediate step in the formation of alkoxy radicals that further decompose to inorganic sulfate and carbonyl compounds. However, considering real atmospheric conditions (with O2 concentrations much higher than O3 concentrations), this formation will be kinetically driven by the reaction with O2, whereas the reaction with O3 may exclusively become relevant in environments highly enriched with ozone.
Overall, the kinetics results show a moderate difference between the rate constants of HO• reactions with methyl sulfate and glycolic acid sulfate. The slightly high rate constant of the reaction of glycolic acid sulfate indicates the enhancing effect of the -COOH group in the hydrogen abstraction by HO•. Moreover, we found that the hydrogen abstraction from the -COOH group in glycolic acid sulfate leads to decarboxylation and eventually forms similar products as methyl sulfate. It can be inferred that for organosulfates having a carboxyl substituent at the β-position relative to the sulfate group, decarboxylation would take place, leading to the formation of the corresponding alkyl sulfate radical. This highlights the potential role that chemical substitution on the carbon chain of organosulfates may play during their decomposition. Given the high variety of organosulfates detected in atmospheric particles, it is necessary to deeply evaluate the role of molecular structures in their chemical transformation in order to guarantee proper understanding of their impact on the chemical composition of aerosols. A general trend of the effect of chemical substitution can be obtained from segregated studies of the chemical transformation of different classes of organosulfates derived from anthropogenic and biogenic precursors.
All data from this research can be obtained upon request by contacting the corresponding author.
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-8575-2025-supplement.
Conceptualization: NTT; funding acquisition: NTT and LD; investigation: LX and NTT; supervision: LD; writing – original draft preparation: LX and NTT. writing – review and editing: SNT, JNG, and LD.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors acknowledge the Wuxi Hengding Supercomputing Center Co., LTD and National Supercomputer Center in Tianjin for providing the computational resources. We thank Kristian H. Møller and Rasmus V. Otkjær of the University of Copenhagen for providing the script to calculate the Eckart tunneling correction.
This research has been supported by the National Key Research and Development Program of China (grant no. 2023YFC3706203), the National Natural Science Foundation of China (grant no. 22376121), and the Natural Science Foundation of Shandong Province (grant no. ZR2023MD041).
This paper was edited by Theodora Nah and reviewed by three anonymous referees.
An, G.-C.: Enhancement of atmospheric nucleation precursors on formic sulfuric anhydride induced nucleation: Theoretical mechanism, Chemosphere, 368, 143684, https://doi.org/10.1016/j.chemosphere.2024.143684, 2024.
Bennett, J. E. and Summers, R.: Product Studies of the Mutual Termination Reactions of sec-Alkylperoxy Radicals: Evidence for Non-Cyclic Termination, Can. J. Chem., 52, 1377–1379, https://doi.org/10.1139/v74-209, 1974.
Bork, N., Kurtén, T., and Vehkamäki, H.: Exploring the atmospheric chemistry of O2SO and assessing the maximum turnover number of ion-catalysed H2SO4 formation, Atmos. Chem. Phys., 13, 3695–3703, https://doi.org/10.5194/acp-13-3695-2013, 2013.
Bork, N., Kurtén, T., Enghoff, M. B., Pedersen, J. O. P., Mikkelsen, K. V., and Svensmark, H.: Structures and reaction rates of the gaseous oxidation of SO2 by an O(H2O)0-5 cluster – a density functional theory investigation, Atmos. Chem. Phys., 12, 3639–3652, https://doi.org/10.5194/acp-12-3639-2012, 2012.
Buehler, R. E., Staehelin, J., and Hoigne, J.: Ozone decomposition in water studied by pulse radiolysis. 1. Perhydroxyl (HO2)/hyperoxide (O2-) and HO3/O as intermediates, J. Phys. Chem., 88, 2560–2564, https://doi.org/10.1021/j150656a026, 1984.
Buhler, R., Staehelin, J., and Hoigne, J.: Ozone Decomposition in Water Studied by Pulse Radiolysis 1. HO2 O and HO3 O as Intermediates – Correction, J. Phys. Chem., 8, 5450–5450, https://doi.org/10.1021/j150666a600, 1984.
