the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 10 Sep 2025
| 10 Sep 2025
Ozonolysis of primary biomass burning organic aerosol particles: insights into reactivity and phase state
Sophie Bogler
Jun Zhang
Rico K. Y. Cheung
André S. H. Prévôt
Imad El Haddad
Biomass burning organic aerosol (BBOA) particles are a major contributor to atmospheric particulate matter, with various effects on climate and public health. Quantifying these effects is limited by our understanding of the BBOA particles' evolving chemical composition during atmospheric aging, driven by their exposure to atmospheric oxidants. This study explores the role of ozone (O3) as an atmospheric oxidant in processing primary BBOA particles. We exposed particulate emissions from beech, spruce, and pine wood fires to O3 in an oxidative flow reactor, monitoring their chemical evolution using high-resolution time-of-flight aerosol mass spectrometry (HR-ToF-AMS) and extractive electrospray ionization time-of-flight mass spectrometry (EESI-ToF-MS). We found that the oxidative state of the particles increased with O3 exposure, as shown by the consistent, albeit minor, rise in ratios. Analysis of the EESI-ToF-MS data revealed specific molecular groups containing 18 and 20 carbon atoms, likely mainly abietic, linoleic, and oleic acids, as highly reactive towards O3 and driving the increase in oxidative state. At higher relative humidity, increased oxidation and loss of reactive species indicate that enhanced O3 diffusion into particles allows the ozonolysis to progress further, highlighting humidity's role in overcoming diffusion barriers that limit ozonolysis in dry conditions. This study provides qualitative insights into the oxidative processing of primary BBOA particles in different phase states, presenting O3 as a selective oxidant. Further research could focus on quantifying the progression of the ozonolysis, in particular, the change in diffusion rates depending on relative humidity conditions or particle sizes.
- Article
(1278 KB) - Full-text XML
-
Supplement
(1798 KB) - BibTeX
- EndNote




Atmospheric particulate matter (PM) affects our climate and public health. The importance of PM for human health is based on its damaging effects on the respiratory and cardiovascular systems after extended exposure to atmospheric PM. For the year 2015, an estimated 8.9 million premature deaths worldwide were linked to this exposure (Burnett et al., 2018), and air pollution was listed in the top five global mortality risk factors (Lelieveld et al., 2015). Given this ranking, it is evident that advancing the understanding of toxic PM sources and their mitigation is crucial to reducing future health impacts. One key contributor to atmospheric submicron PM is organic aerosol (OA) particles, which account for up to 90 % of PM mass (Jimenez et al., 2009).
One of the primary sources of OA particles is combustion processes, such as vehicular exhaust and biomass burning. Biomass burning organic aerosol (BBOA), makes up 15 %–68 % of the total OA mass estimated in Europe during winter in rural areas (Puxbaum et al., 2007), when most of the BBOA is generated for residential heating. During summer, BBOA comes predominantly from prescribed burnings and wildfires. Both fire suppression and climate change have resulted in an increase in the frequency of wildfires and thus increased the BBOA contribution to atmospheric PM (Iglesias et al., 2022; Westerling, 2016).
To assess the impacts of atmospheric BBOA, it is required to characterize the chemical composition and its evolution through various atmospheric aging pathways. As an analytical method, aerosol mass spectrometry has been used to characterize BBOA either offline (e.g., Daellenbach et al., 2016; Liang et al., 2022), online in the field (e.g., Saarnio et al., 2013; Hayden et al., 2022), or in laboratory experiments (e.g., Yazdani et al., 2021). Still, resolving BBOA's chemical composition remains an ongoing analytical challenge, given the presence of millions of individual species (Zhang et al., 2023). Each compound has unique physicochemical properties, which determine its reactivity towards different atmospheric oxidants and its partitioning behavior, both of which control its atmospheric lifetime.
When exposed to sunlight and to atmospheric oxidants, such as the hydroxyl radical (OH), ozone (O3), and the nitrate radical, the chemical composition of the primary BBOA particles evolves in the atmosphere to form aged BBOA. Aged BBOA generally has a higher oxidation state and has lost mass due to evaporation (Yazdani et al., 2021). Given the importance of these atmospheric processing pathways, knowing only the chemical composition of primary BBOA and a few marker species is not sufficient to assess the overall impact of BBOA during its atmospheric lifetime. Also, the use of markers as efficient tracers is limited depending on their stability towards the various oxidants. For example, several studies have shown how levoglucosan decays by oxidation with OH over the course of hours (Bertrand et al., 2018b; Hennigan et al., 2010; Hoffmann et al., 2010; Lai et al., 2014). Beyond conventional gas-phase oxidants, UV light can result in efficient formation of triplet-state chemistry and secondary oxidant formation within the aerosol (Bogler et al., 2021; Liang et al., 2024; Vasilakopoulou et al., 2023).
Laboratory studies targeting specific aging mechanisms with high chemical detail remain scarce. First results from Fortenberry et al. (2018) show how photochemical aging depletes primary BBOA emissions overall while simultaneously enhancing certain marker signals that are likely produced through secondary oxidation processes. In a similar experimental setup, Bertrand et al. (2018a) highlighted how the majority of primary markers from woodstove emissions are lost through aging by OH. Most secondary compounds with potential use as tracers remain unidentified. Yazdani et al. (2021) used mid-infrared spectroscopy to analyze the functional group composition in BBOA aged with OH or nitrate radicals compared to freshly emitted BBOA from wood burning. Here, the authors illustrate the importance of oxygenated functional groups, like carboxylic acids, in aged compounds, while the backbone of the parent compound structure is retained.
One of the ubiquitous oxidants in the atmosphere is O3, in which O3 reacts predominantly with alkenes (Ziemann, 2005; Zelenyuk et al., 2017). Previous work showed limited changes in the oxygen-to-carbon (O:C) ratio of BBOA upon O3 exposure as measured by an aerosol mass spectrometer (AMS) (Browne et al., 2019). However, it is unknown whether these limited changes with O3 exposure were due to diffusion limitations or the limited reactivity of BBOA. It is also unknown whether BBOA reactivity depends on its chemical composition, hence the type of fuel burned and combustion conditions. These experimental findings were also limited by the chemical resolution of the AMS. Because O3 is thought to react predominantly with alkenes, many studies have probed the reactions of alkenes in the aerosol with O3. A common alkene studied is oleic acid, and aging in the presence of O3 has shown that the reaction of O3 only takes place at the surface of the aerosol, which creates a shell of reactive products and limits further reactions (Berkemeier et al., 2021). Given the limited uptake of O3 into oleic acid particles, similar diffusion limitations could play an important role in the heterogeneous reactions of BBOA (Gerrebos et al., 2024). However, this has not definitively been shown in previous work on the heterogeneous reactions of BBOA and warrants further investigation (Hems et al., 2021). Recent work has also shown that BBOA can undergo a liquid–liquid phase separation, which could limit heterogeneous chemistry (Gerrebos et al., 2024; Gregson et al., 2023). These limitations could arise through differences in Henry's law coefficient in the different layers or through the development of a high-viscosity shell.
Here, we provide insight into the role of oxidative processing of BBOA with O3 in the chemical composition of BBOA at a wide range of RHs. Experiments were performed with a series of BBOA sources from residential stoves (beech and spruce wood), as well as open burning of spruce branches. These sources were chosen to represent major biomass burning emissions in Europe from residential wood stoves and to elucidate changes associated with open burning, which are major biomass burning sources in boreal forest regions. We present data both from residential stoves and from open burning to highlight the consistencies between the two emission sources. We have used two complementary online mass spectrometric techniques, the AMS and the extractive electrospray mass spectrometer (EESI-MS), to probe the changes in the bulk and molecular composition of primary BBOA particles, respectively. Specific molecular formulas observed in the BBOA rapidly decayed when exposed to O3, while a large portion of the BBOA constituents were relatively unreactive. We discuss how particle size and relative humidity alter the extent of reactivity.
The ozonolysis experiments of BBOA particles (Table 1) were conducted in the oxidative flow reactor (OFR; Li et al., 2019) and the 8 m3 Teflon smog chamber, which are part of the atmospheric simulation chamber and burning platform located at the Paul Scherrer Institute in Villigen, Switzerland (Platt et al., 2013). In both setups, continuous measurements of the particles were performed with a high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS, Aerodyne Inc.), an extractive electrospray ionization time-of-flight mass spectrometer (EESI-ToF-MS, Tofwerk), and a scanning mobility particle sizer (SMPS, TSI). Sketches of the complete setup are provided in the Supplement (Figs. S1 and S2).
Table 1Overview of experiments.
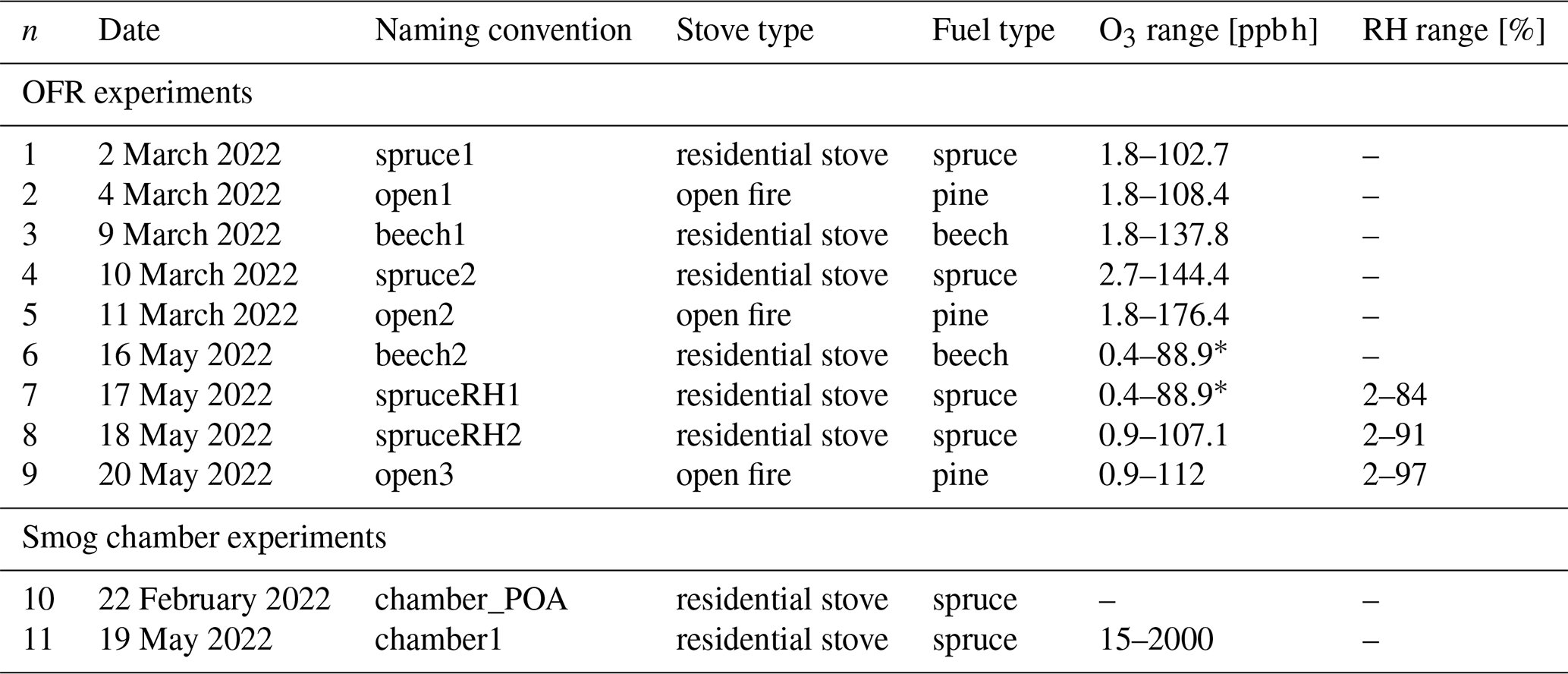
∗ In these experiments, the upper O3 limit is an underestimation, as the O3 monitor's upper measurement limit was restricted. The true value is close to experiment n = 8 or n = 9 (empty entries marked with –). In these experiments, the O3 concentration was at a background level or RH conditions were dry (∼ 2 % RH), respectively.
