the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 Feb 2025
| 28 Feb 2025
The lifetimes and potential change in planetary albedo owing to the oxidation of thin surfactant organic films extracted from atmospheric aerosol by hydroxyl (OH) radicals at the air–water interface of particles
Rosalie H. Shepherd
Andrew D. Ward
Edward J. Stuckey
Rebecca J. L. Welbourn
Neil Brough
Adam Milsom
Christian Pfrang
Thomas Arnold
Water-insoluble organic material extracted from atmospheric aerosol samples collected in urban (Royal Holloway, University of London, UK) and remote (Halley Research Station, Antarctica) locations were shown to form stable thin surfactant films at an air–water interface. These organic films reacted quickly with gas-phase OH radicals and may impact planetary albedo. The X-ray reflectivity measurements additionally indicate that the film may be consistent with having a structure with increased electron density of film molecules towards the water, suggesting amphiphilic behaviour. Assuming the material extracted from atmospheric aerosol produces thin films on aqueous particles and cloud droplets, modelling the oxidation kinetics with a kinetic model of aerosol surface and bulk chemistry (KM-SUB) suggests half-lives of minutes to an hour and values of ksurf of and cm2 s−1 for urban and remote aerosol film extracts, respectively. The superfluous half-lives calculated at typical OH atmospheric ambient mixing ratios are smaller than the typical residence time of atmospheric aerosols; thus, oxidation of organic material should be considered in atmospheric modelling. Thin organic films at the air–water interface of atmospheric aerosol or cloud droplets may alter the light-scattering properties of the aerosol. X-ray reflectivity measurements of atmospheric aerosol film material at the air–water interface resulted in calculated film thickness values to be either ∼10 or ∼17 Å for remote or urban aerosol extracts, respectively, and oxidation did not remove the films completely. One-dimensional radiative transfer modelling suggests the oxidation of thin organic films on atmospheric particles by OH radicals may reduce the planetary albedo by a small, but potentially significant, amount.
- Article
(3032 KB) - Full-text XML
- BibTeX
- EndNote




Atmospheric aerosols significantly contribute to the Earth’s climate (IPCC, 2021) and directly influence the proportion of solar energy reaching the Earth’s surface by scattering or absorbing incoming solar radiation (IPCC, 2021), as well as indirectly through cloud condensation nuclei (Bréon et al., 2002; Lohmann and Feichter, 2005; Ramanathan et al., 2001; Haywood et al., 2023; Merikanto et al., 2009; Rosenfeld et al., 2008; Seinfeld et al., 2016). The current understanding of the effect of atmospheric aerosols on climate is considered low (IPCC, 2021; Burkholder et al., 2017; Prather et al., 2008). The complex chemical composition and variable physical properties associated with atmospheric aerosol make it difficult to obtain complete chemical information on aerosols (Jacobson et al., 2000), and these are consequently contributory factors to the uncertainty surrounding atmospheric aerosols. An atmospheric aerosol may be composed of several different substituents that may either phase separate or form surfactant films in the individual aerosol (Reid et al., 2011; Russell et al., 2002), often resulting in the formation of a thin organic film around a core aerosol droplet or particle (Donaldson and Vaida, 2006; Ellison et al., 1999; Gill et al., 1983; Tervahattu et al., 2002; Wyslouzil et al., 2006; Jones et al., 2015, 2017; Kirpes et al., 2019; Barker et al., 2023), a so-called core–shell aerosol. The presence of a thin organic film may alter the chemical and physical properties of the core–shell aerosol through (a) altering the transport of chemicals from the gas to liquid phase and vice versa (Donaldson and Anderson, 1999; Enami et al., 2014a), (b) reducing the rate of evaporation from the core aerosol (Davies et al., 2013; Eliason et al., 2003; Kaiser et al., 1996; McFiggans et al., 2006), (c) altering the cloud condensation nuclei activation potential (Cruz and Pandis, 1998; Ruehl and Wilson, 2014; King et al., 2009; Ovadnevaite et al., 2017), (d) reducing the scavenging of the core aerosol by larger cloud and ice particles (Andreae and Rosenfeld, 2008; Feingold and Chuang, 2002), and (e) changing the aerosol optical properties and thus the light-scattering properties of the aerosol (Jones et al., 2015; Shepherd et al., 2022; Barker et al., 2023).
The organic-coated aerosol is susceptible to atmospheric oxidation (Eliason et al., 2003; Mmereki and Donaldson, 2003; Jones et al., 2017, 2023; Milsom et al., 2022b; Shepherd et al., 2022; King et al., 2009, 2020) and, as a result, may change both chemically and physically (King et al., 2004). Many oxidant species exist in the atmosphere, and hydroxyl (OH) radicals are one of the dominant oxidising chemicals in the lower atmosphere (Prinn et al., 2001). There has been recent evidence that OH radicals are produced spontaneously in the dark at the surface of aqueous droplets (Li et al., 2023). Understanding the behaviour of organic aerosol upon exposure to OH radicals is paramount for determining the atmospheric chemical lifetime of the aerosol; subsequently, a number of studies have focused on the oxidation of atmospheric aerosols when exposed to OH radicals. These studies include the oxidation of submicron aqueous aerosols when exposed to OH radicals (McNeill et al., 2007), the heterogeneous reaction between OH radicals and submicron unsaturated fatty acid particles (Nah et al., 2013), the oxidation of alkanoic acids at the air–water interface by OH radicals (Enami et al., 2014b), and the effect of sulfur dioxide on the oxidation rate of organic aerosol by OH radicals (Richards-Henderson et al., 2016). A more exhaustive list of reactions of gas-phase OH radicals with surface-bound organic compounds can be found in Table 3 of Shepherd et al. (2022). The thin films described in the work presented here are typically monolayers or a few molecules thick and are formed by molecules with a surfactant behaviour (Davies and Rideal, 1961; Gill et al., 1983), i.e. favouring the air–water interface. The thicker films, such as those formed by phase separation, are not considered.
A proxy for the interface of organic-coated aqueous aerosol can be produced by the use of thin films of proxy atmospheric material at the air–water interface, a method which has been implemented extensively (Chapleski et al., 2016; Cosman et al., 2008; Dennis-Smither et al., 2012; Eliason et al., 2004; George et al., 2007; González-Labrada et al., 2006, 2007; Jones et al., 2023, 2017; King et al., 2009, 2020; Knopf et al., 2007; Pfrang et al., 2014; Nakayama et al., 2013; Slade and Knopf, 2014; Sebastiani et al., 2018, 2022; Vieceli et al., 2004; Voss et al., 2006; Woden et al., 2021; Shepherd et al., 2022). One approach to measuring the structure of a thin film at the air–water interface is X-ray (or neutron) reflectometry, which enables the physical characterisation of a thin film at ångström resolution, including observing structural changes to the interface in real time. A number of studies that focused on organic films at the air–water interface have employed reflection techniques, e.g. the oxidation of a film of oleic acid at the air–water interface with gas-phase ozone (King et al., 2009, 2020) or gas-phase nitrogen dioxide (King et al., 2010), the oxidation of methyl oleate thin films with gas-phase ozone (Pfrang et al., 2014), changes owing to the chemical denaturation of β-lectoglobulin (Perriman et al., 2007), the structure of mixed monolayers with varying concentrations of phospholipids and lipids with polymer head groups (Majewski et al., 1997), adsorption behaviours of DNA and sodium polystyrene sulfonate when exposed to cationic gemini surfactants (Vongsetskul et al., 2009), the self-assembly of conjugated polymers at the air–water interface (Reitzel et al., 2000), the formation of peptide sheets from the denaturation of proteins at the air–water interface (Gidalevitz et al., 1999), and the oxidation of lung lining (Hemming et al., 2015, 2022; Thompson et al., 2013, 2010).
Other techniques have also been successfully applied to study thin films at the air–water interface: González-Labrada et al. (2006) recorded the change in surface tension of a film composed of oleic acid when exposed to ozone using a surface tensiometer, whilst Wadia et al. (2000) studied unsaturated and saturated phospholipids by using an atmospheric pressure ionisation mass spectrometer.
Investigations using atmospheric aerosol extracted from the atmosphere are critical to provide insight into the atmospheric process and composition compared to proxies; however, these studies are sparse. Examples of such studies include Zhou et al. (2014), who studied the heterogeneous oxidation of seawater polyunsaturated fatty acids at the air–water interface, and a study relevant to aerosol research, owing to the material at the surface of the ocean becoming incorporated into atmospheric aerosol (Blanchard, 1964; Donaldson and George, 2012; Marty and Saliot, 1976). Jones et al. (2017) applied X-ray reflection techniques to study the oxidation of thin films extracted from atmospheric aerosol extracts collected at Royal Holloway, University of London, and seawater samples collected from the English Channel by aqueous-phase OH oxidation and gas-phase ozone oxidation. Shepherd et al. (2022) exposed thin films of atmospheric aerosol extracts collected from Royal Holloway, Antarctica, and wood smoke burning to OH radicals and estimated the atmospheric chemical half-life of such films based on a KM SUB kinetic model analysis (Shiraiwa et al., 2010).
The purpose of the study described here is fourfold: firstly to confirm that organic films extracted from atmospheric aerosol can form stable thin films at the air–water interface, secondly to determine the film thickness for organic matter extracted from atmospheric aerosol at the air–water interface, thirdly to determine the chemical lifetime of a thin film of atmospheric aerosol extract at the air–water interface, and lastly to estimate the relevance of the oxidation of thin organic films on aqueous and mineral aerosol to planetary albedo. The study presented here will yield morphological data about the thin surfactant film at the air–water interface through calculating film thickness to ångström precision: accurate estimation of film thickness is paramount to model the light scattering from particles with thin films likely to be found in the atmosphere, owing to the importance of the film thickness in the ability of core–shell aerosol to heat or cool the atmosphere (Barker et al., 2023). The chemical lifetime with respect to the gas-phase OH radical will be estimated by applying a kinetic model (KM SUB) to the experimental data, extrapolating to a range of atmospherically relevant OH radical concentrations. Lastly, the thickness of the thin organic film before and after oxidation by OH radicals will be used to estimate the top-of-atmosphere albedo change, owing to chemical oxidation by the OH radical for a series of exemplary aerosol distributions.
X-ray reflectivity was applied to study a thin surfactant film extracted from atmospheric aerosol at the air–water interface upon exposure to gas-phase OH radicals; the following section very briefly describes the X-ray reflectivity technique and the kinetic model employed to determine the chemical lifetime of these films. Additionally, the method applied to collect the insoluble surface-active atmospheric aerosol extracts for use on the X-ray beamline is described.
2.1 Extraction of atmospheric aerosol
Organic material from atmospheric aerosols was extracted from quartz fibre filters. The samples collected on the filters were from two locations: the campus of Royal Holloway, University of London, during the months of September 2015 and January 2016, and at the Halley Clean Air Sector Laboratory, operated by the British Antarctic Survey over the Antarctic summers of 2015 and 2016. The aerosol extract collected from Royal Holloway, University of London, has been classified as urban, owing to the proximity of London; major motorways such as the M25, M3, and M4; and Heathrow international airport, all lying within a radius of 30 km. In contrast, because of its physical remoteness, Antarctica is considered a clean environment; therefore, for the purpose of the study presented here, the Antarctic aerosol extract is described as remote. The urban samples were each collected over an approximately 30 d period, whilst the remote samples were collected over approximately 60 d.
For the urban sampling sites, the aerosol was extracted from the atmosphere through an air pump that pulled air through clean stainless-steel pipelines at a flow rate of 30 L min−1, whilst remote aerosol was sampled by pulling air through a short length (10 cm) of in. o.d. perfluoroalkoxy (PFA) Teflon tubing by applying a low-volume air sampler (model VM-4) with a flow rate of 20 L min−1. In both sampling sites, the pipelines led to a PFA Savillex commercial filter holder, in which a pre-combusted 47 mm diameter quartz filter was encased. Atmospheric aerosol extract was collected upon the filter. To separate the insoluble organic material from the filter, the filter was shaken manually and very gently sonicated for 5 min in a solution consisting of 10 mL ultrapure water (<18 MΩ cm) and 10 mL chloroform (Sigma-Aldrich, 0.5 %–1 % ethanol as stabiliser); shaking commonly reduced the filter to a pulp. The pulp was subsequently filtered with another pre-combusted quartz filter to remove the filter paper: the pulp was washed with ultrapure water and chloroform several times to maximise the amount of sample extracted. The sonication was not found to be detrimental to the material being extracted (Jones et al., 2017). The resulting filtrate consisted of a chloroform layer in which the material likely to form insoluble films at an air–water interface resided (Jones et al., 2008) and an aqueous layer. The chloroform layer was separated from the aqueous layer by passing the filtrate through a separating funnel. The chloroform was then removed from the atmospheric aerosol extract by evaporative blowdown, leaving an oily residue behind. The residual material was stored in 2 mL of fresh chloroform at −18 °C in the dark until use for the X-ray reflectivity experiment. All glassware used in the extraction process was thoroughly cleaned with ultrapure water and chloroform prior to use, and the extraction process was carried out in a clean glove bag. Analytical blanks were filters extracted in the same manner as filters used to collect atmospheric aerosol. Travelling filters were analytical blanks that travelled to and from the collection sites under the same conditions as the filters used to collect atmospheric aerosol.
Analytical and travelling filter blanks were likewise extracted in exactly the same manner. The analytical blanks provided a method of determining the level of contamination resulting from the extraction process, whilst for the remote samples the amount of contamination resulting from travelling to Antarctica and back was determined from a travelling filter blank that travelled with the sample.
2.2 Experimental set-up
The water was held using a PTFE trough (Ball et al., 2013), with a volume of ∼90 cm3 and surface area of ∼175 cm2, mounted onto an anti-vibration table. Owing to the small size and geometry of the trough in relation to the X-ray beam footprint on the water surface, the trough was used without barriers or a surface tension sensor. The small size was a compromise between the X-ray beam footprint and the limited sample. A UV lamp containing two fluorescent germicidal lamps, each with an output wavelength centred on 254 nm, was suspended above the trough. The mercury discharge lamps were stated as “ozone-free”, although in practise not all radiation of wavelength 185 nm was removed by the lamp tube. To encase the experimental environment and permit the production of an atmosphere containing OH radicals, the trough and UV lamp were enclosed within a Tedlar bag; two thin Kapton windows at either end of the trough facilitated travel of the X-ray beam into and out of the enclosed environment. The Tedlar bag had a stainless-steel inlet and an exhaust to allow gas to enter and leave. The bag had a 25 L volume and a surface area of 612 cm2. Generation of gas-phase OH radicals by ozone photolysis required a saturated atmosphere of water vapour. To ensure the relative humidity of the experimental environment was maintained, a water reservoir with an approximate volume of ∼50 cm3 was included within the Tedlar bag, and the relative humidity within the bag was ∼100 % For each experimental run, the trough was cleaned with chloroform and ultrapure water before a fresh 90 cm3 volume of ultrapure water (<18 MΩ cm) was poured into the trough. Between 100–200 µL of atmospheric aerosol extract in chloroform was added to the air–water interface using a Hamilton syringe. These solutions were spread onto water using a microlitre syringe to produce an approximate surface pressure of ∼15 mN m−1 as determined in offline experiments with the same trough and a surface tensiometer. An excellent and detailed experimental and theoretical consideration of spreading insoluble surfactants at an air–water interface can be found in Davies and Rideal (1961). A similar surface pressure produced either by a monolayer of fatty acid molecules (like oleic acid) that would give a surface coverage of (King et al., 2010), about 16 µg of material or by using the mass density of the spreading solution of a similar aerosol extract (Shepherd et al., 2018) to those used here demonstrates that about 10 µg of material was added to the trough. The material covers the entire surface of the trough, ∼175 cm2; adding more of the material does not produce a large increase in the thickness of these films as shown by Fig. 4 of Shepherd et al. (2022), which shows that the film thickness of a related urban sample approaches an asymptote as more material is added. Adding too much material to the interface produced a thin film with visible lenses of material. Similar behaviour is shown by piston oils such as oleic acid (Gaines, 1966).
