the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 23 Sep 2025
| 23 Sep 2025
A critical review of the use of iron isotopes in atmospheric aerosol research
Yifan Zhang
Rui Li
Zachary B. Bunnell
Yizhu Chen
Guanhong Zhu
Jinlong Ma
Guohua Zhang
Tim M. Conway
Deposition of atmospheric aerosols is recognized as a major source of iron (Fe) to the surface oceans, where it acts as a key micronutrient for primary productivity and the metabolic functions of marine microbes. Initially, natural desert dust was thought to be the main source of aerosol Fe, albeit largely insoluble; however, in the last few decades, the role of anthropogenic and wildfire sources in providing soluble Fe to aerosols has been increasingly recognized. The stable isotope ratio of Fe (δ56Fe) has emerged as a potential tracer for discriminating and quantifying sources of aerosol Fe. In this review, we examine the state of the field for using δ56Fe as an aerosol source tracer, and constraints on endmember signatures. We begin with an overview of the methodology of δ56Fe analysis for aerosol samples. We then describe knowledge of δ56Fe endmember signatures of different source materials, and review existing knowledge of the δ56Fe signature of ambient aerosols collected from around the globe, and how these measurements can be used to enhance atmospheric Fe deposition modelling. We also examine the various chemical processing mechanisms which might influence δ56Fe source signatures of aerosol Fe during its transport in the atmosphere. This review paper is concluded with a perspective on the state of the field and a call for future work. Overall, we find aerosol δ56Fe to be a promising tracer, but highlight that greater constraints on both source endmembers and processing mechanisms are needed to fully utilize this tracer.
- Article
(4194 KB) - Full-text XML
-
Supplement
(452 KB) - BibTeX
- EndNote




Iron (Fe) is the fourth most abundant element in the upper continental crust (UCC), but concentrations of dissolved Fe are very low over much of the surface ocean (<0.1 nmol kg−1) due to the extremely low solubility of Fe(III) in seawater (Boyd and Ellwood, 2010). Thus, the availability of dissolved Fe may limit marine primary productivity, nitrogen fixation, and ocean-atmosphere carbon exchange over large regions of the surface ocean, especially in High-Nutrient Low Chlorophyll (HNLC) regions (Boyd and Ellwood, 2010; Moore et al., 2013). In early views of the Fe cycle, atmospheric deposition of Fe-bearing aerosols was thought to be the only significant source of dissolved Fe to the surface ocean (Johnson et al., 1997; Tagliabue et al., 2017). However, it is now understood that dissolved Fe is supplied to the oceans by a mixture of external sources that vary in importance by basin, including hydrothermal venting, marine sediment dissolution (both reductive and non-reductive), and atmospheric aerosols (Tagliabue et al., 2017; Conway et al., 2024). Rivers and cryospheric sources may also play a role in some regions. In this view, atmospheric deposition remains a key source of dissolved Fe to the surface ocean, but with great chemical, spatial, and temporal variability that must be understood and characterized.
Atmospheric Fe aerosols reaching the oceans were originally considered to be composed only of natural components, derived through the weathering and erosion of UCC materials; indeed, desert dust, mainly emitted from arid and semi-arid regions, is the dominant source of total aerosol Fe at the global scale (Jickells et al., 2005). More recently, however, it has been shown that anthropogenic aerosols can account for a large fraction of soluble aerosol Fe despite their small contribution to total aerosol Fe (Sholkovitz et al., 2009; Ito et al., 2019; Hamilton et al., 2020a; Rathod et al., 2020; Ito et al., 2021; Chen et al., 2024), meaning that they can play an outsized role in influencing marine primary productivity. Anthropogenic aerosols can include emissions from fossil fuel and biofuel combustion, industry, biomass burning, and transportation (Sedwick et al., 2007; Sholkovitz et al., 2009; Zhu et al., 2022; Chen et al., 2024). In this article, non-dust sources are referred to as anthropogenic sources in order to highlight the impacts of anthropogenic emission. However, we note that wildfire Fe aerosol, composed of a combination of soil (∼ 64 %) and biomass Fe (∼ 36 %) (Hamilton et al., 2022; Bunnell et al., 2025), is a natural source that may be enhanced by anthropogenic land use and climate change.
Natural, anthropogenic and wildfire aerosols may undergo various chemical and physical processes in the atmosphere, which can “solubilize” insoluble Fe minerals to soluble Fe. For example, while Fe in desert dust is “insoluble” with initial solubility (operationally defined as the fraction of total Fe that dissolves) being < 0.5 % (Desboeufs et al., 2005; Shi et al., 2011; Oakes et al., 2012b; Li et al., 2022), several chemical processes can promote dissolution of insoluble Fe, among which acid processing (i.e. proton-promoted dissolution) is likely the most important (Meskhidze et al., 2003; Shi et al., 2012; Baker et al., 2021; Zhang et al., 2023). Indeed, many studies have shown that when compared to desert dust, Fe solubility can be much higher for ambient aerosols (aerosol particles in the troposphere) collected over the oceans (Baker and Jickells, 2006; Kumar et al., 2010; Sholkovitz et al., 2012; Mahowald et al., 2018; Sakata et al., 2022), suggesting that other sources (e.g., anthropogenic Fe) or atmospheric processes contribute significantly to soluble aerosol Fe in the troposphere (and for later deposition to the surface oceans).
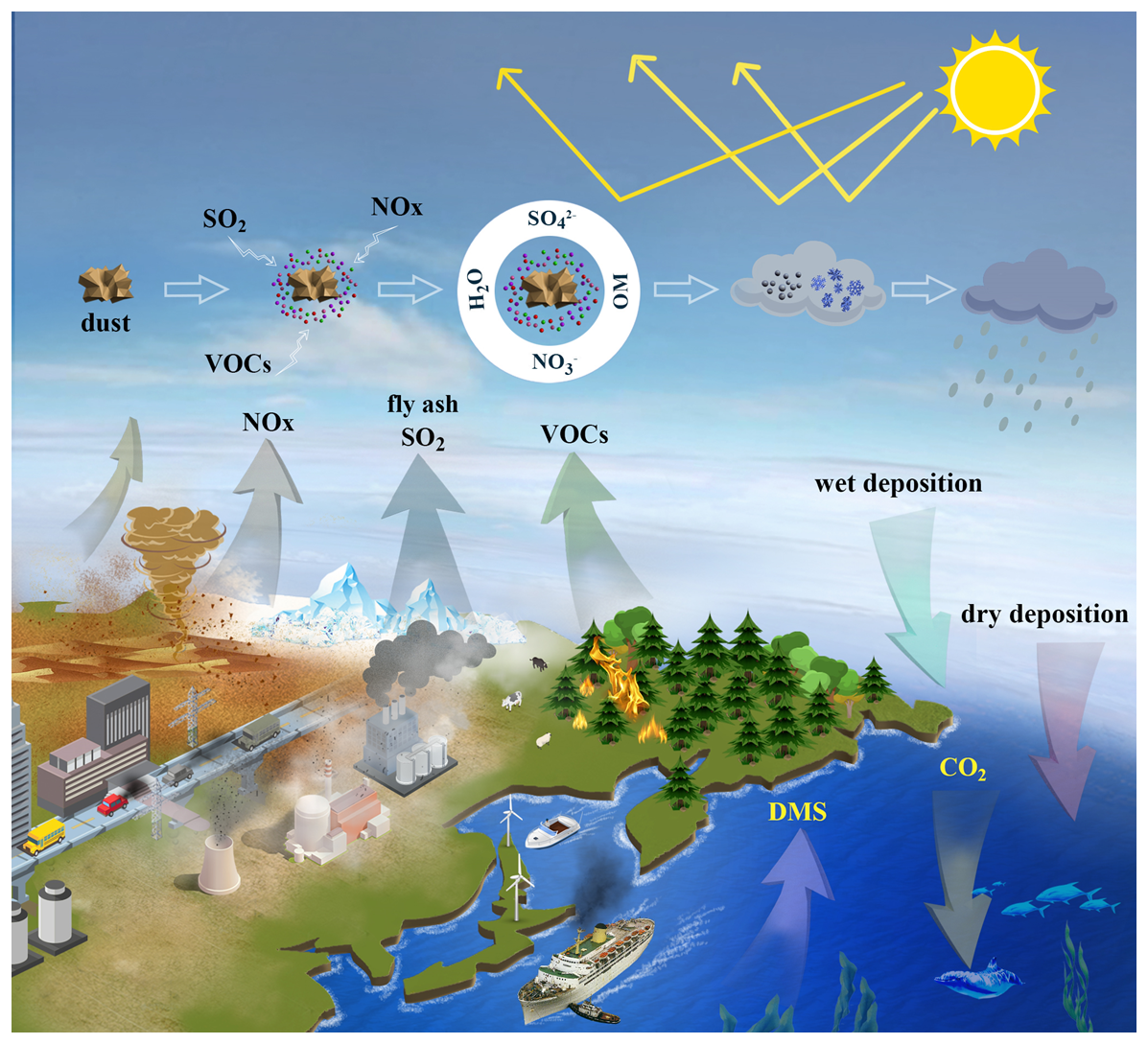
Figure 1Emission, transport, processing and deposition of aerosol Fe. VOCs: volatile organic compounds; OM: organic materials; DMS: dimethyl sulfide.
Figure 1 depicts emission, transport, processing, and deposition of aerosol Fe. Observed variability in aerosol Fe flux, composition, and solubility can, in principle, be explained by a mix of different source emissions and secondary processing in the atmosphere. However, each type of Fe source exhibits large temporal and spatial variations at the local-global scale, and thus it remains difficult to quantitatively disentangle their contributions to a sample or location. Enrichment factors, correlation analysis, and factor analysis (such as positive matrix factorization) are approaches that have been used for source appointment of total and soluble aerosol Fe (Chuang et al., 2005; Sedwick et al., 2007; Sholkovitz et al., 2009; Buck et al., 2010; Desboeufs et al., 2018; Marsay et al., 2022; Zhu et al., 2022; Sakata et al., 2023; Zhang et al., 2023; Chen et al., 2024; Zhang et al., 2024). However, enrichment factors of other “anthropogenically-derived” elements (e.g., Cd, Pb, Ni, and Zn) may not always be indicative of anthropogenic Fe (Chester et al., 1993; Shelley et al., 2015), and a consensus has not been reached on either the relative contribution of various sources to soluble aerosol Fe or the factors which control aerosol Fe solubility (Mahowald et al., 2018; Meskhidze et al., 2019; Baker et al., 2021). Single particle analysis, typically using electron microscopy, X-ray micro-spectroscopy or single particle mass spectrometry, can also be very useful for source identification of aerosol Fe in individual particles (Moffet et al., 2012; Oakes et al., 2012a; Zhang et al., 2014; Li et al., 2017); nevertheless, it can only examine a limited number of aerosol particles, and thus may not be able to provide quantitative information on the source of Fe for an aerosol sample which typically consists of numerous particles.
One tracer that shows recent promise in source quantification of total and soluble Fe within aerosols themselves is the isotopic composition of Fe (δ56Fe). During the last two decades, this parameter has been measured both in potential source materials of Fe to the ocean (Beard et al., 2003a), and later in seawater itself (de Jong et al., 2007), providing very useful information for disentangling the marine Fe cycle (Fitzsimmons and Conway, 2023). The developing application of δ56Fe to aerosols has also been shown to be a promising way to differentiate sources of total and soluble Fe in atmospheric aerosols and to quantify their relative importance (Majestic et al., 2009a; Mead et al., 2013; Conway et al., 2019; Kurisu et al., 2021). Wang et al. (2022) briefly summarized recent advances in isotopic compositions of aerosol Fe, and utilized a Bayesian isotopic mixing model (MIXSIAR) to re-evaluate the sources of aerosol Fe based on Fe isotopic data reported in literature. Wei et al. (2024) compiled a global dataset of aerosol Fe isotope measurements, and identified coal combustion as the major anthropogenic source of aerosol Fe by using MIXSIAR to re-analyze the data they compiled.
Here, we review the application of Fe isotopic analysis to atmospheric aerosol research in a comprehensive and critical manner. We summarize the current consensus, underscore existing discrepancies, and uncertainties/unknowns, as well as outline future research priorities required to improve the use of Fe isotopic analysis in tracing sources of atmospheric aerosol Fe and understanding processes that influence its solubility.
2.1 Natural Fe isotopes
Fe has four stable isotopes which occur naturally, namely 54Fe (5.84 %), 56Fe (91.76 %), 57Fe (2.12 %) and 58Fe (0.28 %). Fe isotopic compositions are typically reported in δ values, as given in Eq. (1) (Beard and Johnson, 2004; Dauphas and Rouxel, 2006):
where x is 56, 57 or 58. While some studies report δ57Fe, the convention is to report δ56Fe, which allows for easy inter-comparability. Here, we use δ56Fe values, although we note that δ57Fe can be converted to δ56Fe by dividing δ57Fe values by 1.475. Fe isotopic data is also typically reported relative to the IRMM-014 standard (provided by the Institute for Reference Materials and Measurements, IRMM), although some early workers have used other standards (Beard and Johnson, 2004). Production of IRMM-014 has ceased and current stocks will likely run out in the future; as a result, new reference materials, such as IRMM-524a, are beginning to be used in place of IRMM-014 (González De Vega et al., 2020; Xu et al., 2022). In this article we use δ56Fe values relative to IRMM014, and for the ease of the reader, have converted published values that were expressed relative to other standards.
2.2 Fe isotopic analysis
Historically, δ56Fe measurements have been made by either multi-collector Thermal Ionization Mass spectrometry (MC-TIMS) or multi-collector Inductively Coupled Plasma Mass Spectrometry (MC-ICP-MS). In early works, TIMS was combined with the double-spike technique, requiring a minimum Fe mass of >1 µg and yielding precision of ±0.5 ‰ (2σ) (Beard and Johnson, 1999; Beard and Johnson, 2004). However, the TIMS-based method, which does not suffer from isobaric interferences, requires a very long time for each measurement (usually 4–8 h), and has low Fe ionization efficiency (Beard et al., 2003b), variable mass bias, and relatively low precision (0.5 ‰) compared to natural variability (typically 1 ‰–3 ‰) in open ocean seawater (Dauphas and Rouxel, 2006; Fitzsimmons and Conway, 2023).
The development of MC-ICP-MS, with higher ionization efficiency, shorter analysis time per measurement, and higher precision (<0.1 ‰) than TIMS, was crucial to application of δ56Fe to natural materials (Beard and Johnson, 2004; Zhang et al., 2022). Currently the precision of δ56Fe measurements via MC-ICP-MS can reach ±0.02 ‰ to ±0.05 ‰ (2σ) (de Jong et al., 2007; Zhu et al., 2018; Conway et al., 2019), around an order of magnitude better than TIMS; therefore, MC-ICP-MS has been much more widely used for Fe isotopic analysis.
Drawbacks of MC-ICP-MS are the need of correction for rapid changes in mass bias, and that measurements can be compromised by isobaric and polyatomic spectral interferences that must be dealt with using several approaches (Beard et al., 2003b; Beard and Johnson, 2004). For example, mass-bias must be corrected for using standard-sample bracketing and/or double-spike techniques; samples must be cleanly purified from interfering isobars (Cr and Ni) and matrix elements (e.g., Ca), and further information on Fe purification can be found elsewhere (Conway et al., 2013; Sieber et al., 2021). A further non-trivial challenge of using MC-ICP-MS is the presence of distinct polyatomic argide interferences (e.g., ArO and ArN) that arise on masses of 54Fe, 56Fe, 57Fe and 58Fe from combination of the plasma gas and solvent matrix, and can be larger than the Fe mass of interest (Weyer and Schwieters, 2003). Furthermore, while elemental isobars and polyatomic interferences, such as Ni, Cr, and CaO, can be dealt with by purification and/or correction, argides must be dealt with by changing instrumental conditions or using “high” resolution instruments which can sufficiently resolve Fe peaks from argide interferences (Weyer and Schwieters, 2003).
The minimum mass of Fe required for isotopic analysis by MC-ICP-MS depends on several factors, including the procedural blank, analytical uncertainty, and minimum concentration at which an accurate isotope ratio can be obtained. As MC-ICPMS Neptune Fe signal intensity decreases, analytical internal standard error increases relatively predictably (John and Adkins, 2010; Conway et al., 2013).
Thus, assuming no inaccuracy at lower concentrations, a minimum mass of Fe depends on instrumental signal sensitivity (volts per ng g−1) and the minimum acceptable uncertainty for a sample (e.g., <0.2 ‰ for Fe). As an example, at the University of South Florida (USF), with a typical sensitivity of 0.1–0.15 V on 56Fe per ng of Fe, we are typically able to achieve acceptable uncertainties (<0.15 ‰, 2σ) in solutions at concentrations of 10 ng/g, a concentration that is > 20–40 times of our chemistry procedural blank which is typically < 0.5 ng g−1 (Conway et al., 2013; Sieber et al., 2021; Sieber et al., 2024). This concentration can be achieved by dissolving 5 ng Fe in 0.5 mL solution. Furthermore, we found that at concentrations below 1V on 56Fe, inaccuracies in our measurement of the zero standard begin to occur. In both cases, 5 ng Fe can therefore be considered an absolute minimum mass for analysis. However, 20–100 ng Fe provides isotopic ratios with smaller uncertainty (< 0.1 ‰), approaching the analytical precision at USF (0.06 ‰). Similarly, Kurisu et al. (2024) used a minimum of 25 ng Fe to obtain Fe isotopic data from aerosol samples with suitable uncertainties, but they preferred ∼ 100 ng where possible. Such precision (<0.1 ‰) is often essential for resolving variability between aerosol samples.
In addition to instrumental analysis, a further consideration for aerosol samples that often include significant filter or digestion blanks is the blank isotopic composition should be established (and subtracted, weighed by concentration, from samples) and/or the effect of blanks minimized by having large sample to blank ratios (Kurisu et al., 2024; Bunnell et al., 2025).
In this section we first present a few examples to illustrate how Fe isotopic analysis may help constrain sources of dissolved Fe to the ocean (Sect. 3.1). After that, we review isotopic compositions of aerosol Fe from relevant sources (Sect. 3.2), Fe isotopic composition of ambient aerosols (Sect. 3.3), modeling studies of isotopic compositions of ambient aerosol Fe (Sect. 3.4), and Fe isotopic fractionation induced by chemical processing (Sect. 3.5).
3.1 Application of Fe isotopic analysis to marine source attribution
Since the first successful application of δ56Fe to seawater samples in 2007–2010 (de Jong et al., 2007; Lacan et al., 2008; John and Adkins, 2010), Fe isotopic analysis has been increasingly deployed in investigating marine biogeochemistry over the last decade, significantly increasing our knowledge of sources and cycles of Fe in the ocean (Conway et al., 2021; Fitzsimmons and Conway, 2023), with a precision of better than ±0.10 ‰. As this article is focused on atmospheric aerosols, we do not intend to provide a comprehensive review of the application of Fe isotopes to marine biogeochemistry; instead, we present a few examples of relevant ocean observational and modeling studies to illustrate how δ56Fe may help constrain sources of dissolved Fe to the ocean, focusing on aerosol deposition case studies. For a more comprehensive discussion of the advances in using δ56Fe to interrogate the marine Fe cycle in recent years, we instead point the reader to Fitzsimmons and Conway (2023).
Although a few marine δ56Fe water column profiles had been measured by 2012 (Lacan et al., 2010; Radic et al., 2011; John and Adkins, 2012), the first ocean “section” of δ56Fe came in 2014 when Conway and John (2014) reported dissolved Fe and δ56Fe in 510 seawater samples at 17 stations along the U.S. GEOTRACES GA03 section of the subtropical North Atlantic Ocean (Fig. 2). A range of δ56Fe values, spanning from −1.35 ‰ to +0.8 ‰, were observed at different regions along this section, reflecting different sources of dissolved Fe. Much of this transect was characterized by δ56Fe > +0.1 ‰, heavier than known sources of Fe to the ocean at the time and attributed to the net dissolution of atmospheric dust (John and Adkins, 2012; Conway and John, 2014). Further, by assigning endmember compositions to each identified Fe source (dust, hydrothermal venting, and sediments), two component mixing was used to quantitatively constrain sources at each station across the basin (Conway and John, 2014): Saharan dust aerosol was found to be the dominant source for dissolved Fe along this section (71 %–87 %), with lesser contribution from non-reductive and reductive sedimentary dissolution (10 %–19 % and 1 %–4 %), and hydrothermal venting (2 %–6 %). However, that study was not able to use δ56Fe as a source constraint at the very surface, due to potential influence of biological uptake, and/or anthropogenic aerosols or sediment sources (Conway and John, 2014; Conway et al., 2019).
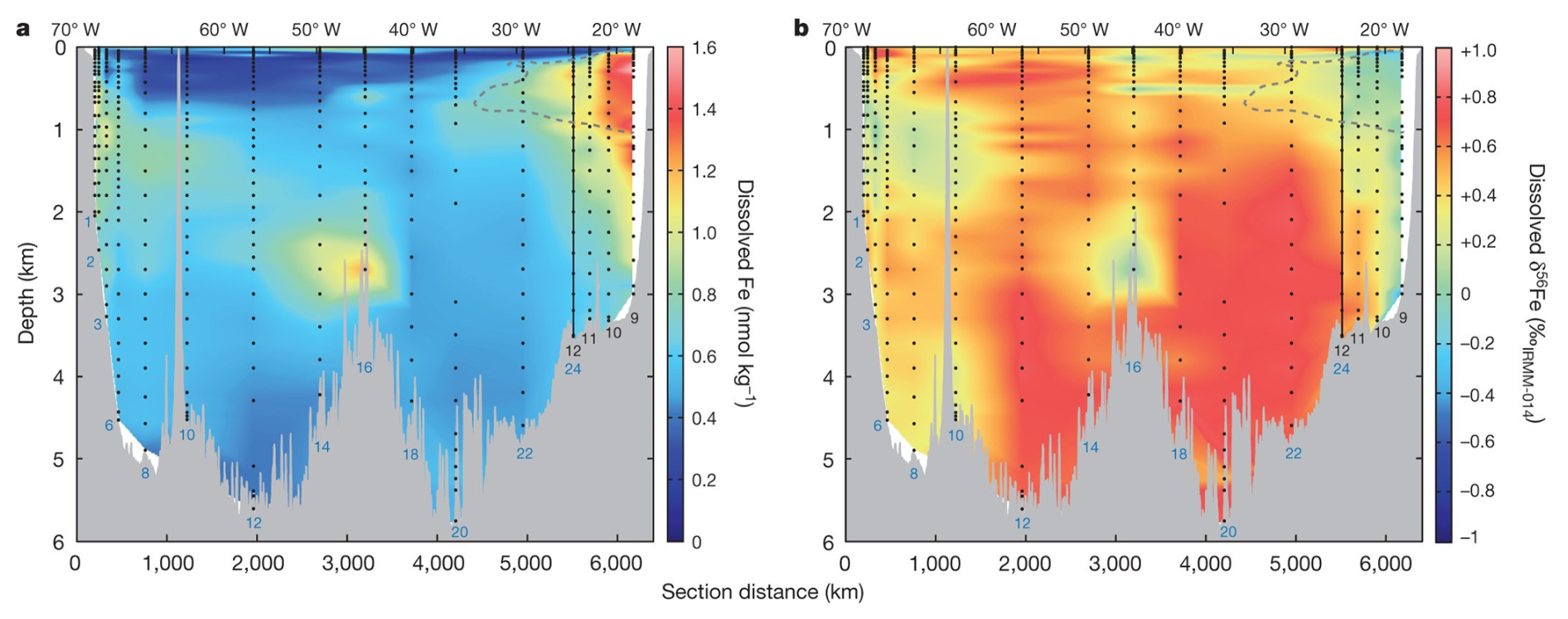
Figure 2Concentrations (a) and δ56Fe values (b) of dissolved Fe in seawater at different depth along a section of the North Atlantic Ocean from Mauritania and Woods Hole. Station numbers are shown in blue or black. Reproduced with permission from Conway and John (2014).
A second example of where δ56Fe has been useful for informing our understanding of atmospheric Fe deposition comes from the North Pacific, where Pinedo-González et al. (2020) analyzed surface seawater samples collected along a latitudinal section at 158° W (from 25 to 42° N) in May 2017. Surface dissolved Fe concentrations peaked at ∼ 35° N and were lower at southern and northern ends of this transect, and a similar latitudinal pattern was observed for dissolved Pb, attributed to deposition of anthropogenic aerosol of both elements. Moreover, the lowest δ56Fe values (from −0.65 ‰ to −0.23 ‰) were observed coincident with the highest concentrations of dissolved Fe (Pinedo-González et al., 2020), consistent with studies that showed anthropogenic aerosols to be isotopically light (Kurisu et al., 2016a; Conway et al., 2019). Using their δ56Fe data and estimates of anthropogenic endmember composition, Pinedo-González et al. (2020) suggested anthropogenic aerosol to be a significant source (21 %–59 %) of dissolved Fe in surface seawater between 35 and 40° N, at least during the high dust season. However, other studies of the North Pacific have shown that the primary δ56Fe signature of aerosol deposition is likely to be strongly attenuated by in situ processing and biological uptake (Kurisu et al., 2024).
Measurements of surface δ56Fe may therefore be useful in investigating atmospheric addition of dissolved Fe, but further work is needed to understand the effect of multiple biogeochemical processes which occur upon dissolution in the ocean. Global biogeochemical modeling may prove useful here, with recent work by König et al. (2021) incorporating Fe isotopes into a global model. This study and a follow-up study focused on the North Pacific (König et al., 2022) found that both external source signatures and fractionation during Fe complexation by organic ligands and uptake by phytoplankton were needed to reproduce oceanic deposition patterns, and that the dissolved δ56Fe in surface waters did not simply follow the footprint of atmospheric deposition. This is confirmed a recent study (Hawco et al., 2025), showing that extensive biological cycling and uptake of dissolved Fe after deposition of “light” Fe from anthropogenic aerosols did fractionate dissolved Fe to “heavy” values.
3.2 Fe isotopic compositions of aerosols from relevant sources
In this section we review Fe isotopic compositions of desert dust (Sect. 3.2.1), raw materials relevant for anthropogenic emission and biomass burning (Sect. 3.2.2), and aerosol particles emitted by various anthropogenic sources (Sect. 3.2.3). In addition, Table S1 in the Supplement summarizes key results reported by previous studies.
3.2.1 Desert dust
Here we discuss Fe isotopic compositions of desert dust reported by previous work. Instead of providing a complete literature survey, we review some representative studies. Fe isotopic compositions appear to be rather homogeneous for UCC, and δ56Fe values fall into a narrow range centered at around ‰ (Beard et al., 2003b; Beard and Johnson, 2004; Poitrasson, 2006).
The average δ56Fe values were determined to be ‰ and ‰ for loess (n=10) and soil samples (n=10) collected from various locations around the world, respectively (Beard et al., 2003a). Waeles et al. (2007) measured Fe isotopic compositions of Chinese and Australian desert dust samples, and the average δ56Fe were determined to be ‰ (n=8) for total Fe and −0.06 ‰ (n=2) for soluble Fe, suggesting that the isotopic composition of soluble Fe in acetate buffer is similar to total Fe. Majestic et al. (2009a) measured Fe isotopic compositions of road dust and agricultural soil in Phoenix (Arizona, USA), and δ56Fe values were found to range from −0.10 ‰ to +0.14 ‰ for road dust and from −0.07 ‰ to +0.04 ‰ for agricultural soil, both similar to the UCC. In another study (Mead et al., 2013), average δ56Fe was reported to be ‰ for three dust samples, namely Arizona Test Dust (ATD), Saharan dust and San Joaquin soil (SRM1709). The δ56Fe values were determined to be ‰, ‰ and ‰ for ATD, China loess and Xinjiang dust (Li et al., 2022), with the average being ‰ .
The δ56Fe values were found to range from +0.06 ‰ to +0.12 ‰ for loss and paleosol samples (n=32) from the China Loess Plateau, with the average being ‰ (Gong et al., 2017). Chen et al. (2020) analyzed isotopic compositions of HCl-leachable Fe in desert dust samples (n=10) from different deserts in northern China: one sample from northeastern China exhibited low δ56Fe (−0.23 ‰), while δ56Fe showed very small variation and had an average value of ‰ for the other nine samples.
Most of the δ56Fe values for desert dust Fe, as reported in previous studies, fall into a small range (−0.1 ‰ to +0.19 ‰); as a result, one may conclude that the Fe isotopic composition of desert dust is very similar to UCC, characterized by an endmember value of around +0.09 ‰. Furthermore, soluble Fe from desert dust appears to be isotopically similar to total Fe (Waeles et al., 2007; Chen et al., 2020). In addition to desert dust, there are other natural sources for aerosol Fe which may have different isotopic composition, such as soil particles entrained into the atmosphere during wildfires (Hamilton et al., 2022). We note from a recent synthesis that isotopic compositions of Fe in soil can range from −0.2 ‰ to +0.95 ‰ (Johnson et al., 2020), showing more variability than in dust, and reflecting the effects of organic matter, mineralogy, and chemistry.
3.2.2 Raw materials relevant for anthropogenic emission and biomass burning
Before we discuss isotopic compositions of aerosol Fe from anthropogenic emission and biomass burning, it is helpful to review Fe isotopic compositions of relevant raw materials, including iron ore, fossil fuel, biomass, and so on.
Iron deposits have been found to display very large variations in Fe isotopic compositions (Johnson et al., 2003; Lobato et al., 2023), with δ56Fe spanning from below −1.5 ‰ to around +2.0 ‰. The δ56Fe value was determined to be ‰ for one commercial gasoline sample in Japan (Kurisu et al., 2016b). To our knowledge, Fe isotopic compositions have not been reported for other important fossil fuels.
Guelke and von Blanckenburg (2007) measured isotopic compositions of Fe in higher plants and plant-available Fe in soils where these plants were grown. Compared to plant-available Fe in soils, Fe in strategy I plants were isotopically lighter by up to −1.6 ‰, and younger parts in these plants were more depleted in heavy Fe. Fe in strategy II plants was isotopically heavier by about +0.2 ‰ than plant-available Fe in soils (Guelke and von Blanckenburg, 2007), and all the parts in these plants had nearly the same isotopic composition.
In a later study (Kurisu and Takahashi, 2019), average δ56Fe were reported to be ‰ for reed and ‰ for reed burning residual ash, both similar to that for nearby soil ( ‰). As a result, they suggested that biomass burning could not explain isotopically lighter Fe observed for ambient aerosols.
Kubik et al. (2021) surveyed Fe isotopic compositions of biological samples and found that in general Fe in strategy I plants is isotopically lighter than strategy II plants. This is because strategy I and II plants use different biochemical mechanisms to absorb Fe from soil (Kubik et al., 2021). While various organic materials have been analyzed for δ56Fe (Guelke and von Blanckenburg, 2007; Guelke-Stelling and von Blanckenburg, 2012; Kurisu and Takahashi, 2019; Kubik et al., 2021), the isotopic signature for biomass burning as a source of ambient aerosol Fe remains unconstrained. Additionally, recent work (Hamilton et al., 2022) showed that soil Fe, rather than biomass Fe, dominates aerosols produced in large scale biomass burning events (wildfires).
In summary, δ56Fe varied greatly from below −1.5 ‰ to around +2.0 ‰ for iron ore; no conclusions can be drawn on δ56Fe for fossil fuel, as observational data are very rare; compared to the UCC, δ56Fe values can be much lower for plants (and especially strategy I plants). However, it should be pointed out that δ56Fe values may not be identical for aerosol particles emitted and their relevant raw materials, because Fe isotopic fractionation may occur during combustion and industrial processes (e.g., iron smelting).
3.2.3 Aerosol particles emitted by anthropogenic sources
Average δ56Fe were determined by Beard et al. (2003a) to be 0.00±0.03 ‰ (n=2) for urban dust (NIST 1649a). Mead et al. (2013) reported the average δ56Fe to be ‰ (n=3) for coal fly ash and ‰ (n=4) for oil fly ash; in addition, δ56Fe values were measured to be ‰ for one diesel particulate matter sample (NIST 1650b) and 0.01±0.12 ‰ for one urban dust sample (NIST 1649a). In another study (Li et al., 2022), δ56Fe were found to be in the range of ‰ to ‰ for three coal fly ash samples (average: ‰) and ‰ for one municipal waste fly ash (BCR-176R).
In one of the largest steelworks in Europe, δ56Fe values displayed larger variations for enriched iron ores, ranging from to ‰ (Flament et al., 2008); for comparison, δ56Fe values were found to be in the range of ‰ to ‰ for sintering fly ash and ‰ for steelwork fly ash. They further suggested that δ56Fe values reported for sintering and steelwork fly ash fell into the range of their raw materials (enriched iron ores), implying no significant Fe isotopic fractionation during steel processes; however, as both enriched iron ores and fly ash displayed large variation in δ56Fe, this conclusion could be uncertain.
In a parking garage, the average δ56Fe was reported to be ‰ for coarse particles (>2.5 µm) (Majestic et al., 2009b), similar to that ( ‰) for fine particles (<2.5 µm) but slightly larger than that for the UCC. Furthermore, δ56Fe were found to be +0.19 ‰ for metallic brake pads, and ranged from +0.42 ‰ to +0.61 ‰ for ceramic brake pads, from −0.08 ‰ to +0.12 ‰ for tire thread, and from +0.04 ‰ to +0.11 ‰ for waste oil.
The aforementioned studies measured isotopic compositions of total Fe in urban dust, coal fly ash, oil fly ash, diesel particulate matter, municipal waste fly ash, sintering and steelwork fly ash, brake pads, tire thread, waste oil of vehicles, and aerosol particles collected in a parking garage. The reported δ56Fe values, which show large variations, are similar to or larger than that for the UCC, and isotopically lighter Fe has hardly been found in these studies.
In contrast, Kurisu et al. (2016b) observed much lower δ56Fe for anthropogenic emissions, as discussed below. For total Fe in aerosol particles collected in a tunnel in Japan, coarse particles (>1 µm) had δ56Fe close to UCC while fine particles (<1 µm) exhibited much lower δ56Fe (as low as −3.2 ‰). Furthermore, δ56Fe were measured to be ‰ , ‰, and ‰ for total Fe in bottom and fly ash of an incinerator in Japan and total suspended particles (TSP) collected close to its chimney, and ‰, ‰, and ‰ for soluble Fe (leached using 1 mol L−1 HCl), respectively. Compared to bottom and fly ash, total Fe in TSP exhibited much lower δ56Fe values; in addition, soluble Fe was isotopically lighter than total Fe. The following mechanism was proposed to explain their observation: the evaporation-condensation process during combustion produced isotopically lighter but more soluble Fe which was enriched in fly ash and especially in emitted aerosol particles, while most Fe in bottom ash did not undergo evaporation-condensation. This argument may explain the discrepancies between the work by Kurisu et al. (2016b) and other studies (Beard et al., 2003a; Flament et al., 2008; Majestic et al., 2009b; Mead et al., 2013; Li et al., 2022), and can also explain isotopic compositions of total and soluble Fe in ambient aerosols reported by field studies (Majestic et al., 2009a; Mead et al., 2013; Kurisu et al., 2016a; Conway et al., 2019; Kurisu et al., 2019, 2021; Zuo et al., 2022).
3.2.4 Discussion
Since the work by Beard et al. (2003a), there have been a small number of studies which measured isotopic compositions of aerosol Fe from relevant sources. As discussed in Sect. 3.2.1, δ56Fe endmember values have been reasonably well understood for total Fe in desert dust, being similar to UCC. On the other hand, previous studies which measured δ56Fe endmember values for anthropogenic aerosols report a range of isotopic compositions (Beard et al., 2003a; Flament et al., 2008; Majestic et al., 2009b; Mead et al., 2013; Kurisu et al., 2016b; Kurisu and Takahashi, 2019; Li et al., 2022), and the endmember values remain poorly constrained for wildfire aerosols (Bunnell et al., 2025). In general, the number of relevant studies of endmembers is very small, and the number of samples covered by each of these studies is also limited; furthermore, compared to total Fe, δ56Fe endmember values of soluble Fe have been much less examined.
The lack of reliable δ56Fe endmember values for total and soluble Fe in non-desert-dust aerosols does limit the use of δ56Fe in tracing and constraining the sources of total and soluble Fe in ambient aerosols. However, we note that the existing studies show much promise. Therefore, we strongly recommend additional measurements of δ56Fe endmember values for total and soluble Fe in non-desert-dust aerosols (aerosols emitted from fossil fuel combustion, biomass burning and metal smelting, for example). In addition, it is very important to continue to explore the dependence of δ56Fe endmember values on particle size, as some previous studies suggested that Fe was isotopically lighter in fine particles when compared to coarse particles (Mead et al., 2013; Kurisu et al., 2016b, 2019, 2024; Bunnell et al., 2025).
3.3 Fe isotopic composition of ambient aerosols
Table S2 summarizes previous studies which measured Fe isotopic compositions of ambient aerosols. Studies published before 2016 and since 2016 are reviewed in Sect. 3.3.1 and 3.3.2, respectively. We choose the year of 2016 as the cutoff, because Kurisu and co-workers who published several papers in this field published the first paper in 2016. Large variations in δ56Fe have been reported for ambient aerosols: δ56Fe ranged from −3.53 ‰ to +0.48 ‰ for total aerosol Fe, and from −4.46 ‰ to +0.47 ‰ for soluble aerosol Fe; by comparison, only a small range of δ56Fe (−0.1 ‰ to +0.2 ‰) has been reported for desert dust, with an average value of +0.1 ‰.
3.3.1 Studies published prior to 2016
Beard et al. (2003a) reported the first measurement of isotopic signatures of ambient aerosol Fe. The average δ56Fe of total Fe were determined to be ‰ for the two TSP samples collected close to the Gobi desert and ‰ for the 12 TSP samples collected at a site in the northwest Pacific (Beard et al., 2003a), both similar to the UCC.
Waeles et al. (2007) measured Fe isotopic compositions of aerosols collected over the Atlantic and in Barbados, and the average δ56Fe was determined to be ‰ for total aerosol Fe and ‰ for soluble aerosol Fe. No significant difference in isotopic composition was observed between total and soluble aerosol Fe, although desert dust aerosol could be greatly aged at Barbados which is ∼ 4000 km away from the dust region.
At an urban site severely affected by a large steel metallurgy plant in France, total aerosol Fe was found to have an average δ56Fe of ‰ (Flament et al., 2008), similar to the UCC. For three aerosol samples collected over the western equatorial Pacific, δ56Fe of total Fe ranged from +0.27 ‰ to +0.38 ‰ with an average value of ‰ (Labatut et al., 2014), slightly higher than the UCC.
Majestic et al. (2009a) investigated isotopic compositions of total Fe in PM2.5 and PM10 simultaneously collected at a mixed suburban/agricultural site (Phoenix, Arizona, USA). The first group of PM10 samples had δ56Fe values (centered at around +0.03 ‰) similar to the UCC, while the other group of PM10 samples exhibited lower δ56Fe values (centered at −0.18 ‰), indicating different Fe sources for the two groups of PM10 samples. Furthermore, the δ56Fe values of total Fe were substantially lower in PM2.5 samples (average: ‰) than PM10 samples (average: ‰), and the difference was correlated with concentrations (in PM2.5) of anthropogenically dominated elements (such as Pb, V and Cr). Therefore, Majestic et al. (2009a) suggested that anthropogenic aerosol Fe may explain the lower δ56Fe for PM2.5 than PM10.
Mead et al. (2013) collected coarse (PM>2.5) and fine (PM2.5) aerosol particles in Bermuda during 2011–2012. As shown in Fig. 3, total Fe concentrations were similar for fine and coarse particles throughout the year, and elevated Fe concentrations in both size fractions were observed during the high dust season (Mead et al., 2013). For total Fe in coarse particles, δ56Fe values were similar to the UCC and exhibited no apparent seasonal variation, with an average value ( ‰, 1σ); in contrast, for total Fe in fine particles, δ56Fe values were significantly higher in the high dust season ‰, (1σ) than the low dust season ( ‰, 1σ), implying significant contribution of non-dust sources to aerosol Fe in fine particles during the low dust season. Furthermore, biomass burning was invoked to potentially explain the lighter Fe in fine particles (Mead et al., 2013) due to lower δ56Fe reported for various plant matters (Guelke and von Blanckenburg, 2007).
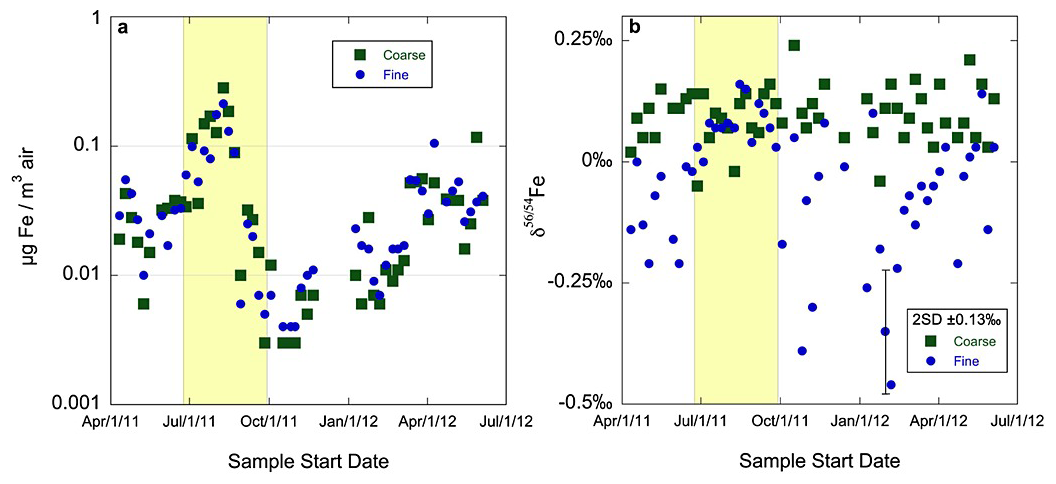
Figure 3Concentrations (a) and δ56Fe values (b) of total Fe in coarse and fine particles collected in Bermuda in 2011–2012. The shaded area represents the high dust season when the sampling site was impacted by Saharan dust aerosol. Reproduced with permission from Mead et al. (2013).
3.3.2 Studies published since 2016
For fine (PM2.5) and coarse (PM>2.5) ambient aerosol particles collected over the northwestern Pacific, Kurisu et al. (2016a) found that the δ56Fe values of total Fe were significantly lower in fine particles ( ‰ and ‰) when compared to coarse particles ( ‰ and ‰). They also measured Fe isotopic compositions for size-fractionated aerosol samples collected at Hiroshima, Japan, and found δ56Fe to be as low as −2.01 ‰ for total aerosol Fe and −3.91 ‰ for soluble aerosol Fe. Figure 4 shows the particle size-dependence of δ56Fe and highlights some important features: (1) δ56Fe of total Fe increased with increasing particle size, and at >1.3 µm were similar to the UCC; (2) at a given particle size, δ56Fe were lower for soluble Fe than total Fe; (3) δ56Fe were lower in August for both total and soluble Fe, when compared to March (when the impact of Asian desert dust was larger). Furthermore, Fe solubility, which appeared to be higher in August than March, increased with a decrease in particle size for both months. Overall, the work by Kurisu et al. (2016a) suggested that anthropogenic Fe, which is more soluble and isotopically lighter than the UCC, was relatively concentrated in finer particles. This conclusion was further supported by Fe speciation via X-ray absorption fine structure (XAFS) spectroscopy analysis.
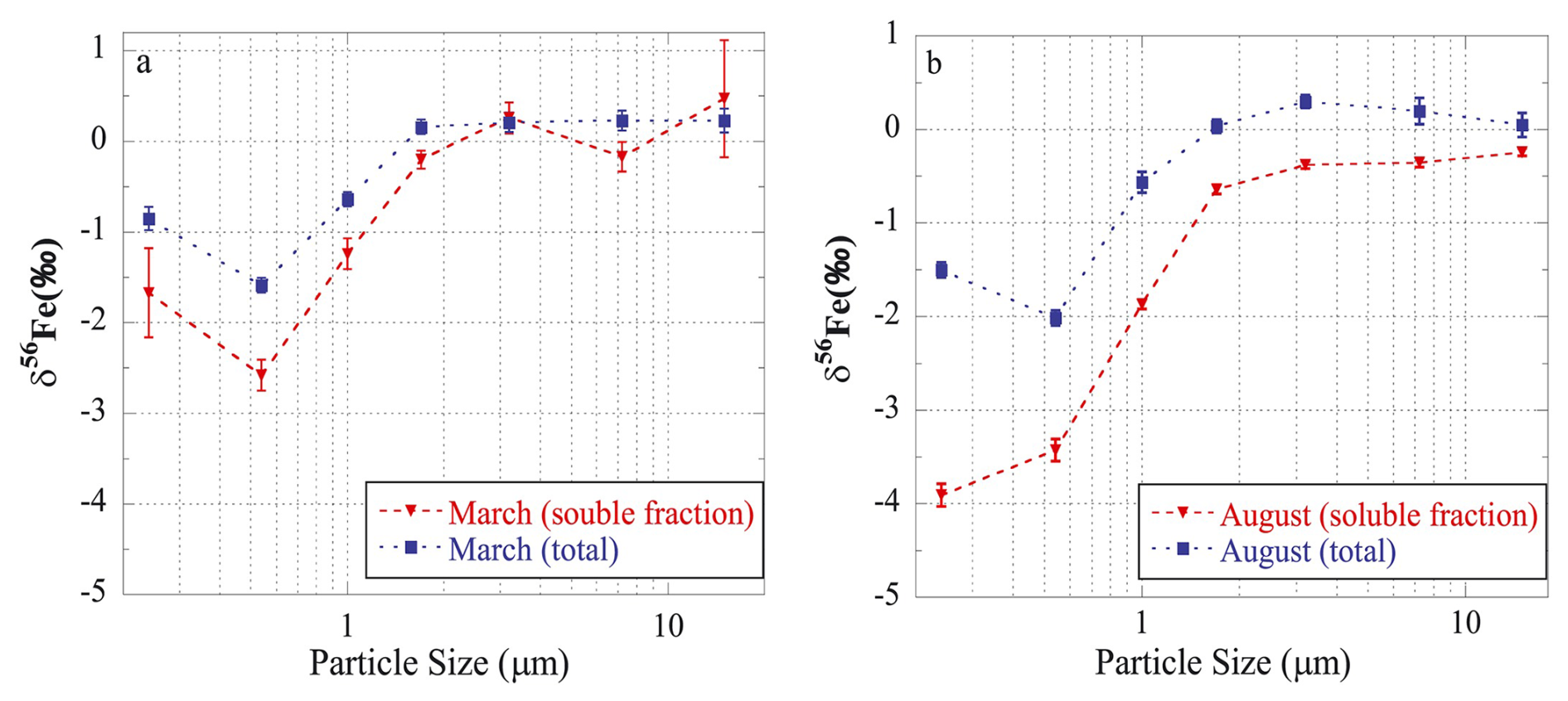
Figure 4Size-resolved δ56Fe of total and soluble Fe for aerosol particles collected at Hiroshima (Japan) in March (a) and August (b). Reproduced with permission from Kurisu et al. (2016a).
Kurisu and Takahashi (2019) measured isotopic compositions of aerosol Fe in ambient air masses affected by a biomass burning event in Tochigi, Japan. Before and after biomass burning, δ56Fe was measured to be ‰ on average for coarse particles (>1 µm), while much lower δ56Fe values were found for fine particles (<1 µm). During the biomass burning event, δ56Fe values of coarse particles were identical to those before and after the event; however, δ56Fe values for fine particles, though still lower than those for coarse particles during the biomass burning event, were found to be higher when compared to those for fine particles before and after the event. As a result, they suggested that the low δ56Fe measured for fine particles before and after the biomass burning event was due to the influence of anthropogenic combustion, and that Fe in biomass burning aerosols did not exhibit low δ56Fe. It was further speculated that combustion temperature during the biomass burning event was not high enough to cause Fe isotopic fractionation (and thus lead to lighter Fe in emitted aerosol particles). In addition, when compared to total Fe, soluble Fe was found to have lower or similar δ56Fe values (Kurisu and Takahashi, 2019).
Another study by Kurisu et al. (2019) examined Fe solubility and isotopic compositions of size-resolved aerosol particles collected in Chiba, Japan, heavily impacted by steel plant emissions, and found higher Fe solubility in fine particles (<1.3 µm) than coarse particles (>1.3 µm). Moreover, δ56Fe ranged from −0.42 ‰ to +0.33 ‰ for coarse particles, likely reflecting Fe isotopic compositions in steel slags and raw materials; Fe in fine particles appeared to be isotopically lighter (δ56Fe in the range of −3.53 ‰ to −0.37 ‰), implying Fe fractionation during evaporation under high temperature. Kurisu et al. (2019) also found that soluble Fe (extracted using ultrapure water or simulated rainwater) was isotopically lighter than total Fe.
Conway et al. (2019) measured solubility and isotopic compositions of aerosol Fe collected over the North Atlantic during winter. Air masses originating from the Saharan region were characterized by higher concentrations of aerosol Fe and lower Fe solubility, and average δ56Fe were reported to be ‰ for total Fe and ‰ for soluble Fe (Fig. 5), similar to the UCC. In contrast, air masses originating from Europe and North America exhibited lower concentrations of aerosol Fe but much higher Fe solubility, and compared to Saharan air masses, δ56Fe in European and North American air masses were much lower for soluble Fe (mean: −0.91 ‰) but only slightly lower for total Fe (mean: ‰). As a result, they suggested that anthropogenic Fe with higher solubility but lower δ56Fe made a large contribution to total Fe and especially soluble Fe in European and North American air masses. This work further utilized a two-component mixing model with assigned endmember δ56Fe values for natural and anthropogenic Fe (+0.09 ‰ and −1.60 ‰ , respectively) to constrain the sources of soluble Fe. Fossil fuel combustion comprised up to ∼ 50 %–100 % of soluble Fe for air masses originating from North America and Europe (Conway et al., 2019), while the Saharan air masses were characterized by ∼ 100 % natural Fe from desert dust.
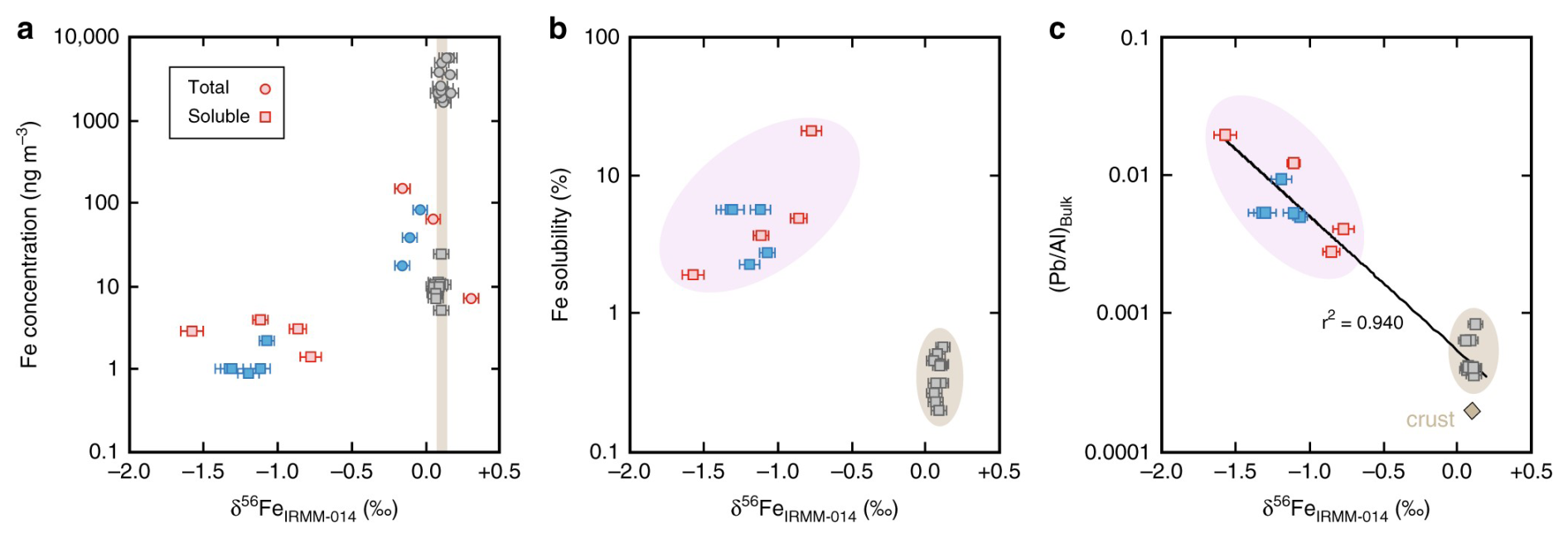
Figure 5Measured δ56Fe of total and soluble Fe versus (a) Fe concentrations and (b) Fe solubility. Aerosol particles from different air masses are separated with colors (gray: Saharan air masses; red: European air masses; blue: North American air masses). Reproduced with permission from Conway et al. (2019).
Kurisu et al. (2021) investigated Fe solubility, speciation, and isotopic compositions of fine (<2.5 µm) and coarse (>2.5 µm) particles collected over the northwestern Pacific. The average δ56Fe value of total Fe was ‰ and ‰ for coarse and fine particles in air masses from central/eastern Pacific, ‰ and ‰ from the North Pacific, and ‰ and ‰ from East Asia. For air masses from East Asia, δ56Fe values of total Fe were found to be negatively correlated with Fe solubility, suggesting that combustion Fe had lower δ56Fe and higher solubility; the relative contribution of combustion to total aerosol Fe could be up to 50 % and 6 % for fine and coarse particles, assuming δ56Fe endmember values to be +0 ‰ and −4.7 ‰ to −3.9 ‰ for dust and combustion Fe. Moreover, for the three samples which contained enough soluble Fe for isotopic analysis, δ56Fe were significantly lower for soluble Fe (as low as ‰) than total Fe (Kurisu et al., 2021), indicating preferential dissolution of Fe with lower δ56Fe (for example, perhaps combustion Fe).
Zuo et al. (2022) analyzed Fe isotopic compositions of magnetic materials in fine (< 2.5 µm) and coarse particles (> 2.5 µm) collected in Beijing during March–May 2021 when dust storms occurred. The average δ56Fe was determined to be ‰ in coarse particles (similar to the UCC), indicating desert dust as the dominant source for magnetic Fe in coarse particles. For fine particles, the average δ56Fe was ‰ during the non-dust-storm period, suggesting important contribution from anthropogenic sources; it increased to ‰ during the dust-storm period, implying an enhanced contribution of desert dust when compared to the non-dust-storm period.
A recent study (Kurisu et al., 2024) measured isotopic composition of total and soluble Fe in TSP and size-resolved aerosol particles collected over the subarctic North Pacific. Overall, for a given sample, δ56Fe was similar or lower for soluble Fe, when compared to total Fe: δ56Fe ranged from −0.5 ‰ to +0.4 ‰ and from −1.9 ‰ to +0.3 ‰ for total and soluble Fe in TSP, and from −2.8 ‰ to +0.5 ‰ for total Fe in the size-resolved aerosol samples. A two-component mixing model was used to estimate the contribution of combustion to total and soluble aerosol Fe, assuming that crustal and combustion Fe had endmembers values of +0.1 ‰ and −4.3 ‰, respectively. The contribution of combustion could reach up to 13 % for total Fe and 45 % for soluble Fe in TSP samples, and was highest in the coastal region of East Asia (Kurisu et al., 2024).
Hsieh and Ho (2024) reported isotopic compositions of total and soluble Fe (ultrapure water leached) for size-resolved aerosol particles collected on a small islet in the East China Sea. The δ56Fe values of total Fe ranged from −0.08 ‰ to +0.16 ‰ for large particles (>1.6 µm), showing no dependence on particle size; however, they decreased to −1.16 ‰ to −0.06 ‰ and further to −3.35 ‰ to −0.45 ‰ for particles in the size range of 1.0–1.6 and 0.57–1.0 µm, respectively. Fe solubility was significantly correlated with δ56Fe of total Fe, suggesting significant contribution of anthropogenic sources to soluble Fe. In addition, soluble Fe was isotopically lighter than total Fe, especially for fine particles (0.57–1.0 and 1.0–1.6 µm). This study reported the lowest δ56Fe values so far for total and soluble Fe in the troposphere (down to −3.35 ‰ and −4.46 ‰, see Table S2). The contribution of anthropogenic sources was estimated to be 3.5 %–7.6 % for total Fe and 22 %–85 % for soluble Fe (Hsieh and Ho, 2024), using a two-component mixing model which assumed δ56Fe endmember values to be +0.1 ‰ and −4.4 ‰ for lithogenic and anthropogenic Fe.
Most recently, Bunnell et al. (2025) measured δ56Fe of TSP and coarse (>0.95 µm) and fine (<0.95 µm) aerosols collected from the North Pacific GEOTRACES GP15 section (along 152° W, from 52° N to 20° S) during a low dust season. In Asian Outflow deployments, total Fe in TSP and coarse particles had δ56Fe values similar to the UCC, ranging from −0.03 ‰ and +0.07 ‰ and +0.02 ‰ to +0.20 ‰, respectively; however, total Fe in fine particles was isotopically lighter (with δ56Fe in the range of −0.39 ‰ to −0.25 ‰), indicative of the contribution of anthropogenic Fe. In the Equatorial Pacific, δ56Fe of total Fe ranged from +0.15 ‰ to +0.41 ‰ for TSP, +0.15 ‰ to +0.27 ‰ for coarse particles, and +0.06 ‰ to +0.36 ‰ for fine particles, similar to or larger than the UCC. Heavier Fe observed in the Equatorial Pacific was attributed to wildfire soil Fe from Californian wildfires, based on the information that up to 64 % of aerosol Fe produced in wildfire is derived from soil due to pyroconvective entrainment (Hamilton et al., 2022) and that δ56Fe are in the range of +0.20 ‰ to +0.95 ‰ for Californian soils (Johnson et al., 2020). Although large differences in δ56Fe of total Fe were observed between Asian Outflow and Equatorial Pacific aerosols, soluble aerosol Fe was generally light (δ56Fe ranging from −1.28 ‰ to +0.02 ‰) throughout the transect (Bunnell et al., 2025), attributed to influence of anthropogenic Fe.
A very recent study (Camin et al., 2025) measured the isotopic composition of total Fe in ten aerosol samples (>1 µm) collected over the Equatorial and Tropical Pacific in August–October 2006. The δ56Fe value was measured to be ‰ for one sample, smaller than that for the UCC, indicating the likely influence of combustion Fe. Total Fe was found to be isotopically heavier than the UCC for the other nine samples, with δ56Fe values ranging from ‰ to ‰. Camin et al. (2025) discussed the possible reasons for the isotopically heavier Fe they observed, and suggested that it could be explained by isotopic fractionation of aerosol Fe during atmospheric transport, as elaborated below. They argue that although aerosol Fe initially had a crustal composition; isotopically lighter Fe was preferentially dissolved and removed during atmospheric transport, and thus the remaining particulate Fe became isotopically heavier; however, this means that the soluble Fe would have had to be removed from the total particles prior to measurement. We also note that Bunnell et al. (2025) attributed heavier aerosol Fe to soil, and that other studies found no fractionation of natural dust Fe during transport.
3.3.3 Discussion
Most field measurements found for total Fe in ambient aerosols, the δ56Fe values were similar to or lower than UCC. From these studies which reported lower δ56Fe for total Fe in ambient aerosols, two features can generally be identified. First, when compared to those dominated by desert dust aerosol, δ56Fe values for total aerosol Fe were lower in air masses severely influenced by anthropogenic emissions. Second, δ56Fe values frequently appeared to be lower for smaller particles than larger particles.
Only a few studies reported isotopic compositions of soluble Fe in ambient aerosols, and some of them reported lower δ56Fe for soluble aerosol Fe (relative to UCC). Similar to total Fe, δ56Fe of soluble Fe were found be lower in air masses with severe impacts by anthropogenic emissions (Kurisu et al., 2016a; Conway et al., 2019), when compared to air masses dominated by desert dust; furthermore, δ56Fe of soluble Fe decreased with decrease in particle size (Kurisu et al., 2016a, 2019). Compared to total aerosol Fe, soluble aerosol Fe was found to be isotopically lighter, especially for smaller particles in air masses severely affected by anthropogenic emissions.
The overall features of isotopic compositions of total and soluble Fe in ambient aerosols, revealed by a limited number of field studies, can be possibly explained by anthropogenic Fe (such as fossil fuel combustion, industrial emission, and etc.) which is isotopically lighter, more soluble, and enriched in fine particles, when compared to desert dust Fe. This explanation is elaborated upon below. First, as anthropogenic Fe is isotopically lighter than desert dust Fe, lower δ56Fe are expected in air masses severely affected by anthropogenic emissions, when compared to air masses dominated by desert dust. Second, since in general anthropogenic Fe is enriched in fine particles while desert dust Fe is enriched in coarse particles, we expect Fe in fine particles to be isotopically lighter than coarse particles. Lastly, anthropogenic Fe is more soluble than desert dust Fe, and therefore the net δ56Fe values are lower for soluble Fe than total Fe.
Some studies have suggested that isotopically lighter δ56Fe in ambient aerosols can be explained by anthropogenic emission (such as fossil fuel combustion and metal smelting). Mead et al. (2013) suggested biomass burning as the likely source of isotopically lighter Fe in ambient aerosols, while more recent studies did not lend support to this idea (Conway et al., 2019; Kurisu and Takahashi, 2019). Furthermore, several studies observed significantly higher δ56Fe (than UCC) for ambient aerosol Fe. This observation has been variably attributed to coal and oil fly ash, to wildfire emissions (Bunnell et al., 2025), or to Fe fractionation due to chemical processing in the atmosphere (Labatut et al., 2014; Camin et al., 2025). Further work to investigate the source signatures of different emissions is required (as discussed in Sect. 3.2).
3.4 Atmospheric modeling studies of isotopic compositions of ambient aerosol Fe
Conway et al. (2019) compared measured solubility and isotopic compositions of aerosol Fe over the Atlantic with those predicted using a 3-D model (Community Atmosphere Model v4, CAM4), in which δ56Fe endmember values were set to +0.09 ‰ for desert dust and −1.6 ‰ for anthropogenic aerosol, as constrained by their observation. Compared to measurements, although the default CAM4 model could reasonably well reproduce Fe solubility for European and North American aerosols, it overestimated Fe solubility for Saharan aerosols; furthermore, it failed to simulate δ56Fe for soluble aerosol Fe (Conway et al., 2019). The model could much better reproduce measured Fe solubility and δ56Fe only if Fe solubility of dust aerosol was reduced from 25 % to 10 % and anthropogenic Fe aerosol emission was increased by a factor of 5. Therefore, it was suggested that the contribution of anthropogenic emission to soluble aerosol Fe might have been significantly underestimated.
Kurisu et al. (2021) employed the integrated massively parallel atmospheric chemical transport (IMPACT) model to simulate isotopic compositions of total Fe in fine (<2.5 µm) and coarse (>2.5 µm) particles over the northwestern Pacific, and in their simulation δ56Fe were set to 0 ‰ for desert dust and −4.7 ‰ to −3.9 ‰ for combustion aerosol. The modelled δ56Fe agreed well with observations for fine particles but were significantly lower for coarse particles. This underestimation may result from using the same endmember δ56Fe values for fine and coarse particles in the model (Kurisu et al., 2021); in other words, if the endmember δ56Fe value of combustion Fe was set to be larger for coarse particles than fine particles, the model may reproduce the observed δ56Fe for both fine and coarse particles.
A very recent study (Bunnell et al., 2025) compared their measured total Fe concentrations and isotopic compositions for aerosols collected from the North Pacific GEOTRACES GP15 section with those simulated by Community Atmosphere Model v6 (CAM6). Based on previous work by Conway et al. (2019), using the initial endmember values (+0.1 ‰ for dust, and −1.60 ‰ for anthropogenic, wildfire and shipping aerosols) in the model caused large underestimations in δ56Fe. When compared to the initial values, the optimal endmember values (which led to best agreement between measured and simulated δ56Fe of ambient total aerosol Fe) remained unchanged for dust and anthropogenic aerosol, but increased to +0.8 ‰ and +0.5 ‰ for wildfire and shipping, respectively (Bunnell et al., 2025).
Overall, modeling studies which simulate isotopic compositions of ambient aerosol Fe are very limited at present. As measurements of isotopic compositions of ambient aerosol Fe and aerosol Fe emitted from various sources are increasing, it is expected that more modeling studies will include isotopic compositions to better constrain sources of aerosol Fe.
3.5 Fe isotopic fractionation induced by chemical processing
Chemical processes which change the speciation of Fe in the environment may also lead to isotopic fractionation (Table S1), and thereby may attenuate Fe isotopic signatures observed in ambient aerosols. However, when using Fe isotopic composition to trace sources of total and soluble aerosol Fe, only a few studies have discussed possible Fe isotopic fractionation induced by atmospheric chemical processing (Labatut et al., 2014; Camin et al., 2025); most previous studies assumed that total and soluble Fe from a given source have the same endmember value, implicitly assuming no Fe isotopic fractionation. On the other hand, Conway et al. (2019) showed that total, water-soluble and seawater-soluble Fe in Saharan dust aerosol over the North Atlantic all had the equivalent δ56Fe to the UCC, suggesting no Fe isotopic fractionation and supporting the use of endmembers without fractionation.
As discussed below, some laboratory studies suggest that chemical processing can lead to significant Fe isotopic fractionation (Table 1). In this section we discuss previous studies which investigated Fe isotopic fractionation induced by a few types of reactions of potential relevance for atmospheric aerosol Fe, namely proton-promoted dissolution, redox reaction, reductive dissolution, ligand complexation and ligand-promoted dissolution. We refer the readers to Yin et al. (2023) for a more comprehensive discussion of Fe isotopic fractionation induced by biogeochemical processes.
3.5.1 Proton-promoted dissolution
For hematite dissolution in 0.9 mol L−1 HCl, no significant equilibrium fractionation of Fe was observed (Skulan et al., 2002). In a later study, Wiederhold et al. (2006) examined goethite dissolution in 0.5 mol L−1 HCl and found that the isotopic composition for dissolved Fe in the solution was not statistically different from that of original goethite over the entire experiment (up to 315 d), suggesting that goethite dissolution in HCl solution did not lead to Fe isotopic fractionation. However, some other studies suggest that proton-promoted dissolution could lead to significant Fe isotopic fractionation. For example, significant Fe isotopic fractionation, with Δ56Fe being as low as −1.8 ‰, was reported for dissolution of granite and basalt in 0.5 mol L−1 HCl (Chapman et al., 2009). Dissolution of biotite and chlorite in HCl solution (pH = 4) also led to significant Fe isotopic fractionation (Kiczka et al., 2010), and Δ56Fe reached as low as −1.4 ‰; furthermore, Kiczka et al. (2010) found the presence of K+ in the solution would facilitate isotopic fractionation.
3.5.2 Redox reaction and reductive dissolution
Johnson et al. (2002) investigated Fe isotopic fractionation for aqueous redox of Fe(III) to Fe(II) at room temperature, and observed similar degrees of Fe isotopic fractionation at pH of 2 and 3: Fe(II) was isotopically lighter than Fe(III), and Δ56Fe was determined to be ‰ when the reaction reached equilibrium. Welch et al. (2003) examined the effects of temperatures (0 and 22 °C) and Cl− concentrations on Fe isotopic fractionation in aqueous redox of Fe(III) to Fe(II) and observed significant enrichment of isotopically lighter Fe in Fe(II). The average Δ56Fe values were ‰, ‰, and ‰ at 22 °C when Cl− concentrations in the solution were 0, 11, and 111 mmol L−1, respectively, suggesting no significant impact of Cl−; moreover, a decrease in temperature from 22 to 0 °C resulted in larger Fe isotopic fractionation, and the average Δ56Fe was ‰ at 0 °C in the absence of Cl− in the aqueous solutions. The two previous studies (Johnson et al., 2002; Welch et al., 2003) both suggested that isotopic fractionation occurred when Fe(III) was reduced to Fe(II) in the solution, with Fe(II) being isotopically lighter than Fe(III).
Wiederhold et al. (2006) explored reductive dissolution of goethite in 0.5 mmol L−1 oxalate solution irradiated using a solar simulator. The dissolved fraction was isotopically lighter in the initial stage, with Δ56Fe being as low as −1.6 ‰; this effect gradually became smaller and the isotopic composition of dissolved Fe eventually was identical to bulk goethite.
3.5.3 Ligand complexation and ligand-promoted dissolution
Wiederhold et al. (2006) showed that at equilibrium Fe isotopic composition was heavier for the Fe(III)-oxalate complex than free Fe(III) in the solution (Δ56Fe = +0.3 ‰). Another study (Dideriksen et al., 2008) found that the formation of the Fe(III)-desferrioxamine B complex in the solution caused Fe isotopic fractionation, and Δ56Fe was determined to be +0.6 ‰ at equilibrium. These experimental results (Wiederhold et al., 2006; Dideriksen et al., 2008) are consistent with theoretical work which predicted enrichment of heavier isotopes in stronger bonding environments under equilibrium fractionation (Schauble, 2004; Dideriksen et al., 2008; Ilina et al., 2013).
Brantley et al. (2001) investigated ligand-promoted dissolution of hornblende in oxalate (0.024 mmol L−1, pH = 7), and found that dissolved Fe was isotopically lighter than that in bulk hornblende (Δ56Fe = −0.3 ‰). In the early stage of ligand-promoted dissolution of goethite in oxalate (5 mmol L−1, pH = 3), significant Fe isotopic fractionation was observed and Δ56Fe could reach as low as −1.2 ‰ (Wiederhold et al., 2006); after that, δ56Fe of dissolved Fe increased gradually with reaction time, and eventually was even slightly larger than bulk goethite due to equilibrium isotope effects. Significant Fe isotopic fractionation occurred during ligand-promoted dissolution (5 mmol L−1 oxalate) of granite and basalt at room temperature (Chapman et al., 2009), and Δ56Fe was down to −1.3 ‰; similarly, Kiczka et al. (2010) found that ligand-promoted dissolution (5 mmol L−1 oxalate) of biotite and chlorite resulted in Fe isotopic fractionation (Δ56Fe as low as −0.5 ‰). In summary, ligand-promoted dissolution resulted in significant Fe isotopic fractionation, and the dissolved Fe appeared to be isotopically lighter due to kinetic fractionation effects.
3.5.4 Fe isotopic fractionation caused by atmospheric chemical processing
Previous studies, as summarized in Table 1, provide important insights into Fe isotopic fractionation induced by chemical processes which may also occur in the atmosphere. However, experimental conditions used in these studies, including minerals examined, may not be of direct relevance for atmospheric aerosols. Recently, some work started to explore the effects of atmospheric chemical processing on Fe isotopic fractionation, as showcased below.
Table 1Fe isotopic fractionation caused by chemical processes relevant for atmospheric aerosol Fe. In this table, pFe(II) represents particulate Fe, dFe(III) and dFe(II) represent dissolved Fe(III) and Fe(II), and dFe(III)-ligand represent dissolved Fe(III) complexed with ligands.

a Skulan et al. (2002), Wiederhold et al. (2006), Chapman et al. (2009), and Kiczka et al. (2010). b Johnson et al. (2002) and Welch et al. (2003). c Wiederhold et al. (2006). d Wiederhold et al. (2006) and Dideriksen et al. (2008). e Brantley et al. (2001), Wiederhold et al. (2006), Chapman et al. (2009), and Kiczka et al. (2010).
Mulholland et al. (2021) studied Fe isotope fractionation during dissolution of Fe-Mn alloy metallurgy fly ash in synthetic cloud water (pH = 2) under UV/VIS radiation from a solar simulator. Compared to Fe in fly ash, dissolved Fe was isotopically lighter in the first stage of dissolution (0–60 min) due to kinetic isotopic effects, with Δ56Fe as low as ‰; in the second stage of dissolution (60–120 min), dissolved Fe was isotopically heavier due to equilibrium isotopic effects, with Δ56Fe as high as ‰. A following study (Maters et al., 2022) further investigated Fe isotope fractionation during the initial stage (0–60 min) of dissolution of Tunisian desert dust and Fe-Mn alloy metallurgy fly ash; compared to Mulholland et al. (2021), the synthetic cloud water used by Maters et al. (2022) additionally contained 1 mmol L−1 oxalate. Compared to Fe in original particles, dissolved Fe was isotopically lighter for both desert dust and fly ash (Maters et al., 2022), and the extent of Fe isotopic fractionation during dissolution appeared to be larger for desert dust.
In the last 10–20 years Fe isotopic analysis has been increasingly used in aerosol research (as discussed in Sect. 3), and has been demonstrated to be a promising way to differentiate sources of total and soluble aerosol Fe, and to quantify the relative importance of different sources. After reviewing and discussing previous studies in a critical manner, we recommend several future research directions below to further enhance the usefulness of Fe isotopes in atmospheric aerosol research.
-
The precision of δ56Fe measurements via MC-ICP-MS can reach ±0.02 to ±0.06 ‰ (2σ) at present, and this should be enough for most applications in atmospheric aerosols. The optimal mass of Fe required for isotopic analysis is around 20–100 ng. It is usually not difficult to collect enough total aerosol Fe for isotopic analysis with acceptable uncertainties, but can be challenging to collect enough soluble aerosol Fe. Therefore, further improvement in analytical methods to reduce the minimum mass of Fe required is warranted to increase the application of Fe isotopic analysis in aerosol research.
-
Using Fe isotopes to constrain the sources of atmospheric aerosol Fe requires constraints on isotopic signatures (δ56Fe endmember values) of aerosol Fe from different sources. The δ56Fe endmember values are reasonably well understood for desert dust Fe, being rather homogeneous and similar to UCC. However, so far only a limited number of studies have measured δ56Fe endmember values of non-desert-dust Fe (Table S1), and the number of samples covered by each of these studies is typically small. The δ56Fe endmember values have large uncertainties for aerosol Fe from various non-desert-dust sources. Therefore, measurements of size-dependent δ56Fe of total and soluble Fe in aerosol particles emitted by various anthropogenic and combustion sources are highly recommended.
-
Up to now only a small number of studies (<20, as summarized in Table S2) have measured isotopic compositions of ambient aerosol Fe, and the isotopic compositions were less measured for soluble aerosol Fe than total aerosol Fe. Further application of Fe isotopes in ambient aerosol research is encouraged, especially in HNLC regions where aerosol Fe deposition may have large impacts on marine primary productivity and in regions where anthropogenic and wildfire emission may have substantial contribution to total and soluble aerosol Fe. Furthermore, additional insights can be gained via comparing source apportionment results constrained using Fe isotopes with those obtained using correlation (Chuang et al., 2005; Sholkovitz et al., 2009; Zhang et al., 2023) and factor analysis (Zhu et al., 2020; Chen et al., 2024; Zhang et al., 2024).
-
In most of previous studies, isotopic compositions are assumed to be identical for total and soluble aerosol Fe from a given source when Fe isotopes are used to constrain the sources of ambient aerosol Fe. This assumption is yet to be verified as some laboratory studies indicate that chemical processing in the atmosphere may lead to Fe isotopic fractionation (Sect. 3.5). As a result, further studies should be carried out to understand isotopic fractionation of aerosol Fe caused by chemical processing under atmospherically relevant conditions. We suggest that dissolution fractions (in %) should also be reported together with isotopic fractionation, in order to facilitate comparisons between different studies.
-
We also encourage modeling studies to include isotopic compositions of aerosol Fe. Comparison of modelled isotopic compositions of total and soluble aerosol Fe with measurements (the number of which is increasing) can provide additional constraints to their sources, in addition to using spatial and temporal variability of their concentrations.
-
Particle size distribution and mineralogy play a critical role in deposition of soluble aerosol Fe, because particle size largely dictates sources, chemical processing and fractional solubility of aerosol Fe (Zhang et al., 2022; Chen et al., 2024) as well as its lifetime, transport and deposition (Liu et al., 2024). A few studies (Mead et al., 2013; Kurisu et al., 2016a, 2019; Kurisu and Takahashi, 2019; Kurisu et al., 2021, 2024) suggest that isotopic compositions of total and soluble aerosol Fe change significantly with particle size, implying variation of their sources with particle size. As a result, size-dependence of isotopic compositions of total and soluble Fe deserves further investigation.
-
Modeling work (Myriokefalitakis et al., 2015; Hamilton et al., 2020b; Hamilton et al., 2020a; Bergas-Massó et al., 2023) suggests that when compared to the preindustrial era, the relative contribution of desert dust, anthropogenic emission and biomass burning to total and soluble aerosol Fe may have changed immensely. Measurements of isotopic compositions of total and dissolved Fe in ice core samples (Conway et al., 2015; Xiao et al., 2020) can provide valuable data to verify historical changes given by modeling simulations.
Enrichment factor: The enrichment factor of an element (X), can be calculated using Eq. (A1) (Gao et al., 2002):
where EF is the enrichment factor of X (to be more precise, the crustal enrichment factor), Y is the reference element dominantly originating from the crustal source, is the mass ratio of X to Y in an aerosol sample, and is the mass ratio of X to Y in the UCC. Usually Al is used as the reference element, although other elements, such as La, Ti and Th, are also be used (Gao et al., 2002; Hird et al., 2024). The non-crustal sources contribute significantly to X in an aerosol sample if its enrichment factor is >10 (or >5).
PM2.5: particulate matters whose aerodynamic diameters do not exceed 2.5 µm. PM>2.5 is frequently used (as in this article) to represent particulate matters with aerodynamic diameters >2.5 µm.
PM10: particulate matters whose aerodynamic diameters do not exceed 10 µm.
Strategy I and II plants: Strategy I plants comprise the dicots and non-grass monocots, while strategy II plants comprise graminaceous plant species. Strategy I and II plants use different strategies to absorb available Fe in soil (Guelke-Stelling and von Blanckenburg, 2012).
Total, dissolved and soluble Fe: Total Fe, which is defined as the total amount of Fe contained in a sample, is typically measured after the sample is digested (Morton et al., 2013). In marine science, dissolved and soluble Fe are typically defined as Fe (contained in a seawater sample) that can pass through a 0.2 or 0.02 µm filter, respectively (Meskhidze et al., 2019). In atmospheric science, soluble Fe is defined as Fe (contained in an aerosol sample) that can be extracted into the solution using a leaching protocol. Please note the definition of soluble Fe in atmospheric science is still inconsistent, as briefly discussed below (Baker and Croot, 2010; Meskhidze et al., 2019; Li et al., 2023). First of all, a wide variety of leaching protocols are used in previous studies to extract soluble Fe in aerosol particles, and different leaching protocols can lead to significant difference in the amount of soluble Fe extracted. Moreover, a few other terms (including dissolved, labile and leachable Fe) are also used interchangeably in the atmospheric science community, and their exact definitions can vary across different studies.
Data used in this review paper all come from previous studies.
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-11067-2025-supplement.
YZ: formal analysis, writing – original draft, writing – review & editing; RL: formal analysis, writing – original draft, writing – review & editing; ZBB: writing – original draft, writing – review & editing; YC: writing – original draft, writing – review & editing; GZhu: writing – review & editing; JM: writing – review & editing; GZha: writing – review & editing; TMC: conceptualization, writing – original draft, writing – review & editing; MT: conceptualization, formal analysis, writing – original draft, writing – review & editing.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors. Also, please note that this paper has not received English language copy-editing. Views expressed in the text are those of the authors and do not necessarily reflect the views of the publisher.
This article is part of the special issue “RUSTED: Reducing Uncertainty in Soluble aerosol Trace Element Deposition (AMT/ACP/AR/BG inter-journal SI)”. It is not associated with a conference.
This work was sponsored by National Natural Science Foundation of China (42321003 and 42277088), Guangzhou Bureau of Science and Technology (2024A04J6533), International Partnership Program of Chinese Academy of Sciences (164GJHZ2024011FN), Guangdong Foundation for Program of Science and Technology Research (2023B1212060049), and Scientific Committee on Oceanic Research (SCOR) Working Group 167 (Reducing Uncertainty in Soluble aerosol Trace Element Deposition, RUSTED). Zachary B. Bunnell and Tim M. Conway were supported by NSF Award OCE-1737136.
This paper was edited by Eliza Harris and reviewed by two anonymous referees.
Baker, A. R. and Croot, P. L.: Atmospheric and marine controls on aerosol iron solubility in seawater, Mar. Chem., 120, 4–13, https://doi.org/10.1016/j.marchem.2008.09.003, 2010.
Baker, A. R. and Jickells, T. D.: Mineral particle size as a control on aerosol iron solubility, Geophys. Res. Lett., 33, L17608, https://doi.org/10.1029/2006GL026557, 2006.
Baker, A. R., Kanakidou, M., Nenes, A., Myriokefalitakis, S., Croot, P. L., Duce, R. A., Gao, Y., Guieu, C., Ito, A., Jickells, T. D., Mahowald, N. M., Middag, R., Perron, M. M. G., Sarin, M. M., Shelley, R., and Turner, D. R.: Changing atmospheric acidity as a modulator of nutrient deposition and ocean biogeochemistry, Science Advances, 7, eabd8800, https://doi.org/10.1126/sciadv.abd8800, 2021.
Beard, B. L. and Johnson, C. M.: High precision iron isotope measurements of terrestrial and lunar materials, Geochim. Cosmochim. Acta, 63, 1653–1660, 1999.
Beard, B. L. and Johnson, C. M.: Fe isotope variations in the modern and ancient earth and other planetary bodies, in: Geochemistry of Non-Traditional Stable Isotopes, edited by: Johnson, C. M., Beard, B. L., and Albarede, F., Reviews in Mineralogy & Geochemistry, 319–357, https://doi.org/10.2138/gsrmg.55.1.319, 2004.
Beard, B. L., Johnson, C. M., Von Damm, K. L., and Poulson, R. L.: Iron isotope constraints on Fe cycling and mass balance in oxygenated Earth oceans, Geology, 31, 629–632, https://doi.org/10.1130/0091-7613(2003)031<0629:IICOFC>2.0.CO;2, 2003a.
Beard, B. L., Johnson, C. M., Skulan, J. L., Nealson, K. H., Cox, L., and Sun, H.: Application of Fe isotopes to tracing the geochemical and biological cycling of Fe, Chemical Geology, 195, 87–117, https://doi.org/10.1016/S0009-2541(02)00390-X, 2003b.
Bergas-Massó, E., Gonçalves Ageitos, M., Myriokefalitakis, S., Miller, R. L., van Noije, T., Le Sager, P., Montané Pinto, G., and Pérez García-Pando, C.: Pre-Industrial, Present and Future Atmospheric Soluble Iron Deposition and the Role of Aerosol Acidity and Oxalate Under CMIP6 Emissions, Earth's Future, 11, e2022EF003353, https://doi.org/10.1029/2022EF003353, 2023.
Boyd, P. W. and Ellwood, M. J.: The biogeochemical cycle of iron in the ocean, Nature Geosci., 3, 675–682, https://doi.org/10.1038/ngeo1964, 2010.
Brantley, S. L., Liermann, L., and Bullen, T. D.: Fractionation of Fe isotopes by soil microbes and organic acids, Geology, 29, 535–538, https://doi.org/10.1130/0091-7613(2001)029<0535:FOFIBS>2.0.CO;2, 2001.
Buck, C. S., Landing, W. M., and Resing, J. A.: Particle size and aerosol iron solubility: A high-resolution analysis of Atlantic aerosols, Mar. Chem., 120, 14–24, https://doi.org/10.1016/j.marchem.2008.11.002, 2010.
Bunnell, Z. B., Sieber, M., Hamilton, D. S., Marsay, C. M., Buck, C. S., Landing, W. M., John, S. G., and Conway, T. M.: The influence of natural, anthropogenic, and wildfire sources on iron and zinc aerosols delivered to the North Pacific Ocean, Geophys. Res. Lett., 52, e2024GL113877, https://doi.org/10.1029/2024GL113877, 2025.
Camin, C., Lacan, F., Pradoux, C., Labatut, M., Johansen, A., and Murray, J. W.: Iron isotopes suggest significant aerosol dissolution over the Pacific Ocean, Atmos. Chem. Phys., 25, 8213–8228, https://doi.org/10.5194/acp-25-8213-2025, 2025.
Chapman, J. B., Weiss, D. J., Shan, Y., and Lemburger, M.: Iron isotope fractionation during leaching of granite and basalt by hydrochloric and oxalic acids, Geochim. Cosmochim. Acta, 73, 1312–1324, https://doi.org/10.1016/j.gca.2008.11.037, 2009.
Chen, T., Li, W., Guo, B., Liu, R., Li, G., Zhao, L., and Ji, J.: Reactive iron isotope signatures of the East Asian dust particles: Implications for iron cycling in the deep North Pacific, Chem. Geol., 531, 119342, https://doi.org/10.1016/j.chemgeo.2019.119342, 2020.
Chen, Y. Z., Wang, Z. Y., Fang, Z. Y., Huang, C. P., Xu, H., Zhang, H. H., Zhang, T. Y., Wang, F., Luo, L., Shi, G. L., Wang, X. M., and Tang, M. J.: Dominant Contribution of Non-dust Primary Emissions and Secondary Processes to Dissolved Aerosol Iron, Environ. Sci. Technol., 58, 17355–17363, https://doi.org/10.1021/acs.est.4c05816, 2024.
Chester, R., Murphy, K. J. T., Lin, F. J., Berry, A. S., Bradshaw, G. A., and Corcoran, P. A.: Factors controlling the solubilities of trace metals from non-remote aerosols deposited to the sea surface by the “dry” deposition mode, Mar. Chem., 42, 107–126, https://doi.org/10.1016/0304-4203(93)90241-F, 1993.
Chuang, P. Y., Duvall, R. M., Shafer, M. M., and Schauer, J. J.: The origin of water soluble particulate iron in the Asian atmospheric outflow, Geophys. Res. Lett., 32, L07813, https://doi.org/10.1029/2004GL021946, 2005.
Conway, T. M. and John, S. G.: Quantification of dissolved iron sources to the North Atlantic Ocean, Nature, 511, 212–215, https://doi.org/10.1038/nature13482, 2014.
Conway, T. M., Rosenberg, A. D., Adkins, J. F., and John, S. G.: A new method for precise determination of iron, zinc and cadmium stable isotope ratios in seawater by double-spike mass spectrometry, Anal. Chim. Acta, 793, 44–52, https://doi.org/10.1016/j.aca.2013.07.025, 2013.
Conway, T. M., Wolff, E. W., Rothlisberger, R., Mulvaney, R., and Elderfield, H. E.: Constraints on soluble aerosol iron flux to the Southern Ocean at the Last Glacial Maximum, Nature Communications, 6, 7850, https://doi.org/10.1038/ncomms8850, 2015.
Conway, T. M., Hamilton, D. S., Shelley, R. U., Aguilar-Islas, A. M., Landing, W. M., Mahowald, N. M., and John, S. G.: Tracing and constraining anthropogenic aerosol iron fluxes to the North Atlantic Ocean using iron isotopes, Nature Comm., 10, 2628, https://doi.org/10.1038/s41467-019-10457-w, 2019.
Conway, T. M., Horner, T. J., Plancherel, Y., and González, A. G.: A decade of progress in understanding cycles of trace elements and their isotopes in the oceans, Chem. Geol., 580, 120381, https://doi.org/10.1016/j.chemgeo.2021.120381, 2021.
Conway, T. M., Middag, R., and Schlitzer, R.: GEOTRACES: Ironing out the details of the oceanic iron sources?, Oceanography, 37, 35–45, https://doi.org/10.5670/oceanog.2024.416, 2024.
Dauphas, N. and Rouxel, O.: Mass spectrometry and natural variations of iron isotopes, Mass Spectrom. Rev., 25, 515–550, https://doi.org/10.1002/mas.20078, 2006.
de Jong, J., Schoemann, V., Tison, J.-L., Becquevort, S., Masson, F., Lannuzel, D., Petit, J., Chou, L., Weis, D., and Mattielli, N.: Precise measurement of Fe isotopes in marine samples by multi-collector inductively coupled plasma mass spectrometry (MC-ICP-MS), Anal. Chim. Acta, 589, 105–119, https://doi.org/10.1016/j.aca.2007.02.055, 2007.
Desboeufs, K., Bon Nguyen, E., Chevaillier, S., Triquet, S., and Dulac, F.: Fluxes and sources of nutrient and trace metal atmospheric deposition in the northwestern Mediterranean, Atmos. Chem. Phys., 18, 14477–14492, https://doi.org/10.5194/acp-18-14477-2018, 2018.
Desboeufs, K. V., Sofikitis, A., Losno, R., Colin, J. L., and Ausset, P.: Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter, Chemosphere, 58, 195–203, https://doi.org/10.1016/j.chemosphere.2004.02.025, 2005.
Dideriksen, K., Baker, J. A., and Stipp, S. L. S.: Equilibrium Fe isotope fractionation between inorganic aqueous Fe(III) and the siderophore complex, Fe(III)-desferrioxamine B, Earth and Planetary Science Letters, 269, 280–290, https://doi.org/10.1016/j.epsl.2008.02.022, 2008.
Fitzsimmons, J. N. and Conway, T. M.: Novel Insights into Marine Iron Biogeochemistry from Iron Isotopes, Annu. Rev. Mar. Sci., 15, 383–406, https://doi.org/10.1146/annurev-marine-032822-103431, 2023.
Flament, P., Mattielli, N., Aimoz, L., Choël, M., Deboudt, K., Jong, J. d., Rimetz-Planchon, J., and Weis, D.: Iron isotopic fractionation in industrial emissions and urban aerosols, Chemosphere, 73, 1793–1798, https://doi.org/10.1016/j.chemosphere.2008.08.042, 2008.
Gao, Y., Nelson, E. D., Field, M. P., Ding, Q., Li, H., Sherrell, R. M., Gigliotti, C. L., Van Ry, D. A., Glenn, T. R., and Eisenreich, S. J.: Characterization of atmospheric trace elements on PM2.5 particulate matter over the New York-New Jersey harbor estuary, Atmos. Environ., 36, 1077–1086, https://doi.org/10.1016/s1352-2310(01)00381-8, 2002.
Gong, Y., Xia, Y., Huang, F., and Yu, H.: Average iron isotopic compositions of the upper continental crust: constrained by loess from the Chinese Loess Plateau, Acta Geochim., 36, 125–131, https://doi.org/10.1007/s11631-016-0131-5, 2017.
González de Vega, C., Chernonozhkin, S. M., Grigoryan, R., Costas-Rodríguez, M., and Vanhaecke, F.: Characterization of the new isotopic reference materials IRMM-524A and ERM-AE143 for Fe and Mg isotopic analysis of geological and biological samples, Journal of Analytical Atomic Spectrometry, 35, 2517–2529, https://doi.org/10.1039/D0JA00225A, 2020.
Guelke, M. and von Blanckenburg, F.: Fractionation of Stable Iron Isotopes in Higher Plants, Environ. Sci. Technol., 41, 1896–1901, https://doi.org/10.1021/es062288j, 2007.
Guelke-Stelling, M. and von Blanckenburg, F.: Fe isotope fractionation caused by translocation of iron during growth of bean and oat as models of strategy I and II plants, Plant and Soil, 352, 217–231, https://doi.org/10.1007/s11104-011-0990-9, 2012.
Hamilton, D. S., Scanza, R. A., Rathod, S. D., Bond, T. C., Kok, J. F., Li, L., Matsui, H., and Mahowald, N. M.: Recent (1980 to 2015) Trends and Variability in Daily-to-Interannual Soluble Iron Deposition from Dust, Fire, and Anthropogenic Sources, Geophys. Res. Lett., 47, e2020GL089688, https://doi.org/10.1029/2020GL089688, 2020a.
Hamilton, D. S., Moore, J. K., Arneth, A., Bond, T. C., Carslaw, K. S., Hantson, S., Ito, A., Kaplan, J. O., Lindsay, K., Nieradzik, L., Rathod, S. D., Scanza, R. A., and Mahowald, N. M.: Impact of Changes to the Atmospheric Soluble Iron Deposition Flux on Ocean Biogeochemical Cycles in the Anthropocene, Glob. Biogeochem. Cycle, 34, e2019GB006448, https://doi.org/10.1029/2019gb006448, 2020b.
Hamilton, D. S., Perron, M. G. G., Bond, T. C., Bowie, A. R., Buchholz, R. R., Guieu, C., Ito, A., Maenhaut, W., Myriokefalitakis, S., Olgun, N., Rathod, S. D., Schepanski, K., Tagliabue, A., Wagner, R., and Mahowald, N. M.: Earth, Wind, Fire, and Pollution: Aerosol Nutrient Sources and Impacts on Ocean Biogeochemistry, Ann. Rev. Mar. Sci., 14, 303–330, https://doi.org/10.1146/annurev-marine-031921-013612, 2022.
Hawco, N. J., Conway, T. M., Coesel, S. N., Barone, B., Seelen, E. A., Yang, S.-C., Bundy, R. M., Pinedo-Gonzalez, P., Bian, X., Sieber, M., Lanning, N. T., Fitzsimmons, J. N., Foreman, R. K., König, D., Groussman, M. J., Allen, J. G., Juranek, L. W., White, A. E., Karl, D. M., Armbrust, E. V., and John, S. G.: Anthropogenic iron alters the spring phytoplankton bloom in the North Pacific transition zone, Proceedings of the National Academy of Sciences, 122, e2418201122, https://doi.org/10.1073/pnas.2418201122, 2025.
Hird, C., Perron, M. M. G., Holmes, T. M., Meyerink, S., Nielsen, C., Townsend, A. T., de Caritat, P., Strzelec, M., and Bowie, A. R.: On the use of lithogenic tracer measurements in aerosols to constrain dust deposition fluxes to the ocean southeast of Australia, Aerosol Research, 2, 315–327, https://doi.org/10.5194/ar-2-315-2024, 2024.
Hsieh, C.-C. and Ho, T.-Y.: Contribution of Anthropogenic and Lithogenic Aerosol Fe in the East China Sea, Journal of Geophysical Research: Oceans, 129, e2024JC021113, https://doi.org/10.1029/2024JC021113, 2024.
Ilina, S. M., Poitrasson, F., Lapitskiy, S. A., Alekhin, Y. V., Viers, J., and Pokrovsky, O. S.: Extreme iron isotope fractionation between colloids and particles of boreal and temperate organic-rich waters, Geochim. Cosmochim. Acta, 101, 96–111, https://doi.org/10.1016/j.gca.2012.10.023, 2013.
Ito, A., Myriokefalitakis, S., Kanakidou, M., Mahowald, N. M., Scanza, R. A., Hamilton, D. S., Baker, A. R., Jickells, T., Sarin, M., Bikkina, S., Gao, Y., Shelley, R. U., Buck, C. S., Landing, W. M., Bowie, A. R., Perron, M. M. G., Guieu, C., Meskhidze, N., Johnson, M. S., Feng, Y., Kok, J. F., Nenes, A., and Duce, R. A.: Pyrogenic iron: The missing link to high iron solubility in aerosols, Science Adv., 5, eaau7671, https://doi.org/10.1126/sciadv.aau7671, 2019.
Ito, A., Ye, Y., Baldo, C., and Shi, Z. B.: Ocean fertilization by pyrogenic aerosol iron, NPJ Clim. Atmos. Sci., 4, 30, https://doi.org/10.1038/s41612-021-00185-8, 2021.
Jickells, T. D., An, Z. S., Andersen, K. K., Baker, A. R., Bergametti, G., Brooks, N., Cao, J. J., Boyd, P. W., Duce, R. A., Hunter, K. A., Kawahata, H., Kubilay, N., laRoche, J., Liss, P. S., Mahowald, N., Prospero, J. M., Ridgwell, A. J., Tegen, I., and Torres, R.: Global Iron Connections between Desert Dust, Ocean Biogeochemistry, and Climate, Science, 308, 67–71, https://doi.org/10.1126/science.1105959, 2005.
John, S. G. and Adkins, J. F.: Analysis of dissolved iron isotopes in seawater, Marine Chemistry, 119, 65–76, https://doi.org/10.1016/j.marchem.2010.01.001, 2010.
John, S. G. and Adkins, J.: The vertical distribution of iron stable isotopes in the North Atlantic near Bermuda, Glob. Biogeochem. Cycle, 26, GB2034, https://doi.org/10.1029/2011GB004043, 2012.
Johnson, C., Beard, B., and Weyer, S.: Iron Geochemistry: An Isotopic Perspective, Springer Cham, Switzerland, https://doi.org/10.1007/978-3-030-33828-2, 2020.
Johnson, C. M., Skulan, J. L., Beard, B. L., Sun, H., Nealson, K. H., and Braterman, P. S.: Isotopic fractionation between Fe(III) and Fe(II) in aqueous solutions, Earth Planet. Sci. Lett., 195, 141–153, 2002.
Johnson, C. M., Beard, B. L., Beukes, N. J., Klein, C., and O'Leary, J. M.: Ancient geochemical cycling in the Earth as inferred from Fe isotope studies of banded iron formations from the Transvaal Craton, Contributions to Mineralogy and Petrology, 144, 523–547, https://doi.org/10.1007/s00410-002-0418-x, 2003.
Johnson, K. S., Gordon, R. M., and Coale, K. H.: What controls dissolved iron concentrations in the world ocean?, Mar. Chem., 57, 137–161, https://doi.org/10.1016/S0304-4203(97)00043-1, 1997.
Kiczka, M., Wiederhold, J. G., Frommer, J., Kraemer, S. M., Bourdon, B., and Kretzschmar, R.: Iron isotope fractionation during proton- and ligand-promoted dissolution of primary phyllosilicates, Geochim. Cosmochim. Acta, 74, 3112–3128, https://doi.org/10.1016/j.gca.2010.02.018, 2010.
König, D., Conway, T. M., Ellwood, M. J., Homoky, W. B., and Tagliabue, A.: Constraints on the Cycling of Iron Isotopes From a Global Ocean Model, Glob. Biogeochem. Cycle, 35, e2021GB006968, https://doi.org/10.1029/2021GB006968, 2021.
König, D., Conway, T. M., Hamilton, D. S., and Tagliabue, A.: Surface Ocean Biogeochemistry Regulates the Impact of Anthropogenic Aerosol Fe Deposition on the Cycling of Iron and Iron Isotopes in the North Pacific, Geophys. Res. Lett., 49, e2022GL098016, https://doi.org/10.1029/2022GL098016, 2022.
Kubik, E., Moynier, F., Paquet, M., and Siebert, J.: Iron Isotopic Composition of Biological Standards Relevant to Medical and Biological Applications, Frontiers in Medicine, 8, 696367, https://doi.org/10.3389/fmed.2021.696367, 2021.
Kumar, A., Sarin, M. M., and Srinivas, B.: Aerosol iron solubility over Bay of Bengal: Role of anthropogenic sources and chemical processing, Mar. Chem., 121, 167–175, https://doi.org/10.1016/j.marchem.2010.04.005, 2010.
Kurisu, M. and Takahashi, Y.: Testing Iron Stable Isotope Ratios as a Signature of Biomass Burning, Atmosphere, 10, 76, https://doi.org/10.3390/atmos10020076, 2019.
Kurisu, M., Adachi, K., Sakata, K., and Takahashi, Y.: Stable isotope ratios of combustion iron produced by evaporation in a steel plant, ACS Earth and Space Chem., 3, 588–598, https://doi.org/10.1021/acsearthspacechem.8b00171, 2019.
Kurisu, M., Takahashi, Y., Iizuka, T., and Uematsu, M.: Very low isotope ratio of iron in fine aerosols related to its contribution to the surface ocean, J. Geophys. Res.-Atmos., 121, 11119–11136, 2016a.
Kurisu, M., Sakata, K., Miyamoto, C., Takaku, Y., Iizuka, T., and Takahashi, Y.: Variation of Iron Isotope Ratios in Anthropogenic Materials Emitted through Combustion Processes, Chem. Lett., 45, 970–972, https://doi.org/10.1246/cl.160451, 2016b.
Kurisu, M., Sakata, K., Uematsu, M., Ito, A., and Takahashi, Y.: Contribution of combustion Fe in marine aerosols over the northwestern Pacific estimated by Fe stable isotope ratios, Atmos. Chem. Phys., 21, 16027–16050, https://doi.org/10.5194/acp-21-16027-2021, 2021.
Kurisu, M., Sakata, K., Nishioka, J., Obata, H., Conway, T. M., Hunt, H. R., Sieber, M., Suzuki, K., Kashiwabara, T., Kubo, S., Takada, M., and Takahashi, Y.: Source and fate of atmospheric iron supplied to the subarctic North Pacific traced by stable iron isotope ratios, Geochim. Cosmochim. Acta, 378, 168–185, https://doi.org/10.1016/j.gca.2024.06.009, 2024.
Labatut, M., Lacan, F., Pradoux, C., Chmeleff, J., Radic, A., Murray, J. W., Poitrasson, F., Johansen, A. M., and Thil, F.: Iron sources and dissolved-particulate interactions in the seawater of the Western Equatorial Pacific, iron isotope perspectives, Glob. Biogeochem. Cycle, 28, 1044–1065, https://doi.org/10.1002/2014GB004928, 2014.
Lacan, F., Radic, A., Jeandel, C., Poitrasson, F., Sarthou, G., Pradoux, C., and Freydier, R.: Measurement of the isotopic composition of dissolved iron in the open ocean, Geophys. Res. Lett., 35, L24610, https://doi.org/10.1029/2008GL035841, 2008.
Lacan, F., Radic, A., Labatut, M., Jeandel, C., Poitrasson, F., Sarthou, G., Pradoux, C., Chmeleff, J., and Freydier, R.: High-Precision Determination of the Isotopic Composition of Dissolved Iron in Iron Depleted Seawater by Double Spike Multicollector-ICPMS, Analytical Chemistry, 82, 7103–7111, https://doi.org/10.1021/ac1002504, 2010.
Li, R., Zhang, H. H., Wang, F., He, Y. T., Huang, C. P., Luo, L., Dong, S. W., Jia, X. H., and Tang, M. J.: Mass fractions, solubility, speciation and isotopic compositions of iron in coal and municipal waste fly ash, Sci. Total Environ., 838, 155974, https://doi.org/10.1016/j.scitotenv.2022.155974, 2022.
Li, R., Dong, S. W., Huang, C. P., Yu, F., Wang, F., Li, X. F., Zhang, H. H., Ren, Y., Guo, M. X., Chen, Q. C., Ge, B. Z., and Tang, M. J.: Evaluating the effects of contact time and leaching solution on measured solubilities of aerosol trace metals, Appl. Geochem., 148, 105551, https://doi.org/10.1016/j.apgeochem.2022.105551, 2023.
Li, W. J., Xu, L., Liu, X. H., Zhang, J. C., Lin, Y. T., Yao, X. H., Gao, H. W., Zhang, D. Z., Chen, J. M., Wang, W. X., Harrison, R. M., Zhang, X. Y., Shao, L. Y., Fu, P. Q., Nenes, A., and Shi, Z. B.: Air pollution–aerosol interactions produce more bioavailable iron for ocean ecosystems, Science Adv., 3, e1601749, https://doi.org/10.1126/sciadv.1601749, 2017.
Liu, M., Matsui, H., Hamilton, D. S., Rathod, S. D., Lamb, K. D., and Mahowald, N. M.: Representation of iron aerosol size distributions of anthropogenic emissions is critical in evaluating atmospheric soluble iron input to the ocean, Atmos. Chem. Phys., 24, 13115–13127, https://doi.org/10.5194/acp-24-13115-2024, 2024.
Lobato, L. M., Figueiredo e Silva, R. C., Angerer, T., de Cássia Oliveira Mendes, M., and Hagemann, S. G.: Iron Isotopes Applied to BIF-Hosted Iron Deposits, in: Isotopes in Economic Geology, Metallogenesis and Exploration, edited by: Huston, D., and Gutzmer, J., Springer International Publishing, Cham, 399–432, https://doi.org/10.1007/978-3-031-27897-6_13, 2023.
Mahowald, N. M., Hamilton, D. S., Mackey, K. R. M., Moore, J. K., Baker, A. R., Scanza, R. A., and Zhang, Y.: Aerosol trace metal leaching and impacts on marine microorganisms, Nature Comm., 9, 2614, https://doi.org/10.1038/s41467-018-04970-7, 2018.
Majestic, B. J., Anbar, A. D., and Herckes, P.: Stable Isotopes as a Tool to Apportion Atmospheric Iron, Environ. Sci. Technol., 43, 4327–4333, https://doi.org/10.1021/es900023w, 2009a.
Majestic, B. J., Anbar, A. D., and Herckes, P.: Elemental and iron isotopic composition of aerosols collected in a parking structure, Sci. Total Environ., 407, 5104–5109, https://doi.org/10.1016/j.scitotenv.2009.05.053, 2009b.
Marsay, C. M., Kadko, D., Landing, W. M., and Buck, C. S.: Bulk Aerosol Trace Element Concentrations and Deposition Fluxes During the U.S. GEOTRACES GP15 Pacific Meridional Transect, Glob. Biogeochem. Cycle, 36, e2021GB007122, https://doi.org/10.1029/2021GB007122, 2022.
Maters, E. C., Mulholland, D. S., Flament, P., de Jong, J., Mattielli, N., Deboudt, K., Dhont, G., and Bychkov, E.: Laboratory study of iron isotope fractionation during dissolution of mineral dust and industrial ash in simulated cloud water, Chemosphere, 299, 134472, https://doi.org/10.1016/j.chemosphere.2022.134472, 2022.
Mead, C., Herckes, P., Majestic, B. J., and Anbar, A. D.: Source apportionment of aerosol iron in the marine environment using iron isotope analysis, Geophys. Res. Lett., 40, 5722–5727, https://doi.org/10.1002/2013GL057713, 2013.
Meskhidze, N., Chameides, W. L., Nenes, A., and Chen, G.: Iron mobilization in mineral dust: Can anthropogenic SO2 emissions affect ocean productivity?, Geophys. Res. Lett., 30, 2085, https://doi.org/10.1029/2003GL018035, 2003.
Meskhidze, N., Volker, C., Al-Abadleh, H. A., Barbeau, K., Bressac, M., Buck, C., Bundy, R. M., Croot, P., Feng, Y., Ito, A., Johansen, A. M., Landing, W. M., Mao, J. Q., Myriokefalitakis, S., Ohnemus, D., Pasquier, B., and Ye, Y.: Perspective on identifying and characterizing the processes controlling iron speciation and residence time at the atmosphere-ocean interface, Mar. Chem., 217, 103704, https://doi.org/10.1016/j.marchem.2019.103704, 2019.
Moffet, R. C., Furutani, H., Rödel, T. C., Henn, T. R., Sprau, P. O., Laskin, A., Uematsu, M., and Gilles, M. K.: Iron speciation and mixing in single aerosol particles from the Asian continental outflow, J. Geophys. Res.-Atmos., 117, D07204, https://doi.org/10.1029/2011JD016746, 2012.
Moore, C. M., Mills, M. M., Arrigo, K. R., Berman-Frank, I., Bopp, L., Boyd, P. W., Galbraith, E. D., Geider, R. J., Guieu, C., Jaccard, S. L., Jickells, T. D., La Roche, J., Lenton, T. M., Mahowald, N. M., Maranon, E., Marinov, I., Moore, J. K., Nakatsuka, T., Oschlies, A., Saito, M. A., Thingstad, T. F., Tsuda, A., and Ulloa, O.: Processes and patterns of oceanic nutrient limitation, Nature Geosci., 6, 701–710, https://doi.org/10.1038/NGEO1765, 2013.
Morton, P. L., Landing, W. M., Hsu, S.-C., Milne, A., Aguilar-Islas, A. M., Baker, A. R., Bowie, A. R., Buck, C. S., Gao, Y., Gichuki, S., Hastings, M. G., Hatta, M., Johansen, A. M., Losno, R., Mead, C., Patey, M. D., Swarr, G., Vandermark, A., and Zamora, L. M.: Methods for the sampling and analysis of marine aerosols: results from the 2008 GEOTRACES aerosol intercalibration experiment, Limnol. Oceanogr. Methods, 11, 62–78, https://doi.org/10.4319/lom.2013.4311.4362, 2013.
Mulholland, D. S., Flament, P., de Jong, J., Mattielli, N., Deboudt, K., Dhont, G., and Bychkov, E.: In-cloud processing as a possible source of isotopically light iron from anthropogenic aerosols: New insights from a laboratory study, Atmos. Environ., 259, 118505, https://doi.org/10.1016/j.atmosenv.2021.118505, 2021.
Myriokefalitakis, S., Daskalakis, N., Mihalopoulos, N., Baker, A. R., Nenes, A., and Kanakidou, M.: Changes in dissolved iron deposition to the oceans driven by human activity: a 3-D global modelling study, Biogeosciences, 12, 3973–3992, https://doi.org/10.5194/bg-12-3973-2015, 2015.
Oakes, M., Weber, R. J., Lai, B., Russell, A., and Ingall, E. D.: Characterization of iron speciation in urban and rural single particles using XANES spectroscopy and micro X-ray fluorescence measurements: investigating the relationship between speciation and fractional iron solubility, Atmos. Chem. Phys., 12, 745–756, https://doi.org/10.5194/acp-12-745-2012, 2012a.
Oakes, M., Ingall, E. D., Lai, B., Shafer, M. M., Hays, M. D., Liu, Z. G., Russell, A. G., and Weber, R. J.: Iron Solubility Related to Particle Sulfur Content in Source Emission and Ambient Fine Particles, Environ. Sci. Technol., 46, 6637–6644, https://doi.org/10.1021/es300701c, 2012b.
Pinedo-González, P., Hawco, N. J., Bundy, R. M., Armbrust, E. V., Follows, M. J., Cael, B. B., White, A. E., Ferrón, S., Karl, D. M., and John, S. G.: Anthropogenic Asian aerosols provide Fe to the North Pacific Ocean, Proc. Natl. Acad. Sci. U.S.A., 117, 27862–27868, https://doi.org/10.1073/pnas.2010315117, 2020.
Poitrasson, F.: On the iron isotope homogeneity level of the continental crust, Chem. Geol., 235, 195–200, https://doi.org/10.1016/j.chemgeo.2006.06.010, 2006.
Radic, A., Lacan, F., and Murray, J. W.: Iron isotopes in the seawater of the equatorial Pacific Ocean: New constraints for the oceanic iron cycle, Earth Planet. Sci. Lett., 306, 1–10, https://doi.org/10.1016/j.epsl.2011.03.015, 2011.
Rathod, S. D., Hamilton, D. S., Mahowald, N. M., Klimont, Z., Corbett, J. J., and Bond, T. C.: A Mineralogy-Based Anthropogenic Combustion-Iron Emission Inventory, J. Geophys. Res.-Atmos., 125, e2019JD032114, https://doi.org/10.1029/2019JD032114, 2020.
Sakata, K., Kurisu, M., Takeichi, Y., Sakaguchi, A., Tanimoto, H., Tamenori, Y., Matsuki, A., and Takahashi, Y.: Iron (Fe) speciation in size-fractionated aerosol particles in the Pacific Ocean: The role of organic complexation of Fe with humic-like substances in controlling Fe solubility, Atmos. Chem. Phys., 22, 9461–9482, https://doi.org/10.5194/acp-22-9461-2022, 2022.
Sakata, K., Sakaguchi, A., Yamakawa, Y., Miyamoto, C., Kurisu, M., and Takahashi, Y.: Measurement report: Stoichiometry of dissolved iron and aluminum as an indicator of the factors controlling the fractional solubility of aerosol iron – results of the annual observations of size-fractionated aerosol particles in Japan, Atmos. Chem. Phys., 23, 9815–9836, https://doi.org/10.5194/acp-23-9815-2023, 2023.
Schauble, E. A.: Applying stable isotope fractionation theory to new systems, in: Geochemistry of Non-Traditional Stable Isotopes, edited by: Johnson, C. M., Beard, B. L., and Albarede, F., Reviews in Mineralogy & Geochemistry, 65–111, https://doi.org/10.2138/gsrmg.55.1.65, 2004.
Sedwick, P. N., Sholkovitz, E. R., and Church, T. M.: Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: Evidence from the Sargasso Sea, Geochem. Geophys. Geosyst., 8, Q10Q06, https://doi.org/10.1029/2007GC001586, 2007.
Shelley, R. U., Morton, P. L., and Landing, W. M.: Elemental ratios and enrichment factors in aerosols from the US-GEOTRACES North Atlantic transects, Deep Sea Research Part II: Topical Studies in Oceanography, 116, 262–272, https://doi.org/10.1016/j.dsr2.2014.12.005, 2015.
Shi, Z. B., Woodhouse, M. T., Carslaw, K. S., Krom, M. D., Mann, G. W., Baker, A. R., Savov, I., Fones, G. R., Brooks, B., Drake, N., Jickells, T. D., and Benning, L. G.: Minor effect of physical size sorting on iron solubility of transported mineral dust, Atmos. Chem. Phys., 11, 8459–8469, https://doi.org/10.5194/acp-11-8459-2011, 2011.
Shi, Z. B., Krom, M. D., Jickells, T. D., Bonneville, S., Carslaw, K. S., Mihalopoulos, N., Baker, A. R., and Benning, L. G.: Impacts on iron solubility in the mineral dust by processes in the source region and the atmosphere: A review, Aeolian Res., 5, 21–42, https://doi.org/10.1016/j.aeolia.2012.03.001, 2012.
Sholkovitz, E. R., Sedwick, P. N., and Church, T. M.: Influence of anthropogenic combustion emissions on the deposition of soluble aerosol iron to the ocean: Empirical estimates for island sites in the North Atlantic, Geochim. Cosmochim. Acta, 73, 3981–4003, https://doi.org/10.1016/j.gca.2009.04.029, 2009.
Sholkovitz, E. R., Sedwick, P. N., Church, T. M., Baker, A. R., and Powell, C. F.: Fractional solubility of aerosol iron: Synthesis of a global-scale data set, Geochim. Cosmochim. Acta, 89, 173–189, https://doi.org/10.1016/j.gca.2012.04.022, 2012.
Sieber, M., Conway, T. M., de Souza, G. F., Hassler, C. S., Ellwood, M. J., and Vance, D.: Isotopic fingerprinting of biogeochemical processes and iron sources in the iron-limited surface Southern Ocean, Earth and Planetary Science Letters, 567, 116967, https://doi.org/10.1016/j.epsl.2021.116967, 2021.
Sieber, M., Lanning, N. T., Steffen, J. M., Bian, X., Yang, S.-C., Lee, J. M., Weiss, G., Hunt, H. R., Charette, M. A., Moore, W. S., Hautala, S. L., Hatta, M., Lam, P. J., John, S. G., Fitzsimmons, J. N., and Conway, T. M.: Long Distance Transport of Subsurface Sediment-Derived Iron From Asian to Alaskan Margins in the North Pacific Ocean, Geophys. Res. Lett., 51, e2024GL110836, https://doi.org/10.1029/2024GL110836, 2024.
Skulan, J. L., Beard, B. L., and Johnson, C. M.: Kinetic and equilibrium Fe isotope fractionation between aqueous Fe(III) and hematite, Geochim. Cosmochim. Acta, 66, 2995–3015, https://doi.org/10.1016/S0016-7037(02)00902-X, 2002.
Tagliabue, A., Bowie, A. R., Boyd, P. W., Buck, K. N., Johnson, K. S., and Saito, M. A.: The integral role of iron in ocean biogeochemistry, Nature, 543, 51–59, https://doi.org/10.1038/nature21058, 2017.
Waeles, M., Baker, A. R., Jickells, T., and Hoogewerff, J.: Global dust teleconnections: aerosol iron solubility and stable isotope composition, Environ. Chem., 4, 233–237, https://doi.org/10.1071/EN07013, 2007.
Wang, Y., Wu, L., Hu, W., Li, W., Shi, Z., Harrison, R. M., and Fu, P.: Stable iron isotopic composition of atmospheric aerosols: An overview, npj Climate and Atmospheric Science, 5, 75, https://doi.org/10.1038/s41612-022-00299-7, 2022.
Wei, T., Dong, Z., Zong, C., Liu, X., Kang, S., Yan, Y., and Ren, J.: Global-scale constraints on the origins of aerosol iron using stable iron isotopes: A review, Earth-Sci. Rev., 258, 104943, https://doi.org/10.1016/j.earscirev.2024.104943, 2024.
Welch, S. A., Beard, B. L., Johnson, C. M., and Braterman, P. S.: Kinetic and equilibrium Fe isotope fractionation between aqueous Fe(II) and Fe(III), Geochim. Cosmochim. Acta, 67, 4231–4250, https://doi.org/10.1016/S0016-7037(03)00266-7, 2003.
Weyer, S. and Schwieters, J. B.: High precision Fe isotope measurements with high mass resolution MC-ICPMS, Int. J. Mass Spectrom., 226, 355–368, https://doi.org/10.1016/S1387-3806(03)00078-2, 2003.
Wiederhold, J. G., Kraemer, S. M., Teutsch, N., Borer, P. M., Halliday, A. N., and Kretzschmar, R.: Iron Isotope Fractionation during Proton-Promoted, Ligand-Controlled, and Reductive Dissolution of Goethite, Environ. Sci. Technol., 40, 3787–3793, https://doi.org/10.1021/es052228y, 2006.
Xiao, C., Du, Z., Handley, M. J., Mayewski, P. A., Cao, J., Schüpbach, S., Zhang, T., Petit, J.-R., Li, C., Han, Y., Li, Y., and Ren, J.: Iron in the NEEM ice core relative to Asian loess records over the last glacial–interglacial cycle, National Science Review, 8, nwaa144, https://doi.org/10.1093/nsr/nwaa144, 2020.
Xu, L., Yang, J.-H., Wang, H., Xie, L.-W., Yang, Y.-H., Huang, C., and Wu, S.-T.: Analytical feasibility of a new reference material (IRMM-524A Fe metal) for the in situ Fe isotopic analysis of pyrite and ilmenite without matrix effects by femtosecond LA-MC-ICP-MS, Journal of Analytical Atomic Spectrometry, 37, 1835–1845, https://doi.org/10.1039/D2JA00151A, 2022.
Yin, N.-H., Louvat, P., Thibault-De-Chanvalon, A., Sebilo, M., and Amouroux, D.: Iron isotopic fractionation driven by low-temperature biogeochemical processes, Chemosphere, 316, 137802, https://doi.org/10.1016/j.chemosphere.2023.137802, 2023.
Zhang, G. H., Bi, X. H., Lou, S. R., Li, L., Wang, H. L., Wang, X. M., Zhou, Z., Sheng, G. Y., Fu, J. M., and Chen, C. H.: Source and mixing state of iron-containing particles in Shanghai by individual particle analysis, Chemosphere, 95, 9–16, https://doi.org/10.1016/j.chemosphere.2013.04.046, 2014.
Zhang, H., Li, R., Huang, C., Li, X., Dong, S., Wang, F., Li, T., Chen, Y., Zhang, G., Ren, Y., Chen, Q., Huang, R., Chen, S., Xue, T., Wang, X., and Tang, M.: Seasonal variation of aerosol iron solubility in coarse and fine particles at an inland city in northwestern China, Atmos. Chem. Phys., 23, 3543–3559, https://doi.org/10.5194/acp-23-3543-2023, 2023.
Zhang, H. H., Li, R., Dong, S. W., Wang, F., Zhu, Y. J., Meng, H., Huang, C. P., Ren, Y., Wang, X. F., Hu, X. D., Li, T. T., Peng, C., Zhang, G. H., Xue, L. K., Wang, X. M., and Tang, M. J.: Abundance and Fractional Solubility of Aerosol Iron During Winter at a Coastal City in Northern China: Similarities and Contrasts Between Fine and Coarse Particles, J. Geophys. Res.-Atmos., 127, e2021JD036070, https://doi.org/10.1029/2021JD036070, 2022.
Zhang, T., Liu, J., Xiang, Y., Liu, X., Zhang, J., Zhang, L., Ying, Q., Wang, Y., Wang, Y., Chen, S., Chai, F., and Zheng, M.: Quantifying anthropogenic emission of iron in marine aerosol in the Northwest Pacific with shipborne online measurements, Sci. Total Environ., 912, 169158, https://doi.org/10.1016/j.scitotenv.2023.169158, 2024.
Zhu, C., Lu, W., He, Y., Ke, S., Wu, H., and Zhang, L.: Iron isotopic analyses of geological reference materials on MC-ICP-MS with instrumental mass bias corrected by three independent methods, Acta Geochimica, 37, 691–700, https://doi.org/10.1007/s11631-018-0284-5, 2018.
Zhu, Y., Li, W., Wang, Y., Zhang, J., Liu, L., Xu, L., Xu, J., Shi, J., Shao, L., Fu, P., Zhang, D., and Shi, Z.: Sources and processes of iron aerosols in a megacity in Eastern China, Atmos. Chem. Phys., 22, 2191–2202, https://doi.org/10.5194/acp-22-2191-2022, 2022.
Zhu, Y. H., Li, W. J., Lin, Q. H., Yuan, Q., Liu, L., Zhang, J., Zhang, Y. X., Shao, L. Y., Niu, H. Y., Yang, S. S., and Shi, Z. B.: Iron solubility in fine particles associated with secondary acidic aerosols in east China, Environ. Pollut., 264, 114769, https://doi.org/10.1016/j.envpol.2020.114769, 2020.
Zuo, P., Huang, Y., Liu, P., Zhang, J., Yang, H., Liu, L., Bi, J., Lu, D., Zhang, Q., Liu, Q., and Jiang, G.: Stable Iron Isotopic Signature Reveals Multiple Sources of Magnetic Particulate Matter in the 2021 Beijing Sandstorms, Environ. Sci. Tech. Lett., 9, 299–305, https://doi.org/10.1021/acs.estlett.2c00144, 2022.
- Abstract
- Introduction
- Introduction to Fe isotopes and their analysis
- Application of Fe isotopic analysis in aerosol research
- Perspectives
- Appendix A: Glossary
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Introduction to Fe isotopes and their analysis
- Application of Fe isotopic analysis in aerosol research
- Perspectives
- Appendix A: Glossary
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Special issue statement
- Financial support
- Review statement
- References
- Supplement