Buxton, G. V., Greenstock, C. L., Helman, W. P., and Ross, A. B.: Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH O−) in Aqueous Solution, J. Phys. Chem. Ref. Data, 17, 513–886, https://doi.org/10.1063/1.555805, 1988.
Cai, D., Wang, X., Chen, J., and Li, X.: Molecular Characterization of Organosulfates in Highly Polluted Atmosphere Using Ultra-High-Resolution Mass Spectrometry, J. Geophys. Res.-Atmos., 125, e2019JD032253, https://doi.org/10.1029/2019JD032253, 2020.
Chan, M. N., Surratt, J. D., Chan, A. W. H., Schilling, K., Offenberg, J. H., Lewandowski, M., Edney, E. O., Kleindienst, T. E., Jaoui, M., Edgerton, E. S., Tanner, R. L., Shaw, S. L., Zheng, M., Knipping, E. M., and Seinfeld, J. H.: Influence of aerosol acidity on the chemical composition of secondary organic aerosol from β-caryophyllene, Atmos. Chem. Phys., 11, 1735–1751, https://doi.org/10.5194/acp-11-1735-2011, 2011.
Cheng, J., Yue, L., Hua, J., Dong, H., Li, Y.-Y., Zhou, J., and Lin, R.: Hydrothermal heating with sulphuric acid contributes to improved fermentative hydrogen and methane co-generation from Dianchi Lake algal bloom, Energ. Convers. Manage., 192, 282–291, https://doi.org/10.1016/j.enconman.2019.04.003, 2019.
Cheng, Y., Wang, R., Chen, Y., Tian, S., Gao, N., Zhang, Z., and Zhang, T.: Hydrolysis of SO3 in Small Clusters of Sulfuric Acid: Mechanistic and Kinetic Study, ACS Earth and Space Chem., 6, 3078–3089, https://doi.org/10.1021/acsearthspacechem.2c00290, 2022.
Collins, F. C. and Kimball, G. E.: Diffusion-controlled reaction rates, J. Coll. Sci., 4, 425–437, https://doi.org/10.1016/0095-8522(49)90023-9, 1949.
Ding, C., Cheng, Y., Wang, H., Yang, J., Li, Z., Lily, M., Wang, R., and Zhang, T.: Determination of the influence of water on the SO3+ CH3OH reaction in the gas phase and at the air–water interface, Phys. Chem. Chem. Phys., 25, 15693–15701, https://doi.org/10.1039/D3CP01245J, 2023.
Eckart, C.: The Penetration of a Potential Barrier by Electrons, Phys. Rev. 35, 1303–1309, https://doi.org/10.1103/PhysRev.35.1303, 1930.
Ehn, M., Junninen, H., Petäjä, T., Kurtén, T., Kerminen, V.-M., Schobesberger, S., Manninen, H. E., Ortega, I. K., Vehkamäki, H., Kulmala, M., and Worsnop, D. R.: Composition and temporal behavior of ambient ions in the boreal forest, Atmos. Chem. Phys., 10, 8513–8530, https://doi.org/10.5194/acp-10-8513-2010, 2010.
Einstein, A.: Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen, Ann. Phys., 322, 549–560, https://doi.org/10.1002/andp.19053220806, 1905.
Enghoff, M. B. and Svensmark, H.: The role of atmospheric ions in aerosol nucleation – a review, Atmos. Chem. Phys., 8, 4911–4923, https://doi.org/10.5194/acp-8-4911-2008, 2008.
England, C. and Corcoran, W. H.: Kinetics and Mechanisms of the Gas-Phase Reaction of Water Vapor and Nitrogen Dioxide, Ind. Eng. Chem. Fund., 13, 373–384, https://doi.org/10.1021/i160052a014, 1974.
Estillore, A. D., Hettiyadura, A. P., Qin, Z., Leckrone, E., Wombacher, B., Humphry, T., Stone, E. A., and Grassian, V. H.: Water Uptake and Hygroscopic Growth of Organosulfate Aerosol, Environ. Sci. Technol, 50, 4259–4268, https://doi.org/10.1021/acs.est.5b05014, 2016.
