the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 28 May 2024
| 28 May 2024
Observation-constrained kinetic modeling of isoprene SOA formation in the atmosphere
Chuanyang Shen
Xiaoyan Yang
Joel Thornton
John Shilling
Chenyang Bi
Gabriel Isaacman-VanWertz
Isoprene has the largest global non-methane hydrocarbon emission, and the oxidation of isoprene plays a crucial role in the formation of secondary organic aerosol (SOA). Two primary processes are known to contribute to SOA formation from isoprene oxidation: (1) the reactive uptake of isoprene-derived epoxides on acidic or aqueous particle surfaces and (2) the absorptive gas–particle partitioning of low-volatility oxidation products. In this study, we developed a new multiphase condensed isoprene oxidation mechanism that includes these processes with key molecular intermediates and products. The new mechanism was applied to simulate isoprene gas-phase oxidation products and SOA formation from previously published chamber experiments under a variety of conditions and atmospheric observations during the Southern Oxidant and Aerosol Studies (SOAS) field campaign. Our results show that SOA formation from most of the chamber experiments is reasonably reproduced using our mechanism, except when the concentration ratios of initial nitric oxide to isoprene exceed ∼ 2, the formed SOA is significantly underpredicted. The SOAS simulations also reasonably agree with the measurements regarding the diurnal pattern and concentrations of different product categories, while the total isoprene SOA remains underestimated. The molecular compositions of the modeled SOA indicate that multifunctional low-volatility products contribute to isoprene SOA more significantly than previously thought, with a median mass contribution of ∼ 57 % to the total modeled isoprene SOA. However, this contribution is intricately intertwined with IEPOX-derived SOA (IEPOX: isoprene-derived epoxydiols), posing challenges for their differentiation using bulk aerosol composition analysis (e.g., the aerosol mass spectrometer with positive matrix factorization). Furthermore, the SOA from these pathways may vary greatly, mainly dependent on the volatility estimation and treatment of particle-phase processes (i.e., photolysis and hydrolysis). Our findings emphasize that the various pathways to produce these low-volatility species should be considered in models to more accurately predict isoprene SOA formation. The new condensed isoprene chemical mechanism can be further incorporated into regional-scale air quality models, such as the Community Multiscale Air Quality Modelling System (CMAQ), to assess isoprene SOA formation on a larger scale.
- Article
(3438 KB) - Full-text XML
-
Supplement
(13798 KB) - BibTeX
- EndNote





Isoprene (2-methyl-1,3-butadiene, C5H8) is a highly reactive hydrocarbon that is widely recognized as the most abundant biogenic volatile organic compound (BVOC) emitted into the atmosphere (Guenther et al., 2006). Given its large flux globally and high reactivity, isoprene plays a key role in affecting the balance of atmospheric trace species such as O3, NOx (= NO + NO2), and HOx radicals (= OH + HO2) and is also a significant source of secondary organic aerosol (SOA) (Edney et al., 2005; Kroll and Seinfeld, 2005; Dommen et al., 2006; Henze and Seinfeld, 2006; Kroll et al., 2006; Surratt et al., 2006; Lewandowski et al., 2008; Fu et al., 2009; Jaoui et al., 2010; Surratt et al., 2010). Field measurements indicate high SOA mass concentrations from isoprene, which can reach 4 µg m−3 or even higher in summertime (Claeys et al., 2004; Kourtchev et al., 2005; Budisulistiorini et al., 2013; Hu et al., 2015). Therefore, understanding the isoprene atmospheric oxidation and the corresponding SOA formation mechanisms is of crucial importance for accurate estimation of ambient PM2.5 mass concentrations, especially in BVOC-dominant regions.
The gas-phase oxidation of isoprene initiated by both hydroxyl radicals (OH) and nitrate radicals (NO3) has been extensively investigated in laboratory and computational studies (Mcgivern et al., 2000; Paulot et al., 2009; Peeters et al., 2009; Crounse et al., 2011; Teng et al., 2017; Wennberg et al., 2018; Berndt et al., 2019; Guo et al., 2020; Vereecken et al., 2021; Tsiligiannis et al., 2022; Zhang et al., 2022), with many different chemical mechanisms having been proposed (Paulson and Seinfeld, 1992; Carter, 1996; Pöschl et al., 2000; Fan and Zhang, 2004; Taraborrelli et al., 2009; Zhang et al., 2011; Wennberg et al., 2018). Likewise, in the particle phase, many isoprene-oxidation-derived molecular species have also been reported in aerosol samples from field and laboratory studies that can provide insight into the SOA formation mechanisms (Claeys et al., 2004; Edney et al., 2005; Kourtchev et al., 2005; Carlton et al., 2009; Surratt et al., 2010; Lin et al., 2012, 2013; Liu et al., 2016; Schwantes et al., 2019). While great advances have been made in both the gas and particle phases, a comprehensive molecular-level isoprene SOA model is lacking, partly because of the highly complex processes that can contribute to isoprene SOA under different conditions. In general, two primary pathways contribute to SOA formation from isoprene oxidation: (1) the reactive uptake of epoxides on acidic or aqueous particle surfaces (Surratt et al., 2010; Lin et al., 2012, 2013; Nguyen et al., 2014; Y. Zhang et al., 2018) and (2) the gas–particle absorptive partitioning of multifunctional low-volatility compounds, which are usually formed via multigenerational oxidation (Krechmer et al., 2015; Liu et al., 2016; Schwantes et al., 2019).
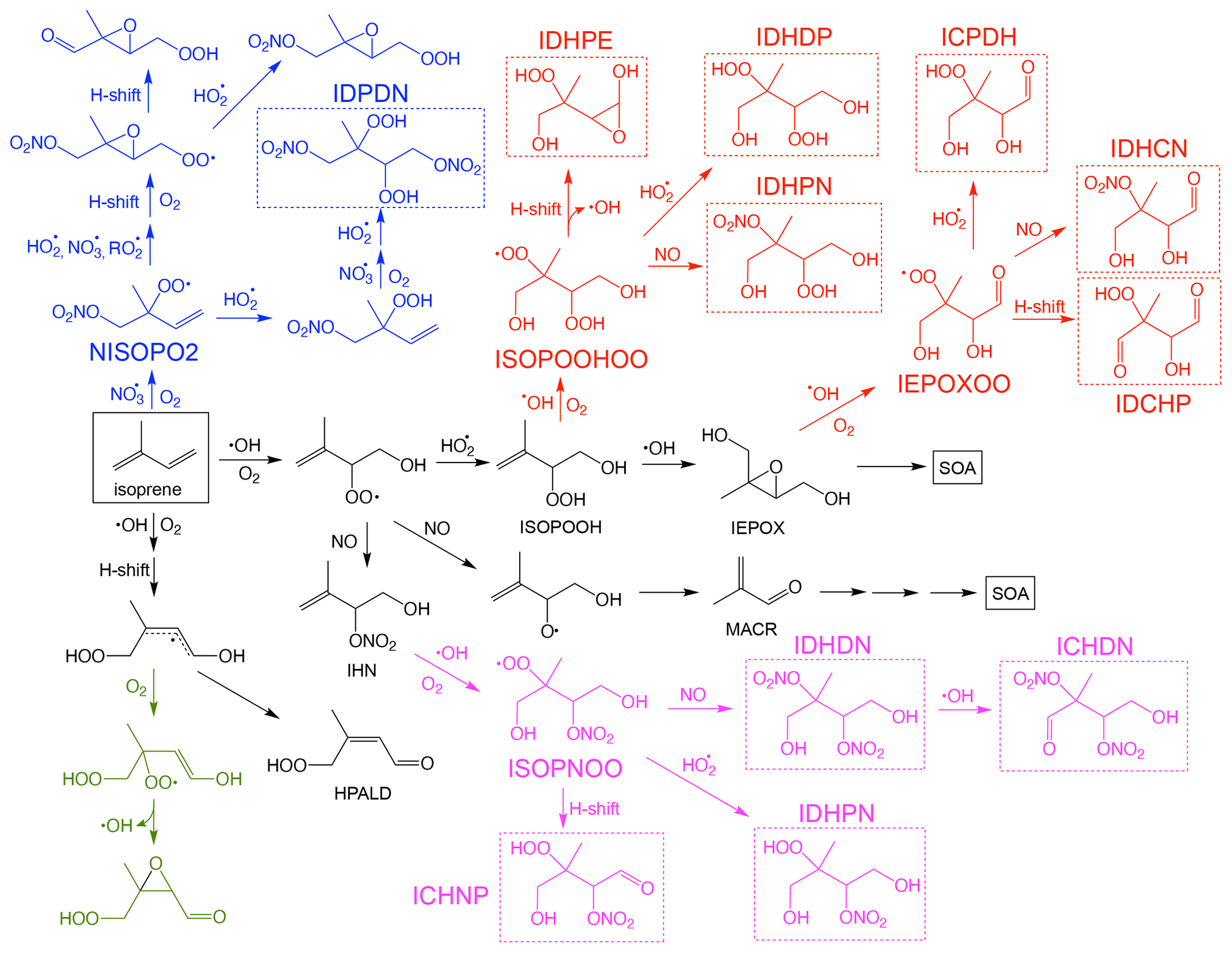
Figure 1Simplified reaction scheme for isoprene oxidation by OH and NO3. The major low-volatility species that may contribute to SOA formation are highlighted in dashed boxes. For simplicity, further reactions of the low-volatility species with OH and photolysis, as well as RO2 + RO2 reactions, are not shown in the scheme, but they are included in the mechanism.
SOA formation from the reactive uptake of epoxides generally refers to the ring-opening reactions of isoprene-derived epoxydiols (IEPOX) onto aerosols catalyzed by acidity or water. The main SOA products from this pathway include 2-methyltetrols (2-MT), C5-alkenetriols, and isoprene-derived organosulfates (IEPOX-OS) (Surratt et al., 2010; Lin et al., 2012). Both SOA yield and composition from this pathway vary greatly and depend on many factors, such as particle surface area, particle acidity, and particle phase state (Gaston et al., 2014; Nguyen et al., 2014; Xu et al., 2015; Y. Zhang et al., 2018; Yee et al., 2020). Owing to its reported substantial contribution to ambient PM2.5, this pathway has been extensively studied in prior laboratory research (Lin et al., 2012; Nguyen et al., 2014). Many model studies have also attempted to explicitly model SOA production from IEPOX reactive uptake (Pye et al., 2013; Budisulistiorini et al., 2015). In addition to IEPOX, other epoxide and lactone species, such as methacrylic acid epoxide (MAE) and hydroxymethel-methyl-α-lactone (HMML), have also been suggested as significant contributors to isoprene SOA in the presence of NOx (Lin et al., 2013; Nguyen et al., 2015b; Riedel et al., 2015). Meanwhile, low-volatility products from multigenerational oxidation may also contribute to isoprene SOA, especially for those that maintain the five-carbon moiety (C5-LV). A well-studied example is the fact that isoprene hydroxy hydroperoxide (ISOPOOH), the direct precursor of IEPOX, was found to undergo OH oxidation and form highly oxidized low-volatility products that form SOA (see Fig. 1) (Krechmer et al., 2015; Liu et al., 2016; D'ambro et al., 2017; Mettke et al., 2023). In addition, N-containing multifunctional low-volatility species (C5-NLV) from chamber and field measurements suggest that further oxidation of isoprene nitrates could be another SOA source (Lee et al., 2016; Schwantes et al., 2019). These low-volatility isoprene nitrates could be formed from NOx-involved pathways during OH oxidation or during nighttime NO3 oxidation (Fig. 1) (Ng et al., 2008; Schwantes et al., 2015, 2019).
While the basic understanding of these pathways and their contribution to isoprene SOA formation have been established, they have not been fully incorporated into chemical models, especially for ambient SOA simulations. In early studies, most regional and global models used highly simplified gas-phase isoprene oxidation condensed mechanisms such as the Carbon Bond mechanism (Gery et al., 1989; Yarwood et al., 2005, 2010) and the SAPRC mechanisms (Carter, 2000; Carter, 2010; Carter, 2023). Most of these mechanisms are too condensed to comprehensively represent these low-volatility products that are important for SOA formation (Perring et al., 2009; Archibald et al., 2011). Furthermore, the SOA formation in these early models used volatility-based yield parameterizations (Odum et al., 1996; Donahue et al., 2006). Because these parameterizations are derived from laboratory SOA mass concentrations formed under specific oxidation conditions, it may bring large uncertainties to the SOA estimation under realistic conditions (Marais et al., 2016). In order to better interpret and predict isoprene SOA, more detailed representations of the isoprene gas-phase oxidation and multiphase processes are needed in chemical mechanisms, especially for the products that are relevant to SOA formation. In a prior work, Thornton et al. (2020) incorporated and modified the near-explicit Master Chemical Mechanism (MCM) into a dimensionless (0-D) model to simulate isoprene SOA formation from chamber studies. Their model estimations agree well with the observations in SOA mass concentrations. However, such near-explicit mechanisms are too large to be applied to regional or global models. To overcome these issues, a few recent studies have attempted to apply intermediate-size mechanisms in large-scale models which include the isoprene gas-phase oxidation scheme to a certain extent of detail (Stadtler et al., 2018; Bates and Jacob, 2019; Müller et al., 2019). This implementation allowed for simulations of the key gas-phase products such as ISOPOOH and IEPOX, which turned out to be very similar to the MCM (Müller et al., 2019). The model investigated by Bates and Jacob (2019) estimated that the global production of isoprene SOA is about one-third from each of IEPOX, C5-NLV, and C5-LV. Nevertheless, not all the important SOA species and formation pathways were included in these mechanisms; gas–particle partitioning and particle-phase chemistry were not always considered and simplified parameterizations were still used in some of these models. Systematic validation of these mechanisms against laboratory and field measurements was also lacking.
In this study, we developed a new condensed multiphase isoprene oxidation chemical mechanism adapted to the SAPRC structure (Carter, 2010). The new mechanism represents isoprene chemistry with an intermediate level of chemical details to include the major SOA species. It was also made flexible for the inclusion of new isoprene chemistry that is reported in laboratory, mechanistic, and field studies (e.g., Wennberg et al., 2018; Vasquez et al., 2020; Mettke et al., 2022; Carlsson et al., 2023). Lastly, this mechanism is also implementable into regional or global air quality models to better represent isoprene chemistry and SOA formation. This mechanism was incorporated into a box model to simulate existing isoprene oxidation chamber experiments under various initial conditions (e.g., OH oxidation, NO3 oxidation, and different NOx levels). The key gas-phase products from all the pathways described above and SOA mass concentrations were compared with laboratory observations (where available) and other chemical mechanisms to evaluate the mechanism's performance. We also applied the new mechanism to model the 2013 Southern Oxidant and Aerosol Studies (SOAS) field campaign at the Centreville, AL, site (Lee et al., 2016; H. Zhang et al., 2018) to elucidate the relative importance of the various pathways in SOA formation under real atmospheric conditions. To the best of our knowledge, this is the first time that a comprehensive molecular-level isoprene SOA mechanism is evaluated using field observations. Lastly, we also discuss the major uncertainties in current mechanistic understandings and the needed future research directions regarding isoprene SOA chemistry.
2.1 F0AM-WAM
We use the Framework for zero-dimensional Atmospheric Modelling (F0AM v3.2) (Wolfe et al., 2016) coupled to the Washington Aerosol Module (WAM), denoted as F0AM-WAM, to simulate the isoprene oxidation processes and predict SOA formation and evolution (Thornton et al., 2020). F0AM is a flexible and efficient MATLAB-based framework for modeling zero-dimensional atmospheric chemistry, and it allows for easy incorporation of new and modified chemical mechanisms to simulate a variety of typical problems, including photochemical chambers and field observations from the ground and aircraft (Brune et al., 2021; Lyu et al., 2022). The WAM is a specialized module designed to simulate the formation and evolution of SOA by explicitly treating the condensation and evaporation of low-volatility compounds. In combination, F0AM-WAM provides a comprehensive tool for studying the interactions between atmospheric gas-phase chemistry and aerosol processes.
2.2 Gas-phase chemistry
A new isoprene oxidation gas-phase kinetic mechanism was developed in this study, named UCR-ISOP. It was developed on top of a version of the SAPRC07 mechanism that is currently used in the CMAQ model (i.e., SAPRC07tic) (Carter, 2010; Xie et al., 2013). The other chemical mechanisms discussed in this work include the MCMv3.3.1 (denoted as “MCM” below), the Caltech isoprene mechanism (the “reduced_plus_v5” version denoted as “Caltech” below) summarized by Wennberg et al. (2018) and reduced by Bates and Jacob (2019), the modified MCM by Thornton et al. (2020) (denoted as “MCM-UW” below), and the isoprene mechanism proposed by the Forschungszentrum Jülich (denoted as “FZJ” below) (Vereecken et al., 2021; Tsiligiannis et al., 2022; Carlsson et al., 2023).
