the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 06 Jan 2025
| 06 Jan 2025
Widespread trace bromine and iodine in remote tropospheric non-sea-salt aerosols
Gregory P. Schill
Karl D. Froyd
Daniel M. Murphy
Christina J. Williamson
Charles A. Brock
Tomás Sherwen
Mat J. Evans
Eric A. Ray
Eric C. Apel
Rebecca S. Hornbrook
Alan J. Hills
Jeff Peischl
Thomas B. Ryerson
Chelsea R. Thompson
Ilann Bourgeois
Donald R. Blake
Joshua P. DiGangi
Glenn S. Diskin
Reactive halogens catalytically destroy O3 and therefore affect (1) stratospheric O3 depletion and (2) the oxidative capacity of the troposphere. Reactive halogens also partition into the aerosol phase, but what governs halogen-aerosol partitioning is poorly constrained in models. In this work, we present global-scale measurements of non-sea-salt aerosol (nSSA) bromine and iodine taken during the NASA Atmospheric Tomography Mission (ATom). Using the Particle Analysis by Laser Mass Spectrometry instrument, we found that bromine and iodine are present in 8 %–26 % (interquartile range, IQR) and 12 %–44 % (IQR) of accumulation-mode nSSA, respectively. Despite being commonly found in nSSA, the concentrations of bromine and iodine in nSSA were low but potentially important, at 0.11–0.57 pmol mol−1 (IQR) and 0.04–0.24 pmol mol−1 (IQR), respectively. In the troposphere, we find two distinct sources of bromine and iodine for nSSA: (1) a primary source from biomass burning and (2) a pervasive secondary source. In the stratosphere, nSSA bromine and iodine concentrations increased with increasing O3 concentrations; however, higher concentrations of stratospheric nSSA bromine and iodine were found in organic-rich particles that originated in the troposphere. Finally, we compared our ATom nSSA iodine measurements to the global chemical transport model GEOS-Chem (Goddard Earth Observing System); nSSA bromine concentrations could not be compared because they were not tracked in the model. We found that the model compared well to our ATom nSSA iodine measurements in the background atmosphere but not in the marine boundary layer, biomass burning plumes, or stratosphere.
- Article
(18179 KB) - Full-text XML
- BibTeX
- EndNote




Reactive halogens, including the reactive forms of bromine and iodine, affect O3 loss in both the troposphere and stratosphere and therefore contribute to uncertainties in global radiative forcing and stratospheric O3 loss, respectively. In the troposphere, incorporating halogen chemistry into global chemical transport models decreases global tropospheric O3 burdens by ∼ 17 %–19 %, with > 70 %–95 % of the global odd oxygen loss from halogens coming from bromine and iodine (Saiz-Lopez et al., 2014; Sherwen et al., 2016b). In the stratosphere, bromine and iodine destroy O3 analogous to chlorine, but bromine is approximately 45× more effective than chlorine at destroying stratospheric O3 on a per-atom basis (Daniel et al., 1999); iodine is even more efficient than bromine on a per-atom basis (∼ 10×, Koenig et al., 2020), suggesting that even a few tenths of a picomole per mole (pmol mol−1; equal to parts per trillion by volume, pptv) of reactive iodine in the lower stratosphere can be significant for mid-latitude, lower-stratosphere O3 loss (Solomon et al., 1994; Saiz‐Lopez et al., 2015).
Gas-phase bromine and iodine compounds are sourced from both anthropogenic and natural emissions, with natural oceanic emissions being the largest contributors. Once photolyzed, these gas-phase compounds form atomic Br and I radicals, which can catalytically destroy O3. Both biotic and abiotic processes in the ocean form volatile organohalogens, including CH3Br (reactive lifetime of τ∼ 0.8 years) and CH3I (τ∼ 7 d), which are emitted directly into the marine boundary layer (MBL). Additionally, inorganic iodine (HOI, I2) formed from the reaction of O3 with iodine compounds in the ocean surface has been shown to be the dominant contributor to tropospheric reactive iodine budgets (Carpenter et al., 2013). Since 1950, this inorganic iodine source has been increasing, which can be attributed to enhanced O3 concentrations from anthropogenic activity (Cuevas et al., 2018; Legrand et al., 2018) and enhanced sub-ice biological activity due to Arctic sea ice thinning (Cuevas et al., 2018). In addition to CH3Br, long-lived halons sourced primarily from fire extinguishers and fire retardants are well-known contributors to reactive bromine concentrations; furthermore, modeling studies have shown that very short-lived bromine species (τ< 6 months) can also contribute significantly to reactive bromine concentrations, even in the stratosphere (Stachnik et al., 2013; Keber et al., 2020).
In addition to gas-phase chemistry, the multi-phase chemistry of aerosol and clouds can be a source of reactive bromine and iodine. For example, sea-salt aerosol (SSA), which is produced from wave breaking or bubble bursting at the ocean surface, contains bromine at saltwater concentrations, and models suggest that debromination of SSA can be the largest source of reactive bromine to the troposphere (Zhu et al., 2019). Consequently, heterogeneous reactions of aerosols and clouds are needed to correctly model reactive bromine concentrations (Schmidt et al., 2016; Badia et al., 2019). Iodine is often enriched in SSA (Murphy et al., 1997), but it is unclear whether that is from iodine enrichment in the sea-surface microlayer (Dean, 1963) or from fast gas- or multi-phase iodine chemistry (Duce et al., 1983) or both.
While multi-phase chemistry can be a source of reactive halogens in the gas phase, gas-phase bromine and iodine can also react to form secondary aerosol in non-SSA (nSSA). For example, bromine radicals are known to react with volatile organic compounds and can form low-volatility products that partition into the aerosol phase (Ofner et al., 2012; Badia et al., 2019). Thus, the bromine aerosol contribution needs reactive organic species to contribute to secondary aerosol; iodine, however, can contribute to secondary aerosol through self-reactions. The formation of higher iodine oxides (, x=2, 3, 4) and/or iodine oxoacids (HIOX, x=2, 3) from reactive iodine precursors helps partition iodine to nSSA and can even cause new particle formation at coastal sites (O'Dowd et al., 2002) and in the Arctic (Allan et al., 2015) when reactive iodine concentrations are sufficient. Global chemistry models estimate that 15.3 % of iodine emissions are converted into aerosol (Sherwen et al., 2016c), but this is dependent on the fate of higher iodine oxides, which is poorly understood (Saiz-Lopez et al., 2014; Sherwen et al., 2016b). More recently, studies in the CERN (European Organization for Nuclear Research) cloud chamber have also shown that reactive iodine emissions can react to form iodic acid (Finkenzeller et al., 2023), which is the major iodine species driving both nucleation and growth of iodine oxoacid particles in pristine regions. The efficacy of iodine oxoacids to form new particles has been shown to exceed that of the H2SO4 ⋅ NH3 system at the same acid concentrations (He et al., 2021).
Quantitative nSSA bromine measurements are sparse and often difficult to separate from SSA; while nSSA differs from SSA in that it is typically non-refractory and smaller in diameter (fine mode), it is difficult to cleanly separate nSSA from SSA based on size alone. Nonetheless, filter-based measurements suggest that fine-mode aerosol is enhanced in Br− concentrations relative to sea salt (Sturges and Barrie, 1988; Hara et al., 2002). Single-particle techniques can separate nSSA from SSA based on their chemical composition. Qualitative measurements from the single-particle mass spectrometer Particle Analysis by Laser Mass Spectrometry (PALMS) instrument also suggest that bromine is present in nSSA in the upper troposphere/lower stratosphere (UT/LS), in roughly equal amounts to iodine (Murphy and Thomson, 2000). nSSA iodine measurements are more prevalent than bromine measurements but are still sparse, especially in the free troposphere. A compilation of near-surface measurements suggest that aerosol iodine can range from 0.013–4.62 pmol mol−1 (0.07–24 ng sm−3, standard meter; Saiz-Lopez et al., 2012). High-latitude UT/LS aerosol iodine measurements are of a similar magnitude (Koenig et al., 2020). Combined, these measurements suggest that nSSA bromine and iodine may be pervasive in the lower atmosphere, akin to their gas-phase, reactive-halogen counterparts; however, more measurements are needed across a wider range of latitudes, longitudes, and altitudes.
In this work, we present global-scale measurements of bromine and iodine in nSSA using the NOAA PALMS instrument. PALMS is a single-particle mass spectrometer which measures both the number fraction of halogen-containing aerosol and the absolute mass of halogen in the aerosol. Absolute mass measurements were constrained by new laboratory calibrations of PALMS and independently measured particle size distributions. Measurements were made primarily during the NASA Atmospheric Tomography Mission (ATom). ATom consisted of four flight circuits that flew southward over the Pacific Ocean basin and northward over the Atlantic Ocean basin. Each circuit consisted of a series of 8–10 h flights, with each flight conducting 6–10 vertical profiles from ∼ 0.15 to 12 km. Additional measurements were made during two flight campaigns over the continental US (Deep Convective Clouds and Chemistry (DC3) and Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys (SEAC4RS)) and another flight campaign in the tropical Atlantic (Costa Rica Aura Validation Experiment (CR-AVE)).
In these measurements, trace levels of iodine and bromine were ubiquitous in remote tropospheric nSSA, regardless of season. Our measurements suggest that nSSA bromine and iodine have two sources in the troposphere – a primary source from biomass burning and a pervasive secondary source. We also found that nSSA iodine and bromine concentrations increased in the stratosphere, but higher concentrations of bromine and iodine were measured in particles that originated in the troposphere. Single-particle measurements suggest this is due to their higher organic mass fraction, implying that organics help bind iodine in nSSA or that organics serve as a proxy for another physico-chemical attribute that helps partition halogens to nSSA. Finally, we compared our nSSA iodine observations to a global chemical transport model; nSSA bromine was not explicitly tracked in the model. Outside of the MBL, biomass burning plumes, and the stratosphere, the model did well in reproducing nSSA iodine vertical profiles in both shape and magnitude. Iodine aerosol in the model is formed from the production of higher iodine oxides and HI, which form in the model via multi-step reactions involving Ix (I and IO radicals). Thus, while we do not believe that our model–measurement comparison implicates higher iodine oxides and HI as the only iodine aerosol contributors during ATom, it does suggest that Ix is involved in the widespread, trace nSSA iodine concentrations we observed in the remote atmosphere.
2.1 PALMS
The PALMS instrument configuration during NASA's ATom has been described in detail in several publications (e.g., Schill et al., 2020). Briefly, PALMS operated inside the cabin of the NASA DC-8. Aerosol from outside the plane was sampled from the University of Hawai'i's (UH) shrouded, near-isokinetic inlet (McNaughton et al., 2007), which has been maintained by the NASA Langley Aerosol Research Group Experiment (LARGE). The inlet was mounted to two window plates on the starboard side of the DC-8. Aerosol transmission through the inlet is essentially unity at small sizes. The transmission of large particles through the UH-LARGE inlet is pressure dependent. The large size cutoff (D50) at low altitudes was measured to have a 5.0 µm aerodynamic diameter; theoretical losses at 12 km have been calculated from these near-surface measurements, and the D50,aerodynamic is expected to be 3.2 µm. During ATom, PALMS detected particles between approximately 100 nm and 5 µm in geometric diameter, but the vast majority (99 %) were 152 nm to 2.3 µm in geometric diameter. The lower limit for size detection in PALMS was set by noise in the optical system, which varied by flight due to differences in instrumental conditions. The upper size limit was mainly set by transmission through the pressure-dependent, UH-LARGE aircraft inlet, the tubing from the aircraft inlet to the PALMS instrument, and the PALMS aerosol focusing lens.
An exemplary detection efficiency curve for PALMS, calculated for ATom-1, is presented in Fig. 6 in Froyd et al. (2019); however, in this work, we calculated the number fraction and mass concentrations by coupling PALMS to an independently measured, quantitative size distribution (Froyd et al., 2019; Brock et al., 2021). For the size distribution measurements, we used the Aerosol Microphysical Properties (AMP; Brock et al., 2019). AMP-sampled air from the same UH-LARGE inlet and downstream losses in tubing bends and due to gravitational settling were calculated, and the data were corrected based on loss calculations (Brock et al., 2019). Using this coupling method, the PALMS detection efficiencies do not matter as long as a sufficient number of particles within each size bin are sampled and that PALMS measures each particle type within each size bin with similar detection efficiencies. To couple PALMS to the size distribution measurements, single-particle mass spectra were first classified into broad particle types such as biomass burning, sulfate–organic–nitrate mixtures (without biomass burning material), mineral dust, sea salt, and others. We then mapped the PALMS particle type fractions to the size distribution measurements using four bins (bin start/stop: 100, 252, 504, 1128, and 5036 nm), which generated mass concentrations for each particle type. The coupled PALMS–AMP size distributions required at least five particles in each bin. An inherent assumption of this technique is that the particle type fractions are constant within each bin. For ATom, we have found this assumption to be reasonable for the four bins described above (Froyd et al., 2019). Sub-components of each particle type, e.g., organics and sulfate, can also be calculated from laboratory calibrations. For all nSSA particle types, we calculated the organic and sulfate mass concentrations (Froyd et al., 2019); the relative statistical error associated with this technique is ∼ 10 %–25 %, for sulfate and organic mass concentrations, if those concentrations are > 0.01 µg sm−3. These errors are comparable to the estimated error of the size distributions (Brock et al., 2019). For this study, we generated nSSA bromine and iodine mass concentrations (and volume mixing ratios) using the same method (see Sect. 2.2).
Mass concentrations and mixing ratios for particle types and for halogens/chemical constituents were computed at 3 min intervals, which strikes a balance between time resolution and particle statistics. All curtain plots and vertical profiles were the average or median of these 3 min values.
2.2 PALMS bromine and iodine mass calibration
To calibrate PALMS for bromine and iodine mass in nSSA, we spiked trace amounts of bromine and iodine in an nSSA proxy. In this work, we define nSSA as non-refractory particles that PALMS classifies into four broad chemical types: sulfate–organic–nitrate, biomass burning, vanadium-containing (largely sulfate and organic and generally from sea-shipping activity), and meteoric-sulfuric-acid particles (stratospherically sourced). The nSSA proxy consisted of ammonium sulfate and a mixture of equal parts adipic and succinic acid. The organic mass fraction (∼ 0.6) and organic O:C ratio (0.8) were chosen to represent campaign averages from ATom. We performed separate calibrations for positive and negative spectra. For bromine and iodine, ammonium bromide and ammonium iodide were used. Positive and negative calibration spectra are similar to typical nSSA mass spectra from ATom (Fig. A1).
Trace amounts of bromine or iodine (mass fraction of 0.00055–0.10) were calibrated to a single peak in a single particle's positive or negative mass spectrum (Fig. A1). The area under 79 (A79) was chosen for bromine, and the area under 127 (A127) was chosen for iodine. Both A79 and A127 were modified to exclude spectra with confounding, non-halogen A79 or A127. For example, modified A79 (MA79) excluded spectra where A81 was less than 2× A79, which is consistent with bromine isotopic ratios. Particles that contain methanesulfonic acid (MSA) have large A95 and A97 peaks but will also have large a large A79 peak from MSA fragmentation; thus MA79 excludes negative and positive spectra where A79 was less than 5× A95 and 5× A97 to omit false positives from MSA. MA79 also excludes negative spectra where A79 is less than 5× A63, which excludes particles that contain phosphorous (PO). Finally, MA79 excludes spectra where A79 is less than 5× A77 + A78, which excludes spectra that contain a large number of organic peaks out to 79. Modified A127 excluded positive and negative spectra where A129 was larger than A127. Like MA79, this filtered out rare spectra with a large number of organic peaks out to 127.
A calibration curve was made by taking the average of MA79 or MA127 at a given mass fraction and fitting the natural log of those averages to a fourth-order polynomial (Fig. A2). For both MA79 and MA127, a separate curve was made for positive and negative spectra. We did not extrapolate below the lowest values in each calibration curve; instead those values were automatically set to 0. We did, however, add an artificial point where the bromine/iodine mass fraction equals 1 (MA79 or MA127 = 1) to ensure that the polynomial fit behaves well at higher mass fractions. The highest mass fractions in the bromine and iodine calibrations were 0.077 and 0.044, respectively. During ATom, 99.98 % of nSSA contained < 0.077 bromine by mass, and 99.97 % of nSSA contained < 0.044 by iodine by mass. Furthermore, the interquartile range of nSSA mass fractions during ATom was 0.0012–0.0049 for bromine and 0.0011–0.0036 for iodine; thus, the vast majority of the observations are within the calibration range.
For a given mass fraction of bromine or iodine, a higher ion signal from a particle corresponds to the higher probabilities that enough bromine or iodine ions arrive at the microchannel plate (MCP) to compete with instrumental noise. Thus, we used two calibration curves each for both positive and negative spectra: one curve was made from calibration mass spectra with a high total ion signal (high MCP output, units: Coulomb (C)) and one for a low total ion signal (low MCP output, Fig. A2). The low/high MCP output cutoffs for positive and negative spectra are C and C, respectively. These cutoffs were chosen from histograms of single-particle nSSA MCP outputs during all four ATom instances.
In order to couple PALMS data to an independently measured optical particle counter (Froyd et al., 2019), we took a 3 min average of MA79 or MA127 prior to applying the calibration. MCP output for each particle was averaged in that 3 min period to determine if the low or high calibration curve was used. The calibration-normalized root mean square error (RMSE) shrinks with higher bromine or iodine mass fractions and/or as more particles are collected (Fig. S3). Fortunately, it takes as few as 5–10 single particles for the normalized RMSE to plateau; thus, having > 10 particles in a 3 min period only marginally reduces the error of these measurements. Both positive and negative mass spectra were used for bromine and iodine mass calculations. If a given 3 min period has both positive and negative mass spectra, a time-weighted average was calculated.
Despite the similarity between the calibration spectra and typical bromine- and iodine-containing nSSA mass spectra, bromide and iodide are not the only forms of nSSA bromine and iodine. For iodine aerosol, the main species are iodide, iodate, and organo-iodine (Saiz-Lopez et al., 2012), but their ratios are not universal. While we did not use a proxy for organo-iodines, similar calibrations were done with ammonium iodate and yielded the same relationships as those shown in Fig. A2. The majority of nSSA bromine is expected to be in soluble bromide form (Sander et al., 2003). Nonetheless, the nSSA bromine and iodine measured in this study were often found at latitudes, longitudes, and altitudes where detailed nSSA halogen speciation has not been determined. Because we do not know the speciation of the ATom nSSA bromine and iodine, we also do not know their ionization efficiencies relative to our laboratory-generated nSSA with bromide and iodide. Thus, the measurements reported here are susceptible to systematic errors. A factor of 2 would be a generous estimate of these systematic errors. There are several lines of evidence for this: (1) bromide and iodide + iodate likely make up more than 50 % of the nSSA bromine and iodine that we measured during ATom (Gómez Martín et al., 2022), (2) the nSSA bromine and iodine in the ATom and calibration mass spectra were predominately attributable to only the atomic bromine and atomic iodine peaks, and (3) a comparison of PALMS nSSA iodine mass to aerosol mass spectrometer (AMS) iodine mass measurements during ATom-1 and ATom-2 from Koenig et al. (2020) show a median bias of only +8 % for PALMS. The 25th and 75th percentile biases are within a factor of 2 from the median bias. The AMS and PALMS measurements were co-located, the AMS transmission efficiency curve was applied to the PALMS data, and the data were binned by latitude (2° bins) and altitude (0.5 km bins). The AMS measurements were calibrated to three different iodine species: iodide, iodate, and an organo-iodine proxy (Koenig et al., 2020).
The total ion signal dependence was built into the bromine and iodine mass calibrations; thus, a significant correlation between MA79 or MA127 and MCP output does not exist (Fig. A4). However, when calculating number fractions (i.e., the number of nSSA in an air mass with MA79 or MA127 > 0), a positive relationship between the bromine or iodine number fraction and MCP output does exist. Thus, our calculated number fractions are likely a lower limit to the true number fractions of bromine- or iodine-containing nSSA. Considering this lower limit, we only used negative mass spectra for bromine number fraction calculations (i.e., MA79 > 0); likewise, we only used positive mass spectra for iodine number fraction calculations (i.e., MA127 > 0). In these two scenarios, using only the most sensitive mass spectrum polarity calculates our highest estimates of bromine- or iodine-containing nSSA without gratuitous filtering.
2.3 Regional and altitude boundary definitions
To separate vertical profiles into tropical, mid-latitude, and polar regions, we used the same latitude cuts found in Table S1 in Schill et al. (2020). Briefly, the latitude cuts were based on changes in the zonal winds as well as concentration gradients of gas-phase and aerosol products. We separated the UT from the LS using a combination of altitude, O3, and CO concentrations. Specifically, we defined the stratosphere as air that is above 8 km and has [O3] [CO] > 3. Finally, the identification of the top of the marine boundary layer (MBL) is described in Brock et al. (2021). Briefly, the top of the MBL is defined by an abrupt change in temperature; dew point; wind speed/direction; and gas-phase tracers such as O3, NO2, and CO. Each MBL top was inspected by hand as oftentimes the location of the MBL top was ambiguous, especially in regions where the MBL was not uniformly mixed.
2.4 Auxiliary gas-phase measurements
In this work, we used O3, HCN, CH3CN, and H2O gas-phase measurements to support the PALMS aerosol measurements. O3 was used as a proxy for the depth into the stratosphere. During ATom, O3 was measured with the NOAA NOyO3 chemiluminescence instrument (Bourgeois et al., 2020). During CR-AVE, O3 was measured using the NOAA O3 Photometer (Gao et al., 2008).
HCN and CH3CN were used as a qualitative measure for both biomass burning influence and the age of biomass-burning-influenced air. In situ HCN and CH3CN were measured from the NASA DC-8 using the National Science Foundation (NSF) National Center for Atmospheric Research (NCAR) Trace Organic Gas Analyzer (TOGA) during ATom-1 to ATom-4. TOGA is an online fast gas chromatograph with a quadrupole mass spectrometer (GC/MS) that sampled and analyzed for 35 s every 2 min during ATom flights (Apel et al., 2015). TOGA was calibrated in flight for CH3CN using several different and volatile organic compound (VOC) mixtures at the level of parts per billion (ppb-level) or million (ppm-level) including CH3CN that is dynamically diluted with zero air from an integrated clean-air generator (CAG) to ambient mixing ratio levels. TOGA was calibrated for both HCN and CH3CN before and after each ATom deployment in the laboratory using ppb- and ppm-level standard mixes diluted to ambient mixing ratios using the CAG. HCN and CH3CN during DC3 and SEAC4RS were measured with the University of Innsbruck proton-transfer-reaction mass spectrometer (PTR-MS; Wisthaler et al., 2002).
Water vapor was measured using the NASA Diode Laser Hygrometer (DLH) instrument (Diskin et al., 2002). DLH measures water vapor mixing ratio along an external path using near-infrared diode laser absorption, providing a fast, high-precision measurement relatively free of sampling artifacts. Relative humidities with respect to liquid water and ice are calculated from the mixing ratio using static temperature and pressure data. Water vapor was used to qualitatively assess the potential effects of RHwater (relative humidity) on the aerosol phase, water available in the gas phase, and the age of air (see Sect. 3.1).
The CH3I measurements aboard the NASA DC-8 during ATom were measured with the University of California, Irvine, Whole Air Sample (WAS; Colman et al., 2001). The WAS CH3I measurements were used to validate the GEOS-Chem (Goddard Earth Observing System) CH3I measurements, as a rough estimation of the model's skill in reproducing reactive iodine precursors.
2.5 Back trajectory calculations
Back trajectories were used to estimate the number of days since an air mass sampled during ATom had encountered a fire (Schill et al., 2020). The data product “days since most recent fire influence” was an average of 245 back trajectories. The cluster of 245 back trajectories was initialized every minute along the ATom flight tracks and was projected back 30 d using the TRAJ3D model (Bowman, 1993; Bowman and Carrie, 2002). The model used the National Center for Environmental Prediction's meteorological fields at high resolution (0.5° × 0.5°).
Fire locations were taken from the MODIS fire radiative power (FRP) (Collection 6); Visible Infrared Imaging Radiometer Suite (VIIRS) 375 m FRP; and Global Fire Emissions Database, Version 2 (GFEDv2). Fire plume injection heights were estimated from MODIS and VIIRS, based on FRP (Sofiev et al., 2012). GFED2, which does not report FRP, was assumed to inject smoke into a well-mixed boundary layer. A back trajectory was considered to “cross a fire” if it coincided with the latitude and longitude of a MODIS, VIIRS, or GFED2 fire and was also below the calculated or assumed fire height.
3.1 Geographic and vertical distribution of non-sea-salt aerosol bromine and iodine
Bromine and iodine were commonly found in nSSA during ATom, regardless of season (Figs. 1 and 2). The median number fraction (and interquartile range, IQR) of nSSA containing bromine and iodine was 18 % (8 %–26 %) and 25 % (12 %–44 %), respectively. The trace amounts of bromine and iodine means that they can easily fall below the detection limit (mass fraction of < 0.00055) when the total numbers of ions from a particle is small because, for example, a particle was near the edge of the ionizing laser beam. While this was accounted for in the iodine and bromine mass calibrations, it causes a low bias in their number fractions. If we only use those particles whose MCP output was in the top 50th percentile, the number fraction IQR shifts from 8 %–26 % to 18 %–45 % for bromine and from 12 %–44 % to 33 %–72 % for iodine. Such high number fractions, especially in the free troposphere, are indicative of a pervasive secondary source of nSSA bromine and iodine, whereby gas-phase bromine and iodine condense onto particles.