Fehsenfeld, F. and Ferguson, E.: Laboratory studies of negative ion reactions with atmospheric trace constituents, J. Chem. Phys., 61, 3181–3193, https://doi.org/10.1063/1.1682474, 1974.
Ford, P. C. and Miranda, K. M.: The solution chemistry of nitric oxide and other reactive nitrogen species, Nitric Oxide, 103, 31-46, https://doi.org/10.1016/j.niox.2020.07.004, 2020.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams, Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 09 Rev. E.01, Wallingford, CT, http://www.gaussian.com (last access: 31 March 2025), 2013.
George, I. J. and Abbatt, J. P. D.: Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals, Nat. Chem., 2, 713–722, https://doi.org/10.1038/nchem.806, 2010.
Ghigo, G., Maranzana, A., and Tonachini, G.: Combustion and atmospheric oxidation of hydrocarbons: Theoretical study of the methyl peroxyl self-reaction, J. Chem. Phys., 118, 10575–10583, https://doi.org/10.1063/1.1574316, 2003.
Gweme, D. T. and Styler, S. A.: OH Radical Oxidation of Organosulfates in the Atmospheric Aqueous Phase, J. Phys. Chem. A, 128, 9462–9475, https://doi.org/10.1021/acs.jpca.4c02877, 2024.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Heindel, J. P., Yu, Q., Bowman, J. M., and Xantheas, S. S.: Benchmark Electronic Structure Calculations for H3O+(H2O)n, n = 0–5, Clusters and Tests of an Existing 1,2,3-Body Potential Energy Surface with a New 4-Body Correction, J. Chem. Theory Comput., 14, 4553–4566, https://doi.org/10.1021/acs.jctc.8b00598, 2018.
Hettiyadura, A. P. S., Stone, E. A., Kundu, S., Baker, Z., Geddes, E., Richards, K., and Humphry, T.: Determination of atmospheric organosulfates using HILIC chromatography with MS detection, Atmos. Meas. Tech., 8, 2347–2358, https://doi.org/10.5194/amt-8-2347-2015, 2015.
Hettiyadura, A. P. S., Jayarathne, T., Baumann, K., Goldstein, A. H., de Gouw, J. A., Koss, A., Keutsch, F. N., Skog, K., and Stone, E. A.: Qualitative and quantitative analysis of atmospheric organosulfates in Centreville, Alabama, Atmos. Chem. Phys., 17, 1343–1359, https://doi.org/10.5194/acp-17-1343-2017, 2017.
Hofmann, M. and Schleyer, P. v. R.: Acid Rain: Ab Initio Investigation of the H2OSO3 Complex and Its Conversion to H2SO4, J. Am. Chem. Soc., 116, 4947–4952, https://doi.org/10.1021/ja00090a045, 1994.
Hofmann-Sievert, R. and Castleman, A. W., Jr.: Reaction of sulfur trioxide with water clusters and the formation of sulfuric acid, J. Phys. Chem., 88, 3329–3333, https://doi.org/10.1021/j150659a038, 1984.
Huang, D. D., Li, Y. J., Lee, B. P., and Chan, C. K.: Analysis of Organic Sulfur Compounds in Atmospheric Aerosols at the HKUST Supersite in Hong Kong Using HR-ToF-AMS, Environ. Sci. Technol., 49, 3672–3679, https://doi.org/10.1021/es5056269, 2015.
Huang, R.-J., Cao, J., Chen, Y., Yang, L., Shen, J., You, Q., Wang, K., Lin, C., Xu, W., Gao, B., Li, Y., Chen, Q., Hoffmann, T., O'Dowd, C. D., Bilde, M., and Glasius, M.: Organosulfates in atmospheric aerosol: synthesis and quantitative analysis of PM2.5 from Xi'an, northwestern China, Atmos. Meas. Tech., 11, 3447–3456, https://doi.org/10.5194/amt-11-3447-2018, 2018.
Hughes, D. D. and Stone, E. A.: Organosulfates in the Midwestern United States: abundance, composition and stability, J. Environ. Chem., 16, 312–322, https://doi.org/10.1071/EN18260, 2019.