2.1 Ozonolysis procedure in the oxidation flow reactor (OFR)
For each experiment, we generated fresh BBOA emissions from fires of either spruce, beech, or pine wood and injected the sample through heated stainless steel lines (150 °C) into a 1 m3 insulated stainless steel holding chamber. Logs of spruce and beech wood were burned in a residential stove, while the pine wood branches and needles were burned in an open stainless steel cylinder (65 cm diameter, 35 cm height) to mimic wildfire conditions with high oxygen content. Typical temperature conditions present during a wildfire can vary from 500–1500 °C (Dennison et al., 2006). The holding chamber was filled with the desired emissions until the particle mass concentration reached 5 to 10 mg m−3 (50–150 µg m−3 in the sampling lines after dilution with an ejector dilutor, DI-1000, Dekati Ltd). While filling the holding chamber, a mix of emissions from flaming and smoldering conditions of the fires was sampled. The holding chamber served as a reservoir in which the primary BBOA and gaseous emissions were stored for the duration of the experiments at room temperature (20 °C) and from which we continuously sampled into the OFR. The emissions in the holding chamber were sampled at ∼ 0.6 L min−1 (detailed flow conditions in the Supplement, Tables S1 and S2) for the duration of the experiment, with an equal makeup flow of clean air (clean air generator 737–250, AADCO instrument Inc.) to maintain a constant volume and pressure. The slow wall loss was corrected in order to separate the decrease in particle mass concentration from changes due to O3 exposure (see Sect. 2.3.3). In between experiments, the holding chamber was flushed with clean air to minimize carryover of OA.
The holding chamber held both the primary BBOA and gas-phase volatile organic compounds (VOCs) emitted by the combustion sources, and to probe the reaction of BBOA with O3, the VOCs were scrubbed with a charcoal denuder (Ionicon) at a flow rate of 0.6 L min−1. In between experiments, the denuder was regenerated by heating it to 200 °C while passing N2 through the denuder to ensure a stable gas removal efficiency during the experiment. Following denuding, the BBOA was exposed to O3 in the OFR, a 50.8 cm long quartz-fused cylinder tube with a total volume of 16 L. More details on the OFR design are described elsewhere (Li et al., 2019). To achieve optimal flow conditions through the OFR of about 6 L min−1, a flow of clean air into the OFR was added to the sample flow of ∼ 0.6 L min−1. An ozone generator provided the O3 in the OFR, generating O3 in situ by the photolysis of O2, with a constant input flow of 0.1 L min−1 clean air. By splitting the exiting flow from the ozone generator into an exhaust and a line into the OFR, the O3-containing flow in this line could be set between 0.01 and 0.1 L min−1. Note that the total flow through the OFR was kept constant at 6 L min−1 by adjusting the dilution flow of clean air. With the O3 flow controlled between 0.01 and 0.1 L min−1, the O3 concentration inside the OFR varied between 0.02 and 4 ppm, as was continuously measured by an ozone gas monitor (Thermo 49C). The average residence time of the particles in the OFR was 160 s (); therefore, this O3 concentration range results in equivalent O3 exposures between 1.4 and 176.3 ppb h. To control the exposure time, we installed a second denuder directly after the OFR particle sampling outlet. In all experiments, the particles were sampled with an AMS, SMPS, and EESI-ToF-MS.
In each experiment, we exposed the primary BBOA particles to 8 to 11 different O3 concentration steps (Tables S1 and S2). Three out of nine experiments included steps at different RHs, which were increased compared to the background RH level of ∼ 2 %. To increase RH in the OFR, the clean air supply was split into a dry air line and a wet air line, which bubbled through a water bottle. At each step, the particles' chemical composition and size distribution after exposure to O3 were monitored for at least 10 min after stable O3 and RH conditions, respectively, were reached. Additional steps included sampling primary BBOA particles with no O3 exposure to monitor the wall loss (Sect. 2.3.3).
2.2 Smog chamber ozonolysis procedure
A subset of experiments with longer O3 exposures were accomplished by filling an atmospheric simulation chamber with the primary organic aerosol (POA) from a residential wood stove. The emissions, again, were passed through heated lines (180 °C), and prior to entry to the chamber, they were passed through charcoal denuders in parallel to strip the gaseous aerosol species (VOCs). For all measurements, the non-methane VOCs were less than 10 ppbC from measurements with a total hydrocarbon (THC) analyzer. After injection into the chamber, O3 was steadily ramped to 1 ppm, and the experiment was allowed to proceed for +6 h before the chamber was cleaned by flushing with zero air.
Sampling from the chamber for these measurements included an AMS, SMPS, and gas monitors (O3 – Thermo 49C, THC – Horiba APHA, CO and CO2 – PICARRO).
2.3 Particle instruments
2.3.1 High-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS)
The HR-ToF-AMS provides online quantitative measurements of the BBOA particle chemical composition (DeCarlo et al., 2006). Here, we operated the HR-ToF-AMS at a mass resolution of 2000 to 4000 m Δm−1 in V-mode using either a high-resolution ToF (H-ToF, lower resolution limit) or a long ToF (L-ToF, upper resolution limit). The time resolution for 1 measurement cycle was 1 min. For the conversion of ion counts into particle mass concentration of µg m−3, we determined the ionization efficiency based on a calibration with 300 nm ammonium nitrate particles and simultaneous SMPS (model 3938, TSI) measurements. Data analysis of raw mass spectra was conducted in Igor Pro 8.0.4.2, using the SQUIRREL data analysis toolkit (version 1.63l) for unit mass resolution data up to 600 and the PIKA module (version 1.23I) for high-resolution peak fitting in the range 12 to 170 . To represent the composition at each O3 step, lasting a minimum of 10 min, we averaged the mass spectra of 11 to 13 measurement runs and reported the standard error (standard deviation/(number of runs averaged)) at each .
2.3.2 Extractive electrospray ionization time-of-flight mass spectrometer (EESI-ToF-MS)
The EESI-ToF-MS provides real-time, high-resolution measurement of the OA on a near-molecular level (details in Lopez-Hilfiker et al., 2019). Incoming particles are sampled through a charcoal denuder to remove any gas-phase species and collide with electrospray droplets, which extract water-soluble molecular constituents of the particles. Here, we used an aqueous solution of pure Milli-Q water doped with 100 ppm of sodium iodide (NaI). Components of OA that are insoluble in water or those that do not effectively bind to Na+ are not detected, restricting the overall performance and sensitivity of the molecular analysis.
The solvent is evaporated rapidly from the droplets by passing the heated (∼ 270 °C) capillary inlet to the mass spectrometer, and analytes are ionized by forming adducts with Na+. These ions are then sampled into an atmospheric pressure interface time-of-flight mass spectrometer (APi-ToF-MS) for separation at a mass resolution of ∼ 10 500 m Δm−1 (Tofwerk, Thun, Switzerland). Mass spectra were recorded in the positive mode with a time resolution of 1 s. For data processing, the mass spectra were pre-averaged over 10 s. After averaging, the HR data analysis was performed using the Tofware software version 3.2.3 for Igor Pro (8.0.4.2), fitting ions up to 420 .
The continuous measurement alternated between sampling the incoming aerosol for 6 min and recording the ambient background signal for 90 s by sampling the incoming flow through a high-volume HEPA filter. To retrieve the background-corrected average intensity of each ion with a time resolution of 7 to 8 min, the average intensity during a filter period was subtracted from the average intensity during the preceding signal period. For the composition representing one O3 exposure condition, we averaged the ion intensities over 2 to 4 signal periods and reported the error propagated from the standard errors of all signal periods in that O3 period. Species were considered for the analysis if their intensity was > 10 cps in all relevant signal periods.
2.3.3 Wall loss correction
There are two loss mechanisms (dilution and wall loss) for the particles in the holding chamber. Additionally, gaseous species are absorbed to the walls, driving their partitioning out of the particulate phase at a rate depending on their volatility (Fig. S4). To separate these decreases in particle concentration from potential mass loss due to O3 exposure, we applied a wall loss correction to all measured particle concentrations (HR-ToF-AMS, SMPS) and individual ion intensities (EESI-ToF-MS) for the OFR experiments (Eq. 1, Fig. S4).
In Eq. (1), the subscript “corr” refers to the final corrected value for any variable at time point t, and “measured” refers to the value measured on the instrument. The 0 is the average concentration or intensity during the first stable period of POA, and “fit” refers to the value retrieved from a linear fit through periods of POA. This linear fit was adequate to capture both the particle loss dynamics and repartitioning losses for the duration of the ozonolysis experiments. For experiments where a higher flow demand was required to supply additional particle instruments, we applied an exponential fit instead of a linear fit (experiments beech2 and spruceRH2, Fig. S3).
3.1 Primary BBOA particle composition
3.1.1 HR-ToF-AMS data
The HR-ToF-AMS mass spectra of primary BBOA particles exhibit consistent dominating features across burning and fuel types (representative example from experiment spruce2 in Figs. 1a and S6) and match qualitatively with HR-ToF-AMS measurements of primary BBOA from previous studies (Alfarra et al., 2007; Fortenberry et al., 2018; He et al., 2010; Zhang et al., 2023). The chemical class containing zero (CH) fragments, characterized by ion series of CnH2n−1, makes up the largest fraction of the total particle mass concentration, accounting for 45.9 ± 0.1 % to 58.2 ± 0.1 % across all experiments. Other prominent peaks include 29, 43, 44, and 60, resulting from the contribution of oxygen-containing ions CHO+, C2H3O+, , and C2H4O+ that are typical fragments of BBOA compounds (Alfarra et al., 2007; Fine et al., 2002; Lin et al., 2016; Ortega et al., 2013; Zhang et al., 2023). We also consistently found a clear signal up to 2.7 % for the marker species of primary emitted BBOA at 60, . This ion results from the pyrolysis of anhydrous sugars like levoglucosan and is commonly labeled as f60 (Aiken et al., 2009; Cubison et al., 2011; Lee et al., 2010; Simoneit et al., 1999). Though we do observe variations in relative fractions of f60 up to 0.8 % between experiments (Fig. 2c–e), these are likely based on variations in the fuel material and burning conditions and are within the variability for complex burning experiments.
