the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 04 Apr 2024
| 04 Apr 2024
Reaction of SO3 with H2SO4 and its implications for aerosol particle formation in the gas phase and at the air–water interface
Rui Wang
Yang Cheng
Shasha Chen
Rongrong Li
Yue Hu
Xiaokai Guo
Tianlei Zhang
Fengmin Song
Hao Li
The reactions between SO3 and atmospheric acids are indispensable in improving the formation of aerosol particles. However, relative to those of SO3 with organic acids, the reaction of SO3 with inorganic acids has not received much attention. Here, we explore the atmospheric reaction between SO3 and H2SO4, a typical inorganic acid, in the gas phase and at the air–water interface using quantum chemical (QC) calculations and Born–Oppenheimer molecular dynamics simulations. We also report the effect of H2S2O7, the product of the reaction between SO3 and H2SO4, on new particle formation (NPF) in various environments using the Atmospheric Cluster Dynamics Code (ACDC) kinetic model and QC calculations. The present findings show that the gas-phase reactions of SO3 + H2SO4 without and with water molecules are both low-energy-barrier processes. With the involvement of interfacial water molecules, H2O induced the formation of the ion pair, mediated the formation of the ion pair, and the deprotonation of H2S2O7 was observed and proceeded on the picosecond timescale. The present findings suggest the potential contribution of the SO3–H2SO4 reaction to NPF and aerosol particle growth, showing that (i) although H2S2O7 is easily hydrolyzed with water to form H2SO4, it can directly participate in H2SO4–NH3-based cluster formation and can present a more obvious enhancement effect on SA–A-based cluster formation, and (ii) the formed interfacial can attract candidate species from the gas phase to the water surface and, thus, accelerate particle growth.
- Article
(3748 KB) - Full-text XML
-
Supplement
(6793 KB) - BibTeX
- EndNote





Sulfur trioxide (SO3) is a major air pollutant (Zhuang and Pavlish, 2012; Chen and Bhattacharya, 2013; Cao et al., 2010; Kikuchi, 2001; Mitsui et al., 2011) and can be considered the most important oxidation product of SO2 (Starik et al., 2004). As an active atmospheric species, SO3 can lead to the formations of acid rain and atmospheric aerosol (Sipilä et al., 2010; Mackenzie et al., 2015; England et al., 2000; Li et al., 2016; Renard et al., 2004) and thus plays a well-documented role in regional climate and human health (Zhang et al., 2012, 2015; Pöschl, 2005; Pöschl and Shiraiwa, 2015; Haywood and Boucher, 2000; Lohmann and Feichter, 2005). In the atmosphere, the hydrolysis of SO3 to the H2SO4 product (SA) is a major loss route of SO3 (Morokuma and Muguruma, 1994; Akhmatskaya et al., 1997; Larson et al., 2000; Hazra and Sinha, 2011; Long et al., 2013a; Torrent-Sucarrat et al., 2012; Ma et al., 2020). As a complement to the loss of SO3, the ammonolysis reaction of SO3 in polluted areas of NH3 can form H2NSO3H, which not only can be competitive with the formation of SA from the hydrolysis reaction of SO3 but also can enhance the formation rates of sulfuric acid (SA)–dimethylamine (NH(CH3)2; DMA) clusters by about 2 times. Similarity, the reactions of SO3 with CH3OH and organic acids (such as HCOOH) were reported (Liu et al., 2019; Hazra and Sinha, 2011; Long et al., 2012; Mackenzie et al., 2015; Huff et al., 2017; Smith et al., 2017; H. Li et al., 2018), and both processes can provide a mechanism for incorporating organic matter into aerosol particles. However, the reaction mechanisms between SO3 and inorganic species are still unclear.
As a major inorganic acidic air pollutant (Tilgner et al., 2021), SA can play an important role in new particle formation (Weber et al., 1995, 1996, 2001; Sihto et al., 2006; Riipinen et al., 2007; Sipilä et al., 2010; Zhang et al., 2012) and acid rain (Calvert et al., 1985; Finlayson-Pitts and Pitts, 1986; Wayne, 2000). The source of gas-phase SA is mainly the gas-phase hydrolysis reaction of SO3. The direct reaction between SO3 and H2O hardly takes place in the atmosphere due to a high energy barrier (Chen and Plummer, 1985; Hofmann and Schleyer, 1994; Morokuma and Muguruma, 1994; Steudel, 1995). However the addition of a second water molecule (Morokuma and Muguruma, 1994; Larson et al., 2000; Loerting and Liedl, 2000), the hydroperoxyl radical (Gonzalez et al., 2010), formic acid (Hazra and Sinha, 2011; Long et al., 2012), sulfuric acid (Torrent-Sucarrat et al., 2012), nitric acid (Long et al., 2013a), oxalic acid (Lv et al., 2019), and ammonia (Bandyopadhyay et al., 2017) has been reported to catalyze the formation of SA from the hydrolysis reaction of SO3 as these compounds can promote atmospheric proton transfer reactions. Similarly, as SA can give out protons more readily than H2O, which in turn is more conducive to the proton transfer, we predict that the addition reaction involving the proton transfer between SO3 and SA is much easier under atmospheric conditions than that between SO3 and H2O. However, this gas-phase reaction has not been investigated as far as we know. Previous studies have shown that the concentration of water vapor decreases significantly with increasing altitude (Anglada et al., 2013), leading to longer atmospheric lifetimes of SO3. The gas-phase reaction of SO3 with H2SO4 may contribute significantly to the loss of SO3 in dry areas where [H2SO4] is relatively high (especially at lower temperatures) and at a higher altitude. So, it is important to study the reaction mechanism of SO3 with H2SO4 and its competition with H2O-assisted hydrolysis of SO3. Meanwhile, in many gas-phase reactions, a single water molecule can play a catalyst role by increasing the stability of pre-reactive complexes and reducing the activation energy of transition states (Kanno et al., 2006; Stone and Rowley, 2005; Chen et al., 2014; Viegas and Varandas, 2012, 2016). For example, a single water molecule in the H2O⋯HO2 + SO3 reaction can catalyze the formation of HSO5 (Gonzalez et al., 2010). Thus, it is equally important to study the SO3 + SA reaction without and with H2O. In addition to the gas-phase reactions, many new atmospheric processes and new reaction pathways have been observed at the air–water interface (Zhong et al., 2017a, b; Kumar et al., 2017, 2018; Zhu et al., 2016; Li et al., 2016; Zhu et al., 2017). For example, the organic acids reacting with SO3 can form the ion pair of carboxylic sulfuric anhydride and hydronium at the air–water interface (Zhong et al., 2019). This mechanism is different from the gas-phase reaction in which the organic acid either serves as a catalyst for the hydrolysis of SO3 or acts as a reactant reacting with SO3 directly. So, water droplets may play important roles in atmospheric behaviors between SO3 and SA. Thus, it is also important to study the interfacial mechanism between SO3 and SA and to compare its difference with the corresponding gas-phase reaction.