Iinuma, Y., Müller, C., Böge, O., Gnauk, T., and Herrmann, H.: The formation of organic sulfate esters in the limonene ozonolysis secondary organic aerosol (SOA) under acidic conditions, Atmos. Environ., 41, 5571–5583, https://doi.org/10.1016/j.atmosenv.2007.03.007, 2007.
Jensen, S. K., Keiding, S. R., and Thøgersen, J.: The hunt for HCO(aq), Phys. Chem. Chem. Phys., 12, 8926–8933, https://doi.org/10.1039/B924902H, 2010.
Kourtchev, I., Godoi, R. H. M., Connors, S., Levine, J. G., Archibald, A. T., Godoi, A. F. L., Paralovo, S. L., Barbosa, C. G. G., Souza, R. A. F., Manzi, A. O., Seco, R., Sjostedt, S., Park, J.-H., Guenther, A., Kim, S., Smith, J., Martin, S. T., and Kalberer, M.: Molecular composition of organic aerosols in central Amazonia: an ultra-high-resolution mass spectrometry study, Atmos. Chem. Phys., 16, 11899–11913, https://doi.org/10.5194/acp-16-11899-2016, 2016.
Kwong, K. C., Chim, M. M., Davies, J. F., Wilson, K. R., and Chan, M. N.: Importance of sulfate radical anion formation and chemistry in heterogeneous OH oxidation of sodium methyl sulfate, the smallest organosulfate, Atmos. Chem. Phys., 18, 2809–2820, https://doi.org/10.5194/acp-18-2809-2018, 2018.
Larson, L. J., Kuno, M., and Tao, F.-M.: Hydrolysis of sulfur trioxide to form sulfuric acid in small water clusters, J. Chem. Phys., 112, 8830–8838, 2000.
Le Breton, M., Wang, Y., Hallquist, Å. M., Pathak, R. K., Zheng, J., Yang, Y., Shang, D., Glasius, M., Bannan, T. J., Liu, Q., Chan, C. K., Percival, C. J., Zhu, W., Lou, S., Topping, D., Wang, Y., Yu, J., Lu, K., Guo, S., Hu, M., and Hallquist, M.: Online gas- and particle-phase measurements of organosulfates, organosulfonates and nitrooxy organosulfates in Beijing utilizing a FIGAERO ToF-CIMS, Atmos. Chem. Phys., 18, 10355–10371, https://doi.org/10.5194/acp-18-10355-2018, 2018.
Lee, Y. N. and Schwartz, S. E.: Reaction kinetics of nitrogen dioxide with liquid water at low partial pressure, J. Phys. Chem., 85, 840–848, https://doi.org/10.1021/j150607a022, 1981.
Liang, Y.-N., Li, J., Wang, Q.-D., Wang, F., and Li, X.-Y.: Computational Study of the Reaction Mechanism of the Methylperoxy Self-Reaction, J. Phys. Chem. A, 115, 13534–13541, https://doi.org/10.1021/jp2048508, 2011.
Liao, J., Froyd, K. D., Murphy, D. M., Keutsch, F. N., Yu, G., Wennberg, P. O., St. Clair, J. M., Crounse, J. D., Wisthaler, A., Mikoviny, T., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Ryerson, T. B., Pollack, I. B., Peischl, J., Anderson, B. E., Ziemba, L. D., Blake, D. R., Meinardi, S., and Diskin, G.: Airborne measurements of organosulfates over the continental U.S, J. Geophys. Res.-Atmos., 120, 2990–3005, https://doi.org/10.1002/2014JD022378, 2015.
Lin, Y.-H., Budisulistiorini, S. H., Chu, K., Siejack, R. A., Zhang, H., Riva, M., Zhang, Z., Gold, A., Kautzman, K. E., and Surratt, J. D.: Light-Absorbing Oligomer Formation in Secondary Organic Aerosol from Reactive Uptake of Isoprene Epoxydiols, Environ. Sci. Technol., 48, 12012–12021, https://doi.org/10.1021/es503142b, 2014.