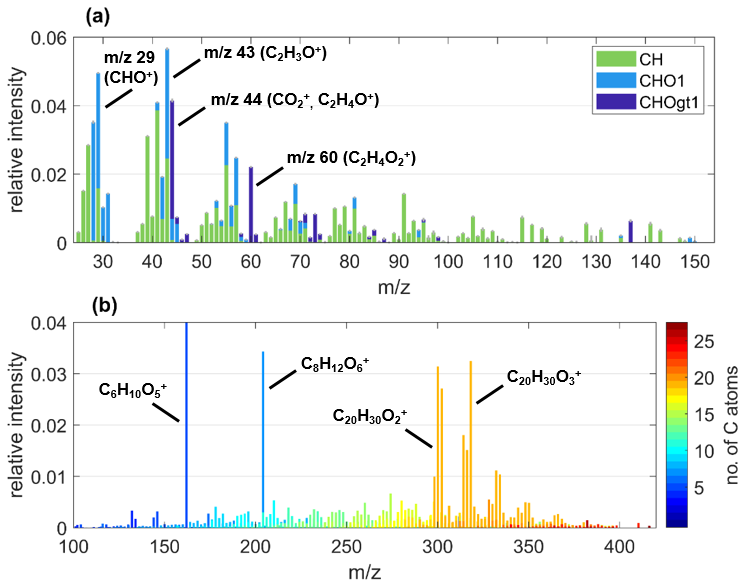
Figure 1Exemplary (a) AMS and (b) EESI-MS under POA conditions of BBOA particles from the experiment spruce2, highlighting important ions. In panel (a), the colors differentiate the groups of ions containing zero (CH), one (CHO1), or more than one O atom (CHOgt1). In panel (a), the y-scale intensity is relative to the average POA mass concentration summed over each nominal mass up to 170 and, in panel (b), it is relative to the total average intensity at POA conditions shown in panel (b). Note that, in panel (b), the most intense fraction of C6H10O5 would expand the y scale by a factor of 3 (relative intensity of 0.17), so this ion was not considered for setting the scale.
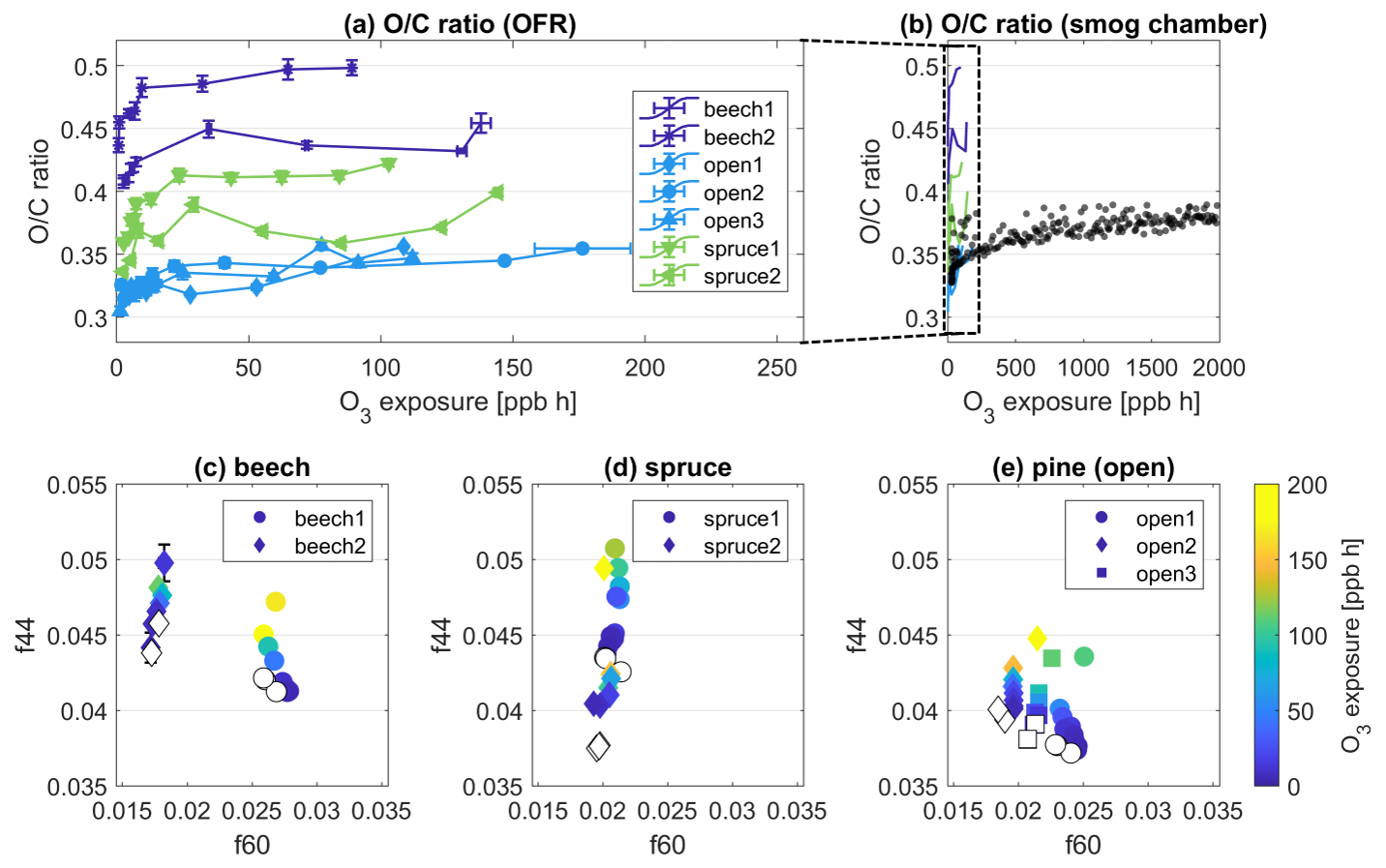
Figure 2Evolution of the ratio measured by the AMS in primary BBOA particles as a function of O3 exposure in (a) OFR experiments, (b) a smog chamber experiment with spruce wood. (c–e) Evolutions of the fraction 44 (f44) vs. 60 (f60) from OFR experiments measured by the AMS and color-coded by O3 exposure. Each panel (c–e) combines data from all experiments for each fuel type given in the subtitle. Empty markers represent POA conditions of the respective experiment.
3.1.2 EESI-ToF-MS data
In the EESI-ToF-MS mass spectra of primary BBOA particles, we identified a small number of distinct, outstanding peaks recurring across fuel and burning types in the fitted mass range up to 420 (example from experiment spruce2 in Fig. 1b). As a particular characteristic for BBOA, the most intense peak was consistently measured for C6H10O5 with relative intensities up to 30.2 %. This molecular formula corresponds to the marker species levoglucosan and its isomers (162.0523 ), which have been frequently matched with biomass burning sources before (Kumar et al., 2022; Qi et al., 2019; Stefenelli et al., 2019; Zhang et al., 2023). In all but one experiment (open2), the second-highest contributions are found for C8H12O6, which can likely be attributed to derivatives of syringol, a monomeric subunit of the biomass polymer lignin (Yee et al., 2013). Lastly, groups of C20 species around 302 and 318 are highlights of the EESI-ToF-MS POA mass spectra. Possible sources of these C20 species are discussed in more detail in Sect. 3.3.1. Also note that although polyaromatic hydrocarbons (PAHs) are another unique group of molecules typically present in POA (Bozzetti et al., 2017), the current configuration of the EESI does not measure these molecules because either their solubility or their binding dynamics with Na+ preclude their detection.
This group of dominating ions (C6H10O5, C8H12O6, and C20's) remained largely the same despite overall variability in the intensities of individual ions between each experiment's POA spectra (Fig. S7). This POA variability is likely the result of inherent variations between experiments, driven by the diversity in the fuel material (age of the logs, amount of resin, etc.) and specifics of the burning conditions such as the exact duration of flaming and smoldering stages. As extensive fragmentation occurs with the HR-ToF-AMS, this POA variability is only reliably detected with the EESI-ToF-MS, highlighting the complementary use of both instruments (Zhang et al., 2023).
3.2 Bulk composition changes of primary BBOA upon O3 exposure
3.2.1 HR-ToF-AMS data
Without O3 exposure, the oxygen-to-carbon () ratios of primary BBOA particles range from 0.30 to 0.43, increasing from pine to spruce to beech (Fig. 2a). The increase in with O3 exposure is reproducible across all experiments performed, and thus the ratio is useful as a characteristic bulk feature for each fuel type and highlights the consistency of this process regardless of the combustion source (residential stove vs. open burning). The highest ratios for beech wood POA also coincide with the highest mass fractions of oxygenated chemical classes CxHyO1 and CxHyO2+ of 40.6 ± 0.1 % for this fuel, while the average of all experiments was at 33.5 ± 0.1 %. The range of ratios measured here is comparable to previous measurements of primary BBOA (e.g., Aiken et al., 2009; Xu et al., 2020).
Upon exposure to O3, the ratio increases for all conducted experiments, which is indicative of oxidative aging in the particle phase (Fig. 2a). The increases in ratio from POA conditions to the highest O3 exposure of each experiment are similar throughout, even though the experiments differed in maximum O3 exposure tested. The range of increase is between 0.03 (experiment open2) and 0.06 (experiment spruce2), which is equivalent to an increase of 15 % in the particles' oxygen content. For all experiments, the increase in is rapid, occurring at O3 exposures below 50 ppb h (equivalent to 1 h of a typical summer day in the atmosphere), which is well below the maximum O3 exposure of any experiment (176 ppb h).
In experiments performed in the smog chamber, we tested O3 exposures up to 10 times the OFR range, i.e., up to 2000 ppb h. Similar to the OFR measurements, there was a fast increase in the in the first minutes of exposure, and an upper limit was reached after ∼ 3 h of O3 exposure. In comparison, during a background POA experiment with no O3, the increased slowly over 2–3 h to +10 % of the initial value (chamber_POA, Table 1, Fig. S8), presumably from evaporation of semi-volatile species. After a 10-fold increase in O3 exposures in the smog chamber, the ratio increase remains similar to that in the OFR experiments (Fig. 2b). After a steep increase of up to ∼ 50 ppb h, the slope gets shallow, which could mean either that the reactive species present in the POA are fully consumed after this period of time or that there could be a diffusion limitation governing the maximum degree of oxidation in the ozonolysis experiments at dry conditions, slowing down the oxidation process. These possibilities will be explored below in the “Atmospheric implications” section.
Figure 2c–e show scatter plots of f44 vs. f60, which is an established method to visualize the evolution of oxidative aging in the BBOA: f44 is the mass fraction of 44, which is mostly , i.e., the end of an oxidative chain. The fraction at 60, f60, is used as a marker species for primary BBOA (Sect. 3.1.1). Upon oxidation, it is expected that f44 increases and f60 decreases or stays the same. Indeed, Fig. 2c–e shows an increase in f44, thereby illustrating an overall increase in the oxidative state of the particles upon O3 exposure. Across fuel types and repeated experiments, f44 increases by a maximum of 1.2 % on the scale of total mass, comparing the fraction at each experiment's maximum O3 exposure to the fraction in fresh POA. In contrast, f60 () remains largely stable throughout all O3 exposures, which is not surprising because of the low uptake coefficient observed in previous studies (Knopf et al., 2011). As this ion is largely derived from levoglucosan, this stability implies that levoglucosan is not reactive towards O3 and is consistent with its lack of double bonds on the carbon chain. The increase in f44 and O:C must therefore result from the oxidation of other chemical moieties within the primary BBOA particles that contain double bonds, which are reactive with O3.