Previous experimental studies (Otto and Steudel, 2001; Abedi and Farrokhpour, 2013) found that disulfuric acid (H2S2O7, DSA) is the product of the reaction between SO3 and SA. From the perspective of structure, DSA possesses two HO functional groups. Both HO groups can act as hydrogen donors and acceptors to interact with atmospheric particle precursors. It has been shown that the reaction between SO3 and some important atmospheric species (H. Li et al., 2018; Yang et al., 2021; Liu et al., 2019; Rong et al., 2020) not only can cause appreciable consumption of SO3 and thus reduce the abundance of SA from the hydrolysis of SO3 in the atmosphere but also can promote the new particle formation (NPF) process by their products. For example, the products of NH2SO3H, HOOCOOSO3H, CH3OSO3H, and HOCCOOSO3H from the reactions of SO3 with NH3 (H. Li et al., 2018), H2C2O4 (Yang et al., 2021), CH3OH (Liu et al., 2019) and HOOCCHO (Rong et al., 2020) all have a catalytic effect on the formation of new particles in aerosols. However, whether DSA produced by the reaction between SO3 and SA contributes to aerosol formation or not is still unclear. Thus, another main question that we intend to address here is the role of DSA in atmospheric SA–NH3 (A) nucleation. These chemicals have been recognized as dominant precursors in highly polluted areas, especially in some megacities in Asia.
In this work, using quantum chemical calculations and the master equation, we first studied the gas-phase reaction between SO3 and SA to produce DSA, with H2O acting as a catalyst. Then, we use the Born–Oppenheimer molecular dynamics (BOMD) simulations to evaluate the reaction mechanism of SO3 with SA at the air–water interface. Finally, we used the Atmospheric Clusters Dynamic Code (ACDC) kinetic model and quantum chemical calculations to investigate atmospheric implications of the SO3–SA reaction for the atmospheric particle formation. Particular attention in this work is given to the study of (i) the mechanism difference of the SO3 + SA reaction in the gas phase and at the air–water interface and (ii) the fate of DSA in atmospheric NPF and its influence in various environmental conditions.
2.1 Quantum chemical calculation
The M06-2X functional has been proved to be one of the best functionals to describe the noncovalent interactions and estimate the thermochemistry and equilibrium structures for atmospheric reactions (Elm et al., 2012; Mardirossian and Head-Gordon, 2016). So, for the SO3 + SA reaction without and with water molecules in the gas phase, the optimized geometries and vibrational frequencies of reactants, pre-reactive complexes, transition states (TSs), post-reactive complexes, and products were calculated using the M06-2X method (Zhao and Truhlar, 2008; Elm et al., 2012) with the 6-311G(2df,2pd) basis set by Gaussian 09 packages (Hratchian et al., 2009). It is noted that the calculated bond distances and bond angles at the M06-2X/6-311G(3df,2pd) level (Fig. S1 in the Supplement) agree well with the available experimental values (Kuczkowski et al., 1981). At the same level, the connectivity between the TSs and the suitable pre- and post-reactant complexes was performed by intrinsic reaction coordinate (IRC) calculations. Then, single-point energy calculations were calculated at the CCSD(T)-F12/cc-pVDZ-F12 level (Adler et al., 2007; Knizia et al., 2009) using ORCA (Neese, 2012).
A multistep global minimum sampling technique was used to search for the global minima of the (DSA)x(SA)y(A)z () molecular clusters. Specifically, a multistep global minimum sampling technique was used to search for the global minima of the (SA)x(A)y(DSA)z () clusters. Specifically, the initial n × 1000 (1 < n < 5) configurations for each cluster were systematically generated by the ABCluster program (Zhang and Dolg, 2015, 2016) and were optimized at the semi-empirical PM6 (Stewart, 2013) methods using MOPAC 2016 (Stewart, 2016, 2013, 2007). Then, up to n × 100 structures with relatively lowest energy among the n × 1000 (1 < n < 5) structures were selected and re-optimized at the M06-2X/6-31+G(d,p) level. Finally, n × 10 lowest-lying structures were optimized by the M06-2X/6-311G(2df,2pd) level to determine the global minimum. To obtain the reliable energies, single-point energy calculations were refined at the DLPNO-CCSD(T)/aug-cc-pVTZ level based on the optimized geometries at the M06-2X/6-311G(2df,2pd) level. The optimized structures and the formation Gibbs free energy of the stable clusters are summarized in Fig. S9 and Table S8 in the Supplement, respectively.
2.2 Rate constant calculations
Using the Rice–Ramsperger–Kassel–Marcus-based master equation (ME/RRKM) (Miller and Klippenstein, 2006), the kinetics for the SO3 + SA reaction without and with water molecule were calculated by adopting a Master Equation Solver for Multi Energy-well Reactions (MESMER) code (Glowacki et al., 2012). In the MESMER calculation, the rate coefficients for the bimolecular barrier-less association step (from reactants to pre-reactive complexes) were evaluated by the inverse Laplace transform (ILT) method (Horváth et al., 2020); meanwhile the unimolecular step was performed by the RRKM theory combined with the asymmetric Eckart model. The ILT method and RRKM theory can be represented in Eqs. (1) and (2), respectively.
where h is denoted as Planck's constant, ρ(E) is denoted as the active density of state of the reactant at energy level E, E0 is denoted as the reaction threshold energy, and W(E−E0) is denoted as the sum of the rovibrational states of the transition state (TS) geometry (excluding the degree of freedom related to passing the transition state). The input parameters for electronic geometries, vibrational frequencies, and rotational constants were calculated at the M06-2X/6-311G(2df,2pd) level, and single-point energy calculations were refined at the CCSD(T)-F12/cc-pVDZ-F12 level for the modeling.
