the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 05 Jun 2024
| 05 Jun 2024
Dual roles of the inorganic aqueous phase on secondary organic aerosol growth from benzene and phenol
Jiwon Choi
Spencer Blau
Benzene, emitted from automobile exhaust and biomass burning, is ubiquitous in ambient air. Benzene is a precursor hydrocarbon (HC) that forms secondary organic aerosol (SOA), but its SOA formation mechanism is not well studied. To accurately predict the formation of benzene SOA, it is important to understand the gas mechanisms of phenol, which is one of the major products formed from the atmospheric oxidation of benzene. Laboratory data presented herein highlight the impact of the aqueous phase on SOA generated through benzene and phenol oxidation. The roles of the aqueous phase consist of (1) suppression of the aging of hydrocarbon and (2) conventional acid-catalyzed reactions in the inorganic phase. To explain this unusual effect, it is hypothesized that a persistent phenoxy radical (PPR) effectively forms via a heterogeneous reaction of phenol and phenol-related products in the presence of wet inorganic aerosol. These PPR species are capable of catalytically consuming ozone during an NOx cycle and negatively influencing SOA growth. In this study, explicit gas mechanisms were derived to produce the oxygenated products from the atmospheric oxidation of phenol or benzene. Gas mechanisms include the existing Master Chemical Mechanism (MCM v3.3.1), the reaction path for peroxy radical adducts originating from the addition of an OH radical to phenols forming low-volatility products (e.g., multi-hydroxy aromatics), and the mechanisms to form heterogeneous production of PPR. The simulated gas products were classified into volatility- and reactivity-based lumped species and incorporated into the Unified Partitioning Aerosol Reaction (UNIPAR) model that predicts SOA formation via multiphase reactions of phenol or benzene. The predictability of the UNIPAR model was examined using chamber data, which were generated for the photooxidation of phenol or benzene under controlled experimental conditions (NOx levels, humidity, and inorganic seed types). The SOA formation from both phenol and benzene still increased in the presence of wet inorganic seed because of the oligomerization of reactive organic species in the aqueous phase. However, model simulations show a significant suppression of ozone, the oxidation of phenol or benzene, and SOA growth compared with those without PPR mechanisms. The production of PPR is accelerated in the presence of acidic aerosol and this weakens SOA growth. In benzene oxidation, up to 53 % of the oxidation pathway is connected to phenol formation in the reported gas mechanism. Thus, the contribution of PPR to gas mechanisms is less than that of phenol. Overall, SOA growth in phenol or benzene is negatively related to NOx levels in the high-NOx region (HC ppbC / NOx ppb < 5). However, the simulation indicates that the significance of PPR rises with decreasing NOx levels. Hence, the influence of NOx levels on SOA formation from phenol or benzene is complex under varying temperature and seed type conditions. Adding the comprehensive reaction of phenolic compounds will improve the prediction of SOA formation from aromatic HCs due to the missing mechanisms in the current air quality model.
- Article
(6767 KB) - Full-text XML
-
Supplement
(1111 KB) - BibTeX
- EndNote





Hydrocarbons (HCs) are emitted from both anthropogenic (e.g., fuel combustion, vehicle exhaust, and industrial activities) and biogenic (e.g., vegetation) sources (Carlton et al., 2010). The photochemical oxidation of these HCs can produce ozone via incorporation with the NOx cycle. In addition, the formation of semi-volatile or nonvolatile oxygenated products through a series of chemical reactions of precursor HCs in the atmospheric environment can yield secondary organic aerosol (SOA). This SOA constitutes a large proportion of organic aerosol in the ambient air, ranging from 20 % at midlatitudes to 90 % in tropical forested area (Jimenez et al., 2009; Zhang et al., 2007; Kanakidou et al., 2005). Hence, the estimation of SOA formation potential is important to accurately evaluate the impact of atmospheric organic aerosol on health and climate formation.
Benzene, the simplest aromatic HC, is emitted mainly from automobile exhaust, and it can be found in gases from biomass burning. As it is a solvent, benzene is also emitted from industrial processes such as chemical synthesis, construction, and pharmaceutical facilities (Verma and Tombe, 2002; Wang et al., 2014). In addition, benzene is known to be emitted from oceans, including the remote Southern Ocean and the Arctic marginal ice zone (Wohl et al., 2023). The major atmospheric oxidation path of benzene is the reaction with OH radicals. Benzene's oxidation rate (i.e., cm3 molecule−1 s−1 at 298 K) (Borrás and Tortajada-Genaro, 2012) is relatively slow, but its SOA yield is high. According to laboratory studies (Ng et al., 2007), benzene's SOA yields can be even greater than high-yield aromatic HCs (i.e., toluene) under given experimental conditions. However, the prediction of benzene SOA has not been well studied due to uncertainties in both gas oxidation mechanisms and aerosol-phase reactions that form oligomeric species under various environmental conditions.
In benzene oxidation with an OH radical, about 53 % of pathways are linked to the formation of phenol, while the remaining oxidation paths are connected to ring-opening products in the current Master Chemical Mechanism (MCM v3.3.1) (Bloss et al., 2005). In addition, a significant fraction (20 %) of oxygenated aromatic, emitted from biomass burning, is phenol (Akherati et al., 2020). Hence, understanding phenol oxidation mechanisms is a key to predicting benzene oxidation and SOA formation. Phenol gas oxidation includes the reaction path for peroxy radical adducts originating from the addition of an OH radical to phenols to form low-volatility products (e.g., multi-hydroxy aromatics). Laboratory studies report that the gas oxidation of phenol forms low-volatility highly oxygenated organic molecules (HOMs) (Nakao et al., 2011; Ji et al., 2017; Yee et al., 2013). Phenols are highly reactive to the addition of OH radicals to an aromatic ring due to the electron-receiving characteristics of the phenolic OH group. Phenols effectively produce multi-hydroxybenzenes compared with conventional alkyl-substituted monocyclic aromatic HCs (Hansch et al., 2000).
In addition to gas mechanisms to form HOMs, phenol and phenolic products (e.g., catechols and nitrophenols) can yield persistent phenoxy radical (PPR) species, which can potentially suppress the atmospheric oxidation capability and SOA growth. Unlike aliphatic alkoxy radicals, which can react with an oxygen molecule to form a hydroperoxyl (HO2) radical, PPR species have no aliphatic hydrogen at the carbon attached to the O radical. A p–π conjugated system can help stabilize phenoxy radicals. Thus, these phenoxy radicals have a relatively long lifetime in ambient air and can catalytically deplete ozone (Tao and Li, 1999).
In a recent modeling study, Choi and Jang (2022) explicitly predicted phenol gas oxidation, including the formation of HOMs and other multi-hydroxyphenols (Pillar-Little et al., 2015; Choi and Jang, 2022; Yee et al., 2013; Yu et al., 2016). The resulting oxidation products were categorized into volatility- and reactivity-based lumped species and integrated into the Unified Partitioning Aerosol Reaction (UNIPAR) model, developed by Im et al. (2014), which simulated SOA formation via multiphase reactions of HCs. Choi and Jang showed the importance of HOMs to predict the formation of phenol SOA through the simulation of chamber data using the UNIPAR model (Choi and Jang, 2022). In the absence of wet inorganic seed aerosol, where the relative humidity (RH) was kept above efflorescence relative humidity (ERH) to avoid the crystallization of seed, their model successfully simulated SOA formation; however, in the presence of wet inorganic seed, the model failed to predict both gas oxidation (ozone, NOx, and decay of phenol) and SOA formation. In the last 2 decades, the impact of aerosol acidity on SOA has been studied in numerous laboratories (Jang et al., 2002; Garland et al., 2006; Hallquist et al., 2009; Deng et al., 2021; Surratt et al., 2007a). The reactive oxygenated products, formed from the oxidation of biogenic and aromatic HCs, undergo acid-catalyzed reactions (e.g., hydration, oligomerization, formation of hemiacetal/acetal/trioxane, aldol condensation, and cationic rearrangement) in the presence of acidic inorganic aerosol and accelerate SOA formation. However, Choi and Jang (2022) observed an unexpected impact of aerosol acidity on SOA formation, which suppressed SOA formation more than neutral, wet ammonium sulfate (AS) seed. As the wet inorganic aerosol suppresses gas oxidation (ozone and the decay of phenol), it is unlikely to be associated with the inaccuracy of the oligomerization of reactive oxygenated species in the aqueous phase. This finding is distinct from the typical SOA formation from alkyl-substituted aromatics or biogenic HCs, which are positively correlated with aerosol acidity (Jang et al., 2002; Hallquist et al., 2009).
Table 1Chamber experimental conditions for SOA formation from the photooxidation of phenol and benzene under various conditions.
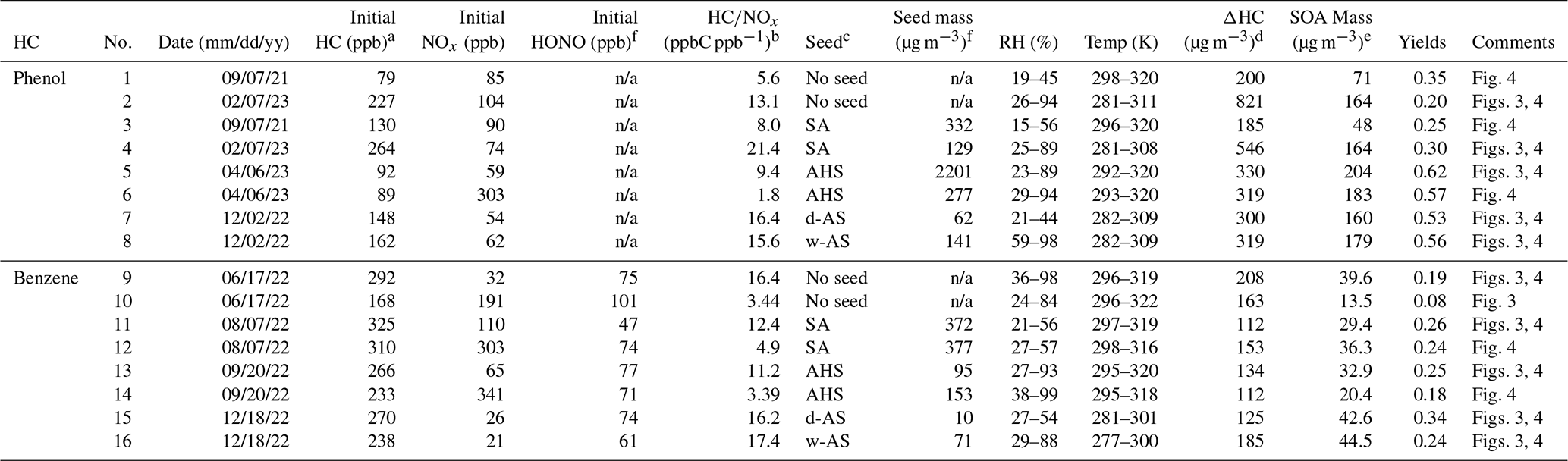
a High NOx: HC ppbC / NOx ppb < 5; low NOx: HC/NOx > 5 ppbC ppb−1. b The seed condition refers to the injected electrolytic seed: sulfuric acid (SA), ammonium hydrogen sulfate (AHS), wet ammonium sulfate (w-AS), and dry ammonium sulfate (d-AS). c ΔHC is difference between the initial HC concentration and the HC concentration at 16:00 EST. d The SOA mass is determined with OC data at 16:00 EST. e The reported SOA mass was corrected for the particle loss to the chamber wall based on the first-order deposition rate at 64 particle size beans. The dilution rate of SOA is estimated with the gas dilution factor determined using trace gas (CCl4). f n/a denotes not applicable.
In this study, we hypothesize, based on chamber experiments and complex model data, that the production of PPR species from the atmospheric oxidation of phenol and its phenolic products (e.g., catechols and nitrophenols) can be modulated via heterogeneous reactions in wet inorganic aerosol. Increased PPR production via the heterogeneous oxidation of phenolic compounds in the presence of wet inorganic aerosol catalytically consumes ozone during an NOx cycle and, ultimately, influences SOA formation from both phenol and benzene. A gas mechanism, named the Heterogeneous Phenoxy Radical model (H-PPR model), was derived to improve the prediction of both the gas oxidation of phenol and benzene and their SOA formation in presence of the aqueous phase. The prediction of phenol oxidation was improved by integrating HOMs and the H-PPR into explicit gas mechanisms. Similarly, representation of benzene gas oxidation was improved using updated phenol mechanisms. Ultimately the resulting gas mechanisms of phenol or benzene were then applied to the UNIPAR model to predict SOA formation via the multiphase reactions of phenol and benzene. The suitability of the UNIPAR model was demonstrated by comparing simulations and chamber data obtained from the photooxidation of phenol and benzene under different experimental conditions in a large outdoor photochemical reactor.