In UCR-ISOP, for the species that were already included in the SAPRC07tic mechanism, we have preserved their nomenclature. For other species, the naming convention is the same as in the Caltech mechanism, which lumps the isomers with the same functional groups into one compound. It is to be noted that our new mechanism includes many multifunctional C5 species which were not included in the SAPRC07 mechanism but are pivotal as SOA precursors. For example, isomers with two hydroxy (–OH) and two hydroperoxide (–OOH) groups are now represented by a single species, IDHDP. Thus, each of the low-volatility species shown in Fig. 1 is described as an individual compound that could represent the sum of several isomers. On the contrary, certain isomers are individually represented (with some extent of lumping in certain cases) for several major species, including the isoprene hydroxyl peroxy radicals (ISOPOHOO, two isomers), hydroperoxyl aldehydes (HPALD, two isomers), ISOPOOH (three isomers), IEPOX (two isomers), and isoprene hydroxynitrates (IHN, three isomers). These species have been extensively studied in the literature and distinct reactivities and reaction products have been reported (Wennberg et al., 2018). Maintaining some of the lumped isomers for these species permits more accurate representations of their further product distributions. All the abbreviated names in the UCR-ISOP mechanism are described in Table S1 in the Supplement. Compared to SAPRC07tic (38 species and 124 reactions), UCR-ISOP adds 39 additional species and 118 additional gas-phase reactions. In comparison, the MCM has 610 species and 1974 reactions (related to isoprene); the Caltech mechanism has 155 species and 429 reactions. In addition, our mechanism also incorporates many up-to-date theoretical or experimental findings on isoprene oxidation and SOA formation, including (1) temperature and pressure dependence of organic nitrate yield from peroxy radical (RO2) + NO reactions as suggested by the Caltech mechanism, (2) isomerization reactions for the major RO2 based on recent studies (D'ambro et al., 2017; Wennberg et al., 2018; Vereecken et al., 2021; Mettke et al., 2023), (3) dimer formation from several RO2 + RO2 reactions that were supported by prior chamber experiments (Ng et al., 2008; Mettke et al., 2023), and (4) the loss pathways of C5-LV and C5-NLV species via reactions with OH and photolysis as suggested by either the Caltech mechanism or extrapolated from analogous reactions in MCM v3.3.1. For the isoprene + NO3 reactions, the new FZJ mechanism proposed by recent studies was also incorporated to some extent as discussed later (Vereecken et al., 2021; Tsiligiannis et al., 2022; Carlsson et al., 2023).
This mechanism was implemented into F0AM-WAM to simulate published chamber experimental data under different conditions. The model outputs are compared with both the available measurements and other existing mechanisms (i.e., the Caltech mechanism and the MCM). It should be noted that the mechanism is highly condensed and simplified for potential application in regional or global models. Thus, it does not capture all the known chemical reactions in isoprene oxidation.
2.3 Gas–particle partitioning
In this work, partitioning of low-volatility or semi-volatility species into the particle phase is parameterized to include two separate processes: absorptive equilibrium partitioning into an organic phase (Pankow, 1994; Odum et al., 1996) and aqueous uptake by liquid water (Wania et al., 2015; Isaacman-Vanwertz et al., 2016). In general, the condensation kinetics to particle is calculated as
where Kcond is the condensation rate (s−1), rp is the particle radius (cm) obtained from particle size measurements, Dg is the gas-phase diffusivity (cm2 s−1), α represents the mass accommodation coefficient, ω is the molecular mean thermal velocity (cm s−1), and SA is the aerosol surface area per volume (cm2 cm−3). In ideal gas–particle partitioning assumptions, α=1 is used, while for nonideal partitioning (e.g., in the presence of diffusion limitation), α in the range of 0.1–1 has been used (Saleh et al., 2013; Zhang et al., 2015). In Thornton et al. (2020), it was suggested that this range of α value has little impact on simulated isoprene SOA under chamber experimental conditions. In this work, we will test the influence of α in the SOAS simulations in sensitivity analysis.
The evaporation rate back to the gas phase (s−1) is calculated as
where Haq is the estimated Henry's law constant (M atm−1), LWC is the aerosol liquid water content (µg m−3), R is the ideal gas constant, T is the temperature (K), COA is the organic aerosol mass concentration (µg m−3), and C∗ is the saturation concentration (µg m−3). The C∗ value is calculated from the vapor pressure, which can be estimated using EVAPORATION (Compernolle et al., 2011) and SIMPOL.1 (Pankow and Asher, 2008). The calculated C∗ values for the major low-volatility and semi-volatility species are listed in Table S2. When different C∗ values are estimated for different isomers of each low-volatility species, we used the lowest C∗, and the uncertainty of this treatment is tested and discussed later. When the gas–particle equilibrium is established, the fraction of species in the particle phase Fp will be estimated as
Under dry conditions, Fp will be simplified into given that LWC = 0 µg m−3.
2.4 Reactive uptake
All the chamber experiments used in this work to evaluate the isoprene multiphase mechanism were performed in the absence of aqueous and acidic sulfate seed aerosols (Kroll et al., 2006; Ng et al., 2008; Schwantes et al., 2015; Liu et al., 2016; D'ambro et al., 2017; Shilling et al., 2019). Therefore, the reactive uptake of IEPOX (and other epoxides) is not expected to occur in these chamber experiments. However, in the SOAS field campaign where aqueous particles containing sulfate were ubiquitous, IEPOX-derived SOA was reported to be an important contributor to total organic aerosol (Hu et al., 2015; Xu et al., 2015; H. Zhang et al., 2018). Therefore, in the application of the model to the field measurements, in addition to the absorptive partitioning of semi-volatility and low-volatility oxidation products, we also consider the SOA formation from IEPOX reactive uptake onto acidic and aqueous aerosols. For modeling simplicity, only 2-MT and IEPOX-OS are assumed to be formed from this process. Therefore, the “2-MT” in the model is likely a summation of 2-MT, C5-alkenetriols, and other minor IEPOX-derived non-OS species. Measurements report C5-alkenetriols as a tracer for IEPOX-derived SOA (Lin et al., 2012), but no formation pathway is known for these compounds. Some work has indicated that they may partly be analytical products of other tracers such as IEPOX-OS (Rattanavaraha et al., 2016; Cui et al., 2018), but their origin remains highly uncertain, so no formation mechanism is included in the mechanism examined here. The IEPOX-SOA formation is parameterized in the model as follows:
where khet is the heterogeneous reaction rate of IEPOX (s−1), SA is the surface area of the aerosol that IEPOX is taken up onto (cm2 cm−3), ω is the mean molecular speed of IEPOX in the gas phase (cm s−1), and γ is the reactive uptake coefficient, which can be parameterized using a resistor model from previous studies (Gaston et al., 2014). This resistor model can be calculated as
where α is the unitless accommodation coefficient (0.02), rp is the aerosol particle's radius (cm), Dgas is the gas-phase diffusion of IEPOX (cm2 s−1), and the aqueous term, Γaq, is calculated from the following equation:
where V is the particle volume concentration (cm3 cm−3), R is the ideal gas constant, T is the ambient temperature (K), ω is the gas-phase mean molecular speed (cm s−1) of IEPOX, and kaq is the pseudo-first-order rate constant (s−1) defined in Sect. S1 in the Supplement. The parameter with the largest uncertainty regarding IEPOX reactive uptake is Haq, for which prior studies have used values ranging from 1.9 × 107–4 × 108 M atm−1 (Chan et al., 2010; Gaston et al., 2014; Schmedding et al., 2020). Here, we choose to use a median Haq value of 1.3 × 108 M atm−1, which was predicted by Eddingsaas et al. (2010). In model sensitivity analysis, it turns out that the IEPOX-SOA concentration is not very sensitive to the chosen Haq value in this range.
An additional description of this process can be found in Sect. S1. In the model, the aerosols were assumed to be a homogeneous mixture of organic and inorganic constituents. However, the influence of core–shell particle morphology on reactive uptake is simulated based on the method reported by Zhang et al. (2018) in sensitivity analyses. The LWC is predicted using the thermodynamic equilibrium model ISORROPIA II based on the measured concentrations of inorganic species, including ammonium, nitrate, and sulfate (Fountoukis and Nenes, 2007). The IEPOX concentration is from the output of the gas-phase oxidation reactions. In addition to IEPOX, several other products from isoprene oxidation may also undergo reactive uptake, such as HMML (Nguyen et al., 2015b), MAE (Lin et al., 2013), 1,2-IHN (Vasquez et al., 2020), glyoxal (Kroll et al., 2005; Carlton et al., 2007), and the other epoxide products included in the mechanism (several examples shown in Fig. 1). The reactive uptake of these species is included in the model. For HMML and MAE, the major reactive uptake products are 2-methylglyceric acid (MGA) and its corresponding organosulfate. The other epoxides which have not been studied in prior research are assumed to undergo a similar process as IEPOX in the model that form ring-open alcohols and organosulfates. In the case of 1,2-IHN, the reactive uptake product is expected to be a diol (IDH) via hydrolysis that is expected to quickly evaporate back to the gas phase. The reaction rate is calculated from LWC, Haq, and the aqueous hydrolysis rate used in Vasquez et al. (2020). For the reactive uptake of glyoxal, because the equilibrium state is quickly established between aqueous and hydrated glyoxal and the hydrated state is strongly favored, we adopt the Haq of 2.6 × 107 M atm−1 to calculate the glyoxal-derived aqueous SOA (Hastings et al., 2005).
2.5 Particle-phase reactions
After low-volatility compounds partition to the particle phase, they likely continue to undergo chemical evolution processes. These processes can either decrease organic aerosol mass such as particle-phase photolysis and hydrolysis (Pye et al., 2015; Krapf et al., 2016; Zawadowicz et al., 2020) or promote SOA formation like accretion reactions (Kroll and Seinfeld, 2008). Thus, it is necessary to include or parameterize these particle-phase reactions in models in order to better predict SOA's evolving mass concentration and chemical composition.
In prior work, Surratt et al. (2006) reported substantial formation of peroxides in isoprene SOA formed under NOx-free conditions, which exhibited a pronounced decrease with extended radiation time. Consistently, organic peroxides are believed to be susceptible to photolysis (Chacon-Madrid et al., 2013) with lifetimes of about 6 d in Los Angeles. Zawadowicz et al. (2020) found that the SOA produced from isoprene oxidation under low-NOx conditions underwent photolysis-induced mass loss at rates between 1.5 % and 2.2 % of NO2 photolysis (). In order to simulate the SOA decay in the NOx-free chamber experiments (Kroll et al., 2006; Liu et al., 2016), we apply a first-order photolysis rate coefficient that is 2 % of to all the products with one or more –OOH groups formed under NOx-free conditions, as proposed by Thornton et al. (2020). However, under high-NOx conditions, prior studies did not observe such a rapid SOA mass decay (Kroll et al., 2006; Schwantes et al., 2019). Thus, we assume that particle-phase organic nitrate products photolyze at similar rate coefficients as for the known gas-phase alkylnitrates, which is much slower.
In the SOAS campaign simulations, because of the high relative humidity (RH) and LWC at the field site, we also apply hydrolysis reactions for the organic nitrate species in the simulated isoprene SOA. We assume that their average lifetime against hydrolysis is 3 h, through which the –ONO2 group is converted to the –OH group (Pye et al., 2015). Other particle-phase processes such accretion and heterogeneous OH oxidation are not included in the current model because the detailed kinetics and mechanisms are highly uncertain.
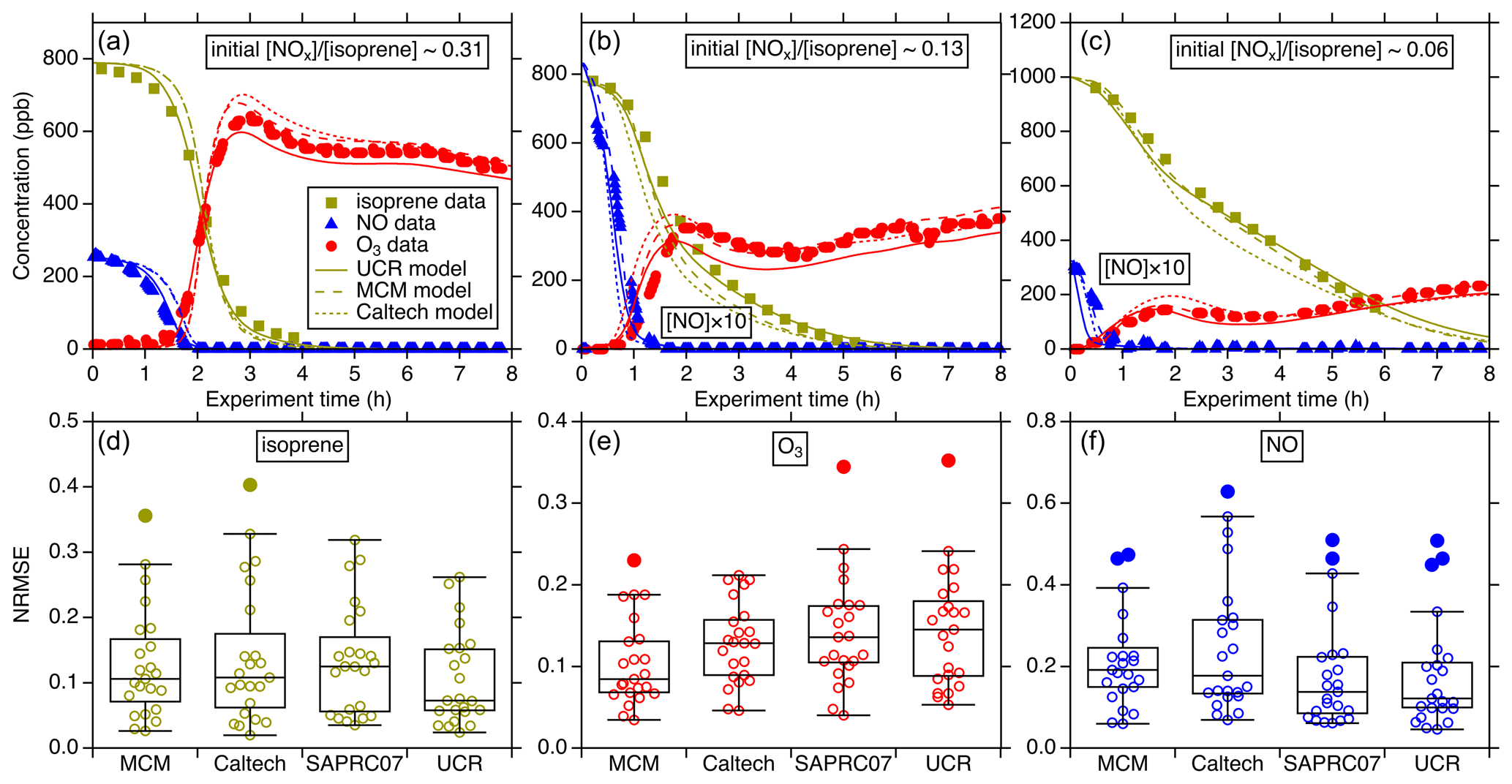
Figure 2Intercomparison of simulated isoprene, O3, and NO using different chemical mechanisms (MCM, Caltech, and UCR-ISOP, denoted as UCR) for the UNC-2010/2012 experiments: (a) 20101021N with an initial ratio of 0.31, (b) 20120603N with an initial ratio of 0.13, and (c) 20100904N with an initial ratio of 0.06. The three plots in the lower panel are the NRMSE for all the simulated UNC-2010/2012 and Kroll-2006 experiments using different chemical mechanisms: panel (d) is the comparison for isoprene, panel (e) is for O3, and panel (f) is for NO. NRMSE is defined as . denotes the mean of the observed values. On the x axis, SAPRC07 denotes the SAPRC07tic mechanism. For each box, the central horizontal line in the box indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers, and the outliers are plotted using the solid circle markers (other data points plotted using open circle markers).
3.1 Simulations of chamber isoprene oxidation experiments
3.1.1 Description of chamber experiments
Isoprene oxidation chamber experiments from previously published datasets were used here to test UCR-ISOP's performance in simulating trace gas species and SOA formation under different conditions. These experiments were designated as UNC-2010/2012 (Zhang et al., 2011, 2013), Kroll-2006 (Kroll et al., 2006), PNNL-2014 (Liu et al., 2016), PNNL-2018 (Thornton et al., 2020; Zawadowicz et al., 2020), Schwantes-2019 (Schwantes et al., 2019), Ng-2008 (Ng et al., 2008), Carlsson-2023 (Carlsson et al., 2023), and Schwantes-2015 (Schwantes et al., 2015). Kroll-2006 (Run 1–9) and PNNL-2018 were performed under NOx-free conditions; UNC-2010/2012, Kroll-2006 (Run 9–14), PNNL-2014, and Schwantes-2019 experiments were performed under high-NOx conditions; and Ng-2008, Carlsson-2023, and Schwantes-2015 experiments were performed under NO3 oxidation conditions. Some of these experiments were only used for gas-phase mechanism evaluation due to unavailable particle measurements, and some were also used to evaluate the SOA simulations. The conditions for these chamber experiments can be found in Tables S3–S7, and additional details can be found in the corresponding literature.