Loerting, T. and Liedl, K. R.: Toward elimination of discrepancies between theory and experiment: The rate constant of the atmospheric conversion of SO3 to H2SO4, P. Natl Acad. Sci. USA, 97, 8874–8878, https://doi.org/10.1073/pnas.97.16.8874, 2000.
Lovato, M. E., Martín, C. A., and Cassano, A. E.: A reaction kinetic model for ozone decomposition in aqueous media valid for neutral and acidic pH, Chem. Eng. J., 146, 486–497, https://doi.org/10.1016/j.cej.2008.11.001, 2009.
Lu, T. and Chen, F.: Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 33, 580–592, https://doi.org/10.1002/jcc.22885, 2012.
Markovitch, O. and Agmon, N.: Structure and Energetics of the Hydronium Hydration Shells, J. Phys. Chem. A, 111, 2253–2256, https://doi.org/10.1021/jp068960g, 2007.
Morokuma, K. and Muguruma, C.: Ab initio Molecular Orbital Study of the Mechanism of the Gas Phase Reaction SO3+ H2O: Importance of the Second Water Molecule, J. Am. Chem. Soc., 116, 10316–10317, https://doi.org/10.1021/ja00101a068, 1994.
Nangia, P. S. and Benson, S. W.: The kinetics of the interaction of peroxy radicals. II. Primary and secondary alkyl peroxy, Int. J. Chem. Kinet., 12, 43–53, https://doi.org/10.1002/kin.550120105, 1980.
Nguyen, Q. T., Christensen, M. K., Cozzi, F., Zare, A., Hansen, A. M. K., Kristensen, K., Tulinius, T. E., Madsen, H. H., Christensen, J. H., Brandt, J., Massling, A., Nøjgaard, J. K., and Glasius, M.: Understanding the anthropogenic influence on formation of biogenic secondary organic aerosols in Denmark via analysis of organosulfates and related oxidation products, Atmos. Chem. Phys., 14, 8961–8981, https://doi.org/10.5194/acp-14-8961-2014, 2014.
Nozière, B., Ekstrom, S., Alsberg, T., and Holmstrom, S.: Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols, Geophys. Res. Lett., 37, L05806, https://doi.org/10.1029/2009gl041683, 2010.
Nozière, B., Kalberer, M., Claeys, M., Allan, J., D'Anna, B., Decesari, S., Finessi, E., Glasius, M., Grgić, I., Hamilton, J. F., Hoffmann, T., Iinuma, Y., Jaoui, M., Kahnt, A., Kampf, C. J., Kourtchev, I., Maenhaut, W., Marsden, N., Saarikoski, S., Schnelle-Kreis, J., Surratt, J. D., Szidat, S., Szmigielski, R., and Wisthaler, A.: The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 115, 3919–3983, https://doi.org/10.1021/cr5003485, 2015.
Ostovari, H., Zahedi, E., Sarvi, I., and Shiroudi, A.: Kinetic and mechanistic insight into the formation of amphetamine using the Leuckart–Wallach reaction and interaction of the drug with GpC• CpG base-pair step of DNA: a DFT study, Monatsh. Chem., 149, 1045–1057, https://doi.org/10.1007/s00706-018-2145-7, 2018.
Passananti, M., Kong, L., Shang, J., Dupart, Y., Perrier, S., Chen, J., Donaldson, D. J., and George, C.: Organosulfate Formation through the Heterogeneous Reaction of Sulfur Dioxide with Unsaturated Fatty Acids and Long-Chain Alkenes, Angew. Chem. Int. Edit., 55, 10336–10339, https://doi.org/10.1002/anie.201605266, 2016.
Peng, C., Razafindrambinina, P. N., Malek, K. A., Chen, L., Wang, W., Huang, R.-J., Zhang, Y., Ding, X., Ge, M., Wang, X., Asa-Awuku, A. A., and Tang, M.: Interactions of organosulfates with water vapor under sub- and supersaturated conditions, Atmos. Chem. Phys., 21, 7135–7148, https://doi.org/10.5194/acp-21-7135-2021, 2021.
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 034106, https://doi.org/10.1063/1.4773581, 2013.
