the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 25 Mar 2025
| 25 Mar 2025
Kinetics of the reactions of OH with CO, NO, and NO2 and of HO2 with NO2 in air at 1 atm pressure, room temperature, and tropospheric water vapour concentrations
Michael Rolletter
Andreas Hofzumahaus
Anna Novelli
Andreas Wahner
The termolecular reactions of hydroxyl radicals (OH) with carbon monoxide (CO), nitric oxide (NO), and nitrogen dioxides (NO2) and the termolecular reaction of hydroperoxy radicals (HO2) with NO2 greatly impact the atmospheric oxidation efficiency. Few studies have directly measured the pressure-dependent rate coefficients in air at 1 atm pressure and water vapour as third-body collision partners. In this work, rate coefficients were measured with a high accuracy (<5 %) at 1 atm pressure, at room temperature, and in humidified air using laser flash photolysis and detection of the radical decay by laser-induced fluorescence. The rate coefficients derived in dry air are cm3 s−1 for the OH reaction with CO, cm3 s−1 for the OH reaction with NO, cm3 s−1 for the OH reaction with NO2, and cm3 s−1 for the HO2 reaction with NO2. For the OH reactions with CO and NO, no dependence on water vapour was observed for the range of water partial pressures tested (3 to 22 hPa), and for NO2, only a weak increase of 3 % was measured, in agreement with the study by Amedro et al. (2020). For the rate coefficient of HO2 with NO2 an enhancement of up to 25 % was observed. This can be explained by a faster rate coefficient of the reaction of the HO2–water complex with NO2 having a value of cm3 s−1.
- Article
(4012 KB) - Full-text XML
- BibTeX
- EndNote




The inorganic pressure-dependent reactions of the OH radical with CO, NO, and NO2 and of HO2 with NO2 link the chemistry of HOx (the sum of OH and HO2) and NOx (NO and NO2) in the atmosphere and affect largely the chemical transformation of pollutants (Newsome and Evans, 2017). The OH radical is the most important oxidant, reacting with most volatile compounds. Its reaction with pollutants initiates radical chain reactions in which HO2 radicals are often formed and in which OH can be eventually regenerated. In the troposphere, for example, CO, which is emitted from combustion processes, is oxidised in the termolecular reaction with OH.
M is a third-body collision partner. The reaction of OH with CO has been studied experimentally and theoretically over a wide range of temperatures and pressures because of its general importance in the planetary atmospheres of Earth and Mars (Atkinson et al., 2004; Burkholder et al., 2020).
Nitrogen oxides are mainly emitted by combustion processes and produced in the atmosphere by lightning. They play an important role in atmospheric radical chemistry in several ways. The reactions of NO with peroxy radicals are responsible for the regeneration of OH radicals. Conversely, the reactions of OH with NO and NO2 and of HO2 with NO2 form products that terminate the cyclic chain reactions between OH and HO2 and can produce long-lived compounds that can act as radical reservoirs. In addition, the oxidation reaction of NO to NO2 by peroxy radicals followed by NO2 photolysis is the only relevant chemical source of tropospheric ozone (Ehhalt, 1999).
The termolecular reaction of OH with NO produces nitrous acid (HONO).
HONO can be rapidly photolysed so that OH, NO, and HONO concentrations are in a photochemical equilibrium at daytime (Kleffmann et al., 2005).
The reaction of OH with NO2 is a termolecular reaction leading to the formation of nitric acid (HNO3) or pernitrous acid (HOONO).
In the lower troposphere, HNO3 is mainly lost by surface deposition due to its long chemical lifetime. The reaction channel leading to its formation is therefore a net loss of OH radicals and nitrogen oxides. In contrast, HOONO is thermally unstable and decomposes mainly in the boundary layer at mid-latitude temperatures so that there is no net loss of the reactants. If HOONO underwent other atmospheric reactions, its formation would be a radical and NO2 sink, but such reactions have not been reported. The branching ratio between Reactions (R4) and (R5) increases with pressure and is approximately 14 % at atmospheric pressure and room temperature (Mollner et al., 2010).
Pernitric acid (HO2NO2), formed by the termolecular reaction of hydroperoxyl radicals (HO2) and NO2, can decompose thermally in the troposphere so that their concentrations are in a thermal equilibrium (Gierczak et al., 2005).
In the upper troposphere and lower stratosphere, where HO2NO2 is thermally stable due to the cold temperatures, its subsequent reaction with OH is an important sink for HOx radicals (Kim et al., 2007). Measurements of HO2NO2 can also be used to diagnose HO2 and NO2 concentrations, but accurate rate coefficients are required to calculate steady-state concentrations.
The reactions of OH with CO, NO, and NO2 and the reaction of HO2 with NO2 are termolecular reactions, in which an activated association complex is formed. The rates of dissociation and collisional stabilisation of the activated complex determine the rate coefficients of the overall reaction. Therefore, the rate coefficients are pressure-dependent (expressed as the number density concentration of the bath gas molecules, M), which can be parameterised by the Troe formalism (Troe, 1983). The Troe expression parameterises the rate using high-pressure (k∞) and low-pressure (k0) limiting rate coefficients. A “fall-off” transition is described by the broadening factor F. The expression used e.g. by IUPAC is (Atkinson et al., 2004)
where m and n are dimensionless temperature exponents. The broadening factor F is
with and Fc being the broadening factor at the centre of the fall-off transition. The parameterisation by NASA–JPL is only slightly different.
Despite the importance of these reactions for the atmospheric cycle of radicals and nitrogen oxides, there have only been a few studies that have directly measured their rate coefficients in air at 1 atm pressure.
The evaluations of rate coefficients are based on the limited data reported in the literature, resulting in notable differences in the values recommended by NASA–JPL (Burkholder et al., 2020) and IUPAC (Atkinson et al., 2004) at 1 atm. For example, the recommendations differ by a factor of 1.3 for the OH reaction with NO (within the stated accuracies) and by a factor of 1.8 for the HO2 reaction with NO2 (higher than the stated accuracies). Consequently, the predictions of atmospheric chemistry models that rely on recommendations in databases may be subject to considerable uncertainties, emphasising the need for further laboratory studies to reduce the uncertainties (Burkholder et al., 2017; Fiore et al., 2024; Ervens et al., 2024).
Previous studies have shown that the presence of water vapour can affect the rate coefficients of OH and HO2 reactions through the formation of a hydrogen-bonded complex between HO2 and a water molecule (Cox and Burrows, 1979; Aloisio et al., 2000; Kanno et al., 2005; Buszek et al., 2011) or by collisional stabilisation of the activated association complex by water molecules (Amedro et al., 2020; Sun et al., 2022).
For example, significantly increased rate coefficients have been observed in the self-reaction of HO2 (e.g. Lii et al., 1981; Kircher and Sander, 1984) or in the reaction of HO2 with NO2 (Sander and Peterson, 1984) at low pressure in the presence of water vapour. However, with a few exceptions, such as the self-reaction of HO2, possible water vapour dependencies have not been considered in the NASA–JPL and IUPAC recommendations due to the lack of sufficient experimental data.
In this work, a laser flash photolysis laser-induced fluorescence (LP-LIF) method was used to generate OH radicals by ozone photolysis in a flow tube and to observe the rate of their chemical decay. Unlike with many pump-and-probe instruments, the radical detection does not take place in the reaction volume but in a low-pressure cell, which allows an extremely sensitive OH fluorescence detection.
The instrument was originally developed to measure the chemical OH lifetime in ambient air at tropospheric conditions (Hofzumahaus et al., 2009; Lou et al., 2010). In atmospheric studies, the measured OH lifetime is a valuable kinetic parameter which can be used to determine the production and destruction rates of atmospheric OH, allowing the quantification of potentially unknown sources and sinks (Hofzumahaus et al., 2009; Lou et al., 2010; Kovacs and Brune, 2001; Martinez et al., 2003; Sadanaga et al., 2004a; Whalley et al., 2011; Fuchs et al., 2013, 2014; Griffith et al., 2016; Yang et al., 2016).
The inverse atmospheric OH lifetime is called the total OH reactivity (kOH) and is equal to the pseudo-first-order loss rate coefficient. Its value depends on the concentrations of all atmospheric reactants i (e.g. CO, NOx, hydrocarbons) and their second-order rate coefficients (kOH+i).
In the lower troposphere, observed OH reactivity values are in the range from 1 to 100 s−1 for conditions ranging from very clean to extremely polluted air (Hofzumahaus et al., 2009; Yang et al., 2016).
In this work, the instrument was used to determine the rate coefficients of the reaction of OH with CO, NO, and NO2 and of HO2 with NO2 in air at atmospheric pressure and room temperature and in the presence of water vapour. Similar reactivity instruments have been used previously for kinetic studies of OH (Sadanaga et al., 2004b; Amedro et al., 2012; Nakashima et al., 2012; Stone et al., 2016; Speak et al., 2020; Berg et al., 2024; Chao et al., 2019; Sheps and Au, 2024). The method can also be used to study the kinetics of HO2 radicals by adding excess CO in the flow tube to convert all initially produced OH to HO2 (Nehr et al., 2011, 2012; Zhou et al., 2019). In this work, the reaction of HO2 with NO2 was studied using this approach.
2.1 Measurement of pseudo-first-order rate coefficients
The central components of the laser flash photolysis laser-induced fluorescence (LP-LIF) instrument used in this work to determine OH and HO2 rate coefficients are a laminar flow tube reactor, in which OH radicals are produced by flash photolysis, and an attached fluorescence detection cell for measuring the OH decay (Fig. 1). The flow tube has a total length of 80 cm and an internal diameter of 40 mm. It is made of black anodised aluminium and is sealed at both ends by fused silica quartz windows with an antireflective coating for 266 nm (Laser Optics). The distance between the entrance of the flow tube and the sampling point of the detection cell is 50 cm. The air is replaced every 1.8 s using a mass flow controller (Bronkhorst, low Δp series, flow rate: 21 L min−1) backed by a vacuum pump (Vacuubrand, MD4C). For the experiments in this work, the flow tube was kept at room temperature and ambient pressure. Sensors monitor the pressure (Honeywell, PPT), the temperature, and the relative humidity (Vaisala, HUMICAP) of the gas at the outlet of the flow tube. The flow in the flow tube is laminar with a Reynolds number of 710.
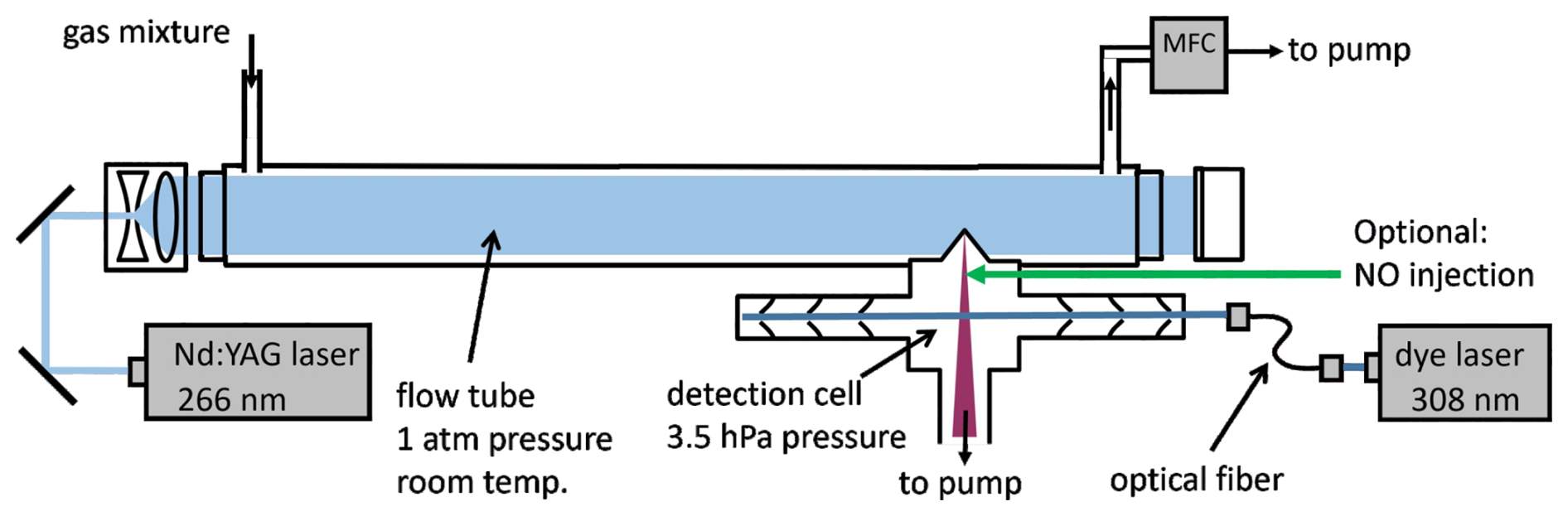
Figure 1Schematic diagram of the instrument. The air mixture in the flow tube is exposed to laser pulses at 266 nm to generate OH radicals by flash photolysis of ozone. Air is sampled through the flow tube using a mass flow controller (MFC) backed by a pump. The decay of OH is measured by laser-induced fluorescence in a low-pressure detection cell. Optional injection of NO into the low-pressure detection cell allows the detection of HO2 radicals after their chemical conversion to OH.
Laser flash photolysis of added ozone is used to generate excited oxygen atoms (O(1D)) which react with the water molecules to form OH on a timescale of nanoseconds in the flow tube.
A frequency-quadrupled Nd:YAG laser (Quantel, Ultra) delivers short pulses (10 ns) of 266 nm radiation at a repetition rate of 1 Hz and pulse energies of 20 to 27 mJ. The laser beam is expanded by an optical telescope to a diameter of 30 mm. Since the collimated photolysis laser beam is not attenuated as it passes through the reaction volume (the optical density is less than 10−3), the same initial OH concentration is obtained along the axis of the flow tube. Depending on the water vapour concentration and the laser pulse energy, the mean initial OH number concentrations range from 2×109 to 9×109 cm−3. To produce HO2 radicals, 80 ppmv CO can be added to the gas in the flow tube for a rapid conversion of the initially produced OH. The time constant of the conversion is 2 ms.
Once formed, the OH or HO2 radicals react with the reactive components in the air mixture. The concentration decreases following pseudo-first-order kinetics for all experimental conditions in this study:
where [OH]0 and are the initial radical concentrations and k′ is the first-order rate coefficient of the exponential decay, which is the sum of the first-order rate coefficients of the loss in the reaction with the gaseous reactant and the wall loss.
The time-resolved decay of the radical concentration is measured in a low-pressure detection cell (3.5 hPa), which continuously draws gas from the reaction volume through a conical nozzle (Beam Dynamics, nickel, 0.6 mm orifice, 3.6 L min−1 flow rate; Fig. 1). For the experiments with NO2, a gold-plated nozzle was used to prevent corrosion of the inlet.
In the detection cell, the OH is excited by pulsed laser radiation at a wavelength of 308 nm matching the rotational absorption line Q1(3) of the OH(A2Σ, ← X2Π, ) band transition. The UV radiation is generated by a custom-built, tunable, frequency-doubled dye laser (Strotkamp et al., 2013), which is pumped by a pulsed frequency-doubled Nd:YAG laser (Spectra-Physics, Navigator). The laser pulse repetition rate is 8.5 kHz, and the typical UV output power is 20 mW.
This system is also capable of detecting HO2 if the radical is chemically converted to OH before passing through the 308 nm probing laser beam (Fig. 1). This is achieved by injecting pure NO (Air Liquide, purity 99.5 %, flow rate 5 cm3 min−1 at standard conditions) into the sampled gas flow in the detection cell (Nehr et al., 2011, 2012; Miyazaki et al., 2013).
The added NO was purified by passing it through a cartridge filled with sodium-hydroxide-coated silica (Sigma-Aldrich, Ascarite) to avoid spurious OH background signals from the 308 nm photolysis of NO impurities. This method can give almost the same detection sensitivity (within 5 %) for HO2 as for OH (Fuchs et al., 2011).
The OH fluorescence is recorded by a multi-channel scaler photon counting system (Becker & Hickl, PMS-400A) with a time resolution of 1 ms over a time period of 1 s. In this instrument, the reaction time is determined by the electronic clock of the multi-channel scaler in contrast to flow tube experiments with sliding injectors, where the reaction time is determined from the flow rate of the gas in the reaction volume. The radical detection method using OH fluorescence is extremely sensitive and allows the measurement of atmospheric OH concentrations on the order of 106 cm−3 with a measurement time of 1 min (Hofzumahaus et al., 2009; Lou et al., 2010). In the present study, in which OH reactivity is measured, typically 10 000 OH decays were averaged, resulting in an integration time for photon counting of about 1 s for each 1 ms time bin. The resulting limit of detection (<107 cm−3) made it possible to follow the decays over 2 to 3 orders of magnitude, allowing an accurate fit of the decay time.
The separation of the detection in a low-pressure cell from the high-pressure reaction volume has several advantages for the study of OH and HO2 reactions at tropospheric pressures:
-
The low-pressure OH detection minimises the loss of sensitivity due to quenching of the OH fluorescence, which is particularly efficient for water and O2 (relative rate coefficients in units of 10−10 cm3 s−1 at 298 K: kcoll(H2O), kcoll(O2), kcoll(N2), and kcoll(Ar) equal 6.6, 1.4, 0.31, and 0.00036, respectively; Heard and Henderson, 2000). In contrast, previous fluorescence-based studies have often used Ar or N2 as a buffer gas to reduce fluorescence quenching.
-
The high detection sensitivity by the fluorescence method allows the use of low initial radical concentrations (a few 109 cm−3) and thereby makes the influence of interfering radical–radical reactions and subsequent reactions with products including the potential photolysis of the OH reaction products by the 266 nm radiation negligible on the timescale of the measured decays.
-
The method can be used to determine rate coefficients under typical tropospheric conditions of pressure and temperature and concentrations of water vapour and reactants. This is of particular interest for the study of termolecular reactions, whose rate coefficients depend on the pressure and the properties of the bath gas molecules.