3.1.2 Gas-phase modeling
To evaluate the new UCR-ISOP gas-phase mechanism, we compared its simulations against the above-described chamber experimental data and other existing mechanisms. The gas-phase species concentrations that are typically available for model evaluations are O3, NOx, and isoprene. Quantitative organic product concentrations, although crucial, are usually unavailable. Therefore, we evaluated the UCR-ISOP mechanism against isoprene–O3–NOx measurements from prior high-NOx (UNC-2010/2012) and low-NOx chamber experiments (Kroll-2006) and against simulated oxidation products between different mechanisms, including the MCM (Jenkin et al., 2015), the Caltech mechanism (Wennberg et al., 2018), and the SAPRC07tic mechanism (Carter, 2010; Xie et al., 2013).
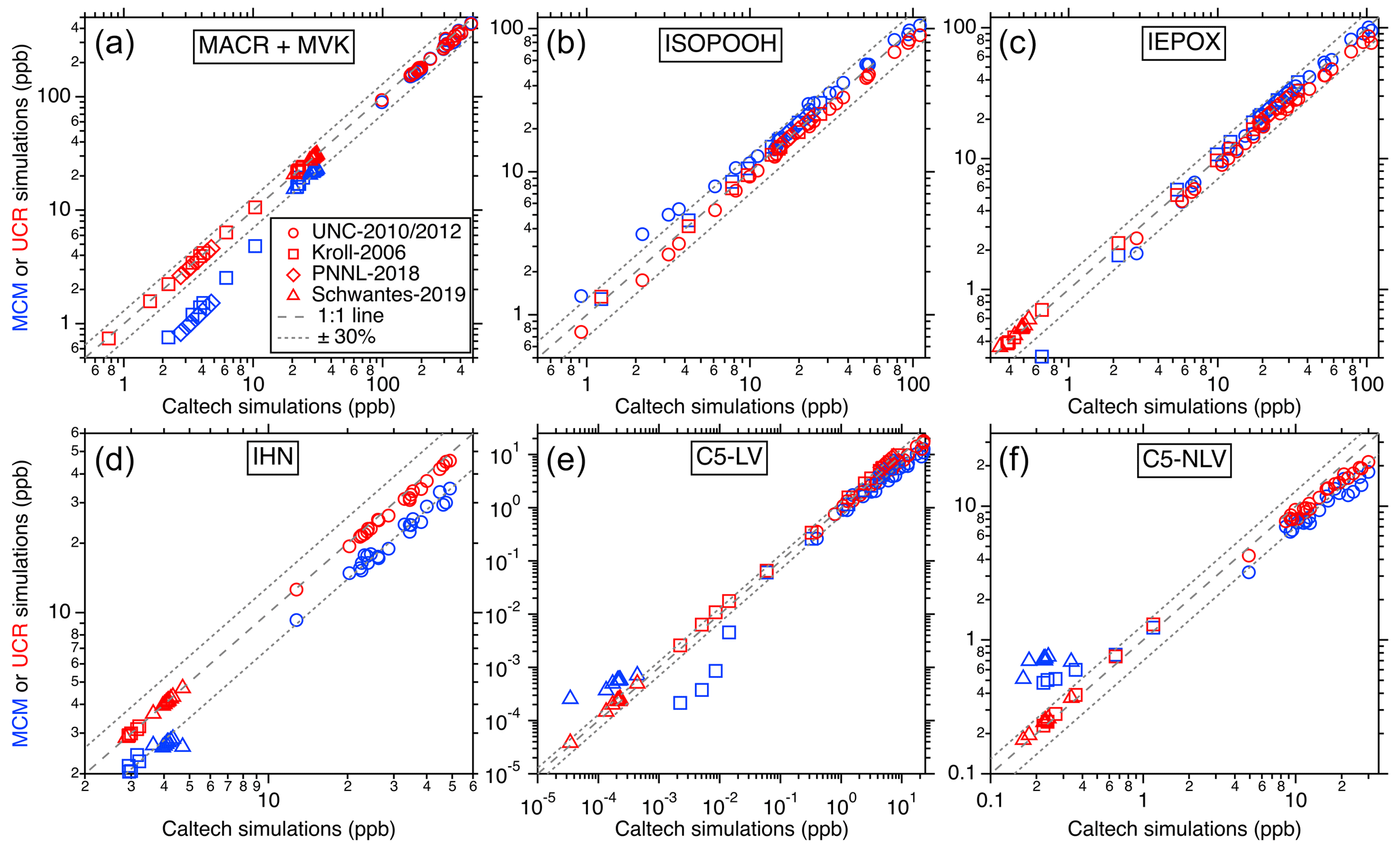
Figure 3Comparisons of the simulated isoprene oxidation products' maximum concentrations in each laboratory experiment between different chemical mechanisms. The x axes represent the predictions using the Caltech mechanism, and y axes represent values from UCR-ISOP (red markers) and MCM (blue markers). Different marker types represent different chamber studies. In all panels, the dashed line indicates a 1:1 correspondence, and the dotted line delineates a 30 % uncertainty boundary.
Figure 2a–c show representative examples of the simulation performance of the various gas-phase mechanisms for three UNC-2010/2012 high-NOx experiments with varied initial NOx isoprene concentration ratios. The results indicate that UCR-ISOP can reasonably predict the temporal evolutions of isoprene, O3, and NO concentrations under a wide range of initial conditions. The statistical evaluation for all the simulated experiments is summarized in Fig. 2d–f using the normalized root mean square error (NRMSE) as the metric. NRMSE can provide a harmonized assessment of the average magnitude of errors between the predicted and the observed values. It appears that the isoprene and NO decay is modeled very well by UCR-ISOP compared to other mechanisms with a median NRMSE of 0.07 and 0.12, respectively. The O3 concentrations are modeled reasonably well by all four mechanisms with median errors less than 0.15. For the NO prediction, the MCM and the Caltech mechanism predicted relatively larger bias against measurements with median errors around 0.2. The simulation–measurement comparison for NO2 was not examined because the measured NO2 is interfered with by other NOy species (e.g., NO3, HNO3, alkyl nitrates, N2O5) (Zhang et al., 2011). The performances of all the compared mechanisms are in general similar to each other. These results support the fact that with the new additions of species and pathways key to isoprene SOA formation on top of the SAPRC07 mechanism as well as reduction of the more explicit mechanisms into UCR-ISOP, the capability to accurately predict isoprene, O3, and NOx is not negatively affected.
For the other important gas-phase products such as ISOPOOH, IEPOX, methacrolein (MACR), methyl vinyl ketone (MVK), IHN, glyoxal, methylglyoxal, and all the multifunctional low-volatility compounds, comparisons were made only among different mechanisms since real-time and quantitative measurements were not available. The results for the major products and categories (i.e., C5-LV and C5-NLV) from isoprene OH oxidation can be found in Fig. 3, and additional comparisons for individual species can be found in Fig. S1 in the Supplement. In these comparisons, the predictions for all these products are generally consistent between the UCR-ISOP mechanism, Caltech mechanism, and MCM with some species and conditions more scattered than the others. Specifically, the six major individual and groups of products presented in Fig. 3 show very good agreement between the three mechanisms, especially under the lower concentration ranges, suggesting that UCR-ISOP does not sacrifice model performance during mechanism reduction and can simulate the most important products very well under most OH oxidation conditions. However, the mechanism comparisons for some individual species exhibit larger differences, as illustrated in Fig. S1. For example, the two low-volatility products from ISOPOOH + OH oxidation, IDHPE and IDHDP, exhibit opposite trends when comparing between the UCR-ISOP and Caltech mechanisms. This is because we adopted a slower isomerization rate coefficient for ISOPOOHOO (C5H11O6, peroxy radical from ISOPOOH + OH, see Fig. 1) than that used in the Caltech mechanism (Wennberg et al., 2018) and other models (D'ambro et al., 2017; Thornton et al., 2020). Recent work by Mettke et al. (2023) suggested that the ISOPOOHOO isomerization is ∼ 0.002 s−1 rather than the order of 0.1 s−1 reported by D'ambro et al. (2017) and Wennberg et al. (2018). Thus, in the UCR-ISOP mechanism, we chose to use a rate coefficient 10 times slower than the Caltech mechanism (i.e., 0.01 s−1), which is between the two very different suggested rate coefficients. This change greatly affects the yields of IDHPE vs. IDHDP. In addition, we consider the rapid ISOPOOHOO self-reaction to form the corresponding carbonyl (C5H10O6), alcohol (C5H12O6), and dimer (C10H22O10), suggested by Mettke et al. (2023), with a rate coefficient of 1 × 10−11 . This dimer formation pathway could partly explain the slightly lower C5-LV simulation using the UCR-ISOP mechanism under high concentrations (Fig. 3e).
Moreover, the higher ICPDH predictions in the MCM simulations are due to the fact that the IEPOX-derived RO2 reacting with HO2 is assumed to completely form ICPDH, while in the Caltech and UCR-ISOP mechanisms, a much smaller branching ratio (∼ 35 %) leads to this hydroperoxide, and the rest leads to RO that subsequently decomposes. The latter treatment is likely more reasonable because several isomers of IEPOX-derived RO2 are tertiary RO2, which were suggested to form RO at high branching ratios by reacting with HO2 (Kurtén et al., 2017). Another very different simulated product is glyoxal, for which the UCR-ISOP and Caltech mechanisms predict lower glyoxal concentrations than the MCM by a factor of ∼ 3. This result is driven by the combined influence of many reactions. One of the major contributors to glyoxal formation in the MCM is from RO decomposition of C527O, which stems from isomerization of a first-generation RO from isoprene + OH (CISOPCO, see Fig. S2). However, we think this is a less likely pathway considering the multiple complex H shifts involved. Two other major glyoxal contributors in the MCM are from the NO3 oxidation pathway that will be discussed later.
The gas-phase isoprene + NO3 mechanism has a large uncertainty and is not consistent among different experimental studies. The key discrepancy lies in the fates of the alkoxy radicals (INO) from the primary nitrate RO2 (NISOPO2) reacting with NO3, HO2, and RO2. In the Caltech mechanism (Wennberg et al., 2018), the β-isomers of INO exclusively undergo dissociation to form MACR+MVK, formaldehyde, and NO2, while the δ-isomers of INO mainly isomerize or add O2 to form isoprene carbonyl nitrates (ICN) and HO2. This mechanism was constrained by prior laboratory measurements by Schwantes et al. (2015). In contrast, the recent FZJ mechanism suggests that all the INO isomers predominantly undergo ring-closure reactions to form epoxide-containing RO2. The two different mechanisms could lead to significant discrepancies in the concentrations of MACR/MVK, ICN, HO2, and NO2. Carlsson et al. (2023) suggested that the FZJ mechanism is likely more accurate by showing that the Caltech mechanism significantly overpredicts MACR+MVK measured in experiments due to INO fragmentation. However, Carlsson et al. (2023) also stated that the FZJ mechanism underpredicts HO2, suggesting missing sources. In the UCR-ISOP mechanism, we seek to reach a balance between the two isoprene + NO3 mechanisms. We tentatively choose to have 50 % of the β-1,2-INO isomer (a major isomer) undergo the ring-closure reaction, while the rest of the INO does not form epoxides. This leads to much better agreement with the measured MACR+MVK than the Caltech mechanism (but still slightly worse than the FZJ simulations). Figures S3 and S4 show the model results in comparison to the experiments in Carlsson et al. (2023) and Schwantes et al. (2015). Nevertheless, we regard this as a simplified solution and there are still large uncertainties in this pathway. Future mechanistic studies are needed to better understand the fates of INO. The lower isoprene carbonyl nitrates (ICN) and hydroperoxy nitrates (IPN) in the UCR-ISOP simulations than the other two mechanisms are generally found in UNC-2010/2012 conditions (OH oxidation with high NOx, under which NO3 oxidation of isoprene is also occurring) and the Schwantes-2015 conditions (NO3 oxidation, Fig. S4). This is in large part due to the differing treatment of the NO3+ isoprene pathway in UCR-ISOP. In particular, the MCM assumes that INO exclusively forms ICN, which is also a major source of glyoxal via ozonolysis and OH oxidation (Fig. S2). Thus, the higher simulated ICN in the MCM is also a major reason for the higher glyoxal predictions. Nevertheless, UCR-ISOP appears to agree with the Schwantes-2015 experimental data slightly better for ICN and IPN compared to the other two mechanisms (Fig. S4). Furthermore, the simulated IDHDN and ICHDN in UCR-ISOP are both lower than those predicted by the Caltech mechanism because the INO fates are treated differently. However, it should be noted that these differences stem largely from conditions where later-generation RO2 from NO3 + isoprene reacts with NO. Such conditions are only prominent in the UNC-2010/2012 experiments. In the real atmosphere, NO3 + isoprene is minimal during daytime when NO may be present. Thus, these differences are less likely to be as significant as found under the laboratory experiments. But as stated above, this pathway is still highly uncertain and requires future investigation.
Furthermore, we considered that NISOPO2 from isoprene + NO3 undergoes rapid self-reactions at a rate coefficient of 5 × 10−12 , suggested by Schwantes et al. (2015). In a previous study, Kwan et al. (2012) proposed dimer formation from NISOPO2 + NISOPO2 with a branching ratio of 3 %–4 % based on gas-phase measurements. However, Ng et al. (2008) observed a substantial quantity of dimers in the SOA from the same experiments, suggesting that the actual dimer formation branching ratio could be much higher given their very low volatility. In UCR-ISOP, we assume this branching ratio to be 10 %, which leads to good agreement with the SOA simulation (see next section). This dimer formation pathway from NISOPO2 + NISOPO2 could also partly explain the slightly lower C5-NLV simulation using the UCR-ISOP mechanism under high concentrations (Fig. 3f).
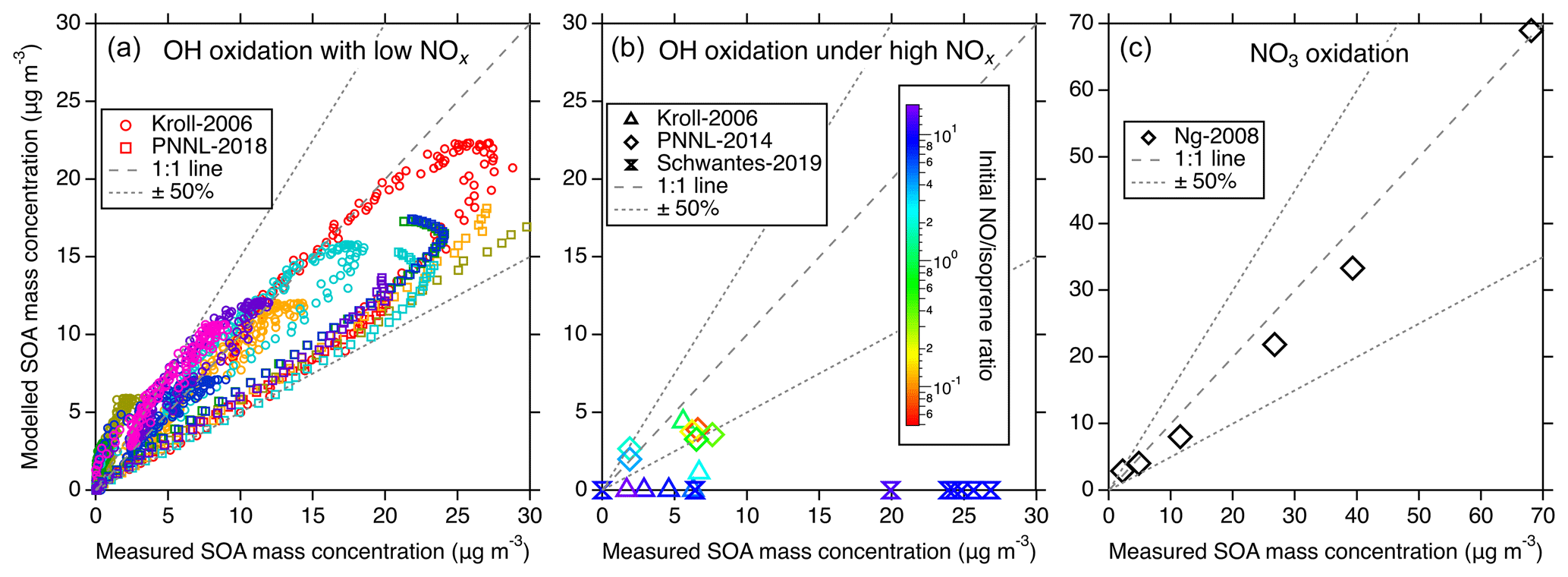
Figure 4Comparisons of the modeled and measured SOA in different chamber studies. (a) Modeled vs. measured SOA from the Kroll-2006 (Run 1–9) and PNNL-2018 chamber studies under low-NOx OH oxidation conditions. Different colors represent different experimental runs. Each marker represents the comparison of real-time SOA mass concentrations. (b) Modeled vs. measured SOA mass concentrations for the Kroll-2006 (Run 9–14), Schwantes-2019, and PNNL-2014 chamber studies under high-NOx OH oxidation conditions. Each marker represents the maximum SOA mass concentration from a single experiment run and the color scheme represents the initial concentration ratio. (c) Simulation–measurement comparisons from the Ng-2008 chamber study under NO3 oxidation conditions. Each marker represents the maximum SOA mass concentration from a single experiment run. In all panels, the dashed line indicates a 1:1 correspondence, and the dotted line delineates a 50 % uncertainty boundary.