Importantly, both phenol and benzene are abundant in biomass burning smoke, and they can largely contribute to SOA formation (Majdi et al., 2019). Despite this recognition, however, several unresolved issues persist, such as understanding the gas-phase mechanisms involved and the role of the aqueous phase in SOA formation. The SOA model of this study can augment the evaluation of the impact of NOx levels on SOA formation during wildfires under the rural set (low NOx) and the urban set (high NOx). The wildfire plume can transport emissions thousands of kilometers from the wildfire source (Edwards et al., 2006; Wotawa and Trainer, 2000), influencing background atmosphere air quality (Hudson et al., 2004; Schill et al., 2020). For example, in the European Mediterranean area, annual biomass burning emission have been found to account for 19 %–21 % of organic carbon levels in particulate matter in Barcelona, Spain (Reche et al., 2012). Outdoor chamber data from this study and gas mechanisms of phenols can augment a better understanding of the impact of biomass burning smoke on the atmospheric oxidation ability of hydrocarbons and SOA formation in the city.
To generate SOA, the University of Florida Atmospheric PHotochemical Outdoor Reactor (UF-APHOR) was used. The reactor consists of dual Teflon film chambers, each with a volume of 52 m3 and a surface area of 86 m2, that are exposed to ambient sunlight. SOA is produced by the photooxidation of phenol (99 %; Acros Organics) or benzene (≥ 99 %; Sigma-Aldrich) under varying NOx levels and using different inorganic seed types, namely, no seed; sulfuric acid – SA; ammonium hydrogen sulfate - AHS; and ammonium sulfate – AS. Detailed descriptions of the chamber experiment procedure have been reported in previous studies (Beardsley and Jang, 2016; Choi and Jang, 2022; Han and Jang, 2022, 2023; Im et al., 2014; Yu et al., 2021b; Zhou et al., 2019). In summary, NO (2 % in N2; Airgas, USA), HCs, nonreactive CCl4 (≥ 99.5 %; Sigma-Aldrich), and inorganic seed aerosol were injected into the chamber before sunrise. HCs (phenol or benzene) were vaporized into the chamber using a glass manifold under clean-air streams. For seeded SOA experiments, 0.05M SA or AHS or 0.05M AS aqueous solution was atomized into the chamber using a nebulizer (LC STAR; PARI, Starnberg, Germany). For dry-AS-seeded experiments, the RH was maintained below ERH, whereas the RH was maintained above ERH for wet-AS-seeded experiments in order to prevent crystallization of the seed.
A photometric ozone analyzer (Model 106-L; 2B Technologies, MA, USA) and a chemiluminescence NO/NOx analyzer (Model 405; 2B Technologies, MA, USA) were used to monitor the concentrations of ozone and NOx, respectively. The error associated with NO, NO2, and O3 was 2 %. A gas chromatograph with a flame ionization detector (GC-FID) (7820A; Agilent Technologies, CA, USA) was employed to measure the concentrations of phenol or benzene. A proton-transfer-reaction time-of-flight mass spectrometer (PTR-ToF-MS) (PTR 3C; Kore Technology, Cambridgeshire, UK) was also utilized to monitor the decay of phenol or benzene. To monitor air dilution in the chamber, CCl4 was introduced into the chamber and measured using the GC-FID.
A scanning mobility particle sizer (SMPS) comprising an aerosol classifier (Model 3082; TSI, MN, USA) and a condensation particle counter (Model 3750; TSI) was used to measure the particle population and volume concentration. The aerosol chemical speciation monitor (ACSM) was used to measure the quantity of sulfate, ammonium, and nitrate ions in aerosol phase. An organic carbon/elemental carbon (OC/EC) aerosol analyzer (Sunset Laboratory, OR, USA) was employed to measure the concentration of organic carbon in the aerosol. An ion chromatograph (Compact IC 761) was used to measure the concentration of water-soluble inorganic species (sulfate, nitrate, and ammonium ions) coupled to a particle-into-liquid sampler (PILS-IC) (ADISO 2081). All gas data were corrected for chamber air dilution. All aerosol data were corrected for gas dilution and aerosol loss to the chamber wall, as performed in previous chamber studies (Beardsley and Jang, 2016; Choi and Jang, 2022; Han and Jang, 2022, 2023; Im et al., 2014; Yu et al., 2021b; Zhou et al., 2019).
A hygrometer (CR1000 measurement and control system; Campbell Scientific, UT, USA) was used to measure temperature and RH within the chamber. Sunlight intensity was monitored with a total UV radiometer (TUVR; Eppley Laboratory, RI, USA). Table 1 provides a summary of the experimental conditions used in the chamber for this study. Figure S1 displays the profiles of sunlight, temperature, and humidity on 19 October 2022.
3.1 UNIPAR SOA model
The UNIPAR model simulates SOA formation via the multiphase reaction of phenol or benzene. Figure 1 displays the overall structure of the UNIPAR model. Briefly, the key components of the UNIPAR model are described as follows:
-
As seen in Fig. 1, the oxidized products predicted from near-explicit mechanisms of phenol or benzene in the gas phase (g) are categorized into 50 lumped groups based on their reactivity and volatility. Table S1 displays the physicochemical parameters (i.e., molecular weight – MWi; oxygen-to-carbon ratio – O:Ci; and hydrogen bonding – HBi) of lumped species, which are used to process multiphase partitioning and aerosol chemistry. The predetermined mathematical equations (Tables S2–S7) dynamically build the stoichiometric coefficient arrays for each precursor (benzene and phenol). Tables S8 and S9 illustrate major products placed in lumping arrays. No difference appears between Tables S8 and S9 with respect to the major product of each lumped group. The estimated stoichiometric coefficient reflects the influence of NOx levels and gas aging on gas product distributions. The distribution of products was also influenced by H-PPR as a function of the amount of sulfuric acid. H-PPR increases the contribution of the fresh-product distribution.
-
The concentration of lumped species is distributed into the gas (Cg), organic (Cor), and inorganic phases (Cin) using partitioning coefficients estimated based on Pankow's absorptive partitioning model (Pankow, 1994) with the vapor pressure, the estimated activity coefficients of lumped species in both the organic and inorganic phases, and aerosol's average molecular weight in each phase.
-
The resulting Cor and Cin of each lumped species are applied to process SOA formation via their multiphase partitioning (OMP) and aerosol-phase reactions (OMAR) in both the organic and inorganic phases.
-
The kinetic parameters to calculate the aerosol-phase reaction rate constants in the organic and inorganic phases, such as lumped species' reactivity scales and their basicity constants, are reported Sect. S2. Both organic-phase oligomerization and aqueous reactions of reactive species in the inorganic phase yield nonvolatile organic matter (OM) in the model.
-
The SOA mass formed from gas–organic partitioning (OMP) is estimated using the Newtonian method (Schell et al., 2001) based on a mass balance of organic compounds between the gas and particle phases governed by Raoult's law. OMAR is considered to be a preexisting absorbing material for gas–particle partitioning (Cao and Jang, 2007; Im et al., 2014).
-
The inorganic composition and aerosol acidity, which are predicted using the inorganic thermodynamic model, are incorporated into the UNIPAR model. The deliquescence RH (DRH), which is predicted using the equation derived from the inorganic thermodynamic model, and the ERH, which is predicted using a pretrained neural network model based on the inorganic composition (Yu et al., 2021a), are used to determine the aerosol state: wet (organic phase + inorganic aqueous phase) or dry (organic phase + solid dry inorganic phase).
-
In the model, the formation of dialkylsulfate (Liggio et al., 2005; Surratt et al., 2007b; Li et al., 2015) is simulated using a Hinshelwood-type reaction (Im et al., 2014). The decreased acidic sulfate due to the dialkylsulfate formation is applied to inorganic compositions to calculate aerosol acidity and aerosol water content for the next step.
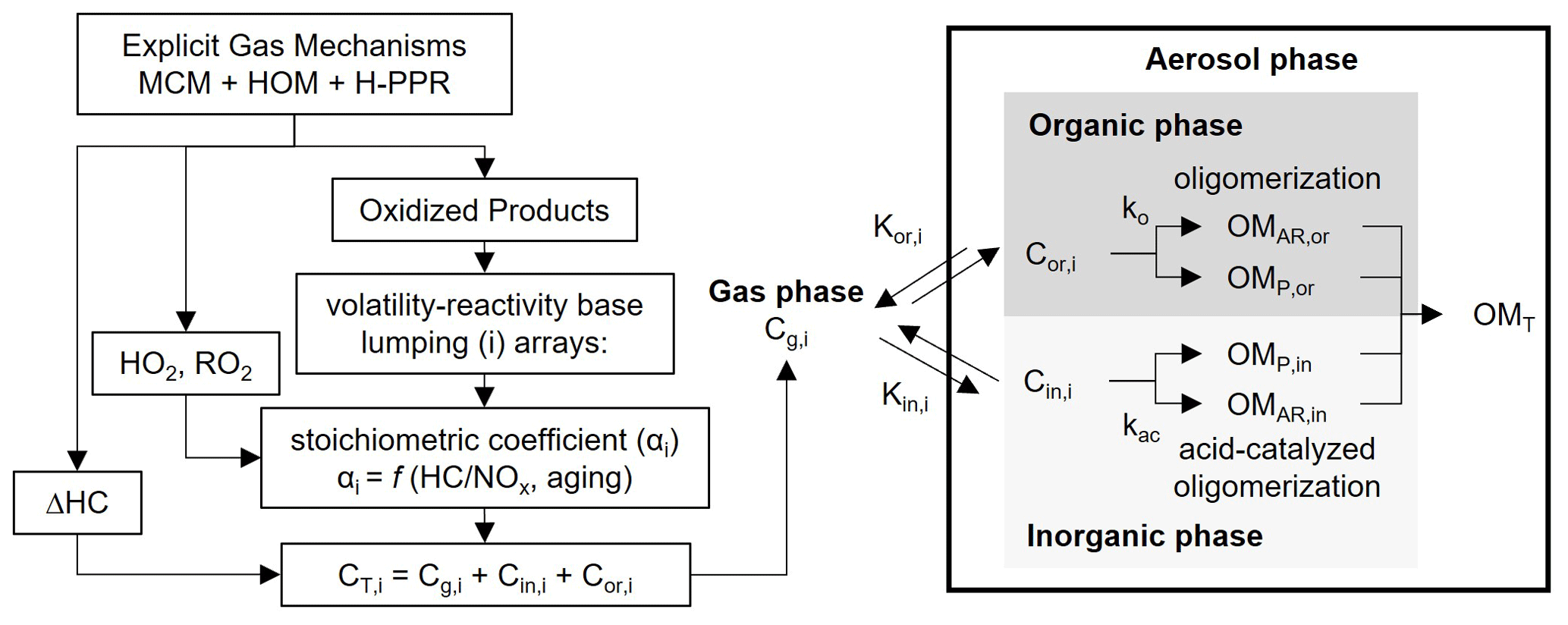
Figure 1Scheme of the UNIPAR model to predict SOA formation from the multiphase oxidation of phenol or benzene. The oxidized products predicted from the modified explicit gas mechanism (MCM v.3.3.1) integrated with the H-PPR model are classified into 50 lumped species (i) based on volatility and reactivity. The consumption of hydrocarbons (ΔHCs) and the concentration of hydroperoxy radical (HO2), alkylperoxy radical (RO2), and the organic products are simulated using explicit gas mechanisms. These values are integrated with stoichiometric coefficients in UNIPAR to estimate concentrations of lumping species. The stoichiometric coefficient is dynamically constructed as a function of the HC ppbC / NOx ratio and the aging scale, which is estimated with the concentrations of HO2 and RO2 radicals. “C” denotes the concentration of an organic compound and “K” denotes the partitioning coefficient of an organic compound. Subscripts “g”, “or”, and “in” represent the gas, organic, and inorganic phases, respectively. OM refers to the organic matter in aerosol. Subscripts “AR”, “P”, and “T” denote aerosol-phase reaction, partitioning, and total.
3.2 Explicit gas mechanisms
The gas oxidation of benzene and phenols was processed with the Master Chemical Mechanism (MCM v3.3.1) (Jenkin et al., 2003) coupled with the reaction path for the formation of HOM products (e.g., multi-hydroxy aromatics) and the mechanisms to form PPR via heterogeneous acid-catalyzed reactions of phenolic compounds. The simulation of the atmospheric oxidation of HCs is performed using a box model platform equipped with the Dynamically Simple Model of Atmospheric Chemical Complexity (DSMACC) incorporated with the Kinetic PreProcessor (KPP) (Emmerson and Evans, 2009). The description of the mechanism to form HOMs and PPR is shown in the following sections.
3.2.1 HOM formation
The gas mechanism to form HOMs in phenol oxidation has been reported in a recent study by Choi and Jang (2022). Figure S2 depicts the pathways to form HOMs from the oxidation of phenol. The resulting phenol gas mechanisms are integrated with benzene gas oxidation. The oxidation of phenol or benzene begins with the reaction with an OH radical and forms HOMs via multi-generation oxidation (Nakao et al., 2011; Yee et al., 2013; Sun et al., 2010; Garmash et al., 2020; Calvert et al., 2002; Atkinson, 2000; Olmez-Hanci and Arslan-Alaton, 2013). HOMs include multi-hydroxy benzenes, phenolic compounds, and the products derived from peroxy radical adducts of multi-hydroxy benzenes. The gas-phase reaction rate constants for the addition of the OH radical to different aromatic compounds were calculated with the structure–reactivity relationship using Hammett parameters (Kwok and Atkinson, 1995; Brown and Okamoto, 1958). This estimation method can be reliable when used within its database, but extrapolation to organic compounds outside of the database can result in a lack of assurance with respect to its accuracy.