-
Another advantage is the minimal radical wall loss due to the slow diffusion at atmospheric pressure. This allows for the reactions to be studied on a timescale of 1 s, which is comparable to the typical timescales of HOx reactions in the lower troposphere. The timescale and radical concentrations employed allow experiments to be carried out under pseudo-first-order conditions with lower reactant concentrations than those used in previous studies. Consequently, the impact of potentially interfering reactions of the reactants (e.g. the reaction of NO or NO2 with ozone or the formation of dinitrogen tetroxide (N2O4) from the self-reaction of NO2) is suppressed. However, unimolecular reactions, such as the reaction of an association product, may become important.
2.2 Gas mixtures
The gas mixtures overflowing the instrument inlet were prepared in two steps: first by combining flows of dry synthetic air (flow rate 23 L min−1), humidified synthetic air (3 L min−1), and air containing ozone (0.1 L min−1). From the combined mixed flow, 1 L min−1 was continuously sampled by a hygrometer (Vaisala, HUMICAP) and an ozone analyser (Environment SA, O341M). The remaining flow was combined with a small flow (<2 L min−1) of a reactant gas premixed in N2. All gas flows were controlled by mass flow controllers (Bronkhorst EL Flow, Bronkhorst IQ Flow, Brooks 5850). Each time, when one of the flow controller settings was changed, the flow rates were measured using a primary volumetric standard (Drycal, Definer 220), which has an accuracy of 0.75 % of the reading.
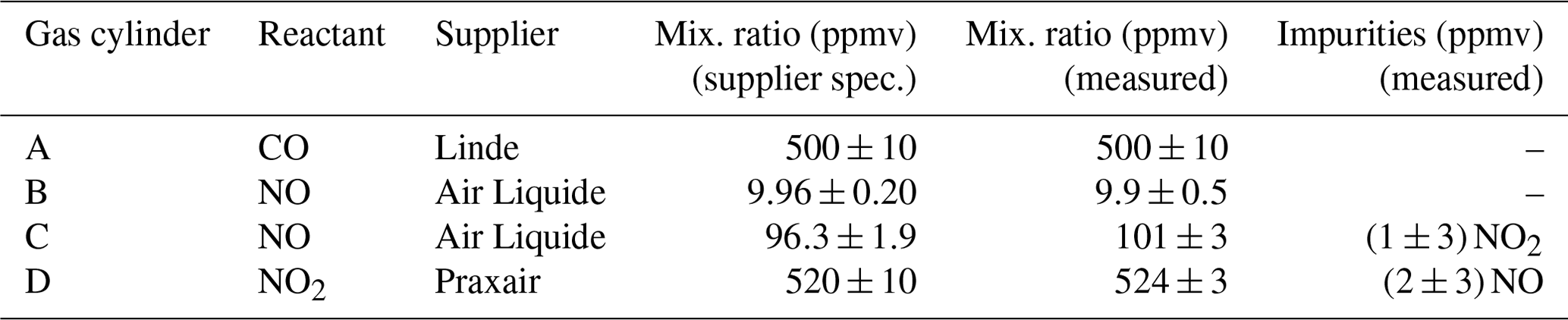
Table 1Mixing ratios of the reactants in the gas mixtures as specified by the suppliers and measured in this work. All reactants were mixed in N2.
Synthetic air (79 % N2 and 21 % O2) was produced from evaporated high-purity liquid N2 and O2 (Linde, purities >99.9999 %). Impurities in the synthetic air supply are generally below the detection limits of analytical instruments (e.g. CO <10 ppbv, NO+NO2 <10 pptv, hydrocarbons <50 pptv). Water vapour was produced by a controlled evaporation and mixing system (Bronkhorst, CEM) using pure water (Milli-Q). Ozone was produced by oxygen photolysis in synthetic air using the 185 nm radiation from a low-pressure mercury lamp. In the flow tube, typical ozone mixing ratios were 35 ppbv and the partial pressures of water vapour were in the range of 2.0 to 22.5 hPa, equivalent to relative humidities of 10 % to 98 %.
The reactant gases CO, NO, and NO2 were supplied as certified mixtures in N2 from commercial suppliers. The concentrations of the mixtures were controlled independently (Table 1). To measure CO concentrations, a small flow (cylinder A; Table 1) was diluted with a synthetic airflow, both controlled by mass flow controllers, and the resulting CO concentration was measured using a near-infrared cavity ring-down spectrometer (Picarro, G2401). This instrument has a high precision of a few parts per billion by volume and high linearity (Zellweger et al., 2012) and was calibrated against a CO standard from NPL (National Physical Laboratory, UK).
Two cylinders with different NO concentrations were used in this work (cylinders B and C; Table 1). For the analysis of the NO concentration in cylinder B, a flow of the gas mixture was further diluted with N2 using mass flow controllers. The resulting NO concentration was measured using a chemiluminescence instrument (Ecophysics, CLD770) for mixing ratios up to 100 ppbv. The instrument was calibrated using an NPL standard with a stated uncertainty of 0.8 %. The mixing ratios of NO and NO2 in cylinders C and D (Table 1) were measured directly using a UV-VIS photometer (ABB, Limas 11HW) which is suitable for measurements up to 1000 ppmv. For all gas cylinders, the derived mixing ratios were found to be in agreement with the suppliers' specifications within the experimental uncertainties (Table 1). A weighted average of the measured and the supplier values was used to calculate the concentrations in the reaction kinetics experiments.
2.3 Kinetic analysis
The measured radical decay curves are expected to follow pseudo-first-order kinetics (Eqs. 4, 5). The corresponding time-dependent OH fluorescence signals (photon counts N(t)) include a constant background signal which is caused by scattered radiation from the probe laser and detector noise:
where N0 is the initial fluorescence count and B is the background.
The parameters N0, B, and k′ were determined for the measured decay curves using a non-linear, least-square Levenberg–Marquardt fitting algorithm. The counts were weighted in the fit by their statistical errors, which follow Poisson statistics. The first 10 ms of the measured OH decay was generally discarded. The signal in this time period showed deviations from a single exponential behaviour due to inhomogeneities in the initial OH concentration, and this time was necessary for the conversion of OH to HO2 in the experiments with HO2.
Experiments with zero air, which contained additionally only water vapour and ozone, were performed to determine the zero rate coefficient (k0) of the OH and HO2 decays caused by wall loss and potential gas-phase reactions in the zero gas (Eq. 7). For both radicals, the values were in the range of (1.8±0.1) s−1 for water vapour partial pressures between 2.0 and 22.5 hPa.
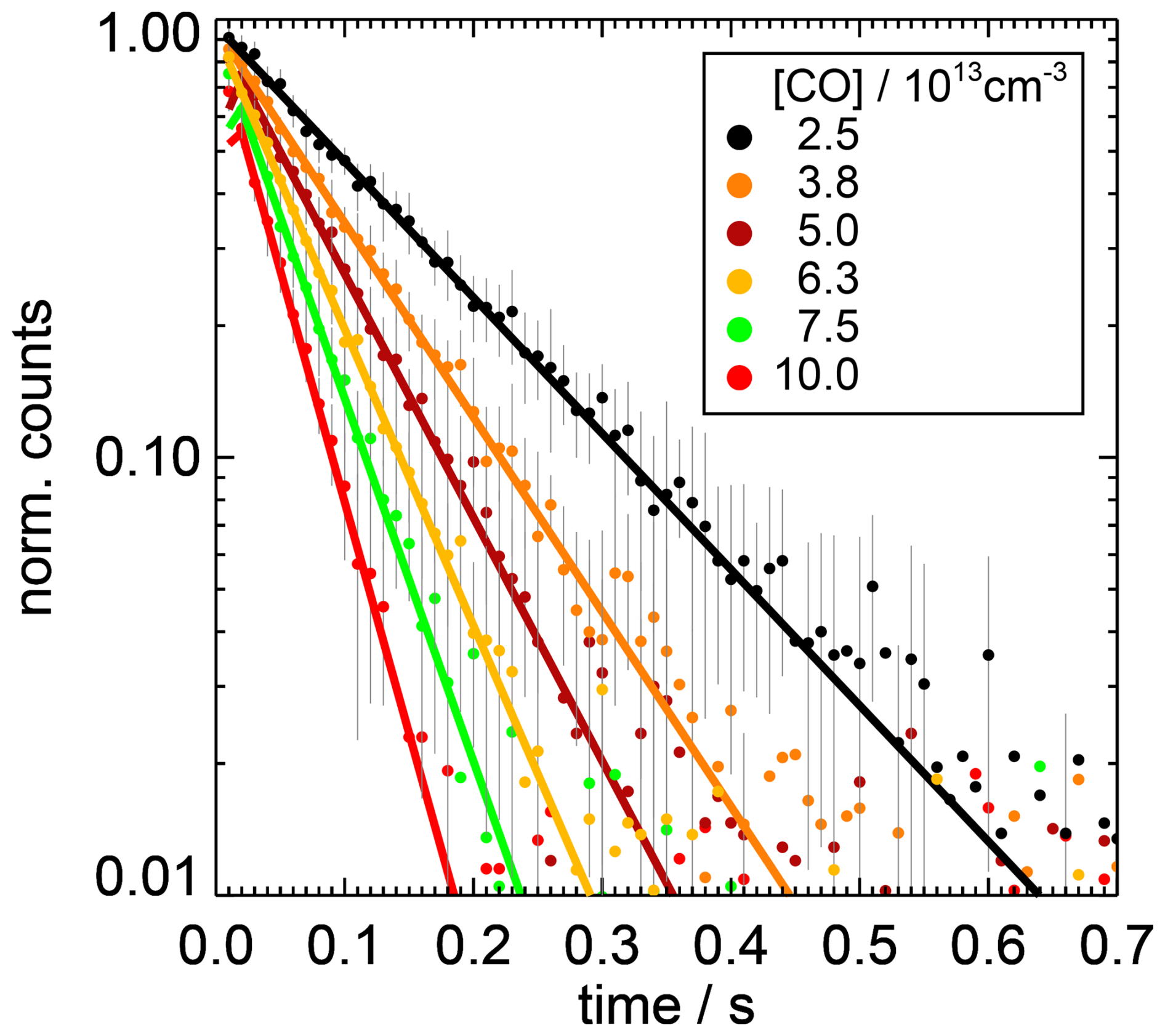
Figure 2Examples of measured (dots) and fitted (lines) OH concentration decays (normalised to the fitted amplitude) for different CO concentrations measured at a temperature of 297 K and a pressure of 1 atm in this work. The background values determined by the fit are subtracted. For clarity, the measured decays are shown in the figure with a time resolution of 10 ms. Error bars are 1σ statistical errors in the measurements.
The calculated, known contributions to the zero rate coefficient from gas-phase reactions were very small. The reaction of the added ozone (mixing ratio 35 ppbv) with OH and HO2 contributed only 0.06 and 0.0017 s−1, respectively, to the reactivity. The reactivity of self-reactions of OH and HO2 radicals is also less than 0.07 and 0.04 s−1, respectively. The variability in the zero rate coefficient over the range of added water vapour concentrations gives an upper limit of 0.1 s−1 for the reactivity from potentially co-evaporated impurities of the water supply. The reactivity from potential impurities (e.g. CO, NOx, hydrocarbons) in the synthetic air supply can be estimated to have an upper limit of 0.1 s−1.
For these reasons, the zero rate coefficient was mainly determined by the lateral transport of radicals to the wall of the flow tube, where radicals are lost. This assumption is consistent with the diffusion of radicals, for which the mean quadratic displacement (〈Δr2〉) can be calculated by Einstein's relation (〈Δr2〉=4 D t, diffusion coefficients: D(OH)=0.217 cm2s−1, D(HO2)=0.141 cm2s−1; Ivanov et al., 2007).
In the experiments, the concentrations of the reactants were varied to determine the rate coefficients (Fig. 2). The reactant concentrations gave reactivities between 0 and 40 s−1. Approximately 10 decay curves, each integrating 100 to 250 photolysis laser shots, were accumulated for each reactant concentration. The slope of a linear regression of the measured first-order rate coefficients against the reactant concentration [i] gives the second-order reaction rate coefficient kOH+i. The intercept is the zero rate coefficient k0.
3.1 Rate coefficient of the OH reaction with CO
The rate coefficient of the reaction of carbon monoxide (CO) with OH was studied in four experiments at room temperature (296 to 298 K; Table A1), in each of which the OH reactivity was measured for eight CO concentrations (Fig. 3). As CO is transparent at a wavelength of 266 nm (Okabe, 1978), effects from the photolysis of CO in the flow tube by the photolysis laser can be excluded.
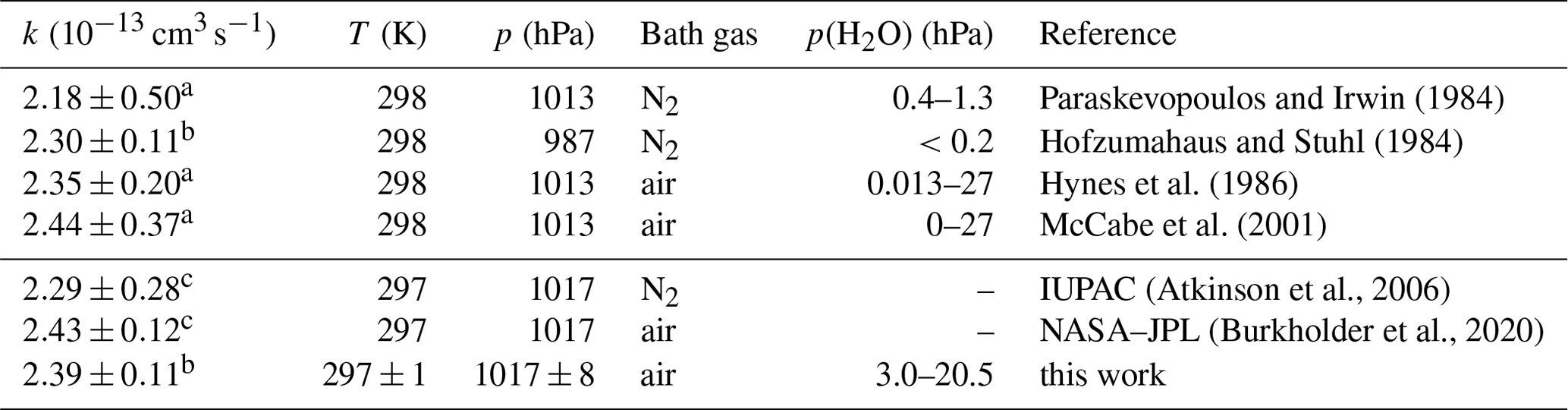
The experiments differed in the water vapour content with water vapour partial pressures between 3 and 20.5 hPa equivalent to relative humidities between 13 % and 93 %. Since the rate coefficients agreed within 4 % and showed no trend with the presence of water vapour (Fig. 3), a water vapour independent value of cm3 s−1 is determined from the weighted average of the rate coefficients determined at the different humidities (Table A1). The uncertainty is the total 2σ error, which is mainly due to the uncertainty in the CO concentration.
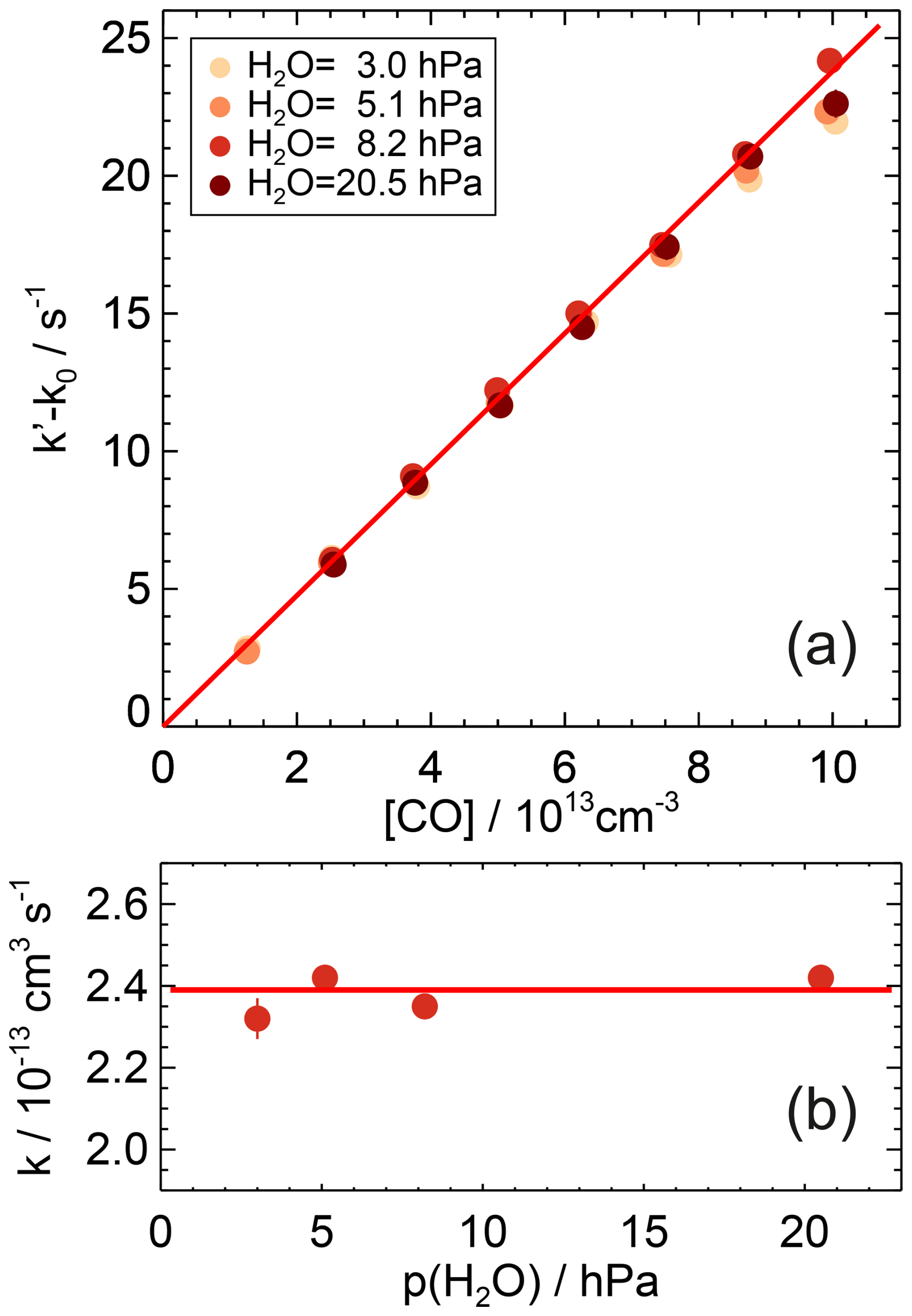
Figure 3First-order rate coefficients of the OH reaction with CO at room temperature ((297±1) K) and ambient pressure ((1017±8) hPa) in air and partial water vapour pressures between 3 and 20.5 hPa (a). The zero rate coefficient k0 is subtracted from the linear fit of the measured OH reactivity (Eq. 7). The slope of the red line is the weighted average of the second-order rate coefficients determined at the different humidities. No dependence of the rate coefficient on water vapour is observed (b). Error bars (1σ statistical errors) are partly smaller than the size of the symbols.