2.3 Born–Oppenheimer molecular dynamics (BOMD) simulation
The CP2K code (Hutter et al., 2014) was used in the BOMD simulations. The Becke–Lee–Yang–Parr (BLYP) functional (Becke, 1988; Lee et al., 1988) was chosen to look at the exchange and correlation interactions, and Grimme's dispersion was carried out to account for the weak dispersion interaction (Grimme et al., 2010). The Goedecker–Teter–Hutter (GTH) conservation pseudopotential (Goedecker et al., 1996; Hartwigsen et al., 1998) with the Gaussian DZVP basis set (VandeVondele and Hutter, 2007) and the auxiliary plane wave basis set was applied to correct the system valence electrons and the core electrons, respectively. For the plane wave basis set and Gaussian basis set, the energy cutoff levels (Zhong et al., 2017a, b, 2018, 2019) were set to 280 and 40 Ry, respectively. For each simulation in the gas phase, a 15 × 15 × 15 Å3 supercell with periodic boundary condition was adopted with a time step of 0.5 fs. As the droplet system with 191 water molecules is sufficient to describe the interfacial mechanism (Zhong et al., 2017a), the air–water interfacial system here included 191 water molecules, SO3, and SA in the BOMD simulation. It is pointed out that the droplet system with 191 water molecules had been equilibrated before SO3 and H2SO4 were added at the water surface. The details of the equilibrium process for the droplet system with 191 water molecules are shown in the Supplement Sect. S4. To avoid periodic interactions between adjacent water droplets, the size of the simulation box (Kumar et al., 2017, 2018; Ma et al., 2020) was set as 35 × 35 × 35 Å3 with a time step of 1.0 fs. Notably, the time step of 1.0 fs has been proved to achieve sufficient energy conservation for the water system (Zhong et al., 2015; Li et al., 2016; Zhu et al., 2016; Kumar et al., 2017). For all the simulations in the gas phase and at the air–water interface, the Nosé–Hoover thermostat (Zhong et al., 2017a, b, 2018, 2019; Kumar et al., 2017, 2018; Ma et al., 2020) was selected in the NVT ensemble to control the temperature around 300 K. To eliminate the influence of the initial configuration on the simulation results of interfacial reaction, 40 BOMD simulations for the air–water interface reactions were carried out.
2.4 Atmospheric Clusters Dynamic Code (ACDC) model
The Atmospheric Cluster Dynamics Code (ACDC) (McGrath et al., 2012) model was used to simulate the cluster formation rates and mechanisms of (DSA)x(SA)y(A)z () clusters at different temperatures and monomer concentrations. The thermodynamic data of quantum chemical calculation at the DLPNO-CCSD(T)/aug-cc-pVTZ//M06-2X/6-311G(2df,2pd) level of theory can be used as the input of the ACDC. The birth–death equation (Eq. 3) for clusters solves the time development of cluster concentrations by numerical integration using the ode15s solver in MATLAB (Shampine and Reichelt, 1997).
where ci is the concentration of cluster i, βi,j is the collision coefficient between clusters i and j, is the evaporation coefficient of cluster i+j evaporating into clusters i and j, and Qi is all other source term of cluster i (see more details of β and γ in SI Appendix Part 4). In addition, a constant coagulation sink coefficient 2 × 10−2 s−1 (corresponding to the median observed in contaminated areas) was used for taking into account external losses (Yao et al., 2018; Zhang et al., 2022; L. Liu et al., 2021). The boundary conditions in the ACDC require that the smallest clusters outside of the simulated system should be very stable so that they do not evaporate back immediately (McGrath et al., 2012). Based on cluster volatilization rate (shown in Table S10 in the Supplement) and the formation Gibbs free energy of the clusters (shown in Table S8), the cluster boundary conditions simulated in this study were set as (SA)4⋅(A)3, (SA)4⋅(A)4, , , and . According to field observations, the concentration of SA and A was, respectively, set in a range of 106–108 and 107–1011 (Almeida et al., 2013; Kuang et al., 2008; Bouo et al., 2011; Zhang et al., 2018). As the prediction in Table S7 in the Supplement shows, the concentration of DSA is set to 104–108 . However, DSA is easily hydrolyzed with abundant water in the troposphere to form H2SO4; the concentration of DSA listed in Fig. S9 was overestimated. So, the maximum concentration of DSA (108 ) was not included in the effects of H2S2O7 on new particle formation (NPF) in various environments. In addition, the temperature was set to be in the range 218.15–298.15 K, which spans most regions of the troposphere and the polluted atmospheric boundary layer.
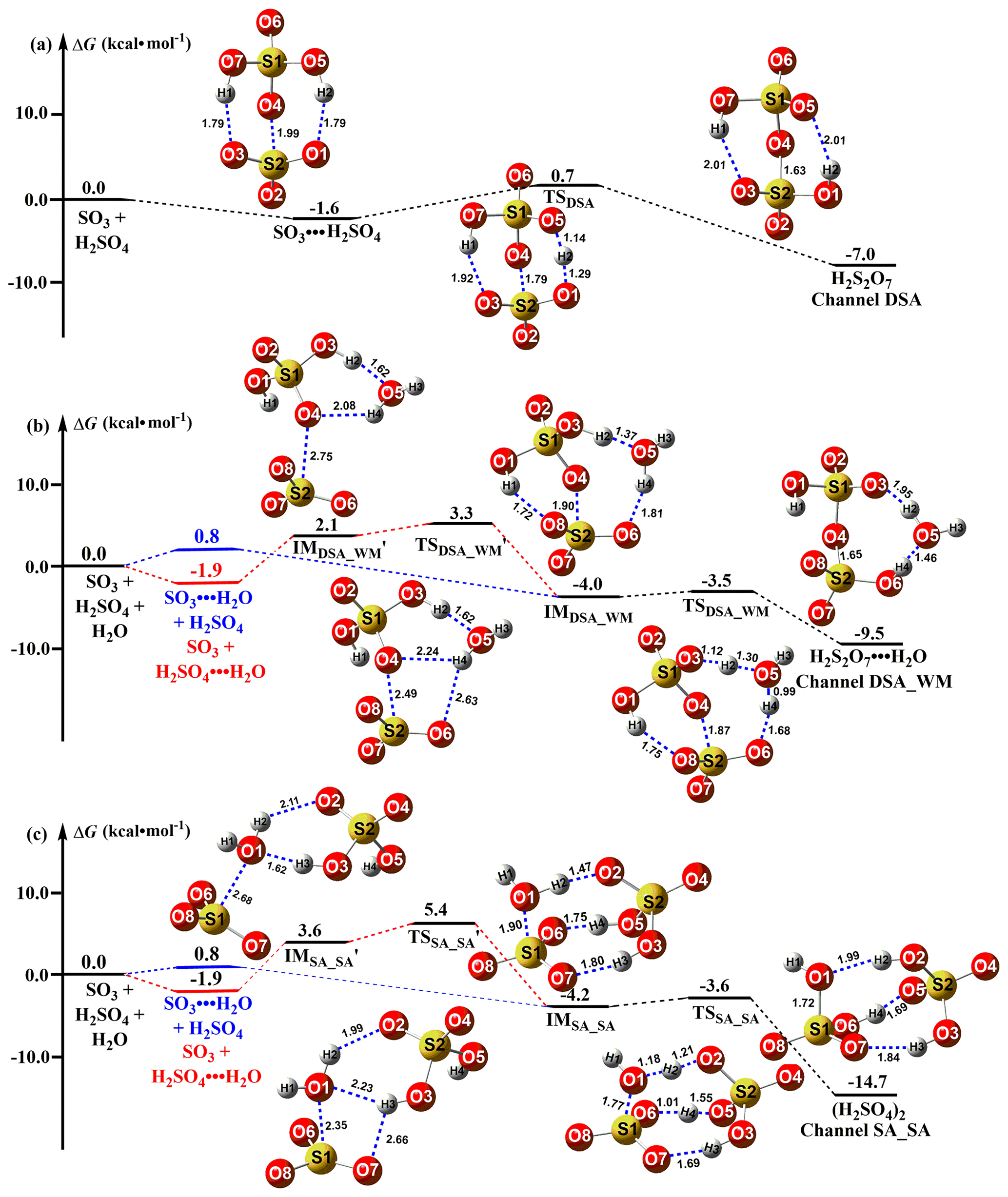
Figure 1Schematic potential energy surface for the SO3 + H2SO4 → H2S2O7 reaction. Distances are given in units of Ångström at the M06-2X/6-311G(2df,2pd) level, while the energy values correspond to the calculations at the CCSD(T)-F12/cc-pVDZ-F12//M06-2X/6-311G(2df,2pd) level. The pre-reactive complex and TS for the route of DSA formation from the SO3 + H2SO4 reaction with H2O were denoted by “IMDSA_WM” and “TSDSA_WM”, respectively, while the corresponding pre-reactive complex and TS for the process of SA formation from the hydrolysis of SO3 with H2SO4 were respectively labeled as “IMSA_SA” and “TSSA_SA”.