Rudziński, K. J., Gmachowski, L., and Kuznietsova, I.: Reactions of isoprene and sulphoxy radical-anions – a possible source of atmospheric organosulphites and organosulphates, Atmos. Chem. Phys., 9, 2129–2140, https://doi.org/10.5194/acp-9-2129-2009, 2009.
Russell, G. A.: Deuterium-isotope Effects in the Autoxidation of Aralkyl Hydrocarbons. Mechanism of the Interaction of PEroxy Radicals1, J. Am. Chem. Soc., 79, 3871–3877, https://doi.org/10.1021/ja01571a068, 1957.
Salo, V.-T., Valiev, R., Lehtola, S., and Kurtén, T.: Gas-Phase Peroxyl Radical Recombination Reactions: A Computational Study of Formation and Decomposition of Tetroxides, J. Phys. Chem. A, 126, 4046–4056, https://doi.org/10.1021/acs.jpca.2c01321, 2022.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, New York, 88 pp., https://doi.org/10.1063/1.882420, 1998.
Shang, J., Passananti, M., Dupart, Y., Ciuraru, R., Tinel, L., Rossignol, S., Perrier, S., Zhu, T., and George, C.: SO2 Uptake on Oleic Acid: A New Formation Pathway of Organosulfur Compounds in the Atmosphere, Environ. Sci. Tech. Let., 3, 67–72, https://doi.org/10.1021/acs.estlett.6b00006, 2016.
Smoluchowski, M. V.: Mathematical Theory of the Kinetics of the Coagulation of Colloidal Solutions, Z. Phys. Chem., 92, 129–168, 1917.
Staehelin, J. and Hoigne, J.: Decomposition of ozone in water: rate of initiation by hydroxide ions and hydrogen peroxide, Environ. Sci. Technol., 16, 676–681, https://doi.org/10.1021/es00104a009, 1982.
Staehelin, J., Buehler, R. E., and Hoigne, J.: Ozone decomposition in water studied by pulse radiolysis. 2. Hydroxyl and hydrogen tetroxide (HO4) as chain intermediates, J. Phys. Chem., 88, 5999–6004, https://doi.org/10.1021/j150668a051, 1984.
Stone, E. A., Yang, L., Yu, L. E., and Rupakheti, M.: Characterization of organosulfates in atmospheric aerosols at Four Asian locations, Atmos. Environ., 47, 323–329, https://doi.org/10.1016/j.atmosenv.2011.10.058, 2012.
Surratt, J. D., Gomez-Gonzalez, Y., Chan, A. W. H., Vermeylen, R., Shahgholi, M., Kleindienst, T. E., Edney, E. O., Offenberg, J. H., Lewandowski, M., Jaoui, M., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Organosulfate formation in biogenic secondary organic aerosol, J. Phys. Chem. A, 112, 8345–8378, https://doi.org/10.1021/jp802310p, 2008.
Svensmark, H., Pedersen, J. O. P., Marsh, N. D., Enghoff, M. B., and Uggerhoj, U. I.: Experimental evidence for the role of ions in particle nucleation under atmospheric conditions, P. R. Soc. A, 463, 385–396, https://doi.org/10.1098/rspa.2006.1773, 2007.
Tan, S. P. and Piri, M.: Modeling the Solubility of Nitrogen Dioxide in Water Using Perturbed-Chain Statistical Associating Fluid Theory, Ind. Eng. Chem. Res., 52, 16032–16043, https://doi.org/10.1021/ie402417p, 2013.
Truhlar, D. G.: Nearly encounter-controlled reactions: The equivalence of the steady-state and diffusional viewpoints, J. Chem. Educ., 62, 104, https://doi.org/10.1021/ed062p104, 1985.
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J.: Current Status of Transition-State Theory, J. Phys. Chem., 100, 12771–12800, https://doi.org/10.1021/jp953748q, 1996.
Tsona, N. T. and Du, L.: Hydration of glycolic acid sulfate and lactic acid sulfate: Atmospheric implications, Atmos. Environ., 216, 116921, https://doi.org/10.1016/j.atmosenv.2019.116921, 2019a.