Table 2Second-order rate coefficients (k) of the OH reaction with CO measured absolutely in air or N2 at ambient total pressure (p) and temperature (T). In addition, recommended values by IUPAC and NASA–JPL are given for the conditions used in this work. Errors in the rate coefficients are 2σ uncertainties.
a Linear fit of measured data to ambient conditions. b Measurement for stated conditions. c Parameterisation based on the literature.
The reaction of OH with CO has been studied experimentally and theoretically over a wide range of conditions (e.g. Fulle et al., 1996; Atkinson et al., 2004; Johnson et al., 2014; Burkholder et al., 2020; Barker et al., 2020). It shows a complex non-Arrhenius temperature and pressure dependence. This can be explained by the formation of an activated radical intermediate, HOCO∗ (Smith and Zellner, 1973), which can be collisionally stabilised to HOCO or can decompose to CO2 and an H atom (Reactions R1, R2).
At high temperatures (>600 K), HOCO becomes thermally unstable and forms OH and CO (Fulle et al., 1996), while in the atmosphere (200 to 300 K) it reacts mainly with O2 to form HO2.
The corresponding lifetime of HOCO is 130 ns in 1 atm pressure in air at a temperature of 298 K (Miyoshi et al., 1994). Similarly, the H atom produced in the decomposition of HOCO∗ (Reaction R2) reacts with O2 to form HO2 at a similar rate (Burkholder et al., 2020).
Consistent with this mechanism, the OH decays measured in the present work showed a single exponential behaviour without regeneration of OH.
Previous experimental studies at atmospheric temperatures have shown that the OH+CO reaction (Reaction R1) does not depend on the temperature at low pressure and shows only a small decrease (about 10 %) as the temperature increases from 200 to 300 K at a pressure of 1 atm (McCabe et al., 2001; Liu and Sander, 2015). The rate coefficient has a linear pressure dependence and increases by a factor of 1.6 in the pressure range from 0 to 1 atm (Atkinson et al., 2004; Burkholder et al., 2020).
Despite its importance in atmospheric chemistry, only two absolute measurements of the rate coefficient have been reported in previous studies at room temperature and 1 atm pressure of air (Table 2; Hynes et al., 1986; McCabe et al., 2001). The data from these two studies are in very good agreement within 2 % with the results in this work. This is better than would be expected from the reported uncertainties of the pressure-dependent expressions of the rate coefficient in Hynes et al. (1986) (12 %) and in McCabe et al. (2001) (15 %) and the measurement error of 5 % in this work.
Other studies have investigated the rate coefficient in N2 at ambient pressure. The values agree within 5 % to 10 % with measurements in air (Table 2). This is consistent with the experiments of Hynes et al. (1986) and McCabe et al. (2001), which show that the collisional stabilisation of the reactive complex is the same for N2 and O2 within the experimental uncertainties. However, experiments in N2 require great care to avoid oxygen impurities, as H atoms (Reaction R2) could react not only with molecular oxygen to form HO2 (Reaction R10), but also with HO2, thereby regenerating OH. This can lead to an apparent reduction in the effective rate coefficient (Hofzumahaus and Stuhl, 1984; Paraskevopoulos and Irwin, 1984; Liu and Sander, 2015). Measurements in air, as in this work, avoid this potential problem because the oxygen concentration is high and any H atoms react exclusively with O2.
The results of this work are in good agreement with all previously reported absolute measurements in N2 and air (Table 2) and are well within the uncertainties of recent recommendations from IUPAC (Atkinson et al., 2004) and NASA–JPL (Burkholder et al., 2020). The IUPAC-recommended value is only 5 % lower, and the NASA–JPL-recommended value is 3 % higher than the value in this work (Fig. 3). The small discrepancies suggest that the uncertainties of the recommended values are likely overestimated by a factor of 2 at atmospheric pressure, although the uncertainty over the full range of the fall-off region may be higher.
No water vapour dependence of the rate coefficient was observed. The variability in the values ((3±3) %) gives an upper limit for the collisional stabilisation by water relative to air of 10. This agrees with previous measurements, where the efficiency was found to be a factor of 10 higher than that of N2 at low pressures (up to 27 hPa) in pure water and helium (Paraskevopoulos and Irwin, 1984). At atmospheric pressure, however, no significant effect of water vapour on the rate coefficient could be detected for partial water vapour pressures up to 27 hPa (McCabe et al., 2001). Based on these studies and results in this work, relevant water vapour effects due to clustering of water molecules with OH as assumed in previous experimental work (Beno et al., 1985) or with HOCO as discussed in a theoretical study (Aloisio and Francisco, 2000) can be ruled out as being significant for atmospheric conditions at room temperature.
3.2 Rate coefficient of the OH reaction with NO
The rate coefficient of the OH reaction with NO (Reaction R3) was measured in air at a pressure of 1019 hPa and a temperature of 297 K. In addition, the partial pressure of water vapour was varied between 3.1 and 22.5 hPa equivalent to relative humidities between 14 % and 98 %. NO was provided by two gas standards (Table 1).
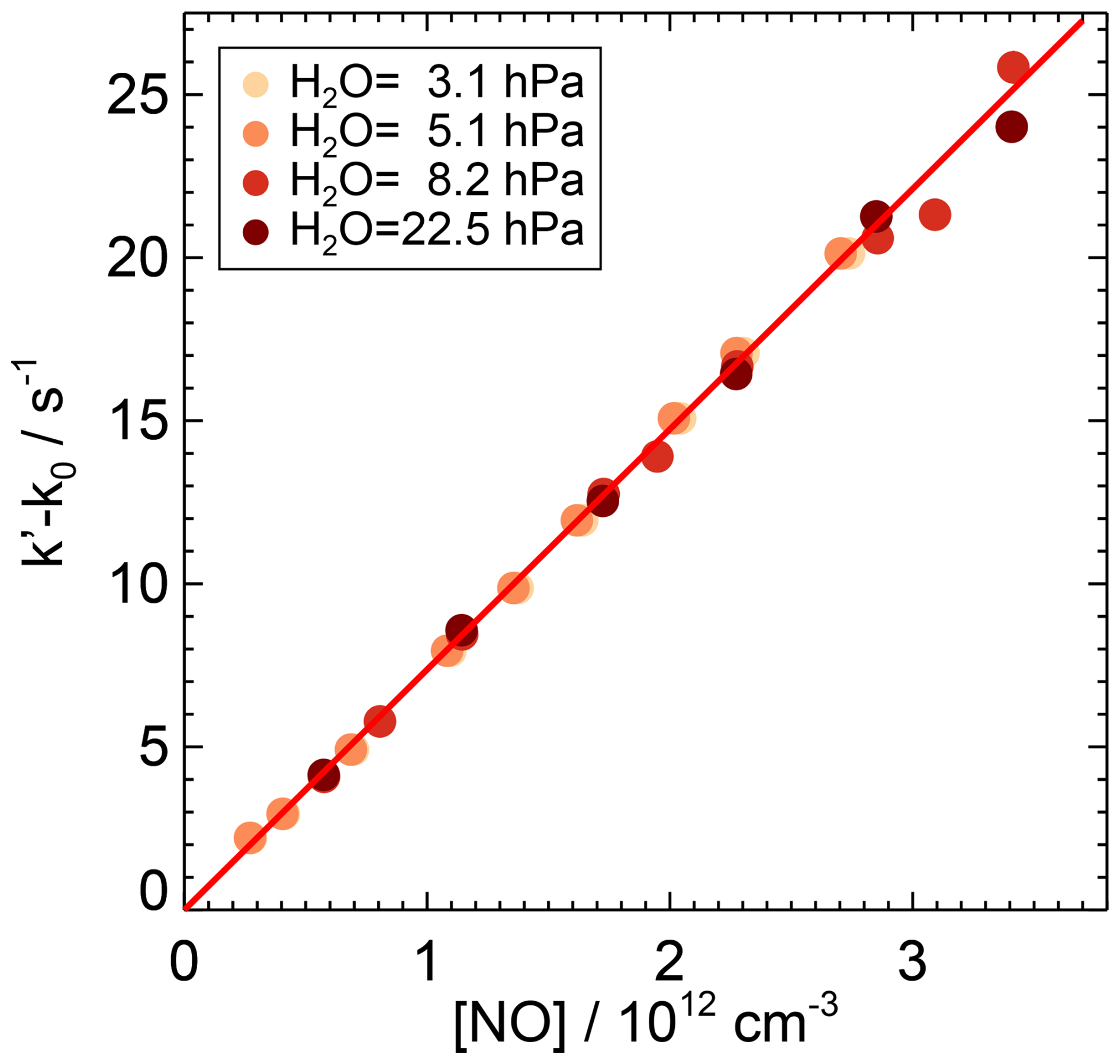
Figure 4First-order rate coefficients of the OH reaction with NO at ambient temperature ((297±1) K) and pressure ((1019±3) hPa) in air and various partial pressures of water vapour. The zero rate coefficient k0 is subtracted from the linear fit of the measured OH reactivity (Eq. 7). The slope of the red line is the weighted average of the second-order rate coefficients determined at the different humidities, as there is no observed dependence of the rate coefficient on water vapour. Error bars (1σ statistical errors) are partly smaller than the size of the symbols.
In the evaluation of the rate coefficient, systematic errors due to side reactions of the NO reactant need consideration. First, an influence of the photolysis laser on the NO concentration can be excluded because NO does not absorb at 266 nm (Okabe, 1978). However, a small effect is expected from the reaction of NO with ozone forming NO2 in the reaction volume. Under the experimental conditions in this work, a gradual decrease in NO of 1.6 % is expected in the flow tube before the air is sampled by the inlet of the LIF detection cell, using a rate coefficient of the NO reaction with O3 of cm3 s−1 at a temperature of 298 K (Burkholder et al., 2020). As the rate coefficient for the reaction of OH with NO2 is 1.6 times faster than that with NO, a small bias of +1 % can be estimated for the determination of the OH reaction rate with NO due to the formation of NO2. Measured values (Table A1) are corrected for this bias.
The rate coefficient of the OH reaction with NO was derived from the slope of the measured OH reactivity when the NO concentration was varied between 0.2 and 3.5×1012 cm−3 (Fig. 4, Table A1). The partial water vapour pressure was changed between 3.1 and 22.5 hPa. No significant effect of water vapour on the rate coefficient was observed. The weighted average of the slopes derived from measurements at four different water vapour concentrations gives a rate coefficient of cm3 s−1 at a pressure of (1019±3) hPa and a temperature of (297±1) K in air. The total uncertainty is mainly due to the uncertainty in the NO concentrations.
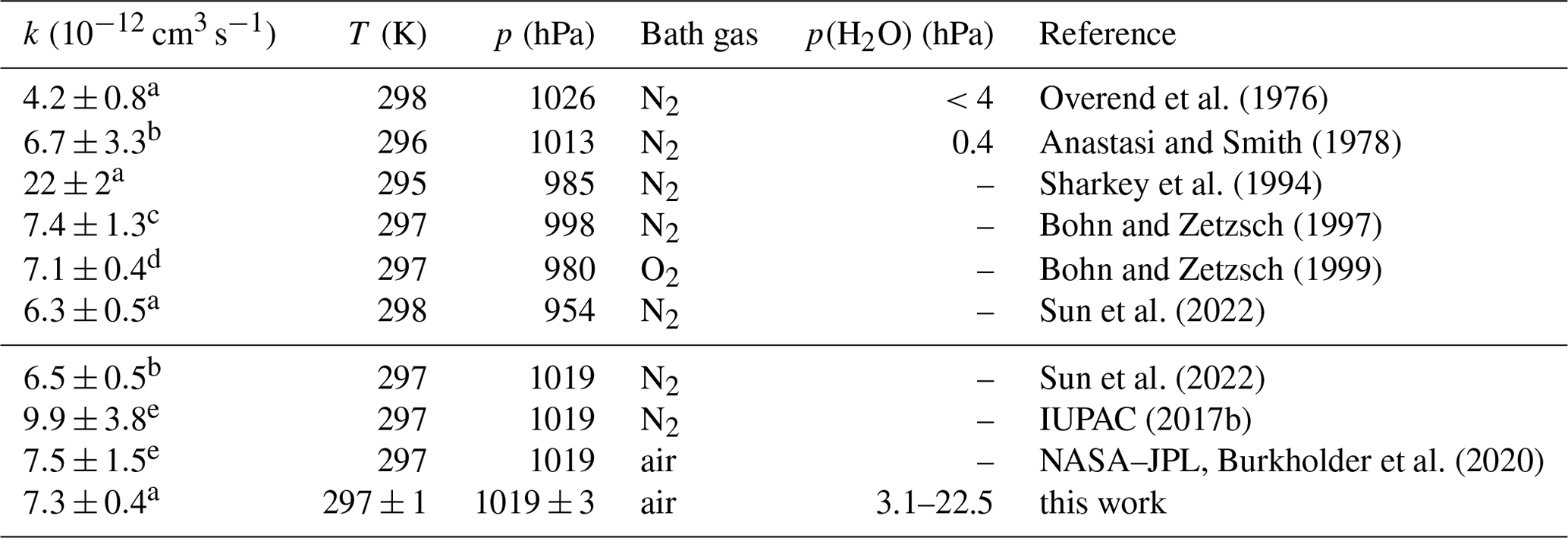
Overend et al. (1976)Anastasi and Smith (1978)Sharkey et al. (1994)Bohn and Zetzsch (1997)Bohn and Zetzsch (1999)Sun et al. (2022)Sun et al. (2022)IUPAC (2017b)Burkholder et al. (2020)Table 3Second-order rate coefficient (k) of the OH reaction with NO in air or N2 at ambient total pressure (p) and temperature (T). In addition, values calculated from the parameterisations in the IUPAC and NASA–JPL recommendations and reported in the literature are given for the conditions in this work. Errors in the rate coefficients are 2σ uncertainties.
a Measurement for stated conditions. b Parameterisation based on measured data. c Derived from bi-exponential OH decays in a complex reaction system containing H2O2 and NO. d Derived from bi-exponential OH decays in a complex reaction system containing H2O2 and benzene. e Parameterisation based on the literature.
The differences between the rate coefficients in this work and the values recommended by NASA–JPL (Burkholder et al., 2020) and determined by Bohn and Zetzsch (1997) and Bohn and Zetzsch (1999) are less than 4 % (Table 3). Recent work by Sun et al. (2022) provides a parameterisation of the rate coefficient from measurements over a broad pressure range (15 to 990 hPa) at different temperatures (273, 298, 333 K). Their parameterisation gives values which are approximately 13 % lower than those recommended by NASA–JPL for the experimental conditions in this work but agrees better at lower pressures. The measurements in Sun et al. (2022) were carried out in N2. The authors assume that the collisional stabilisation of the activated association complex by N2 and O2 is similar so that their parameterisation can also be used for air.
The values recommended by IUPAC (2017b) are 35 % to 50 % higher than the measurements by Sun et al. (2022), Bohn and Zetzsch (1997, 1999), and this work (Fig. 5), suggesting that the IUPAC recommendation may need to be revised.