Based on the evolution of ratio and f44 vs. f60, the effect of O3 on the bulk composition of the primary BBOA particles could be clearly and reproducibly observed. Under dry conditions, the degree of oxidation increases in the particles upon exposure to O3, although this change is minor. These findings are consistent with previous measurements performed in Browne et al. (2019), where limited changes occur under relatively dry conditions (∼ 2 % RH from our results vs. 30 % RH).
3.2.2 EESI-ToF-MS data
To identify the groups of species on a molecular level that drive the observed bulk reactivity towards O3 (Sect. 3.2.1), we binned the species detected by the EESI-ToF-MS by the number of C and O atoms. The intensity of each bin was scaled relative to the total intensity in Figs. 3 and S9, and this classification shows that most species contain 5 to 20 C atoms and 0 to 7 O atoms. The dominant peaks in the mass spectra (Fig. 1b) stand out at 6 and 8 C atoms, with the largest contributions between the single species C6H10O5 and C8H12O6, respectively.
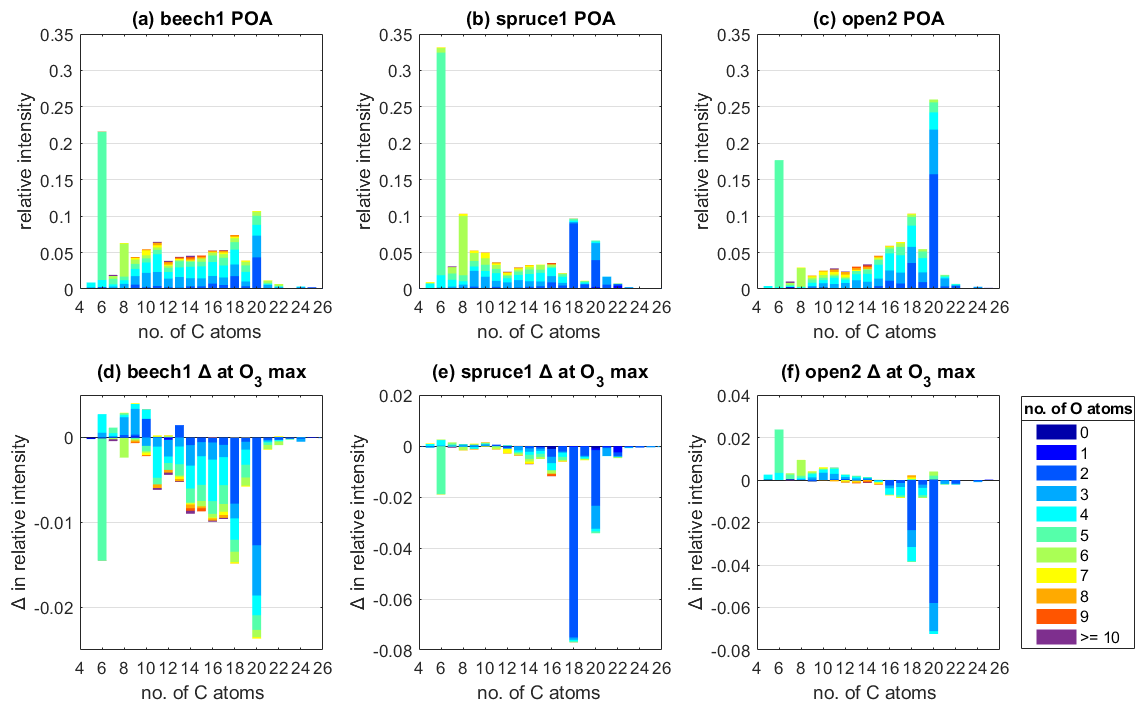
Figure 3Average POA composition measured by the EESI for the experiments (a) beech1, (b) spruce1, and (c) open2, classified by the number of C atoms, color-coded by the number of O atoms, and scaled as relative intensity compared to total intensity measured at POA conditions. (d–f) Change in relative intensity at the highest O3 exposure of each experiment compared to POA conditions. The highest O3 exposure varied between (d) 137 ppb h for beech1, (e) 102 ppb h for spruce1, and (f) 176 ppb h for open2.
Contrasting POA conditions (Fig. 3a–c) to the changes in relative intensity at the maximum O3 exposure (102–176 ppb h) of each experiment (Fig. 3d–f) reveals that the same groups of species for all fuel types are strongly reactive towards O3. Most prominently, species containing 18 and 20 C atoms with 1 to 3 O atoms decay significantly upon exposure to O3. In particular, the intensity of C18HxxO2 species decreased by 44.2 ± 0.3, 83.5 ± 0.3, and 66.2 ± 0.1 % compared to their initial intensity in the experiments beech, spruce, and open2, respectively. Similarly, C20HxxO2 species decayed by 29.2 ± 0.1 (beech), 58.4 ± 0.1 (spruce), and 36.9 ± 0.1 % (open2). As a third important group, C16 species are among the highest decaying species; for example, in the experiments spruce1 and open3 (Fig. S9). Most species that are not C16, C18, or C20 each contribute less than 1 % in relative change to the total POA intensity at maximum O3 exposure. The higher variation of species like levoglucosan, C6H10O5, is considered to be within errors related to wall loss correction.
When comparing the experiments further, three observations are especially evident:
-
The experiment with the highest O3 exposure overall was open2 (176 ppb h, Fig. 3c), but the strongest decrease in the reactive species groups was observed in experiment spruce, where maximum O3 was at 102 ppb h (Fig. 3b). These observations point at parameters other than the O3 exposure (Sect. 3.3) determining the progression of the ozonolysis, and this limitation starts at an O3 exposure threshold below the maximum exposures tested in the experiments here, regardless of the fuel type.
-
The beech experiments exhibited the lowest change upon O3 exposure (Fig. 3a), which matches the POA composition being more oxygenated than spruce or pine and, therefore, the compounds therein being the least reactive. The higher oxygenated state under POA conditions is shown in the order of ratios under POA conditions (Fig. 2a) and the above average abundance of the chemical classes CxHyO1 and CxHyO2+. For example, species containing 4 or more O atoms (excluding C6H10O5) make up 49.1 % of the total POA intensity in beech, compared to 24.5 % in experiment spruce1 and 36.8 % in open2 (Fig. 3). Therefore, the different fuel types have different emissions that react more or less with O3 (beech emitting species that react less and spruce/pine emitting species that react more).
-
A significant number of product species were only observed in experiment open2 (Fig. 3c). One possible reason for the overall lack of detection could be that many ozonolysis products are volatile, evaporate from the particle, and therefore evade detection. For instance, the ozonolysis of oleic acid results in carbon double bond scission and C9 molecules (Gallimore et al., 2017).
Overall, the EESI-ToF-MS bulk analysis of the dry OFR experiments reveals that groups of low-oxygenated species containing 16, 18, and 20 C atoms drive the O3 reactivity within primary BBOA particles. These identified groups of species consistently decay upon O3 exposure and are present across all tested fuel types, albeit with varying relative abundance (higher abundance in pine and spruce combustion emissions). The exclusivity of O3 reactivity to a small subset within the total POA composition matches well with the small degree of overall changes that were observed in the bulk HR-ToF-AMS characteristics (Sect. 3.2.1).
3.3 Reactivity of primary BBOA during ozonolysis on a molecular level
3.3.1 Key decaying species
Within the groups of C20HxxO2 and C18HxxO2, the most important contributors in all dry OFR experiments to the reactivity towards O3 are the species C20H28O2, C20H30O2, C18H32O2, and C18H34O2. These species have the highest absolute signal intensity and the highest intensity decrease at maximum O3 exposure within their groups. Analyzing their individual decay as a function of O3 exposure further mirrors the trend in the ratio observations (Fig. 2a). The intensities of these species drop steeply up to 50 ppb h followed by a decrease with a shallow negative slope at higher O3 exposures (Fig. 4). At maximum O3 exposures and across experiments, fractions down to 29.0 %, 12.6 %, and 12.2 % remain for C20H30O2, C18H32O2, and C18H34O2, respectively. The remaining fraction of C20H28O2 at the highest O3 exposure is on average ∼ 20 % higher than for C20H30O2.
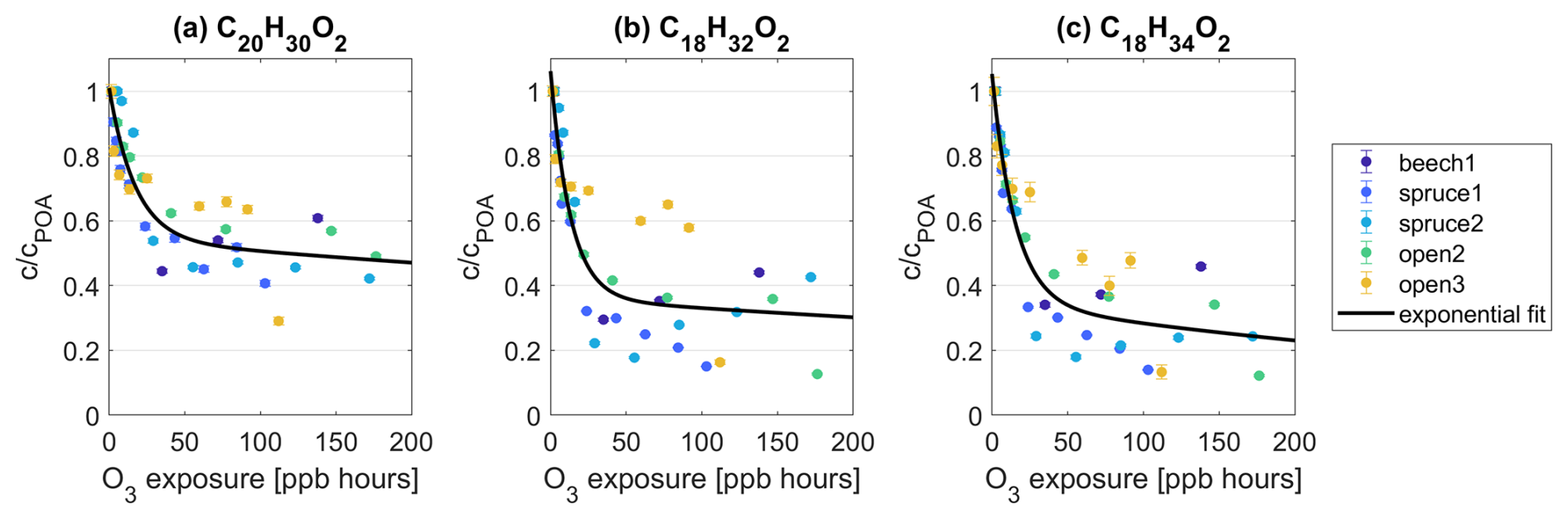
Figure 4Intensity c of species (a) C20H30O2, (b) C18H32O2, and (c) C18H34O2 as a fraction of their intensity at POA conditions cPOA and as a function of O3 exposure. The data points from all experiments were combined and are fitted with an exponential function of = . The fit coefficients for C20H30O2 (C18H32O2, C18H34O2) are a = 0.46 (0.70, 0.70), b = (ppb h)−1, c = 0.54 (0.36, 0.35), and d = (ppb h)−1. is the O3 concentration in ppb. t = 160 s is the reaction time in the OFR.
