the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 02 Oct 2025
| 02 Oct 2025
Stratospheric δ13CO2 observed over Japan and its governing processes
Satoshi Sugawara
Shinji Morimoto
Shigeyuki Ishidoya
Taku Umezawa
Shuji Aoki
Takakiyo Nakazawa
Sakae Toyoda
Kentaro Ishijima
Daisuke Goto
Hideyuki Honda
Due to very few reports of δ13CO2 (the stable carbon isotopic ratio of CO2) observations in the stratosphere, its variations are not well understood. In order to elucidate stratospheric δ13CO2 variations and their governing mechanisms, we have collected stratospheric air samples using balloon-borne cryogenic samplers over Japan since 1985 and analyzed them for δ13CO2. To obtain precise δ13CO2 values, we incorporated the mass-independent fractionation of 17O and 18O in the δ13CO2 calculation. δ13CO2 has decreased through time in the mid-stratosphere with an average rate of change of −0.026 ± 0.001 ‰ yr−1 for the period 1985–2020, consistent with that in the troposphere. However, mid-stratospheric δ13CO2 values did not show a time delay compared to the tropical tropospheric values. This could be explained by the production of CO2 by CH4 oxidation and the gravitational separation of 13CO2 and 12CO2. To confirm this hypothesis, we used a two-dimensional model to simulate the stratospheric δ13CO2 values while accounting for these processes. The results indicate that these two effects strongly impact the vertical distribution of δ13CO2. We newly defined “stratospheric potential δ13C” (δ13CP) as a quasi-conservative parameter incorporating the kinetic isotope effect of CH4 oxidation and gravitational separation, and we found that δ13CP in the mid-latitude mid-stratosphere decreases over time with about a 5 yr lag relative to the tropical upper troposphere. This fact strongly supports that stratospheric δ13CO2 variations are governed by the airborne production of 13C-depleted CO2 by CH4 oxidation, the gravitational separation, and the propagation of the decreasing tropospheric δ13CO2 trend into the stratosphere.
- Article
(10897 KB) - Full-text XML
-
Supplement
(511 KB) - BibTeX
- EndNote




The isotopic ratio of carbon in CO2 (here, we use “δ13CO2” to distinguish it from that in CH4, “δ13CH4”) is thought to provide information on the carbon cycle, and observations have been carried out mainly using ground stations, ships, and aircraft in the troposphere (e.g., Keeling et al., 1995; Francey et al., 1995; Nakazawa et al., 1993; Morimoto et al., 2000). However, there have been very few reports of δ13CO2 observations in the mid-stratosphere, mainly because of the difficulty in collecting air samples at such altitudes; the only available reports are balloon observations over Japan and Scandinavia (Gamo et al., 1995; Aoki et al., 2003). It is well known that tropospheric δ13CO2 values have decreased with time due to anthropogenic emissions with low δ13CO2 values resulting from fossil fuel consumption. Gamo et al. (1995) reported that δ13CO2 values observed in the mid-stratosphere were higher than those in the troposphere over the same period, and that the average stratospheric δ13CO2 value during 1986–1992 was about −7.5 ‰ and decreasing with time. Aoki et al. (2003) reported that the mid-stratosphere (25–32 km altitude) δ13CO2 value over Japan in 1997 was approximately −7.7 ‰; this value is consistent with the expected value extrapolated from the results of Gamo et al. (1995), suggesting that their estimated secular decreasing trend is plausible.
Anomalous 18O enrichment in stratospheric CO2 was first reported in balloon observations (Gamo et al., 1989), which spurred extended studies of the mass-independent isotopic effect (MIE) on the triple oxygen isotope system (e.g., Thiemens, 1999; Kawagucci et al., 2008). The isotopic fractionation is usually caused by the effect of mass differences between isotopologues (i.e., the mass-dependent isotopic effect). In this case, the fractionation between 18O and 16O is almost twice as large as that between 17O and 16O. However, the isotopic fractionation that does not follow this relation occurs mainly in photochemical processes. These MIE studies have clarified that the relationship between 17O and 18O enrichment in stratospheric CO2 is quite different from that in the troposphere. This fact is very important for stratospheric δ13CO2 measurements because mass spectrometry measures the ratios 45COCO2 and 46COCO2, and an assumed relationship between 17O and 18O enrichment is needed to calculate δ13CO2. The influence of the MIE on oxygen in stratospheric δ13CO2 has not been taken into account in previous studies. Although Aoki et al. (2003) found that δ13CO2 values increase with increasing altitude in the stratosphere over Japan and suggested that the MIE on 17O and 18O should be considered when interpreting such a vertical distribution, no quantitative investigation of these factors has been conducted.
Stratospheric methane (CH4) is destroyed by reactions with hydroxyl (OH), excited singlet oxygen (O(1D)), and chlorine (Cl) radicals, and thus plays an important role in stratospheric chemistry. However, the quantitative contributions of these chemical processes to CH4 loss and their temporal variations remain poorly understood. It is thought that δ13CH4 provides useful information about CH4 destruction in the stratosphere because characteristic isotopic fractionations occur in each reaction (e.g., Saueressig et al., 1995; Sugawara et al., 1997; Rice et al., 2003; Röckmann et al., 2011). However, only a few measurements have been made so far in the mid-stratosphere. Oxidation of CH4 produces CO, which is eventually oxidized to CO2. In general, the isotopic fractionations in the above reactions cause the faster destruction of 12CH4 relative to 13CH4, making the chemical products depleted in 13C (decreased δ13C) while the remaining CH4 becomes enriched in 13C (increased δ13CH4). Indeed, Brenninkmeijer et al. (1996) found that CO in the lower Antarctic stratosphere is extremely depleted in 13C (decreased δ13CO) mainly because of the local production of CO via the reaction of CH4 with Cl. Recently, Gromov et al. (2018) and Röckmann et al. (2024) have discussed δ13CO variations associated with the CH4+ Cl reaction. From these studies, it is expected that the CH4–CO–CO2 chain reaction could be an airborne source of CO2 with extremely low δ13C values, depressing stratospheric δ13CO2 values. However, no study has yet quantitatively assessed this effect.
The age of stratospheric air is the transit time of air from around the tropical tropopause to a certain location in the stratosphere, which is a powerful tool for diagnosing stratospheric transport (see Garny et al., 2024 for a review of the latest developments in air age studies). It is expected that possible changes to the Brewer–Dobson circulation induced by climate change are detectable as long-term changes in the mean age of air. For this purpose, age tracers, such as mole fractions of CO2 and SF6, have been measured in air samples collected by scientific balloons and satellites (e.g., Engel et al., 2009, 2017; Stiller et al., 2012). CO2 mole fractions observed in the mid-stratosphere over Japan have been combined with SF6 data to provide the longest record of the mean age of air, as reported by Engel et al. (2009, 2017) and Ray et al. (2014). Engel et al. (2017) reported that the mean age of air in the mid-stratosphere does not indicate a significant trend over the period 1975–2016, with only a slightly positive increase of +0.15 ± 0.18 yr decade−1. However, recent models predict accelerating circulation throughout the stratosphere (e.g., Butchart, 2014), which will decrease the mean age of air. Such a decreasing trend is consistent with observational results in the lower stratosphere (Ray et al., 2014). Despite many efforts to elucidate the trend of age of air, a discrepancy remains between mid-stratosphere age trends in models and observations. One reason for this discrepancy is the large uncertainties on the observational trends. To resolve this problem, it is necessary to increase the frequency of high-altitude balloon observations and observe additional age tracers beyond CO2 and SF6. Perfluorocarbons and hydrofluorocarbons with extremely long lifetimes could be alternative age tracers (Leedham Elvidge et al., 2018; Laube et al., 2025; Umezawa et al., 2025), and observations of such multiple age tracers should reduce the uncertainties on mean age of air estimates.
The gravitational separation (hereafter, “GS”) of major atmospheric constituents in the stratosphere was first reported from balloon observations (Ishidoya et al., 2006, 2008a, b, 2013). They showed that vertical gradients of the isotopic and elemental ratios of major atmospheric components (N2, O2, and Ar) in the stratosphere are caused by molecular diffusion and depend on molecular mass. Stratospheric GS has now been observed in polar and equatorial regions (Ishidoya et al., 2018; Sugawara et al., 2018) and reproduced in numerical models (Belikov et al., 2019; Birner et al., 2020). Alongside age of air, stratospheric GS can be used to diagnose stratospheric transport processes (Ishidoya et al., 2013; Sugawara et al., 2018; Birner et al., 2020). These findings also suggest that GS could affect the mole fractions and isotopic ratios of all stratospheric constituents, although no studies have addressed the impact of GS on stratospheric δ13CO2 values.
The main objective of this study is to clarify the mechanisms governing δ13CO2 variations in the stratosphere and understand the long-term variations. A comprehensive understanding of the variations of stratospheric δ13CO2 requires that the MIE on oxygen, CH4 oxidation and associated isotopic fractionation, mean age of air, and GS be considered. Balloon observations provide a unique opportunity to evaluate these effects by studying a variety of atmospheric constituents using high-precision measurements. In this study, we constructed a new δ13CO2 record for the stratosphere over Japan spanning the 35 yr from 1985 to 2020, primarily by newly analyzing archived CO2 samples and in part by including previously published data (Gamo et al., 1995; Aoki et al., 2003). We then examined vertical and temporal variations in the record. Furthermore, we investigated in detail the various mechanisms affecting the vertical distribution of stratospheric δ13CO2 and propose herein a new concept called “stratospheric potential δ13C.” We also investigated the possibility of estimating the age of stratospheric air from the δ13CO2 record.
2.1 Air sample collection
We have collected stratospheric air samples over Japan since 1985 using balloon-borne cryogenic samplers (e.g., Nakazawa et al., 1995). A large scientific balloon equipped with a sampler was launched nearly once a year during 1985–2002 from the Sanriku Balloon Center (SBC; 39° 10′ N, 141° 50′ E). The frequency of observations has decreased since 2004, with the longest interval between observations being 5 yr. During this period, the balloon launch site was relocated from SBC to the Taiki Aerospace Research Field in Hokkaido (TARF; 42°30′ N 143°26′ E) in 2008. Balloon observations conducted so far over Japan are summarized in Table S1 of the Supplement. The cryogenic air sampler consists mainly of a liquid helium dewar, stainless steel bottles, motor-driven valves, and a control unit (Honda et al., 1996). Liquid helium was used as a refrigerant for collecting stratospheric air cryogenically under low atmospheric pressures, and 20–30 L STP of stratospheric air was collected in each bottle, depending on the altitude. Air sampling was performed typically at 11 different altitudes, spaced about 2 km apart, between 14 and 35 km altitude. Occasionally, anomalously low CO2 mole fractions were observed, probably due to sample deterioration in the bottle. In such cases, both CO2 mole fraction and δ13CO2 data were excluded from the data record. In 2020, the CO2 mole fraction and δ13CO2 value at 28.8 and 30.9 km altitude were not available due to water contamination of the air samples.
2.2 Analytical methods for mole fractions and isotopic ratios
Collected air samples were distributed to several institutes in Japan for analysis of the mole fractions and isotopic compositions of various gases. The CO2 mole fraction was measured using a nondispersive infrared gas analyzer at Tohoku University (TU) with an analytical precision of <0.02 µmol mol−1; the CO2 measurement protocol has been described in detail in previous studies (Nakazawa et al., 1995; Aoki et al., 2003; Sugawara et al., 2018). N2O and CH4 mole fractions were measured using gas chromatographs equipped with an electron capture detector and flame ionization detector, respectively, at TU. The N2O mole fraction was used both for mass spectrometry analyses to determine δ13CO2 and to investigate the MIE between 17O and 18O in CO2, as described below. The analytical precisions for the N2O and CH4 mole fractions were 0.3 and 3 nmol mol−1, respectively (Nakazawa et al., 2002; Aoki et al., 2003; Ishijima et al., 2007).
The analytical procedures for δ13CO2 in air samples collected after 1988 have already been described in previous studies (Nakazawa et al., 1993, 1997a; Morimoto et al., 2000), and is briefly summarized here. An aliquot of each air sample (about 550 mL STP) was introduced into a CO2 extraction system, and the pure extracted CO2 was divided and sealed in small glass tubes; some tubes were archived for future study. The carbon and oxygen isotopic ratios (δ13C and δ18O) of the sample CO2 were measured using a mass spectrometer (Finnigan MAT-δS) installed at TU against an in-house reference CO2 calibrated to Vienna Peedee Belemnite (VPDB). The analytical precision of δ13CO2 was ±0.02 ‰ (Morimoto et al., 2000). Air samples collected in 1985, 1986, 1988, and 1989 were analyzed for δ13CO2 using a mass spectrometer (Finnigan MAT250) with a reproducibility of less than ±0.02 ‰ at the University of Tokyo (Gamo et al., 1989, 1995).
In this study, we modified the calculation of isotopic ratios to take into account the stratospheric MIE and GS. Hereafter, the isotopic ratios of 13C to 12C, 17O and 18O to 16O, and 45CO2 and 46CO2 to 44CO2 are expressed as 13R=n(13C)C), 17R=n(17O)O), 18R=n(18O)O), 45R=n(45CO2)CO2), and 46R=n(46CO2)CO2), respectively, where n is the amount of each isotopologue. δ13CO2 is defined as
where 13RVPDB is the known 13R value of VPDB (Coplen and Shrestha, 2016). The isotopic ratios 45R and 46R are expressed as (Santrock et al., 1985)
To calculate δ13C and δ18O values from 45R and 46R, the relationship between 17O and 18O should be known, because 45CO2 contains 12C17O16O, whereas 46CO2 contains both 13C17O16O and 12C17O2. The relationship between 17R and 18R can be expressed as (e.g., Meijer and Li, 1998)
where 17RVSMOW and 18RVSMOW are the known ratios of Vienna standard mean ocean water (VSMOW; Coplen and Shrestha, 2016). In measurements of tropospheric δ13C and δ18O, the mass-dependent relationship between 17O and 18O can be assumed, i.e., β=0.528 (Meijer and Li, 1998; Assonov and Brenninkmeijer, 2003). However, the MIE between 17O and 18O is non-negligible in the stratosphere (Gamo et al., 1995; Thiemens, 1999). Therefore, the δ17O value is essential for accurately determining the δ13C value in the stratosphere. Our air samples were also measured for triple oxygen isotopes, as reported by Kawagucci et al. (2008). However, δ17O measurements were not performed for all of our stratospheric samples. In such cases, the following method was applied to obtain δ17O values. The values of 17R and 18R calculated from δ17O and δ18O observed by Kawagucci et al. (2008) were fitted to Eq. (2c), and the average value of β was calculated to be 1.7 ± 0.2 using the least squares method. For samples for which δ17O was not analyzed, we used Eq. (2c) with this β value to calculate δ13C. By substituting Eqs. (2a) and (2b) into (2c), we obtained the following equation:
where γ is defined as
Equation (3a) was solved for 17R numerically using the bisection method and the measured 45R and 46R values, and then 13R and δ13C were calculated by Eqs. (2a) and (1), respectively. We note that GS occurs in CO2 molecules with different mass numbers (44, 45, 46). Therefore, it is not accurate to correct δ13C by assuming a mass number difference of uniquely Δm=1. To obtain the correct δ13C of stratospheric CO2, it is necessary to first apply the GS correction to the measured values of 45R and 46R, and then to calculate δ13C using the method described above. The method used to correct 45R and 46R for GS is discussed in Sect. 3.3.
The MIE calculation described above was not considered in Gamo et al. (1995) and Aoki et al. (2003), and it is therefore likely that their δ13C values are not correct, especially at higher altitudes. δ17O and δ18O in CO2 increase rapidly with increasing altitude in the stratosphere (Gamo et al., 1995; Kawagucci et al., 2008), and the deviations from mass-dependent values also increase with altitude, meaning that the overestimation of δ13C also increases with altitude. The influence of the oxygen MIE on δ13C is almost zero near the tropopause, but at an altitude of about 35 km over Japan, it is approximately 0.6 ‰. Therefore, this effect significantly impacts the vertical distributions of δ13C. We reanalyzed some of the CO2 samples used by Gamo et al. (1995) and Aoki et al. (2003) to determine the differences between their results (not accounting for the oxygen MIE) and ours to allow us to correct their data and add them to our long-term record. The MIE on δ17O and δ18O occurs through photochemical reactions in O2, O3, and CO2 (e.g., Thiemens, 1999), which are relatively tightly correlated with the mole fraction of N2O, which is also destroyed through photochemical processes. Figure 1 shows the relationships between the N2O mole fraction and δ17O and δ18O in CO2 in the stratosphere over Japan. These results show that δ17O and δ18O values calculated using β=1.7 are consistent with the values observed by Kawagucci et al. (2008).
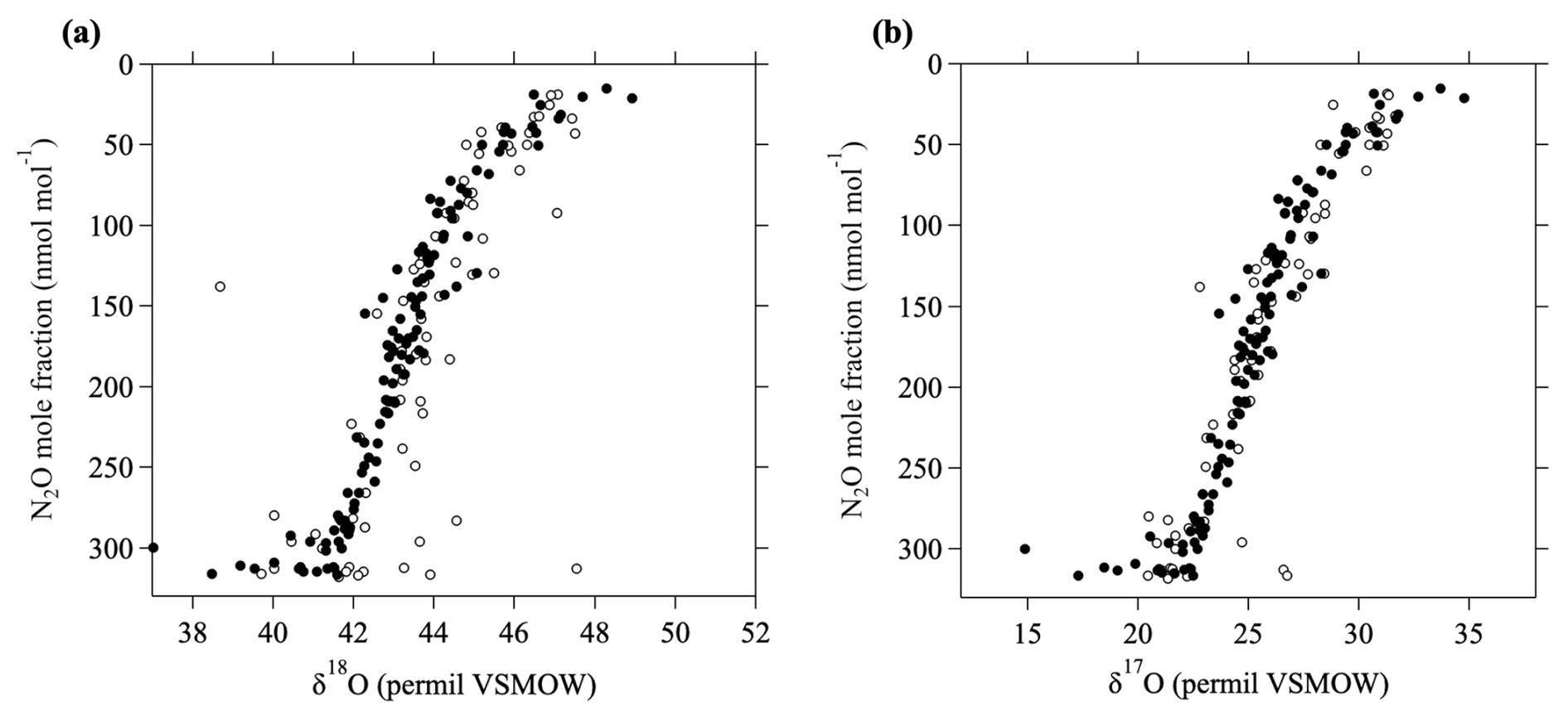
Figure 1The relationship between the N2O mole fraction and (a) δ18O in CO2 and (b) δ17O in CO2. δ18O and δ17O values measured by Kawagucci et al. (2008) are shown by open circles. δ18O and δ17O values calculated assuming an MIE factor of 1.7 (see text) in this study are shown by closed circles.