2.3 X-ray reflectivity
The surface specular X-ray reflectometer at Diamond Light Source, I07, was applied to study the film of atmospheric aerosol extract at the air–water interface. I07 is described in detail by Nicklin et al. (2016) and Arnold et al. (2012), and the X-ray reflectivity technique is described in detail by Chateigner (2013); however, a brief description will be given here. The X-ray beam had a wavelength of 0.992 Å and was specularly reflected off the interface and onto a PILATUS detector, where the beam was measured as a function of momentum transfer, Q, defined as
versus specular reflectivity, R, which is defined as
From Eq. (1), λ is defined as the wavelength of the specular X-ray, and θ is the incident angle of the specular X-ray beam onto the interface of study. From Eq. (2), δ is the film thickness, and ρ is the X-ray scattering length density. The X-ray scattering length density is related to the electron density of the interface; therefore, heavier atoms that have a greater number of electrons will have a larger electron density and thus a greater X-ray scattering length density (Jones et al., 2017). The profile of momentum transfer versus reflectivity will be called an X-ray reflection profile henceforth in the paper. To optimise signal relative to time, X-ray reflection profiles were collected over a time period of 8 min. X-ray reflection as a technique is well established as a method that averages the microscopic structure perpendicular to the surface over macroscopic length scales in the plane of the surface. The X-ray footprint was comparable to the length of the trough (200 mm) at low angles and approx. 200 µm wide. It was moved horizontally across the trough (perpendicular to the beam scattering plane) between measurements of the X-ray reflectivity profile to prevent beam damage. Further information on the beam footprint is clearly described in Salah et al. (2007). To determine precise values of X-ray scattering length density and film thickness, the experimentally collected X-ray reflection profiles were simulated by an Abelès formalism (Abelès, 1950) in the computer software Motofit (Nelson, 2006). The Abelès formalism used a model of the atmospheric aerosol extract at the air–water interface, including parameters for the X-ray scattering length density, film thickness and film roughness to simulate the experimentally determined X-ray reflectivity profile. The values were initially estimated and then refined until a fit between experimental, RExperimental(Q), and simulated, RCalculated(Q), data was determined: application of a fitting metric χ2 ensured the final value provided the best fit, where
Typically for a film of known composition at an air–water interface, the surface coverage (number of molecules per unit area) can be calculated by , where b is the scattering length of a molecule. However, in the study presented here the organic film is (a) a complex mixture of unknown materials and (b) a reaction system producing unknown surface-active product molecules at the air–water interface whose relative amounts change with time. Thus, the value of the scattering length, b, of the molecules in the film is not known. For the atmospheric aerosol extract films exposed to gas-phase OH radicals, scattering length per unit area, ρδ, was plotted as a function of time (Jones et al., 2017) instead. Such plots allowed for the change in the film to be monitored and for the quantity ρδ to be used as a kinetic variable (Jones et al., 2017). Each point on the plots corresponds to a new X-ray reflection profile; continuous collection of subsequent profiles allowed a temporal graph of ρδ as a function of time to be plotted. In contrast to the preliminary study by Jones et al. (2017), the study described here split the organic layer into two separate layers with differing scattering length densities and thicknesses to better reproduce the experimental X-ray reflection profiles. Upgrades in the beamline allowed for this refinement of the reflectivity profile since the work of Jones et al. (2017). In the study presented here, the quantity was followed as a function of time, t.
2.4 Gas-phase OH radical generation
Gas-phase OH radicals were formed from the photolysis of ozone in the presence of water vapour; oxygen saturated with water vapour flowed at 1 L min−1 through a photolytic ozoniser (Ultra-Violet Products Ltd) to produce ozone at a mixing ratio of 0.85 ppm . The oxygen was saturated with water vapour by bubbling through water. The gas-phase concentration of OH radicals in the Tedlar bag was estimated to be . The concentration of OH radicals was calculated by kinetic modelling (i.e. solution of first-order differential equations using a Runge–Kutta solver (Press et al., 1987)) of reactions 1–30 in Atkinson et al. (2004), which cover the basic HOx and Ox reactions occurring in the photolysis of ozone in the presence of water vapour. A first-order wall loss of OH radicals, 2 s−1, was added using the method outlined in Dilbeck and Finlayson-Pitts (2013) based on a 25 L Tedlar bag with a surface area of 0.612 m2 and assuming the reaction probability for OH radicals on Tedlar will be similar to halocarbon wax, (Bertram et al., 2001). The photolysis rate constant for the photolysis of ozone, J(O(1D)) and hydrogen peroxide, were determined with an intensity-calibrated Metcon radiometer (normally used for measurements of atmospheric photolysis reactions, (e.g. Kukui et al., 2014)), and the value of the rate constants for the photolysis of molecular oxygen was the value for ozone scaled by the product of the absorption cross-sections and the quantum yields. The concentration of ozone within the bag was measured by UV–Vis spectrometry after sampling in a 10 cm glass cell. The concentration of water vapour was based on the vapour pressure of water (Haynes, 2016) to be 2.34 kPa at 20 °C.
The reflectivity of an atmospheric aerosol extract at the air–water interface was recorded three times prior to oxidation (a) to establish that the film was initially stable at the air–water interface and (b) to provide good signal-to-noise X-ray reflection profiles to allow for precise determination of X-ray scattering length density and film thickness. After the three measurements, the film was exposed to gas-phase OH radicals. Continuous collection of X-ray reflection profiles during the exposure of the atmospheric aerosol extract to gas-phase OH radicals allowed the change in the film to be analysed as a function of time.
Three “ozone-only” control kinetic runs were performed in the absence of UV light to demonstrate no apparent loss of film material owing to reaction with gas-phase ozone (as opposed to the hydroxyl radical). A control kinetic run with oxygen only (no ozone or UV light present) was not required as no loss of film was observed with ozone/oxygen mixtures. A blank run with UV illumination only (i.e. no ozone) is not reported as the UV lamps generate ozone and subsequently OH radicals (Shepherd et al., 2022) from the photolysis of molecular oxygen.
2.5 Kinetic modelling of the reaction
The reaction system was modelled using a kinetic model of aerosol surface and bulk chemistry (KM-SUB) (Shiraiwa et al., 2010), which is based on the Pöschl–Rudich–Ammann framework (Pöschl et al., 2007) for the following reaction:
Hydroxy radical surface adsorption and desorption are resolved along with the surface reaction between OH radicals and the insoluble film material. The inverse square of the film thickness was used to estimate the average initial surface concentration of film molecules, assuming the film thickness is the average molecular length from the X-ray reflectivity measurements. The KM-SUB model constructed here is analogous to previous modelling work for reactions with insoluble films (Shepherd et al., 2022), which employs the reaction scheme presented in Eq. (4) with the addition of a residual film that may be present. A differential evolution (Storn and Price, 1997) method was used to optimise the model, with only the surface reaction coefficient (ksurf) varied. Other parameters were held constant (see the model described in our previous study on aerosol extract oxidation kinetics (Shepherd et al., 2022)). The Langmuir–Hinshelwood surface reaction mechanism is assumed due to the evidence that OH radicals favour this mechanism (Arangio et al., 2015; Bagot et al., 2008; Enami et al., 2014b). The uncertainty in the value of ksurf was estimated using a Markov chain Monte Carlo (MCMC) algorithm (Foreman-Mackey et al., 2013; Hogg and Foreman-Mackey, 2018). A description of the model and details of the MCMC sampling procedure are presented in a previous study on a similar system (Shepherd et al., 2022). The models were constructed and optimised using MultilayerPy, a framework for building kinetic multilayer models (Milsom et al., 2022a). Jupyter Notebook files along with the model code are available alongside this publication (see the “Code availability” section).
2.6 The 1-D radiative transfer and Mie modelling
A combination of Mie light-scattering modelling and 1-D radiative transfer modelling were undertaken to quantify the effects of the oxidation-caused change in film thicknesses observed in this work on the top-of-atmosphere albedo. The models used were adapted from Bohern and Huffman’s core–shell model “bhcoat” (Bohern and Huffman, 1998) and the TUV (Tropospheric Ultraviolet-Visible) radiation transfer model (Madronich and Flocke, 1999). The core–shell Mie model was used to produce the wavelength-dependent single-scattering albedos and the asymmetry parameter of mineral spheres coated in organics averaged over different aerosol populations. The values of the real refractive indexes of silica and water were taken from Kitamura et al. (2007) and Schiebener et al. (1990), respectively, and the values of the imaginary refractive indexes were taken from Khashan and Nassif (2001) and Hale and Querry (1973). The temperature of silica and water was assumed to be 293 K for calculating optical properties. Values of the complex refractive indexes of urban and remote organic thin film material were taken as a combination of real components produced by Shepherd et al. (2018) and imaginary components by Kirchstetter et al. (2004) and Virkkula et al. (2022). Values of single-scattering albedo and asymmetry parameter were calculated over a range of core sizes of 10–1000 nm using the characteristic size distribution functions of aerosol from the respective environments as published by Jaenicke (1993). The calculation was repeated for film thicknesses between 0.05 to 50 nm, noting that the experimental measurements of thickness presented here are of the order of 1 nm, i.e. towards the smaller end of the range studied. The wavelength-dependent single-scattering albedo (SSA) and the asymmetry parameter, g, were used in an atmospheric 1-D radiative transfer model to estimate the impact of the oxidation of films on top-of-atmosphere albedo. The TUV model was used with minimal adjustments. Three consecutive 1 km layers were placed on the surface to form a 3 km thick planetary boundary layer of aerosol, which was given the optical properties generated from Mie modelling with an aerosol optical depth for each layer of 0.235. The rest of the atmosphere was described by an Elterman aerosol profile with no clouds present. The top–of-atmosphere albedo was calculated as a ratio of upwelling and downwelling irradiance at an altitude of 80 km with a solar zenith angle of 60°. The change in top-of-atmosphere albedo owing to the oxidation of organic films, ΔAlbedo, was determined by subtracting the top-of-atmosphere albedo calculated using aerosol with a given film thickness from that of a model using the same aerosol with films of half that thickness. The changes owing to oxidation for single-scattering albedo, ΔSSA, and the asymmetrical light scattering parameter, Δgsca, were also produced. Four profiles of ΔAlbedo, Δgsca and ΔSSA versus initial film thickness were produced which were representative of organic-coated silica and water aerosol whose size and film optical properties were representative of urban and remote environments from Jaenicke (1993).
The results and discussion of the X-ray reflectivity of thin films of material extracted from atmospheric aerosol at the air–water interface are presented, followed by a kinetic analysis of its oxidation with gas-phase OH radicals.
3.1 X-ray reflection and properties of the thin film
Organic thin films composed of material extracted from the atmospheric aerosol were studied at the air–water interface. Figure 1 depicts the X-ray reflection profiles for aerosol extracts sourced from (a) urban and (b) remote locations along with the corresponding analytical (or travelling) blank and clean air–water interface. The figure demonstrates a definite change in the reflectivity profile with and without the atmospheric aerosol extract sample. The figure also demonstrates that the travelling and analytical blanks are effectively indistinguishable from the clean air–water interface. Very close inspection of Fig. 1b suggests that the travelling blank may have received a tiny quantity of contamination on its long return journey from Antarctica. However, the changes in the X-ray reflectivity profile relative to the clean air–water interface are many orders of magnitude smaller than the sample. The agreement is remarkable. Figure 1 demonstrates the essential requirement for these experiments to be performed at X-ray synchrotrons as the difference between sample and no sample at the air–water interface requires a large signal-to-noise ratio in reflectivity at large values of Q, i.e. momentum transfer values. A commercial X-ray reflectometer is generally unlikely to achieve sufficient signal-to-noise ratios at the larger values of Q (plotted in Fig. 1) in timescales important for studying kinetics.
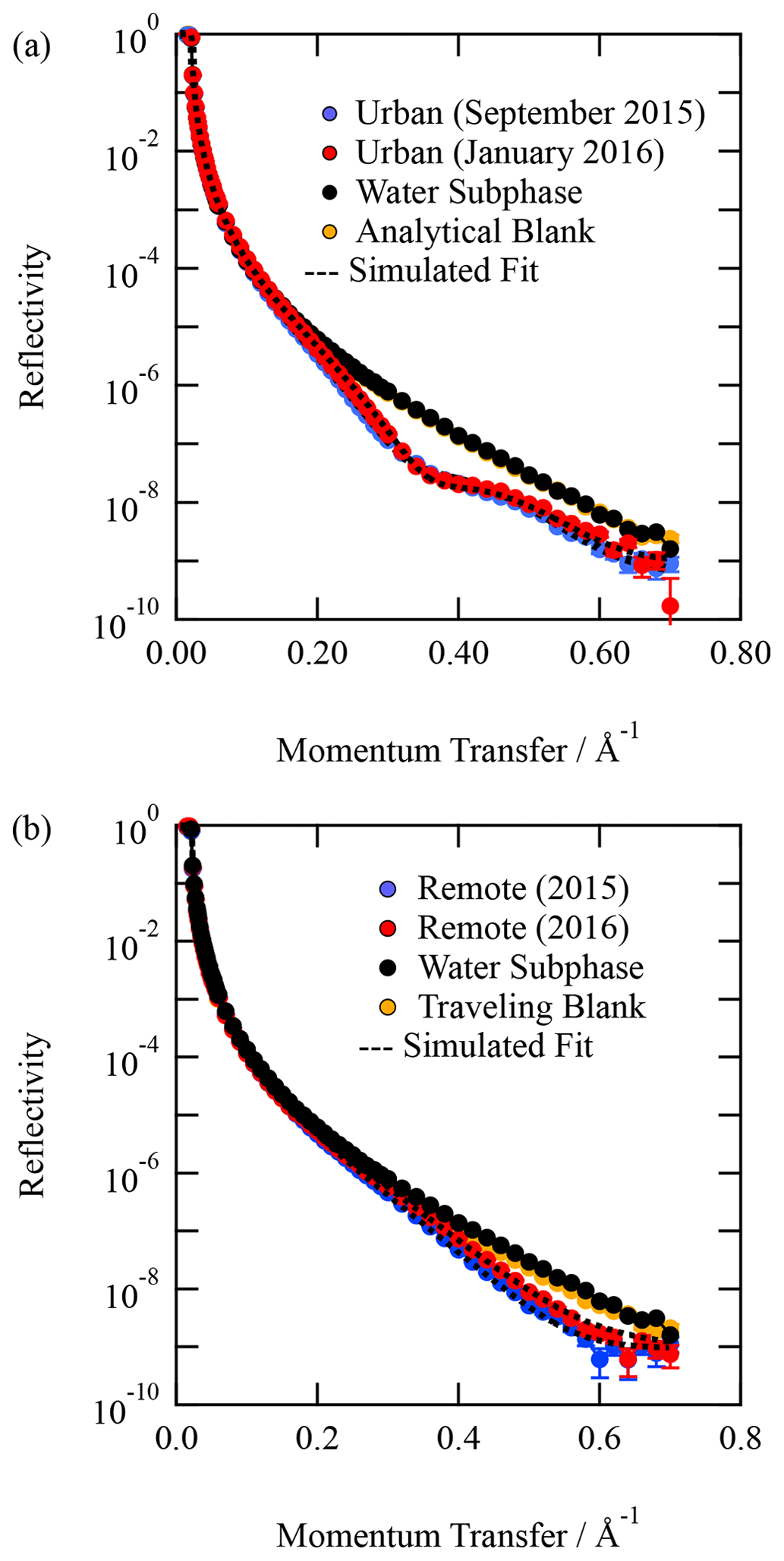
Figure 1Typical X-ray reflection profiles for (a) urban atmospheric aerosol extract and (b) remote atmospheric aerosol extract. A corresponding analytical (or travelling blank) is displayed in each figure. In both panels, the analytical blank is indistinguishable from the water sub-phase with a clean air–water interface within error. The error bars represent statistical counting errors from accumulation and binning of photons, and they are generally smaller than the points depicted.