3.1.3 SOA formation modeling
To evaluate the UCR-ISOP mechanism for SOA formation, we applied the mechanism to model several chamber experiments and compared the simulated SOA with measurements. The chamber experiments include Kroll-2006 (Kroll et al., 2006), PNNL-2018 (Thornton et al., 2020; Zawadowicz et al., 2020), PNNL-2014 (Liu et al., 2016), Schwantes-2019 (Schwantes et al., 2019), and Ng-2008 (Ng et al., 2008). The detailed model setup can be found in Sect. S2.
Figure 4 shows the comparisons between modeled isoprene SOA and observations under different conditions. Under low-NOx OH oxidation conditions presented in Fig. 4a, a noteworthy consistency between the modeled and measured SOA is evident. Despite a slightly lower bias in the modeled SOA derived from the PNNL-2018 chamber, it lies within a reasonable 50 % uncertainty range. This bias can be perceived as reasonable given the underlying uncertainty encapsulating the estimation of C∗, measurement and experimental errors, uncertainties in the assumed particle density, and the high sensitivity of SOA mass production to the starting ratio in these types of experiments (Chen et al., 2023). In comparison to the simulations from Thornton et al. (2020) for the same experiments, our results show slightly larger discrepancies compared to the data. This is caused by two main factors. First, we used the ISOPOOH + OH rate coefficients from the Caltech mechanism, which were obtained from carefully performed chamber measurements (Paulot et al., 2009; St. Clair et al., 2016). These rate coefficients are slower than those used by Thornton et al. (2020) by 40 % and 6 % for the major ISOPOOH isomers. The second reason was described above regarding the different treatments of the ISOPOOHOO isomerization rate coefficient and hence the product distributions. These differences together determine the slightly worse model performance by UCR-ISOP, but as shown in Fig. 4a, the overall model uncertainty within 50 % is still reasonable.
Contrastingly, the high-NOx OH oxidation conditions outlined in Fig. 4b present a complicated case in the SOA simulations. When the initial concentration ratio is relatively low (< 2), which corresponds to PNNL-2014 and some of the Kroll-2006 experiments, the model-predicted SOA is generally consistent with the measurements within 50 %, similar to the results under low-NOx conditions shown in Fig. 4a. Under these conditions, the predicted SOA composition is a mixture of both the low-NOx products like IDHDP and ICPDH and high-NOx products like IDHPN and IDHDN. A lower initial ratio enhances the contribution from low-NOx products, leading to the overall reasonable simulation–measurement agreement. However, when the initial ratio is relatively high (> 2), which corresponds to Schwantes-2019 and some of the Kroll-2006 experiments, there is a discernible underestimation in the modeled SOA formation, with the simulations showing in principle negligible SOA formation. To rule out the possibility that this may be due to the uncertainties in volatility calculations, we estimate the upper limit of the SOA mass concentrations by assuming that all the included low-volatility products (e.g., IDHDN, IDHPN, ICHNP, IDHCN, and ICHDN) can entirely partition to the particle phase (Fig. S5). But this still significantly underestimates the SOA mass concentrations. This stark deviation from measurements is not only for the UCR-ISOP mechanism but is a common theme observed in other compared chemical mechanisms, as shown in Fig. S5. This pronounced discrepancy unveils a substantial gap in our current understanding of isoprene oxidation under high-NOx conditions, necessitating a concerted focus by future studies to unravel the complexities therein.
For the NO3 oxidation of isoprene, as the simulations shown in Fig. 4c, there is reasonable consistency between the modeled and measured SOA mass concentration, with dimers, IDHDN, and IHPDN as the primary contributors (Fig. S6). For the Caltech and MCM-UW mechanisms (Thornton et al., 2020), the dominant contributors are IDHPN, IDHDN, and ICHNP. The difference in SOA species largely comes from the fates of NISOPO2. In the Caltech mechanism, the dominant sink of NISOPO2 is to react with NO3 and itself to form INO, IHN, and ICN, which will later be oxidized into IDHPN, IDHDN, or ICHNP. A small fraction of NISOP2 is consumed by HO2 to form hydroperoxide nitrates and other species like MVK and MACR. In the UCR-ISOP mechanism, the formation of dimers from NISOPO2 + NISOPO2, as described above, also greatly contributes to SOA under this experimental condition. In a recent study, Graham et al. (2023) showed that SOA from isoprene + NO3 exhibits lower volatility than that from α-pinene + NO3, supporting the fact that dimers are largely present in isoprene + NO3 SOA.
In addition to the gas-phase formation mechanisms for the low-volatility products, the other potential major uncertainty in simulating isoprene SOA from these chamber experiments lies in the C∗ calculations. As described in Sect. 2.3, the C∗ values of the low-volatility species are calculated from the vapor pressure, which can be estimated using EVAPORATION (Compernolle et al., 2011) and SIMPOL.1 (Pankow and Asher, 2008). Both EVAPORATION and SIMPOL.1 are group contribution structure–activity relationships, but EVAPORATION includes proximity-based functional group interactions, so it responds to differences in the locations of functional groups, while SIMPOL.1 does not vary based on functional group locations. In the results shown in Fig. 4, we used EVAPORATION to estimate the vapor pressures, using the lowest vapor pressure for all the possible isomers of each low-volatility species. This introduces some uncertainty, as the lowest-volatility isomers are not necessarily the most dominant isomers, which is not always obvious due to mechanism reduction. To investigate how the selections of isomeric structures and vapor pressure estimation methods could affect the simulated SOA, we compare the simulated maximum SOA mass concentrations for the experiments shown in Fig. 4 (experiments with > 2 excluded for this comparison) using different vapor pressure estimation methods and (in the case of EVAPORATION) isomers with the higher-bound C∗ vs. those with the lower-bound C∗. The comparison results shown in Fig. S7 suggest that the model-predicted SOA is generally lower than the measured values, especially when the higher-bound C∗ values are adopted. When the lower-bound C∗ values are used (as used in the simulations shown in Fig. 4), the model prediction is within 50 % compared to the measurements. Using higher-bound vapor pressures in EVAPORATION or using SIMPOL.1, simulated SOA is lowered by ∼ 20 %. This highlights the needs to better estimate the vapor pressures of multifunctional oxidation products in SOA as they may lead to uncertainties as great as those from the less constrained chemical mechanisms.
3.2 Simulation of field observations using the multiphase isoprene chemical mechanism
3.2.1 Model setup
To further evaluate the multiphase isoprene mechanism and understand the impacts of the various pathways on isoprene SOA formation under atmospheric conditions, we performed 0-D kinetic box model simulations for the 2013 SOAS campaign and compared our model results with the field observations. The field site information can be found in previous literature (Nguyen et al., 2015a; Xu et al., 2015; Lee et al., 2016; H. Zhang et al., 2018). In the F0AM-WAM setup, meteorological parameters such as temperature, pressure, RH, and boundary layer height were directly obtained from the measurements. Photolysis rates in the model were calculated from real-time solar zenith angle (not adjusted by cloud coverage). Model inputs including the gas-phase concentrations of isoprene, OH, HO2, NO, NO2, NO3, and O3, as well as the mass concentrations of total submicron organic aerosols, LWC, and inorganic ions were all constrained by measurements that were averaged hourly throughout the campaign. In dealing with missing data, for instances where data were missing for less than 6 h, linear interpolation was applied. In cases where the missing data spanned longer, we used an average diurnal profile derived from measurements taken throughout the entire field campaign. The submicron organic aerosol mass concentrations measured by time-of-flight aerosol mass spectrometry (AMS) were used to calculate gas–particle partitioning based on organic absorptive equilibrium (Pankow, 1994); the inorganic ion concentrations, also from AMS measurements, were used to estimate aerosol acidity (Song et al., 2018), and the LWC data were used to calculate aqueous uptake of water-soluble compounds (Wania et al., 2015). The AMS-derived positive matrix factorization (PMF) for IEPOX-SOA was used to compare with our simulated isoprene SOA (Hu et al., 2015). Furthermore, molecular-level measurements of gas-phase isoprene products measured in real time by time-of-flight chemical ionization mass spectrometry with CF3O− ionization (CF3O−-CIMS) (Nguyen et al., 2015a) and particle-phase isoprene oxidation products measured by offline thermal desorption two-dimensional gas chromatography time-of-flight mass spectrometry (TD-GC × GC-MS), in situ semi-volatile thermal desorption aerosol gas chromatography mass spectrometry (TAG-MS), and the iodide-adduct CIMS with a filter inlet for gases and aerosols (FIGAERO) were also used to compare with the model simulations (Isaacman-Vanwertz et al., 2016; Lee et al., 2016; H. Zhang et al., 2018).
Both dry and wet depositions are parameterized and incorporated in the box model. For each species, the dry deposition velocity (cm s−1) is assumed to follow a diurnal pattern proportional to the cosine of the solar zenith angle, with the peak value estimated using the parameterization method illustrated in Nguyen et al. (2015a). The dry deposition velocity for particles is assumed to be 0.2 cm s−1 (Farmer et al., 2021). The dry deposition rate (s−1) for each species is the ratio of its dry deposition velocity to boundary layer height. For the wet deposition, Bi and Isaacman-Vanwertz (2022) illustrated that the wet deposition lifetime for one species can be simply estimated only from its Henry's law constant, Haq. The detailed precipitation information like droplet distribution and precipitation intensity has little influence on the wet deposition lifetime. Thus, in our model, we calculated the wet deposition lifetime for all species based on Haq values and apply the corresponding wet deposition rate (i.e., first-order loss at a rate of 5.5 × 10−5 s−1 for the most soluble gases) when precipitation is observed. The Haq values were estimated from EPI Suite (Card et al., 2017). The estimated Haq values were also used for modeling aqueous-phase uptake. Given the difficulty in quantifying the influence of advection on local concentrations in the 0-D model, a first-order dilution rate was added to all species to account for potential mixing and ventilation. The diurnal variation of the dilution rate was scaled based on boundary layer height (Kaiser et al., 2016) with the time-dependent scaling factors determined such that the modeled MACR + MVK concentrations could approximately agree with the measurements. This is based on the assumption that the MACR and MVK can be reasonably simulated by the Caltech isoprene mechanism (Zhang et al., 2022). However, as discussed above, this treatment could have larger uncertainties for nighttime dilution rate estimation owing to the potential overprediction of MACR + MVK from isoprene NO3 oxidation by UCR-ISOP. Nevertheless, the overprediction is only within a factor of 2 (Fig. S3), and the nighttime isoprene concentration is very low for SOAS. Thus, this is likely less critical than the other uncertainties discussed in this work.
During the SOAS campaign, because RH is usually at a moderate to high level (50 %–100 %) and many oxidation products from isoprene are relatively water-soluble (Fig. S8), the aqueous uptake of soluble compounds was considered for the species with Haq larger than 1 × 107 M atm−1, including all the abovementioned low-volatility species as well as other smaller water-soluble products, such as glyoxal. Furthermore, because the particle phase state is very important for gas–particle partitioning of low-volatility species and the reactive uptake of IEPOX (Y. Zhang et al., 2018), the average O:C ratio (derived from the AMS measurements), organic mass to sulfate ratio, and ambient RH were used to determine the particle phase and occurrence of phase separation behavior (Schmedding et al., 2020). Aerosols were found to be in liquid-like phase and internally mixed most of the time during the SOAS campaign at ground level (Fig. S9), suggesting that the usage of α=1 and homogeneous mixing of inorganic and organic species are valid. However, as described above, we still examined the influence of nonideal partitioning and core–shell morphology on simulated isoprene SOA in sensitivity analyses.
3.2.2 SOAS simulation results
With the hourly constraints of meteorological conditions, isoprene, and major oxidants (Fig. S10), the model is well suited to simulate isoprene chemistry at the SOAS ground site. Because a portion of the HO2 concentrations were from prior CMAQ simulations rather than measurements, extra verification was conducted by comparing the modeled gas-phase H2O2 (mostly from HO2 + HO2) with the measurements (Fig. S11), which shows reasonable agreement. This suggests that the initial isoprene oxidation chemistry and ISOPOHOO's various unimolecular and bimolecular fates (e.g., reacting with NO and HO2) are expected to be well represented. This setup should also lead to reasonable simulations of the first- and second-generation major products whose chemistry has been well studied. However, when comparing these gas-phase products between the simulations and quantitative measurements, especially for ISOPOOH+IEPOX and IHN, the model overpredicts their concentrations by factors of 1.8 and 1.9 on average, reaching 2.7 and 3.3 at the daily peak, respectively (Figs. S12 and S13). This overprediction is not only produced by UCR-ISOP, but also by the Caltech mechanism because these two mechanisms predict almost identical gas-phase concentrations. It should be pointed out that the loss of these species via reactive uptake onto acidic and aqueous particles (for IEPOX and IHN) has already been considered in UCR-ISOP using kinetic information from the literature (Pye et al., 2013; Vasquez et al., 2020). For IEPOX reactive uptake, we tested different Haq values that have been reported in prior studies (ranging from 1.9 × 107–4 × 108 M atm−1), but the amount of IEPOX taken up is not very sensitive to this value and this uncertainty does not help resolve the differences. Thus, the discrepancy must suggest additional loss pathways for these species, such as loss via cloud interactions, which is not considered in the current model. Alternatively, better quantification of these key gas-phase intermediates is needed.
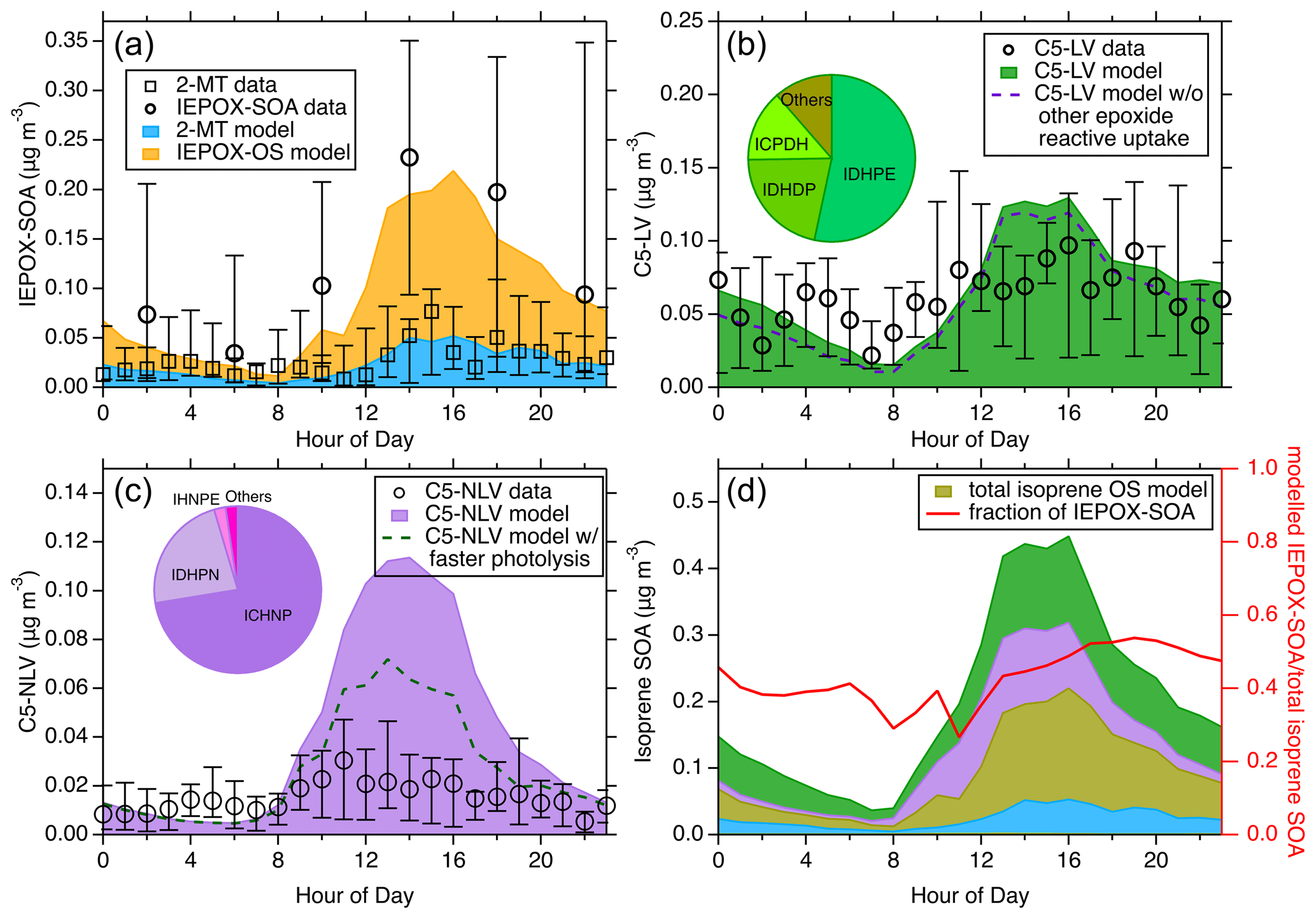
Figure 5The diurnal trend of the modeled and measured isoprene SOA from different pathways during the 2013 SOAS campaign. (a) The modeled diurnal trends of IEPOX-SOA from the reactive uptake pathway. The measured filter-based isoprene SOA by TD-GC × GC-MS and the measured 2-MT by FIGAERO-CIMS are presented. (b) Comparison between the modeled and measured total C5-LV, the latter of which is derived from a detailed summation of specific molecular species measured by FIGAERO-CIMS. The dashed line corresponds to the model results when the reactive uptake of other epoxides is not considered. (c) Comparison between the modeled and measured total C5-NLV. The dashed line corresponds to the model results when rapid photolysis of C5-NLV species containing a –OOH group is assumed. The inserted pie charts in (b) and (c) show the relative contributions of several major species to the respective SOA categories. (d) The total modeled isoprene SOA from different pathways (left y axis). “ISOP-OS” includes both the IEPOX- and other epoxide-derived OS; the colors blue, purple, and green represent the same categories as shown in (a–c). The diurnal fraction of IEPOX-SOA in total modeled isoprene SOA is shown with the right y axis. For all the measurement data, the whiskers indicate the 25th and 75th percentiles.