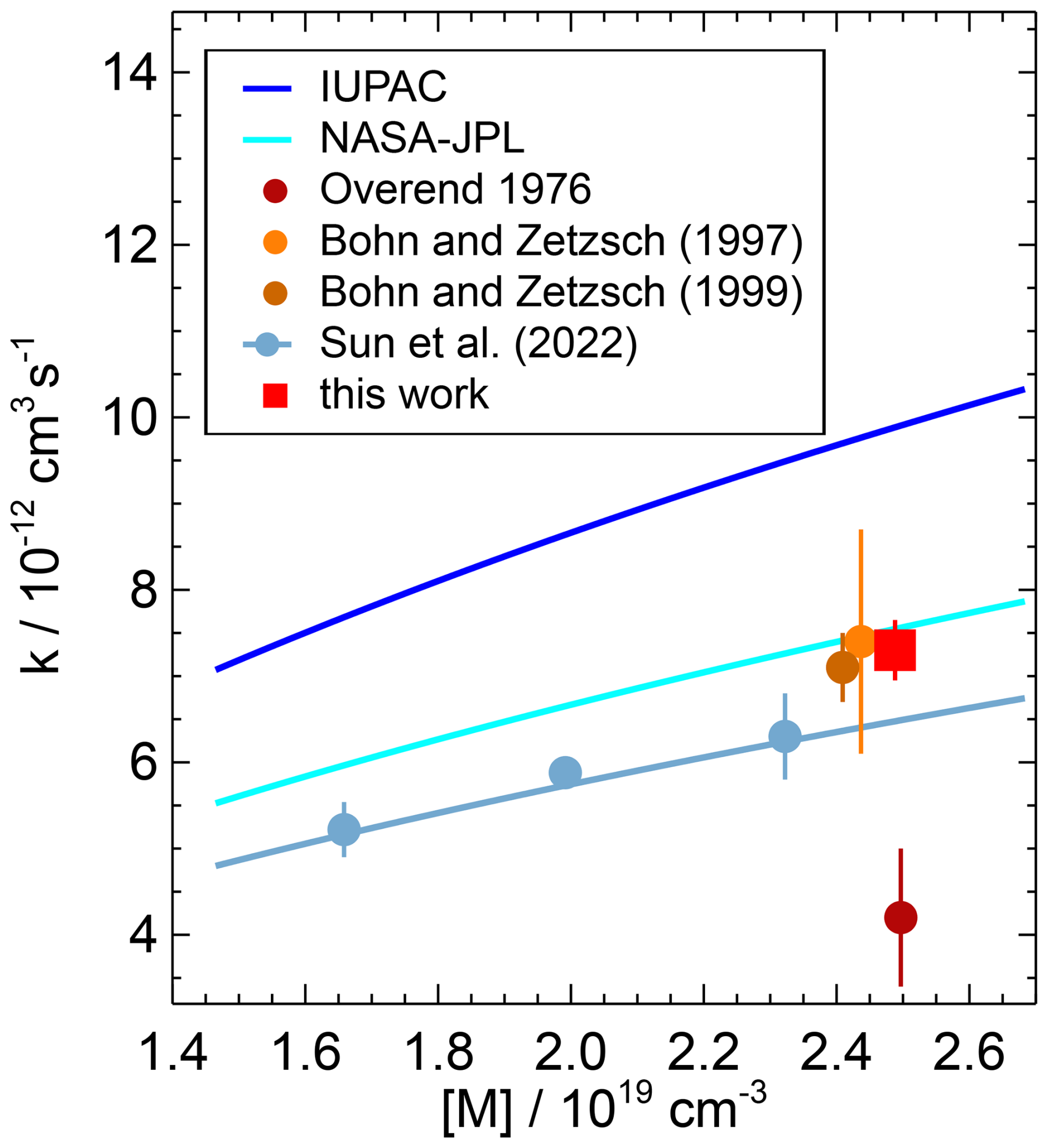
Figure 5Pressure dependence of the second-order rate coefficient of the OH reaction with NO reported in the literature. Data points represent measured values, and solid lines represent parameterisations using Troe equations. The parameterisation of Sun et al. (2022) (line) is calculated for pure N2 at 297 K, while the corresponding data points were measured at 298 K. Error bars are total errors. The high value measured by Sharkey et al. (1994) (Table 3) is not shown.
Sun et al. (2022) also investigated the effect of water vapour on the rate coefficient. Measurements at various water concentrations at low pressure (66 hPa) and room temperature showed a rate coefficient at a water vapour partial pressure of 12 hPa that was 60 % higher than in pure N2. The authors explained this behaviour by the more efficient collisional stabilisation of the activated association complex by water molecules, which was estimated to be a factor of 5 to 6 more efficient than that of N2.
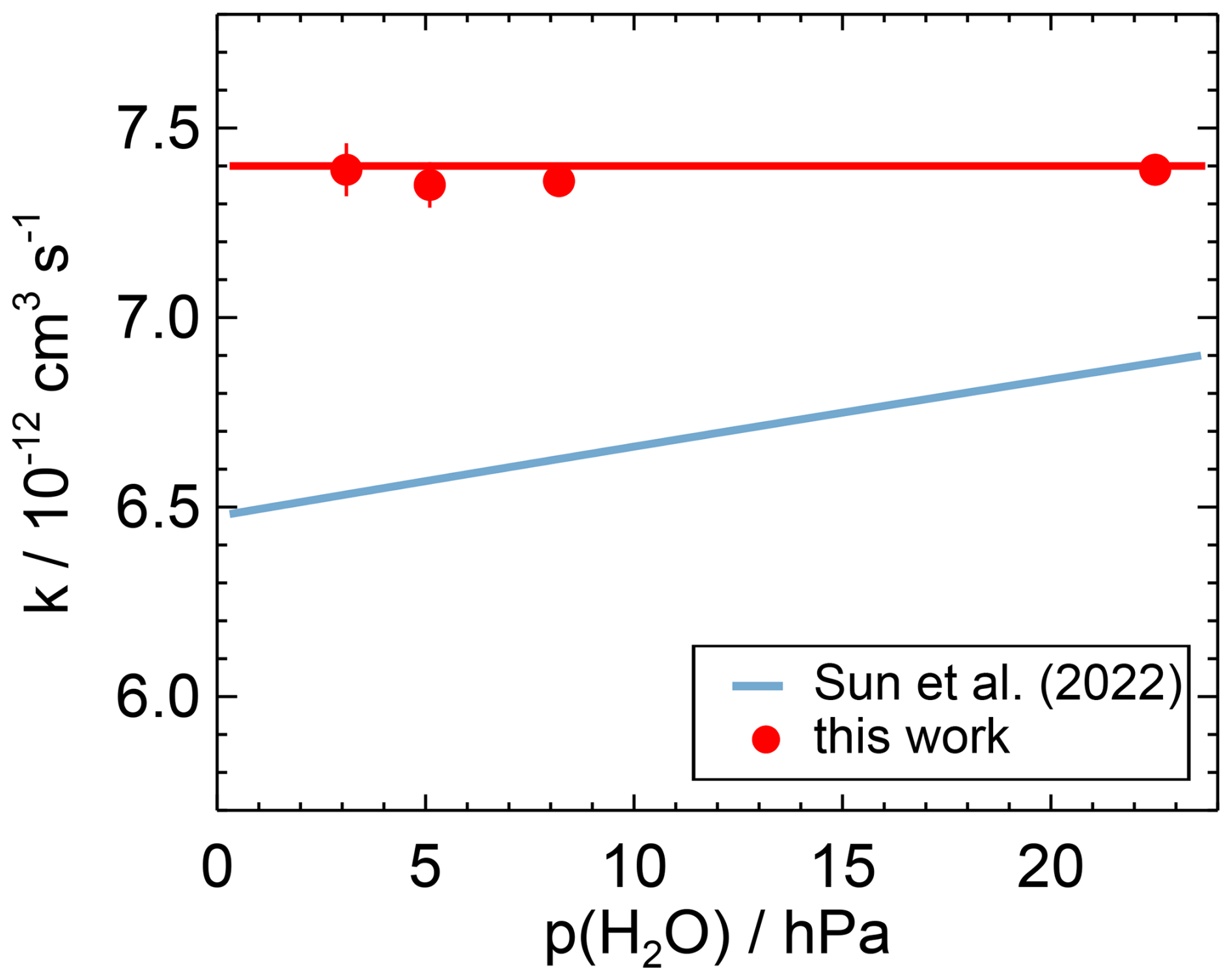
Figure 6Water vapour dependence of the second-order rate coefficient of the OH reaction with NO measured in this work and calculated from the parameterisation derived in Sun et al. (2022) for the conditions of this work (T=297 K, p=1019 hPa). The horizontal red line is the weighted average of the data measured in this work (Table A1). Error bars (1σ statistical errors) are partly smaller than the size of the symbols.
Sun et al. (2022) derived a Troe equation using different low-pressure rate coefficients for N2 and water vapour following the approach described in Amedro et al. (2020). Using the values in Sun et al. (2022), the difference between the rate coefficients for the lowest and highest water vapour concentrations in the experiments in this work is 5 % (Fig. 6). This is higher than the variability in the measured rate coefficients (1 %) determined at the different water vapour concentrations in this work and is a significant discrepancy.
Liessmann et al. (2011) studied the influence of water on the reaction of OH with NO at low total pressures (<10 hPa) and low temperatures (60 to 300 K). They observed a strong enhancement of the rate coefficient of up to 40 % at a water vapour mixing ratio of 3 % at temperatures below 135 K in a Laval nozzle gas expansion, but the enhancement disappeared at room temperature and became hardly detectable.
The results of this work suggest that the efficiency of the collisional stabilisation by water at the conditions in the lower troposphere is smaller than predicted by the parameterisation in Sun et al. (2022). The different behaviour may be due to invalid assumptions in the determination of the parameterisation or due to undetected measurement errors in the data of Sun et al. (2022) or in the present work. More studies are required to resolve this discrepancy.
3.3 Rate coefficient of the OH reaction with NO2
The rate coefficient of the OH reaction with NO2 was measured at a pressure of 1034 hPa, a temperature of 295 K, and two water vapour partial pressures (6.2 and 17.6 hPa, relative humidities 32 % and 90 %) (Table A1). The values were determined from the slope of OH reactivity measurements with varying NO2 concentrations.
The reaction of NO2 with OH can produce either nitric acid (HNO3, Reaction R4) or pernitrous acid (HOONO, Reaction R5). The ratio of the products, HOONO to HNO3, increases with pressure and is (14.2±1.2) % for the experimental conditions of this work (Mollner et al., 2010). Pernitrous acid is thermally unstable and decomposes back to OH and NO2. Its chemical lifetime is approximately 1.2 s at room temperature calculated using the NASA–JPL rate coefficients of the forward reaction and the equilibrium constant (Burkholder et al., 2020). This is a factor of 6 to 36 longer than the OH lifetimes in the experiments in this work. Therefore, the OH decays are expected to represent the sum of the two OH loss reaction channels with little influence of the OH regeneration by the re-dissociation of HOONO. In agreement with the expectation, the observed OH decays showed no obvious deviation from a single exponential behaviour and were first fitted with the expression in Eq. (4).
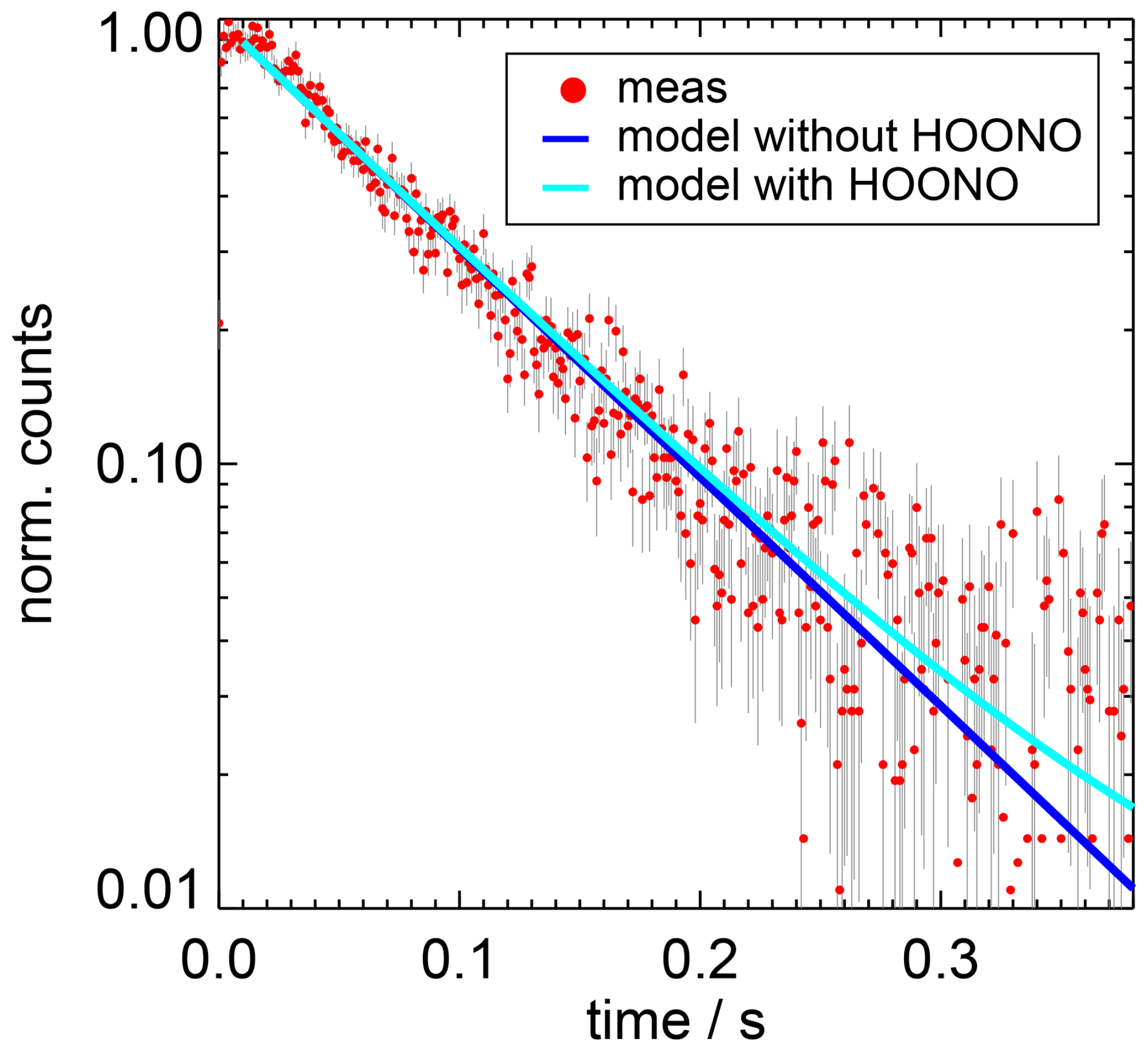
Figure 7Example of the measured OH decay (normalised counts) and results of simulated decays including either only the OH loss in the reaction with NO2 (“without HOONO”) or additionally the OH production from the HOONO decomposition (“with HOONO”). In the example, the corrected OH loss rate is 12.8 s−1 and the measurement was performed with an NO2 concentration of 1×1012 cm−3 and a water vapour partial pressure of 6.2 hPa.
In order to estimate the small effect of the HOONO decomposition on the derived rate coefficients, the OH decay curves were simulated for two cases using a model that either included or excluded the HOONO decomposition (Fig. 7). The model uses the value of the equilibrium constant (Reaction R5) by NASA–JPL ( cm−3; Burkholder et al., 2020) and the branching ratio determined by Mollner et al. (2010). The results in Fig. 7 show that the two simulations agree well with the measured OH decay over the 1st order of magnitude and start to diverge from each other only after more than three OH lifetimes, when the noise of the measured decay curve becomes large.
The ratio of the two simulated OH decay curves was used to correct the measured OH decay for the OH production from the HOONO decompositions. Fitting the corrected decay curves to a single exponential function (Eq. 4) gave 3 % to 5 % higher decay rates than without the corrections. The largest effects are obtained for the lowest NO2 concentrations. The corrected OH decay rates (Table A1) were used to calculate the rate coefficients of the OH reaction with NO2 (Fig. 8).
Some other possible systematic errors in the determination of the rate coefficient can be ruled out. (1) The reaction of NO2 with O3 is far too slow ( cm3 s−1; Burkholder et al., 2020) to cause a significant change in the NO2 concentration in the flow tube at the given experimental conditions. (2) Although NO2 absorbs at the wavelength of the photolysis laser (266 nm), the effect is negligible since less than 10−4 of the NO2 molecules are photodissociated (absorption cross section cm2; Vandaele et al., 1998) at unity quantum yield. (3) Impurities of NO in the NO2 gas mixture in the gas cylinder showed no detectable impurity of NO (Table 1) and therefore did not affect the determination of the rate coefficient. (4) The formation of NO2 dimers (N2O4) was insignificant as their estimated concentration was about 1.4×106 cm−3 at the maximum NO2 concentration (2.5×1012 cm−3) used in the experiments.
The type of bath gas may also affect the results, as the relative efficiency of the collisional stabilisation of the activated association complex by O2 to N2 is in the range of 0.67 (Mollner et al., 2010) and 0.74 (Amedro et al., 2019). As the experiments in this work were carried out in humidified synthetic air, the measured values refer to rate coefficients in a mixture of 79 % N2 and 21 % O2 and variable traces of water vapour.
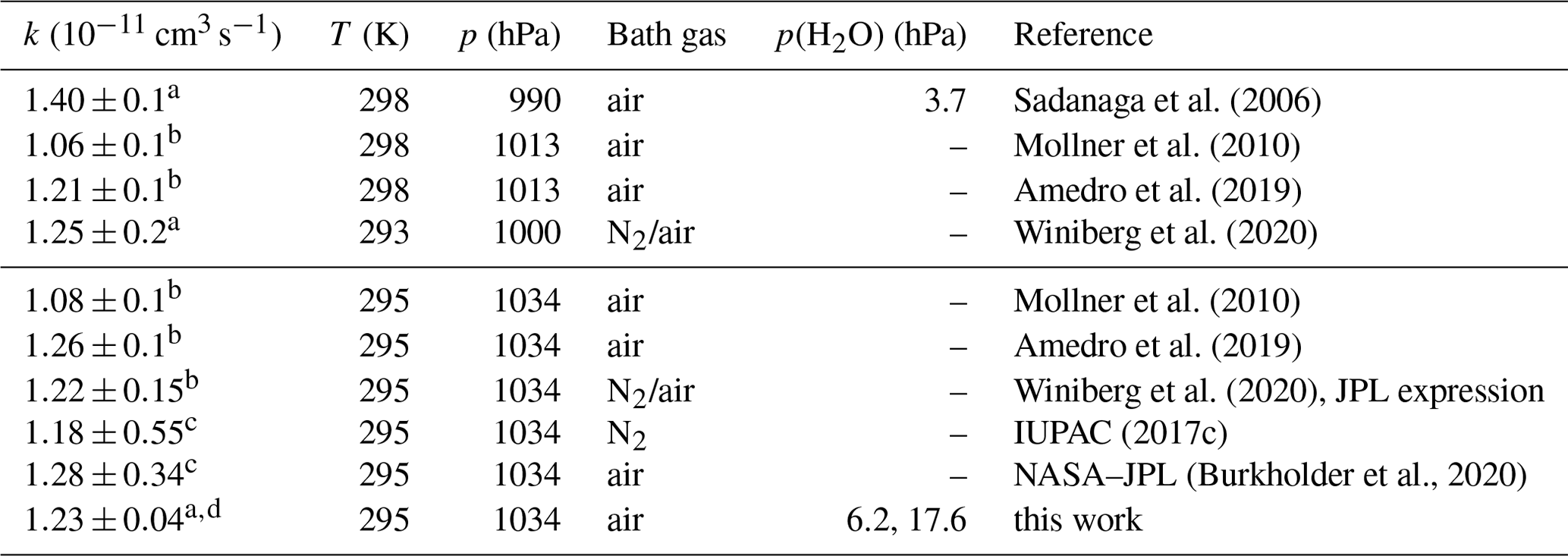
Sadanaga et al. (2006)Mollner et al. (2010)Amedro et al. (2019)Winiberg et al. (2020)Mollner et al. (2010)Amedro et al. (2019)Winiberg et al. (2020)IUPAC (2017c)(Burkholder et al., 2020)Table 4Second-order rate coefficient (k) of the OH reaction with NO2 in air or N2 at ambient total pressure (p) and temperature (T). In addition, values calculated from the parameterisation in the IUPAC and NASA–JPL recommendations and reported in the literature are given for the conditions in this work. Errors in the rate coefficients are 2σ uncertainties.
a Measurement for stated conditions. b Parameterisation based on measured data. c Parameterisation based on the literature. d Decay curves corrected for HOONO decomposition.