Table 1The rate constant () for the SO3 + H2SO4 reaction and the effective rate constant () for the SO3 + H2SO4 reaction with H2O (100 % RH) within the temperature range of 280–320 K.
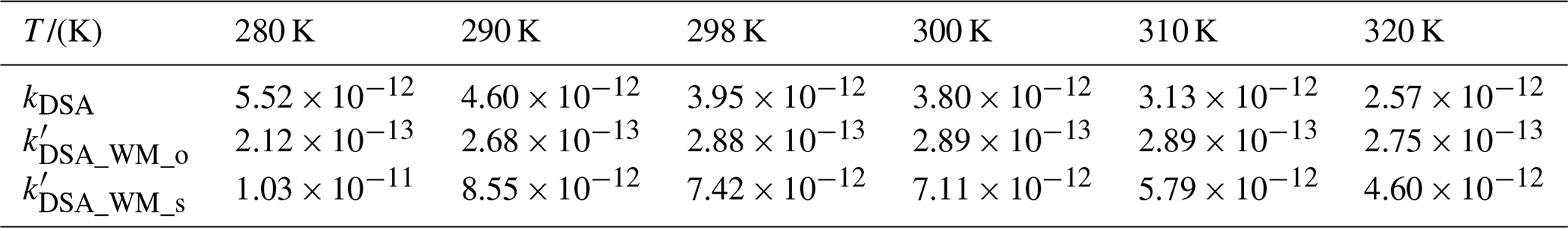
kDSA is the rate constant for the SO3 + H2SO4 reaction. and are, respectively, the effective rate constants for the H2O-assisted SO3 + H2SO4 reaction occurring through one-step and stepwise routes.
3.1 Reactions in the gas phase
The addition reaction involving the proton transfer between SO3 and SA (Channel DSA) proceeded through the formation of the SO3⋯H2SO4 complex followed by unimolecular transformation through the transition state TSDSA to form H2S2O7 (Fig. 1a). The reactant complex SO3⋯H2SO4 was a double six-membered ring complex with a relative Gibbs free energy of −1.6 kcal mol−1. After the formation of the SO3⋯H2SO4 complex, Channel DSA overcame a Gibbs free energy barrier of 2.3 kcal mol−1, which was lower than that of H2O-catalyzed hydrolysis of SO3 by 4.2 kcal mol−1 (Fig. S1). The rate constant for the SO3 + SA reaction was calculated at various temperatures (Table 1). Within the temperature range of 280–320 K, the rate constants for the SO3 + SA reaction were calculated to be 2.57 × 10−12–5.52 × 10−12 , which were larger than the corresponding values of H2O-catalyzed hydrolysis of SO3 by 3.43–4.03 times. Therefore, it can be said that the direct reaction between SO3 and SA occurs easily under atmospheric conditions.
The SO3 + H2SO4 reaction with H2O produced two distinct products, labeled (i) H2S2O7 (DSA; Channel DSA_WM) and (ii) H2SO4 (SA; Channel SA_SA). A single water molecule in (i) acted as a catalyst, while it played the role of a reactant in (ii). The schematic potential energy surface for the SO3 + H2SO4 reaction with H2O was shown in Fig. 1. As the probability of simultaneous collision (Pérez-Ríos et al., 2014; Elm et al., 2013) of three molecules of SO3, SA, and H2O was quite low under realistic conditions, both Channel DSA_WM and Channel SA_SA can be considered sequential bimolecular processes. In other words, both Channel DSA_WM and Channel SA_SA occurred via the collision between SO3 (or H2SO4) and H2O to form the dimer (SO3⋯H2O and H2SO4⋯H2O) first and then the dimer encountered with the third reactant H2SO4 or SO3. The computed Gibbs free energies of dimer complexes SO3⋯H2O and H2SO4⋯H2O were, respectively, 0.8 and −1.9 kcal mol−1, which were, respectively, consistent with the previous values (the range from −0.2 to 0.62 kcal mol−1 for the SO3⋯H2O complex (Bandyopadhyay et al., 2017; Long et al., 2012) and the range from −1.82 to −2.63 kcal mol−1 for the H2SO4⋯H2O complex (Long et al., 2013b; Tan et al., 2018)). The Gibbs free energy of H2SO4⋯H2O was lower than that of SO3⋯H2O by 2.7 kcal mol−1, thus leading to the equilibrium constant of the former complex being larger than that of the latter one in Table S2 in the Supplement by 1–2 orders of magnitude. Additionally, the larger equilibrium constant of the H2SO4⋯H2O complex led to its higher concentration in the atmosphere. For example, when the concentrations of SO3 (Yao et al., 2020), H2SO4 (Liu et al., 2015), and H2O (Anglada et al., 2013) were 106, 108, and 1017 , respectively, the concentrations of SO3⋯H2O and H2SO4⋯H2O were 2.41 × 103–2.01 × 104 and 5.01 × 105–3.01 × 108 within the temperature range of 280–320 K (see Table S3 in the Supplement), respectively. So, we predict that Channel DSA_WM and Channel SA_SA mainly take place via the collision of H2SO4⋯H2O with SO3. In order to check this prediction, the effective rate constants for two bimolecular reactions of H2SO4⋯H2O + SO3 and SO3⋯H2O + H2SO4 were calculated, and the details are shown in SI Appendix, Part 3 and Table 1. As seen in Table 1, the SO3⋯H2O + H2SO4 reaction in both Channel DSA_WM and Channel SA_SA can be neglected as their effective rate constants were smaller than the corresponding values in the H2SO4⋯H2O + SO3 reaction by 16.7–48.5 and 1.02–3.05 times within the temperature range of 280–320 K, respectively. Therefore, we only considered the H2SO4⋯H2O + SO3 bimolecular reaction in both Channel DSA_WM and Channel SA_SA.