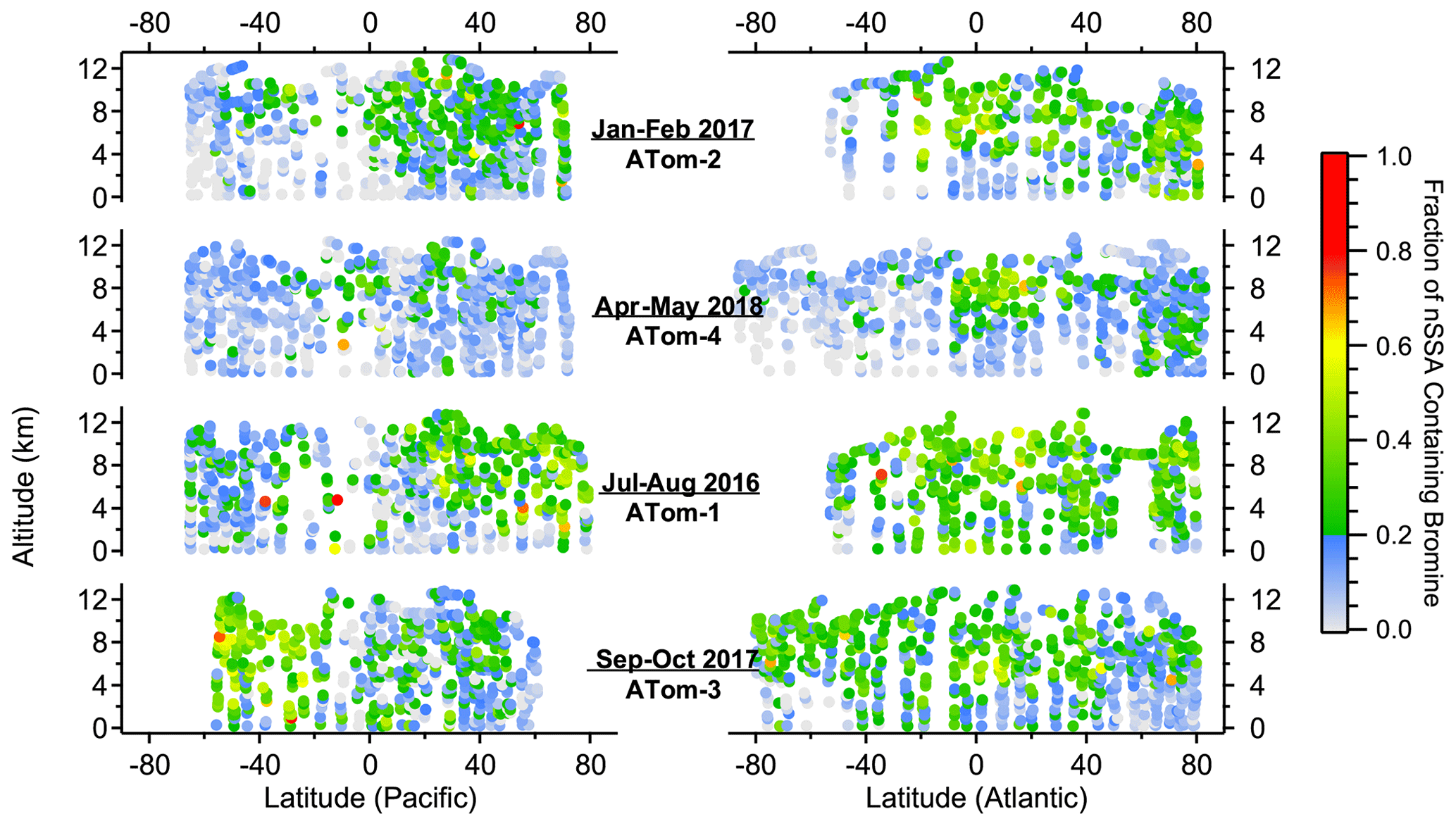
Figure 1Altitude–latitude curtain plots of the number fraction of nSSA that contains bromine (i.e., MA79 > 0) from ATom-1 to ATom-4. The color bar was chosen to visually delineate when more than 20 % of nSSA in an air mass contains bromine. Trace amounts of bromine in particles can lead to undercounting in particles with a low total ion signal (see Sect. 3.1); thus, the number fractions presented in this figure are considered to be lower limits.
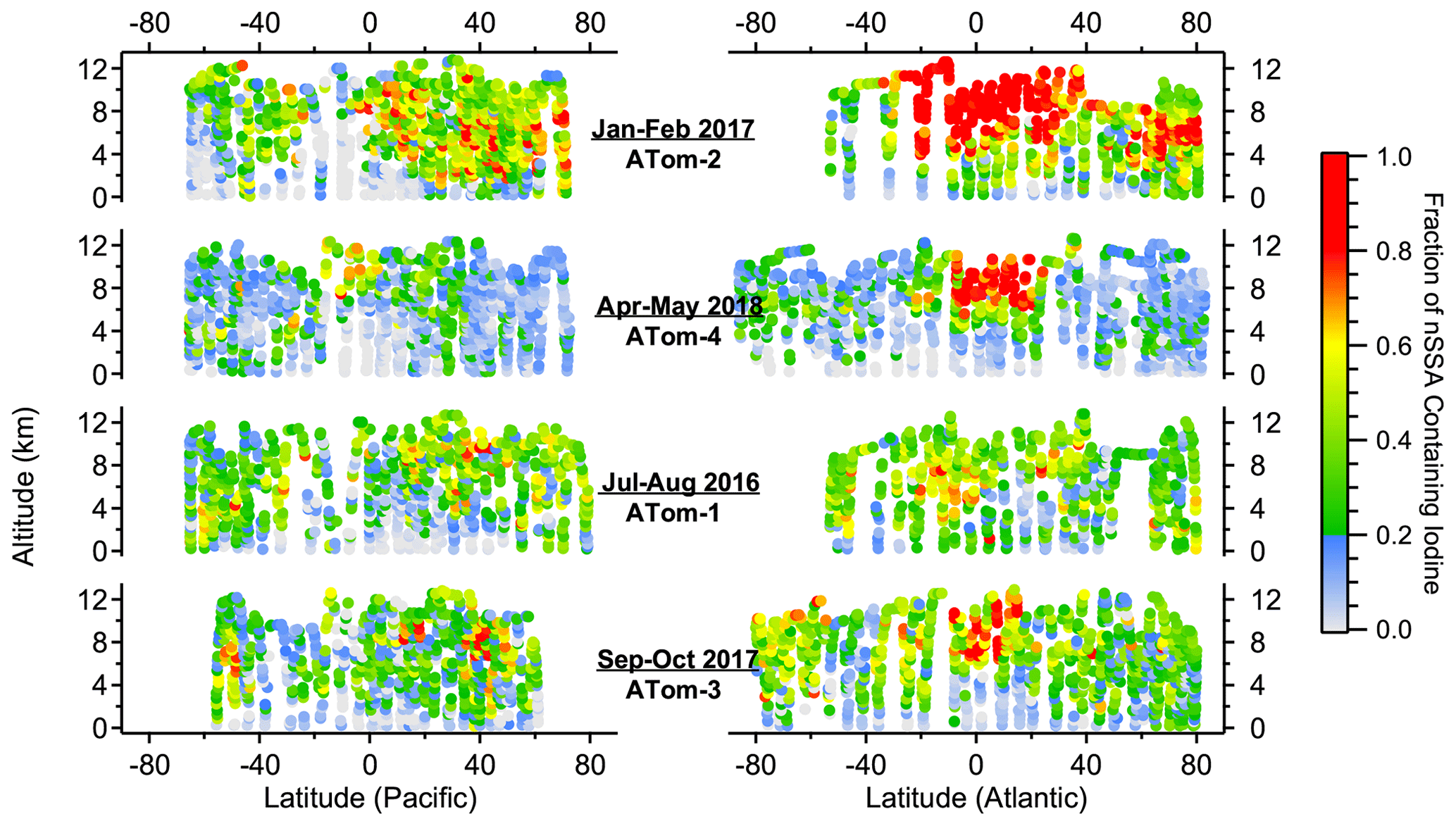
Figure 2Altitude–latitude curtain plots of the number fraction of nSSA that contains iodine (i.e., MA127 > 0) from ATom-1 to ATom-4. The color bar was chosen to visually delineate when more than 20 % of nSSA in an air mass contains iodine. Trace amounts of iodine in particles can lead to undercounting in particles with a low total ion signal (see Sect. 3.1); thus, the number fractions presented in this figure are considered to be lower limits.
The pervasive nature of nSSA iodine and bromine is highlighted by a general lack of geographic and vertical trends. For example, the north–south, Atlantic–Pacific, and vertical gradients of nSSA bromine and iodine during ATom were weak, unlike many primary and secondary aerosol species. There were, however, some geographical highlights. The tropical Atlantic UT, which was typically deficient in primary aerosol, was especially rich in halogen-containing nSSA. For example, during ATom-2, the number fraction of nSSA containing iodine in the tropical Atlantic UT was generally over 80 %. In contrast, nSSA in the remote MBL and remote tropical Pacific were typically deficient in nSSA bromine and iodine. The MBL and remote tropical Pacific also contained the lowest O3 concentrations measured during ATom (Bourgeois et al., 2020). We found that the number fraction of nSSA that contains bromine and/or iodine monotonically increased with increasing tropospheric O3 up to 50–60 ppb, where it levels out (Figs. A5 and A6). Thus, low O3 may be an important condition for time periods when the conversion of reactive bromine and iodine to their aerosol-bound forms is inhibited or if aerosol-bound bromine and iodine titrate O3 through heterogeneous reactions that liberate these halogens from the aerosol phase; however, it is difficult to draw a quantitative conclusion from these correlations because number fraction and picomole per mole (pmol mol−1) are inherently different units that scale differently with pressure.
In addition to O3, we also found that the number fraction of nSSA that contains bromine and iodine correlated monotonically with both temperature and RHwater in tropospheric air (Figs. A5 and A6). We believe these correlations were the result of other processes. The correlation with temperature was weak (Spearman's rho (ρ): −0.42 to −0.47) and became insignificant below 273 K. The correlation with RHwater was stronger (Spearman's rho (ρ): −0.48 to −0.78) and only got stronger at RHwater<75 %. The driest air sampled during ATom also corresponds to air that has been descending; thus, drier air contains more aged aerosol, allowing for more time for particles to gain a detectable amount of secondary iodine or bromine.
Although bromine and iodine were ubiquitous in nSSA, their mass concentrations were small relative to major aerosol constituents like organics and sulfate (Figs. 3 and 4). Median concentrations of bromine and iodine in nSSA were 0.7 (IQR: 0.3–1.5) and 0.4 (IQR: 0.16–1.0) ng sm−3, respectively. This corresponds to 0.25 (0.11–0.57) pmol mol−1 and 0.10 (0.04–0.24) pmol mol−1, respectively. We convert between picomole per mole (pmol mol−1) and nanogram per standard cubic meter (ng sm−3) by assuming that the atmosphere is an ideal gas and that the MW of bromine and iodine are 79.9 and 126.9 g mol−1, respectively. These IQRs are lower than the range of total iodine concentrations that Gómez Martín et al. (2021) calculated for the remote MBL cruises TransBrom and AMT21 (Atlantic Meridional Transect) (0.57–1.05 pmol mol−1), but it should be noted that these cruise measurements are not able to separate nSSA from SSA. This bias will be exacerbated for total bromine, since the natural seawater concentration of bromine is higher than iodine by orders of magnitude and much more bromine will be found naturally in sea-salt aerosol. Thus, we will not attempt to make a comparison of bulk measurements to our bromine measurements here. For comparison, during ATom, the median concentrations of organic and sulfate aerosol measured by PALMS were ∼ 100 ng sm−3. Thus, while halogens were ubiquitous in nSSA during ATom, they were only ubiquitous at trace mass concentrations.
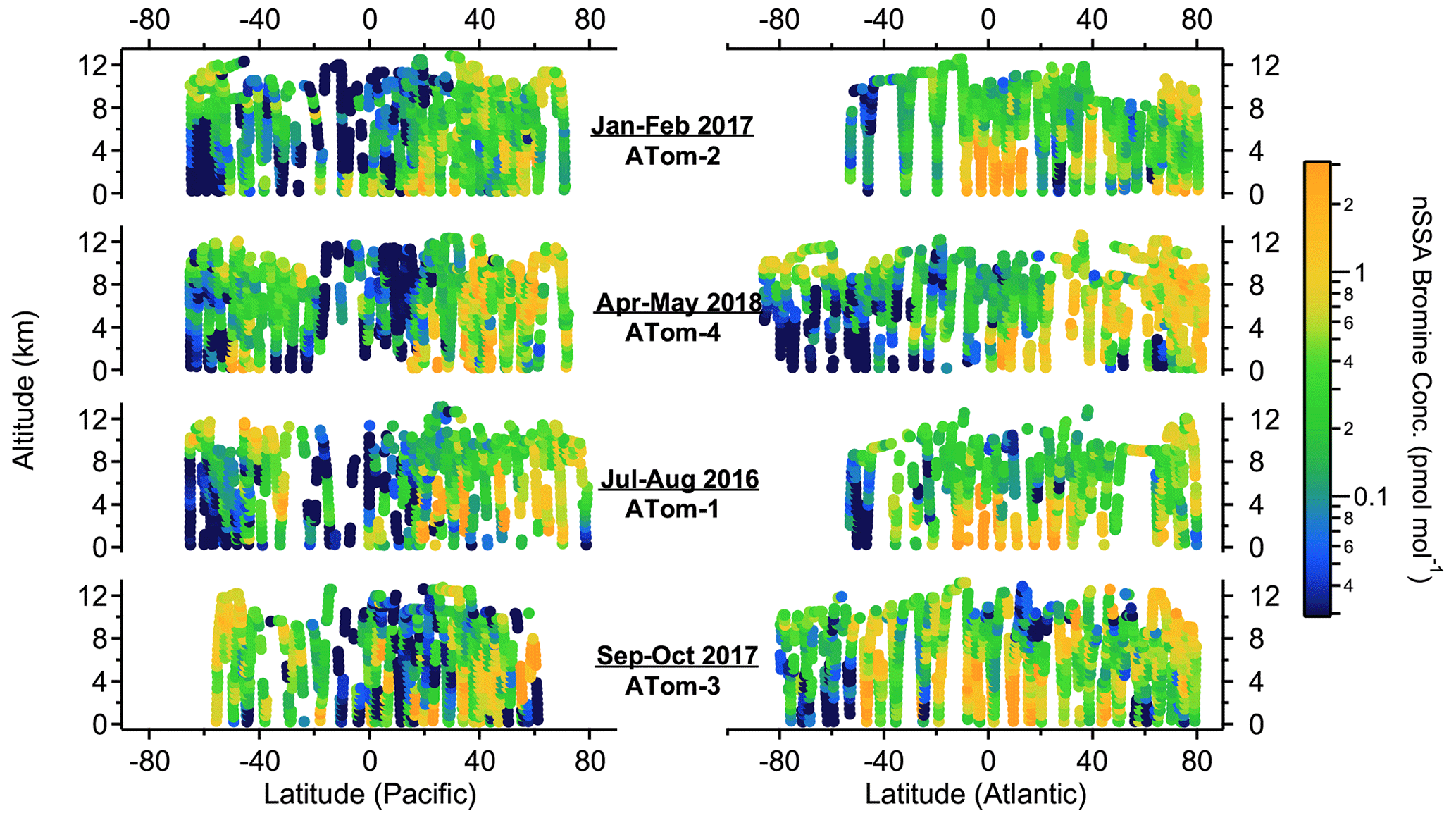
Figure 3Altitude–latitude curtain plots of nSSA bromine concentrations (pmol mol−1) from ATom-1 to ATom-4.
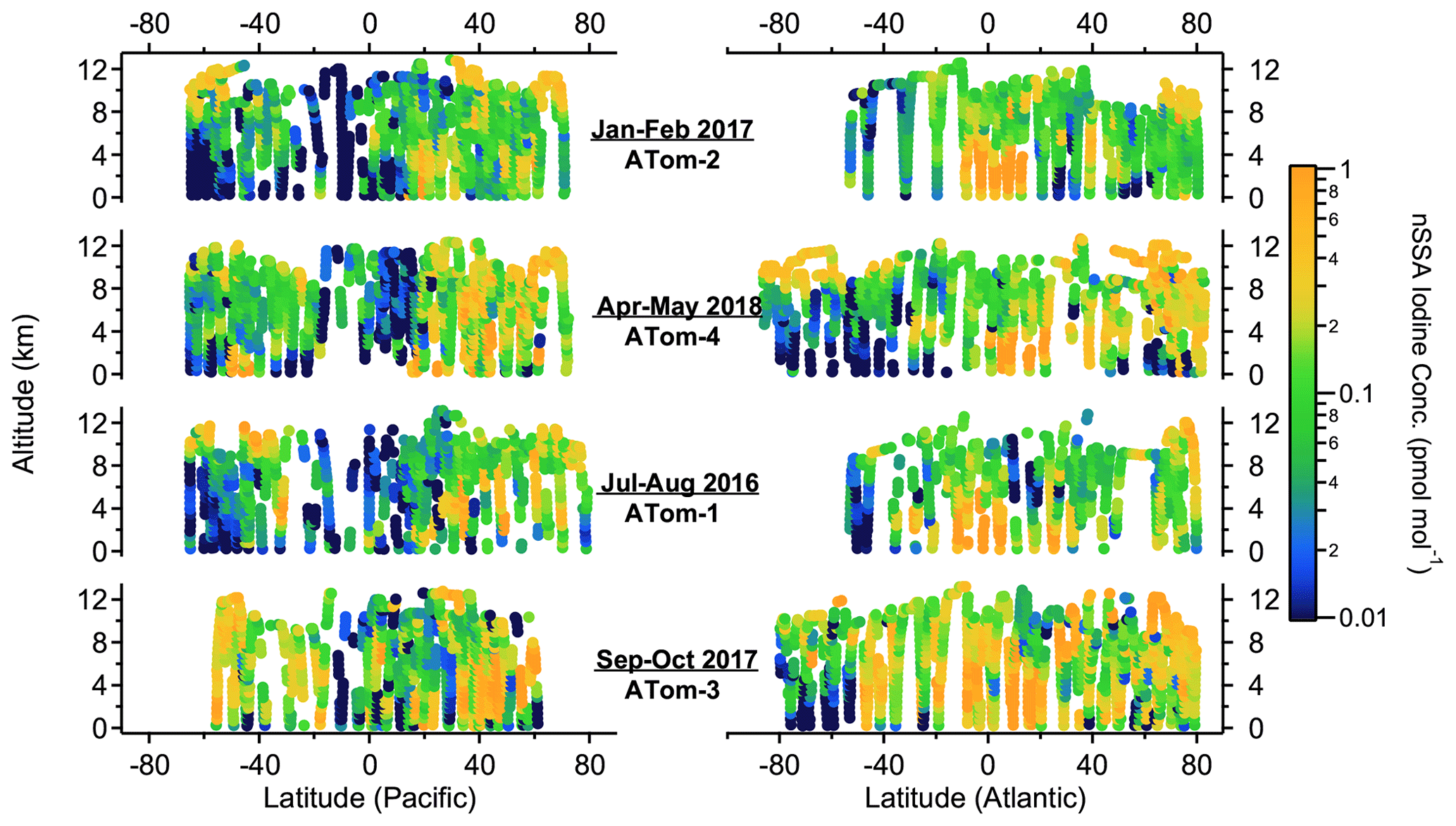
Figure 4Altitude–latitude curtain plots of nSSA iodine concentrations (pmol mol−1) from ATom-1 to ATom-4.
High nSSA iodine and bromine concentrations (> 1 pmol mol−1) were consistently found in the tropical Atlantic at altitudes below ∼ 6 km (Figs. 3 and 4). In these areas, we also encountered biomass burning plumes from the African continent, where biomass burning particles were often > 80 % of the accumulation-mode aerosol number and biomass burning aerosol mass concentrations were ∼ 1 µg sm−3 (Schill et al., 2020). In Sect. 3.2.1, we explore biomass burning as a source of primary nSSA bromine and iodine.
Elevated nSSA bromine and iodine concentrations (∼ 0.3–0.4 pmol mol−1) were also found in the lowermost stratosphere (defined as air masses where the altitude was > 8 km and the ratio of [O3] [CO] was > 3). Because of the ∼ 12–13 km ceiling of the DC-8, the lowermost stratosphere was sampled during ATom only at polar latitudes and in mid-latitude tropopause folds (Murphy et al., 2021). Elevated nSSA iodine concentrations in the lower stratosphere are consistent with the results from Koenig et al. (2020), who measured an increase in aerosol iodine concentrations when transitioning from the UT to the LS using an aerosol mass spectrometer. These elevated concentrations suggest that the stratosphere may be a source of nSSA bromine and iodine; however, we found that most of the nSSA bromine and iodine in the lowermost stratosphere are found on tropospherically sourced particles. We explore this further in Sect. 3.2.3.
Finally, the vertical profiles of bromine and iodine in nSSA were similar in both their shapes and magnitudes (Fig. 5). Similar vertical profile shapes suggest that there are similar sources and sinks of nSSA bromine and iodine. In many cases, both bromine and iodine were found in the same particle (Fig. A1). Of all the negative spectra sampled during ATom, 47 % of nSSA that contained bromine also contained detectable amounts of iodine; similarly, 59 % of nSSA that contained iodine also contained bromine. Similar bromine and iodine concentrations in nSSA suggest that a higher fraction of reactive iodine partitions into the aerosol phase than reactive bromine. Estimates of total reactive bromine (Bry) in the global troposphere are ∼ 3 pmol mol−1 (Sherwen et al., 2016b). Conversely, estimates from the same model suggest that total reactive iodine (Iy) in the free troposphere is only ∼ 0.5 pmol mol−1. Thus, in the global troposphere, nSSA bromine is only ∼ 8 % of the total reactive bromine concentration, whereas nSSA iodine makes up ∼ 20 % of the total reactive iodine concentration.
3.2 Sources of bromine and iodine in non-sea-salt aerosol
From both the curtain plots and vertical profiles of nSSA bromine and iodine number fractions and mass, we suggest that there are two sources of bromine and iodine for tropospheric nSSA: (1) a primary biomass burning source and (2) a pervasive secondary source from reactive uptake of gas-phase species. Additionally, we found that nSSA bromine and iodine concentrations increased in the lower stratosphere. Further classification of nSSA into stratospherically sourced and tropospherically sourced particles suggests that most of the stratospheric nSSA bromine and iodine were found on the tropospherically sourced particles and that organics may aid in binding bromine and iodine in nSSA. In the following sections, we will explore these sources in detail.
3.2.1 Biomass burning
One advantage of using single-particle mass spectrometry instead of a bulk measurement is that we can separate nSSA halogen contributions from different aerosol types (e.g., biomass burning vs. sulfate–organic–nitrate). While biomass burning and sulfate–organic–nitrate mixtures are similar in their bulk composition (i.e., they are both largely organics and sulfate by mass), biomass burning particles are distinct due to their potassium content (Schill et al., 2020). From the shape of each aerosol type's vertical profile, we can surmise the sources and sinks of nSSA iodine and bromine. For example, the concentration of nSSA iodine and bromine at low to middle altitudes was large in the tropical Atlantic. Separating nSSA iodine and bromine contributions from different particle types confirms that these high concentrations at 0–4 km in the tropical Atlantic were due to iodine and bromine in biomass burning aerosol (Figs. 6 and A7). Furthermore, as these biomass burning plumes were convectively lofted and underwent wet removal, biomass burning aerosol decreased in mass, as did the associated iodine and bromine concentrations.