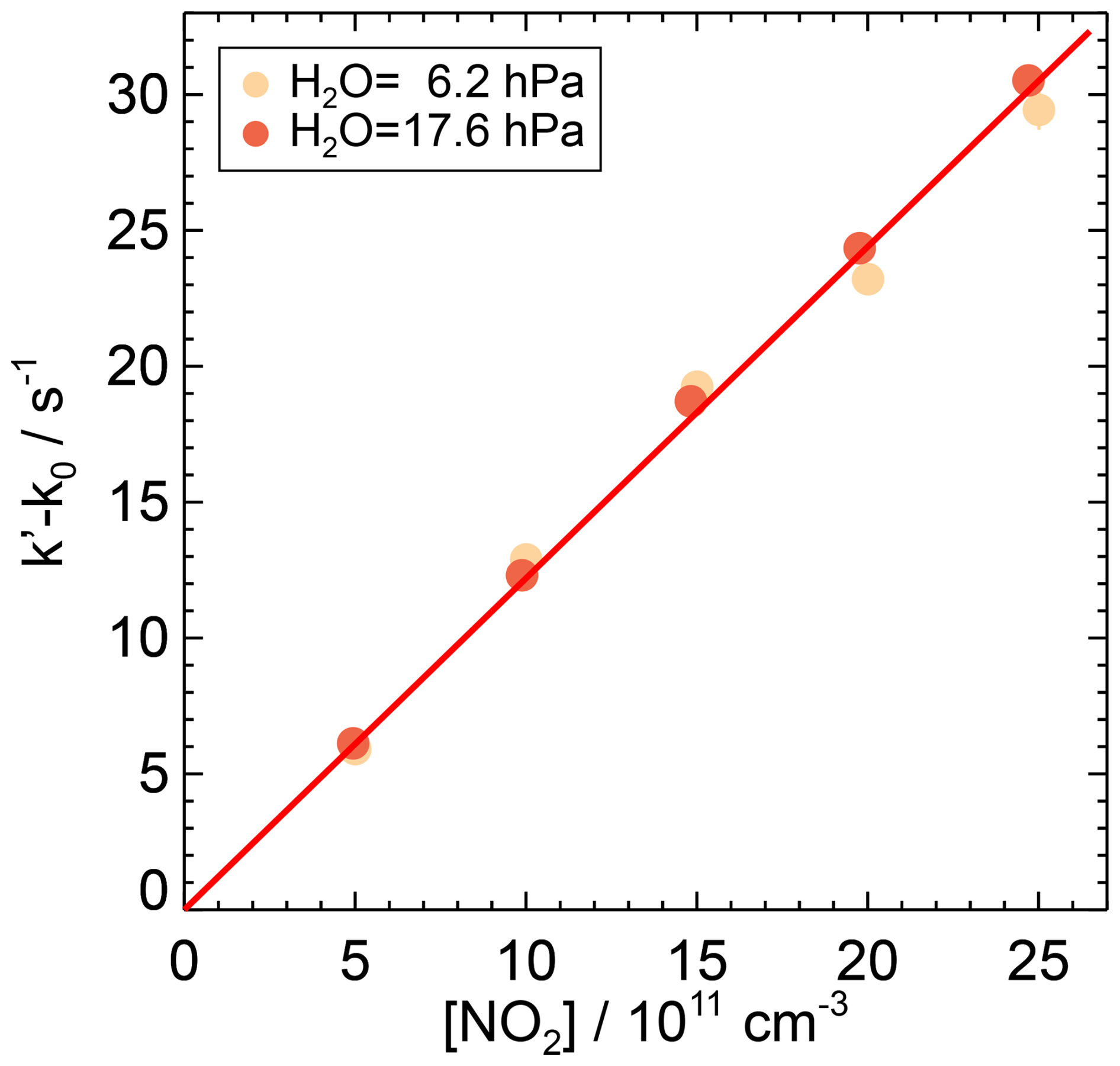
Figure 8First-order rate coefficients of the OH reaction with NO2 from OH reactivity measurements at ambient temperature (295 K) and pressure (1034 hPa) in air at two partial pressures of water vapour. The zero rate coefficient k0 is subtracted from the linear fit of the measured OH reactivity (Eq. 7). The slope of the red line is the weighted average of the second-order rate coefficients determined at the different humidities, as there is no observed dependence of the rate coefficient on water vapour. Error bars (1σ statistical errors) are partly smaller than the size of the symbols.
The second-order rate coefficients of the OH reaction with NO2 obtained in this work for water vapour partial pressures of 6.2 and 17.2 hPa differed by only 3.3 %, which is slightly higher than the combined statistical errors (±2.2 %). The weighted average gives a value of cm3 s−1. The total error includes the uncertainty of the reactant concentration.
The second-order rate constant in air measured in this work is 4 % higher than the value recommended by IUPAC (2017c) and is 4 % lower than the value recommended by NASA–JPL (Burkholder et al., 2020) (Table 4). The differences between the IUPAC and NASA–JPL recommendations become much larger at low pressure and low temperature as discussed in Amedro et al. (2019). The IUPAC-recommended values are given for N2 as a bath gas, whereas the NASA–JPL recommendation takes into account the differences in the collisional stabilisation of the activated association complex by N2 and O2. If this effect was taken into account in the IUPAC recommendation, the rate coefficient would be approximately 3 % lower (Amedro et al., 2019), further increasing the difference between the values of the IUPAC and NASA–JPL recommendations. This also affects the agreement with the value determined in this work. However, the resulting difference is still smaller than the uncertainty of the recommendations (Table 4).
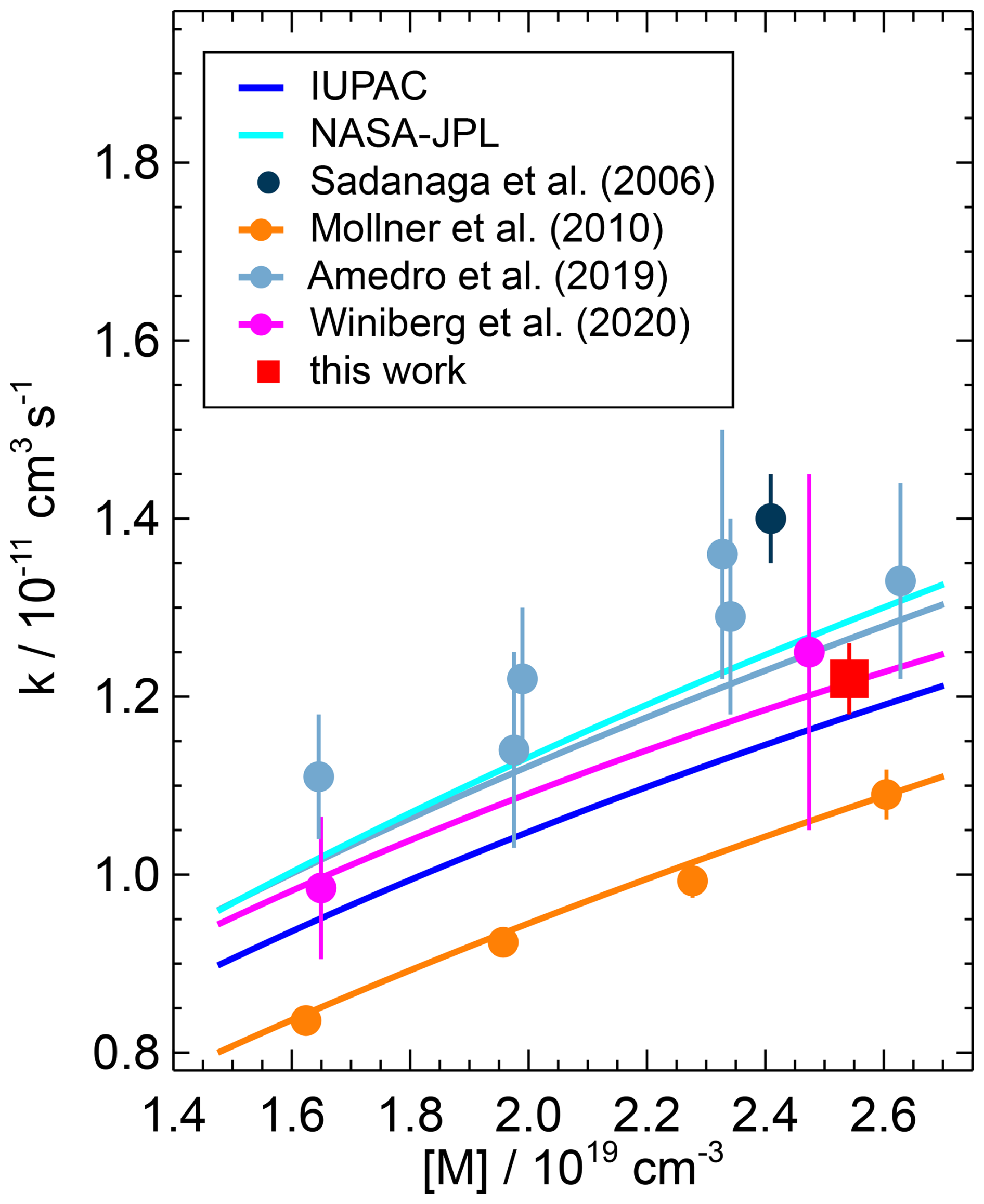
Figure 9Pressure dependence of the second-order rate coefficient for the OH reaction with NO2 reported in the literature. Data points represent measured values, and solid lines represent parameterisations using Troe equations. The parameterisation of Winiberg et al. (2020) is the NASA–JPL-type Troe equation given in their work. The parameterisations are calculated for the temperature used in the present study (295 K), but the measured data points shown were obtained at slightly different conditions (Mollner et al. (2010): T=298 K; Amedro et al. (2019): T=298 K, N2 bath gas; Winiberg et al. (2020): T=293 K). Error bars are total errors.
Few other studies have measured the rate coefficient at ambient pressure, which is in the fall-off region (Fig. 9). The rate coefficients in two recent studies (Amedro et al., 2019; Winiberg et al., 2020) that derived Troe expressions (Eq. 1) agree well to within ±2 % (Table 4). The value obtained in the study by Mollner et al. (2010) is approximately 15 % lower than the values obtained in the more recent studies. Possible reasons for the lower value in Mollner et al. (2010) are discussed in Amedro et al. (2019), including possible systematic errors in the determination of the NO2 concentration, but the exact reason remains unclear.
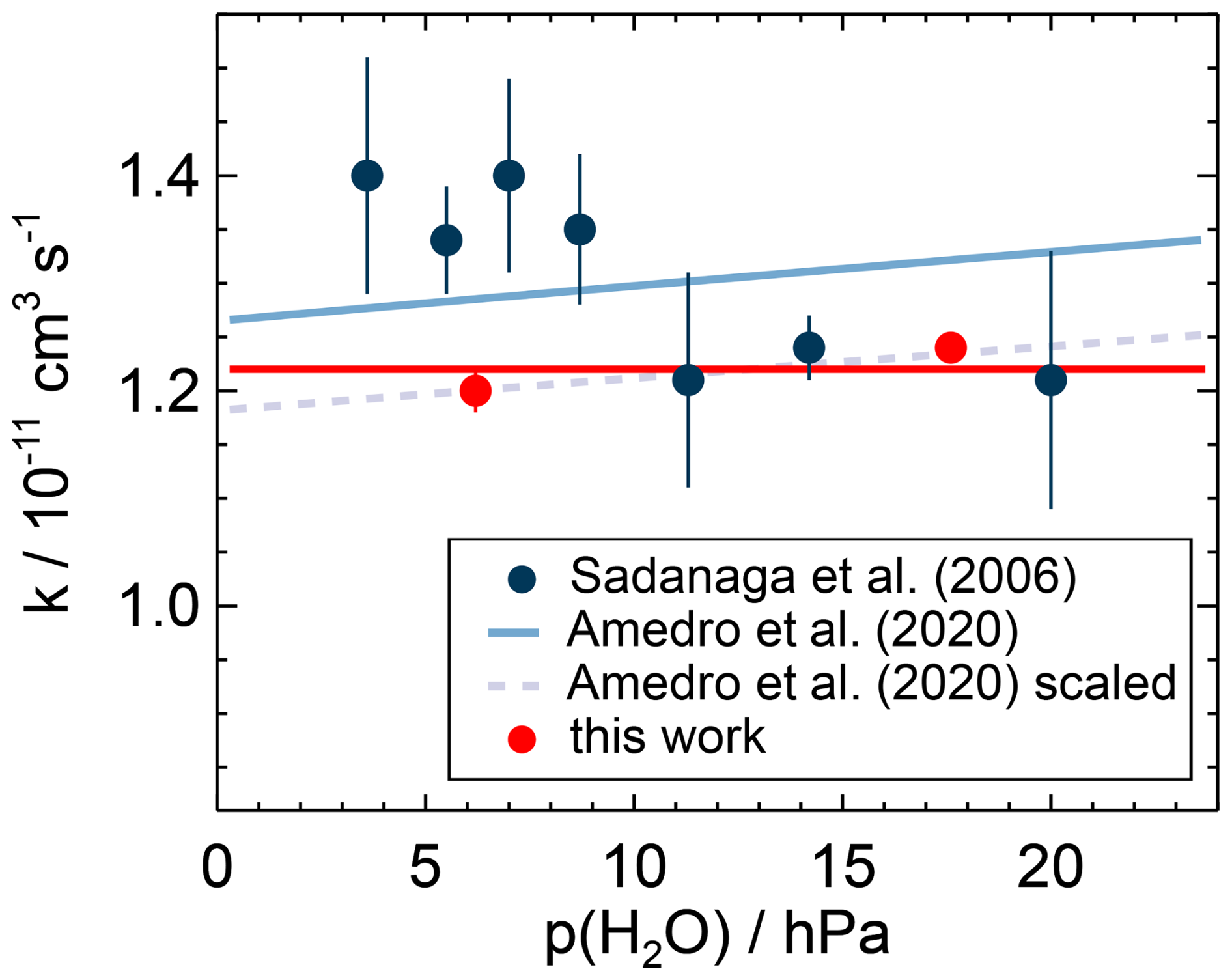
Figure 10Water vapour dependence of the second-order rate coefficient of the OH reaction with NO2 measured in this work and calculated from the parameterisation of the measurements in Amedro et al. (2020) for conditions of this work (air, T=295 K, p=1034 hPa). The parameterisation fits the measured data of this work when scaled by a factor of 0.93. The measurements of Sadanaga et al. (2006) were made at a temperature of 298 K and a pressure of 990 hPa in air.
Amedro et al. (2020) determined the effect of collisional stabilisation of the association complex by water molecules in experiments at low pressure and high partial pressures of water vapour. These experiments show that the collision efficiency is 6 times higher than for N2, similar to the effect on the OH reaction with NO (Sun et al., 2022). Using the Troe equation determined by Amedro et al. (2020) for N2–H2O mixtures, the rate coefficient increases by 2.7 % for water vapour partial pressures of 6.2 and 17.2 hPa tested in this work (Fig. 10). The prediction is in good agreement with the increase in the rate coefficients of (3.3±2.2) % observed in this work (Fig. 10).
Sadanaga et al. (2006) found that the reaction rate coefficient decreases by 18 % when the partial pressure of water vapour is increased from 4 to 29 hPa. The experimental conditions were similar to those in this work in terms of bath gas, temperature, and pressure, and similar OH reactivity instruments were used. It is worth noting that the rate coefficients determined in the work by Sadanaga et al. (2006) for water vapour partial pressures higher than 10 hPa are in good agreement with the rate coefficients in this work (Fig. 10). However, the increase in the rate coefficient at lower water vapour pressure contradicts the results in the present work and in Amedro et al. (2020). Theoretical calculations in Sadanaga et al. (2006) could not explain their observed water vapour dependence. Therefore, the discrepancies with the recent studies remain unexplained.
3.4 Rate coefficient of the HO2 reaction with NO2
The reaction of HO2 with NO2 was studied in this work in air at a total pressure of 1031 hPa at a temperature of 297 K for different water vapour partial pressures between 2.0 hPa and 17.5 hPa equivalent to relative humidities between 11 % and 90 %. For these measurements, the instrument was operated to produce HO2 in the flow and to detect the HO2 decay (Sect. 2.1). The HO2 reaction with NO2 forms pernitric acid (HO2NO2) in a termolecular reaction (Reaction R8).
