the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Jul 2024
| 12 Jul 2024
Secondary reactions of aromatics-derived oxygenated organic molecules lead to plentiful highly oxygenated organic molecules within an intraday OH exposure
Yuwei Wang
Chuang Li
Ying Zhang
Yueyang Li
Gan Yang
Xueyan Yang
Yizhen Wu
Hefeng Zhang
Highly oxygenated organic molecules (HOMs) can participate in new particle formation (NPF) and enhance growth of newly formed particles partially because of their low volatility. Previous studies have shown formation of HOMs via autoxidation reactions of RO2 intermediates generated by OH-initiated oxidation of anthropogenic volatile organic compounds (VOCs). It was also suggested that multi-generation OH oxidation could be an important source for aromatics-derived HOMs. However, our understanding of the generation of aromatics-derived HOMs is still insufficient, especially of their formation mechanisms, which determine molar yields of HOMs and are essential to the establishment of global chemical box models related to HOMs. In this study, with a potential aerosol mass oxidation flow reactor (PAM OFR), two series of OH-initiated oxidation experiments of 1,3,5-trimethylbenzene (1,3,5-TMB) were conducted to investigate the formation of aromatics-derived HOMs. In the first series, the evolution of oxidation products of 1,3,5-TMB in an OH exposure range of (0.5–5.0) × 1010 molecules cm−3 s, equivalent to an OH exposure of 0.7–6.9 h at an OH concentration ([OH]) of 2×106 molecules cm−3, was investigated by a nitrate-based chemical ionization mass spectrometer and a Vocus proton-transfer-reaction mass spectrometer, indicating significant secondary OH chemistry during the aging of stabilized first-generation oxygenated products within an intraday OH exposure and formation of various HOMs with lower double-bond equivalence (DBE). In addition, organonitrates, formed after the introduction of NOx into the reaction systems, further confirmed the existence of such secondary reactions. The second series of experiments was conducted with same residence time but much lower [OH], which also shows the generation of multi-generation HOMs with an [OH] as low as 1.06×107 molecules cm−3 for 53 s, i.e., an OH exposure of around 5.86×108 molecules cm−3 s. Our study suggests the important role of secondary OH chemistry in the oxidation of aromatics if these oxygenated products survived long enough in the ambient atmosphere and elucidates detailed formation mechanisms of certain HOM products.
- Article
(3304 KB) - Full-text XML
-
Supplement
(875 KB) - BibTeX
- EndNote





OH radicals can react with volatile organic compounds (VOCs) in the atmosphere, converting primary pollutants to secondary ones. Generated from oxidation of VOCs, oxygenated organic molecules (OOMs) are crucial in a variety of atmospheric chemical processes, contributing efficiently to the formation of secondary organic aerosols (SOAs) and ground-level O3 (Ng et al., 2010; Wang et al., 2022; Qu et al., 2021). Among the enormous number of oxygenated VOCs (OVOCs), highly oxygenated organic molecules (HOMs) have recently attracted significant attention (Bianchi et al., 2019). Most HOMs are low-volatility organic compounds (LVOCs) or extremely low volatility organic compounds (ELVOCs) and thus are able to drive the initial formation of nucleated particles under certain conditions and contribute to the subsequent growth of newly formed particles, which finally enhance SOA formation (Tröstl et al., 2016; Lehtipalo et al., 2018; Stolzenburg et al., 2018; Mohr et al., 2019; Qiao et al., 2021).
Formation of HOMs is triggered by oxidation of VOCs in the gas phase. Peroxy radicals (RO2) are generated in the initial step and will undergo an intramolecular hydrogen atom shift, forming a hydroperoxide functionality and an alkyl radical. A molecular oxygen will rapidly attach to this alkyl radical and form a new and more oxidized RO2. This reaction is called autoxidation, and the newly formed RO2 can go through another autoxidation or bimolecular termination reaction to form a stabilized product (Crounse et al., 2013). Autoxidation is suggested to be responsible for widely detected HOMs in the atmosphere because it can form highly oxygenated RO2 on a short timescale. In terms of biomolecular reactions, RO2 only reacts appreciably with the hydroperoxyl radical (HO2), NO, and another RO2. The RO2 reaction chain in polluted areas is largely terminated by NO, which prohibits generation of compounds with high oxidation levels and reduces yields of HOMs (Bianchi et al., 2019).
Nevertheless, autoxidation reactions alone are not enough to explain the large numbers of oxygen atoms and low double-bond equivalence (DBE; calculated as , where nC, nH, and nN stand for the number of carbon, hydrogen, and nitrogen atoms, respectively, in a molecule) for HOMs observed in laboratory experiments and ambient campaigns. Take alkylbenzenes as an example: previous studies suggest that the main products of OH-initiated oxidation of alkylbenzenes (CxH2x−6, x=7, 8, or 9), i.e., bicyclic peroxy radicals (BPRs; CxH2x−5O5•, x=7, 8, or 9) (Jenkin et al., 2003), can undergo an autoxidation reaction and form a new RO2 (CxH2x−5O7•, x=7, 8, or 9) (Wang et al., 2017). Autoxidation of BPRs could be very fast if it has a favorable structure, as found in a previous study (Wang et al., 2017). On the other hand, the structure of the resulting CxH2x−5O7• is strongly different to that of BPRs, whose autoxidation reaction rate can be as low as the order of 0.001 s−1, since it lacks enhancements from favorable transition state geometries and substitutes or resonance structures (Bianchi et al., 2019; Otkjær et al., 2018). Such a slow autoxidation reaction rate cannot explain the extensive existence of HOM monomers with more than 7 oxygen atoms and HOM dimers with more than 10 oxygen atoms, which are the maximum numbers of oxygen atoms in stabilized first generation monomer and dimer products, respectively, formed from CxH2x−5O7• (Molteni et al., 2018; Y. Wang et al., 2020; Mentel et al., 2015; Berndt et al., 2018b). Another possibility is the formation of a second oxygen bridge after the hydrogen shift of BPRs (Molteni et al., 2018), but this reaction pathway would not allow a further oxygenation reaction without a breakage of the carbon ring, which is also unpromising. A very recent investigation offers new insights into the formation mechanism of these products, indicating that the molecular rearrangement of BPRs can initiate a series of autoxidation, but its applicability to aromatics containing more than 9 carbon atoms remains to be demonstrated (Iyer et al., 2023). Meanwhile, the formation mechanism of HOMs with a large hydrogen atom number, i.e., low DBE, is still vague. For example, monomer products with 16 hydrogen atoms in the OH-initiated oxidation of TMB and those with 14 hydrogen atoms in the OH-initiated oxidation of xylene were observed in the laboratory, both with a DBE of 2 lower than their precursors' (Molteni et al., 2018), but their formation mechanisms cannot be explained by any known mechanisms with only one OH attack.
Multi-generation reactions of VOCs complicate HOMs' formation. Previous studies indicate that HOMs can also be formed by sequential oxidation of stabilized first-generation products of benzene and toluene (Garmash et al., 2020; Cheng et al., 2021). Garmash et al. (2020) conducted OH oxidation experiments of benzene and toluene with an OH exposure equivalent to atmospheric oxidation times of 10 h–15 d at OH concentrations of ∼ 106 molecules cm−3. Cheng et al. (2021) simulated oxidation of benzene and toluene with an OH exposure equivalent to 2.4–19.4 d of atmospheric photochemical aging. Certainly, such extremely high OH exposures favor secondary OH chemistry and help to facilitate our understanding of product distributions, but such a long timescale limits the atmospheric implications of their results, given the complex physical and chemical processes that occur at night.
Compared to benzene and toluene, trimethylbenzene (TMB) is a compound characterized by much larger HOM molar yields when reacted with OH, and the abundance of TMB in the atmosphere cannot be ignored (Molteni et al., 2018; Yuan et al., 2012). Previous laboratory experiments on TMB-derived HOMs mainly focused on the autoxidation reactions of BPRs and the influences of NOx, and the quantity of experiments was very finite, restricting the applicability of their conclusions to atmospherically relevant conditions (Tsiligiannis et al., 2019; Y. Wang et al., 2020). From the mechanism perspective, a number of HOM monomers with more than 7 oxygen atoms detected in the OH-initiated oxidation of TMB were previously assumed to be generated via multiple autoxidation reactions (Molteni et al., 2018). Nevertheless, a subsequent OH oxidation of the first-generation oxygenated products might be more plausible for the formation of HOM monomers with more than 7 oxygen atoms from the present point of view. Indeed, laboratory experiments showed that RO2 formed during the second-generation OH oxidation of the stabilized first-generation oxidation products can also undergo autoxidation reactions, which entangles reaction mechanisms potentially involved in the formation of those HOMs and justifies more investigations on the multi-generation OH oxidation of aromatics (Y. Wang et al., 2020). Atmospheric OH concentration ([OH]) up to 6×106–2.6×107 molecule cm−3, which is several times higher than the typical average atmospheric [OH], 1.2×106 molecule cm−3 (Jacob, 1999), has been frequently observed in both urban and suburban environments in China (Tan et al., 2019; Lu et al., 2012), leading to a realistic implication of multi-generation OH oxidation. Therefore, it is imperative to study chemical characteristics of aromatics-derived HOMs at different OH exposures, especially those that are less than or equivalent to 1 d of atmospheric oxidation.
In this study, two series of laboratory experiments on OH-initiated oxidation of 1,3,5-TMB, selected as an example of anthropogenic VOCs, were conducted. One was conducted with [OH] ranging from 9.32×107 to 1.03×109 molecule cm−3, corresponding to an OH exposure equivalent to atmospheric oxidation times of roughly 0.7–6.9 h at an average daytime [OH] of 2.0×106 molecules cm−3. A nitrate-based chemical ionization mass spectrometer (nitrate CIMS) and a Vocus proton-transfer-reaction mass spectrometer (Vocus PTR) were deployed to measure the oxidation products and the precursor, respectively. We explored the evolution of oxidation products to investigate the secondary OH chemistry of stabilized first-generation oxidation products generated by the oxidation of 1,3,5-TMB. Furthermore, the influence of NO on the formation of HOMs was investigated by introducing N2O into the reaction system. In addition, another series of experiments under atmospherically relevant [OH] was conducted to confirm the applicability of the above-developed multi-generation OH oxidation mechanisms in the ambient atmosphere.
OH-initiated oxidation of 1,3,5-TMB was investigated in a potential aerosol mass oxidation flow reactor (PAM OFR) system at K and a pressure of 1 atm (Lambe et al., 2015). Two series of experiments were conducted, one under high-[OH] conditions and the other under low-[OH] conditions. Hereafter, we refer to the series of high-[OH] experiments as the “first-round” experiments and the low-[OH] ones as the “second-round” experiments, respectively. The ith experiment in the first-round experiments is labeled 1-i and the one in the second-round experiments as 2-i, where i stands for its serial number. The experimental settings in this study differed slightly from what were used previously (Y. Wang et al., 2020). In the first-round experiments, 40 OH experiments without NOx (Exp. 1-1–1-40) and 28 experiments with NOx (Exp. 1-41–1-68) were performed. A total of 7 experiments were conducted in the second round, 4 without NOx (Exp. 2-1–2-4) and 3 with NOx (Exp. 2-5–2-7). The experimental conditions are summarized in Table S1 in the Supplement, including concentrations of the precursor, ozone, and NO and NO2. The equivalent OH exposure in the OFR for each experiment was estimated according to the precursor consumption, and also listed in Table S1. OH exposures in the OFR were in the range of (5.2–48.7) ×109 and (0.6–5.5) ×109 molecules cm−3 s in the first-round and second-round experiments, respectively.
A homemade 1,3,5-TMB–N2 cylinder was used as a stable gaseous precursor source in the experiments, from which the flow rate of 1,3,5-TMB–N2 varied between 1–3 sccm (standard cubic centimeters per minute), leading to 7.08×1011–1.54×1012 molecule cm−3 of 1,3,5-TMB in the first-round experiments and 7.55×1011 or 8.45×1011 molecule cm−3 of 1,3,5-TMB in the second-round experiments, respectively (Table S1). A total flow of 15 slpm (standard liters per minute) zero gas generated by a zero-gas generator (model 737-13, Aadco Instruments Inc.), together with the 1,3,5-TMB–N2 flow, was introduced into the OFR. The reaction time in both series of experiments was kept at around 53 s, and the flow reactor was kept as a plug flow one in both series. The flow in the PAM OFR is laminar with very low axial mixing, as characterized by a Taylor dispersion model in a previous study (Lambe et al., 2011). Out of the 15 slpm zero gas, 6 slpm was initially passed through a Nafion humidifier (Perma Pure Model FC100-80-6MSS) filled with ultra-pure water and finally converged with the main flow into the OFR to achieve and keep a desired RH of 20.0±2.5 % in the OFR throughout all the experiments, and 2 slpm was initially passed through a separate ozone chamber, resulting in an initial ozone concentration of around 1.05×1013–2.16×1013 molecule cm−3 in the OFR in the first-round experiments and 3.01×1012–3.72×1012 molecule cm−3 in the second-round experiments, respectively. The OFR was operated with only the 254 nm lights on, under which the primary oxidant production reactions in the OFR were O3+hv (254 nm) and . After turning on of UV lights, a HOM compound is believed to be generated if its signal is more than 3 standard deviations of its background signal. If the fluctuations in the 1 min averaged signals of both TMB in the Vocus PTR and typical HOMs (i.e., C9H14O7 (NO3)−) in the nitrate CIMS are within 2 % during a 10 min period, a steady state was assumed to be reached. It usually took around no more than 2 min for the signals of HOMs to stabilize after the adjustment of UV lights. We typically monitored the reaction products for around 20 min for each experiment. An ozone monitor (Model 106-M, 2B technologies) and a trace-gas analyzer for NO-NO2-NOx (Thermo, 42i-TL) were placed at the exit of the OFR to measure concentrations of ozone and NOx, respectively.
Non-tropospheric VOC and OVOC photolysis is a typical issue that should be taken into account when evaluating the OFR settings, especially under the high UV light dose settings in the first-round experiments. Our evaluation on photolysis of the precursor and HOMs shows that photolysis was not a contributor to our observation on C9 and C18 HOM formation. The photolysis rate of 1,3,5-TMB can be estimated based on the absorption cross-sections of 1,3,5-TMB at 254 nm (Keller-Rudek et al., 2013) and UV photon fluxes estimated by a chemistry model discussed in the following sections. The ratio of photolysis-to-OH reaction for 1,3,5-TMB in our first-round experiments was merely 0.010–0.033. Hence, photolysis of 1,3,5-TMB was insignificant in the OFR. For stabilized products such as C9 and C18 HOMs, the cross sections of organic molecules are usually – cm2 (Peng et al., 2016), while the reaction rate between OH and the stabilized first-generation products is estimated to be around molecule−1 cm3 s−1, as suggested by the Master Chemical Mechanism (MCM) (Jenkin et al., 2003). Hence, the ratio of photolysis rates of C9 and C18 HOMs to their secondary OH oxidation rates is estimated to be around 0.020–0.056 in the first-round experiments. In the second round, the influences of photolysis should be even lower due to the much lower light intensity.