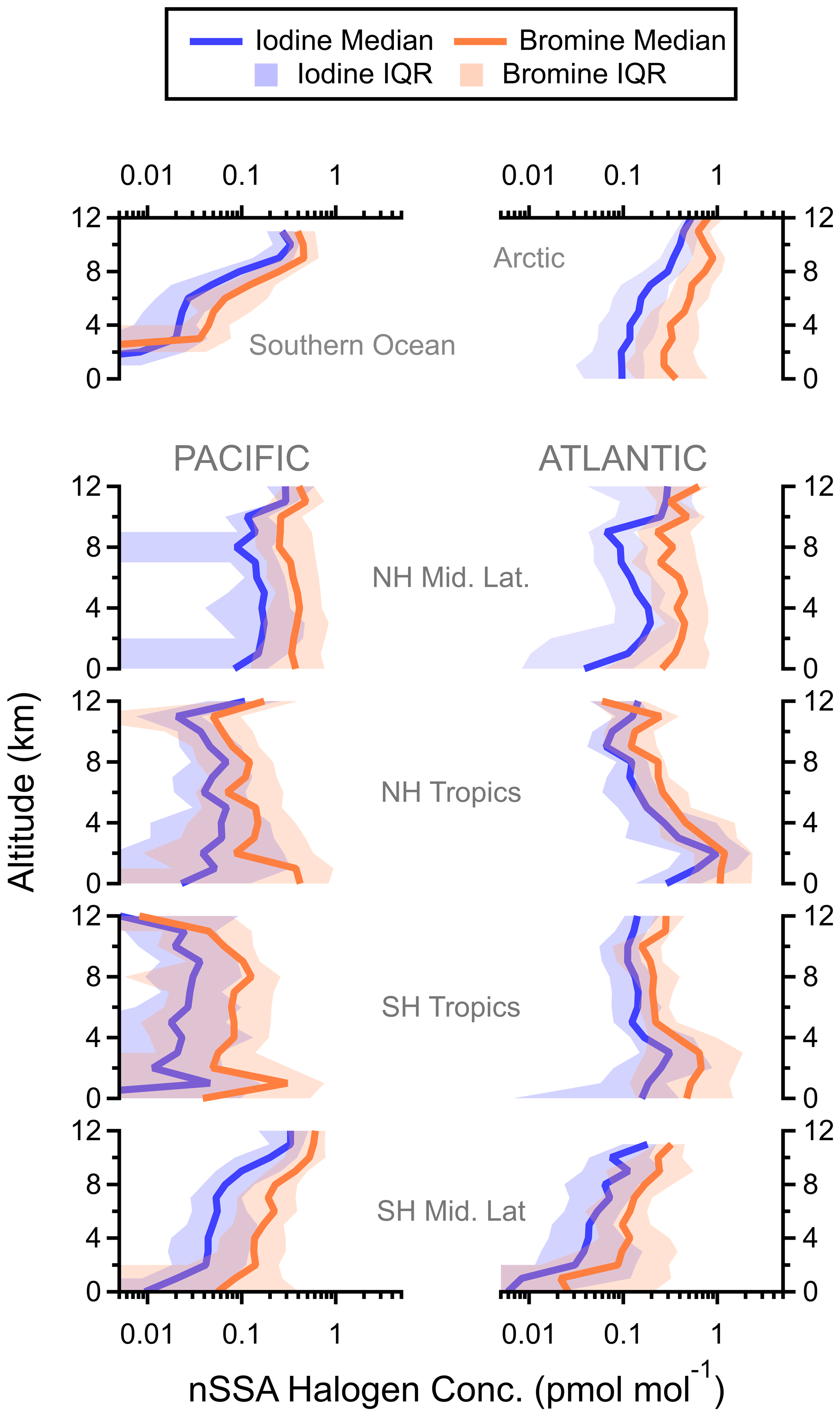
Figure 5Regional vertical profiles of nSSA halogen (pmol mol−1) from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020). NH: Northern Hemisphere, SH: Southern Hemisphere.
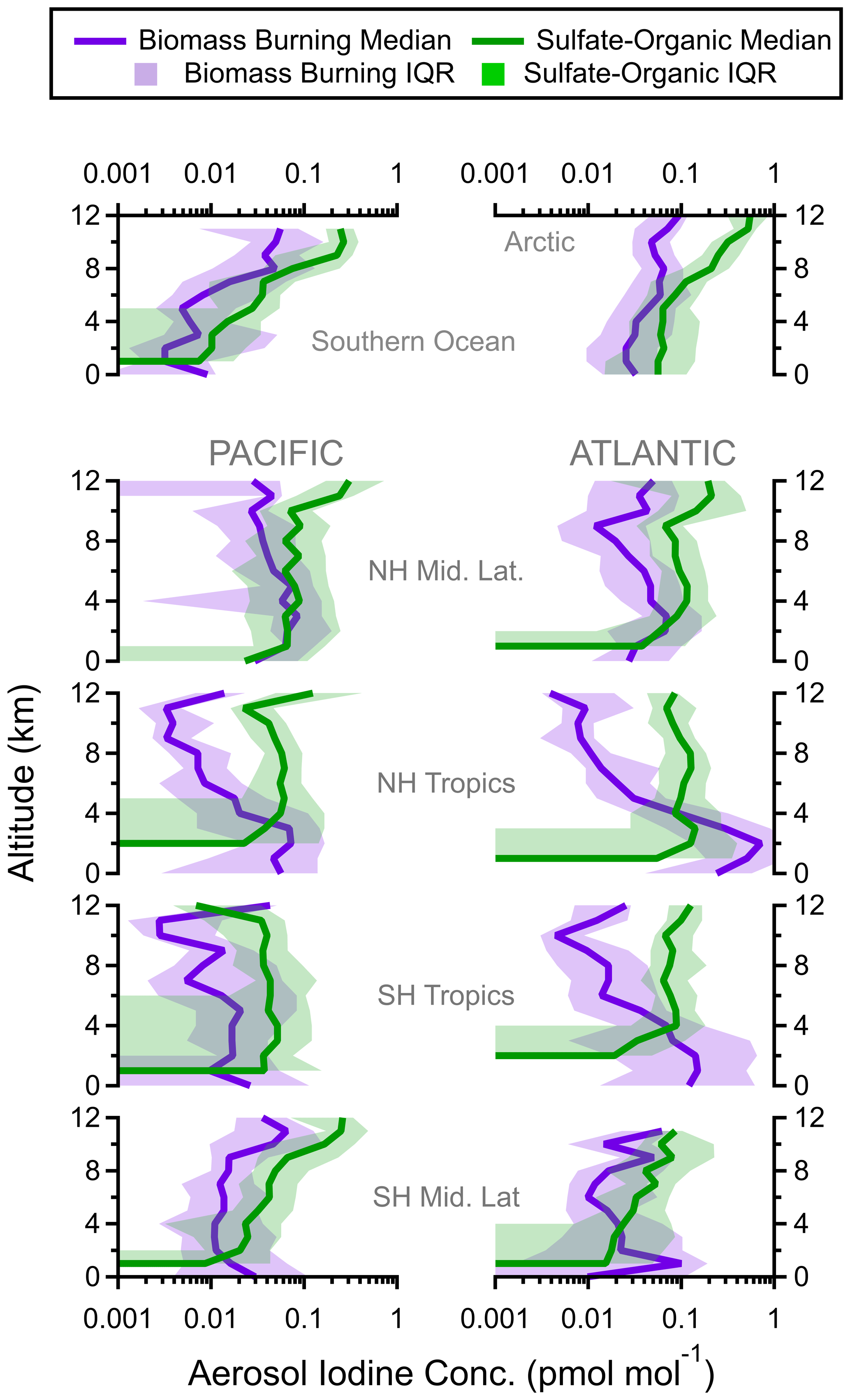
Figure 6Regional vertical profiles of iodine (pmol mol−1) in biomass burning and sulfate–organic–nitrate particles. Median values (solid lines) and interquartile ranges (shading) are from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020).
Although the biomass-burning-associated iodine and bromine aerosol mass was high in these plumes (global max of ∼ 1 pmol mol−1), the mass fraction of iodine and bromine in individual biomass burning particles was low, at ∼ 0.3 % (Fig. A8). Thus, fresh biomass burning particles have a small amount of iodine and bromine per particle, but high biomass burning aerosol mass concentrations in a plume amount to large absolute iodine and bromine concentrations.
Plotting nSSA iodine and bromine concentrations against several biomass burning tracers measured during ATom (Figs. 7 and A9) confirms a primary source of nSSA iodine and bromine from biomass burning. To extend these results to higher biomass burning tracer concentrations, we also include data from DC3 (Barth et al., 2015) and SEAC4RS (Toon et al., 2016). Several large biomass burning events were encountered in both campaigns; however, all of the data from each campaign were used regardless of tracer concentrations. We find that there was a near-monotonic increase in iodine and bromine in nSSA with increasing concentrations of both CH3CN and HCN (Fig. 7). From these plots, if we encountered at least 300 pptv of CH3CN (i.e., if we are in a biomass burning plume), then we generally found that median values of nSSA iodine and bromine were > 1 pmol mol−1; further, as gas-phase biomass burning tracers diluted and dissipated, so did nSSA iodine and bromine concentrations, corroborating the vertical profile analyses above.
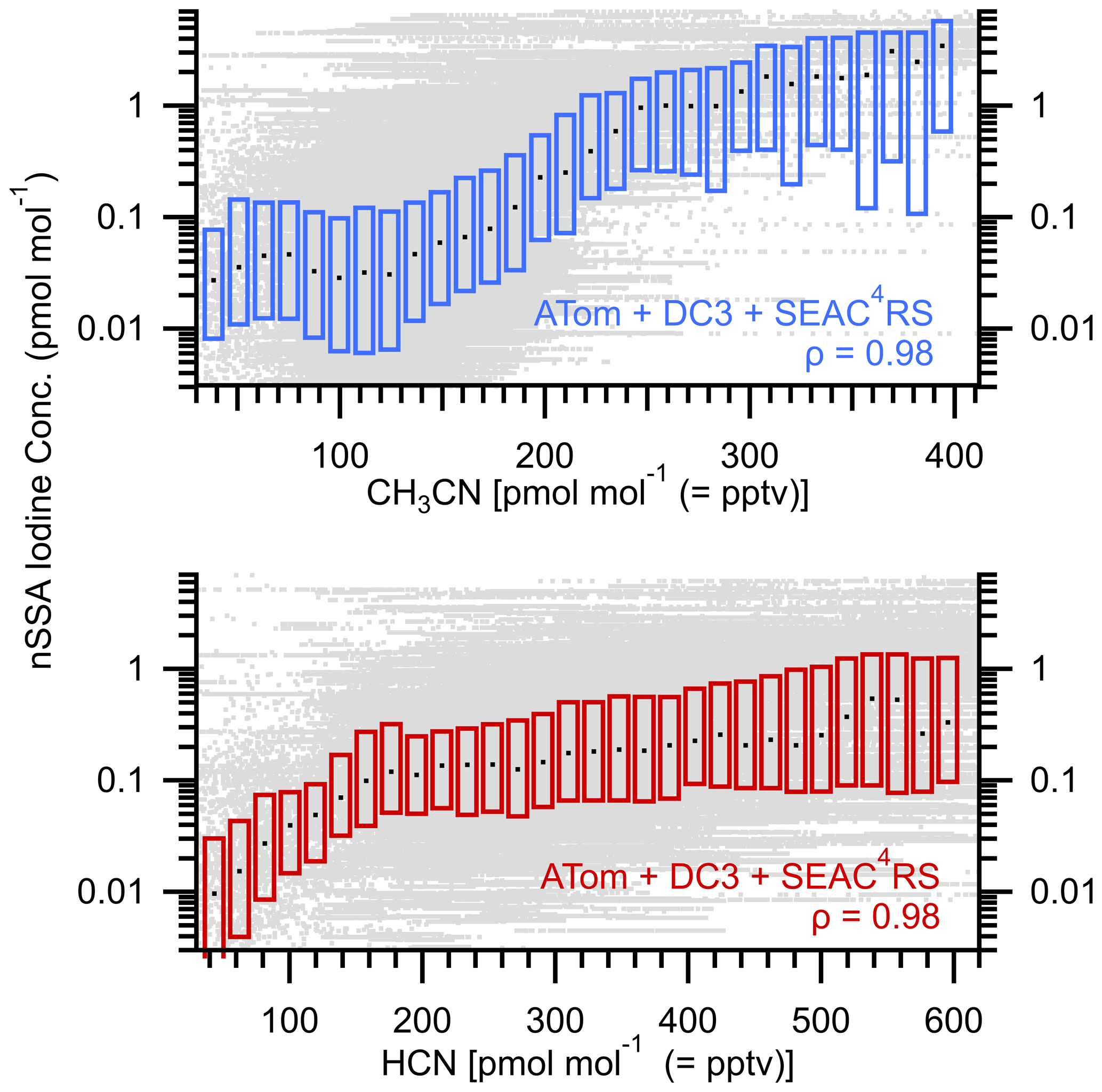
Figure 7Box plots of nSSA iodine (pmol mol−1) vs. gas-phase biomass burning tracers CH3CN (pptv) and HCN (pptv). Raw data are gray dots, median values are horizontal black dashes, and the interquartile range is bound by the colored boxes. Median value of Spearman's rho (ρ) for each is 0.98, which indicates a near-monotonic relationship and suggests that biomass burning is a primary source of nSSA iodine.
The above discussion could imply that nSSA bromine and iodine are always emitted from biomass burning, with little source variation; however, the amount of nSSA bromine and iodine emitted from biomass burning is likely dependent on both the fuel type and flaming conditions. This has been shown for methyl chloride and particulate chloride in laboratory burns of Douglas fir and duff beds (Reinhardt and Ward, 1995), where smoldering conditions favor methyl chloride emission and flaming conditions promote chlorine formation in fine particulate matter. Similar studies for nSSA bromine and iodine, to the authors' knowledge, do not exist. Furthermore, while there is guidance for fuel-type-specific emissions of methyl halons (Andreae, 2019), nSSA bromine and iodine emission factors specific to fuel types and fire condition are largely unknown. Finally, fires that affect aerosol populations in the remote atmosphere, which are sometimes days or weeks old according to back trajectory calculations, are likely a mixture of different fires that have different fuels and flaming conditions. This will cause a smearing effect and give us the general picture shown in Figs. 7 and A9.
3.2.2 Pervasive secondary source
Above the MBL, vertical profiles of iodine and bromine concentrations within sulfate–organic–nitrate particles were constant with altitude in the tropics and increased with altitude in the extratropical and polar regions (Figs. 6 and A7). Sulfate–organic–nitrate particles are a general class of particles that are generated from a variety of primary and secondary sources but are not from biomass burning or shipping-oil combustion, and they do not contain stratospherically sourced meteoric material. The shape of these vertical profiles were similar to vertical profiles of the average number of days since a back trajectory ensemble encountered the boundary layer (“days since boundary layer influence”), which suggests a pervasive secondary source of both iodine and bromine for nSSA in the free troposphere whose precursors are sourced from the surface. By plotting the mass fraction of iodine and bromine as an individual sulfate–organic–nitrate aerosol vs. days since boundary layer influence, we confirm that the iodine and bromine mass fractions monotonically increased in sulfate–organic–nitrate particles as their average time since last encountering boundary layer air increased (Figs. 8 and A10).
As discussed in Sect. 3.2.1, biomass burning aerosol were a primary source of nSSA iodine and bromine; however, despite a large absolute mass concentration of iodine and bromine, the freshest biomass burning particles had low halogen mass fractions (Fig. A8). As biomass burning plumes aged and diluted into the background atmosphere, they accumulated iodine and bromine and their iodine and bromine mass fractions increased (Figs. 8 and A10). Along with the shapes of the sulfate–organic–nitrate aerosol iodine and bromine vertical profiles, this suggests two non-exclusive lines of evidence for a pervasive, secondary source of nSSA iodine or bromine: (1) iodine and bromine partition to nSSA and accumulate as the particles age over long time periods, and (2) aged air masses, which have undergone more aerosol removal, have lower absolute nSSA concentrations and will accumulate more iodine and bromine per particle per time in a pervasive background of precursors.
Figures 8 and A10 show that particles accumulate iodine as they age, but we do not suggest that the halogens are inert in aerosol after uptake. For example, it is known that iodide in aerosol can react heterogeneously with O3 to form I2, which is then liberated from the particles. This was traditionally thought to deplete all iodide in aerosol particles (Vogt, 1999), but field measurements consistently show non-negligible iodide in fine-mode marine aerosol (Gómez Martín et al., 2022). Furthermore, iodate was traditionally thought to accumulate in particles, but several pathways to reduce aerosol iodate have been proposed (e.g., Pechtl et al., 2007). Finally, organically bound iodine in aerosol makes up a significant fraction of the fine-mode iodine aerosol (Gómez Martín et al., 2022), but its participation in heterogenous reactions that liberate iodine from aerosol is largely unexplored. Less is known about bromine liberation in nSSA. Unfortunately, in this study, PALMS did not determine what form the aerosol iodine or bromine is in. Thus, we are unable to quantitatively discuss the impact of heterogeneous recycling reactions in this work.
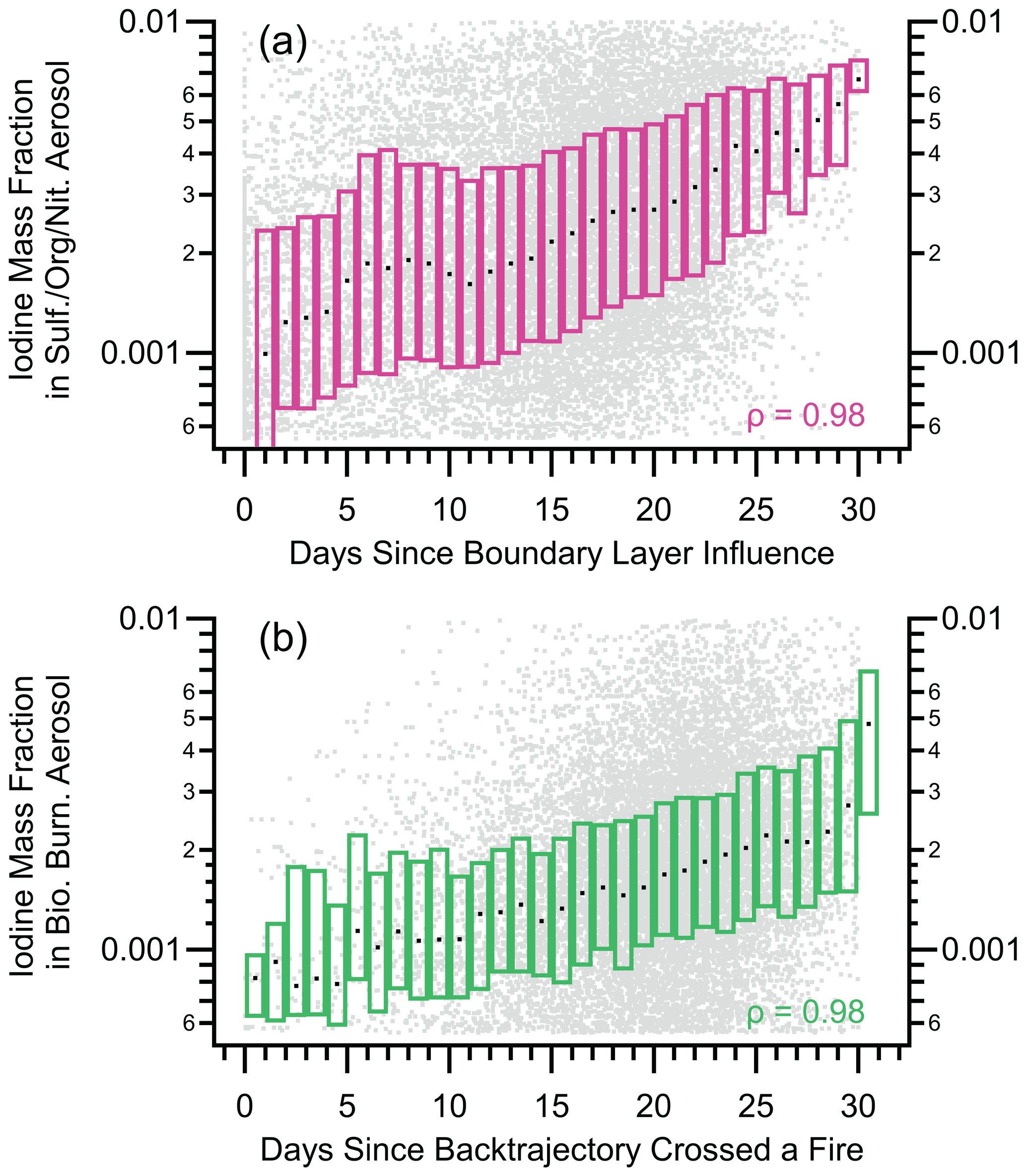
Figure 8Box plots of nSSA iodine mass fraction vs. two back trajectory products: (a) the average number of days since a back trajectory was in the boundary layer and (b) the number of days since a back trajectory crossed a fire. Raw data are gray dots, median values are horizontal black dashes, and the interquartile range is bound by the colored boxes. Median value of Spearman's rho (ρ) for each is 0.98, which indicates a near-monotonic relationship and suggests that biomass burning is a primary source of nSSA iodine mass.
In the MBL, iodine and bromine mass in sulfate–organic–nitrate particles was often below our limit of detection (mass fraction of < 0.00055). The remote MBL, however, is a known source of reactive iodine and bromine precursors (e.g., Carpenter et al., 2013). Previous measurements suggest that the highest concentrations of reactive iodine radicals are found in the MBL (Volkamer et al., 2015); furthermore, debromination from acidic SSA is known to occur in the MBL and may be one of the largest sources of reactive bromine (Sander et al., 2003). Thus, we expect high nSSA halogen concentrations in the MBL; however, from our measurements, it seems that the emission/formation of reactive-halogen precursors in the MBL is not the limiting factor to forming nSSA iodine and bromine in the MBL. One possibility is that low O3 or high water vapor concentrations are preventing the formation of iodine or bromine aerosol or its precursors, directly affecting their partitioning from the gas phase to the aerosol phase. Additionally, it is likely that dry particles are less efficient than wet particles at heterogeneously recycling aerosol iodine species (Sect. 3.1). Another possibility is that wet removal in the humid/wet MBL is faster than the slow accumulation of secondary bromine and iodine.
3.2.3 Stratosphere
During ATom, the DC-8 sampled lower-stratospheric air, but this was limited to the high-latitude flights due to the DC-8's ∼ 12 km ceiling (Murphy et al., 2021). In these flights, at altitudes greater than 8 km, we found that iodine and bromine concentrations in nSSA monotonically increased with O3 concentrations until it plateaued near 0.3–0.5 pmol mol−1 (Figs. 9 and A11). If we use O3 concentration at altitudes greater than 8 km as a proxy for penetration depth into the lower stratosphere, we see that nSSA iodine and bromine increased from the UT to the LS. This is similar to the results found in Koenig et al. (2020), who showed an increase in nSSA aerosol iodine in the lower stratosphere was concomitant with a decrease in iodine oxide radical (IO) concentrations. The IO measurements by Koenig et al. (2020), however, were taken primarily in the tropical UT/LS. Thus, in addition to the high-latitude UT/LS data from ATom, we also include data from the CR-AVE mission, which sampled the UT/LS in the tropics. Above ∼ 8 km in altitude and ∼ 25 ppbv O3, nSSA iodine and bromine concentrations increased from the UT to the LS in both polar (ATom) and tropical (CR-AVE) regions. The stratosphere is an interesting foil for the MBL in that it is generally dry, and this may be further evidence that RH is important for aerosol iodine retention. This indicates that the enhanced partitioning of halogens to aerosol in the stratosphere may be a global phenomenon, but further measurements at other locations in the UT/LS are needed.
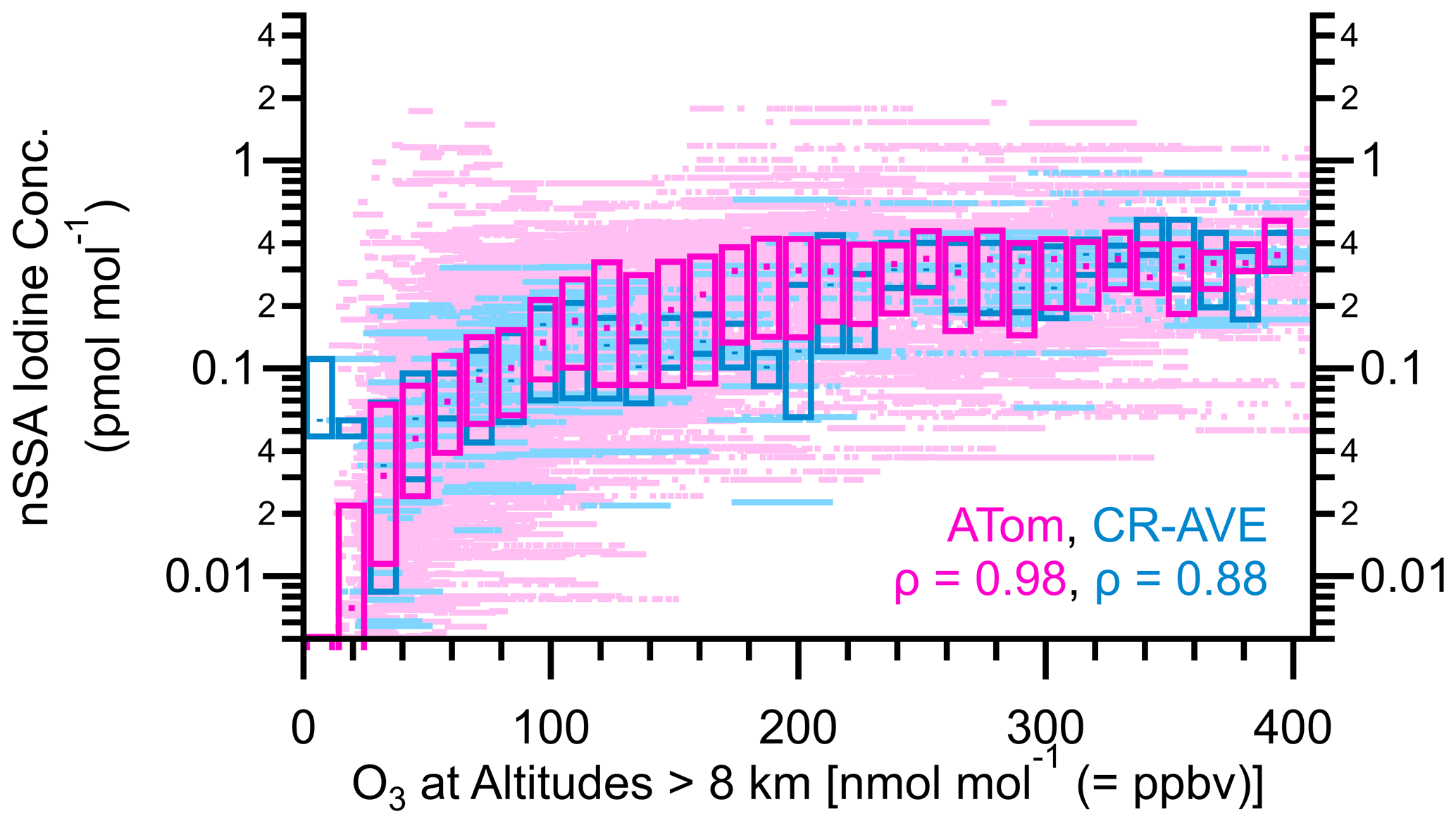
Figure 9Box plots of nSSA iodine (pmol mol−1) vs. O3 for ATom (yellow) and CR-AVE (magenta). The median values of Spearman's rho (ρ) for ATom and CR-AVE are 0.98 and 0.88, respectively, which indicates a near-monotonic relationship and suggests that the stratosphere may be a source of nSSA iodine globally.