Although Fig. 1 demonstrates that the material extracted from atmospheric aerosol produces stable thin films at the air–water interface and thus may form these films on aqueous droplets in the atmosphere, it does not necessarily imply that the aerosol from which these insoluble surfactants were extracted had such a monolayer and may have had a more complex morphology.
Application of Abelès formalism allowed the X-ray scattering length density and film thickness of the aerosol extract film to be calculated. Additionally, more detailed structural information of the extract at the air–water interface was determined; the urban and remote aerosol extracts both demonstrated a good fit between experimental and calculated X-ray reflectivity profiles to a two-layer system (as opposed to a one- or three-layer system). It was not possible to achieve a realistic reproduction of the experimental reflectivity profile with one layer at the air–water interface, and although a three-layer system provided a good visual fit, it did not lower the value of the fitting metric, χ2. The two-layer system may be indicative of a film that has a structure with increased electron density of film molecules towards the water, suggesting amphiphilic, lipid-like, behaviour, e.g. a phosphocholine lipid molecule like 1,2-dipalmitoylphosphatidylcholine having a head group of phosphatidylcholine ((CH3)3NCH2CH2OP(O2)O−) and a tail of two palmitic acids (C16H32O2).
The layers of the two-layer system will be described as a layer beside the air, “air interface”, and a layer at the water interface, “water interface”. The X-ray scattering length densities and film thickness determined for urban and remote atmospheric aerosol extracts are displayed in Table 1. Uncertainties in the values displayed in Table 1 were determined by adjusting the scattering length density or film thickness in turn until the simulated data no longer resembled the experimental data, as determined by eye. Application of a χ2 test (see Eq. 3) ensured the X-ray scattering length density and film thickness values displayed were the values that gave the best fit. A demonstration of the robustness of the fitting procedure is given in Fig. 2. Figure 2 is a plot of the value of the fitting metric, χ2, presented as a function of the X-ray scattering length density for three different film thickness. Figure 2 demonstrates well-defined almost symmetrical minima. Note that the minima of these curves are independent of thickness near the best fit. The upper panel is for a layer of aerosol extract at the air interface, and the lower panel is for the layer at the water interface. Each layer was investigated independently, and the data presented in Fig. 3 are from the urban sample. Figure 6 suggests that the X-ray scattering length density can easily be fitted to .
Table 1Table of the X-ray scattering length density (ρ) and film thickness (δ) for urban and remote atmospheric aerosol extract thin films at the air–water interface.
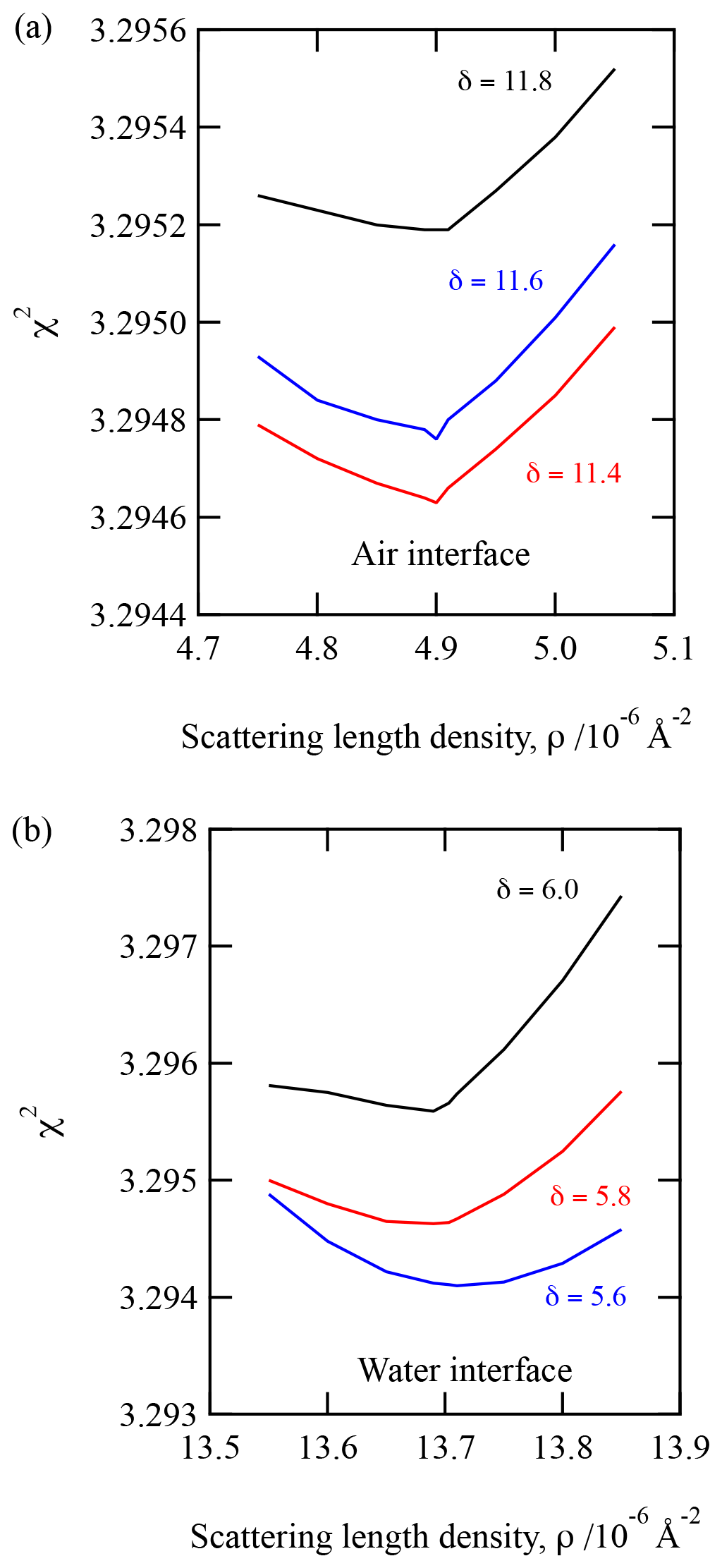
Figure 2A figure of merit for reproducing the X-ray reflectivity profile by a computed profile relative to the experimental data as a function of the value of the computed scattering length density for different film thicknesses. Note that the minima of the figure of merit for different scattering length densities seem insensitive to the thickness in the range of values presented. Both panels are for the urban September 2015 sample. Panel (a) corresponds to the air interface, and panel (b) corresponds to the water interface.
The number of samples studied is small, owing to limited access to the X-ray synchrotron source and limited sample from the atmospheric aerosol (the Antarctic aerosol sample was completely consumed in these studies). However, inspection of Table 1 demonstrates that the thickness and scattering length density are consistent between samples from the same locale but different between locales. The remote sample is indicative of background polar/marine aerosol (Wolff, 1990) and is a thinner film with a smaller electron density than the polluted urban sample. The polluted urban sample is characterised by its proximity to the London megacity, a major international airport (Heathrow) and the junction of three major arterial motorways. Shepherd et al. (2022) studied the oxidation of very similar organic films extracted from the atmosphere at the air–water interface in their work but used neutron reflectometry instead of X-ray reflectometry. The Antarctic samples were the same as used here, but the urban samples were the same location and year but collected in different months; a comparison is shown in Table 3. The thicknesses were broadly comparable: the films were 10–20 Å thick. The film thickness is sensitive to the amount of material successfully added to the air–water interface, and more material was added in the X-ray work relative to the neutron work. Note that not all material attempted to be added to an air–water interface is successfully placed there, and the concentrations and chemical identities of the materials are unknown.
The separation of the atmospheric aerosol extract into two layers indicates that the extract may contain hydrophobic and hydrophilic components. For both urban and remote extracts, the X-ray scattering length density and film thickness for the layer at the water interface are larger than the X-ray scattering length density for the layer at the air interface. The literature reports similar trends in X-ray scattering length density of thin films composed of molecules containing hydrophobic and hydrophilic regions; for example, Dabkowska et al. (2013) studied monolayer films of the lipid distearoylphosphatidylcholine (DSPC) at the air–water interface and reported an X-ray scattering length density that was larger by Å−2 for the hydrophilic head group region than for the hydrophobic chain region of the lipid.
Previous neutron and X-ray studies have used a film thickness ranging from 10–20 Å for a thin film at the air–water interface (Jones et al., 2017; King et al., 2009; Pfrang et al., 2014) and 24–51 nm at the air–solid interface (Milsom et al., 2022b). The air–solid films are spin-coated onto the solid surface. In the study presented here, the total thickness of the atmospheric aerosol extract films did not exceed 18 Å for urban aerosol extracts or 10 Å for remote aerosol extracts, agreeing with previous measurements. These thicknesses are important for atmospheric and core–shell morphology modelling (Jones et al., 2015; Shepherd et al., 2022; McGrory et al., 2022); see Sect. 2.6.
As evidenced by the control experiments, labelled “ozone blank” in Figs. 4 and 5, the X-ray reflectivity profile did not change, demonstrating that there was little or no reaction with ozone and that the film on the trough is horizontally homogenous. The trough is moved between the measurements of X-ray reflectivity to prevent beam damage.
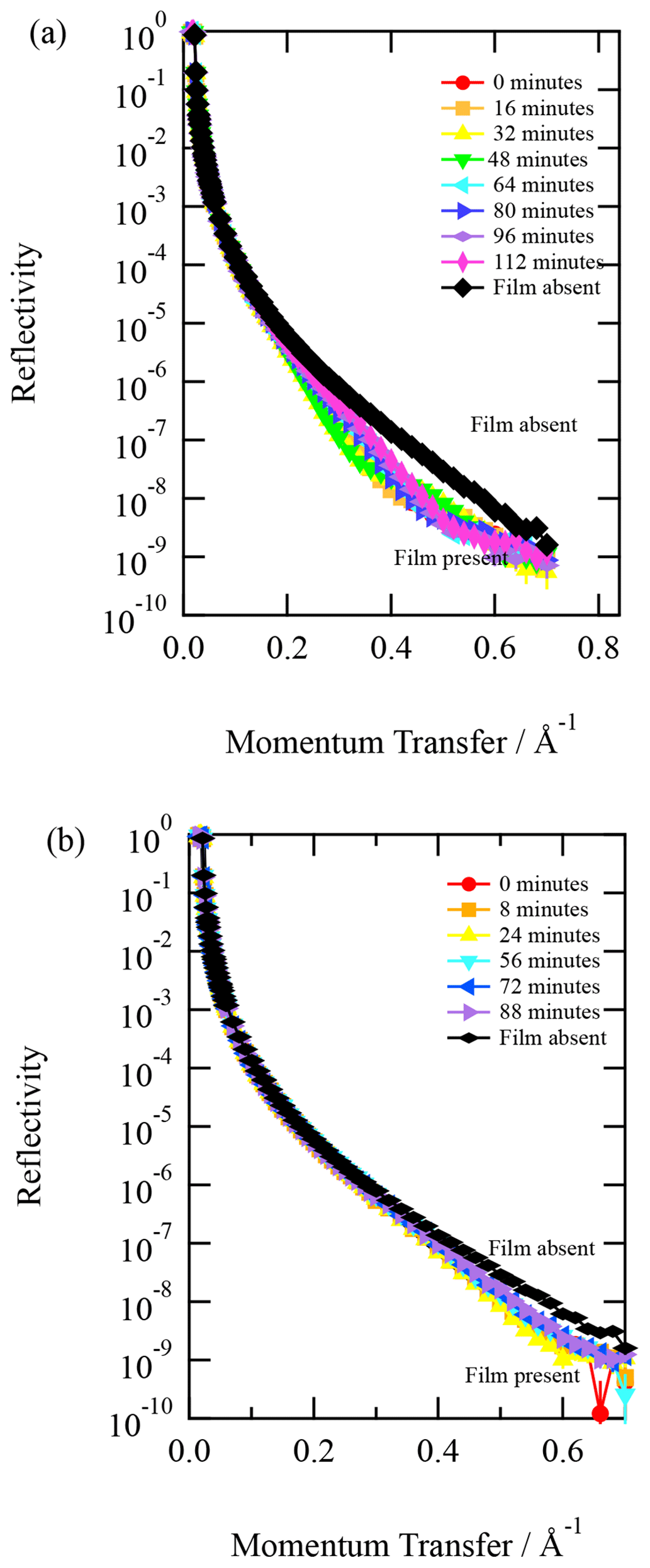
Figure 3X-ray reflection profiles of a film of urban atmospheric aerosol (collected September 2015) as the film was exposed to gas-phase OH radicals. Upon exposure to gas-phase OH radicals, the reflectivity of the film increases, indicating a reaction is occurring at the air–water interface.