As for Channel DSA_WM, the H2SO4⋯H2O + SO3 reaction occurred in a stepwise process as displayed in Fig. 1b, which was similar to the favorable routes in the hydrolysis of COS, HCHO, and CH3CHO catalyzed by sulfuric acid (Long et al., 2013b; K. Li et al., 2018; Tan et al., 2018). When the H2SO4⋯H2O complex and SO3 served as reactants, the reaction was initiated by the complex , where a van der Waals interaction (S2⋯O4, 2.75 Å) was found between the O4 atom of SA moiety in H2SO4⋯H2O and the S atom of SO3. After the complex , the ring enlargement from to the SO3⋯H2SO4⋯H2O complex occurred through the transition state with a Gibbs free energy barrier of 1.2 kcal mol−1. The IMDSA_WM complex was 6.1 kcal mol−1 lower in energy than . In IMDSA_WM, SO3 acted as a double donor of the hydrogen bond to form a cage-like hydrogen bonding network with H2SO4⋯H2O. Then, starting with the IMDSA_WM complex, the H2SO4⋯H2O + SO3 reaction occurred through the transition state TSDSA_WM with a Gibbs free barrier energy of 0.5 kcal mol−1 to form a quasi-planar network complex, H2S2O7⋯H2O. TSDSA_WM was in the middle of a double proton transfer, where H2O played the role of a bridge for proton transfer, along with the simultaneous formation of the O4⋯S2 bond. In order to estimate the catalytic ability of H2O in the SO3 + SA reaction, the effective rate constant () of the H2SO4⋯H2O + SO3 reaction was compared with the rate constant (kDSA) of the SO3 + H2SO4 reaction. As seen in Table 1, under the experimental concentration (Anglada et al., 2013) ([H2O] = 5.20 × 1016–2.30 × 1018 ) within the temperature range of 280–320 K, the calculated was 1.03 × 10−11–4.60 × 10−12 , which was larger than that of kDSA by 1.79–1.86 times. This result shows that H2O exerts a catalytic role in promoting the rate of the SO3 + H2SO4 reaction.
Regarding Channel SA_SA, the stepwise reaction occurred firstly via the ring enlargement from the six-membered ring complex to a cage-like hydrogen bonding network IMSA_SA and then took place by going through a transition state, TSSA_SA, to form the product complex (H2SO4)2. TSDSA_WM was in the middle of a double-hydrogen transfer, where H2SO4 acted as a bridge of the hydrogen atom from the H2O to SO3 along with the O1 atom of the H2O addition to the S atom of SO3. It is worth noting that the energy barriers of two elementary reactions involved in the stepwise route of Channel SA_SA were only 1.8 and 0.6 kcal mol−1, respectively, showing that the occurrence of the stepwise route of Channel SA_SA is feasible from an energetic point of view. To check whether Channel DSA_WM is more favorable than Channel SA_SA or not, their rate ratios listed in Eq. (4) have been calculated and are shown in Table 1. The calculated rate ratio shows that Channel DSA_WM is more important than Channel SA_SA because the rate ratio is 1.53–3.04 within the temperature range of 280–320 K. So, we predicted that the SO3 + H2SO4 reaction with H2O producing H2S2O7 is more favorable than that forming H2SO4.
3.2 Reactions at the air–water interface
The mechanism for the SO3 + SA reaction at the air–water interface was lacking. Notably, SO3, SA, and DSA molecules can stay at the interface for 35.8 %, 30.1 %, and 39.2 % of the time in the 150 ns simulation (Fig. S2 in the Supplement), respectively, revealing that the existence of SO3, SA, and DSA at the air–water interface cannot be negligible. So, the BOMD simulations were used to evaluate the reaction mechanism of SO3 with SA at the aqueous interfaces. Similar to the interfacial reaction of SO3 with organic and inorganic acids (Cheng et al., 2023; Zhong et al., 2019), the reaction between SO3 and SA at the aqueous interface may occur in three ways: (i) SO3 colliding with adsorbed SA at the air–water interface, (ii) SA colliding with adsorbed SO3 at the aqueous interface, or (iii) the SO3–SA complex reacting at the aqueous interface. However, due to the high reactivity of both SO3 and SA at the air–water interface, the lifetimes of SO3 (Zhong et al., 2019) and SA (Fig. S3) (on the order of a few picoseconds) on the water droplet were extremely short, and the SA− ion can be formed quickly. In addition, as shown by the result calculated above, the SO3⋯H2SO4 complex can generate DSA easily before it approaches the air–water interface. So, two possible models were mainly considered for the SO3 + SA reaction on the water surface: (i) gaseous SO3 colliding with SA− at the air–water interface and (ii) the DSA (the gas-phase product of SO3 and SA) dissociating on the water droplet.
3.2.1 Gaseous SO3 colliding with SA− at the air–water interface
At the water droplet's surface, the interaction between SO3 and SA− included two main channels: (i) H2O induced the formation of the ion pair (Fig. 2 and Fig. S4 and Movie S1 in the Supplement), and (ii) SA− mediated the formation of the ion pair (Fig. 3 and Figs. S5 and S6 and Movies S2 and S3 in the Supplement). The BOMD simulations for the H2O-induced formation of the ion pair are illustrated in Fig. 2; the H1 atom of the SA− ion can combine with a nearby interfacial water molecule at 8.18 ps via a hydrogen bond (d(O3–H1) = 1.17 Å) interaction, thus forming the hydrated hydrogen sulfate ion (SA−⋯H2O). Then, the H1 atom of the SA− ion was moved to the O3 atom of the interfacial water molecule at 8.28 ps, revealing the formation of the ion pair. Additionally, gradually approached the SO3 molecule, with a shortening of the S1–O1 bond. At 9.26 ps, the S1–O1 bond length was 1.84 Å, which was close to the length of the S–O1 (1.65 Å) bond in the ion (Fig. S8), revealing the formation of the ion pair.