3.2.2 PPR formation
The first-generation products from phenol gas oxidation include a phenoxy radical (fraction in oxidation paths: 0.06), catechol (0.657), bicyclic peroxy radical (0.183), and monocyclic peroxy radical (0.1) (Bloss et al., 2005; Jenkin et al., 2003). The resulting phenoxy radical (C6H5O⋅) in the gas phase can catalytically react with ozone (Tao and Li, 1999).
Studies have shown that phenol and benzene have the relatively low minimal incremental reactivity (MIR) or photochemical ozone creation potential (POCP) values (Carter, 1994; Jenkin et al., 2017; Zhang et al., 2021). Such low ozone formation potential can be explained by the catalytic consumption of ozone by PPR. Phenyl peroxy radical (C6H5OO⋅) is able to regenerate C6H5O⋅ via reactions in the gas phase with NO, NO2, and NO3 (Carter and Atkinson, 1989; Jagiella and Zabel, 2007).
The catalytic decay of ozone influences the production of the OH radical that can be created via the reaction of water vapor with O(1D), a photolysis product of ozone (Finlayson-Pitts and Pitts, 2000). Furthermore, the oxidation of phenol or coexisting HCs can be retarded due to PPR. Figure 2 illustrates the time series of gas simulations and observations in the UF-APHOR chamber. The gas mechanisms reasonably simulate the low ozone formation and the retarded oxidation of phenol or benzene in the absence of inorganic seed. However, the suppression of the gas oxidation capability increased in the presence of wet AS aerosol and was further amplified with increasing aerosol acidity. This surprising discovery suggests that there should be a reaction path to heterogeneously form PPR.
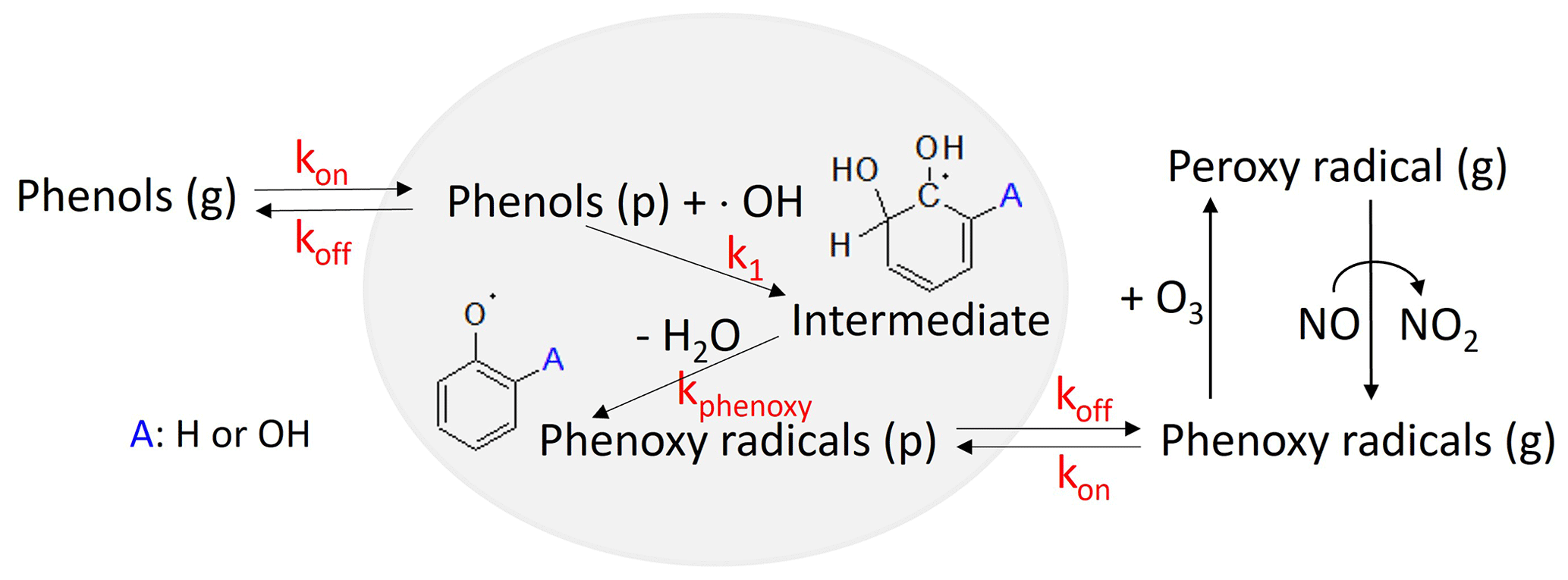
Figure 2An overview of the kinetic mechanisms that streamlines the heterogeneous phenoxy radical formation (H-PPR) of gaseous phenols in the presence of acidic aerosol. kon and koff denote the respective uptake rate constant and desorption rate constant for phenols in the presence of acidic aerosol. k1 is the rate constant to heterogeneously form intermediates (hydroxyphenols) and kphenoxy is the rate constant to form an intermediate to a phenoxy radical.
The formation of phenoxy radicals has been reported in condensed matrixes at low temperatures (Sun et al., 1990), strong liquid acids (Dixon and Murphy, 1976), or gas-phase clusters under specific conditions (Steadman and Syage, 1991). The existence of phenoxy radicals has been detected in strong acidic solutions using electron spin resonance (ESR) spectroscopy (Holton and Murphy, 1979). Phenol-like compounds, such as phenol, catechol, and pyrogallol (1,2,3-trihydroxybenzene), partition to salted aqueous aerosol and can heterogeneously react with the aqueous-phase OH radical. Various oxidants, such as OH, HO2, and ozone, can partition into the aqueous phase and oxidize hydrophilic organic species. Similar to the gas phase, the OH radical can be added into a phenolic aromatic ring to form an OH-added intermediate phenol (HO-C6H5•OH) (phenol_OH_int) in the aqueous phase (Mvula et al., 2001). The resulting phenol_OH_int can form the phenoxy radical, yielding a water molecule (Das, 2005; Mvula et al., 2001). This reaction step is accelerated by acid catalysis.
In this study, the H-PPR mechanism was integrated into the explicit gas mechanisms accounting for the impact of the aqueous phase on SOA formation from phenol and benzene, as illustrated in Fig. 2. In this mechanism, the partitioning of phenols between the g and inorganic (in) phases is kinetically expressed using the absorption rate constant (kon_phenol) and the desorption rate constant (koff_phenol) as follows:
To describe the production of PPR, phenol is used, but various phenolic compounds can also be involved in the formation of H-PPR. kon_phenol is calculated as follows:
where fS_M is the aerosol surface area concentration (m2 m−3), i.e., m2 µg−1 for a particle size near 100 nm; fabs is the coefficient for the uptake process and is set to 2 in order to gear fast gas–particle partitioning in the model; and ω is the mean molecular velocity (m s−1) of each chemical species. ω is calculated as follows:
where MW is the molecular weight (kg mol−1) of organic species, R is a gas constant (8.314 J mol−1 K−1), and T (K) is an absolute temperature.
In the presence of wet inorganic seed aerosol, the lumped species produced from the oxidation of HCs are split into the g, organic, and inorganic phases by the gas–aerosol absorptive partitioning model (Pankow, 1994). The g–in partitioning coefficient (Kin,i) (m3 µg−1) is expressed as follows:
where MWin represents the average molecular weight (g mol−1) of inorganic-phase aerosol and is the liquid vapor pressure (in mm Hg) of the product i, which is calculated using the group contribution method (Jang and Kamens, 1997, 1998; Zhou et al., 2019). The activity coefficient (γor,i) of organic species i in the organic phase is treated as 1 (Im et al., 2014). The activity coefficient of i in the inorganic phase (γin,i) is predicted using a semiempirical regression equation (Zhou et al., 2019). The theoretical estimation of γin,i was conducted using the thermodynamic Aerosol Inorganic–Organic Mixtures Functional groups Activity Coefficients (AIOMFAC; Zuend et al., 2011) for the given set of conditions and aerosol parameters. γin,i is a function of the aerosol environment variables (RH ranging from 0 to 1 and fractional sulfate (FS = [SO]/[SO]+[NH])) and the physicochemical parameters of lumped species i (MWi, O:Ci, and HBi):
Phenol can be both a donor and an accepter for hydrogen bonding, while a phenoxy radical can be only a hydrogen bonding accepter. We assumed that the oxygen radical in the phenoxy radical is treated as a ketone functional group to calculate its vapor pressure and activity coefficient. koff_phenol is inversely related to the partitioning coefficient (Kin,i; Fig. 1) of species i (phenol) in the inorganic phase via the following relation:
The g–in partitioning coefficient of atmospheric oxidant, ox (i.e., H2O2, HONO, HO2, OH, O3, NO, NO2, CH3OOH, and CH3CO3H), is estimated using Henry's constant (KH,i):
where Min (g L−1) is the mass concentration of the inorganic phase and Vin (cm−3 L−1) is the volume mixing ratio of the inorganic phase.
Phenol (in) in Reaction (R6) further reacts with the OH radical (OH(in)) in the inorganic phase to form an intermediate adduct of phenol (phenol_OH_int(in)) at reaction rate constant k1, a second-order rate constant which is determined empirically and very quickly as seen below.
Intermediate adduct phenol_OH_int (in) can effectively generate a phenoxy radical, C6H5O⋅(in) via an acid-catalyzed reaction:
where kphenoxy=k1 ⋅ . is the proton concentration (mol L−1) in the aqueous phase.
In a similar manner to phenols (Reactions R6 and R7), the resulting phenoxy radical can also partition between the gas and particle phases as follows:
The formation of phenol_OH_int(in) is the rate-determining step in the mechanism. In the absence of an acid catalyst, phenol_OH_int(in) can be further oxidized via the reaction with an oxygen molecule to form catechol and other multifunctional carbonyls (Xu and Wang, 2013). The produced C6H5O⋅(g) is involved in the reaction with ozone (Reaction R2), as seen in Fig. 2, and catalytically reduces the atmospheric oxidation capacity.
4.1 Simulation of gas oxidation and SOA formation
Figure 3 illustrates the gas simulations (phenol, benzene, ozone, NO, and NO2) for chamber data obtained in the UF-APHOR chamber. Overall, explicit gas simulations including HOMs and H-PPR agree well with observations. In the presence of wet inorganic seed (i.e., wet AS, AHS, and SA), both simulations and chamber data show a significant suppression of gas oxidation (i.e., ozone formation and the decay of phenol or benzene) compared with the gas oxidation in non-seeded conditions. For example, simulations deviate considerably from observation in both Fig. 3b and h when simulations are performed with gas mechanisms excluding the H-PPR mechanism for phenol and benzene oxidation in the presence of SA seed. Under the same experimental conditions, simulations with H-PPR in Fig. 3c and h predict chamber data well, showing the importance of H-PPR (Reactions R3–R5). In addition to phenol, catechols and nitrophenols, which are major products of phenol oxidation, can also undergo PPR formation. The suppressed ozone can lessen the production of OH radical and further retard the aging of organic products.
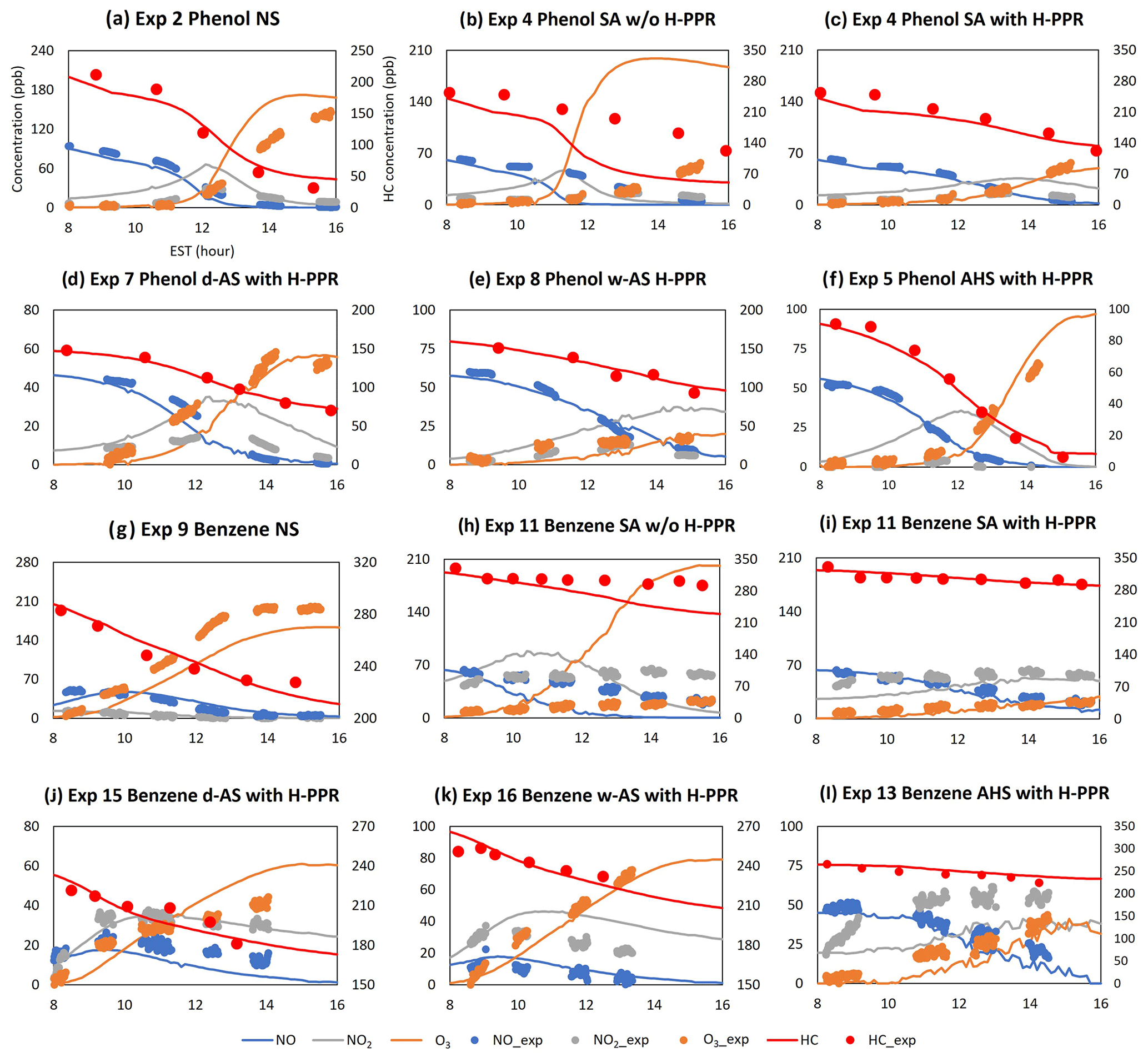
Figure 3The time profiles of observations and the prediction for concentrations of NO, NO2, O3, and hydrocarbons (Table 1). The x axis represents time (EST), the first y axis represents the concentration (ppb) of gas species, and the second y axis represents the hydrocarbon concentration in the gas phase (ppb) as shown in panel (a). “HC” and “HCexp” denote the gas simulation of hydrocarbons used in experiment and the measurement of hydrocarbon used in experiment, respectively. The error associated with NO, NO2, and O3 is 2 % and is not visible in this figure.
Figure 4a–p show chamber-generated SOA mass from the photooxidation of phenol (Fig. 4a–h) or benzene (Fig. 4i–p) under different inorganic seed conditions (Table 1) and the simulations of SOA formation using the UNIPAR model. Non-seed phenol SOA (Fig. 4a, b) and non-seed benzene (Fig. 4i, j) are shown. SOA masses produced in the presence of inorganic seed (SA, AHS, wet AS, or dry AS) are depicted in Fig. 4c–h for phenol and Fig. 4k–p for benzene. Overall, the improved SOA simulation of phenol or benzene was also found with the improved gas simulation incorporating with HOM and H-PPR.
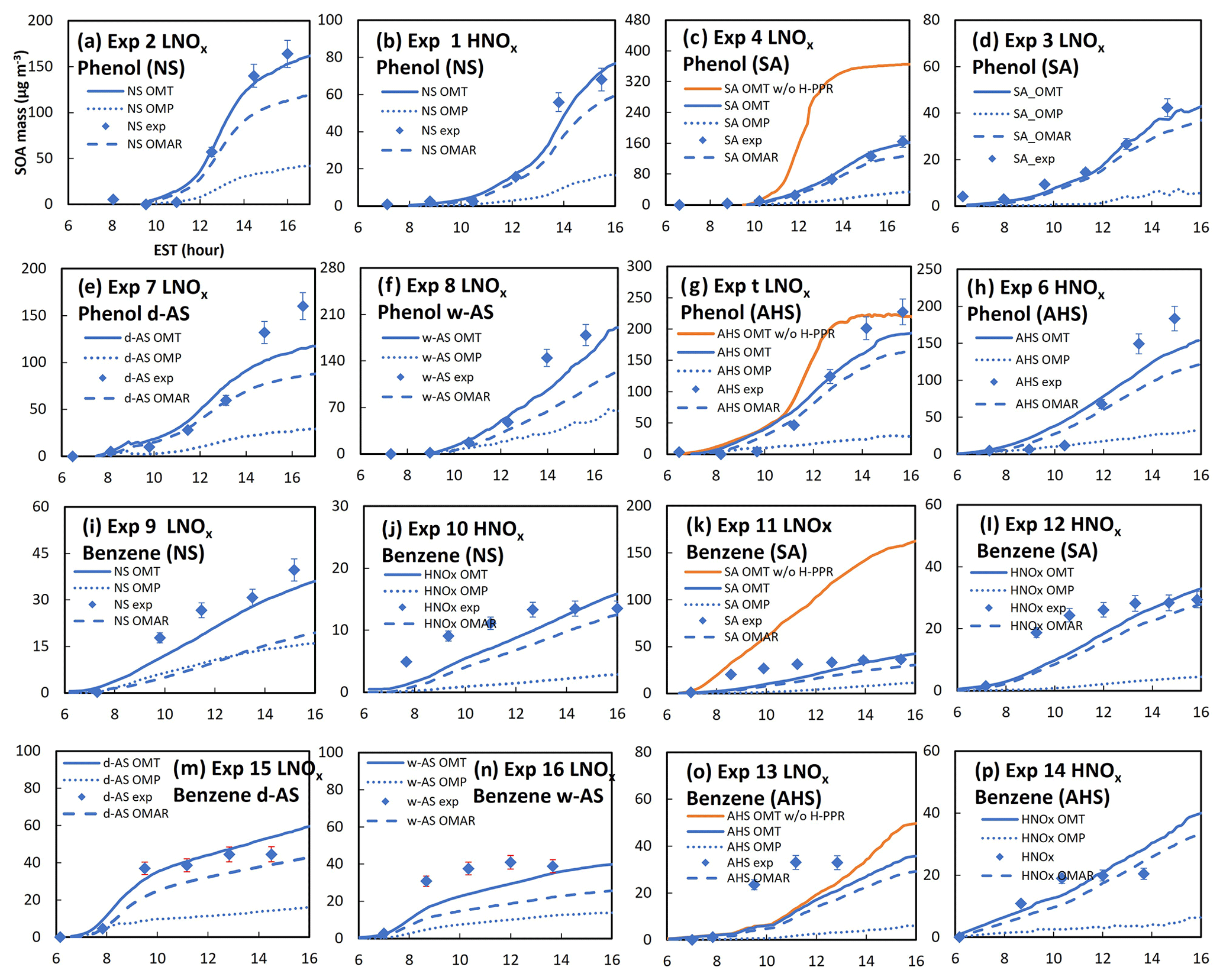
Figure 4The measured and simulated SOA mass under different seed conditions and various NOx concentrations. Solid lines indicate the simulated SOA mass and points indicate the experimentally measured mass. The SOA mass simulated with the H-PPR model (solid red line) was compared with that simulated without the H-PPR model (solid blue line). The UNIPAR-predicted OMAR (heterogeneous reaction in the aerosol phase) and OMp (partitioning) are also included. The error associated with SOA data was 9 % according to the uncertainty in data.
The importance of H-PPR mechanisms for SOA prediction is demonstrated in Fig. 4c and g for phenol and in Fig. 4k and o for benzene by comparing simulations with H-PPR with those without H-PPR. The suppression of SOA formation was effective with acidic aerosol. Additionally, the formation rate of PPR can be influenced by the chemical properties of the aerosol medium. For example, Mitroka et al. (2010) reported that the reactivity of the OH radical is considerably higher in polar, protic solvent than in dipolar, aprotic solvent. Protic solvent is a hydrogen bond donor that stabilizes the transition state of the OH radical addition reaction. Thus, the reaction of phenols with OH radical is more favorable in the inorganic phase than in the organic phase. The reported radical-scavenging ability of phenols by forming PPR is as follows: 1,2,3-trihydroxybenzene > 1,2,4-trihydroxybenzene > catechol (1,2-dihydroxybenzene) > 1.4-dihydroxybenzene > 1,3-dihydroxybenzene ≈ 1,3,5-trihydroxybenzene (Thavasi et al., 2009). Phenol oxidation can produce various multifunctional phenols. As shown in Reaction (R8), phenol in salted aqueous media reacts with OH(in) in a similar way, with the OH addition to the aromatic ring in the gas phase to form an intermediate product – phenol_OH_int(in) (Fig. 2). Figure S3 is the proposed mechanism to form phenoxy radical via the acid-catalyzed reaction. In addition, a quinone can yield catechol in aqueous acidic media. Quinones are well recognized for their ability to promote superoxide formation (Guin et al., 2011). Lowering the pH increases the redox potential (Walczak et al., 1997) of quinone–hydroquinone. However, the reduction potential of oxygen can be lower under acidic conditions and is advantageous for O/HO2• formation (Wei et al., 2022) (Sect. S4).
The importance of HOMs on phenol SOA has been demonstrated in a previous study by Choi and Jang (Choi and Jang, 2022). For example, a large fraction of OMP in Fig. 4a is contributed by HOMs. The contribution of HOMs to SOA mass increases with decreasing NO levels. The systematic evaluation of the UNIPAR model integrated with the explicit gas mechanisms is performed via the model sensitivity to various environmental variables (i.e., NOx levels, seed, temperature, and humidity) in Sect. 4.2.
4.2 Sensitivity of SOA formation to environmental variables
4.2.1 Evaluation of the impact of H-PPR on SOA formation: aerosol acidity
In order to assess the impact of H-PPR on phenolic SOA and benzene SOA, the UNIPAR simulation was performed as a function of aerosol acidity (FS value), as seen in Fig. 5. This sensitivity was performed under the following given conditions: temperature = 298 K, NOx level = 7.5 ppbC ppb−1, and RH = 0.6 with the sunlight conditions on 19 October 2022 (Fig. S1 in the Supplement). All seeds are wet under this RH level (Peng et al., 2022). The concentration of the initial HCs for each simulation was set to 30 ppb for four different HC compositions (see Fig. 5): phenol (panel a); benzene (panel b); phenol:benzene = 3:1 ppb ppb−1 (panel c), and phenol:benzene = 1:1 ppb ppb−1 (panel d). The concentrations of consumed phenol or consumed benzene varied according to the different rate constant of each HC with the OH radical and the H-PPR degree.
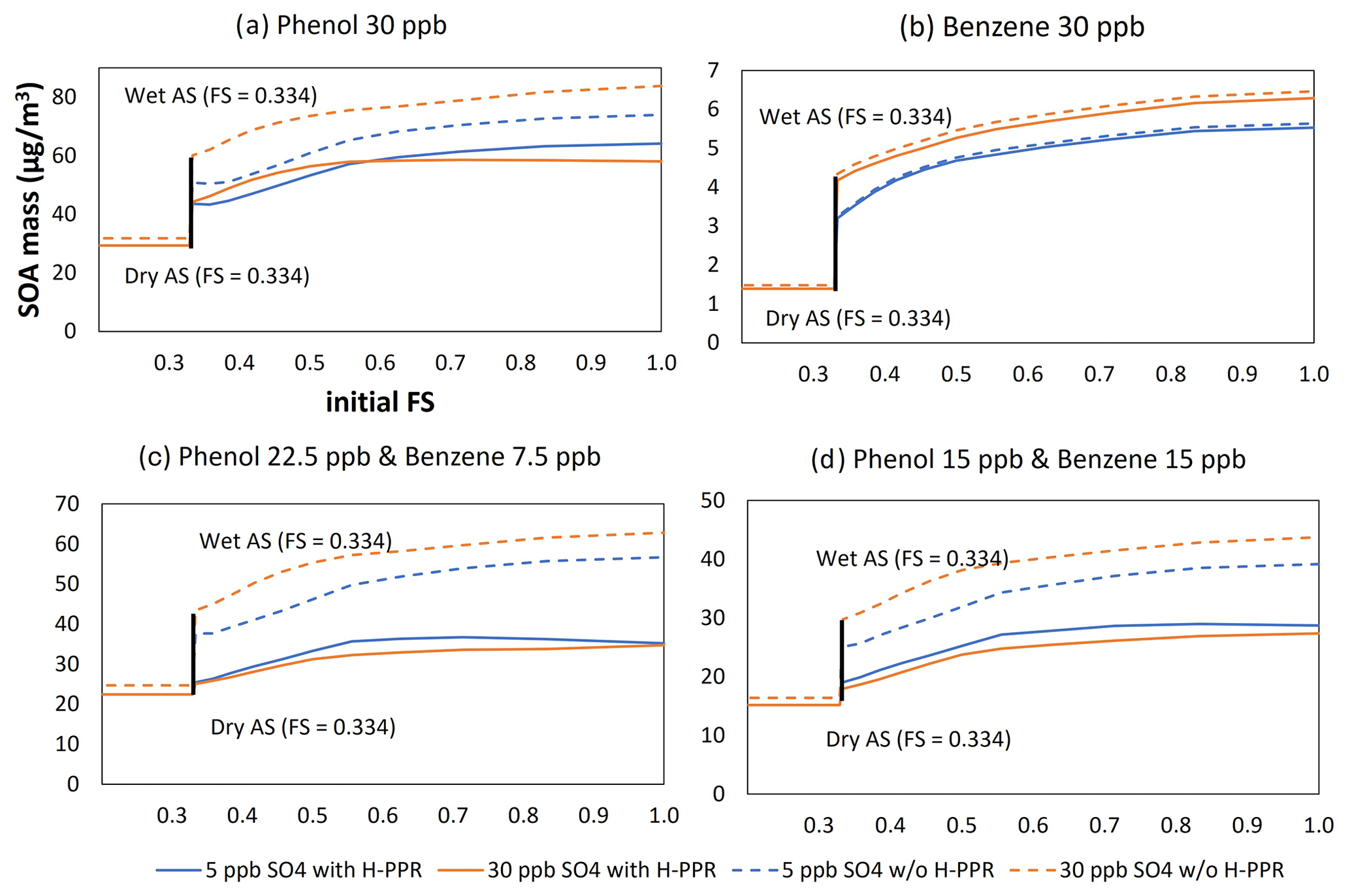
Figure 5Sensitivity of SOA production to aerosol acidity (FS value) for different precursor mixes using the UNIPAR model: phenol (a), benzene (b), phenol : benzene = 3:1 ppb ppb−1 (c), and phenol : benzene = 1:1 ppb ppb−1 (d). The initial precursor hydrocarbon concentrations were 30 ppb. The initial concentrations of HONO, formaldehyde, and acetaldehyde were 5, 6, and 2 ppb, respectively. The simulation was performed under ambient sunlight on 19 October 2022. RH was set to 0.6, temperature was set to 298 K, and HC ppbC / NOx ppb was 6.5 ppb sulfate = 20 µg m−3 and 30 ppb sulfate = 120 µg m−3.