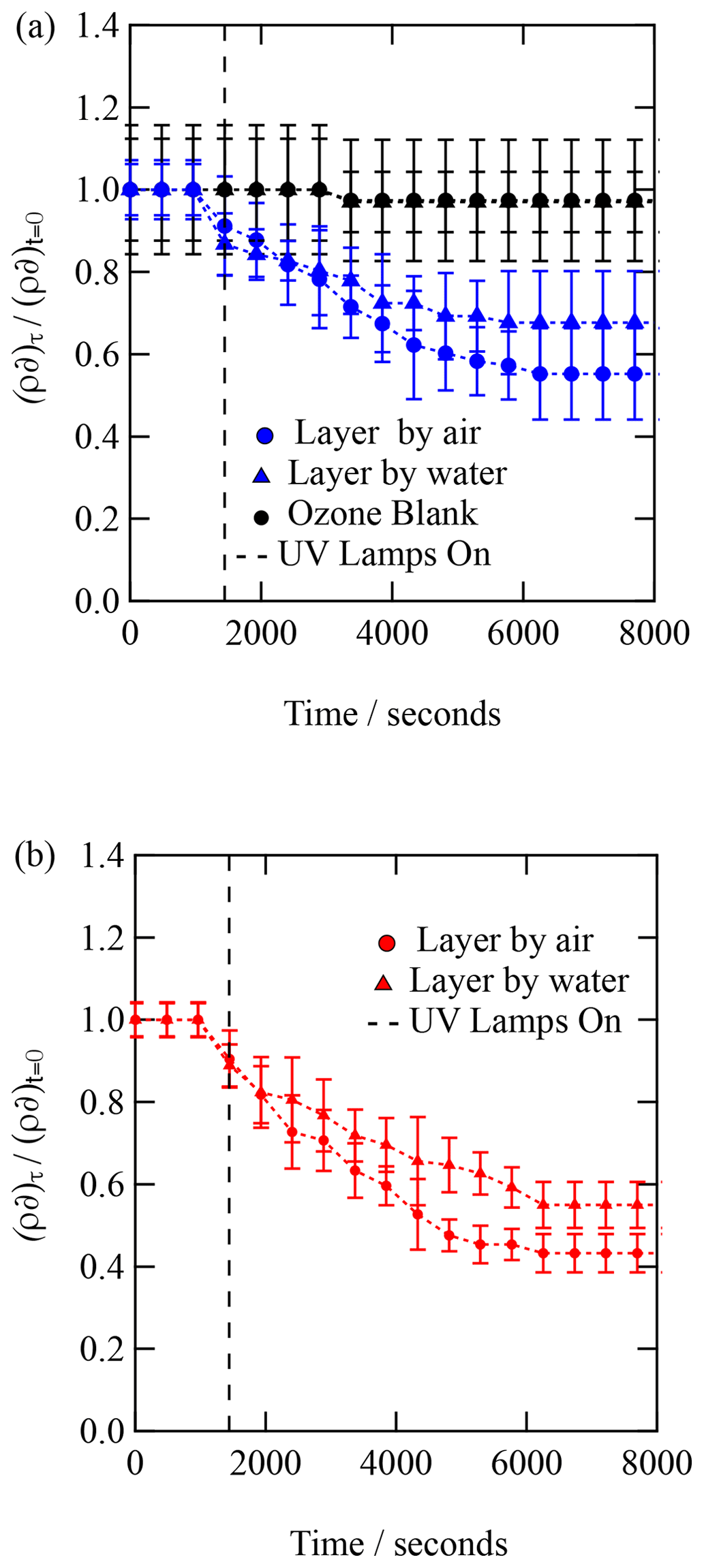
Figure 4The kinetic decay of the scattering length per unit area for an organic film or urban atmospheric aerosol extract collected during (a) September 2015 and (b) January 2016 when exposed to hydroxyl radicals. .
3.2 Oxidation of atmospheric aerosol
The films of atmospheric aerosol extract at the air–water interface were exposed to gas-phase OH radicals with a concentration of . The change in X-ray reflectivity was followed continuously in 8 min intervals. Figure 3 depicts the change in the X-ray reflection profile with time as the atmospheric aerosol extract at the air–water interface was exposed to gas-phase OH radicals. Figure 3 clearly shows changes to the interface during oxidation and that oxidation does not completely remove all material from the interface back to just water. Results from the neutron reflectivity study of similar samples (Shepherd et al., 2022) also demonstrated some film remaining after oxidation, although perhaps less than shown in the study presented here.
Each X-ray reflection profile collected was simulated and fitted with a calculated reflection profile to determine the X-ray scattering length density and film thickness as a function of time. The experimental X-ray reflectivity profiles of the reacting system were calculated and fitted as a function of time as one- and two-layer systems. The two-layer calculation of the X-ray reflectivity profile gave a superior fit to the experimental data throughout the oxidation reaction. The values of the scattering length density, ρ, and thickness, δ, are plotted as versus time in Fig. 4. Figure 4 depicts the kinetic decay observed for the films composed of urban aerosol extract upon exposure to gas-phase OH radicals, whilst Fig. 5 depicts the kinetic decay observed for the films composed of remote aerosol extracts upon exposure to gas-phase OH radicals. Uncertainty bars in Figs. 3, 4 and 5 represent the propagated uncertainty (Bevington et al., 1993) from the standard deviations in X-ray scattering length density and film thickness from data fitting.
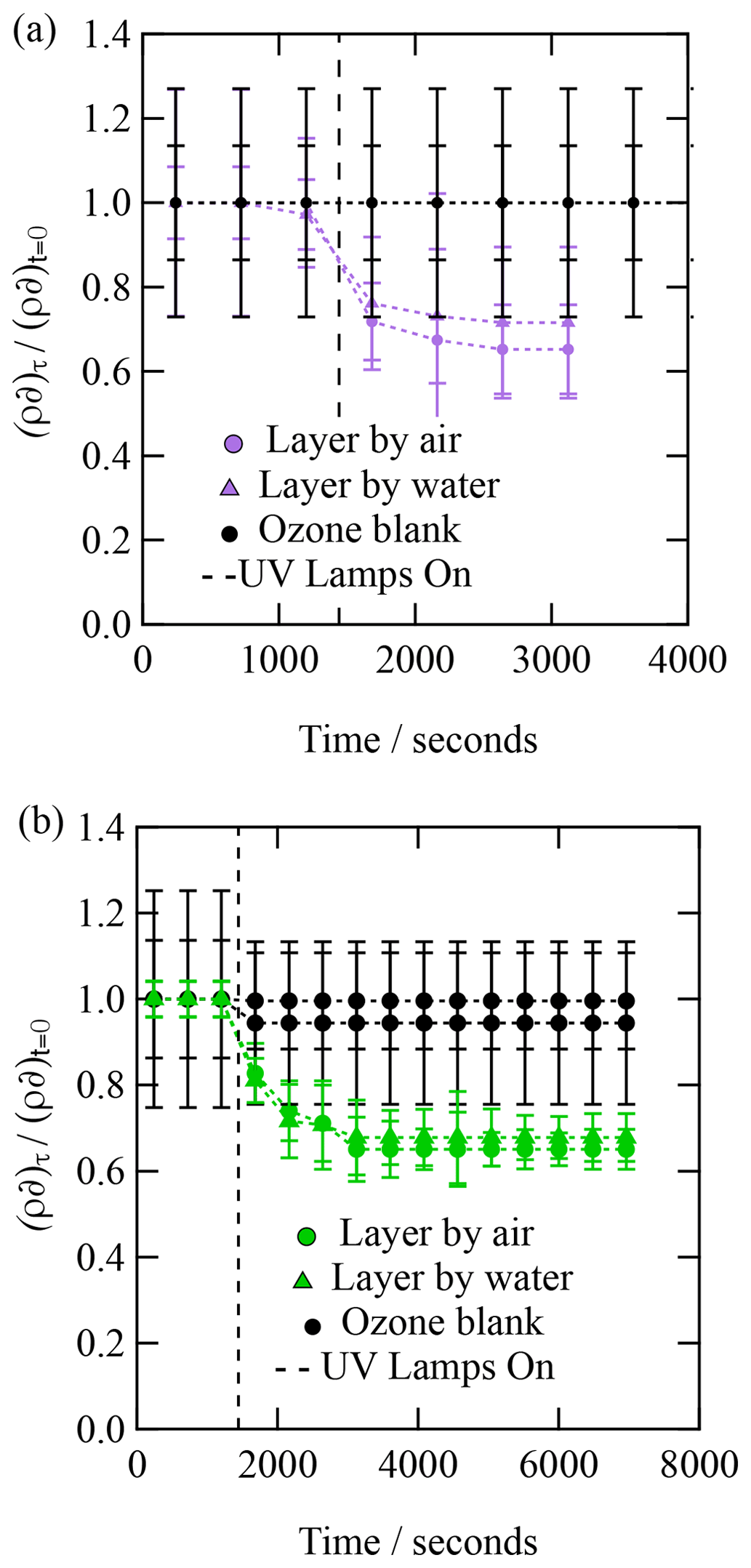
Figure 5The kinetic decay of the scattering length per unit area for an organic film of Antarctic atmospheric aerosol extract collected during (a) 2015 Antarctic summer and (b) 2016 Antarctic summer when exposed to hydroxyl radicals.
. UV lamp switched on at 24 min.
Figures. 4 and 5 both demonstrate an exponential decay in the scattering length per unit area, , when each thin film was exposed to OH radicals, and it can be observed that the rate of decay for the two layers (irrespective of sample) lies within the error of each other. The decay in as a function of time shows that the interface is becoming more like pure water upon exposure to OH radicals, suggesting that either the interface is becoming hydrated or material is being lost from the interface to the water sub-phase of the gas phase, owing to reactions with OH radicals. Figures 4 and 5 both demonstrate that urban and remote aerosol extracts do not change when exposed to ozone only; therefore, the reaction observed is consistent with the reaction with gas-phase OH radical. Jones et al. (2017) also observed no reactivity to gas-phase ozone when films of seawater extracts and atmospheric aerosol extracts were also exposed to ozone.
The kinetic decay profiles, , versus time are the same for the two layers, so the values of ρδ for the two layers were averaged and the decay fitted to an exponential curve of the form . Where k is a decay constant, t is the reaction time and c represents the material remaining at the interface. Figure 6 depicts the kinetic decay profiles for the reaction of the two urban and two remote atmospheric aerosol extracts with gas-phase OH radicals.
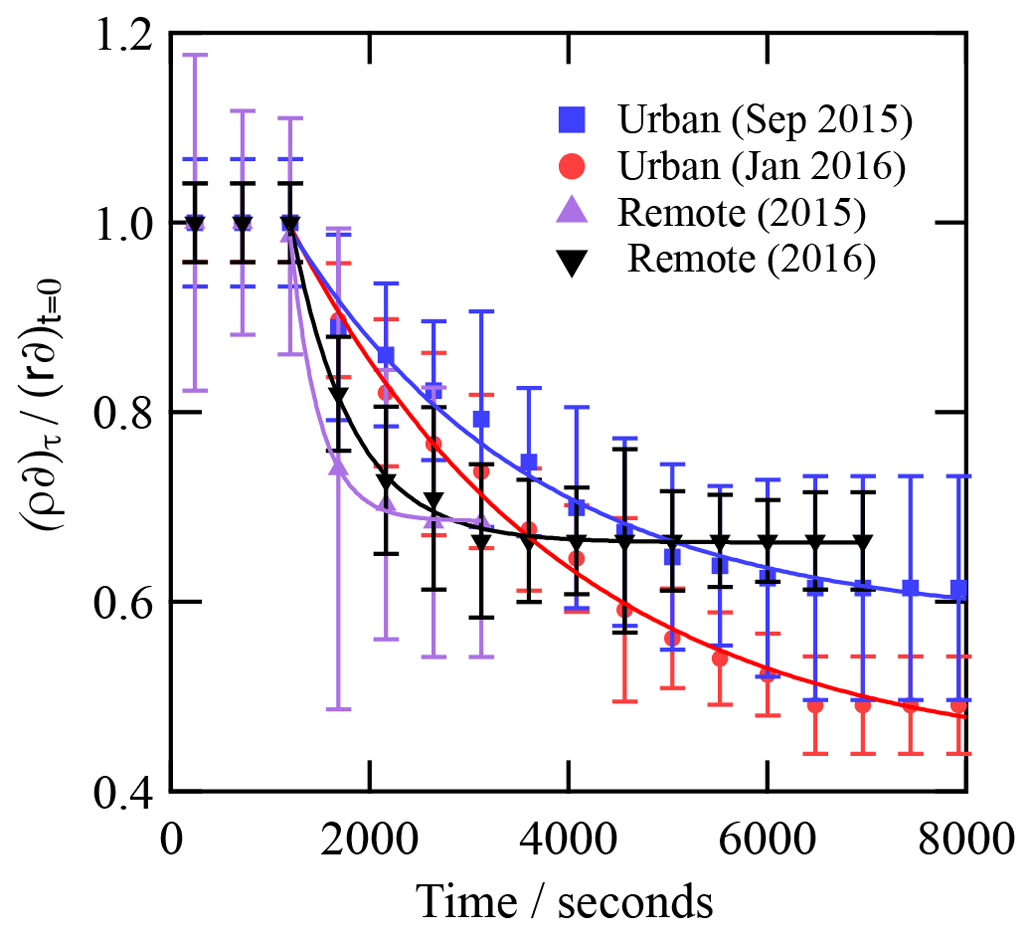
Figure 6The kinetic decay of the scattering length per unit area for an organic film or urban atmospheric aerosol extract collected during (a) September 2015 and (b) January 2016 when exposed to hydroxyl radicals. .
It should be noted that the work presented here confirms that these extracted samples form stable thin films at the air–water interface whose thickness does not increase notably with the addition of more material but results in the formation of lenses (Gaines, 1966) like the related films in other studies (Shepherd et al., 2022; Stuckey et al., 2024). However, it is useful to contrast the films presented here with the wood smoke sample of Stuckey et al. (2024), which is unusual in producing much thicker films stabilised by differentiating the thick film into three separate layers. The work of Stuckey et al. (2024) demonstrates that reflection techniques would be sensitive to thick film formation if it were occurring in the work presented here.
3.3 Kinetic KM SUB model results
Values of ksurf are larger for the remote samples compared with those of the urban samples (Table 2). The low number of data points fitted for both remote aerosol extracts in the decaying portion of the decay curves reduces the precision of the model fit (Fig. 7). However, the general trend is consistent with the trend in k′ from the analytical fits to the decays (Table 2). Note that the KM SUB kinetic model also accounted for the initial average surface concentration of film molecules, estimated as the inverse square of the initial film thickness, resulting in different initial surface concentrations of film molecules for each model run, and the model was sensitive to this. Fitted values of ksurf for reaction of the OH radicals with the remote samples are generally larger in the study presented here compared to identical samples measured in our neutron study (Shepherd et al., 2022) and are compared in Table 3. The values of ksurf for the urban sample are pleasingly close to the values presented by Shepherd et al. (2022). The fitting of the KM SUB model to the data in Fig. 7 is not ideal for the remote samples, and the authors have more confidence in the values in the previous study (Shepherd et al., 2022), where more temporal resolution was achieved.
Table 2Values of ksurf for the reaction between gas-phase OH radicals and the named organic thin film at the air–water interface.
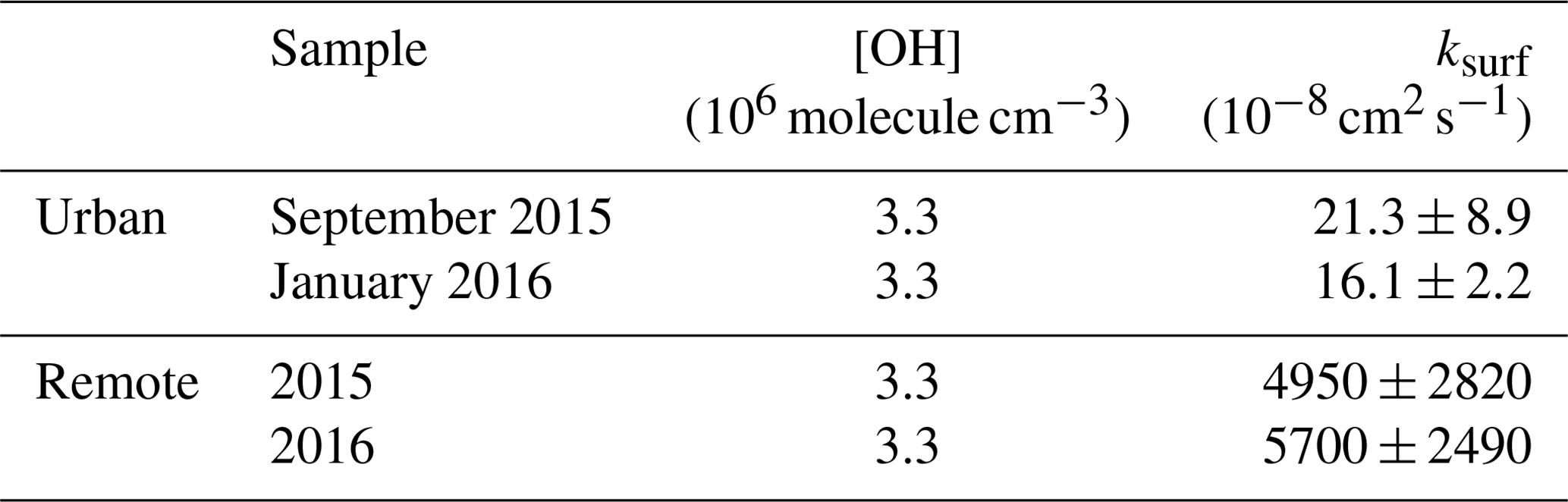
Table 3A comparison of the properties of similar films analysed in the work presented here and taken from Shepherd et al. (2022). Film thickness and ksurf from the KM SUB kinetic modelling are shown. These films are comparable but not identical. The values of the quantities from the X-ray reflection work (presented here) are marked with XR, and the values from the neutron reflection (NR) work and from Shepherd et al. (2022) are also shown.
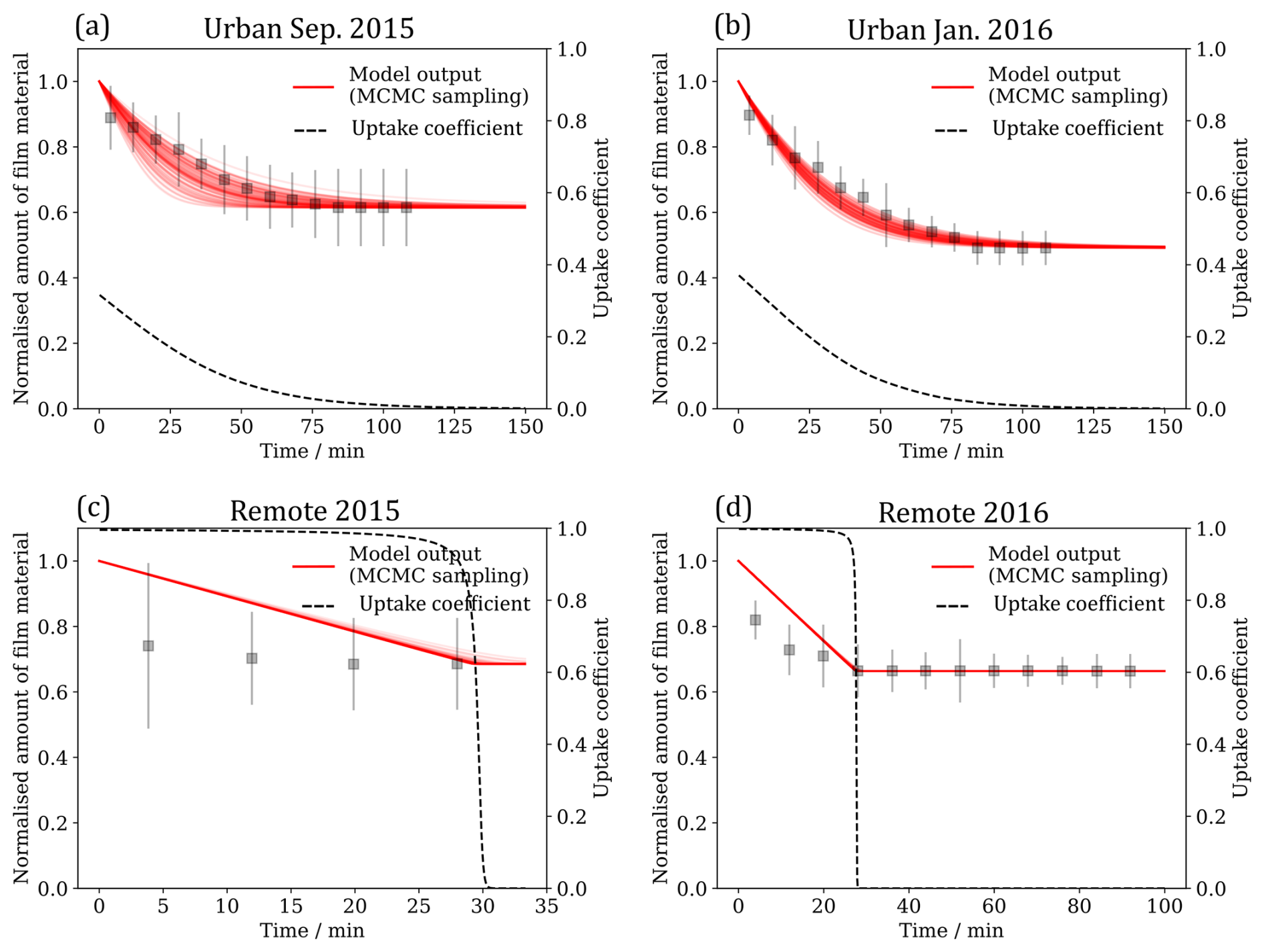
Figure 7Results from the fitting the kinetic model (red) described in Sect. 3.3 to the experimental data (grey). The range of model outputs from the MCMC sampling procedure consistent with the data are presented. The evolution of the uptake coefficient over time is extracted from the best fitting model run for each sample.
It is possible to calculate the uptake coefficient γ from the kinetic model output (Shiraiwa et al., 2010).
γ is a function of the rate of adsorption, JAds; rate of desorption, JDes; and rate of OH radical collision with the surface, JColl (note that JAds,JDes and JColl are defined in Shiraiwa et al., 2010). The uptake coefficient indicates the rate of OH radical replacement at the surface. As JAds and JDes are dependent on the surface concentration of OH radicals and JColl is constant, γ indirectly indicates the probability that an OH radical will react at a surface. Generally, the value of γ is initially larger for the samples collected at remote locations, whereas urban samples have smaller initial values of γ (Fig. 7).
3.4 Material remaining at the air–water interface
There is evidence for a residual film after oxidation with OH radicals (Fig. 7). All kinetic decays appear to level off after a certain degree of OH radical exposure and after the calculated uptake coefficient approaches zero. The kinetic model fitted to these decay data was adapted to incorporate such a residual film. It is unclear whether the residual film is an unreactive portion of the original film or an oxidised unreactive product (or even a mixture of both). Oxidised material that has left the interface has either dissolved in the aqueous sub-phase or is volatile and entered the gas phase. What is certain is that oxidation has changed the film composition, which itself can affect the lifetime of atmospheric aerosol (Gilman et al., 2004).
3.5 Atmospheric implications: chemical lifetime of organic extracts
It is useful to compare the chemical lifetime of the extracted material with respect to the OH radical oxidation to typical lifetimes of atmospheric aerosol. Here, the chemical lifetime of the film is defined as the chemical half-life extracted from the optimised kinetic models for each film at an atmospherically relevant [OH] range (Fig. 8). If the chemical lifetime of the film is short compared to aerosol atmospheric lifetime, oxidation would need to be considered in atmospheric models. The lifetime of an atmospheric aerosol can be governed by the relative humidity (Li et al., 2020; Li and Knopf, 2021) of the environment, by the presence (Gill et al., 1983) and composition (Gilman et al., 2004) of a film, and by the oxidants present in the surrounding environment (Knopf et al., 2011). Williams et al. (2002) calculated a lifetime ranging from 1–10 d for an aerosol with a diameter of 0.1 µm at an altitude ranging from 0–8 km. George et al. (2008) sampled ambient aerosol from Toronto, Canada, and oxidised it in the lab with OH radicals in a flow tube and found that OH can oxidise this matter on timescales of 4 d. Likewise, Robinson et al. (2006) predicted the timescale for OH oxidation of organic aerosol to be 1–9 d. The chemical lifetimes reported here are very short, but in contrast to the studies of George et al. (2008) and Robinson et al. (2006), the reaction is a surface reaction only. There is no doubt that the reaction is fast and should be included in models. The lifetime of the atmospheric aerosol extract in the study presented here was calculated to lie between 48–110 min at [OH]atm (Fig. 8). The modelled lifetime is very short in comparison to typical aerosol lifetimes of 1–10 d (Kanakidou et al., 2005). Hence, it can be concluded that atmospheric aerosol films composed of similar materials may react very quickly in the atmosphere and consequently need to be considered in atmospheric aerosol models.
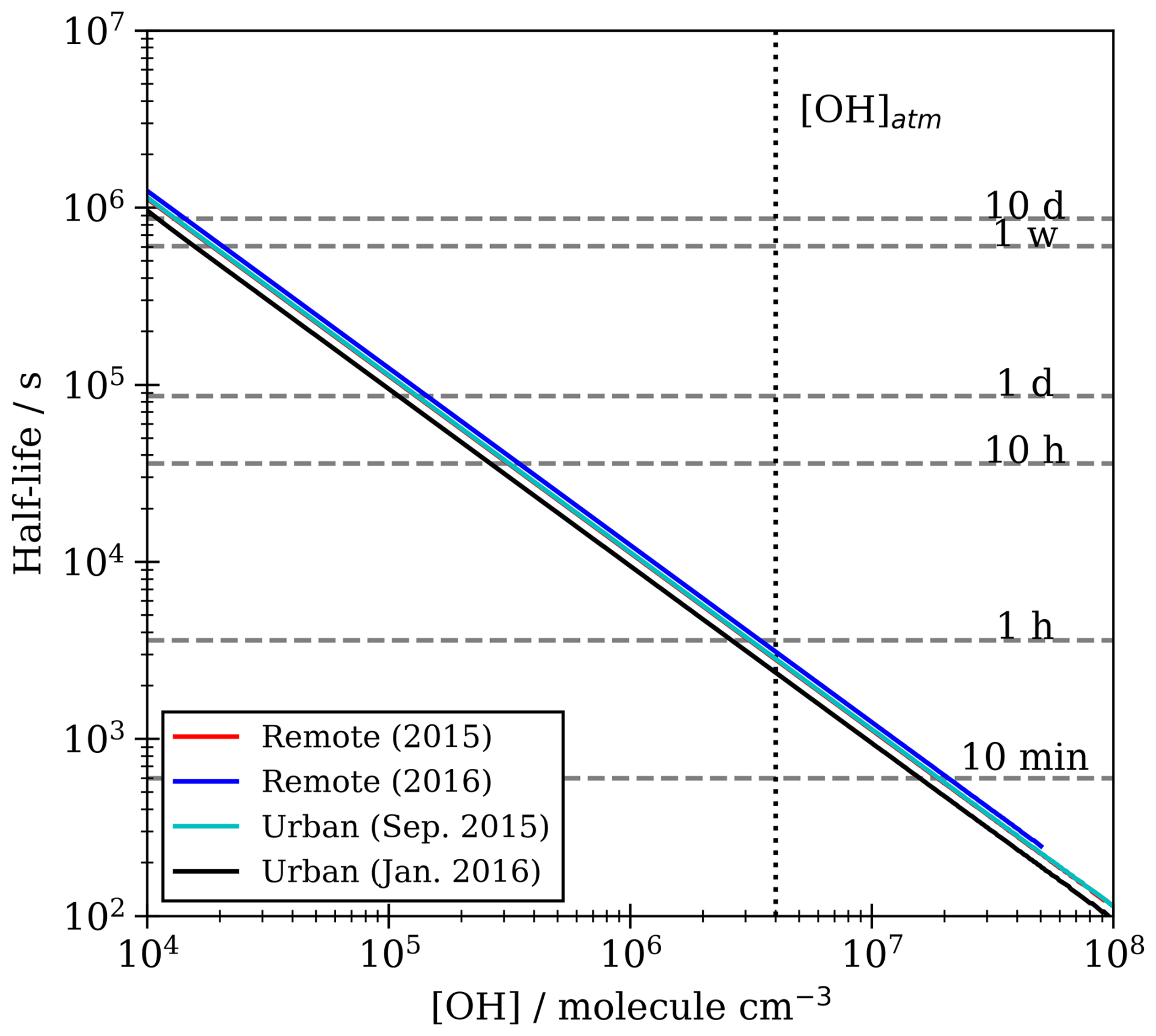
Figure 8The half life of the film owing to chemicals oxidation by OH radical for different OH radical mixing ratios. A typical hydroxyl radical mixing ratio [OH]atm is shown as a vertical dotted line.

Figure 9Change in aerosol single-scattering albedo (ΔSSA), asymmetry parameter (Δgsca) and top-of-atmosphere albedo (ΔAlbedo; 80 km altitude, 60° solar zenith angle) calculated using a combination of Mie and 1-D radiative transfer modelling for the impact of the oxidation (represented by a halving of film thickness) of organic-coated spherical silica and aqueous aerosol from urban and remote environments for wavelengths of light between 320–785 nm. Core and film materials were given complex refractive indexes representative of the material/environment. The single-scattering albedo and asymmetrical light scattering of these four types of aerosol were calculated for aerosol with core sizes between 10–1000 nm with initial film thicknesses of 0.1–10 nm, normalised by the size distribution of aerosol from their respective environments. The single-scattering albedo and asymmetry parameter were calculated for aerosol before and after oxidation. Radiative transfer models were produced for each aerosol distribution for each film thickness before and after oxidation. Values of single-scattering albedo and asymmetry parameter were applied to a 3 km thick layer of aerosol at the planetary boundary layer, and top-of-atmosphere albedo was determined for a given model using aerosol with a given film thickness. ΔSSA, Δgsca and ΔAlbedo are the differences between the post-oxidation models and the pre-oxidation models.
3.6 Atmospheric implications: change in top-of-atmosphere albedo owing to oxidation of the thin film
The presence of a thin organic film can alter the optical properties of an atmospheric core–shell aerosol, and this effect is determined in part by the real and complex refractive indexes and sizes of the core and film (Barker et al., 2023; Donaldson and Vaida, 2006; Forrister et al., 2015; Gill et al., 1983; Jones et al., 2017; Lack and Cappa, 2010; McGrory et al., 2022; Moffet and Prather, 2009; Schnaiter et al., 2005; Shepherd et al., 2018). The accumulation of an organic film will alter the ratio of light that is scattered/absorbed by a particulate as well as the direction in which this light is scattered (Bond et al., 2013; Jacobson et al., 2000; Lack and Cappa, 2010; Moffet and Prather, 2009; Shiraiwa et al., 2010; Wu et al., 2014). Furthermore, owing to substantial differences between the refractive indexes and size distributions of aerosols between environments, the optical contributions of films are dependent on the environment (Barker et al., 2023; Kirchstetter et al., 2004; Kirpes et al., 2019; Shepherd et al., 2018; Virkkula et al., 2022). As changes are made to organic films, i.e. film thickness reduction through oxidation, the optical properties of the aerosol will also vary; thus, the nature and intensity of these variations are also likely to be dependent on the environment. A combination of core–shell Mie and 1-D radiative transfer modelling has previously been used to estimate the change in top-of-atmosphere albedo from uncoated aerosol to coated aerosol of varying film thicknesses and environments (Barker et al., 2023). Barker et al. (2023) showed that while there were significant differences in how albedo varied with increasing film thickness between environments, the presence of films on mineral aerosol in both urban and remote environments caused a decrease in albedo regardless of film thickness for films between 0.1–100 nm. The oxidation-induced changes to the optical properties of aerosol, as well as the effect this would have on top-of-atmosphere albedo, are estimated here by a combination of Mie and 1-D radiative transfer modelling. The aim was to quantify the albedo impact of changes to film thicknesses observed in this work (i.e., the oxidation of an organic film is simplified to a halving of film thickness). Particular attention was made towards films with thickness of 0.1–10 nm, owing to the atmospheric relevance of this film thickness range. Calculations were made using both aqueous and mineral cores and using optical properties of organics and size distributions of aerosols in urban and remote environments. Variation in the source environment of aerosol resulted in substantial differences in ΔAlbedo between urban and remote aerosols, with effects ranging between +0.001 to +0.014 for urban aerosol and for remote aerosol, with less dependence on the film thickness for remote aerosol. Changes to ΔAlbedo are driven by an interplay of scattering and absorption effects, which were made evident in the variation of the single-scattering albedo and asymmetrical light scattering as initial film thickness was increased. The oxidation of urban aerosol films with increasing thicknesses resulted in an increase in the single-scattering albedo of 0.004 to 0.067 and a decrease of the asymmetry parameter from 0.000 to 0.022, whereas the oxidation of remote films caused variation in the single-scattering albedo of +0.09 to −0.003 and variation of the asymmetry parameter of +0.004 to −0.010. While the core size, core refractive index, initial film thickness and film refractive index all had some impact on ΔAlbedo, the environment-dependent differences in ΔAlbedo versus initial film thickness are driven predominantly by two factors: the imaginary refractive index of material from urban environments is significantly greater than that from remote environments, and the film thicknesses are of greater size relative to the core size of urban aerosol than remote aerosol. There is a distinct difference in changes to the asymmetry parameter and the single-scattering albedo between environments and a lack of significant differences when comparing aerosols with aqueous and mineral cores. Owing to the optical and physical differences in these aerosols, it appears that, typically, the oxidation of a highly absorbing film on a smaller core (urban) has a greater effect on top-of-atmosphere albedo than the oxidation of a less-absorbing film of the same thickness on a larger core (remote). However, the effects of oxidation of films that are <1 nm in thickness on ΔAlbedo are similar in intensity between urban and remote environments. Barker et al. (2023) demonstrated that adding a film to mineral aerosol resulted in a decreasing top-of-atmosphere albedo; hence, the removal of the film would cause an increase in albedo. The effect is also demonstrated for the removal of half the film from the mineral as well as aqueous aerosol; however, the intensity of the effect is much smaller and may be negligible for remote aerosol and an important, but small, effect for urban aerosol.
The study presented here demonstrates the successful formation of a thin surfactant film of atmospheric aerosol at the air–water interface from material extracted from atmospheric aerosol. The film was determined to comprise a two-layer system, indicating aerosol extracts may produce a film thickness of ∼10 Å for urban extracts and ∼17 Å for remote extracts. In the absence of other data sources, the film thickness determined in the study could be used in atmospheric radiative transfer models for aqueous core–shell systems. The two-layer system indicates that the atmospheric aerosol extracts may contain hydrophobic and hydrophilic regions. The atmospheric aerosol extracts were exposed to gas-phase OH radicals at a concentration of and demonstrated an exponential decrease. Optimised KM SUB kinetic models suggested that the atmospheric lifetime of the reactive component of the films studied can vary between minutes and days, depending on the ambient hydroxyl radical concentration assuming aqueous particles and cloud droplets. The determined film thickness and film lifetimes presented in this work represent values for a large ensemble of molecules, and we give an estimate of the values. However, these values do not represent an average; the average macroscopic-scale film properties at the air–water interface of a trough may not be statistically applicable to individual aerosol droplets. The removal of half of the organic thin film from the air–water interface may have an increasing effect on the top-of-atmosphere albedo for urban thin films and a negligible effect for remote thin films.
The code for the kinetics modelling is available https://doi.org/10.5281/zenodo.11962921 (King et al., 2024) as a series of Jupyter Notebook files. The MultilayerPy software used in this work, including tutorials and documentation, is available at https://github.com/tintin554/multilayerpy (Milsom et al., 2022a) and https://doi.org/10.5281/zenodo.7034729 (Milsom, 2022). The code is released under the GPL v3.0 license.
Data used in this paper are available at https://doi.org/10528/zenodo.11962921 (King et al., 2024).
All samples were consumed in the experiment.
RHS conducted all experiments, extracted all atmospheric aerosol extracts, analysed and interpreted data collected, and wrote the paper. MDK and ADW conceived the experiment idea and assisted during the experiment. NB collected samples from Antarctica. TA was the synchrotron beamline scientist who assisted with the experiment and reduced all data. AM performed the surface kinetic modelling, which was analysed and interpreted with input from CP. ES, MK and RW conceived the modelling, and it was performed by ES.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors would also like to thank Jerry Morris (RHUL) for the manufacture of the Langmuir trough.
This research has been supported by the Natural Environment Research Council (grant no. NE/T00732X/1), the Science and Technology Facilities Council (grant no. SI13493), and the Science and Technology Facilities Council (grant no. ST/L504279/1).
This paper was edited by Daniel Knopf and reviewed by two anonymous referees.
Abelès, F.: La détermination de l'indice et de l'épaisseur des couches minces transparentes, J. Phys. Radium, 11, 310–314, https://doi.org/10.1051/jphysrad:01950001107031000ff. ffjpa00234262f, 1950. a
Andreae, M. and Rosenfeld, D.: Aerosol–cloud–precipitation interactions. Part 1. The nature and sources of cloud-active aerosols, Earth-Sci. Rev., 89, 13–41, 2008. a
Arangio, A. M., Slade, J. H., Berkemeier, T., Pöschl, U., Knopf, D. A., and Shiraiwa, M.: Multiphase chemical kinetics of OH radical uptake by molecular organic markers of biomass burning aerosols: humidity and temperature dependence, surface reaction, and bulk diffusion, J. Phys. Chem. A, 119, 4533–4544, 2015. a
Arnold, T., Nicklin, C., Rawle, J., Sutter, J., Bates, T., Nutter, B., McIntyre, G., and Burt, M.: Implementation of a beam deflection system for studies of liquid interfaces on beamline I07 at Diamond, J. Synchro. Ra., 19, 408–416, https://doi.org/10.1107/S0909049512009272, 2012. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004. a
Bagot, P. A. J., Waring, C., Costen, M. L., and McKendrick, K. G.: Dynamics of Inelastic Scattering of OH Radicals from Reactive and Inert Liquid Surfaces, The J. Phys. Chem. C, 112, 10868–10877, 2008. a
Ball, C. P., Levick, A. P., Woolliams, E. R., Green, P. D., Dury, M. R., Winkler, R., Deadman, A. J., Fox, N. P., and King, M. D.: Effect of polytetrafluoroethylene (PTFE) phase transition at 19 °C on the use of Spectralon as a reference standard for reflectance, Appl. Opt., 52, 4806, https://doi.org/10.1364/AO.52.004806, 2013. a
Barker, C. R., Poole, M. L., Wilkinson, M., Morison, J., Wilson, A., Little, G., Stuckey, E. J., Welbourn, R. J. L., Ward, A. D., and King, M. D.: Ultraviolet refractive index values of organic aerosol extracted from deciduous forestry, urban and marine environments, Environ. Sci.-Atmos., 3, 1008–1024, https://doi.org/10.1039/D3EA00005B, 2023. a, b, c, d, e, f, g, h
Bertram, A. K., Ivanov, A. V., Hunter, M., Molina, L. T., and Molina, M. J.: The Reaction Probability of OH on Organic Surfaces of Tropospheric Interest, The J. Phys. Chem. A, 105, 9415–9421, 2001. a
Bevington, P. R., Robinson, D. K., Blair, J. M., Mallinckrodt, A. J., and McKay, S.: Data Reduction and Error Analysis for the Physical Sciences, Comput. Phys., 7, 415–416, 1993. a
Blanchard, D. C.: Sea-to-Air Transport of Surface Active Material, Science, 146, 396–397, 1964. a
Bohern, C. and Huffman, D.: Absorption and Scattering of Light by Small Particles, Wiley-VCH Verlag GmbH & Co. KGaA, 544 pp., https://doi.org/10.1002/9783527618156, 1998. a
Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T., DeAngelo, B. J., Flanner, M. G., Ghan, S., Kärcher, B., Koch, D., Kinne, S., Kondo, Y., Quinn, P. K., Sarofim, M. C., Schultz, M. G., Schulz, M., Venkataraman, C., Zhang, H., Zhang, S., Bellouin, N., Guttikunda, S. K., Hopke, P. K., Jacobson, M. Z., Kaiser, J. W., Klimont, Z., Lohmann, U., Schwarz, J. P., Shindell, D., Storelvmo, T., Warren, S. G., and Zender, C. S.: Bounding the role of black carbon in the climate system: A scientific assessment, J. Geophys. Res.-Atmos., 118, 5380–5552, https://doi.org/10.1002/jgrd.50171, 2013. a
Bréon, F.-M., Tanré, D., and Generoso, S.: Aerosol Effect on Cloud Droplet Size Monitored from Satellite, Science, 295, 834–838, 2002. a
Burkholder, J. B., Abbatt, J. P. D., Barnes, I., Roberts, J. M., Melamed, M. L., Ammann, M., Bertram, A. K., Cappa, C. D., Carlton, A. G., Carpenter, L. J., Crowley, J. N., Dubowski, Y., George, C., Heard, D. E., Herrmann, H., Keutsch, F. N., Kroll, J. H., McNeill, V. F., Ng, N. L., Nizkorodov, S. A., Orlando, J. J., Percival, C. J., Picquet-Varrault, B., Rudich, Y., Seakins, P. W., Surratt, J. D., Tanimoto, H., Thornton, J. A., Tong, Z., Tyndall, G. S., Wahner, A., Weschler, C. J., Wilson, K. R., and Ziemann, P. J.: The Essential Role for Laboratory Studies in Atmospheric Chemistry, Environ. Sci. Technol., 51, 2519–2528, 2017. a
Chapleski, R. C., Zhang, Y., Troya, D., and Morris, J. R.: Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces, Chem. Soc. Rev., 45, 3731–3746, 2016. a
Chateigner, D.: X-Ray Reflectivity, chap. 8, 235–255 pp., John Wiley & Sons, Ltd, ISBN 9781118622506, 2013. a
Cosman, L. M., Knopf, D. A., and Bertram, A. K.: N2O5 Reactive Uptake on Aqueous Sulfuric Acid Solutions Coated with Branched and Straight-Chain Insoluble Organic Surfactants, The J. Phys. Chem. A, 112, 2386–2396, https://doi.org/10.1021/jp710685r, 2008. a
Cruz, C. N. and Pandis, S. N.: The effect of organic coatings on the cloud condensation nuclei activation of inorganic atmospheric aerosol, J. Geophys. Res.-Atmos., 103, 13111–13123, 1998. a
Dabkowska, A. P., Talbot, J. P., Cavalcanti, L., Webster, J. R. P., Nelson, A., Barlow, D. J., Fragneto, G., and Lawrence, M. J.: Calcium mediated interaction of calf-thymus DNA with monolayers of distearoylphosphatidylcholine: a neutron and X-ray reflectivity study, Soft Matter, 9, 7095–7105, 2013. a
Davies, J. F., Miles, R. E. H., Haddrell, A. E., and Reid, J. P.: Influence of organic films on the evaporation and condensation of water in aerosol, P. Natl. Acad. Sci. USA, 110, 8807–8812, 2013. a
Davies, J. T. and Rideal, R. K.: Interfacial Phenomena, Academic Press, 480 pp., https://doi.org/10.1016/B978-0-12-206056-4.X5001-2, 1961. a, b