The simulated isoprene SOA diurnal medians from three general categories (i.e., IEPOX-SOA, C5-LV, and C5-NLV) are shown in Fig. 5a–c along with the respective molecular-level measurements during the 2013 SOAS campaign. The C5-LV and C5-NLV represent C5 low-volatility species without and with nitrogen, respectively. The detailed time series comparison throughout the field campaign can be seen in Fig. S14. As shown in Fig. 5a, the measurements from FIGAERO-CIMS (the “2-MT data”) are the summation of the estimated concentrations for chemical formulas of C5H12O4, assumed to represent 2-MT and C5H10O3, assumed to represent C5-alkenetriols; those from the filter-based TD-GC × GC-MS are the non-OS IEPOX-SOA data, including 2-MT, C5-alkenetriols, and other species that correlate well with them (H. Zhang et al., 2018). However, it has been suggested that the IEPOX-OS may partly decompose to 2-MT and C5-alkenetriols during the thermal desorption process in GC (Rattanavaraha et al., 2016; Cui et al., 2018). Thus, the TD-GC × GC-MS measurements could represent the total IEPOX-SOA to some extent. The diurnal variations from both measurements are characterized by a peak in the afternoon and a nadir in the morning. Our modeled IEPOX-SOA nicely replicates this diurnal trend and the magnitude, demonstrating its proficient capacity in mirroring the IEPOX reactive uptake pathway. In contrast to the comparison shown in Fig. 5a, the TAG-MS measurements exhibit much higher concentrations, especially during nighttime and morning (Fig. S15), despite the fact that the TAG-MS analysis principle is essentially the same as that by the TD-GC × GC-MS. The discrepancies in these measurements suggest that the 2-MT and C5-alkenetriol tracers are likely partly decomposed from other tracers (e.g., IEPOX-OS) to different extents, highlighting the needs to better quantify these important IEPOX-SOA tracers in future work.
Figure 5b and c show the median diurnal variations of modeled C5-LV and C5-NLV, respectively. The measured C5-LV and C5-NLV are determined from the sum of many molecular species measured by the FIGAERO-CIMS with chemical formulas of C5H8–12O5–7 (C5-LV) and C5H7–11O5–9N (C5-NLV) (Lee et al., 2016; H. Zhang et al., 2018). It should be noted that some of these formulas may not all be from isoprene oxidation. For instance, C5H8O5 could also be 3-hydroxyglutaric acid from monoterpene oxidation (Claeys et al., 2007). In addition, the quantification of these species in the FIGAERO-CIMS data could have high uncertainties. For example, the C5-NLV species were quantified using an IHN isomer with the highest sensitivity as the surrogate standards (Lee et al., 2016). Thus, the quantified C5-NLV is considered the lower limit. For C5-NLV species which have lower sensitivity such as carbonyl nitrates, this quantification approach will underestimate the concentrations. These abovementioned issues could lead to uncertainties in quantifying the C5-LV and C5-NLV mass concentrations. Nevertheless, these measurements are likely still the best quantitative data available from field measurements. The simulated C5-LV shown in Fig. 5b exhibits similar diurnal trends and magnitude as the measurements, while the C5-NLV shows a more significant discrepancy between the modeled and measured mass concentration (Fig. 5c), in part due to the quantification uncertainties mentioned above. It is interesting to note that in the C5-LV simulations, ICPDH, ITHC, and IDHPN in these species can partly be contributed by the reactive uptake of epoxides other than IEPOX. As shown in the gas-phase simulations (Fig. S16), although the other epoxides are smaller than IEPOX, they may also be important, with the largest epoxides being ICPE from ISOPOHOO isomerization and ICHE from HPALD oxidation. This underscores the need to further study the multiphase fates of these previously less-studied epoxides. In addition, the C5-NLV simulations may also be greatly affected if hydrolysis and photolysis rates are treated differently. For example, as shown in Fig. 5c, if C5-NLV species containing −OOH groups are allowed to undergo rapid photolysis like those formed in low-NOx conditions, the simulated C5-NLV may be largely reduced. To gain further insights into the roles of the low-volatility pathways in isoprene SOA formation, the modeled molecular contributions for each SOA category throughout the SOAS field campaign are investigated. In the C5-LV category, IDHPE and IDHDP are the two largest contributors (see the pie chart insert in Fig. 5b), both originating from the oxidation of ISOPOOH under low-NOx conditions. Interestingly, despite the fact that IDHPE and IDHDP are predicted to have large concentrations in SOAS isoprene SOA, their chemical formula (C5H12O6, assuming IDHPE opens the epoxide ring in the particle phase to form hydroperoxyltetrols) was found to be very low in the FIGAERO-CIMS measurements (D'ambro et al., 2017). It should be noted that rapid photolysis of these hydroperoxide compounds have already been considered in the model. Thus, we suspect that additional multiphase or bulk-phase reactions also readily take place that further transform these labile species into more stable oxygenated compounds (e.g., products reported by Jaoui et al., 2019). ICPDH (C5H10O5) is the third-largest C5-LV species that is formed from IEPOX + OH oxidation, followed by bimolecular reaction with HO2. In the C5-NLV category, ICHNP (C5H9NO7) primarily from IHN + OH and subsequent RO2 (ISOPNOO) isomerization emerges as the largest contributor (> 70 %, see the pie chart insert in Fig. 5c). This chemical formula has been shown to be a major particle-phase organic nitrate with time series consistent with isoprene SOA during SOAS (Lee et al., 2016). The other main C5-NLV species, IDHPN (C5H11NO7), can be formed from both ISOPNOO + HO2 and ISOPOOHOO + NO (see Fig. 1).
In Fig. 5d, we present the diurnal variations of the total isoprene SOA derived from both the low-volatility and reactive uptake pathways, offering a holistic perspective on the isoprene SOA composition and concentrations stemming from different formation mechanisms. This comparison suggests that the explainable non-IEPOX fraction accounts for ∼ 57 % of total simulated isoprene SOA during SOAS throughout the day, which is unexpected and highlights the importance of better understanding the reaction pathways in more detail. Notably, this fraction is approximately consistent with a previous laboratory study (Liu et al., 2015), but the results shown in the present work are for realistic atmospheric conditions. In addition, we also compare the simulated low-volatility SOA (C5-LV and C5-NLV) in correlation with the IEPOX-SOA with hourly resolution (Fig. S17). It turns out that the isoprene SOA from these two formation pathways correlated reasonably well (R2 = 0.80). In particular, calculated R2 is 0.82 for IEPOX-SOA with C5-LV and 0.35 for C5-NLV, which highlights a stronger convolution of IEPOX-SOA with C5-LV than C5-NLV. Interpreting from this strong time series correlation and based on the way PMF works in deconvoluting organic aerosol sources (Lanz et al., 2007; Zhang et al., 2007; Ulbrich et al., 2009), we suggest that the AMS-PMF analysis may not always effectively separate the IEPOX-SOA from the other isoprene SOA, despite the fact that prior studies have reported a specific AMS-PMF factor for the ISOPOOH-derived SOA which is quite different than the well-known IEPOX-SOA factor (Riva et al., 2016). Therefore, the IEPOX-SOA factor from AMS-PMF, which was previously considered to represent SOA only from the IEPOX reactive uptake pathway, could partly include isoprene SOA from the LV pathways.
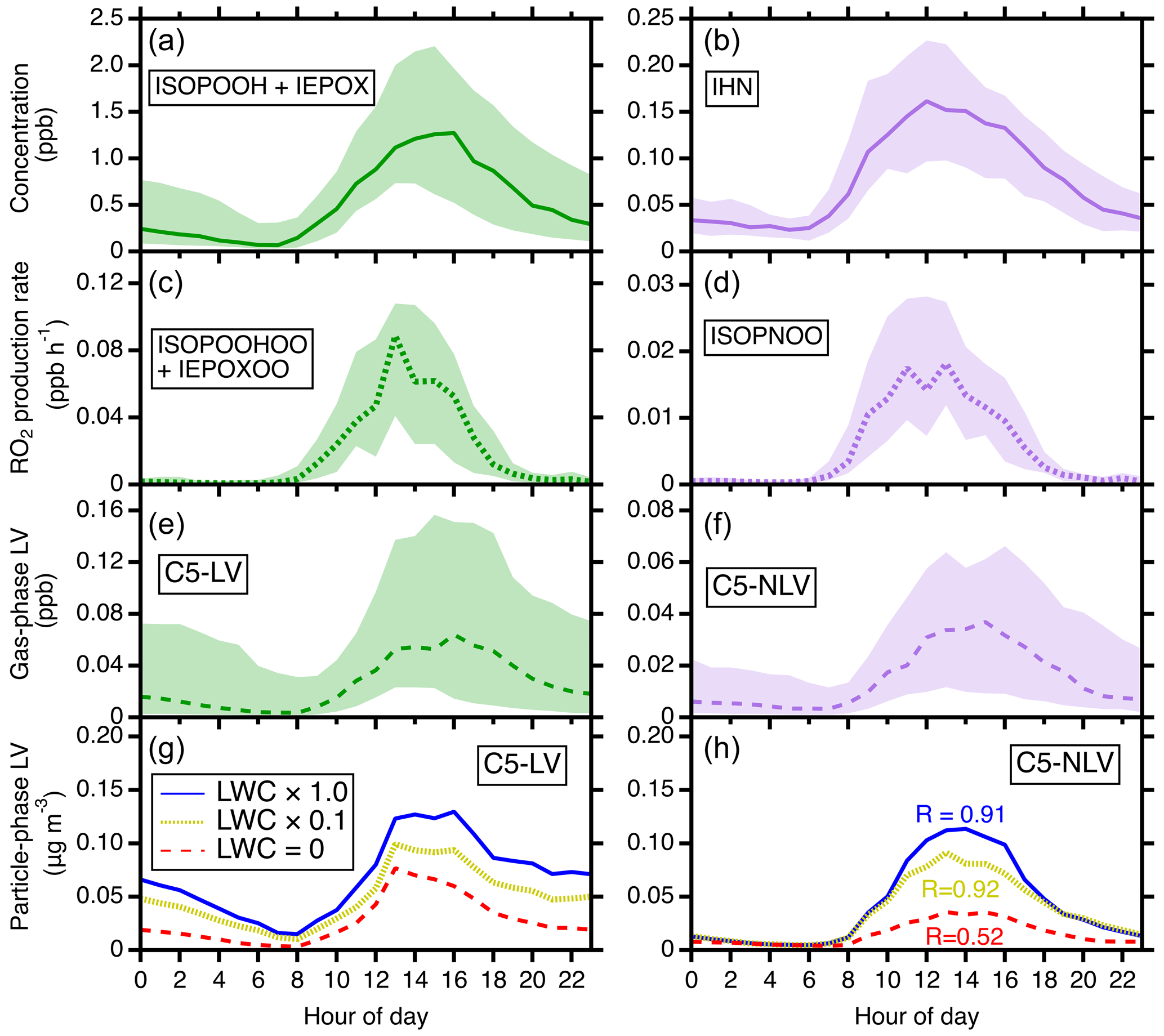
Figure 6The comparisons of the C5-LV and C5-NLV formation pathways. (a, b) The modeled diurnal concentrations of ISOPOOH + IEPOX (precursors for C5-LV) and IHN (precursors for C5-NLV). (c, d) The modeled diurnal RO2 production rates for C5-LV RO2 (i.e., ISOPOOHOO and IEPOXOO) and C5-NLV RO2 (i.e., ISOPNOO). (e, f) The modeled diurnal concentrations of gas-phase C5-LV and C5-NLV. In (a–f), the simulations are for the gas phase only. (g, h) The influence of LWC on particle-phase C5-LV and C5-NLV mass concentrations. Simulated scenarios include LWC = 0 µg m−3 (red), LWC set to be a factor of 10 lower than actual concentrations (olive), and LWC from actual concentrations (blue). In (h), “R” represents the ratio of C5-LV and C5-NLV at the daily maximum (averages from 12:00 to 16:00 LT, local time). Throughout the figure from top to bottom, the ratios of the corresponding N-containing over non-N-containing products increase from 0.1 to 0.91 under SOAS conditions.
However, a discernible underestimation of our modeled total isoprene SOA is present when compared to the IEPOX-SOA factor (Fig. S15), suggesting additional SOA formation pathways from isoprene oxidation in the atmosphere that our mechanism does not include. Furthermore, the observed discrepancy between modeled results and actual measurements could be exacerbated if slow gas–particle partitioning and core–shell particle morphology are considered. In the sensitivity test shown in Fig. S18, such an adaptation in the model could lead to a further decrease in peak IEPOX-SOA estimates by ∼ 40 %, consistent with Y. Zhang et al. (2018), but negligible change for non-IEPOX SOA due to slow particle diffusion and partitioning, consistent with Thornton et al. (2020). In chamber experiment simulations discussed above, we failed to simulate SOA formation under initial ratios higher than 2. But during the SOAS campaign, the Centreville site is always under low-NOx conditions with this ratio usually lower than ∼ 0.1 (Fig. S10). Thus, it is unlikely that this unrepresented SOA formation explains the model underestimation. In our model, we extensively included pathways that lead to multifunctional low-volatility products which retain the isoprene C5 backbone. Thus, it is possible that some fragmentation products (<C5) may also contain multiple functional groups through further oxidation and contribute to SOA formation. For these species, we only included SOA from HMML+MAE as they are well-studied isoprene SOA precursors (Lin et al., 2013; Nguyen et al., 2015b). This could partly explain the observed model–observation discrepancy. Moreover, although dimer formation from RO2 + RO2 reactions is considered in the model in both the daytime and nighttime pathways with rapid reaction rate coefficients and high branching ratios, it is predicted to be low under the SOAS conditions. However, prior studies suggested that the formation of dimers from isoprene RO2 and monoterpene RO2 may be prominent under conditions like the SOAS site (Tiszenkel and Lee, 2023). This process is not included in our mechanism because the monoterpene chemistry is not explicitly described. To better understand the isoprene SOA molecular composition from these pathways, especially in atmospheric aerosols, future research is certainly warranted.
It is also remarkable to note that the predicted C5-NLV mass concentration is nearly as high as (ratio ∼ 0.91) that from the C5-LV category at daily maximum during SOAS, despite the fact that the field site is an isoprene-dominant forest area with low NOx. For reference, the major gas-phase intermediates for C5-NLV (IHN) are about a factor of 10 lower than those for C5-LV (i.e., ISOPOOH + IEPOX) (Fig. 6a and b). The strong contrast suggests that the IHN oxidation pathway is much more efficient in producing LV SOA than the ISOPOOH + OH and IEPOX + OH pathways. We suggest that this results from several effects. First, the OH oxidation of ISOPOOH and IEPOX produces C5-LV RO2 (i.e., ISOPOOHOO and IEPOXOO) at smaller branching ratios than C5-NLV RO2 (ISOPNOO) production from IHN + OH (Wennberg et al., 2018), and IEPOX has a lifetime ∼ 5 times longer than IHN against OH. These kinetic differences lead to a reduced difference between the production rates of the C5-LV RO2 and the C5-NLV RO2 to a factor of ∼ 4 (Fig. 6c and d). Furthermore, ISOPNOO is more effective to be converted to ICHNP (dominant C5-NLV species) under the SOAS conditions in comparison with ISOPOOHOO and IEPOXOO to IDHPE, IDHDP, and ICPDH. As shown in Fig. S19, the pseudo-first-order rate of RO2 against bimolecular reactions () during SOAS estimated using HO2 and NO measurements is < 0.02 s−1 during most times of day. Thus, the unimolecular isomerization of ISOPNOO (∼ 0.04–0.08 s−1) outcompetes its bimolecular reactions and directly produces the most abundant C5-NLV species, ICHNP at 100 % yield. In contrast, the unimolecular isomerization rate constant of ISOPOOHOO is similar to , while that of IEPOXOO is fast but produces C5-LV products at much lower yields (Wennberg et al., 2018). As a result, the produced C5-NLV C5-LV ratio in the gas phase is further reduced to a factor of only ∼ 2 (Fig. 6e and f). Lastly, the presence of LWC brings particle-phase C5-NLV and C5-LV concentrations even closer because LWC more prominently enhances SOA formation for the C5-NLV species. As shown in Fig. S8, the C5-NLV species have the highest Haq values. This leads to the high sensitivity of this category to LWC. In contrast, the LWC allowing for IEPOX reactive uptake diminishes formation of some of C5-LV species from IEPOX oxidation. As a result, in the sensitivity analysis shown in Fig. 6g and h, one can see that the C5-NLV mass concentration and ratio over C5-LV both substantially increase as LWC is enhanced from 0 to the SOAS ambient level. These comparisons indicate that formation of organic nitrates in SOA can be important even for low-NOx environments.