While cryogenically extracting CO2 from the air sample, N2O was simultaneously trapped with the CO2. Because N2O and CO2 have the same mass number, we must correct for N2O to obtain the true δ13C value. The correction method was described in detail by Nakazawa et al. (1993), and we summarize it here. The ionization efficiency of N2O was measured in advance, and the measured δ13C value was corrected using the mole fraction ratio of the air sample. In the troposphere, the mole fraction ratio does not change significantly, and the N2O correction is almost constant (approximately +0.2 ‰; Nakazawa et al., 1997a). However, because the N2O mole fraction decreases rapidly with increasing altitude in the stratosphere (e.g., Toyoda et al., 2001), the mole fraction ratio decreases with altitude, and the magnitude of the N2O correction also varies vertically. Therefore, the N2O correction also strongly influences the observed vertical distribution of δ13C in the stratosphere.
Stratospheric air samples were also measured for δ13CH4 using a gas chromatograph-combustion-isotope ratio spectrometer developed at the National Institute of Polar Research (NIPR), Japan. The method has been described in detail by Morimoto et al. (2006) and is briefly summarized here. An air sample was flushed by pure helium and passed through a trap containing HayeSep D at a temperature of −120 °C to concentrate CH4 in the sample. Then, the CH4 was released and transferred into a PoraBOND Q cryofocusing trap at −197 °C. The CH4 released from the trap when brought to room temperature was separated from the residual components using a PoraPLOT Q capillary column and combusted into CO2 in a furnace with a Pt-Ni-Cu catalyst at 940 °C. The converted CO2 was analyzed using a continuous-flow mass spectrometer (Finnigan MAT252). The precision was reported to be 0.06 ‰ (Morimoto et al., 2006). Before 1996, δ13CH4 measurements were performed using off-line oxidation equipment (Sugawara et al., 1997), in which CH4 in the sample was converted to CO2 using a catalyst (0.5 % Pt supported on alumina pellets) at 750 °C, and the pure CO2 sample thus prepared was analyzed using the mass spectrometer installed at TU in the same way as for δ13CO2, except using a micro-volume inlet system. The overall analytical precision was 0.07 ‰ (Sugawara et al., 1997). To produce a high-quality data record, we needed to verify that these two methods produced the same results. Therefore, several sets of stratospheric air samples were analyzed using both methods, and the δ13CH4 results differed by about 0.85 ‰. To correct for this difference, the off-line measurement results were adjusted to match the on-line measurements. In this study, the δ13CH4 value is reported relative to VPDB. The international comparison of δ13CH4 measurements described in detail by Umezawa et al. (2018) revealed a difference of 0.2 ‰ between NIPR and the Institute of Arctic and Alpine Research (INSTAAR) results. Therefore, all δ13CH4 values observed by the National Oceanic and Atmospheric Administration Global Monitoring Laboratory (NOAA/GML) and INSTAAR were shifted to match the NIPR scale.
The method used to analyze GS is described in Appendix A.
3.1 Vertical profiles of CO2 mole fraction and δ13CO2
Figure 2 shows vertical profiles of the CO2 mole fraction and δ13CO2 over Japan throughout the study period. CO2 mole fractions and δ13CO2 values have increased and decreased, respectively, from 1985 to 2020. Vertical δ13CO2 profiles observed by Gamo et al. (1995) in 1985 and 1986 are included in this figure after MIE correction. Previous studies reported that the CO2 mole fraction decreases with increasing altitude from the tropopause to around 20–25 km, and then it becomes almost constant at higher altitudes (Nakazawa et al., 1995, 2002). Our CO2 mole fraction data show a similar vertical distribution during the observation period. Because our δ13CO2 data are more scattered with respect to altitude than the CO2 mole fraction, we calculated detrended vertical δ13CO2 profiles as follows. First, the long-term trend of the tropospheric δ13CO2 at Mauna Loa, Hawaii (hereafter, MLO) (Michel et al., 2025) was calculated as a representative tropospheric trend, and all our stratospheric δ13CO2 observation data were shifted up or down based on that trend to produce values corresponding to August 2016 for comparison with the model results described below. Then, the δ13CO2 values thus obtained for the entire study period were grouped into 11 2 km vertical bins, and the average value for each bin was calculated (Fig. 2c). The results showed that δ13CO2 values increased with increasing altitude from near the tropopause to around 25 km and then decreased slightly above that altitude. Gamo et al. (1989) originally reported that δ13CO2 values increased by approximately 0.4 ‰ from the tropopause to 25 km altitude in September 1985, whereas our recalculated results showed an increase of about 0.2 ‰ (Fig. 2b). The average increase shown in Fig. 2c was even smaller, only about 0.1 ‰. This difference between the vertical profiles originally reported by Gamo et al. (1989) and our present results is due to our incorporation of the MIE on 17O and 18O (Sect. 2.2).
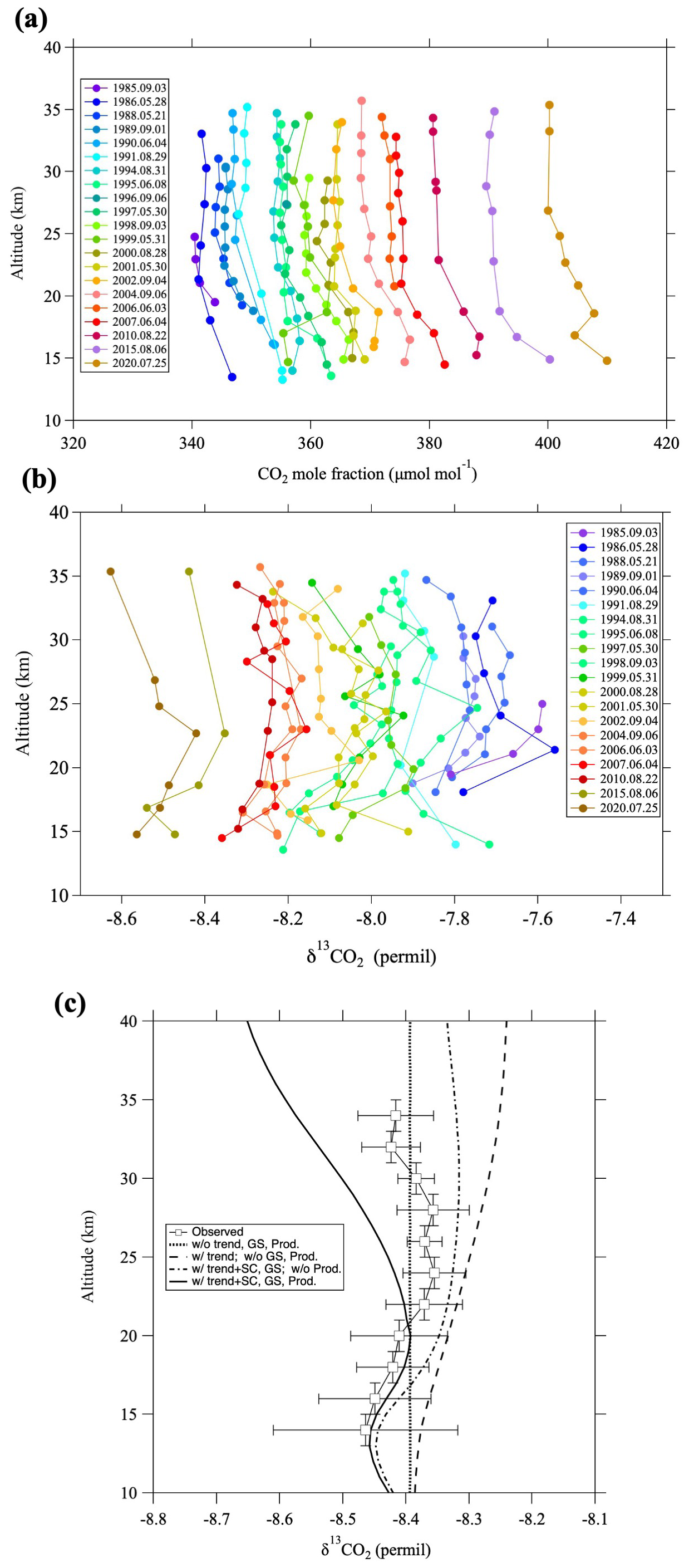
Figure 2Vertical profiles of (a) CO2 mole fraction and (b) δ13CO2 over Japan during the period 1985–2020. (c) δ13CO2 values averaged over the study period and grouped in 11 2 km vertical bins (open squares). Curves show 2-D modeling results without the tropospheric trend, seasonal cycle, gravitational separation (GS), or airborne sources (thick dotted line), including only the tropospheric trend (dashed line), including the tropospheric trend, seasonal cycle, and GS (dotted-dashed line), and including the tropospheric trend, seasonal cycle, GS, and airborne sources (solid line).
Below an altitude of ∼ 17 km, δ13CO2 values are quite variable, often being irregularly high or low depending on the observation year, which is exhibited by the large error bars in Fig. 2c. Based on detailed aircraft observations of δ13CO2 variations in the troposphere over Japan, Nakazawa et al. (1993) reported that the seasonal cycle of δ13CO2 between 8 km altitude and the tropopause show a minimum around May and a maximum around September, with a peak-to-peak amplitude of 0.36 ‰. Indeed, our balloon observations were conducted in either spring (late May to early June) or summer (late July to early September), and the observed δ13CO2 values at lower altitudes tended to be lower in spring and higher in summer. Accordingly, we attribute the large δ13CO2 variations below an altitude of 17 km to the upper tropospheric air itself, or stratospheric air directly influenced by the troposphere.
Our data clearly reveal a secular decrease in mid-stratospheric δ13CO2 values. To compare the temporal variations of δ13CO2 in the mid-stratosphere with those in the troposphere, the δ13CO2 values obtained by averaging mid-stratospheric data above 24 km altitude (30 hPa) from each year and monthly average MLO data are shown in Fig. 3. When comparing our data with the NOAA/GML and INSTAAR data at MLO (Michel et al., 2025), any possible offset of δ13CO2 measurements should be taken into account. The inter-comparison results of the WMO/IAEA Round Robin Comparison Experiment (https://gml.noaa.gov/ccgg/wmorr/, last access: 20 February 2025) show that our values are approximately 0.07 ‰ lower than INSTAAR values. In this study, we directly compared δ13CO2 data at Ny-Ålesund, Svalbard observed by Goto et al. (2017) with the INSTAAR data at the Zeppelin Observatory (ZEP) in Ny-Ålesund because we used the same δ13CO2 measurement system and standard scale as Goto et al. (2017). Monthly average δ13CO2 values at Ny-Ålesund from both studies were calculated by applying a curve-fitting procedure (Nakazawa et al., 1997b) over the years compared, 1996–2014. As a result, we estimated the difference between this study and INSTAAR data to be insignificant (−0.005 ± 0.067 ‰) and did not apply any further correction to our δ13CO2 data. The average rate of change of δ13CO2 in the mid-stratosphere was calculated to be −0.026 ± 0.001 ‰ yr−1 for the period 1985–2020, consistent with the average rate of change at MLO from 1990 to 2024 (−0.026 ± 0.001 ‰ yr−1) despite the slightly different time periods. Tropospheric δ13CO2 values are known to decrease over time due to the emission of CO2 with low δ13CO2 by anthropogenic fossil fuel consumption, and our results show that a similar decreasing trend is observable in the stratosphere. It is also well known that mid-stratospheric air over the mid-latitudes is “aged,” mainly because of the slow transport of air from the tropical tropopause to the mid-latitude mid-stratosphere. Indeed, the age of mid-stratospheric air estimated from CO2 mole fractions was approximately 4.4 yr older on average than that of tropical tropospheric air (see Sect. 3.5). Nevertheless, the δ13CO2 values observed in the mid-stratosphere over Japan did not seem to show any significant time delay compared with the observed values in the tropical troposphere. Although systematic observations of δ13CO2 have not yet been performed in the tropical upper troposphere where stratospheric air originates, Assonov et al. (2010) found from CARIBIC aircraft observations that δ13CO2 variations in the tropical upper troposphere are close to those at MLO. Therefore, if we assume no difference between the δ13CO2 values at MLO and those in the tropical upper troposphere, the lack of a delay between the values in the mid-stratosphere over Japan and those at MLO clearly contradicts the concept of the age of air. Simply following the logic of the age of air, we would expect the decrease of stratospheric δ13CO2 to lag behind the tropospheric trend, such that the δ13CO2 value at any given time should be higher in the mid-stratosphere than in the troposphere. This discrepancy suggests that δ13CO2 is not a conserved quantity in the stratosphere and is instead modified not only by air transport but also by other mechanisms. In the following sections we discuss possible mechanisms in detail.
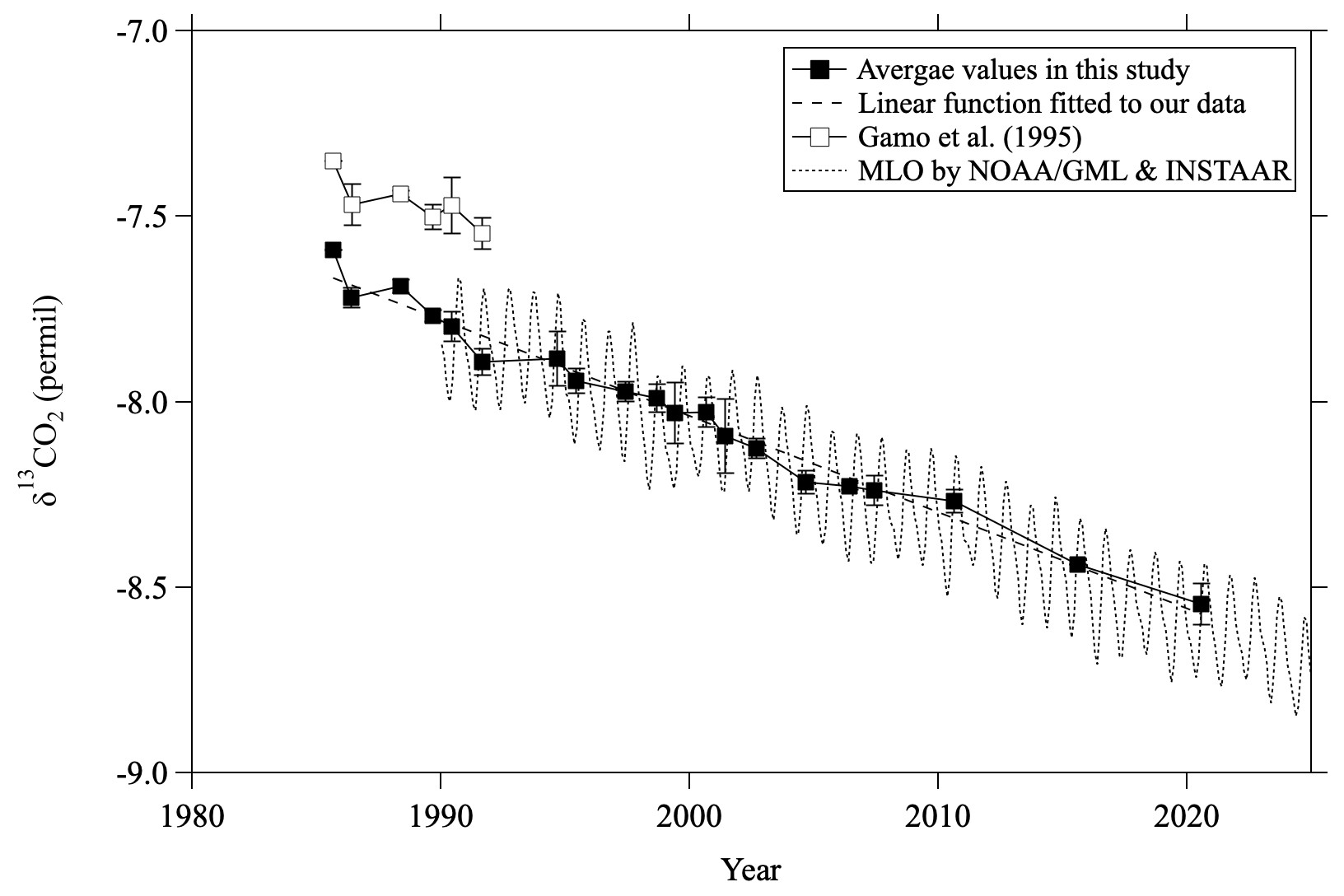
Figure 3Average mid-stratospheric (>24 km altitude) δ13CO2 values over Japan (closed squares). The linear function fitted to the average values using the least-squares method is shown by the dashed line. Values reported by Gamo et al. (1995) are shown by open squares. Monthly average δ13CO2 observed at Mauna Loa by NOAA/GML and INSTAAR (Michel et al., 2025) is also shown by the dotted line.
3.2 Airborne CO2 source from CH4 oxidation
In the stratosphere, CH4 is destroyed by reactions with OH, O(1D), and Cl, which produce intermediate products such as CH3O2 and CH2O. These products are immediately converted to CO because of their short lifetimes. However, CO is also destroyed by reaction with OH. Therefore, the chemical budget of CO in the stratosphere is expressed as (Minschwaner et al., 2010)
where , , , and kCO+OH are the reaction rates for the respective chemical reactions, and is the rate of photodissociation of CO2. The last term on the right-hand side of this equation represents the oxidation of CO to CO2. Although the amount of CO2 produced this way is small compared to the stratospheric abundance, it cannot be ignored as an airborne source, especially in terms of isotopic signatures. The timescales for the photochemical loss and production of CO in the mid-latitude mid-stratosphere (∼ 35 km altitude) are about 30 d (Minschwaner et al., 2010), quite short compared to the timescale of the stratospheric mean meridional circulation (more than several years). If we assume that the chemical budget of CO is in a steady state and that CO2 photodissociation is negligibly small below the mid-stratosphere, the rate of CH4 loss should equal the rate of CO2 production. Indeed, to estimate the age of stratospheric air from the CO2 mole fraction, a small amount of CO2 produced by the chemical destruction of CH4 was taken into account (e.g., Engel et al., 2002; Sugawara et al., 2018). To correct for this effect, each observed CO2 mole fraction was adjusted using the CH4 mole fraction measured from the corresponding air sample prior to age calculation (see Appendix C). The adjustment increases with increasing altitude and becomes larger than 1 µmol mol−1 at the highest altitudes. As with the CO2 mole fraction, 13C can be corrected by assuming a steady-state 13CO budget in which 13C is added to 13CO2 from 13CH4 through a series of oxidation reactions from CH4 to CO2. Indeed, it is well known that isotopic fractionation occurs during CH4 oxidation (Saueressig et al., 1995; Sugawara et al., 1997; Rice et al., 2003; Röckmann et al., 2011).
Figure 4a, b shows the vertical CH4 mole fraction and δ13CH4 profiles observed over Japan, respectively. A common feature of both profiles is their rapid change with altitude, although δ13CH4 values increase, whereas the mole fractions decrease with increasing altitude. Such vertical distributions have been attributed to large kinetic isotopic effects (KIEs) associated with the chemical destruction of CH4 in the stratosphere (Sugawara et al., 1997; Rice et al., 2003; Röckmann et al., 2011). KIEs on CH4 have also been studied using numerical models (Saueressig et al., 2001; Wang et al., 2002; McCarthy et al., 2003). In previous studies, the apparent fractionation factor was derived from the Rayleigh distillation model by assuming that the chemical destruction of CH4 occurs in a closed system (Sugawara et al., 1997; Rice et al., 2003). In general, the Rayleigh distillation model for the CH4 mole fraction and δ13CH4 is expressed as
where δ13CH4_0, µ, and α are the isotopic ratio before CH4 is destroyed, the ratio of mole fractions (μ=), and the apparent fractionation factor, respectively. The apparent fractionation factor can be estimated by applying Eq. (5) to observed data. We note that [CH4]0 and δ13CH4_0 change with time and should be taken here as the values in the tropical upper troposphere at the time when air entered the stratosphere through the tropical tropopause layer (TTL). Therefore, the tropical upper tropospheric records of CH4 and δ13CH4 were used to give [CH4]0 and δ13CH4_0 values going back in time based on the mean age estimated from CO2 mole fractions (see Appendix C). Details of the tropical upper tropospheric records of CH4 and δ13CH4 are described in Sect. 3.5. Figure 4c shows that the relationship between ln {(δ13CHCH4_0+1)} and ln([CH4] [CH4]0) can be approximated by linear functions if we divided the data between the lower (<24 km) and mid-stratosphere (>24 km), with the upper layer showing larger isotopic fractionation effects. The apparent fractionation factors calculated using the least-squares method were 0.9889 ± 0.0003 and 0.9866 ± 0.0007 for the lower and mid-stratosphere, respectively. We attributed these differences in apparent fractionation factors to differences in the relative contributions of the CH4 reactions with OH, O(1D), or Cl over the air transport pathway, and/or the effect of air mixing (Röckmann et al., 2011).