The molecular formulas C20H30O2, C18H32O2, and C18H34O2 could be connected to abietic acid, linoleic acid, and oleic acid, respectively (Fig. S10). Abietic-acid-like structures are known components of resins and needles in conifers (Iinuma et al., 2007; Mofikoya et al., 2020; Schauer et al., 2001; Liang et al., 2021, 2022), which is consistent with their higher abundance in pine and spruce combustion emissions. Linoleic and oleic acid have also been detected in emissions from pine wood burnings (Nolte et al., 2001), but they have been measured in other emissions, e.g., from cooking, too (Robinson et al., 2006). All three compounds are unsaturated, explaining their observed reactivity with O3. Oleic acid, in particular, has often been applied as a model compound for ozonolysis (Berkemeier et al., 2021). We also detected, presumably, dehydroabietic acid (C20H28O2) in similar abundance to C20H30O2, which showed some reactivity towards O3 but which was smaller than that of the other molecules noted in this section. One explanation could be the presence of aromatic systems in C20H28O2 with delocalized electrons, where O3 is less prone to attack the molecule compared to the double bonds as in C20H30O2.
Based on the experiments conducted under dry conditions, we have outlined the reactivity of BBOA particles from spruce, pine, and beech fires towards O3 and identified the drivers of this reactivity at the molecular level. In all analyzed parameters, most of the oxidative changes by O3 were reached at exposures around 50 ppb h in the OFR for both the bulk changes (Fig. 2) and molecular changes (Figs. 3 and 4), with slow progression at higher O3 exposures. We hypothesize that the diminished effect at O3 exposures exceeding that threshold could be due to either the diffusion of O3 through the BBOA particle starting to limit the further progression of the ozonolysis on the timescale of OFR experiments or different isomers present in the POA, with some isomers reactive to O3 and others that are unreactive. The diffusion limitation of molecules in BBOA could be either related to the diffusivity of the primary BBOA itself or via reaction products of O3 creating a shell with low diffusivity, which has been observed in pure oleic acid aerosol (Berkemeier et al., 2021). Section 3.3.2 and 3.3.3 discuss the effect of relative humidity and the relevance of particle size, supporting the hypothesis of the diffusion limitation for the progression of the ozonolysis.
3.3.2 The effect of relative humidity on the reactivity of primary BBOA
To test the hypothesis that diffusion of O3 is limiting the progression of the ozonolysis, we performed a subset of experiments comparing dry conditions to 50 % and > 80 % relative humidity at distinct exposures of O3. Increasing RH should enhance the diffusion within the POA particles, as they become less viscous due to the uptake of water (e.g., Asa-Awuku et al., 2009; Gerrebos et al., 2024; Reid et al., 2018), but Gregson et al. (2023) showed that a phase-separated aerosol can have different diffusivity associated with the different layers, with the hydrophobic layer maintaining low diffusivity to high relative humidities (> 90 %). Diffusion rates of small molecules (e.g., O3) through a particle increase from ∼ 10−10 cm2 s−1 in glasses/solids to ∼ 10−5 cm2 s−1 in liquid particles (Shiraiwa et al., 2011). If diffusion limits the extent of ozonolysis, increasing the RH will likely mitigate that limit and enable the ozonolysis to advance further.
Figure 5 compares the bulk ratio change for each type of POA for different O3 exposures and RHs. Indeed, the increases more during periods with increased RH than under dry conditions and the additional increase was largest at 90 ppb h O3 exposure. Comparing the difference in ratio change between low and high O3 exposure, the difference with changing RH was negligible (experiment spruceRH1) in the low O3 exposure range, while at high exposures there is a greater change in the under high-humidity conditions. At high exposures of O3, potentially all POA molecules that are reactive to O3 and are at the surface of the particle do react quickly with it. Then the diffusion limitation becomes relevant; for the ozonolysis of the POA particle to progress further, O3 needs to diffuse further into the particle to reach more POA reactive molecules or these molecules need to diffuse to the surface to react with O3. The observation is consistent with diffusivity (for either O3 or reactive organic molecules) being increased at elevated RH.
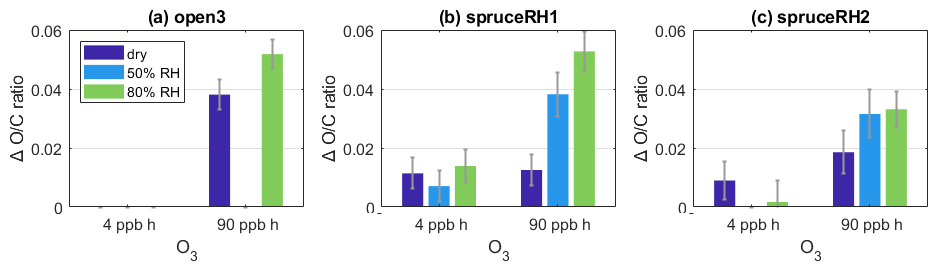
Figure 5Change in ratio compared to POA conditions at low (0.1 ppm/4 ppb h−1) and high (2 ppm/90 ppb h−1) O3 exposure at varying RHs for the experiments (a) open3, (b) spruceRH1, and (c) spruceRH2. The exact O3 and RH conditions are given in Tables S1 and S2.
Figure 6a–c show the EESI-ToF-MS data for C20H30O2, C18H32O2, and C18H34O2 for the experiment spruceRH1. For these species that react away with O3 in the particles (Sect. 3.3.1), the increase in loss at elevated RH is especially evident and illustrates that ozonolysis proceeds further under wet conditions. For example, at 90 ppb h O3 exposure, the remaining fraction of C18H32O2 at 50 % and 80 % RH is reduced to 11.1 % and 16.9 % compared to POA conditions, whereas 80 % remains at the same O3 exposure under dry conditions. A similar pattern is observed for C20H30O2 and C18H34O2. Note that there is some experimental variability in the extent of loss shown in Fig. 6 between 50 % and 80 %, where the 50 % data point is occasionally lower than the 80 % data point. These differences are of a similar order to the variability shown in Fig. 4. Overall, the changes in the extent of ozonolysis of BBOA with increasing RH observed by the EESI-ToF-MS reinforce the observations of the ratio changes in Fig. 5, supporting the diffusion limitation hypothesis.
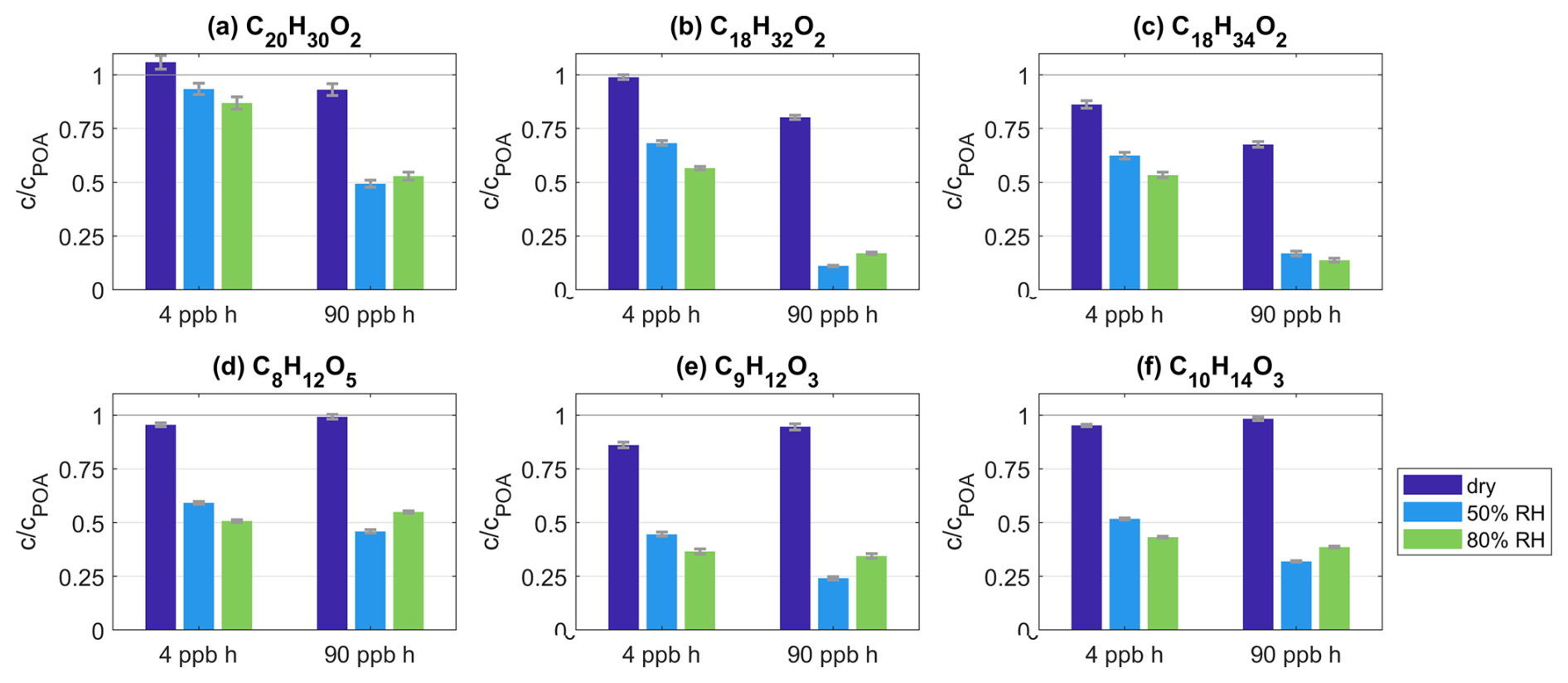
Figure 6(a–f) Intensity c as a fraction of intensity under POA conditions cPOA and as a function of varying O3 exposure and RH level. Each subplot shows the data for one species as given in the subtitle from experiment spruceRH1. The exact O3 and RH conditions are given in Tables S1 and S2.
In addition to increasing the diffusion of O3 into the BBOA particle, increased water content will increase the bulk diffusivity of the molecules present within the BBOA, allowing molecules to diffuse to the surface faster and evaporate out of the particle to achieve equilibrium between gas and condensed phases under wet conditions. As a result, a large fraction of volatile, lower-molecular-weight species containing 6–14 C atoms are lost. In the experiment spruceRH1, the overall EESI intensity at 50 % and 80 % RH decreased by 39.7 % and 29.8 %, respectively, compared to dry conditions at 90 ppb h O3 exposure (Fig. S12). As the current background reference at 4 ppb h O3 exposure also shows a similar decay under wet conditions, the observed loss is likely not connected to ozonolysis. Additionally, this effect is illustrated by the species C8H12O5, C9H12O3, and C10H14O3 from experiment spruceRH1 as the largest contributors to the intensity of the lower-molecular-weight species (Fig. 6d–f). Their intensity decreases by up to 76 % compared to POA conditions when conditions are wet. The difference in the remaining fractions for all three species is negligible between 4 and 90 ppb h O3 exposure. Therefore, the loss is the result not of ozonolysis but rather of enhanced evaporation. Further, enhanced evaporation under humid conditions additionally supports the hypothesis that molecular diffusion within primary BBOA is limited under dry conditions but that these limitations ease at elevated RH.
3.3.3 Particle size dependence
Investigating the particle size dependence of the POA ozonolysis was possible as the SMPS measured continuously and as the average POA particle size increases with increasing residence time in the holding chamber. This increase in average size is predominantly due to wall loss of small particles and coagulation of smaller particles to form larger particles.
As diffusion timescales increase with particle size, we expect the heterogeneous ozonolysis of smaller particles to progress further than that of larger particles for the same reaction duration, that is, if there is indeed a diffusion limitation.
Figure 7 supports this hypothesis based on changes in ratio measured by the AMS for the experiments open2, open3, and spruce2. In all three experiments, the ratio increased more in the smaller particles than in the larger particles upon O3 exposure. Comparing the experiments, the difference in ratio change further increased with the difference in particle size. In the experiment open3, the size difference was smallest (5 nm) and the difference in change remained within the range of uncertainty. By contrast, in the experiment spruce2, the diameter increased by 32 %, and here, the increase in ratio compared to POA conditions was double (0.06) for the smaller particles than for the larger particles (0.03).
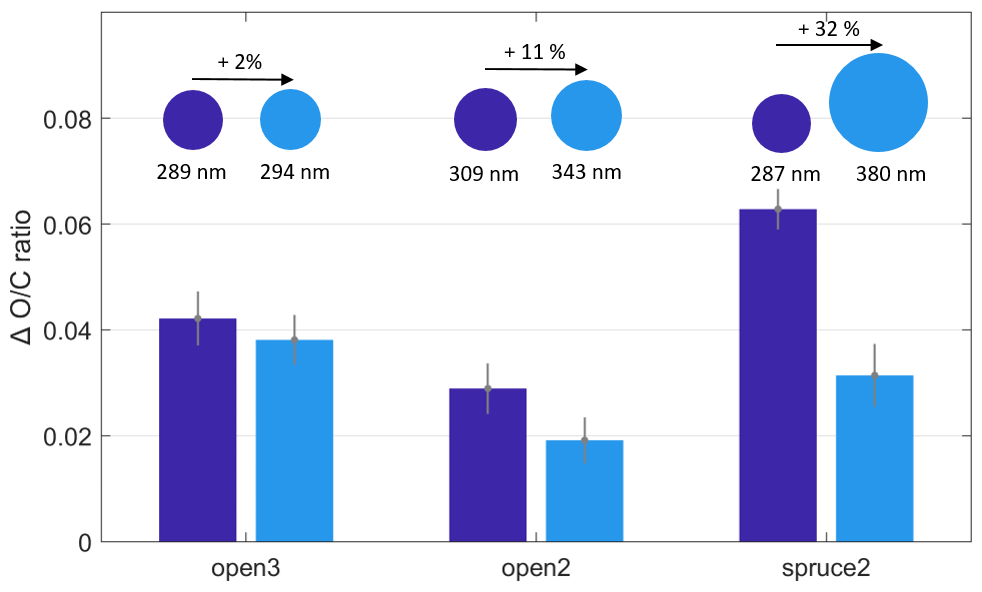
Figure 7Change in ratio compared to POA conditions at varying particle size and O3 exposure. The O3 exposure varied between 91 and 112 ppb h (open3), 146 and 176 ppb h (open2), and 144 and 173 ppb h (spruce2). The particle size is given as the average particle geometric mean diameter.
Notably, the O3 exposures of the experimental steps compared in Fig. 7 were not constant, ranging up to a maximum of 30 ppb h (see caption of Fig. 7). Therefore, the differences in change are driven both by a changing O3 exposure and by a changing particle size. However, all exposures were above 50 ppb h (minimum 91 ppb h), the threshold previously demonstrated as the limit for O3 exposure dependence for the ozonolysis under dry conditions in the OFR. Based on this result, we argue that the driving factor for the differences in ratio change in Fig. 7 is indeed the differences in particle size.
Overall, these trends support the role of a diffusion limitation in the dry, heterogeneous ozonolysis of POA, with smaller particles exhibiting greater increases in ratio compared to larger particles under the same reaction conditions.
The results presented herein provide comprehensive chemical measurements of the heterogeneous reactions of O3 with authentic primary biomass burning organic aerosol (BBOA) generated from residential stoves or open burning processes. Overall, these results show that the ozonolysis proceeds only to a limited extent under dry conditions for all combustion sources investigated here, consistent with previous results. In all cases, the O3 uptake into primary BBOA is limited to the surface under dry conditions at 20 °C but is eased when exposed to elevated RH. The change in ratio because of heterogeneous uptake is always very minor. This means one cause of the limitation is the lack of reactive species towards O3. The other point driving the limitation under dry conditions is the molecular diffusion limitation. Both the diffusion of O3 into the particle and the diffusion of species towards the surface are limited within BBOA under dry conditions. In the atmosphere, there are limited places where strong BBOA emissions occur with low relative humidity at ∼ 20 °C, but BBOA emitted from wildfires can frequently reach the troposphere and, on rare occasions, the stratosphere (Wilkins et al., 2020). When strong convection is coupled with the emissions from wildfires, the ambient temperatures will decrease below 10 °C, creating conditions where BBOA is most likely a glassy solid with viscosities above 1012 Pa s (Arangio et al., 2015). In these atmospheric conditions, the uptake of O3 will be even more limited to the surface because of the higher viscosities and lower diffusivity.
These results should also extend to heterogeneous reactivity with OH or NO3 radicals. Although OH radicals will react with more species present within the aerosol than O3, the limited diffusivity observed with O3 should also limit OH radicals under similar dry and cold conditions. Further, given the surface reactivity of OH radicals, they could also result in the build-up of a high-viscosity medium at the surface, which would further exacerbate limited molecular diffusion. Of course, the results here are focused on the heterogeneous reactivity of gases at the interface with BBOA, and consequently, the formation of radicals and oxidants within BBOA could be considered a more important source of BBOA oxidation than heterogeneous reactions (Bogler et al., 2021; Liang et al., 2024).
Previous work has shown that the reactions of oleic acid with O3 result in a high-viscosity shell on the surface of particles, which then limits the diffusion of molecules reaching the surface (Berkemeier et al., 2021). Because the oxidation state changes observed here are small, and these species make up a small fraction of the measured aerosol, we hypothesize that there are insufficient amounts of oxidation products formed capable of creating the low-diffusivity layer that limits molecules' diffusion to the surface. Future work will focus on investigating the dynamics across a wide range of O3 exposures to elucidate molecular changes under different relative humidities (e.g., 0 %, 50 %, and 90 %). Within this work, the changes with different O3 exposures were only investigated in detail under dry conditions, but specific experiments to target changes with RHs were performed to determine the effect of RH on the extent of the reactivity of primary BBOA. Having detailed chemical changes at a series of RHs will make it possible to model the diffusivity of O3 and its changes with RH, similar to the modeling performed within Berkemeier et al. (2021), which will provide constraints on the diffusivity within primary BBOA. Within this work, we focus only on qualitative terms regarding diffusion limitations because not enough data are available to provide insight at different RHs. This work provides important insight into the diffusion limitations present within authentic primary BBOA and the chemical changes associated with heterogeneous reactions of O3. This work answers important questions about the phase state of primary BBOA, and future work should investigate quantitative details regarding molecular diffusion rates.
Data from the figures may be found at https://doi.org/10.5281/zenodo.14721551 (Bell, 2025).
The supplement related to this article is available online at https://doi.org/10.5194/acp-25-10229-2025-supplement.
Flow tube experiments were performed by SB and JZ with support from DMB. Chamber measurements were performed by SB, RC, and DMB. Conceptualization of the measurements was achieved by DMB, KL, ASHP, and IEH. The data were analyzed by SB. The paper was written by SB and DMB. All authors read and agreed with the submission of the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We are grateful for our technician, Pascal Schneider, who built the holding chamber and the associated equipment with the flow tube.
This work was supported by the Swiss National Science Foundation (SNSF) grant MOLORG (grant no. 200020_188624), SNSF joint research project (grant no. 189883), and ATMO-ACCESS Integrating Activity (grant no. 101008004). The facility is part of ACTRIS ERIC and receives funding from the Swiss State Secretariat for Education, Research and Innovation (SERI).
This paper was edited by Theodora Nah and reviewed by two anonymous referees.
Aiken, A. C., Salcedo, D., Cubison, M. J., Huffman, J. A., DeCarlo, P. F., Ulbrich, I. M., Docherty, K. S., Sueper, D., Kimmel, J. R., Worsnop, D. R., Trimborn, A., Northway, M., Stone, E. A., Schauer, J. J., Volkamer, R. M., Fortner, E., de Foy, B., Wang, J., Laskin, A., Shutthanandan, V., Zheng, J., Zhang, R., Gaffney, J., Marley, N. A., Paredes-Miranda, G., Arnott, W. P., Molina, L. T., Sosa, G., and Jimenez, J. L.: Mexico City aerosol analysis during MILAGRO using high resolution aerosol mass spectrometry at the urban supersite (T0) – Part 1: Fine particle composition and organic source apportionment, Atmos. Chem. Phys., 9, 6633–6653, https://doi.org/10.5194/acp-9-6633-2009, 2009.
Alfarra, M. R., Prevot, A. S. H., Szidat, S., Sandradewi, J., Weimer, S., Lanz, V. A., Schreiber, D., Mohr, M., and Baltensperger, U.: Identification of the Mass Spectral Signature of Organic Aerosols from Wood Burning Emissions, Environ. Sci. Technol., 41, 5770–5777, https://doi.org/10.1021/es062289b, 2007.
Arangio, A. M., Slade, J. H., Berkemeier, T., Pöschl, U., Knopf, D. A., and Shiraiwa, M.: Multiphase Chemical Kinetics of OH Radical Uptake by Molecular Organic Markers of Biomass Burning Aerosols: Humidity and Temperature Dependence, Surface Reaction, and Bulk Diffusion, J. Phys. Chem. A, 119, 4533–4544, https://doi.org/10.1021/jp510489z, 2015.
Asa-Awuku, A., Miracolo, M. A., Kroll, J. H., Robinson, A. L., and Donahue, N. M.: Mixing and phase partitioning of primary and secondary organic aerosols, Geophys. Res. Lett., 36, L15827, https://doi.org/10.1029/2009GL039301, 2009.
Bell, D.: Data for Main Text Figures, Zenodo [data set], https://doi.org/10.5281/zenodo.14721551, 2025.
Berkemeier, T., Mishra, A., Mattei, C., Huisman, A. J., Krieger, U. K., and Pöschl, U.: Ozonolysis of Oleic Acid Aerosol Revisited: Multiphase Chemical Kinetics and Reaction Mechanisms, ACS Earth Space Chem., 5, 3313–3323, https://doi.org/10.1021/acsearthspacechem.1c00232, 2021.
Bertrand, A., Stefenelli, G., Jen, C. N., Pieber, S. M., Bruns, E. A., Ni, H., Temime-Roussel, B., Slowik, J. G., Goldstein, A. H., El Haddad, I., Baltensperger, U., Prévôt, A. S. H., Wortham, H., and Marchand, N.: Evolution of the chemical fingerprint of biomass burning organic aerosol during aging, Atmos. Chem. Phys., 18, 7607–7624, https://doi.org/10.5194/acp-18-7607-2018, 2018a.
Bertrand, A., Stefenelli, G., Pieber, S. M., Bruns, E. A., Temime-Roussel, B., Slowik, J. G., Wortham, H., Prévôt, A. S. H., El Haddad, I., and Marchand, N.: Influence of the vapor wall loss on the degradation rate constants in chamber experiments of levoglucosan and other biomass burning markers, Atmos. Chem. Phys., 18, 10915–10930, https://doi.org/10.5194/acp-18-10915-2018, 2018b.
Bogler, S., Borduas-Dedekind, N., el Haddad, I., Bell, D., and Dällenbach, K.: How quality and quantity of brown carbon influence singlet oxygen production in aqueous organic aerosols, EGU General Assembly 2021, online, 19–30 Apr 2021, EGU21-10743, https://doi.org/10.5194/egusphere-egu21-10743, 2021.
Bozzetti, C., El Haddad, I., Salameh, D., Daellenbach, K. R., Fermo, P., Gonzalez, R., Minguillón, M. C., Iinuma, Y., Poulain, L., Elser, M., Müller, E., Slowik, J. G., Jaffrezo, J.-L., Baltensperger, U., Marchand, N., and Prévôt, A. S. H.: Organic aerosol source apportionment by offline-AMS over a full year in Marseille, Atmos. Chem. Phys., 17, 8247–8268, https://doi.org/10.5194/acp-17-8247-2017, 2017.
Browne, E. C., Zhang, X., Franklin, J. P., Ridley, K. J., Kirchstetter, T. W., Wilson, K. R., Cappa, C. D., and Kroll, J. H.: Effect of heterogeneous oxidative aging on light absorption by biomass burning organic aerosol, Aerosol Sci. Tech., 53, 663–674, https://doi.org/10.1080/02786826.2019.1599321, 2019.
Burnett, R., Chen, H., Szyszkowicz, M., Fann, N., Hubbell, B., Pope, C. A., Apte, J. S., Brauer, M., Cohen, A., Weichenthal, S., Coggins, J., Di, Q., Brunekreef, B., Frostad, J., Lim, S. S., Kan, H., Walker, K. D., Thurston, G. D., Hayes, R. B., Lim, C. C., Turner, M. C., Jerrett, M., Krewski, D., Gapstur, S. M., Diver, W. R., Ostro, B., Goldberg, D., Crouse, D. L., Martin, R. V., Peters, P., Pinault, L., Tjepkema, M., van Donkelaar, A., Villeneuve, P. J., Miller, A. B., Yin, P., Zhou, M., Wang, L., Janssen, N. A. H., Marra, M., Atkinson, R. W., Tsang, H., Quoc Thach, T., Cannon, J. B., Allen, R. T., Hart, J. E., Laden, F., Cesaroni, G., Forastiere, F., Weinmayr, G., Jaensch, A., Nagel, G., Concin, H., and Spadaro, J. V.: Global estimates of mortality associated with long-term exposure to outdoor fine particulate matter, P. Natl. Acad. Sci. USA, 115, 9592–9597, https://doi.org/10.1073/pnas.1803222115, 2018.
Cubison, M. J., Ortega, A. M., Hayes, P. L., Farmer, D. K., Day, D., Lechner, M. J., Brune, W. H., Apel, E., Diskin, G. S., Fisher, J. A., Fuelberg, H. E., Hecobian, A., Knapp, D. J., Mikoviny, T., Riemer, D., Sachse, G. W., Sessions, W., Weber, R. J., Weinheimer, A. J., Wisthaler, A., and Jimenez, J. L.: Effects of aging on organic aerosol from open biomass burning smoke in aircraft and laboratory studies, Atmos. Chem. Phys., 11, 12049–12064, https://doi.org/10.5194/acp-11-12049-2011, 2011.
Daellenbach, K. R., Bozzetti, C., Křepelová, A., Canonaco, F., Wolf, R., Zotter, P., Fermo, P., Crippa, M., Slowik, J. G., Sosedova, Y., Zhang, Y., Huang, R.-J., Poulain, L., Szidat, S., Baltensperger, U., El Haddad, I., and Prévôt, A. S. H.: Characterization and source apportionment of organic aerosol using offline aerosol mass spectrometry, Atmos. Meas. Tech., 9, 23–39, https://doi.org/10.5194/amt-9-23-2016, 2016.
DeCarlo, P. F., Kimmel, J. R., Trimborn, A., Northway, M. J., Jayne, J. T., Aiken, A. C., Gonin, M., Fuhrer, K., Horvath, T., Docherty, K. S., Worsnop, D. R., and Jimenez, J. L.: Field-Deployable, High-Resolution, Time-of-Flight Aerosol Mass Spectrometer, Anal. Chem., 78, 8281–8289, https://doi.org/10.1021/ac061249n, 2006.
Dennison, P. E., Charoensiri, K., Roberts, D. A., Peterson, S. H., and Green, R. O.: Wildfire temperature and land cover modeling using hyperspectral data, Remote Sens. Environ., 100, 212–222, 2006.
Fine, P. M., Cass, G. R., and Simoneit, B. R. T.: Organic compounds in biomass smoke from residential wood combustion: Emissions characterization at a continental scale, J. Geophys. Res.-Atmos., 107, ICC 11-1–ICC 11-9, https://doi.org/10.1029/2001JD000661, 2002.
Fortenberry, C. F., Walker, M. J., Zhang, Y., Mitroo, D., Brune, W. H., and Williams, B. J.: Bulk and molecular-level characterization of laboratory-aged biomass burning organic aerosol from oak leaf and heartwood fuels, Atmos. Chem. Phys., 18, 2199–2224, https://doi.org/10.5194/acp-18-2199-2018, 2018.
Gallimore, P. J., Griffiths, P. T., Pope, F. D., Reid, J. P., and Kalberer, M.: Comprehensive modeling study of ozonolysis of oleic acid aerosol based on real-time, online measurements of aerosol composition, J. Geophys. Res.-Atmos., 122, 4364–4377, https://doi.org/10.1002/2016JD026221, 2017.
Gerrebos, N. G. A., Zaks, J., Gregson, F. K. A., Walton-Raaby, M., Meeres, H., Zigg, I., Zandberg, W. F., and Bertram, A. K.: High Viscosity and Two Phases Observed over a Range of Relative Humidities in Biomass Burning Organic Aerosol from Canadian Wildfires, Environ. Sci. Technol., 58, 21716–21728, https://doi.org/10.1021/acs.est.4c09148, 2024.
Gregson, F. K. A., Gerrebos, N. G. A., Schervish, M., Nikkho, S., Schnitzler, E. G., Schwartz, C., Carlsten, C., Abbatt, J. P. D., Kamal, S., Shiraiwa, M., and Bertram, A. K.: Phase Behavior and Viscosity in Biomass Burning Organic Aerosol and Climatic Impacts, Environ. Sci. Technol., 57, 14548–14557, https://doi.org/10.1021/acs.est.3c03231, 2023.
Hayden, K. L., Li, S.-M., Liggio, J., Wheeler, M. J., Wentzell, J. J. B., Leithead, A., Brickell, P., Mittermeier, R. L., Oldham, Z., Mihele, C. M., Staebler, R. M., Moussa, S. G., Darlington, A., Wolde, M., Thompson, D., Chen, J., Griffin, D., Eckert, E., Ditto, J. C., He, M., and Gentner, D. R.: Reconciling the total carbon budget for boreal forest wildfire emissions using airborne observations, Atmos. Chem. Phys., 22, 12493–12523, https://doi.org/10.5194/acp-22-12493-2022, 2022.
He, L.-Y., Lin, Y., Huang, X.-F., Guo, S., Xue, L., Su, Q., Hu, M., Luan, S.-J., and Zhang, Y.-H.: Characterization of high-resolution aerosol mass spectra of primary organic aerosol emissions from Chinese cooking and biomass burning, Atmos. Chem. Phys., 10, 11535–11543, https://doi.org/10.5194/acp-10-11535-2010, 2010.
Hems, R. F., Schnitzler, E. G., Liu-Kang, C., Cappa, C. D., and Abbatt, J. P. D.: Aging of Atmospheric Brown Carbon Aerosol, ACS Earth Space Chem., 5, 722–748, https://doi.org/10.1021/acsearthspacechem.0c00346, 2021.
Hennigan, C. J., Sullivan, A. P., Collett Jr., J. L., and Robinson, A. L.: Levoglucosan stability in biomass burning particles exposed to hydroxyl radicals, Geophys. Res. Lett., 37, L09806, https://doi.org/10.1029/2010GL043088, 2010.
Hoffmann, D., Tilgner, A., Iinuma, Y., and Herrmann, H.: Atmospheric Stability of Levoglucosan: A Detailed Laboratory and Modeling Study, Environ. Sci. Technol., 44, 694–699, https://doi.org/10.1021/es902476f, 2010.
Iglesias, V., Balch, J. K., and Travis, W. R.: U. S. fires became larger, more frequent, and more widespread in the 2000s, Science Advances, 8, eabc0020, https://doi.org/10.1126/sciadv.abc0020, 2022.
Iinuma, Y., Brüggemann, E., Gnauk, T., Müller, K., Andreae, M. O., Helas, G., Parmar, R., and Herrmann, H.: Source characterization of biomass burning particles: The combustion of selected European conifers, African hardwood, savanna grass, and German and Indonesian peat, J. Geophys. Res.-Atmos., 112, D08209, https://doi.org/10.1029/2006JD007120, 2007.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Knopf, A., Forrester, S. M., and Slade, J. H.: Heterogeneous oxidation kinetics of organic biomass burning aerosol surrogates by O3, NO2, N2O5, and NO3, Phys. Chem. Chem. Phys., 13, 21050–21062, https://doi.org/10.1039/C1CP22478F, 2011.
Kumar, V., Giannoukos, S., Haslett, S. L., Tong, Y., Singh, A., Bertrand, A., Lee, C. P., Wang, D. S., Bhattu, D., Stefenelli, G., Dave, J. S., Puthussery, J. V., Qi, L., Vats, P., Rai, P., Casotto, R., Satish, R., Mishra, S., Pospisilova, V., Mohr, C., Bell, D. M., Ganguly, D., Verma, V., Rastogi, N., Baltensperger, U., Tripathi, S. N., Prévôt, A. S. H., and Slowik, J. G.: Highly time-resolved chemical speciation and source apportionment of organic aerosol components in Delhi, India, using extractive electrospray ionization mass spectrometry, Atmos. Chem. Phys., 22, 7739–7761, https://doi.org/10.5194/acp-22-7739-2022, 2022.
Lai, C., Liu, Y., Ma, J., Ma, Q., and He, H.: Degradation kinetics of levoglucosan initiated by hydroxyl radical under different environmental conditions, Atmos. Environ., 91, 32–39, https://doi.org/10.1016/j.atmosenv.2014.03.054, 2014.
Lee, T., Sullivan, A. P., Mack, L., Jimenez, J. L., Kreidenweis, S. M., Onasch, T. B., Worsnop, D. R., Malm, W., Wold, C. E., Hao, W. M., and Collett, J. L.: Chemical Smoke Marker Emissions During Flaming and Smoldering Phases of Laboratory Open Burning of Wildland Fuels, Aerosol Sci. Tech., 44, i–v, https://doi.org/10.1080/02786826.2010.499884, 2010.
Lelieveld, J., Evans, J. S., Fnais, M., Giannadaki, D., and Pozzer, A.: The contribution of outdoor air pollution sources to premature mortality on a global scale, Nature, 525, 367–371, https://doi.org/10.1038/nature15371, 2015.
Li, K., Liggio, J., Lee, P., Han, C., Liu, Q., and Li, S.-M.: Secondary organic aerosol formation from α-pinene, alkanes, and oil-sands-related precursors in a new oxidation flow reactor, Atmos. Chem. Phys., 19, 9715–9731, https://doi.org/10.5194/acp-19-9715-2019, 2019.
Liang, Y., Jen, C. N., Weber, R. J., Misztal, P. K., and Goldstein, A. H.: Chemical composition of PM2.5 in October 2017 Northern California wildfire plumes, Atmos. Chem. Phys., 21, 5719–5737, https://doi.org/10.5194/acp-21-5719-2021, 2021.
Liang, Y., Stamatis, C., Fortner, E. C., Wernis, R. A., Van Rooy, P., Majluf, F., Yacovitch, T. I., Daube, C., Herndon, S. C., Kreisberg, N. M., Barsanti, K. C., and Goldstein, A. H.: Emissions of organic compounds from western US wildfires and their near-fire transformations, Atmos. Chem. Phys., 22, 9877–9893, https://doi.org/10.5194/acp-22-9877-2022, 2022.
Liang, Z., Zhou, L., Chang, Y., Qin, Y., and Chan, C. K.: Biomass-burning organic aerosols as a pool of atmospheric reactive triplets to drive multiphase sulfate formation, P. Natl. Acad. Sci. USA, 121, e2416803121, https://doi.org/10.1073/pnas.2416803121, 2024.
Lin, P., Aiona, P. K., Li, Y., Shiraiwa, M., Laskin, J., Nizkorodov, S. A., and Laskin, A.: Molecular Characterization of Brown Carbon in Biomass Burning Aerosol Particles, Environ. Sci. Technol., 50, 11815–11824, https://doi.org/10.1021/acs.est.6b03024, 2016.
Lopez-Hilfiker, F. D., Pospisilova, V., Huang, W., Kalberer, M., Mohr, C., Stefenelli, G., Thornton, J. A., Baltensperger, U., Prevot, A. S. H., and Slowik, J. G.: An extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF) for online measurement of atmospheric aerosol particles, Atmos. Meas. Tech., 12, 4867–4886, https://doi.org/10.5194/amt-12-4867-2019, 2019.
Mofikoya, O. O., Mäkinen, M., and Jänis, J.: Chemical Fingerprinting of Conifer Needle Essential Oils and Solvent Extracts by Ultrahigh-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry, ACS Omega, 5, 10543–10552, https://doi.org/10.1021/acsomega.0c00901, 2020.
Nolte, C. G., Schauer, J. J., Cass, G. R., and Simoneit, B. R.: Highly polar organic compounds present in wood smoke and in the ambient atmosphere, Environ. Sci. Technol., 35, 1912–1919, 2001.
Ortega, A. M., Day, D. A., Cubison, M. J., Brune, W. H., Bon, D., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosol formation and primary organic aerosol oxidation from biomass-burning smoke in a flow reactor during FLAME-3, Atmos. Chem. Phys., 13, 11551–11571, https://doi.org/10.5194/acp-13-11551-2013, 2013.
Platt, S. M., El Haddad, I., Zardini, A. A., Clairotte, M., Astorga, C., Wolf, R., Slowik, J. G., Temime-Roussel, B., Marchand, N., Ježek, I., Drinovec, L., Močnik, G., Möhler, O., Richter, R., Barmet, P., Bianchi, F., Baltensperger, U., and Prévôt, A. S. H.: Secondary organic aerosol formation from gasoline vehicle emissions in a new mobile environmental reaction chamber, Atmos. Chem. Phys., 13, 9141–9158, https://doi.org/10.5194/acp-13-9141-2013, 2013.
Puxbaum, H., Caseiro, A., Sánchez-Ochoa, A., Kasper-Giebl, A., Claeys, M., Gelencsér, A., Legrand, M., Preunkert, S., and Pio, C.: Levoglucosan levels at background sites in Europe for assessing the impact of biomass combustion on the European aerosol background, J. Geophys. Res.-Atmos., 112, D23S05, https://doi.org/10.1029/2006JD008114, 2007.
Qi, L., Chen, M., Stefenelli, G., Pospisilova, V., Tong, Y., Bertrand, A., Hueglin, C., Ge, X., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 2: Biomass burning influences in winter, Atmos. Chem. Phys., 19, 8037–8062, https://doi.org/10.5194/acp-19-8037-2019, 2019.
Reid, J. P., Bertram, A. K., Topping, D. O., Laskin, A., Martin, S. T., Petters, M. D., Pope, F. D., and Rovelli, G.: The viscosity of atmospherically relevant organic particles, Nat. Commun., 9, 956, https://doi.org/10.1038/s41467-018-03027-z, 2018.
Robinson, A. L., Subramanian, R., Donahue, N. M., Bernardo-Bricker, A., and Rogge, W. F.: Source Apportionment of Molecular Markers and Organic Aerosol. 3. Food Cooking Emissions, Environ. Sci. Technol., 40, 7820–7827, https://doi.org/10.1021/es060781p, 2006.
Saarnio, K., Teinilä, K., Saarikoski, S., Carbone, S., Gilardoni, S., Timonen, H., Aurela, M., and Hillamo, R.: Online determination of levoglucosan in ambient aerosols with particle-into-liquid sampler – high-performance anion-exchange chromatography – mass spectrometry (PILS–HPAEC–MS), Atmos. Meas. Tech., 6, 2839–2849, https://doi.org/10.5194/amt-6-2839-2013, 2013.
Schauer, J. J., Kleeman, M. J., Cass, G. R., and Simoneit, B. R. T.: Measurement of Emissions from Air Pollution Sources. 3. C1-C29 Organic Compounds from Fireplace Combustion of Wood, Environ. Sci. Technol., 35, 1716–1728, https://doi.org/10.1021/es001331e, 2001.
Shiraiwa, M., Ammann, M., Koop, T., and Pöschl, U.: Gas uptake and chemical aging of semisolid organic aerosol particles, P. Natl. Acad. Sci. USA, 108, 11003–11008, 2011.
Simoneit, B. R. T., Schauer, J. J., Nolte, C. G., Oros, D. R., Elias, V. O., Fraser, M. P., Rogge, W. F., and Cass, G. R.: Levoglucosan, a tracer for cellulose in biomass burning and atmospheric particles, Atmos. Environ., 33, 173–182, https://doi.org/10.1016/S1352-2310(98)00145-9, 1999.
Stefenelli, G., Pospisilova, V., Lopez-Hilfiker, F. D., Daellenbach, K. R., Hüglin, C., Tong, Y., Baltensperger, U., Prévôt, A. S. H., and Slowik, J. G.: Organic aerosol source apportionment in Zurich using an extractive electrospray ionization time-of-flight mass spectrometer (EESI-TOF-MS) – Part 1: Biogenic influences and day–night chemistry in summer, Atmos. Chem. Phys., 19, 14825–14848, https://doi.org/10.5194/acp-19-14825-2019, 2019.
Vasilakopoulou, C. N., Matrali, A., Skyllakou, K., Georgopoulou, M., Aktypis, A., Florou, K., Kaltsonoudis, C., Siouti, E., Kostenidou, E., Błaziak, A., Nenes, A., Papagiannis, S., Eleftheriadis, K., Patoulias, D., Kioutsioukis, I., and Pandis, S. N.: Rapid transformation of wildfire emissions to harmful background aerosol, npj Clim. Atmos. Sci., 6, 1–9, https://doi.org/10.1038/s41612-023-00544-7, 2023.
Westerling, A. L.: Increasing western US forest wildfire activity: sensitivity to changes in the timing of spring, Philos. T. R. Soc. B, 371, 20150178, https://doi.org/10.1098/rstb.2015.0178, 2016.
Wilkins, J. L., de Foy, B., Thompson, A. M., Peterson, D. A., Hyer, E. J., Graves, C., Fishman, J., and Morris, G. A.: Evaluation of Stratospheric Intrusions and Biomass Burning Plumes on the Vertical Distribution of Tropospheric Ozone Over the Midwestern United States, J. Geophys. Res.-Atmos., 125, e2020JD032454, https://doi.org/10.1029/2020JD032454, 2020.
Xu, W., He, Y., Qiu, Y., Chen, C., Xie, C., Lei, L., Li, Z., Sun, J., Li, J., Fu, P., Wang, Z., Worsnop, D. R., and Sun, Y.: Mass spectral characterization of primary emissions and implications in source apportionment of organic aerosol, Atmos. Meas. Tech., 13, 3205–3219, https://doi.org/10.5194/amt-13-3205-2020, 2020.
Yazdani, A., Dudani, N., Takahama, S., Bertrand, A., Prévôt, A. S. H., El Haddad, I., and Dillner, A. M.: Characterization of primary and aged wood burning and coal combustion organic aerosols in an environmental chamber and its implications for atmospheric aerosols, Atmos. Chem. Phys., 21, 10273–10293, https://doi.org/10.5194/acp-21-10273-2021, 2021.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Ziemann, P. J.: Aerosol products, mechanisms, and kinetics of heterogeneous reactions of ozone with oleic acid in pure and mixed particles, Faraday Discuss., 130, 469–490, https://doi.org/10.1039/B417502F, 2005.
Zelenyuk, A., G. Imre, D., Wilson, J., M. Bell, D., J. Suski, K., Shrivastava, M., Beránek, J., Lizabeth Alexander, M., L. Kramer, A., and Simonich, S. L. M.: The effect of gas-phase polycyclic aromatic hydrocarbons on the formation and properties of biogenic secondary organic aerosol particles, Faraday Discuss., 200, 143–164, https://doi.org/10.1039/C7FD00032D, 2017.
Zhang, J., Li, K., Wang, T., Gammelsæter, E., Cheung, R. K. Y., Surdu, M., Bogler, S., Bhattu, D., Wang, D. S., Cui, T., Qi, L., Lamkaddam, H., El Haddad, I., Slowik, J. G., Prevot, A. S. H., and Bell, D. M.: Bulk and molecular-level composition of primary organic aerosol from wood, straw, cow dung, and plastic burning, Atmos. Chem. Phys., 23, 14561–14576, https://doi.org/10.5194/acp-23-14561-2023, 2023.