Several potential systematic errors in the determination of the rate coefficients can be excluded:
-
HO2NO2 is thermally unstable (Gierczak et al., 2005) and could affect the HO2 decay by producing HO2. The chemical lifetime of HO2NO2 was approximately 10 s calculated using the NASA–JPL equilibrium constant and reaction rates (Burkholder et al., 2020). This is much longer than the timescale of the experiments in this work. Consequently, no deviations from single-exponent behaviour were observed.
-
As discussed for the OH reaction with NO2 (Sect. 3.3), laser photolysis of NO2 was negligible, since less than 10−4 of the NO2 molecules were photolysed, and thus the NO2 concentration did not change. Although NO produced in the photolysis of NO2 can react with HO2, this does not affect the observed HO2 decay because the OH produced reacts back to HO2 in the reaction with excess CO.
-
The reaction of HO2 with NO from impurities in the NO2 mixture of the air supply (Table 1) or from NO2 photolysis could have contributed to the total HO2 loss. However, the expected NO concentrations were very low and the NO reaction with HO2 produces OH, which immediately reacted back to HO2 in the reaction with excess CO (Sect. 2.1).
-
Systematic errors due to the self-reaction of HO2, as reported in previous studies (e.g. Kurylo and Ouellette, 1986; Christensen et al., 2004), were negligible due to the very low initial HO2 concentrations used in this work (Sect. 2.3).
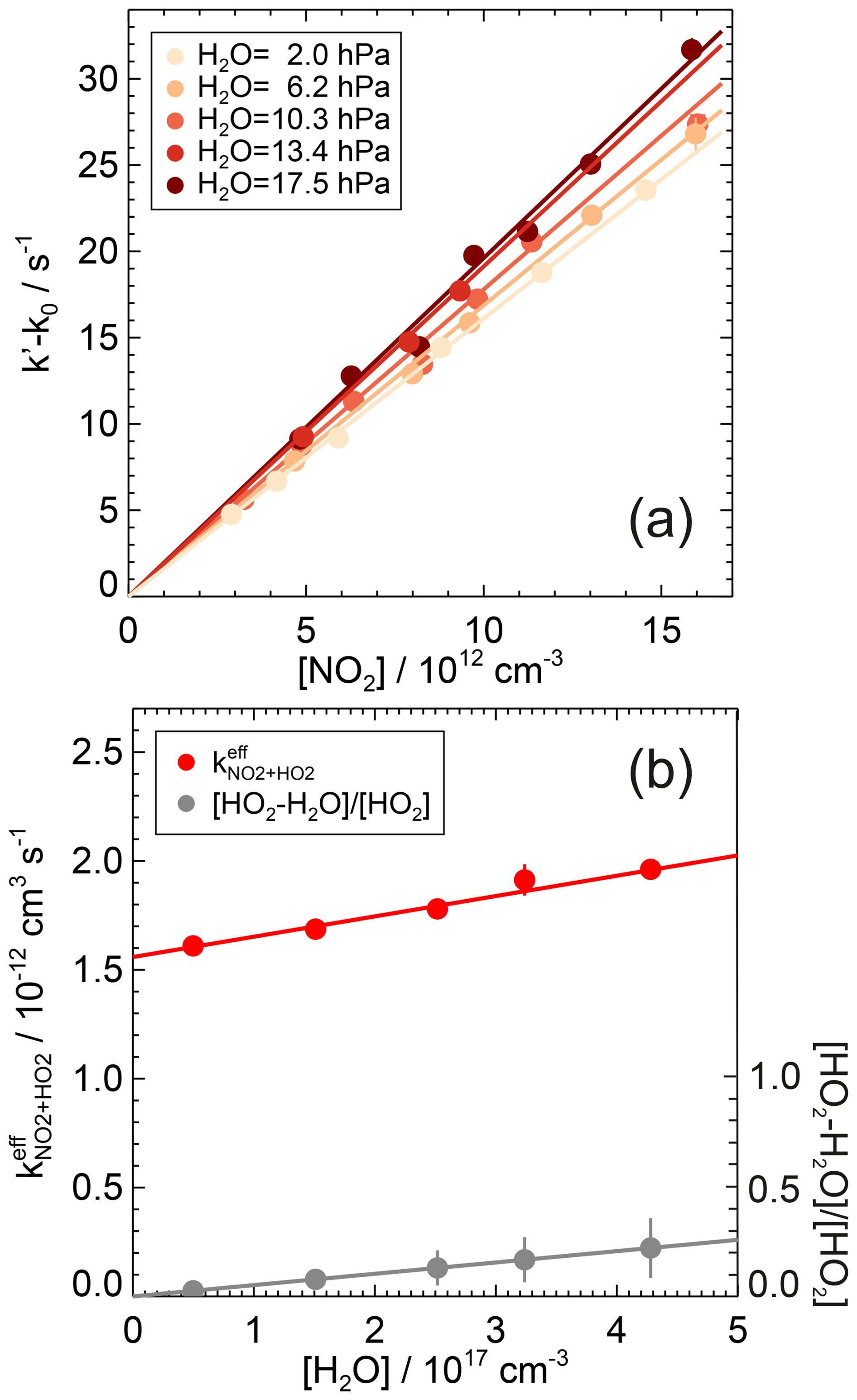
Figure 11First-order rate coefficients for the reaction of HO2 with NO2 at different humidities and ambient temperature ((297±1) K) and pressure ((1026±5) hPa) in air (a). The zero rate coefficient k0 is subtracted from the linear fit of the measured HO2 reactivity (Eq. 7). The lines are the results of a linear fit to the measurements at each humidity. The rate coefficients show a linear increase with the water vapour concentration, which scales with the concentration ratio of HO2 complexed with H2O to free HO2 radicals (b). Error bars (1σ statistical errors) are partly smaller than the size of the symbols.
Measurements were performed at different water vapour partial pressures. The rate coefficients increased linearly by approximately 20 % as the water vapour partial pressure increased from 2.0 to 17.5 hPa (Fig. 11, Table A1). The observed linear dependence on water concentration can be empirically described by
where is the measured second-order rate coefficient determined from the observed HO2 decays. The rate coefficient represents the value in dry air at 1 atm, and is a third-order rate coefficient that describes the enhancement of the observed rate coefficient by water vapour. A linear fit of the measurements (Fig. 11) yields values of cm3 s−1 and cm6 s−1. The errors are total uncertainties that include the measurement errors and the uncertainties in the NO2 and H2O concentrations.
Sander and Peterson (1984) observed a similar behaviour and proposed an enhanced rate coefficient of the NO2 reaction with the hydrogen-bonded HO2⋅H2O complex.
The bimolecular rate coefficient of this reaction can be determined from the observed water vapour dependence, taking into account the chemical equilibrium between the free HO2 radical and the HO2⋅H2O complexes, which are in a fast equilibrium (e.g. Cox and Burrows, 1979; Aloisio et al., 2000).
Kanno et al. (2005) determined the value of the equilibrium constant at room temperature to be cm3, which is in good agreement with the results of other studies (Cox and Burrows, 1979; Lii et al., 1981; Aloisio et al., 2000) and is also the value recommended by NASA–JPL (Burkholder et al., 2020), where the uncertainty is estimated to be a factor of 2. The equilibrium can be assumed to be instantaneous on the timescale of the HO2 decay in the flow tube of the instrument used in this work. The fraction f of the free HO2 radicals can be estimated by the following approach:
where Keq[H2O] is a small number (<0.2) for the range of water vapour concentrations used in this work and represents the concentration ratio of the complexed HO2 to free HO2 radicals (Fig. 11).
The detection of HO2 in the low-pressure detection cell of the instrument can be assumed to be equally sensitive to the free HO2 radical and the HO2–water complex. Calibration measurements show that the HO2 detection sensitivity decreases slightly by 15 % with increasing water vapour concentrations in the range used in this work. This can be quantitatively explained by fluorescence quenching by water molecules (Fuchs et al., 2011), providing evidence for the same instrument sensitivity for the free HO2 radical and the HO2–water complex.
The observed radical decay using Eq. (9) is then given by
This approach gives a linear dependence of the effective rate coefficient, , on water vapour, as observed (Fig. 11).
Table 5Second-order rate coefficients for the reaction of the free HO2 radical and the HO2–water complex with NO2 and the third-order rate coefficient describing the water dependence at ambient pressure (p) and temperature (T), as well as varying humidity, determined in this work. In addition to the IUPAC and NASA–JPL recommendations (values calculated for the experimental conditions), the results of experiments reported in the literature are given. The errors in the rate coefficients are 2σ uncertainties.
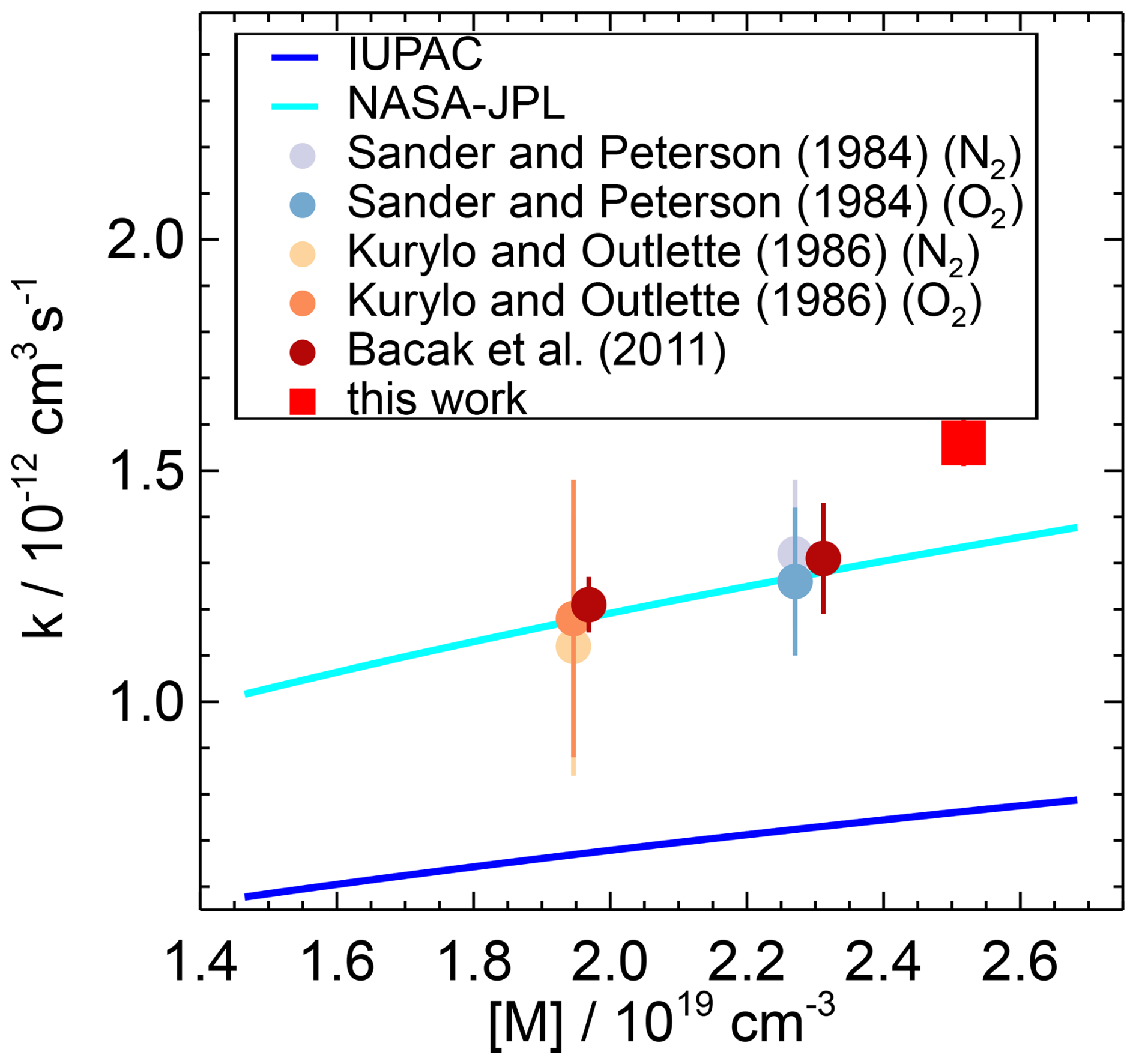
Figure 12Pressure dependence of the second-order rate coefficient of the HO2 reaction with NO2 reported in the literature and measured in this work. IUPAC and NASA–JPL values are calculated for the conditions of this work (T=297 K), and measurements of Bacak et al. (2011) were performed in N2 at room temperature (298 K) and a pressure of 933 hPa. Error bars are total errors.
Comparing the empirical expression of the H2O dependence (Eq. 8) with Eq. (11) and using the observed values (Table 5) allow us to calculate the value of the second-order rate coefficient for the NO2 reaction with the HO2–water complex (Reaction R11). This yields a value of cm3 s−1. The uncertainty is higher than for the other rate coefficients due to the uncertainty of the equilibrium constant (Kanno et al., 2005).
The enhancement of the rate coefficient for the HO2–water complex can be explained by the Chaperone mechanism, where the water molecule bonded to HO2 acts as a third body that removes energy and stabilises the association product of the reaction between HO2 and NO2 similar to the mechanism discussed in Christensen et al. (2004) for the methanol-bonded HO2 radical.
To the best of our knowledge, our study provides the first experimental data on the HO2 reaction with NO2 in 1 atm air. Previous studies were mostly carried out at lower total pressures, and only the study by Bacak et al. (2011) was carried out under conditions close to those of this work (950 hPa in N2 in a turbulent flow tube; Table 4, Fig. 12).
The rate coefficient derived in this work for the NO2 reaction with the free HO2 radical is a factor of 2 higher than the IUPAC (2017a) recommendation and 17 % higher than the NASA–JPL recommendation (Burkholder et al., 2020) (Table 5, Fig. 12). The NASA–JPL recommendation for room temperature is based on measurements by Sander and Peterson (1984), Kurylo and Ouellette (1986), Christensen et al. (2004), and Bacak et al. (2011). In these studies, the rate coefficients were measured at lower-than-ambient pressures between 250 and 950 hPa and temperatures between 277 and 298 K. A recent re-analysis of the rate coefficients available in the literature using a master equation analysis by McKee et al. (2022) gave a parameterisation which agrees well with the values recommended by NASA–JPL.
The IUPAC recommendation is based only on measurements by Christensen et al. (2004), where experiments were performed at much lower-than-ambient pressures (<270 hPa). IUPAC excludes the studies by Sander and Peterson (1984) and Kurylo and Ouellette (1986) from their analysis because in these studies HO2 was produced using methanol as a precursor. This can affect the results by the formation of a hydrogen-bonded adduct with HO2, as the adduct can introduce a systematic error at temperatures below 250 K due to an increased rate of the NO2 reaction with the methanol–HO2 complex (Christensen et al., 2004). The large discrepancy between the NASA–JPL and IUPAC predictions in the fall-off region around 1 atm may therefore be caused by the different data sets parameterised over different pressure ranges and/or by the use of different broadening factors in the Troe equations. Both recommendations underestimate the value determined in this work, indicating the need for more extensive experimental studies covering a wider pressure range around 1 atm.
The effect of an increasing effective reaction rate in the presence of water was also observed in the experiments of Sander and Peterson (1984), which were carried out at room temperature and a low pressure of 467 hPa with water vapour partial pressures between 0 and 21 hPa in N2. They determined a third-order rate coefficient (Eq. 8) of cm6 s−1 which is very close to the value of this work in 1 atm air (Table 5). It should be noted, however, that the expression for the rate coefficient includes the pressure-dependent second-order rate coefficient , so the agreement is not necessarily expected. However, the pressure sensitivity is small at room temperature because the value is mainly determined by the pressure-independent rate coefficient , which is a factor 2 to 3.5 higher than the pressure-dependent rate coefficient for the conditions of the two studies.
Sander and Peterson (1984) also derived a relationship similar to that in Eq. (10) to determine the second-order rate coefficient for the NO2 reaction with the HO2–water complex but did not substitute [HO2] with in their rate equation. Using Eq. (11) and the latest recommendation for Keq (Kanno et al., 2005; Burkholder et al., 2020), the re-calculation of the second-order rate coefficient gives a value of m3 s−1 for their data. The difference with the value in this work is 14 %, but this is well within the measurement errors in both studies. The similarity of the values obtained in Sander and Peterson (1984) and in this work at different total pressures (467 and 1031 hPa) supports the assumption that the reaction follows a Chaperone mechanism that is independent of the buffer gas (N2, air).
An increase in the HO2 reactivity due to the complexation with water molecules has been observed for other reactions, such as the self-reaction of HO2, which can be enhanced by a factor of up to 2 in the moist troposphere (Lii et al., 1981; Kircher and Sander, 1984). Christensen et al. (2004) reported a similar effect for methanol, which also forms an adduct with HO2 and increases the reaction rate between HO2 and NO2.
A temperature dependence of the rate coefficient of the NO2 reaction with the HO2–water complex can be estimated from measured data in Sander and Peterson (1984), which supports the assumption that the reaction of complexed HO2 with NO2 follows a bimolecular mechanism. In their work, they measured an increase in the value by a factor of 1.3 and 1.6 when the temperature was reduced from 298 to 286 K and 275 K, respectively. A re-calculation of their values (see above) gives a temperature trend with a positive Arrhenius activation energy K. In contrast, the pressure-dependent NO2 reaction with the free HO2 radical shows a negative temperature dependence. The water dependence of the HO2 reaction with NO2 is therefore expected to increase in warmer regions. For example, increasing the temperature by 10 degrees at 298 K and 1 atm increases the ratio of the rate coefficients : from 2.1 to 2.6.
Higher temperatures also mean higher concentrations of water vapour in the atmosphere. However, this does not necessarily increase the concentration of the HO2⋅H2O complex because the equilibrium (Reaction R12) is shifted towards free HO2 radicals at higher temperatures. Overall, the influence of water vapour on the reaction of HO2 with NO2 is complex and remains uncertain mainly because the equilibrium constant has a large uncertainty and because of the general lack of reaction kinetic measurements with water vapour over the tropospheric temperature range.