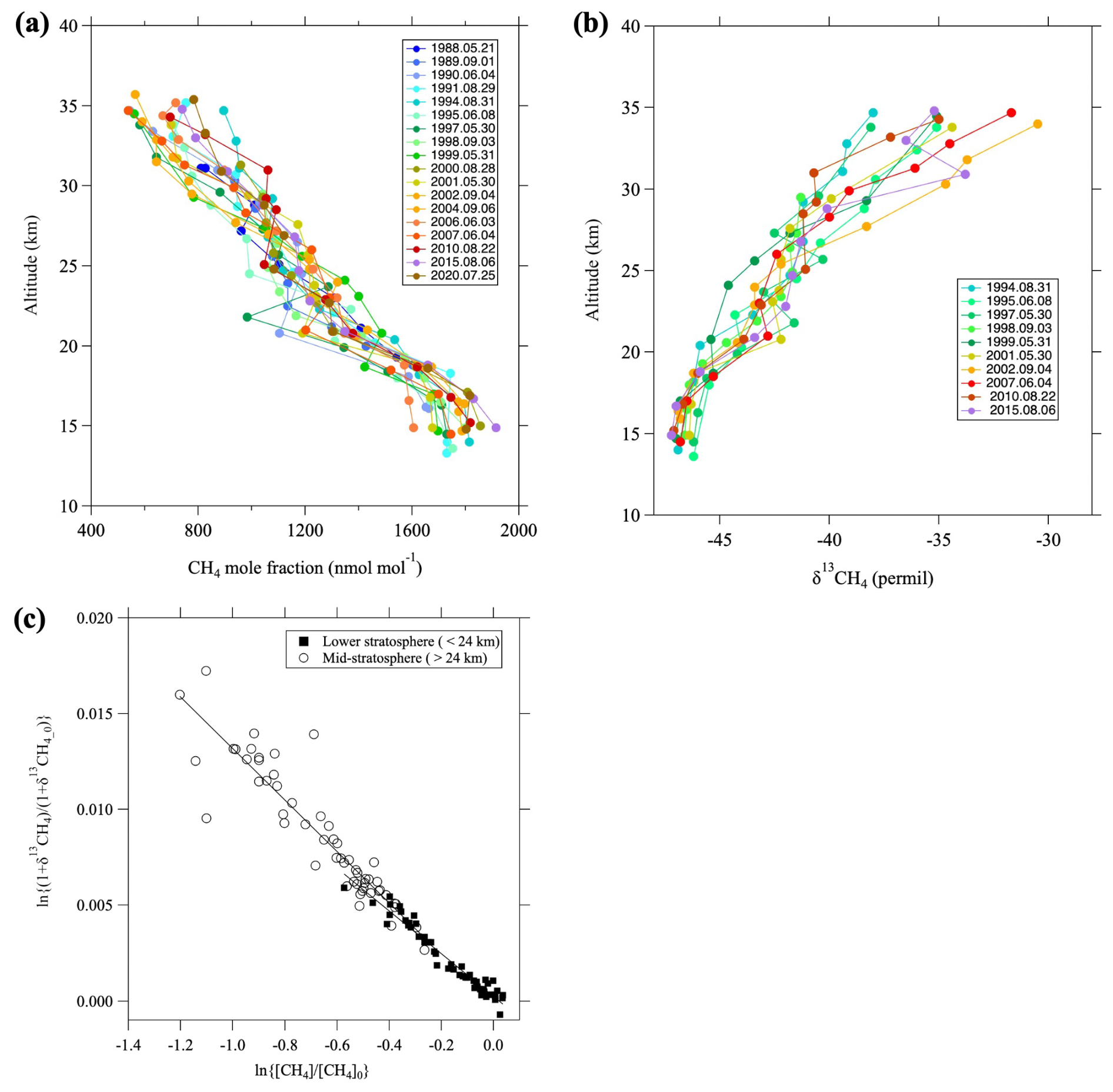
Figure 4Vertical profiles of (a) CH4 mole fraction and (b) δ13CH4 over Japan, and (c) the relationship between ln{(δ13CHCH4_0+1)} and ln([CH4] [CH4]0). In (c), the data are divided into the lower (<24 km, closed squares) and mid-stratosphere (>24 km, open circles), and the linear functions fitted to each dataset using the least-squares method are shown by lines.
Equation (5) expresses the relationship between the mole fraction and isotopic ratio of the remaining CH4. Similarly, focusing on the CH4 lost via the CH4–CO–CO2 chain reaction, the relationship between the CH4 mole fraction and isotopic ratio of lost CH4 (hereafter, δ13CH4_L) is derived from Eq. (5) as
Here, the removed CH4 includes the total amount of CH4 loss integrated over the air transport pathway. Based on this equation, we calculated δ13CH4_L for each year's data from the observed CH4 mole fractions and apparent fractionation factors by grouping them into altitudes below and above 24 km. Typical δ13CH4_L values were about −57 ‰, −56 ‰, and −55 ‰ around altitudes of 20, 30, and 35 km, respectively. The stratospheric δ13CO2 values were corrected for airborne sources simply by assuming that carbon with the isotopic ratio of δ13CH4_L was added to ambient CO2. We note that the amount of CO2 added by CH4 oxidation was estimated from the difference between the CH4 mole fraction observed in the mid-latitude stratosphere and that estimated at the tropical upper troposphere; this does not represent chemical production at the observed location, but apparent production, because CH4 oxidation occurs along the transport pathway of the air mass and is accompanied by air mixing. Consequently, the depression of CH4 integrated over the air transport pathway is observed at a certain location in the stratosphere. Because we assumed the simple Rayleigh distillation model, the δ13CH4_L estimated here is also an apparent value in a similar sense as described above. Accordingly, our assumption described above was a first-order approximation to correct δ13CO2 for airborne CO2 sources.
As an example, we corrected the vertical distributions of the CO2 mole fraction and δ13CO2 (Fig. 5a) and compared with the CH4 mole fraction and δ13CH4 (Fig. 5b) observed on 22 August 2010. The results indicated that the δ13CO2 value decreased by 0.14 ‰ and the CO2 mole fraction increased by approximately 1.1 µmol mol−1 at the highest altitude (∼ 34 km) when corrected for airborne CO2 sources. Therefore, such a correction for the airborne CO2 source with low δ13CO2 values produced by CH4 oxidation is essential for understanding CO2 mole fraction and δ13CO2 variations in the mid-stratosphere. This effect is particularly strong on δ13CO2 and significantly impacted the shape of the vertical distribution.
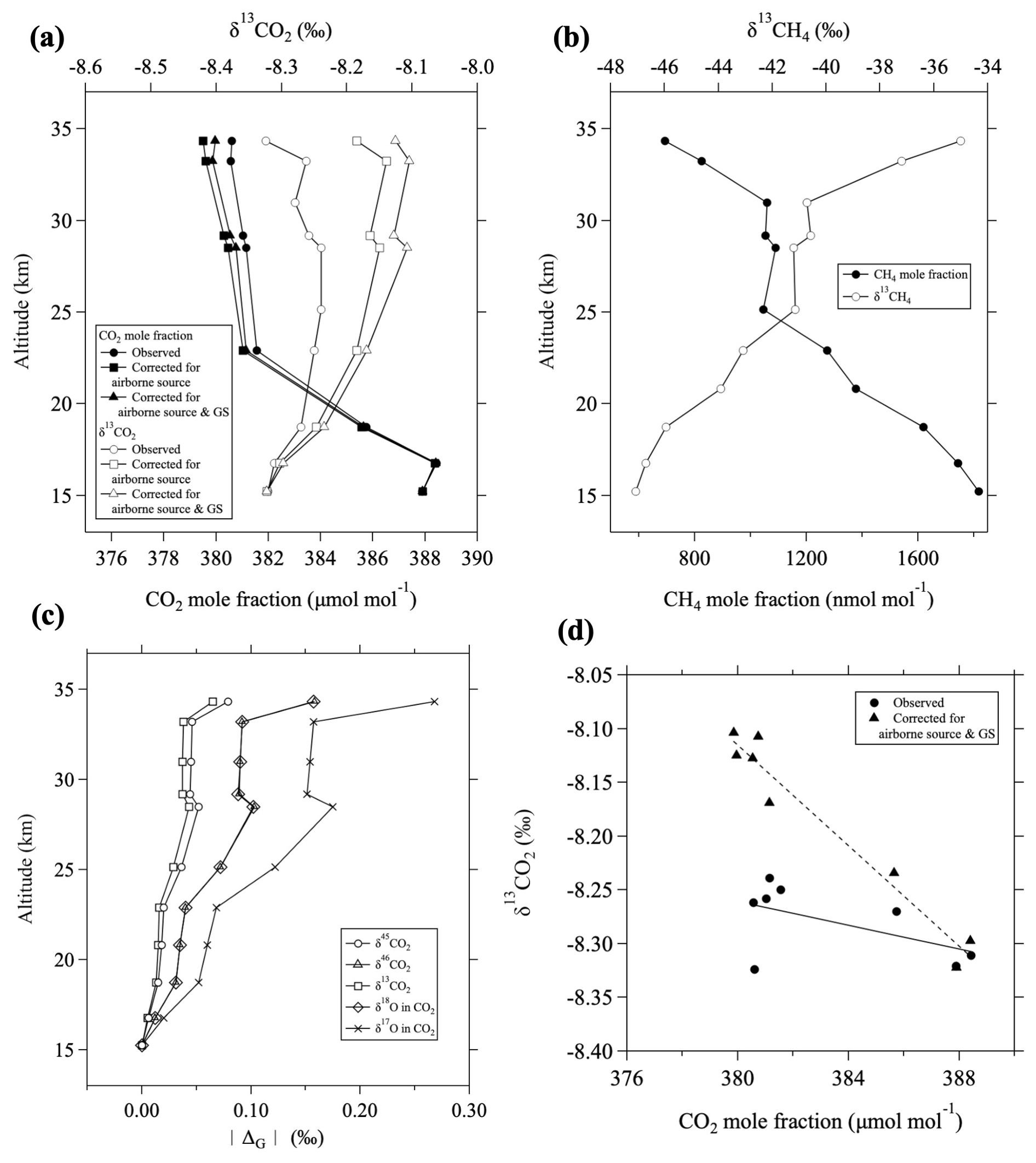
Figure 5(a) Vertical profiles of the CO2 mole fraction (closed circles) and δ13CO2 (open circles) observed over Japan on 22 August 2010. Values corrected for airborne CO2 sources (squares) and additionally for gravitational separation (GS; triangles) are also shown. (b) Vertical profiles of the CH4 mole fraction (closed circles) and δ13CH4 (open circles). (c) Vertical profiles of the magnitude of GS corrections, , for δ45CO2 (circles), δ46CO2 (triangles), δ13CO2 (squares), δ18O (diamonds), and δ17O (crosses) in CO2. (d) Observed δ13CO2 plotted against CO2 mole fraction (closed circles) and those corrected for airborne CO2 sources and GS (triangles). Lines are linear functions fitted to each trend by the least-squares method.
We note that the transport of CO from the troposphere into the stratosphere and its oxidation in the lower stratosphere are not considered in the above discussion. Because the photochemical timescales of CO are longer in the lower stratosphere than in the mid-stratosphere, tropospheric CO could be a stratospheric CO2 source associated with troposphere–stratosphere exchange and may be important in the lower stratospheric CO budget. Observational results on the stable carbon isotopic ratio of CO, δ13CO, have been reported not only for the troposphere (Brenninkmeijer, 1993; Röckmann and Brenninkmeijer, 1997; Röckmann et al., 1999, 2002; Kato et al., 2000) but also for the lower stratosphere (Brenninkmeijer et al., 1996; Röckmann et al., 2024). In particular, Brenninkmeijer et al. (1996) found that extremely low δ13CO values in the southern high-latitude lower stratosphere are accompanied by increasing δ13CH4 values and suggested that the large KIE of CH4 destruction by Cl plays an important role. The local production of CO by CH4-destruction-depleted 13C in stratospheric CO is consistent with the above discussion if CO is rapidly oxidized to CO2 depleted in 13C. However, tropospheric CO could be an additional airborne CO2 source if it is transported into the lower stratosphere and oxidized to CO2. Previous studies have reported that δ13CO values in the free troposphere are around −30 ‰ to −25 ‰ (Brenninkmeijer, 1993; Röckmann and Brenninkmeijer, 1997; Röckmann et al., 1999, 2002; Kato et al., 2000), and the KIE on carbon in the reaction CO + OH depends on atmospheric pressure (Stevens and Wagner, 1989; Bergamaschi et al., 2000). Its fractionation factor is 0.994 (expressed as the ratio of rate constants, at 1013 hPa, but increases to about 1.000 at 400 hPa and further to 1.003–1.005 above the tropopause. Therefore, CO2 produced from the reaction CO + OH should be slightly enriched in 13C in the stratosphere, contrary to CO2 produced in the lower troposphere. Accordingly, tropospheric CO could be an airborne CO2 source with δ13C values lower than around −25 ‰ to −20 ‰ in the lower stratosphere. Assuming that tropospheric air containing 250 nmol mol−1 CO with δ13CO 30 ‰ is transported to the stratosphere where 200 nmol mol−1 CO is lost by reaction with OH, δ13CO2 values should decrease by only about 0.01 ‰ when 0.2 µmol mol−1 CO2 with δ13CO ‰ is added to 390 µmol mol−1 CO2 with δ13CO ‰. Therefore, this effect would be negligibly small compared with the influence of CH4 oxidation. Unfortunately, we could not further evaluate the effect of tropospheric CO from our observations, and further study including measurements of δ13CO is needed.
3.3 Gravitational separation of δ13CO2
The standardized GS is defined as 〈δG〉, which is normalized by the mass number differences of corresponding molecules (see Appendix A). With respect to δ13CO2, the upward decrease due to GS is equal to 〈δG〉 for 13C16O2 and 12C17O16O (Δm=1), and is twice as large for 12C18O16O, 13C17O16O, and 12C17O2 (Δm=2). Therefore, GS equal to and twice the magnitude of 〈δG〉 can be considered to act on δ45CO2 and δ46CO2, respectively:
Here, ΔG(x) and 〈δG〉0 represent the change in isotopic value x due to GS and the value of 〈δG〉 before GS occurs (i.e., corresponding to the relevant value in the upper troposphere), respectively. Because 〈δG〉−〈δG〉0 is negative in the stratosphere (see Appendix A), ΔG(δ45CO2) and ΔG(δ46CO2) are also negative. When δ13CO2 was corrected for GS, Eqs. (7a) and (7b) were first applied to δ45CO2 and δ46CO2, respectively, and then δ13CO2 was determined using the MIE algorithm described in Sect. 2.2. As shown in Fig. 5c, |ΔG(δ13CO2)| was slightly smaller than (about 82 % of) |ΔG(δ45CO2)| in the mid-stratosphere, mainly because of 12C17O16O enrichment due to the MIE in the stratosphere.
The same method can be applied to the mole fraction of a specific molecule (Ishidoya et al., 2006):
Here, C0 and C denote the mole fractions of the molecule before and after GS, respectively, and m and mair are the respective mass numbers of the molecule and air. The maximum depression of the CO2 mole fraction amounts to about 0.6 µmol mol−1 at the highest altitude, assuming C0=400 µmol mol−1 and per meg (10−6). As an example, the vertical profile of the CO2 mole fraction obtained by correcting the 22 August 2010 observations for GS is shown in Fig. 5a. Compared to the profiles corrected for airborne CO2 sources, GS correction increases the CO2 mole fraction by 0.4 µmol mol−1 and the δ13CO2 by approximately 0.06 ‰ at 34 km altitude. Although the effect of GS on the CO2 mole fraction is small, that on δ13CO2 is non-negligible, significantly influencing the vertical profile along with CH4 oxidation. In this regard, the mean age estimated from the CO2 mole fraction without GS correction has an older age bias that increases with increasing altitude. The age bias is negligibly small (<1 month) in the lower stratosphere but not negligible in the mid-stratosphere (>2 months at 35 km altitude). Therefore, we applied the GS correction to the CO2 mole fraction to estimate the CO2 age, as described in Appendix C.
Correcting for both CH4 oxidation and GS, δ13CO2 values increase with altitude and become nearly constant above ∼ 25 km; they are clearly anticorrelated with the CO2 mole fraction. Assuming a linear relationship between the CO2 mole fraction and δ13CO2 (Fig. 5d), the slope calculated by least squares is −0.005 ± 0.003 ‰ (µmol mol for the raw observational data and −0.023 ± 0.002 ‰ (µmol mol for the corrected data. This rate of change for the corrected data is less negative than approximately −0.05 ‰ (µmol mol for the seasonal cycles in the troposphere (Nakazawa et al., 1993) and is near the rate of change for their secular trends (Morimoto et al., 2000; Goto et al., 2017). Generally, because of the Brewer–Dobson circulation, air at higher altitudes is older. Therefore, we consider that this rate of change for the corrected data basically reflects different ages of air.
3.4 Two-dimensional model of stratospheric δ13CO2
The mechanisms governing the vertical δ13CO2 profile in the stratosphere are shown schematically in Fig. 6, including the influence of the age of air as well as the effects of CH4 oxidation and GS discussed in Sects. 3.2 and 3.3. Tropospheric δ13CO2 values have been decreasing through time due to the combustion of fossil fuels. If it is assumed that 12CO2 and 13CO2 have no sinks or sources in the stratosphere, the air that intrudes from the tropical upper troposphere is slowly transported to the mid-latitude stratosphere by the Brewer–Dobson circulation, conserving the δ13CO2 value. Therefore, based on the concept of age of air, it is expected that δ13CO2 values should also decrease with time in the stratosphere after a certain lag time. Accordingly, δ13CO2 values should be higher in the mid-stratosphere than in the troposphere (dotted line in Fig. 6). In contrast, GS decreases δ13CO2 values at higher altitudes because 13C16O2 is heavier than 12C16O2 (dashed line in Fig. 6). In addition, 13C-depleted CO2 produced by CH4 oxidation further decreases δ13CO2 values at high altitudes (solid line in Fig. 6). The observed stratospheric vertical δ13CO2 profile should therefore be formed by a combination of these effects.