Figures 9 and A11 suggest that there is a stratospheric source of nSSA halogens; however, iodine and bromine were not evenly distributed among tropospheric and stratospheric particle types, which are easily differentiated by PALMS (Murphy et al., 2021). Calculating nSSA iodine and bromine mass fractions for different PALMS particle types reveals that most of the nSSA iodine and bromine in the stratosphere resided on tropospherically sourced particles (Figs. 10 and A12). For example, stratospheric acidic sulfate particles with trace meteoric compounds (meteoric particles) sourced from the upper stratosphere contained ∼ 0.1 % iodine and ∼ 0.5 % bromine, while the tropospheric particles (biomass burning and tropospheric sulfate–organic–nitrate) contained 3–10× as much iodine and 1.5–2.5× as much bromine. In terms of absolute mass, there were more sulfate–organic–nitrate particles than meteoric particles in the LS (Fig. A13), so the lower mass fractions were not due to the same amount of secondary iodine and bromine being spread out over more meteoric particles. Thus, while nSSA bromine and iodine increase in the LS due to a more efficient conversion of gas-phase reactive bromine and iodine to their aerosol-bound products (e.g., Koenig et al., 2020), these products preferentially form on the tropospherically sourced particles.
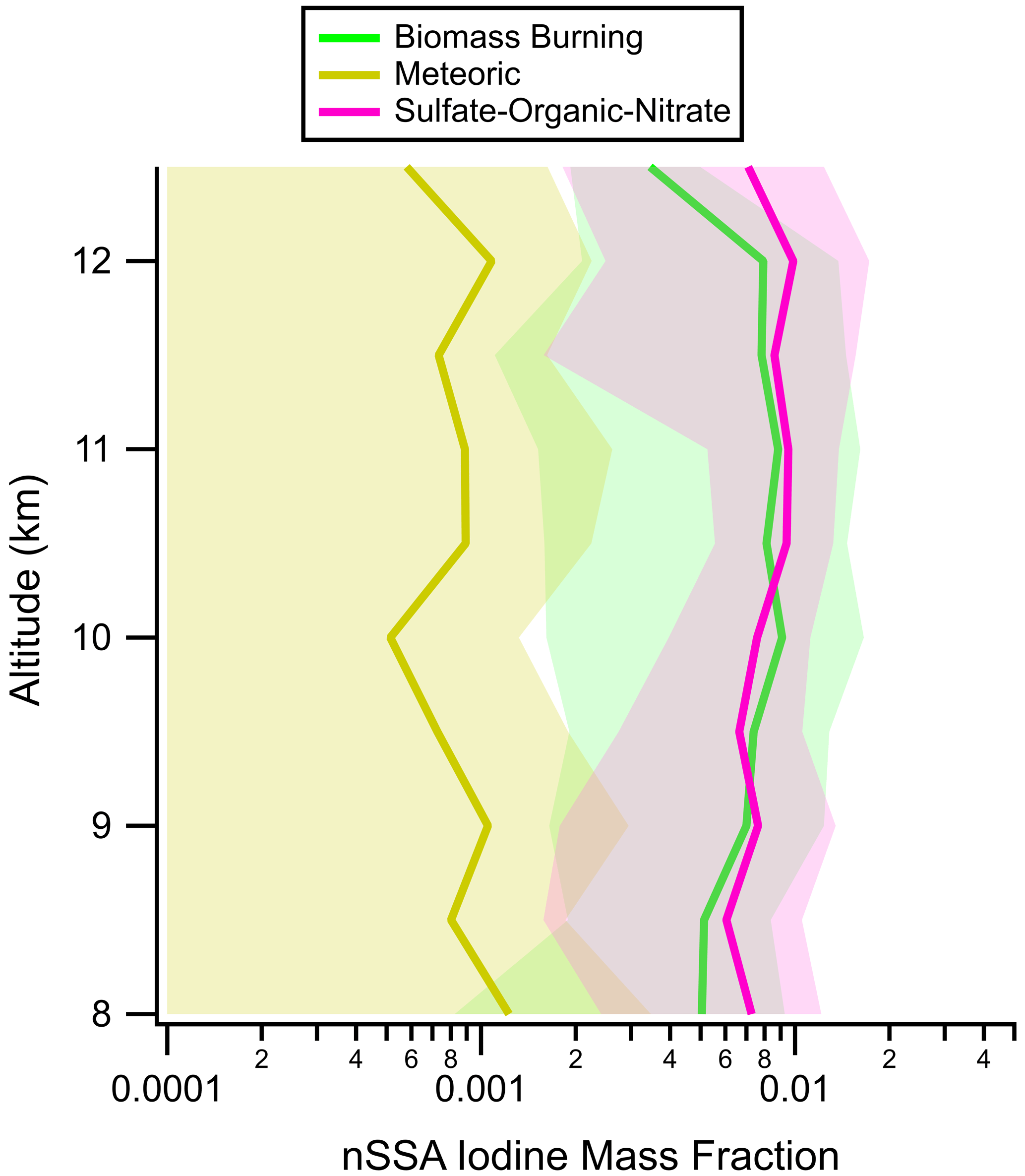
Figure 10Vertical profiles of nSSA iodine mass fractions in the most common particle types found in the lower stratosphere during ATom: tropospherically sourced biomass burning and sulfate–organic–nitrate particles and stratospherically sourced meteoric-sulfuric-acid particles. Average values (solid lines) and standard deviations (shading) are from all four ATom instances. In this work, the lower stratosphere is defined as air masses above 8 km where the [O3] [CO] ratio is greater than 3.
Tropospherically sourced particles generally contain much more organic material than stratospherically sourced particles (Murphy et al., 2021), and previous PALMS studies have shown that mass spectra with higher iodine peaks were found in particles with a higher organic content (Murphy et al., 1997). Furthermore, a comprehensive data set of field observations of iodine speciation in marine aerosol has shown that the soluble iodine content of fine aerosol (PM1) is approximately 50 % soluble organic iodine by mass (Gómez Martín et al., 2022), with one study at the Mace Head Atmospheric Research Station showing that over 90 % of soluble aerosol iodine is organically bound, with the rest being iodide or iodate (Gilfedder et al., 2008). Additionally, HCl has been shown to be soluble in stratospheric wildfire particles with significant organic mass fractions, presumably organic acids (Solomon et al., 2023); HI may be similarly dissolved in these organics.
To explore the idea of organics retaining iodine in aerosol, we plotted the iodine mass fraction against organic mass fractions for four different PALMS particle types in the lower stratosphere – two stratospherically sourced particles, meteoric and sulfuric acid, and two tropospherically sourced particles, biomass burning and sulfate–organic–nitrate (Fig. 11). As shown, higher mass fractions of iodine were found in particles with higher organic content. It should be noted that the organic mass fraction may be a proxy for or correlate to other physico-chemical properties (e.g., particle neutrality) and that iodine preferably partitions into these organic-rich particles because of these proxied properties. One such proxy may be metal content, as meteoric particles are known to have smaller organic mass fractions (Murphy et al., 2021) and some meteoric metals like Fe can convert aerosol-bound iodine back to I2 (Fudge and Sykes, 1952). The proxied properties may even be secondary effects; for example, more neutralized particles may affect organic acid gas–particle partitioning, which then may affect HI solubility in stratospheric particles. The importance of organics, either directly through organic binding/HI dissolution or as a proxy, likely extends into the troposphere, but it is difficult to unearth this relationship because of the two competing primary and secondary sources.
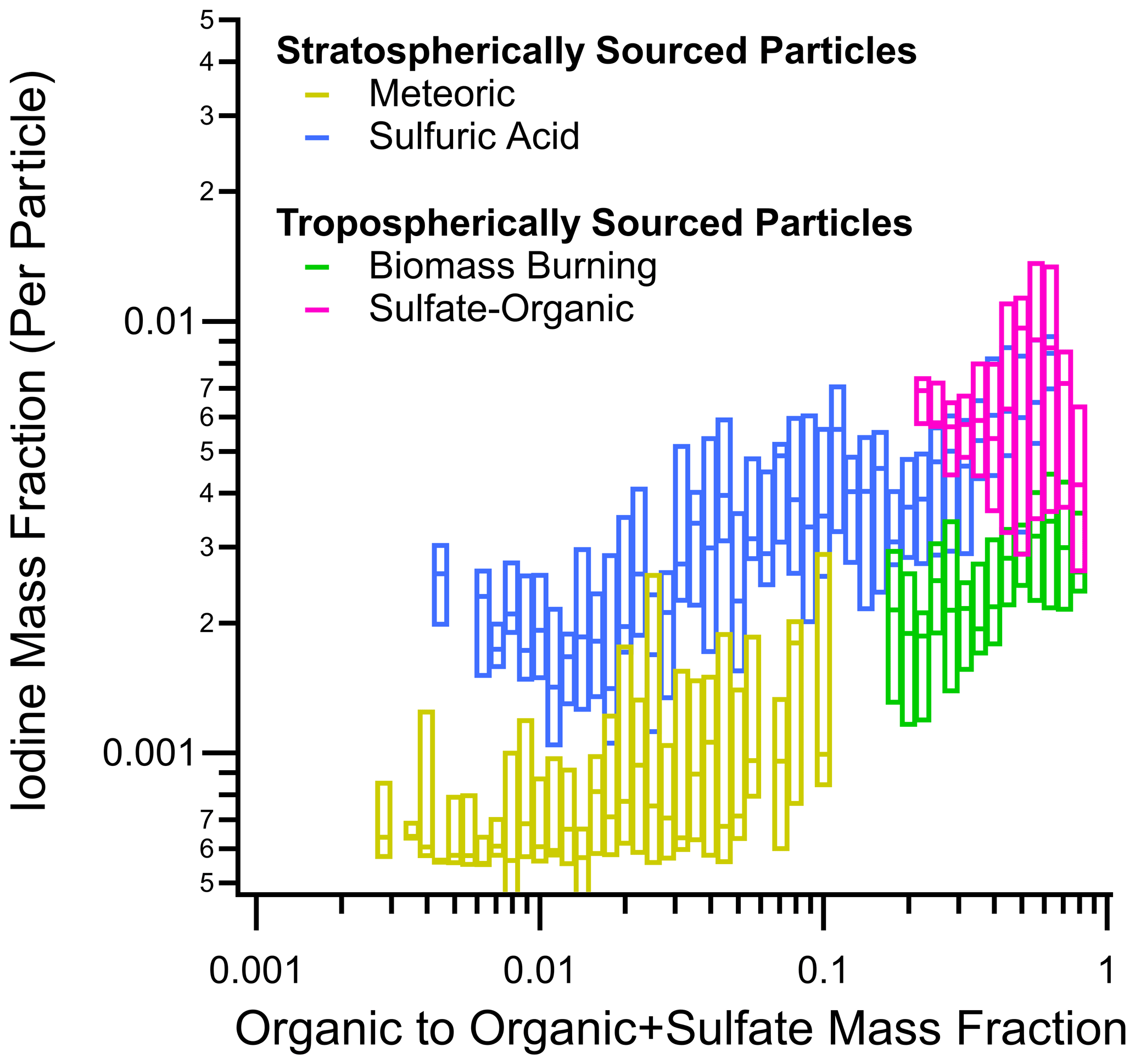
Figure 11Box plots of iodine mass fraction in particles observed in the lower stratosphere as function of the organic-to-organic-plus-sulfate mass ratio from all four ATom instances. Of the four dominant particle types, two are tropospherically sourced (biomass burning and sulfate–organic–nitrate) and two are stratospherically sourced (meteoric-sulfuric-acid and sulfuric-acid particles). Horizontal dashes are median values, and the boxes are interquartile ranges. In this work, the lower stratosphere is defined as air masses above 8 km where the [O3] [CO] ratio is greater than 3.
For bromine, there was a weaker correlation between organic mass fractions and nSSA bromine concentrations in stratospheric particles (Fig. A14). This suggests that organic binding or HBr dissolution in small organic acids is not the only and perhaps not even the primary mechanism to retain nSSA bromine. Regardless, even though the relationship is less clear for bromine than it is for iodine, lower-stratospheric particles with higher organic content did generally contain more bromine. Thus, organics are also likely playing some role in retaining nSSA bromine or acting as a proxy for some physico-chemical properties that help retain nSSA bromine.
3.3 Iodine modeling with GEOS-Chem
In this work, we compare the ATom measurements to the GEOS-Chem chemical transport model (https://geoschem.github.io/, last access: 7 October 2020, version 12.9.1), which has an online NOx–VOC–HOx–Ox–BrOx–ClOx–IOx chemistry scheme (Sherwen et al., 2016b, 2017; Wang et al., 2019) computed for all 72 levels up to 0.01 hPa (Eastham et al., 2014). In GEOS-Chem 12.9.1, nSSA bromine is not tracked, but nSSA iodine is accounted for. The formation of iodine aerosol occurs through the uptake of gas-phase iodine on existing fine- and coarse-mode aerosol (Sherwen et al., 2016b, c), which is then transported and deposited following the model's treatment of its parent aerosol. In GEOS-Chem 12.9.1, iodine partitions into nSSA from the irreversible uptake of HI and higher iodine oxides (I2OX, x=2, 3, 4). It can also partition into aqueous nSSA through several water-soluble gas-phase iodine species, which either react to form new gas-phase species or are released upon drying (see Table B5 in Sherwen et al., 2016b). The model was run at a horizontal resolution of 4×5°, and transport was driven by offline meteorological fields from Goddard Earth Observing System Modern-Era Retrospective analysis for Research and Applications, Version 2 (MERRA-2; Gelaro et al., 2017). Global anthropogenic emissions were from the Community Emissions Data System (CEDS; Hoesly et al., 2018), and biomass burning emissions were from the Global Fire Emissions Database (GFED4; Mu et al., 2011). Inorganic halogen emissions followed Carpenter et al. (2013), and organic halogen emissions followed Ordóñez et al. (2012). The model was run for the entire period of the NASA ATom campaign, following a discarded year of “spinup”, with data extracted for the nearest point in space and time along the route taken by the aircraft.
Iodine aerosol formation in the model is missing many known processes (e.g., heterogeneous/multi-phase reactions, Saiz-Lopez et al., 2012) and some that have been recently proposed (e.g., iodine oxoacid new particle formation, He et al., 2021, and halogen solvation by organics, Solomon et al., 2023); nonetheless, these model–measurement comparisons can help inform the model that their reactive iodine concentrations and the processes that transfer reactive iodine to the aerosol phase are roughly correct. A global surface evaluation of the model's iodine aerosols is described in Sherwen et al. (2016c).
Vertical profiles of nSSA iodine from ATom measurements and GEOS-Chem 12.9.1 agree well in both magnitude and shape (Fig. 12). The agreement was best in the background free troposphere (Fig. A15), i.e., outside of air masses influenced by biomass burning aerosol (“influenced” is defined here as accumulation-mode biomass burning aerosol number fractions > 0.5), below the lower stratosphere, and outside of the MBL. Thus, the model seems to capture the pervasive secondary formation of nSSA iodine well. In the model, higher iodine oxides are directly or eventually formed from self-reactions of IO. HI is formed from the reaction of I and HO2. This suggests that IO and I (Ix) are important precursors to the pervasive secondary nSSA iodine and that the net flux of iodine to nSSA is similar to the reactive uptake of I2Ox with γ=0.02 and HI with γ=0.1.
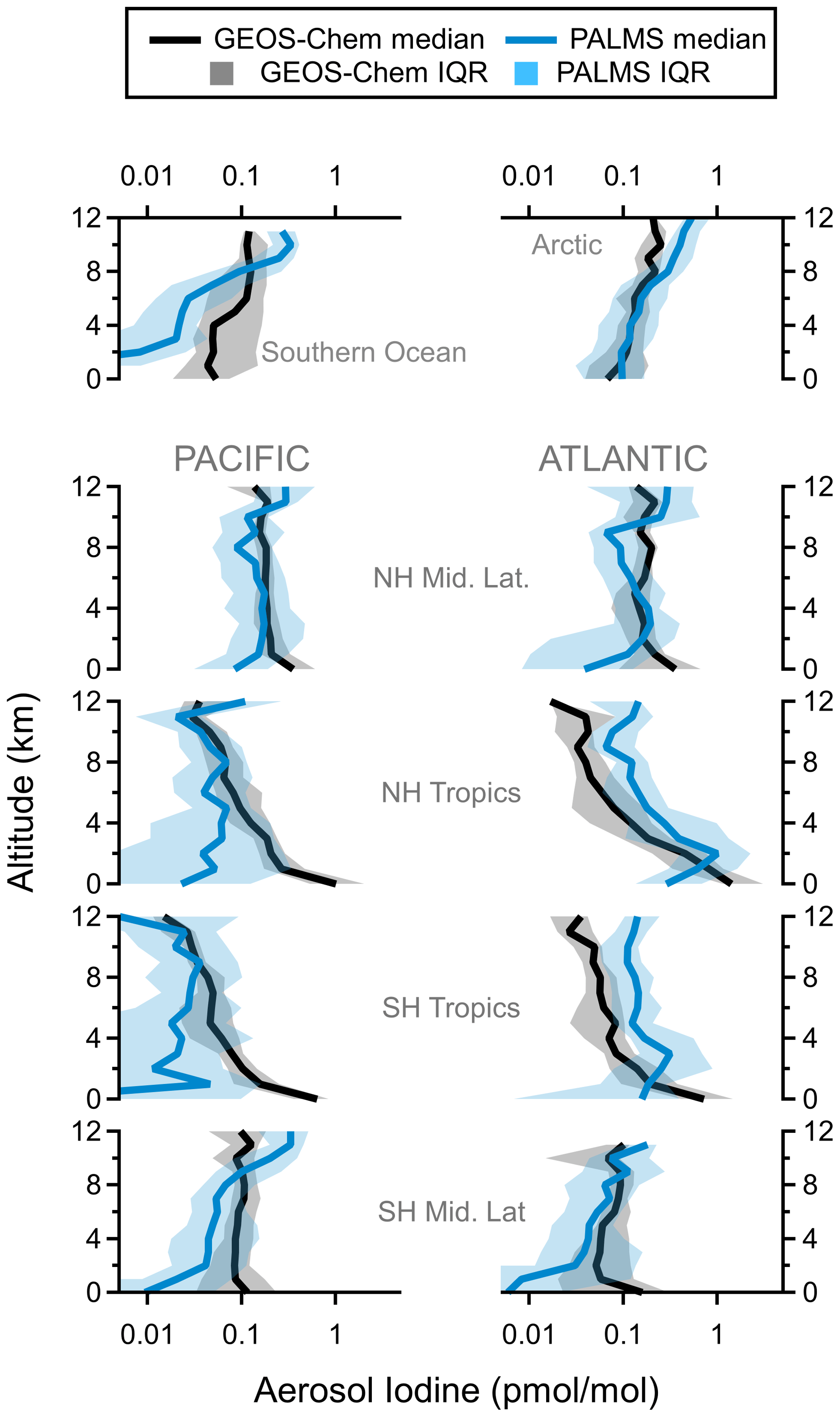
Figure 12Regional vertical profiles of nSSA iodine (pmol mol−1) from PALMS observations (blue) and GEOS-Chem 12.9.1 model output (black). Median values (solid lines) and interquartile ranges (shading) are from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020).
The agreement between GEOS-Chem 12.9.1 output and the ATom measurements suggests that Ix represents important intermediates, or important tracers of the true intermediates, to nSSA iodine formation. The I radical is not routinely measured, and no vertical profiles are known to the authors for comparison to the model. IO, however, has been measured from aircraft platforms (e.g., Volkamer et al., 2015), and the I and IO concentrations are inexorably related as these species rapidly cycle between each other. The model does generally have a high bias to IO (Sherwen et al., 2016a); for example, a comparison to the TORERO (Tropical Ocean tRoposphere Exchange of Reactive halogen species and Oxygenated VOC) aircraft IO measurements (Wang et al., 2015) shows that the model has a bias of +83 % on average, with the greatest bias in the MBL (+125 %). This could explain some of the biases with the ATom measurements; unfortunately, a direct measurement of IO was not made on the DC-8 during ATom.
The dominant source of Ix in the atmosphere is the photolysis of primary inorganic iodine (e.g., HOI, I2) emitted from the ocean, although the photolysis of organic iodine species, primarily CH3I, contributes in the free troposphere. During ATom, we also did not have direct measurements of HOI or I2, but the model does perform well against ATom measurements of CH3I (Fig. A16). Thus, while nSSA iodine measurements suggest that Ix is an important proxy for nSSA iodine precursors, more measurements of I, IO, IxOy, HI, and other short-lived inorganic iodine species are needed to better constrain the mechanism of aerosol formation. These measurements, ideally, need to be accurate to the sub-0.1 pmol mol−1 level in order to be a useful comparison to the nSSA iodine concentrations in this work.
As mentioned above, the model underestimated nSSA iodine in areas of high biomass burning aerosol influence, underestimated nSSA iodine in the stratosphere, and overestimated nSSA iodine in the MBL. Air masses strongly influenced by biomass burning aerosol, defined as air masses where biomass burning aerosol make up over half of the accumulation-mode aerosol by number, exhibited a slight model underestimation, approximately −66 % on average when giving each ATom ocean basin equal weight. This is because the model does not have an nSSA iodine source from biomass burning. Despite not having a biomass burning source of nSSA iodine, there were large variations in the nSSA iodine mean log bias and centered root mean square error (CRMSE; Fig. A15). For example, in the ATom-2 Atlantic, the model underestimated nSSA iodine by an order of magnitude, and the spread of biases is also almost an order of magnitude. This variation suggests that different fuel types and flaming conditions may be important for forming iodine aerosol from biomass burning. Furthermore, in some air masses influenced by biomass burning, the mean log bias was nearly zero; however, as shown in Fig. 12, this may be a convolution of the underestimation of nSSA iodine in biomass burning plumes and overestimation of nSSA iodine in or just above the MBL.
Inside the MBL, the model overestimated nSSA iodine by a factor of 7, on average. In GEOS-Chem 12.9.1, aerosol iodine can condense on both nSSA and also fine-mode and coarse-mode sea salt. It is possible that the partitioning of iodine between nSSA and sea-salt aerosol is incorrect in the model. Also, the model does not include any heterogeneous recycling reactions that would react with HI or higher iodine oxides once they are in the aerosol phase. As mentioned above, the number fraction of iodine-containing aerosol was positively correlated with O3 below 50–60 ppb. Thus, if the model was overestimating O3 concentrations in the MBL, then both IO production and nSSA iodine may be overestimated; however, we find that, in the MBL, the model had a minimal low bias (approximately −8 %) compared to the ATom measurements of O3. Water vapor has been shown to reduce the cluster formation of IxOy (Gómez Martín et al., 2020), which may prevent secondary iodine formation in the humid MBL, although this has been shown to not be the case for iodine oxoacid nucleation (Rörup et al., 2024). Additionally, in the humid and wet MBL, there may be increased wet deposition and rain out of both soluble gas-phase iodine species and hygroscopic aerosol that are underestimated in the model.
In the high-latitude LS, the model underestimated nSSA iodine by a factor of ∼ 2.4. Some portion of this missing nSSA iodine may be the contribution from biomass burning aerosol that was often present in the LS (e.g., Fig. 6). In addition to this, other processes in the model may account for missing nSSA iodine, including incorrect photolysis rates of key gas-phase species like IxOy; incorrect transport of very short-lived (VSL) iodine to the UT/LS; or increased uptake on cold, dry, and/or acidic particles. Additionally, iodine in the form of HI may be efficiently dissolved in the organic phase of stratospheric aerosol particles akin to HCl (Solomon et al., 2023). Missing chemistry in the model could also be driving model–measurement discrepancies in the stratosphere. During the TORERO and CONTRAST campaigns, IO was reported to decrease from the tropical UT to the LS as aerosol iodine increased in a similar magnitude, suggesting that an additional source of iodine from the upper stratosphere is not needed to explain the increase in aerosol iodine (Koenig et al., 2020). Measurements from the CU HR-ToF-AMS (University of Colorado high-resolution time-of-flight aerosol mass spectrometer) during ATom show that aerosol iodine in the stratosphere was consistent with iodate, while most of the aerosol iodine found in the troposphere was iodide or organic iodine (Koenig et al., 2020). In GEOS-Chem 12.9.1, nSSA iodine is formed through the same mechanisms in the stratosphere as it is in the troposphere (irreversible uptake by HI and I2OX, x=2, 3, 4), which is inconsistent with the HR-ToF-AMS iodate results. Unfortunately, the PALMS data presented here did not confirm if the iodine LS aerosol was in fact iodate, but the aerosol iodine measurements between AMS and PALMS agree to within 8 % (median bias) for ATom-1 and ATom-2. The PALMS measurements also suggest that the iodine is organically bound, which is also not directly accounted for in the model. Thus, because low RH, iodate, and organically bound iodine would suppress heterogeneous chemical reactions that liberate iodine back to the gas phase and because the model does not include these reactions and still underestimates aerosol iodine concentrations, a missing chemical pathway is more likely. Finally, the total available iodine may be limiting nSSA iodine concentrations in the LS, as nSSA iodine concentrations were similar between ATom instances despite highly variable aerosol environments (Murphy et al., 2021).