The second-order rate coefficients of the termolecular reactions of OH with CO, NO, and NO2 and of the termolecular reaction of HO2 with NO2 were measured at tropospheric conditions of 1 atm pressure, at room temperature, and using humidified air as bath gas. The water vapour partial pressure was varied between 2.0 and 22.5 hPa. An instrument, which was developed for the measurement of atmospheric OH reactivity in field and chamber experiments (Lou et al., 2010), was used. This instrument measures the decay of OH radicals produced by laser flash photolysis of ozone using laser-induced fluorescence with a high sensitivity. The accuracies of the rate coefficients obtained in this work are better than 5 %, mainly limited by the uncertainty of the certified commercial reactant gas standards, whose concentrations were checked using independent reference instruments.
Except for the rate coefficient of the HO2 reaction with NO2, the measured values are within the range of the recommendations of IUPAC and NASA–JPL evaluations, which partly specify large uncertainties. The experimental method used in this work yields rate coefficients which are among the most accurate values reported so far for atmospheric conditions. It is worth noting that the kinetic decays were carried out on a timescale similar to that of OH reactions in the lower troposphere. The initial OH concentrations were a factor of 10 to 10 000 lower than in all previous studies, and the corresponding reactant concentrations were about a factor of 1000 lower, greatly reducing the potential for perturbation by secondary chemistry.
Measurements of the rate coefficient for the OH reaction with CO are in very good agreement with the NASA–JPL and IUPAC values. The differences are less than the 5 % uncertainty of the value determined in this work. The rate coefficient of the OH reaction with NO agrees within 3 % with the NASA–JPL value, whereas the IUPAC value is 35 % higher than in this work and 50 % higher than the recently measured value by Sun et al. (2022). This suggests that the IUPAC recommendation may need to be revised. The rate coefficient for the reaction of the OH reaction with NO2 measured in this work in air is 4 % lower than the NASA–JPL-recommended value in air and is 4 % higher than the IUPAC-recommended value. Since the collisional stabilisation of the activated association complex is different for N2 and O2 (Amedro et al., 2019), the IUPAC value which is given for N2 would be additionally 3 % lower in air.
Due to the large abundance of water vapour in the troposphere, it is an important question to what extent H2O influences atmospheric reactions by acting as a third-body collision partner or by forming a complex with OH or HO2. In agreement with the literature, no significant influence of H2O was found for the reaction of OH with CO for water vapour partial pressures up to 22.5 hPa on water in 1 atm air at room temperature.
The activated association complexes formed in the OH reactions with NO and NO2 have been shown to be better stabilised by water molecules than by N2 and O2 (Paraskevopoulos and Irwin, 1984; Amedro et al., 2020; Sun et al., 2022). However, the effect becomes small at pressures of 1 atm and water concentrations typically found in the lower troposphere and tested in this work.
For the reaction of OH with NO, recent work by Sun et al. (2022) predicts an increase in the rate constant of up to 5 % for the range of water concentrations in this work. However, the observed variability in this work is only 1 %, suggesting that the effect is smaller than expected from the results in Sun et al. (2022). The rate coefficients of the reaction of OH with NO2 were measured for two water vapour partial pressures (6 and 17 hPa). The small increase in the values of (3.3±2.2) % with increasing water vapour agrees with a prediction of 2.7 % from the Troe equation determined by Amedro et al. (2020). A negative dependence on water vapour reported for atmospheric conditions in air by Sadanaga et al. (2006) could not be confirmed.
A strong water vapour dependence of the effective reaction rate coefficient was found for the reaction of HO2 with NO2 giving a second-order rate coefficient for dry air at 1 atm pressure that is a factor of 2 larger than the recommendation by IUPAC for N2 and 17 % higher than the NASA–JPL-recommended value for air. The measured rate coefficient shows a linear increase of 25 % at a water vapour partial pressure of 17.5 hPa. Similar to the well-known water-dependent HO2 self-reaction, the increased reactivity is presumably caused by HO2 radicals that form a complex with water molecules which reacts faster with NO2 than free HO2 radicals.
The observed increase can be explained by the known thermal equilibrium between free HO2 radicals and HO2 radicals complexed with H2O (Cox and Burrows, 1979; Aloisio et al., 2000). This can be used to determine the second-order rate coefficient for the reaction of the HO2⋅H2O complex with NO2 resulting in a value that is a factor of 2 faster than that of the reaction of the free HO2 radical.
A re-analysis of the data of Sander and Peterson (1984), who studied the water vapour dependence of this reaction at a pressure of 467 hPa and room temperature, gives a good agreement of the rate coefficients of the HO2⋅H2O complex with NO2 with the value determined in this work at 1031 hPa. This agreement supports the assumption that the reaction of the HO2⋅H2O complex with NO2 behaves like a pressure-independent bimolecular reaction. Although the rate coefficient of this Chaperone-type reaction has a large uncertainty, the results suggest that the water effect should be included in atmospheric chemistry models. It also demonstrates the general need to consider potential water effects of reactions relevant in the atmosphere, as discussed in the review by Buszek et al. (2011) and shown in global chemical transport models (Khan et al., 2015).
Overall, the measurements in this work provide highly accurate rate coefficients that can serve as reference values at tropospheric conditions and could be used to improve the parameterisation of termolecular rate coefficients (Burkholder et al., 2017; Fiore et al., 2024). The method of using an OH reactivity instrument for kinetic studies can be extended to also measure the temperature dependence of the rate coefficient, as successfully shown by Berg et al. (2024) for the OH reactions with alkanes, aromatics, and monoterpenes.
Table A1Second-order rate coefficients (k) determined in this study. Errors are 1σ statistical errors and do not include the uncertainty of the reactant's concentration.
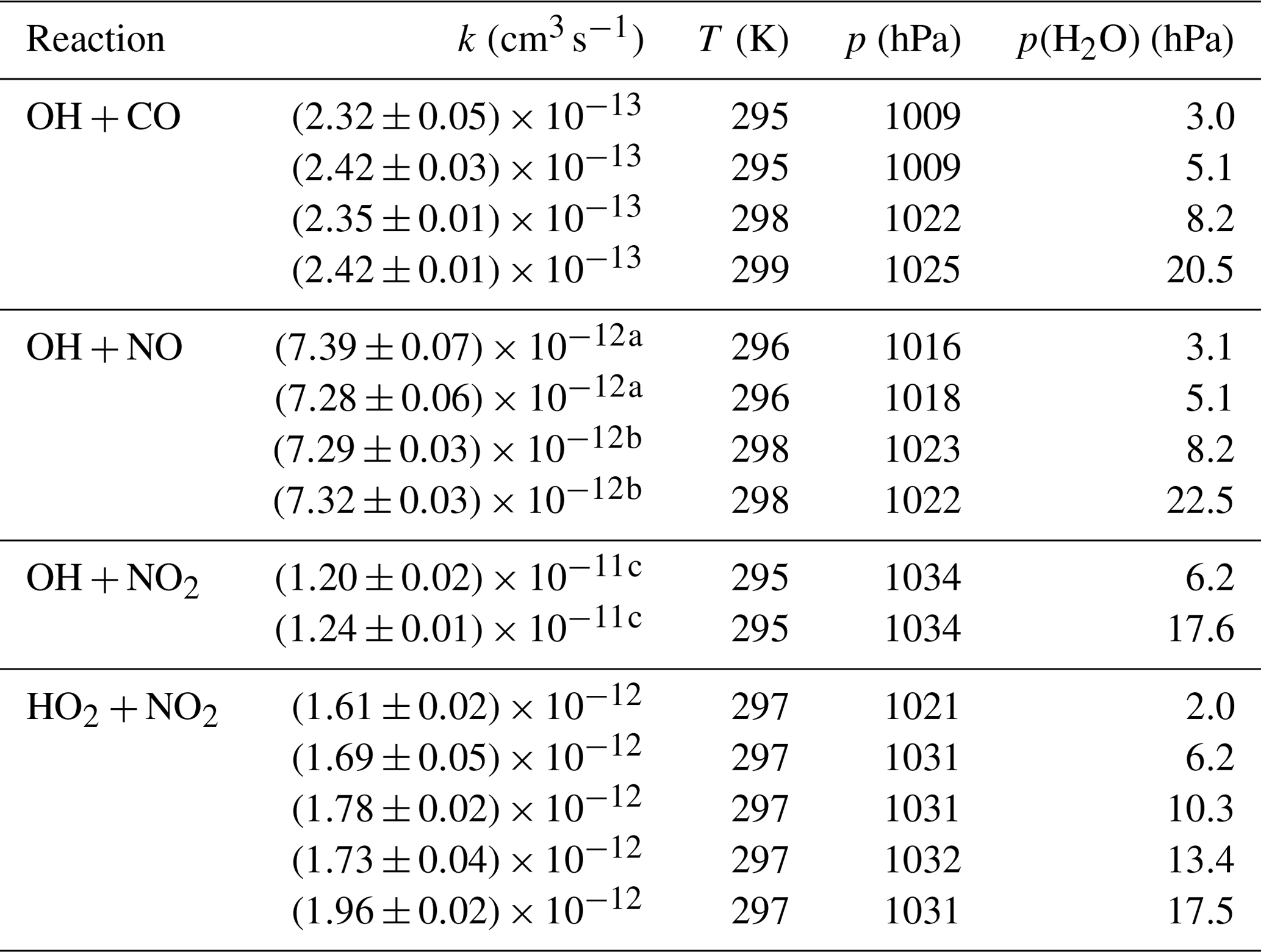
The data are listed in the table in the Appendix.
HF and AH wrote the manuscript, MR performed the measurements and evaluated them to determine the rate coefficients, and AN and AW supported the measurements. All co-authors discussed the results.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank Christian Ehlers and Danielle Barnett for the support of the measurements of concentrations of the gas standards used in this work.
The article processing charges for this open-access publication were covered by the Forschungszentrum Jülich.
This paper was edited by Lisa Whalley and reviewed by two anonymous referees.
Aloisio, S. and Francisco, J. S.: A density functional study of the H2O−-HOCO complex, J. Phys. Chem. A, 104, 404–407, https://doi.org/10.1021/jp9928356, 2000. a
Aloisio, S., Francisco, J. S., and Friedl, R. R.: Experimental evidence for the existence of the HO2–H2O complex, J. Phys. Chem. A, 104, 6597–6601, https://doi.org/10.1021/jp0006330, 2000. a, b, c, d
Amedro, D., Miyazaki, K., Parker, A., Schoemaecker, C., and Fittschen, C.: Atmospheric and kinetic studies of OH and HO2 by the FAGE technique, J. Environ. Sci., 24, 78–86, https://doi.org/10.1016/S1001-0742(11)60723-7, 2012. a
Amedro, D., Bunkan, A. J. C., Berasategui, M., and Crowley, J. N.: Kinetics of the OH + NO2 reaction: rate coefficients (217–333 K, 16–1200 mbar) and fall-off parameters for N2 and O2 bath gases, Atmos. Chem. Phys., 19, 10643–10657, https://doi.org/10.5194/acp-19-10643-2019, 2019. a, b, c, d, e, f, g, h, i
Amedro, D., Berasategui, M., Bunkan, A. J. C., Pozzer, A., Lelieveld, J., and Crowley, J. N.: Kinetics of the OH + NO2 reaction: effect of water vapour and new parameterization for global modelling, Atmos. Chem. Phys., 20, 3091–3105, https://doi.org/10.5194/acp-20-3091-2020, 2020. a, b, c, d, e, f, g, h, i
Anastasi, C. and Smith, I. W. M.: Rate measurements of reactions of OH by resonance absorption. Part 6. Rate constants for over a wide range of temperature and pressure, J. Chem. Soc. Farad. Trans., 74, 1056–1064, https://doi.org/10.1039/F29787401056, 1978. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004. a, b, c, d, e, f
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a
Bacak, A., Cooke, M. C., Bardwell, M. W., McGillen, M. R., Archibald, A. T., Huey, L. G., Tanner, D., Utembe, S. R., Jenkin, M. E., Derwent, R. G., Shallcross, D. E., and Percival, C. J.: Kinetics of the HO2+NO2 reaction: On the impact of new gas-phase kinetic data for the formation of HO2NO2 on HOX, NOX and HO2NO2 levels in the troposphere, Atmos. Environ., 45, 6414–6422, https://doi.org/10.1016/j.atmosenv.2011.08.008, 2011. a, b, c, d
Barker, J. R., Stanton, J. F., and Nguyen, T. L.: Semiclassical transition state theory/master equation kinetics of HO+CO: Performance evaluation, Int. J. Chem. Kin., 52, 1022–1045, https://doi.org/10.1002/kin.21420, 2020. a
Beno, M. F., Jonah, C. D., and Mulac, W. A.: Rate constants for the reaction OH+CO as functions of temperature and water concentration, Int. J. Chem. Kin., 17, 1091–1101, https://doi.org/10.1002/kin.550171006, 1985. a
Berg, F., Novelli, A., Dubus, R., Hofzumahaus, A., Holland, F., Wahner, A., and Fuchs, H.: Temperature-dependent rate coefficients for the reactions of OH radicals with selected alkanes, aromatic compounds, and monoterpenes, Atmos. Chem. Phys., 24, 13715–13731, https://doi.org/10.5194/acp-24-13715-2024, 2024. a, b
Bohn, B. and Zetzsch, C.: Rate Constants of HO2+NO Covering Atmospheric Conditions. 1. HO2 Formed by OH+H2O2, J. Phys. Chem. A, 101, 1488–1493, https://doi.org/10.1021/jp961396x, 1997. a, b, c
Bohn, B. and Zetzsch, C.: Gas-phase reaction of the OH–benzene adduct with O2: reversibility and secondary formation of HO2, Phys. Chem. Chem. Phys., 1, 5097–5107, https://doi.org/10.1039/A904887A, 1999. a, b, c
Burkholder, J. B., Abbatt, J. P. D., Barnes, I., Roberts, J. M., Melamed, M. L., Ammann, M., Bertram, A. K., Cappa, C. D., Carlton, A. G., Carpenter, L. J., Crowley, J. N., Dubowski, Y., George, C., Heard, D. E., Herrmann, H., Keutsch, F. N., Kroll, J. H., McNeill, V. F., Ng, N. L., Nizkorodov, S. A., Orlando, J. J., Percival, C. J., Picquet-Varrault, B., Rudich, Y., Seakins, P. W., Surratt, J. D., Tanimoto, H., Thornton, J. A., Tong, Z., Tyndall, G. S., Wahner, A., Weschler, C. J., Wilson, K. R., and Ziemann, P. J.: The essential role for laboratory studies in atmospheric chemistry, Environ. Sci. Technol., 51, 2519–2528, https://doi.org/10.1021/acs.est.6b04947, 2017. a, b
Burkholder, J. B., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies–evaluation number 19, Nasa panel for data evaluation technical report, 19-5, 1–1610, https://jpldataeval.jpl.nasa.gov/pdf/NASA-JPL Evaluation 19-5.pdf (last access: 25 October 2024), 2020. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s, t
Buszek, R. J., Francisco, J. S., and Anglada, J. M.: Water effects on atmospheric reactions, Int. Rev. Phys. Chem., 30, 335–369, https://doi.org/10.1080/0144235X.2011.634128, 2011. a, b
Chao, W., Jr-Min Lin, J., Takahashi, K., Tomas, A., Yu, L., Kajii, Y., Batut, S., Schoemaecker, C., and Fittschen, C.: Water vapor does not catalyze the reaction between methanol and OH radicals, Angew. Chem. Int. Ed., 58, 5013–5017, https://doi.org/10.1002/anie.201900711, 2019. a
Christensen, L. E., Okumura, M., Sander, S. P., Friedl, R. R., Miller, C. E., and Sloan, J. J.: Measurements of the rate constant of using near-infrared wavelength-modulation spectroscopy and UV−visible absorption spectroscopy, J. Phys. Chem. A, 108, 80–91, https://doi.org/10.1021/jp035905o, 2004. a, b, c, d, e, f
Cox, R. A. and Burrows, J. P.: Kinetics and mechanism of the disproportionation of HO2 in the gas phase, J. Chem. Phys., 20, 2560–2588, https://doi.org/10.1021/j100483a002, 1979. a, b, c, d
Ehhalt, D. H.: Photooxidation of trace gases in the troposphere, Phys. Chem. Chem. Phys., 1, 5401–5408, https://doi.org/10.1039/a905097c, 1999. a
Ervens, B., Rickard, A., Aumont, B., Carter, W. P. L., McGillen, M., Mellouki, A., Orlando, J., Picquet-Varrault, B., Seakins, P., Stockwell, W. R., Vereecken, L., and Wallington, T. J.: Opinion: Challenges and needs of tropospheric chemical mechanism development, Atmos. Chem. Phys., 24, 13317–13339, https://doi.org/10.5194/acp-24-13317-2024, 2024. a
Fiore, A. M., Mickley, L. J., Zhu, Q., and Baublitz, C. B.: Climate and tropospheric oxidizing capacity, Annu. Rev. Earth Planet. Sci., 52, 321–349, https://doi.org/10.1146/annurev-earth-032320-090307, 2024. a, b
Fuchs, H., Bohn, B., Hofzumahaus, A., Holland, F., Lu, K. D., Nehr, S., Rohrer, F., and Wahner, A.: Detection of HO2 by laser-induced fluorescence: calibration and interferences from RO2 radicals, Atmos. Meas. Tech., 4, 1209–1225, https://doi.org/10.5194/amt-4-1209-2011, 2011. a, b
Fuchs, H., Hofzumahaus, A., Rohrer, F., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Holland, F., Kaminski, M., Li, X., Lu, K., Nehr, S., Tillmann, R., Wegener, R., and Wahner, A.: Experimental evidence for efficient hydroxyl radical regeneration in isoprene oxidation, Nat. Geosci., 6, 1023–1026, https://doi.org/10.1038/NGEO1964, 2013. a
Fuchs, H., Acir, I.-H., Bohn, B., Brauers, T., Dorn, H.-P., Häseler, R., Hofzumahaus, A., Holland, F., Kaminski, M., Li, X., Lu, K., Lutz, A., Nehr, S., Rohrer, F., Tillmann, R., Wegener, R., and Wahner, A.: OH regeneration from methacrolein oxidation investigated in the atmosphere simulation chamber SAPHIR, Atmos. Chem. Phys., 14, 7895–7908, https://doi.org/10.5194/acp-14-7895-2014, 2014. a