While the molecular-level understanding of isoprene oxidation chemistry has improved significantly, it is still challenging to include all the process in a multiphase chemical mechanism for laboratory and atmospheric SOA predictions. This work first presents such a condensed chemical mechanism for modeling gas-phase isoprene oxidation chemistry as well as SOA with the major molecular products represented. Our condensed mechanism provides a substantial step toward improved model estimation of isoprene-derived SOA, including both multigenerational oxidation leading to low-volatility products and the reactive uptake pathways. In the process of developing and evaluating the new mechanism by comparing with other mechanisms and data from chamber studies and field measurements, it is recognized that significant uncertainties remain in understanding isoprene oxidation chemistry and SOA formation. Thus, we consider this mechanism to be a starting point with flexibilities for future updates.
Among the many uncertainties, a few major ones are summarized here. First, there are large discrepancies in isomerization rate coefficients for some key isoprene RO2 and RO species between different measurements and between experiments and computational calculations. These rate coefficients may be crucial for determining the RO2 and RO fates and hence product distributions, especially under atmospheric conditions where unimolecular isomerization (for RO2) can be more important. An example we show in this work is that the difference in RO2 fates plays a significant role in controlling the C5-NLV composition and formation efficiency: in the SOAS simulations, the ISOPNOO unimolecular isomerization outcompetes its bimolecular reactions to directly produce the most abundant C5-NLV species, ICHNP at 100 % yield. Instead, under laboratory experimental conditions, bimolecular reactions of ISOPNOO likely dominate its fate and produce other C5-NLV species at smaller yields. These striking differences highlight the challenge to mimic atmospheric oxidation conditions in laboratory experiments and the fact that distinct RO2 fates can significantly shift the product distributions. Besides, gas-phase dimer formation from RO2 + RO2 reactions should be better understood. This is a need not only for the isoprene chemistry, but also for other VOC systems (e.g., monoterpenes). It can be even more complex but important when RO2 + RO2 reactions occur across different VOC systems. Moreover, we suggest that the gas-phase mechanisms for isoprene + NO3 as well as the high-NOx pathways are not well understood in terms of how and what low-volatility products are formed. In addition, we show that our model does not accurately predict the atmospheric concentrations of major gas-phase products such as ISOPOOH, IEPOX, and IHN in the SOAS field campaign. We regard this to be a lack of understanding of their missing sinks, rather than their formation chemistry, because the mechanisms and kinetics for their production are likely well understood from prior laboratory studies. Investigating the missing sinks could be crucial for improving our understandings of areas such as cloud processes.
Regarding isoprene SOA formation, a major uncertainty lies in the gap between the predicted total SOA and the measurements from a variety of techniques (e.g., AMS, FIGAERO-CIMS, and GC-based techniques). It is certainly crucial to resolve the differences between these measurements and examine the possible decomposition processes during analyses. We also suggested that part of the gap is due to the SOA species with carbon numbers smaller than 5 not being represented in the current mechanism. But it should be noted that the gap is more significant during nighttime and morning, especially when comparing the simulations with measurements by TAG-MS and AMS, which exhibit a smaller diurnal pattern than the model (e.g., comparing Figs. S15 and 5d). The source of this nighttime SOA is not well described by the model, warranting future investigation. In addition, the vapor pressure estimation of the low-volatility species could also introduce uncertainty. This uncertainty may be a greater challenge in cases where isomers are lumped into a condensed mechanism. Furthermore, prior studies have extensively studied the reactive uptake of IEPOX (Surratt et al., 2010; Lin et al., 2012, 2013; Nguyen et al., 2014; Y. Zhang et al., 2018). Here, we show that if the other epoxides formed from isoprene oxidation undergo similar reactive uptake reactions, they may also contribute to SOA formation. These epoxides should be more thoroughly studied in future research. Lastly, significant uncertainties remain for the particle-phase reactions. In the current mechanism, we simplified the photolysis for hydroperoxides and hydrolysis for organic nitrates by using the same photolysis and hydrolysis rate coefficients, respectively. But we also show that the total isoprene SOA mass concentrations and compositions could be greatly affected by these parameters. In addition, other particle-phase reactions such as oxidative aging and accretion are not well constrained but are important to bridge molecular-level measurements and model predictions.
Despite the uncertainties, the model can reasonably predict the mass concentration and composition of isoprene SOA in the SOAS field campaign, estimating the contributions from different pathways in the ambient environment. Our model results also highlight that the low-volatility pathways contribute greatly to isoprene SOA formation. We expect that their importance could be even higher in the future given that the emissions of sulfur and nitrogen are reduced. Implementing the multiphase mechanism into air quality models in future studies may provide new insights into isoprene SOA chemistry at regional and global scales.
The condensed mechanism (ISOP-UCR) and associated codes in the F0AM format can be found at https://github.com/zhangucr/UCR-ISOP.git (Shen and Zhang, 2024).
All the datasets used in this work are from previous publications, as mentioned in Sect. 3.1.1. The data are available to the public from the corresponding publications.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-6153-2024-supplement.
CS and HZ designed and performed the simulations. JT and JS provided insights into the laboratory experiments and chemical mechanisms. The F0AM-WAM model was originally developed by JT's research group. XY prepared part of the mechanism development. CB and GIV discussed the results and contributed part of the model simulation. CS and HZ prepared the paper with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This work was supported by the US National Science Foundation (AGS-2037698) and DOE Office of Biological and Environmental Research (DE-SC0023330). Pacific Northwest National Laboratory (PNNL) experiments and John Shilling were supported by the Atmospheric System Research (ASR) program as part of the DOE Office of Biological and Environmental Research under PNNL project 57131. PNNL is operated for the DOE by the Battelle Memorial Institute under contract DEA06-76RLO1830. Gabriel Isaacman-VanWertz was supported by the National Science Foundation (AGS-2046367) and DOE Office of Biological and Environmental Research (DE-SC0022020). We thank Paul Wennberg (Caltech), Philip Carlsson (Forschungszentrum Jülich), and William Vizuete (University of North Carolina, Chapel Hill) for useful discussions.
This work has been supported by the US National Science Foundation (AGS-2037698 and AGS-2046367) and US DOE (DE-SC0023330, DE-A06-76RLO1830, and DE-SC0022020).
This paper was edited by Thomas Berkemeier and reviewed by two anonymous referees.
Archibald, A., Levine, J. G., Abraham, N., Cooke, M., Edwards, P., Heard, D., Jenkin, M., Karunaharan, A., Pike, R., and Monks, P.: Impacts of HOx regeneration and recycling in the oxidation of isoprene: Consequences for the composition of past, present and future atmospheres, Geophys. Res. Lett., 38, 2011.
Bates, K. H. and Jacob, D. J.: A new model mechanism for atmospheric oxidation of isoprene: global effects on oxidants, nitrogen oxides, organic products, and secondary organic aerosol, Atmos. Chem. Phys., 19, 9613–9640, https://doi.org/10.5194/acp-19-9613-2019, 2019.
Berndt, T., Hyttinen, N., Herrmann, H., and Hansel, A.: First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2, 21, https://doi.org/10.1038/s42004-019-0120-9, 2019.
Bi, C. and Isaacman-VanWertz, G.: Estimated timescales for wet deposition of organic compounds as a function of Henry's law constants, Environmental Science: Atmospheres, 2, 1526–1533, https://doi.org/10.1039/D2EA00091A, 2022.
Brune, W. H., McFarland, P. J., Bruning, E., Waugh, S., MacGorman, D., Miller, D. O., Jenkins, J. M., Ren, X., Mao, J., and Peischl, J.: Extreme oxidant amounts produced by lightning in storm clouds, Science, 372, 711–715, https://doi.org/10.1126/science.abg0492, 2021.
Budisulistiorini, S. H., Canagaratna, M. R., Croteau, P. L., Marth, W. J., Baumann, K., Edgerton, E. S., Shaw, S. L., Knipping, E. M., Worsnop, D. R., and Jayne, J. T.: Real-time continuous characterization of secondary organic aerosol derived from isoprene epoxydiols in downtown Atlanta, Georgia, using the Aerodyne Aerosol Chemical Speciation Monitor, Environ. Sci. Technol., 47, 5686–5694, 2013.
Budisulistiorini, S. H., Li, X., Bairai, S. T., Renfro, J., Liu, Y., Liu, Y. J., McKinney, K. A., Martin, S. T., McNeill, V. F., Pye, H. O. T., Nenes, A., Neff, M. E., Stone, E. A., Mueller, S., Knote, C., Shaw, S. L., Zhang, Z., Gold, A., and Surratt, J. D.: Examining the effects of anthropogenic emissions on isoprene-derived secondary organic aerosol formation during the 2013 Southern Oxidant and Aerosol Study (SOAS) at the Look Rock, Tennessee ground site, Atmos. Chem. Phys., 15, 8871–8888, https://doi.org/10.5194/acp-15-8871-2015, 2015.
Card, M. L., Gomez-Alvarez, V., Lee, W.-H., Lynch, D. G., Orentas, N. S., Lee, M. T., Wong, E. M., and Boethling, R. S.: History of EPI Suite™ and future perspectives on chemical property estimation in US Toxic Substances Control Act new chemical risk assessments, Environmental Science: Processes & Impacts, 19, 203–212, 2017.
Carlsson, P. T. M., Vereecken, L., Novelli, A., Bernard, F., Brown, S. S., Brownwood, B., Cho, C., Crowley, J. N., Dewald, P., Edwards, P. M., Friedrich, N., Fry, J. L., Hallquist, M., Hantschke, L., Hohaus, T., Kang, S., Liebmann, J., Mayhew, A. W., Mentel, T., Reimer, D., Rohrer, F., Shenolikar, J., Tillmann, R., Tsiligiannis, E., Wu, R., Wahner, A., Kiendler-Scharr, A., and Fuchs, H.: Comparison of isoprene chemical mechanisms under atmospheric night-time conditions in chamber experiments: evidence of hydroperoxy aldehydes and epoxy products from NO3 oxidation, Atmos. Chem. Phys., 23, 3147–3180, https://doi.org/10.5194/acp-23-3147-2023, 2023.
Carlton, A. G., Turpin, B. J., Altieri, K. E., Seitzinger, S., Reff, A., Lim, H.-J., and Ervens, B.: Atmospheric oxalic acid and SOA production from glyoxal: Results of aqueous photooxidation experiments, Atmos. Environ., 41, 7588–7602, https://doi.org/10.1016/j.atmosenv.2007.05.035, 2007.
Carlton, A. G., Wiedinmyer, C., and Kroll, J. H.: A review of Secondary Organic Aerosol (SOA) formation from isoprene, Atmos. Chem. Phys., 9, 4987–5005, https://doi.org/10.5194/acp-9-4987-2009, 2009.
Carter, W. P.: Documentation of the SAPRC-99 chemical mechanism for VOC reactivity assessment, Contract 92, 95–308, https://intra.engr.ucr.edu/~carter/pubs/s99doc.pdf (last access: 17 September 2023), 2000.
Carter, W. P.: Documentation of the SAPRC-22 Mechanism, https://intra.engr.ucr.edu/~carter/pubs/s99doc.pdf (last access: 17 September 2023), 2023.
Carter, W. P. L.: Condensed atmospheric photooxidation mechanisms for isoprene, Atmos. Environ., 30, 4275–4290, https://doi.org/10.1016/1352-2310(96)00088-X, 1996.
Carter, W. P. L.: Development of the SAPRC-07 chemical mechanism, Atmos. Environ., 44, 5324–5335, https://doi.org/10.1016/j.atmosenv.2010.01.026, 2010.
Chacon-Madrid, H. J., Henry, K. M., and Donahue, N. M.: Photo-oxidation of pinonaldehyde at low NOx: from chemistry to organic aerosol formation, Atmos. Chem. Phys., 13, 3227–3236, https://doi.org/10.5194/acp-13-3227-2013, 2013.
Chan, M. N., Surratt, J. D., Claeys, M., Edgerton, E. S., Tanner, R. L., Shaw, S. L., Zheng, M., Knipping, E. M., Eddingsaas, N. C., Wennberg, P. O., and Seinfeld, J. H.: Characterization and Quantification of Isoprene-Derived Epoxydiols in Ambient Aerosol in the Southeastern United States, Environ. Sci. Technol., 44, 4590–4596, https://doi.org/10.1021/es100596b, 2010.
Chen, Y., Zaveri, R. A., Vandergrift, G. W., Cheng, Z., China, S., Zelenyuk, A., and Shilling, J. E.: Nonequilibrium Behavior in Isoprene Secondary Organic Aerosol, Environ. Sci. Technol., 57, 14182–14193, https://doi.org/10.1021/acs.est.3c03532, 2023.
Claeys, M., Graham, B., Vas, G., Wang, W., Vermeylen, R., Pashynska, V., Cafmeyer, J., Guyon, P., Andreae, M. O., Artaxo, P., and Maenhaut, W.: Formation of Secondary Organic Aerosols Through Photooxidation of Isoprene, Science, 303, 1173–1176, https://doi.org/10.1126/science.1092805, 2004.
Claeys, M., Szmigielski, R., Kourtchev, I., Van der Veken, P., Vermeylen, R., Maenhaut, W., Jaoui, M., Kleindienst, T. E., Lewandowski, M., Offenberg, J. H., and Edney, E. O.: Hydroxydicarboxylic Acids: Markers for Secondary Organic Aerosol from the Photooxidation of α-Pinene, Environ. Sci. Technol., 41, 1628–1634, https://doi.org/10.1021/es0620181, 2007.
Compernolle, S., Ceulemans, K., and Müller, J.-F.: EVAPORATION: a new vapour pressure estimation methodfor organic molecules including non-additivity and intramolecular interactions, Atmos. Chem. Phys., 11, 9431–9450, https://doi.org/10.5194/acp-11-9431-2011, 2011.
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, 2011.
Cui, T., Zeng, Z., dos Santos, E. O., Zhang, Z., Chen, Y., Zhang, Y., Rose, C. A., Budisulistiorini, S. H., Collins, L. B., Bodnar, W. M., de Souza, R. A. F., Martin, S. T., Machado, C. M. D., Turpin, B. J., Gold, A., Ault, A. P., and Surratt, J. D.: Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environmental Science: Processes & Impacts, 20, 1524–1536, https://doi.org/10.1039/C8EM00308D, 2018.
D'Ambro, E. L., Møller, K. H., Lopez-Hilfiker, F. D., Schobesberger, S., Liu, J., Shilling, J. E., Lee, B. H., Kjaergaard, H. G., and Thornton, J. A.: Isomerization of Second-Generation Isoprene Peroxy Radicals: Epoxide Formation and Implications for Secondary Organic Aerosol Yields, Environ. Sci. Technol., 51, 4978–4987, https://doi.org/10.1021/acs.est.7b00460, 2017.
Dommen, J., Metzger, A., Duplissy, J., Kalberer, M., Alfarra, M., Gascho, A., Weingartner, E., Prévôt, A. S., Verheggen, B., and Baltensperger, U.: Laboratory observation of oligomers in the aerosol from isoprene/NOx photooxidation, Geophys. Res. Lett., 33, L13805, https://doi.org/10.1029/2006GL026523, 2006.
Donahue, N. M., Robinson, A., Stanier, C., and Pandis, S.: Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 40, 2635–2643, 2006.
Eddingsaas, N. C., VanderVelde, D. G., and Wennberg, P. O.: Kinetics and Products of the Acid-Catalyzed Ring-Opening of Atmospherically Relevant Butyl Epoxy Alcohols, J. Phys. Chem. A, 114, 8106–8113, https://doi.org/10.1021/jp103907c, 2010.
Edney, E., Kleindienst, T., Jaoui, M., Lewandowski, M., Offenberg, J., Wang, W., and Claeys, M.: Formation of 2-methyl tetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NOx/SO2/air mixtures and their detection in ambient PM2. 5 samples collected in the eastern United States, Atmos. Environ., 39, 5281–5289, 2005.