In this work, we have provided global-scale measurements of the accumulation-mode number fraction, mass fraction, and absolute mass of nSSA bromine and iodine. We find that these halogens were commonly found in free-tropospheric nSSA, and, as a lower estimate, bromine and iodine were present (i.e., have mass fractions > 0.00055) in 0.08–0.26 (IQR) and 0.12–0.44 (IQR) of the nSSA sampled during ATom, respectively. Despite being commonly found in nSSA, the concentration of these halogens was low, approximately 0.11–0.57 pmol mol−1 (IQR) and 0.04–0.24 pmol mol−1 (IQR) for bromine and iodine, respectively. The normalized root mean square error for these measurements was approximately 20 %–60 % for most of the atmosphere (Fig. A3), and systematic biases due to unknown iodine speciation is likely below a factor of 2 (Sect. 2.2).
Using the capabilities of single-particle mass spectrometry, we have attributed several sources to the ubiquitous, trace bromine and iodine in nSSA. First, we find that there is a primary source of bromine and iodine in nSSA from biomass burning. Fresh biomass burning aerosol contains lower mass fractions of bromine and iodine than background particles, but higher aerosol mass concentrations in plumes suggest that > 1 pmol mol−1 of bromine and iodine can be found in nSSA in aged biomass burning plumes. The second source of bromine and iodine to nSSA is a pervasive secondary source. Here, organic and inorganic bromine and iodine sources from the oceans are converted into a pervasive background of reactive species that can form low-volatility bromine and iodine products that partition into the aerosol phase. It is unclear what the ultimate fate of these aerosol-bound halogens are, but plotting mass fractions in individual particles as a function of back trajectory age suggests that individual nSSA generally accumulates bromine and iodine as they age. Finally, we also find that there was an increase in bromine and iodine in stratospheric nSSA but that these are concentrated on tropospherically sourced organic–sulfate mixtures, not the stratospherically sourced sulfuric-acid particles that sometimes contain trace meteoric metals.
We have compared our results to output from the global chemical transport model GEOS-Chem 12.9.1, which has an online NOx–VOC–HOx–Ox–BrOx–ClOx–IOx chemistry scheme. Iodine aerosol is formed in the model, but there are no mechanisms for nSSA bromine to form. We find that the model compares well to our nSSA iodine measurements in the background troposphere, which corroborates that nSSA iodine is formed through secondary processes, likely involving Ix (I and IO). While the agreement in the background atmosphere is good, the model underestimated nSSA iodine in biomass burning plumes and in the stratosphere. Additionally, the model overestimated the concentration of nSSA bromine and iodine in the marine boundary layer by a factor of 7. Thus, while the agreement outside of biomass burning plumes, the stratosphere, and the MBL suggests that the iodine aerosol schemes are relevant to background aerosol, some sources and chemistry in the iodine aerosol schemes are missing. Thus, global chemistry climate models that wish to explore the full extent of the impact of ubiquitous, trace bromine and iodine in nSSA on atmospheric constituents, like odd oxygen, may first need to update their emission inventories and chemical schemes.
The following section includes figures that support the paper but would disrupt the flow of descriptions or demonstrations. These figures support the PALMS bromine and iodine mass calibrations, show box plots that outline general trends of nSSA bromine and iodine with gas-phase and meteorological tracers, are the nSSA bromine companions figures to the nSSA iodine figures in the main text, or help us understand the biases and trends in the GEOS-Chem model.
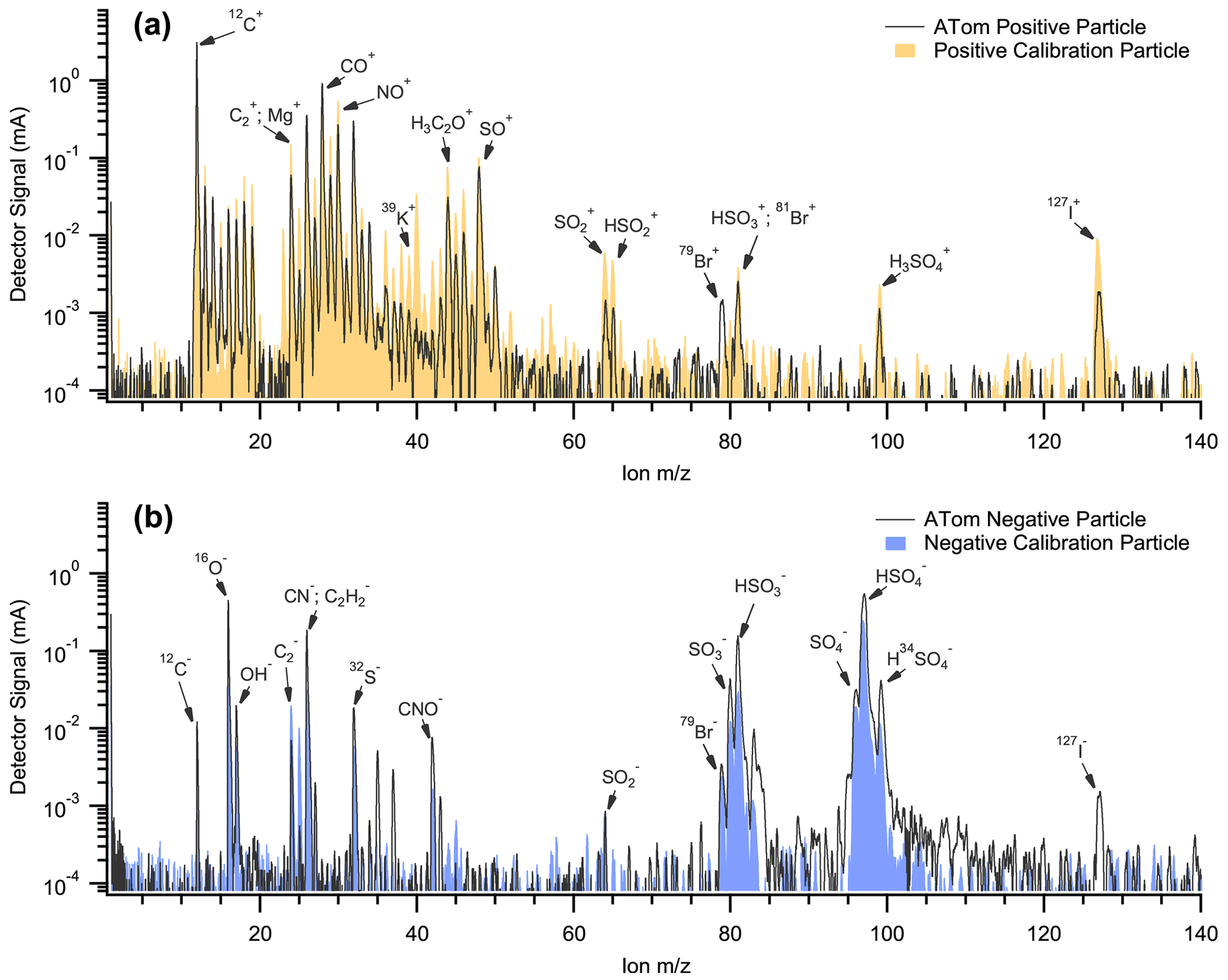
Figure A1Example mass spectra of (a) a positive iodine calibration particle and an iodine-containing nSSA from ATom and (b) a negative bromine calibration particle and a bromine-containing nSSA from ATom.
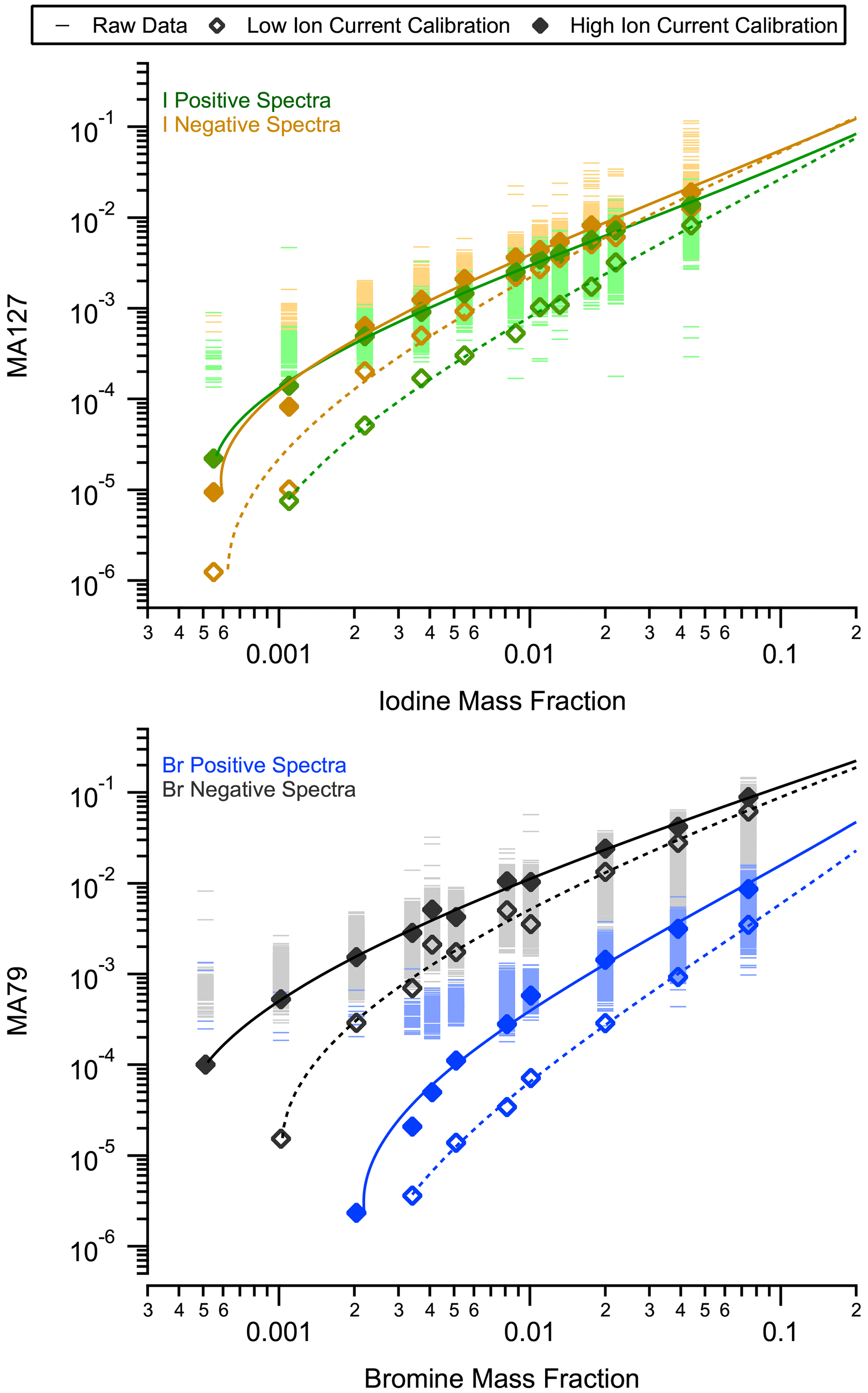
Figure A2MA79 (MA127) as a function of the bromine (iodine) mass fraction in a proxy for nSSA. Raw data are shown as horizontal bars, average signals per mass fraction are shown as markers, and calibration curves are shown as lines. Zero values are not shown because of the log scale but are included when calculating the average values.
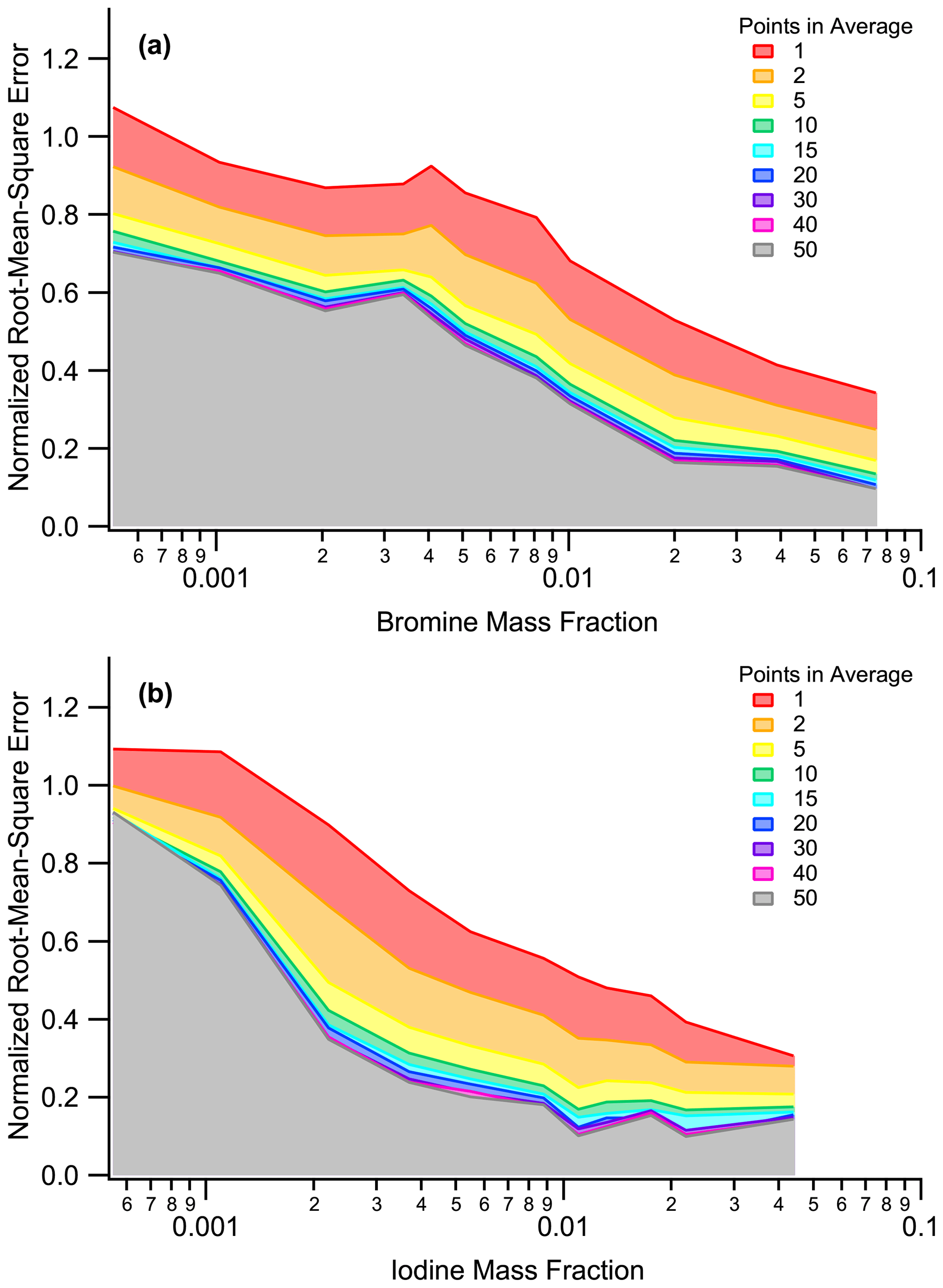
Figure A3Normalized root mean square error as a function of (a) the bromine mass fraction and (b) the iodine mass fraction. The number of particles used in an average are shown as overlaid graphs. Normalized root mean square errors are reduced with higher halogen mass fractions and with more particles used in an average.
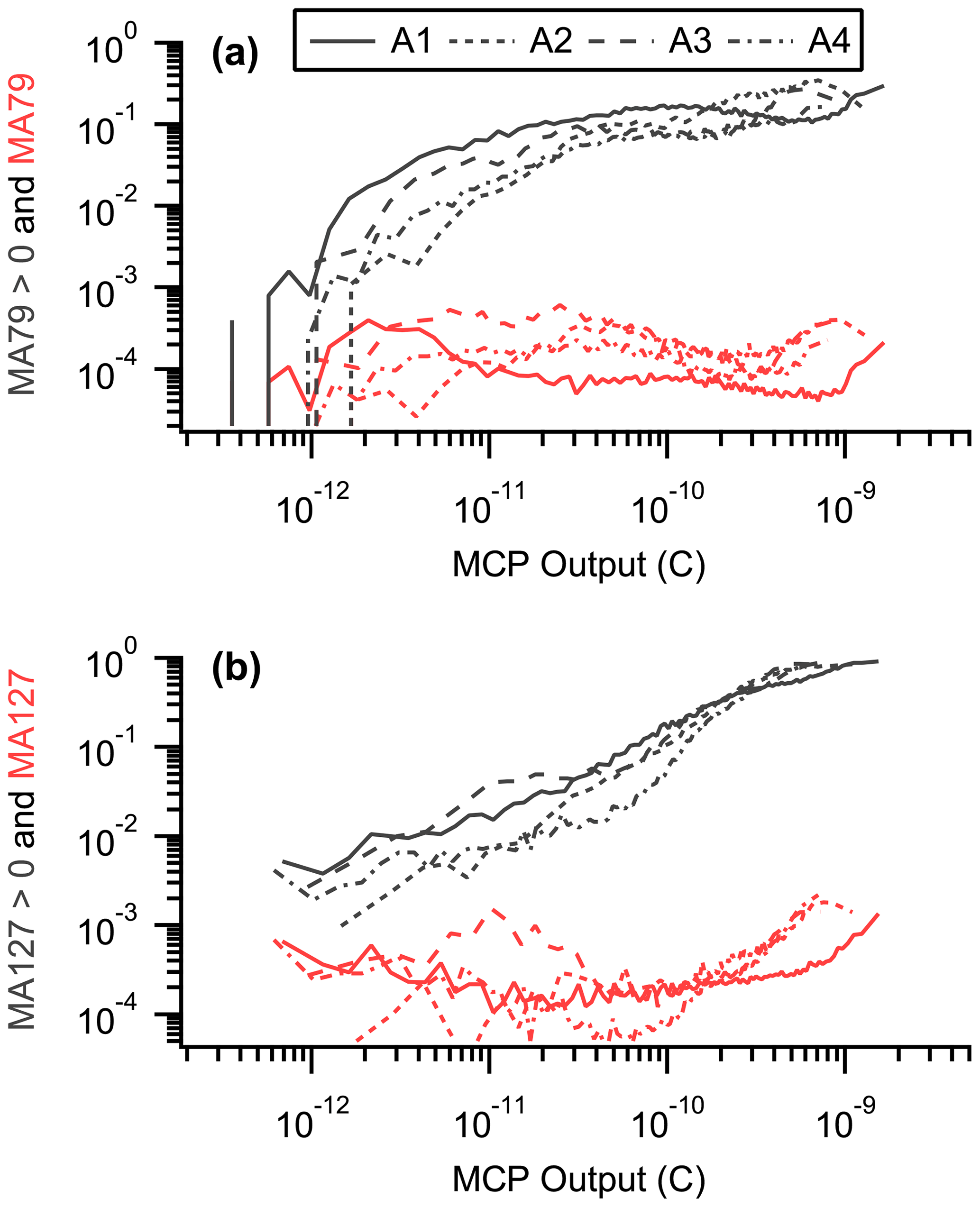
Figure A4Number fraction of halogen-containing particles (black) and halogen peak area (red) vs. MCP output for (a) bromine (MA79) and (b) iodine (MA127). Number fractions are calculated as MA79 > 0 for bromine and MA127 > 0 for iodine. In this example, only positive spectra were used. Each line corresponds to a different ATom (e.g., A1 is ATom-1).
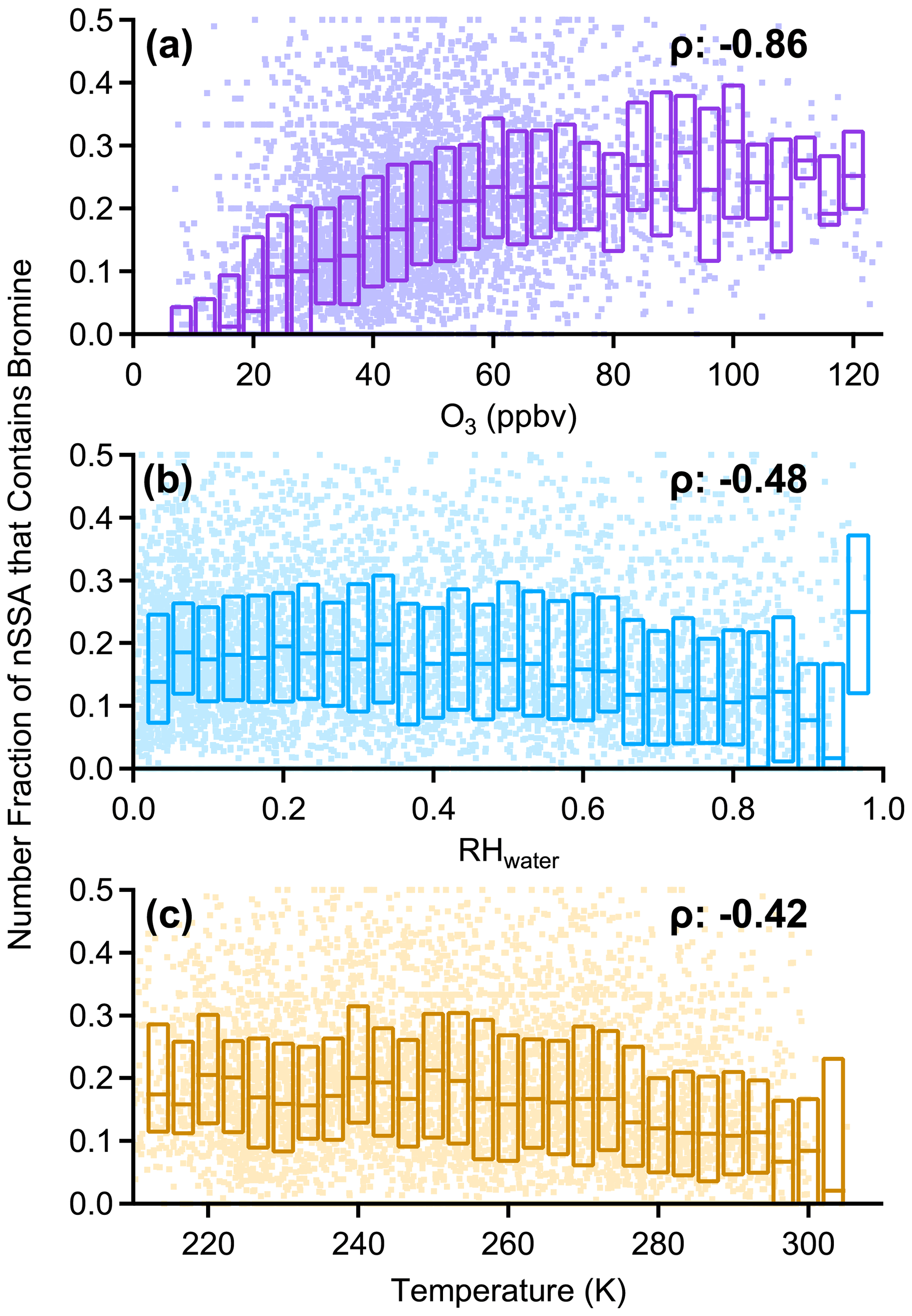
Figure A5Box plots of the number fraction of nSSA that contains bromine (MA79 > 0) vs. (a) tropospheric O3, (b) RHwater, and (c) temperature. Tropospheric air was defined as (altitudes > 8 km) ∧ (O3 > 100 ppbv) = 0. Raw data are dots, median values are horizontal dashes, and the interquartile range is bound by the boxes. Median value of Spearman's rho (ρ) is reported in each panel.