Fulle, D., Hamann, H. F., Hippler, H., and Troe, J.: High pressure range of addition reactions of HO. II. Temperature and pressure dependence of the reaction , J. Chem. Phys., 105, 983–1000, https://doi.org/10.1063/1.471991, 1996. a, b
Gierczak, T., Jiménez, E., Riffault, V., Burkholder, J. B., and Ravishankara, A. R.: Thermal decomposition of HO2NO2 (peroxynitric acid, PNA): Rate coefficient and determination of the enthalpy of formation, J. Phys. Chem. A, 109, 586–596, https://doi.org/10.1021/jp046632f, 2005. a, b
Griffith, S. M., Hansen, R. F., Dusanter, S., Michoud, V., Gilman, J. B., Kuster, W. C., Veres, P. R., Graus, M., de Gouw, J. A., Roberts, J., Young, C., Washenfelder, R., Brown, S. S., Thalman, R., Waxman, E., Volkamer, R., Tsai, C., Stutz, J., Flynn, J. H., Grossberg, N., Lefer, B., Alvarez, S. L., Rappenglueck, B., Mielke, L. H., Osthoff, H. D., and Stevens, P. S.: Measurements of hydroxyl and hydroperoxy radicals during CalNex-LA: Model comparisons and radical budgets, J. Geophys. Res., 121, 4211–4232, https://doi.org/10.1002/2015JD024358, 2016. a
Heard, D. E. and Henderson, D. A.: Quenching of OH(A, ) several collision partners between 200 and 344 K. Cross-section measurements and model comparisons, Phys. Chem. Chem. Phys., 2, 67–72, 2000. a
Hofzumahaus, A. and Stuhl, F.: Rate constant of the reaction HO+CO in the presence of N2 and O2, Ber. Bunsenges. Phys. Chem., 88, 557–561, https://doi.org/10.1002/bbpc.19840880612, 1984. a, b
Hofzumahaus, A., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C.-C., Fuchs, H., Holland, F., Kita, K., Kondo, Y., Li, X., Lou, S., Shao, M., Zeng, L., Wahner, A., and Zhang, Y.: Amplified trace gas removal in the troposphere, Science, 324, 1702–1704, https://doi.org/10.1126/science.1164566, 2009. a, b, c, d
Hynes, A. J., Wine, P. H., and Ravishankara, A. R.: Kinetics of the OH+CO reaction under atmospheric conditions, J. Geophys. Res., 91, 11815–11820, https://doi.org/10.1029/JD091iD11p11815, 1986. a, b, c, d
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, Data sheets for gas phase reactions of NOx species, Data Sheet I.A3.46 NOx16, https://iupac.aeris-data.fr/ (last access: 24 October 2024), 2017a. a, b
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, Data sheets for gas phase reactions of NOx species, Data Sheet I.A3.42 NOx12, https://iupac.aeris-data.fr/ (last access: 24 October 2024), 2017b. a, b
IUPAC: Task Group on Atmospheric Chemical Kinetic Data Evaluation, Data sheets for gas phase reactions of NOx species, Data Sheet I.A3.43 NOx13, https://iupac.aeris-data.fr/ (last access: 24 October 2024), 2017c. a, b
Ivanov, A. V., Trakhtenberg, S., Bertram, A. K., Gershenzon, Y. M., and Molina, M. J.: OH, HO2, and ozone gaseous diffusion coefficients, J. Phys. Chem. A, 111, 1632–1637, https://doi.org/10.1021/jp066558w, 2007. a
Johnson, C. J., Otto, R., and Continetti, R. E.: Spectroscopy and dynamics of the HOCO radical: insights into the reaction, Phys. Chem. Chem. Phys., 16, 19091–19105, https://doi.org/10.1039/C4CP02593H, 2014. a
Kanno, N., Tonokura, K., Tezaki, A., and Koshi, M.: Water dependence of the HO2 self reaction: Kinetics of the HO2–HO2 complex, J. Phys. Chem. A, 109, 3153–3158, https://doi.org/10.1021/jp044592+, 2005. a, b, c, d, e
Khan, M. A. H., Cooke, M. C., Utembe, S. R., Archibald, A. T., Derwent, R. G., Jenkin, M. E., Morris, W. C., South, N., Hansen, J. C., Francisco, J. S., Percival, C. J., and Shallcross, D. E.: Global analysis of peroxy radicals and peroxy radical-water complexation using the STOCHEM-CRI global chemistry and transport model, Atmos. Environ., 106, 278–287, https://doi.org/10.1016/j.atmosenv.2015.02.020, 2015. a
Kim, S., Huey, L. G., Stickel, R. E., Tanner, D. J., Crawford, J. H., Olson, J. R., Chen, G., Brune, W. H., Ren, X., Lesher, R., Wooldridge, P. J., Bertram, T. H., Perring, A., Cohen, R. C., Lefer, B. L., Shetter, R. E., Avery, M., Diskin, G., and Sokolik, I.: Measurement of HO2NO2 in the free troposphere during the Intercontinental Chemical Transport Experiment–North America 2004, J. Geophys. Res., 112, D12S01, https://doi.org/10.1029/2006JD007676, 2007. a
Kircher, C. C. and Sander, S. P.: Kinetics and mechanism of HO2 and DO2 disproportionations, J. Phys. Chem., 88, 2082–2091, https://doi.org/10.1021/j150654a029, 1984. a, b
Kleffmann, J., Gavriloaiei, T., Hofzumahaus, A., Holland, F., Koppmann, R., Rupp, L., Schlosser, E., Siese, M., and Wahner, A.: Daytime formation of nitrous acid: A major source of OH radicals in a forest, Geophys. Res. Lett., 32, L05818, https://doi.org/10.1029/2005GL022524, 2005. a
Kovacs, T. A. and Brune, W. H.: Total OH loss rate measurement, J. Atmos. Chem., 39, 105–122, 2001. a
Kurylo, M. J. and Ouellette, P. A.: Pressure dependence of the rate constant for the reaction (M = N2, O2) at 298 K, J. Phys. Chem., 90, 441–444, https://doi.org/10.1021/j100275a018, 1986. a, b, c
Liessmann, M., Miller, Y., Gerber, B., and B., A.: Reaction of OH and NO at Low Temperatures in the Presence of Water: the Role of Clusters, Zeitschrift für physikalische Chemie, 225, 1129–1144, https://doi.org/10.1524/zpch.2011.0181, 2011. a
Lii, R.-R., Sauer, Myran C., J., and Gordon, S.: Temperature dependence of the gas-phase self-reaction of hydroperoxo in the presence of water, J. Phys. Chem., 85, 2833–2834, https://doi.org/10.1021/j150619a027, 1981. a, b, c
Liu, Y. and Sander, S. P.: Rate constant for the OH+CO reaction at low temperatures, J. Chem. Phys. A, 119, 10060–10066, https://doi.org/10.1021/acs.jpca.5b07220, 2015. a, b
Lou, S., Holland, F., Rohrer, F., Lu, K., Bohn, B., Brauers, T., Chang, C. C., Fuchs, H., Häseler, R., Kita, K., Kondo, Y., Li, X., Shao, M., Zeng, L., Wahner, A., Zhang, Y., Wang, W., and Hofzumahaus, A.: Atmospheric OH reactivities in the Pearl River Delta – China in summer 2006: measurement and model results, Atmos. Chem. Phys., 10, 11243–11260, https://doi.org/10.5194/acp-10-11243-2010, 2010. a, b, c, d
Martinez, M., Harder, H., Kovacs, T. A., Simpas, J. B., Bassis, J., Lesher, R., Brune, W. H., Frost, G. J., Williams, E. J., Stroud, C. A., Jobson, B. T., Roberts, J. M., Hall, S. R., Shetter, R. E., Wert, B., Fried, A., Alicke, B., Stutz, J., Young, V. L., White, A. B., and Zamora, R. J.: OH and HO2 concentrations, sources, and loss rates during the Southern Oxidants Study in Nashville, Tennessee, summer 1999, J. Geophys. Res., 108, 4617, https://doi.org/10.1029/2003JD003551, 2003. a
McCabe, D. C., Gierczak, T., Talukdar, R. K., and Ravishankara, A. R.: Kinetics of the reaction OH+CO under atmospheric conditions, Geophys. Res. Lett., 28, 3135–3138, https://doi.org/10.1029/2000GL012719, 2001. a, b, c, d, e, f
McKee, K., Blitz, M. A., Shannon, R. J., and Pilling, M. J.: HO2+NO2: Kinetics, Thermochemistry, and evidence for a bimolecular product channel, J. Phys. Chem. A, 126, 7514–7522, https://doi.org/10.1021/acs.jpca.2c04601, 2022. a
Miyazaki, K., Nakashima, Y., Schoemaecker, C., Fittschen, C., and Kajii, Y.: Note: A laser-flash photolysis and laser-induced fluorescence detection technique for measuring total HO2 reactivity in ambient air, Rev. Sci. Instrum., 84, 076106, https://doi.org/10.1063/1.4812634, 2013. a
Miyoshi, A., Matsui, H., and Washida, N.: Detection and reactions of the HOCO radical in gas phase, J. Chem. Phys., 100, 3532–3539, https://doi.org/10.1063/1.466395, 1994. a
Mollner, A. K., Valluvadasan, S., Feng, L., Sprague, M. K., Okumura, M., Milligan, D. B., Bloss, W. J., Sander, S. P., Martien, P. T., Harley, R. A., McCoy, A. B., and Carter, W. P. L.: Rate of gas phase association of hydroxyl radical and nitrogen dioxide, Science, 330, 646–649, https://doi.org/10.1126/science.1193030, 2010. a, b, c, d, e, f, g, h, i
Nakashima, Y., Tsurumaru, H., Imamura, T., Bejan, I., Wenger, J. C., and Kajii, Y.: Total OH reactivity measurements in laboratory studies of the photooxidation of isoprene, Atmos. Environ., 62, 243–247, https://doi.org/10.1016/j.atmosenv.2012.08.033, 2012. a
Nehr, S., Bohn, B., Fuchs, H., Hofzumahaus, A., and Wahner, A.: HO2 formation from the OH+benzene reaction in the presence of O2, Phys. Chem. Chem. Phys., 13, 10699–10708, https://doi.org/10.1039/C1CP20334G, 2011. a, b
Nehr, S., Bohn, B., and Wahner, A.: Prompt HO2 formation following the reaction of OH with aromatic compounds under atmospheric conditions, J. Phys. Chem. A, 116, 6015–6026, https://doi.org/10.1021/jp210946y, 2012. a, b
Newsome, B. and Evans, M.: Impact of uncertainties in inorganic chemical rate constants on tropospheric composition and ozone radiative forcing, Atmos. Chem. Phys., 17, 14333–14352, https://doi.org/10.5194/acp-17-14333-2017, 2017. a
Okabe, H.: Photochemistry of small molecules, John Wiley and Sons, https://doi.org/10.1002/nadc.19790270512, 1978. a, b
Overend, R., Paraskevopoulos, G., and Black, C.: Rates of OH radical reactions. II. The combination reaction , J. Chem. Phys., 64, 4149–4154, https://doi.org/10.1063/1.431984, 1976. a
Paraskevopoulos, G. and Irwin, R. S.: The pressure dependence of the rate constant of the reaction of OH radicals with CO, J. Phys. Chem., 80, 259–266, https://doi.org/10.1063/1.446488, 1984. a, b, c, d
Sadanaga, Y., Yoshino, A., Kato, S., Watanaba, K., Miyakawa, Y., Hayashi, I., and Kajii, Y.: The importance of NO2 and volatile organic compounds in the urban air from the viewpoint of the OH reactivity, Geophys. Res. Lett., 31, L08102, https://doi.org/10.1029/2004GL019661, 2004a. a
Sadanaga, Y., Yoshino, A., Watanaba, K., Yoshioka, A., Wakazono, Y., Kanaya, Y., and Kajii, Y.: Development of a measurement system of peroxy radicals using a chemical amplification/laser-induced fluorescence technique, Rev. Sci. Instrum., 75, 864–872, https://doi.org/10.1063/1.1666985, 2004b. a
Sadanaga, Y., Kondo, S., Hashimoto, K., and Kajii, Y.: Measurement of the rate coefficient for the OH+NO2 reaction under the atmospheric pressure: Its humidity dependence, Chem. Phys. Lett., 419, 474–478, https://doi.org/10.1016/j.cplett.2005.12.026, 2006. a, b, c, d, e, f
Sander, S. P. and Peterson, M. E.: Kinetics of the reaction , J. Phys. Chem., 88, 1566–1571, https://doi.org/10.1021/j150652a025, 1984. a, b, c, d, e, f, g, h, i, j, k
Sharkey, P., Sims, I. R., Smith, I. W. M., Bocherel, P., and Rowe, B. R.: Pressure and temperature dependence of the rate constants for the association reaction of OH radicals with NO between 301 and 23 K, J. Chem. Soc. Faraday Trans., 90, 3609–3616, https://doi.org/10.1039/FT9949003609, 1994. a, b
Sheps, L. and Au, K.: New instrument for time-resolved OH and HO2 quantification in high-pressure laboratory kinetics studies, J. Phys. Chem. A, 128, 3916–3925, https://doi.org/10.1021/acs.jpca.4c00994, 2024. a
Smith, I. W. M. and Zellner, R.: Rate measurements of reactions of OH by resonance absorption. Part 2. Reactions of OH with CO, C2H4 and C2H2, J. Chem. Soc., Faraday Trans. 2, 69, 1617–1627, https://doi.org/10.1039/F29736901617, 1973. a
Speak, T. H., Blitz, M. A., Stone, D., and Seakins, P. W.: A new instrument for time-resolved measurement of HO2 radicals, Atmos. Meas. Tech., 13, 839–852, https://doi.org/10.5194/amt-13-839-2020, 2020. a
Stone, D., Blitz, M., Ingham, T., Onel, L., Medeiros, D. J., and Seakins, P. W.: An instrument to measure fast gas phase radical kinetics at high temperatures and pressures, Rev. Sci. Instr., 87, 054102, https://doi.org/10.1063/1.4950906, 2016. a
Strotkamp, M., Munk, A., Jungbluth, B., Dahlhoff, K., Jansen, P., Broch, S., Gomm, S., Bachner, M., Fuchs, H., Holland, F., and Hofzumahaus, A.: Design of a rugged 308 nm tunable UV laser for airborne LIF measurements on top of Zeppelin NT, Solid State Lasers Xxii: Technology and Devices, Proceedings of SPIE, 8599, 85990L, https://doi.org/10.1117/12.2000408, 2013. a
Sun, W., Lelieveld, J., and Crowley, J. N.: Rate coefficients for in the fall-off regime and the impact of water vapor, J. Phys. Chem. A, 126, 3863–3872, https://doi.org/10.1021/acs.jpca.2c02369, 2022. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r
Troe, J.: Theory of thermal unimolecular reactions in the fall-off range. I, Strong collision rate constants, Ber. Bunsen Phys. Chem., 87, 161–169, https://doi.org/10.1002/bbpc.19830870217, 1983. a
Vandaele, A. C., Hermans, C., Simon, P. C., Carleer, M., Colin, R., Fally, S., M´erienne, M. F., Jenouvrier, A., and Coquart, B.: Measurements of the NO2 absorption cross-section from 42 000 cm−1 to 10 000 cm−1 (238–1000 nm) at 220 K and 294 K, J. Quant. Spe. Rad. Trans., 59, 171–184, https://doi.org/10.1016/S0022-4073(97)00168-4, 1998. a
Whalley, L. K., Edwards, P. M., Furneaux, K. L., Goddard, A., Ingham, T., Evans, M. J., Stone, D., Hopkins, J. R., Jones, C. E., Karunaharan, A., Lee, J. D., Lewis, A. C., Monks, P. S., Moller, S. J., and Heard, D. E.: Quantifying the magnitude of a missing hydroxyl radical source in a tropical rainforest, Atmos. Chem. Phys., 11, 7223–7233, https://doi.org/10.5194/acp-11-7223-2011, 2011. a
Winiberg, F. A. F., Zuraski, K., Liu, Y., Sander, S. P., and Percival, C. J.: Pressure and temperatured dependencies of rate coefficients for the reaction products, J. Phys. Chem. A, 124, 10121–10131, https://doi.org/10.1021/acs.jpca.0c08920, 2020. a, b, c, d, e
Yang, Y., Shao, M., Wang, X., Nölscher, A. C., Kessel, S., Guenther, A., and Williams, J.: Towards a quantitative understanding of total OH reactivity: A review, Atmos. Environ., 134, 147–161, https://doi.org/10.1016/j.atmosenv.2016.03.010, 2016. a, b
Zellweger, C., Steinbacher, M., and Buchmann, B.: Evaluation of new laser spectrometer techniques for in-situ carbon monoxide measurements, Atmos. Meas. Tech., 5, 2555–2567, https://doi.org/10.5194/amt-5-2555-2012, 2012. a
Zhou, J., Murano, K., Kohno, N., Sakamoto, Y., and Kajii, Y.: Real-time quantification of the total HO2 reactivity of ambient air and HO2 uptake kinetics onto ambient aerosols in Kyoto (Japan), Atmos. Environ., 223, 117189, https://doi.org/10.1016/j.atmosenv.2019.117189, 2019. a