Tsona, N. T. and Du, L.: A potential source of atmospheric sulfate from O2−-induced SO2 oxidation by ozone, Atmos. Chem. Phys., 19, 649–661, https://doi.org/10.5194/acp-19-649-2019, 2019b.
Wang, R., Kang, J., Zhang, S., Shao, X., Jin, L., Zhang, T., and Wang, Z.: Catalytic effect of (H2O)n (n=1–2) on the hydrogen abstraction reaction of H2O2+HS → H2S+HO2 under tropospheric conditions, Comput. Theor. Chem., 1110, 25-34, https://doi.org/10.1016/j.comptc.2017.03.045, 2017.
Wang, R., Cheng, Y., Chen, S., Li, R., Hu, Y., Guo, X., Zhang, T., Song, F., and Li, H.: Reaction of SO3 with H2SO4 and its implications for aerosol particle formation in the gas phase and at the air–water interface, Atmos. Chem. Phys., 24, 4029–4046, https://doi.org/10.5194/acp-24-4029-2024, 2024.
Wang, R., Yao, Q., Wen, M., Tian, S., Wang, Y., Wang, Z., Yu, X., Shao, X., and Chen, L.: Catalytic effect of (H2O)n (n= 1–3) clusters on the HO2+ SO2 → HOSO +3O2 reaction under tropospheric conditions, RSC Adv., 9, 16195–16207, https://doi.org/10.1039/C9RA00169G, 2019.
Wang, Y., Zhao, Y., Wang, Y., Yu, J.-Z., Shao, J., Liu, P., Zhu, W., Cheng, Z., Li, Z., Yan, N., and Xiao, H.: Organosulfates in atmospheric aerosols in Shanghai, China: seasonal and interannual variability, origin, and formation mechanisms, Atmos. Chem. Phys., 21, 2959–2980, https://doi.org/10.5194/acp-21-2959-2021, 2021.
Worton, D. R., Goldstein, A. H., Farmer, D. K., Docherty, K. S., Jimenez, J. L., Gilman, J. B., Kuster, W. C., de Gouw, J., Williams, B. J., Kreisberg, N. M., Hering, S. V., Bench, G., McKay, M., Kristensen, K., Glasius, M., Surratt, J. D., and Seinfeld, J. H.: Origins and composition of fine atmospheric carbonaceous aerosol in the Sierra Nevada Mountains, California, Atmos. Chem. Phys., 11, 10219–10241, https://doi.org/10.5194/acp-11-10219-2011, 2011.
Xu, L. and Coote, M. L.: Methods To Improve the Calculations of Solvation Model Density Solvation Free Energies and Associated Aqueous pKa Values: Comparison between Choosing an Optimal Theoretical Level, Solute Cavity Scaling, and Using Explicit Solvent Molecules, J. Phys. Chem. A, 123, 7430–7438, https://doi.org/10.1021/acs.jpca.9b04920, 2019.
Xu, R., Ng, S. I. M., Chow, W. S., Wong, Y. K., Wang, Y., Lai, D., Yao, Z., So, P.-K., Yu, J. Z., and Chan, M. N.: Chemical transformation of α-pinene-derived organosulfate via heterogeneous OH oxidation: implications for sources and environmental fates of atmospheric organosulfates, Atmos. Chem. Phys., 22, 5685–5700, https://doi.org/10.5194/acp-22-5685-2022, 2022.
Zhang, P., Wang, W., Zhang, T., Chen, L., Du, Y., Li, C., and Lü, J.: Theoretical Study on the Mechanism and Kinetics for the Self-Reaction of C2H5O2 Radicals, J. Phys. Chem. A, 116, 4610–4620, https://doi.org/10.1021/jp301308u, 2012.
Zhang, X., Lian, Y., Tan, S., and Yin, S.: Organosulfate produced from consumption of SO3 speeds up sulfuric acid–dimethylamine atmospheric nucleation, Atmos. Chem. Phys., 24, 3593–3612, https://doi.org/10.5194/acp-24-3593-2024, 2024.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, https://doi.org/10.1007/s00214-007-0310-x, 2008.