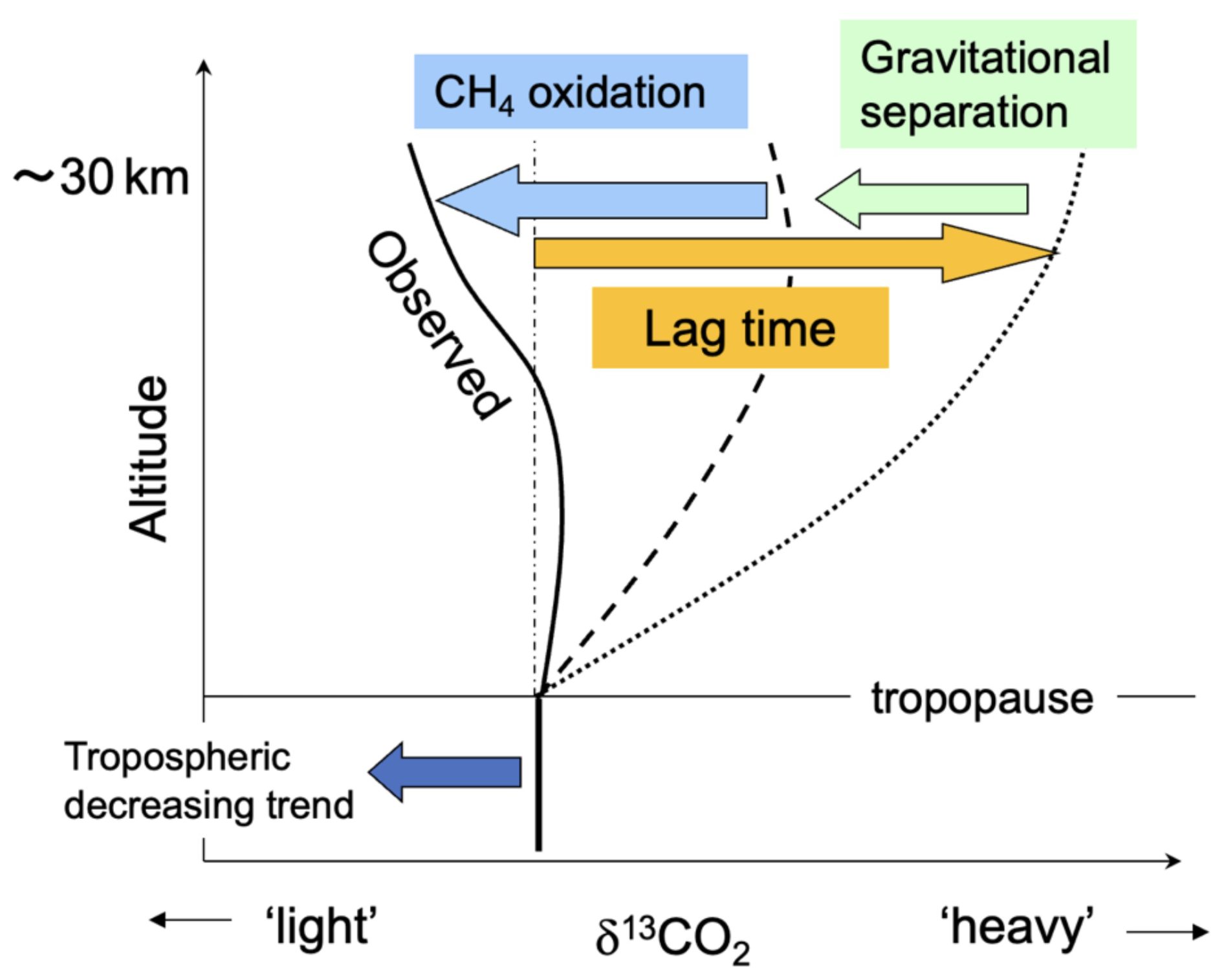
Figure 6Schematic representation of the mechanisms influencing the vertical profile of stratospheric δ13CO2 (see text).
To verify this hypothesis, we performed numerical simulations using a two-dimensional model of the middle atmosphere (SOCRATES) developed by the National Center for Atmospheric Research (NCAR; Huang et al., 1998; Park et al., 1999; Khosravi et al., 2002). Details of the model calculations are described in Appendix B. The monthly average meridional δ13CO2 distribution calculated by SOCRATES for August 2016, which includes the effects of CH4 oxidation and GS, is shown in Fig. 7. The effect of the CO2 production associated with the KIE on CH4 destruction is included in a simplified manner (see Appendix B). The δ13CO2 values decrease with increasing altitude up to about 14 km and then increase up to about 20 km in the mid-latitudes of the northern hemisphere during summer. The distribution of δ13CO2 from the troposphere to the lower stratosphere should therefore be closely related to the propagation of its seasonal cycle in the troposphere. δ13CO2 values reach a maximum at around 20 km and then gradually decrease. At high latitudes in the southern (winter) hemisphere, the vertical decrease of δ13CO2 is significant, suggesting that the effects of CH4 oxidation and GS are enhanced by the descending upper atmosphere.
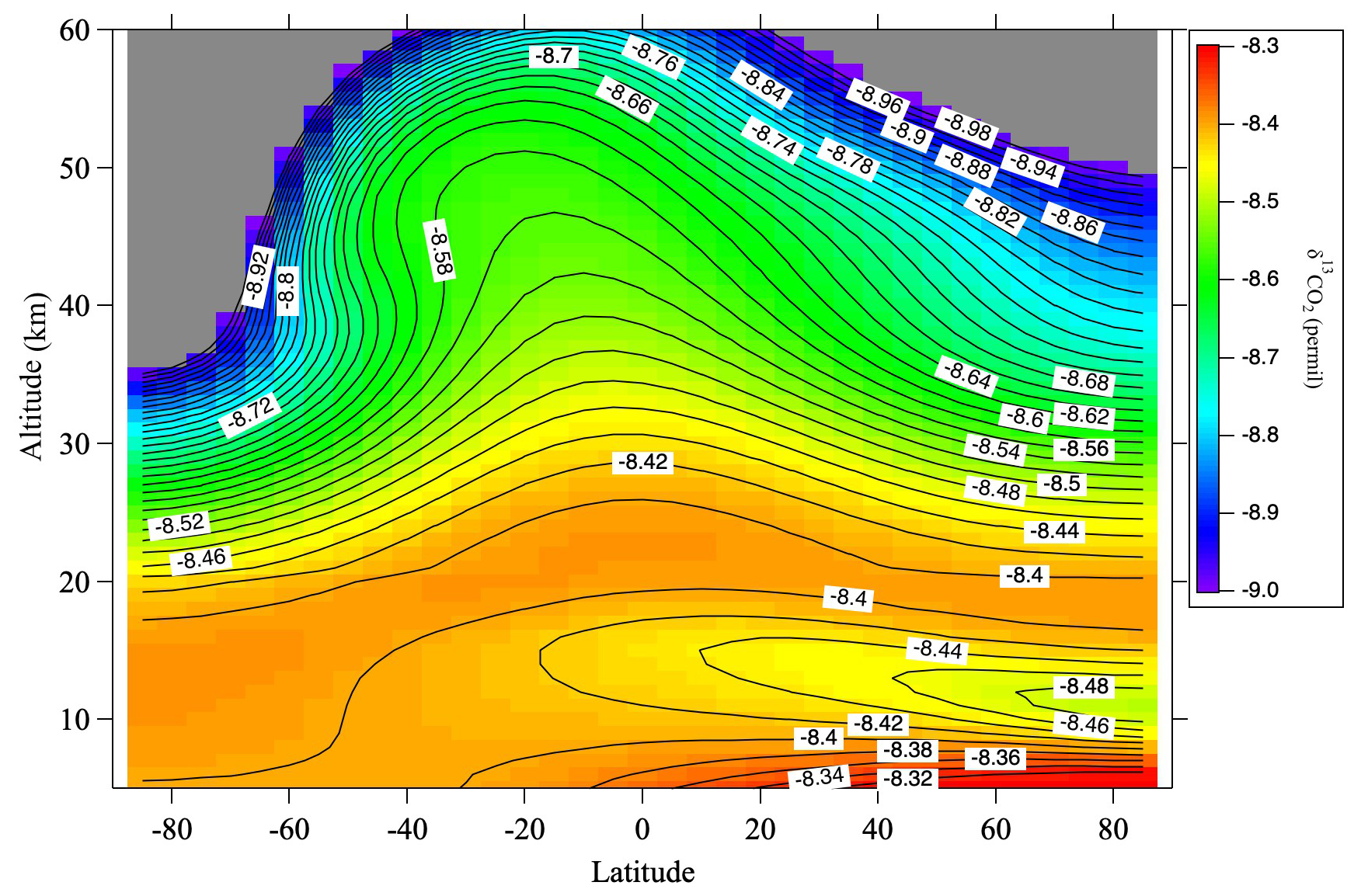
Figure 7Monthly average meridional distribution of δ13CO2 for August 2016 calculated using the SOCRATES model. Values lower than −9.0 ‰ are colored gray.
To investigate the vertical δ13CO2 profile in detail, we compared this model with our observations in Fig. 2c. The modeled δ13CO2 profile calculated without accounting for CH4 oxidation and GS increased with increasing altitude, and it was 0.14 ‰ higher at altitudes around 35 km than near the tropopause. Because the average rate of change of the long-term δ13CO2 record at MLO is −0.026 ‰ yr−1 for the period 1990–2022, the lag time at 35 km (0.14 ‰ divided by 0.026 ‰ yr−1) should be approximately 5.4 yr, nearly consistent with the calculated mean age of air (see Appendix C). If the effect of GS is further taken into account, the δ13CO2 decreased by approximately 0.07 ‰ at 35 km. The most significant impact on the modeled vertical profile was the effect of CH4 oxidation, which imparted a decrease of 0.26 ‰ at around 35 km. The vertical profile calculated including all effects exhibited a maximum at an altitude of about 20 km and then decreased monotonically at greater altitudes, mainly because of the effect of CH4 oxidation. However, the observed δ13CO2 profile increased to about 25 km altitude, indicating that the model results clearly overestimated the 13C depletion at higher altitudes. We note, though, that this model calculation included the seasonal cycle of tropospheric δ13CO2 and nearly reproduced the vertical distribution observed at 14–20 km altitude, which is mainly due to the upward propagation of the seasonal cycle from the upper troposphere to the lower stratosphere. Therefore, the difference between the observed and modeled results above 20 km was likely due mainly to an overestimation of the effect of CH4 oxidation.
One possible reason for this difference was an overestimation of CH4 loss at high altitudes. The observed CH4 mole fraction at ∼ 35 km altitude ranged between about 500 and 900 nmol mol−1 (Fig. 4), but the model results indicated a monthly average of about 600 nmol mol−1 for August, which was lower than the observed average value (see Fig. B1c in Appendix B). This discrepancy may be related to the fact that we weakened the mass stream function to reproduce a realistic age of air (see Appendix B). Another reason may be that the model simulation set the KIE on CH4 destruction in a simplified manner, with the apparent fractionation factors set uniformly at 0.9889 and 0.9866 for the lower and mid-stratosphere, respectively. In reality, the apparent fractionation factor for CH4 oxidation varies with altitude and latitude, and it is determined not only by a complex process that depends on the paths of air masses, but also by the chemical reactions that occur along the pathway (e.g., Röckmann et al., 2011). In this study, the KIEs on the CH4–CO–CO2 reaction processes were not calculated explicitly, but rather in a simplified manner, which may have resulted in an overestimation of the effect on δ13CO2. However, the present model simulation was able to prove our hypothesis that three factors – CH4 oxidation, GS, and air age – are essential to determining vertical δ13CO2 profiles.
3.5 Stratospheric potential δ13C
From the above discussion, we considered that the total amount of 13C in both CO2 and CH4, δ13CT, was conserved. We defined δ13CT as
which is quasi-conservative with respect to the KIEs associated with CH4 oxidation and consequent CO2 production in the stratosphere. Furthermore, as mentioned above, the effect of GS cannot be ignored in the stratosphere. Taking this into account, we defined the stratospheric potential δ13C, δ13CP, by correcting δ13CT for GS:
where ΔG(δ13CO2) denotes the GS correction for δ13CO2. Normally, ΔG(δ13CO2) gives a negative value, which means that it has a positive effect on δ13CP. This equation does not include the effect of GS on δ13CH4, but that contribution to δ13CP is negligibly small (about 10−4 ‰). Equations (9) and (10) were applied to the observed results to calculate stratospheric δ13CT and δ13CP profiles. For some air samples without δ13CH4 measurements, values were estimated from the CH4 mole fraction using a linear function fitted to the log–log plot of the observed data shown in Fig. 4. The 〈δG〉 values were also partially complemented by applying the average vertical profile. The average vertical profile of δ13CP thus calculated is shown in Fig. 8, alongside the CO2 mole fraction. The data plotted in this figure were detrended and normalized to August 2016. The CO2 mole fractions were also corrected for CH4 oxidation and GS, although the magnitude of these corrections was relatively small. δ13CP increased with increasing altitude from the tropopause to about 25 km, then was almost constant with increasing altitude. Unlike δ13CO2 (Fig. 2c), δ13CP was inversely correlated with the CO2 mole fraction. Figure 9 shows the temporal variations of the averaged δ13CO2, δ13CT, and δ13CP values above 24 km altitude. δ13CT and δ13CP changed almost in parallel with δ13CO2, showing a similar decreasing trend through time. The average difference from δ13CO2 was −0.079 ± 0.025 ‰ for δ13CT and −0.034 ± 0.027 ‰ for δ13CP.
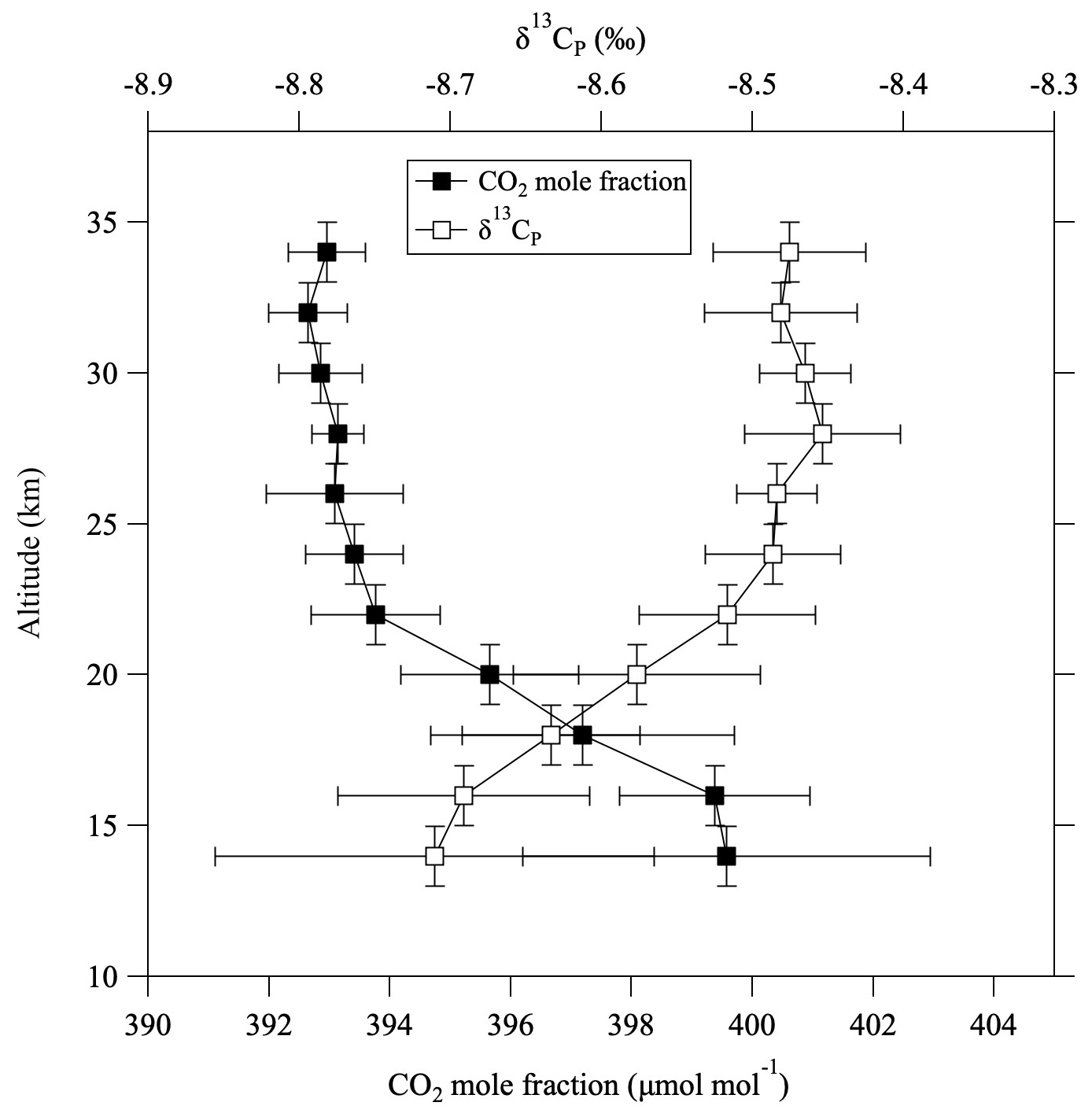
Figure 8Vertical profiles of CO2 mole fraction (closed squares) and stratospheric potential δ13C (δ13CP, open squares) based on their averages in 11 vertical bins over the period 1985–2020. All values were detrended and normalized to values in August 2016.
If δ13CP is truly a conserved quantity in the stratosphere, it should decrease over time with a time lag relative to the variation in the tropical upper troposphere. However, there are no long-term data records of δ13C values in CO2 and CH4 in the tropical upper troposphere. Therefore, we hypothetically synthesized a δ13CP record in the tropical upper troposphere based on surface data observed at MLO and in the equatorial Pacific region by INSTAAR (Michel et al., 2025, 2023). In our previous study, we employed CO2 mole fraction data obtained in the tropical upper troposphere using the Automatic Air Sampling Equipment in the Comprehensive Observation Network for TRace gases by AIrLiner (CONTRAIL) program (Machida et al., 2008; Sawa et al., 2008; Matsueda et al., 2015) to estimate the mean age of stratospheric air (Sugawara et al., 2018). A similar method was used in this study. We first estimated the average seasonal cycle of δ13CO2 in the tropical upper troposphere from the observed seasonal cycle of the CO2 mole fraction, assuming that the correlation between the seasonal cycles of the CO2 mole fraction and δ13CO2 in the upper troposphere was approximately −0.05 ‰ (µmol mol (Nakazawa et al., 1993). The long-term δ13CO2 trend was calculated from the observation data at MLO and then adjusted to match the observations in the equatorial Pacific region. The average seasonal cycles were then added to the long-term δ13CO2 trend estimated in the equatorial Pacific region. The difference between δ13CO2 values at the surface and those in the upper troposphere in the equatorial region is currently unknown. However, Sugawara et al. (2018) reported that the CO2 and SF6 ages in the TTL are 0.5–0.6 yr, consistent with the SF6 age of 0–1.5 yr in the TTL based on Michelson Interferometer for Passive Atmospheric Sounding (Stiller et al., 2008). Taking this into account, we assumed the difference between surface and upper troposphere δ13CO2 values in the equatorial region to be equivalent to 0.5 yr. Because systematic measurements of CH4 mole fraction and δ13CH4 in the upper troposphere were performed in the CONTRAIL program (Umezawa et al., 2012), the MLO data obtained by INSTAAR were adjusted to fit the CONTRAIL data in the equatorial upper troposphere to produce long-term records of CH4 mole fraction and δ13CH4 in the equatorial upper troposphere. Because the seasonal cycle of δ13CH4 in the equatorial upper troposphere is not clear (Umezawa et al., 2012), the seasonal cycle was ignored in the calculation of the upper tropospheric δ13CP. Since GS, i.e., 〈δG〉−〈δG〉0, should be zero in the equatorial upper troposphere, tropospheric δ13CP=δ13CT.
As seen in Fig. 9, δ13CO2 values did not differ significantly between the troposphere and the mid-stratosphere. However, the δ13CP values calculated from observed values in the mid-stratosphere were, on average, about 0.14 ± 0.03 ‰ higher than those calculated for the tropical troposphere. This means that δ13CP is conservative in the stratosphere and that the decreasing tropospheric δ13CP trend through time propagates into the stratosphere with a time delay. Roughly calculated, this lag time is about 5 yr, consistent with the age of air estimated from CO2 mole fractions (Appendix C) and the model calculation. If the mid-stratospheric δ13CP values were shifted by −5 yr, they were in good agreement with the tropospheric δ13CP trend (Fig. 9).