The DSMACC box model platform in this study is equipped with the integrated reaction rate (IRR) analysis technique, which can show the chemical reaction flow in the oxidation mechanisms. Based on the IRR analysis, the production of PPR is contributed mainly by the reaction of phenol with OH radicals in the gas phase and the catechol H-PPR mechanism. Phenoxy radical production via phenol's H-PPR path is trivial due to the low partitioning of phenol into the aqueous phase. Unlike phenol, catechol, a major product of phenol oxidation, can yield the semiquinone radical (PPR of catechol) via the heterogeneous reaction mechanism. For example, in the presence of SA seed (Fig. 5a), the contribution of catechol's H-PPR path is 1.22 times greater than that of the gas-phase reaction of phenol with OH radicals. In the presence of wet AS seed, the contribution of catechol's H-PPR path is 20 % that of the gas-phase reaction of phenol with OH radicals.
In Fig. 5, a considerable difference was identified between the SOA mass simulated with H-PPR (solid line) and that without H-PPR (dashed line). SOA growth is suppressed with H-PPR. This gap gradually increases with increasing aerosol acidity (FS). The reduction in SOA mass with acidic seed is mainly caused by the retardation of gas oxidation, which is linked to the catalytic consumption of ozone and, consequently, connected to the slow oxidation of phenol (i.e., Fig. 3b, c) or benzene (i.e., Fig. 3h, i). The simulation also depicts the impact of the amount of inorganic seed on the SOA formation of phenol or benzene. Surprisingly, phenol SOA mass is lower with 30 µg m−3 of inorganic seed than that with 5 µg m−3 of inorganic seed, in contrast to the typical tendency of SOA formation from non-phenolic HCs.
Compared with phenol (Fig. 5a), the impact of H-PPR on benzene SOA formation is small, as seen in Fig. 5b. Benzene is more affected by acid-catalyzed oligomerization than phenol. Benzene oxidation yields various products other than phenol. Some reactive organic products are involved in oligomerization in the aqueous phase. As seen in Fig. 5c and d, when benzene is mixed with phenol in the presence of a high concentration of wet seed, SOA growth shows more suppression, suggesting the impact of PPR from phenol. The SOA mass difference due to the H-PPR mechanism is large in the mixture of phenol and benzene. The smaller amount of phenol consumption in the gas mixture, as seen in Fig. 5c and d, yields less SOA mass, which impacts the partitioning of products and their oligomerization in aerosol. Thus, the impact of H-PPR on SOA mass in Fig. 5c and d is relatively more significant than that in Fig. 5a due to the difference in SOA yields. These simulation results suggest that phenolic compounds in biomass burning smoke gases might impact the atmospheric oxidation ability of urban air.
4.2.2 Sensitivity of SOA formation to NOx level, temperature, and RH
Figure 6a–d display the sensitivity of the SOA mass to NOx levels at three different temperatures (278, 288, and 298 K) and two different seed conditions (no seed and AHS seed at RH = 0.6). Figure 7 illustrates the sensitivity of phenol or benzene SOA mass to RH (0.3 and 0.6) for two different seed conditions (AHS and AS seed). The UNIPAR simulation was evaluated under the given sunlight conditions on 19 October 2022 (between 06:30 to 17:30 EST; Fig. S1). The initial concentration of HCs in each simulation in Figs. 6 and 7 was set to 30 ppb to mimic real-world conditions.
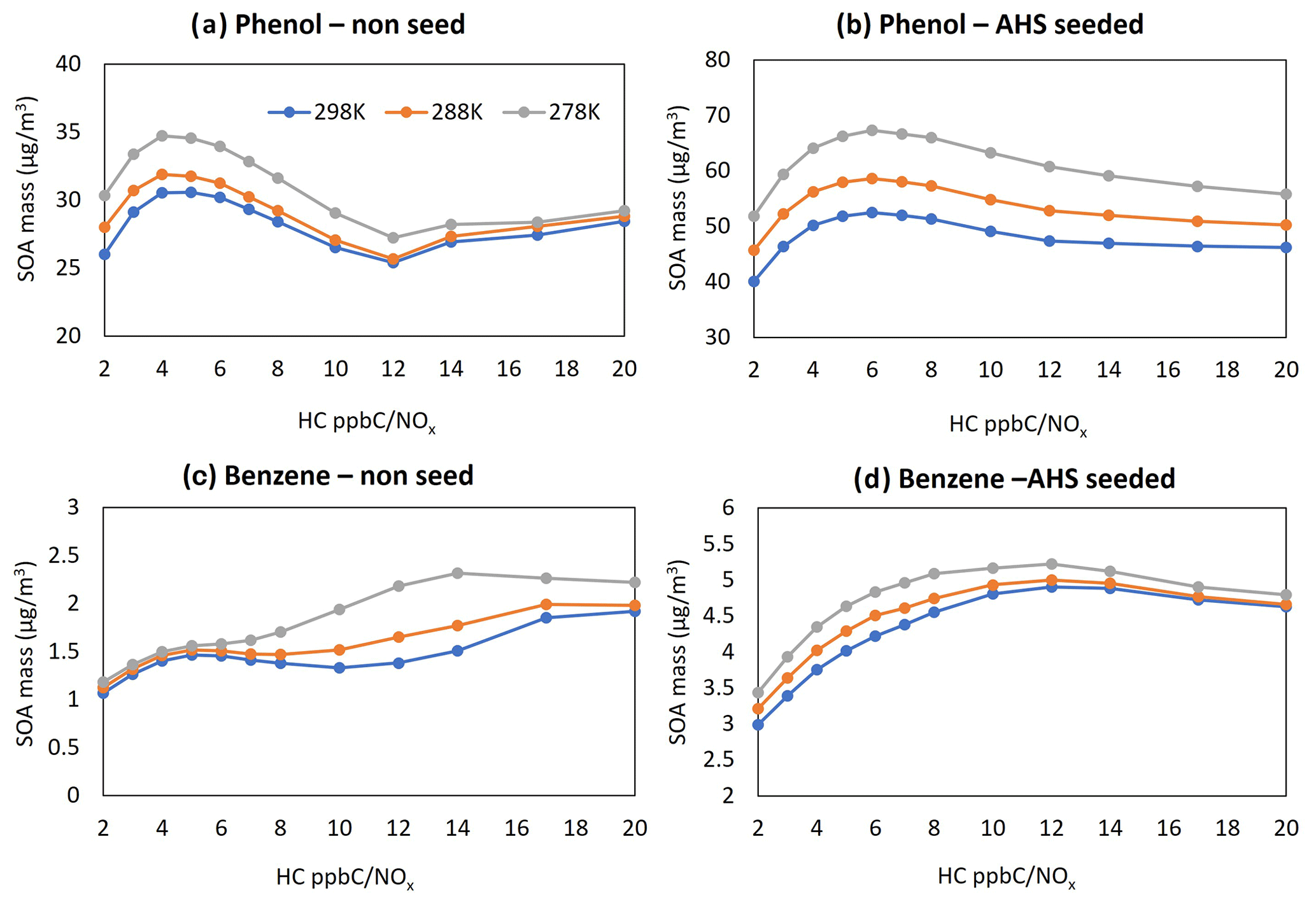
Figure 6Sensitivity of SOA mass to major model variables. SOA simulations were performed using a given sunlight profile on 19 October 2022 (from 06:30 to 17:30 EST). All simulations were performed with an initial HC concentration of 30 ppb. Panel (a) presents the predicted phenol SOA mass to NOx levels at three different temperatures in the presence of ASH seed; panel (b) shows the predicted phenol SOA mass to NOx levels at three different temperatures in the absence of seed; panel (c) displays the predicted benzene SOA mass to NOx levels at three different temperatures in the presence of ASH seed; and panel (d) shows the predicted benzene SOA mass to NOx levels at three different temperatures in the absence of seed. Temperature levels were set to 298, 288, and 278 K. NOx levels (HC ppbC / NOx ppb) ranged from 2 to 20.
A typical negative correlation appears between SOA mass and temperature (Fig. 6). Regardless of seed conditions, a typical NOx impact on SOA formation appears in high-NOx regions (HC ppbC / NOx≤ 5), showing a negative correlation between NOx levels and SOA formation. At the higher NOx level, more organonitrate forms via the reaction of peroxy radicals with NO. Organonitrates contribute to SOA formation mainly via the partitioning process, and their volatility is relatively high (Choi and Jang, 2022). Phenol SOA is more sensitive to the NOx level than benzene SOA. As the NOx level decreases in low-NOx regions (HC ppbC / NOx>5), SOA production gradually increases because of the increased contribution of low-volatility multi-hydroxyl products (HOM products that form via multiple additions of OH radicals into phenol) in the absence of wet seed (Fig. 6a, c). Thus, the increase in HOM products at the low NOx level decrease the sensitivity of SOA formation to temperature. In the presence of acidic seed, the production of phenoxy radicals increases with decreasing NOx levels due to increased oxidants in the aerosol phase, and it negatively influences SOA formation.
Phenol rapidly reacts with OH radicals (rate constant of cm3 molecule−1 s−1 at 298 K) (Kwok and Atkinson, 1995; Yee et al., 2013) in the gas phase and quickly produces low-volatility products compared with benzene ( cm3 molecule−1 s−1 at 298 K) (Kwok and Atkinson, 1995). Thus, phenol produces a higher SOA mass than benzene under given simulation conditions (under the same initial HC concentration, NOx levels, and sunlight conditions). The reaction rate of benzene with the OH radical is even slower than that of NO2 with the OH radical. Nearly 47 % of benzene oxidation would form gas oxidation products other than phenol, and some of them can be involved in aerosol-phase oligomerization. Benzene SOA yields can be significant, although benzene decay is slow (Figs. 3, 4).
Of the total PPR production, the contribution of daytime phenol oxidation with nitrate radicals is only 0.1 % of that from the phenol oxidation with OH radicals under the high-NOx condition (VOC ppbC / NOx ppb = 2, where VOC denotes volatile organic compound) in Fig. 6b. However, the contribution of the nitrate radical mechanism to form PPR increases in the absence of sunlight. For example, the contribution of nitrate radicals on PPR is nearly 30 % of that with OH radicals under a given simulation condition under the high-NOx condition between 16:00 and 17:00 EST (Fig. 6a). This simulation result suggests that nitrate radical oxidation with phenol is important to form PPR during nighttime.
AS seed in Fig. 7a can be effloresced at RH = 0.35–0.4. Thus, AS seed is dry at RH = 0.3 but wet at RH = 0.6. Figure 7a displays a higher SOA formation at RH = 0.6 than at RH = 0.3 because of aqueous-phase oligomerization (Choi and Jang, 2022; Han and Jang, 2022, 2023; Im et al., 2014; Yu et al., 2021a, b; Zhou et al., 2019). Overall, both phenol SOA and benzene SOA are slightly sensitive to RH conditions. In the presence of AHS seed that is wet under both RH conditions (0.3 and 0.6), both phenol and benzene exhibit an unusual tendency: higher SOA mass with lower RH. SOA growth from both benzene and phenol can increase due to acid-catalyzed oligomerization, but it can also be partly suppressed via the H-PPR mechanism. Figure 7b and c display OMAR (oligomeric SOA mass) and OMP (partitioning mass) contributions to OMT. Both benzene SOA and phenol SOA are dominated by OMAR under the given simulation conditions.
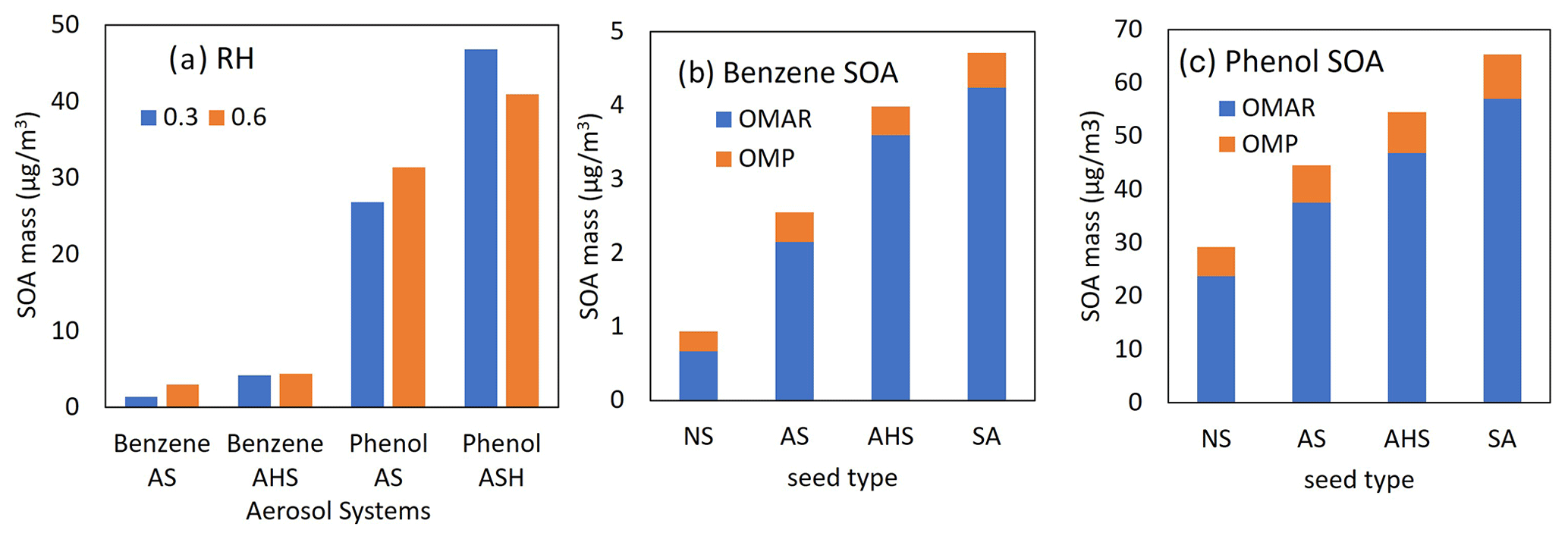
Figure 7(a) The predicted SOA mass from the photooxidation of benzene or phenol at two different RH levels (0.3 and 0.6) in the presence of AS or AHS seed (HC ppbC / NOx=6.64). OMAR (oligomeric mass) and OMP (partitioning mass) in benzene SOA (b) and phenol SOA (c). For panels (b) and (c), SOA production is simulated with an initial HC concentration = 30 ppb (HC ppbC / NOx= 6.64), RH = 0.6, temperature = 298 K, and sulfate = 20 µg m−3 under the sunlight conditions on 19 October 2022.
4.3 Uncertainty in SOA formation to model parameters
Figure 8 illustrates the uncertainties in important model parameters with respect to predicting SOA mass using the UNIPAR model. The partitioning process (OMP) is most impacted by the lumped species' vapor pressure (VP). OMAR is influenced by the reaction rates of oligomerization of the reactive lumped species (koligomerization) and H-PPR (kphenoxy). The uncertainties in the estimated VP are set to the reported value associated with the group contribution method (Zhao et al., 1999; Yu et al., 2021b). The corresponding change in the SOA mass owing to VP uncertainties ranges from −20 % to 24 % in benzene SOA simulation with wet AS seed under the given simulation conditions (Fig. 8a). Under the same condition, the change in the phenol SOA mass due to VP uncertainties ranges from −13 % to 14 %. Benzene SOA (Fig. 8a) is more sensitive to the VP uncertainty than phenol SOA (Fig. 8b). As seen in Fig. 8c and d, SOA mass is more significantly impacted by the uncertainty associated with the oligomerization rate constant than that of the H-PPR rate constant at a given uncertainty range. The variation in SOA formation with the change in kphenoxy is trivial. For H-PPR, the amount of available oxidants (i.e., OH radicals, HONO, and H2O2) is more critical than kphenoxy in our model.
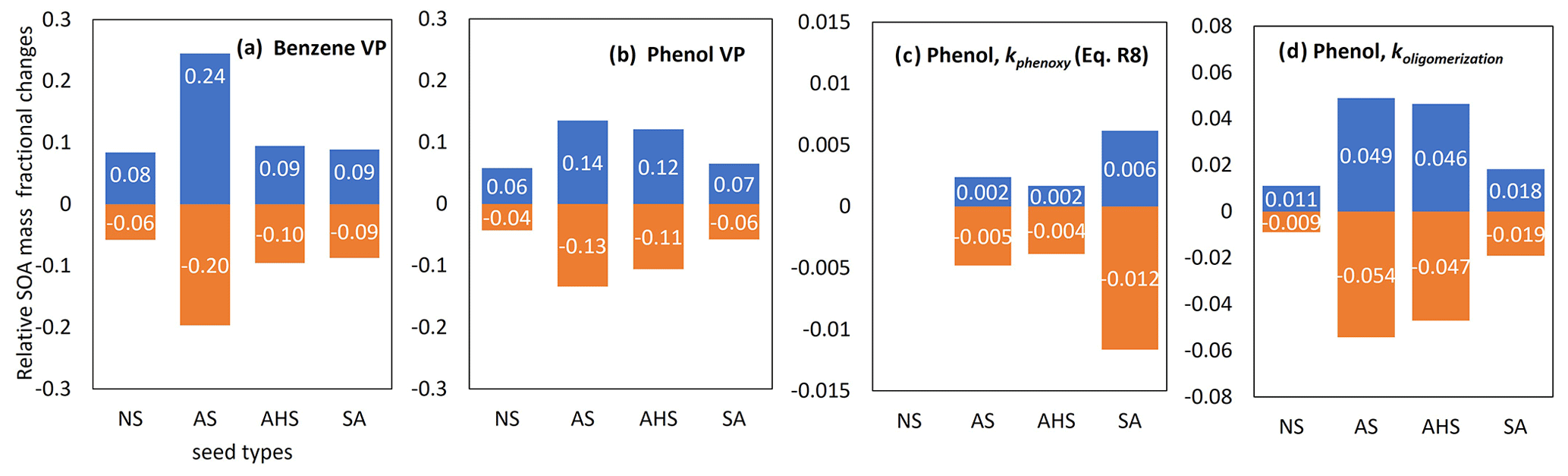
Figure 8Uncertainties in the VP of benzene (a), the VP of phenol (b), the koligomerization of phenol SOA (c), and the kphenoxy of phenol SOA (d). Their variations at the lower and upper boundaries are set to a factor of 0.5 and 2, respectively. SOA production is simulated with the following initial conditions: HC concentration = 30 ppb (HC ppbC / NOx=6), RH = 0.6, temperature = 298 K, and sulfate = 20 µg m−3 under the sunlight conditions on 19 October 2022.
Hitherto, the typical role of inorganic acids has been as follows: an acid catalyst that can accelerate the oligomerization of reactive organic species (i.e., aldehydes and epoxide) in the aerosol phase (Jang et al., 2002; Garland et al., 2006; Hallquist et al., 2009). SOA growth accelerated by an acid catalyst has been reported in various chamber studies including both aromatic and biogenic HCs. Surprisingly, acidic aerosol along with phenol and benzene suppressed the oxidation of air, including ozone formation and the decay of HCs (Fig. 3), due to the heterogeneous formation of PPR species.
The gas mechanisms including the formation of HOMs and H-PPR simulated gas oxidation well (i.e., ozone, NOx, and the decay of benzene or phenol) and improved the prediction of SOA formation using UNIPAR (Fig. 4). As seen in Fig. 5, phenol SOA still increases via heterogeneous oligomerization, showing a difference in SOA mass between dry AS and wet AS seed. As discussed in Sect. 3.2.2, the heterogeneously produced PPR production occurs via the reaction of phenolic compounds with the aerosol-phase OH radical that is available during the daytime due to the photolysis of H2O2 and HONO. Thus, the heterogeneously produced PPR is effective with wet inorganic aerosol during the daytime.
Fundamentally, biomass burning under an open flame is performed at low temperatures and produces very low NO concentrations (Simoneit, 2002; Mebust and Cohen, 2013; Xu et al., 2021). The chemistry slows to a standstill without NOx and, thus, halts ozone formation, although gaseous HCs are abundant. When these fire plumes mix into urban atmospheres abundant in NOx, ozone formation becomes active, impacting the air quality of the city. Chamber data from this study mimic phenol oxidation in the presence of NOx. In addition, hygroscopic inorganic aerosols comprising nitrate, sulfate, and ammonium ions are available in the city environment, which is rich in NOx, SO2, and NH3. When wildfire plumes mix in city air, their phenolic compounds interact with NOx and hygroscopic inorganic aerosol. The results from this study suggest that PPR produced during the atmospheric process of phenolic compounds in wildfire plumes can temporarily retard atmospheric oxidation in urban environments. For example, as seen in Fig. 5, the SOA simulation with low concentrations of phenol and typical atmospheric tracer gases (formaldehyde and acetaldehyde) shows that phenol SOA is considerably suppressed, even with a small amount of wet inorganic aerosol ranging from weak to neutral acidity. For example, phenol SOA mass decreases by 12 % with 5 ppb of ammonium hydrogen sulfate (FS = 0.5) (Fig. 5a), while the SOA mass from the mixture of phenol and benzene decreased by 28 % (Fig. 5c).
The impact of NOx on SOA formation appears to be negative under high NOx levels, as shown in Fig. 6. A significant fraction of phenolic SOA forms due to HOMs and oligomeric matter. The contribution of HOMs and oligomeric matter to SOA formation is generally higher at lower NOx levels. Thus, phenol or benzene SOA under the NOx-limited condition is relatively insensitive to temperature (Fig. 6) due to the formation of nonvolatile products. Similarly, SOA from biomass burning might be insignificantly affected by temperature under low-NOx regimes. When the concentrations of NOx drop in the high-NOx regime, SOA formation increases. The role of PPR with respect to the atmospheric oxidation capacity in the blending of wildfire smoke and urban pollutants needs to be studied under different NOx levels.
A variety of phenolic compounds, including phenol, cresol, catechol, methoxyphenols, and dimethylphenols (Akherati et al., 2020; Bruns et al., 2016), can consist of more than 80 % of the precursor HCs in wildfire smoke. These multifunctional phenolic compounds can also yield PPR as active scavengers for ozone (Sect. 3.2.2). To date, the impact of phenolic compounds on the retardation of atmospheric aging of HCs in the city air has not been sufficiently studied. It is important to comprehend the formation mechanisms of PPR-like chemical species and their role in the atmospheric oxidation capability to accurately predict the elevation of ozone and SOA as well as their peaks.
Several unresolved issues need to be addressed in order to accurately predict SOA formation from biomass burning smoke, including the gas mechanisms of unidentified HCs in wildfire smoke, the role of the aqueous phase in the formation of PPR, and unidentified SOA formation mechanisms. For example, there are still missing mechanisms, including cross-reactions of RO2 radicals and the distribution of oxidation paths. This study focused on the oxidation of phenol and benzene through their reaction with OH radicals. Ozone, which mainly forms during daytime, often persists through the night and reacts with NO2 to form nitrate radicals at night. Phenol can react with this nitrate radical during the night, although its reaction is slower than that with the OH radical. The reaction of phenols with the nitrate radical can also produce PPR-like species and can catalytically scavenge ozone. Nighttime humidity, which increases as temperature decreases, can surpass the delinquent RH (DRH) of hygroscopic inorganic constituents and form wet inorganic aerosol. The resulting aerosol can be wet until humidity drops below ERH. To better predict SOA formation, the atmospheric processes of biomass burning smoke needs to be studied under different climate conditions, such as temperature, humidity, and sunlight levels, and air pollutant emission levels.
Phenol is the most abundant first-generation product from the oxidation of benzene (Smith et al., 2014; Johnson et al., 2004). Additionally, the formation of cresol is involved in 18 % of toluene–OH reactions. Xu et al. (2015) reported that toluene SOA formation was underestimated by about 20 %, proposing an importance of phenolic routes in aromatic oxidation. Dimethylphenols can also be produced from the oxidation of xylenes. Comprehension of the atmospheric process of phenolic compounds can improve the prediction of SOA formation from aromatic HCs. A large fraction of phenolic compounds are directly emitted, but their impact on SOA mass is missing in the current air quality model (Pye et al., 2023).
Chamber simulation data used in this study are available upon request.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-6567-2024-supplement.
JC and MJ conducted the chamber experiments and simulated the UNIPAR SOA model. JC, MJ, and SB processed the chamber data.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This research was supported by the National Institute of Environmental Research (grant no. NIER2020-01-01-010); the National Science Foundation (grant no. AGS1923651); LG Electronics Inc. (grant no. C2023007555); and the Fine Particle Research Initiative in East Asia Considering National Differences (FRIEND) project through the National Research Foundation of Korea (NRF), funded by the Ministry of Science and ICT (grant no. 2020M3G1A1114556).
This research has been supported by the Directorate for Geosciences (grant no. AGS1923651), the National Institute of Environmental Research (grant no. NIER2020-01-01-010), the National Research Foundation of Korea (grant no. 2020M3G1A1114556), and LG Electronics Inc. (grant no. C2023007555).
This paper was edited by Ivan Kourtchev and reviewed by three anonymous referees.
Akherati, A., He, Y., Coggon, M. M., Koss, A. R., Hodshire, A. L., Sekimoto, K., Warneke, C., de Gouw, J., Yee, L., Seinfeld, J. H., Onasch, T. B., Herndon, S. C., Knighton, W. B., Cappa, C. D., Kleeman, M. J., Lim, C. Y., Kroll, J. H., Pierce, J. R., and Jathar, S. H.: Oxygenated Aromatic Compounds are Important Precursors of Secondary Organic Aerosol in Biomass-Burning Emissions, Environ. Sci. Technol., 54, 8568–8579, https://doi.org/10.1021/acs.est.0c01345, 2020.
Atkinson, R.: Atmospheric chemistry of VOCs and NOx, Atmos. Environ., 34, 2063–2101, 2000.
Beardsley, R. L. and Jang, M.: Simulating the SOA formation of isoprene from partitioning and aerosol phase reactions in the presence of inorganics, Atmos. Chem. Phys., 16, 5993–6009, https://doi.org/10.5194/acp-16-5993-2016, 2016.
Bloss, C., Wagner, V., Jenkin, M. E., Volkamer, R., Bloss, W. J., Lee, J. D., Heard, D. E., Wirtz, K., Martin-Reviejo, M., Rea, G., Wenger, J. C., and Pilling, M. J.: Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons, Atmos. Chem. Phys., 5, 641–664, https://doi.org/10.5194/acp-5-641-2005, 2005.
Borrás, E. and Tortajada-Genaro, L. A.: Secondary organic aerosol formation from the photo-oxidation of benzene, Atmos. Environ., 47, 154–163, https://doi.org/10.1016/j.atmosenv.2011.11.020, 2012.
Brown, H. C. and Okamoto, Y.: Electrophilic Substituent Constants, J. Am. Chem. Soc., 80, 4979–4987, https://doi.org/10.1021/ja01551a055, 1958.
Bruns, E. A., El Haddad, I., Slowik, J. G., Kilic, D., Klein, F., Baltensperger, U., and Prévôt, A. S. H.: Identification of significant precursor gases of secondary organic aerosols from residential wood combustion, Sci. Rep., 6, 27881, https://doi.org/10.1038/srep27881, 2016.
Calvert, J. G., Atkins, R., Becker, K. H., Kamens, R. M., Seinfeld, J. H., Wallington, T. J., and Yarwood, G.: Mechanisms of atmospheric oxidation of aromatic hydrocarbons, Reactions of aromatic compounds with OH radicals (Chap. 2), Oxford University Press, New York, https://doi.org/10.1093/oso/9780195146288.001.0001, 2002.
Cao, G. and Jang, M.: Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx, Atmos. Environ., 35, 7603–7613, 2007.
Carlton, A. G., Bhave, P. V., Napelenok, S. L., Edney, E. O., Sarwar, G., Pinder, R. W., Pouliot, G. A., and Houyoux, M.: Model Representation of Secondary Organic Aerosol in CMAQv4.7, Environ. Sci. Technol., 44, 8553–8560, https://doi.org/10.1021/es100636q, 2010.
Carter, W. P. and Atkinson, R.: Alkyl nitrate formation from the atmospheric photoxidation of alkanes; a revised estimation method, J. Atmos. Chem., 8, 165–173, 1989.
Carter, W. P. L.: Development of Ozone Reactivity Scales for Volatile Organic Compounds, Air Waste, 44, 881–899, https://doi.org/10.1080/1073161X.1994.10467290, 1994.
Choi, J. and Jang, M.: Suppression of the Phenolic SOA formation in the Presence of Electrolytic Inorganic Seed, Sci. Total Environ., 851, 158082, https://doi.org/10.1016/j.scitotenv.2022.158082, 2022.
Das, T. N.: Oxidation of Phenol in Aqueous Acid: Characterization and Reactions of Radical Cations vis-à-vis the Phenoxyl Radical, J. Phys. Chem. A, 109, 3344–3351, https://doi.org/10.1021/jp050015p, 2005.
Deng, Y., Inomata, S., Sato, K., Ramasamy, S., Morino, Y., Enami, S., and Tanimoto, H.: Temperature and acidity dependence of secondary organic aerosol formation from α-pinene ozonolysis with a compact chamber system, Atmos. Chem. Phys., 21, 5983–6003, https://doi.org/10.5194/acp-21-5983-2021, 2021.
Dixon, W. T. and Murphy, D.: Determination of the acidity constants of some phenol radical cations by means of electron spin resonance, J. Chem. Soc. Perk. T. 2, 72, 1221–1230, https://doi.org/10.1039/F29767201221, 1976.
Edwards, D. P., Emmons, L. K., Gille, J. C., Chu, A., Attié, J.-L., Giglio, L., Wood, S. W., Haywood, J., Deeter, M. N., Massie, S. T., Ziskin, D. C., and Drummond, J. R.: Satellite-observed pollution from Southern Hemisphere biomass burning, J. Geophys. Res.-Atmos., 111, D14312, https://doi.org/10.1029/2005JD006655, 2006.
Emmerson, K. M. and Evans, M. J.: Comparison of tropospheric gas-phase chemistry schemes for use within global models, Atmos. Chem. Phys., 9, 1831–1845, https://doi.org/10.5194/acp-9-1831-2009, 2009.
Finlayson-Pitts, B. J. and Pitts, J. N.: Chemistry of the upper and lower atmosphere: Theory, Experiments, and Applications, Academic Press, New York, 176 pp., https://doi.org/10.1016/B978-0-12-257060-5.X5000-X, 2000.
Garland, R. M., Elrod, M. J., Kincaid, K., Beaver, M. R., Jimenez, J. L., and Tolbert, M. A.: Acid-catalyzed reactions of hexanal on sulfuric acid particles: Identification of reaction products, Atmos. Environ., 40, 6863–6878, 2006.
Garmash, O., Rissanen, M. P., Pullinen, I., Schmitt, S., Kausiala, O., Tillmann, R., Zhao, D., Percival, C., Bannan, T. J., Priestley, M., Hallquist, Å. M., Kleist, E., Kiendler-Scharr, A., Hallquist, M., Berndt, T., McFiggans, G., Wildt, J., Mentel, T. F., and Ehn, M.: Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 20, 515–537, https://doi.org/10.5194/acp-20-515-2020, 2020.
Guin, P. S., Das, S., and Mandal, P. C.: Electrochemical Reduction of Quinones in Different Media: A Review, Int. J. Electrochem., 2011, 816202, https://doi.org/10.4061/2011/816202, 2011.
Hallquist, M., Wenger, J. C., Baltensperger, U., Rudich, Y., Simpson, D., Claeys, M., Dommen, J., Donahue, N. M., George, C., Goldstein, A. H., Hamilton, J. F., Herrmann, H., Hoffmann, T., Iinuma, Y., Jang, M., Jenkin, M. E., Jimenez, J. L., Kiendler-Scharr, A., Maenhaut, W., McFiggans, G., Mentel, Th. F., Monod, A., Prévôt, A. S. H., Seinfeld, J. H., Surratt, J. D., Szmigielski, R., and Wildt, J.: The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 9, 5155–5236, https://doi.org/10.5194/acp-9-5155-2009, 2009.
Han, S. and Jang, M.: Modeling daytime and nighttime secondary organic aerosol formation via multiphase reactions of biogenic hydrocarbons, Atmos. Chem. Phys., 23, 1209–1226, https://doi.org/10.5194/acp-23-1209-2023, 2023.
Hansch, C., McKarns, S. C., Smith, C. J., and Doolittle, D. J.: Comparative QSAR evidence for a free-radical mechanism of phenol-induced toxicity, Chem.-Biol. Interact., 127, 61–72, https://doi.org/10.1016/s0009-2797(00)00171-x, 2000.
Holton, D. M. and Murphy, D.: Determination of acid dissociation constants of some phenol radical cations. Part 2, J. Chem. Soc. Perk. T. 2, 75, 1637–1642, https://doi.org/10.1039/F29797501637, 1979.
Hudson, P. K., Murphy, D. M., Cziczo, D. J., Thomson, D. S., de Gouw, J. A., Warneke, C., Holloway, J., Jost, H.-J., and Hübler, G.: Biomass-burning particle measurements: Characteristic composition and chemical processing, J. Geophys. Res.-Atmos., 109, D23S27, https://doi.org/10.1029/2003JD004398, 2004.
Im, Y., Jang, M., and Beardsley, R. L.: Simulation of aromatic SOA formation using the lumping model integrated with explicit gas-phase kinetic mechanisms and aerosol-phase reactions, Atmos. Chem. Phys., 14, 4013–4027, https://doi.org/10.5194/acp-14-4013-2014, 2014.
Jagiella, S. and Zabel, F.: Reaction of phenylperoxy radicals with NO2 at 298 K, Phys. Chem. Chem. Phys., 9, 5036–5051, 2007.
Jang, M. and Kamens, R. M.: Application of group contribution methods to the partitioning of semi-volatile organic compounds on atmospheric particulate matter, 771–781, Specialty conference on measurement of toxic and related air pollutants Research Triangle Park, NC (United States), 29 April–1 May 1997, 1997.
Jang, M. and Kamens, R. M.: A Thermodynamic Approach for Modeling Partitioning of Semivolatile Organic Compounds on Atmospheric Particulate Matter: Humidity Effects, Environ. Sci. Technol., 32, 1237–1243, 1998.
Jang, M., Czoschke, N. M., Lee, S., and Kamens, R. M.: Heterogeneous Atmospheric Aerosol Production by Acid- Catalyzed Particle-Phase Reactions, Science, 298, 814–817, 2002.
Jenkin, M. E., Saunders, S. M., Wagner, V., and Pilling, M. J.: Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds, Atmos. Chem. Phys., 3, 181–193, https://doi.org/10.5194/acp-3-181-2003, 2003.
Jenkin, M. E., Derwent, R. G., and Wallington, T. J.: Photochemical ozone creation potentials for volatile organic compounds: Rationalization and estimation, Atmos. Environ., 163, 128–137, https://doi.org/10.1016/j.atmosenv.2017.05.024, 2017.
Ji, Y., Zhao, J., Terazono, H., Misawa, K., Levitt, N. P., Li, Y., Lin, Y., Peng, J., Wang, Y., Duan, L., Pan, B., Zhang, F., Feng, X., An, T., Marrero-Ortiz, W., Secrest, J., Zhang, A. L., Shibuya, K., Molina, M. J., and Zhang, R.: Reassessing the atmospheric oxidation mechanism of toluene, P. Natl. Acad. Sci. USA, 114, 8169–8174, https://doi.org/10.1073/pnas.1705463114, 2017.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of Organic Aerosols in the Atmosphere, Science (Washington, DC, United States), 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Johnson, D., Jenkin, M. E., Wirtz, K., and Martin-Reviejo, M.: Simulating the Formation of Secondary Organic Aerosol from the Photooxidation of Toluene, Environ. Chem., 1, 150–165, https://doi.org/10.1071/EN04069, 2004.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kwok, E. S. C. and Atkinson, R.: Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: an update, Atmos. Environ., 29, 1685–1695, https://doi.org/10.1016/1352-2310(95)00069-b, 1995.
Li, J., Jang, M., and Beardsley, R.: Dialkylsulfate Formation in Sulfuric Acid Seeded Secondary Organic Aerosol Produced Using an Outdoor Chamber Under Natural Sunlight, Environ. Chem., 13, 590–601, https://doi.org/10.1071/EN15129, 2015.
Liggio, J., Li, S.-M., and McLaren, R.: Reactive uptake of glyoxal by particulate matter, J. Geophy. Res., 110, D10304-10301–D10304/10313, https://doi.org/10.1029/2004JD005113, 2005.
Majdi, M., Sartelet, K., Lanzafame, G. M., Couvidat, F., Kim, Y., Chrit, M., and Turquety, S.: Precursors and formation of secondary organic aerosols from wildfires in the Euro-Mediterranean region, Atmos. Chem. Phys., 19, 5543–5569, https://doi.org/10.5194/acp-19-5543-2019, 2019.
Mebust, A. K. and Cohen, R. C.: Observations of a seasonal cycle in NOx emissions from fires in African woody savannas, Geophys. Res. Lett., 40, 1451–1455, https://doi.org/10.1002/grl.50343, 2013.
Mitroka, S., Zimmeck, S., Troya, D., and Tanko, J. M.: How solvent modulates hydroxyl radical reactivity in hydrogen atom abstractions, J. Am. Chem. Soc., 132, 2907–2913, https://doi.org/10.1021/ja903856t, 2010.
Mvula, E., Schuchmann, M. N., and von Sonntag, C.: Reactions of phenol-OH-adduct radicals. Phenoxyl radical formation by water elimination vs. oxidation by dioxygen, J. Chem. Soc., Perkin Transactions 2, 264–268, https://doi.org/10.1039/B008434O, 2001.
Nakao, S., Clark, C., Tang, P., Sato, K., and Cocker III, D.: Secondary organic aerosol formation from phenolic compounds in the absence of NOx, Atmos. Chem. Phys., 11, 10649–10660, https://doi.org/10.5194/acp-11-10649-2011, 2011.
Ng, N. L., Chhabra, P. S., Chan, A. W. H., Surratt, J. D., Kroll, J. H., Kwan, A. J., McCabe, D. C., Wennberg, P. O., Sorooshian, A., Murphy, S. M., Dalleska, N. F., Flagan, R. C., and Seinfeld, J. H.: Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes, Atmos. Chem. Phys., 7, 5159–5174, https://doi.org/10.5194/acp-7-5159-2007, 2007.
Olmez-Hanci, T. and Arslan-Alaton, I.: Comparison of sulfate and hydroxyl radical based advanced oxidation of phenol, Chem. Eng. J., 224, 10–16, https://doi.org/10.1016/j.cej.2012.11.007, 2013.
Pankow, J. F.: An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol, Atmos. Environ., 28, 189–193, 1994.
Peng, C., Chen, L., and Tang, M.: A database for deliquescence and efflorescence relative humidities of compounds with atmospheric relevance, Fundamental Research, 2, 578–587, https://doi.org/10.1016/j.fmre.2021.11.021, 2022.
Pillar-Little, E. A., Zhou, R., and Guzman, M. I.: Heterogeneous Oxidation of Catechol, J. Phys. Chem. A, 119, 10349–10359, https://doi.org/10.1021/acs.jpca.5b07914, 2015.
Pye, H. O. T., Place, B. K., Murphy, B. N., Seltzer, K. M., D'Ambro, E. L., Allen, C., Piletic, I. R., Farrell, S., Schwantes, R. H., Coggon, M. M., Saunders, E., Xu, L., Sarwar, G., Hutzell, W. T., Foley, K. M., Pouliot, G., Bash, J., and Stockwell, W. R.: Linking gas, particulate, and toxic endpoints to air emissions in the Community Regional Atmospheric Chemistry Multiphase Mechanism (CRACMM), Atmos. Chem. Phys., 23, 5043–5099, https://doi.org/10.5194/acp-23-5043-2023, 2023.
Reche, C., Viana, M., Amato, F., Alastuey, A., Moreno, T., Hillamo, R., Teinilä, K., Saarnio, K., Seco, R., Peñuelas, J., Mohr, C., Prévôt, A. S. H., and Querol, X.: Biomass burning contributions to urban aerosols in a coastal Mediterranean City, Sci. Total Environ., 427–428, 175–190, https://doi.org/10.1016/j.scitotenv.2012.04.012, 2012.
Schell, B., Ackermann, I. J., Hass, H., Binkowski, F. S., and Ebel, A.: Modeling the formation of secondary organic aerosol within a comprehensive air quality model system, J. Geophys. Res.-Atmos., 106, 28275–28293, 2001.
Schill, G. P., Froyd, K. D., Bian, H., Kupc, A., Williamson, C., Brock, C. A., Ray, E., Hornbrook, R. S., Hills, A. J., Apel, E. C., Chin, M., Colarco, P. R., and Murphy, D. M.: Widespread biomass burning smoke throughout the remote troposphere, Nat. Geosci., 13, 422–427, https://doi.org/10.1038/s41561-020-0586-1, 2020.
Simoneit, B. R. T.: Biomass burning – a review of organic tracers for smoke from incomplete combustion, Appl. Geochem., 17, 129–162, https://doi.org/10.1016/S0883-2927(01)00061-0, 2002.
Smith, J. D., Sio, V., Yu, L., Zhang, Q., and Anastasio, C.: Secondary Organic Aerosol Production from Aqueous Reactions of Atmospheric Phenols with an Organic Triplet Excited State, Environ. Sci. Technol., 48, 1049–1057, https://doi.org/10.1021/es4045715, 2014.
Steadman, J. and Syage, J. A.: Picosecond studies of proton transfer in clusters. 2. Dynamics and energetics of solvated phenol cation, J. Am. Chem. Soc., 113, 6786–6795, https://doi.org/10.1021/ja00018a011, 1991.
Sun, Q., Tripathi, G. N. R., and Schuler, R. H.: Time-resolved resonance Raman spectroscopy of p-aminophenol radical cation in aqueous solution, J. Phys. Chem., 94, 6273–6277, https://doi.org/10.1021/j100379a023, 1990.
Sun, Y. L., Zhang, Q., Anastasio, C., and Sun, J.: Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry, Atmos. Chem. Phys., 10, 4809–4822, https://doi.org/10.5194/acp-10-4809-2010, 2010.
Surratt, J. D., Lewandowski, M., Offenberg, J. H., Jaoui, M., Kleindienst, T. E., Edney, E. O., and Seinfeld, J. H.: Effect of acidity on secondary organic aerosol formation from isoprene, Environ. Sci. Technol., 41, 5363–5369, https://doi.org/10.1021/es0704176, 2007a.
Surratt, J. D., Kroll, J. H., Kleindienst, T. E., Edney, E. O., Claeys, M., Sorooshian, A., Ng, N. L., Offenberg, J. H., Lewandowski, M., Jaoui, M., Flagan, R. C., and Seinfeld, J. H.: Evidence for Organosulfates in Secondary Organic Aerosol, Environ. Sci. Technol., 41, 517–527, 2007b.
Tao, Z. and Li, Z.: A kinetics study on reactions of C6H5O with C6H5O and O3 at 298 k, Int. J. Chem. Kinet., 31, 65–72, https://doi.org/10.1002/(SICI)1097-4601(1999)31:1<65::AID-KIN8>3.0.CO;2-J, 1999.
Thavasi, V., Bettens, R. P. A., and Leong, L. P.: Temperature and Solvent Effects on Radical Scavenging Ability of Phenols, J. Phys. Chem. A, 113, 3068–3077, https://doi.org/10.1021/jp806679v, 2009.
Verma, D. K. and Tombe, K. D.: Benzene in Gasoline and Crude Oil: Occupational and Environmental Implications, AIHA J., 63, 225–230, https://doi.org/10.1080/15428110208984708, 2002.
Walczak, M. M., Dryer, D. A., Jacobson, D. D., Foss, M. G., and Flynn, N. T.: pH Dependent Redox Couple: An Illustration of the Nernst Equation, J. Chem. Educ., 74, 1195, https://doi.org/10.1021/ed074p1195, 1997.
Wang, T., Bo, P., Bing, T., Zhaoyun, Z., Liyu, D., and Yonglong, L.: Benzene homologues in environmental matrixes from a pesticide chemical region in China: Occurrence, health risk and management, Ecotox. Environ. Safe., 104, 357–364, https://doi.org/10.1016/j.ecoenv.2014.01.035, 2014.
Wei, J., Fang, T., and Shiraiwa, M.: Effects of Acidity on Reactive Oxygen Species Formation from Secondary Organic Aerosols, ACS Environ. Au., 2, 336–345, https://doi.org/10.1021/acsenvironau.2c00018, 2022.
Wohl, C., Li, Q., Cuevas, C. A., Fernandez, R. P., Yang, M., Saiz-Lopez, A., and Simó, R.: Marine biogenic emissions of benzene and toluene and their contribution to secondary organic aerosols over the polar oceans, Sci. Adv., 9, eadd9031, https://doi.org/10.1126/sciadv.add9031, 2023.
Wotawa, G. and Trainer, M.: The Influence of Canadian Forest Fires on Pollutant Concentrations in the United States, Science (New York, N.Y.), 288, 324–328, https://doi.org/10.1126/science.288.5464.324, 2000.
Xu, C. and Wang, L.: Atmospheric Oxidation Mechanism of Phenol Initiated by OH Radical, J. Phys. Chem. A, 117, 2358–2364, https://doi.org/10.1021/jp308856b, 2013.
Xu, J., Griffin, R. J., Liu, Y., Nakao, S., and Cocker, D. R.: Simulated impact of NOx on SOA formation from oxidation of toluene and m-xylene, Atmos. Environ., 101, 217–225, https://doi.org/10.1016/j.atmosenv.2014.11.008, 2015.
Xu, L., Crounse, J. D., Vasquez, K. T., Allen, H., Wennberg, P. O., Bourgeois, I., Brown, S. S., Campuzano-Jost, P., Coggon, M. M., Crawford, J. H., DiGangi, J. P., Diskin, G. S., Fried, A., Gargulinski, E. M., Gilman, J. B., Gkatzelis, G. I., Guo, H., Hair, J. W., Hall, S. R., Halliday, H. A., Hanisco, T. F., Hannun, R. A., Holmes, C. D., Huey, L. G., Jimenez, J. L., Lamplugh, A., Lee, Y. R., Liao, J., Lindaas, J., Neuman, J. A., Nowak, J. B., Peischl, J., Peterson, D. A., Piel, F., Richter, D., Rickly, P. S., Robinson, M. A., Rollins, A. W., Ryerson, T. B., Sekimoto, K., Selimovic, V., Shingler, T., Soja, A. J., St. Clair, J. M., Tanner, D. J., Ullmann, K., Veres, P. R., Walega, J., Warneke, C., Washenfelder, R. A., Weibring, P., Wisthaler, A., Wolfe, G. M., Womack, C. C., and Yokelson, R. J.: Ozone chemistry in western U.S. wildfire plumes, Sci. Adv., 7, eabl3648, https://doi.org/10.1126/sciadv.abl3648, 2021.
Yee, L. D., Kautzman, K. E., Loza, C. L., Schilling, K. A., Coggon, M. M., Chhabra, P. S., Chan, M. N., Chan, A. W. H., Hersey, S. P., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from biomass burning intermediates: phenol and methoxyphenols, Atmos. Chem. Phys., 13, 8019–8043, https://doi.org/10.5194/acp-13-8019-2013, 2013.
Yu, L., Smith, J., Laskin, A., George, K. M., Anastasio, C., Laskin, J., Dillner, A. M., and Zhang, Q.: Molecular transformations of phenolic SOA during photochemical aging in the aqueous phase: competition among oligomerization, functionalization, and fragmentation, Atmos. Chem. Phys., 16, 4511–4527, https://doi.org/10.5194/acp-16-4511-2016, 2016.
Yu, Z., Jang, M., and Madhu, A.: Prediction of Phase State of Secondary Organic Aerosol Internally Mixed with Aqueous Inorganic Salts, J. Phys. Chem. A, 125, 10198–10206, https://doi.org/10.1021/acs.jpca.1c06773, 2021a.
Yu, Z., Jang, M., Zhang, T., Madhu, A., and Han, S.: Simulation of Monoterpene SOA Formation by Multiphase Reactions Using Explicit Mechanisms, ACS Earth Space Chem., 5, 1455–1467, 2021b.
Zhang, Q., Jimenez, J. L., Canagaratna, M. R., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M. R., Takami, A., Middlebrook, A. M., Sun, Y. L., Dzepina, K., Dunlea, E., Docherty, K, DeCarlo, P. F., Salcedo, D., Onasch, T., Jayne, J. T., Miyoshi, T., Shimono, A, Hatakeyama, S., Takegawa, N., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S, Weimer, S., Demerjian, K., Williams, P., Bower, K., Bahreini, R., Cottrell, L., Griffin, R. J., Rautiainen, J., Sun, J. Y., and Zhang, Y. M.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophy. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhang, Y., Xue, L., Carter, W. P. L., Pei, C., Chen, T., Mu, J., Wang, Y., Zhang, Q., and Wang, W.: Development of ozone reactivity scales for volatile organic compounds in a Chinese megacity, Atmos. Chem. Phys., 21, 11053–11068, https://doi.org/10.5194/acp-21-11053-2021, 2021.
Zhao, L., Li, P., and Yalkowsky, S. H.: Predicting the entropy of boiling for organic compounds, J. Chem. Info. Comput. Sci., 39, 1112–1116, 1999.
Zhou, C., Jang, M., and Yu, Z.: Simulation of SOA formation from the photooxidation of monoalkylbenzenes in the presence of aqueous aerosols containing electrolytes under various NOx levels, Atmos. Chem. Phys., 19, 5719–5735, https://doi.org/10.5194/acp-19-5719-2019, 2019.
Zuend, A., Marcolli, C., Booth, A. M., Lienhard, D. M., Soonsin, V., Krieger, U. K., Topping, D. O., McFiggans, G., Peter, T., and Seinfeld, J. H.: New and extended parameterization of the thermodynamic model AIOMFAC: calculation of activity coefficients for organic-inorganic mixtures containing carboxyl, hydroxyl, carbonyl, ether, ester, alkenyl, alkyl, and aromatic functional groups, Atmos. Chem. Phys., 11, 9155–9206, https://doi.org/10.5194/acp-11-9155-2011, 2011.