Fan, J. and Zhang, R.: Atmospheric oxidation mechanism of isoprene, Environ. Chem., 1, 140–149, 2004.
Farmer, D. K., Boedicker, E. K., and DeBolt, H. M.: Dry Deposition of Atmospheric Aerosols: Approaches, Observations, and Mechanisms, Annu. Rev. Phys. Chem., 72, 375–397, https://doi.org/10.1146/annurev-physchem-090519-034936, 2021.
Fountoukis, C. and Nenes, A.: ISORROPIA II: a computationally efficient thermodynamic equilibrium model for K+–Ca2+–Mg2+––Na+–––Cl−–H2O aerosols, Atmospheric Chemistry and Physics, 7, 4639–4659, https://doi.org/10.5194/acp-7-4639-2007, 2007.
Fu, P., Kawamura, K., Chen, J., and Barrie, L. A.: Isoprene, monoterpene, and sesquiterpene oxidation products in the high Arctic aerosols during late winter to early summer, Environ. Sci. Technol., 43, 4022–4028, 2009.
Gaston, C. J., Riedel, T. P., Zhang, Z., Gold, A., Surratt, J. D., and Thornton, J. A.: Reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles, Environ. Sci. Technol., 48, 11178–11186, 2014.
Gery, M. W., Whitten, G. Z., Killus, J. P., and Dodge, M. C.: A photochemical kinetics mechanism for urban and regional scale computer modeling, J. Geophys. Res.-Atmos., 94, 12925–12956, 1989.
Graham, E. L., Wu, C., Bell, D. M., Bertrand, A., Haslett, S. L., Baltensperger, U., El Haddad, I., Krejci, R., Riipinen, I., and Mohr, C.: Volatility of aerosol particles from NO3 oxidation of various biogenic organic precursors, Atmos. Chem. Phys., 23, 7347–7362, https://doi.org/10.5194/acp-23-7347-2023, 2023.
Guenther, A., Karl, T., Harley, P., Wiedinmyer, C., Palmer, P. I., and Geron, C.: Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 6, 3181–3210, https://doi.org/10.5194/acp-6-3181-2006, 2006.
Guo, X., Ma, F., Liu, C., Niu, J., He, N., Chen, J., and Xie, H.-B.: Atmospheric oxidation mechanism and kinetics of isoprene initiated by chlorine radicals: A computational study, Sci. Total Environ., 712, 136330, https://doi.org/10.1016/j.scitotenv.2019.136330, 2020.
Hastings, W. P., Koehler, C. A., Bailey, E. L., and De Haan, D. O.: Secondary Organic Aerosol Formation by Glyoxal Hydration and Oligomer Formation: Humidity Effects and Equilibrium Shifts during Analysis, Environ. Sci. Technol., 39, 8728–8735, https://doi.org/10.1021/es050446l, 2005.
Henze, D. K. and Seinfeld, J. H.: Global secondary organic aerosol from isoprene oxidation, Geophys. Res. Lett., 33, L09812, https://doi.org/10.1029/2006GL025976, 2006.
Hu, W. W., Campuzano-Jost, P., Palm, B. B., Day, D. A., Ortega, A. M., Hayes, P. L., Krechmer, J. E., Chen, Q., Kuwata, M., Liu, Y. J., de Sá, S. S., McKinney, K., Martin, S. T., Hu, M., Budisulistiorini, S. H., Riva, M., Surratt, J. D., St. Clair, J. M., Isaacman-Van Wertz, G., Yee, L. D., Goldstein, A. H., Carbone, S., Brito, J., Artaxo, P., de Gouw, J. A., Koss, A., Wisthaler, A., Mikoviny, T., Karl, T., Kaser, L., Jud, W., Hansel, A., Docherty, K. S., Alexander, M. L., Robinson, N. H., Coe, H., Allan, J. D., Canagaratna, M. R., Paulot, F., and Jimenez, J. L.: Characterization of a real-time tracer for isoprene epoxydiols-derived secondary organic aerosol (IEPOX-SOA) from aerosol mass spectrometer measurements, Atmos. Chem. Phys., 15, 11807–11833, https://doi.org/10.5194/acp-15-11807-2015, 2015.
Isaacman-VanWertz, G., Yee, L. D., Kreisberg, N. M., Wernis, R., Moss, J. A., Hering, S. V., de Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Riva, M., Surratt, J. D., Viegas, J., Manzi, A., Edgerton, E., Baumann, K., Souza, R., Artaxo, P., and Goldstein, A. H.: Ambient Gas-Particle Partitioning of Tracers for Biogenic Oxidation, Environ. Sci. Technol., 50, 9952–9962, https://doi.org/10.1021/acs.est.6b01674, 2016.
Jaoui, M., Corse, E. W., Lewandowski, M., Offenberg, J. H., Kleindienst, T. E., and Edney, E. O.: Formation of organic tracers for isoprene SOA under acidic conditions, Atmos. Environ., 44, 1798–1805, 2010.
Jaoui, M., Szmigielski, R., Nestorowicz, K., Kolodziejczyk, A., Sarang, K., Rudzinski, K. J., Konopka, A., Bulska, E., Lewandowski, M., and Kleindienst, T. E.: Organic hydroxy acids as highly oxygenated molecular (HOM) tracers for aged isoprene aerosol, Environ. Sci. Technol., 53, 14516–14527, 2019.
Jenkin, M. E., Young, J. C., and Rickard, A. R.: The MCM v3.3.1 degradation scheme for isoprene, Atmos. Chem. Phys., 15, 11433–11459, https://doi.org/10.5194/acp-15-11433-2015, 2015.
Kaiser, J., Skog, K. M., Baumann, K., Bertman, S. B., Brown, S. B., Brune, W. H., Crounse, J. D., de Gouw, J. A., Edgerton, E. S., Feiner, P. A., Goldstein, A. H., Koss, A., Misztal, P. K., Nguyen, T. B., Olson, K. F., St. Clair, J. M., Teng, A. P., Toma, S., Wennberg, P. O., Wild, R. J., Zhang, L., and Keutsch, F. N.: Speciation of OH reactivity above the canopy of an isoprene-dominated forest, Atmos. Chem. Phys., 16, 9349–9359, https://doi.org/10.5194/acp-16-9349-2016, 2016.
Kourtchev, I., Ruuskanen, T., Maenhaut, W., Kulmala, M., and Claeys, M.: Observation of 2-methyltetrols and related photo-oxidation products of isoprene in boreal forest aerosols from Finland, Atmos. Chem. Phys., 5, 2761–2770, https://doi.org/10.5194/acp-5-2761-2005, 2005.
Krapf, M., El Haddad, I., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prévôt, A. S., Baltensperger, U., and Dommen, J.: Labile peroxides in secondary organic aerosol, Chem, 1, 603–616, 2016.
Krechmer, J. E., Coggon, M. M., Massoli, P., Nguyen, T. B., Crounse, J. D., Hu, W., Day, D. A., Tyndall, G. S., Henze, D. K., Rivera-Rios, J. C., Nowak, J. B., Kimmel, J. R., Mauldin, R. L., III, Stark, H., Jayne, J. T., Sipilä, M., Junninen, H., St. Clair, J. M., Zhang, X., Feiner, P. A., Zhang, L., Miller, D. O., Brune, W. H., Keutsch, F. N., Wennberg, P. O., Seinfeld, J. H., Worsnop, D. R., Jimenez, J. L., and Canagaratna, M. R.: Formation of Low Volatility Organic Compounds and Secondary Organic Aerosol from Isoprene Hydroxyhydroperoxide Low-NO Oxidation, Environ. Sci. Technol., 49, 10330–10339, https://doi.org/10.1021/acs.est.5b02031, 2015.
Kroll, J. H. and Seinfeld, J. H.: Representation of Secondary Organic Aerosol Laboratory Chamber Data for the Interpretation of Mechanisms of Particle Growth, Environ. Sci. Technol., 39, 4159–4165, https://doi.org/10.1021/es048292h, 2005.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, 2008.
Kroll, J. H., Ng, N. L., Murphy, S. M., Varutbangkul, V., Flagan, R. C., and Seinfeld, J. H.: Chamber studies of secondary organic aerosol growth by reactive uptake of simple carbonyl compounds, J. Geophys. Res.-Atmos., 110, D23207, https://doi.org/10.1029/2005JD006004, 2005.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary Organic Aerosol Formation from Isoprene Photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Kurtén, T., Møller, K. H., Nguyen, T. B., Schwantes, R. H., Misztal, P. K., Su, L., Wennberg, P. O., Fry, J. L., and Kjaergaard, H. G.: Alkoxy Radical Bond Scissions Explain the Anomalously Low Secondary Organic Aerosol and Organonitrate Yields From α-Pinene + NO3, J. Phys. Chem. Lett., 8, 2826–2834, https://doi.org/10.1021/acs.jpclett.7b01038, 2017.
Kwan, A. J., Chan, A. W. H., Ng, N. L., Kjaergaard, H. G., Seinfeld, J. H., and Wennberg, P. O.: Peroxy radical chemistry and OH radical production during the NO3-initiated oxidation of isoprene, Atmos. Chem. Phys., 12, 7499–7515, https://doi.org/10.5194/acp-12-7499-2012, 2012.
Lanz, V. A., Alfarra, M. R., Baltensperger, U., Buchmann, B., Hueglin, C., and Prévôt, A. S. H.: Source apportionment of submicron organic aerosols at an urban site by factor analytical modelling of aerosol mass spectra, Atmos. Chem. Phys., 7, 1503–1522, https://doi.org/10.5194/acp-7-1503-2007, 2007.
Lee, B. H., Mohr, C., Lopez-Hilfiker, F. D., Lutz, A., Hallquist, M., Lee, L., Romer, P., Cohen, R. C., Iyer, S., Kurtén, T., Hu, W., Day, D. A., Campuzano-Jost, P., Jimenez, J. L., Xu, L., Ng, N. L., Guo, H., Weber, R. J., Wild, R. J., Brown, S. S., Koss, A., de Gouw, J., Olson, K., Goldstein, A. H., Seco, R., Kim, S., McAvey, K., Shepson, P. B., Starn, T., Baumann, K., Edgerton, E. S., Liu, J., Shilling, J. E., Miller, D. O., Brune, W., Schobesberger, S., D'Ambro, E. L., and Thornton, J. A.: Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets, P. Natl. Acad. Sci. USA, 113, 1516–1521, https://doi.org/10.1073/pnas.1508108113, 2016.
Lewandowski, M., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Edney, E. O., Sheesley, R. J., and Schauer, J. J.: Primary and secondary contributions to ambient PM in the midwestern United States, Environ. Sci. Technol., 42, 3303–3309, 2008.
Lin, Y.-H., Zhang, Z., Docherty, K. S., Zhang, H., Budisulistiorini, S. H., Rubitschun, C. L., Shaw, S. L., Knipping, E. M., Edgerton, E. S., and Kleindienst, T. E.: Isoprene epoxydiols as precursors to secondary organic aerosol formation: acid-catalyzed reactive uptake studies with authentic compounds, Environ. Sci. Technol., 46, 250–258, 2012.
Lin, Y.-H., Zhang, H., Pye, H. O., Zhang, Z., Marth, W. J., Park, S., Arashiro, M., Cui, T., Budisulistiorini, S. H., and Sexton, K. G.: Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, P. Natl. Acad. Sci. USA, 110, 6718–6723, 2013.
Liu, J., D'Ambro, E. L., Lee, B. H., Lopez-Hilfiker, F. D., Zaveri, R. A., Rivera-Rios, J. C., Keutsch, F. N., Iyer, S., Kurten, T., and Zhang, Z.: Efficient isoprene secondary organic aerosol formation from a non-IEPOX pathway, Environ. Sci. Technol., 50, 9872–9880, 2016.
Liu, Y., Kuwata, M., Strick, B. F., Geiger, F. M., Thomson, R. J., McKinney, K. A., and Martin, S. T.: Uptake of Epoxydiol Isomers Accounts for Half of the Particle-Phase Material Produced from Isoprene Photooxidation via the HO2 Pathway, Environ. Sci. Technol., 49, 250–258, https://doi.org/10.1021/es5034298, 2015.
Lyu, X., Guo, H., Zou, Q., Li, K., Xiong, E., Zhou, B., Guo, P., Jiang, F., and Tian, X.: Evidence for Reducing Volatile Organic Compounds to Improve Air Quality from Concurrent Observations and In Situ Simulations at 10 Stations in Eastern China, Environ. Sci. Technol., 56, 15356–15364, https://doi.org/10.1021/acs.est.2c04340, 2022.
Marais, E. A., Jacob, D. J., Jimenez, J. L., Campuzano-Jost, P., Day, D. A., Hu, W., Krechmer, J., Zhu, L., Kim, P. S., Miller, C. C., Fisher, J. A., Travis, K., Yu, K., Hanisco, T. F., Wolfe, G. M., Arkinson, H. L., Pye, H. O. T., Froyd, K. D., Liao, J., and McNeill, V. F.: Aqueous-phase mechanism for secondary organic aerosol formation from isoprene: application to the southeast United States and co-benefit of SO2 emission controls, Atmos. Chem. Phys., 16, 1603–1618, https://doi.org/10.5194/acp-16-1603-2016, 2016.
McGivern, W. S., Suh, I., Clinkenbeard, A. D., Zhang, R., and North, S. W.: Experimental and Computational Study of the OH-Isoprene Reaction: Isomeric Branching and Low-Pressure Behavior, J. Phys. Chem. A, 104, 6609–6616, 2000.
Mettke, P., Mutzel, A., Böge, O., and Herrmann, H.: Synthesis and Characterization of Atmospherically Relevant Hydroxy Hydroperoxides, Atmosphere, 13, 507, https://doi.org/10.3390/atmos13040507, 2022.
Mettke, P., Brüggemann, M., Mutzel, A., Gräfe, R., and Herrmann, H.: Secondary Organic Aerosol (SOA) through Uptake of Isoprene Hydroxy Hydroperoxides (ISOPOOH) and its Oxidation Products, ACS Earth and Space Chemistry, 7, 1025–1037, https://doi.org/10.1021/acsearthspacechem.2c00385, 2023.
Müller, J.-F., Stavrakou, T., and Peeters, J.: Chemistry and deposition in the Model of Atmospheric composition at Global and Regional scales using Inversion Techniques for Trace gas Emissions (MAGRITTE v1.1) – Part 1: Chemical mechanism, Geosci. Model Dev., 12, 2307–2356, https://doi.org/10.5194/gmd-12-2307-2019, 2019.
Ng, N. L., Kwan, A. J., Surratt, J. D., Chan, A. W. H., Chhabra, P. S., Sorooshian, A., Pye, H. O. T., Crounse, J. D., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3), Atmos. Chem. Phys., 8, 4117–4140, https://doi.org/10.5194/acp-8-4117-2008, 2008.
Nguyen, T. B., Coggon, M. M., Bates, K. H., Zhang, X., Schwantes, R. H., Schilling, K. A., Loza, C. L., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Organic aerosol formation from the reactive uptake of isoprene epoxydiols (IEPOX) onto non-acidified inorganic seeds, Atmos. Chem. Phys., 14, 3497–3510, https://doi.org/10.5194/acp-14-3497-2014, 2014.
Nguyen, T. B., Crounse, J. D., Teng, A. P., St. Clair, J. M., Paulot, F., Wolfe, G. M., and Wennberg, P. O.: Rapid deposition of oxidized biogenic compounds to a temperate forest, P. Natl. Acad. Sci. USA, 112, E392–E401, https://doi.org/10.1073/pnas.1418702112, 2015a.
Nguyen, T. B., Bates, K. H., Crounse, J. D., Schwantes, R. H., Zhang, X., Kjaergaard, H. G., Surratt, J. D., Lin, P., Laskin, A., Seinfeld, J. H., and Wennberg, P. O.: Mechanism of the hydroxyl radical oxidation of methacryloyl peroxynitrate (MPAN) and its pathway toward secondary organic aerosol formation in the atmosphere, Phys. Chem. Chem. Phys., 17, 17914–17926, https://doi.org/10.1039/C5CP02001H, 2015b.
Odum, J. R., Hoffmann, T., Bowman, F., Collins, D., Flagan, R. C., and Seinfeld, J. H.: Gas/Particle Partitioning and Secondary Organic Aerosol Yields, Environ. Sci. Technol., 30, 2580–2585, https://doi.org/10.1021/es950943+, 1996.
Pankow, J. F.: An absorption model of gas/particle partitioning of organic compounds in the atmosphere, Atmos. Environ., 28, 185–188, https://doi.org/10.1016/1352-2310(94)90093-0, 1994.
Pankow, J. F. and Asher, W. E.: SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 8, 2773–2796, https://doi.org/10.5194/acp-8-2773-2008, 2008.
Paulot, F., Crounse, J. D., Kjaergaard, H. G., Kürten, A., St. Clair, J. M., Seinfeld, J. H., and Wennberg, P. O.: Unexpected Epoxide Formation in the Gas-Phase Photooxidation of Isoprene, Science, 325, 730–733, https://doi.org/10.1126/science.1172910, 2009.