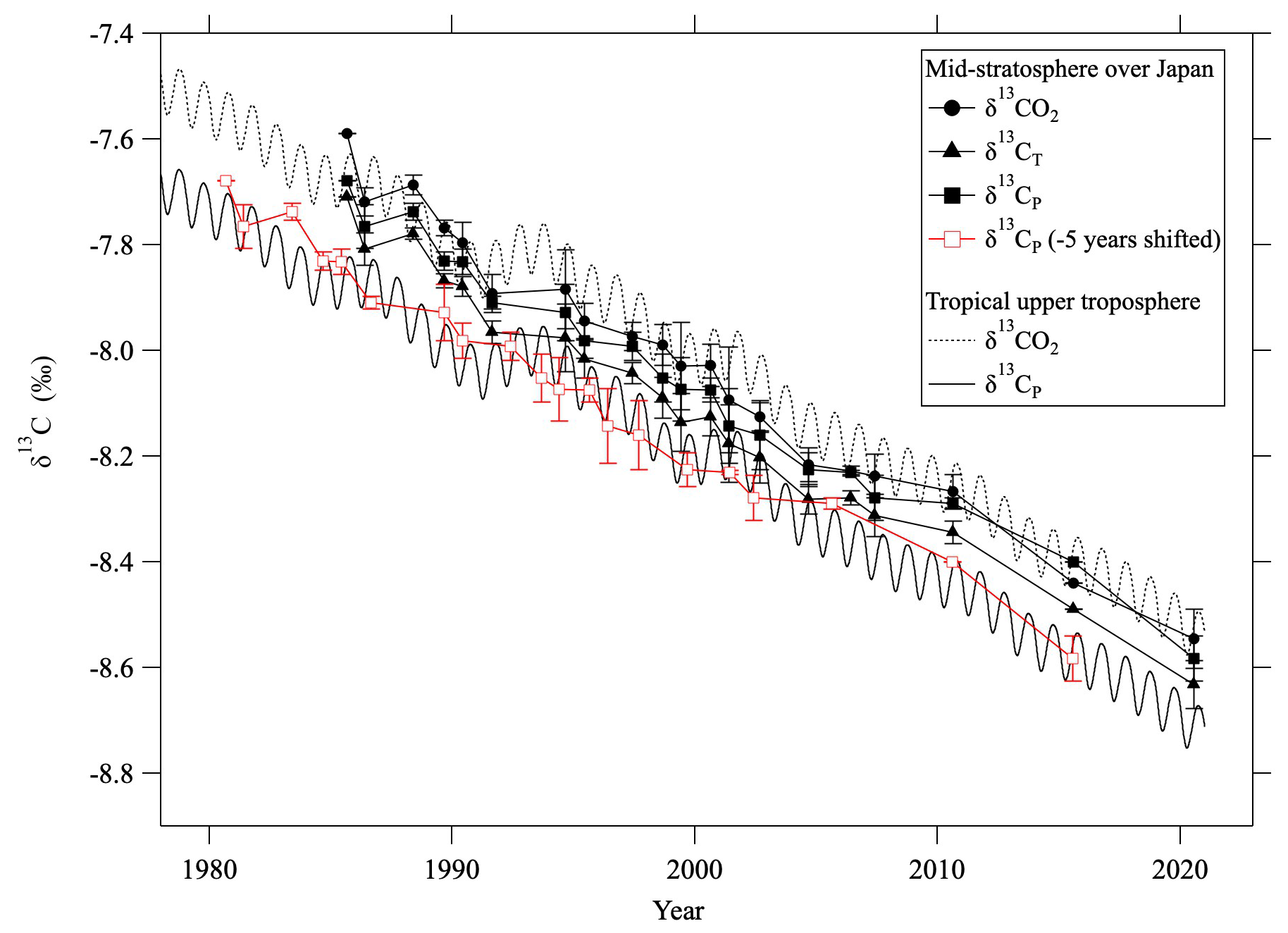
Figure 9Average mid-stratospheric (>24 km altitude) values of δ13CO2 (closed circles), δ13CT (closed triangles), and δ13CP (closed squares) over Japan. δ13CO2 and δ13CP in the tropical upper troposphere are shown by dashed and solid lines, respectively. Red squares show the mid-stratospheric δ13CP shifted by −5.0 yr.
It is thus possible to estimate the mean age of air using δ13CP, which can serve as an additional air age tracer. Therefore, we estimated the mean age of air not only from the CO2 mole fraction but also from δ13CP using a convolution method. Details of the age estimation method are described in Appendix C. The total uncertainty on individual δ13CP ages was estimated to be ± 1.9 yr. This rather large uncertainty means that δ13CP, with its smaller tropospheric secular change compared to other age tracers, was somewhat disadvantageous for age estimations. Because some of the δ13CP values in the lower stratosphere showed high variability, with values lower than the tropospheric reference record, the convolution method could not provide reasonable age estimations for those data. We attributed these irregularities and low values to the large seasonal variations in the upper troposphere and the stratosphere–troposphere air exchange in the lower stratosphere. Therefore, only mid-stratospheric (>24 km altitude) data were taken into account for age estimations, and the average values of the mean age were calculated.
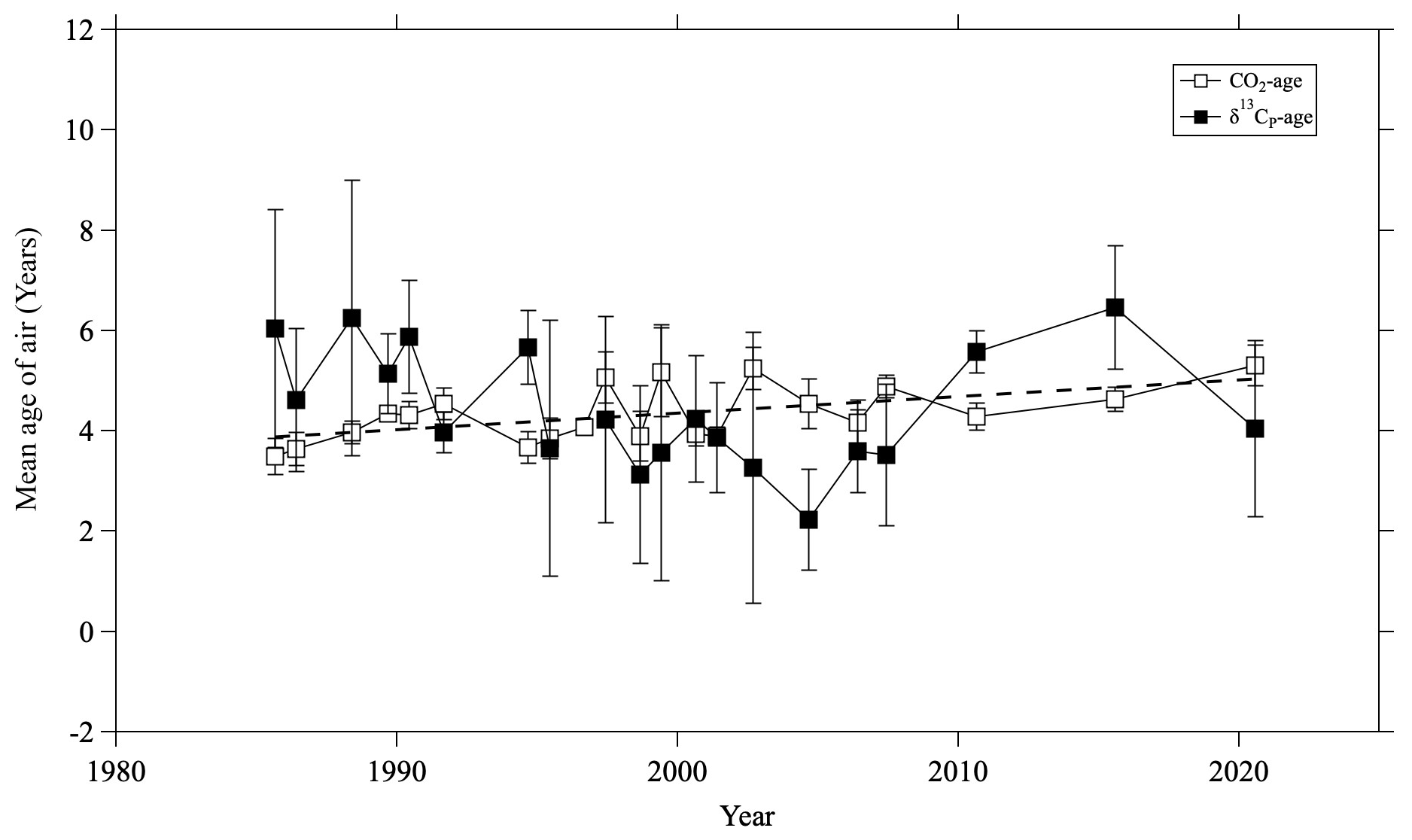
Figure 10Temporal variations of averaged δ13CP ages (closed squares) and CO2 ages (open squares) in the mid-stratosphere (>24 km altitude) over Japan. The dashed line is a linear least-squares fit to the CO2 ages. The linear fit to the δ13CP age is not shown because the trend is not significant.
The results are shown in Fig. 10, along with the mean age calculated from the CO2 mole fraction using the same method. The average δ13CP and CO2 ages were 5.5 ± 1.6 and 4.4 ± 0.6 yr, respectively. Because the uncertainty of the δ13CP age was large, no significant difference was apparent between the two values. To examine the long-term trend of mean age, a linear function was fitted to both the CO2 and δ13CP age data using the least squares method. A slightly positive trend (0.04 ± 0.01 yr yr−1) was observed for the CO2 age, but no specific trend was observed for the δ13CP age. It is important to note that our data were unevenly distributed over time, with sparse observations in recent years and more frequent observations before 2007. This made it difficult to clarify the exact 35 yr trend from our data alone. At present, the δ13CP age has a relatively large uncertainty, making it unsuitable for use as an additional constraint for the age estimation. However, considering that the average δ13CP age was estimated to be 5.5 ± 1.6 yr, the concept of stratospheric δ13CP itself would be valid.
Previous studies on δ13CO2 have mainly focused on observations in the troposphere and on elucidating the global CO2 budget based on monitoring programs. However, little research has been performed on δ13CO2 in the stratosphere, and the mechanisms of its distribution and variations are not fully understood. This study presented a comprehensive analysis of stratospheric δ13CO2 obtained from balloon observations over Japan during the period 1985–2020. The following four factors were found to be important for understanding the variations of δ13CO2 in the stratosphere. (1) Appropriate corrections for the MIE on 17O and 18O in isotopic analyses are essential. However, even after applying this correction, we found that the average values of δ13CO2 in the northern mid-latitude mid-stratosphere and the tropical troposphere were almost identical, despite the secular decreasing δ13CO2 trend and the expected time lag (age of air). We attributed this strange agreement to the other three factors: (2) airborne production of 13C-depleted CO2 by the oxidation of CH4 in the stratosphere, (3) GS of 12CO2 and 13CO2 in the stratosphere, and (4) propagation of the decreasing tropospheric δ13CO2 trend into the stratosphere. Based on these considerations, we introduced “stratospheric potential δ13C” (δ13CP) as a new concept and inspected δ13CP in terms of the age of air. The average δ13CP age was calculated to be 5.5 ± 1.6 yr for the period of 1985–2020. Because the δ13CP age has larger uncertainties than the CO2 age at present, it is difficult to refine the mean age estimation. However, δ13CP in the mid-latitude mid-stratosphere decreased over time with a time delay and it was found to be quasi-conservative in the stratosphere.
The observation of numerous stratospheric air constituents, as in this study, requires the collection of a large amount of stratospheric air with a high-quality method and multi-component gas analyses of each air sample. GS of atmospheric constituents in the stratosphere was first revealed by our previous balloon observations, and this knowledge is indispensable for a better understanding of the variations of stratospheric δ13CO2. The quality of the air samples is particularly important for observing GS in the mid-stratosphere, because even slight separations due to molecular diffusion during air sampling could prevent GS observations. At present, the only method capable of such sampling is our cryogenic air sampler.
The 2-D model reproduced the basic structure of stratospheric δ13CO2 and elucidated the mechanisms governing δ13CO2 behavior. However, our numerical model simulations remained insufficient because the mean age of stratospheric air could not be reproduced adequately without arbitrarily tuning the model. Accordingly, our model overestimated the effect of airborne CO2 production via CH4 oxidation because the reaction process was treated in a very coarse manner. To improve upon this model, it will be necessary to incorporate KIEs into all reactions related to stratospheric carbon. Although a two-dimensional model was used in this proof-of-concept study, a three-dimensional model is desirable for more realistic simulations in the future.
The gravitational separation (GS) of atmospheric components in the stratosphere was first reported from our balloon observations in Japan (Ishidoya et al., 2006, 2008a, 2013), in which the isotopic and elemental ratios of major atmospheric compositions, such as δ15N in N2, δ18O in O2, and δ(Ar N2), were measured by mass spectrometry at the National Institute of Advanced Industrial Science and Technology (AIST; e.g., Ishidoya et al., 2006, 2008a; Sugawara et al., 2018). Technical aspects of our mass spectrometry analyses are described in detail in Ishidoya and Murayama (2014). In this study, we used δ15N, δ18O, and δ(Ar N2) to evaluate GS, which are respectively defined as
and
where “sp” and “st” denote the sample and standard gases, respectively. The 1σ reproducibilities of our δ15N, δ18O, and δ(Ar N2) measurements were ±2, ±5, and ±7 per meg, respectively, which was sufficient to detect GS in the stratosphere. Ishidoya et al. (2013) concluded that vertical variations of the isotopic ratios of major atmospheric components are dominated by differences in molecular mass, based on mass-dependent relationships among related molecules such as δ15N in N2, δ18O in O2, and the Ar N2 ratio. Such GS in the stratosphere is known to be proportional to the mass difference, as would be expected for pure molecular diffusion, even though eddy diffusion obviously far exceeds molecular diffusion in the stratosphere. Therefore, GS can be expressed as
where Δm is the mass number difference, the suffix “0” refers to the value before GS occurs (i.e., corresponding to the relevant value in the upper troposphere), and 〈δG〉 is the average of δ values normalized to Δm=1 as reported by Ishidoya et al. (2013) and Sugawara et al. (2018). 〈δG〉 is defined by
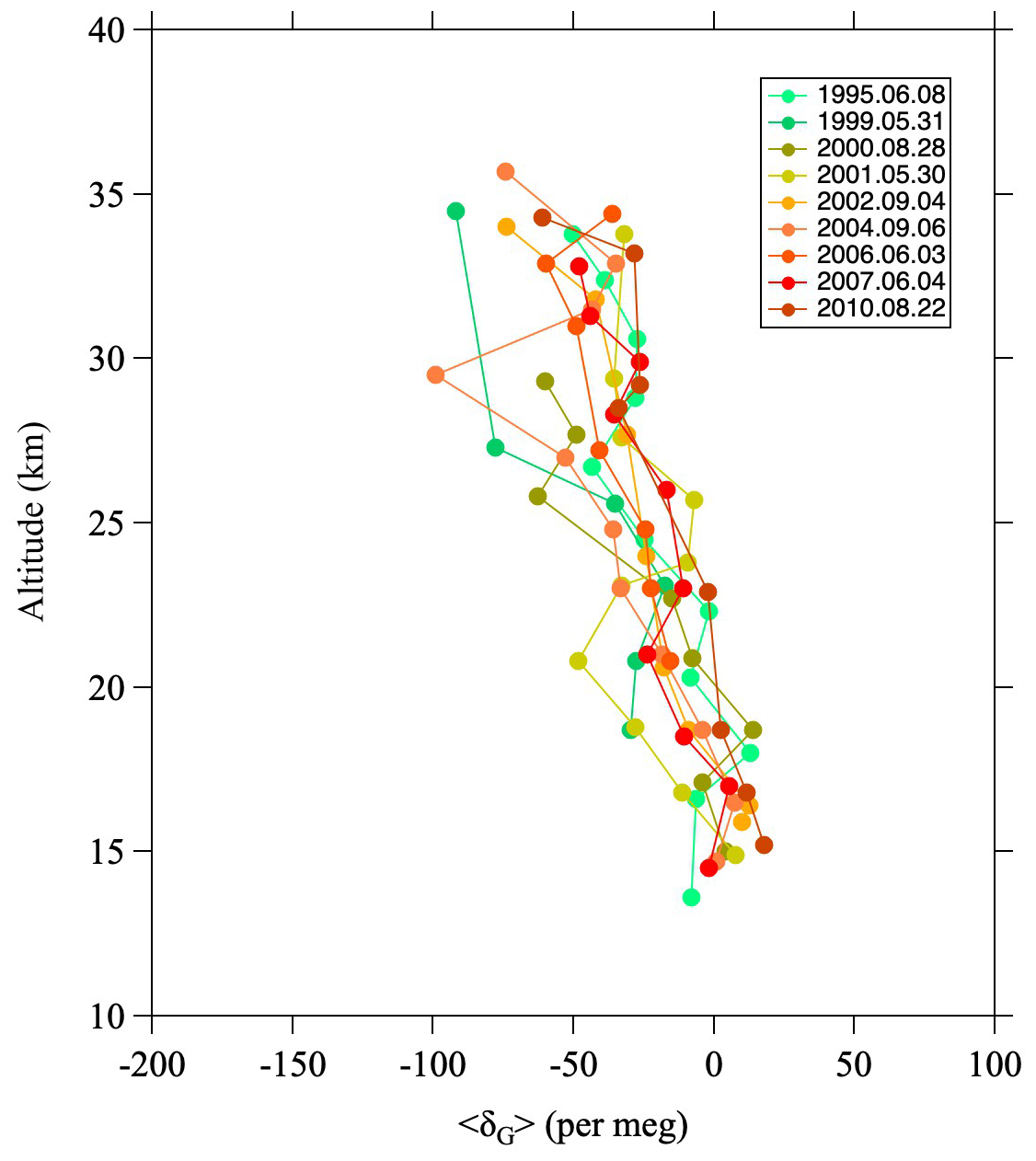
Figure A1Vertical profiles of GS, 〈δG〉, observed over Japan (Ishidoya et al., 2013).
Figure A1 shows vertical profiles of 〈δG〉 obtained from balloon observations over Japan (Ishidoya et al., 2013). The value of 〈δG〉 decreases gradually with increasing altitude. By fitting a linear function to the vertical profiles, the average difference of 〈δG〉 between the lowermost and uppermost altitudes was found to be −66 ± 17 per meg. Here, it is important to mention the relationship between the GS of δ18O and photochemical processes. As mentioned in Sect. 2.2., in the stratosphere, O3 and CO2 are significantly enriched in 17O and 18O by MIEs associated with photochemical reaction processes. Because stratospheric O2 molecules are the source of the 17O and 18O enrichment, δ17O and δ18O values of O2 are thought to be lower in the stratosphere than in the troposphere due to photochemical processes. This assumption is crucial not only for the Dole–Morita effect, but also for the 17O budget of tropospheric O2 (Bender et al., 1994; Luz et al., 1999; Luz and Barkan, 2011; Ishidoya et al., 2025). However, δ18O values of stratospheric O2 measured in our balloon observations show a very large decrease, i.e., by approximately −130 per meg, at around 35 km that is almost entirely dominated by GS. This decrease is probably also due in part to photochemical effects, but their contribution should be negligible, amounting to only a few per meg (Ishidoya et al., 2025).