For experiments with NOx in the first-round experiments, 350 sccm N2O (99.999 %, Air Liquid) was added into the OFR to produce and sustain NOx mixing ratios at levels that were sufficiently high to be a competitive sink for RO2 radicals. NO and NO2 were produced via the reaction , followed by the reaction . Two sets of irradiance intensities were chosen for NOx experiments, generally resulting in two NOx levels, 4.41×1010 molecule cm−3 NO + 1.72×1012 molecule cm−3 NO2 (Exp. 1-41–1-54) and 1.18×1011 molecule cm−3 NO + 2.94×1012 molecule cm−3 NO2 (Exp. 1-55–1-68) at the exit of the OFR. With the aim to slightly modify OH exposure but keep NOx concentrations constant among each set of experiments, the initial concentrations of 1,3,5-TMB were adjusted in a large range from 4.09×1011 to 2.06×1012 molecule cm−3, while RH and irradiances were not changed, as an increase in the precursor concentration corresponds to a larger sink for OH. In the second-round experiments, due to the lower O(1D) in the PAM OFR, 2.5 slpm pure N2O was utilized instead, whereas the total flow rate was kept the same as that in the first round. We lowered the light intensity to obtain a lower [OH] in the PAM OFR, which also resulted in fluctuations in the NO concentrations ([NO]) from 3.19×1010 to 1.74×1011 molecule cm−3 and the NO2 concentrations ([NO2]) from 2.70×1011 to 9.31×1011 molecule cm−3.
A nitrate CIMS (Ehn et al., 2014; Eisele and Tanner, 1993) and a Vocus PTR (Krechmer et al., 2018) were deployed at the exit of the OFR to measure the oxidation products of 1,3,5-TMB in the first-round experiments. These two mass spectrometers have been well characterized in a previous study (Y. Wang et al., 2020).
The sample flow rate for the nitrate CIMS in the first-round experiments was 8 slpm through a Teflon tube with an outer diameter (OD) of in. and a length of 70 cm. The sheath flow for the nitrate CIMS was supplied by a zero-gas generator at a flow rate of 15 slpm. Mass resolution was approximately 8000 for ions with larger than 200 Th. HOMs generated from TMB oxidation were charged in the ambient pressure interface region by collisions with nitrate clusters, (HNO3)x⋅ NO (x=0–2), and detected by the nitrate CIMS as clusters with NO, i.e., HOM • NO (Hyttinen et al., 2015). In addition, HOMs' signals were corrected with relative transmission efficiencies of our nitrate CIMS (Heinritzi et al., 2016). We followed the same sampling method of the PAM OFR as those in previous studies, in order to obtain a similar flow tube residence time distributions (RTDs) and thus validate usage of a modified PAM_chem_v8 model to estimate concentrations of radicals in the OFR as discussed below.
The Vocus PTR was applied to quantify precursor concentrations. The focusing ion–molecule reactor (FIMR) was heated up, and its temperature was maintained at 100 °C during the experiments. The FIMR can be operated under 2.0 mbar without a strong interference from corresponding water clusters when ionizing the neutral compounds. The Vocus front and back voltages were 650 and 15 V, respectively, forming an axial voltage of 635V and a reduced electrical field (, where E is the electric field strength and N is the number density of the buffer gas in FIMR) of 180 Td. The radio frequency (RF) voltages and frequency were set to be 450 V and 1.3 MHz, respectively. The sample flow was introduced to the Vocus PTR through a Teflon tube with an OD of 1/4 in. and a length of 120 cm from the OFR. A total sample flow of 1.4 slpm was maintained by a pump with an orifice to minimize the delay time of sampling, from which approximately 125 sccm was sampled into the FIMR through a capillary tube.
In the second-round experiments, a Vocus CI-TOF (Towerk AG, Switzerland) equipped with a Vocus Aim inlet and the same nitrate-ion chemical ionization source as adopted in the first-round experiments were utilized to measure oxidation products, hereafter referred as nitrate CI-TOF. The nitrate CI-TOF was characterized by a flat transmission efficiency between 60 and 500 Th, as well as a mass resolution of 10 000 at 200 Th. In this series of experiments, the reaction products were sampled from the PAM OFR via a 30 cm-long Teflon tube with a 1/2 in. OD to our nitrate CI-TOF. The Vocus PTR and the ozone monitor were connected to the PAM OFR from a separate port via a 120 cm-long Teflon tube with a 1/4 in. OD.
We did not quantify HOMs' concentrations. Since the inner diameters of the PAM OFR, the sampling tube, and the nitrate CIMS inlet were different, and two reducing unions were used during sampling, the estimation of the penetration efficiency and the sampling efficiency of HOMs are thus of a significant uncertainty. The initial concentrations of TMB utilized in both sets of experiments fluctuated slightly, which resulted from sample preparation processes and were more obvious in the first-round experiments. Therefore, in the discussion on the data of the first-round experiments, we tried to minimize potential influences of the differences in the initial TMB concentrations on the signals of HOMs by normalizing the HOMs' signals with the initial TMB signal. To precisely illustrate changes in the abundance of HOMs at different OH exposures, a normalized signal was chosen to present the abundance of detected HOMs, which is defined as the ratio of the signals of HOMs in the nitrate CIMS normalized by the reagent ions and the initial signal of 1,3,5-TMB, i.e., . S(HOMs) is the signal of HOM detected by the nitrate CIMS normalized with the signal of reagent ions, whereas S(TMB) is the initial signal of 1,3,5-TMB detected by the Vocus PTR.
To compare chemical regimes of two series of experiments and the ambient atmosphere, a PAM chemistry model (PAM_chem_v8), utilized widely in previous studies, was chosen with the latest updates to calculate radical profiles in our OFR (Li et al., 2015; Cheng et al., 2021; Y. Wang et al., 2020; Mehra et al., 2020; Lambe et al., 2015, 2017, 2019; Peng and Jimenez, 2020). This model is based on a photochemical box model that includes chemistry of photolysis of oxygen, water vapor, and other trace gases by the primary wavelengths of mercury lamps and simplified VOC and RO2 chemistry, but further reactions of the first-generation stabilized products and the second-generation organic radicals are not considered. The reactions and corresponding kinetics utilized in this model are summarized in Table S2. In this work, autoxidation and accretion of 1,3,5-TMB-derived BPRs, as well as subsequent reactions of the autoxidation product of BPRs, i.e., C9H13O7•, are newly implemented or modified in this model (Reactions R46–62 in Table S2). These two radicals were the most significant RO2 in the system and represented the whole RO2 pool in the PAM chemistry model simulation. The pathways of peroxy radicals and their kinetics are discussed below. NOx-related reactions are also included in the model. When experiments without NOx are simulated, these NOx-related reactions do not contribute to the simulation results.
The detailed reactions involved with RO2 include
Reactions (R1), (R2), and (R3) are reactions of RO2 + RO2, forming alkoxy radicals, carbonyl termination products, and hydroxyl termination products, respectively. Reaction (R4) is the accretion reaction, forming dimers via the combination of two monomeric RO2. Reaction (R5) is the reaction between RO2 and HO2, forming hydroperoxyl radicals. The reaction rate constants for RO2 in Reactions (R1)–(R5) are obtained by MCM or previous investigations (e.g., Jenkin et al., 2003; Berndt et al., 2018b; Peng and Jimenez, 2020). We treat Reactions (R1)–(R3) as a total reaction with a reaction rate constant of molecule−1 cm3 s−1 and branching ratios of Reactions (R1)–(R3) of 0.6, 0.2, and 0.2, respectively, as suggested by the MCM (Jenkin et al., 2003). The reaction rate constants of BPRs and C9H13O7• for Reaction (R4) are and molecule−1 cm3 s−1, respectively (Berndt et al., 2018b). The reaction rate constant for Reaction (R5) is molecule−1 cm3 s−1 (Jenkin et al., 2003).
Reaction (R6) is the reaction between OH and RO2, whose reaction rate constant is molecule−1 cm3 s−1 according to previous studies (Bossolasco et al., 2014; Yan et al., 2016; Assaf et al., 2016, 2017; Peng and Jimenez, 2020). Current knowledge on the reaction products for the reaction of CH3O2• + OH, the most studied RO2+ OH reaction, is summarized in Table S3. The products of this reaction are suggested to include a Criegee intermediate (CH2O2•), a stabilized methylhydrotrioxide (CH3OOOH), an alkoxy radical (CH3O•), and methanol (CH3OH) (Yan et al., 2016; Fittschen, 2019; Caravan et al., 2018; Müller et al., 2016). Müller et al. (2016) and Caravan et al. (2018) suggested that the formation of CH2O2• is actually infeasible, and Yan et al. (2016) estimated an upper limit branching ratio of 5 % for this pathway. The branching ratios of stabilized products CH3OH and CH3OOOH are 6 %–7 % (Caravan et al., 2018; Müller et al., 2016) and 7 % (Müller et al., 2016), respectively. The most significant product of this reaction is the alkoxy radical (CH3O•), with a branching ratio of more than 86 % (Müller et al., 2016). In the absence of NOx, CH3OH and CH3O• can also be formed via the traditional unimolecular reaction between CH3O2• and RO2, i.e., Reactions (R1) and (R3). The possible role of this reaction of large RO2, i.e., BPRs and other C9-RO2, with OH has not yet been investigated. However, according to the branching ratios for the reaction of CH3O2• + OH, this reaction is likely to form RO instead of stabilized C9 products. Hence, we assume that the branching ratios of hydrotrioxide (ROOOH), RO, and ROH are 0.07, 0.86, and 0.07, respectively, for BPR + OH and C9-RO2+ OH.
Reaction (R7) is the unimolecular reactions of RO2 in the PAM OFR. RO2 isomerization rate coefficients are highly dependent on their structures, spanning from 10−3–106 s−1 (Bianchi et al., 2019; Crounse et al., 2013; Knap and Jørgensen, 2017; Praske et al., 2018). However, only some substituted acyl RO2 can undergo rapid isomerization at a reaction rate of 106 s−1 (Knap and Jørgensen, 2017). The 1,3,5-TMB-derived BPR and its autoxidation product, C9H13O7•, do not belong to this group of substituted acyl RO2 (Molteni et al., 2018; Tsiligiannis et al., 2019). The most important unimolecular reaction for the 1,3,5-TMB-derived BPR is likely autoxidation, while the precise autoxidation reaction rates of the 1,3,5-TMB-derived BPR and other RO2 in this system are currently unclear (Bianchi et al., 2019; Molteni et al., 2018). Previous theoretical investigations suggest that more than 90 % BPRs generated by the oxidation of 1,3,5-TMB possess a structure favoring autoxidation, and thus their overall autoxidation reaction rate is relatively fast (Wang et al., 2017). We follow quantum calculation results on the autoxidation reaction of a methyl group adjacent to the RO2 functionality group (Wang et al., 2017) and multiply the suggested rate (0.026 s−1) by 3 due to the symmetry with three methyl groups in our parent compound. The obtained autoxidation reaction rate is 0.078 s−1.
Reactions (R8) and (R9) are the reactions between NO and RO2, generating alkoxy radicals and organonitrates, respectively. The reaction rate for the sum of these two reactions is molecule−1 cm3 s−1. The branching ratios of these two reactions are 0.843 and 0.157, respectively, according to MCM (Jenkin et al., 2003).
Alkoxy radicals, RO, will be generated in Reactions (R1), (R6), and (R8). The widely used near-explicit mechanism, MCM, assumes that RO formed via the alkoxy channel of BPRs (Reaction R1) will decompose into small molecules. Recently, Xu et al. (2020) probed the chemical fates of BPR-derived RO, hereafter referred to as bicyclic alkoxy radical (BCP-oxy), in the oxidation of benzene by laboratory experiments and model calculations, which can be taken as a reference to induce the mechanism of 1,3,5-TMB-derived BCP-oxy. BCP-oxy can undergo two reactions, i.e., ring breakage and ring closure, and a new calculation result suggests that the branching ratio of ring-breakage reaction is larger than 98 % (Wang et al., 2013). 56 % of ring-breakage reactions will break benzene-derived BCP-oxy into butenedial and glyoxal, and the remaining 44 % will generate a C6 alkyl radical by a 1,5-aldehydic H shift. The latter C6 alkyl radical will further undergo other reactions, including a 93 % branching ratio for decomposition reactions that results in a reduction of carbon atom number (Xu et al., 2020). Therefore, most of the benzene-derived BCP-oxy will likely decompose into compounds with fewer carbon atoms. We assume that 1,3,5-TMB-derived BCP-oxy will undertake these decomposition reactions with a similar branching ratio, which means that these radicals cannot form a large number of stabilized products that can influence the distributions of stabilized C9 products in the nitrate CIMS.
Reaction (R10) is the physical loss of RO2. The physical loss of RO2 in the PAM OFR consists of the condensation loss to the aerosol particles and the diffusion loss to the OFR walls. In our experiments, measurement results by a long-SMPS show that the aerosol particles presented in the PAM OFR were few. The long SMPS consisted of a long-DMA (TSI model 3081) and a CPC (TSI model 3787), covering a particle number size distribution from 13.6 to 736.5 nm. Thus, though not detected in this study, we cannot absolutely deny the possibility that particles might have been generated, resulting in a larger physical loss of HOMs. This part of physical loss might be underestimated. The first-order loss rate of HOMs to the OFR walls, kwall, is limited by eddy diffusion and can be calculated with the following function (Cheng et al., 2021; Palm et al., 2016; McMurry and Grosjean, 1985):
where the OFR surface-area-volume ratio () is 25 m−1, and the coefficient of eddy diffusion (ke) is 0.0042 s−1, as estimated by the method utilized in a previous study (Brune, 2019) and given in Eq. (2):
where V is the enclosure volume (m3). The molecular diffusion coefficient, Dg, is estimated with the method as described by Fuller et al. (1966) and is around m2 s−1, with the 1,3,5-TMB-derived BPR as an example. Hence, kwall is around 0.0023 s−1 in the PAM OFR.
Other kinetic data in the modified PAM_chem_v8 model are obtained from the IUPAC (International Union of Pure and Applied Chemistry) dataset (https://iupac-aeris.ipsl.fr, last access: 26 October 2023) and the MCM dataset (MCM v3.3.1, https://mcm.york.ac.uk/MCM/, last access: 9 October 2023).
For the first-round experiments, the input parameters of temperature, mean residence time, water vapor concentration, O3 concentration, and the initial 1,3,5-TMB concentration are 25°, 53 s, 0.63 %, 1.23×1013, and 1.23×1012 molecule cm−3, respectively, as measured directly. For the second-round experiments, the input parameters of O3 concentration and initial 1,3,5-TMB concentration were updated as 3.68×1012 and 7.55×1011 molecule cm−3, respectively. In the NOx experiments, the input flow rate of N2O is 350 sccm in the first-round experiments and 2.5 slpm in the second-round experiments, respectively. The actinic flux at 254 nm, I254, is constrained by comparing OH exposures by model output and OH exposures estimated by the consumption of 1,3,5-TMB as measured by the Vocus PTR. Consumption of O3 estimated by the model agrees well with the measured results, with discrepancies being always within 10 % at different OH exposures.