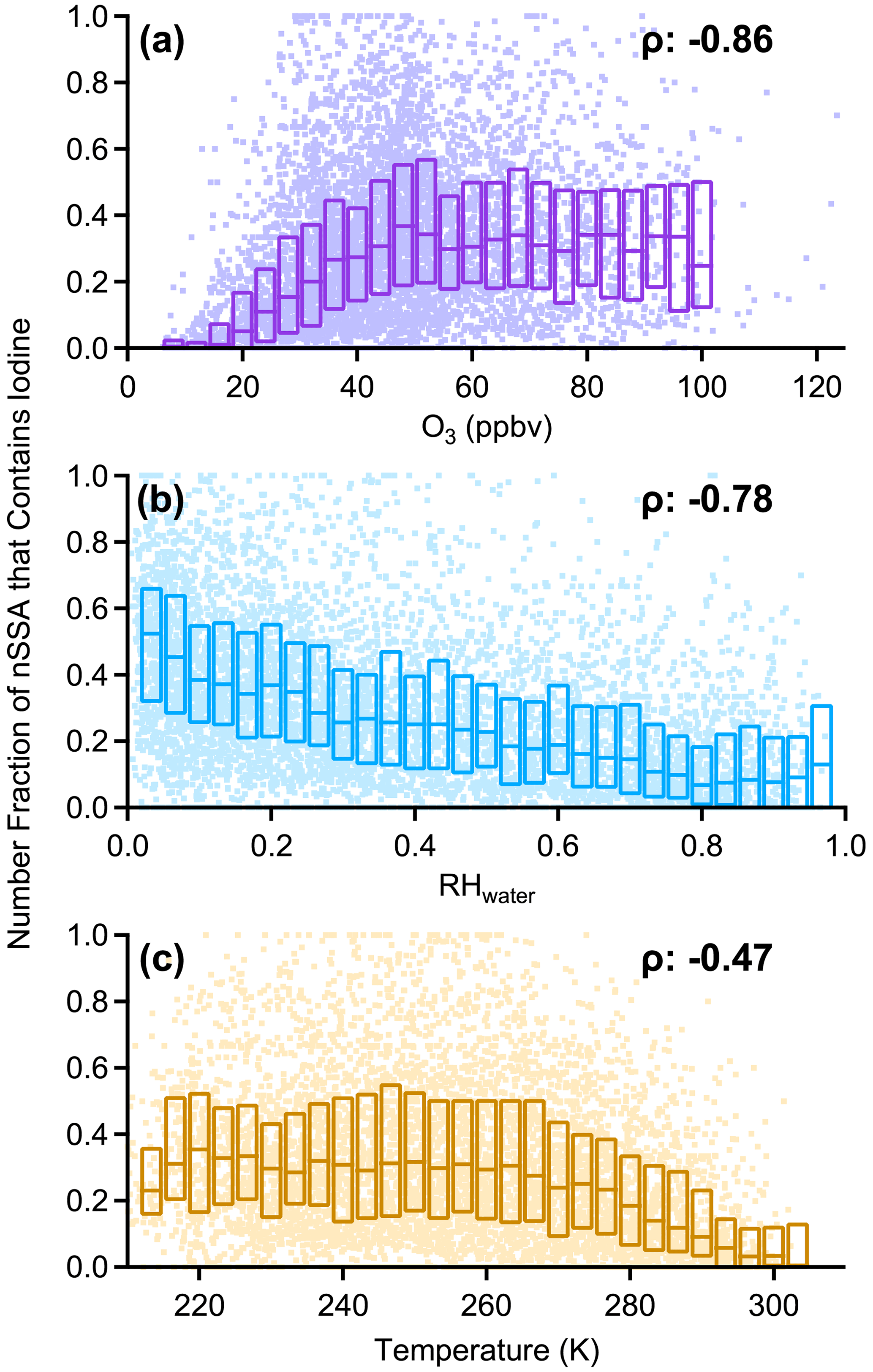
Figure A6Box plots of the number fraction of nSSA that contains iodine (MA127 > 0) vs. (a) tropospheric O3, (b) RHwater, and (c) temperature. Tropospheric air was defined as (altitudes > 8 km) ∧ (O3 > 100 ppbv) = 0. Raw data are dots, median values are horizontal dashes, and the interquartile range is bound by the boxes. Median value of Spearman's rho (ρ) is reported in each panel.
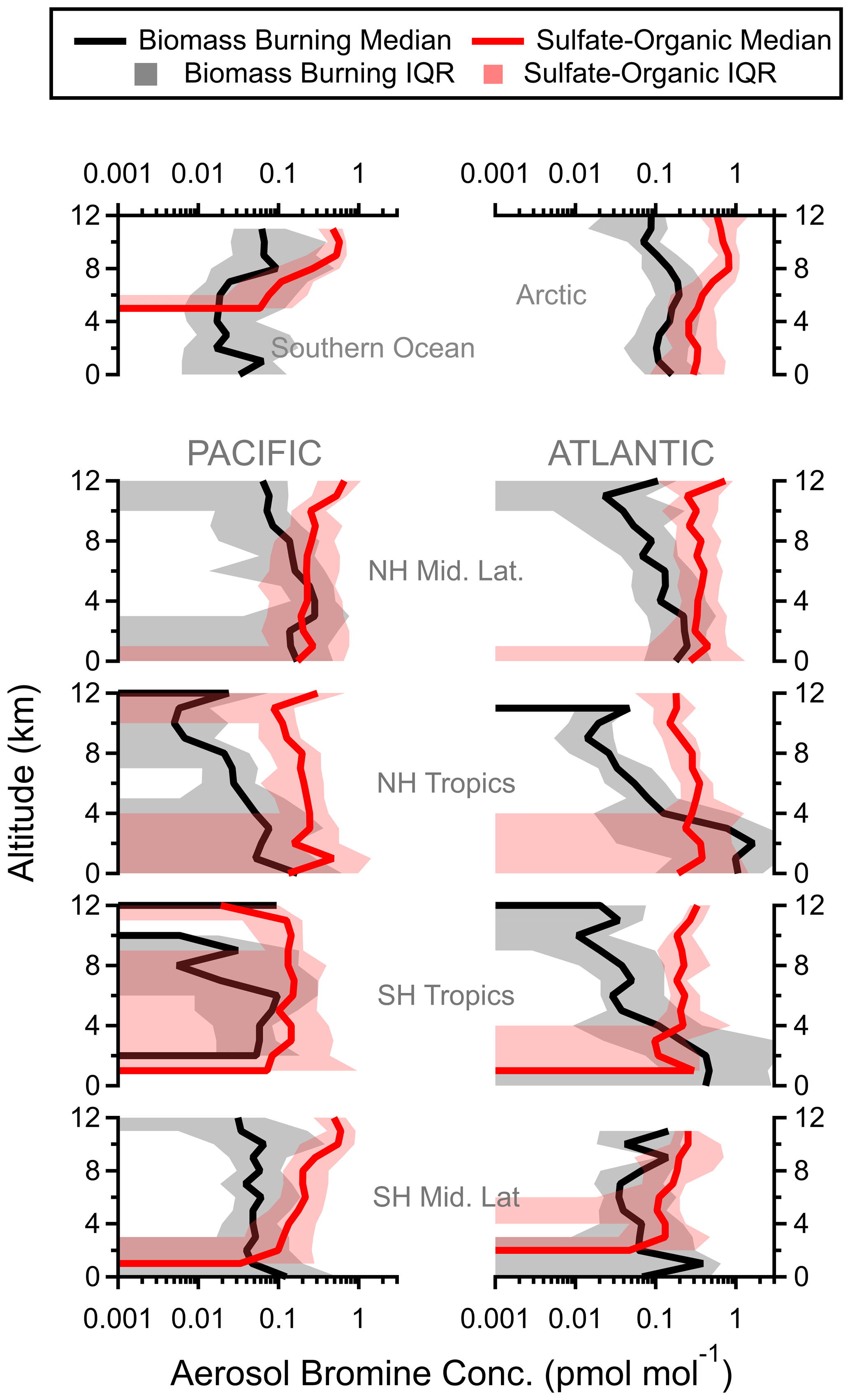
Figure A7Regional vertical profiles of bromine (pmol mol−1) in biomass burning and sulfate–organic–nitrate particles. Median values (solid lines) and interquartile ranges (shading) are from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020).
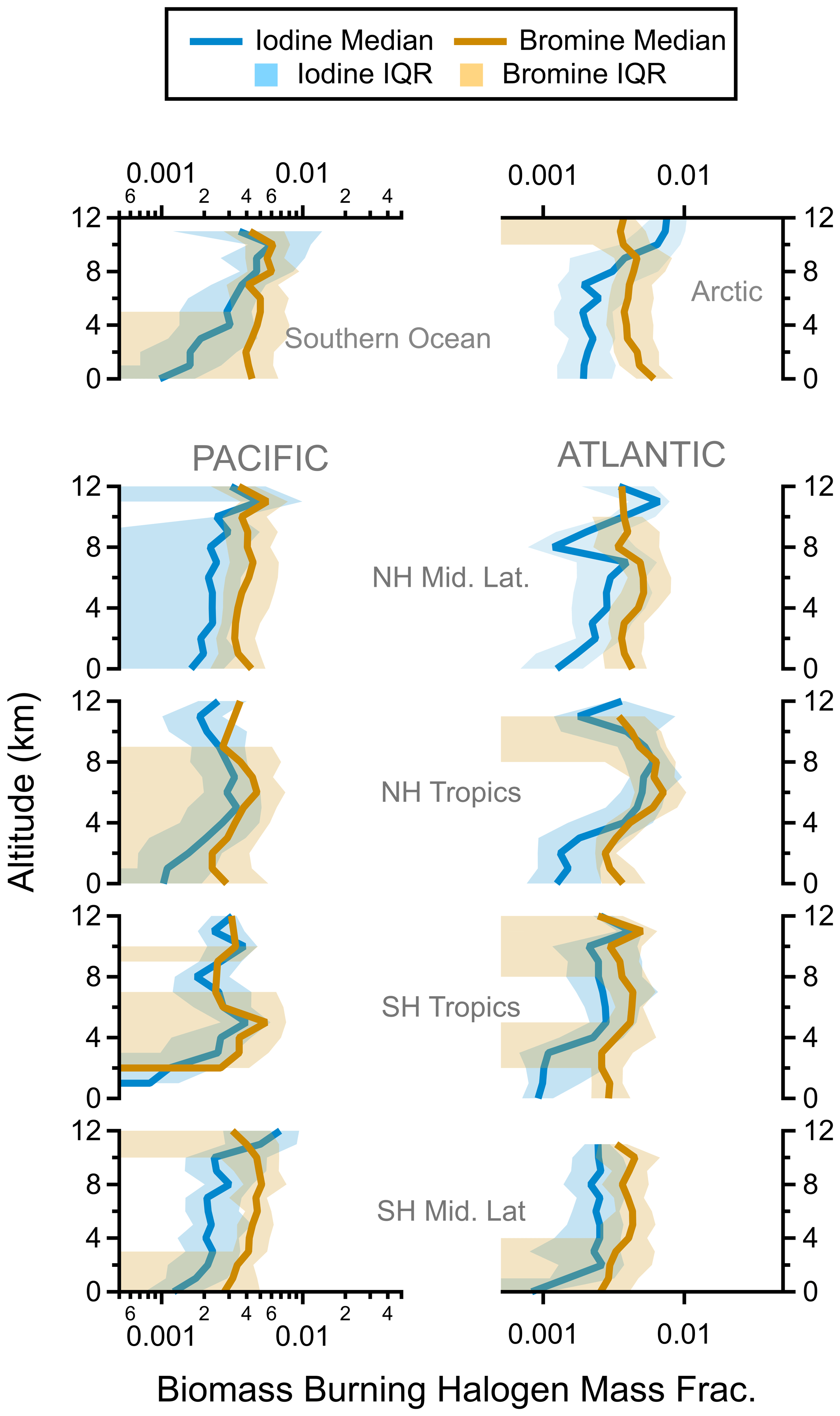
Figure A8Regional vertical profiles of the iodine and bromine mass fraction in biomass burning particles. Median values (solid lines) and interquartile ranges (shading) are from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020).
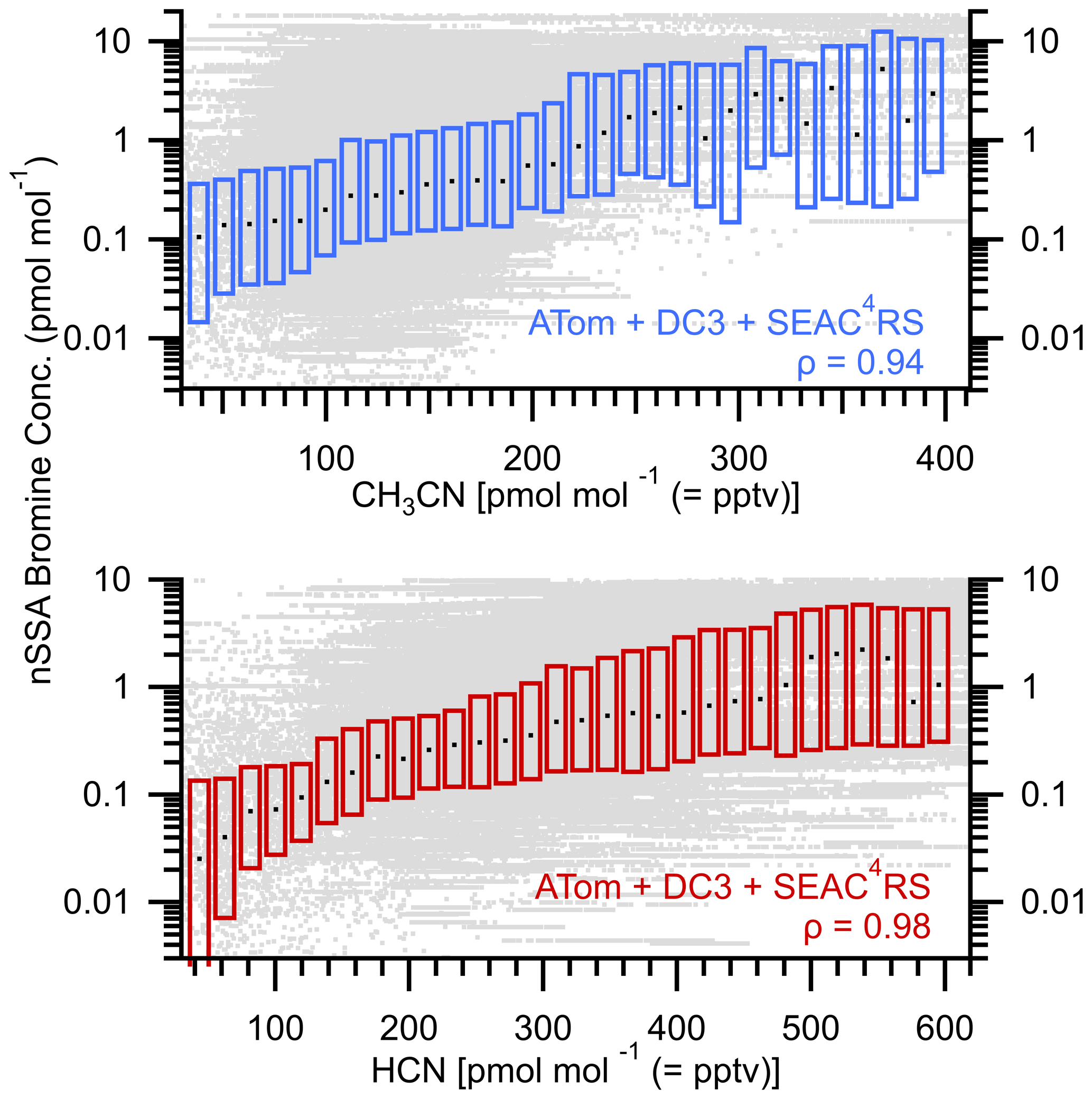
Figure A9Box plots of nSSA bromine (pmol mol−1) vs. gas-phase biomass burning tracers CH3CN (pptv) and HCN (pptv). Raw data are gray dots, median values are horizontal black dashes, and the interquartile range is bound by the colored boxes. Median value of Spearman's rho (ρ) is > 0.94, which indicates a near-monotonic relationship and suggests that biomass burning is a primary source of nSSA bromine.
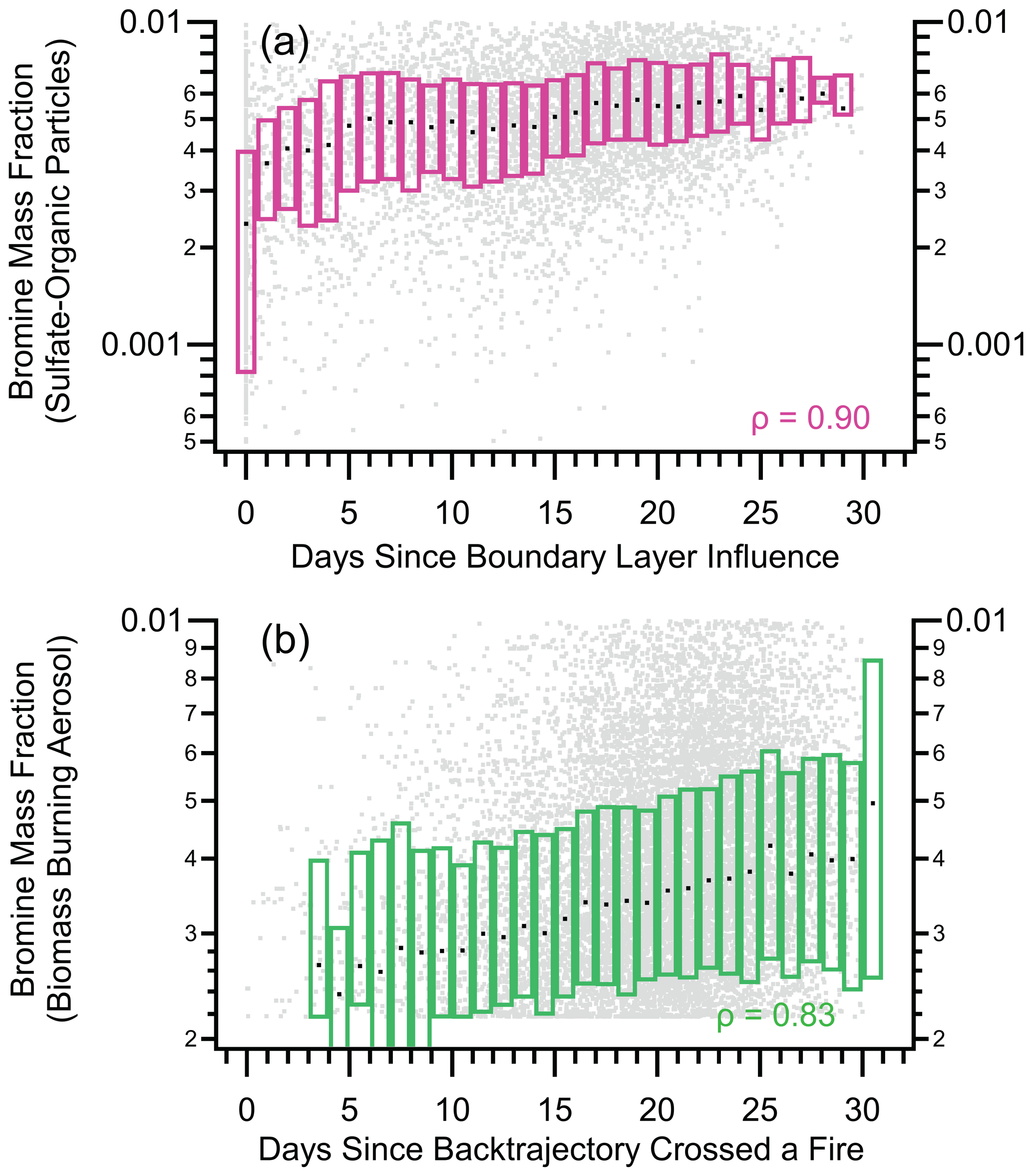
Figure A10Box plots of nSSA bromine mass fractions vs. two back trajectory products, (a) the average number of days since a back trajectory was in the boundary layer and (b) the number of days since a back trajectory crossed a fire. Raw data are gray dots, median values are horizontal black dashes, and the interquartile range is bound by the colored boxes. Median value of Spearman's rho (ρ) is > 0.83, which indicates a near-monotonic relationship and suggests that biomass burning is a primary source of nSSA bromine mass.
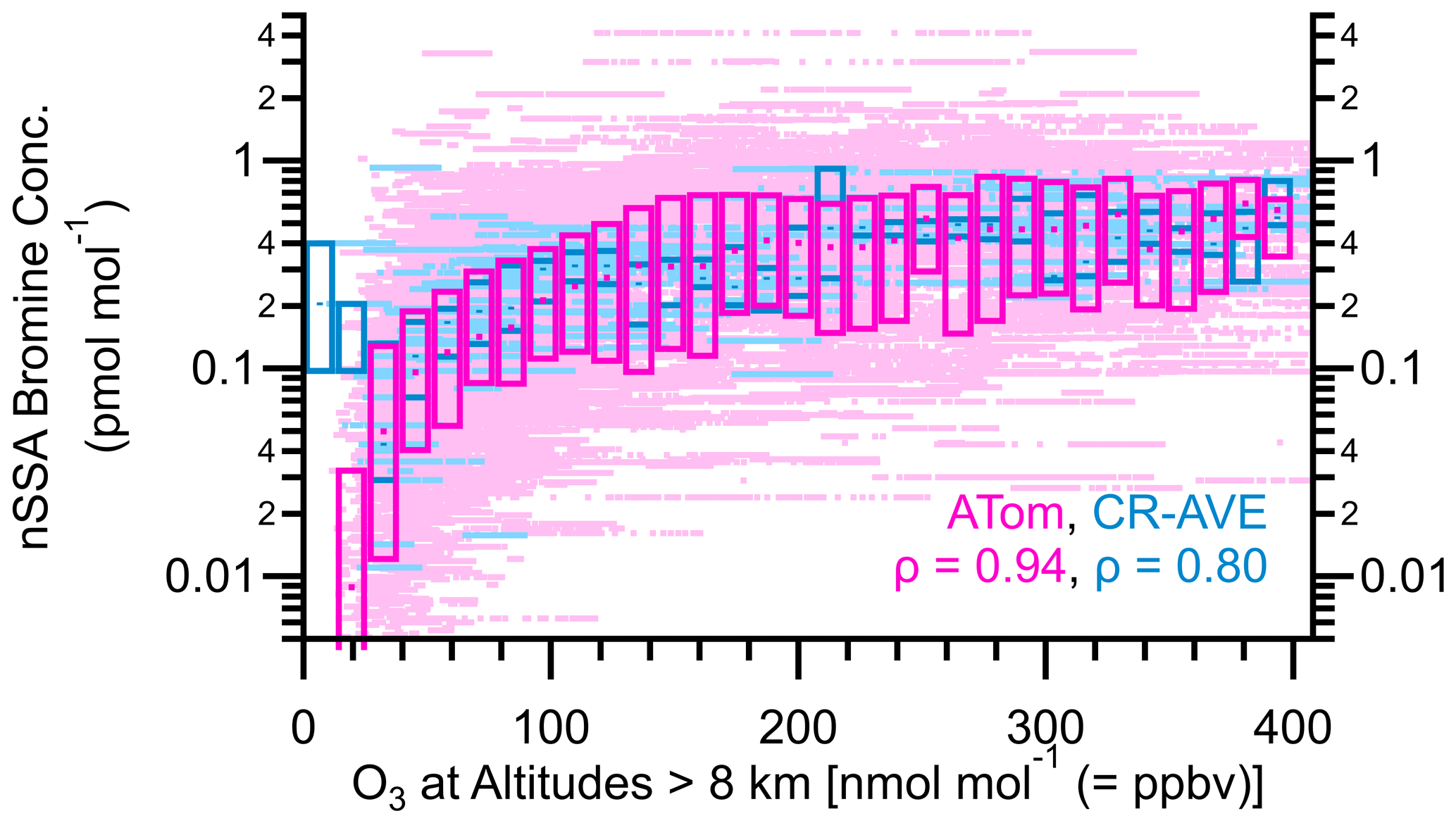
Figure A11Box plots of nSSA bromine (pmol mol−1) vs. O3 for ATom (yellow) and CR-AVE (magenta). The median values of Spearman's rho (ρ) for ATom and CR-AVE are 0.94 and 0.80, respectively, which indicates a near-monotonic relationship and suggests that the stratosphere may be a source of nSSA bromine globally.