We used a two-dimensional model of the middle atmosphere (SOCRATES) developed by the National Center for Atmospheric Research (NCAR; Huang et al., 1998; Park et al., 1999; Khosravi et al., 2002). The advantage of using this model is that we have already calculated 13C16O2 and 12C16O2 and examined stratospheric GS in our previous studies (Ishidoya et al., 2013; Sugawara et al., 2018). Therefore, we give only a brief description of the model calculation here. To reproduce GS, the molecular diffusion flux must be calculated. The vertical component of the molecular diffusion flux () is given by
where ni, , mi, and are the number density, molecular diffusion coefficient, molecular mass, and thermal diffusion factor of atmospheric constituent i, respectively, and g, R, and T are the gravitational acceleration, gas constant, and temperature, respectively (Banks and Kockarts, 1973). However, because the thermal diffusion flux is not important in this case, we ignored it in the model calculations. Because it is cumbersome to include all CO2 isotopologues in the model calculations, only 12C16O2 and 13C16O2 were calculated independently in the model, and δ13CO2 was approximated using the respective mole fractions of n(13C16O2) and n(12C16O2) as
Because δ13CO2 has a large seasonal cycle in the troposphere, we used NOAA/GML and INSTAAR data to give latitude-dependent seasonal cycles as boundary conditions at the model surface. In addition, a secular decrease of δ13CO2 in the troposphere was given by the MLO data (Michel et al., 2025). SOCRATES originally included a series of carbon reactions starting with CH4 oxidation. Model calculations were repeated with and without GS, the secular trend and seasonal cycle of tropospheric δ13CO2, and airborne CO2 production, and comparisons were made between them (Fig. 2c). The chemical budget of CO2 in SOCRATES is calculated as
where kCO+OH and are the reaction rates of their respective chemical reactions, and M is the third body which carries off excess energy in a termolecular reaction. Because CO2 photodissociation is important only above the mesosphere (e.g., Garcia et al., 2014), it was ignored in this study. KIEs in the CH4–CO–CO2 chain reaction were not included explicitly in these simulations but were rather treated in a coarse manner as follows. Simulations were performed with and without the chemical production of CO2. Taking the CO2 mole fractions simulated with (“wP”) and without chemical production (“nP”) to be n(CO2)wP and n(CO2)nP, respectively, the CO2 mole fraction difference between the two simulations, n(CO2)wP−n(CO2)nP, was calculated as the apparent CO2 production in the stratosphere. The apparent CO2 production is distinct from the local production calculated in Eq. (B3) because it is an apparent value influenced by the accumulated chemical processes along the pathway of the air mass, including transport and mixing. A typical value of the apparent CO2 production was simulated to be about 1.5 µmol mol−1 at 35 km altitude in the mid-latitudes. Similarly, the δ13CO2 value simulated excluding chemical CO2 production was expressed by δ13CO2,nP. After both these simulations, the δ13CO2 value including airborne CO2 sources from the CH4 destruction, δ13CO2,wP, was calculated as
In this equation, δ13CH4_L was calculated using Eq. (6) assuming apparent fractionation factors of 0.9889 and 0.9866 at lower (<24 km) and upper (>24 km) altitudes, respectively (see Sect. 3.2). Note that δ13CH4_L values were estimated from the apparent fractionation factors by assuming Rayleigh distillation relationships between vertical profiles of CH4 mole fraction and δ13CH4. Therefore, these apparent fractionation factors differed from fractionation factors sensu stricto and were influenced by the accumulated KIEs along the pathway of the air mass, as with the apparent CO2 production described above. Equation (B4) means that δ13CO2 was simulated by combining the apparent fractionation factors estimated from observations with the apparent CO2 production simulated by the model.
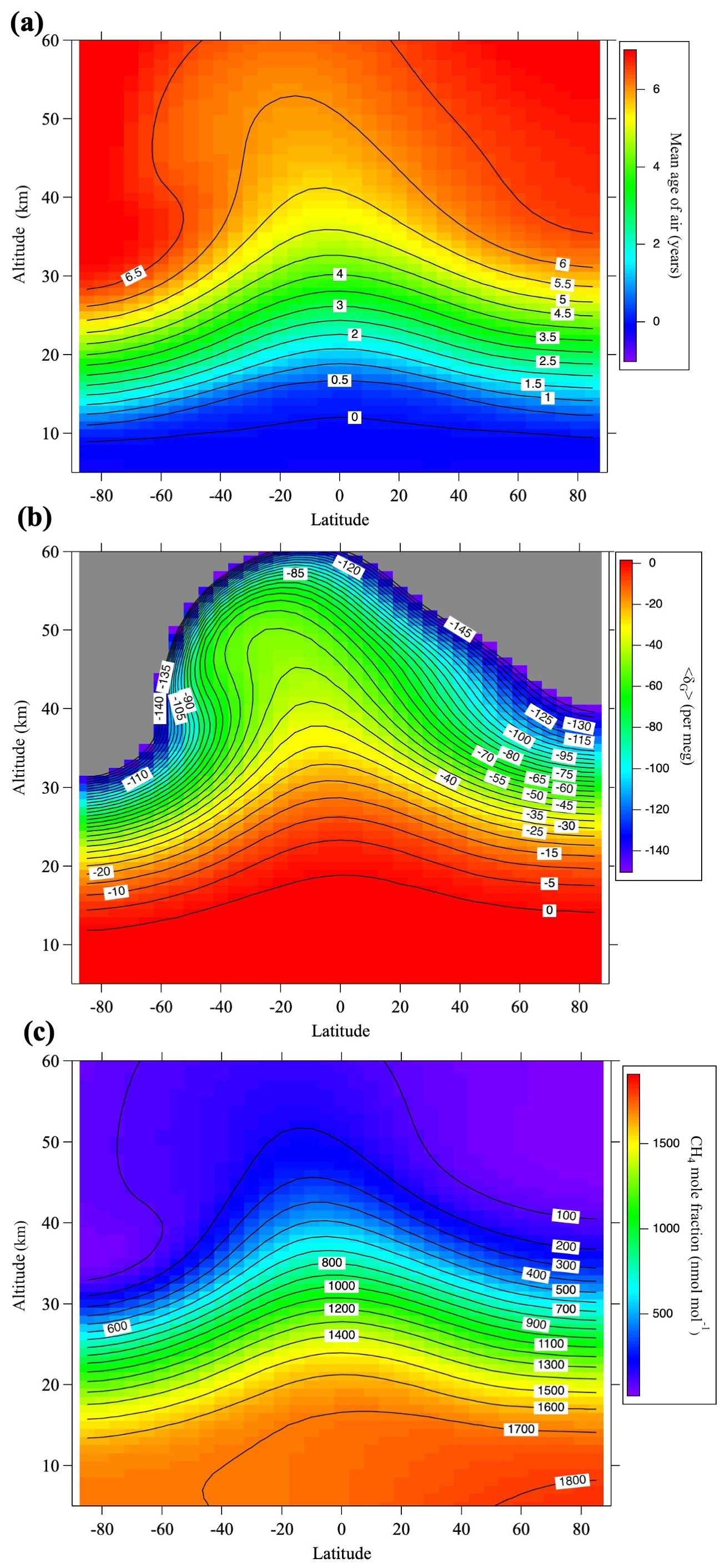
Figure B1(a) Monthly average meridional distributions of the mean age of air for August 2016 calculated using the SOCRATES model. (b) As in (a), but for gravitational separation, 〈δG〉. Values lower than −150 per meg are shown in gray. (c) As in (a), but for the CH4 mole fraction.
A virtual clock tracer was used to calculate the mean age of air. However, previous research has shown that the speed of the Brewer–Dobson circulation in SOCRATES is fast, leading to an underestimation of age of air. Therefore, similar to Sugawara et al. (2018), we reduced the mass stream function to approximate a realistic mean age, which also improved the reproduction of GS. The model results of GS reproduced well the almost linear decrease with increasing altitude above the tropopause in the observational results (Fig. A1). The model calculations were carried out over the 25 yr from 1995 to 2020. The monthly average meridional distribution of the mean air age, GS, and CH4 mole fraction calculated by SOCRATES for August 2016 is shown in Fig. B1.
We estimated the mean age of air from δ13CP using the same method that was previously applied to the CO2 age estimation (Ray et al., 2017; Sugawara et al., 2018). The calculation of CO2 age based on our balloon observations has been partly reported in previous studies (Engel et al., 2002; Ishidoya et al., 2013; Umezawa et al., 2025). The CO2 ages estimated using air samples collected from 1995 to 2010 were published as supplementary data in Ishidoya et al. (2013). However, the method of age estimation was subsequently updated in Sugawara et al. (2018). In this study, we further improved the estimation method by correcting the δ13CP and CO2 mole fraction data for GS, creating a new 35 yr record. We estimated the mean age of air using the convolution method (Ray et al., 2017; Leedham Elvidge et al., 2018; Sugawara et al., 2018). This method has been described in detail by Sugawara et al. (2018), so only a brief explanation is given here. Hypothetical age spectra were used in the convolution method to estimate the mean age (Waugh and Hall, 2002). Expected temporal variations in the age tracer in the stratosphere, x(Γ,t), were calculated by convolution of the tropospheric reference curve, x0(t), and the age spectrum, G(Γ,t):
where TB is the integration time interval. TB should theoretically be ∞, but we truncated it to 20 yr for the calculations. Actual age spectra, G(Γ,t), are usually unknown. Therefore, we used the inverse Gaussian distribution (Waugh and Hall, 2002):
Here, Δ denotes the width of the age spectrum, and it was parameterized using the mean age (Γ), i.e., the ratio of moments, . This value has usually been assumed to be 0.7 yr, as suggested by Hall and Plumb (1994) based on results from a general circulation model calculation, and it was used to estimate SF6- and CO2-derived mean ages in the northern mid- and high-latitude stratosphere (Engel et al., 2002). Recently, however, Fritsch et al. (2020) reported that a value of 1.25 yr is better for estimating ages from the SF6 mole fraction. Therefore, we used 1.25 yr for the ratio of moments in this study. After calculating the convolutions, the mean age was determined by substituting the observed values, xobs, into the inverse function, Γ (x, t). The convolutions are shown in Fig. C1 along with the calculated δ13CP values.
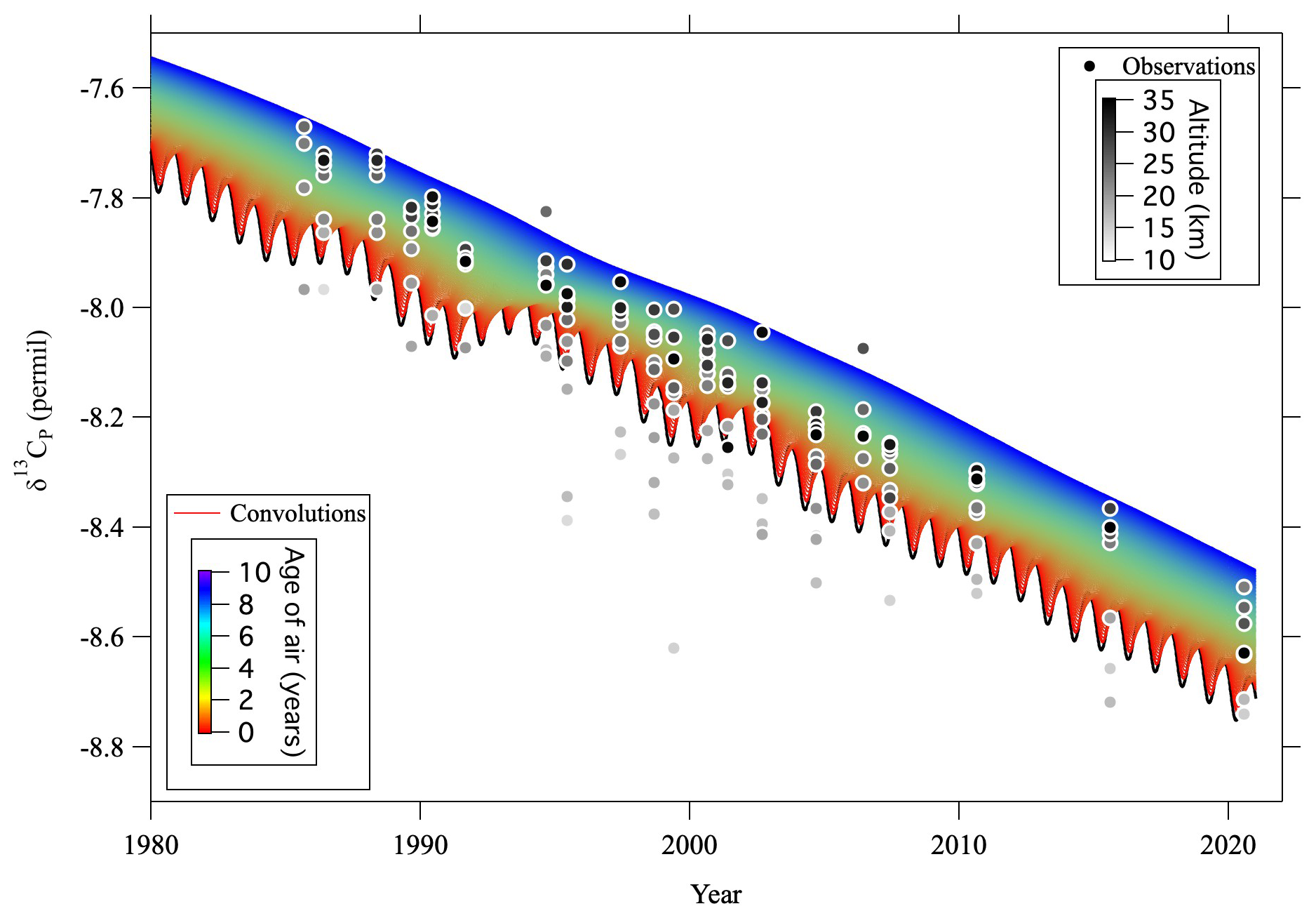
Figure C1δ13CP observed over Japan (circles) and convolutions of δ13CP (colored lines) calculated from the tropospheric reference record and age spectrum. Observation altitudes of δ13CP are indicated by the gray scale of the symbol colors.
It should be noted that uncertainties of individual mole fraction or isotopic ratio measurements, as well as of the tropospheric reference record, lead to uncertainties of individual age estimates (e.g., Leedham Elvidge et al., 2018). Umezawa et al. (2025) estimated a total uncertainty of 0.72 yr for our CO2 ages. Similarly, we estimated the uncertainty for δ13CP ages by adding normal pseudo-random numbers to all measured values and then calculating δ13CP values using Eqs. (9) and (10). The uncertainty of the individual δ13CP values was nearly identical to that of δ13C, ±0.02 ‰. On the other hand, the uncertainty of the tropospheric reference record for δ13CP was calculated to be ±0.03 ‰. This value is in close agreement with the standard deviation obtained by applying curve fitting to the MLO δ13C data used as the tropospheric reference record. Taking into account all these uncertainties and the tropospheric trend of −0.026 ‰ yr−1, the total uncertainty of the individual δ13CP ages was estimated to be ±1.9 yr. The ratio of moments and its influence on age estimates have been examined in previous studies (e.g., Hauck et al., 2019; Nguyen et al., 2021). In this study, the mean age calculation was repeated as a sensitivity test for different ratios of moments ranging from 0.05 to 2.00 yr. As a result, the mean ages derived from δ13CP were found to be within the observational uncertainties of the δ13CP age. The δ13CP age is larger than the CO2 age (4.4 ± 0.6 yr) by about 1.1 yr on average. In this regard, δ13CT was calculated from the observed CH4 mole fraction and its δ13C, assuming a closed system (Eq. (9)). However, actual chemical processes do not occur in a closed system, and atmospheric mixing processes always result in apparent fractionation being smaller than true fractionation (Rahn et al., 1998; Kaiser et al., 2006; Toyoda et al., 2018). In this study, the isotopic effect of CH4 oxidation was calculated based on the apparent fractionation factor. This would result in an underestimation of the CH4 oxidation effect in Eq. (9) and an overestimation of the δ13CP age. To solve this problem, it is necessary to explicitly incorporate the isotope effect of CH4 into the model, which will be a future challenge.
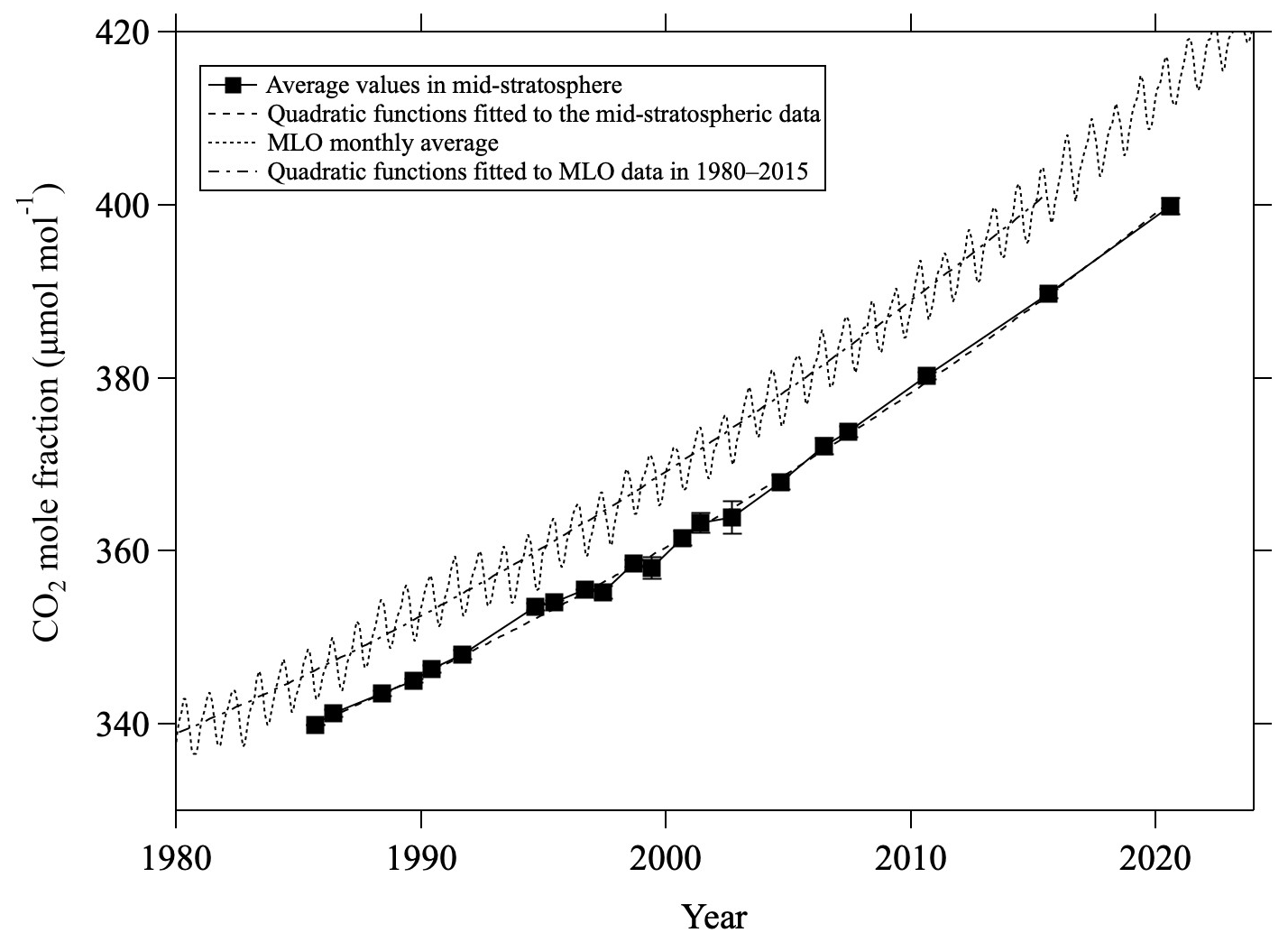
Figure C2Average values of mid-stratospheric CO2 mole fraction (>24 km altitude) over Japan (closed squares). Monthly average CO2 mole fractions at Mauna Loa observed by NOAA/GML are shown by the dotted line. Quadratic functions fitted to the mid-stratospheric data (dashed line) and annual average MLO data from 1980 to 2015 (dashed-dotted line) are also shown.
Corresponding to the secular decrease of δ13CO2, an increase in the CO2 mole fraction was also clearly observed in the mid-stratosphere. To compare the temporal variations of the CO2 mole fraction in the mid-stratosphere with those in the troposphere, the average values above 24 km altitude were calculated for each year, shown in Fig. C2 alongside the monthly average data obtained by NOAA/GML at MLO. The results of the sixth WMO/IAEA Round Robin Comparison Experiment showed that our CO2 mole fraction values were approximately 0.2 µmol mol−1 higher than the NOAA/GML values on the WMO-CO2-X2007 scale; accordingly, our CO2 mole fraction values are plotted in Fig. C2 only after subtracting 0.2 µmol mol−1. The CO2 mole fraction in the mid-stratosphere over Japan increased monotonically and reached about 400 µmol mol−1 in 2020, lagging behind that at MLO by about 5 yr. The average rate of change of the CO2 mole fraction in the mid-stratosphere was calculated to be 1.68 ± 0.04 µmol mol−1 yr−1 by applying the least-squares method to the observed values. However, this increasing trend was not linear, gradually steepening over the last 40 yr. Therefore, a quadratic function of the form n(CO) would better represent the observed variations. Here, t (years) is the elapsed time since 1980. The coefficients of the CO2 mole fraction in the mid-stratosphere, K0, K1, and K2, were calculated to be 333.7, 1.0448, and 0.0147 55, respectively. The standard deviation of residuals was 0.8 µmol mol−1. The same procedure was applied to the annual mean CO2 mole fractions at MLO for the period 1980–2015, considering a time lag of 5 yr between the mid-stratosphere and the troposphere. The coefficients K0, K1, and K2 for the MLO data were calculated to be 338.85, 1.2307, and 0.0145 77, respectively. As shown in Fig. C2, the two quadratic functions thus obtained agreed well to within ±0.7 µmol mol−1. This result suggests that even temporal changes in the rate of increase in the troposphere propagate to the mid-stratosphere with a certain time delay.