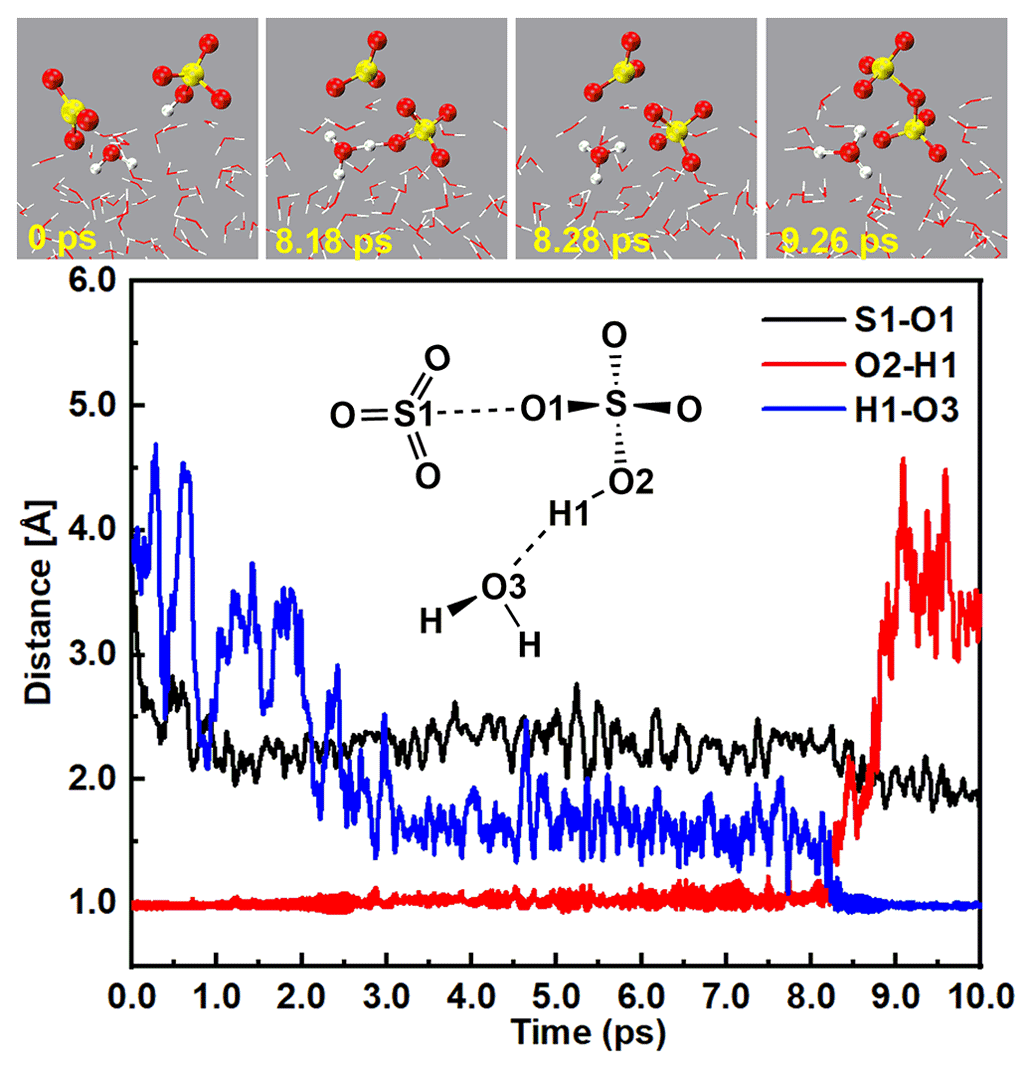
Figure 2Top panel: snapshot structures taken from the BOMD simulations, which illustrate that H2O induced the formation of the ion pair as a result of the reaction of SO3 with at the air–water interface. Lower panel: time evolution of key bond distances (S–O1, O2–H1, and O3–H1) involved in the induced mechanism.
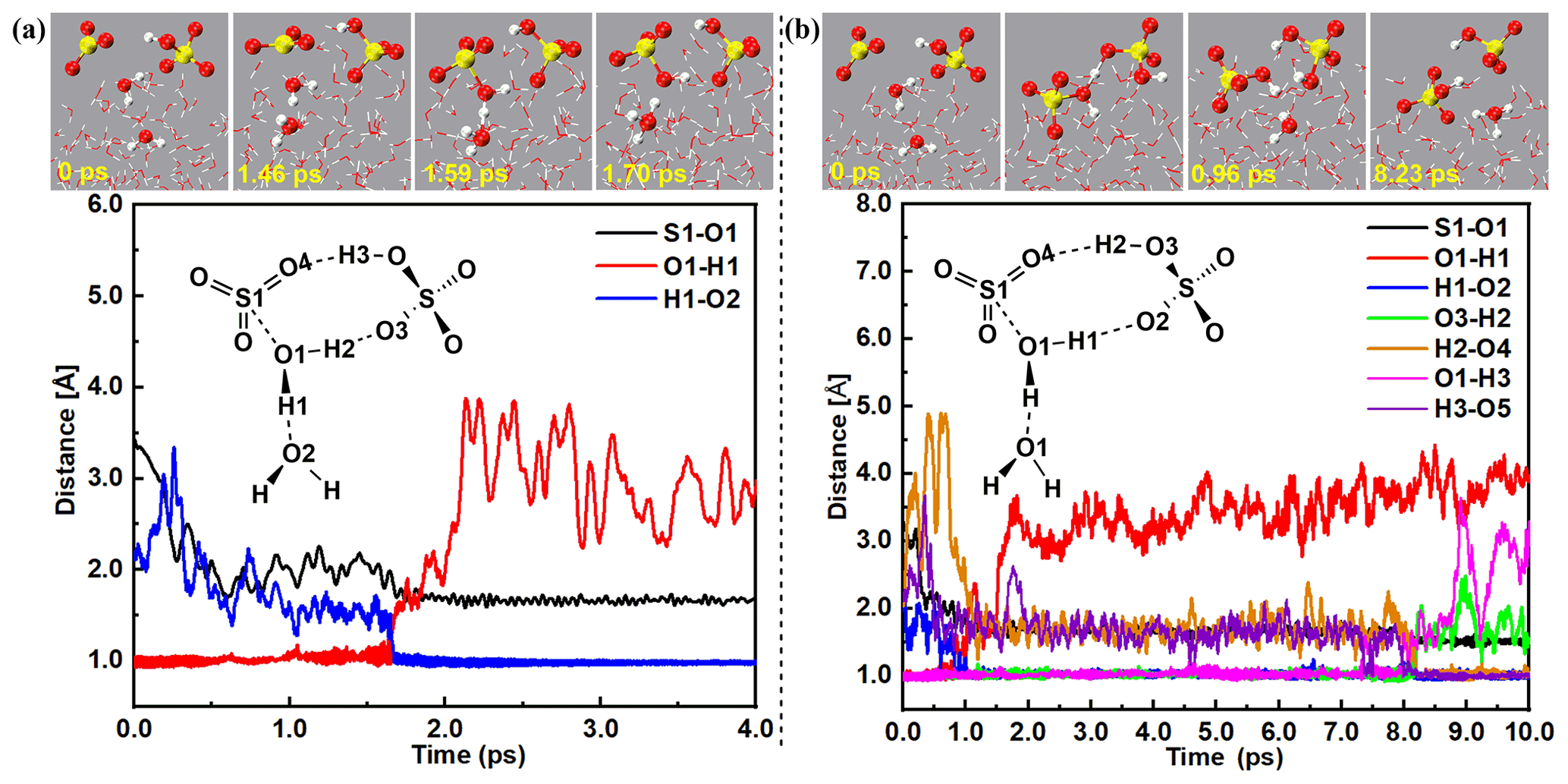
Figure 3The top parts of (a) and (b) show snapshot structures taken from the BOMD simulations, which illustrate the hydration reaction mechanism of SO3 mediated by at the air water interface. The bottom parts of (a) and (b) show the time evolution of key bond distances (S–O1, O1–H2, O5–H2, O2–H1, O3–H4, and O4–H3) involved in the hydration mechanism.
Both direct (Figs. 3a and S5 and Movie S2) and indirect (Figs. 3b and S6 and Movie S3) forming mechanisms were observed in SA−-mediated formation of the ion pair. The direct SA−-mediated formation of the ion pair was a loop structure mechanism, which was consistent with gas-phase hydrolysis of SO3 assisted by acidic catalysts of HCOOH, HNO3, H2C2O4, and SA in previous works (Long et al., 2012, 2013a; Torrent-Sucarrat et al., 2012; Lv et al., 2019) and the interfacial reactions of HNO3-mediated Criegee hydration (Kumar et al., 2018) and the hydration of SO3 via the loop-structure formation (Lv and Sun, 2020). As for the direct formation mechanism of the ion pair seen in Fig. 3a and Movie S2, an eight-membered loop complex, SO3⋯H2O(1)⋯SA−, was found at 1.46 ps, with the formation of two hydrogen bonds (d(O3⋯H2) = 2.13 Å; d(O4⋯H3) = 2.18 Å) and a van der Waals interaction (d(S1⋯O1) = 2.14 Å). Subsequently, SO3 and interfacial H2O(1) were close to each other. At 1.59 ps, a transition-state-like loop structure was observed, and proton transfer from interfacial H2O(1) to another suspended H2O(2) was found, where the bond lengths of S1–O1, O1–H1, and H1–O2 were 1.94, 1.19, and 1.32 Å, respectively. At 1.70 ps, the bond lengths of S–O1 and H1–O2 were reduced to 1.73 and 1.01 Å, while the bond length of H1–O2 was extended to 1.61 Å, showing the formation of the ion pair. During the direct formation route of the ion pair, SA− played the role of a spectator, while interfacial water molecules acted as both a reactant and a proton acceptor. As compared with the hydration reaction mechanism of SO3 at the air–water interface reported by Lv et al. (Lv and Sun, 2020), the loop-structure formation with proton transferred in the loop was not observed in the direct mechanism of SA−-mediated formation of the ion pair. This was probably because the SA− ion was more difficult to give the proton.
As seen in Fig. 3b and Movie S3, the indirect forming process of ion pair contained two steps: (i) SO3 hydration along with SA formation and (ii) SA deprotonation. Specifically, as for step (i), at 0.70 ps, a transition-state-like structure of SO3 hydration was observed with SO3, SA−, and an interfacial water molecule involved. Note that at this time the H1 atom in the interfacial H2O molecule migrated to the O2 atom of the SA− ion instead of the surrounding water molecule. At 0.96 ps, the O1–H1 bond of H2O was broken with the length of 1.56 Å, while the S1–O1 bond was formed with the length of 1.75 Å, demonstrating the completion of the hydrolysis reaction of SO3 and the formation of the SA molecule. Then, at 8.08 ps, the H2 proton transferred from SA to the O4 atom of SA− ion and to the O5 atom of the nearby water molecule was occurred, where the O3–H2 and O1–H3 bonds extended to 1.13 and 1.22 Å, and the length of O4–H2 and O5–H3 bonds shortened to 1.45 and 1.20 Å. Finally, SA deprotonation was completed at 8.23 ps with the formation of the ion pair. During the whole indirect formation process of the ion pair, SA− played the role of protons' donor and acceptor, and water molecules acted as hydration reactants and proton acceptors. Compared with the direct mechanism of the ion pair, the indirect formation process of the ion pair required more time. This was consistent with the interfacial reactions of CH2OO + HNO3 (Kumar et al., 2018) and the hydration of SO3 (Lv and Sun, 2020), where the direct forming mechanism needed less time than indirect forming mechanism.