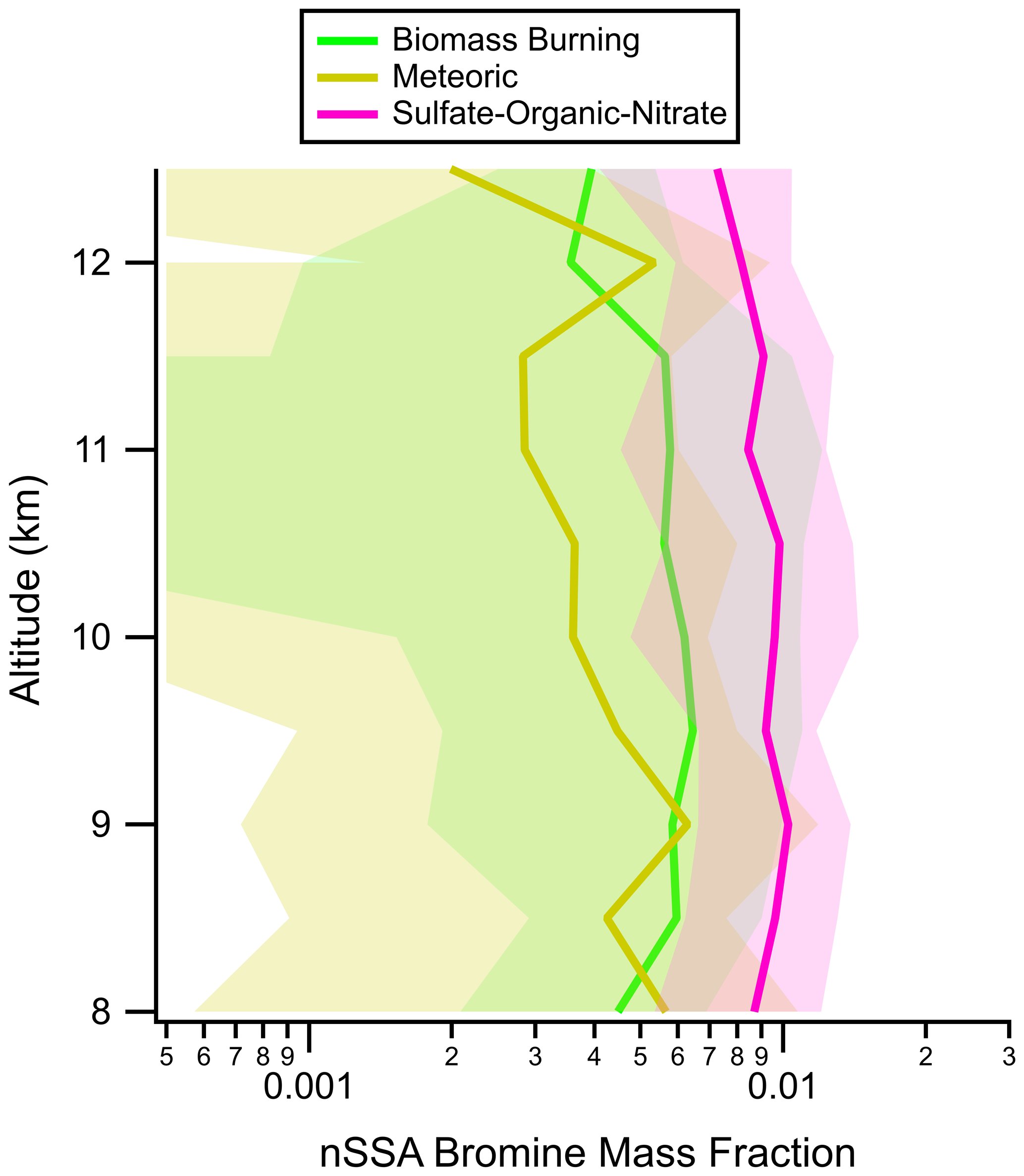
Figure A12Vertical profiles of nSSA bromine mass fractions in the most common particle types found in the lower stratosphere during ATom: tropospherically sourced biomass burning and sulfate–organic–nitrate particles and stratospherically sourced meteoric-sulfuric-acid particles. Average values (solid lines) and standard deviations (shading) are from all four ATom instances. In this work, the lower stratosphere is defined as air masses above 8 km where the [O3] [CO] ratio is greater than 3.
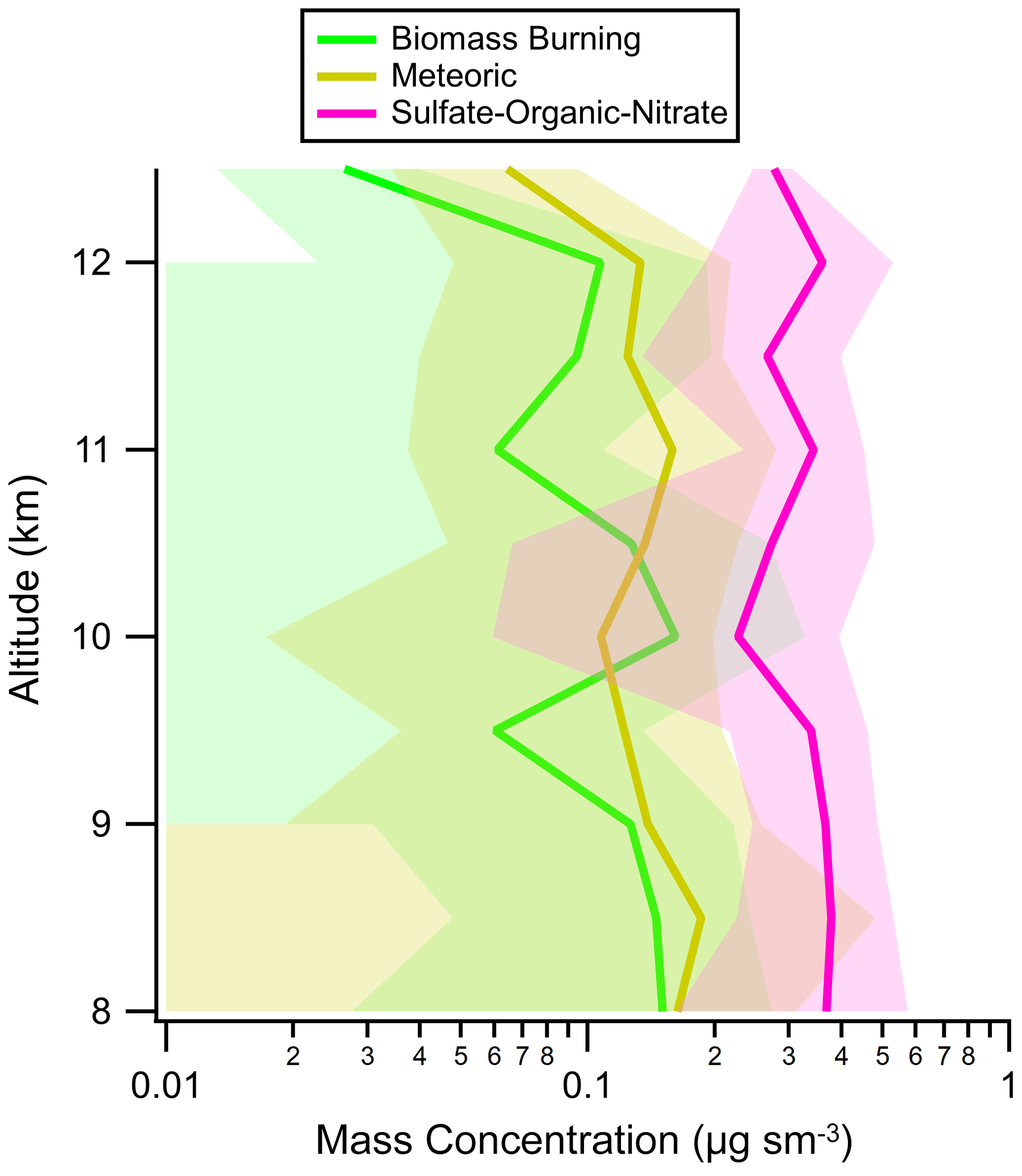
Figure A13Vertical profiles of aerosol mass concentrations of biomass burning, sulfate–organic–nitrate, and meteoric-sulfuric-acid aerosol in the lower stratosphere during ATom. Average values (solid lines) and standard deviations (shading) are from all four ATom instances. In this work, the lower stratosphere is defined as air masses above 8 km where the [O3] [CO] ratio is greater than 3.
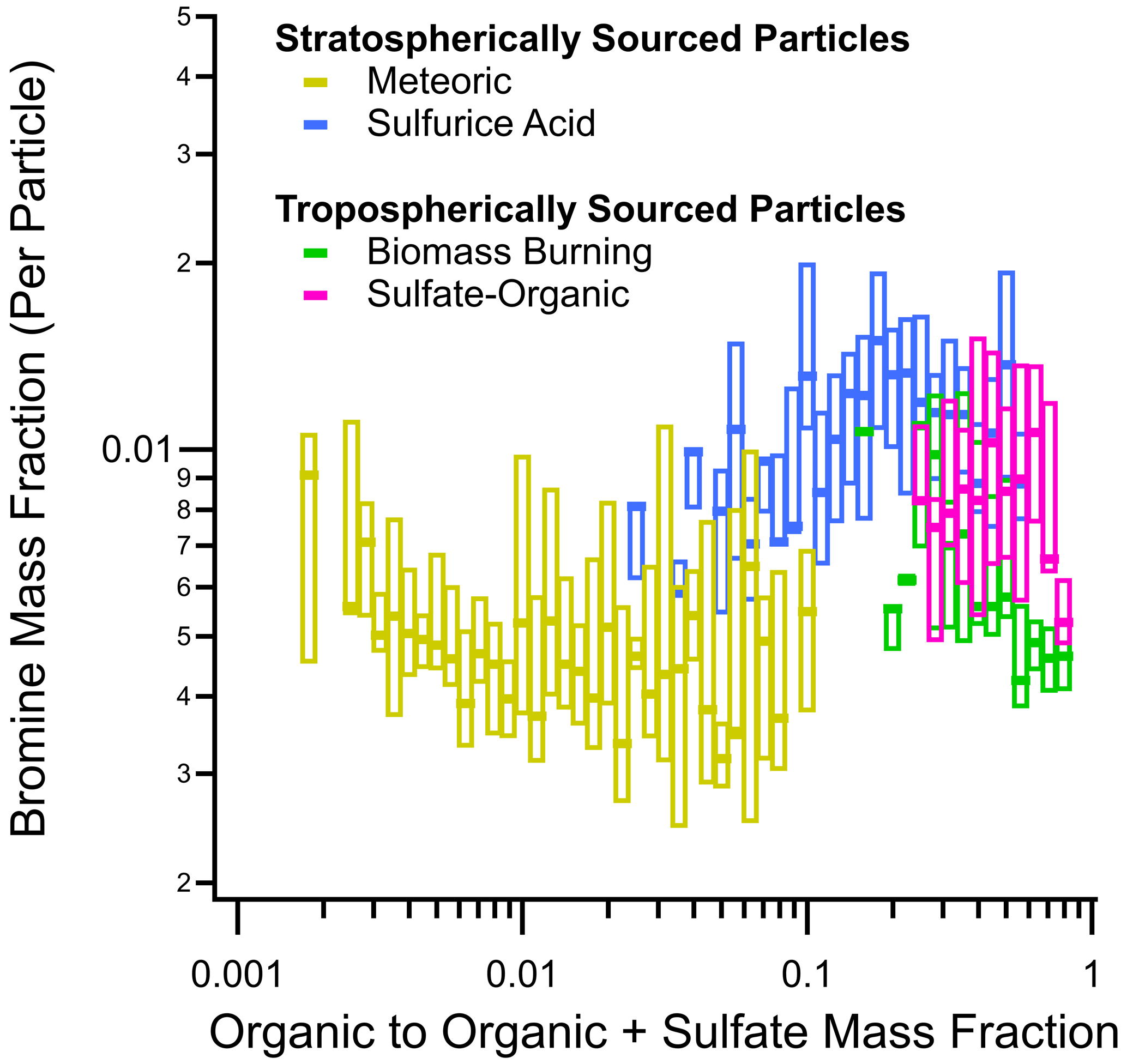
Figure A14Box plots of the bromine mass fraction in different particle types found in the lower stratosphere as a function of the organic-to-organic-plus-sulfate mass ratio from all four ATom instances. Of the four dominant particle types, two are tropospherically sourced (biomass burning and sulfate–organic–nitrate) and two are stratospherically sourced (meteoric-sulfuric-acid and sulfuric-acid particles). Horizontal dashes are median values, and the boxes are interquartile ranges. In this work, the lower stratosphere is defined as air masses above 8 km where the [O3] [CO] ratio is greater than 3.
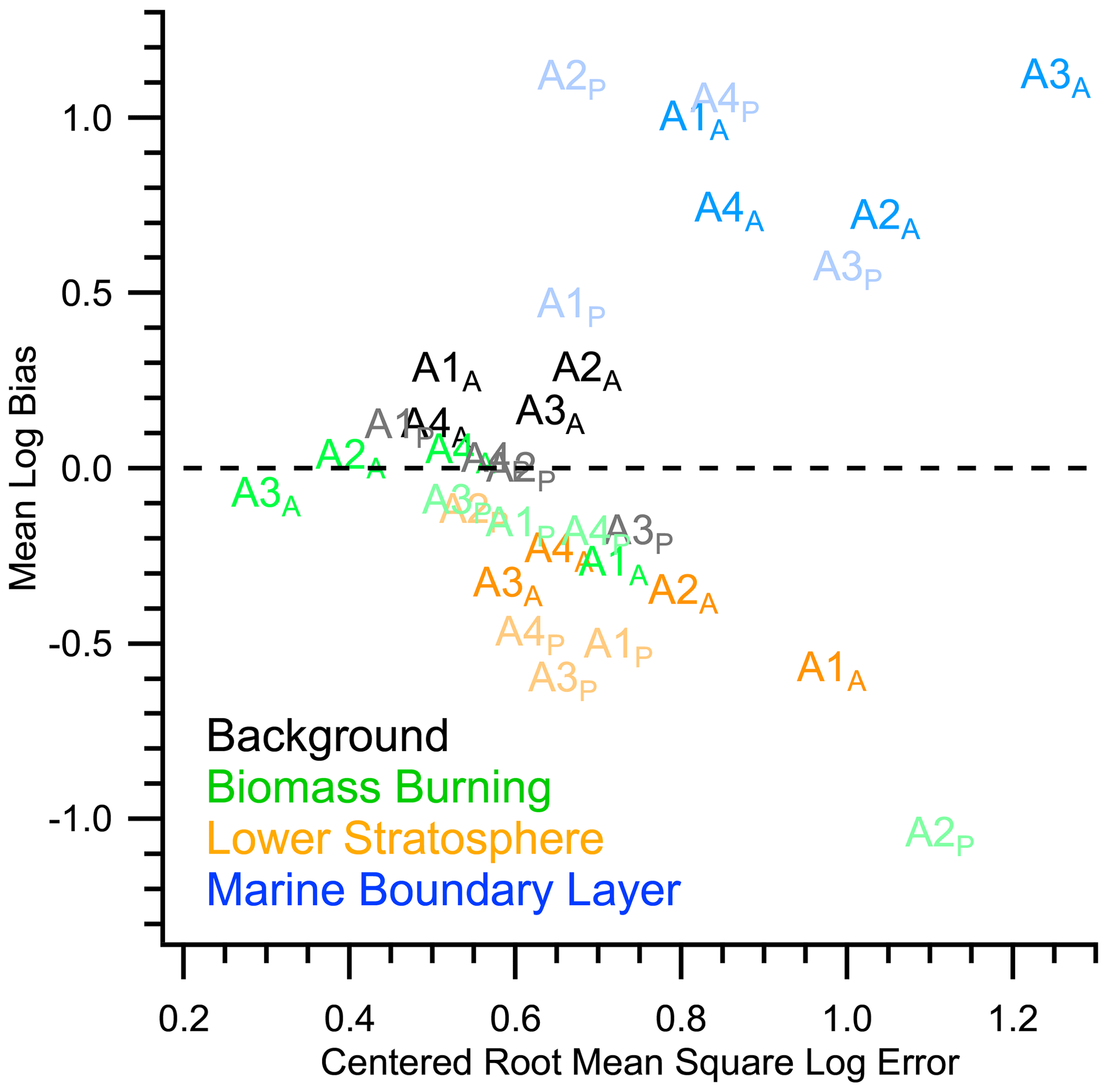
Figure A15Mean log bias vs. centered root mean square log error (CRMSLE) of modeled nSSA iodine from GEOS-Chem 12.9.1. Both the mean log bias and CRMSLE are calculated for each ATom and ocean basin (e.g., A1P is ATom-1 for the Pacific Ocean, and A1A is ATom-1 for the Atlantic Ocean). While the model is minimally biased in the background troposphere, the model tends to underestimate nSSA iodine in air influenced by biomass burning aerosol and in the lower stratosphere. Conversely, the model tends to overestimate the nSSA iodine in the marine boundary layer. Large biases are often associated with large CRMSLEs, which means that biases are not consistently offset and could be due to missing processes instead of systematic errors.
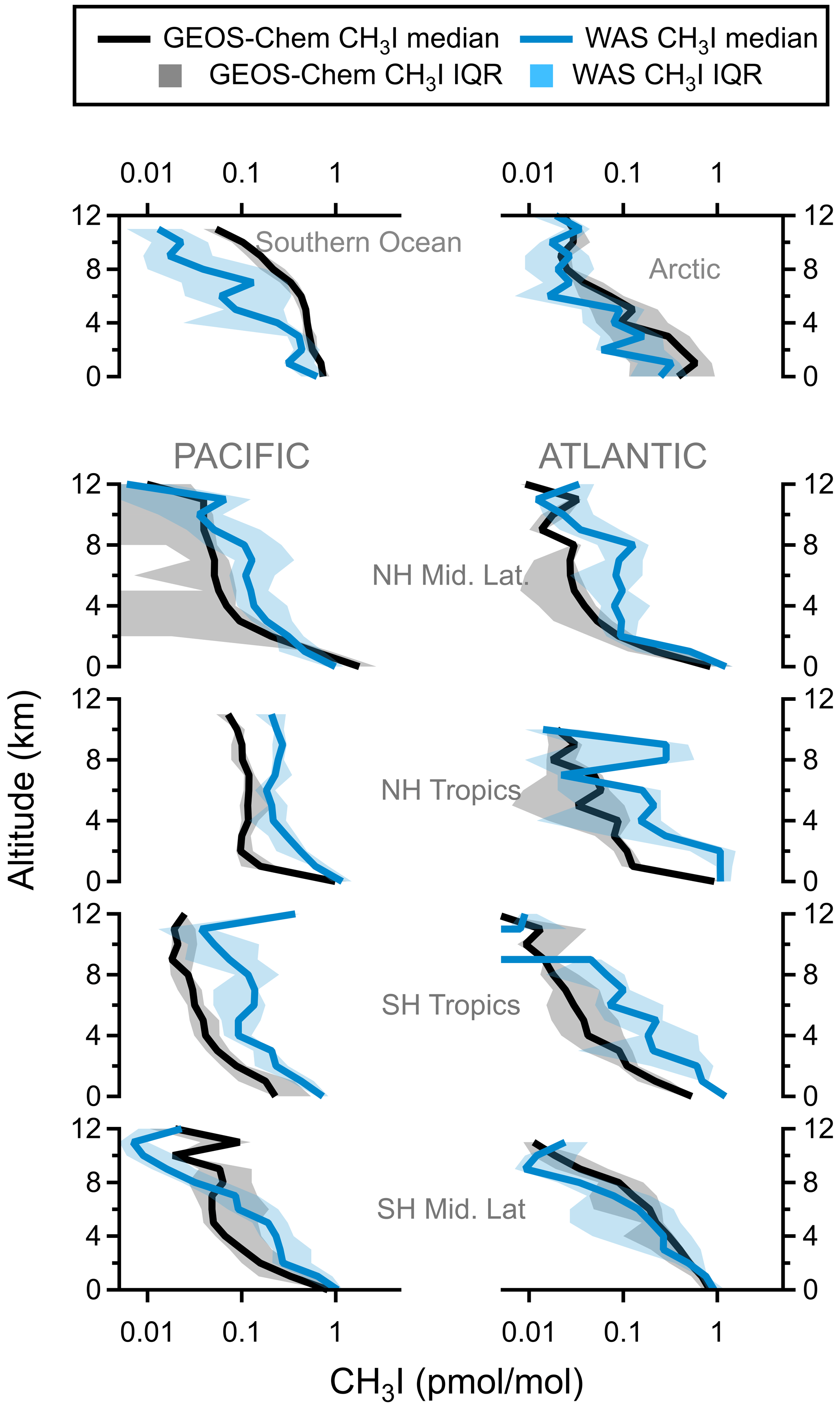
Figure A16Regional vertical profiles of CH3I (pmol mol−1). Median values (solid lines) and interquartile ranges (shading) are from all four ATom instances. Latitude cuts for each region are in Table S1 in Schill et al. (2020).
The data used in this paper are publicly available in online repositories. The ATom data can be found at https://doi.org/10.3334/ORNLDAAC/1925 (Wofsy et al., 2021). The CR-AVE data can be found at https://espoarchive.nasa.gov/archive/browse/cr_ave/WB57 (NASA, 2023a). The DC3 data can be found at https://www-air.larc.nasa.gov/cgi-bin/ArcView/dc3 (NASA, 2023b). Finally, the SEAC4RS data can be found at https://www-air.larc.nasa.gov/cgi-bin/ArcView/seac4rs (NASA, 2023c).
GPS, KDF, and DMM performed the PALMS measurements. CJW and CAB performed the size distribution measurements. TS and MJE performed the GEOS-Chem modeling. EAR performed the back trajectory analysis. ECA, RSS, and AJH performed the TOGA measurements. JP, TBR, CRT, and IB performed the NOyO3 measurements. DRB performed the WAS measurements. JPD and GSD performed the DLH measurements. All measurement performers analyzed their data. GPS wrote the manuscript draft. All authors reviewed and edited the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The mission as a whole was supported by NASA's Earth System Science Pathfinder Program EVS-2 (Earth Venture) funding. Participation in ATom flights by Gregory P. Schill, Karl D. Froyd, Christina J. Williamson, Charles A. Brock, and Daniel M. Murphy was supported by NOAA climate funding (grant no. NNH15AB12I). Gregory P. Schill, Karl D. Froyd, Christina J. Williamson, Jeff Peischl, Thomas B. Ryerson, Chelsea R. Thompson, and Ilann Bourgeois were supported by NOAA (cooperative agreement no. NA17OAR4320101). This material is based upon work supported by the NSF National Center for Atmospheric Research, which is a major facility sponsored by the US National Science Foundation (cooperative agreement no. 1852977).
This paper was edited by Anoop Mahajan and reviewed by Xu-Cheng He and one anonymous referee.
Allan, J. D., Williams, P. I., Najera, J., Whitehead, J. D., Flynn, M. J., Taylor, J. W., Liu, D., Darbyshire, E., Carpenter, L. J., Chance, R., Andrews, S. J., Hackenberg, S. C., and McFiggans, G.: Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA, Atmos. Chem. Phys., 15, 5599–5609, https://doi.org/10.5194/acp-15-5599-2015, 2015. a
Andreae, M. O.: Emission of trace gases and aerosols from biomass burning – an updated assessment, Atmos. Chem. Phys., 19, 8523–8546, https://doi.org/10.5194/acp-19-8523-2019, 2019. a
Apel, E. C., Hornbrook, R. S., Hills, A. J., Blake, N. J., Barth, M. C., Weinheimer, A., Cantrell, C., Rutledge, S. A., Basarab, B., Crawford, J., Diskin, G., Homeyer, C. R., Campos, T., Flocke, F., Fried, A., Blake, D. R., Brune, W., Pollack, I., Peischl, J., Ryerson, T., Wennberg, P. O., Crounse, J. D., Wisthaler, A., Mikoviny, T., Huey, G., Heikes, B., O'Sullivan, D., and Riemer, D. D.: Upper tropospheric ozone production from lightning NOx-impacted convection: Smoke ingestion case study from the DC3 campaign, J. Geophys. Res.-Atmos., 120, 2505–2523, https://doi.org/10.1002/2014JD022121, 2015. a
Badia, A., Reeves, C. E., Baker, A. R., Saiz-Lopez, A., Volkamer, R., Koenig, T. K., Apel, E. C., Hornbrook, R. S., Carpenter, L. J., Andrews, S. J., Sherwen, T., and von Glasow, R.: Importance of reactive halogens in the tropical marine atmosphere: a regional modelling study using WRF-Chem, Atmos. Chem. Phys., 19, 3161–3189, https://doi.org/10.5194/acp-19-3161-2019, 2019. a, b
Barth, M. C., Cantrell, C. A., Brune, W. H., Rutledge, S. A., Crawford, J. H., Huntrieser, H., Carey, L. D., MacGorman, D., Weisman, M., Pickering, K. E., Bruning, E., Anderson, B., Apel, E., Biggerstaff, M., Campos, T., Campuzano-Jost, P., Cohen, R., Crounse, J., Day, D. A., Diskin, G., Flocke, F., Fried, A., Garland, C., Heikes, B., Honomichl, S., Hornbrook, R., Gregory Huey, L., Jimenez, J. L., Lang, T., Lichtenstern, M., Mikoviny, T., Nault, B., O'Sullivan, D., Pan, L. L., Peischl, J., Pollack, I., Richter, D., Riemer, D., Ryerson, T., Schlager, H., St. Clair, J., Walega, J., Weibring, P., Weinheimer, A., Wennberg, P., Wisthaler, A., Wooldridge, P. J., and Ziegler, C.: The deep convective clouds and chemistry (DC3) field campaign, Bull. Am. Meteorol. Soc., 96, 1281–1310, https://doi.org/10.1175/BAMS-D-13-00290.1, 2015. a
Bourgeois, I., Peischl, J., Thompson, C. R., Aikin, K. C., Campos, T., Clark, H., Commane, R., Daube, B., Diskin, G. W., Elkins, J. W., Gao, R. S., Gaudel, A., Hintsa, E. J., Johnson, B. J., Kivi, R., McKain, K., Moore, F. L., Parrish, D. D., Querel, R., Ray, E., Sánchez, R., Sweeney, C., Tarasick, D. W., Thompson, A. M., Thouret, V., Witte, J. C., Wofsy, S. C., and Ryerson, T. B.: Global-scale distribution of ozone in the remote troposphere from the ATom and HIPPO airborne field missions, Atmos. Chem. Phys., 20, 10611–10635, https://doi.org/10.5194/acp-20-10611-2020, 2020. a, b
Bowman, K. P.: Large-scale isentropic mixing properties of the Antarctic polar vortex from analyzed winds, J. Geophys. Res., 98, 23013, https://doi.org/10.1029/93JD02599, 1993. a
Bowman, K. P. and Carrie, G. D.: The Mean-Meridional Transport Circulation of the Troposphere in an Idealized GCM, J. Atmos. Sci., 59, 1502–1514, https://doi.org/10.1175/1520-0469(2002)059<1502:TMMTCO>2.0.CO;2, 2002. a
Brock, C. A., Williamson, C., Kupc, A., Froyd, K. D., Erdesz, F., Wagner, N., Richardson, M., Schwarz, J. P., Gao, R.-S., Katich, J. M., Campuzano-Jost, P., Nault, B. A., Schroder, J. C., Jimenez, J. L., Weinzierl, B., Dollner, M., Bui, T., and Murphy, D. M.: Aerosol size distributions during the Atmospheric Tomography Mission (ATom): methods, uncertainties, and data products, Atmos. Meas. Tech., 12, 3081–3099, https://doi.org/10.5194/amt-12-3081-2019, 2019. a, b, c
Brock, C. A., Froyd, K. D., Dollner, M., Williamson, C. J., Schill, G., Murphy, D. M., Wagner, N. J., Kupc, A., Jimenez, J. L., Campuzano-Jost, P., Nault, B. A., Schroder, J. C., Day, D. A., Price, D. J., Weinzierl, B., Schwarz, J. P., Katich, J. M., Wang, S., Zeng, L., Weber, R., Dibb, J., Scheuer, E., Diskin, G. S., DiGangi, J. P., Bui, T., Dean-Day, J. M., Thompson, C. R., Peischl, J., Ryerson, T. B., Bourgeois, I., Daube, B. C., Commane, R., and Wofsy, S. C.: Ambient aerosol properties in the remote atmosphere from global-scale in situ measurements, Atmos. Chem. Phys., 21, 15023–15063, https://doi.org/10.5194/acp-21-15023-2021, 2021. a, b
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013. a, b, c
Colman, J. J., Swanson, A. L., Meinardi, S., Sive, B. C., Blake, D. R., and Rowland, F. S.: Description of the Analysis of a Wide Range of Volatile Organic Compounds in Whole Air Samples Collected during PEM-Tropics A and B, Anal. Chem., 73, 3723–3731, https://doi.org/10.1021/ac010027g, 2001. a
Cuevas, C. A., Maffezzoli, N., Corella, J. P., Spolaor, A., Vallelonga, P., Kjær, H. A., Simonsen, M., Winstrup, M., Vinther, B., Horvat, C., Fernandez, R. P., Kinnison, D., Lamarque, J.-F., Barbante, C., and Saiz-Lopez, A.: Rapid increase in atmospheric iodine levels in the North Atlantic since the mid-20th century, Nat. Commun., 9, 1452, https://doi.org/10.1038/s41467-018-03756-1, 2018. a, b
Daniel, J. S., Solomon, S., Portmann, R. W., and Garcia, R. R.: Stratospheric ozone destruction: The importance of bromine relative to chlorine, J. Geophys. Res.-Atmos., 104, 23871–23880, https://doi.org/10.1029/1999JD900381, 1999. a
Dean, G. A.: The iodine content of some New Zealand drinking waters with a note on the contribution from sea spray to the iodine in rain, New Zeal. J. Sci., 6, 208–214, 1963. a
Diskin, G. S., Podolske, J. R., Sachse, G. W., and Slate, T. A.: Open-path airborne tunable diode laser hygrometer, in: Proc.SPIE, edited by: Fried, A., Vol. 4817, Diode Lasers and Applications in Atmospheric Sensing, https://doi.org/10.1117/12.453736, 2002. a
Duce, R. A., Arimoto, R., Ray, B. J., Unni, C. K., and Harder, P. J.: Atmospheric trace elements at Enewetak Atoll: 1. Concentrations, sources, and temporal variability, J. Geophys. Res.-Ocean., 88, 5321–5342, https://doi.org/10.1029/JC088iC09p05321, 1983. a
Eastham, S. D., Weisenstein, D. K., and Barrett, S. R.: Development and evaluation of the unified tropospheric–stratospheric chemistry extension (UCX) for the global chemistry-transport model GEOS-Chem, Atmos. Environ., 89, 52–63, https://doi.org/10.1016/j.atmosenv.2014.02.001, 2014. a
Finkenzeller, H., Iyer, S., He, X.-C., Simon, M., Koenig, T. K., Lee, C. F., Valiev, R., Hofbauer, V., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bell, D. M., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dada, L., Duplissy, J., Heinritzi, M., Kemppainen, D., Kim, C., Krechmer, J., Kürten, A., Kvashnin, A., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mauldin, R. L., Mentler, B., Müller, T., Petäjä, T., Philippov, M., Ranjithkumar, A., Rörup, B., Shen, J., Stolzenburg, D., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, D. S., Wang, M., Wang, Y., Weber, S. K., Nie, W., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Hansel, A., Baltensperger, U., Brioude, J., Curtius, J., Donahue, N. M., Haddad, I. E., Flagan, R. C., Kulmala, M., Kirkby, J., Sipilä, M., Worsnop, D. R., Kurten, T., Rissanen, M., and Volkamer, R.: The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 15, 129–135, https://doi.org/10.1038/s41557-022-01067-z, 2023. a
Froyd, K. D., Murphy, D. M., Brock, C. A., Campuzano-Jost, P., Dibb, J. E., Jimenez, J.-L., Kupc, A., Middlebrook, A. M., Schill, G. P., Thornhill, K. L., Williamson, C. J., Wilson, J. C., and Ziemba, L. D.: A new method to quantify mineral dust and other aerosol species from aircraft platforms using single-particle mass spectrometry, Atmos. Meas. Tech., 12, 6209–6239, https://doi.org/10.5194/amt-12-6209-2019, 2019. a, b, c, d, e
Fudge, A. J. and Sykes, K. W.: 25. The reaction between ferric and iodide ions. Part I. Kinetics and mechanism, J. Chem. Soc., 1952, 119–124, https://doi.org/10.1039/jr9520000119, 1952. a
Gao, R. S., Hall, S. R., Swartz, W. H., Schwarz, J. P., Spackman, J. R., Watts, L. A., Fahey, D. W., Aikin, K. C., Shetter, R. E., and Bui, T. P.: Calculations of solar shortwave heating rates due to black carbon and ozone absorption using in situ measurements, J. Geophys. Res.-Atmos., 113, D14203, https://doi.org/10.1029/2007JD009358, 2008. a
Gelaro, R., McCarty, W., Suárez, M. J., Todling, R., Molod, A., Takacs, L., Randles, C. A., Darmenov, A., Bosilovich, M. G., Reichle, R., Wargan, K., Coy, L., Cullather, R., Draper, C., Akella, S., Buchard, V., Conaty, A., da Silva, A. M., Gu, W., Kim, G.-K., Koster, R., Lucchesi, R., Merkova, D., Nielsen, J. E., Partyka, G., Pawson, S., Putman, W., Rienecker, M., Schubert, S. D., Sienkiewicz, M., and Zhao, B.: The Modern-Era Retrospective Analysis for Research and Applications, Version 2 (MERRA-2), J. Clim., 30, 5419–5454, https://doi.org/10.1175/JCLI-D-16-0758.1, 2017. a
Gilfedder, B. S., Lai, S. C., Petri, M., Biester, H., and Hoffmann, T.: Iodine speciation in rain, snow and aerosols, Atmos. Chem. Phys., 8, 6069–6084, https://doi.org/10.5194/acp-8-6069-2008, 2008. a
Gómez Martín, J. C., Lewis, T. R., Blitz, M. A., Plane, J. M. C., Kumar, M., Francisco, J. S., and Saiz-Lopez, A.: A gas-to-particle conversion mechanism helps to explain atmospheric particle formation through clustering of iodine oxides, Nat. Commun., 11, 4521, https://doi.org/10.1038/s41467-020-18252-8, 2020. a
Gómez Martín, J. C., Saiz-Lopez, A., Cuevas, C. A., Fernandez, R. P., Gilfedder, B., Weller, R., Baker, A. R., Droste, E., and Lai, S.: Spatial and Temporal Variability of Iodine in Aerosol, J. Geophys. Res.-Atmos., 126, e2020JD034410, https://doi.org/10.1029/2020JD034410, 2021. a
Gómez Martín, J. C., Saiz‐Lopez, A., Cuevas, C. A., Baker, A. R., and Fernández, R. P.: On the Speciation of Iodine in Marine Aerosol, J. Geophys. Res.-Atmos., 127, e2021JD036081, https://doi.org/10.1029/2021JD036081, 2022. a, b, c, d
Hara, K., Osada, K., Matsunaga, K., Iwasaka, Y., Shibata, T., and Furuya, K.: Atmospheric inorganic chlorine and bromine species in Arctic boundary layer of the winter/spring, J. Geophys. Res.-Atmos., 107, 4361, https://doi.org/10.1029/2001JD001008, 2002. a
He, X.-c., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L. J., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., Donahue, N. M., Volkamer, R., Kirkby, J., Worsnop, D. R., and Sipilä, M.: Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 371, 589–595, https://doi.org/10.1126/science.abe0298, 2021. a, b
Hoesly, R. M., Smith, S. J., Feng, L., Klimont, Z., Janssens-Maenhout, G., Pitkanen, T., Seibert, J. J., Vu, L., Andres, R. J., Bolt, R. M., Bond, T. C., Dawidowski, L., Kholod, N., Kurokawa, J.-i., Li, M., Liu, L., Lu, Z., Moura, M. C. P., O'Rourke, P. R., and Zhang, Q.: Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emissions Data System (CEDS), Geosci. Model Dev., 11, 369–408, https://doi.org/10.5194/gmd-11-369-2018, 2018. a
Keber, T., Bönisch, H., Hartick, C., Hauck, M., Lefrancois, F., Obersteiner, F., Ringsdorf, A., Schohl, N., Schuck, T., Hossaini, R., Graf, P., Jöckel, P., and Engel, A.: Bromine from short-lived source gases in the extratropical northern hemispheric upper troposphere and lower stratosphere (UTLS), Atmos. Chem. Phys., 20, 4105–4132, https://doi.org/10.5194/acp-20-4105-2020, 2020. a
Koenig, T. K., Baidar, S., Campuzano-Jost, P., Cuevas, C. A., Dix, B., Fernandez, R. P., Guo, H., Hall, S. R., Kinnison, D., Nault, B. A., Ullmann, K., Jimenez, J. L., Saiz-Lopez, A., and Volkamer, R.: Quantitative detection of iodine in the stratosphere, P. Natl Acad. Sci. USA, 117, 1860–1866, https://doi.org/10.1073/pnas.1916828117, 2020. a, b, c, d, e, f, g, h, i, j
Legrand, M., McConnell, J. R., Preunkert, S., Arienzo, M., Chellman, N., Gleason, K., Sherwen, T., Evans, M. J., and Carpenter, L. J.: Alpine ice evidence of a three-fold increase in atmospheric iodine deposition since 1950 in Europe due to increasing oceanic emissions, P. Natl Acad. Sci. USA, 115, 12136–12141, https://doi.org/10.1073/pnas.1809867115, 2018. a
, McNaughton, C. S., Clarke, A. D., Howell, S. G., Pinkerton, M., Anderson, B., Thornhill, L., Hudgins, C., Winstead, E., Dibb, J. E., Scheuer, E., and Maring, H.: Results from the DC-8 Inlet Characterization Experiment (DICE): Airborne Versus Surface Sampling of Mineral Dust and Sea Salt Aerosols, Aerosol Sci. Tech., 41, 136–159, https://doi.org/10.1080/02786820601118406, 2007. a
Mu, M., Randerson, J. T., van der Werf, G. R., Giglio, L., Kasibhatla, P., Morton, D., Collatz, G. J., DeFries, R. S., Hyer, E. J., Prins, E. M., Griffith, D. W. T., Wunch, D., Toon, G. C., Sherlock, V., and Wennberg, P. O.: Daily and 3-hourly variability in global fire emissions and consequences for atmospheric model predictions of carbon monoxide, J. Geophys. Res.-Atmos., 116, D24303, https://doi.org/10.1029/2011JD016245, 2011. a
Murphy, D. M. and Thomson, D. S.: Halogen ions and NO+ in the mass spectra of aerosols in the upper troposphere and lower stratosphere, Geophys. Res. Lett., 27, 3217–3220, https://doi.org/10.1029/1999GL011267, 2000. a
Murphy, D. M., Thomson, D. S., and Middlebrook, A. M.: Bromine, iodine, and chlorine in single aerosol particles at Cape Grim, Geophys. Res. Lett., 24, 3197–3200, https://doi.org/10.1029/97GL03195, 1997. a, b
Murphy, D. M., Froyd, K. D., Bourgeois, I., Brock, C. A., Kupc, A., Peischl, J., Schill, G. P., Thompson, C. R., Williamson, C. J., and Yu, P.: Radiative and chemical implications of the size and composition of aerosol particles in the existing or modified global stratosphere, Atmos. Chem. Phys., 21, 8915–8932, https://doi.org/10.5194/acp-21-8915-2021, 2021. a, b, c, d, e, f
NASA: ESPO Data Archive: CR-AVE, NASA [data set], https://espoarchive.nasa.gov/archive/browse/cr_ave/WB57, last access: 1 November 2023a. a
NASA: ESPO Data Archive: DC-3, NASA [data set], https://www-air.larc.nasa.gov/cgi-bin/ArcView/dc3, last access: 1 November 2023b. a
NASA: ESPO Data Archive: CR-AVE, NASA [data set], https://www-air.larc.nasa.gov/cgi-bin/ArcView/seac4rs, last access: 1 November 2023c. a
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämeri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002. a
Ofner, J., Balzer, N., Buxmann, J., Grothe, H., Schmitt-Kopplin, P., Platt, U., and Zetzsch, C.: Halogenation processes of secondary organic aerosol and implications on halogen release mechanisms, Atmos. Chem. Phys., 12, 5787–5806, https://doi.org/10.5194/acp-12-5787-2012, 2012. a
Ordóñez, C., Lamarque, J.-F., Tilmes, S., Kinnison, D. E., Atlas, E. L., Blake, D. R., Sousa Santos, G., Brasseur, G., and Saiz-Lopez, A.: Bromine and iodine chemistry in a global chemistry-climate model: description and evaluation of very short-lived oceanic sources, Atmos. Chem. Phys., 12, 1423–1447, https://doi.org/10.5194/acp-12-1423-2012, 2012. a
Pechtl, S., Schmitz, G., and von Glasow, R.: Modelling iodide – iodate speciation in atmospheric aerosol: Contributions of inorganic and organic iodine chemistry, Atmos. Chem. Phys., 7, 1381–1393, https://doi.org/10.5194/acp-7-1381-2007, 2007. a
Reinhardt, T. E. and Ward, D. E.: Factors Affecting Methyl Chloride Emissions from Forest Biomass Combustion, Environ. Sci. Technol., 29, 825–832, https://doi.org/10.1021/es00003a034, 1995. a
Rörup, B., He, X.-C., Shen, J., Baalbaki, R., Dada, L., Sipilä, M., Kirkby, J., Kulmala, M., Amorim, A., Baccarini, A., Bell, D. M., Caudillo-Plath, L., Duplissy, J., Finkenzeller, H., Kürten, A., Lamkaddam, H., Lee, C. P., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mentler, B., Onnela, A., Philippov, M., Scholz, C. W., Simon, M., Stolzenburg, D., Tham, Y. J., Tomé, A., Wagner, A. C., Wang, M., Wang, D., Wang, Y., Weber, S. K., Zauner-Wieczorek, M., Baltensperger, U., Curtius, J., Donahue, N. M., El Haddad, I., Flagan, R. C., Hansel, A., Möhler, O., Petäjä, T., Volkamer, R., Worsnop, D., and Lehtipalo, K.: Temperature, humidity, and ionisation effect of iodine oxoacid nucleation, Environ. Sci.-Atmos., 4, 531–546, https://doi.org/10.1039/D4EA00013G, 2024. a
Saiz-Lopez, A., Plane, J. M. C., Baker, A. R., Carpenter, L. J., von Glasow, R., Gómez Martín, J. C., McFiggans, G., and Saunders, R. W.: Atmospheric Chemistry of Iodine, Chem. Rev., 112, 1773–1804, https://doi.org/10.1021/cr200029u, 2012. a, b, c
Saiz-Lopez, A., Fernandez, R. P., Ordóñez, C., Kinnison, D. E., Gómez Martín, J. C., Lamarque, J.-F., and Tilmes, S.: Iodine chemistry in the troposphere and its effect on ozone, Atmos. Chem. Phys., 14, 13119–13143, https://doi.org/10.5194/acp-14-13119-2014, 2014. a, b
Saiz‐Lopez, A., Baidar, S., Cuevas, C. A., Koenig, T. K., Fernandez, R. P., Dix, B., Kinnison, D. E., Lamarque, J.-F., Rodriguez‐Lloveras, X., Campos, T. L., and Volkamer, R.: Injection of iodine to the stratosphere, Geophys. Res. Lett., 42, 6852–6859, https://doi.org/10.1002/2015GL064796, 2015. a
Sander, R., Keene, W. C., Pszenny, A. A. P., Arimoto, R., Ayers, G. P., Baboukas, E., Cainey, J. M., Crutzen, P. J., Duce, R. A., Hönninger, G., Huebert, B. J., Maenhaut, W., Mihalopoulos, N., Turekian, V. C., and Van Dingenen, R.: Inorganic bromine in the marine boundary layer: a critical review, Atmos. Chem. Phys., 3, 1301–1336, https://doi.org/10.5194/acp-3-1301-2003, 2003. a, b
Schill, G. P., Froyd, K. D., Bian, H., Kupc, A., Williamson, C., Brock, C. A., Ray, E., Hornbrook, R. S., Hills, A. J., Apel, E. C., Chin, M., Colarco, P. R., and Murphy, D. M.: Widespread biomass burning smoke throughout the remote troposphere, Nat. Geosci., 13, 422–427, https://doi.org/10.1038/s41561-020-0586-1, 2020. a, b, c, d, e, f, g, h, i, j, k
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 121, 11819–11835, https://doi.org/10.1002/2015JD024229, 2016. a
Sherwen, T., Evans, M. J., Carpenter, L. J., Andrews, S. J., Lidster, R. T., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Iodine's impact on tropospheric oxidants: a global model study in GEOS-Chem, Atmos. Chem. Phys., 16, 1161–1186, https://doi.org/10.5194/acp-16-1161-2016, 2016a. a
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016b. a, b, c, d, e, f
Sherwen, T., Evans, M. J., Sommariva, R., Hollis, L. D. J., Ball, S. M., Monks, P. S., Reed, C., Carpenter, L. J., Lee, J. D., Forster, G., Bandy, B., Reeves, C. E., and Bloss, W. J.: Effects of halogens on European air-quality, Faraday Discuss., 200, 75–100, https://doi.org/10.1039/C7FD00026J, 2017. a
Sherwen, T. M., Evans, M. J., Spracklen, D. V., Carpenter, L. J., Chance, R., Baker, A. R., Schmidt, J. A., and Breider, T. J.: Global modeling of tropospheric iodine aerosol, Geophys. Res. Lett., 43, 10012–10019, https://doi.org/10.1002/2016GL070062, 2016c. a, b, c
Sofiev, M., Ermakova, T., and Vankevich, R.: Evaluation of the smoke-injection height from wild-land fires using remote-sensing data, Atmos. Chem. Phys., 12, 1995–2006, https://doi.org/10.5194/acp-12-1995-2012, 2012. a
Solomon, S., Garcia, R. R., and Ravishankara, A. R.: On the role of iodine in ozone depletion, J. Geophys. Res., 99, 20491, https://doi.org/10.1029/94JD02028, 1994. a
Solomon, S., Stone, K., Yu, P., Murphy, D. M., Kinnison, D., Ravishankara, A. R., and Wang, P.: Chlorine activation and enhanced ozone depletion induced by wildfire aerosol, Nature, 615, 259–264, https://doi.org/10.1038/s41586-022-05683-0, 2023. a, b, c
Stachnik, R. A., Millán, L., Jarnot, R., Monroe, R., McLinden, C., Kühl, S., Puķīte, J., Shiotani, M., Suzuki, M., Kasai, Y., Goutail, F., Pommereau, J. P., Dorf, M., and Pfeilsticker, K.: Stratospheric BrO abundance measured by a balloon-borne submillimeterwave radiometer, Atmos. Chem. Phys., 13, 3307–3319, https://doi.org/10.5194/acp-13-3307-2013, 2013. a
Sturges, W. and Barrie, L.: Chlorine, Bromine AND Iodine in arctic aerosols, Atmos. Environ., 22, 1179–1194, https://doi.org/10.1016/0004-6981(88)90349-6, 1988. a
Toon, O. B., Maring, H., Dibb, J., Ferrare, R., Jacob, D. J., Jensen, E. J., Luo, Z. J., Mace, G. G., Pan, L. L., Pfister, L., Rosenlof, K. H., Redemann, J., Reid, J. S., Singh, H. B., Thompson, A. M., Yokelson, R., Minnis, P., Chen, G., Jucks, K. W., and Pszenny, A.: Planning, implementation, and scientific goals of the Studies of Emissions and Atmospheric Composition, Clouds and Climate Coupling by Regional Surveys (SEAC 4 RS) field mission, J. Geophys. Res.-Atmos., 121, 4967–5009, https://doi.org/10.1002/2015JD024297, 2016. a
Vogt, R.: Iodine Compounds in the Atmosphere, Springer-Verlag, Berlin/Heidelberg, 113–128, ISBN 978-3-540-69690-2, https://doi.org/10.1007/10628761_4, 1999. a
Volkamer, R., Baidar, S., Campos, T. L., Coburn, S., DiGangi, J. P., Dix, B., Eloranta, E. W., Koenig, T. K., Morley, B., Ortega, I., Pierce, B. R., Reeves, M., Sinreich, R., Wang, S., Zondlo, M. A., and Romashkin, P. A.: Aircraft measurements of BrO, IO, glyoxal, NO2, H2O, O2–O2 and aerosol extinction profiles in the tropics: comparison with aircraft-/ship-based in situ and lidar measurements, Atmos. Meas. Tech., 8, 2121–2148, https://doi.org/10.5194/amt-8-2121-2015, 2015. a, b
Wang, S., Schmidt, J. A., Baidar, S., Coburn, S., Dix, B., Koenig, T. K., Apel, E., Bowdalo, D., Campos, T. L., Eloranta, E., Evans, M. J., DiGangi, J. P., Zondlo, M. A., Gao, R.-S., Haggerty, J. A., Hall, S. R., Hornbrook, R. S., Jacob, D., Morley, B., Pierce, B., Reeves, M., Romashkin, P., ter Schure, A., and Volkamer, R.: Active and widespread halogen chemistry in the tropical and subtropical free troposphere, P. Natl. Acad. Sci. USA, 112, 9281–9286, https://doi.org/10.1073/pnas.1505142112, 2015. a
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019. a
Wisthaler, A., Hansel, A., Dickerson, R. R., and Crutzen, P. J.: Organic trace gas measurements by PTR‐MS during INDOEX 1999, J. Geophys. Res.-Atmos., 107, 8024, https://doi.org/10.1029/2001JD000576, 2002. a
Wofsy, S. C., Afshar, S., Allen, H. M., Apel, E. C., Asher, E. C., Barletta, B., Bent, J., Bian, H., Biggs, B. C., Blake, D. R., Blake, N., Bourgeois, I., Brock, C. A., Brune, W. H., Budney, J. W., Bui, T. P., Butler, A., Campuzano-Jost, P., Chang, C. S., Chin, M., Commane, R., Correa, G., Crounse, J. D., Cullis, P. D., Daube, B. C., Day, D. A., Dean-Day, J. M., Dibb, J. E., DiGangi, J. P., Diskin, G. S., Dollner, M., Elkins, J. W., Erdesz, F., Fiore, A. M., Flynn, C. M., Froyd, K. D., Gesler, D. W., Hall, S. R., Hanisco, T. F., Hannun, R. A., Hills, A. J., Hintsa, E. J., Hoffman, A., Hornbrook, R. S., Huey, L. G., Hughes, S., Jimenez, J. L., Johnson, B. J., Katich, J. M., Keeling, R. F., Kim, M. J., Kupc, A., Lait, L. R., McKain, K., Mclaughlin, R. J., Meinardi, S., Miller, D. O., Montzka, S. A., Moore, F. L., Morgan, E. J., Murphy, D. M., Murray, L. T., Nault, B. A., Neuman, J. A., Newman, P. A., Nicely, J. M., Pan, X., Paplawsky, W., Peischl, J., Prather, M J., Price, D. J., Ray, E. A., Reeves, J. M., Richardson, M., Rollins, A. W., Rosenlof, K. H., Ryerson, T. B., Scheuer, E., Schill, G. P., Schroder, J. C., Schwarz, J. P., St.Clair, J. M., Steenrod, S. D., Stephens, B. B., Strode, S. A., Sweeney, C., Tanner, D., Teng, A. P., Thames, A. B., Thompson, C. R., Ullmann, K., Veres, P. R., Wagner, N. L., Watt, A., Weber, R., Weinzierl, B. B., Wennberg, P. O., Williamson, C. J., Wilson, J. C., Wolfe, G. M., Woods, C. T., Zeng, L. H., and Vieznor, N.: ATom: Merged Atmospheric Chemistry, Trace Gases, and Aerosols, Version 2, ORNL Distributed Active Archive Center [data set], https://doi.org/10.3334/ORNLDAAC/1925, 2021. a
Zhu, L., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Wang, X., Sherwen, T., Evans, M. J., Chen, Q., Alexander, B., Koenig, T. K., Volkamer, R., Huey, L. G., Le Breton, M., Bannan, T. J., and Percival, C. J.: Effect of sea salt aerosol on tropospheric bromine chemistry, Atmos. Chem. Phys., 19, 6497–6507, https://doi.org/10.5194/acp-19-6497-2019, 2019. a