the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Nov 2025
| 18 Nov 2025
Diurnal aging of biomass burning emissions: impacts on secondary organic aerosol formation and oxidative potential
Maria P. Georgopoulou
Kalliopi Florou
Angeliki Matrali
Georgia Starida
Christos Kaltsonoudis
Athanasios Nenes
Spyros N. Pandis
Residential biomass burning is an important wintertime source of aerosols. These particles are subjected to complex diurnal aging processes in the atmosphere, contributing to urban and regional air pollution. The cumulative impact of these aging cycles on aerosol composition and oxidative potential, a key toxicity metric, remains unclear. This study examined the oxidation cycles of biomass burning emissions during day-to-night and night-to-day transitions in the FORTH (Foundation for Research and Technology – Hellas) atmospheric simulation chamber, focusing on emissions from burning of olive wood. The final high-resolution AMS spectra of biomass burning organic aerosol (bbOA) after either oxidation cycle were almost identical (R2 > 0.99, θ= 3°). This indicates transformation into similar biomass burning secondary organic aerosol (bbSOA) regardless of the initial step of the diurnal cycle. A 56 % average increase in the bbOA oxygen-to-carbon (O:C) ratio was observed during both cycle cases (from 0.38 ± 0.06 for the fresh to 0.59 ± 0.07 after aging). Additional OA mass was produced after the two cycles, varying from 35 % to 90 % of the initial OA. The aging of the emissions led to a final water-soluble oxidative potential (WS-OP) increase of 60 % to 68 ± 18 pmol min−1 µg−1 for both cycles, but with notably different transient values that depend on the order of the oxidation regimes. The effect of each oxidation regime on the WS-OP of the bbOA depends on the airmass history. The evolution of the WS-OP was not well correlated with that of the O:C.
- Article
(4090 KB) - Full-text XML
-
Supplement
(3408 KB) - BibTeX
- EndNote




Biomass burning for residential heating has significantly increased over the past two decades in several countries, primarily driven by rising energy costs and efforts to reduce the use of fossil fuels (Alper et al., 2020). Alongside contributions from wildfires, residential biomass burning has emerged as a major source of urban and regional pollution worldwide (Zauli-Sajani et al., 2024). Solid biomass currently represents nearly 45 % of the total bioenergy supply in the EU, 40 % of which is allocated to residential heating, with an anticipated 20 % increase projected by 2050 (IEA, 2019, 2021; Reid et al., 2020). This upward trend in the residential burning of solid biomass, particularly wood, has raised serious concerns regarding air quality and human health (Cincinelli et al., 2019; Guercio et al., 2021; Pardo et al., 2024).
Particles emitted from biomass burning consist of organic compounds, elemental carbon (EC), sulfates, nitrates, ammonium, and ash (Jiang et al., 2024). Biomass burning emissions also include a range of gases; carbon monoxide (Shen et al., 2020), volatile organic compounds (VOCs) such as aldehydes, ketones, and organic acids (Zhang et al., 2021; Huang et al., 2022), carcinogenic polycyclic aromatic hydrocarbons (PAHs and oxy-PAHs) (Tsiodra et al., 2021, 2024; Lim et al., 2022), as well as nitrogen oxides and ammonia (Bray et al., 2021). The emitted VOCs contribute to the formation of biomass burning secondary organic aerosol (bbSOA) and can have direct health effects (Fang et al., 2021). The emission profile of these pollutants is variable, influenced by factors such as fuel type and quality (e.g., logs vs. pellets; hardwood vs. softwood; certified vs. non-certified wood, moisture content etc.), burning conditions (e.g., flaming vs. smoldering, air/oxygen supply, and dilution), and the type of combustion appliance (Fachinger et al., 2017; Nyström et al., 2017; Price-Allison et al., 2021; Trubetskaya et al., 2021).
After their release, biomass burning emissions are subject to chemical transformations through homogeneous or heterogeneous reactions, that differ between daytime and nighttime (Donahue et al., 2012; Hodshire et al., 2019; Yazdani et al., 2023). During these reactions, a significant amount of SOA (Yazdani et al., 2023) and reactive oxygen species (ROS) (Wang et al., 2023) can be generated. Hennigan et al. (2011) reported significant variability in bbSOA formation during the photo-oxidation of different emissions. Yazdani et al. (2023) reported that after 6 to 10 h of daytime exposure, up to 30 % (with an average of 15 %) of the primary bbOA (bbPOA) mass was oxidized, forming bbSOA that was predominantly composed of acids. The coupled gas-particle partitioning, and reaction of semi-volatile vapors (SVOCs) may play an important role in the processing of bbPOA (Hennigan et al., 2011; Srivastava et al., 2022). Li et al. (2024) demonstrated that intermediate volatility species (IVOCs) can contribute approximately 70 % of the formed bbSOA, more than twice the contribution from VOCs.
The nighttime oxidation of biomass burning emissions by the nitrate radical (NO3) also leads to rapid aerosol changes (Kodros et al., 2020), but to a lesser extent compared to OH oxidation (Yazdani et al., 2023). In some cases, a doubling of bbOA levels compared to the initial primary bbOA has been observed. This increase has been attributed to gas-phase reactions between the NO3 radical and mainly phenolic compounds or furanic aldehydes (Hartikainen et al., 2018). Moreover, a substantial increase (7 %–100 %) in the aerosol oxygen-to-carbon (O:C) ratio, as well as in the mass of organic nitrates in bbOA has been reported, as result of nocturnal aging (Kiendler-Scharr et al., 2016; Kodros et al., 2022; Yazdani et al., 2023).
To date, field and atmospheric simulation chamber studies have focused on the oxidation of biomass burning emissions during either daytime or nighttime oxidation regimes, driven respectively by OH and NO3 radicals (Hennigan et al., 2011; Fry et al., 2014; Hodshire et al., 2019; Jorga et al., 2021; Kodros et al., 2022; Wang et al., 2023; Yazdani et al., 2023). While such investigations have significantly advanced our understanding of the individual effects of these oxidation regimes, they do not fully capture the real-world evolution of biomass burning aerosols, which undergo multiple repeated cycles of daytime and nighttime chemistry during their atmospheric lifetime. Studies on successive aging from daytime and nighttime cycling do exist, but have focused on the changes of the optical and chemical properties of bbOA and the gas-particle phase partitioning of semi- and intermediate-volatility organic compounds (Tiitta et al., 2016; Hartikainen et al., 2018; Cappa et al., 2020; Che et al., 2022; Desservettaz et al., 2023; Yazdani et al., 2023). These alternating oxidation regimes cause successive changes in chemical composition, reactivity, and toxicity (Li et al., 2021, 2023; Tomlin et al., 2022; He et al., 2024) that are not well understood. Consequently, the timing of atmospheric BB emissions, being released during the day or night, may also influence the chemical trajectory of BB aerosol aging and therefore affect its composition and properties, including toxicity.
Biomass burning particles are significant sources of reactive oxygen species (ROS), including free radicals (e.g., OH, RO2, ) and non-radicals (e.g., 1O2, H2O2). Upon inhalation, these species interact with biological tissues and can disrupt cellular redox balance, triggering (or propagating) oxidative stress and systemic health effects (Costabile et al., 2023). The ability of particulate matter (PM) to catalyze ROS production, known as oxidative potential (OP), is a critical metric linking aerosol exposure to health outcomes (Zhang et al., 2022; Dominutti et al., 2025). Among the various in vivo and in vitro methods developed to quantify OP (Ng et al., 2019), the abiotic dithiothreitol (DTT) assay is the most well established one, providing a measure of the water-soluble OP (WS-OP) of aerosols through the depletion of surrogate DTT in aerosol extracts (Cho et al., 2005). In this assay, DTT acts as a surrogate biological reducing agent that is oxidized by redox-active PM components. The remaining DTT is determined via a colorimetric reaction with DTNB (5,5'-dithiobis-(2-nitrobenzoic acid)), producing a light-absorbing compound measured spectrophotometrically at 412 nm. Blank-corrected depletion rates are typically normalized to aerosol mass or organic carbon content to provide a per mass health-relevant measure of WS-OP. The broad sensitivity of this method to diverse sources of ROS in aerosols with long lifetimes (Gao et al., 2020a; Rao et al., 2020), along with its optimization over the years (Fang et al., 2015; Puthussery et al., 2020) to provide more rapid measurements of water-soluble OP (WS-OP), makes it highly suitable for large-scale studies. In this study, we focus on WS-OP as a partial measure of aerosol toxicity, acknowledging that the DTT assay reflects only one aspect of oxidative potential, and its direct link to health outcomes remains uncertain. Recent studies emphasize that no single OP assay can fully represent particle toxicity and that complementary approaches (e.g., thiol- and hydroxyl radical–based assays) are needed to capture the full range of oxidative mechanisms and to strengthen links with health-relevant outcomes (Dominutti et al., 2025). Studies using the DTT assay have identified bbOA and SOA as dominant contributors to DTT activity, accounting respectively for 35 % and 30 % of total OP in ambient aerosols in the Southeastern USA (Verma et al., 2015). More recent studies confirm that biomass burning is a significant source of OP in diverse environments, highlighting the importance of understanding diurnal variations in OP from biomass burning (Paraskevopoulou et al., 2019, 2022; Mylonaki et al., 2024).
Photochemical aging during daytime oxidation promotes particle-bound ROS production, enhancing the OP of the aged aerosols (Li et al., 2021; Wang et al., 2023). For bbOA, the OP was found to increase by a factor of two (2.1 ± 0.9) after multiple days (68 h) of atmospheric aging (Wong et al., 2019). This implies that the health impacts of bbOA may extend far from its sources, as it ages and becomes part of the background aerosol (Vasilakopoulou et al., 2023; Mylonaki et al., 2024).
While it is well-established that bbOA ages rapidly at night, the effects of its nocturnal aging on aerosol OP are poorly understood. Moreover, to our knowledge no studies have yet investigated how the oxidation sequence (day-to-night and night-to-day) affects aerosol chemical composition, aging trajectory, and toxicity (i.e., evolution of OP). This study aims to address these knowledge gaps through controlled chamber experiments simulating realistic diurnal oxidation cycles. In these experiments, fresh biomass burning emissions undergo sequential aging, either through daytime oxidation followed by nighttime oxidation or the reverse. By comparing day-to-night and night-to-day sequences, we aim to elucidate the interplay of oxidation regimes on aerosol chemical evolution and OP, providing novel insights into the health impacts of diurnally aged biomass burning aerosols.
2.1 Atmospheric simulation chamber experiments
Emission aging experiments took place at the FORTH-ASC chamber facility at Patras, Greece. Figure 1 illustrates the setup used for conducting the experiments. Fresh biomass burning emissions were produced in the combustion facility beneath FORTH-ASC by a residential wood stove, fed with commercially available olive wood logs and branches. This type of hardwood is widely used as a fuel in Greece. The emissions were diluted before their injection into the smog chamber, using a custom-made dilution device that was located at the chamber inlet.
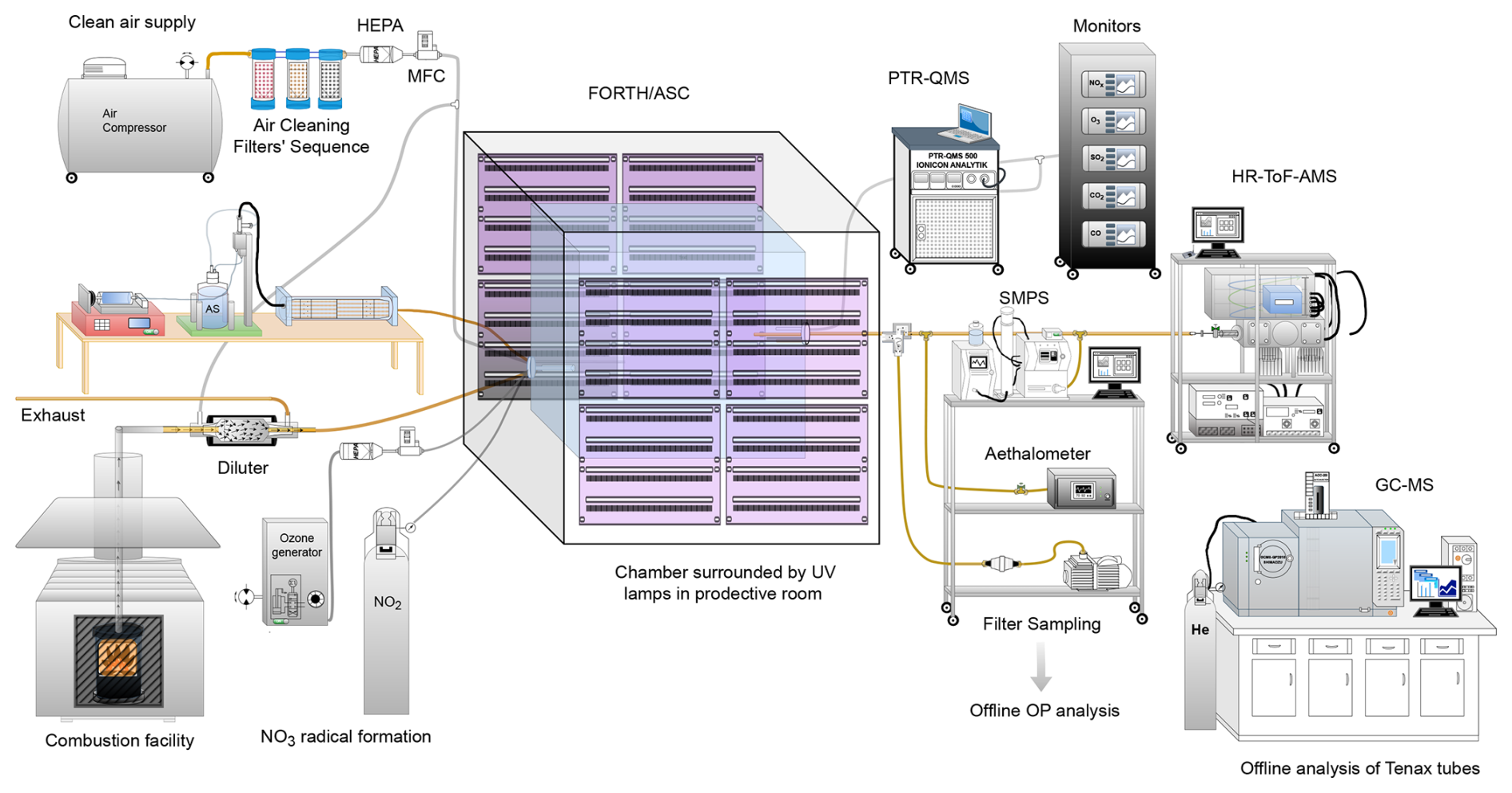
Figure 1Experimental setup of the FORTH-ASC facility, illustrating the surrounding instrumentation and the combustion facility.
The FORTH-ASC consists of 10 m3 squared Teflon chamber, located inside a 30 m3 reflective room (polished interior aluminium walls), which is temperature-regulated and equipped with ultraviolet lights (Osram, L 36W/73 UV lamps). This setup yields a maximum NO2 photo-dissociation rate coefficient (JNO2) of 0.5 min−1 when all lights are on. In this study to of the ultraviolet lights were used during photooxidation, resulting in a NO2 photo-dissociation rate coefficient (JNO2) of 0.17 to 0.33 min−1.
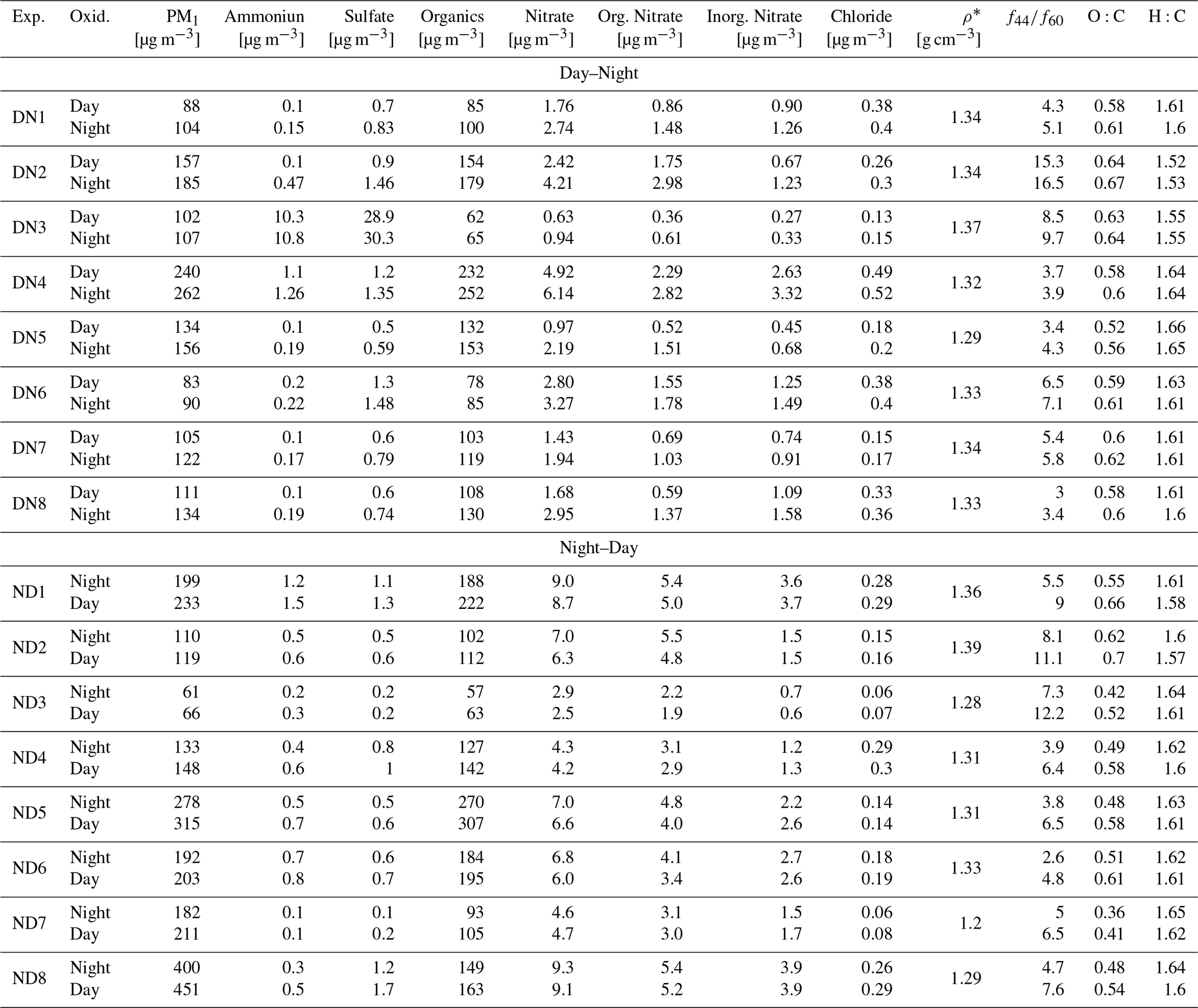
Eight day-to-night (denoted as DN) and eight night-to-day (denoted as ND) aging experiments were performed under dry (12 %–24 % RH) conditions. Because a small amount of water vapor is inherently present in biomass burning emissions, achieving extremely low RH (< 5 %) would require complete removal of this water, which would result in losses of organic vapors and particles and compromise experimental quality. The selected RH range therefore ensured stable experimental conditions and is consistent with previous chamber studies (Kodros et al., 2022; Li et al., 2023). Table 1 summarizes the initial aerosol composition and experimental conditions for all the conducted experiments. To investigate the impact of fire starter on biomass burning emissions characteristics, pine kindling mixed with olive logs was used in two of the ND experiments (ND7, ND8). Pine, which is a softwood, has chemically distinct characteristics compared to olive wood (hardwood) and is used as a kindling material because it burns quickly due to its high resin content. While all night-to-day (ND) and day-to-night (DN) experiments were conducted under the same general initial chamber conditions (temperature, relative humidity, and sampling protocol), there were the unavoidable in these chamber experiments differences in the initial aerosol and gas-phase composition, including the starting organic aerosol (OA) mass, black carbon (BC) content, and oxidant concentrations (e.g., O3, NO2) (Table 1).