As described in the main text, the influences on CO2 production of CH4 oxidation and GS should be considered to precisely estimate the CO2 age. Therefore, the CO2 mole fraction was corrected as follows. At first, the tropical upper tropospheric CH4 data, , were created by adjusting the annual mean data at MLO to fit the CONTRAIL data in the tropical upper troposphere (Umezawa et al., 2012). Then, we corrected xobs as
where xcor and denote the corrected CO2 mole fraction and observed CH4 mole fraction, respectively. ΔG is the correction for GS (see Sect. 3.3.). The CO2 mole fractions corrected for CH4 oxidation and GS are shown with the convolutions in Fig. C3. Finally, the CO2 age was determined as . We note that Eq. (C3) contains the mean age of air, Γ, because the CH4 mole fraction in the troposphere has increased with time and CH4 is destroyed over the air transport pathway from the tropical upper troposphere to the observation altitude in the stratosphere. Therefore, was solved iteratively by starting from . This iteration converged sufficiently after the second time of calculation. The vertical profiles of , corresponding to CO2 age, are shown in Fig. C4.
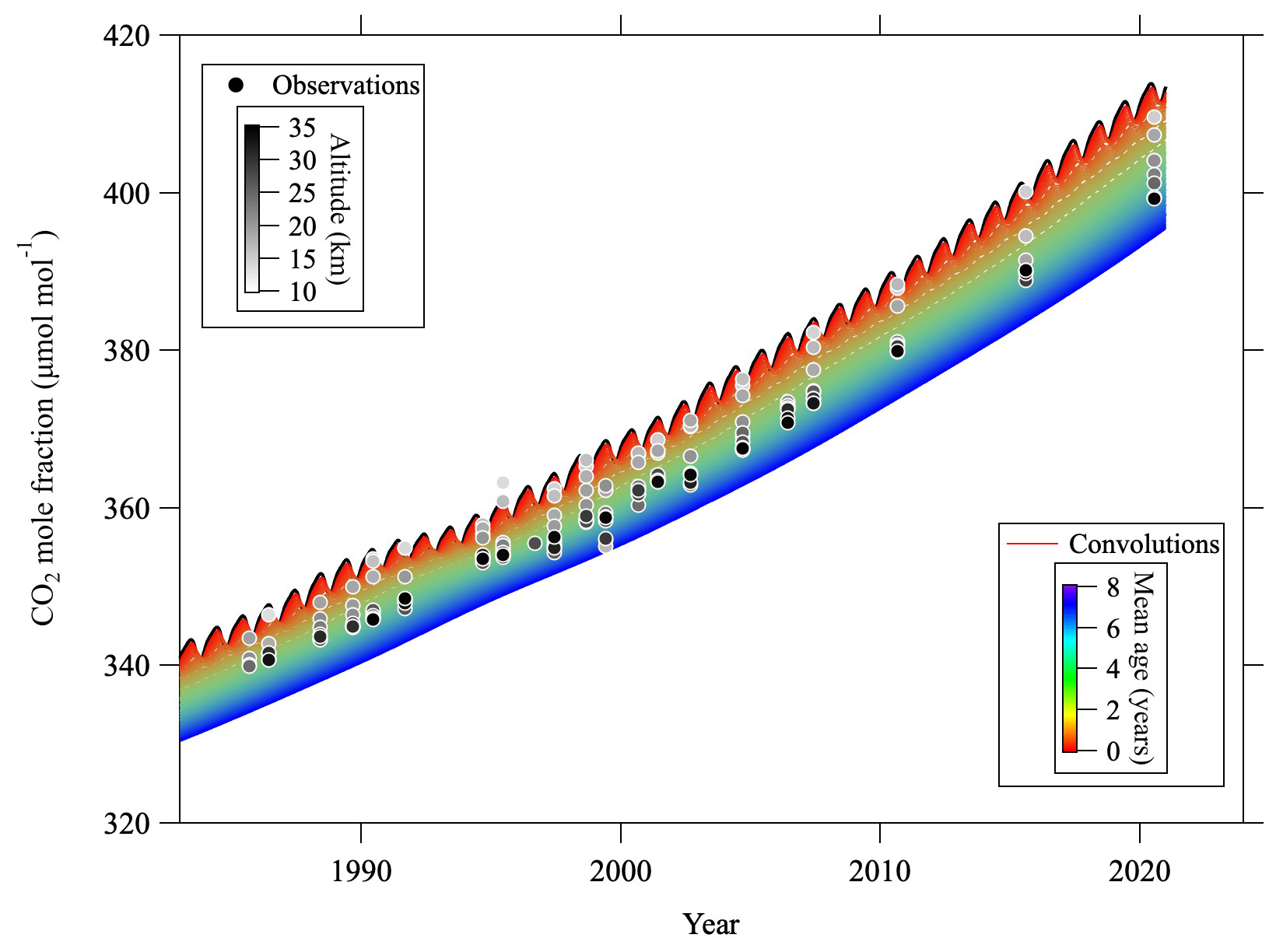
Figure C3CO2 mole fractions observed over Japan (circles) and convolutions (colored lines) calculated from the tropospheric reference record and age spectrum. Observation altitudes of the CO2 mole fraction are indicated by the gray scale of the symbol colors. Note that the CO2 mole fractions plotted have been corrected for CH4 oxidation and GS.
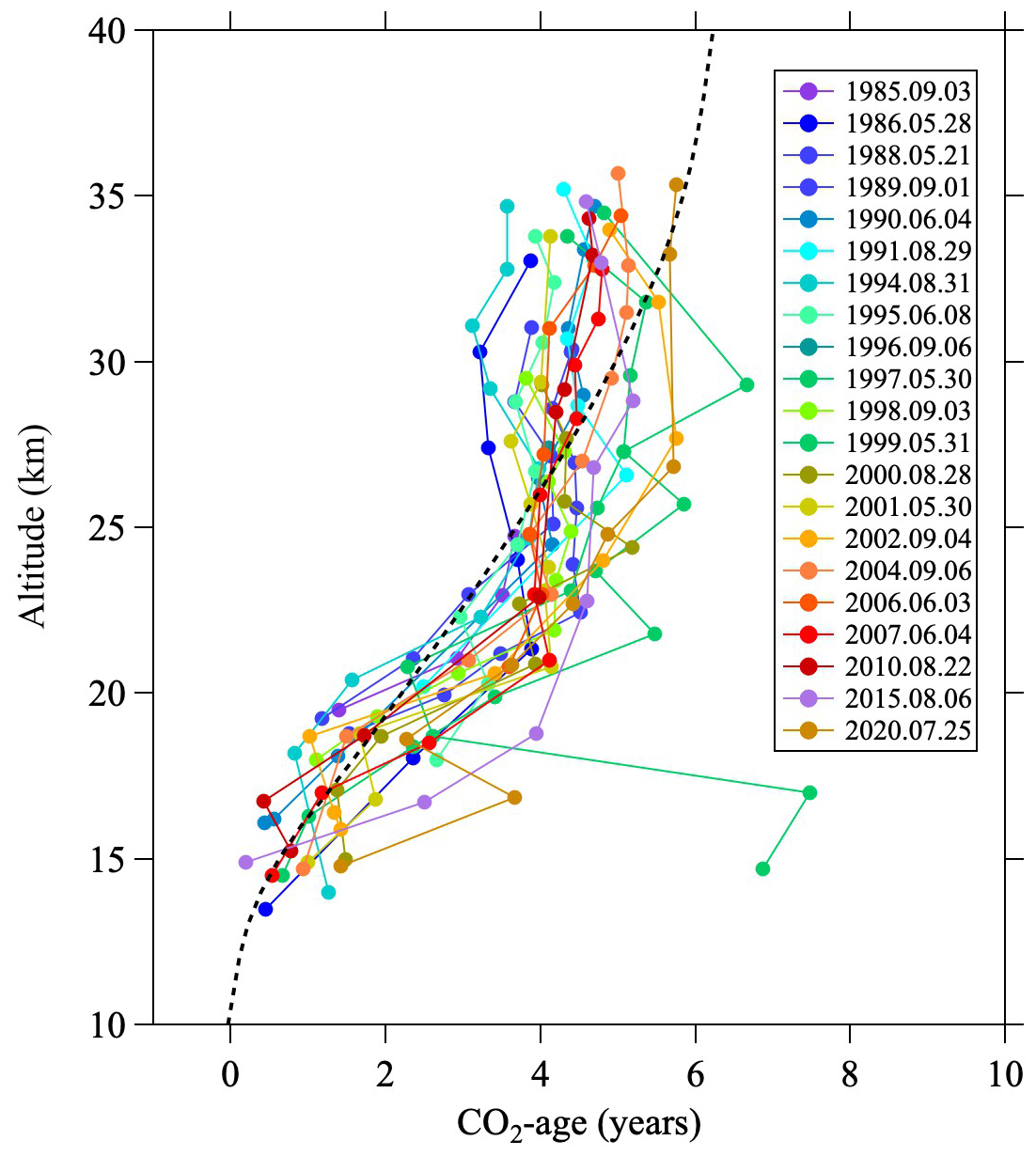
Figure C4Vertical profiles of the CO2 age calculated from CO2 mole fractions from 1985 to 2020. The monthly average vertical profile of the mean age of air for August 2016 calculated using the SOCRATES model is shown by the dashed line.
The observational data obtained by our balloon measurements are included as a Supplement to the paper.
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-11895-2025-supplement.
SS designed the study, conducted the measurements of mole fractions of greenhouse gases, and drafted the manuscript. SM, TN, SA, and SS conducted the measurements of carbon isotopic ratios of CO2. SM, SS, and TU conducted the measurements of carbon isotopic ratios of CH4. SI conducted the measurements of isotopic and elemental ratios of atmospheric major compositions. KI conducted the measurements of N2O mole fraction. HH, TN, SA, SM, and SS conducted the balloon experiments. SI, ST, and DG participated in balloon experiments. All authors approved the final paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We deeply thank the Scientific Ballooning (DAIKIKYU) Research and Operation Group of the Institute of Space and Astronautical Science (ISAS), JAXA, Japan. We also deeply thank Sylvia E. Michel, Kerstin Braun, Justin Reid Clark, and John Ortega at INSTAAR, University of Colorado, and John Mund at NOAA GML for providing us their isotopic data.
This study was supported by Japan Society for the Promotion of Science KAKENHI grants (grant nos. 22H05006, 23H00513, and 23K11396).
This paper was edited by Gabriele Stiller and reviewed by two anonymous referees.
Aoki, S., Nakazawa, T., Machida, T., Sugawara, S., Morimoto, S., Hashida, G., Yamanouchi, T., Kawamura, K., and Honda, H.: Carbon dioxide variations in the stratosphere over Japan, Scandinavia and Antarctica, Tellus B, 55, 178–186, https://doi.org/10.1034/j.1600-0889.2003.00059.x, 2003.
Assonov, S. S. and Brenninkmeijer, C. A. M.: A redetermination of absolute values for 17RVPDB-CO2 and 17RVSMOW, Rapid Commun Mass Spectrom., 17, 1017–1029, https://doi.org/10.1002/rcm.10111, 2003.
Assonov, S. S., Brenninkmeijer, C. A. M., Schuck, T. J., and Taylor, P.: Analysis of 13C and 18O isotope data of CO2 in CARIBIC aircraft samples as tracers of upper troposphere/lower stratosphere mixing and the global carbon cycle, Atmos. Chem. Phys., 10, 8575–8599, https://doi.org/10.5194/acp-10-8575-2010, 2010.
Belikov, D., Sugawara, S., Ishidoya, S., Hasebe, F., Maksyutov, S., Aoki, S., Morimoto, S., and Nakazawa, T.: Three-dimensional simulation of stratospheric gravitational separation using the NIES global atmospheric tracer transport model, Atmos. Chem. Phys., 19, 5349–5361, https://doi.org/10.5194/acp-19-5349-2019, 2019.
Banks, P. M. and Kockarts, G.: Aeronomy, Parts A and B, Academic Press, Inc. New York, 1973.
Bender, M., Sowers, T., and Labeyrie, L.: The Dole effect and its variations during the last 130,000 years as measured in the Vostok ice core, Global Biogeochem. Cy., 8, 363–376, 1994.
Bergamaschi, P., Hein, R., Brenninkmeijer, C. A. M., and Crutzen P. J.: Inverse modeling of the global CO cycle: 2. Inversion of 13C/12C and 18O/16O isotope ratios, J. Geophys. Res., 105, 1929–1945, https://doi.org/10.1029/1999JD900819, 2000.
Birner, B., Chipperfield, M. P., Morgan, E. J., Stephens, B. B., Linz, M., Feng, W., Wilson, C., Bent, J. D., Wofsy, S. C., Severinghaus, J., and Keeling, R. F.: Gravitational separation of Ar/N2 and age of air in the lowermost stratosphere in airborne observations and a chemical transport model, Atmos. Chem. Phys., 20, 12391–12408, https://doi.org/10.5194/acp-20-12391-2020, 2020.
Brenninkmeijer, C. A. M.: Measurement of the abundance of 14CO in the atmosphere and the 13C/12C and 18O/16O ratio of atmospheric CO, with application in New-Zealand and Antarctica, J. Geophys. Res., 98, 10595–10614, 1993.
Brenninkmeijer, C. A. M., Müller, R., Crutzen, P. J., Lowe, D. C., Manning, M. R., Sparks, R. J., and Velthoven, P. J. V.: A large 13CO deficit in the lower Antarctic stratosphere due to “ozone hole” chemistry: part 1, observations, Geophys. Res. Lett., 23, 2125–2128, https://doi.org/10.1029/96GL01471, 1996.
Butchart, N.: The Brewer-Dobson circulation, Rev. Geophys., 52, 157–184, https://doi.org/10.1002/2013RG000448, 2014.
Coplen, T. B. and Shrestha, Y.: Isotope-abundance variations and atomic weights of selected elements: 2016 (IUPAC Technical Report), Pure Appl. Chem., 88, 1203–1224, https://doi.org/10.1515/pac-2016-0302, 2016.
Engel, A., Strunk, M., Müller, M., Haase, H.-P., Poss, C., Levin, I., and Schmidt, U.: Temporal development of total chlorine in the high-latitude stratosphere based on reference distributions of mean age derived from CO2 and SF6, J. Geophys. Res., 107, https://doi.org/10.1029/2001JD000584, 2002.
Engel, A., Möbius, T., Bönisch, H., Schmidt, U., Heinz, R., Levin, I., Atlas, E., Aoki, S., Nakazawa, T., Sugawara, S., Moore, F., Hurst, D., Elkins, J., Schauffler, S., Andrews, A., and Boering, K.: Age of stratospheric air unchanged within uncertainties over the past 30 years, Nat. Geosci., 2, 28–31, https://doi.org/10.1038/Ngeo388, 2009.
Engel, A., Bönisch, H., Ullrich, M., Sitals, R., Membrive, O., Danis, F., and Crevoisier, C.: Mean age of stratospheric air derived from AirCore observations, Atmos. Chem. Phys., 17, 6825–6838, https://doi.org/10.5194/acp-17-6825-2017, 2017.
Francey, R. J., Tans, P. P., Allison, C. E., Enting, I. G., White, J. W. C., and Trolier, M.: Changes in oceanic and terrestrial carbon uptake since 1982, Nature, 373, 326–330, https://doi.org/10.1038/373326a0, 1995.
Fritsch, F., Garny, H., Engel, A., Bönisch, H., and Eichinger, R.: Sensitivity of age of air trends to the derivation method for non-linear increasing inert SF6, Atmos. Chem. Phys., 20, 8709–8725, https://doi.org/10.5194/acp-20-8709-2020, 2020.
Gamo, T., Tsutsumi, M., Sakai, H., Nakazawa, T., Tanaka, M., Honda, H., Kubo, H., and Itoh, T.: Carbon and oxygen isotopic ratios of carbon dioxide of a stratospheric profile over Japan, Tellus B, 41, 127–133, https://doi.org/10.1111/j.1600-0889.1989.tb00130.x, 1989.
Gamo, T., Tsutsumi, M., Sakai, H., Nakazawa, T., Machida, T., Honda H., and Itoh, T.: Long-term monitoring of carbon and oxygen isotope ratios of stratospheric CO2 over Japan, Geophys. Res. Lett., 22, 397–400, 1995.
Garcia, R. R., López-Puertas, M., Funke, B., Marsh, D. R., Kinnison, D. E., Smith, A. K., and González-Galindo, F.: On the distribution of CO2 and CO in the mesosphere and lower thermosphere, J. Geophys. Res.-Atmos., 119, 5700–5718, https://doi.org/10.1002/2013JD021208, 2014.
Garny, H., Ploeger, F., Abalos, M., Bönisch, H., von Clarmann, T., Diallo, M., Engel, A., Laube, J. C., Linz, M., Neu, J. L., Podglajen, A., Ray, E., Rivoire, L., Saunders, L. N., Stiller, G., Voet, F., Wagenhäuser, T., and Walker, K. A.: Age of stratospheric air: Progress on processes, observations, and long-term trends, Rev. Geophys., 62, e2023RG000832, https://doi.org/10.1029/2023RG000832, 2024.
Goto, D., Morimoto, S., Ishidoya, S., Aoki, S., and Nakazawa, T.: Terrestrial biospheric and oceanic CO2 uptakes estimated from long-term measurements of atmospheric CO2 mole fraction, δ13C, and δ(O2/N2) at Ny-Ålesund, Svalbard, J. Geophys. Res.-Biogeosci., 122, 1192–1202, https://doi.org/10.1002/2017JG003845, 2017.
Gromov, S., Brenninkmeijer, C. A. M., and Jöckel, P.: A very limited role of tropospheric chlorine as a sink of the greenhouse gas methane, Atmos. Chem. Phys., 18, 9831–9843, https://doi.org/10.5194/acp-18-9831-2018, 2018.
Hall, T. M. and Plumb, R. A.: Age as a diagnostic of stratospheric transport, J. Geophys. Res., 99, 1059–1070, 1994.
Hauck, M., Fritsch, F., Garny, H., and Engel, A.: Deriving stratospheric age of air spectra using an idealized set of chemically active trace gases, Atmos. Chem. Phys., 19, 5269–5291, https://doi.org/10.5194/acp-19-5269-2019, 2019.
Honda, H., Aoki, S., Nakazawa, T., Morimoto, S., and Yajima, N.: Cryogenic air sampling system for measurements of the concentrations of stratospheric trace gases and their isotopic ratios over Antarctica, J. Geomagn. Geoelectr., 48, 1145–1155, 1996.
Huang, T. Walters, S., Brasseur, G., Hauglustaine, D., and Wu, W.: Description of SOCRATES – A chemical dynamical radiative two-dimensional model, NCAR/TN-440+EDD NCAR TECHNICAL NOTE, 1998.
Ishidoya, S. and Murayama, S.: Development of high precision continuous measuring system of the atmospheric O2/N2 and Ar N2 ratios and its application to the observation in Tsukuba, Japan, Tellus, 66B, 22574, https://doi.org/10.3402/tellusb.v66.22574, 2014.
Ishidoya, S., Sugawara, S., Hashida, G., Morimoto, S., Aoki, S., Nakazawa, T., and Yamanouchi, T.: Vertical profiles of the O2/N2 ratio in the stratosphere over Japan and Antarctica, Geophys. Res. Lett., 33, L13701, https://doi.org/10.1029/2006GL025886, 2006.
Ishidoya, S., Sugawara, S., Morimoto, S., Aoki, S., and Nakazawa, T.: Gravitational separation of major atmospheric components of nitrogen and oxygen in the stratosphere, Geophys. Res. Lett., 35, L03811, https://doi.org/10.1029/2007GL030456, 2008a.