3.1 Comparison of chemical regimes
Concentration profiles of OH, RO2, and HO2 as a function of OH exposures in our high-[OH] experiments without NOx, i.e., the first-round experiments, are illustrated in Fig. S1a in the Supplement. According to the modified PAM_chem_v8 model, when [OH] increased from 9.32×107 to 1.03×109 molecule cm−3, [HO2] increased from 7.25×108 to 2.79×109 molecule cm−3, whereas [RO2] concentrations increased from 5.17×109 to 9.5×109 molecule cm−3. The radical concentrations in high-[OH] experiments with NOx (Fig. S1b) varied in a similar range, with [RO2] ranging from 4.38×109 to 9.13×109 molecule cm−3, HO2 ranging from 4.47×109 to 6.47×109 molecule cm−3, and OH ranging from 3.86×108 to 7.82×108 molecule cm−3, respectively. The ratios of between and in the first-round experiments were generally on the same order of magnitude as those in the ambient atmosphere (Whalley et al., 2021).
Radical concentrations were also estimated by the PAM_chem_v8 model to illustrate the chemical regimes in the second-round experiments (Table S4). The average [HO2], [OH], and [RO2] were 9.7×107, 1.64×107, and 1.69×109 molecule cm−3, respectively, in Exp. 2-3 and were 6.7×107, 1.04×107, and 1.34×109 molecule cm−3, respectively, in Exp. 2-4, both of which generally differ by no more than a factor of 3 from the summer daytime ambient ones in polluted atmospheres (Tan et al., 2017, 2018, 2019; Whalley et al., 2021; Lu et al., 2012). The average [HO2], [OH], and [RO2], as well as the NO and NO2 concentrations in Exp. 2-7, are generally very close to those in the same environment (Tan et al., 2019).
We take Exp. 1-12 ([OH] = ∼ 8.47×108 molecule cm−3 and NOx=0) and Exp. 2-3 ([OH] = ∼ 1.64×107 molecule cm−3 and NOx=0) as representative examples and compare simulation results with those from the ambient atmosphere, since NOx in the ambient is believed not to impact relative ratios for Reactions (R1)–(R3), (R5), and (R6). In the ambient atmosphere, the average [HO2], [OH], and [RO2] were 2.7×108, 8.0×106, and 1.4×109 molecule cm−3, respectively, at around noon in the summertime in urban Beijing (Whalley et al. 2021), and (4–28) ×108, (0.8–2.4) ×107, and 1.2×109 molecule cm−3 (modeled) at a suburban site in the Yangtze River Delta (Ma et al., 2022). As shown in Fig. 1a, for the most important RO2, the BPR, the fractions of monomeric termination reactions of RO2+ RO2 (Reactions R1–R3), RO2+ HO2 (Reaction R5), and RO2+ OH (Reaction R6) were 6.2 %, 29.3 %, and 64.5 %, respectively, in Exp. 1-12. In contrast, the fractions were 32.5 %, 31.8 %, and 35.7 %, respectively, in Exp. 2-3, whereas the values were 20.3 %, 66.6 %, and 13.2 %, respectively, for summertime in urban Beijing.

Figure 1(a) The fraction of monomeric termination reactions and (b) monomeric termination products of the BPR in a representative high-[OH] experiment without NOx (Exp. 1–12), a representative low-[OH] experiment without NOx (Exp. 2–3), and summertime urban Beijing (Whalley et al. 2021). NOx-related reactions and products for the Beijing study are not included for a better comparison. (c) The fraction of monomeric reactions (Reactions R1–R3 and R5–R9) and (d) monomeric termination products of the BPR in a representative high-[OH] experiment with NOx (Exp. 1–48), a representative low-[OH] experiment with NOx (Exp. 2–7), and summertime urban Beijing (Whalley et al., 2021). Reactions and kinetic rate coefficients used in the calculations are provided in Table S2.
Our NOx-free experiments are characterized by an inherent drawback that the proportion of the HO2 termination pathway (Reaction R5) is actually lower than that under ambient conditions, which is similar to most other laboratory experiments (Bianchi et al., 2019). In our high-[OH] experiments without NOx, the reaction rates of unimolecular reactions, e.g., autoxidation reaction (Reaction R7) and condensation (Reaction R10), did not change with [OH] that increased in our experiments relative to that in the ambient. As a result, relative proportions of autoxidation and condensation were lowered. On the other hand, the 1,3,5-TMB-derived BPR was suggested to undergo autoxidation (Reaction R7) at a reaction rate of 0.078 s−1 (Wang et al., 2017), which represented 36.8 %, 94.4 %, and 92.8 % of the overall rates of Reactions (R1)–(R3) and (R5)–(R7) in Exp. 1-12, Exp. 2-3, and summertime urban Beijing, respectively. Because of its dominant proportion in Exp. 2-3 and the ambient atmosphere, the autoxidation channel is not included for clarity in Fig. 1a. Autoxidation did possess a lower significance in our high-[OH] experiments due to the other accelerated bimolecular reactions. However, it would only influence the oxygen content of our products but would not change the DBE. Both the accretion reaction (Reaction R4) and the condensation (Reaction R10) have been taken into account in the model, but they would not influence the distributions of monomeric stabilized products. We will specifically discuss these two pathways in the following sections because of their complexity between the laboratory and ambient conditions.
RO2 radicals other than the BPR and C9H13O7• existed in the PAM OFR, and these were not included in the model simulation. Their reaction rates of the accretion reaction (Reaction R4) and the autoxidation reaction (Reaction R7) should be different from the BPR and C9H13O7• due to the strong dependence of these two reaction rates on the molecular structure. Rates for the other reaction channels, on the other hand, should be the same as those of the BPR and C9H13O7•. Therefore, their fates in terms of the monomeric termination reactions (Reactions R1–R3, R5–R6, and R8–R9) should be similar to those of the BPR and C9H13O7•.
Calculated from yields of stabilized monomeric termination products of the BPR, the fractions of monomeric termination reaction products in Exp. 1-12, Exp. 2-3, and summertime urban Beijing (Whalley et al., 2021) are presented in Fig. 1b, showing a lot of similarities between these conditions. The fractions of R = O, ROH, ROOH, and ROOOH in Exp. 1-12 were 3.1 %, 14.1 %, 71.7 %, and 11.1 %, respectively. These fractions were 13.0 %, 18.1 %, 63.9 %, and 5.0 %, respectively, in Exp. 2-3 and were 5.3 %, 6.5 %, 87.0 %, and 1.2 %, respectively, in the summertime Beijing case. Among them, the majority of products are always ROOH and ROH, with ROOH being the most abundant. Therefore, the monomeric termination products of the BPR in our experiments are atmospherically relevant. In addition, only the R = O product has a DBE higher than the reacted RO2, but it merely accounted for a limited proportion. All the other stabilized termination products have a DBE that is 1 lower than the precursor and are the majority in both laboratory and ambient conditions. This indicates that the majority of the first-generation products typically have a DBE that is 1 lower than that of 1,3,5-TMB, whereas the majority of subsequent-generation products typically have a DBE that is 2 lower than that of 1,3,5-TMB. Once a monomeric compound with a DBE that is at least 2 lower than that of 1,3,5-TMB is observed, multi-generation OH reactions have happened in the system.
In laboratory experiments in the absence of NOx (e.g., Exp. 1-12), the proportions of Reaction (R8)–(R90), i.e., the NO channel in the urban atmosphere, were attributed to termination reactions of Reactions (R1)–(R6), i.e., RO2+ RO2, the accretion reaction, RO2+ HO2, and RO2+ OH. By expanding proportions of these termination reactions, laboratory investigations on product distributions can be facilitated, as the detection of certain HOM products became more precise and the mass spectra became simplified.
In experiments with NOx, the chemical fates of the BPR in high-[OH] experiments (Exp. 1–48 as an example, [OH] = ∼ 6.77×108 molecule cm−3, [NO] = ∼ 4.73×1010 molecule cm−3. [NO2] = ∼ 1.67×1012 molecule cm−3), low-[OH] experiments (Exp. 2–7 as an example, [OH] = ∼ 1.69×107 molecule cm−3, NO = ∼ 3.19×1010 molecule cm−3. NO2= ∼ 2.70×1011 molecule cm−3), and summertime urban Beijing are compared. As shown in Figure 1c, in all three conditions, RO2 reactions with NO were always the most significant pathway, with autoxidation being the second most significant.
Accounting for at least 52 % of monomeric termination products under all conditions, organonitrates were always the most important termination products, as shown in Fig. 1d. On the other hand, based on the formulae of organonitrates, the detailed formulae of monomer RO2 could be probed, which can help us better understand the chemical reactions inside the system. Alkoxy radicals generated in the NO termination channel will unlikely influence the distributions of C9 stabilized products since they tend to get decomposed in the subsequent reactions, as discussed in our previous discussion on the fate of alkoxy radicals in Sect. 2.
Due to the complexity of the ambient RO2 pool, it is difficult to estimate the detailed fraction of the accretion reaction, Reaction (R4). In the laboratory experiments, the RO2 pool mainly consists of the BPR and its autoxidation reaction product C9H13O7•, which can both undergo the accretion reaction rapidly (Berndt et al., 2018b). The concentrations of these two radicals were estimated by PAM_chem_v8. The reaction rate of accretion (Reaction R4) for the BPR was around 1.61 s−1 in Exp. 1-12, which is 88.4 % of Reactions (R1)–(R7), and was 0.29 s−1 in Exp. 2-3, equivalent to 77.7 % of Reactions (R1)–(R7). Certain uncertainties exist in the estimation of the proportions of accretion reactions, as the PAM_chem_v8 model only includes the first-generation reactions of precursors, whereas the subsequential fragmentation and re-initiation of stabilized products can generate a series of new RO2 that will influence the proportions of accretion reactions. We are only certain that the significance of accretion reactions in both Exp. 1-12 and Exp. 2-3 is larger than the ambient atmosphere. The much-expanded proportion of HOM dimers through accretion reactions makes it inadequate to compare yields of HOM dimers and HOM monomers. However, this deviation will not influence our conclusion on multi-generation OH oxidation, and identification of HOM dimers can help us identify the exact RO2 in the OFR and confirm the conditions of secondary OH oxidation according to the number of hydrogen atoms in the molecules.
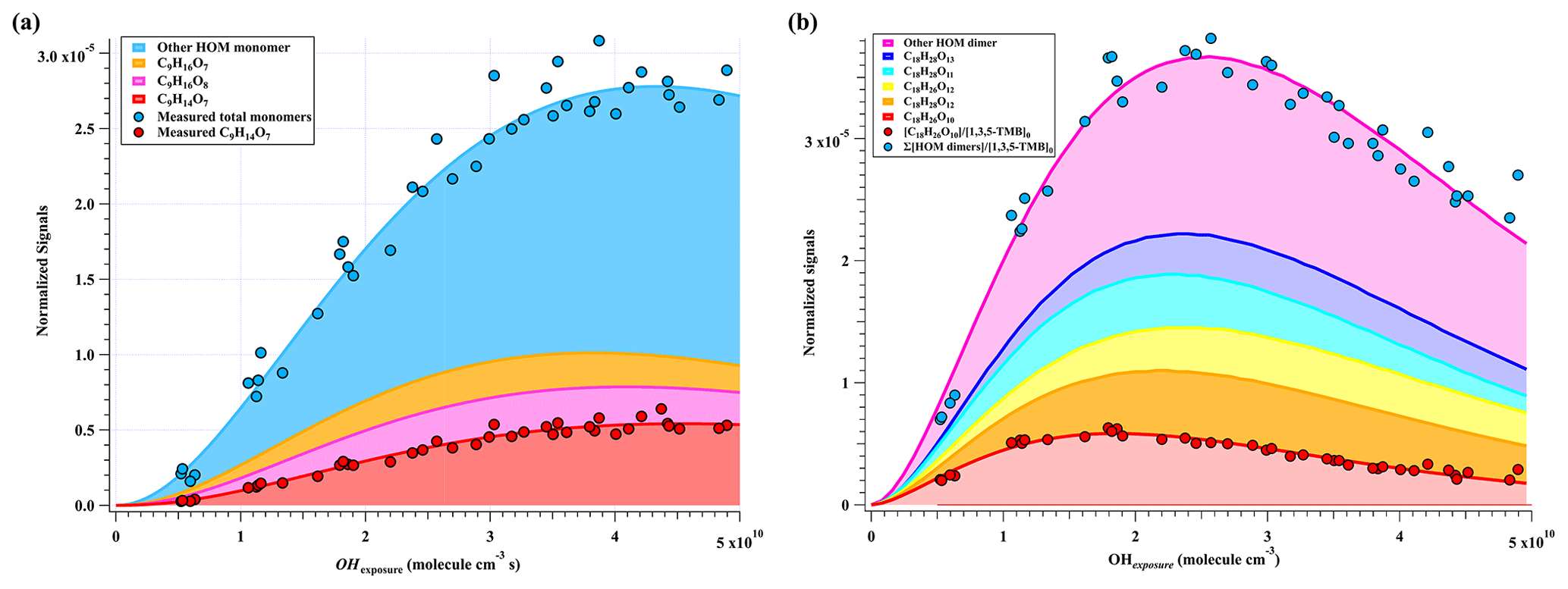
Figure 2Normalized signals of (a) HOM monomers and (b) HOM dimers versus OH exposure in the high-[OH] experiments, which are fitted via a gamma function and shown in stack.
In addition, certain compounds might have condensed onto pre-existing particles in the real atmosphere before an appreciable fraction of such compounds undergoes the re-initiated OH oxidation. Therefore, even if the same product can be generated in both the laboratory experiments and the ambient atmosphere, the relative significance of this product is not completely identical. Though OOMs might have the potential to undergo multi-generation OH oxidation, the exact proportion of this reaction in the ambient atmosphere strongly depends on their volatility, in other words, the condensation sink of these OOMs. The typical monomeric termination products of the 1,3,5-TMB-derived BPR, C9H12O4, C9H14O4, C9H14O5, and C9H13NO6, are estimated to have saturation vapor concentrations (C*) of 30.20, 30.20, 0.85, and 3.39 µg m−3 at 300 K, respectively, with the volatility parameterization developed in the CLOUD chamber oxidation experiments of aromatics (M. Wang et al., 2020). From the perspective of volatility, they all belong to semi-volatile organic compounds (SVOCs; 0.3 < C* < 300 µg m−3) and are expected to exist in both the condensed phase and the gas phase at equilibrium in the atmosphere (Bianchi et al., 2019). Compared to ambient conditions, the proportion of their condensation in the laboratory was biased to be lower due to the accelerated bimolecular reactions. However, this will not prevent the high-[OH] experiments from showing the potential and ability of these compounds to go through re-initiated OH oxidation, as these compounds would exist in significant fractions in the gas phase in the real atmosphere.
However, the conditions are completely different for other HOM monomer products and HOM dimer products with much lower volatility. It is difficult for a HOM dimer, e.g., C18H26O10, estimated with a C* of µg m−3 at 300 K, to survive long enough to experience an appreciable re-initiated photochemical aging. The lifetime of HOMs that can be classified as LVOCs ( < C* < 0.3 µg m−3) and ELVOCs (C* < µg m−3) can be estimated according to the condensation sink (CS) in the atmosphere, as they are lost irreversibly onto surfaces. The median value of CS in urban Beijing was reported to be around 0.019 and 0.057 s−1 during NPF days and non-NPF days, respectively, whereas the values in Shanghai were reported to be around 0.013 and 0.017 s−1, respectively (Deng et al., 2020; Yao et al., 2018); these values are all much higher than the physical loss in our PAM OFR, i.e., 0.0023 s−1. LVOCs and ELVOCs are believed to be lost irreversibly to the surface in both the laboratory and the ambient atmosphere because of their low volatility. By assuming a similar diffusion coefficient of LVOCs and ELVOCs to that of sulfuric acid, the lifetimes of LVOCs and ELVOCs in the ambient atmosphere still can still be as high as 77 s for the condensation loss, which is close to the residence time of our PAM OFR. Therefore, if they were generated by oxidation of aromatics in the ambient, these LVOCs and ELVOCs should at least have the potential to experience the same OH exposures as those in our low-[OH] experiments, i.e., at least 5.86×108 molecule cm−3 s. On the other hand, the detailed proportions of LVOCs and ELVOCs after a large OH exposure should be lower than those in the lab due to their magnified physical loss in the ambient. This means that if the multi-generation products of those compounds were observed in the ambient air, they should have been generated via a reaction that happened very recently.
3.2 Oxidation products in high-[OH] experiments
A total of 33 HOM monomers with formulae of C7−9H8−16O6−11 and 22 HOM dimers with formulae of C17−18H24−30O8−14 were observed in the first-round experiments of gas phase OH-initiated oxidation of 1,3,5-TMB in the OFR, i.e., high-[OH] experiments, as listed in Table S5. The relative signal contributions of HOMs to the total signals of all HOMs at an OH exposure of 2.38×1010 molecules cm−3 s are listed as an example in Table S5. The most abundant HOM products were also shown in stack in Fig. 2, whose relationships with OH exposures are superimposed by a gamma function ) simulation line to guide the eyes. The sum of normalized HOM monomers' abundance increased monotonically up to the highest OH exposure of 5×1010 molecule cm−3 s, whereas that of HOM dimers showed a non-monotonic dependence on OH exposure. The observed faster increase in accretion products than that of HOM monomers can be explained jointly by the fast second-order kinetics for accretion reactions of RO2 (Berndt et al., 2018b) and the high concentrations of relevant radicals in this work. On the other hand, most of the first-generation HOM dimers formed from accretion reactions contain at least one C=C bond and have more functionalities than HOM monomers and thus should be more reactive to OH radicals, which, together with a faster deposition loss of dimers, results in a faster consumption of HOM dimers than monomers in the OFR. The faster production and consumption of HOM dimers allowed their concentrations to summit at middle levels of OH exposures. As stated in Sect. 3.1, because of the inherent disadvantage of laboratory experiments, [RO2] is always too high in the OFR, which has been pointed out in a previous study (Bianchi et al., 2019). The accretion reactions in the OFR are relatively more significant than they should be in the ambient atmosphere. We do not mean to compare the total abundance of HOM monomers with that of HOM dimers here but to pay attention to the molecular characterization.
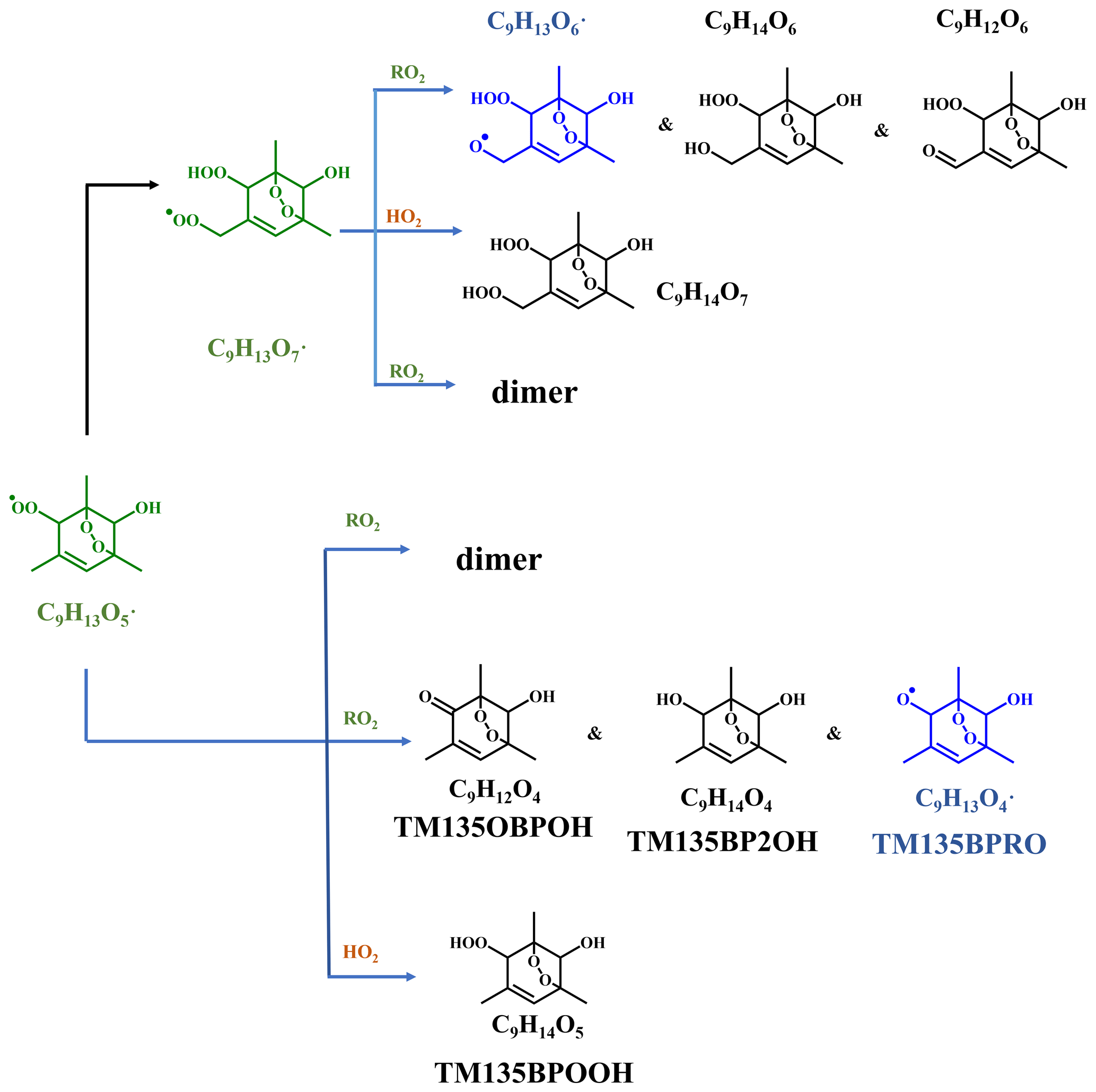
Scheme 1Oxidation pathways of the bicyclic peroxy radical C9H13O5• (MCM name: TM135BPRO2) in the OH-initiated oxidation of 1,3,5-TMB. Green, blue, and black formulae denote alkyl peroxy radicals, alkoxy radicals, and stabilized products, respectively. Black arrows denote the autoxidation pathway. MCM names for HO2 and RO2 termination products of TM135BPRO2 are present.
Theoretically, at a given RH and UV (i.e., a given [OH]), an increase in the initial TMB would lead to formation of more RO2, which corresponds to a larger . However, under our high-[OH] experimental conditions, the channels of RO2 radicals are always minor, and thus an increase in would not have a significant impact on the relative distribution of products formed from these channels. We compared product mass spectra for experiments with a similar OH exposure but different initial concentrations of TMB (e.g., Exp. 1-3 vs. Exp. 1-19, and Exp. 1-12 vs. Exp. 1–22). The OH exposures of Exp. 1-3 and Exp. 1-19 were estimated by the consumption of precursors to be 5.2×109 and 5.3×109 molecule cm−3 s, respectively, but the initial concentration of TMB of Exp. 1-3 was 25 % more than that in Exp. 1-19. Meanwhile, the OH exposures of Exp. 1-12 and Exp. 1-22 were 4.5×1010 and 4.4×1010 molecule cm−3 s, respectively, but the initial concentration of TMB of Exp. 1-12 was 48 % more than that in Exp. 1-22. Figure S2 shows comparisons between the product mass spectra of Exp. 1-3 and Exp. 1-19, as well as of Exp. 1-12 and Exp. 1-22, indicating that increase in the initial concentration of precursors generally resulted in a minor increment in the absolute signals of HOMs. Clearly, the relative distributions of products in these experiments are quite similar, indicating a minor difference in the relative distributions of products caused by fluctuations of initial concentrations of TMB.
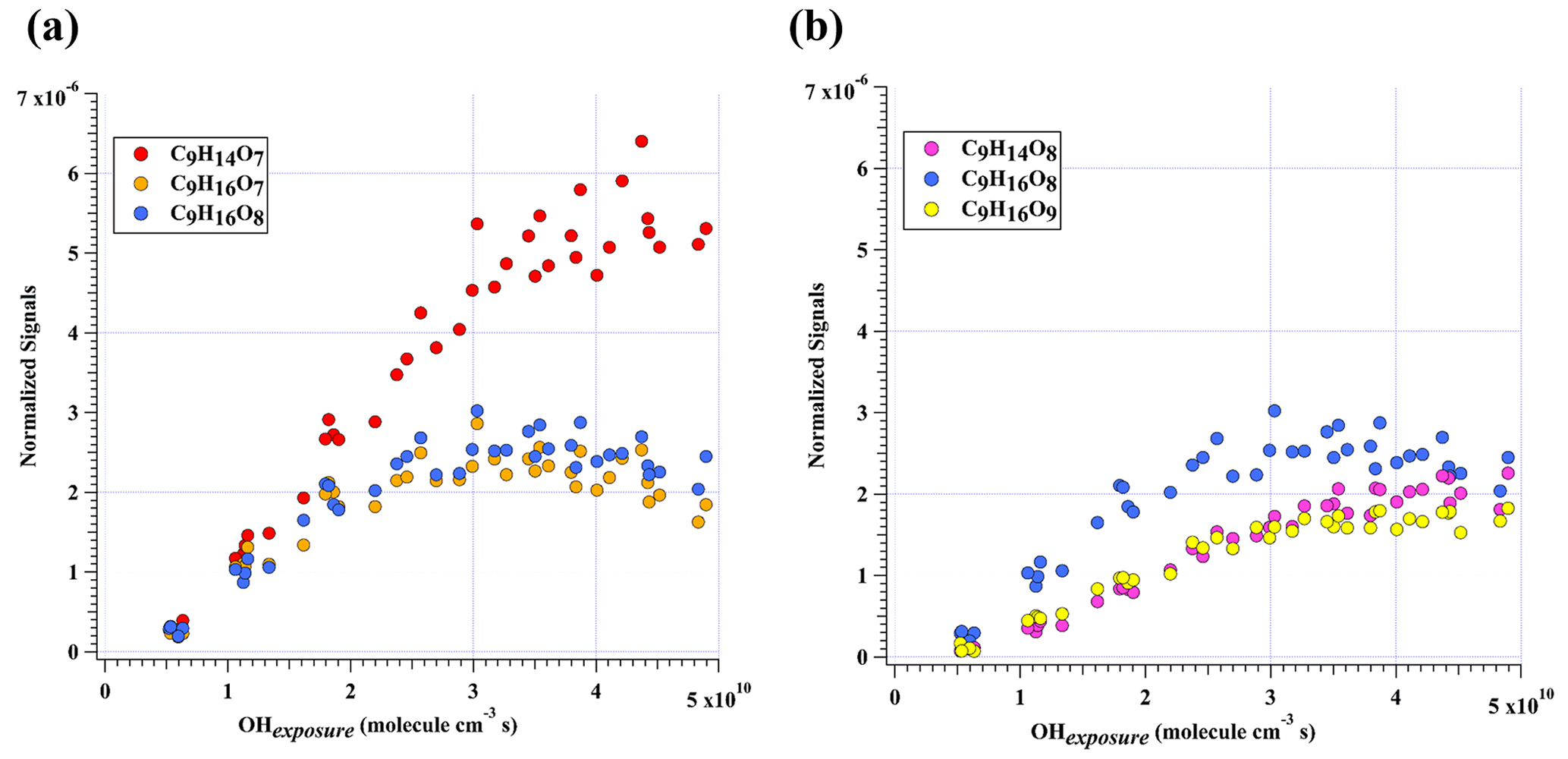
Figure 3Normalized signals of (a) C9H14O7, C9H16O7, and C9H16O8 and (b) C9H14O8, C9H16O8, and C9H16O9 measured at the exit of OFR in our high-[OH] experiments without NOx as a function of OH exposure. C9H16O8 is shown in both plots to better illustrate the chemical profiles of different compound groups.
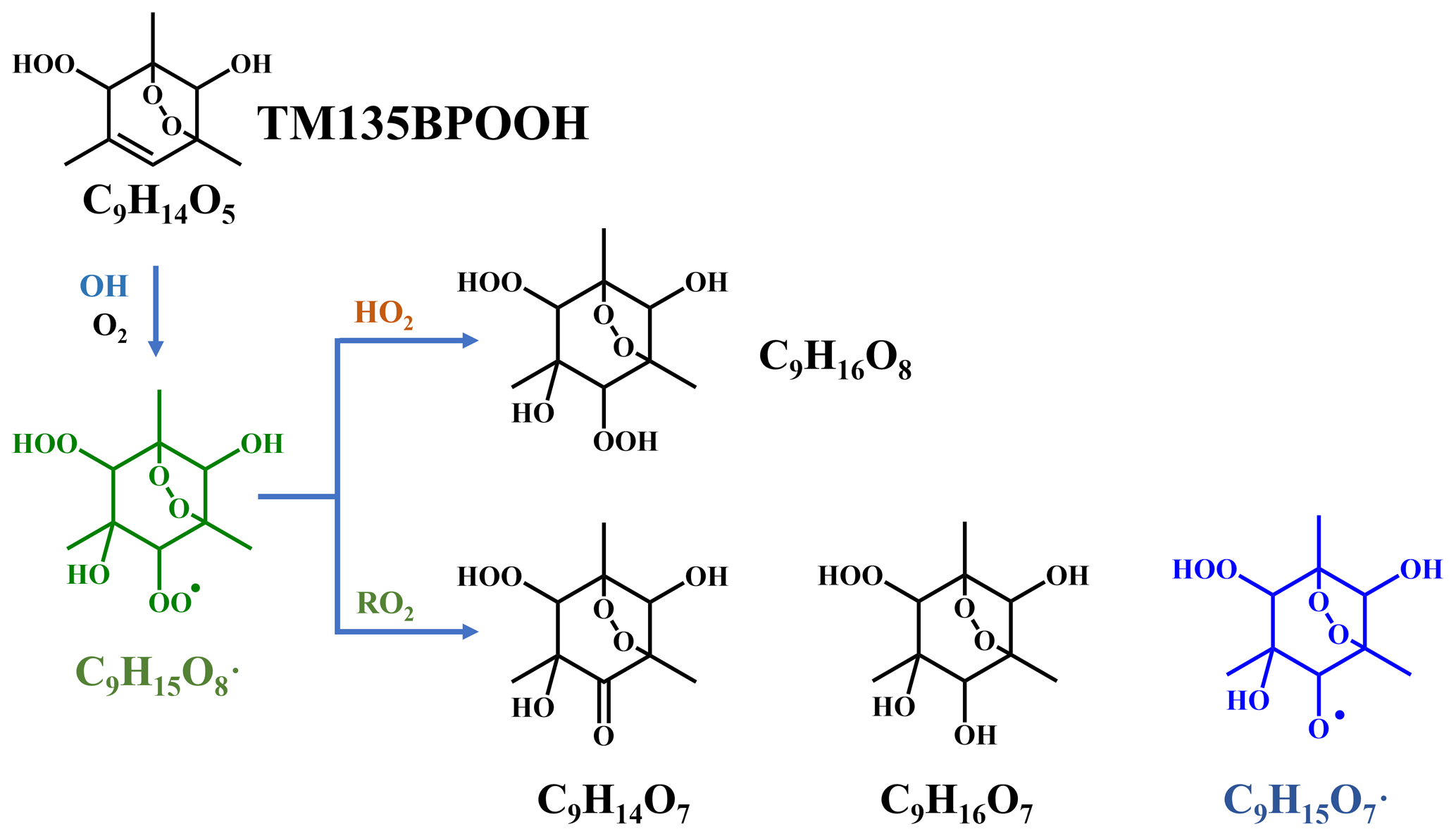
Scheme 2Proposed formation pathways of C9H14O7, C9H16O7, and C9H16O8 via the secondary OH oxidation of TM135BPOOH.
3.2.1 HOM monomers
Previous studies indicate that oxidation products derived from the peroxide–bicyclic pathway represent a main fraction of HOMs (Wang et al., 2017; Zaytsev et al., 2019). For 1,3,5-TMB, this pathway, as recommended by MCM, starts from a BPR, C9H13O5• (MCM name: TM135BPRO2) (Molteni et al., 2018). According to MCM and Molteni et al. (2018), Scheme 1 has been proposed to provide a good understanding of this reaction system and the structures of oxidation products. Molteni et al. (2018) suggested that C9H13O7•, i.e., the peroxy radical formed from autooxidation of C9H13O5•, has two isomers. A second step of endo-cyclization is required in the formation of one of the isomer, which is extremely slow and not competitive, as shown in several previous studies using both experimental and theoretical approaches (Wang et al., 2017; Xu et al., 2020). Even if such a second O2 bridging to a double bond is assumed to be possible, the abundance of this isomer should be significantly smaller than the other one because of the much faster reaction rate of H-shift reaction. Therefore, we do not take the C9H13O7• isomer containing a double endo-cyclization into consideration in this work. The majority of HOM monomers are generated from subsequent reactions of C9H13O5• and newly formed C9H13O7•, both of which contain one C=C bond in the carbon backbone and thus have a feasible site for OH addition. Meanwhile, the autoxidation reaction rate for newly formed C9H13O7• should be significantly smaller than C9H13O5•, as there is no hydrogen atom in C9H13O7• that is able to undergo a hydrogen atom shift at an appreciable rate based on our current understanding. Therefore, the subsequent autoxidation reaction should not be able to generate large amounts of more oxidized RO2.

Scheme 3Proposed formation pathways of C9H14O8, C9H16O8, and C9H16O9 via the secondary OH oxidation of TM135BPOOH.
Monomeric termination products of the BPR, as shown in Scheme 1, were not detected by the nitrate CIMS in this round of experiments, which might be due to the fast sub-sequential OH oxidation of these products under high-[OH] environment since they were observed under low-[OH] environments, as shown in Sect. 3.3. Monomeric termination products of C9H13O7• were all observed clearly, including C9H12O6, C9H14O6, and C9H14O7. In particular, C9H14O7 was the most abundant one among all of the HOM monomer products (Fig. 2a). As proved by a previous study, these three species should be typical first-generation stabilized products derived from autoxidation (Y. Wang et al., 2020). These HOM monomers should consist of several isomers bearing the same formula, because products from the secondary reactions cannot share the same structure as that of the one from the first-generation reaction. However, limited by the inherent disadvantages of mass spectrometers, we could not distinguish isomers here and further illustrate their different chemical behaviors.

Scheme 4NO termination reactions of the bicyclic peroxy radical C9H13O5• (MCM name: TM135BPRO2) and its autoxidation reaction products. Green, blue, and black formulae denote alkyl peroxy radicals, alkoxy radicals, and stabilized products, respectively. Black arrows denote the autoxidation pathway. MCM names of NO termination products of TM135BPRO2 are present.
In addition to these three ones, the next most prominent products to C9H14O7 were C9H16O7 and C9H16O8 (Fig. 3a), which are produced from multi-generation oxidation according to their DBE. Based on the formulae of these three HOM monomers, they (C9H14O7, C9H16O7, and C9H16O8) could be formed from the bimolecular termination reactions of C9H15O8•, which can be generated by an OH attack to C9H14O5 (Scheme 2), the hydroperoxyl termination product of the BPR, C9H13O5•. The other HOM monomers characterized by high signals were C9H14O8 and C9H16O9 (Fig. 3b). These two HOM monomers (C9H14O8 and C9H16O9), together with C9H16O8, correspond to the monomeric termination products of C9H15O9•, which is highly likely the peroxy radical generated by an OH attack to C9H14O6 (Scheme 3), i.e., the hydroxyl termination product of C9H13O7•. As discussed earlier, C9H13O7• is a typical autoxidation reaction product of the BPR of C9H13O5•. Therefore, detected signals of C9H16O8 should be the sum of two isomers' signals at least. Other HOM monomers were generally observed at much lower signals and thus were not plotted individually.
It is worth noting that HOM monomers with 18 hydrogen atoms, i.e., a DBE of 1, were never observed in our experiments, including potential stabilized hydroperoxyl products formed from C9H17Om•. This is expected, since C9H17Om• should be in really low concentrations, if at all. As indicated by its hydrogen number, a C9H17Om• was formed by at least two OH additions to the C=C bond of a C9H13Om•, but the main BPR, C9H13O5•, and its autoxidation product (C9H13O7•) are characterized by one C=C bond on the ring, which makes this formation pathway impossible. Other ring-breakage pathways should not contribute to the formation of this radical (C9H17Om•) because of their low branching ratio, as determined by recent studies (Zaytsev et al., 2019; Xu et al., 2020).
Proposed according to MCM and Molteni et al. (2018), Scheme 4 shows the NO termination pathways of the main BPR C9H13O5• and its autoxidation product, C9H13O7•. After introducing N2O into PAM OFR, quantities of organonitrates were generated, including both C9 and C18 organonitrates. The average mass spectrometry of the nitrate CIMS in the 4.41×1010 molecule cm−3 NO experiment and 1.18×1011 molecule cm−3 NO experiment is shown in Fig. S3. Organonitrates were formed via the NO + RO2 reaction, called NO termination reactions. The distribution of oxidation products under these two NO settings was similar.
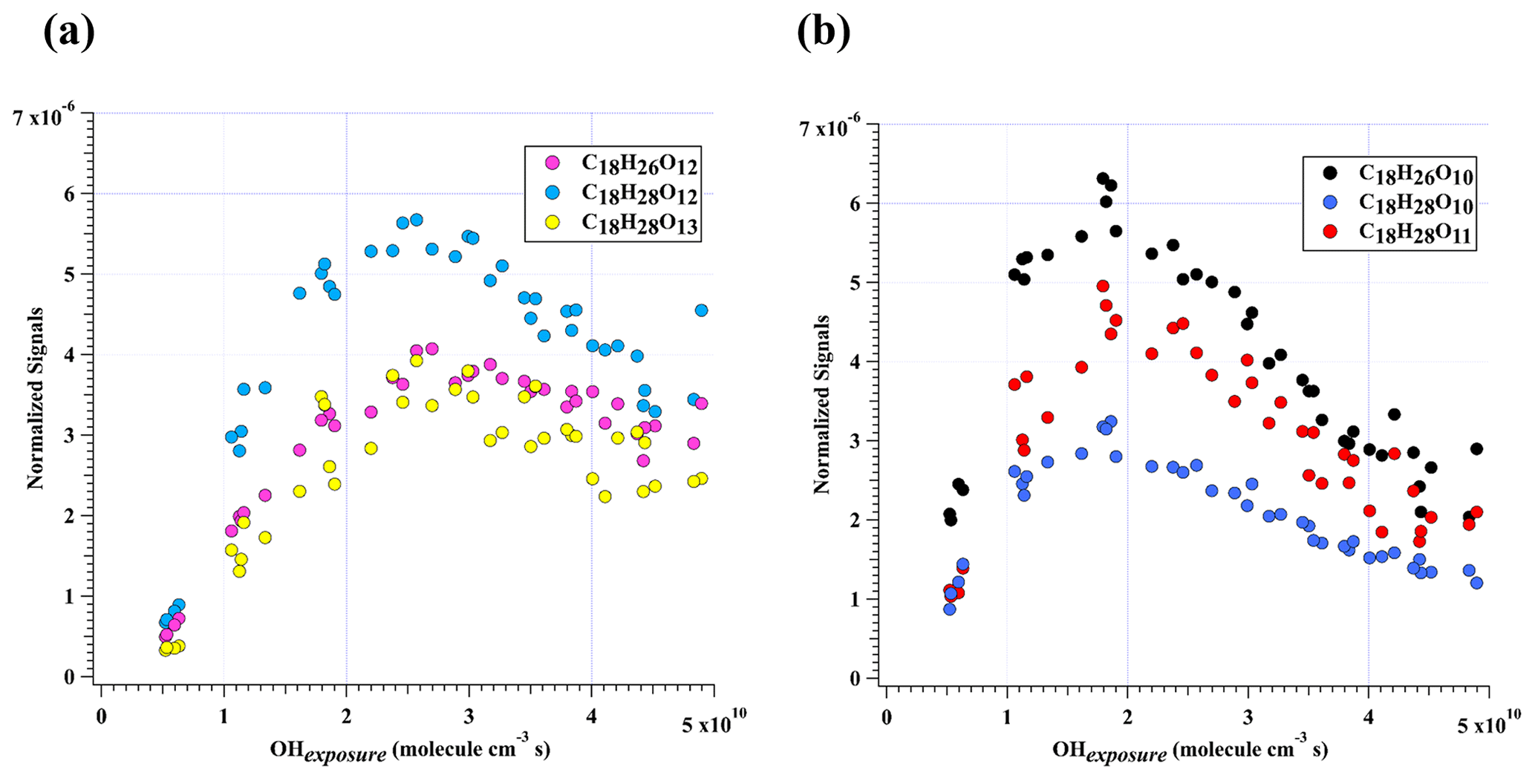
Figure 4Normalized signals of (a) C18H26O12, C18H28O12, and C18H28O13 and (b) C18H26O10, C18H28O10, and C18H28O11 measured at the exit of OFR in our high-[OH] experiments without NOx as a function of OH exposure.
As discussed above, most of the first-generation HOMs should contain a C=C bond in the carbon backbone. The ubiquitous existence of organonitrates that contain two nitrogen atoms confirms the extensive secondary OH oxidation in the systems exactly because the NO termination reaction of RO2 is the only pathway that can generate organonitrates in our experiments, and this pathway can only introduce one nitrogen atom at a time, as indicated in Scheme 4. RO2 can react with NO2 to form peroxynitrates (ROONO2), but these species are thermally unstable except at very low temperatures or when the RO2 is an acylperoxy radical (Orlando and Tyndall, 2012), neither of which were met in our experiments. The concentrations of NO3 were estimated to be lower than 2.45×107 molecule cm−3 by our modified PAM_chem_v8 because of the existence of sufficient concentrations of NO, which would consume NO3 at a rapid reaction rate, i.e., molecule−1 cm3 s−1 (IUPAC dataset, https://iupac-aeris.ipsl.fr, last access: 26 October 2023). Therefore, NO2 and NO3 were not likely to react with RO2 to form large amounts of organonitrates in our experiments. Taking the most abundant organonitrate, C9H14N2O10, as an example, it was exactly the NO termination product of C9H14NO9•, which was generated from an OH attack and a subsequent O2 addition to C9H13NO6, the NO termination product of C9H13O5•. For other organonitrates, C9H13NO8, the second-most-abundant organonitrate, could either be a NO termination product of C9H13O7• or, together with the other most abundant organonitrates, C9H15NO7 and C9H15NO8, classical termination products of C9H14NO9•. C9H14N2O10, C9H15NO7, and C9H15NO8 all have a DBE of 2 lower than the precursor and thus are typical multi-generation OH oxidation products.
The NO:RO2 ratio in the PAM OFR in this series of experiments is lower than typical values in the ambient atmosphere, which is due to the existence of O3 that was utilized to generate O(1D) in the OFR and its rapid reaction rate with NO. However, due to rapid reaction rate constants between NO and RO2, i.e., around molecule−1 cm3 s−1, the reaction rate for the NO termination channel of RO2 was as fast as around 0.3–1.0 s−1. Large amounts of organonitrates would still be formed, as discussed in Sect. 3.1. Our conclusion is also valid because of the detection of compounds with multiple nitrogen atoms.
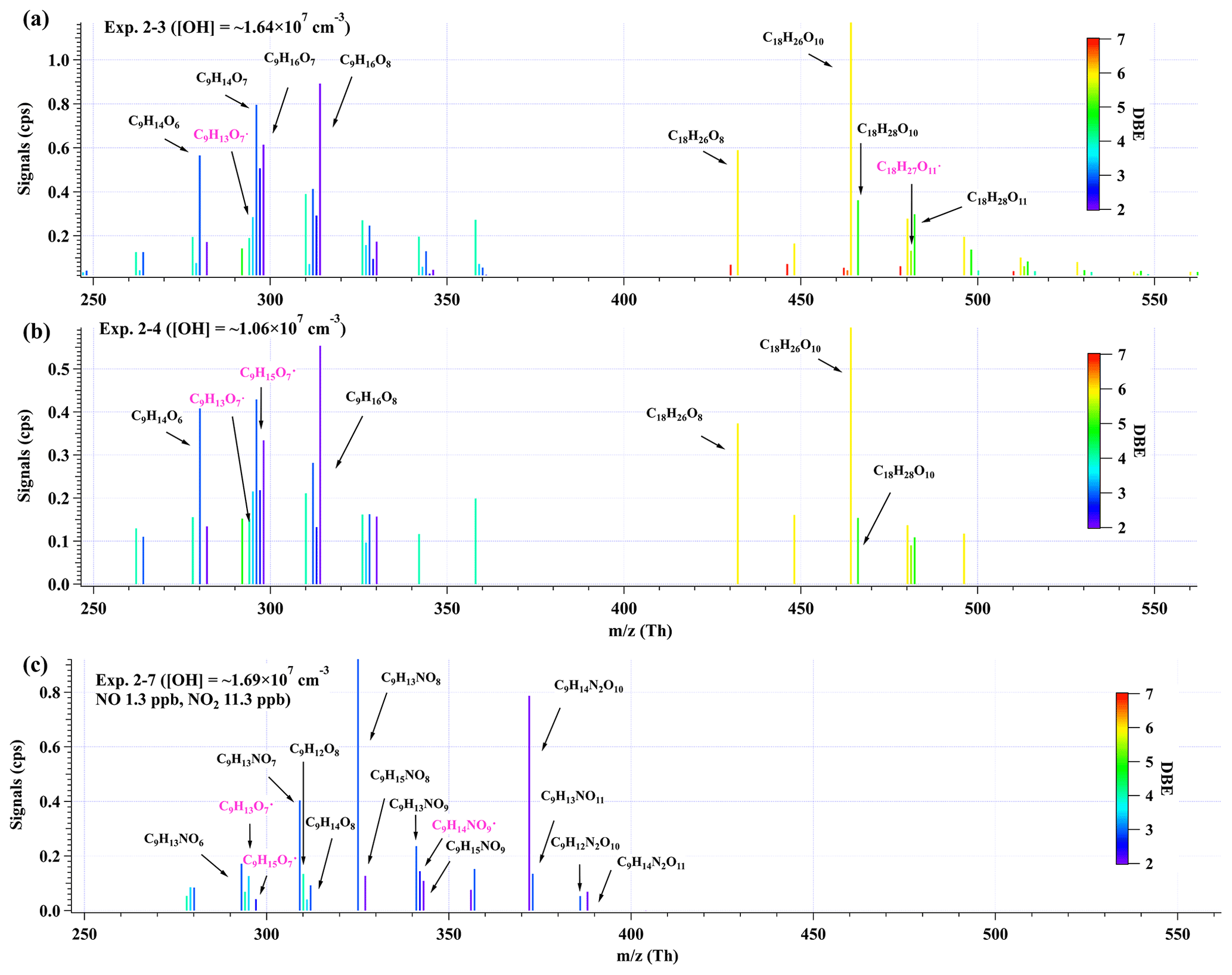
Figure 5Distributions of C9 and C18 products detected by nitrate CI-TOF in (a) Exp. 2–3, (b) Exp. 2–4, and (c) Exp. 2–7. The reagent ion, NO, is omitted in the label for the molecular formula. Important radicals were labeled in pink. Note that no convinced signals of HOM dimers were observed in the second-round experiments with NOx.
3.2.2 HOM dimers
The accretion reaction, to , is a source of gas-phase dimer compounds from highly oxidized, functional RO2 radicals (Ehn et al., 2014; Berndt et al., 2018b; Zhao et al., 2018; Berndt et al., 2018a). C18H26O8 and C18H26O10 are two typical accretion reaction products in the 1,3,5-TMB + OH system, whose formation pathways have been elucidated (Berndt et al., 2018b). C18H26O8 can only be formed via the accretion reaction of two C9H13O5• radicals. C9H13O3• is not likely to react with C9H13O7• to form large amounts of C18H26O8. C9H13O3• can only be formed after addition of a hydroxyl radical to the aromatic ring of 1,3,5-TMB and a subsequent O2 addition to the newly formed hydroxyl-substituted cyclohexadienyl radical (Vereecken, 2019). However, the lifetime of this radical is extremely short, as C9H13O3• will undertake a ring-closure reaction and get attached by a O2 very rapidly, forming the BPR, C9H13O5•. Its short lifetime and low concentration, as indicated by Berndt et al. (2018b), lead to its insignificant role in the accretion reactions. In contrast, C18H26O10 can be formed either by the accretion reaction between C9H13O5• and C9H13O7• or via a second OH attack to C18H26O8. These two HOM dimers are so far the only ones that are confirmed to be formed via the accretion reactions (Berndt et al., 2018b; Bianchi et al., 2019).
C18H26O10 was characterized by the highest dimer signals for experiments with OH exposures under 3.5×1010 molecule cm−3 s. Nevertheless, C18H26O10, together with C18H28O12, C18H26O12, C18H28O11, C18H28O13, and C18H28O10, contributed more than 50 % of total HOM dimer signals at all OH exposure levels (Fig. 2b). These six most abundant HOM dimers correspond exactly to the hydroperoxyl, hydroxyl, and carbonyl termination products of C18H27O11• and C18H27O13•, respectively. These two RO2 radicals (C18H27O11• and C18H27O13•), on the other hand, could be generated by OH attacks to C18H26O8 and C18H26O10, respectively, which strongly suggests the significant role of secondary OH chemistry in the formation of HOMs in our experiments. In addition, C18H28Ox can also be formed through accretion of a C9H13Om• radical and a C9H15Om• radical, as suggested by previous studies (Molteni et al., 2018; Tsiligiannis et al., 2019). However, since a C9H15Om• radical, as suggested by its hydrogen atom number, can only be formed via an OH addition to the stabilized C9H14Om products through multi-generation OH reactions, our conclusion that C18H28Ox species are multi-generation OH oxidation products still holds. Figure 4 shows the normalized signals of these abundant HOM dimers at different OH exposures.
This decrease in dimers at relatively high OH exposures is likely due to the accelerated accretion reactions in the OFR, resultant of the high RO2 concentrations. The HOM dimers are formed earlier compared to those under ambient conditions and can then go through further oxidation reactions. Note that this does not mean the maximum concentrations of HOM dimers will also accurately occur at the same OH exposures in the atmosphere because the detailed appearance time of the maximum concentrations of HOM dimers is dependent on their formation rate and loss rate. In our experiments, the formation rate and loss rate were not accelerated equally. On the other hand, the loss pathways of HOM dimers were not exactly the same as the ambient. This series of experiments is not meant to specifically find out the detailed OH exposures whereby the maximum concentrations of HOM dimers will occur but to try to indicate how HOM dimers evolve with the increase in OH exposures. This work can be regarded as an indicator for the potential chemical fates of HOM dimers in the atmosphere if their survival time permitted. It should be noted that the gas-phase chemistry in the PAM OFR cannot be exactly the same as that in the ambient. Reactions of OH with OVOCs often lead to HO2 formation, resulting in a HO2:RO2 ratio larger than 1 in the real atmosphere (Bianchi et al., 2019). A recent campaign conducted at a rural site in the Yangtze River Delta estimated that the local ratio of HO2:RO2, the latter of which was presumably derived from longer chain alkanes (> C3), alkenes, and aromatic compounds, was around 1.66 (Ma et al., 2022). Such a high HO2:RO2 ratio condition is typically difficult to simulate in the laboratory experiments, as the precursors are usually hydrocarbons without any OVOCs (Peng and Jimenez, 2020). This is exactly the case for our experiments, but its influences on our conclusion were tiny, as has been discussed in the Sect. 3.1. Therefore, the difference in the distribution of products will not change our conclusion.

Figure 6The changes in normalized abundance of C9 and C18 products observed by nitrate CI-TOF in (a) Exp. 2-3 relative to Exp. 2-1, and (b) Exp. 2-3 relative to Exp. 1-12. The reagent ion, NO, is omitted in the label. The normalized abundance was obtained by normalizing all the products to the most abundant one in each experiment, i.e., C18H26O10 in Exp. 2-1 and Exp. 2-3 and C9H14O7 in Exp. 1–12.
Such an active secondary OH chemistry is consistent with the fast OH reaction rates of HOMs. We take C18H26O8 whose plausible structure is shown in Fig. S4 as an example, which is the accretion product of two C9H13O5• radicals. Its OH reaction rate constant is estimated to be around 2.07 × 10−10 cm3 molecule−1 s−1 according to the structure–activity relationship (Jenkin et al., 2018b, a), whose details are provided in Sect. S1 in the Supplement. This rate is several times larger than that of 1,3,5-TMB, which enables a very active secondary OH chemistry in the system. MCM recommended an OH reaction rate of 1.28 × 10−10 cm3 molecule−1 s−1 for TM135BPOOH (C9H14O5) and 1.00 × 10−10 cm3 molecule−1 s−1 for TM135OBPOH (C9H12O4) (Jenkin et al., 2003). The OH reaction rate for C18H26O8 should also be fast due to the C=C bonds in its structure, which is activated by the adjacent functionalities. Our calculation result is consistent with this estimation.
The distributions of C18 organonitrates also verified the extensive secondary reactions. The most abundant C18 organonitrate, C18H27NO12, was a NO termination product of radical C18H27O11•, which, as mentioned above, was the radical generated from the OH reaction with C18H26O8. C18H27NO12 can also be formed either by accretion between a C9H15Om• radical and a C9H12NOm• radical or by accretion between a C9H13Om• radical and a C9H14NOm• radical. Both C9H15Om• and C9H14NOm• radicals are typical multi-generation RO2 products and thus prove C18H27NO12 is a multi-generation OH oxidation product. Other C18 organonitrates are believed to be formed via a similar pathway. Hence, plenty of organonitrates have been formed via the multi-generation OH reactions of first-generation stabilized products.
3.3 Oxidation products in low-[OH] experiments
Given the larger sampling port, lower initial ozone concentrations, lower UV light intensities, and a better performance of mass spectrometer in this series of low-[OH] experiments, a number of new species were detected in the second-round experiments, including three typical termination reaction products of the BPR, i.e., C9H14O4, C9H14O5, and C9H13NO6, and a number of low-volatility compounds, e.g., C9HxO11 (x=12–15). The distributions of oxidation products detected by nitrate CI-TOF in Exp. 2-3, 2-4, and 2-7, representative low-[OH] experiments, are displayed in Fig. 5. The detailed molecular formula and their contributions to total HOMs' signals are provided in Tables S6 and S7.
In addition, certain C9 and C18 HOMs with lower DBE than typical first-generation products predicted by MCM (Saunders et al., 2003) or reported by previous studies (Berndt et al., 2018b) were detected in Exp. 2-3, 2-4, and 2-7, although [OH] in these experiments is much lower than in the first-round experiments.
Observation of compounds with lower DBE in Exp. 2-3, 2-4, and 2-7, including HOM monomers with DBE lower than 3 and HOM dimers with DBE lower than 6, as well as monomer radicals with DBE lower than 3, including C9H15Om• (m=7–11) and C9H14NO9•, proves the re-initiation of OH oxidation of the stabilized products in experiments with atmospherically relevant [OH]. All the stabilized products and radicals depicted in the proposed mechanisms (Schemes 2 and 3) were detected in both Exp. 2-3 and 2-4, except for C9H15O9•, which was only detected in Exp. 2-3. This means that the proposed reaction pathways have already happened under atmospheric [OH] conditions with limited OH exposures. However, as we do not know the exact structures of these OOMs and radicals, the proposed reaction pathways are merely based on the chemical formulae detected by the nitrate CIMS and nitrate CI-TOF and proposed according to the general mechanisms of OH addition reactions to the C=C bond. Other reaction pathways to generate these compounds or other isomers generated in these pathways are undoubtedly feasible.
A lot of compounds detected in the experiments without NOx were not observed in the counterpart experiments with NOx. We also did not detect decent signals of HOM dimers in the NOx-present experiments in the second-round experiments. Such a dramatic decrease in the abundance of HOM dimers after the introduction of NOx into the aromatic oxidation system has been reported in several previous studies (Garmash et al., 2020; Y. Wang et al., 2020; Tsiligiannis et al., 2019). This might come from the dominant significance of NO + RO2 (Reactions R8–R9) after the introduction of NOx into the system, making signals of certain HOMs from other channels lower than the detection limit of the instrument. The proportions of other reaction channels decreased and were reassigned to the NO channel, as evidenced by the fact that most of observed oxidation products were organonitrates, which is in excellent agreement with the modeled channel proportions in Sect. 3.1.
Many organonitrates were observed in both series of experiments. In the low-[OH] experiments, the most significant compound was C9H13NO8, whose formula matches the NO termination product of C9H13O7•, i.e., autoxidation product of the BPR. The second-most-important compound, C9H14N2O10 in our low-[OH] experiments, was the most significant product in the high-[OH] experiments in presence of NOx, whose formula matches the NO termination product of C9H14NO9•, i.e., the RO2 formed via an OH addition to C9H13NO6, the NO termination product of the BPR. All of the products and radicals mentioned above were observed in Exp. 2-7, as shown in Fig. 5c. From the perspective of the molecular formula, C9H14N2O10 is also one of the most frequently observed multi-nitrogen-containing compound in polluted atmospheres, whose seasonal variations show a good correlation with [OH] (Guo et al., 2022; Yang et al., 2023).
A comparison of relative abundances of C9 and C18 products under different [OH] levels is helpful for the elucidation of their formation pathways. The difference in product distributions between Exp. 2-3 ([OH] = ∼ 1.69×107 molecule cm−3) and Exp. 2-1 ([OH] = molecule cm−3), as well as between Exp. 2-3 and Exp. 1-12 ([OH] = ∼ 8.47×108 molecule cm−3), is shown in Fig. 6. The normalized abundance was obtained by normalizing all the products to the most abundant one in each experiment, i.e., C18H26O10 in Exp. 2-1 and Exp. 2-3 and C9H14O7 in Exp. 1-12. The changes in the normalized abundance were obtained by subtracting the normalized abundance in Exp. 2-1 from that in Exp. 2-3, and Exp. 1-12 from Exp. 2-3. As the [OH] and OH exposure increased, there was a noticeable rise in the relative abundance of more oxygenated compounds, which can be attributed to the larger proportion of multi-generation OH oxidation in high OH exposure experiments. This comparison demonstrates the capacity and potential of multi-generation OH oxidation to reduce DBE and elevate the oxygenated levels of oxidation products.
In conclusion, observation of the same low DBE compounds, i.e., DBE = 2, in both low-[OH] and high-[OH] experiments confirms the feasibility of the generation of HOMs under atmospherically relevant conditions. The detection of C9H14O5, C9H15O8•, C9H14O7, C9H14O8, C9H15O7•, and C9H16O8 and of C9H14O6, C9H15O9•, C9H14O8, C9H14O9, C9H15O8•, and C9H16O9 in low-[OH] experiments also confirms the potential existence of the proposed mechanisms, i.e., Schemes 2 and 3, respectively. Certainly, other potential formation pathways for these products are possible.
This study highlights the influences of OH exposure on the distribution and evolution of 1,3,5-TMB-derived HOMs. Secondary OH reactions can influence HOMs' composition by directly reacting with the stabilized first-generation oxidation products, leading to enhanced formation of HOMs, if the stabilized, first-generation oxidation products could survive from condensation loss onto pre-existing particles. Observation of organonitrates generated in the NO experiments further confirmed the secondary OH oxidation. Due to the elevated abundance and the reduced volatility of HOMs, growth rates of newly formed nanoparticles in the presence of HOMs could be increased, especially in high-OH environments, which prevails at noon in the summer. Substantially high concentrations of OH have been frequently observed in polluted environments during summer, e.g., megacities in China (Tan et al., 2019), and thus more active secondary OH reactions are expected compared to wintertime. As a plausible consequence, seasonal differences of HOMs and new particle formation (NPF) are evident (Qiao et al., 2021; Yao et al., 2018; Guo et al., 2022). Furthermore, previous studies suggest that high concentrations of NO can suppress the formation of HOMs via the suppression of autoxidation (Pye et al., 2019), but the influences of such a suppression could have been overestimated, since secondary OH reactions can continue to oxidize the stabilized organonitrates. Our conclusions help to explain the existing gap between model prediction and ambient measurement of HOMs' concentrations (Qi et al., 2018) and to build a global HOMs' simulation model.
Data used in this work are available upon request from the corresponding authors.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-7961-2024-supplement.
LW and YW designed the experiments. YW and CL conducted the laboratory experiments. YW analyzed the data. YW and LW wrote the paper. All co-authors discussed the results and commented on the manuscript.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
Yuwei Wang would like to thank Andrew T. Lambe, Peng Zhe, and Jose Jimenez for helpful discussions on PAM experiments.
This work was financially supported by the National Natural Science Foundation of China (grant nos. 21925601, 22127811) and the National Key R&D Program of China (grant no. 2022YFB2602001).
This paper was edited by John Liggio and reviewed by two anonymous referees.
Assaf, E., Song, B., Tomas, A., Schoemaecker, C., and Fittschen, C.: Rate Constant of the Reaction between CH3O2 Radicals and OH Radicals Revisited, J. Phys. Chem. A, 120, 8923–8932, https://doi.org/10.1021/acs.jpca.6b07704, 2016.
Assaf, E., Tanaka, S., Kajii, Y., Schoemaecker, C., and Fittschen, C.: Rate constants of the reaction of C2–C4 peroxy radicals with OH radicals, Chem. Phys. Lett., 684, 245–249, https://doi.org/10.1016/j.cplett.2017.06.062, 2017.
Berndt, T., Mentler, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of α-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018a.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Self- and Cross-Reactions of RO2 Radicals in the Atmosphere, Angew. Chemie – Int. Ed., 57, 3820–3824, https://doi.org/10.1002/anie.201710989, 2018b.
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019.
Bossolasco, A., Faragó, E. P., Schoemaecker, C., and Fittschen, C.: Rate constant of the reaction between CH3O2 and OH radicals, Chem. Phys. Lett., 593, 7–13, https://doi.org/10.1016/j.cplett.2013.12.052, 2014.
Brune, W. H.: The Chamber Wall Index for Gas-Wall Interactions in Atmospheric Environmental Enclosures, Environ. Sci. Technol., 53, 3645–3652, https://doi.org/10.1021/acs.est.8b06260, 2019.
Caravan, R. L., Khan, M. A. H., Zádor, J., Sheps, L., Antonov, I. O., Rotavera, B., Ramasesha, K., Au, K., Chen, M.-W., Rösch, D., Osborn, D. L., Fittschen, C., Schoemaecker, C., Duncianu, M., Grira, A., Dusanter, S., Tomas, A., Percival, C. J., Shallcross, D. E., and Taatjes, C. A.: The reaction of hydroxyl and methylperoxy radicals is not a major source of atmospheric methanol, Nat. Commun., 9, 4343, https://doi.org/10.1038/s41467-018-06716-x, 2018.
Cheng, X., Chen, Q., Jie Li, Y., Zheng, Y., Liao, K., and Huang, G.: Highly oxygenated organic molecules produced by the oxidation of benzene and toluene in a wide range of OH exposure and NOx conditions, Atmos. Chem. Phys., 21, 12005–12019, https://doi.org/10.5194/acp-21-12005-2021, 2021.
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of organic compounds in the atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013.
Deng, C., Fu, Y., Dada, L., Yan, C., Cai, R., Yang, D., Zhou, Y., Yin, R., Lu, Y., Li, X., Qiao, X., Fan, X., Nie, W., Kontkanen, J., Kangasluoma, J., Chu, B., Ding, A., Kerminen, V. M., Paasonen, P., Worsnop, D. R., Bianchi, F., Liu, Y., Zheng, J., Wang, L., Kulmala, M., and Jiang, J.: Seasonal characteristics of new particle formation and growth in urban Beijing, Environ. Sci. Technol., 54, 8547–8557, https://doi.org/10.1021/acs.est.0c00808, 2020.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Eisele, F. L. and Tanner, D. J.: Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.-Atmos., 98, 9001–9010, https://doi.org/10.1029/93JD00031, 1993.
Fittschen, C.: The reaction of peroxy radicals with OH radicals, Chem. Phys. Lett., 725, 102–108, https://doi.org/10.1016/j.cplett.2019.04.002, 2019.
Fuller, E. N., Schettler, P. D., and Giddings, J. C.: A new method for prediction of binary gas-phase diffusion coefficients, Ind. Eng. Chem., 58, 18–27, https://doi.org/10.1021/ie50677a007, 1966.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Guo, Y., Yan, C., Liu, Y., Qiao, X., Zheng, F., Zhang, Y., Zhou, Y., Li, C., Fan, X., Lin, Z., Feng, Z., Zhang, Y., Zheng, P., Tian, L., Nie, W., Wang, Z., Huang, D., Daellenbach, K. R., Yao, L., Dada, L., Bianchi, F., Jiang, J., Liu, Y., Kerminen, V.-M., and Kulmala, M.: Seasonal variation in oxygenated organic molecules in urban Beijing and their contribution to secondary organic aerosol, Atmos. Chem. Phys., 22, 10077–10097, https://doi.org/10.5194/acp-22-10077-2022, 2022.
Heinritzi, M., Simon, M., Steiner, G., Wagner, A. C., Kürten, A., Hansel, A., and Curtius, J.: Characterization of the mass-dependent transmission efficiency of a CIMS, Atmos. Meas. Tech., 9, 1449–1460, https://doi.org/10.5194/amt-9-1449-2016, 2016.
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 2015.
Iyer, S., Kumar, A., Savolainen, A., Barua, S., Daub, C., Pichelstorfer, L., Roldin, P., Garmash, O., Seal, P., Kurtén, T., and Rissanen, M.: Molecular rearrangement of bicyclic peroxy radicals is a key route to aerosol from aromatics, Nat. Commun., 14, 4984, https://doi.org/10.1038/s41467-023-40675-2, 2023.
Jacob, D. J.: Introduction to atmospheric chemistry, Princeton, Princeton University Press, Princeton, NJ, https://doi.org/10.1515/9781400841547, 1999.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aliphatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9297–9328, https://doi.org/10.5194/acp-18-9297-2018, 2018a.
Jenkin, M. E., Valorso, R., Aumont, B., Rickard, A. R., and Wallington, T. J.: Estimation of rate coefficients and branching ratios for gas-phase reactions of OH with aromatic organic compounds for use in automated mechanism construction, Atmos. Chem. Phys., 18, 9329–9349, https://doi.org/10.5194/acp-18-9329-2018, 2018b.
Keller-Rudek, H., Moortgat, G. K., Sander, R., and Sörensen, R.: The MPI-Mainz UV/VIS Spectral Atlas of Gaseous Molecules of Atmospheric Interest, Earth Syst. Sci. Data, 5, 365–373, https://doi.org/10.5194/essd-5-365-2013, 2013.
Knap, H. C. and Jørgensen, S.: Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals, J. Phys. Chem. A, 121, 1470–1479, https://doi.org/10.1021/acs.jpca.6b12787, 2017.
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and De Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion- Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018.
Lambe, A., Massoli, P., Zhang, X., Canagaratna, M., Nowak, J., Daube, C., Yan, C., Nie, W., Onasch, T., Jayne, J., Kolb, C., Davidovits, P., Worsnop, D., and Brune, W.: Controlled nitric oxide production via O(1D) + N2O reactions for use in oxidation flow reactor studies, Atmos. Meas. Tech., 10, 2283–2298, https://doi.org/10.5194/amt-10-2283-2017, 2017.
Lambe, A. T., Krechmer, J. E., Peng, Z., Casar, J. R., Carrasquillo, A. J., Raff, J. D., Jimenez, J. L., and Worsnop, D. R.: HOx and NOx production in oxidation flow reactors via photolysis of isopropyl nitrite, isopropyl nitrite-d7, and 1,3-propyl dinitrite at λ = 254, 350, and 369 nm, Atmos. Meas. Tech., 12, 299–311, https://doi.org/10.5194/amt-12-299-2019, 2019.
Lambe, A. T., Ahern, A. T., Williams, L. R., Slowik, J. G., Wong, J. P. S., Abbatt, J. P. D., Brune, W. H., Ng, N. L., Wright, J. P., Croasdale, D. R., Worsnop, D. R., Davidovits, P., and Onasch, T. B.: Characterization of aerosol photooxidation flow reactors: heterogeneous oxidation, secondary organic aerosol formation and cloud condensation nuclei activity measurements, Atmos. Meas. Tech., 4, 445–461, https://doi.org/10.5194/amt-4-445-2011, 2011.
Lambe, A. T., Chhabra, P. S., Onasch, T. B., Brune, W. H., Hunter, J. F., Kroll, J. H., Cummings, M. J., Brogan, J. F., Parmar, Y., Worsnop, D. R., Kolb, C. E., and Davidovits, P.: Effect of oxidant concentration, exposure time, and seed particles on secondary organic aerosol chemical composition and yield, Atmos. Chem. Phys., 15, 3063–3075, https://doi.org/10.5194/acp-15-3063-2015, 2015.
Lehtipalo, K., Yan, C., Dada, L., Bianchi, F., Xiao, M., Wagner, R., Stolzenburg, D., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Buchholz, A., Mazon, S. B., Chen, D., Chen, X., Dias, A., Dommen, J., Draper, D. C., Duplissy, J., Ehn, M., Finkenzeller, H., Fischer, L., Frege, C., Fuchs, C., Garmash, O., Gordon, H., Hakala, J., He, X., Heikkinen, L., Heinritzi, M., Helm, J. C., Hofbauer, V., Hoyle, C. R., Jokinen, T., Kangasluoma, J., Kerminen, V. M., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lawler, M. J., Mai, H., Mathot, S., Mauldin, R. L., Molteni, U., Nichman, L., Nie, W., Nieminen, T., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Piel, F., Pospisilova, V., Quéléver, L. L. J., Rissanen, M. P., Rose, C., Sarnela, N., Schallhart, S., Schuchmann, S., Sengupta, K., Simon, M., Sipilä, M., Tauber, C., Tomé, A., Tröstl, J., Väisänen, O., Vogel, A. L., Volkamer, R., Wagner, A. C., Wang, M., Weitz, L., Wimmer, D., Ye, P., Ylisirniö, A., Zha, Q., Carslaw, K. S., Curtius, J., Donahue, N. M., Flagan, R. C., Hansel, A., Riipinen, I., Virtanen, A., Winkler, P. M., Baltensperger, U., Kulmala, M., and Worsnop, D. R.: Multicomponent new particle formation from sulfuric acid, ammonia, and biogenic vapors, Sci. Adv., 4, 1–10, https://doi.org/10.1126/sciadv.aau5363, 2018.
Li, R., Palm, B. B., Ortega, A. M., Hlywiak, J., Hu, W., Peng, Z., Day, D. A., Knote, C., Brune, W. H., De Gouw, J. A., and Jimenez, J. L.: Modeling the radical chemistry in an oxidation flow reactor: Radical formation and recycling, sensitivities, and the OH exposure estimation equation, J. Phys. Chem. A, 119, 4418–4432, https://doi.org/10.1021/jp509534k, 2015.
Lu, K. D., Rohrer, F., Holland, F., Fuchs, H., Bohn, B., Brauers, T., Chang, C. C., Häseler, R., Hu, M., Kita, K., Kondo, Y., Li, X., Lou, S. R., Nehr, S., Shao, M., Zeng, L. M., Wahner, A., Zhang, Y. H., and Hofzumahaus, A.: Observation and modelling of OH and HO2 concentrations in the Pearl River Delta 2006: a missing OH source in a VOC rich atmosphere, Atmos. Chem. Phys., 12, 1541–1569, https://doi.org/10.5194/acp-12-1541-2012, 2012.
Ma, X., Tan, Z., Lu, K., Yang, X., Chen, X., Wang, H., Chen, S., Fang, X., Li, S., Li, X., Liu, J., Liu, Y., Lou, S., Qiu, W., Wang, H., Zeng, L., and Zhang, Y.: OH and HO2 radical chemistry at a suburban site during the EXPLORE-YRD campaign in 2018, Atmos. Chem. Phys., 22, 7005–7028, https://doi.org/10.5194/acp-22-7005-2022, 2022.
McMurry, P. H. and Grosjean, D.: Gas and Aerosol Wall Losses in Teflon Film Smog Chambers, Environ. Sci. Technol., 19, 1176–1182, https://doi.org/10.1021/es00142a006, 1985.
Mehra, A., Wang, Y., Krechmer, J. E., Lambe, A., Majluf, F., Morris, M. A., Priestley, M., Bannan, T. J., Bryant, D. J., Pereira, K. L., Hamilton, J. F., Rickard, A. R., Newland, M. J., Stark, H., Croteau, P., Jayne, J. T., Worsnop, D. R., Canagaratna, M. R., Wang, L., and Coe, H.: Evaluation of the chemical composition of gas- and particle-phase products of aromatic oxidation, Atmos. Chem. Phys., 20, 9783–9803, https://doi.org/10.5194/acp-20-9783-2020, 2020.
Mentel, T. F., Springer, M., Ehn, M., Kleist, E., Pullinen, I., Kurtén, T., Rissanen, M., Wahner, A., and Wildt, J.: Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships, Atmos. Chem. Phys., 15, 6745–6765, https://doi.org/10.5194/acp-15-6745-2015, 2015.
Mohr, C., Thornton, J. A., Heitto, A., Lopez-hil, F. D., Lutz, A., Riipinen, I., Hong, J., Donahue, N. M., Hallquist, M., Petäjä, T., Kulmala, M., and Yli-juuti, T.: Molecular identification of organic vapors driving atmospheric nanoparticle growth, Nat. Commun., 10, 4442, https://doi.org/10.1038/s41467-019-12473-2, 2019.
Molteni, U., Bianchi, F., Klein, F., El Haddad, I., Frege, C., Rossi, M. J., Dommen, J., and Baltensperger, U.: Formation of highly oxygenated organic molecules from aromatic compounds, Atmos. Chem. Phys., 18, 1909–1921, https://doi.org/10.5194/acp-18-1909-2018, 2018.
Müller, J. F., Liu, Z., Nguyen, V. S., Stavrakou, T., Harvey, J. N., and Peeters, J.: The reaction of methyl peroxy and hydroxyl radicals as a major source of atmospheric methanol, Nat. Commun., 7, 1–11, https://doi.org/10.1038/ncomms13213, 2016.
Ng, N. L., Canagaratna, M. R., Zhang, Q., Jimenez, J. L., Tian, J., Ulbrich, I. M., Kroll, J. H., Docherty, K. S., Chhabra, P. S., Bahreini, R., Murphy, S. M., Seinfeld, J. H., Hildebrandt, L., Donahue, N. M., DeCarlo, P. F., Lanz, V. A., Prévôt, A. S. H., Dinar, E., Rudich, Y., and Worsnop, D. R.: Organic aerosol components observed in Northern Hemispheric datasets from Aerosol Mass Spectrometry, Atmos. Chem. Phys., 10, 4625–4641, https://doi.org/10.5194/acp-10-4625-2010, 2010.
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: An overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/c2cs35166h, 2012.
Otkjær, R. V., Jakobsen, H. H., Tram, C. M., and Kjaergaard, H. G.: Calculated Hydrogen Shift Rate Constants in Substituted Alkyl Peroxy Radicals, J. Phys. Chem. A, 122, 8665–8673, https://doi.org/10.1021/acs.jpca.8b06223, 2018.
Palm, B. B., Campuzano-Jost, P., Ortega, A. M., Day, D. A., Kaser, L., Jud, W., Karl, T., Hansel, A., Hunter, J. F., Cross, E. S., Kroll, J. H., Peng, Z., Brune, W. H., and Jimenez, J. L.: In situ secondary organic aerosol formation from ambient pine forest air using an oxidation flow reactor, Atmos. Chem. Phys., 16, 2943–2970, https://doi.org/10.5194/acp-16-2943-2016, 2016.
Peng, Z. and Jimenez, J. L.: Radical chemistry in oxidation flow reactors for atmospheric chemistry research, Chem. Soc. Rev., 49, 2570–2616, https://doi.org/10.1039/c9cs00766k, 2020.
Peng, Z., Day, D. A., Ortega, A. M., Palm, B. B., Hu, W., Stark, H., Li, R., Tsigaridis, K., Brune, W. H., and Jimenez, J. L.: Non-OH chemistry in oxidation flow reactors for the study of atmospheric chemistry systematically examined by modeling, Atmos. Chem. Phys., 16, 4283–4305, https://doi.org/10.5194/acp-16-4283-2016, 2016.
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018.
Pye, H. O. T., D'Ambro, E. L., Lee, B. H., Schobesberger, S., Takeuchi, M., Zhao, Y., Lopez-Hilfiker, F., Liu, J., Shilling, J. E., Xing, J., Mathur, R., Middlebrook, A. M., Liao, J., Welti, A., Graus, M., Warneke, C., de Gouw, J. A., Holloway, J. S., Ryerson, T. B., Pollack, I. B., and Thornton, J. A.: Anthropogenic enhancements to production of highly oxygenated molecules from autoxidation, P. Natl. Acad. Sci. USA, 116, 6641–6646, https://doi.org/10.1073/pnas.1810774116, 2019.
Qi, X., Ding, A., Roldin, P., Xu, Z., Zhou, P., Sarnela, N., Nie, W., Huang, X., Rusanen, A., Ehn, M., Rissanen, M. P., Petäjä, T., Kulmala, M., and Boy, M.: Modelling studies of HOMs and their contributions to new particle formation and growth: comparison of boreal forest in Finland and a polluted environment in China, Atmos. Chem. Phys., 18, 11779–11791, https://doi.org/10.5194/acp-18-11779-2018, 2018.
Qiao, X., Yan, C., Li, X., Guo, Y., Yin, R., Deng, C., Li, C., Nie, W., Wang, M., Cai, R., Huang, D., Wang, Z., Yao, L., Worsnop, D. R., Bianchi, F., Liu, Y., Donahue, N. M., Kulmala, M., and Jiang, J.: Contribution of Atmospheric Oxygenated Organic Compounds to Particle Growth in an Urban Environment, Environ. Sci. Technol., 55, 13646–13656, https://doi.org/10.1021/acs.est.1c02095, 2021.
Qu, H., Wang, Y., Zhang, R., Liu, X., Huey, L. G., Sjostedt, S., Zeng, L., Lu, K., Wu, Y., Shao, M., Hu, M., Tan, Z., Fuchs, H., Broch, S., Wahner, A., Zhu, T., and Zhang, Y.: Chemical Production of Oxygenated Volatile Organic Compounds Strongly Enhances Boundary-Layer Oxidation Chemistry and Ozone Production, Environ. Sci. Technol., 55, 13718–13727, https://doi.org/10.1021/acs.est.1c04489, 2021.
Saunders, S. M., Jenkin, M. E., Derwent, R. G., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 161–180, https://doi.org/10.5194/acp-3-161-2003, 2003.
Stolzenburg, D., Fischer, L., Vogel, A. L., Heinritzi, M., Schervish, M., Simon, M., Wagner, A. C., Dada, L., Ahonen, L. R., Amorim, A., Baccarini, A., Bauer, P. S., Baumgartner, B., Bergen, A., Bianchi, F., Breitenlechner, M., Brilke, S., Mazon, S. B., Chen, D., Dias, A., Draper, D. C., Duplissy, J., Haddad, I. El, Finkenzeller, H., Frege, C., Fuchs, C., Garmash, O., Gordon, H., He, X., Helm, J., Hofbauer, V., Hoyle, C. R., Kim, C., Kirkby, J., Kontkanen, J., Kürten, A., Lampilahti, J., Lawler, M., Lehtipalo, K., Leiminger, M., Mai, H., Mathot, S., Mentler, B., Molteni, U., Nie, W., Nieminen, T., Nowak, J. B., Ojdanic, A., Onnela, A., Passananti, M., Petäjä, T., Quéléver, L. L. J., Rissanen, M. P., Sarnela, N., Schallhart, S., Tauber, C., Tomé, A., Wagner, R., Wang, M., Weitz, L., Wimmer, D., Xiao, M., Yan, C., Ye, P., Zha, Q., Baltensperger, U., Curtius, J., Dommen, J., Flagan, R. C., Kulmala, M., Smith, J. N., Worsnop, D. R., Hansel, A., Donahue, N. M., and Winkler, P. M.: Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range, P. Natl. Acad. Sci. USA, 115, 9122–9127, https://doi.org/10.1073/pnas.1807604115, 2018.
Tan, Z., Fuchs, H., Lu, K., Hofzumahaus, A., Bohn, B., Broch, S., Dong, H., Gomm, S., Häseler, R., He, L., Holland, F., Li, X., Liu, Y., Lu, S., Rohrer, F., Shao, M., Wang, B., Wang, M., Wu, Y., Zeng, L., Zhang, Y., Wahner, A., and Zhang, Y.: Radical chemistry at a rural site (Wangdu) in the North China Plain: observation and model calculations of OH, HO2 and RO2 radicals, Atmos. Chem. Phys., 17, 663–690, https://doi.org/10.5194/acp-17-663-2017, 2017.
Tan, Z., Rohrer, F., Lu, K., Ma, X., Bohn, B., Broch, S., Dong, H., Fuchs, H., Gkatzelis, G. I., Hofzumahaus, A., Holland, F., Li, X., Liu, Y., Liu, Y., Novelli, A., Shao, M., Wang, H., Wu, Y., Zeng, L., Hu, M., Kiendler-Scharr, A., Wahner, A., and Zhang, Y.: Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign, Atmos. Chem. Phys., 18, 12391–12411, https://doi.org/10.5194/acp-18-12391-2018, 2018.
Tan, Z., Lu, K., Jiang, M., Su, R., Wang, H., Lou, S., Fu, Q., Zhai, C., Tan, Q., Yue, D., Chen, D., Wang, Z., Xie, S., Zeng, L., and Zhang, Y.: Daytime atmospheric oxidation capacity in four Chinese megacities during the photochemically polluted season: a case study based on box model simulation, Atmos. Chem. Phys., 19, 3493–3513, https://doi.org/10.5194/acp-19-3493-2019, 2019.
Tröstl, J., Chuang, W. K., Gordon, H., Heinritzi, M., Yan, C., Molteni, U., Ahlm, L., Frege, C., Bianchi, F., Wagner, R., Simon, M., Lehtipalo, K., Williamson, C., Craven, J. S., Duplissy, J., Adamov, A., Almeida, J., Bernhammer, A. K., Breitenlechner, M., Brilke, S., Dias, A., Ehrhart, S., Flagan, R. C., Franchin, A., Fuchs, C., Guida, R., Gysel, M., Hansel, A., Hoyle, C. R., Jokinen, T., Junninen, H., Kangasluoma, J., Keskinen, H., Kim, J., Krapf, M., Kürten, A., Laaksonen, A., Lawler, M., Leiminger, M., Mathot, S., Möhler, O., Nieminen, T., Onnela, A., Petäjä, T., Piel, F. M., Miettinen, P., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Sipilä, M., Smith, J. N., Steiner, G., Tomè, A., Virtanen, A., Wagner, A. C., Weingartner, E., Wimmer, D., Winkler, P. M., Ye, P., Carslaw, K. S., Curtius, J., Dommen, J., Kirkby, J., Kulmala, M., Riipinen, I., Worsnop, D. R., Donahue, N. M., and Baltensperger, U.: The role of low-volatility organic compounds in initial particle growth in the atmosphere, Nature, 533, 527–531, https://doi.org/10.1038/nature18271, 2016.
Tsiligiannis, E., Hammes, J., Salvador, C. M., Mentel, T. F., and Hallquist, M.: Effect of NOx on 1,3,5-trimethylbenzene (TMB) oxidation product distribution and particle formation, Atmos. Chem. Phys., 19, 15073–15086, https://doi.org/10.5194/acp-19-15073-2019, 2019.
Vereecken, L.: Reaction Mechanisms for the Atmospheric Oxidation of Monocyclic Aromatic Compounds, Adv. Atmos. Chem., 377–527, https://doi.org/10.1142/9789813271838_0006, 2019.
Wang, L., Wu, R., and Xu, C.: Atmospheric oxidation mechanism of benzene. Fates of alkoxy radical intermediates and revised mechanism, J. Phys. Chem. A, 117, 14163–14168, https://doi.org/10.1021/jp4101762, 2013.
Wang, M., Chen, D., Xiao, M., Ye, Q., Stolzenburg, D., Hofbauer, V., Ye, P., Vogel, A. L., Mauldin, R. L., Amorim, A., Baccarini, A., Baumgartner, B., Brilke, S., Dada, L., Dias, A., Duplissy, J., Finkenzeller, H., Garmash, O., He, X. C., Hoyle, C. R., Kim, C., Kvashnin, A., Lehtipalo, K., Fischer, L., Molteni, U., Petäjä, T., Pospisilova, V., Quéléver, L. L. J., Rissanen, M., Simon, M., Tauber, C., Tomé, A., Wagner, A. C., Weitz, L., Volkamer, R., Winkler, P. M., Kirkby, J., Worsnop, D. R., Kulmala, M., Baltensperger, U., Dommen, J., El-Haddad, I., and Donahue, N. M.: Photo-oxidation of Aromatic Hydrocarbons Produces Low-Volatility Organic Compounds, Environ. Sci. Technol., 54, 7911–7921, https://doi.org/10.1021/acs.est.0c02100, 2020.
Wang, S., Wu, R., Berndt, T., Ehn, M., and Wang, L.: Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 51, 8442–8449, https://doi.org/10.1021/acs.est.7b02374, 2017.
Wang, W., Yuan, B., Peng, Y., Su, H., Cheng, Y., Yang, S., Wu, C., Qi, J., Bao, F., Huangfu, Y., Wang, C., Ye, C., Wang, Z., Wang, B., Wang, X., Song, W., Hu, W., Cheng, P., Zhu, M., Zheng, J., and Shao, M.: Direct observations indicate photodegradable oxygenated volatile organic compounds (OVOCs) as larger contributors to radicals and ozone production in the atmosphere, Atmos. Chem. Phys., 22, 4117–4128, https://doi.org/10.5194/acp-22-4117-2022, 2022.
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020.
Whalley, L. K., Slater, E. J., Woodward-Massey, R., Ye, C., Lee, J. D., Squires, F., Hopkins, J. R., Dunmore, R. E., Shaw, M., Hamilton, J. F., Lewis, A. C., Mehra, A., Worrall, S. D., Bacak, A., Bannan, T. J., Coe, H., Percival, C. J., Ouyang, B., Jones, R. L., Crilley, L. R., Kramer, L. J., Bloss, W. J., Vu, T., Kotthaus, S., Grimmond, S., Sun, Y., Xu, W., Yue, S., Ren, L., Acton, W. J. F., Hewitt, C. N., Wang, X., Fu, P., and Heard, D. E.: Evaluating the sensitivity of radical chemistry and ozone formation to ambient VOCs and NOx in Beijing, Atmos. Chem. Phys., 21, 2125–2147, https://doi.org/10.5194/acp-21-2125-2021, 2021.
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G., and Wennberg, P. O.: New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene, Environ. Sci. Technol., 54, 13467–13477, https://doi.org/10.1021/acs.est.0c04780, 2020.
Yan, C., Kocevska, S., and Krasnoperov, L. N.: Kinetics of the Reaction of CH3O2 Radicals with OH Studied over the 292–526 K Temperature Range, J. Phys. Chem. A, 120, 6111–6121, https://doi.org/10.1021/acs.jpca.6b04213, 2016.
Yang, C., Yao, N., Xu, L., Chen, G., Wang, Y., Fan, X., Zhou, P., Clusius, P., Tham, Y. J., Lin, Z., Chen, Y., Li, M., Hong, Y., and Chen, J.: Molecular Composition of Anthropogenic Oxygenated Organic Molecules and Their Contribution to Organic Aerosol in a Coastal City, Environ. Sci. Technol., 57, 15956–15967, https://doi.org/10.1021/acs.est.3c03244, 2023.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Kontkanen, J., Junninen, H., Mazon, S. B., Ehn, M., Paasonen, P., Sipilä, M., Wang, M., Wang, X., Xiao, S., Chen, H., Lu, Y., Zhang, B., Wang, D., Fu, Q., Geng, F., Li, L., Wang, H., Qiao, L., Yang, X., Chen, J., Kerminen, V.-M., Petäjä, T., Worsnop, D. R., Kulmala, M., and Wang, L.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 80, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
Yuan, B., Chen, W., Shao, M., Wang, M., Lu, S., Wang, B., Liu, Y., Chang, C. C., and Wang, B.: Measurements of ambient hydrocarbons and carbonyls in the Pearl River Delta (PRD), China, Atmos. Res., 116, 93–104, https://doi.org/10.1016/j.atmosres.2012.03.006, 2012.
Zaytsev, A., Koss, A. R., Breitenlechner, M., Krechmer, J. E., Nihill, K. J., Lim, C. Y., Rowe, J. C., Cox, J. L., Moss, J., Roscioli, J. R., Canagaratna, M. R., Worsnop, D. R., Kroll, J. H., and Keutsch, F. N.: Mechanistic study of the formation of ring-retaining and ring-opening products from the oxidation of aromatic compounds under urban atmospheric conditions, Atmos. Chem. Phys., 19, 15117–15129, https://doi.org/10.5194/acp-19-15117-2019, 2019.
Zhao, Y., Thornton, J. A., and Pye, H. O. T.: Quantitative constraints on autoxidation and dimer formation from direct probing of monoterpene-derived peroxy radical chemistry, P. Natl. Acad. Sci. USA, 115, 12142–12147, https://doi.org/10.1073/pnas.1812147115, 2018.