Table 1Initial biomass burning aerosol composition and initial chamber conditions for all the conducted experiments. NA: not available.
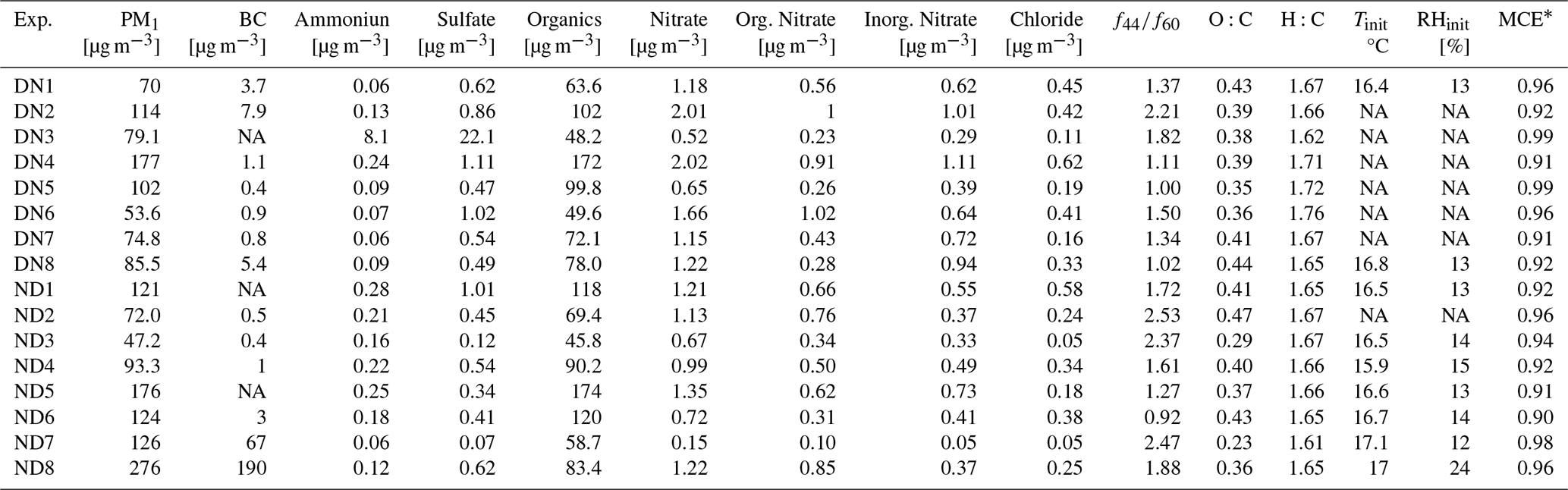
* Modified combustion efficiency (MCE) calculated based on equation: ([ΔCO2]/([ΔCO]+[ΔCO2]).
The smog chamber was first flushed with clean air overnight at a rate of 20 L min−1. Approximately 30 min after the combustion ignition in the wood stove, when flaming conditions had been achieved, a fraction of the fresh emissions was diluted with clean air (dilution ratio ranging from 1:5 to 1:10) and was injected into the chamber, which was pre-filled with clean air and regulated to the desired RH level. This resulted in additional dilution (dilution ratio ∼ 1:30) of the emissions. Two high precision mass flow controllers (Bronkhorst EL-FLOW Prestige FG-201CVP), operating at flow rates ranging from 0 to 20 L min−1, were used; one to supply clean air to the smog chamber during its filling and cleaning stages, and the other to supply clean air to the dilution system. The initial PM1 concentration achieved in the chamber was 112 ± 56 µg m−3 on average (Table 1). The fresh emissions were left to equilibrate and were characterized for about 2 h. 30–90 ppb of d9-butanol (98 %, Cambridge Isotope Laboratories) was also injected in the chamber as a tracer to determine the concentration of OH radicals (Barmet et al., 2012).
Subsequently, in DN experiments, the UV lights were turned on, initiating the daytime aging of fresh biomass burning emissions by OH radicals for at least 2 h, without the addition of further oxidants. This oxidation step was then followed by at least 2 h of aging with NO3 radicals under dark conditions. To initiate NO3 radical formation, NO2 (50–150 ppb) was first injected into the chamber and allowed to mix for approximately 10 min, followed by a 1 min O3 injection (60–280 ppb). This sequence ensured uniform NO2 distribution and prevented its immediate consumption, enabling accurate concentration control. The injected NO2 concentrations are consistent with polluted urban environments, while the O3 levels correspond to those observed during daytime pollution episodes and in the residual layer above the nocturnal boundary layer, from where they can gradually mix downward and react with NO2 to form NO3 radicals (Kodros et al., 2020). Homogeneous mixing was confirmed through real-time gas monitoring, and measurements commenced only after full mixing to minimize artifacts. During ND cycling experiments, the same oxidation steps were performed but in reverse order. In DN experiments, “time zero” was defined as the moment when the UV lights were turned on, whereas in ND experiments, it was the point at which O3 was injected.
Particle wall losses were also characterized for each experiment. After the completion of the two oxidation stages ammonium sulfate ((NH4)2SO4≥ 99 %, Sigma Aldrich) was injected into the chamber and its loss rate was monitored for at least 3 h. The dry seeds were produced by atomizing a (NH4)2SO4 solution using a TSI atomizer (model 3076) and drying the resulting droplets with a diffusion silica gel dryer (Fig. 1), as described in Wang et al. (2018). Particle wall loss corrections were applied to all aerosol data. Organic vapor wall losses were neglected over the corresponding experimental timescales. Wall losses of inorganic gases, such as NO2 and O3, were evaluated during preliminary chamber characterization and found to be minimal, typically only a few percent or less (< 5 %) over the course of the experiments.
2.2 Online instrumentation
A suite of instrumentation was used for the online characterization of both particle and gas-phase pollutants (Fig. 1). A scanning mobility particle sizer (SMPS; Classifier) model 3080; DMA, model 3081, TSI) coupled to a butanol-based condensation particle counter (CPC, model 3775 high, TSI), was used for the measurement of the number and volume size distributions (mobility diameter in the range of 13–700 nm) of the aerosol particles. The SMPS sampled every 3 min with its sheath flow rate set at 3 L min−1 and the sample flow rate at 0.6 L min−1. A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Research Inc.), working in V mode with vaporizer temperature set at 600° and sampling flow rate of approximately 0.1 L min−1, was used for monitoring the time evolution of the non-refractory organic and inorganic PM1 aerosol composition with time resolution of 3 min. Aerosol absorption and black carbon (BC) concentration were measured with a seven-wavelength aethalometer (Magee Scientific, Model AE33-7), sampling at 2 L min−1. VOCs were measured using a proton transfer reaction mass spectrometer (PTR-QMS 500, Ionicon Analytik), sampling at 0.5 L min−1. The drift tube was maintained at 2.3 mbar and operated at 600 V. A detailed explanation of the PTR-MS operational parameters and the calibration procedure using VOC standards can be found in in Kaltsonoudis et al. (2016). Concentrations of carbon monoxide (CO) and dioxide (CO2), sulfur dioxide (SO2), ozone (O3) and nitrogen oxides (NOx) were measured using the corresponding monitors; CO (Teledyne model 300E), CO2 (Teledyne model T360), (Thermo model 43i-TLE), O3 (Teledyne model 400E), NO and NO2 (Teledyne model T201). The total sampling flow rate of all monitors was 3.8 L min−1.
2.3 Online data analysis methodology
The initial combustion conditions in the chamber were characterized by calculating the modified combustion efficiency (MCE) as the ratio of the carbon dioxide (CO2) to the sum of CO2 and carbon monoxide (CO) (Yokelson et al., 1996).
The HR-ToF-AMS data were analyzed using the packages SQUIRREL (Sequential Igor data Retrieval; v1.57) and PIKA (Peak Integration by Key Analysis; v1.16) incorporated in Igor Pro software (WaveMetrics; version 6.37). The method described in Canagaratna et al. (2015) was used to estimate of elemental O:C ratio. The AMS collection efficiency (CE) and the corresponding OA density have been determined using the algorithm proposed by Kostenidou et al. (2007). This approach combines the volume distributions obtained from the SMPS and the mass distributions of the main PM1 components from the AMS. The BC concentration obtained by the aethalometer was also included in the calculation, assuming a size distribution for BC similar to that of OA.
SMPS measurements were corrected using size-dependent wall loss rate constants, estimated by monitoring the decline in the mass concentration of (NH4)2SO4 particles injected into the chamber at the end of each experiment. Practically size independent first-order wall loss rates were observed for particle diameters ranging from 60 to 700 nm. Based on this, the concentrations of the non-refractory PM1 aerosol species measured by the AMS were corrected using one experiment-specific, size-independent wall loss rate constant that was 0.15 ± 0.05 h−1 on average.
The total OA was split into primary (bbPOA) and secondary (bbSOA) following the approach proposed by Jorga et al. (2020) and applied for bbOA by Kodros et al. (2022). To quantify the variation between primary and secondary bbOA mass spectra, obtained by the AMS, the theta angle (θ) was estimated (Kostenidou et al., 2009). This angle represents the inner product of the two spectra (i.e., fresh and aged one), considered as n-dimensional vectors (n is to the number of the mass-to-charge () ratios). Theta angles less than 10° imply high similarity, while major differences between two compared spectra correspond to θ values higher than 25° (Florou et al., 2023). The approach described in Kiendler-Scharr et al. (2016) was used to quantify the particulate organic nitrate (ON). In the present study the minimum measured ratio in all experiments was 0.04. The corresponding measured ratio for pure NH4NO3, determined through calibration, was equal to 0.56.
Prior to each experiment, background VOC levels in the chamber were measured using the PTR-MS for at least 1 h. The PTR-MS was unavailable during experiments DN2–DN7. The PTR-MS measurements of the protonated VOCs were background-corrected and averaged at the end (over the last 1 h) of the fresh emissions' stabilization period, as well as at the end (over the last 1 h) of each oxidation step. The final values are summarized in Table S1 in the Supplement, along with a classification of the identified VOCs by chemical structure and functional groups.
Following the work of Barmet et al. (2012), the average OH radical concentration was estimated from the decline/reduction in the concentration of the 66 (protonated mass of d9-butanol). A d9-butanol reaction rate coefficient equal to 3.4 × 10−12 cm3 molec.−1 s−1 (at 295 K) was assumed (Allani et al., 2021).
2.4 Collection of samples for offline analysis
To investigate the WS-OP of both fresh and aged BB aerosol, as well as to measure their organic (OC) and elemental carbon (EC) content, filter samples (Whatman Tissuquartz 2500QAO-UP, 47 mm, 0.45 pore size) were collected for 1 h at the end of the emissions' equilibration period as well as at the end of each oxidation step. Prior to each experiment, blank filter samples were also collected. Sampling was conducted using a filter holder coupled with a PM2.5 cyclone positioned at the chamber exit. An external vacuum pump (Becker VT 4.10, 150 mbar), operating at a flow rate of 16.7 L min−1, was used, with its exhaust connected to a HEPA filter (Whatman 6702-9500). Prior to sampling, the quartz filters were baked at 500 °C for 10 h and left in the oven overnight, to remove any absorbed organic material. Each filter was wrapped in prebaked aluminum foil and was kept before and after sampling in sterile polystyrene petri dishes (50 mm, Pall Laboratory). After sampling all filters were stored at a temperature of −20 °C, until WS-OP and analysis.
Tenax sorbent tubes (stainless steel 3.5 × in tubes, filled with Tenax TA, Markes International) were used to collect VOC samples at specific time intervals. The custom-made sampling system used included a mass flow controller (Alicat Scientific MC-500SCCM-D/5M), the sampling tube, and a diaphragm vacuum pump (AIRPO, Model D2028B 12VDC), operating at a flow of 0.3 L min−1 for 1 to 1.5 h, resulting in total collected sample volumes ranging from 18–27 L. After sampling all sorbent tubes were capped with long-term storage brass caps containing PTFE ferrules and were stored in a freezer at −18 ° (Harshman et al., 2016).
2.5 TD-GCMS measurements
The offline determination of VOCs/IVOCs involved a two-step desorption process. The compounds adsorbed in the Tenax tubes were first desorbed using a thermal desorber (UNITY–Air Server-xr, Markes International Ltd.). During thermal desorption (TD), the sorbent tube underwent heating up to 280 °C for 10 min to release all its contents. Subsequently, the desorbed VOCs were captured using Helium (as the carrier gas) and then deposited onto a sorption cold trap at 20 °C. Subsequently, the temperature of the cold trap was gradually increased from 20 °C to 300 °C at a rate of 100 °C s−1, where it remained for 6 min. The retained analytes were then injected into a single quadrupole gas chromatograph-mass spectrometer (GSMS, Shimadzu model QP2010, with helium as carrier gas). The GC-MS system was equipped with an inert capillary column (MEGA-5MS, 30 m length, 0.25 mm inner diameter, 0.25 µm film thickness). The oven temperature of the GC column remained at 32 °C for approximately 5 min, increasing to 320 °C at 5 °C min−1. MS data acquisition was conducted in full scan mode, scanning within the range of 35 to 300 amu. After the analysis, both the Tenax tubes and the GC column were cleaned. Calibration of the system was performed using standards of specific VOCs (EPA labelled) loaded in clean tubes. The species detected by TD-GCMS for a typical experiment (DN4) are presented in Table S2.
2.6 Oxidative potential (OP) measurements
The water-soluble oxidative potential (WS-OP) of redox-active aerosol components was measured using a DTT assay system (Fig. S1) at FORTH/ICE-HT in Patras, Greece, which is based on the semi-automated method of Fang et al. (2015). A detailed description of the system components, operation, measurement protocol, and data treatment, is provided in Sect. S1. Briefly, the fresh and aged aerosol samples (1.5 cm2 punches of the collected quartz filters) are extracted, filtered, and incubated with DTT, in excess, under controlled conditions. The DTT is gradually oxidized by ROS in the sample, with its consumption rate (DTT activity, in nmol min−1) determined spectrophotometrically by measuring the absorbance of 2-nitrobenzoic acid (TNB), the derivatization product of DTT with DTNB reagent, at 412 nm at specific time intervals. The WS-OP was calculated by correcting for blank samples and was normalized to the OC mass of the sample, yielding net DTT consumption rates (mass-normalized DTT activity – DTTm) in pmol min−1 µg−1 (Table S3). OC was quantified via thermal-optical analysis (NIOSH-870 protocol), with an estimated relative standard deviation of 15 ± 5 % for replicate measurements.
3.1 Characterization of fresh olive wood emissions
Flaming conditions predominated in all experiments, as indicated by the estimated modified combustion efficiency (MCE) that ranged from 0.91 to 0.99 (Table 1) (Li et al., 2015; Briggs et al., 2016). The initial PM1 concentration of the fresh olive wood burning emissions in the chamber varied from 47 to 177 µg m−3 (considering experiments DN1-DN8 and ND1-ND6). This range of concentrations is representative of light to severe biomass burning pollution episodes in polluted urban areas during wintertime (Chen et al., 2022; Luo et al., 2022; Othman et al., 2022). The average AMS collection efficiency (CE) of the fresh emissions averaged 0.8 ± 0.2, while the mean OA density, calculated following the approach of Kostenidou et al. (2007), was 1.11 ± 0.12 g cm−3. Estimating the OA density from measured O:C and hydrogen-to-carbon (H:C) ratios, following the Kuwata et al. (2012) approach, yielded an average of 1.18 ± 0.03 g cm−3.