Dennis-Smither, B. J., Miles, R. E. H., and Reid, J. P.: Oxidative aging of mixed oleic acid/sodium chloride aerosol particles, J. Geophys. Res.-Atmos., 117, D20204, https://doi.org/10.1029/2012JD018163, 2012. a
Dilbeck, C. W. and Finlayson-Pitts, B. J.: Hydroxyl radical oxidation of phospholipid-coated NaCl particles, Phys. Chem. Chem. Phys., 15, 9833–9844, 2013. a
Donaldson, D. J. and Anderson, D.: Adsorption of Atmospheric Gases at the Air−Water Interface. 2. C1−C4 Alcohols, Acids, and Acetone, The J. Phys. Chem. A, 103, 871–876, 1999. a
Donaldson, D. J. and George, C.: Sea-Surface Chemistry and Its Impact on the Marine Boundary Layer, Environ. Sci. Technol., 46, 10385–10389, 2012. a
Donaldson, D. J. and Vaida, V.: The Influence of Organic Films at the Air−Aqueous Boundary on Atmospheric Processes, Chem. Rev., 106, 1445–1461, 2006. a, b
Eliason, T., Aloisio, S., Donaldson, D., Cziczo, D., and Vaida, V.: Processing of unsaturated organic acid films and aerosols by ozone, Atmos. Environ., 37, 2207–2219, 2003. a, b
Eliason, T., Gilman, J., and Vaida, V.: Oxidation of organic films relevant to atmospheric aerosols, Atmos. Environ., 38, 1367–1378, 2004. a
Ellison, G. B., Tuck, A. F., and Vaida, V.: Atmospheric processing of organic aerosols, J. Geophys. Res.-Atmos., 104, 11633–11641, 1999. a
Enami, S., Hoffmann, M. R., and Colussi, A. J.: In situ mass spectrometric detection of interfacial intermediates in the oxidation of RCOOH(aq) by gas-phase OH-radicals, J.Phys. Chem. A, 118, 4130–4137, 2014a. a
Enami, S., Hoffmann, M. R., and Colussi, A. J.: In situ mass spectrometric detection of interfacial intermediates in the oxidation of RCOOH(aq) by gas-phase OH-radicals, J. Phys. Chem. A, 118, 4130–4137, 2014b. a, b
Feingold, G. and Chuang, P. Y.: Analysis of the Influence of Film-Forming Compounds on Droplet Growth: Implications for Cloud Microphysical Processes and Climate, J. Atmos. Sci., 59, 2006–2018, https://doi.org/10.1175/1520-0469(2002)059<2006:AOTIOF>2.0.CO;2, 2002. a
Foreman-Mackey, D., Hogg, D. W., Lang, D., and Goodman, J.: emcee: The MCMC Hammer, Publications of the Astronomical Society of the Pacific, 125, 306, https://doi.org/10.1086/670067, 2013. a
Forrister, H., Liu, J., Scheuer, E., Dibb, J., Ziemba, L., Thornhill, K. L., Anderson, B., Diskin, G., Perring, A. E., Schwarz, J. P., Campuzano-Jost, P., Day, D. A., Palm, B. B., Jimenez, J. L., Nenes, A., and Weber, R. J.: Evolution of brown carbon in wildfire plumes, Geophys. Res. Lett., 42, 4623–4630, https://doi.org/10.1002/2015GL063897, 2015. a
Gaines, G. L.: Insoluble monolayers at liquid gas interfaces, Interscience Publisers, 386 pp., ISBN 0608103039, 1966. a, b
George, I. J., Vlasenko, A., Slowik, J. G., Broekhuizen, K., and Abbatt, J. P. D.: Heterogeneous oxidation of saturated organic aerosols by hydroxyl radicals: uptake kinetics, condensed-phase products, and particle size change, Atmos. Chem. Phys., 7, 4187–4201, https://doi.org/10.5194/acp-7-4187-2007, 2007. a
George, I. J., Slowik, J., and Abbatt, J. P. D.: Chemical aging of ambient organic aerosol from heterogeneous reaction with hydroxyl radicals, Geophys. Res. Lett., 35, L13811, https://doi.org/10.1029/2008GL033884, 2008. a, b
Gidalevitz, D., Huang, Z., and Rice, S. A.: Protein folding at the air–water interface studied with x-ray reflectivity, P. Natl. Acad. Sci. USA, 96, 2608–2611, https://doi.org/10.1073/pnas.96.6.2608, 1999. a
Gill, P. S., Graedel, T. E., and Weschler, C. J.: Organic films on atmospheric aerosol particles, fog droplets, cloud droplets, raindrops, and snowflakes, Rev. Geophys., 21, 903–920, 1983. a, b, c, d
Gilman, J. B., Eliason, T. L., Fast, A., and Vaida, V.: Selectivity and stability of organic films at the air-aqueous interface, J. Colloid Interface Sci., 280, 234–243, 2004. a, b
González-Labrada, E., Schmidt, R., and DeWolf, C. E.: Real-time monitoring of the ozonolysis of unsaturated organic monolayers, Chem. Commun., 23, 2471–2473 pp., https://doi.org/10.1039/b603501a, 2006. a, b
González-Labrada, E., Schmidt, R., and DeWolf, C. E.: Kinetic analysis of the ozone processing of an unsaturated organic monolayer as a model of an aerosol surface, Phys. Chem. Chem. Phys., 9, 5814–5821, 2007. a
Hale, G. M. and Querry, M. R.: Optical Constants of Water in the 200-nm to 200-µm Wavelength Region, Appl. Opt., 12, 555–563, https://doi.org/10.1364/AO.12.000555, 1973. a
Haynes, W. M. (Ed.): CRC Handbook of Chemistry and Physics, Taylor and Francis group CRC press, 97th edn., 1670 pp., https://doi.org/10.1201/9781315380476, 2016. a
Haywood, J. M., Jones, A., Jones, A. C., Halloran, P., and Rasch, P. J.: Climate intervention using marine cloud brightening (MCB) compared with stratospheric aerosol injection (SAI) in the UKESM1 climate model, Atmos. Chem. Phys., 23, 15305–15324, https://doi.org/10.5194/acp-23-15305-2023, 2023. a
Hemming, J. M., Hughes, B. R., Rennie, A. R., Tomas, S., Campbell, R. A., Hughes, A. V., Arnold, T., Botchway, S. W., and Thompson, K. C.: Environmental Pollutant Ozone Causes Damage to Lung Surfactant Protein B (SP-B), Biochemistry, 54, 5185–5197, https://doi.org/10.1021/acs.biochem.5b00308, 2015. a
Hemming, J. M., Szyroka, J., Shokano, G., Arnold, T., Skoda, M. W. A., Rennie, A. R., and Thompson, K. C.: Changes to lung surfactant monolayers upon exposure to gas phase ozone observed using X-ray and neutron reflectivity, Environ. Sci.-Atmos., 2, 753–760, https://doi.org/10.1039/D2EA00032F, 2022. a
Hogg, D. W. and Foreman-Mackey, D.: Data Analysis Recipes: Using Markov Chain Monte Carlo*, The Astrophys. J. Supplement Series, 236, 11, https://doi.org/10.3847/1538-4365/aab76e, 2018. a
IPCC: Climate Change 2021: The Physical Science Basis. Contribution of Working Group I to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, editedy by: Masson-Delmotte, V., Zhai, P., Pirani, A., Connors, S. L., Péan, C., Berger, S., Caud, N., Chen, Y., Goldfarb, L., Gomis, M. I., Huang, M., Leitzell, K., Lonnoy, E., Matthews, J. B. R., Maycock, T. K., Waterfield, T., Yelekçi, O., Yu, R., and Zhou, B., Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2021, https://doi.org/10.1017/9781009157896, in press, 2021. a, b, c
Jacobson, M. C., Hansson, H. C., Noone, K. J., and Charlson, R. J.: Organic atmospheric aerosols: Review and state of the science, Rev. Geophys., 38, 267–294, 2000. a, b
Jaenicke, R.: Chapter 1 Tropospheric Aerosols, in: Aerosol–Cloud–Climate Interactions, edited by: Hobbs, P. V., Vol. 54 of International Geophysics, 1–31 pp., Academic Press, https://doi.org/10.1016/S0074-6142(08)60210-7, 1993. a, b
Jones, A. E., Wolff, E. W., Salmon, R. A., Bauguitte, S. J.-B., Roscoe, H. K., Anderson, P. S., Ames, D., Clemitshaw, K. C., Fleming, Z. L., Bloss, W. J., Heard, D. E., Lee, J. D., Read, K. A., Hamer, P., Shallcross, D. E., Jackson, A. V., Walker, S. L., Lewis, A. C., Mills, G. P., Plane, J. M. C., Saiz-Lopez, A., Sturges, W. T., and Worton, D. R.: Chemistry of the Antarctic Boundary Layer and the Interface with Snow: an overview of the CHABLIS campaign, Atmos. Chem. Phys., 8, 3789–3803, https://doi.org/10.5194/acp-8-3789-2008, 2008. a
Jones, S. H., King, M. D., and Ward, A. D.: Atmospherically relevant core–shell aerosol studied using optical trapping and Mie scattering, Chem. Commun., 51, 4914–4917, 2015. a, b, c
Jones, S. H., King, M. D., Ward, A. D., Rennie, A. R., Jones, A. C., and Arnold, T.: Are organic films from atmospheric aerosol and sea water inert to oxidation by ozone at the air-water interface?, Atmos. Environ., 161, 274–287, https://doi.org/10.1016/j.atmosenv.2017.04.025, 2017. a, b, c, d, e, f, g, h, i, j, k, l, m
Jones, S. H., King, M. D., Rennie, A. R., Ward, A. D., Campbell, R. A., and Hughes, A. V.: Aqueous Radical Initiated Oxidation of an Organic Monolayer at the Air–Water Interface as a Proxy for Thin Films on Atmospheric Aerosol Studied with Neutron Reflectometry, The J. Phys. Chem. A, 127, 8922–8934, https://doi.org/10.1021/acs.jpca.3c03846, 2023. a, b
Kaiser, T., Roll, G., and Schweiger, G.: Investigation of coated droplets in an optical trap: Raman-scattering, elastic-light-scattering, and evaporation characteristics, Appl. Opt., 35, 5918–5924, 1996. a
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005. a
Khashan, M. and Nassif, A.: Dispersion of the optical constants of quartz and polymethyl methacrylate glasses in a wide spectral range: 0.2–3 µm, Opt. Commun., 188, 129–139, https://doi.org/10.1016/S0030-4018(00)01152-4, 2001. a
King, M. D., Thompson, K. C., and Ward, A. D.: Laser Tweezers Raman Study of Optically Trapped Aerosol Droplets of Seawater and Oleic Acid Reacting with Ozone: Implications for Cloud-Droplet Properties, J. Am. Chem. Soc., 126, 16710–16711, https://doi.org/10.1021/ja044717o, 2004. a
King, M. D., Rennie, A. R., Thompson, K. C., Fisher, F. N., Dong, C. C., Thomas, R. K., Pfrang, C., and Hughes, A. V.: Oxidation of oleic acid at the air–water interface and its potential effects on cloud critical supersaturations, Phys. Chem. Chem. Phys., 11, 7699–7707, 2009. a, b, c, d, e
King, M. D., Rennie, A. R., Pfrang, C., Hughes, A. V., and Thompson, K. C.: Interaction of nitrogen dioxide (NO2) with a monolayer of oleic acid at the air–water interface – A simple proxy for atmospheric aerosol, Atmos. Environ., 44, 1822–1825, https://doi.org/10.1016/j.atmosenv.2010.01.031, 2010. a, b
King, M. D., Jones, S. H., Lucas, C. O. M., Thompson, K. C., Rennie, A. R., Ward, A. D., Marks, A. A., Fisher, F. N., Pfrang, C., Hughes, A. V., and Campbell, R. A.: The reaction of oleic acid monolayers with gas-phase ozone at the air water interface: the effect of sub-phase viscosity, and inert secondary components, Phys. Chem. Chem. Phys., 22, 28032–28044, https://doi.org/10.1039/D0CP03934A, 2020. a, b, c
King, M., Shepherd, R., Ward, A., Stuckey, E., Rebecca, W., Brough, N., Milsom, A., Pfrang, C., and Arnold, T.: The lifetimes and potential change in planetary albedo owing to the oxidation of organic films extracted from atmospheric aerosol by hydroxyl (OH) radical at the air-water interface of aerosol particles, Zenodo [data set, code], https://doi.org/10.5281/zenodo.11962921, 2024. a, b
Kirchstetter, T. W., Novakov, T., and Hobbs, P. V.: Evidence that the spectral dependence of light absorption by aerosols is affected by organic carbon, J. Geophys. Res.-Atmos., 109, D21208, https://doi.org/10.1029/2004JD004999, 2004. a, b
Kirpes, R. M., Bonanno, D., May, N. W., Fraund, M., Barget, A. J., Moffet, R. C., Ault, A. P., and Pratt, K. A.: Wintertime Arctic Sea Spray Aerosol Composition Controlled by Sea Ice Lead Microbiology, ACS Central Science, 5, 1760–1767, https://doi.org/10.1021/acscentsci.9b00541, 2019. a, b
Kitamura, R., Pilon, L., and Jonasz, M.: Optical constants of silica glass from extreme ultraviolet to far infrared at near room temperature, Appl. Opt., 46, 8118–8133, https://doi.org/10.1364/AO.46.008118, 2007. a
Knopf, D. A., Cosman, L. M., Mousavi, P., Mokamati, S., and Bertram, A. K.: A Novel Flow Reactor for Studying Reactions on Liquid Surfaces Coated by Organic Monolayers: Methods, Validation, and Initial Results, The J. Phys. Chem. A, 111, 11021–11032, https://doi.org/10.1021/jp075724c, 2007. a
Knopf, D. A., Forrester, S. M., and Slade, J. H.: Heterogeneous oxidation kinetics of organic biomass burning aerosol surrogates by O3, NO2, N2O5, and NO3, Phys. Chem. Chem. Phys., 13, 21050–21062, 2011. a
Kukui, A., Legrand, M., Preunkert, S., Frey, M. M., Loisil, R., Gil Roca, J., Jourdain, B., King, M. D., France, J. L., and Ancellet, G.: Measurements of OH and RO2 radicals at Dome C, East Antarctica, Atmos. Chem. Phys., 14, 12373–12392, https://doi.org/10.5194/acp-14-12373-2014, 2014. a
Lack, D. A. and Cappa, C. D.: Impact of brown and clear carbon on light absorption enhancement, single scatter albedo and absorption wavelength dependence of black carbon, Atmos. Chem. Phys., 10, 4207–4220, https://doi.org/10.5194/acp-10-4207-2010, 2010. a, b
Li, J. and Knopf, D. A.: Representation of Multiphase OH Oxidation of Amorphous Organic Aerosol for Tropospheric Conditions, Environ. Sci. Technol., 55, 7266–7275, https://doi.org/10.1021/acs.est.0c07668, 2021. a
Li, J., Forrester, S. M., and Knopf, D. A.: Heterogeneous oxidation of amorphous organic aerosol surrogates by O3, NO3, and OH at typical tropospheric temperatures, Atmos. Chem. Phys., 20, 6055–6080, https://doi.org/10.5194/acp-20-6055-2020, 2020. a
Li, K., Guo, Y., Nizkorodov, S. A., Rudich, Y., Angelaki, M., Wang, X., An, T., Perrier, S., and George, C.: Spontaneous dark formation of OH radicals at the interface of aqueous atmospheric droplets, P. Natl. Acad. Sci. USA, 120, e2220228120, https://doi.org/10.1073/pnas.2220228120, 2023. a
Lohmann, U. and Feichter, J.: Global indirect aerosol effects: a review, Atmos. Chem. Phys., 5, 715–737, https://doi.org/10.5194/acp-5-715-2005, 2005. a
Madronich, S. and Flocke, S.: The Role of Solar Radiation in Atmospheric Chemistry, 1–26 pp., Springer Berlin Heidelberg, Berlin, Heidelberg, ISBN 978-3-540-69044-3, https://doi.org/10.1007/978-3-540-69044-3_1, 1999. a
Majewski, J., Kuhl, T. L., Gerstenberg, M. C., Israelachvili, J. N., and Smith, G. S.: Structure of Phospholipid Monolayers Containing Poly(ethylene glycol) Lipids at the Air−Water Interface, The J. Phys. Chem. B, 101, 3122–3129, https://doi.org/10.1021/jp962623y, 1997. a
Marty, J. C. and Saliot, A.: Hydrocarbons (normal alkanes) in the surface microlayer of seawater, Deep Sea Res.-Oceanographic Abstracts, 23, 863–873, 1976. a
McFiggans, G., Artaxo, P., Baltensperger, U., Coe, H., Facchini, M. C., Feingold, G., Fuzzi, S., Gysel, M., Laaksonen, A., Lohmann, U., Mentel, T. F., Murphy, D. M., O'Dowd, C. D., Snider, J. R., and Weingartner, E.: The effect of physical and chemical aerosol properties on warm cloud droplet activation, Atmos. Chem. Phys., 6, 2593–2649, https://doi.org/10.5194/acp-6-2593-2006, 2006. a