Ishidoya, S., Morimoto, S., Sugawara, S., Watai, T., Machida, T., Aoki, S., Nakazawa, T., and Yamanouchi, T.: Gravitational separation suggested by O2/N2, δ15N of N2, δ18O of O2, Ar N2 observed in the lowermost part of the stratosphere at northern middle and high latitudes in the early spring of 2002, Geophys. Res. Lett., 35, L03812, https://doi.org/10.1029/2007GL031526, 2008b.
Ishidoya, S., Sugawara, S., Morimoto, S., Aoki, S., Nakazawa, T., Honda, H., and Murayama, S.: Gravitational separation in the stratosphere – a new indicator of atmospheric circulation, Atmos. Chem. Phys., 13, 8787–8796, https://doi.org/10.5194/acp-13-8787-2013, 2013.
Ishidoya, S., Sugawara, S., Inai, Y., Morimoto, S., Honda, H., Ikeda, C., Hashida, G., Machida, T., Tomikawa, Y., Toyoda, S., Goto, D., Aoki, S., and Nakazawa, T.: Gravitational separation of the stratospheric air over Syowa, Antarctica and its connection with meteorological fields, Atmos. Sci. Lett., 19, e857, https://doi.org/10.1002/asl.857, 2018.
Ishidoya, S., Sugawara, S., and Okazaki, A.: Diurnal, seasonal, and interannual variations in δ(18O) of atmospheric O2 and its application to evaluate natural and anthropogenic changes in oxygen, carbon, and water cycles, Atmos. Chem. Phys., 25, 1965–1987, https://doi.org/10.5194/acp-25-1965-2025, 2025.
Ishijima, K., Sugawara, S., Kawamura, K., Hashida, G., Morimoto, S., Murayama, S., Aoki, S., and Nakazawa, T.: Temporal variations of the atmospheric nitrous oxide concentration and its δ15N and δ18O for the latter half of the 20th century reconstructed from firn air analyses, J. Geophys. Res., 112, D03305, https://doi.org/10.1029/2006JD007208, 2007.
Kaiser, J., Engel, A., Borchers, R., and Röckmann, T.: Probing stratospheric transport and chemistry with new balloon and aircraft observations of the meridional and vertical N2O isotope distribution, Atmos. Chem. Phys., 6, 3535–3556, https://doi.org/10.5194/acp-6-3535-2006, 2006.
Kato, S., Kajii, Y., Akimoto, H., Bräunlich, M., Röckmann, T., and Brenninkmeijer, C. A. M.: Observed and modeled seasonal variation of 13C, 18O, and 14C of atmospheric CO at Happo, a remote site in Japan, and a comparison with other records, J. Geophys. Res., 105, 8891–8900, https://doi.org/10.1029/1999JD901144, 2000.
Kawagucci, S., Tsunogai, U., Kudo, S., Nakagawa, F., Honda, H., Aoki, S., Nakazawa, T., Tsutsumi, M., and Gamo, T.: Long-term observation of mass-independent oxygen isotope anomaly in stratospheric CO2, Atmos. Chem. Phys., 8, 6189–6197, https://doi.org/10.5194/acp-8-6189-2008, 2008.
Keeling, C. D., Wholf, T. P., Wahlen, M., and van der Plichtt, J.: Interannual extremes in the rate of rise of atmospheric carbon dioxide since 1980, Nature, 375, 666–670, https://doi.org/10.1038/375666a0, 1995.
Khosravi, R., Brasseur, G., Smith, A., Rusch, D., Walters, S., Chabrillat, S., and Kockarts, G.: Response of the mesosphere to human-induced perturbations and solar variability calculated by a 2-D model, J. Geophys. Res., 107, 4358, https://doi.org/10.1029/2001JD001235, 2002.
Laube, J. C., Schuck, T. J., Chen, H., Geldenhuys, M., van Heuven, S., Keber, T., Popa, M. E., Tuffnell, E., Vogel, B., Wagenhäuser, T., Zanchetta, A., and Engel, A.: Vertical distribution of halogenated trace gases in the summer Arctic stratosphere determined by two independent in situ methods, EGUsphere [preprint], https://doi.org/10.5194/egusphere-2024-4034, 2025.
Leedham Elvidge, E. C., Bönisch, H., Brenninkmeijer, C. A. M., Engel, A., Fraser, P. J., Gallacher, E., Langenfelds, R., Mühle, J., Oram, D. E., Ray, E. A., Ridley, A. R., Röckmann, T., Sturges, W. T., Weiss, R. F., and Laube, J. C.: Evaluation of stratospheric age of air from CF4, C2F6, C3F8, CHF3, HFC-125, HFC-227ea and SF6; implications for the calculations of halocarbon lifetimes, fractional release factors and ozone depletion potentials, Atmos. Chem. Phys., 18, 3369–3385, https://doi.org/10.5194/acp-18-3369-2018, 2018.
Luz, B. and Barkan, E.: The isotopic composition of atmospheric oxygen, Global Biogeochem. Cy., 25, GB3001, https://doi.org/10.1029/2010GB003883, 2011.
Luz, B., Barkan, E., Bender, M. L., Thiemens, M. H., and Boering, K. A.: Triple-isotope composition of atmospheric oxygen as a tracer of biosphere productivity, Nature, 400, 547–550, https://doi.org/10.1038/22987, 1999.
Machida, T., Matsueda, H., Sawa, Y., Nakagawa, Y., Hirotani, K., Kondo, N., Goto, K., Nakazawa, N., Ishikawa, K., and Ogawa, T.: Worldwide measurements of atmospheric CO2 and other trace gas species using commercial airlines, J. Atmos. Ocean. Tech., 25, 1744–1754, https://doi.org/10.1175/2008JTECHA1082.1, 2008.
Matsueda, H., Machida, T., Sawa, Y., and Niwa, Y.: Long-term change of CO2 latitudinal distribution in the upper troposphere, Geophys. Res. Lett., 42, https://doi.org/10.1002/2014GL062768, 2015.
McCarthy, M. C., Boering, K. A., Rice, A. L., Tyler, S. C., Connell P., and Atlas, E.: Carbon and hydrogen isotopic compositions of stratospheric methane: 2. Two-dimensional model results and implications for kinetic isotope effects, J. Geophys. Res., 108, https://doi.org/10.1029/2002JD003183, 2003.
Meijer, H. A. J. and Li, W. J.: The use of electrolysis for accurate δ17O and δ18O isotope measurements in water, Isotopes in Environmental and Health Studies, 34, 349–369, https://doi.org/10.1080/10256019808234072, 1998.
Michel, S., Braun, K., Ortega, J., Tans, P., Li, J., Baugh, K., Crotwell, M., Madronich, M., Moglia, E., Mund, J., NOAA Global Monitoring Laboratory, and University Of Colorado Institute Of Arctic And Alpine Research (INSTAAR): Monthly means of Stable Isotopic Composition of Atmospheric Carbon Dioxide (d13C-CO2) from the NOAA GML Carbon Cycle Cooperative Global Air Sampling Network, 1990–2024, NOAA Global Monitoring Laboratory [data set], https://doi.org/10.15138/6E9X-QA28, 2025.
Michel, S. E., Clark, J. R., Vaughn, B. H., Crotwell, M., Madronich, M., Moglia, E., Neff, D., and Mund, J., University of Colorado, Institute of Arctic and Alpine Research (INSTAAR), Stable Isotopic Composition of Atmospheric Methane (13C) from the NOAA GML Carbon Cycle Cooperative Global Air Sampling Network, 1998–2022 Version: 2023-09-21 [data set], https://doi.org/10.15138/9p89-1x02, 2023.
Minschwaner, K., Manney, G. L., Livesey, N. J., Pumphrey, H. C., Pickett, H. M., Froidevaux, L., Lambert, A., Schwartz, M. J., Bernath, P. F., Walker, K. A., and Boone, C. D.: The photochemistry of carbon monoxide in the stratosphere and mesosphere evaluated from observations by the Microwave Limb Sounder on the Aura satellite, J. Geophys. Res., 115, D13303, https://doi.org/10.1029/2009JD012654, 2010.
Morimoto, S., Nakazawa, T., Higuchi, K., and Aoki, S.: Latitudinal distribution of atmospheric CO2 sources and sinks inferred by δ13C measurements from 1985 to 1991, J. Geophys. Res., 105, 24315–24326, https://doi.org/10.1029/2000JD900386, 2000.
Morimoto, S., Aoki, S., Nakazawa T., and Yamanouchi, T.: Temporal Variations of the Carbon Isotopic Ratio of Atmospheric Methane Observed at Ny Ålesund, Svalbard from 1996 to 2004, Geophys. Res. Lett., 33, https://doi.org/10.1029/2005GL024648, 2006.
Nakazawa, T., Morimoto, S., Aoki, S., and Tanaka, M.: Time and space variations of the carbon isotopic ratio of tropospheric carbon dioxide over Japan, Tellus B, 45, 258–274, https://doi.org/10.3402/tellusb.v45i3.15728, 1993.
Nakazawa, T., Machida, T., Sugawara, S., Murayama, S., Morimoto, S., Hashida, G., Honda, H., and Itoh, T.: Measurements of the stratospheric carbon dioxide concentration over Japan using a balloon-borne cryogenic sampler, Geophys. Res. Lett., 22, 1229–1232, https://doi.org/10.1029/95GL01188, 1995.
Nakazawa, T., Morimoto, S., Aoki, S., and Tanaka, M.: Temporal and spatial variations of the carbon isotopic ratio of atmospheric carbon dioxide in the western Pacific region, J. Geophys. Res., 102, 1271–1285, 1997a.
Nakazawa, T., Ishizawa, M., Higuchi, K., and Trivett, N. B. A.: Two curve fitting methods applied to CO2 flask data, Environmetrics, 8, 197–218, https://doi.org/10.1002/(SICI)1099-095X(199705)8:3<197::AID-ENV248>3.0.CO;2-C, 1997b.
Nakazawa, T., Aoki, S., Kawamura, K., Saeki, T., Sugawara, S., Honda, H., Hashida, G., Morimoto, S., Yoshida, N., Toyoda, S., Makide, Y., and Shirai, T.: Variations of stratospheric trace gases measured using a balloon-borne cryogenic sampler, Adv. Space Res., 30, 1349–1357, https://doi.org/10.1016/S0273-1177(02)00551-3, 2002.
Nguyen, T. H., Ishijima, K., Sugawara, S., and Hasebe, F.: Application of a nudged general circulation model to the interpretation of the mean age of air derived from stratospheric samples in the tropics, J. Meteorol. Soc. Jpn. II, 99, 1149–1167, https://doi.org/10.2151/jmsj.2021-056, 2021.
Park, J. H., Ko, M. K. W., Jackman, C. H., Plumb, R. A., Kaye, J. A., and Sage, K. H.: Models and Measurements Intercomparison II, NASA/TM-1999-209554, available at: http://www.cs.odu.edu/~mln/ltrs-pdfs/NASA-99-tm209554.pdf (last access: 19 July 2025), 1999.
Rahn, T., Zhang, H., Wahlen, M., and Blake, G. A.: Stable isotope fractionation during ultraviolet photolysis of N2O, Geophys. Res. Lett., 25, 4489–4492, https://doi.org/10.1029/1998GL900186, 1998.
Ray, E. A., Moore, F. L., Rosenlof, K. H., Davis, S. M., Sweeney, C., Tans, P., Wang, T., Elkins, J. W., Bönisch, H., Engel, A., Sugawara, S., Nakazawa, T., and Aoki, S.: Improving stratospheric transport trend analysis based on SF6 and CO2 measurements, J. Geophys. Res.-Atmos., 119, 14110–114128, https://doi.org/10.1002/2014JD021802, 2014.
Ray, E. A., Moore, F. L., Elkins, J. W., Rosenlof, K. H., Laube, J. C., Röckmann, T., Marsh, D. R., and Andrews, A. E.: Quantification of the SF6 lifetime based on mesospheric loss measured in the stratospheric polar vortex, J. Geophys. Res.-Atmos., 122, 4626–4638, https://doi.org/10.1002/2016JD026198, 2017.
Rice, A. L., Tyler, S. C., McCarthy, M. C., Boering K. A., and Atlas, E.: Carbon and hydrogen isotopic compositions of stratospheric methane: 1. High-precision observations from the NASA ER-2 aircraft, J. Geophys. Res., 108, 4460, https://doi.org/10.1029/2002JD003042, 2003.
Röckmann, T. and Brenninkmeijer, C. A. M.: CO and CO2 isotopic composition in Spitsbergen during the 1995 ARCTOC campaign, Tellus, 49B, 455–465, 1997.
Röckmann, T., Brenninkmeijer, C. A. M., Crutzen, P. J., and Platt, U.: Short-term variations in the 13C/12C ratio of CO as a measure of Cl activation during tropospheric ozone depletion events in the Arctic, J. Geophys. Res., 104, 1691–1697, https://doi.org/10.1029/1998JD100020, 1999.
Röckmann, T., Jöckel, P., Gros, V., Bräunlich, M., Possnert, G., and Brenninkmeijer, C. A. M.: Using 14C, 13C, 18O and 17O isotopic variations to provide insights into the high northern latitude surface CO inventory, Atmos. Chem. Phys., 2, 147–159, https://doi.org/10.5194/acp-2-147-2002, 2002.
Röckmann, T., Brass, M., Borchers, R., and Engel, A.: The isotopic composition of methane in the stratosphere: high-altitude balloon sample measurements, Atmos. Chem. Phys., 11, 13287–13304, https://doi.org/10.5194/acp-11-13287-2011, 2011.
Röckmann T., van Herpen, M., Brashear, C., van der Veen, C., Gromov, S., Li, Q., Saiz-Lopez, A., Meidan, D., Barreto, A., Prats, N., Mármol, I., Ramos, R., Baños, I., Arrieta, J. M., Zaehnle, S., Jordan, A., Moossen, H., Timas, H., Young, D., Sperlich, P., Moss, R., and Johnson, M. S.: The use of δ13C in CO to determine removal of CH4 by Cl radicals in the atmosphere, Environ. Res. Lett., 19, 064054, https://doi.org/10.1088/1748-9326/ad4375, 2024.
Santrock, J., Studley, S. A., and Hayes, J. M.: Isotopic analyses based on the mass spectra of carbon dioxide, Anal. Chem., 57, 1444–1448, https://doi.org/10.1021/ac00284a060, 1985.
Saueressig, G., Bergamaschi, P., Crowley, J. N., and Fischer, H.: Carbon KIE in the reaction of CH4 with Cl atoms, Geophys. Res. Lett., 22, 1225–1228, 1995.
Saueressig, G., Crowley, J. N., Bergamaschi, P., Brühl, C., Brenninkmeijer, C. A. M., and Fischer, H.: Carbon-13 and D kinetic isotope effects in the reactions of CH4 with O(1D) and OH: New laboratory measurements and their implications for the isotopic composition of stratospheric methane, J. Geophys. Res., 106, 23127–23138, 2001.
Sawa, Y., Machida, T., and Matsueda, H.: Seasonal variations of CO2 near the tropopause observation by commercial aircraft, J. Geophys. Res., 113, D23301, https://doi.org/10.1029/2008JD010568, 2008.
Stevens, C. M. and Wagner, A. F.: The Role of isotope fractionation effects in atmospheric chemistry, Zeitschrift fur Naturforschung A, 44, 376–384, https://doi.org/10.1515/zna-1989-0505, 1989.
Stiller, G. P., von Clarmann, T., Höpfner, M., Glatthor, N., Grabowski, U., Kellmann, S., Kleinert, A., Linden, A., Milz, M., Reddmann, T., Steck, T., Fischer, H., Funke, B., López-Puertas, M., and Engel, A.: Global distribution of mean age of stratospheric air from MIPAS SF6 measurements, Atmos. Chem. Phys., 8, 677–695, https://doi.org/10.5194/acp-8-677-2008, 2008.
Stiller, G. P., von Clarmann, T., Haenel, F., Funke, B., Glatthor, N., Grabowski, U., Kellmann, S., Kiefer, M., Linden, A., Lossow, S., and López-Puertas, M.: Observed temporal evolution of global mean age of stratospheric air for the 2002 to 2010 period, Atmos. Chem. Phys., 12, 3311–3331, https://doi.org/10.5194/acp-12-3311-2012, 2012.
Sugawara, S., Nakazawa, T., Shirakawa, Y., Kawamura, K., Aoki, S., Machida, T., and Honda, H.: Vertical profile of the carbon isotopic ratio of stratospheric methane over Japan, Geophys. Res. Lett., 24, 2989–2992, 1997.
Sugawara, S., Ishidoya, S., Aoki, S., Morimoto, S., Nakazawa, T., Toyoda, S., Inai, Y., Hasebe, F., Ikeda, C., Honda, H., Goto, D., and Putri, F. A.: Age and gravitational separation of the stratospheric air over Indonesia, Atmos. Chem. Phys., 18, 1819–1833, https://doi.org/10.5194/acp-18-1819-2018, 2018.
Thiemens, M. H.: Mass-independent isotope effects in planetary atmospheres and the early solar system, Science, 283, 341–345, https://doi.org/10.1126/science.283.5400.341, 1999.
Toyoda, S., Yoshida, N., Urabe, T., Aoki, S., Nakazawa, T., Sugawara, S., and Honda H.: Fractionation of N2O isotopomers in the stratosphere, J. Geophys. Res., 106, 7515–7522, https://doi.org/10.1029/2000JD900680, 2001.
Toyoda, S., Yoshida, N., Morimoto, S., Aoki, S., Nakazawa, T., Sugawara, S., Ishidoya, S., Uematsu, M., Inai, Y., Hasebe, F., Ikeda, C., Honda, H., and Ishijima, K.: Vertical distributions of N2O isotopocules in the equatorial stratosphere, Atmos. Chem. Phys., 18, 833–844, https://doi.org/10.5194/acp-18-833-2018, 2018.
Umezawa, T., Machida, T., Ishijima, K., Matsueda, H., Sawa, Y., Patra, P. K., Aoki, S., and Nakazawa, T.: Carbon and hydrogen isotopic ratios of atmospheric methane in the upper troposphere over the Western Pacific, Atmos. Chem. Phys., 12, 8095–8113, https://doi.org/10.5194/acp-12-8095-2012, 2012.
Umezawa, T., Brenninkmeijer, C. A. M., Röckmann, T., van der Veen, C., Tyler, S. C., Fujita, R., Morimoto, S., Aoki, S., Sowers, T., Schmitt, J., Bock, M., Beck, J., Fischer, H., Michel, S. E., Vaughn, B. H., Miller, J. B., White, J. W. C., Brailsford, G., Schaefer, H., Sperlich, P., Brand, W. A., Rothe, M., Blunier, T., Lowry, D., Fisher, R. E., Nisbet, E. G., Rice, A. L., Bergamaschi, P., Veidt, C., and Levin, I.: Interlaboratory comparison of δ13C and δ13C and δD measurements of atmospheric CH4 for combined use of data sets from different laboratories, Atmos. Meas. Tech., 11, 1207–1231, https://doi.org/10.5194/amt-11-1207-2018, 2018.
Umezawa, T., Sugawara, S., Hikichi, S., Morimoto, S., Saito, T., Krummel, P. B., Fraser, P. J., and Weiss, R. F.: Evaluation of stratospheric age of air estimated from halocarbon measurements of air samples collected by a balloon-borne cryogenic air sampler over Japan, SOLA, 21, 237–243, https://doi.org/10.2151/sola.2025-029, 2025.
Wang, J. S., McElroy, M. B., Spivakovsky C. M., and Jones, D. B. A.: On the contribution of anthropogenic Cl to the increase in δ13C of atmospheric methane, Global Biogeochem. Cy., 16, 1047, https://doi.org/10.1029/2001GB001572, 2002.
Waugh, D. W. and Hall, T. M.: Age of stratospheric air: Theory, observations, and models, Rev. Geophys., 40, 1010, https://doi.org/10.1029/2000RG000101, 2002.
- Abstract
- Introduction
- Experimental procedures
- Results and discussion
- Conclusions
- Appendix A: Gravitational separation of major atmospheric components
- Appendix B: Two-dimensional model
- Appendix C: Age calculation
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Experimental procedures
- Results and discussion
- Conclusions
- Appendix A: Gravitational separation of major atmospheric components
- Appendix B: Two-dimensional model
- Appendix C: Age calculation
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement