the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 18 Sep 2025
| 18 Sep 2025
Evaluating NOx fate and organic nitrate chemistry from α-pinene oxidation using stable oxygen and nitrogen isotopes
Wendell W. Walters
Masayuki Takeuchi
Danielle E. Blum
Gamze Eris
David Tanner
Weiqi Xu
Jean Rivera-Rios
Fobang Liu
Tianchang Xu
Greg Huey
Justin B. Min
Rodney Weber
Nga L. Ng
Meredith G. Hastings
The oxidation of biogenic volatile organic compounds (BVOCs) such as α-pinene in the presence of nitrogen oxides (NOx= NO + NO2) initiates complex photochemical processes that produce organic nitrates (RONO2) and influence atmospheric oxidation capacity, air quality, and the fate of reactive nitrogen. However, tracking the chemical fate of RONO2 remains challenging as it includes pathways such as renoxification, aerosol partitioning, deposition, and/or hydrolysis to nitric acid (HNO3). Stable oxygen (Δ17O, δ18O) and nitrogen (δ15N) isotope measurements can provide a unique tool to probe these processes, as NOy species can exhibit distinct isotopic signatures due to characteristic oxygen-transfer dynamics and isotope fractionation. Here, we present chamber experiments of α-pinene oxidation in the presence of NOx under a range of oxidant and photochemical conditions, reporting the Δ17O, δ18O, and δ15N values of simultaneously collected NO2, HNO3, and particulate nitrate (pNO3), the latter of which derived predominantly from RONO2 in the conducted experiments. A strong linear relationship between δ18O and Δ17O across all NOy species (r=0.992; p<0.01) supports a two-endmember mixing model, in which oxygen atoms are transferred from isotopically distinct sources that include ozone (O3) with high δ18O and Δ17O as well as peroxy and hydroxyl radicals (RO2, HO2, OH) with lower values. Nitrogen isotope fractionation, quantified as the difference in δ15N values (Δδ15N), revealed consistently positive Δδ15N(HNO3–NO2) values (+28.9 ± 13.4 ‰ in daytime experiments; +22.2 ± 1.4 ‰ at night) and negative Δδ15N(pNO3–NO2) values (−13.6 ± 5.8 ‰ in daytime experiments). This reflected distinct formation pathways and isotope effects including NOx photochemical cycling, thermal dinitrogen pentoxide (N2O5)–nitrate radical (NO3)–NO2 equilibrium, and HNO3 production mechanisms. Box-model simulations based on Δ17O values as a constraint were conducted using a newly developed gas-phase mechanism, which reproduced Δ17O(NO2) and Δ17O(pNO3) (compared to simulated Δ17O(RONO2)) accurately, with an average model bias of 0.9 ± 2.4 ‰ (R2=0.98) and −1.4 ± 2.4 ‰ (R2= 0.55 and R2= 0.97 when excluding one outlier), respectively. We further empirically derived important isotopic parameters such as the Δ17O value transferred from O3 through comparison of model-simulated oxygen atom source contributions with observed Δ17O values for NO2 and pNO3 across experiments. This yielded best-fit slopes of 39.4 ± 0.6 ‰ for NOx photochemical cycling and 41.7 ± 1.2 ‰ for RONO2 formation, consistent with near-surface observations of Δ17O in the terminal oxygen atom of O3. Despite the agreement with NO2 and RONO2, accurately simulating Δ17O(HNO3) proved challenging. Sensitivity tests revealed that model biases likely stemmed from a combination of factors including background HNO3 chamber blanks affecting low-NOx experiments, missing N2O5 heterogeneous hydrolysis under nighttime conditions, and an overestimation in the Δ17O(HNO3) mass balance resulting from the NO2+ OH reaction, which was improved by adjusting the contribution from ()Δ17O(NO2) to ()Δ17O(NO2). These adjustments reduced the average model bias in Δ17O(HNO3) from 6.7 ± 3.3 ‰ (R2= 0.39) in the base mechanism to 1.6 ± 1.3 ‰ (R2= 0.48) in the modified mechanism. These findings demonstrate the utility of Δ17O and δ15N for disentangling nitrate formation mechanisms, while also highlighting critical gaps in our understanding of the isotope dynamics involving HNO3 formation. Future experimental work targeting isolated HNO3 pathways is essential to refine isotopic mass balance assumptions and nitrogen isotope fractionation.
- Article
(2037 KB) - Full-text XML
-
Supplement
(3237 KB) - BibTeX
- EndNote




The oxidation of biogenic volatile organic compounds (BVOCs) in the presence of nitrogen oxides (NOx= NO + NO2) plays a central role in atmospheric chemistry, linking anthropogenic emissions to the formation of ozone (O3), secondary organic aerosol (SOA), and the cycling of reactive nitrogen (Hoyle et al., 2011; Ng et al., 2017; Romer et al., 2016; Sato et al., 2022; Takeuchi and Ng, 2019; Xu et al., 2015b, 2020; Zare et al., 2018). A key product of this interaction is organic nitrate (RONO2), which can be produced during both daytime and nighttime oxidation reactions involving monoterpenes (Ng et al., 2017). The RONO2 product can act either as a temporary NOx reservoir or a permanent sink, depending on its atmospheric fate (Fisher et al., 2016). Once formed, gas-phase RONO2 can photolyze or oxidize, leading to the release of NOx (“renoxification”); partition into the particle phase, resulting in particulate RONO2 (pRONO2) (Beaver et al., 2012; Browne et al., 2014; Browne and Cohen, 2012; Fisher et al., 2016; Wang et al., 2023); and/or undergo dry and wet deposition, leading to the removal of reactive nitrogen from the atmosphere. Hydrolysis has emerged as an important pathway that directly converts RONO2 into HNO3, which acts as a permanent sink of NOx, but the efficiency of this process remains challenging to constrain. Model investigations have suggested a lifetime of RONO2 with respect to hydrolysis on the order of a few hours (Fisher et al., 2016; Zare et al., 2018). Experimental measurements have indicated a complex picture, in which not all monoterpene-derived RONO2 hydrolyzes, and the lifetime of α-pinene-derived RONO2 due to hydrolysis ranges on the order of minutes to hours and even days (Rindelaub et al., 2015; Takeuchi and Ng, 2019; Wang et al., 2021). Due to these complexities, the contributions of RONO2 to HNO3 and particulate nitrate (pNO3) budgets remain poorly constrained. These uncertainties hinder our ability to predict NOx lifetime and oxidant budgets, particularly in regions where high BVOC emissions coincide with anthropogenic NOx such as forested areas near urban locations. Here, we develop new tools aimed at tracking RONO2 contributions to HNO3 and pNO3 budgets using stable isotope measurements (Δ17O, δ18O, δ15N) in controlled α-pinene + NOx chamber experiments, providing mechanistic insights into RONO2 formation and loss pathways.
The natural variations of stable oxygen and nitrogen isotopes in various reactive nitrogen (NOy= NOx+ HNO3+ RONO2+ peroxy nitrate (RO2NO2)+ etc.) molecules offer a powerful diagnostic tool to investigate NOx and oxidation chemistry (Michalski et al., 2012; Walters et al., 2018). Stable isotope constraints may also serve as quantitative tracers to distinguish daytime (e.g., RO2+ NO) versus nighttime (e.g., NO3 + BVOC) RONO2 production, quantify the extent of RONO2 hydrolysis to HNO3, and constrain the relative contributions of organic versus inorganic pathways to the pNO3 budget. Variations in oxygen (O) isotope ratios (i.e., 18O 16O and 17O 16O), commonly quantified using isotope delta notation (Δ17O and δ18O), offer a powerful proxy for assessing oxidation pathways involving NOx photochemical cycling and nitrate formation (Albertin et al., 2021; Alexander et al., 2020; Michalski et al., 2003; Morin et al., 2011; Walters et al., 2024b). This is owing to distinct Δ17O and δ18O values exhibited by key atmospheric oxidants, which are proportionally transferred to NOx during oxidation in the atmosphere (Hastings et al., 2003; Michalski et al., 2003). For instance, tropospheric O3 has an elevated Δ17O with a mean value near 26 ‰ and the transferable terminal oxygen atom of O3 (O) exhibiting a Δ17O of 39 ± 2 ‰ and elevated δ18O near 126 ± 12 ‰ based on recent near-surface observations (Ishino et al., 2017; Vicars and Savarino, 2014). In contrast, other atmospheric oxidants such as RO2 HO2 and OH have Δ17O values assumed to be near 0 ‰ (Lyons, 2001; Walters et al., 2024a). The δ18O values of RO2, HO2, and OH have not been directly determined but are anticipated to be lower than the δ18O(O) (Michalski et al., 2012).
The Δ17O isotopic composition provides a quantitative framework to evaluate NOx photochemical cycling and the formation pathways of nitrate-containing species. This tracer has been widely used to assess NOx oxidation and secondary product formation, as different atmospheric reactions impart distinct Δ17O signatures based on mass balance relationships (Alexander et al., 2020; Michalski et al., 2003; Morin et al., 2011; Walters et al., 2024b) (Table 1). For instance, mass balance calculations predict that Δ17O should differ between HNO3 produced via the daytime NO2+ OH reaction and that derived from the hydrolysis of daytime-formed RONO2, where the Δ17O of the nitrooxy (−NO3) functional group reflects a combination of NO + RO2 oxidation. These differences make Δ17O a potentially powerful diagnostic tool for quantifying RONO2 contributions to HNO3 formation. Moreover, substantial Δ17O differences are expected between RONO2 formed through daytime RO2+ NO reactions and those formed via nighttime NO3+ BVOC reactions. Thus, Δ17O could also be a useful tool for distinguishing between daytime and nighttime formation pathways of RONO2, potentially aiding our ability to accurately predict the atmospheric burden of RONO2. Despite major advances in applying Δ17O to constrain atmospheric nitrogen oxidation chemistry, a significant limitation remains: most Δ17O pathway estimates rely on theoretical mass balance assumptions that have only been empirically validated for a limited number of reactions (e.g., NO + O3 and NO2+ O3) (Berhanu et al., 2012; Savarino et al., 2008). Further, we have yet to measure the Δ17O of RONO2 directly. Expanding direct Δ17O measurements of key NOy compounds under various oxidant conditions is critical for testing these assumptions.
Table 1Expected Δ17O values for reactive nitrogen species formed via different oxidation pathways based on oxygen isotope mass balance. Each pathway is expressed as a weighted average of the Δ17O values of precursor oxidants based on the proposed reaction mechanism. Species include NO2, HNO3, and RONO2.
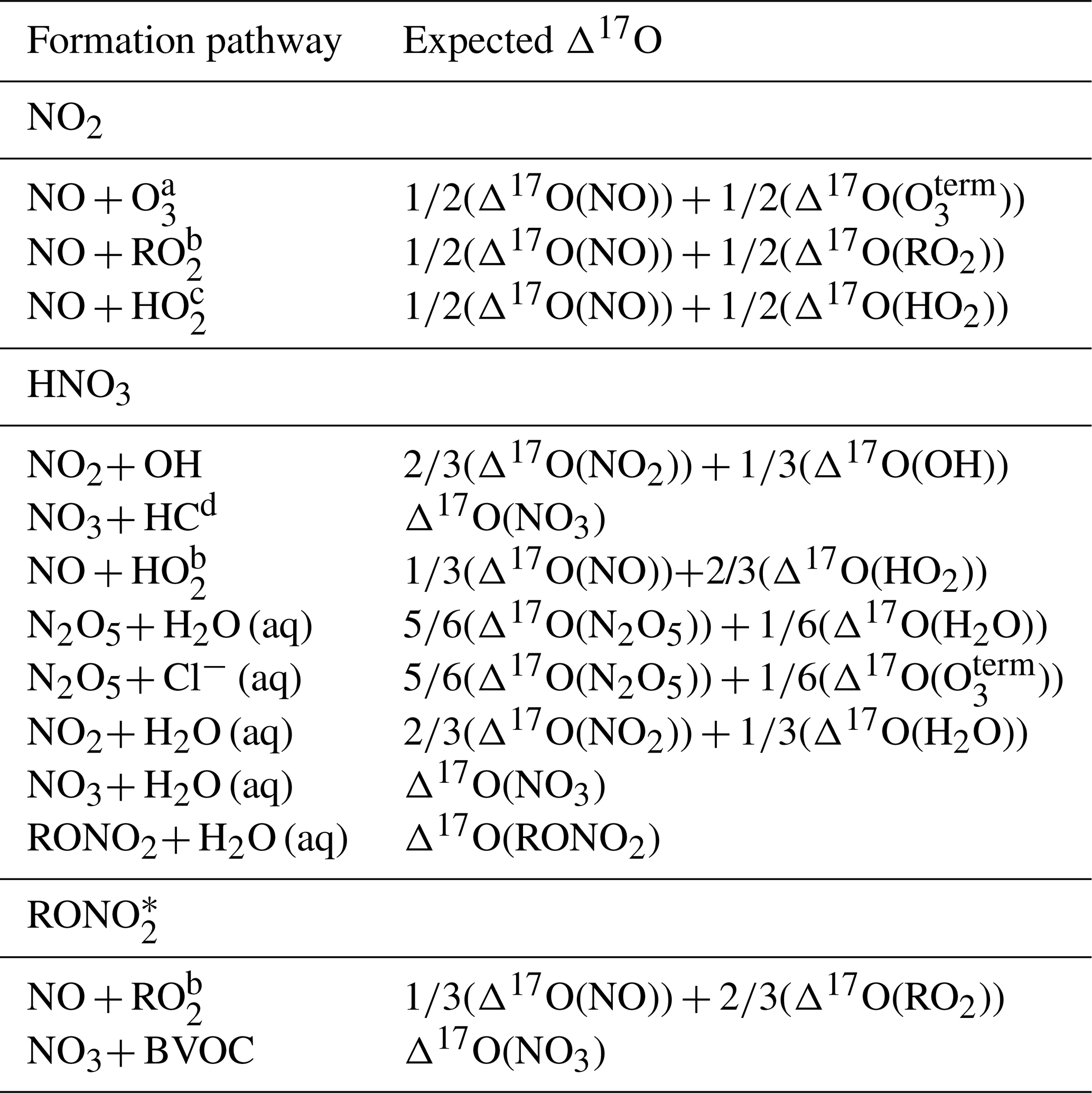
a O terminal oxygen in ozone. b RO2= organic peroxy radical. c HO2= hydroperoxyl radical. d HC = hydrocarbon. * Δ17O calculated from the nitrooxy (−NO3) functional group.
The stable nitrogen (N) isotope ratio variations (δ15N) of NOx and atmospheric nitrate have long served as a valuable proxy for evaluating precursor emission sources because of the preserved N mass between the precursor and oxidized end products (Elliott et al., 2019; Hastings et al., 2013). However, it is essential to consider that NOx photochemical cycling and atmospheric nitrate formation processes can also induce significant mass-dependent fractionation effects (Freyer et al., 1993; Li et al., 2020; Walters et al., 2016; Walters and Michalski, 2015, 2016a). Field δ15N observations of NO2 and nitrate have demonstrated the potential of these fractionation effects to offer additional valuable information concerning NOx photochemical cycling and atmospheric nitrate formation (Albertin et al., 2021; Bekker et al., 2023; Li et al., 2021; Walters et al., 2018). Recently, a novel chemical mechanism was devised to model the nitrogen isotope fractionation associated with NOx chemistry, called incorporating 15N into the Regional Atmospheric Chemistry Mechanism (iNRACM) (Fang et al., 2021). Leveraging these advancements, we may utilize δ15N to gather supplementary quantitative insights into BVOC and NOx interactions and their impact on RONO2 and contributions to HNO3 formation. Importantly, nitrogen isotope fractionation may lead to distinct δ15N values in HNO3 and RONO2 due to differences in their formation pathways. If characterized, these δ15N signatures could offer an additional tracer for quantifying RONO2 contributions to the overall HNO3 budget. While strides have been made in understanding δ15N fractionation during NOx oxidation and equilibrium partitioning (Freyer et al., 1993; Li et al., 2020; Walters et al., 2016; Walters and Michalski, 2015), the specific nitrogen fractionation factors associated with HNO3 and RONO2 formation remain poorly constrained. Targeted laboratory and field studies are needed to directly measure these values and validate their use as diagnostic tools in atmospheric reactive nitrogen chemistry.
This study presents the first simultaneous measurements of Δ17O, δ18O, and δ15N in key NOy species that included NO2, HNO3, and RONO2 generated under controlled laboratory conditions involving α-pinene oxidation. By varying oxidant regimes to probe distinct RO2 fates, we aimed to (1) determine the Δ17O and δ15N values of simultaneously collected HNO3, NO2, and RONO2 under a range of atmospheric oxidation conditions; (2) evaluate the validity of oxygen isotope mass balance assumptions used in the formation of NO2, HNO3, and RONO2; (3) characterize nitrogen isotope fractionation patterns across NOy species; and (4) assess whether stable isotope measurements can provide meaningful constraints on the fate of RONO2 in the atmosphere. Further, by incorporating recent isotope modeling frameworks (Walters et al., 2024a) we simulated the Δ17O values of various NOy components and compared them against observations, yielding insights into the oxidative formation and Δ17O transfer dynamics during NOx oxidation.
2.1 Chamber experiments
Photochemical and nighttime oxidation chamber experiments were conducted involving α-pinene, NOx, and oxidant precursors at the Georgia Institute of Technology Environmental Chamber Facility that houses two 12 m3 Teflon reactors (Boyd et al., 2015). A total of six different initial experimental conditions were targeted, including five photochemical and one nighttime condition as previously reported (Blum et al., 2023) (Table 2). The experiments varied in their precursor concentrations and oxidant types, which were utilized to probe different α-pinene oxidation reactions involving OH, O3, and NO3 and RO2 fates. Replicates were conducted in two of the targeted experimental conditions. The conducted chamber experiments follow previous laboratory protocols (Boyd et al., 2015; Nah et al., 2016; Takeuchi and Ng, 2019; Tuet et al., 2017). Briefly, photochemical experiments were conducted by injecting dry ammonium sulfate seed aerosol and precursor (i.e., α-pinene (99 % Sigma-Aldrich)), NO (Matheson), hydrogen peroxide (H2O2), or nitrous acid (HONO)) into the chamber, where either H2O2 or HONO was used as an OH precursor to simulate different extents of the RO2+ NO pathway. Once the levels of precursors stabilized, the chamber lights were turned on, signifying the start of the photochemical experiments. The procedure used to generate HONO (e.g., the reaction of sodium nitrite with sulfuric acid) also leads to the generation of significant NO and NO2 as a reaction by-product (Kroll et al., 2005; Tuet et al., 2017). For simulated nighttime conditions, dry ammonium sulfate seed aerosol and α-pinene were injected into the chamber, followed by flowing dinitrogen pentoxide (N2O5) for 15 min (Boyd et al., 2015; Takeuchi and Ng, 2019). The N2O5 injection corresponded to the start of the nighttime experiments. The N2O5 was generated by reacting NO2 from a gas cylinder (Matheson) with O3 in a flow tube prior to the introduction to the chamber at a ratio of 2:1 to minimize O3 concentrations in the chamber to avoid ozonolysis. All experiments were conducted at a relative humidity (RH) and temperature of 30 % and 22 °C, respectively. Before each experiment, the chamber was flushed with zero air and irradiated for at least 24 h.
Table 2Summary of measured NOy precursor concentrations and targeted H2O2 concentrations for the chamber experiments. All experiments were conducted using dry ammonium sulfate seed at a fixed temperature (22 °C) and relative humidity (30 %).
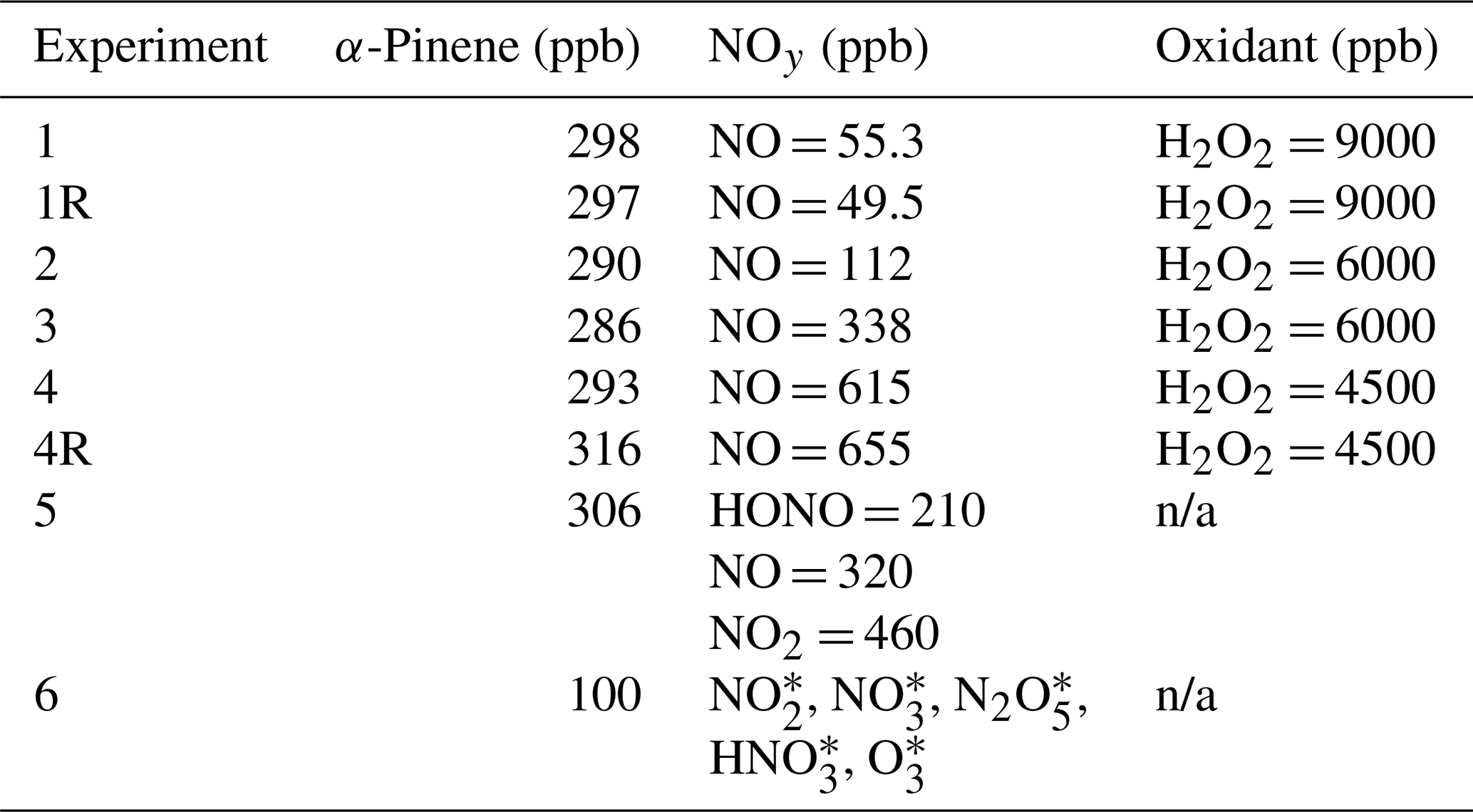
* The emission rates of NO2, NO3, N2O5, HNO3, and O3 into the chamber for a 20 min injection period were modeled based on a flow tube simulation of the reaction of NO2 with O3 with a residence time of 70 s.
n/a: not applicable.
Continuous online measurements of NO, NO2, and O3 were conducted using chemiluminescence (Teledyne 200EU), cavity-attenuated phase shift (CAPS), and an O3 monitor (Teledyne T400). A chemical ionization mass spectrometer (CIMS) was used for various NOy measurements including HONO and HNO3 (Huey et al., 1998). The α-pinene decay was monitored using a gas chromatography flame ionization detector (GC-FID; Agilent 7890A). Gaseous organic nitrate was monitored using a filter inlet for gases and aerosols (FIGAERO) coupled to a high-resolution time-of-flight iodide chemical ionization mass spectrometer (HR-ToF-I-CIMS) with particles collected on a Teflon filter (Lopez-Hilfiker et al., 2014; Nah et al., 2016; Takeuchi et al., 2022; Takeuchi and Ng, 2019; Wang et al., 2023). Aerosol composition was measured using a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS) that included measurement of non-refractory organics (Org), sulfate (SO4), nitrate (NO3), and ammonium (NH4) (DeCarlo et al., 2006; Farmer et al., 2010). Water-soluble aerosol components were also measured using a particle-into-liquid sampler (PILS) coupled to ion chromatography (IC) (Orsini et al., 2003). This method differs from the HR-ToF-AMS measurements, which quantify total aerosol composition, including both water-soluble and water-insoluble components. For example, nitrate measured by the AMS represents total pNO3, whereas nitrate measured by the PILS-IC system corresponds only to the water-soluble fraction of pNO3.
Collections of various NOy gaseous and aerosol components, including HNO3, NO2, and pNO3, were conducted using a modified version of the ChemComb Speciation Cartridge (CCSC) with an extended denuder body for offline concentration and isotope composition analysis (Blum et al., 2020, 2023). Briefly, the CCSC collections began when the aerosol mass spectrometer data indicated the nitrate and secondary organic aerosol mass concentrations had peaked. The CCSC samples were collected at 8 L min−1 for up to 4 h. To maintain the chamber integrity, zero air was used to dilute at 25 L min−1 once the aerosol peak was reached and CCSC sample collection initiated. The CCSC denuder bodies were replaced one to four times depending on the concentration of NOx in the chamber. For each experiment, a single filter was used in the CCSC. In addition to the chamber experiments, samples were collected directly from the NO2 tank (Matheson), which was used in the generation of N2O5 for the nighttime oxidation experiments.
Honeycomb denuders were coated for the selective collection of HNO3 (captured as nitrate (NO)) and NO2 (captured as nitrite (NO)). A detailed description of the coating solutions, denuder preparation, and denuder extraction was previously described, and the pooled isotope reproducibility for both HNO3 and NO2 was ± 1.7 ‰, ± 1.8 ‰, and ± 0.7 ‰ for δ15N, δ18O, and Δ17O for these chamber experiments (Blum et al., 2023). The collection of PM2.5 was conducted using a quartz filter (Cytiva Whatman, Grade QM-A; 47 mm diameter) that was housed in the ChemComb filter cartridge positioned downstream of the denuders. Prior to use, filters were pre-combusted at 550 °C overnight and stored in an airtight container. Quartz filters were selected because they facilitate efficient water-based extraction of collected material and tolerate high-temperature pre-cleaning to remove organic contaminants. The filter samples were extracted in 20 mL of Milli-Q water (> 18.2 MΩ) and allowed to leach for at least 1 week at room temperature. This method was conducted to enable hydrolysis of collected organic nitrate particles as previous studies have shown organic nitrate derived from α-pinene oxidation to hydrolyze to NO in water with a lifetime of 8.8 h at pH = 6.9 (Rindelaub et al., 2016) and 2.5 h at pH = 7.44 (Wang et al., 2021). Other types of organic nitrate, such as secondary nitrates, have been reported to be stable in water, especially at a neutral pH (Wang et al., 2021). The efficiency of our filter extraction technique for facilitating organic nitrate hydrolysis was evaluated using the online AMS and PILS data. After the filters were leached, the filters were removed, and the samples were shipped to Brown University where they were placed in a freezer until subsequent concentration and isotope analysis. For all sample media types, including denuders and filters, lab blanks were frequently taken. These blanks were prepared, handled, and analyzed the same way as all samples.
2.2 Concentration and isotope analysis
The denuder and filter extracts were analyzed for their NO and NO content using a standardized colorimetric technique (e.g., EPA Methods 353.2) or ion chromatography, as previously described (Blum et al., 2020, 2023). The limits of detection (LOD) were approximately 0.1 µmol L−1 and 0.3 µmol L−1 for NO and NO via colorimetric analysis and 3.0 µmol L−1 for NO via ion chromatography. For all analyses, the average percent relative standard deviation was below 5 %. The NO and NO concentrations from denuder blank extractions used for NO2 (n= 5) and HNO3 (n= 5) collection were below the detection limits. Significant blanks were observed in the quartz filter extracts (1.5 ± 0.2 µmol L−1; n= 5), which we refer to hereinafter as a method blank.
The δ15N, δ18O, and Δ17O isotope compositions were analyzed using the denitrifier method for NO samples (e.g., from HNO3 denuder and aerosol filter extracts) following Casciotti et al. (2002), Kaiser et al. (2007), and Sigman et al. (2001) and the sodium azide and acetic acid buffer method for NO samples (from NO2 denuder extracts), following McIlvin and Altabet (2005) and Walters and Hastings (2023). Briefly, 20 nmol of NO or NO samples was converted to N2O, which is extracted, purified, concentrated, and injected into a continuous-flow isotope ratio mass spectrometer (IRMS) for δ15N and δ18O determination from measurement at 44, 45, and 46. In a separate batch analysis, 50 nmol of NO or NO was converted to N2O, decomposed to O2 using a gold tube heated at 770 °C, and analyzed at 32, 33, and 34 for Δ17O determination (Kaiser et al., 2007; Walters and Hastings, 2023). To minimize potential memory effects from residual O2 in the gold tube and headspace trapping system, samples were grouped and analyzed in separate batches based on their expected Δ17O values. Specifically, NO2 samples (high Δ17O), HNO3 (moderate Δ17O), and pNO3 (low Δ17O) were each analyzed in separate batches. Analytical blanks associated with the conversion of NO and/or NO to N2O or O2 for subsequent IRMS analysis were assessed for each batch and were always below the detection limit (∼ 0.2 nmol). These blanks are not anticipated to affect the analytical precision of the reported isotope values. The samples were calibrated with respect to internationally recognized NO standards (IAEA-NO-3, USGS35, USGS34) or NO reference materials (RSIL-N7373 and RSIL-10219) (Böhlke et al., 2003, 2007). In line with the identical treatment principle, the reference materials are treated the same as samples, including matching concentrations, sample amounts, and reagent additions, to ensure analytical consistency. The pooled standard deviations of the reference materials were ± 0.1 ‰ and ± 0.6 ‰ for δ15N and δ18O of the NO standards (n = 78) and ± 0.3 ‰ and ± 0.3 ‰ for δ15N and δ18O of the NO reference materials (n= 15), respectively. The Δ17O had a pooled standard deviation of ± 0.6 ‰ (n= 53).
All isotope measurements are reported relative to reference standards using delta (δ) notation (Eq. 1):
where R is the ratio of the abundance of the heavy to the light isotope (i.e., 15N 14N, 18O 16O, or 17O 16O), x denotes the sample, and SD is an abbreviation for standard (Sharp, 2017). The nitrogen and oxygen reference material includes atmospheric air and Vienna Standard Mean Ocean Water (VSMOW), respectively. Oxygen isotope mass independence (Δ17O) was quantified using the linear definition with a mass-dependent coefficient of 0.52, which is approximately representative of oxygen mass-dependent coefficients expected and observed in nature (Young et al., 2002) (Eq. 2):
The linear Δ17O approximation is commonly used to describe large mass-independent fractionation such as those related to O3 reactions, and this definition is commonly used in the atmospheric chemistry community to track the influence of O3 oxidation and Δ17O propagation into reactive components (Alexander et al., 2020; Kim et al., 2023; Michalski et al., 2003; Savarino et al., 2013).
2.3 Data reduction and corrections
Due to significant NO blanks found in the pNO3 filter extracts (i.e., method blank), the measured nitrate isotope values (δ15N, δ18O, and Δ17O) were corrected using a mass balance approach to isolate the isotopic composition of NO generated within the chamber experiments (Eqs. 3–4):
where [NO] corresponds to the concentration of NO in either the blank or sample, and f(blank) corresponds to the NO method blank fraction. The quartz filter method blanks had measured δ15N, δ18O, and Δ17O values of 1.6 ± 1.1 ‰, 16.6 ± 1.4 ‰, and 3.4 ± 0.5 ‰, respectively (n= 3). Blank corrections were made for all samples when f(blank) was less than 30 %. Samples with an f(blank) that exceeded 30 % were not reported for their isotope compositions, which included one out of eight quartz filter extracts. The uncertainty in the blank-corrected isotope deltas was calculated using a Monte Carlo simulation for 10 000 iterations and assuming a normal distribution using MATLAB. For the quality assurance criterion of an f(blank) < 30 %, the uncertainties were calculated to be less than 4.1 ‰, 1.4 ‰, and 0.9 ‰ for δ15N, δ18O, and Δ17O, respectively. These values are small compared to the observed ranges for pNO3, which spanned 67.8 ‰ for δ15N, 29.3 ‰ for δ18O, and 10.4 ‰ for Δ17O and thus do not significantly affect the interpretation of isotope patterns. These uncertainties reflect the standard deviation of the 10 000 Monte Carlo iterations for each pNO3 sample, which account for uncertainty in both the sample and method blank concentrations and isotope values. These reported uncertainties for chamber-derived pNO3 isotope values represent total propagated error after blank correction, not the raw instrumental precision.
2.4 Aerosol nitrate composition
The relative contribution of organic aerosol nitrate (pNO3(Org)) to the total pNO3 was determined from two approaches. First, the relative proportion of pNO3(Org), was calculated based on NO+ and NO HR-ToF-AMS fragmentation as previously described (Farmer et al., 2010; Fry et al., 2009; Kiendler-Scharr et al., 2016; Xu et al., 2015a) (Eq. 5):
where f(pNO3, Org) refers to the fraction of pNO3(Org) to the total pNO3, R refers to NO+ NO fragments, and obs, AN, and ON refer to the observations, ammonium nitrate, and organic nitrate, respectively. The RAN was obtained from routine ionization efficiency calibration of the HR-ToF-AMS using 300 nm ammonium nitrate aerosols and was 1.37. The RON was calculated based on the measured RAN and the ratio of previously reported for similar conducted experiments (Takeuchi and Ng, 2019), resulting in an RON of 2.70 ± 0.29 and 3.86 ± 0.34 for photochemical and nighttime oxidation experiments, respectively. The second method for qualitatively determining f(pNO3, Org) involved evaluating the relative change in the molar ratio of NH4 SO4, as an increase in NH4 SO4 has been observed to be associated with inorganic pNO3 formation (Takeuchi and Ng, 2019).
2.5 Box-model simulations
The chamber experiments were simulated using the Framework for 0-D Atmospheric Modeling (F0AM) box model (Wolfe et al., 2016). The conducted model simulations primarily focus on accurately representing initial precursor oxidation (i.e., NO, α-pinene) and simulation of Δ17O observations. We chose not to include δ15N model simulations in this study due to several key uncertainties. These include incomplete knowledge of the δ15N values of all initial NOy sources, limited constraints on isotope effects during NOy oxidation and photolysis reactions, the potential for unquantified, species-specific fractionation due to chamber wall interactions, and potential for chamber blanks with unknown δ15N values. Given these limitations, δ15N modeling will be deferred to future work focused specifically on nitrogen isotope dynamics.
The model was initiated for each experiment using the measured precursor concentrations for NO, NO2, HONO, and α-pinene before chamber lights were turned on or N2O5 was injected and using the targeted H2O2 concentrations. The pressure, temperature, and relative humidity were fixed at 1013 mbar, 295 K, and 30 %, respectively. The measured chamber light flux data were used. The model was run in two parts for the photochemical reactions, including from “lights on” to peak SOA mass concentration (part 1) and from aerosol decay and chamber dilution to the end of NOy collections (part 2). For the nighttime experiments, the model simulations were conducted in three parts, including from the start of N2O5 injection to the end of N2O5 injection (part 1), from the end of N2O5 injection to peak SOA mass concentrations (part 2), and from the decay of organic aerosol and chamber dilution to the end of NOy collection (part 3). The N2O5 injection was simulated by first modeling the NO2 reaction with O3 in the flow tube, considering a flow tube residence time of 70 s. The nighttime experiment was then simulated by allowing the flow tube products (i.e., NO2, O3, NO3, HNO3, and N2O5) to emit into the chamber for 20 min (part 1). Next, the experiment was modeled without the flow tube emission to the start of aerosol decay and chamber dilution (part 2) and from aerosol decay and chamber dilution to the end of NOy collections (part 3). For both photochemical and nighttime experiments, the model simulations from the decay of organic aerosol to the end of NOy collection included a chamber dilution rate constant (kdil) of s−1, which was calculated based on the dilution flow rate (25 LPM) and chamber volume (12 m3). Sensitivity tests were performed to assess the impact of the selected kdil on model results by evaluating a range of kdil values.
A new chemical mechanism was developed, termed NOx-α-pinene (NOx-API), to accurately model the oxidation of NOx and α-pinene (Tables S1–6). This mechanism was developed due to difficulties in simulating the initial decay of the aerosol precursors including α-pinene, NO, NO2, and HONO for the various experimental conditions using either the Regional Atmospheric Chemical Mechanism, v2 (RACM2) (Goliff et al., 2013) or the Master Chemical Mechanism v3.3.1 (Jenkin et al., 1997; Saunders et al., 2003). The NOx-API mechanism focuses on simulating α-pinene and NO decay along with NOx oxidation but does not intend to accurately simulate SOA production and later-generation chemistry. The mechanism has the inorganic reactions in RACM2, including 16 species and 45 reactions. It also incorporates 29 organic species and 61 reactions to detail organic chemistry up to one generation past pinonaldehyde formation as well as the formation of pinonaldehyde-derived peroxyacetyl nitrate formation, with subsequent chemistry represented by a lumped approach. The α-pinene oxidation pathways involving OH, O3, and NO3, along with specific reactions of the resulting RO2 with HO2, NO, NO3, and other RO2 radicals, are also included. The photochemical oxidation of α-pinene largely follows the MCMv3.3.1 (Master Chemical Mechanism) (Saunders et al., 2003), incorporating two hydroxyl-nitrate isomers from OH/O2/NO, including one tertiary (ONITa) and one secondary (ONITb) and the formation of a tertiary pinene carbonyl nitrate (ONITc). Nighttime oxidation chemistry integrates a recent mechanism for organic nitrate formation, producing pinene nitrate hydroperoxide, including one tertiary (ONITOOHa) and one secondary (ONITOOHb) via HO2 reactions and dimer/pinene dinitrate (PDN) through RO2 interactions (Bates et al., 2022). The box-model simulation is a gas-phase mechanism that does not explicitly model heterogeneous reactions or aerosol chemistry, such as organic nitrate hydrolysis. The impact of heterogenous reactions is considered in the evaluation of the Δ17O model simulation results compared to observations.
The Δ17O values of NOy compounds were simulated using the newly developed NOx-API mechanism modified using the InCorporating Oxygen Isotopes of oxidized reactive Nitrogen in the Regional Atmospheric Chemistry Mechanism, Version 2 (ICOIN-RACM2) model framework (Walters et al., 2024a). Briefly, the model framework tracks the transfer and propagation of Δ17O from O3 into NOy and Ox species. This mechanism tags the oxygen atoms transferred from O3 into NOy and Ox considering mass balance and reaction stoichiometry and enables the offline calculation of Δ17O based on the output of concentrations of various NOy and HOx isotopologues (Eq. 6):
where X refers to the various NOy and Ox molecules and f(Q) is the fraction of oxygen atoms deriving from O for a particular molecule. The Δ17O(O) represents the Δ17O value of the terminal and transferrable O atom of O3, which was assumed to be 39 ± 2 ‰ based on recent near-surface collections of O3 (Ishino et al., 2017; Vicars and Savarino, 2014) and O3 generated from O2 NOx photochemical experiments conducted under normal temperature and pressure conditions (Michalski et al., 2014).
All Δ17O model simulations were conducted without considering chamber wall losses. The potential impact of wall loss was evaluated through sensitivity tests that involved comparing simulated Δ17O values. The sensitivity tests included a no-wall-loss case, wall loss involving O3, NO, NO2, HNO3, N2O5, and organic nitrate based on previous reports from chamber experiments (Morales et al., 2021; Wang et al., 2014), and an elevated wall loss scenario, in which the wall loss rate constants were increased by 10 times to account for uncertainty since wall loss rates were not determined in this work (Table S7).
3.1 Isotope observations
The Δ17O, δ18O, and δ15N measurements of NOy species offer insight into the oxidation pathways and sources contributing to their formation. In this study, we focus on interpreting the Δ17O, δ18O, and δ15N of NO2, HNO3, and pNO3 collected under a range of controlled experiments involving NO-pinene oxidation. These observations provide constraints on the relative importance of different oxidants (e.g., O3, OH, RO2) and reaction mechanisms, and they also allow us to test our understanding of oxygen isotope mass balance assumptions, O isotope transfer dynamics, and nitrogen isotope fractionation associated with NOx oxidation. Below, we first discuss the observed patterns in oxygen isotopes (Δ17O and δ18O), followed by an examination of nitrogen isotope (δ15N) trends and their implications.
3.1.1 Δ17O and δ18O of NOy
The observations indicate a significant relationship between δ18O and Δ17O across NOy species (δ18O = 11.1(± 1.0) + 2.42(± 0.04) × Δ17O; r= 0.992 ; p<0.01) (Fig. 1a). The strong linear relationship between δ18O and Δ17O indicates that the oxygen isotopes of the various collected NOy compounds derived from two dominant pools of O sources, with high and low δ18O and Δ17O values that were consistent across all experimental conditions. The observed Δ17O and δ18O values of NOy species are anticipated to reflect a balance between oxygen atom transfer from O3, which has high Δ17O and δ18O values, and oxygen atom transfer from RO2, HO2, OH, and H2O, which have lower Δ17O and δ18O values. Assuming a Δ17O(O) value of 39 ± 2 ‰ (Ishino et al., 2017; Vicars and Savarino, 2014) would indicate a δ18O transferred from O to the NOy products of 106 ± 5.0 ‰. Conversely, the low-Δ17O oxidant endmember at a Δ17O ≈ 0 ‰ implies an associated δ18O value of approximately 11.1 ± 1.0 ‰. This value likely reflects oxygen transfer from oxidants such as RO2, HO2, and OH.
The derived low-end δ18O oxidant value is consistent with a scenario in which these radicals are sourced from atmospheric O2 (δ18O ≈ 23 ‰; Craig, 1957) but undergo isotopic fractionation during their formation and subsequent oxygen atom transfer to NOy. Although the exact δ18O enrichment factors for RO2, HO2, or OH formation and reaction are not well-constrained, a net isotope enrichment factor ∼ −12 ‰ is plausible, particularly for unidirectional reactions involving 18O fractionation (Walters and Michalski, 2016a). Additionally, contributions from OH could further influence the low-δ18O endmember, especially if oxygen atom exchange with ambient water vapor occurs (Dubey et al., 1997). Altogether, the inferred δ18O of 11.1 ‰ for the low-Δ17O oxidant endmember likely represents a composite signal from multiple oxidants (RO2, HO2, OH) originating from O2 and/or H2O, modified by kinetic and equilibrium isotope effects. Despite these uncertainties, the consistent δ18O–Δ17O trend across NOy products supports a two-endmember mixing model governed by oxidants with distinct isotopic values.
The Δ17O and δ18O values increased in the order pNO3 ( ± σ; Δ17O = 10.0 ± 3.4 ‰; δ18O = 35.0 ± 10.1 ‰; n= 7) < HNO3 (Δ17O = 16.7 ± 2.0 ‰; δ18O = 50.2 ± 4.5 ‰; n= 20) < NO2 (Δ17O = 29.6 ± 12.8 ‰; δ18O = 84.1 ± 29.6 ‰; n= 20). The Δ17O values and δ18O of NO2, HNO3, and pNO3 were sensitive to the types of experiments and thus oxidant conditions (Fig. 1b). For example, NO2 samples collected during the photochemical experiments (i.e., Exps. 1–5) indicated that δ18O(NO2) and Δ17O(NO2) increased with the initial [NOy], the ratio of initial [NOy] : [BVOC], and with decreasing initial [H2O2]. These sensitivities of δ18O(NO2) and Δ17O(NO2) reflect the balance between NO branching ratios involving O3 versus RO2 HO2 (Albertin et al., 2021; Walters et al., 2018). Thus, the relative branching ratios of NO + O3 and NO + RO2 HO2 changed with experimental photochemical conditions, favoring a greater proportion of NO + O3 reactions for higher initial NOy and lower [H2O2] conditions. For the nighttime oxidation experiment (Exp. 6), the Δ17O and δ18O reflected the initial production of N2O5 from the oxidation of NO2 (from a gas cylinder) with O3. The expected Δ17O and δ18O values can be calculated assuming N2O5 equilibrium between NO3 and NO2 (i.e., N2O5 NO2+ NO3) and using O isotope mass balance (Eq. 7):
where δ refers to either Δ17O or δ18O, O refers to the O atom at the terminal end of O3, and NO refers to the NO2 from the tank source with measured Δ17O and δ18O values of −0.1 ± 0.1 ‰ (n= 3) and 13.1 ± 0.2 ‰ (n= 3), respectively. Using the assumed Δ17O(O) of 39 ± 2 ‰ and the calculated δ18O(O) incorporated into NOy (106 ± 5.0 ‰) we estimate the Δ17O and δ18O of NO2 for the nighttime oxidation experiment to be 7.7 ± 0.4 ‰ and 31.7 ± 1.0 ‰, respectively, which was near their measured values from the nighttime chamber experiments of (7.2 ± 0.2 ‰) and (31.9 ± 0.7 ‰) (n= 3), respectively.
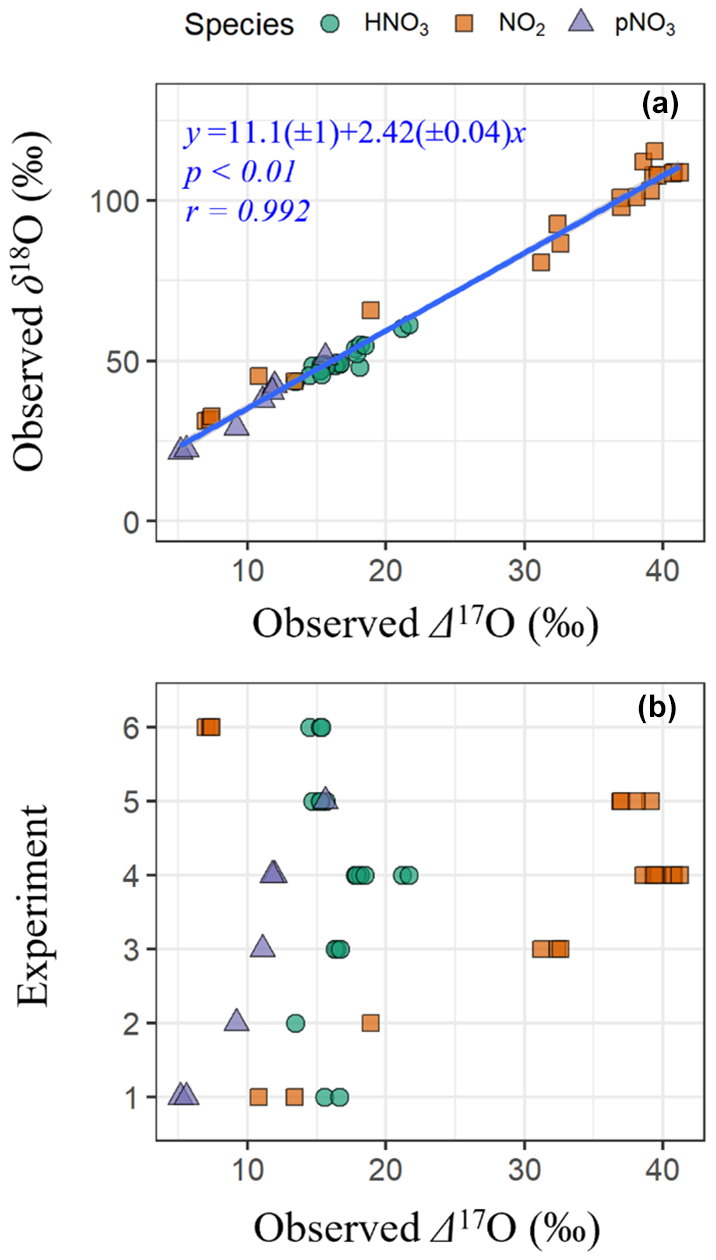
Figure 1The observed oxygen isotope delta values of various NOy species (i.e., HNO3, NO2, and pNO3) from the α-pinene NOy oxidation experiments including (a) linear regression between δ18O and Δ17O and (b) Δ17O values of NOy species sorted by experiment.
3.1.2 δ15N of NOy
The observed δ15N of all NOy species exhibited a large range from −90.3 ‰ to −4.0 ‰ (n = 47) (Fig. 2a). This large range of δ15N values was significantly influenced by the δ15N values of the various initial NOy sources that included tank NO (Exps. 1–4), HONO (Exp. 5), and tank NO2 (Exps. 6). The experiments using tank NO had the lowest δ15N ( ± σ) of (−56.1 ± 21.3 ‰; n = 32), followed by tank NO2 of (−34.7 ± 12.2 ‰; n = 6), and the highest average was for the HONO experiments of (−7.8 ± 5.7 ‰; n = 9). The differences in the observed δ15N values by NOy source likely reflect the isotopic composition of the initial NOy used in each experiment. The trend in observed δ15N across experiments is consistent with either measured or literature-based reports of the δ15N values of the initial NOy species. For example, the δ15N of laboratory-generated HONO, prepared using a similar methodology as in this study, has been reported to be −5.9 ± 0.5 ‰ (Chai and Hastings, 2018). This value is relatively high compared to the δ15N(NO2) from tank NO2 used in these experiments that was measured to be −40.9 ± 0.2 ‰, which is higher than the δ15N(NO) of tank NO previously reported at −70.0 ± 1.4 ‰ (Fibiger et al., 2014). In addition to the source δ15N effects, the experiments also indicate large δ15N fractionation between the various NOy species. Overall, δ15N(HNO3) averaged −25.9 ± 13.0 ‰ (n = 20), which was significantly higher than both δ15N(NO2) (−52.5 ± 25.2 ‰; n = 20) and δ15N(pNO3) (−72.6 ± 22.9 ‰; n = 7) based on a two-sample t test (p < 0.01). While δ15N(NO2) values were higher than those of δ15N(pNO3), this difference was not statistically significant based on a two-sample t test (p > 0.05). This trend suggests that the produced HNO3 was generally associated with a positive nitrogen isotope enrichment factor (15ε) favoring preferential incorporation of 15N relative to NO2, whereas pNO3 formation involved a negative ε, favoring incorporation of 14N.
We quantified the δ15N enrichment of HNO3 and pNO3 relative to NO2 as Δδ15N (defined as δ15N(product) – δ15N(NO2)) (Fig. 2b). Among the photochemical experiments initiated with NO and H2O2 (Exps. 1–4), the average Δδ15N(HNO3–NO2) was consistently high, averaging 33.7 ± 7.1 ‰, while the HONO experiment (Exp. 5) had a much smaller enrichment value of 0.4 ± 1.7 ‰. This difference is somewhat surprising, given that both systems are expected to involve NO2 + OH as a major pathway for HNO3 production. The 15ε associated with NO2+ OH has yet to be directly measured but has been predicted in the literature with large differences in the suggested value. For example, the 15ε for NO2+ OH has been suggested to be −3 ‰ based on the reduced masses of the transition complex (Freyer et al., 1993), while it has been predicted to be +40 ‰ in the iNRACM based on the assumption that NO2 and the excited HNO3 intermediate formed during the NO2+ OH reaction achieve isotopic equilibrium prior to collisional deactivation (Fang et al., 2021). The relatively low Δδ15N(HNO3–NO2) observed in the HONO experiment is more consistent with the former, while the higher Δδ15N(HNO3–NO2) values in the NO H2O2 experiments support the latter interpretation. The cause of this discrepancy remains unclear but may reflect differences in reaction kinetics and environmental conditions. For example, in the HONO experiment, the aerosol formation peak (Fig. 3) and HNO3 production (Fig. S1) occurred relatively rapidly compared to the NO H2O2 experiments. This timing shift may potentially alter the influence of nitrogen isotope fractionation effects such as NOx photochemical equilibrium. Additional isotope effects may also contribute, such as fractionation during HONO photolysis, unknown mass-dependent processes, or experimental artifacts including wall loss or residual background levels. Further investigation is needed to isolate and quantify the influence of these factors.
In the nighttime experiment (Exp. 6), Δδ15N(HNO3–NO2) was also elevated with an average value of 22.2 ± 1.4 ‰ . The cause of the nighttime 15N enrichment in the generated HNO3 relative to NO2 is likely due to isotopic effects associated with the NO2+ NO3 N2O5 equilibrium, which has been predicted to have 15ε of 25.5 ‰ at 300 K (Walters and Michalski, 2016b) falling near the observed value for the nighttime experiments (Fig. 2b). The Δδ15N(pNO3–NO2) values were consistently negative, averaging −13.6 ± 5.8 ‰ across the photochemical experiments. This value suggests that pNO3 formation involved reactions that preferentially favored 14N. Before speculating on the cause of the low observed Δδ15N(pNO3–NO2), it is essential to first determine the chemical composition of the produced pNO3 and whether it originates primarily from inorganic or organic nitrate.
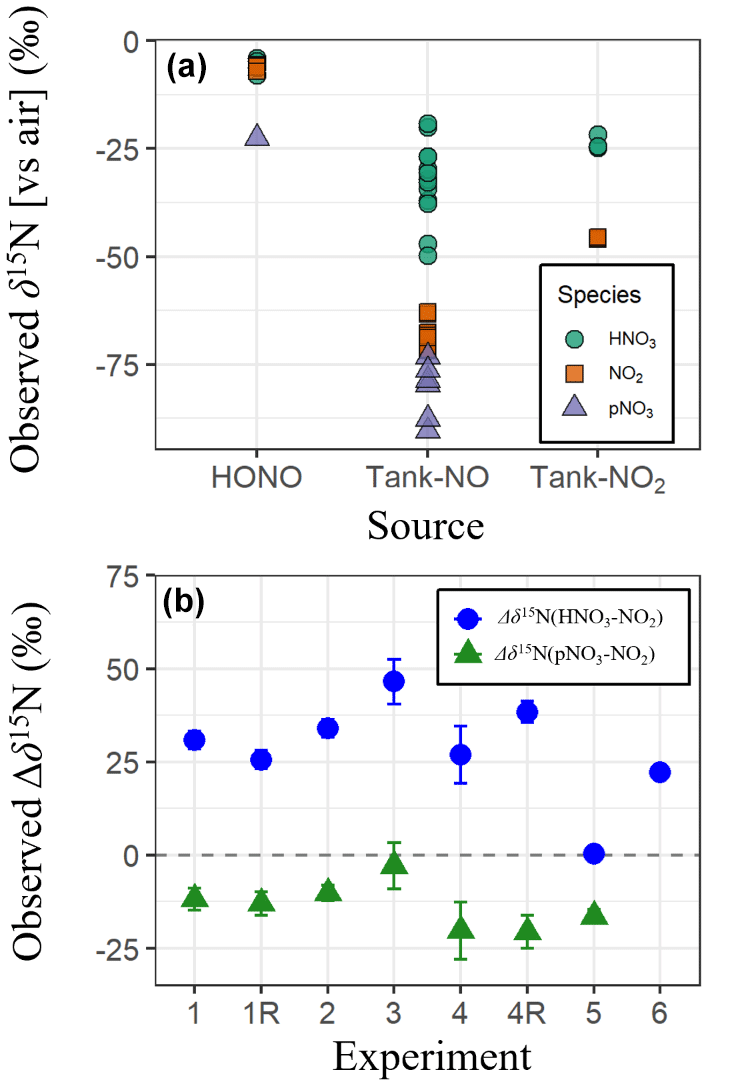
Figure 2(a) The observed δ15N of various NOy species (i.e., HNO3, NO2, and pNO3) collected during the various conducted α-pinene and NOy oxidation experiments. The measured δ15N values were sorted by the various starting NOy sources, including HONO, tank NO, and tank NO2. (b) Calculated Δδ15N values for each experiment, defined as the δ15N difference between HNO3 and NO2 and between pNO3 and NO2. Data represent experiment-specific averages, with error bars reflecting the propagated standard deviation.
3.2 Particle nitrate composition
The generated pNO3 could have both inorganic (i.e., HNO3 condensation) and organic (i.e., organic nitrate condensation) contributions. The δ15N data qualitatively indicate that the pNO3 was derived from a source separate from HNO3 due to their large δ15N differences, which would suggest that pNO3 was derived primarily from organic nitrate. For example, the observed δ15N difference between HNO3 and pNO3 that averaged 46.7 ‰ suggests that these species may originate from distinct sources. Given that inorganic nitrate would typically equilibrate isotopically between HNO3 and NO with an expected offset of only ∼ 1 ‰–3 ‰, and often slightly enriched in pNO3 (Bekker et al., 2023), the substantially lower δ15N values observed in pNO3 imply that the collected nitrate may originate from organic nitrate species or unique NOy formation pathways from HNO3 production.
We utilized the HR-ToF-AMS NO+ and NO data to evaluate the contributions of pNO3 for the experiments. The f(pNO3, Org) was calculated according to Eq. (5) for each of the conducted experiments (Table 3). Overall, f(pNO3, Org) was calculated to have a mean of (1.25 ± 0.04; n=8), indicating that the generated pNO3 derived from organic nitrate. The calculated f(pNO3, Org) was higher than 1 even when considering uncertainty estimates. This could be due to calculating RON values from reported ratios from previously conducted α-pinene oxidation experimental conditions utilizing substantially lower (∼ ×10) initial precursor concentrations (Takeuchi and Ng, 2019). Thus, due to the potential uncertainty in our approach in estimating f(pNO3, Org), the composition of the generated pNO3 was also investigated using a qualitative approach involving evaluating the relative change in the molar ratio of NH4 SO4 from the HR-ToF-AMS (Fig. S2). For each type of experiment, we found the NH4 SO4 molar ratio to be consistently near 1.5. This type of NH4 SO4 profile is consistent with the generated pNO3 deriving from organic nitrate, as the dissolution of HNO3 into aqueous aerosol followed by neutralization with available NH3 would be expected to lead to an abrupt increase in the molar ratio of NH4 SO4 (Takeuchi and Ng, 2019). Furthermore, the acidic nature of the particles and limited availability of NH likely inhibited HNO3 uptake, suppressing condensation pathways and reinforcing the interpretation that pNO3 originated predominantly from organic nitrate formation. Overall, both the quantitative and qualitative analysis of pNO3 composition utilizing the AMS data as well as our δ15N data indicates that pNO3 was mainly derived from organic nitrate. Hereafter, we assume that the NO extracted from the filter collections derived from organic nitrate.
Table 3Summary of the HR-ToF-AMS data including NO+ NO fragmentation data (Robs), calculated f(pNO3, Org), and maximum pNO3 (Max(pNO3)). Additionally, we quantified the amount of pNO3 from the PILS (PILS AMS) and the filter collection relative to the HR-ToF-AMS (filter AMS). Uncertainties for pNO3 quantification and intercomparison ratios are reported in parentheses.
NA: not available.
The pNO3 measured by the HR-ToF-AMS indicated similar profiles for the various types of conducted experiments, in which pNO3 concentrations peaked and subsequently decayed due to wall loss and chamber dilution (Fig. 3). Overall, the maximum pNO3 concentrations ranged from 13.3 to 38.6 µg m−3, depending on the experiment (Table 3). The typical measurement uncertainty for pNO3 quantification using the HR-ToF-AMS is approximately ± 14 % (Bahreini et al., 2009; Takeuchi et al., 2024). Given that we assume 100 % of the particulate nitrate is organic nitrate (i.e., f(pNO3, Org) = 1), the uncertainty in pNO3 concentration is based solely on the AMS nitrate measurement error, estimated at ± 14 %. The lowest maximum pNO3 corresponded to experimental conditions with low initial NOx relative to H2O2 and BVOC conditions (i.e., Exp. 1). In contrast, the highest maximum pNO3 occurred during the nighttime oxidation experiments (i.e., Exp. 6). The pNO3 concentrations determined from the HR-ToF-AMS were compared with additional measurement techniques, including the PILS and the filter collections for offline analysis (Fig. 3; Table 2). The PILS pNO3 measurements were available for four out of the eight conducted experiments and indicated a similar time profile as the HR-ToF-AMS; however, the PILS pNO3 observations were always lower than the HR-ToF-AMS with the amount of pNO3 determined from PILS relative to the HR-ToF-AMS (PILS AMS) ranging between 33.2 %–53.8 %. The uncertainty associated with pNO3 quantification by the PILS system is approximately ± 20 % (Guo et al., 2016). Accordingly, the propagated uncertainty for the PILS AMS ratio is approximately 24 %.
The pNO3 quantified using the filter collection and extraction technique was higher than the PILS and in closer agreement with the HR-ToF-AMS. For the photochemical experiments (Exps. 1–5), the pNO3 determined using the filter collection relative to the HR-ToF-AMS (filter AMS) for the photochemical experiments ranged between 59.5 % and 105.3 % and averaged 86.5 ± 12.4 % (n=7). The filter collection technique has an estimated uncertainty for pNO3 quantification of approximately ± 20 % based on the average percent difference from side-by-side ChemComb filter pack measurements of ambient air using nylon filters (Blum et al., 2020). Although different filters were used in this study, the same collection system and mass flow controllers were employed, and we therefore expect a comparable difference between system replicates. Accordingly, the propagated uncertainty for the filter AMS ratio is ± 24 %. However, the filter collection resulted in nearly negligible pNO3 for the nighttime oxidation experiments (i.e., Exp. 6).
The pNO3 concentrations determined using the PILS were always lower than that determined by the HR-ToF-AMS and the offline filter collection technique, which would indicate that not all collected pNO3, which was shown to mainly derive from organic nitrate, was hydrolyzed to NO within the PILS chamber before quantification via ion chromatography. The filter collection and extraction method (i.e., leach in MQ water for 1 week) enabled the successful hydrolysis of the collected pNO3 to NO from the photochemical experiments, an important pre-requisite for subsequent isotope analysis. The filter collection technique, however, resulted in near-negligible pNO3 for the nighttime oxidation experiments, limiting our ability to measure the isotope composition of pNO3 from this experiment. This difference in the efficacy of the offline filter collection technique for pNO3 characterization between the photochemical and nighttime oxidation experiments could be related to the type of organic nitrate formed during the conducted experiments. The photochemical α-pinene oxidation experiments have been suggested to result in higher relative production of tertiary organic nitrate, while nighttime oxidation leads to a relatively lower fraction of tertiary organic nitrate with estimated values of 62 % and 15 %, respectively (Zare et al., 2018). Recent work has suggested a hydrolysis lifetime of no more than 30 min and a hydrolyzable portion of particulate organic nitrate from α-pinene oxidation experiments between 23 %–32 % and 9 %–17 % for α-pinene + OH and α-pinene + NO3 reactions, respectively (Takeuchi and Ng, 2019).
The offline filter collection and extraction technique follows the observed trend of a greater proportion of pNO3 hydrolyzed to NO in photochemical experiments (82.6 ± 14 %; n= 7) compared to nighttime conditions (7.6 %; n= 1). This is inferred based on the relative amount of quantified NO from the filter extraction solution to the total pNO3 measured by HR-ToF-AMS. However, the filter-based method suggested a higher proportion of potentially hydrolyzable pNO3 in the photochemical experiments than previously reported estimates. From these results and comparisons, we conclude that the pNO3 offline filter measurements encompass the hydrolyzable portion of pNO3 within 1 week, while the HR-ToF-AMS measurements represent the total pNO3, and the PILS measurements correspond to the readily hydrolyzable portion of pNO3. Further, the box-model simulations (see below) of organic nitrate speciation indicate that the nighttime organic nitrate had a high fraction of dimer and pinene dinitrate (Fig. S3). If this assignment is correct, the results imply that these organic nitrate compounds are effectively non-hydrolyzable under aqueous conditions.
Our results demonstrated that organic nitrate aerosols (pRONO2) can hydrolyze and contribute to the NO measured in aerosol extracts. While our chamber experiments were conducted under controlled conditions with low relative humidity (∼ 30 %) and dry aerosol seeds, they highlight the need to consider the fact that NO collected on filters may originate from both inorganic nitrate (derived from HNO3 uptake) and hydrolyzed organic nitrate. However, we caution that these findings do not imply that all pNO3 observed in ambient field measurements is organic in origin. The extent to which organic nitrate contributes to pNO3 in field settings will depend on regional BVOC emissions, which govern precursor availability, as well as environmental factors such as aerosol pH and relative humidity, which influence the lifetime and hydrolysis rates of pRONO2 prior to filter collection. Further, while gas-phase organic nitrates (e.g., RONO2, RO2NO2) can be present and were detected by CIMS measurements during the experiments, the strong agreement between filter-based and AMS-based pNO3 measurements supports the idea that the nitrate extracted from aerosol filters was primarily derived from particle-phase organic nitrate rather than from gas-phase organic nitrate contributions. Finally, given that the collected pNO3 was predominantly derived from RONO2, the negative Δδ15N(pNO3–NO2) values (Fig. 2b) suggest a preferential incorporation of 14N into the RONO2 product. This is consistent with NOx photochemical equilibrium in which 15N is enriched in NO2, leaving NO relatively depleted in 15N (Li et al., 2020; Walters et al., 2016). Subsequent reaction of this 15N-depleted NO with RO2 forms RONO2, thereby transferring the isotopically lighter nitrogen signature into RONO2 that then condenses to the particle phase and is hydrolyzed to NO.
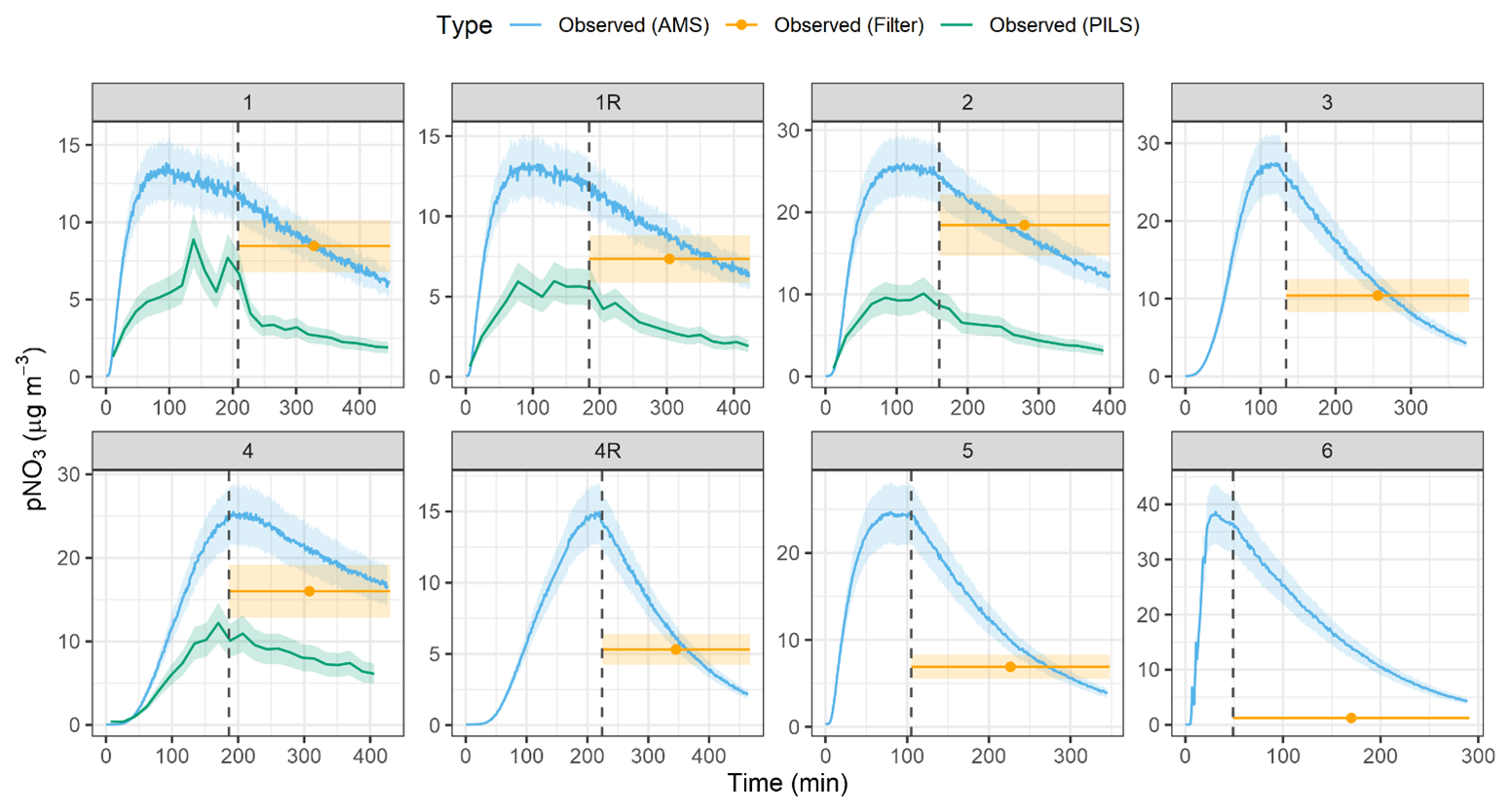
Figure 3The observed pNO3 concentration data are shown for each of the conducted experiments. Concentrations were determined using a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS), a particle-into-liquid sampler (PILS), and filter collection (filter). The start of chamber dilution is indicated by the dashed vertical lines, corresponding to the abrupt decrease in pNO3. The lighter shaded regions correspond to the measurement uncertainty for the various analytical techniques.
3.3 Model simulations
To further interpret the experimental results, we employed a box model to simulate the formation and evolution of NOy species and their Δ17O values. We begin by examining the developed gas-phase chemical mechanism (NOx-API) simulation of the initial aerosol precursor decay including α-pinene and NO. For comparison, simulations were also conducted using established gas-phase chemical mechanisms that included RACM2 and the MCM to evaluate the treatment of α-pinene oxidation and oxidation chemistry across different chemical frameworks for a range of experimental conditions. Model sensitivity tests were then conducted to assess the impact of key physical parameters that included chamber dilution rate and wall loss rates on simulated Δ17O values. Finally, we compare the modeled Δ17O values of NOy species with experimental observations to evaluate the model's ability to reproduce isotopic values under different photochemical and nighttime oxidation conditions.
3.3.1 Precursor decay
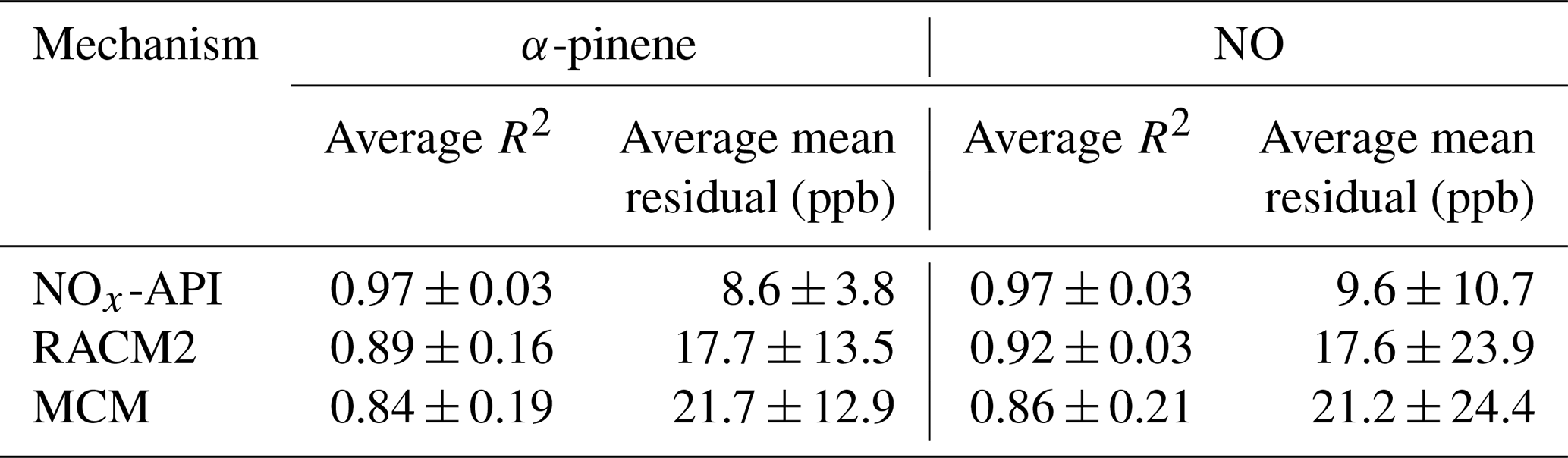
Box-model simulations were conducted to evaluate the oxidation and decay of precursors used in the experiments, ensuring that the correct amount of oxidant was accurately simulated. The MCM, RACM2, and NOx-API were used to simulate the decay of α-pinene and NO, with results compared against experimental observations (Figs. 4–5). Model performance was evaluated by comparing measured and modeled concentrations of NO and α-pinene using one-to-one plots, with corresponding R2 values and quantification of model biases (Figs. S4–5), and Table 4 summarizes these results. Overall, the NOx-API mechanism provided improved model performance, as evidenced by consistently higher R2 values (averaging 0.97 ± 0.03) and lower absolute residuals for both α-pinene and NO decay compared to the other mechanisms. We attribute this enhanced model performance to the simplified treatment of higher-generation products in the NOx-API mechanism. Unlike MCM and RACM2, which allow continued gas-phase reactions of all products through extensive reaction propagation, the NOx-API mechanism terminates the chemistry of these products after a limited number of steps. Given that the box-model simulations do not include an explicit aerosol-phase treatment, continued gas-phase reactivity of condensable species (as implemented in MCM and RACM2) may unrealistically disrupt the oxidant budget. Therefore, the NOx-API mechanism as employed in the box model is better aligned with the experimental design and will be used for subsequent Δ17O simulations of the chamber experiments.
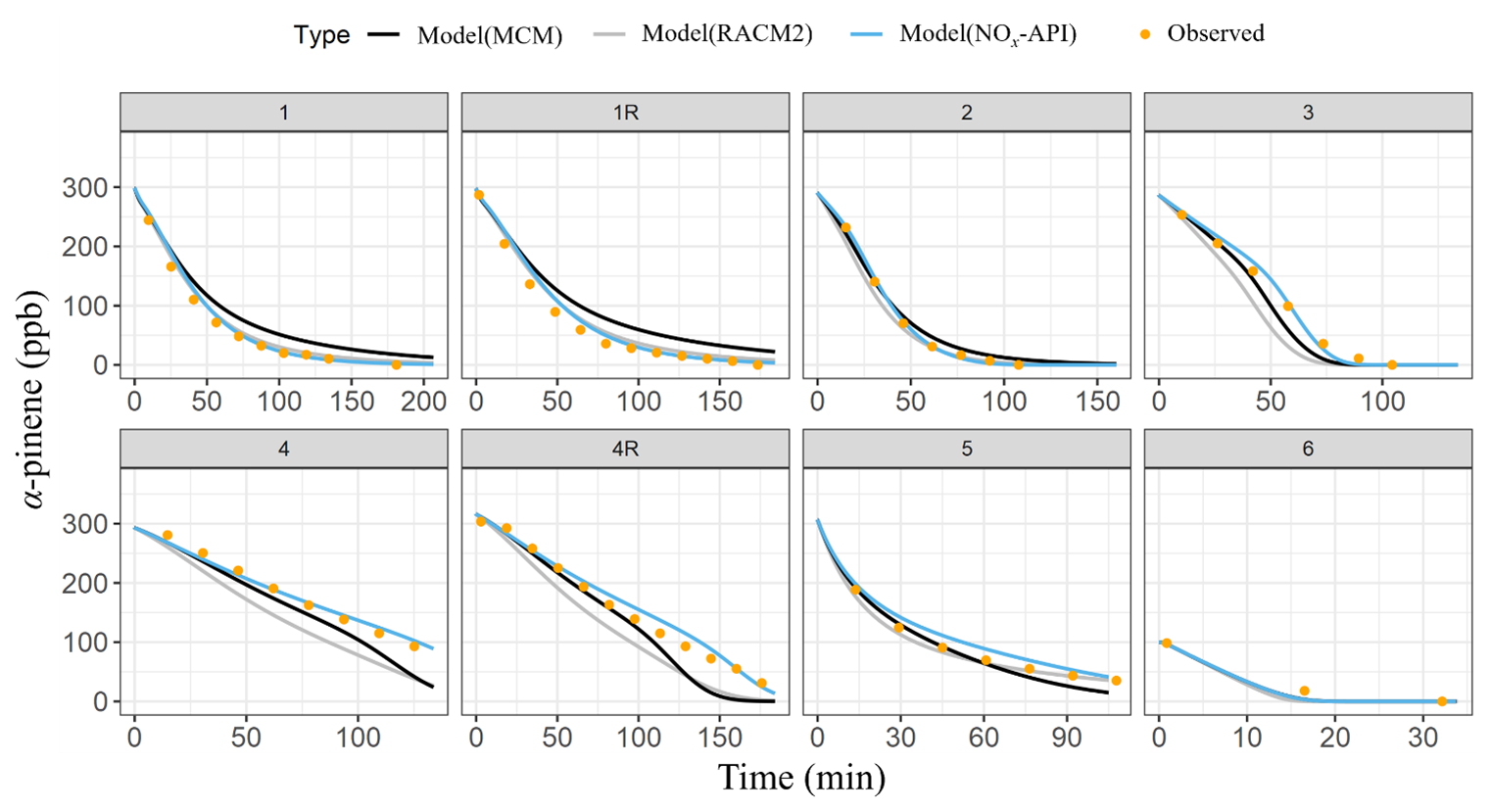
Figure 4The observed (orange data points) and the modeled (lines) α-pinene decay for the various conducted experiments. The modeled results are based on three chemical mechanisms: MCM (black), RACM2 (gray), and NOx-API (light blue).
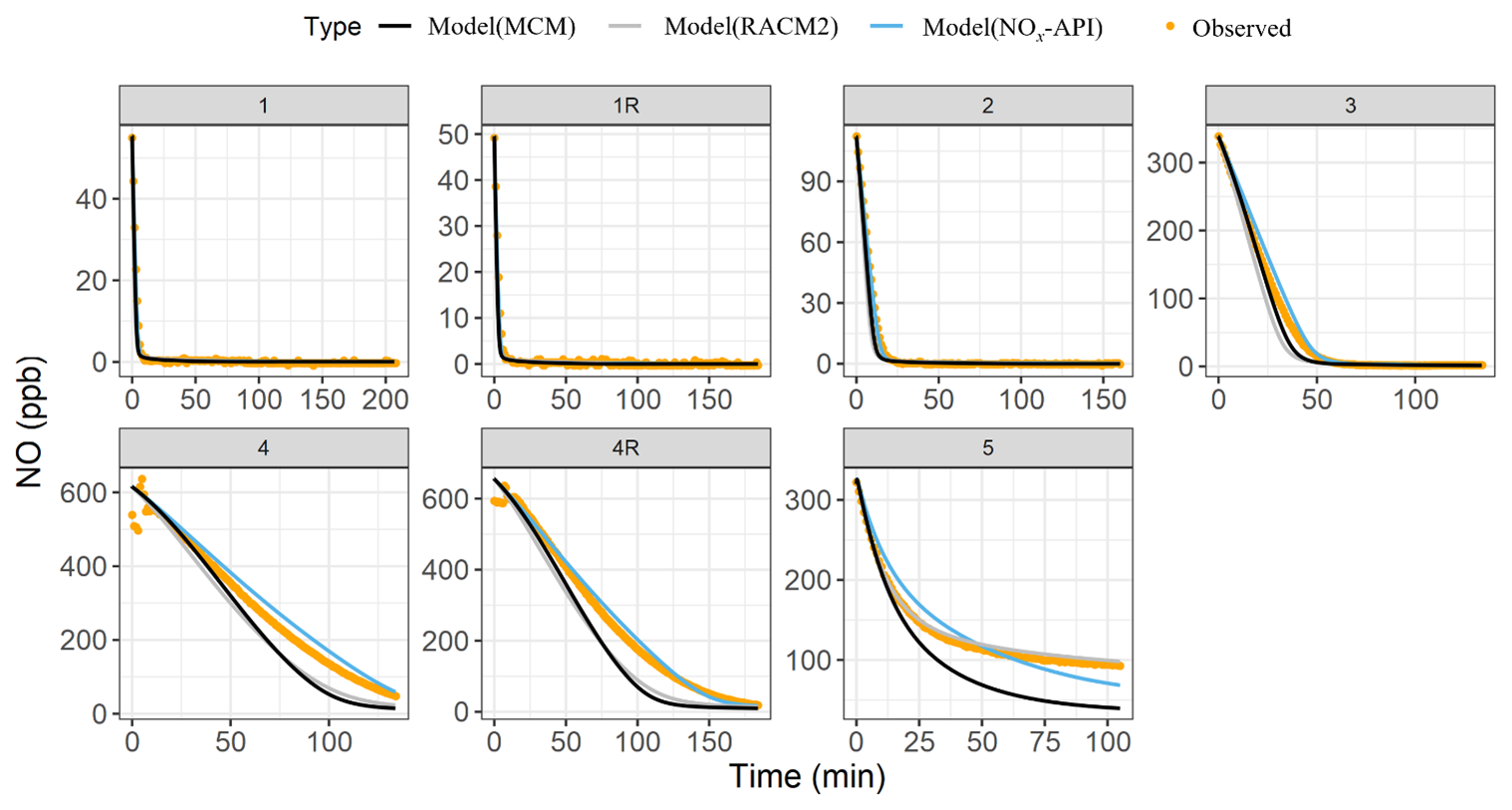
Figure 5The observed (orange data points) and the modeled (lines) NO decay for the various conducted photochemical experiments. The modeled results are based on three chemical mechanisms: MCM (black), RACM2 (gray), and NOx-API (light blue). Experiment 6 (nighttime oxidation) was omitted from the analysis as NO was not among the initial reactants.
Table 4Summary of model performance for α-pinene and NO using the NOx-API, RACM2, and MCM. Values represent the average coefficient of determination (R2) and average mean residuals (in ppb) across all experiments, with associated standard deviations.
3.3.2 Δ17O model sensitivity to dilution and wall loss
Before comparing modeled Δ17O values to observations, we first evaluated the model's sensitivity to our treatment of key physical parameters, including chamber dilution and wall loss, and their potential impact on the simulated Δ17O (Fig. 6). These sensitivity tests focused on Experiment 1, one of the longest-duration experiments, where dilution and wall loss would be expected to exert the strongest influence on gas-phase chemistry and Δ17O values.
The impact of chamber dilution was assessed using four scenarios: no dilution and first-order dilution rate constants (kdil) of , , and s−1 (Fig. 6a). Dilution was initiated at t= 208 min in the model to match the experimental protocol. Across all scenarios, simulated Δ17O values of total organic nitrate (ONIT = ONITa + ONITb + ONITc + ONITOOHa + ONITOOHb + DIMER + PDN) were minimally impacted by dilution. For instance, Δ17O(ONIT) varied by only 0.06 ‰ between the no-dilution and highest dilution rate constant scenario (kdil= s−1), corresponding to a relative difference of −1.1 %. This insensitivity reflects the fact that organic nitrate chemistry was largely completed by the time dilution began. In contrast, simulated Δ17O values for HNO3 and NO2 were more sensitive to the dilution rate. Between the no-dilution and lowest dilution rate constant scenario (kdil= s−1), Δ17O(HNO3) and Δ17O(NO2) changed by only −0.01 ‰ (corresponding to a relative difference of −0.2 %) and −0.11 ‰ (−0.7 %), respectively. However, increasing the dilution rate constant from to s−1 led to additional decreases of 0.32 ‰ (−5.0 %) for HNO3 and 0.88 ‰ (−5.6 %) for NO2. The most extreme dilution rate constant scenario (kdil= s−1) reduced Δ17O by a total of 0.50 ‰ (−7.9 %) for HNO3 and 1.62 ‰ (−10.3 %) for NO2 relative to the no-dilution case. For the main box-model simulations, we adopted a dilution rate constant of s−1, corresponding to a measured flow rate of 25 LPM in a 12 m3 chamber. Assuming no more than ± 20 % uncertainty in the actual flow rate, we estimate that the resulting uncertainty in simulated Δ17O values due to dilution would be approximately ± 2.5 % for HNO3, ± 3 % for NO2, and less than ± 1 % for ONIT. These estimates are based on the observed Δ17O variation between the and s−1 scenarios.
Next, we evaluated the potential influence of chamber wall loss on the simulated Δ17O values (Fig. 6b). Three scenarios were tested: (1) no wall loss, (2) a wall loss scenario incorporating NOy and O3 loss rates from previous studies (Morales et al., 2021; Wang et al., 2014), and (3) an extreme case in which wall loss rate constants were increased by a factor of 10 (Table S7). In the base comparison between the no-wall-loss and reported wall loss scenario, the effect on modeled Δ17O values was minimal. Specifically, Δ17O changed by −0.02 ‰ for HNO3 (−0.3 %), −0.37 ‰ for NO2 (−2.3 %), and +0.03 ‰ for ONIT (+0.5 %). These small differences suggest that moderate wall loss rates, consistent with literature values, would not substantially alter the Δ17O simulations. However, the extreme wall loss scenario revealed a much stronger impact. In this case, Δ17O decreased by 2.5 ‰ for NO2 (−15.8 %) and increased by 0.50 ‰ for HNO3 (+7.8 %) and 0.69 ‰ for ONIT (+11.4 %) relative to the no-wall-loss case. The drop in Δ17O(NO2) arises from the altered oxidant concentrations and rapid photochemical cycling of NOx, which can reset its Δ17O values on short timescales. Conversely, the rise in Δ17O for HNO3 and ONIT reflects preferential removal of early-formed products with lower Δ17O, allowing later-formed products with higher Δ17O values to dominate. Because we lacked experimental constraints on wall loss during the chamber experiments, the box-model simulations presented in this study did not include wall loss. Based on our sensitivity analysis, omitting wall loss introduces an estimated uncertainty of ± 0.3 % for HNO3, ± 2.3 % for NO2, and ± 0.5 % for ONIT assuming a moderate wall loss scenario. If actual wall loss rates in the chamber were significantly higher than those reported in the literature, the model is anticipated to overestimate Δ17O in NO2 and underestimate it in HNO3 and ONIT.
Overall, based on the results of the model sensitivity tests, we estimate that the propagated uncertainty in the simulated Δ17O values due to chamber dilution and wall loss is approximately ± 2.5 % for HNO3, ± 3.8 % for NO2, and ± 1.1 % for ONIT. These values reflect the combined effects of plausible variation in dilution rate (± 20 %) and the difference in simulated Δ17O values from literature-based estimates of NOy and O3 wall loss compared to a case of no wall loss. Further, considering that the uncertainty in the measured Δ17O(O) is approximately ± 5 % (Vicars and Savarino, 2014), we calculate an overall model uncertainty for Δ17O of ± 5.6 %, ± 6.3 %, and ± 5.1 % for HNO3, NO2, and ONIT, respectively. Clearly, the uncertainty in Δ17O(O) is expected to be the largest source of uncertainty in the modeled Δ17O values as opposed to our treatment of dilution and chamber wall loss. Still, we conservatively apply these propagated uncertainty estimates when presenting and interpreting the model results.
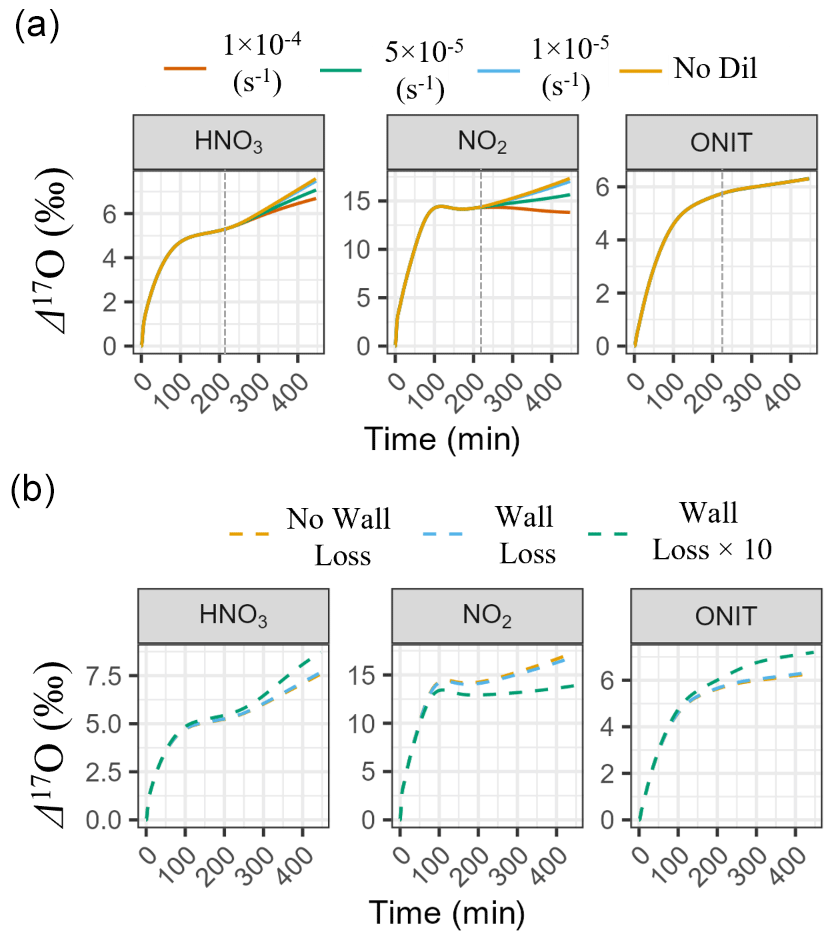
Figure 6Modeled Δ17O sensitivity to physical chamber parameters. (a) Model simulations testing the sensitivity of Δ17O in NO2, HNO3, and ONIT to chamber dilution rates, including scenarios with no dilution and with first-order dilution constants of , , and s−1. (b) Model simulations testing the impact of wall loss, comparing a no-wall-loss case, a wall loss scenario using reaction rates from Wang et al. (2014) and Morales et al. (2021), and a high-loss case in which wall loss rates were increased by a factor of 10. All simulations were performed under the conditions of Experiment 1. The gray dashed line in panel (a) corresponds to the start of chamber dilution.
3.3.3 Δ17O base model simulations
Building on the results of the sensitivity analyses, we next evaluate the performance of the base box model in simulating the Δ17O values of key NOy species, including NO2, HNO3, and ONIT under the various experimental conditions. These simulations incorporate the best-estimate physical parameters (e.g., dilution rate) and chemical mechanism inputs, including a representative Δ17O(O) value of 39 ± 2 ‰ (Fig. 7). Overall, the model simulations for the photochemical experiments indicate a substantial temporal change in Δ17O for all considered NOy compounds. The Δ17O(NO2) initially starts at 0 ‰ and begins to increase due to the production of O3 that elevates Δ17O(NO2) as NO is oxidized by O3. For the nighttime experiment, the box model predicts Δ17O(NO2) to remain steady with a value near 7.6 ‰ due to N2O5 thermal equilibrium with NO2 and NO3, resulting in the Δ17O(NO2)≈Δ17O(N2O5). Generally, the Δ17O simulations of NO2 were in excellent agreement with the observations, as indicated by an average model bias of 0.9 ± 2.4 ‰ (n= 8; Table 5) and a strong correlation indicated by a regression R2 value of 0.98. This strong agreement indicates that the box model and employed chemical mechanism accurately simulated NOx photochemical cycling and NO2 NO3 N2O5 thermal equilibrium. The simulation of Δ17O(NO2) is inherently sensitive to the assumed value of Δ17O(O), which represents the isotopic signature transferred from the terminal O atom of O3 to NO2 during oxidation. The relationship between measured Δ17O(NO2) and the modeled fraction of O atoms in NO2 deriving from O3 (denoted as f(Q)) indicates that Δ17O(O) is 39.4 ± 0.6 ‰ (Fig. S6), which is in excellent agreement with recent independent measurements of tropospheric O3 reporting values of 39 ± 2 ‰ (Ishino et al., 2017; Vicars and Savarino, 2014).
The measured Δ17O(pNO3) was compared with the simulated Δ17O of (ONIT) based on our understanding that pNO3 was apparently dominated by RONO2 contributions. The temporal evolution of the simulated Δ17O(ONIT) closely followed that of Δ17O(NO2) but remained lower due to dilution effects associated with the dominant formation pathway of organic nitrates via NO + RO2 reactions during the photochemical experiments, resulting in ONIT dominated by ONITa, ONITb, and ONITc compounds (Fig. S3). During nighttime oxidation experiments, ONIT formation primarily proceeded via α-pinene + NO3 reactions, leading to ONIT with a higher contribution of DIMER and PDN compounds. Due to NO2 NO3 N2O5 thermal equilibrium that resulted in Δ17O(NO2)≈Δ17O(NO3) ≈Δ17O(N2O5), the simulated nighttime Δ17O(ONIT) values were approximately equal to the simulated Δ17O(NO2). The simulated Δ17O(ONIT) closely matched the Δ17O(pNO3) observations with an average bias of −1.4 ± 2.4 ‰ (n=7), indicating that the relative production routes of organic nitrate (RO2+ NO vs. BVOC + NO3; Table 1) were correctly simulated for the various experimental conditions. The overall correlation was moderate, with R2= 0.55.
The bias for simulated Δ17O(ONIT) compared to the Δ17O(pNO3) observations was within 1.4 ‰ for all experiments except Exp. 5, where a substantially higher bias of −7.0 ‰ was observed. Excluding this outlier increased the correlation to R2= 0.97. Based on Cook's distance and the Studentized residual, Experiment 5 was identified as an outlier in the linear regression analysis. The larger model–data difference for Exp. 5 may reflect different oxidation dynamics or, more likely, uncertainty in the extraction of pNO3. Specifically, Experiment 5 yielded the lowest filter AMS ratio among the photochemical experiments, with the ratio falling below the quantitative range when accounting for propagated uncertainty. This suggests potential under-recovery or sampling artifacts, indicating that the measured Δ17O(pNO3) in this experiment may not accurately represent the pNO3 formed during the chamber experiment (Table 3). Thus, the larger model–measurement Δ17O(ONIT) disagreement for Exp. 5 likely reflects uncertainty in the extracted NO rather than model misrepresentation of ONIT formation pathways. The measured Δ17O(pNO3) was also evaluated relative to the modeled f(Q) for ONIT formation. A linear regression constrained through the origin, excluding Exp. 5, yielded a slope of 41.7 ± 1.2 ‰ (R2 = 0.996; Fig. S7). This derived Δ17O(Q) value is in close agreement with the assumed Δ17O(O) value of 39 ± 2 ‰ and is consistent with the value determined from the measured Δ17O(NO2) comparison with the modeled f(Q) of NO2.
The simulated dynamics of Δ17O(HNO3) closely follow the temporal evolution of Δ17O(NO2), initially starting at 0 ‰ and increasing as O3 production occurs in the chamber during the photochemical experiments (i.e., Exps. 1–5) (Fig. 7). The simulated Δ17O(HNO3) for the photochemical experiments was always lower than Δ17O(NO2) for the photochemical experiments, as the model simulation predicts HNO3 is predominantly produced through the NO2+ OH pathway (Fig. S8). Based on the conventional oxygen isotope mass balance calculations, this pathway results in a dilution factor of relative to Δ17O(NO2) (Table 1). For the nighttime experiment (Exp. 6), Δ17O(HNO3) is predicted to be slightly lower than Δ17O(NO2), primarily due to the contributions from two formation pathways: NO3+ pinonaldehyde and N2O5+ H2O(g), representing approximately 60 % and 40 % of nighttime HNO3 production, respectively. Since Δ17O(NO3)≈Δ17O(NO2) under nighttime conditions due to rapid thermal equilibrium, NO3+ pinonaldehyde should result in Δ17O(HNO3) ≈ Δ17O(NO2). However, the N2O5 hydrolysis pathway incorporates one oxygen atom from water, which has Δ17O ≈ 0 ‰, resulting in an effective dilution of the product Δ17O(HNO3) relative to Δ17O(NO2). Overall, the model exhibits poor agreement with observed Δ17O(HNO3) values with a model bias that ranged from −11.5 ‰ to 7.9 ‰ (Table 4) and a weak correlation (R2= 0.39). The model underpredicts Δ17O(HNO3) for the low-NOx photochemical experiments (Exps. 1 and 2) and the nighttime oxidation experiment (Exp. 6) but overpredicts Δ17O(HNO3) for the high-NOx photochemical experiments (Exps. 3, 4, and 5). If the isotope mass balance assumptions are correct, this pattern suggests that a missing or underrepresented source of high-Δ17O HNO3 may exist under low NOx conditions, and a missing low Δ17O HNO3 source may exist under high-NOx conditions.
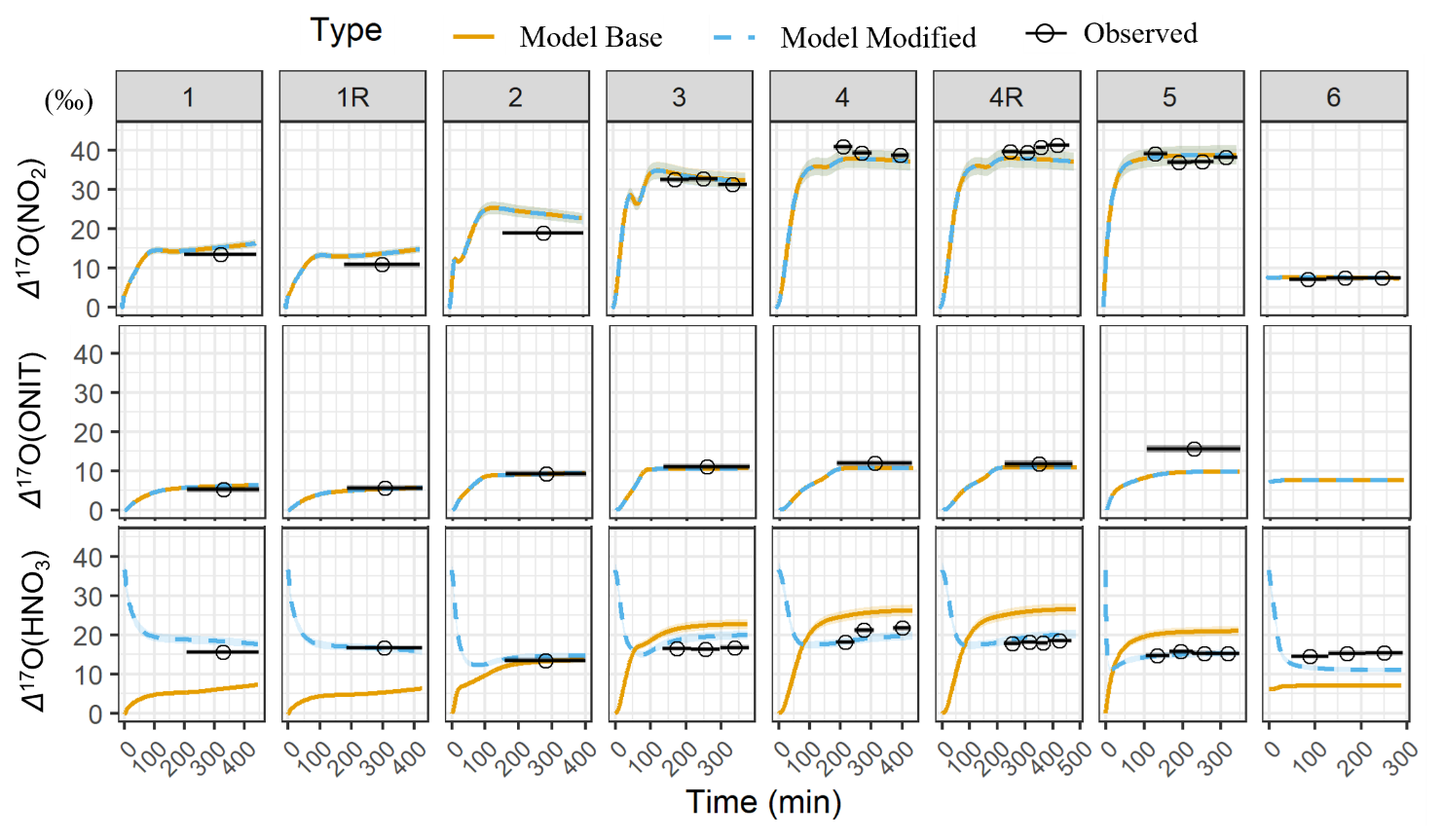
Figure 7Comparison between the modeled (base and modified) and observed Δ17O(NO2), Δ17O(ONIT), and Δ17O(HNO3) values sorted by the various conducted experiments. The data points represent the average time of each sample collection, and the black line spans the duration of the collection period. The measurement uncertainty (± σ) is included as the shaded region. The modeled Δ17O(ONIT) is compared to the observed Δ17O(pNO3).
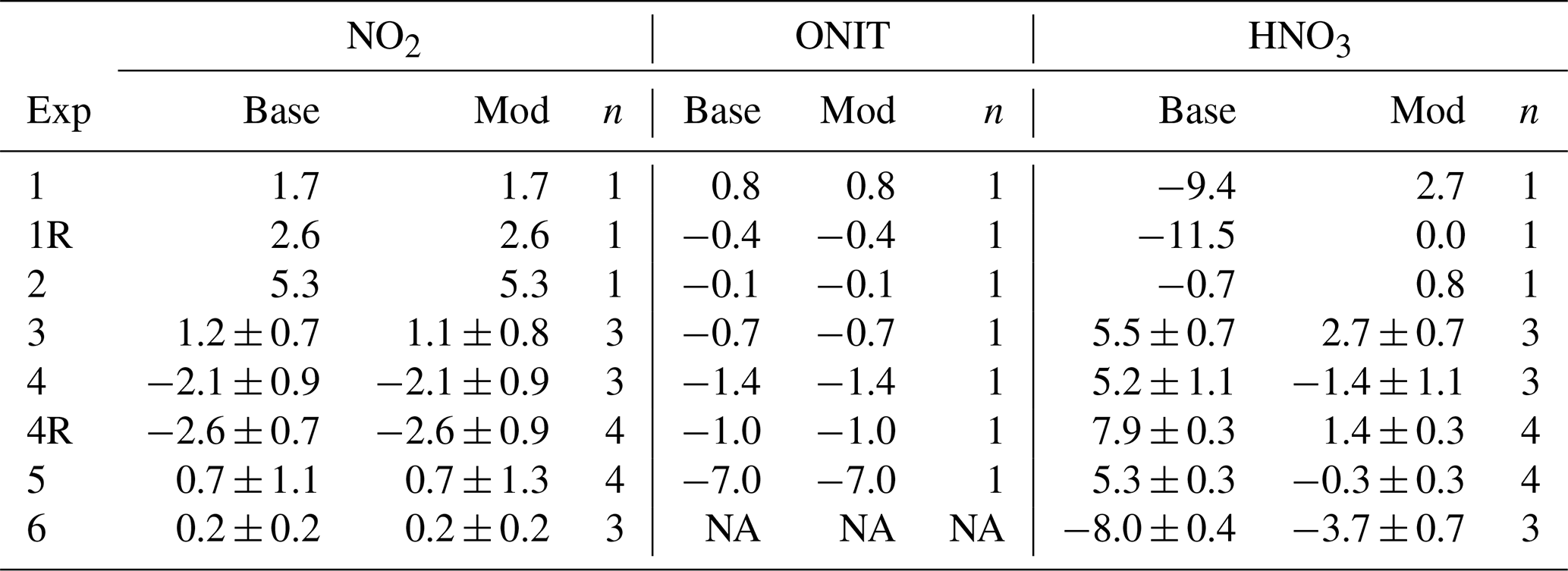
Table 5Summary of the calculated average bias (Δ17O(Model) – Δ17O(Observed)) for the Δ17O model simulations using the NOx-API base and modified (Mod) mechanisms. For experiments with multiple observations, the bias is reported as the mean ± standard deviation.
NA: not available.
3.3.4 Δ17O(HNO3) model sensitivity tests
To investigate the causes of the observed discrepancies between modeled and measured Δ17O(HNO3) values, we conducted a series of model sensitivity tests focused on modifying HNO3 formation pathways. These tests aimed to assess whether adjustments to reaction pathways or rate constants could reconcile the overprediction of Δ17O(HNO3) during the high-NOx photochemical experiments (Exps. 3, 4, and 5) and the underprediction during low-NOx and nighttime experiments. The following sections describe these targeted evaluations and their implications for understanding the Δ17O(HNO3) produced under different chemical regimes.
We first investigated the potential causes of the model overprediction in Δ17O(HNO3) during the high-NOx photochemical experiments (Exps. 3, 4, and 5). Two alternative HNO3 production pathways were identified that could yield lower Δ17O(HNO3) values than the assumed predominant daytime production pathway involving NO2+ OH reactions based on oxygen isotope mass balance assumptions (Table 1). One such pathway is the reaction of NO with HO2, which can form HNO3 with a Δ17O transfer factor of ()Δ17O(NO) (Alexander et al., 2020; Table 1). However, an underestimation of this pathway in the model is unlikely to account for the observed model–measurement discrepancy across all experiments. For instance, HO2 concentrations in Exp. 5 are expected to be low due to the absence of a significant quantity of HO2 precursors, unlike Exps. 3 and 4, which had elevated HO2 precursors from the initial injected H2O2. Moreover, Exps. 1 and 2, which had the highest initial H2O2 concentrations and thus the greatest potential for HO2 formation, exhibited model Δ17O(HNO3) values that were too low, further suggesting that this pathway cannot explain the observed discrepancies. Still, as a sensitivity test, we increased the rate constant for the NO + HO2→ HNO3 reaction (R027 in the NOx-API mechanism; Table S3) by an order of magnitude (Fig. S9). Despite this adjustment in the NO + HO2→ HNO3 reaction rate constant, the impact on modeled Δ17O(HNO3) remained small with a change in Δ17O(HNO3) of less than 1.6 ‰ and as little as 0.3 ‰ for Exp. 5. This change in model Δ17O(HNO3) is too small to account for the observed model bias of 5.2 ‰ to 7.9 ‰ across Exps. 3 to 5 (Table 4).
Next, the hydrolysis of organic nitrates to HNO3 was evaluated as a potential low-Δ17O(HNO3) source, since hydrolysis of photochemically produced RONO2 (R + OH O2 NO) could produce HNO3 with a Δ17O transfer factor of ()(Δ17O(NO)) (Table 1) considering that Δ17O(RO2) is presumably 0 ‰. Based on simulation results (Fig. S10), 21.8 %, 14.8 %, and 13.4 % of the total NOy was present as HNO3 for Exps. 3, 4, and 5, respectively, compared to only 11.0 %, 6.8 %, and 4.0 % as RONO2 (defined as ONIT in our model simulations; ONIT = ONITa + ONITb + ONITc + ONITOOHa + ONITOOHb + PDN + DIMER). Nonetheless, as a sensitivity test, we added ONIT hydrolysis with an assumed lifetime of 30 min (Table S8), consistent with recent experimental determinations (Takeuchi and Ng, 2019); however, this study showed that only 23 %–32 % of organic nitrates derived from α-pinene + OH and 9 %–17 % from α-pinene + NO3 are susceptible to hydrolysis (Takeuchi and Ng, 2019), suggesting that this sensitivity test should overestimate the impact of ONIT hydrolysis as a production route for HNO3. The inclusion of ONIT hydrolysis in the model reduced Δ17O(HNO3) by 1.4 ‰–3.4 ‰ relative to the base model, partially improving agreement with observations (Fig. S11). However, even under the unrealistic assumption of complete organic nitrate hydrolysis, the bias between the model and observed Δ17O(HNO3) values remained between 1.7 ‰ and 4.6 ‰ across experiments, indicating that hydrolysis alone cannot fully explain the model–observation discrepancy. Furthermore, while organic nitrate hydrolysis modestly improved the Δ17O(HNO3) comparison, it worsened the agreement for Δ17O(ONIT) (Fig. S12). The bias for the simulated Δ17O(ONIT) relative to the observed Δ17O(pNO3) increased substantially, ranging from 5.1 ‰–15.0 ‰. These results suggest that organic nitrate hydrolysis is unlikely to be the primary cause of the observed lower Δ17O(HNO3) values relative to model simulations.
Overall, we were unable to identify a missing or underrepresented HNO3 production pathway that could reconcile the observed Δ17O(HNO3) values within the bounds of the assumed Δ17O mass balance framework (Table 1). This suggests that current assumptions regarding oxygen atom transfer during HNO3 formation may need to be revisited. For the high-NOx photochemical experiments (Exps. 3–5), plotting measured Δ17O(HNO3) against the modeled fraction of O3-derived oxygen atoms (f(Q)(HNO3)) gave a slope of 28.9 ± 0.5 ‰, representing the Δ17O of the O (Fig. S13). This slope is lower than the ∼ 39 ‰–41 ‰ slope obtained from similar analyses of Δ17O(NO2) and Δ17O(pNO3) (Fig. S6–7), which are more consistent with recent near-surface observations of Δ17O(O) (Ishino et al., 2017; Vicars and Savarino, 2014). The lower slope in the f(Q) versus Δ17O(HNO3) relationship potentially implies that not all oxygen atoms from NO2 are retained during HNO3 formation in the dominant NO2+ OH reaction under high-NOx photochemical conditions. Traditionally, it is assumed that two-thirds of the oxygen atoms in HNO3 are inherited from NO2 and one-third from OH (with Δ17O ∼ 0 ‰) (Table 1), but this oxygen mass balance may need adjustment. Adjusting the slope for Δ17O(HNO3) versus f(Q) to match that for NO2 would require adjustment of the oxygen mass balance in the NO2+ OH reaction. Specifically, scaling the NO2 contribution by ∼ 0.74 lowers its O atom fractional contribution in HNO3 from 66.7 % to ∼ 49 %, resulting in an effective relationship of Δ17O(HNO3) Δ17O(NO2). This deviation from the expected to an effective contribution of Δ17O(NO2) may reflect partial oxygen atom exchange or isotopic scrambling during the formation of an excited HNO3 intermediate prior to collisional stabilization. Such processes could result in the effective loss or redistribution of the oxygen atoms originally inherited from NO2 in the HNO3 product. Indeed, previous experimental studies using isotopically labeled 18OH in reactions with NO2 have demonstrated that the O atoms in the HNO3 reactive intermediate product can undergo rapid intramolecular scrambling (Donahue et al., 2001). While this specific mechanism alone cannot easily explain the observed loss of approximately one-sixth of the original NO2-derived oxygen atoms in the final HNO3 product, it does suggest that interesting O isotope dynamics occur during the NO2+ OH reaction. While the exact mechanism that could explain the redistribution of ∼ of O atoms derived from NO2 in the HNO3 product remains uncertain and warrants further investigation, we conducted a sensitivity test by updating the NOx-API mechanism to reflect the potential modified mass balance for the NO2 + OH reaction, assuming Δ17O(HNO3)= O(NO2) (Fig. S14). This adjustment substantially improved the model performance for Exps. 3–5, yielding biases that ranged from −2.6 ‰ to +1.0 ‰ relative to the observations.
Next, we examine the cause of the low model bias in Δ17O(HNO3) relative to observations during the nighttime oxidation experiment. In this experiment, the measured Δ17O(HNO3) exceeded that of Δ17O(NO2), implying greater O3 incorporation into HNO3 than is represented in the model. The dominant modeled pathway for HNO3 formation at night was the reaction of NO3 with pinaldehyde (Fig. S8). However, this route yields Δ17O(HNO3) ≈ Δ17O(NO3) because NO2, NO3, and N2O5 rapidly equilibrate thermally overnight, leading to Δ17O(NO2) ≈ Δ17O(NO3) ≈ Δ17O(N2O5). Therefore, this pathway alone cannot account for the elevated observed Δ17O(HNO3) relative to the model simulation. To explain this discrepancy, an additional source of high-Δ17O external to the NOy reservoir must be involved. One possibility is that the heterogeneous uptake of N2O5 on aerosol surfaces could involve incorporation of an oxygen atom from O3 rather than liquid water. In this case, based on our mass balance framework (Table 1), the Δ17O of HNO3 formed via this pathway can be represented as
Since Δ17O(NO2) ≈ Δ17O(N2O5) for our nighttime oxidation experiment conditions, this expression simplifies to
This mass balance equation is similar to that assumed for N2O5 heterogenous reaction involving particulate Cl− (Table 1). While we did not initiate our experiment with a Cl− source, we must have a source of Δ17O(O) external to NOx to explain the underestimate of simulated Δ17O(HNO3) relative to observations.
To test the potential impact of this mechanism, we incorporated N2O5 heterogeneous uptake into the model (Table S9). The uptake rate coefficient (khet) was estimated assuming an aerosol seed number and volume concentration upon atomization of 2×104 cm−3 and 2×1010 nm3 cm−3, respectively, which was taken from previously reported values from similarly conducted α-pinene + NO3 nighttime experiments (Takeuchi and Ng, 2019), and an N2O5 uptake coefficient (γ) of for organic carbon with RH ≥ 30 % (Escorcia et al., 2010). Due to uncertainty in these parameters, we performed a sensitivity analysis across a range of khet values from to s−1, corresponding to N2O5 lifetimes between approximately 0.46 and 4.6 h. Model results show that increasing the N2O5 heterogeneous reaction rate systematically reduces the Δ17O(HNO3) model bias. Specifically, the bias decreased from 8.1 ‰ in the base case (no heterogeneous uptake) to 3.5 ‰ for the highest assumed khet (Fig. S15). This supports the hypothesis that N2O5 heterogeneous reactions incorporating O3-derived oxygen significantly influence the isotopic composition of HNO3 produced under nighttime conditions. However, the highest assumed khet rate led to an increase in the overall model bias for Δ17O(HNO3) simulations the high-NOx photochemical experiments (Exps. 3–5), shifting from −0.8 ± 1.5 ‰ in the modified mass balance simulation to +1.8 ± 2.5 ‰ (Fig. S16). To avoid overcorrection, we selected a khet rate of s−1, which improved agreement in the nighttime oxidation experiments without negatively impacting the Δ17O(HNO3) predictions under high-NOx photochemical conditions.
Lastly, we sought to diagnose the potential underprediction of Δ17O(HNO3) in the model relative to observations from the low-NOx photochemical experiments (Exps. 1–2). We observed a relatively large amount of HNO3 present before the experiments began, ranging from 3.2 to 5.1 ppb, which represented 20.1 %, 18.0 %, and 7.9 % of the maximum HNO3 concentrations measured during Exps. 1, 1R, and 2, respectively (Table S10; Fig. S1). This suggested the possibility of a substantial chamber blank influencing the Δ17O(HNO3) measurements. Unfortunately, we did not directly quantify chamber blanks during these experiments. Thus, to evaluate the potential impact, we conducted a sensitivity analysis by introducing a new model tracer, HNO, which was initialized using pre-experiment CIMS measurements. This tracer was treated identically to HNO3 in the model, undergoing the same reactions and loss processes as HNO3 (Table S11). We then used an isotope mass balance framework to infer the Δ17O of the blank component required to reproduce the observed Δ17O(HNO3) (Eq. 10):
where Δ17O(HNO) is the observed value, Δ17O(HNO) is the value for photochemically produced HNO3 in the model, and fblank is the fractional contribution of the chamber blank from the model simulation:
Using this approach, we calculated an average Δ17O(HNO) of 36.1 ± 4.0 ‰ (n= 3). While elevated, this value is plausible if HNO3 formation occurred predominantly through nighttime N2O5 heterogeneous uptake under dark chamber conditions prior to the start of the experiment and if the precursor NO2 had a high Δ17O value. Such conditions could arise from residual NO2 in the chamber either from previous experiments or background air that underwent oxidation while the chamber remained dark and inactive before the conducted experiments were initiated. It is also important to note that the CIMS HNO3 measurements are subject to a relatively large uncertainty of approximately ± 20 ‰, which may further influence the inferred blank correction.
Overall, to improve the accuracy of simulated Δ17O(HNO3), we implemented a series of modifications to the NOx-API mechanism based on the conducted sensitivity tests, termed NOx-API (modified). First, we revised the oxygen mass balance for Δ17O(HNO3) formation via the NO2+ OH reaction to ()Δ17O(NO2). Second, we included a chamber-derived HNO3 background using a fixed Δ17O value of 36.1 ‰. This background was initialized in all simulations based on CIMS-derived HNO3 concentrations prior to photolysis onset (Table S10). Finally, we added a heterogeneous N2O5 hydrolysis pathway with a first-order loss rate of s−1, using a Δ17O(HNO3) formation mass balance of () ×Δ17O(N2O5) + () ×Δ17O(O). As shown in Fig. 7, while these modifications had a minor effect on Δ17O(NO2) and Δ17O(ONIT), in which the base NOx-API mechanism were already in strong agreement with the observations, they substantially improved the model's performance for Δ17O(HNO3). The average absolute bias across all experiments decreased from 6.7 ± 3.3 ‰ in the base mechanism to 1.6 ± 1.3 ‰ in the modified mechanism (Table 5) with an improved correlation (R2= 0.48). These results demonstrate the difficulty of accurately simulating Δ17O(HNO3) across diverse experimental conditions, highlighting the need for future experiments that more directly constrain oxygen isotope mass balance assumptions in HNO3 formation pathways. At the same time, careful attention to HNO3 collection methods and blank corrections is essential to ensure meaningful comparisons between models and observations.
This study presents the first chamber experiments combining comprehensive NOy and α-pinene oxidation chemistry with stable isotope constraints. Using a suite of observations and box-model simulations, we demonstrate how multi-isotope analyses of Δ17O, δ18O, and δ15N can yield novel insights into atmospheric oxidation pathways, including the formation and transformation of gas-phase and pNO3 species. We observed a strong linear relationship between Δ17O and δ18O values across all collected NOy species. We derived a δ18O value of 106 ± 5 ‰ for oxygen atoms transferred into NOy from O and a value of 11.1 ± 1.0 ‰ for oxygen atoms transferred from other oxidants with an assumed Δ17O ∼ 0 ‰, such as RO2, HO2, and OH. These results provide a new isotopic constraint for disentangling multi-oxidant systems in both chamber and ambient settings. Nitrogen isotope fractionation was evaluated from the observations, which indicated Δδ15N(HNO3–NO2) values were generally enriched, while Δδ15N(pNO3–NO2) values were negative, reflecting preferential 15N incorporation into HNO3 and 14N incorporation into pNO3. However, large differences in the observed Δδ15N(HNO3–NO2) between H2O2 NO (33.7 ± 7.1 ‰) and HONO (0.4 ± 1.7 ‰) photochemical experiments remain unresolved, particularly since these experiments should have produced HNO3 from a similar pathway, namely the NO2+ OH reaction. These discrepancies make it challenging for us to recommend a fractionation value associated with the NO2+ OH reaction and indicate the need for future targeted studies of the isotope effects during the NO2+ OH reaction. Importantly, isotope observations revealed stark differences between HNO3 (medium to high Δ17O and high δ15N) and pNO3 (low Δ17O and low δ15N), with the later predominately derived from organic nitrate. These isotope differences between HNO3 and organic nitrate could serve as a useful qualitative constraint to evaluate inorganic and organic nitrate contributions to HNO3 and pNO3 budgets.
Model simulations of Δ17O were conducted and indicated that our model captured Δ17O(NO2) and Δ17O(ONIT) (compared with observed Δ17O(pNO3) values) well. Simulating Δ17O(HNO3), however, proved more challenging. The model tended to overestimate Δ17O under high NOx conditions, underestimate it in low NOx experiments, and underpredict nighttime values. A series of sensitivity tests suggests that this mismatch likely arises from multiple contributing factors, including potential HNO3 measurement biases (e.g., chamber blank), missing heterogeneous pathways (e.g., N2O5 hydrolysis), and the need to revisit assumptions in the isotopic mass balance of the NO2+ OH reaction. From these experiments, it is evident that we have a solid understanding of oxygen isotope transfer associated with NOx photochemical cycling and the formation of organic nitrates. However, our understanding of the oxygen and nitrogen isotope dynamics of HNO3 remains more uncertain. Our findings indicate the need for future experiments specifically designed to probe the formation pathways of HNO3 and their associated isotope dynamics. This includes chamber studies that isolate individual pathways as well as targeted flow tube experiments. Such efforts are essential to refine oxygen isotope mass balance assumptions and are critical if Δ17O and δ15N are to be used quantitatively to track HNO3 chemistry in both laboratory and ambient environments.
The box-model simulations, including model mechanisms, input files, and output files, have been made publicly available at https://doi.org/10.5281/zenodo.15615851 (Walters, 2025a). The experimental data and figure scripts have been made publicly available at https://doi.org/10.5281/zenodo.15616525 (Walters, 2025b). The chemical mechanism and isotope data are provided in the Supplement.
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-10707-2025-supplement.
WWW, MT, NLN, and MGH designed the conducted experiments. WWW, MT, DEB, GE, PT, WX, JR, FL, GH, and JBM conducted the experiments. WWW and DEB conducted the offline data analysis. WWW conducted the chamber simulations with input from MGH, MT, and NLN. WWW wrote the paper with input from all authors. WWW and MGH secured funding.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank Ruby Ho for laboratory assistance.
This research has been supported by the National Oceanic and Atmospheric Administration (grant no. NA18OAR4310118).
This paper was edited by Arthur Chan and reviewed by Matthew Johnson, Mei-Yi Fan, Yanlin Zhang, and one anonymous referee.
Albertin, S., Savarino, J., Bekki, S., Barbero, A., and Caillon, N.: Measurement report: Nitrogen isotopes (δ15N) and first quantification of oxygen isotope anomalies (Δ17O, δ18O) in atmospheric nitrogen dioxide, Atmos. Chem. Phys., 21, 10477–10497, https://doi.org/10.5194/acp-21-10477-2021, 2021.
Alexander, B., Sherwen, T., Holmes, C. D., Fisher, J. A., Chen, Q., Evans, M. J., and Kasibhatla, P.: Global inorganic nitrate production mechanisms: comparison of a global model with nitrate isotope observations, Atmos. Chem. Phys., 20, 3859–3877, https://doi.org/10.5194/acp-20-3859-2020, 2020.
Bahreini, R., Ervens, B., Middlebrook, A. M., Warneke, C., de Gouw, J. A., DeCarlo, P. F., Jimenez, J. L., Brock, C. A., Neuman, J. A., Ryerson, T. B., Stark, H., Atlas, E., Brioude, J., Fried, A., Holloway, J. S., Peischl, J., Richter, D., Walega, J., Weibring, P., Wollny, A. G., and Fehsenfeld, F. C.: Organic aerosol formation in urban and industrial plumes near Houston and Dallas, Texas, J. Geophys. Res.-Atmos., 114, D00F16, https://doi.org/10.1029/2008JD011493, 2009.
Bates, K. H., Burke, G. J., Cope, J. D., and Nguyen, T. B.: Secondary organic aerosol and organic nitrogen yields from the nitrate radical (NO3) oxidation of alpha-pinene from various RO2 fates, Atmos. Chem. Phys., 22, 1467–1482, https://doi.org/10.5194/acp-22-1467-2022, 2022.
Beaver, M. R., Clair, J. S., Paulot, F., Spencer, K. M., Crounse, J. D., LaFranchi, B. W., Min, K. E., Pusede, S. E., Wooldridge, P. J., and Schade, G. W.: Importance of biogenic precursors to the budget of organic nitrates: observations of multifunctional organic nitrates by CIMS and TD-LIF during BEARPEX 2009, Atmos. Chem. Phys., 12, 5773–5785, https://doi.org/10.5194/acp-12-5773-2012, 2012.
Bekker, C., Walters, W. W., Murray, L. T., and Hastings, M. G.: Nitrate chemistry in the northeast US – Part 1: Nitrogen isotope seasonality tracks nitrate formation chemistry, Atmos. Chem. Phys., 23, 4185–4201, https://doi.org/10.5194/acp-23-4185-2023, 2023.
Berhanu, T. A., Savarino, J., Bhattacharya, S. K., and Vicars, W. C.: 17O excess transfer during the NO2+ O3 → NO3+ O2 reaction, J. Chem. Phys., 136, 044311, https://doi.org/10.1063/1.3666852, 2012.
Blum, D. E., Walters, W. W., and Hastings, M. G.: Speciated Collection of Nitric Acid and Fine Particulate Nitrate for Nitrogen and Oxygen Stable Isotope Determination, Anal. Chem., 92, 16079–16088, https://doi.org/10.1021/acs.analchem.0c03696, 2020.
Blum, D. E., Walters, W. W., Eris, G., Takeuchi, M., Huey, L. G., Tanner, D., Xu, W., Rivera-Rios, J. C., Liu, F., Ng, N. L., and Hastings, M. G.: Collection of Nitrogen Dioxide for Nitrogen and Oxygen Isotope Determination-Laboratory and Environmental Chamber Evaluation, Anal. Chem., 95, 3371–3378, https://doi.org/10.1021/acs.analchem.2c04672, 2023.
Böhlke, J. K., Mroczkowski, S. J., and Coplen, T. B.: Oxygen isotopes in nitrate: New reference materials for 18O: 17O: 16O measurements and observations on nitrate-water equilibration, Rapid Commun. Mass Sp., 17, 1835–1846, https://doi.org/10.1002/rcm.1123, 2003.
Böhlke, J. K., Smith, R. L., and Hannon, J. E.: Isotopic analysis of N and O in nitrite and nitrate by sequential selective bacterial reduction to N2O, Anal. Chem., 79, 5888–5895, https://doi.org/10.1021/ac070176k, 2007.
Boyd, C. M., Sanchez, J., Xu, L., Eugene, A. J., Nah, T., Tuet, W. Y., Guzman, M. I., and Ng, N. L.: Secondary organic aerosol formation from the β-pinene + NO3 system: effect of humidity and peroxy radical fate, Atmos. Chem. Phys., 15, 7497–7522, https://doi.org/10.5194/acp-15-7497-2015, 2015.
Browne, E. C. and Cohen, R. C.: Effects of biogenic nitrate chemistry on the NOx lifetime in remote continental regions, Atmos. Chem. Phys., 12, 11917–11932, https://doi.org/10.5194/acp-12-11917-2012, 2012.
Browne, E. C., Wooldridge, P. J., Min, K.-E., and Cohen, R. C.: On the role of monoterpene chemistry in the remote continental boundary layer, Atmos. Chem. Phys., 14, 1225–1238, https://doi.org/10.5194/acp-14-1225-2014, 2014.
Casciotti, K. L., Sigman, D. M., Hastings, M. G., Böhlke, J. K., and Hilkert, A.: Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method, Anal. Chem., 74, 4905–4912, https://doi.org/10.1021/ac020113w, 2002.
Chai, J. and Hastings, M. G.: Collection Method for Isotopic Analysis of Gaseous Nitrous Acid, Anal. Chem., 90, 830–838, https://doi.org/10.1021/acs.analchem.7b03561, 2018.
Craig, H.: Isotopic standards for carbon and oxygen and correction factors for mass-spectrometric analysis of carbon dioxide, Geochim. Cosmochim. Ac., 12, 133–149, https://doi.org/10.1016/0016-7037(57)90024-8, 1957.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., and Docherty, K. S.: Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Donahue, N. M., Mohrschladt, R., Dransfield, T. J., Anderson, J. G., and Dubey, M. K.: Constraining the Mechanism of OH + NO2 Using Isotopically Labeled Reactants: Experimental Evidence for HOONO Formation, J. Phys. Chem. A, 105, 1515–1520, https://doi.org/10.1021/jp0035582, 2001.
Dubey, M. K., Mohrschladt, R., Donahue, N. M., and Anderson, J. G.: Isotope specific kinetics of hydroxyl radical (OH) with water (H2O): Testing models of reactivity and atmospheric fractionation, J. Phys. Chem. A, 101, 1494–1500, https://doi.org/10.1021/jp962332p, 1997.
Elliott, E. M., Yu, Z., Cole, A. S., and Coughlin, J. G.: Isotopic advances in understanding reactive nitrogen deposition and atmospheric processing, Sci. Total Environ., 662, 393–403, https://doi.org/10.1016/j.scitotenv.2018.12.177, 2019.
Escorcia, E. N., Sjostedt, S. J., and Abbatt, J. P. D.: Kinetics of N2O5 Hydrolysis on Secondary Organic Aerosol and Mixed Ammonium Bisulfate-Secondary Organic Aerosol Particles, J. Phys. Chem. A, 114, 13113–13121, https://doi.org/10.1021/jp107721v, 2010.
Fang, H., Walters, W. W., Mase, D., and Michalski, G.: iNRACM: incorporating 15N into the Regional Atmospheric Chemistry Mechanism (RACM) for assessing the role photochemistry plays in controlling the isotopic composition of NOx, NOy, and atmospheric nitrate, Geosci. Model Dev., 14, 5001–5022, https://doi.org/10.5194/gmd-14-5001-2021, 2021.
Farmer, D. K., Matsunaga, A., Docherty, K. S., Surratt, J. D., Seinfeld, J. H., Ziemann, P. J., and Jimenez, J. L.: Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, P. Natl. Acad. Sci. USA, 107, 6670–6675, https://doi.org/10.1073/pnas.0912340107, 2010.
Fibiger, D. L., Hastings, M. G., Lew, A. F., and Peltier, R. E.: Collection of NO and NO2 for Isotopic Analysis of NOx Emissions, Anal. Chem., 86, 12115–12121, https://doi.org/10.1021/ac502968e, 2014.
Fisher, J. A., Jacob, D. J., Travis, K. R., Kim, P. S., Marais, E. A., Chan Miller, C., Yu, K., Zhu, L., Yantosca, R. M., Sulprizio, M. P., Mao, J., Wennberg, P. O., Crounse, J. D., Teng, A. P., Nguyen, T. B., St. Clair, J. M., Cohen, R. C., Romer, P., Nault, B. A., Wooldridge, P. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Shepson, P. B., Xiong, F., Blake, D. R., Goldstein, A. H., Misztal, P. K., Hanisco, T. F., Wolfe, G. M., Ryerson, T. B., Wisthaler, A., and Mikoviny, T.: Organic nitrate chemistry and its implications for nitrogen budgets in an isoprene- and monoterpene-rich atmosphere: constraints from aircraft (SEAC4RS) and ground-based (SOAS) observations in the Southeast US, Atmos. Chem. Phys., 16, 5969–5991, https://doi.org/10.5194/acp-16-5969-2016, 2016.
Freyer, H. D., Kley, D., Volz-Thomas, A., and Kobel, K.: On the interaction of isotopic exchange processes with photochemical reactions in atmospheric oxides of nitrogen, J. Geophys. Res., 98, 14791–14796, https://doi.org/10.1029/93JD00874, 1993.
Fry, J. L., Kiendler-Scharr, A., Rollins, A. W., Wooldridge, P. J., Brown, S. S., Fuchs, H., Dubé, W., Mensah, A., Dal Maso, M., and Tillmann, R.: Organic nitrate and secondary organic aerosol yield from NO3 oxidation of β-pinene evaluated using a gas-phase kinetics/aerosol partitioning model, Atmos. Chem. Phys., 9, 1431–1449, https://doi.org/10.5194/acp-9-1431-2009, 2009.
Goliff, W. S., Stockwell, W. R., and Lawson, C. V.: The regional atmospheric chemistry mechanism, version 2, Atmos. Environ., 68, 174–185, https://doi.org/10.1016/j.atmosenv.2012.11.038, 2013.
Guo, H., Sullivan, A. P., Campuzano-Jost, P., Schroder, J. C., Lopez-Hilfiker, F. D., Dibb, J. E., Jimenez, J. L., Thornton, J. A., Brown, S. S., Nenes, A., and Weber, R. J.: Fine particle pH and the partitioning of nitric acid during winter in the northeastern United States, J. Geophys. Res.-Atmos., 121, 10355–10376, https://doi.org/10.1002/2016JD025311, 2016.
Hastings, M. G., Sigman, D. M., and Lipschultz, F.: Isotopic evidence for source changes of nitrate in rain at Bermuda, J. Geophys. Res.-Atmos., 108, 4790, https://doi.org/10.1029/2003JD003789, 2003.
Hastings, M. G., Casciotti, K. L., and Elliott, E. M.: Stable isotopes as tracers of anthropogenic nitrogen sources, deposition, and impacts, Elements, 9, 339–344, https://doi.org/10.2113/gselements.9.5.339, 2013.
Hoyle, C. R., Boy, M., Donahue, N. M., Fry, J. L., Glasius, M., Guenther, A., Hallar, A. G., Huff Hartz, K., Petters, M. D., and Petäjä, T.: A review of the anthropogenic influence on biogenic secondary organic aerosol, Atmos. Chem. Phys., 11, 321–343, https://doi.org/10.5194/acp-11-321-2011, 2011.
Huey, L. G., Dunlea, E. J., Lovejoy, E. R., Hanson, D. R., Norton, R. B., Fehsenfeld, F. C., and Howard, C. J.: Fast time response measurements of HNO3 in air with a chemical ionization mass spectrometer, J. Geophys. Res.-Atmos., 103, 3355–3360, https://doi.org/10.1029/97JD02214, 1998.
Ishino, S., Hattori, S., Savarino, J., Jourdain, B., Preunkert, S., Legrand, M., Caillon, N., Barbero, A., Kuribayashi, K., and Yoshida, N.: Seasonal variations of triple oxygen isotopic compositions of atmospheric sulfate, nitrate, and ozone at Dumont d'Urville, coastal Antarctica, Atmos. Chem. Phys., 17, 3713–3727, https://doi.org/10.5194/acp-17-3713-2017, 2017.
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997.
Kaiser, J., Hastings, M. G., Houlton, B. Z., Röckmann, T., and Sigman, D. M.: Triple Oxygen Isotope Analysis of Nitrate Using the Denitrifier Method and Thermal Decomposition of N2O, Anal. Chem., 79, 599–607, https://doi.org/10.1021/ac061022s, 2007.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prévôt, A. S., Äijälä, M., Allan, J., Canonaco, F., and Canagaratna, M.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kim, H., Walters, W. W., Bekker, C., Murray, L. T., and Hastings, M. G.: Nitrate chemistry in the northeast US – Part 2: Oxygen isotopes reveal differences in particulate and gas-phase formation, Atmos. Chem. Phys., 23, 4203–4219, https://doi.org/10.5194/acp-23-4203-2023, 2023.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation under high-NOx conditions, Geophys. Res. Lett., 32, 1869–1877, https://doi.org/10.1021/es0524301, 2005.
Li, J., Zhang, X., Orlando, J., Tyndall, G., and Michalski, G.: Quantifying the nitrogen isotope effects during photochemical equilibrium between NO and NO2: implications for δ15N in tropospheric reactive nitrogen, Atmos. Chem. Phys., 20, 9805–9819, https://doi.org/10.5194/acp-20-9805-2020, 2020.
Li, J., Davy, P., Harvey, M., Katzman, T., Mitchell, T., and Michalski, G.: Nitrogen isotopes in nitrate aerosols collected in the remote marine boundary layer: Implications for nitrogen isotopic fractionations among atmospheric reactive nitrogen species, Atmos. Environ., 245, 118028, https://doi.org/10.1016/j.atmosenv.2020.118028, 2021.
Lopez-Hilfiker, F. D., Mohr, C., Ehn, M., Rubach, F., Kleist, E., Wildt, J., Mentel, T. F., Lutz, A., Hallquist, M., and Worsnop, D.: A novel method for online analysis of gas and particle composition: description and evaluation of a Filter Inlet for Gases and AEROsols (FIGAERO), Atmos. Meas. Tech., 7, 983–1001, https://doi.org/10.5194/amt-7-983-2014, 2014.
Lyons, J. R.: Transfer of mass-independent fractionation in ozone to other oxygen-containing radicals in the atmosphere, Geophys. Res. Lett., 28, 3231–3234, https://doi.org/10.1029/2000GL012791, 2001.
McIlvin, M. R. and Altabet, M. A.: Chemical Conversion of Nitrate and Nitrite to Nitrous Oxide for Nitrogen and Oxygen Isotopic Analysis in Freshwater and Seawater, Anal. Chem., 77, 5589–5595, https://doi.org/10.1021/ac050528s, 2005.
Michalski, G., Scott, Z., Kabiling, M., and Thiemens, M. H.: First measurements and modeling of Δ17O in atmospheric nitrate, Geophys. Res. Lett., 30, 1870, https://doi.org/10.1029/2003GL017015, 2003.
Michalski, G., Bhattacharya, S. K., and Mase, D. F.: Oxygen isotope dynamics of atmospheric nitrate and its precursor molecules, in: Handbook of environmental isotope geochemistry, edited by: Baskaran, M., Springer, New York, 613–635, https://doi.org/10.1007/978-3-642-10637-8, 2012.
Michalski, G., Bhattacharya, S. K., and Girsch, G.: NOx cycle and the tropospheric ozone isotope anomaly: an experimental investigation, Atmos. Chem. Phys., 14, 4935–4953, https://doi.org/10.5194/acp-14-4935-2014, 2014.
Morales, A. C., Jayarathne, T., Slade, J. H., Laskin, A., and Shepson, P. B.: The production and hydrolysis of organic nitrates from OH radical oxidation of β-ocimene, Atmos. Chem. Phys., 21, 129–145, https://doi.org/10.5194/acp-21-129-2021, 2021.
Morin, S., Sander, R., and Savarino, J.: Simulation of the diurnal variations of the oxygen isotope anomaly (Δ17 O) of reactive atmospheric species, Atmos. Chem. Phys., 11, 3653–3671, https://doi.org/10.5194/acp-11-3653-2011, 2011.
Nah, T., Sanchez, J., Boyd, C. M., and Ng, N. L.: Photochemical Aging of α-pinene and β-pinene Secondary Organic Aerosol formed from Nitrate Radical Oxidation, Environ. Sci. Technol., 50, 222–231, https://doi.org/10.1021/acs.est.5b04594, 2016.
Ng, N. L., Brown, S. S., Archibald, A. T., Atlas, E., Cohen, R. C., Crowley, J. N., Day, D. A., Donahue, N. M., Fry, J. L., Fuchs, H., Griffin, R. J., Guzman, M. I., Herrmann, H., Hodzic, A., Iinuma, Y., Jimenez, J. L., Kiendler-Scharr, A., Lee, B. H., Luecken, D. J., Mao, J., McLaren, R., Mutzel, A., Osthoff, H. D., Ouyang, B., Picquet-Varrault, B., Platt, U., Pye, H. O. T., Rudich, Y., Schwantes, R. H., Shiraiwa, M., Stutz, J., Thornton, J. A., Tilgner, A., Williams, B. J., and Zaveri, R. A.: Nitrate radicals and biogenic volatile organic compounds: oxidation, mechanisms, and organic aerosol, Atmos. Chem. Phys., 17, 2103–2162, https://doi.org/10.5194/acp-17-2103-2017, 2017.
Orsini, D. A., Ma, Y., Sullivan, A., Sierau, B., Baumann, K., and Weber, R. J.: Refinements to the particle-into-liquid sampler (PILS) for ground and airborne measurements of water soluble aerosol composition, Atmos. Environ., 37, 1243–1259, https://doi.org/10.1016/S1352-2310(02)01015-4, 2003.
Rindelaub, J. D., McAvey, K. M., and Shepson, P. B.: The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis, Atmos. Environ., 100, 193–201, https://doi.org/10.1016/j.atmosenv.2014.11.010, 2015.
Rindelaub, J. D., Borca, C. H., Hostetler, M. A., Slade, J. H., Lipton, M. A., Slipchenko, L. V., and Shepson, P. B.: The acid-catalyzed hydrolysis of an á-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact, Atmos. Chem. Phys., 16, 15425–15432, https://doi.org/10.5194/acp-16-15425-2016, 2016.
Romer, P. S., Duffey, K. C., Wooldridge, P. J., Allen, H. M., Ayres, B. R., Brown, S. S., Brune, W. H., Crounse, J. D., De Gouw, J., and Draper, D. C.: The lifetime of nitrogen oxides in an isoprene-dominated forest, Atmos. Chem. Phys., 16, 7623–7637, https://doi.org/10.5194/acp-16-7623-2016, 2016.
Sato, K., Ikemori, F., Ramasamy, S., Iijima, A., Kumagai, K., Fushimi, A., Fujitani, Y., Chatani, S., Tanabe, K., and Takami, A.: Formation of secondary organic aerosol tracers from anthropogenic and biogenic volatile organic compounds under varied NOx and oxidant conditions, Atmos. Environ. X, 14, 100169, https://doi.org/10.1016/j.aeaoa.2022.100169, 2022.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Savarino, J., Bhattacharya, S. K., Morin, S., Baroni, M., and Doussin, J.-F.: The NO + O3 reaction: A triple oxygen isotope perspective on the reaction dynamics and atmospheric implications for the transfer of the ozone isotope anomaly, J. Chem. Phys., 128, 194303, https://doi.org/10.1063/1.2917581, 2008.
Savarino, J., Morin, S., Erbland, J., Grannec, F., Patey, M. D., Vicars, W., Alexander, B., and Achterberg, E. P.: Isotopic composition of atmospheric nitrate in a tropical marine boundary layer, P. Natl. Acad. Sci. USA, 110, 17668–17673, https://doi.org/10.1073/pnas.1216639110, 2013.
Sharp, Z.: Principles of Stable Isotope Geochemistry, 2nd Edn., Open Textbooks, https://doi.org/10.25844/h9q1-0p82, 2017.
Sigman, D. M., Casciotti, K. L., Andreani, M., Barford, C., Galanter, M., and Böhlke, J. K.: A bacterial method for the nitrogen isotopic analysis of nitrate in seawater and freshwater, Anal. Chem., 73, 4145–4153, https://doi.org/10.1021/ac010088e, 2001.
Takeuchi, M. and Ng, N. L.: Chemical composition and hydrolysis of organic nitrate aerosol formed from hydroxyl and nitrate radical oxidation of α-pinene and β-pinene, Atmos. Chem. Phys., 19, 12749–12766, https://doi.org/10.5194/acp-19-12749-2019, 2019.
Takeuchi, M., Berkemeier, T., Eris, G., and Ng, N. L.: Non-linear effects of secondary organic aerosol formation and properties in multi-precursor systems, Nat. Commun., 13, 7883, https://doi.org/10.1038/s41467-022-35546-1, 2022.
Takeuchi, M., Wang, Y., Nault, B. A., Chen, Y., Canagaratna, M. R., and Ng, N. L.: Evaluating the response of the Aerodyne aerosol mass spectrometer to monoterpene- and isoprene-derived organic nitrate standards, Aerosol Sci. Technol., 58, 1371–1388, https://doi.org/10.1080/02786826.2024.2389183, 2024.
Tuet, W. Y., Chen, Y., Xu, L., Fok, S., Gao, D., Weber, R. J., and Ng, N. L.: Chemical oxidative potential of secondary organic aerosol (SOA) generated from the photooxidation of biogenic and anthropogenic volatile organic compounds, Atmos. Chem. Phys., 17, 839–853, https://doi.org/10.5194/acp-17-839-2017, 2017.
Vicars, W. C. and Savarino, J.: Quantitative constraints on the 17 O-excess (Δ17O) signature of surface ozone: Ambient measurements from 50° N to 50° S using the nitrite-coated filter technique, Geochim. Cosmochim. Ac., 135, 270–287, https://doi.org/10.1016/j.gca.2014.03.023, 2014.
Walters, W.: Box Model Simulations for NO-pinene Chamber Experiments (v2.0), Zenodo [data set], https://doi.org/10.5281/zenodo.15615851, 2025a.
Walters, W.: Data and Figure Codes for, “Evaluating NOx Fate and Organic Nitrate Chemistry from α-Pinene Oxidation Using Stable Oxygen and Nitrogen Isotopes” (v1.1), Zenodo [data set], https://doi.org/10.5281/zenodo.15616525, 2025b.
Walters, W. W. and Hastings, M. G.: Triple oxygen stable isotope analysis of nitrite measured using continuous flow isotope ratio mass spectrometry, MethodsX, 11, 102413, https://doi.org/10.1016/j.mex.2023.102413, 2023.
Walters, W. W. and Michalski, G.: Theoretical calculation of nitrogen isotope equilibrium exchange fractionation factors for various NOy molecules, Geochim. Cosmochim. Ac., 164, 284–297, https://doi.org/10.1016/j.gca.2015.05.029, 2015.
Walters, W. W. and Michalski, G.: Ab initio study of nitrogen and position-specific oxygen kinetic isotope effects in the NO + O3 reaction, J. Chem. Phys., 145, 224311, https://doi.org/10.1063/1.4968562, 2016a.
Walters, W. W. and Michalski, G.: Theoretical calculation of oxygen equilibrium isotope fractionation factors involving various NOy molecules, OH, and H2O and its implications for isotope variations in atmospheric nitrate, Geochim. Cosmochim. Ac., 191, 89–101, https://doi.org/10.1016/j.gca.2016.06.039, 2016b.
Walters, W. W., Simonini, D. S., and Michalski, G.: Nitrogen isotope exchange between NO and NO2 and its implications for δ15N variations in tropospheric NOx and atmospheric nitrate, Geophys. Res. Lett., 43, 2015GL066438, https://doi.org/10.1002/2015GL066438, 2016.
Walters, W. W., Fang, H., and Michalski, G.: Summertime diurnal variations in the isotopic composition of atmospheric nitrogen dioxide at a small midwestern United States city, Atmos. Environ., 179, 1–11, https://doi.org/10.1016/j.atmosenv.2018.01.047, 2018.
Walters, W. W., Takeuchi, M., Ng, N. L., and Hastings, M. G.: Incorporating Oxygen Isotopes of Oxidized Reactive Nitrogen in the Regional Atmospheric Chemistry Mechanism, version 2 (ICOIN-RACM2), Geoscientific Model Development, 17, 4673–4687, https://doi.org/10.5194/gmd-17-4673-2024, 2024a.
Walters, W. W., Pye, H. O. T., Kim, H., and Hastings, M. G.: Modeling the Oxygen Isotope Anomaly (Δ17O) of Reactive Nitrogen in the Community Multiscale Air Quality Model: Insights into Nitrogen Oxide Chemistry in the Northeastern United States, ACS ES&T Air, 1, 451–463, https://doi.org/10.1021/acsestair.3c00056, 2024b.
Wang, X., Liu, T., Bernard, F., Ding, X., Wen, S., Zhang, Y., Zhang, Z., He, Q., Lü, S., and Chen, J.: Design and characterization of a smog chamber for studying gas-phase chemical mechanisms and aerosol formation, Atmos. Meas. Tech., 7, 301–313, https://doi.org/10.5194/amt-7-301-2014, 2014.
Wang, Y., Piletic, I., Takeuchi, M., Xu, T., France, S., and Ng, N. L.: Synthesis and hydrolysis of atmospherically relevant monoterpene-derived organic nitrates, Environ. Sci. Technol., 55, 14595–14606, https://doi.org/10.1021/acs.est.1c05310, 2021.
Wang, Y., Takeuchi, M., Wang, S., Nizkorodov, S. A., France, S., Eris, G., and Ng, N. L.: Photolysis of Gas-Phase Atmospherically Relevant Monoterpene-Derived Organic Nitrates, J. Phys. Chem. A, 127, 987–999, https://doi.org/10.1021/acs.jpca.2c04307, 2023.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Xu, L., Suresh, S., Guo, H., Weber, R. J., and Ng, N. L.: Aerosol characterization over the southeastern United States using high-resolution aerosol mass spectrometry: spatial and seasonal variation of aerosol composition and sources with a focus on organic nitrates, Atmos. Chem. Phys., 15, 7307–7336, https://doi.org/10.5194/acp-15-7307-2015, 2015a.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015b.
Xu, L., Tsona, N. T., You, B., Zhang, Y., Wang, S., Yang, Z., Xue, L., and Du, L.: NOx enhances secondary organic aerosol formation from nighttime α-terpinene ozonolysis, Atmos. Environ., 225, 117375, https://doi.org/10.1016/j.atmosenv.2020.117375, 2020.
Young, E. D., Galy, A., and Nagahara, H.: Kinetic and equilibrium mass-dependent isotope fractionation laws in nature and their geochemical and cosmochemical significance, Geochim. Cosmochim. Ac., 66, 1095–1104, https://doi.org/10.1016/S0016-7037(01)00832-8, 2002.
Zare, A., Romer, P. S., Nguyen, T., Keutsch, F. N., Skog, K., and Cohen, R. C.: A comprehensive organic nitrate chemistry: insights into the lifetime of atmospheric organic nitrates, Atmos. Chem. Phys., 18, 15419–15436, https://doi.org/10.5194/acp-18-15419-2018, 2018.