The fresh aerosol primarily consisted of organics (95 ± 3 %) with OA concentrations ranging from 46 up to 174 µg m−3 (Table 1). The rest of the aerosol consisted of BC (2.4 ± 2.4 %), nitrates (1.4 ± 0.7 %), sulfates (0.7 ± 0.4 %), chloride (0.4 ± 0.2 %) and ammonium (0.2 ± 0.1 %). In experiment DN3, ammonium sulfate seeds were also present explaining the higher initial sulfate (28 %) and ammonium (10 %) content.
The initial mass ratio of the organic aerosol to black carbon () ranged from 13 to 263. The differs significantly depending on the combustion conditions. When MCE values exceed 0.9, the ratio can range between 0.3 to 105 (McClure et al., 2020), with higher values indicating more efficient combustion (Novakov et al., 2005). The relatively low primary BC concentrations in experiments DN3, DN5, DN6, and ND2, despite high MCEs of 0.96–0.99, likely reflect variability in combustion conditions and fuel composition during our biomass burning experiments. Such variability can lead to differences in ratios even under high MCE conditions. Overall, our values indicate relatively efficient wood stove operation.
The average initial oxygen to carbon ratio (O:C) of the bbOA in all olive wood burning experiments was 0.39 ± 0.04. The average initial hydrogen to carbon ratio (H:C) was 1.67 ± 0.04 ranging from 1.62 to 1.76. These values are consistent with previously reported field and smog chamber O:C and H:C observations for fresh biomass burning aerosols (Ng et al., 2010; Sun et al., 2016; Lim et al., 2019; Kodros et al., 2020; He et al., 2024). The relatively low AMS ratios (1.56 ± 0.52) observed in the experiments are representative of fresh biomass burning emissions from wildfires and laboratory wood burning chamber studies (Li et al., 2023).
The average high-resolution (HR) fresh bbOA mass spectrum obtained by AMS for the olive wood burning experiments (Fig. S2a) showed predominant fragments at 29 (CHO+, C2H), 41 (C2HO+, C2H3N+, C3H), 43 (C2H3O+, C3H), 55 (C3H3O+, C4H), 57 (C3H5O+), 69 (C5H, C4H5O+) and 73 (C3H5O), suggesting a significant presence of alkenes, alkanes, and fatty acids. The observed signals at 44 (CO) and 60 (C2H4O), are typical tracer fragments for OOA and bbOA, respectively. The obtained fresh bbOA spectrum profile is quite similar to those reported in previous biomass burning chamber studies that examined wood or pellets burning (He et al., 2010; Kodros et al., 2020, 2022; Florou et al., 2023). The average theta angle θ of the fresh bbOA spectra, calculated for all possible pairs of the olive wood burning experiments in the present study, was on average 9° ± 7° (Fig. S3), indicating a generally similar composition of fresh bbOA.
Based on PTR-MS measurements, oxygen-containing compounds contributed the largest portion of the protonated VOCs identified in the fresh emissions (Fig. 2a). Aldehydes, including acetaldehyde ( 45; 12.9 ± 3.7 ppb), formaldehyde ( 31; 1.6 ± 0.7 ppb), acrolein ( 57; 3.5 ± 1.5 ppb), and hexenal ( 99; 2.1 ± 1.5 ppb), along with saturated ketones like acetone ( 59; 4.7 ± 2.0 ppb) and unsaturated ones such as ethyl vinyl ketone ( 85; 2.1 ± 1.4 ppb), contributed a total of 32.5 ppb, accounting for 19.7 % of the measured VOCs. Carboxylic acids, such as formic ( 47) and acetic ( 61) acids, averaged a total concentration of 8.2 ppb, comprising 5 % of the total VOCs (Fig. 2a). The main identified alcohol was 1-butanol ( 75), which accounted for 3 % of the VOC composition, with concentrations varying from 2.1 to 9.5 ppb across experiments (Table S1). Furans and their derivatives ( 69, 83, 113, 147) had an average concentration of 7.9 ppb, accounting for 5 % of the total measured VOCs (Fig. 2a).
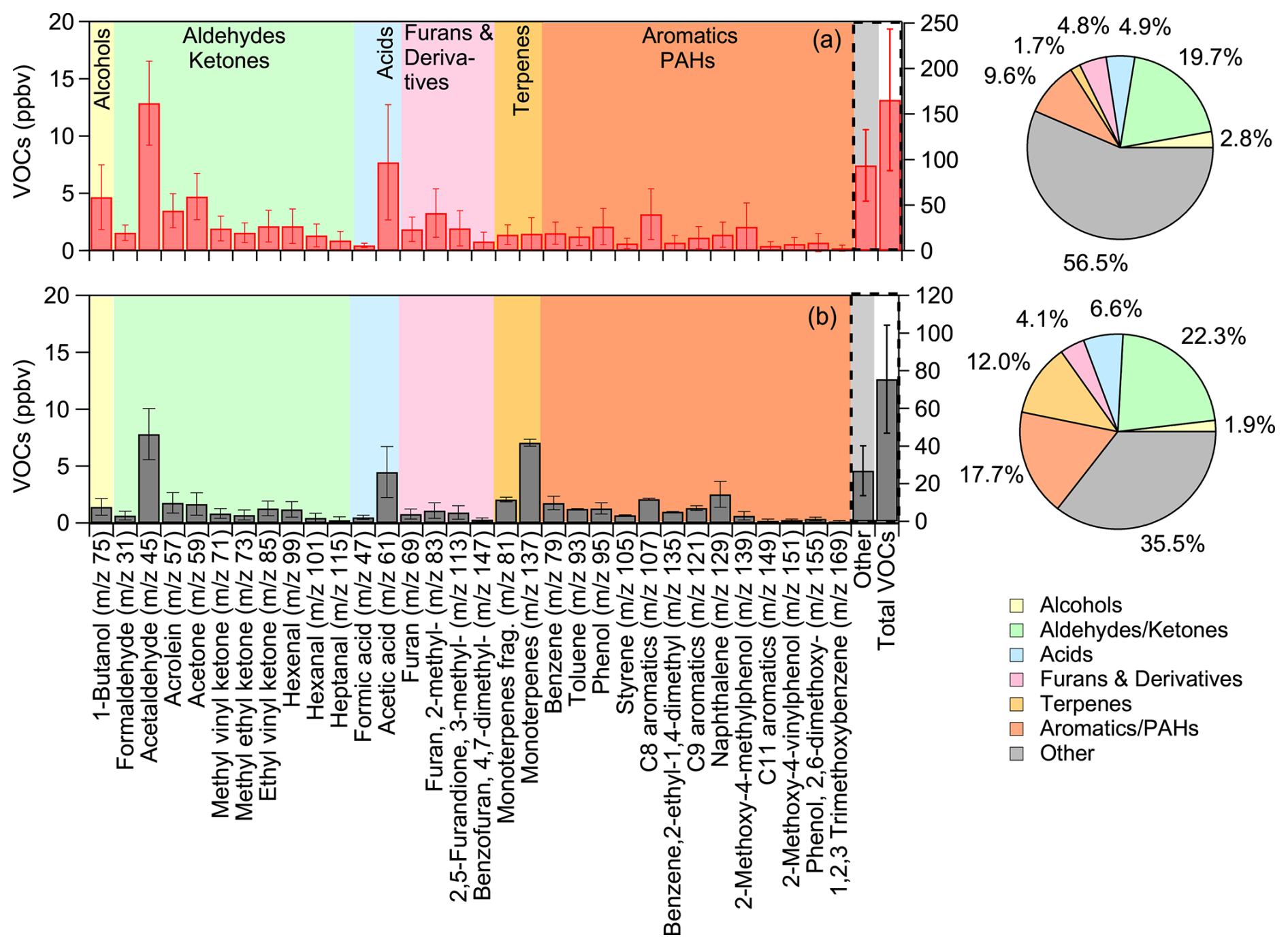
Figure 2Average concentrations, in ppb, of the identified VOCs in (a) fresh olive wood burning emissions (red bars) and (b) fresh olive-pine mixed emissions (grey bars), along with their percentage contribution to the total VOCs concentration measured by PTR-QMS. The protonated for each compound is shown in parentheses on the x axis. The left y axis shows the concentrations of identified VOCs, while the right y axis displays the concentrations of the sum of the unidentified (other) and the total measured VOCs.
Cyclic and heterocyclic aromatic compounds (with 1-ring or 2-ring structure) contributed approximately 10 % to the total VOCs. This includes benzene ( 79; 1.5 ± 1.0 ppb) and its substituted forms ( 139, 151, 155; 3.3 ppb), toluene ( 93; 1.2 ± 0.8 ppb), phenol ( 95; 2.1 ± 1.7 ppb) and its substituted forms ( 121, 135, 149, 169; 2.4 ppb in total), and C8 aromatics, including xylenes, ( 107; 3.2 ± 2.4 ppb). Other minor contributors, with varying concentrations across experiments, included terpenes and terpenoids ( 81 and 137), averaging 2.9 ppb, and naphthalene ( 129), averaging 1.4 ± 1.1 ppb. The presence of these aromatic species is corroborated by the Tenax samples, along with compounds like benzonitrile, trimethoxy- benzene, methylindene and benzofurans. For a typical sample of fresh emissions, chromatographic analysis yielded a variety of phenolic species other than phenol, with functional groups including several alkyl groups (methyl-, dimethyl-, ethyl-), but also with oxygenated functional groups (methoxy-, dimethoxy-) as presented in Table S2. Furans comprised approximately 11 % of the identifiable VOCs in the offline analysis, with the most prominent being furfural, followed by methyl-furans and methyl- furancarboxaldehyde. In terms of polyaromatic species, similarly to the PTR-QMS observations, the most abundant was naphthalene, while there were several alkyl-substituted naphthalenes present in comparable concentrations. Trace amounts of higher ring number PAHs (e.g., phenanthrene) were also observed. Most of these compounds have been previously reported in biomass burning ambient and laboratory studies (Stockwell et al., 2014; Bruns et al., 2017; Sun et al., 2019; Desservettaz et al., 2023; Florou et al., 2023).
The average WS-OP of the fresh olive wood burning aerosol was 42.9 ± 16.1 pmol min−1 µg−1, comparable to toxicity levels reported in literature for the water- and methanol-soluble portion of freshly emitted bbOA, which were also estimated using the acellular DTT assay protocol (Cao et al., 2021; Wang et al., 2023). The WS-OP values ranged from 21.2 ± 5.7 pmol min−1 µg−1 (in DN3) to 79 ± 11.3 pmol min−1 µg−1 (in DN7) (Table S3).
3.2 Effect of pine kindling on fresh olive wood emissions
In experiments ND7 and ND8, where pine kindling sticks were mixed with olive wood logs, the PM1 concentration during the characterization period was 126 and 276 µg m−3, respectively (Table 1). High amounts of BC (67 and 190 µg m−3) were produced in these experiments, constituting more than half (53 % and 69 %) of the total fresh PM1 mass. Given the efficient combustion conditions (MCE ranged from 0.96 to 0.98), these elevated BC levels were likely related to the properties of the pine (e.g., higher moisture, ash, and carbon content) compared to the olive logs (Nyström et al., 2017; Trubetskaya et al., 2021). The initial O:C of the fresh bbOA was 0.23 in experiment ND7 and 0.36 in ND8. The O:C in ND7 was the lowest of all experiments.
Comparison of the average fresh bbOA mass spectrum from olive-pine mixed emissions with that of olive logs burning (Fig. S4a) reveals significantly higher peaks at 28 (CO+; +69 %), 41 (+36 %), 44 (+40 %), and 73 (+39 %), indicating an increase in certain oxygenated organic species. Additionally, the stronger fractional signals at 91 (C7H; 104 %), and at higher masses, such as 105 (C8H; 154 %), 129 (C10H; +166 %), suggest a higher relative contribution of cyclic hydrocarbons, PAHs, and other aromatic compounds. The theta angle of the two average fresh spectra was approximately 20°, implying distinct chemical composition of olive-pine mixed emissions. For the VOCs, while most aromatic compound concentrations were lower in the mixed fuel emissions, their relative contribution to the total VOCs was higher (17.7 % vs. 9.6 % in olive wood alone), suggesting differences in pyrolysis pathways and thermal degradation mechanisms between the two wood types (Fig. 2a, b). Additionally, monoterpenes ( 137 and their fragment 81) showed a significant increase in the mixed emissions, rising from 2.9 to 9.1 ppb, highlighting the influence of pine higher terpene content on VOC composition (Fig. 2). The variations observed in aldehydes, ketones, and heavier PAHs were within the experimental uncertainty. A more detailed breakdown of the absolute and CO2-normalized VOC concentrations, including experiment-specific observations and comparisons, is provided in the Supplement (Fig. S5, Table S1).
No changes were observed in the WS-OP of the fresh olive-pine mixed emissions compared to fresh olive wood emissions. The corresponding DTTm values in experiments ND7 and ND8 were 44.7 ± 4.0 and 41.1 ± 3.4 pmol min−1 µg−1, respectively (Table S3). Similar WS-OP values (25 to 45 pmol min−1 µg−1) were reported by Wang et al. (2023) for fresh bbOA from pine combustion under smoldering conditions (MCE = 0.61). These values are comparable to the average WS-OP measured in this study for olive wood emissions (42.9 ± 16.1 pmol min−1 µg−1).
3.3 Typical day-to-night (DN) aging experiment
During a typical dry DN oxidation experiment (DN1), two hours before the start of oxidation (at t= −2 h), 70 ± 0.4 µg m−3 of fresh olive wood burning PM1 (91 % OA) were injected into the chamber along with approximately 14 ppb of O3 (Fig. 3). During the emissions equilibration period (−2 to 0 h), the average O:C was 0.43, H:C was 1.67, was 17, and the ratio was 1.37 (Table 1), and remained quite stable. The WS-OP of the fresh aerosol was estimated at 51.4 ± 4.7 pmol min−1 µg−1 (Table S3).
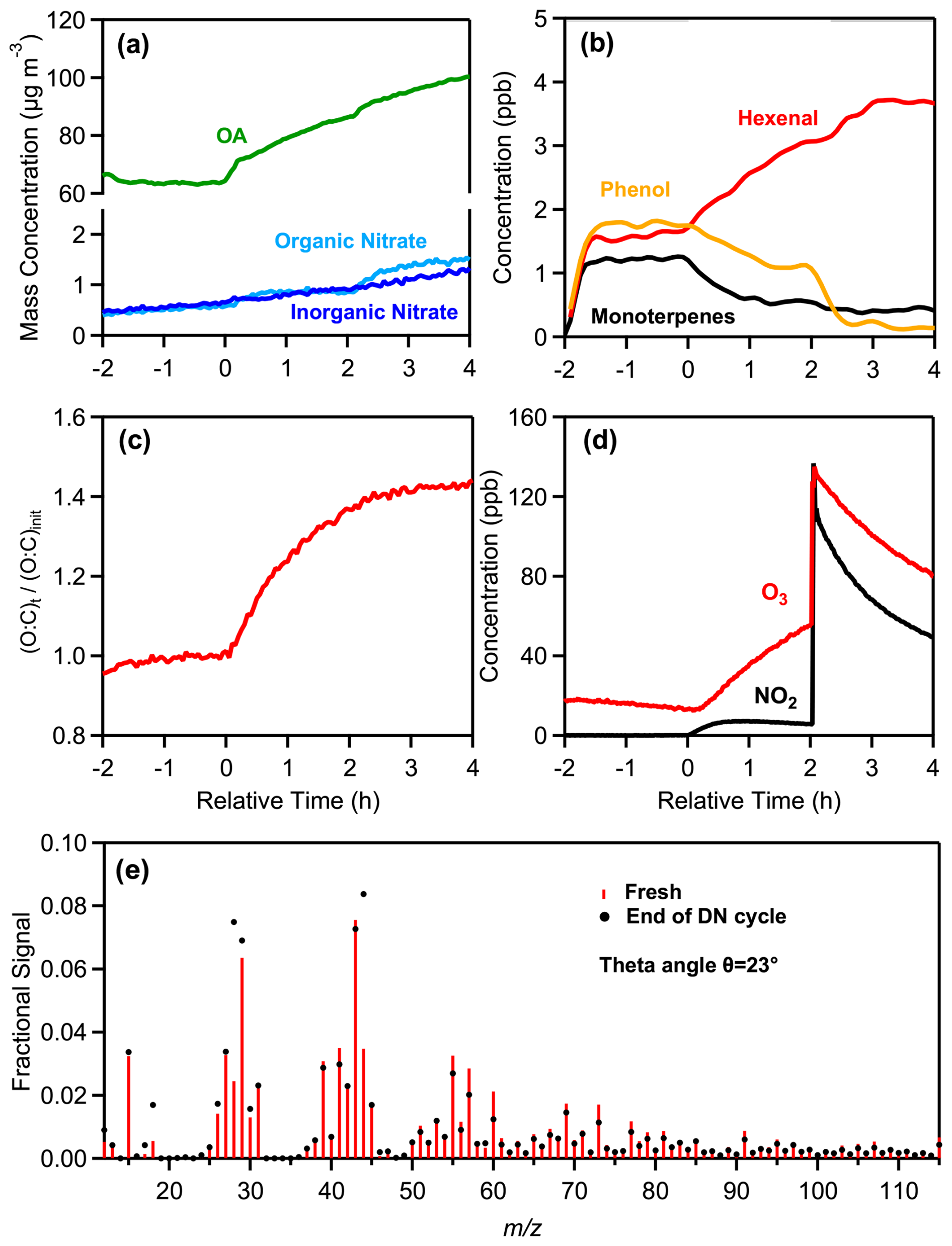
Figure 3Measurements from the experiment DN1, showing the time evolution of: (a) wall-loss-corrected organic aerosol, particulate organic and inorganic nitrate, (b) selected VOCs, including monoterpenes ( 137), hexenal ( 99), and phenol ( 95), (c) normalized O:C ratio, (d) O3 and NO2, and (e) a comparison of the fresh (red sticks) and nighttime (black markers) oxidized aerosol mass spectra at the end of the DN oxidation cycle.