McGrory, M. R., Shepherd, R. H., King, M. D., Davidson, N., Pope, F. D., Watson, I. M., Grainger, R. G., Jones, A. C., and Ward, A. D.: Mie scattering from optically levitated mixed sulfuric acid–silica core–shell aerosols: observation of core–shell morphology for atmospheric science, Phys. Chem. Chem. Phys., 24, 5813–5822, https://doi.org/10.1039/D1CP04068E, 2022. a, b
McNeill, V. F., Wolfe, G. M., and Thornton, J. A.: The Oxidation of Oleate in Submicron Aqueous Salt Aerosols: Evidence of a Surface Process, The J. Phys. Chem. A, 111, 1073–1083, 2007. a
Merikanto, J., Spracklen, D. V., Mann, G. W., Pickering, S. J., and Carslaw, K. S.: Impact of nucleation on global CCN, Atmos. Chem. Phys., 9, 8601–8616, https://doi.org/10.5194/acp-9-8601-2009, 2009. a
Milsom, A.: tintin554/multilayerpy: v1.0.2 (v1.0.2), Zenodo [data set], https://doi.org/10.5281/zenodo.7034729, 2022. a
Milsom, A., Lees, A., Squires, A. M., and Pfrang, C.: MultilayerPy (v1.0): a Python-based framework for building, running and optimising kinetic multi-layer models of aerosols and films, Geosci. Model Dev., 15, 7139–7151, https://doi.org/10.5194/gmd-15-7139-2022, 2022a. a, b
Milsom, A., Squires, A. M., Skoda, M. W. A., Gutfreund, P., Mason, E., Terrill, N. J., and Pfrang, C.: The evolution of surface structure during simulated atmospheric ageing of nano-scale coatings of an organic surfactant aerosol proxy, Environ. Sci.-Atmos., 2, 964–977, https://doi.org/10.1039/D2EA00011C, 2022b. a, b
Mmereki, B. T. and Donaldson, D. J.: Direct Observation of the Kinetics of an Atmospherically Important Reaction at the Air−Aqueous Interface, The J. Phys. Chem. A, 107, 11038–11042, 2003. a
Moffet, R. C. and Prather, K. A.: In-situ measurements of the mixing state and optical properties of soot with implications for radiative forcing estimates, P. Natl. Acad. Sci. USA, 106, 11872–11877, https://doi.org/10.1073/pnas.0900040106, 2009. a, b
Nah, T., Kessler, S. H., Daumit, K. E., Kroll, J. H., Leone, S. R., and Wilson, K. R.: OH-initiated oxidation of sub-micron unsaturated fatty acid particles, Phys. Chem. Chem. Phys., 15, 18649–18663, 2013. a
Nakayama, T., Sato, K., Matsumi, Y., Imamura, T., Yamazaki, A., and Uchiyama, A.: Wavelength and NOx dependent complex refractive index of SOAs generated from the photooxidation of toluene, Atmos. Chem. Phys., 13, 531–545, https://doi.org/10.5194/acp-13-531-2013, 2013. a
Nelson, A.: Co-refinement of multiple-contrast neutron/X-ray reflectivity data using MOTOFIT, J. Appl. Crystallogr., 39, 273–276, 2006. a
Nicklin, C., Arnold, T., Rawle, J., and Warne, A.: Diamond beamline I07: a beamline for surface and interface diffraction, J. Synchrotron. Ra., 23, 1245–1253, 2016. a
Ovadnevaite, J., Zuend, A., Laaksonen, A., Sanchez, K. J., Roberts, G., Ceburnis, D., Decesari, S., Rinaldi, M., Hodas, N., Facchini, M. C., Seinfeld, J. H., and O'Dowd, C.: Surface tension prevails over solute effect in organic-influenced cloud droplet activation, Nature, 546, 637–641, https://doi.org/10.1038/nature22806, 2017. a
Perriman, A. W., Henderson, M. J., Holt, S. A., and White, J. W.: Effect of the Air−Water Interface on the Stability of β-Lactoglobulin, The J. Phys. Chem. B, 111, 13527–13537, https://doi.org/10.1021/jp074777r, 2007. a
Pfrang, C., Sebastiani, F., Lucas, C. O. M., King, M. D., Hoare, I. D., Chang, D., and Campbell, R. A.: Ozonolysis of methyl oleate monolayers at the air–water interface: oxidation kinetics, reaction products and atmospheric implications, Phys. Chem. Chem. Phys., 16, 13220–13228, 2014. a, b, c
Pöschl, U., Rudich, Y., and Ammann, M.: Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions – Part 1: General equations, parameters, and terminology, Atmos. Chem. Phys., 7, 5989–6023, https://doi.org/10.5194/acp-7-5989-2007, 2007. a
Prather, K. A., Hatch, C. D., and Grassian, V. H.: Analysis of Atmospheric Aerosols, Annu. Rev. Anal. Chem., 1, 485–514, 2008. a
Press, W. H., Flannery, B. P., Teukolsky, S. A., Vetterling, W. T., and Kramer, P. B.: Numerical Recipes: The Art of Scientific Computing, Phys. Today, 40, 120–122, 1987. a
Prinn, R. G., Huang, J., Weiss, R. F., Cunnold, D. M., Fraser, P. J., Simmonds, P. G., McCulloch, A., Harth, C., Salameh, P., O'Doherty, S., Wang, R. H., Porter, L., and Miller, B. R.: Evidence for substantial variations of atmospheric hydroxyl radicals in the past two decades, Science, 292, 1882–1888, 2001. a
Ramanathan, V., Crutzen, P. J., Kiehl, J. T., and Rosenfeld, D.: Aerosols, Climate, and the Hydrological Cycle, Science, 294, 2119–2124, https://doi.org/10.1126/science.1064034, 2001. a
Reid, J. P., Dennis-Smither, B. J., Kwamena, N.-O. A., Miles, R. E. H., Hanford, K. L., and Homer, C. J.: The morphology of aerosol particles consisting of hydrophobic and hydrophilic phases: hydrocarbons, alcohols and fatty acids as the hydrophobic component, Phys. Chem. Chem. Phys., 13, 15559–15572, 2011. a
Reitzel, N., Greve, D. R., Kjaer, K., Howes, P. B., Jayaraman, M., Savoy, S., McCullough, R. D., McDevitt, J. T., and Bjørnholm, T.: Self-Assembly of Conjugated Polymers at the Air/Water Interface. Structure and Properties of Langmuir and Langmuir−Blodgett Films of Amphiphilic Regioregular Polythiophenes, J. Am. Chem. Soc., 122, 5788–5800, https://doi.org/10.1021/ja9924501, 2000. a
Richards-Henderson, N. K., Goldstein, A. H., and Wilson, K. R.: Sulfur Dioxide Accelerates the Heterogeneous Oxidation Rate of Organic Aerosol by Hydroxyl Radicals, Environ. Sci. Technol., 50, 3554–3561, 2016. a
Robinson, A. L., Donahue, N. M., and Rogge, W. F.: Photochemical oxidation and changes in molecular composition of organic aerosol in the regional context, J. Geophys. Res., 111, D03302, https://doi.org/10.1029/2005JD006265, 2006. a, b
Rosenfeld, D., Lohmann, U., Raga, G. B., O'Dowd, C. D., Kulmala, M., Fuzzi, S., Reissell, A., and Andreae, M. O.: Flood or Drought: How Do Aerosols Affect Precipitation?, Science, 321, 1309–1313, https://doi.org/10.1126/science.1160606, 2008. a
Ruehl, C. R. and Wilson, K. R.: Surface Organic Monolayers Control the Hygroscopic Growth of Submicrometer Particles at High Relative Humidity, The J. Phys. Chem. A, 118, 3952–3966, 2014. a
Russell, L. M., Maria, S. F., and Myneni, S. C. B.: Mapping organic coatings on atmospheric particles, Geophys. Res. Lett., 29, 26-1–26-4, 2002. a
Salah, F., Harzallah, B., and van der Lee, A.: Data reduction practice in X-ray reflectometry, J. Appl. Crystallogr., 40, 813–819, https://doi.org/10.1107/S0021889807030403, 2007. a
Schiebener, P., Straub, J., Levelt Sengers, J. M. H., and Gallagher, J. S.: Refractive index of water and steam as function of wavelength, temperature and density, J. Phys. Chem. Ref. Data, 19, 677–717, https://doi.org/10.1063/1.555859, 1990. a
Schnaiter, M., Linke, C., Möhler, O., Naumann, K.-H., Saathoff, H., Wagner, R., Schurath, U., and Wehner, B.: Absorption amplification of black carbon internally mixed with secondary organic aerosol, J. Geophys. Res.-Atmos., 110, D19204, https://doi.org/10.1029/2005JD006046, 2005. a
Sebastiani, F., Campbell, R. A., Rastogi, K., and Pfrang, C.: Nighttime oxidation of surfactants at the air–water interface: effects of chain length, head group and saturation, Atmos. Chem. Phys., 18, 3249–3268, https://doi.org/10.5194/acp-18-3249-2018, 2018. a
Sebastiani, F., Campbell, R. A., and Pfrang, C.: Night-time oxidation at the air–water interface: co-surfactant effects in binary mixtures, Environ. Sci.-Atmos., 2, 1324–1337, https://doi.org/10.1039/D2EA00056C, 2022. a
Seinfeld, J. H., Bretherton, C., Carslaw, K. S., Coe, H., DeMott, P. J., Dunlea, E. J., Feingold, G., Ghan, S., Guenther, A. B., Kahn, R., Kraucunas, I., Kreidenweis, S. M., Molina, M. J., Nenes, A., Penner, J. E., Prather, K. A., Ramanathan, V., Ramaswamy, V., Rasch, P. J., Ravishankara, A. R., Rosenfeld, D., Stephens, G., and Wood, R.: Improving our fundamental understanding of the role of aerosol−cloud interactions in the climate system, P. Natl. Acad. Sci. USA, 113, 5781–5790, https://doi.org/10.1073/pnas.1514043113, 2016. a
Shepherd, R. H., King, M. D., Marks, A. A., Brough, N., and Ward, A. D.: Determination of the refractive index of insoluble organic extracts from atmospheric aerosol over the visible wavelength range using optical tweezers, Atmos. Chem. Phys., 18, 5235–5252, https://doi.org/10.5194/acp-18-5235-2018, 2018. a, b, c, d
Shepherd, R. H., King, M. D., Rennie, A. R., Ward, A. D., Frey, M. M., Brough, N., Eveson, J., Del Vento, S., Milsom, A., Pfrang, C., Skoda, M. W. A., and Welbourn, R. J. L.: Measurement of gas-phase OH radical oxidation and film thickness of organic films at the air–water interface using material extracted from urban, remote and wood smoke aerosol, Environ. Sci.-Atmos., 2, 574–590, 2022. a, b, c, d, e, f, g, h, i, j, k, l, m, n, o, p, q, r, s
Shiraiwa, M., Pfrang, C., and Pöschl, U.: Kinetic multi-layer model of aerosol surface and bulk chemistry (KM-SUB): the influence of interfacial transport and bulk diffusion on the oxidation of oleic acid by ozone, Atmos. Chem. Phys., 10, 3673–3691, https://doi.org/10.5194/acp-10-3673-2010, 2010. a, b, c, d, e
Slade, J. H. and Knopf, D. A.: Multiphase OH oxidation kinetics of organic aerosol: The role of particle phase state and relative humidity, Geophys. Res. Lett., 41, 5297–5306, 2014. a
Storn, R. and Price, K.: Differential Evolution –A Simple and Efficient Heuristic for global Optimization over Continuous Spaces, J. Global Optim., 11, 341–359, 1997. a
Stuckey, E. J., Welbourn, R. J. L., Jones, S. H., Armstrong, A. J., Wilkinson, M., Morison, J. I. L., and King, M. D.: Does gas-phase sulfur dioxide remove films of atmosphere-extracted organic material from the aqueous aerosol air–water interface?, Environ. Sci.-Atmos., 4, 1309–1321, https://doi.org/10.1039/D4EA00098F, 2024. a, b, c
Tervahattu, H., Juhanoja, J., and Kupiainen, K.: Identification of an organic coating on marine aerosol particles by TOF-SIMS, J. Geophys. Res.-Atmos., 107, ACH 18-1–ACH 18-7, 2002. a
Thompson, K. C., Rennie, A. R., King, M. D., Hardman, S. J. O., Lucas, C. O. M., Pfrang, C., Hughes, B. R., and Hughes, A. V.: Reaction of a Phospholipid Monolayer with Gas-Phase Ozone at the Air-Water Interface: Measurement of Surface Excess and Surface Pressure in Real Time, Langmuir, 26, 17295–17303, https://doi.org/10.1021/la1022714, 2010. a
Thompson, K. C., Jones, S. H., Rennie, A. R., King, M. D., Ward, A. D., Hughes, B. R., Lucas, C. O. M., Campbell, R. A., and Hughes, A. V.: Degradation and rearrangement of a lung surfactant lipid at the air-water interface during exposure to the pollutant gas ozone, Langmuir, 29, 4594–4602, 2013. a
Vieceli, J., Roeselová, M., and Tobias, D. J.: Accommodation coefficients for water vapor at the air/water interface, Chem. Phys. Lett., 393, 249–255, 2004. a
Virkkula, A., Grythe, H., Backman, J., Petäjä, T., Busetto, M., Lanconelli, C., Lupi, A., Becagli, S., Traversi, R., Severi, M., Vitale, V., Sheridan, P., and Andrews, E.: Aerosol optical properties calculated from size distributions, filter samples and absorption photometer data at Dome C, Antarctica, and their relationships with seasonal cycles of sources, Atmos. Chem. Phys., 22, 5033–5069, https://doi.org/10.5194/acp-22-5033-2022, 2022. a, b
Vongsetskul, T., Taylor, D. J. F., Zhang, J., Li, P. X., Thomas, R. K., and Penfold, J.: Interaction of a Cationic Gemini Surfactant with DNA and with Sodium Poly(styrene sulphonate) at the Air/Water Interface: A Neutron Reflectometry Study, Langmuir, 25, 4027–4035, https://doi.org/10.1021/la802816s, 2009. a
Voss, L. F., Hadad, C. M., and Allen, H. C.: Competition between atmospherically relevant fatty acid monolayers at the air/water interface., J. Phys. Chem. B, 110, 19487–19490, 2006. a
Wadia, Y., Tobias, D. J., Stafford, A., and Finlayson‐Pitts, B. J.: Real-Time Monitoring of the Kinetics and Gas-Phase Products of the Reaction of Ozone with an Unsaturated Phospholipid at the Air−Water Interface, Langmuir, 16, 9321–9330, 2000. a
Williams, J., de Reus, M., Krejci, R., Fischer, H., and Ström, J.: Application of the variability-size relationship to atmospheric aerosol studies: estimating aerosol lifetimes and ages, Atmos. Chem. Phys., 2, 133–145, https://doi.org/10.5194/acp-2-133-2002, 2002. a
Woden, B., Skoda, M. W. A., Milsom, A., Gubb, C., Maestro, A., Tellam, J., and Pfrang, C.: Ozonolysis of fatty acid monolayers at the air–water interface: organic films may persist at the surface of atmospheric aerosols, Atmos. Chem. Phys., 21, 1325–1340, https://doi.org/10.5194/acp-21-1325-2021, 2021. a
Wolff, E.: Signals of atmospheric pollution in polar snow and ice, Antarct. Sci., 2, 189–205, 1990. a
Wu, Y., Cheng, T., Gu, X., Zheng, L., Chen, H., and Xu, H.: The single scattering properties of soot aggregates with concentric core–shell spherical monomers, J. Quant. Spectrosc. Ra., 135, 9–19, https://doi.org/10.1016/j.jqsrt.2013.11.009, 2014. a
Wyslouzil, B. E., Wilemski, G., Strey, R., Heath, C. H., and Dieregsweiler, U.: Experimental evidence for internal structure in aqueous–organic nanodroplets, Phys. Chem. Chem. Phys., 8, 54–57, 2006. a
Zhou, S., Gonzalez, L., Leithead, A., Finewax, Z., Thalman, R., Vlasenko, A., Vagle, S., Miller, L. A., Li, S.-M., Bureekul, S., Furutani, H., Uematsu, M., Volkamer, R., and Abbatt, J.: Formation of gas-phase carbonyls from heterogeneous oxidation of polyunsaturated fatty acids at the air–water interface and of the sea surface microlayer, Atmos. Chem. Phys., 14, 1371–1384, https://doi.org/10.5194/acp-14-1371-2014, 2014. a