Paulson, S. E. and Seinfeld, J. H.: Development and evaluation of a photooxidation mechanism for isoprene, J. Geophys. Res.-Atmos., 97, 20703–20715, 1992.
Peeters, J., Nguyen, T. L., and Vereecken, L.: HOx radical regeneration in the oxidation of isoprene, Phys. Chem. Chem. Phys., 11, 5935–5939, 2009.
Perring, A. E., Wisthaler, A., Graus, M., Wooldridge, P. J., Lockwood, A. L., Mielke, L. H., Shepson, P. B., Hansel, A., and Cohen, R. C.: A product study of the isoprene+NO3 reaction, Atmos. Chem. Phys., 9, 4945–4956, https://doi.org/10.5194/acp-9-4945-2009, 2009.
Pöschl, U., von Kuhlmann, R., Poisson, N., and Crutzen, P. J.: Development and intercomparison of condensed isoprene oxidation mechanisms for global atmospheric modeling, J. Atmos. Chem., 37, 29–52, 2000.
Pye, H. O. T., Pinder, R. W., Piletic, I. R., Xie, Y., Capps, S. L., Lin, Y.-H., Surratt, J. D., Zhang, Z., Gold, A., Luecken, D. J., Hutzell, W. T., Jaoui, M., Offenberg, J. H., Kleindienst, T. E., Lewandowski, M., and Edney, E. O.: Epoxide Pathways Improve Model Predictions of Isoprene Markers and Reveal Key Role of Acidity in Aerosol Formation, Environ. Sci. Technol., 47, 11056–11064, https://doi.org/10.1021/es402106h, 2013.
Pye, H. O. T., Luecken, D. J., Xu, L., Boyd, C. M., Ng, N. L., Baker, K. R., Ayres, B. R., Bash, J. O., Baumann, K., Carter, W. P. L., Edgerton, E., Fry, J. L., Hutzell, W. T., Schwede, D. B., and Shepson, P. B.: Modeling the Current and Future Roles of Particulate Organic Nitrates in the Southeastern United States, Environ. Sci. Technol., 49, 14195–14203, https://doi.org/10.1021/acs.est.5b03738, 2015.
Rattanavaraha, W., Chu, K., Budisulistiorini, S. H., Riva, M., Lin, Y.-H., Edgerton, E. S., Baumann, K., Shaw, S. L., Guo, H., King, L., Weber, R. J., Neff, M. E., Stone, E. A., Offenberg, J. H., Zhang, Z., Gold, A., and Surratt, J. D.: Assessing the impact of anthropogenic pollution on isoprene-derived secondary organic aerosol formation in PM2.5 collected from the Birmingham, Alabama, ground site during the 2013 Southern Oxidant and Aerosol Study, Atmos. Chem. Phys., 16, 4897–4914, https://doi.org/10.5194/acp-16-4897-2016, 2016.
Riedel, T. P., Lin, Y.-H., Budisulistiorini, S. H., Gaston, C. J., Thornton, J. A., Zhang, Z., Vizuete, W., Gold, A., and Surratt, J. D.: Heterogeneous reactions of isoprene-derived epoxides: reaction probabilities and molar secondary organic aerosol yield estimates, Environ. Sci. Tech. Lett., 2, 38–42, 2015.
Riva, M., Budisulistiorini, S. H., Chen, Y., Zhang, Z., D'Ambro, E. L., Zhang, X., Gold, A., Turpin, B. J., Thornton, J. A., Canagaratna, M. R., and Surratt, J. D.: Chemical Characterization of Secondary Organic Aerosol from Oxidation of Isoprene Hydroxyhydroperoxides, Environ. Sci. Technol., 50, 9889–9899, https://doi.org/10.1021/acs.est.6b02511, 2016.
Saleh, R., Donahue, N. M., and Robinson, A. L.: Time Scales for Gas-Particle Partitioning Equilibration of Secondary Organic Aerosol Formed from Alpha-Pinene Ozonolysis, Environ. Sci. Technol., 47, 5588–5594, https://doi.org/10.1021/es400078d, 2013.
Schmedding, R., Rasool, Q. Z., Zhang, Y., Pye, H. O. T., Zhang, H., Chen, Y., Surratt, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Goldstein, A. H., and Vizuete, W.: Predicting secondary organic aerosol phase state and viscosity and its effect on multiphase chemistry in a regional-scale air quality model, Atmos. Chem. Phys., 20, 8201–8225, https://doi.org/10.5194/acp-20-8201-2020, 2020.
Schwantes, R. H., Teng, A. P., Nguyen, T. B., Coggon, M. M., Crounse, J. D., St. Clair, J. M., Zhang, X., Schilling, K. A., Seinfeld, J. H., and Wennberg, P. O.: Isoprene NO3 Oxidation Products from the RO2 + HO2 Pathway, J. Phys. Chem. A, 119, 10158–10171, https://doi.org/10.1021/acs.jpca.5b06355, 2015.
Schwantes, R. H., Charan, S. M., Bates, K. H., Huang, Y., Nguyen, T. B., Mai, H., Kong, W., Flagan, R. C., and Seinfeld, J. H.: Low-volatility compounds contribute significantly to isoprene secondary organic aerosol (SOA) under high-NOx conditions, Atmos. Chem. Phys., 19, 7255–7278, https://doi.org/10.5194/acp-19-7255-2019, 2019.
Shen, C. and Zhang, H.: zhangucr/UCR-ISOP, GitHub [code], https://github.com/zhangucr/UCR-ISOP.git (last access: 21 May 2024), 2024.
Shilling, J. E., Zawadowicz, M. A., Liu, J., Zaveri, R. A., and Zelenyuk, A.: Photochemical Aging Alters Secondary Organic Aerosol Partitioning Behavior, ACS Earth and Space Chemistry, 3, 2704–2716, https://doi.org/10.1021/acsearthspacechem.9b00248, 2019.
Song, S., Gao, M., Xu, W., Shao, J., Shi, G., Wang, S., Wang, Y., Sun, Y., and McElroy, M. B.: Fine-particle pH for Beijing winter haze as inferred from different thermodynamic equilibrium models, Atmos. Chem. Phys., 18, 7423–7438, https://doi.org/10.5194/acp-18-7423-2018, 2018.
Stadtler, S., Kühn, T., Schröder, S., Taraborrelli, D., Schultz, M. G., and Kokkola, H.: Isoprene-derived secondary organic aerosol in the global aerosol–chemistry–climate model ECHAM6.3.0–HAM2.3–MOZ1.0, Geosci. Model Dev., 11, 3235–3260, https://doi.org/10.5194/gmd-11-3235-2018, 2018.
St. Clair, J. M., Rivera-Rios, J. C., Crounse, J. D., Knap, H. C., Bates, K. H., Teng, A. P., Jørgensen, S., Kjaergaard, H. G., Keutsch, F. N., and Wennberg, P. O.: Kinetics and Products of the Reaction of the First-Generation Isoprene Hydroxy Hydroperoxide (ISOPOOH) with OH, J. Phys. Chem. A, 120, 1441–1451, https://doi.org/10.1021/acs.jpca.5b06532, 2016.
Surratt, J. D., Murphy, S. M., Kroll, J. H., Ng, N. L., Hildebrandt, L., Sorooshian, A., Szmigielski, R., Vermeylen, R., Maenhaut, W., Claeys, M., Flagan, R. C., and Seinfeld, J. H.: Chemical Composition of Secondary Organic Aerosol Formed from the Photooxidation of Isoprene, J. Phys. Chem. A, 110, 9665–9690, https://doi.org/10.1021/jp061734m, 2006.
Surratt, J. D., Chan, A. W., Eddingsaas, N. C., Chan, M., Loza, C. L., Kwan, A. J., Hersey, S. P., Flagan, R. C., Wennberg, P. O., and Seinfeld, J. H.: Reactive intermediates revealed in secondary organic aerosol formation from isoprene, P. Natl. Acad. Sci. USA, 107, 6640–6645, 2010.
Taraborrelli, D., Lawrence, M. G., Butler, T. M., Sander, R., and Lelieveld, J.: Mainz Isoprene Mechanism 2 (MIM2): an isoprene oxidation mechanism for regional and global atmospheric modelling, Atmos. Chem. Phys., 9, 2751–2777, https://doi.org/10.5194/acp-9-2751-2009, 2009.
Teng, A. P., Crounse, J. D., and Wennberg, P. O.: Isoprene Peroxy Radical Dynamics, J. Am. Chem. Soc., 139, 5367–5377, https://doi.org/10.1021/jacs.6b12838, 2017.
Thornton, J. A., Shilling, J. E., Shrivastava, M., D'Ambro, E. L., Zawadowicz, M. A., and Liu, J.: A Near-Explicit Mechanistic Evaluation of Isoprene Photochemical Secondary Organic Aerosol Formation and Evolution: Simulations of Multiple Chamber Experiments with and without Added NOx, ACS Earth and Space Chemistry, 4, 1161–1181, https://doi.org/10.1021/acsearthspacechem.0c00118, 2020.
Tiszenkel, L. and Lee, S. H.: Synergetic effects of isoprene and HOx on biogenic new particle formation, Geophys. Res. Lett., 50, e2023GL103545, https://doi.org/10.1029/2023GL103545, 2023.
Tsiligiannis, E., Wu, R., Lee, B. H., Salvador, C. M., Priestley, M., Carlsson, P. T., Kang, S., Novelli, A., Vereecken, L., and Fuchs, H.: A four carbon organonitrate as a significant product of secondary isoprene chemistry, Geophys. Res. Lett., 49, e2021GL097366, https://doi.org/10.1029/2021GL097366, 2022.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Vasquez, K. T., Crounse, J. D., Schulze, B. C., Bates, K. H., Teng, A. P., Xu, L., Allen, H. M., and Wennberg, P. O.: Rapid hydrolysis of tertiary isoprene nitrate efficiently removes NOx from the atmosphere, P. Natl. Acad. Sci. USA, 117, 33011–33016, https://doi.org/10.1073/pnas.2017442117, 2020.
Vereecken, L., Carlsson, P., Novelli, A., Bernard, F., Brown, S., Cho, C., Crowley, J., Fuchs, H., Mellouki, W., and Reimer, D.: Theoretical and experimental study of peroxy and alkoxy radicals in the NO3-initiated oxidation of isoprene, Phys. Chem. Chem. Phys., 23, 5496–5515, 2021.
Wania, F., Lei, Y. D., Wang, C., Abbatt, J. P. D., and Goss, K.-U.: Using the chemical equilibrium partitioning space to explore factors influencing the phase distribution of compounds involved in secondary organic aerosol formation, Atmos. Chem. Phys., 15, 3395–3412, https://doi.org/10.5194/acp-15-3395-2015, 2015.
Wennberg, P. O., Bates, K. H., Crounse, J. D., Dodson, L. G., McVay, R. C., Mertens, L. A., Nguyen, T. B., Praske, E., Schwantes, R. H., Smarte, M. D., St Clair, J. M., Teng, A. P., Zhang, X., and Seinfeld, J. H.: Gas-Phase Reactions of Isoprene and Its Major Oxidation Products, Chem. Rev., 118, 3337–3390, https://doi.org/10.1021/acs.chemrev.7b00439, 2018.
Wolfe, G. M., Marvin, M. R., Roberts, S. J., Travis, K. R., and Liao, J.: The Framework for 0-D Atmospheric Modeling (F0AM) v3.1, Geosci. Model Dev., 9, 3309–3319, https://doi.org/10.5194/gmd-9-3309-2016, 2016.
Xie, Y., Paulot, F., Carter, W. P. L., Nolte, C. G., Luecken, D. J., Hutzell, W. T., Wennberg, P. O., Cohen, R. C., and Pinder, R. W.: Understanding the impact of recent advances in isoprene photooxidation on simulations of regional air quality, Atmos. Chem. Phys., 13, 8439–8455, https://doi.org/10.5194/acp-13-8439-2013, 2013.
Xu, L., Guo, H., Boyd, C. M., Klein, M., Bougiatioti, A., Cerully, K. M., Hite, J. R., Isaacman-VanWertz, G., Kreisberg, N. M., Knote, C., Olson, K., Koss, A., Goldstein, A. H., Hering, S. V., de Gouw, J., Baumann, K., Lee, S.-H., Nenes, A., Weber, R. J., and Ng, N. L.: Effects of anthropogenic emissions on aerosol formation from isoprene and monoterpenes in the southeastern United States, P. Natl. Acad. Sci. USA, 112, 37–42, https://doi.org/10.1073/pnas.1417609112, 2015.
Yarwood, G., Rao, S., Yocke, M., and Whitten, G.: Updates to the carbon bond chemical mechanism: CB05, Tech. rep., US Environmental Protection Agency, Novato, CA, USA, https://www.camx.com/Files/CB05_Final_Report_120805.pdf (last access: 22 May 2024), 2005.
Yarwood, G., Jung, J., Whitten, G. Z., Heo, G., Mellberg, J., and Estes, M.: Updates to the Carbon Bond mechanism for version 6 (CB6), in: 9th Annual CMAS Conference, 11–13 October 2010, Chapel Hill, NC, https://www.cmascenter.org/conference/2010/abstracts/emery_updates_carbon_2010.pdf (last access: 22 May 2024), 2010.
Yee, L. D., Isaacman-VanWertz, G., Wernis, R. A., Kreisberg, N. M., Glasius, M., Riva, M., Surratt, J. D., de Sá, S. S., Martin, S. T., Alexander, M. L., Palm, B. B., Hu, W., Campuzano-Jost, P., Day, D. A., Jimenez, J. L., Liu, Y., Misztal, P. K., Artaxo, P., Viegas, J., Manzi, A., de Souza, R. A. F., Edgerton, E. S., Baumann, K., and Goldstein, A. H.: Natural and Anthropogenically Influenced Isoprene Oxidation in Southeastern United States and Central Amazon, Environ. Sci. Technol., 54, 5980–5991, https://doi.org/10.1021/acs.est.0c00805, 2020.
Zawadowicz, M. A., Lee, B. H., Shrivastava, M., Zelenyuk, A., Zaveri, R. A., Flynn, C., Thornton, J. A., and Shilling, J. E.: Photolysis Controls Atmospheric Budgets of Biogenic Secondary Organic Aerosol, Environ. Sci. Technol., 54, 3861–3870, https://doi.org/10.1021/acs.est.9b07051, 2020.
Zhang, H., Rattanavaraha, W., Zhou, Y., Bapat, J., Rosen, E. P., Sexton, K. G., and Kamens, R. M.: A new gas-phase condensed mechanism of isoprene-NOx photooxidation, Atmos. Environ., 45, 4507–4521, https://doi.org/10.1016/j.atmosenv.2011.04.011, 2011.
Zhang, H., Parikh, H. M., Bapat, J., Lin, Y.-H., Surratt, J. D., and Kamens, R. M.: Modelling of secondary organic aerosol formation from isoprene photooxidation chamber studies using different approaches, Environ. Chem., 10, 194–209, https://doi.org/10.1071/EN13029, 2013.
Zhang, H., Worton, D. R., Shen, S., Nah, T., Isaacman-VanWertz, G., Wilson, K. R., and Goldstein, A. H.: Fundamental Time Scales Governing Organic Aerosol Multiphase Partitioning and Oxidative Aging, Environ. Sci. Technol., 49, 9768–9777, https://doi.org/10.1021/acs.est.5b02115, 2015.
Zhang, H., Yee, L. D., Lee, B. H., Curtis, M. P., Worton, D. R., Isaacman-VanWertz, G., Offenberg, J. H., Lewandowski, M., Kleindienst, T. E., and Beaver, M. R.: Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States, P. Natl. Acad. Sci. USA, 115, 2038–2043, 2018.
Zhang, Q., Jimenez, J. L., Canagaratna, M., Allan, J. D., Coe, H., Ulbrich, I., Alfarra, M., Takami, A., Middlebrook, A., and Sun, Y.: Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes, Geophys. Res. Lett., 34, L13801, https://doi.org/10.1029/2007GL029979, 2007.
Zhang, X., Wang, S., Apel, E. C., Schwantes, R. H., Hornbrook, R. S., Hills, A. J., DeMarsh, K. E., Moo, Z., Ortega, J., and Brune, W. H.: Probing isoprene photochemistry at atmospherically relevant nitric oxide levels, Chem, 8, 3225–3240, 2022.
Zhang, Y., Chen, Y., Lambe, A. T., Olson, N. E., Lei, Z., Craig, R. L., Zhang, Z., Gold, A., Onasch, T. B., Jayne, J. T., Worsnop, D. R., Gaston, C. J., Thornton, J. A., Vizuete, W., Ault, A. P., and Surratt, J. D.: Effect of the Aerosol-Phase State on Secondary Organic Aerosol Formation from the Reactive Uptake of Isoprene-Derived Epoxydiols (IEPOX), Environ. Sci. Tech. Lett., 5, 167–174, https://doi.org/10.1021/acs.estlett.8b00044, 2018.