At time zero (t= 0 h), daytime oxidation of the emissions was initiated by turning on the UV lights of the chamber, without adding any oxidants, and allowing the process to proceed for 2 h. Under the given experimental conditions, each hour of UV exposure in the simulation chamber corresponds to approximately 2 h of atmospheric photochemical oxidation, assuming an average OH concentration of 1.5 × 106 molec. cm−3 (Liu et al., 2018; Nault et al., 2018). In DN1, the average OH concentration during this 2 h oxidation period, estimated from the decay of d9-butanol, was 3.2 × 106 molec. cm−3, corresponding to an equivalent daytime exposure of 4.3 h. The average O3 concentration was 33 ± 14 ppb.
During this 2 h period the OA (wall loss corrected) increased by 22 µg m−3 (34 %). Organic nitrates also increased by 54 % and O3 reached 56 ppb. The H:C decreased by 4 % while the more than doubled (3 times higher). The O:C increased from 0.43 to 0.58 (35 %), consistent with previous observations (Tiitta et al., 2016). The change in the HR-AMS spectrum of the day-aged OA was modest (θ= 8°). The photochemical processing resulted in an 51% increase of WS-OP (77.6 ± 6.3 pmol min−1 µg−1) of the bbOA (Table S3). Similar increases of OP have also been reported in previous studies (Wong et al., 2019; Lei et al., 2023; Wang et al., 2023).
Furans, terpenes and cyclic aromatic hydrocarbons, major precursors of SOA production, were significantly reduced during daytime (Fig. S6). Aromatic hydrocarbons including toluene ( 93), phenol ( 95), styrene ( 105), C8 aromatics ( 107), C9 aromatics ( 121), and creosol/2-methoxy-4-methylphenol ( 139) reacted and their levels were reduced (Fig. S6b). Daytime aging also led to small changes (1 ppb or less) in the concentrations of formaldehyde, acetaldehyde, acetone, acetic acid, and heptanal which however could also be attributed to chamber background effects. According to TD-GCMS analysis, maleic anhydride was also identified at 99 in the aged emissions (Table S2).
Reaction with OH radicals was estimated to be the dominant daytime oxidation pathway for most of the examined VOC species. For methyl vinyl ketone ( 71), benzene ( 79), monoterpenes fragment ( 81), methyl furan ( 83), toluene ( 93), phenol ( 95), and C8 aromatics ( 107, assuming o-xylene), the observed reductions in concentration were close to the theoretically expected values (Table S4). Lower than predicted reductions, due to OH oxidation, were observed for furan/isoprene ( 69; 32 % less), ethyl vinyl ketone ( 85; 21 % less), styrene ( 105; ∼ 30 % less), C9 aromatics ( 121; assuming 1,2,3 trimethylbenzene; 23 % less), monoterpenes ( 137; assuming a-pinene; 58 % less), and creosol ( 139; 60 % less). This discrepancy from theoretical predictions is likely due to the presence of other compounds at the same signal, including isomers, that react more slowly. Ozone-induced oxidation was a minor consumption mechanism for most of the VOCs ( ranged from 10−17 to 10−22 molec.−1 cm3 s−1) (Table S5), with the exception of monoterpenes and their fragments ( 137 and 81).
At the end of the daytime oxidation (t= 2 h), the UV lights were turned off, and nighttime oxidation of the already aged emissions was conducted for two hours (2–4 h) by injecting additional 80 ppb of O3 and 130 ppb of NO2 into the chamber. The reaction of NO2 and O3 resulted in the decrease of their levels along with production of NO3 radical (Fig. 3d). Although the NO3 radical concentration was not directly measured in this study, it was estimated to range between 1 and 5 × 108 molec. cm−3 (typical for nighttime urban environments) based on previous dark aging experiments conducted with the same chamber setup under similar conditions (Kodros et al., 2022; Florou et al., 2023). This corresponds to approximately 4–7 h of equivalent atmospheric exposure.
Nighttime aging led to further bbSOA production, with OA increasing by 17 %, reaching 100 µg m−3 (Fig. 3a). Organic nitrate increased by 0.62 µg m−3 (72 %) and total nitrate by 0.94 µg m−3 (53 %) compared to their daytime levels. Nighttime enhancement of organic nitrate has been also reported in other studies (Kodros et al., 2020, 2022; Florou et al., 2023). The O:C ratio sightly increased from 0.58 to 0.61 (5 %). A small decrease (< 1 %) in H:C was observed, while the increased further by 19 % due to the nighttime oxidation. At the end of the DN oxidation cycle the theta angle of the aged aerosol compared to the fresh one was 23° (Fig. 3e), suggesting significant differences. The final DTTm of the aged emissions was 68.7 ± 6.0 pmol min−1 µg−1, higher by 11 % compared to the daytime measured WS-OP and 34 % higher than the fresh one (Table S3).
The most notable VOC increases were observed for formaldehyde ( 31), which increased from 1.8 to 2.2 ppb (22 %); hexenal/maleic anhydride ( 99), which increased from 2.7 to 3.7 ppb (37 %), and 2,3-benzofurandione ( 149), that increased from 0.2 to 0.3 ppb (50 %) (Fig. S6). Further decreases in the gas-phase concentrations of furan ( 69) by 0.4 ppb (61 %), methylfuran ( 95) by 0.35 ppb (26 %), phenol ( 95) by 1.1 ppb (86 %), and styrene ( 105) by 0.4 ppb (56 %), were observed (Fig. S6).
3.4 Typical night-to-day (ND) aging experiment
ND1 is as a typical night to day oxidation experiment (Fig. 4). The initial PM1 concentration injected into the chamber was 121 µg m−3, with OA contributing 97 %. The transition from fresh emissions to nighttime (0–2 h) and then daytime (2–4 h) oxidation resulted in significant changes in both the particle and gas phase. OA concentration increased by 78 µg m−3 (65 % increase) during the nighttime oxidation and by 34 µg m−3 (an additional 17 % increase) during daytime oxidation. During nighttime, total nitrate increased from 0.66 to 5.4 µg m−3, driven by production of organic nitrate. During daytime, organic nitrate levels decreased slightly (8 %) compared to nighttime. The ND cycle also led to increases in ammonium levels first by 0.9 µg m−3 (from 0.3 to 1.2 µg m−3) and then by 0.3 µg m−3 (from 1.2 to 1.5 µg m−3).
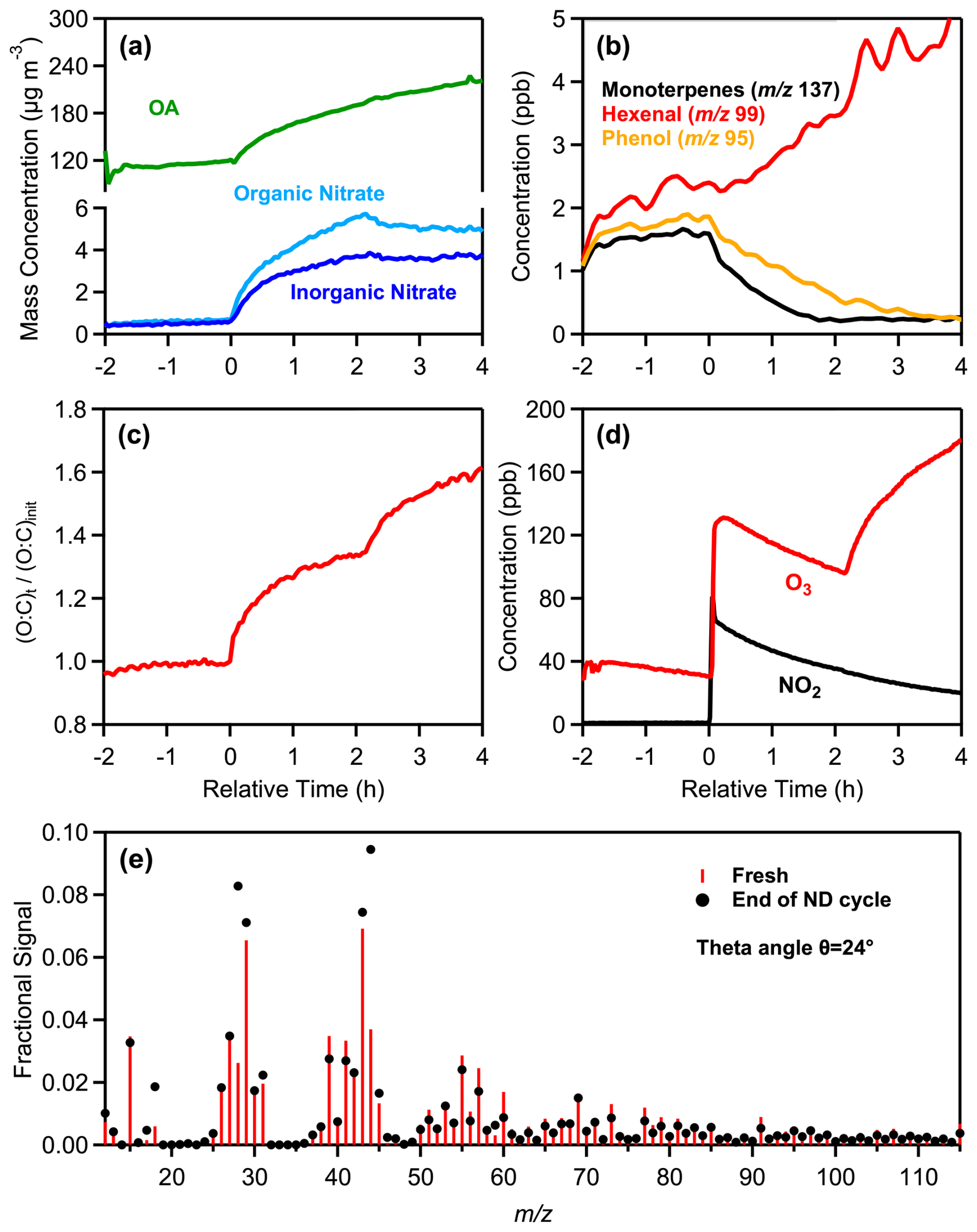
Figure 4Measurements from the experiment ND1, showing the time evolution of: (a) wall-loss-corrected organic aerosol, particulate organic and inorganic nitrate, (b) selected VOCs, including monoterpenes ( 137), hexenal ( 99), and phenol ( 95), (c) normalized O:C ratio, (d) O3 and NO2, and (e) a comparison of the fresh (red sticks) and daytime (black markers) oxidized aerosol mass spectra at the end of the ND oxidation cycle.
The theta angle between the HR-AMS spectra of fresh and night-aged OA was 13° while at the end of the ND oxidation cycle the overall change of spectrum of the aged aerosol compared to the fresh one was 24° (Fig. 4e), similar to that observed during the DN cycle. The ratio increased from 1.7 to 5.5 during night and from 5.5 to 9 during the day, while the H:C decreased from 1.67 to 1.61 and 1.58, respectively (Tables 1 and 2). The O:C increased by 34 % (from 0.41 to 0.55) at night with a further 20 % enhancement (from 0.55 to 0.66) observed after the day aging. DTTm increased from 31.8 ± 2.8 pmol min−1 µg−1 for the fresh aerosol to 42.5 ± 3.1 pmol min−1 µg−1 for night-aged aerosol (33 % increase) and to 71.0 ± 5.7 pmol min−1 µg−1 for day-aged aerosol (67 % increase) (Table S3). Unlike experiment DN1, which exhibited an initial increase (daytime) followed by a decrease (nighttime) in oxidative potential, experiment ND1 showed a monotonic increase with aging (Fig. 5).
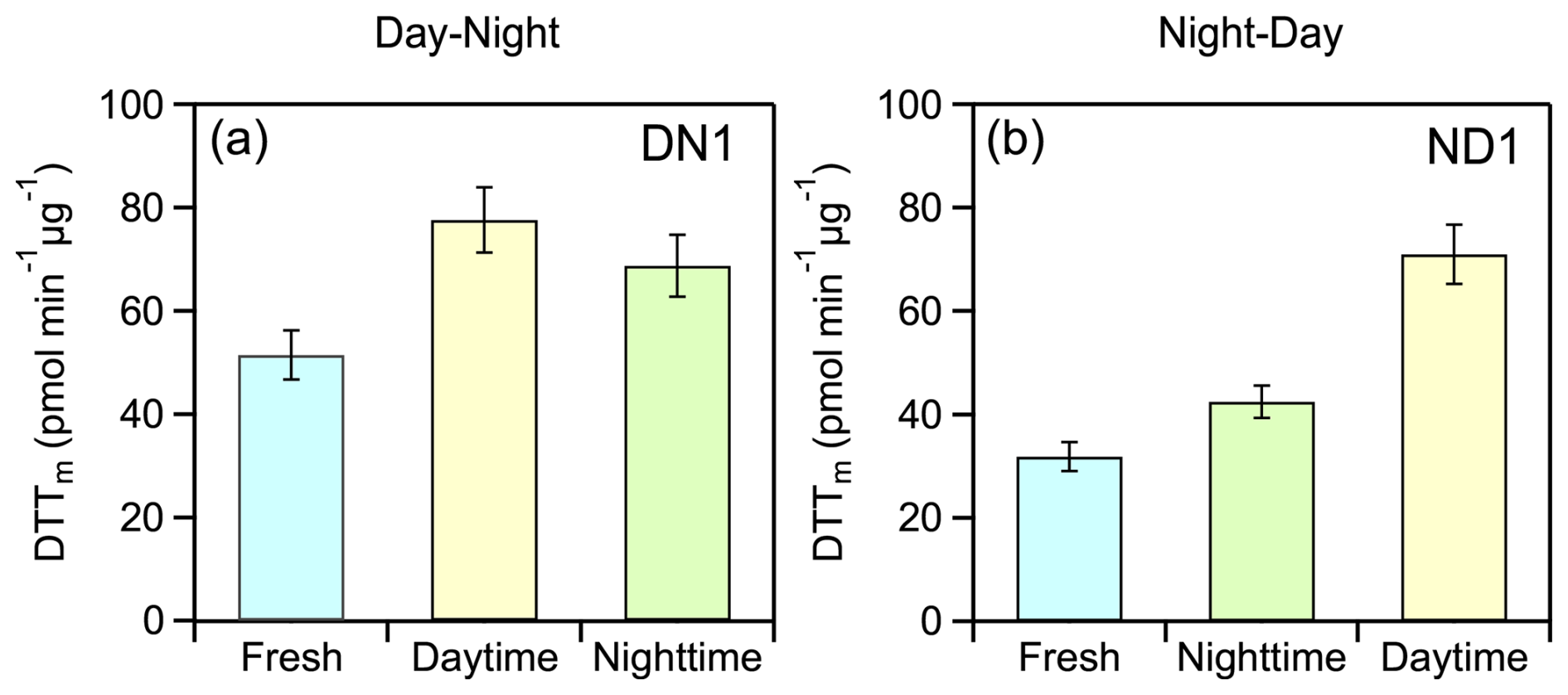
Figure 5Category plots illustrating the evolution in water-soluble oxidative potential (WS-OP), expressed as per OC mass normalized DTTm activity (pmol min−1 µg−1), in case of typical experiment (a) DN1 and (b) ND1.
During daytime oxidation, the OH concentration was 4.2 × 106 molec. cm−3, which corresponds to approximately 5.3 h of equivalent photochemical atmospheric aging. O3 levels increased by 88 ppb, rising from 96 to 184 ppb by the end of daytime oxidation.
Similar trends and concentration levels were observed for most identified VOCs in experiments ND1 and DN1 (Figs. S6a and S7a). The observed differences in the percentage reduction of key bbSOA precursors, such as aromatic compounds and furans, between DN1 and ND1 (Figs. S6b and S7b) suggest that the variability in precursor depletion dynamics is primarily influenced by differences in oxidant availability, photochemical reactivity, and the chemical composition of the emissions.
3.5 Results of all dry DN and ND experiments
The average OA production (including organic nitrate) observed in the DN (DN1-DN8) and ND (ND1-ND6) experiments at the end of a complete diurnal aging cycle was 51 ± 22 µg m−3, ranging from 19 to 136 µg m−3 (Fig. 6a). These values correspond to a total OA mass increase ranging from 35 % to 91 % compared to the fresh OA. In both oxidation cycles the majority of the produced OA was formed during the first stage of oxidation. This is consistent with the higher availability of precursor VOCs initially. No significant differences were observed in the levels of OA produced in experiments ND7 and ND8 compared to the rest (Fig. 6a). This suggests that, although the use of pine kindling resulted in a different composition of fresh wood emissions, its overall impact on SOA production was less significant compared to the influence of oxidation conditions.
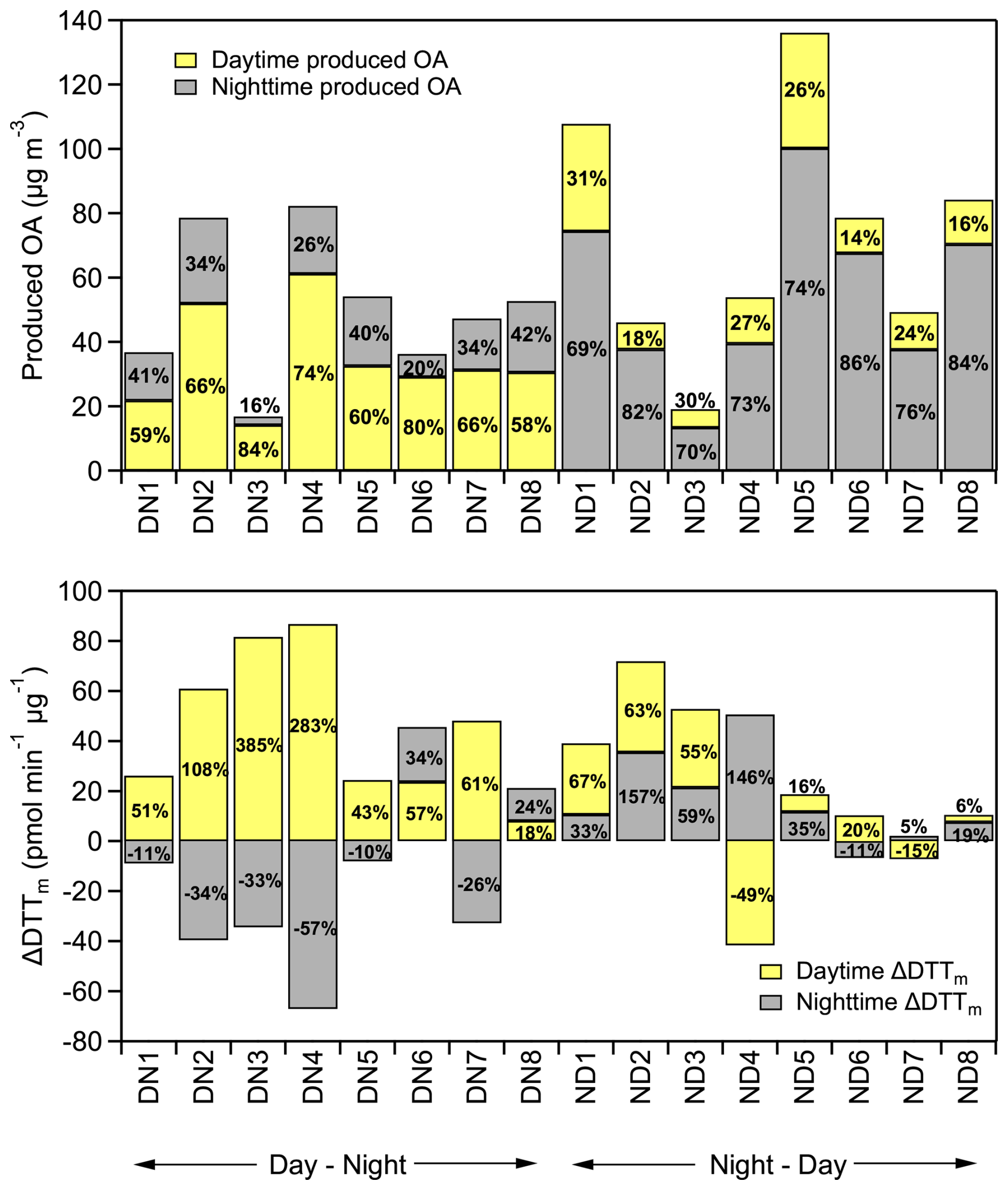
Figure 6(a) Absolute mass (in µg m−3) and percentage increase (%) of OA (including organic nitrate) per oxidation regime (daytime, nighttime); (b) absolute change in DTTm activity (ΔDTTm, in pmol min−1 µg−1) and percentage change (%) per oxidation regime (daytime, nighttime) for both DN and ND cycles, for all conducted experiments.
In some cases, the ND oxidation cycle resulted in higher SOA production (Figs. 6a and 7b). For instance, in experiment ND1, an OA mass increase of over 90 % was observed at the end of the ND cycle. Similarly, in ND5, the OA increased by 78 % compared to fresh. The enhanced SOA observed in ND1 and ND5 appears to be linked to their experimental conditions. Both experiments had high initial OA concentration in the fresh emissions (Table 1) and higher initial O3 levels (at around 30 ppb) compared to the rest of the ND experiments, which had an average level of 14 ± 2 ppb. These conditions suggest that the enhanced SOA formation in ND1 and ND5 was driven by the higher availability of precursors, oxidants, and existing OA mass, which together facilitated higher secondary production. However, statistical analysis across the full dataset did not confirm that the ND oxidation cycle generally leads to higher SOA production compared to the DN cycle.
The experiments in this study were performed under relatively low RH conditions (12 %–24 %) to minimize condensation and artifacts during sampling. While this approach enables comparison across oxidation scenarios, it does not capture the effects of higher RH and associated aerosol liquid water content, which are often significant during atmospheric nighttime aging. Aerosol water can enhance multiphase chemistry and SOA formation, and thus our results should be interpreted in the context of this limitation.
During both DN and ND oxidation cycles, the average density of the aged aerosol increased from 1.17 to 1.33 g cm−3, corresponding to approximately 13 % increase in both cases (Table 2). Similar increases in bbSOA density, in the range of 1.31–1.34 g cm−3, have been also reported in other chamber studies during dark aging (Li et al., 2015; Florou et al., 2023).
Table 2Composition of aged biomass burning aerosol, averaged over the last 30 min of each oxidation state, for both DN and ND experiments.
* Density calculated based on O:C and H:C ratios, following the approach of Kuwata et al. (2012).
At the end of the DN oxidation cycle, the average O:C was 0.61 ± 0.04, 56 % higher than the average O:C (0.39 ± 0.03) of the fresh bbOA in our experiments. Almost 90 % of this increase occured during daytime (O:C increased from 0.39 to 0.59; ΔO:C = 0.2) (Fig. 7c), while the subsequent nighttime oxidation resulted in an additional 10 % increase in O:C (from 0.59 to 0.61). For the ND cycle, the O:C increased from 0.40 ± 0.06 for the fresh emissions to 0.61 ± 0.06 (a 54 % increase) at the end of the cycle (Fig. 7d). In this case, the contributions of the nighttime and daytime oxidation stages to the increase in O:C were almost equal, at 55 % and 45 %, respectively. In both oxidation cycles the final O:C is similar, but the importance of each oxidation stage depends on the order (oxidation sequence).
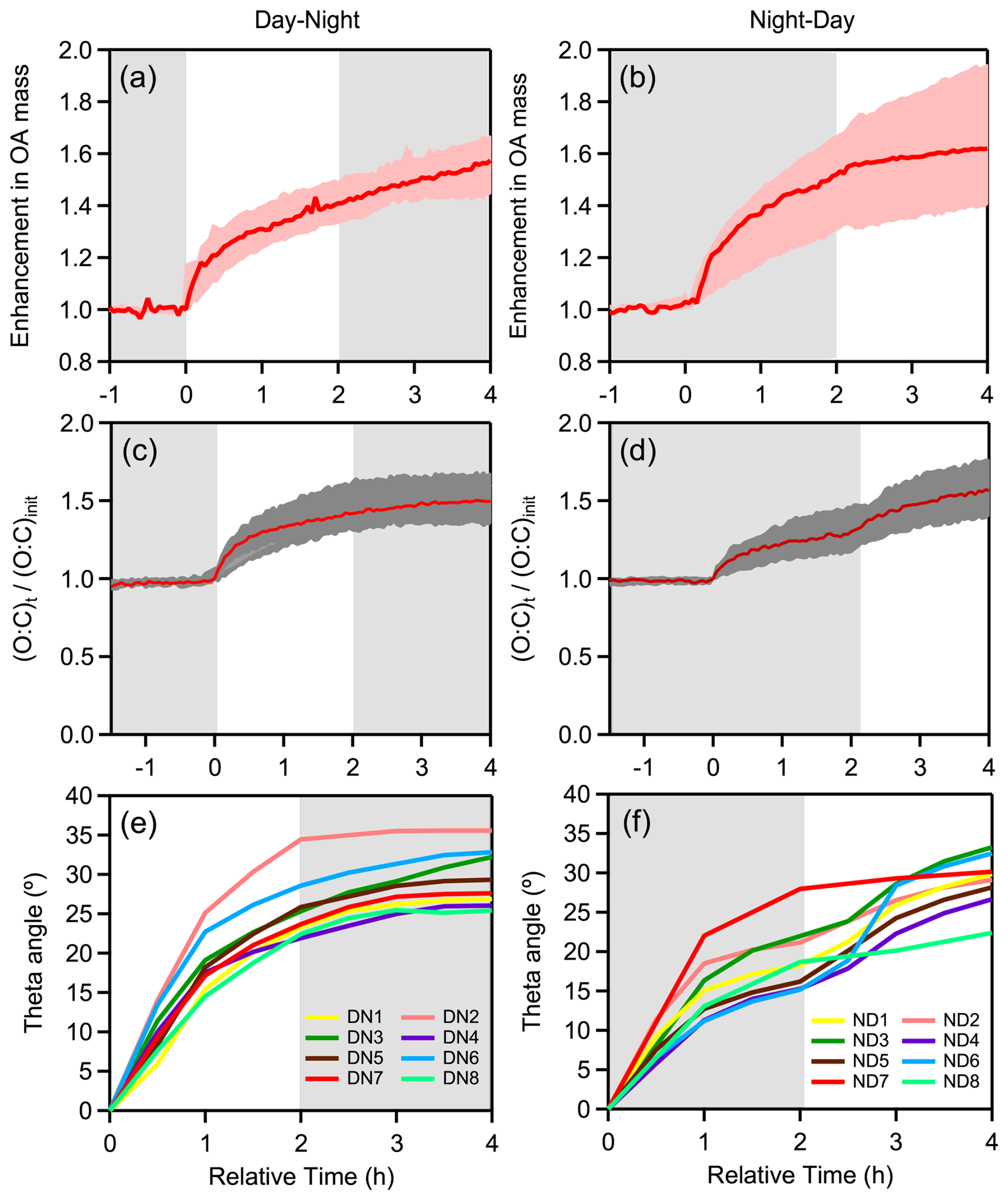
Figure 7Evolution over time of: OA enhancement during (a) DN and (b) ND oxidation cycle; O:C ratio enhancement during (c) DN and (d) ND oxidation cycle; theta angle during (e) DN and (f) ND oxidation for experiments conducted under dry initial conditions using only olive wood logs as burning fuel (DN1-DN8, ND1-ND6). In experiments ND3, ND4, and ND6, the change in spectrum occurred slightly later, as the first-step oxidation extended to 3 h compared to 2 h lasted in the other ND experiments.
In all experiments, the OA AMS spectra changed progressively with aging. The predominant differences between the average fresh and aged bbSOA spectra at the end of DN cycle were found for 28 (more than 2-fold increase) and 44 (1.5-fold increase) (Fig. S2). Significant decreases were observed for 60 (37 %) and 91 (36 %), 115 (38 %) and 137 (42 %). The same changes were observed comparing the fresh and the ND aged bbSOA (Fig. S2). During the DN cycles the main changes in the OA spectrum occurred during the first (daytime) oxidation regime, with a theta angle of 26 ± 4° on average (Fig. 7e). The further change in the second step (nighttime) was 4 ± 2° on average. In contrast, a more balanced change was observed in the evolution of the theta angle over time during the ND cycle (Fig. 7f). The average OA spectrum shifted by 19 ± 4° on average during nighttime, followed by an additional 10° shift during UV exposure. Overall, at the end of both cycles, regardless of the followed oxidation path, the final average bbSOA spectra were almost identical (θ< 3°) (Fig. 8).
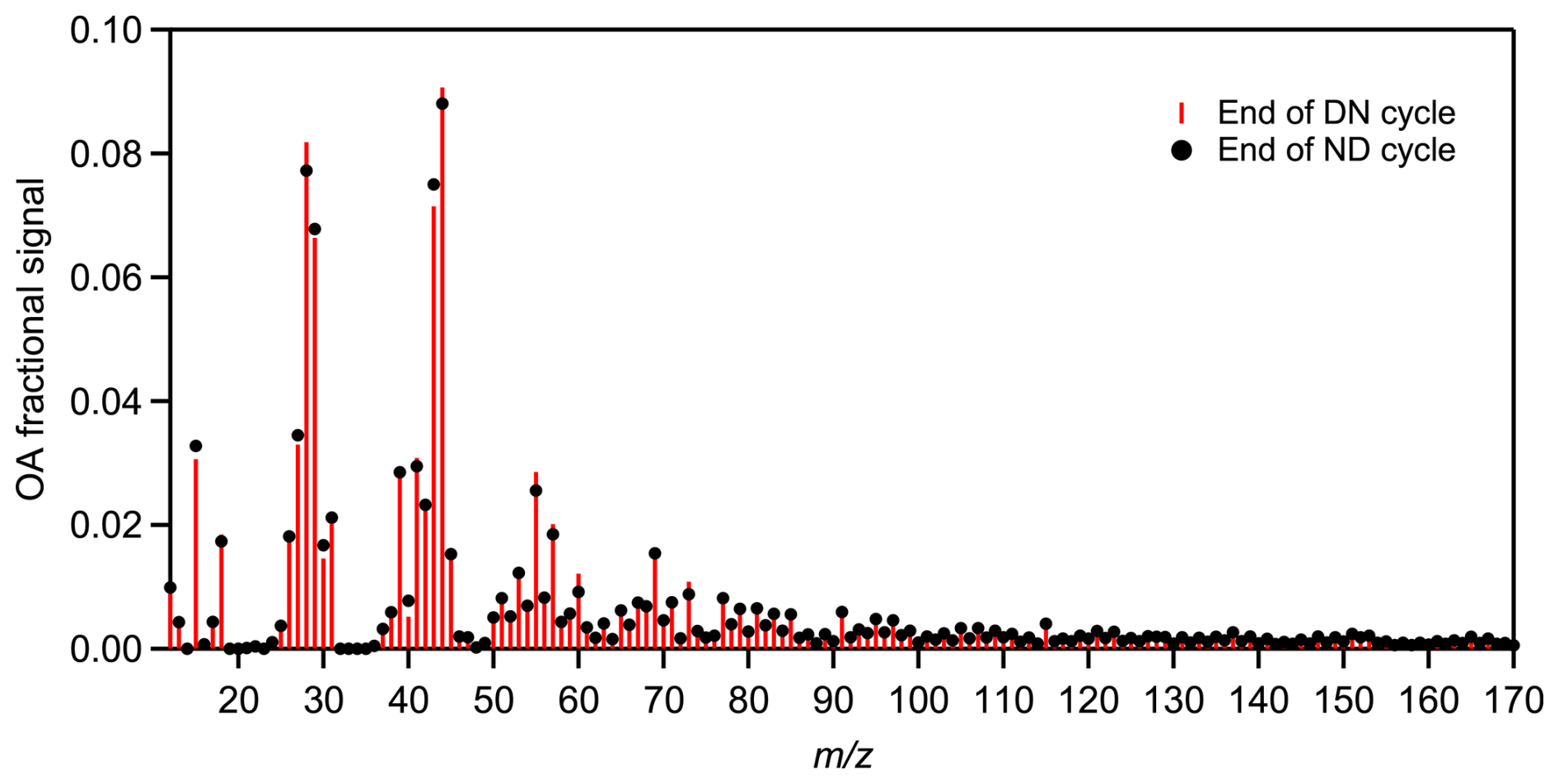
Figure 8Relative differences in the average spectra obtained at the end of DN (red sticks) and ND (black circles) oxidation cycle, respectively, for experiments conducted using olive wood logs as burning fuel. The theta angle between the averaged DN and ND aged spectra was 3° (identical).
To evaluate the environmental relevance of the chamber-produced bbSOA, the final DN and ND bbSOA spectra from this study were compared to the spectra of oxidized OA, that was measured at a remote site in Greece (Pertouli) during the summer of 2022 (Vasilakopoulou et al., 2023). Most of this aged OA was aged emissions of wildfires from different regions of Europe. Two oxygenated OA (OOA) factors; a more-oxidized OOA (MO-OOA) and a less-oxidized OOA (LO-OOA) were needed to reproduce the observed OA spectra. Our final bbSOA spectra showed greater similarity to the LO-OOA factor, with a theta angle of approximately 16°, and were more distinct (θ at around 30°) from the MO-OOA spectra measured in Pertouli. This suggests that our experiments simulated the earlier stages of atmospheric aging, while additional aging processes likely occur under ambient conditions (see also Fig. S8).
Changes in VOC levels of aged emissions across all DN (Fig. S9) and ND (Fig. S10) experiments were consistent with those observed in the typical experiments DN1 and ND1. Both aging cycles resulted in a significant decrease in the concentration of furans and their derivatives, cyclic and polycyclic aromatic hydrocarbons and terpenes. The day aged Tenax samples indicated a moderate decrease in aromatic species like toluene (∼ 20 %) and benzene, which is consistent with their lower reaction rates compared to higher carbon number aromatics. Rapid decrease in concentration was noted for species like phenol (∼ 45 %) and furfural (∼ 75 %), as well as their structurally related compounds. Related products, including 2-nitro-phenol, 4-methyl-2-nitro-phenol, maleic anhydride, and 3-methyl-2,5-furandione, were also detected. p-Benzoquinone was also formed, possibly as a result of the reacted aromatics. Benzofuran was absent from the aged samples; instead, 2,3-benzofurandione was detected. At the same time a progressive increase in aldehydes and ketones was observed, along with significant increases in carboxylic acids, such as formic ( 47) and acetic ( 61). The benzaldehyde concentration increased, accompanied by the formation of benzeneacetaldehyde, 2-hydroxy-benzaldehyde, 3-ethyl-benzaldehyde. A notable increase in butanol was also observed in the Tenax samples, along with the formation of straight-chain aldehydes (hexanal to undecanal). The GC-MS measurements for the night-aged samples following daytime processing were consistent with those of the PTR-MS. Furfural was no longer detected, while a further decrease in phenol and increases in benzaldehyde and butanol were noted. A cumulative depiction of the experiment's progression in terms of oxidation and VOCs detected by the GC-MS, is provided in Fig. S11. Similar results were obtained for the other experiments.
3.6 Effect of DN and ND oxidation cycle on WS-OP
The water-soluble oxidative potential (WS-OP) of fresh emissions ranged from 21 to 79 pmol min−1 µg−1 and that of aged wood-burning emissions from 39 to 127 pmol min−1 µg−1 (Table S3). These values fall within the range reported in literature for fresh bbOA and aged bbOA (Verma et al., 2015; Tuet et al., 2017; Bates et al., 2019; Daellenbach et al., 2020; Wang et al., 2023).
Figure 6b presents the absolute (ΔDTTm, pmol min−1 µg−1) and percent changes in DTTm activity for each experiment. In all DN experiments, WS-OP followed a consistent pattern of increasing during daytime oxidation and decreasing during subsequent nighttime aging, except for DN6 and DN8, where it continued to rise, suggesting experiment-specific chemical variability. Similarly, in the ND experiments, WS-OP generally increased during both nighttime and daytime oxidation, except for ND4 and ND7, which exhibited a reduction during the second daytime step, indicating that the extent of oxidative enhancement can depend on the initial emission composition and oxidation conditions.
The evolution of average WS-OP of fresh and aged emissions, considering all experiments and both oxidation cycles, (Fig. 9) was similar to that observed in experiments DN1 and DN1 (Fig. 5). The average WS-OP values for the DN cycle were 47.9 ± 17.7 pmol min−1 µg−1 for fresh emissions, 93 ± 27 pmol min−1 µg−1 for daytime-aged emissions, representing a 94 % increase compared to fresh aerosol, and 73.4 ± 13.3 pmol min−1 µg−1 for nighttime-aged emissions, indicating a 21 % reduction compared to daytime-aged WS-OP (Fig. 9a). For the ND oxidation cycle, the average WS-OP of the fresh emissions were 37.8 ± 10.6 pmol min−1 µg−1. After nighttime aging, it increased by 44 % to 54.4 ± 13.6 pmol min−1 µg−1, and following daytime aging, it further increased by 62.9 ± 20.4 pmol min−1 µg−1 (Fig. 9b).
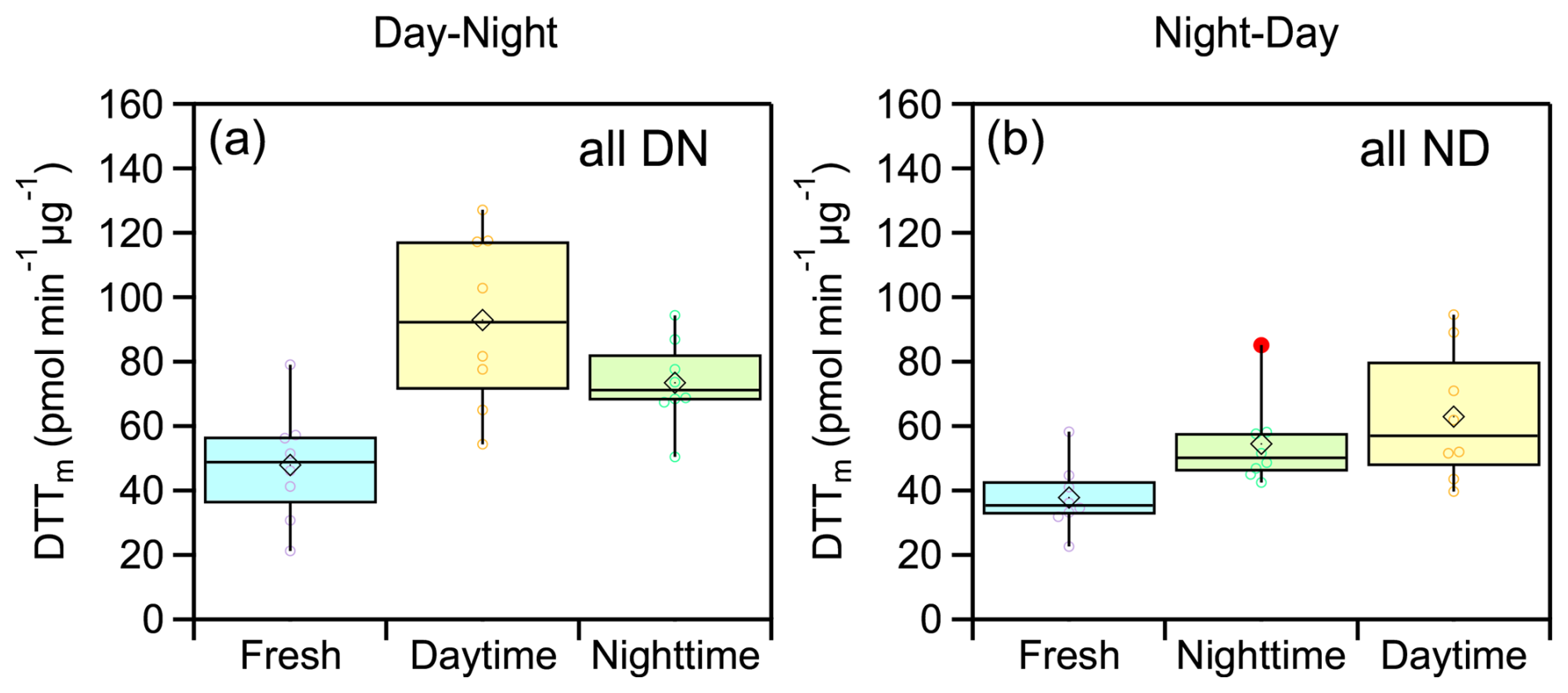
Figure 9Box plots illustrating the changes in WS-OP, expressed as per OC mass normalized DTTm activity (pmol min−1 µg−1), considering all performed experiments, in case of (a) DN oxidation cycle and (b) ND oxidation cycle.
Statistical analysis (t-test) showed that aged WS-OP values were significantly higher than those of fresh emissions in all experiments, for both oxidation cycles. Additionally, for the DN oxidation cycle, a statistically significant difference was observed between the WS-OP of nighttime-aged (NO3-oxidized) and daytime-aged (UV-oxidized) emissions. Further details on the statistical analysis are provided in Sect. S4 and Table S9.
The overall increase in WS-OP at the end of the two oxidation cycles was 53 ± 34 % for the DN cycle and 66 ± 8 % for the ND cycle, indicating that both daytime and nighttime aging of biomass burning emissions consistently enhanced their oxidative potential. Our results suggest that the sequence of chemical processes – whether the emissions are first oxidized by OH or NO3 – can significantly affect the temporal evolution of OP. This, in turn, may also influence the health impacts associated with exposure to biomass burning plumes, depending on the time of day when the emissions occur. Although daytime boundary layer dynamics generally favour mixing and dilution of pollutants, daytime burning in urban environments may actually be as or more aggravating than nighttime burning, owing to the enhanced oxidative processing of the emissions occurring in the former stage of the diurnal cycle.
The correlation of WS-OP with produced OA and degree of oxidation (O:C) were also investigated. Three OA types were considered (fresh, day-aged, and night-aged). WS-OP was not well correlated with either the O:C ratio (R2 < 30 %) of the organic aerosol or its fresh and aged fractions (R2 up to 34 %) (Fig. S12). This implies that the link between bbOA aging, and WS-OP change is complex and cannot be just described by one variable. Weak correlations (R2< 0.3) between WS-OP (both fresh and aged) and most of the rest of the aerosol components (Tables 1 and 2) were also observed (Figs. S13–S17). An exception was the nighttime WS-OP, which showed a stronger correlation with inorganic nitrate (R2 > 0.3) in both oxidation cycles. In addition, after one complete diurnal cycle, WS-OP showed a stronger correlation with OC, with R2 values reaching up to 0.65 (DN cycle) and 0.53 (ND cycle) (Figs. S15 and S17), highlighting the growing influence of organic components. These results point to a complex interplay of chemical processes governing WS-OP variability.
It is important to note that our study focused exclusively on the water-soluble fraction of OP and thus does not capture contributions from relatively water insoluble components (e.g., brown carbon and metals), which can significantly influence total oxidative properties (Gao et al., 2020b). Atmospheric aging usually tends to increase OA solubility through oxidative functionalization, which may move compounds with high OP to the aqueous phase during the extraction. Previous field observations support this hypothesis. Wong et al. (2019) reported that the OP of bbOA increased during the first day of atmospheric transport, even as OC and water soluble organic carbon (WSOC) concentrations decreased, indicating that aging removes less reactive compounds while producing more DTT-active compounds. The majority of this activity (around 80 %) was associated with the water-soluble fraction, highlighting its dominant role in driving the oxidative properties of aged bbOA. These findings emphasize the crucial role of water-soluble aerosol components in driving OP, while also underscoring the need for studies that simultaneously capture both soluble and insoluble contributions.
The observed WS-OP trends could be also linked to the VOC composition and oxidation processes in the DN and ND cycles. The WS-OP of fresh emissions exhibited positive correlations (0.2 < R2 < 0.6) with several VOCs, including aldehydes ( 45, 57), benzene ( 79), monoterpenes ( 81), toluene ( 93), phenol ( 95) and its substituted forms ( 121, 135), styrene ( 105), xylenes ( 107), and naphthalene ( 129) (Fig. S13). Among these, the strongest correlations were observed for benzene (R2= 0.56), phenol (R2= 0.56), and styrene (R2= 0.56), suggesting that the products of aromatic hydrocarbons and phenolic compounds may contribute significantly to the WS-OP of fresh bbOA. The DN cycle exhibits a high daytime WS-OP due to the OH oxidation of VOCs such as furans, aromatics, and phenolic compounds, leading to the formation of reactive species like, 4-methyl-2-nitrophenol, and highly reactive p-benzoquinone. In contrast, the ND cycle shows a gradual increase in WS-OP, with the exception of experiments ND4 and ND7 (Fig. 6b). These contrasting trends likely reflect differences in the subsequent aging of oxidation products. In the DN case, the decrease in intrinsic DTT activity during nighttime oxidation may result from the formation of compounds with lower OP (less redox-active), lower solubility, or both, when daytime OH oxidation products are further processed by NO3 radicals. In some specific cases (DN6, DN8; Fig. 6b), WS-OP continued to increase at night, suggesting potential ongoing nighttime formation of soluble or redox-active compounds under certain oxidation conditions. Conversely, in the ND case, the reverse sequence of reactions appears to generate products with higher OP or solubility or both. Importantly, this divergence does not seem to be controlled by the bulk O:C ratio, but rather by differences in chemical structure of the later-generation products. For example, after nighttime oxidation in the ND cycle the WS-OP was positively correlated with formic acid ( 47; R2= 0.24), toluene ( 93; R2= 0.28), naphthalene ( 129; R2= 0.25) and substituted forms of phenol ( 121, 135; R2= 0.25) and benzene ( 151, 155; R2 up to 0.2) (Fig. S18). After further daytime oxidation WS-OP was only correlated with 121 (trimethylbenzene; R2= 0.20) (Fig. S19). Oxidized derivatives of phenol and benzene formed during atmospheric aging tend to exhibit higher water solubility than their parent compounds, as the functionalization increases molecular polarity and hydrogen-bonding capacity. More detailed work is needed to identify these later generation products and to quantify both their OP and water solubility.
After one complete diurnal cycle, WS-OP values in both cycles converge, indicating that oxidative processes in both pathways ultimately lead to similar levels of oxidation products. This convergence highlights the role of both fast and slow oxidation mechanisms in determining aerosol OP and suggests that even VOCs with lower reactivity can significantly contribute to aerosol toxicity over extended atmospheric aging.
This study investigated how different diurnal oxidation sequences – daytime-first (DN) and nighttime-first (ND) – affect the formation of OA, the gas-phase composition, and the oxidative potential of emissions produced by burning olive wood and olive wood mixed with pine throughout a complete diurnal aging cycle. Both DN and ND oxidation cycles resulted in enhancement in OA levels by 35 %–90 %, demonstrating efficient SOA production under both OH- and NO3-dominated oxidation conditions. The mixture of olive wood with pine kindling resulted in a different composition of fresh emissions, however its overall impact on SOA production was less significant compared to the influence of oxidation conditions.
The DN cycle favoured rapid daytime oxidation, producing highly oxygenated species and increasing the O:C ratio of the fresh emissions from 0.39 ± 0.04 to 0.59 ± 0.04 during daytime, reaching finally at 0.61 ± 0.03 during nighttime. The ND cycle showed a gradual (two-steps) oxidation increase with a similar final O:C ratio of 0.61 ± 0.06. DN cycle exhibited rapid spectral changes during daytime oxidation, while ND cycles showed a more balanced two-step evolution. The convergence of O:C ratios after one diurnal cycle imply that multiple oxidation pathways can yield comparable end products through distinct chemical routes. At the end of both cycles, the final average bbSOA spectra were nearly identical (R2 > 0.99; θ < 3°), indicating that the aerosol was transformed into similar aged OA regardless of the initial oxidation step (daytime or nighttime) at the start of the cycle. The chamber-produced bbSOA resembled the less-oxidized OOA in a field campaign in Greece, with the corresponding OA dominated by aged bbOA, suggesting that the present study has addressed only part of the aging that occurs in the atmosphere.
Both the DN and ND oxidation cycles effectively reduced the concentration of bbSOA precursors (e.g., furans, aromatic hydrocarbons, terpenes). Concurrently, a progressive increase in aldehydes and ketones was observed in both cycles, alongside increases in carboxylic acids, such as formic and acetic acids. The higher daytime WS-OP in the DN cycle likely results from rapid OH oxidation of reactive VOCs (e.g., furans, aromatics, phenolics), producing redox-active species such as nitrophenols and quinones. The subsequent nighttime decrease in DTT activity suggests formation of less soluble, less redox-active products. In contrast, the ND cycle accumulated moderately oxidized intermediates during nighttime NO3 oxidation, which were further transformed into more soluble, higher-OP compounds during daytime processing. Thus, variations in molecular structure and solubility, rather than bulk oxidation state (O:C), primarily drive differences in WS-OP.
From an atmospheric perspective, these findings imply that emissions released at different times of day may contribute differently to local air toxicity, depending on oxidant availability and photochemical activity. For example, daytime burning in urban or suburban environments may enhance the formation of ROS-generating compounds more rapidly than nighttime burning, even if total SOA mass is comparable. This temporal dependence suggests that biomass burning – related health impacts effects may vary with exposure timing, a factor rarely considered in air quality or exposure models. The DN cycle resulted in 53 ± 34 % increase in WS-OP of aerosol while the ND cycle showed a slightly higher increase of 66 ± 8 %. The final WS-OP values of the DN (73 ± 14 pmol min−1 µg−1) and ND (63 ± 20 pmol min−1 µg−1) cycles were statistically similar. This convergence suggests a self-organizing tendency in atmospheric aging toward similar chemical and toxicological endpoints.
Although this study focused on water-soluble OP, insoluble components such as transition metals and brown carbon (not assessed here) can also contribute significantly to total aerosol reactivity. Oxidative functionalization during atmospheric aging generally increases OA solubility, which may shift OP from the insoluble to the soluble phase, either altering or maintaining total OP, depending on chemical composition. Consequently, measuring only the water-soluble fraction likely underestimates the total oxidative capacity of aged bbOA. Future studies combining complementary assays (e.g., ascorbic acid or AA and glutathione or GSH) and phase-resolved analyses are necessary to better constrain the contributions of soluble and insoluble components to overall aerosol toxicity.
A few additional limitations of the present work must be also acknowledged. The experiments were performed at relatively low relative humidity (12 %–24 %) to reduce condensation artifacts, and therefore the potential role of multiphase chemistry enhancement and promotion of SOA formation was not investigated. Furthermore, the aging period examined here corresponds approximately to a single diurnal cycle, representing only the first stages of atmospheric processing, whereas ambient bbOA typically undergoes multi-day oxidation cycles that may further modify its composition and OP. In addition, the controlled combustion setup, while suitable and effective for mechanistic interpretation, inevitably simplifies real-world burning conditions, where variations in fuel moisture, temperature gradients, meteorology, and interactions with co-emitted anthropogenic pollutants play important roles in shaping emissions chemistry.
Despite these constraints, the present study provides a mechanistic framework for understanding how diurnal oxidation sequences regulate the chemical and redox activity evolution of bbOA. The findings underscore the importance of considering oxidation sequences and timing when assessing the environmental fate and health impacts of biomass burning emissions. They highlight the complex and dynamic nature of atmospheric aging processes and emphasize the need for time-resolved approaches to better predict the transformation and OP evolution of bbOA under realistic atmospheric conditions.
The data used in this study are available from the authors upon request (spyros@chemeng.upatras.gr).
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-15835-2025-supplement.
MPG, KF, and AM contributed to investigation, conducted the experiments and performed the laboratory measurements. MPG and GS performed the offline measurement of the water-soluble oxidative potential of the collected aerosol samples. AM performed the offline TD-GCMS analysis of the Tenax samples. CK contributed to chamber set-up optimization. AN conceived and supported the research project. SNP supported and directed this research. MPG and KF interpreted the results and contributed to formal data analysis. MPG wrote the original manuscript with contributions from all co-authors. All authors contributed to the review and editing of the manuscript and have approved the final submitted version.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This work was supported by the project NANOSOMs (grant no. 11504) of the Greek HFRI, the EU Horizon 2020 project REMEDIA (grant agreement no. 874753), and the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement no. 726165, PyroTRACH – Pyrogenic Transformations Affecting Climate and Health).
The article processing charges for this open-access publication were covered by EPFL.
This paper was edited by Mingjin Tang and reviewed by three anonymous referees.
Allani, A., Bedjanian, Y., Papanastasiou, D. K., and Romanias, M. N.: Reaction rate coefficient of OH radicals with d9-butanol as a function of temperature, ACS Omega, 6, 18123–18134, https://doi.org/10.1021/acsomega.1c01942, 2021.
Alper, K., Tekin, K., Karagöz, S., and Ragauskas, A. J.: Sustainable energy and fuels from biomass: a review focusing on hydrothermal biomass processing, Sustain. Energy Fuels, 4, 4390–4414, https://doi.org/10.1039/d0se00784f, 2020.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Bates, J. T., Fang, T., Verma, V., Zeng, L., Weber, R. J., Tolbert, P. E., Abrams, J. Y., Sarnat, S. E., Klein, M., Mulholland, J. A., and Russell, A. G.: Review of Acellular Assays of Ambient Particulate Matter Oxidative Potential: Methods and Relationships with Composition, Sources, and Health Effects, Environ. Sci. Technol., 53, 4003–4019, https://doi.org/10.1021/ACS.EST.8B03430, 2019.
Bray, C. D., Battye, W. H., Aneja, V. P., and Schlesinger, W. H.: Global emissions of NH3, NOx, and N2O from biomass burning and the impact of climate change, J. Air Waste Manage. Assoc., 71, 102–114, https://doi.org/10.1080/10962247.2020.1842822, 2021.
Briggs, N. L., Jaffe, D. A., Gao, H., Hee, J. R., Baylon, P. M., Zhang, Q., Zhou, S., Collier, S. C., Sampson, P. D., and Cary, R. A.: Particulate matter, ozone, and nitrogen species in aged wildfire plumes observed at the mount bachelor observatory, Aerosol Air Qual. Res., 16, 3075–3087, https://doi.org/10.4209/aaqr.2016.03.0120, 2016.
Bruns, E. A., Slowik, J. G., El Haddad, I., Kilic, D., Klein, F., Dommen, J., Temime-Roussel, B., Marchand, N., Baltensperger, U., and Prévôt, A. S. H.: Characterization of gas-phase organics using proton transfer reaction time-of-flight mass spectrometry: fresh and aged residential wood combustion emissions, Atmos. Chem. Phys., 17, 705–720, https://doi.org/10.5194/acp-17-705-2017, 2017.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Cao, T., Li, M., Zou, C., Fan, X., Song, J., Jia, W., Yu, C., Yu, Z., and Peng, P.: Chemical composition, optical properties, and oxidative potential of water- and methanol-soluble organic compounds emitted from the combustion of biomass materials and coal, Atmos. Chem. Phys., 21, 13187–13205, https://doi.org/10.5194/acp-21-13187-2021, 2021.
Cappa, C. D., Lim, C. Y., Hagan, D. H., Coggon, M., Koss, A., Sekimoto, K., de Gouw, J., Onasch, T. B., Warneke, C., and Kroll, J. H.: Biomass-burning-derived particles from a wide variety of fuels – Part 2: Effects of photochemical aging on particle optical and chemical properties, Atmos. Chem. Phys., 20, 8511–8532, https://doi.org/10.5194/acp-20-8511-2020, 2020.
Che, H., Segal-Rozenhaimer, M., Zhang, L., Dang, C., Zuidema, P., Dobracki, A., Sedlacek, A. J., Coe, H., Wu, H., Taylor, J., Zhang, X., Redemann, J., and Haywood, J.: Cloud processing and weeklong ageing affect biomass burning aerosol properties over the south-eastern Atlantic, Commun. Earth Environ., 3, 182, https://doi.org/10.1038/s43247-022-00517-3, 2022.
Chen, G., Canonaco, F., Tobler, A., Aas, W., Alastuey, A., Allan, J., Atabakhsh, S., Aurela, M., Baltensperger, U., Bougiatioti, A., De Brito, J. F., Ceburnis, D., Chazeau, B., Chebaicheb, H., Daellenbach, K. R., Ehn, M., El Haddad, I., Eleftheriadis, K., Favez, O., Flentje, H., Font, A., Fossum, K., Freney, E., Gini, M., Green, D. C., Heikkinen, L., Herrmann, H., Kalogridis, A. C., Keernik, H., Lhotka, R., Lin, C., Lunder, C., Maasikmets, M., Manousakas, M. I., Marchand, N., Marin, C., Marmureanu, L., Mihalopoulos, N., Močnik, G., Nęcki, J., O'Dowd, C., Ovadnevaite, J., Peter, T., Petit, J. E., Pikridas, M., Matthew Platt, S., Pokorná, P., Poulain, L., Priestman, M., Riffault, V., Rinaldi, M., Różański, K., Schwarz, J., Sciare, J., Simon, L., Skiba, A., Slowik, J. G., Sosedova, Y., Stavroulas, I., Styszko, K., Teinemaa, E., Timonen, H., Tremper, A., Vasilescu, J., Via, M., Vodička, P., Wiedensohler, A., Zografou, O., Cruz Minguillón, M., and Prévôt, A. S. H.: European aerosol phenomenology – 8: Harmonised source apportionment of organic aerosol using 22 year-long ACSM/AMS datasets, Environ. Int., 166, 107325, https://doi.org/10.1016/j.envint.2022.107325, 2022.
Cho, A. K., Sioutas, C., Miguel, A. H., Kumagai, Y., Schmitz, D. A., Singh, M., Eiguren-Fernandez, A., and Froines, J. R.: Redox activity of airborne particulate matter at different sites in the Los Angeles Basin, Environ. Res., 99, 40–47, https://doi.org/10.1016/j.envres.2005.01.003, 2005.
Cincinelli, A., Guerranti, C., Martellini, T., and Scodellini, R.: Residential wood combustion and its impact on urban air quality in Europe, Curr. Opin. Environ. Sci. Health, 8, 10–14, https://doi.org/10.1016/j.coesh.2018.12.007, 2019.
Costabile, F., Gualtieri, M., Rinaldi, M., Canepari, S., Vecchi, R., Massimi, L., Di Iulio, G., Paglione, M., Di Liberto, L., Corsini, E., Facchini, M. C., and Decesari, S.: Exposure to urban nanoparticles at low PM1 concentrations as a source of oxidative stress and inflammation, Sci. Rep., 13, 1–18, https://doi.org/10.1038/s41598-023-45230-z, 2023.
Daellenbach, K. R., Uzu, G., Jiang, J., Cassagnes, L. E., Leni, Z., Vlachou, A., Stefenelli, G., Canonaco, F., Weber, S., Segers, A., Kuenen, J. J. P., Schaap, M., Favez, O., Albinet, A., Aksoyoglu, S., Dommen, J., Baltensperger, U., Geiser, M., El Haddad, I., Jaffrezo, J. L., and Prévôt, A. S. H.: Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 587, 414–419, https://doi.org/10.1038/S41586-020-2902-8, 2020.
Desservettaz, M., Pikridas, M., Stavroulas, I., Bougiatioti, A., Liakakou, E., Hatzianastassiou, N., Sciare, J., Mihalopoulos, N., and Bourtsoukidis, E.: Emission of volatile organic compounds from residential biomass burning and their rapid chemical transformations, Sci. Total Environ., 903, 166592, https://doi.org/10.1016/j.scitotenv.2023.166592, 2023.
Dominutti, P. A., Jaffrezo, J.-L., Marsal, A., Mhadhbi, T., Elazzouzi, R., Rak, C., Cavalli, F., Putaud, J.-P., Bougiatioti, A., Mihalopoulos, N., Paraskevopoulou, D., Mudway, I., Nenes, A., Daellenbach, K. R., Banach, C., Campbell, S. J., Cigánková, H., Contini, D., Evans, G., Georgopoulou, M., Ghanem, M., Glencross, D. A., Guascito, M. R., Herrmann, H., Iram, S., Jovanović, M., Jovašević-Stojanović, M., Kalberer, M., Kooter, I. M., Paulson, S. E., Patel, A., Perdrix, E., Pietrogrande, M. C., Mikuška, P., Sauvain, J.-J., Seitanidi, K., Shahpoury, P., Souza, E. J. d. S., Steimer, S., Stevanovic, S., Suarez, G., Subramanian, P. S. G., Utinger, B., van Os, M. F., Verma, V., Wang, X., Weber, R. J., Yang, Y., Querol, X., Hoek, G., Harrison, R. M., and Uzu, G.: An interlaboratory comparison to quantify oxidative potential measurement in aerosol particles: challenges and recommendations for harmonisation, Atmos. Meas. Tech., 18, 177–195, https://doi.org/10.5194/amt-18-177-2025, 2025.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K. H., Möhler, O., Leisner, T., Müller, L., Reinnig, M. C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012.
Fachinger, F., Drewnick, F., Gieré, R., and Borrmann, S.: How the user can influence particulate emissions from residential wood and pellet stoves: Emission factors for different fuels and burning conditions, Atmos. Environ., 158, 216–226, https://doi.org/10.1016/j.atmosenv.2017.03.027, 2017.
Fang, T., Verma, V., Guo, H., King, L. E., Edgerton, E. S., and Weber, R. J.: A semi-automated system for quantifying the oxidative potential of ambient particles in aqueous extracts using the dithiothreitol (DTT) assay: results from the Southeastern Center for Air Pollution and Epidemiology (SCAPE), Atmos. Meas. Tech., 8, 471–482, https://doi.org/10.5194/amt-8-471-2015, 2015.
Fang, Z., Li, C., He, Q., Czech, H., Gröger, T., Zeng, J., Fang, H., Xiao, S., Pardo, M., Hartner, E., Meidan, D., Wang, X., Zimmermann, R., Laskin, A., and Rudich, Y.: Secondary organic aerosols produced from photochemical oxidation of secondarily evaporated biomass burning organic gases: Chemical composition, toxicity, optical properties, and climate effect, Environ. Int., 157, https://doi.org/10.1016/j.envint.2021.106801, 2021.
Florou, K., Kodros, J. K., Paglione, M., Jorga, S., Squizzato, S., Masiol, M., Uruci, P., Nenes, A., and Pandis, S. N.: Characterization and dark oxidation of the emissions of a pellet stove, Environ. Sci. Atmos., 3, 1319–1334, https://doi.org/10.1039/d3ea00070b, 2023.
Fry, J. L., Draper, D. C., Barsanti, K. C., Smith, J. N., Ortega, J., Winkler, P. M., Lawler, M. J., Brown, S. S., Edwards, P. M., Cohen, R. C., and Lee, L.: Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 48, 11944–11953, https://doi.org/10.1021/es502204x, 2014.
Gao, D., Ripley, S., Weichenthal, S., and Godri Pollitt, K. J.: Ambient particulate matter oxidative potential: Chemical determinants, associated health effects, and strategies for risk management, Free Radic. Biol. Med., 151, 7–25, https://doi.org/10.1016/j.freeradbiomed.2020.04.028, 2020a.
Gao, D., Mulholland, J. A., Russell, A. G., and Weber, R. J.: Characterization of water-insoluble oxidative potential of PM2.5 using the dithiothreitol assay, Atmos. Environ., 224, 117327, https://doi.org/10.1016/j.atmosenv.2020.117327, 2020b.
Guercio, V., Pojum, I. C., Leonardi, G. S., Shrubsole, C., Gowers, A. M., Dimitroulopoulou, S., and Exley, K. S.: Exposure to indoor and outdoor air pollution from solid fuel combustion and respiratory outcomes in children in developed countries: a systematic review and meta-analysis, Sci. Total Environ., 755, 142187, https://doi.org/10.1016/j.scitotenv.2020.142187, 2021.
Harshman, S. W., Mani, N., Geier, B. A., Kwak, J., Shepard, P., Fan, M., Sudberry, G. L., Mayes, R. S., Ott, D. K., Martin, J. A., and Grigsby, C. C.: Storage stability of exhaled breath on Tenax TA, J. Breath Res., 10, 046008, https://doi.org/10.1088/1752-7155/10/4/046008, 2016.
Hartikainen, A., Yli-Pirilä, P., Tiitta, P., Leskinen, A., Kortelainen, M., Orasche, J., Schnelle-Kreis, J., Lehtinen, K. E. J., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Volatile organic compounds from logwood combustion: emissions and transformation under dark and photochemical aging conditions in a smog chamber, Environ. Sci. Technol., 52, 4979–4988, https://doi.org/10.1021/acs.est.7b06269, 2018.
He, L.-Y., Lin, Y., Huang, X.-F., Guo, S., Xue, L., Su, Q., Hu, M., Luan, S.-J., and Zhang, Y.-H.: Characterization of high-resolution aerosol mass spectra of primary organic aerosol emissions from Chinese cooking and biomass burning, Atmos. Chem. Phys., 10, 11535–11543, https://doi.org/10.5194/acp-10-11535-2010, 2010.
He, Y., Zhao, B., Wang, S., Valorso, R., Chang, X., Yin, D., Feng, B., Camredon, M., Aumont, B., Dearden, A., Jathar, S. H., Shrivastava, M., Jiang, Z., Cappa, C. D., Yee, L. D., Seinfeld, J. H., Hao, J., and Donahue, N. M.: Formation of secondary organic aerosol from wildfire emissions enhanced by long-time ageing, Nat. Geosci., 17, 124–129, https://doi.org/10.1038/s41561-023-01355-4, 2024.
Hennigan, C. J., Miracolo, M. A., Engelhart, G. J., May, A. A., Presto, A. A., Lee, T., Sullivan, A. P., McMeeking, G. R., Coe, H., Wold, C. E., Hao, W.-M., Gilman, J. B., Kuster, W. C., de Gouw, J., Schichtel, B. A., Collett Jr., J. L., Kreidenweis, S. M., and Robinson, A. L.: Chemical and physical transformations of organic aerosol from the photo-oxidation of open biomass burning emissions in an environmental chamber, Atmos. Chem. Phys., 11, 7669–7686, https://doi.org/10.5194/acp-11-7669-2011, 2011.
Hodshire, A. L., Akherati, A., Alvarado, M. J., Brown-Steiner, B., Jathar, S. H., Jimenez, J. L., Kreidenweis, S. M., Lonsdale, C. R., Onasch, T. B., Ortega, A. M., and Pierce, J. R.: Aging effects on biomass burning aerosol mass and composition: a critical review of field and laboratory studies., Environ. Sci. Technol., 53, 10007–10022, https://doi.org/10.1021/acs.est.9b02588, 2019.
Huang, G., Wang, S., Chang, X., Cai, S., Zhu, L., Li, Q., and Jiang, J.: Emission factors and chemical profile of I/SVOCs emitted from household biomass stove in China, Sci. Total Environ., 842, 156940, https://doi.org/10.1016/j.scitotenv.2022.156940, 2022.
IEA: Key World Energy Statistics 2019, OECD Publishing, Paris, https://doi.org/10.1787/71b3ce84-en, 2019.
IEA: Key World Energy Statistics 2021, OECD Publishing, Paris, https://doi.org/10.1787/2ef8cebc-en, 2021.
Jiang, K., Xing, R., Luo, Z., Huang, W., Yi, F., Men, Y., Zhao, N., Chang, Z., Zhao, J., Pan, B., and Shen, G.: Pollutant emissions from biomass burning: A review on emission characteristics, environmental impacts, and research perspectives, Particuology, 85, 296–309, https://doi.org/10.1016/j.partic.2023.07.012, 2024.
Jorga, S. D., Kaltsonoudis, C., Liangou, A., and Pandis, S. N.: Measurement of formation rates of secondary aerosol in the ambient urban atmosphere using a dual smog chamber system, Environ. Sci. Technol., 54, 1336–1343, https://doi.org/10.1021/acs.est.9b03479, 2020.
Jorga, S. D., Florou, K., Kaltsonoudis, C., Kodros, J. K., Vasilakopoulou, C., Cirtog, M., Fouqueau, A., Picquet-Varrault, B., Nenes, A., and Pandis, S. N.: Nighttime chemistry of biomass burning emissions in urban areas: A dual mobile chamber study, Atmos. Chem. Phys., 21, 15337–15349, https://doi.org/10.5194/acp-21-15337-2021, 2021.
Kaltsonoudis, C., Kostenidou, E., Florou, K., Psichoudaki, M., and Pandis, S. N.: Temporal variability and sources of VOCs in urban areas of the eastern Mediterranean, Atmos. Chem. Phys., 16, 14825–14842, https://doi.org/10.5194/acp-16-14825-2016, 2016.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H. C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016gl069239, 2016.
Kodros, J. K., Papanastasiou, D. K., Paglione, M., Masiol, M., Squizzato, S., Florou, K., Skyllakou, K., Kaltsonoudis, C., Nenes, A., and Pandis, S. N.: Rapid dark aging of biomass burning as an overlooked source of oxidized organic aerosol, P. Natl. Acad. Sci. USA, 117, 33028–33033, https://doi.org/10.1073/pnas.2010365117, 2020.
Kodros, J. K., Kaltsonoudis, C., Paglione, M., Florou, K., Jorga, S., Vasilakopoulou, C., Cirtog, M., Cazaunau, M., Picquet-Varrault, B., Nenes, A., and Pandis, S. N.: Secondary aerosol formation during the dark oxidation of residential biomass burning emissions, Environ. Sci. Atmos., 2, 1221–1236, https://doi.org/10.1039/d2ea00031h, 2022.
Kostenidou, E., Pathak, R. K., and Pandis, S. N.: An algorithm for the calculation of secondary organic aerosol density combining AMS and SMPS data, Aerosol Sci. Technol., 41, 1002–1010, https://doi.org/10.1080/02786820701666270, 2007.
Kostenidou, E., Lee, B. H., Engelhart, G. J., Pierce, J. R., and Pandis, S. N.: Mass spectra deconvolution of low, medium, and high volatility biogenic secondary organic aerosol, Environ. Sci. Technol., 43, 4884–4889, https://doi.org/10.1021/es803676g, 2009.
Kuwata, M., Zorn, S. R., and Martin, S. T.: Using elemental ratios to predict the density of organic material composed of carbon, hydrogen, and oxygen, Environ. Sci. Technol., 46, 787–794, https://doi.org/10.1021/es202525q, 2012.
Lei, R., Wei, Z., Chen, M., Meng, H., Wu, Y., and Ge, X.: Aging effects on the toxicity alteration of different types of organic aerosols: a review, Curr. Pollut. Rep., 1, 1–12, https://doi.org/10.1007/s40726-023-00272-9, 2023.
Li, C., Ma, Z., Chen, J., Wang, X., Ye, X., Wang, L., Yang, X., Kan, H., Donaldson, D. J., and Mellouki, A.: Evolution of biomass burning smoke particles in the dark, Atmos. Environ., 120, 244–252, https://doi.org/10.1016/j.atmosenv.2015.09.003, 2015.
Li, J., Li, J., Wang, G., Ho, K. F., Dai, W., Zhang, T., Wang, Q., Wu, C., Li, L., Li, L., and Zhang, Q.: Effects of atmospheric aging processes on in vitro induced oxidative stress and chemical composition of biomass burning aerosols, J. Hazard. Mater., 401, 123750, https://doi.org/10.1016/j.jhazmat.2020.123750, 2021.
Li, K., Zhang, J., Bell, D. M., Wang, T., Lamkaddam, H., Cui, T., Qi, L., Surdu, M., Wang, D., Du, L., Haddad, I. El, Slowik, J. G., and Prevot, A. S. H.: Uncovering the dominant contribution of intermediate volatility compounds in secondary organic aerosol formation from biomass-burning emissions, Natl. Sci. Rev, 11, https://doi.org/10.1093/NSR/NWAE014, 2024.
Li, S., Liu, D., Wu, Y., Hu, K., Jiang, X., Tian, P., Sheng, J., Pan, B., and Zhao, D.: Aging effects on residential biomass burning emissions under quasi-real atmospheric conditions, Environ. Pollut., 337, 122615, https://doi.org/10.1016/j.envpol.2023.122615, 2023.
Lim, C. Y., Hagan, D. H., Coggon, M. M., Koss, A. R., Sekimoto, K., de Gouw, J., Warneke, C., Cappa, C. D., and Kroll, J. H.: Secondary organic aerosol formation from the laboratory oxidation of biomass burning emissions, Atmos. Chem. Phys., 19, 12797–12809, https://doi.org/10.5194/acp-19-12797-2019, 2019.
Lim, H., Silvergren, S., Spinicci, S., Mashayekhy Rad, F., Nilsson, U., Westerholm, R., and Johansson, C.: Contribution of wood burning to exposures of PAHs and oxy-PAHs in Eastern Sweden, Atmos. Chem. Phys., 22, 11359–11379, https://doi.org/10.5194/acp-22-11359-2022, 2022.
Liu, J., Chu, B., Chen, T., Liu, C., Wang, L., Bao, X., and He, H.: Secondary organic aerosol formation from ambient air at an urban site in Beijing: effects of OH exposure and precursor concentrations, Environ. Sci. Technol., 52, 6834–6841, https://doi.org/10.1021/acs.est.7b05701, 2018.
Luo, L., Bai, X., Liu, S., Wu, B., Liu, W., Lv, Y., Guo, Z., Lin, S., Zhao, S., Hao, Y., Hao, J., Zhang, K., Zheng, A., and Tian, H.: Fine particulate matter (PM2.5/PM1.0) in Beijing, China: variations and chemical compositions as well as sources, J. Environ. Sci., 121, 187–198, https://doi.org/10.1016/j.jes.2021.12.014, 2022.
McClure, C. D., Lim, C. Y., Hagan, D. H., Kroll, J. H., and Cappa, C. D.: Biomass-burning-derived particles from a wide variety of fuels – Part 1: Properties of primary particles, Atmos. Chem. Phys., 20, 1531–1547, https://doi.org/10.5194/acp-20-1531-2020, 2020.
Mylonaki, M., Gini, M., Georgopoulou, M., Pilou, M., Chalvatzaki, E., Solomos, S., Diapouli, E., Giannakaki, E., Lazaridis, M., Pandis, S. N., Nenes, A., Eleftheriadis, K., and Papayannis, A.: Wildfire and African dust aerosol oxidative potential, exposure and dose in the human respiratory tract, Sci. Total Environ., 913, 169683, https://doi.org/10.1016/j.scitotenv.2023.169683, 2024.
Nault, B. A., Campuzano-Jost, P., Day, D. A., Schroder, J. C., Anderson, B., Beyersdorf, A. J., Blake, D. R., Brune, W. H., Choi, Y., Corr, C. A., De Gouw, J. A., Dibb, J., Digangi, J. P., Diskin, G. S., Fried, A., Gregory Huey, L., Kim, M. J., Knote, C. J., Lamb, K. D., Lee, T., Park, T., Pusede, S. E., Scheuer, E., Thornhill, K. L., Woo, J. H., and Jimenez, J. L.: Secondary organic aerosol production from local emissions dominates the organic aerosol budget over Seoul, South Korea, during KORUS-AQ, Atmos. Chem. Phys., 18, 17769–17800, https://doi.org/10.5194/acp-18-17769-2018, 2018.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Ng, N. L., Tuet, W. Y., Chen, Y., Fok, S., Gao, D., Rodriguez, M. S. T., Klein, M., Grosberg, A., Weber, R. J., and Champion, J. A.: Cellular and acellular assays for measuring oxidative stress induced by ambient and laboratory-generated aerosols, Res. Rep. Health. Eff. Inst., 1–57, https://pmc.ncbi.nlm.nih.gov/articles/PMC7266377/pdf/hei-2019-197.pdf (last access: 14 November 2025), 2019.
Novakov, T., Menon, S., Kirchstetter, T. W., Koch, D., and Hansen, J. E.: Aerosol organic carbon to black carbon ratios: analysis of published data and implications for climate forcing, J. Geophys. Res. Atmos., 110, 1–12, https://doi.org/10.1029/2005jd005977, 2005.
Nyström, R., Lindgren, R., Avagyan, R., Westerholm, R., Lundstedt, S., and Boman, C.: Influence of wood species and burning conditions on particle emission characteristics in a residential wood stove, Energy & Fuels, 31, 5514–5524, https://doi.org/10.1021/acs.energyfuels.6b02751, 2017.
Othman, M., Latif, M. T., Hamid, H. H. A., Uning, R., Khumsaeng, T., Phairuang, W., Daud, Z., Idris, J., Sofwan, N. M., and Lung, S. C. C.: Spatial–temporal variability and health impact of particulate matter during a 2019–2020 biomass burning event in Southeast Asia, Sci. Rep., 12, 1–11, https://doi.org/10.1038/s41598-022-11409-z, 2022.
Paraskevopoulou, D., Bougiatioti, A., Stavroulas, I., Fang, T., Lianou, M., Liakakou, E., Gerasopoulos, E., Weber, R., Nenes, A., and Mihalopoulos, N.: Yearlong variability of oxidative potential of particulate matter in an urban Mediterranean environment, Atmos. Environ., 206, 183–196, https://doi.org/10.1016/j.atmosenv.2019.02.027, 2019.
Paraskevopoulou, D., Bougiatioti, A., Zarmpas, P., Tsagkaraki, M., Nenes, A., and Mihalopoulos, N.: Impact of COVID-19 lockdown on oxidative potential of particulate matter: case of Athens (Greece), Toxics, 10, 280, https://doi.org/10.3390/toxics10060280, 2022.
Pardo, M., Li, C., Zimmermann, R., and Rudich, Y.: Health impacts of biomass burning aerosols: relation to oxidative stress and inflammation, Aerosol Science and Technology, 58, 1093–1113, https://doi.org/10.1080/02786826.2024.2379551, 2024.
Price-Allison, A., Mason, P. E., Jones, J. M., Barimah, E. K., Jose, G., Brown, A. E., Ross, A. B., and Williams, A.: The impact of fuelwood moisture content on the emission of gaseous and particulate pollutants from a wood stove, Combust. Sci. Technol., 195, 133–152, https://doi.org/10.1080/00102202.2021.1938559, 2021.
Puthussery, J. V., Singh, A., Rai, P., Bhattu, D., Kumar, V., Vats, P., Furger, M., Rastogi, N., Slowik, J. G., Ganguly, D., Prevot, A. S. H., Tripathi, S. N., and Verma, V.: Real-time measurements of PM2.5 oxidative potential using a dithiothreitol assay in Delhi, India, Environ. Sci. Technol. Lett., 7, 504–510, https://doi.org/10.1021/acs.estlett.0c00342, 2020.
Rao, L., Zhang, L., Wang, X., Xie, T., Zhou, S., Lu, S., Liu, X., Lu, H., Xiao, K., Wang, W., and Wang, Q.: Oxidative potential induced by ambient particulate matters with acellular assays: a review, Processes, 8, 1–21, https://doi.org/10.3390/pr8111410, 2020.
Reid, W. V, Ali, M. K., and Field, C. B.: The future of bioenergy, Glob. Chang. Biol., 26, 274–286, https://doi.org/10.1111/gcb.14883, 2020.
Shen, G., Du, W., Luo, Z., Li, Y., Cai, G., Lu, C., Qiu, Y., Chen, Y., Cheng, H., and Tao, S.: Fugitive emissions of CO and PM2.5 from indoor biomass burning in chimney stoves based on a newly developed carbon balance approach, Environ. Sci. Technol. Lett., 7, 128–134, https://doi.org/10.1021/acs.estlett.0c00095, 2020.
Srivastava, D., Vu, T. V., Tong, S., Shi, Z., and Harrison, R. M.: Formation of secondary organic aerosols from anthropogenic precursors in laboratory studies, Npj Clim. Atmos. Sci., 5, 1–30, https://doi.org/10.1038/s41612-022-00238-6, 2022.
Stockwell, C. E., Yokelson, R. J., Kreidenweis, S. M., Robinson, A. L., DeMott, P. J., Sullivan, R. C., Reardon, J., Ryan, K. C., Griffith, D. W. T., and Stevens, L.: Trace gas emissions from combustion of peat, crop residue, domestic biofuels, grasses, and other fuels: configuration and Fourier transform infrared (FTIR) component of the fourth Fire Lab at Missoula Experiment (FLAME-4), Atmos. Chem. Phys., 14, 9727–9754, https://doi.org/10.5194/acp-14-9727-2014, 2014.
Sun, J., Wang, J., Shen, Z., Huang, Y., Zhang, Y., Niu, X., Cao, J., Zhang, Q., Xu, H., Zhang, N., and Li, X.: Volatile organic compounds from residential solid fuel burning in Guanzhong Plain, China: source-related profiles and risks, Chemosphere, 221, 184–192, https://doi.org/10.1016/j.chemosphere.2019.01.002, 2019.
Sun, Y., Du, W., Fu, P., Wang, Q., Li, J., Ge, X., Zhang, Q., Zhu, C., Ren, L., Xu, W., Zhao, J., Han, T., Worsnop, D. R., and Wang, Z.: Primary and secondary aerosols in Beijing in winter: sources, variations and processes, Atmos. Chem. Phys., 16, 8309–8329, https://doi.org/10.5194/acp-16-8309-2016, 2016.
Tiitta, P., Leskinen, A., Hao, L., Yli-Pirilä, P., Kortelainen, M., Grigonyte, J., Tissari, J., Lamberg, H., Hartikainen, A., Kuuspalo, K., Kortelainen, A.-M., Virtanen, A., Lehtinen, K. E. J., Komppula, M., Pieber, S., Prévôt, A. S. H., Onasch, T. B., Worsnop, D. R., Czech, H., Zimmermann, R., Jokiniemi, J., and Sippula, O.: Transformation of logwood combustion emissions in a smog chamber: formation of secondary organic aerosol and changes in the primary organic aerosol upon daytime and nighttime aging, Atmos. Chem. Phys., 16, 13251–13269, https://doi.org/10.5194/acp-16-13251-2016, 2016.
Tomlin, J. M., Weis, J., Veghte, D. P., China, S., Fraund, M., He, Q., Reicher, N., Li, C., Jankowski, K. A., Rivera-Adorno, F. A., Morales, A. C., Rudich, Y., Moffet, R. C., Gilles, M. K., and Laskin, A.: Chemical composition and morphological analysis of atmospheric particles from an intensive bonfire burning festival, Environ. Sci. Atmos., 2, 616–633, https://doi.org/10.1039/d2ea00037g, 2022.
Trubetskaya, A., Lin, C., Ovadnevaite, J., Ceburnis, D., O'Dowd, C., Leahy, J. J., Monaghan, R. F. D., Johnson, R., Layden, P., and Smith, W.: Study of emissions from domestic solid-fuel stove combustion in Ireland, Energy & Fuels, 35, 4966–4978, https://doi.org/10.1021/acs.energyfuels.0c04148, 2021.
Tsiodra, I., Grivas, G., Tavernaraki, K., Bougiatioti, A., Apostolaki, M., Paraskevopoulou, D., Gogou, A., Parinos, C., Oikonomou, K., Tsagkaraki, M., Zarmpas, P., Nenes, A., and Mihalopoulos, N.: Annual exposure to polycyclic aromatic hydrocarbons in urban environments linked to wintertime wood-burning episodes, Atmos. Chem. Phys., 21, 17865–17883, https://doi.org/10.5194/acp-21-17865-2021, 2021.
Tsiodra, I., Grivas, G., Bougiatioti, A., Tavernaraki, K., Parinos, C., Paraskevopoulou, D., Papoutsidaki, K., Tsagkaraki, M., Kozonaki, F. A., Oikonomou, K., Nenes, A., and Mihalopoulos, N.: Source apportionment of particle-bound polycyclic aromatic hydrocarbons (PAHs), oxygenated PAHs (OPAHs), and their associated long-term health risks in a major European city, Sci. Total Environ., 951, https://doi.org/10.1016/j.scitotenv.2024.175416, 2024.
Tuet, W. Y., Chen, Y., Xu, L., Fok, S., Gao, D., Weber, R. J., and Ng, N. L.: Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 17, 839–853, https://doi.org/10.5194/acp-17-839-2017, 2017.
Vasilakopoulou, C. N., Matrali, A., Skyllakou, K., Georgopoulou, M., Aktypis, A., Florou, K., Kaltsonoudis, C., Siouti, E., Kostenidou, E., Błaziak, A., Nenes, A., Papagiannis, S., Eleftheriadis, K., Patoulias, D., Kioutsioukis, I., and Pandis, S. N.: Rapid transformation of wildfire emissions to harmful background aerosol, Npj Clim. Atmos. Sci., 6, 1–9, https://doi.org/10.1038/s41612-023-00544-7, 2023.
Verma, V., Fang, T., Xu, L., Peltier, R. E., Russell, A. G., Ng, N. L., and Weber, R. J.: Organic aerosols associated with the generation of reactive oxygen species (ROS) by water-soluble PM2.5, Environ. Sci. Technol., 49, 4646–4656, https://doi.org/10.1021/es505577W, 2015.
Wang, N., Jorga, S. D., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Particle wall-loss correction methods in smog chamber experiments, Atmos. Meas. Tech., 11, 6577–6588, https://doi.org/10.5194/amt-11-6577-2018, 2018.
Wang, S., Gallimore, P. J., Liu-Kang, C., Yeung, K., Campbell, S. J., Utinger, B., Liu, T., Peng, H., Kalberer, M., Chan, A. W. H., and Abbatt, J. P. D.: Dynamic wood smoke aerosol toxicity during oxidative atmospheric aging, Environ. Sci. Technol., 57, 1246–1256, https://doi.org/10.1021/acs.est.2c05929, 2023.
Wong, J. P. S., Tsagkaraki, M., Tsiodra, I., Mihalopoulos, N., Violaki, K., Kanakidou, M., Sciare, J., Nenes, A., and Weber, R. J.: Effects of atmospheric processing on the oxidative potential of biomass burning organic aerosols, Environ. Sci. Technol., 53, 6747–6756, https://doi.org/10.1021/acs.est.9b01034, 2019.
Yazdani, A., Takahama, S., Kodros, J. K., Paglione, M., Masiol, M., Squizzato, S., Florou, K., Kaltsonoudis, C., Jorga, S. D., Pandis, S. N., and Nenes, A.: Chemical evolution of primary and secondary biomass burning aerosols during daytime and nighttime, Atmos. Chem. Phys., 23, 7461–7477, https://doi.org/10.5194/acp-23-7461-2023, 2023.
Yokelson, R. J., Griffith, D. W. T., and Ward, D. E.: Open-path Fourier transform infrared studies of large-scale laboratory biomass fires, J. Geophys. Res. Atmos., 101, 21067–21080, https://doi.org/10.1029/96jd01800, 1996.
Zauli-Sajani, S., Thunis, P., Pisoni, E., Bessagnet, B., Monforti-Ferrario, F., De Meij, A., Pekar, F., and Vignati, E.: Reducing biomass burning is key to decrease PM2.5 exposure in European cities, Sci. Rep., 14, 1–11, https://doi.org/10.1038/s41598-024-60946-2, 2024.
Zhang, Y., Kong, S., Sheng, J., Zhao, D., Ding, D., Yao, L., Zheng, H., Wu, J., Cheng, Y., Yan, Q., Niu, Z., Zheng, S., Wu, F., Yan, Y., Liu, D., and Qi, S.: Real-time emission and stage-dependent emission factors/ratios of specific volatile organic compounds from residential biomass combustion in China, Atmos. Res., 248, 105189, https://doi.org/10.1016/j.atmosres.2020.105189, 2021.
Zhang, Z.-H., Hartner, E., Utinger, B., Gfeller, B., Paul, A., Sklorz, M., Czech, H., Yang, B. X., Su, X. Y., Jakobi, G., Orasche, J., Schnelle-Kreis, J., Jeong, S., Gröger, T., Pardo, M., Hohaus, T., Adam, T., Kiendler-Scharr, A., Rudich, Y., Zimmermann, R., and Kalberer, M.: Are reactive oxygen species (ROS) a suitable metric to predict toxicity of carbonaceous aerosol particles?, Atmos. Chem. Phys., 22, 1793–1809, https://doi.org/10.5194/acp-22-1793-2022, 2022.