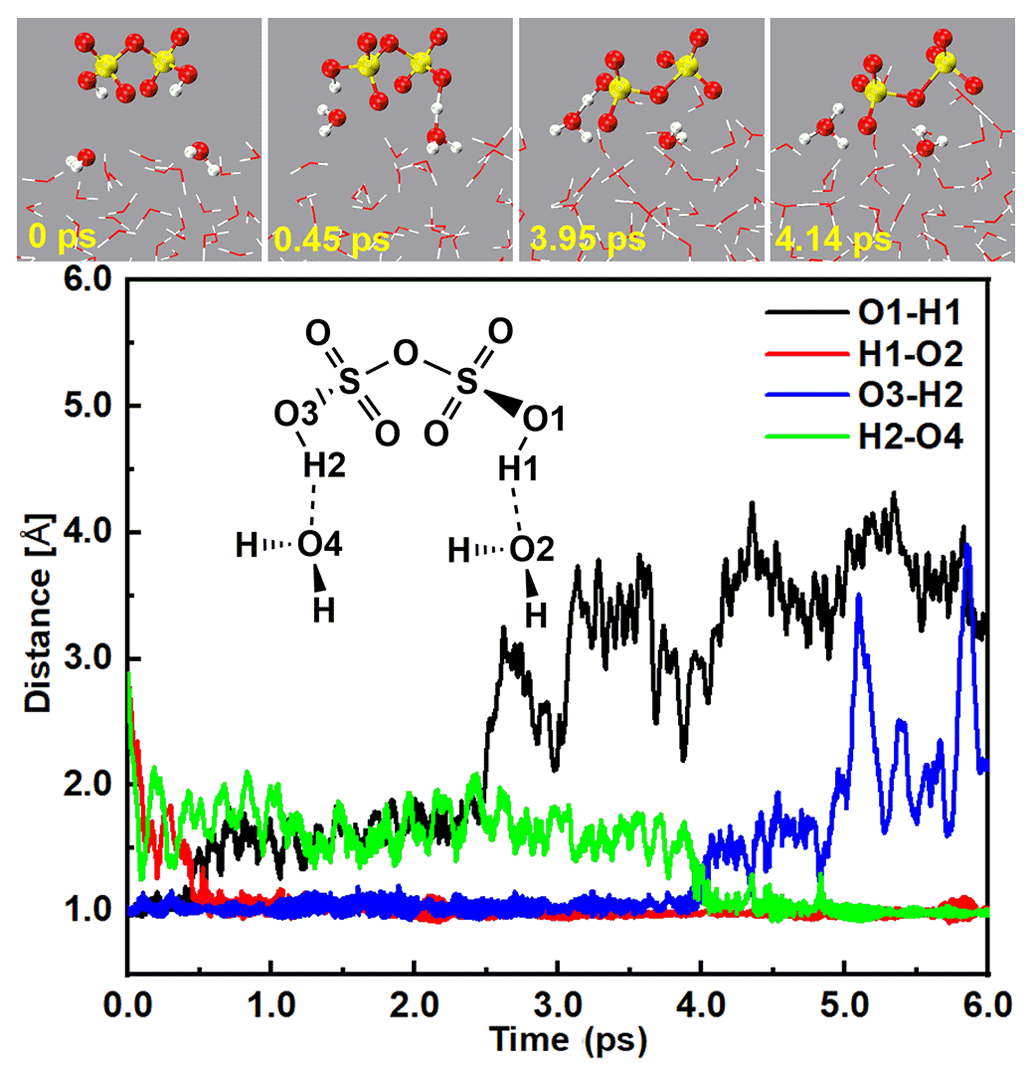
Figure 4Top panel: snapshot structures taken from the BOMD simulations, which illustrate the deprotonation of H2S2O7 at the air water interface. Lower panel: time evolution of key bond distances (O1–HI, O1–H2, O3–H2, and H2–O4) involved in the hydration mechanism.
3.2.2 H2S2O7 dissociating on the water droplet
In addition to the gaseous SO3 colliding with SA− at the air–water interface, DSA, the product of the barrier-less reaction between SO3 and SA can further quickly react with the interfacial water molecule at the air–water interface. As seen in Figs. 4 and S7 and Movie S4, DSA was highly reactive at the air–water interface and can undergo two levels of deprotonation to form the ion. Specifically, the DSA can firstly form a H bond with the interfacial water molecule at 0.45 ps. After that, the H1 atom of DSA transferred to interfacial water and produced and H3O+ ions. The formed ion can survive for ∼ 3 ps on water droplet. At 4.14 ps, the H2 atom of the ion moved to the O4 atom of a nearby interfacial water molecule and formed the ion pair, which was stable at the air–water interface over a simulated timescale of 10 ps. Note that the second deprotonation of DSA indeed needs more time than its first deprotonation as the pKa1 (pKa1 = −16.05) of DSA is much smaller than its pKa2 (pKa2 = −4.81) (Abedi and Farrokhpour, 2013). In brief, at the air–water interface, both of these routes of the formation of the ion pair occurred on the picosecond timescale.
3.3 Atmospheric implications
3.3.1 Application of the SO3 + SA reaction in atmospheric chemistry
In the gas phase, the main sink route of SO3 was H2O-assisted hydrolysis of SO3 (Morokuma and Muguruma, 1994; Akhmatskaya et al., 1997; Larson et al., 2000; Hazra and Sinha, 2011; Long et al., 2013a; Torrent-Sucarrat et al., 2012; Ma et al., 2020). To study the atmospheric importance of the SO3 + SA reaction without and with H2O, the rate ratio () between the SO3 + SA reaction and H2O-assisted hydrolysis of SO3 was compared, which is expressed in Eq. (5).
Table 2The rate ratio between the SO3 + H2SO4 reaction and the hydrolysis of SO3 at different altitudes in the atmosphere of Venus.

kDSA, kDSA_WM_s, and kSA_SA are, respectively, the rate constants for the SO3 + H2SO4 reaction, the H2O-assisted SO3 + H2SO4 reaction occurring through the stepwise route, and the hydrolysis reaction of SO3 + (H2O)2.
In Eq. (5), Keq1 and Keq2 are the equilibrium constants for the formation of H2SO4⋯H2O and SO3⋯H2O complexes shown in Table S2, respectively; kDSA, kDSA_WM_s, and kSA_WM respectively denote the bimolecular rate coefficient for the H2SO4 + SO3, H2SO4⋯H2O + SO3, and SO3⋯H2O + H2O reactions; and [H2O] and [H2SO4] respectively represent the concentration of H2O and SA taken from references (Anglada et al., 2013; Liu et al., 2015). The value of was listed in Table S7 (0 km altitude) and Table S8 (5–30 km altitude). As seen in Table S7, the hydrolysis reaction of SO3 with (H2O)2 dominates over the SO3 + H2SO4 reaction at 0 km altitude as the [H2O] (1016–1018 molec. cm3) was much larger than that of [H2SO4] (104–108 molec. cm3). Although the concentration of water molecules decreases with the increase in altitude in Table S8, the concentration of [H2O] is still much greater than that of [H2SO4], resulting in the SO3 + H2SO4 reaction not being able to compete with the H2O-assisted hydrolysis of SO3 within the altitude range of 5–30 km. Even considering the high H2SO4 concentration at the end and outside the aircraft engine and at a flight altitude of 10 km (Curtius et al., 2002), the SO3 + H2SO4 reaction was not the major sink route of SO3. Notably, as the concentration of sulfuric acid was even greater than that of water vapor in the atmosphere of Venus, the SO3 + SA reaction was probably more favorable than the H2O-assisted hydrolysis of SO3 in Venus' atmosphere. To check whether the SO3 + H2SO4 reaction was more favorable than H2O-assisted hydrolysis of SO3 or not in Venus' atmosphere, the rate ratio of listed in Eq. (4) has been calculated in Table 2. It can be seen from Table 2 that the rate ratio of was 3.24 × 108–5.23 × 1010 within the altitude range of 40–70 km in Venus' atmosphere, which indicates that the SO3 + H2SO4 reaction is significantly more favorable than the hydrolysis reaction of SO3 + (H2O)2 within the altitude range of 40–70 km in Venus' atmosphere.
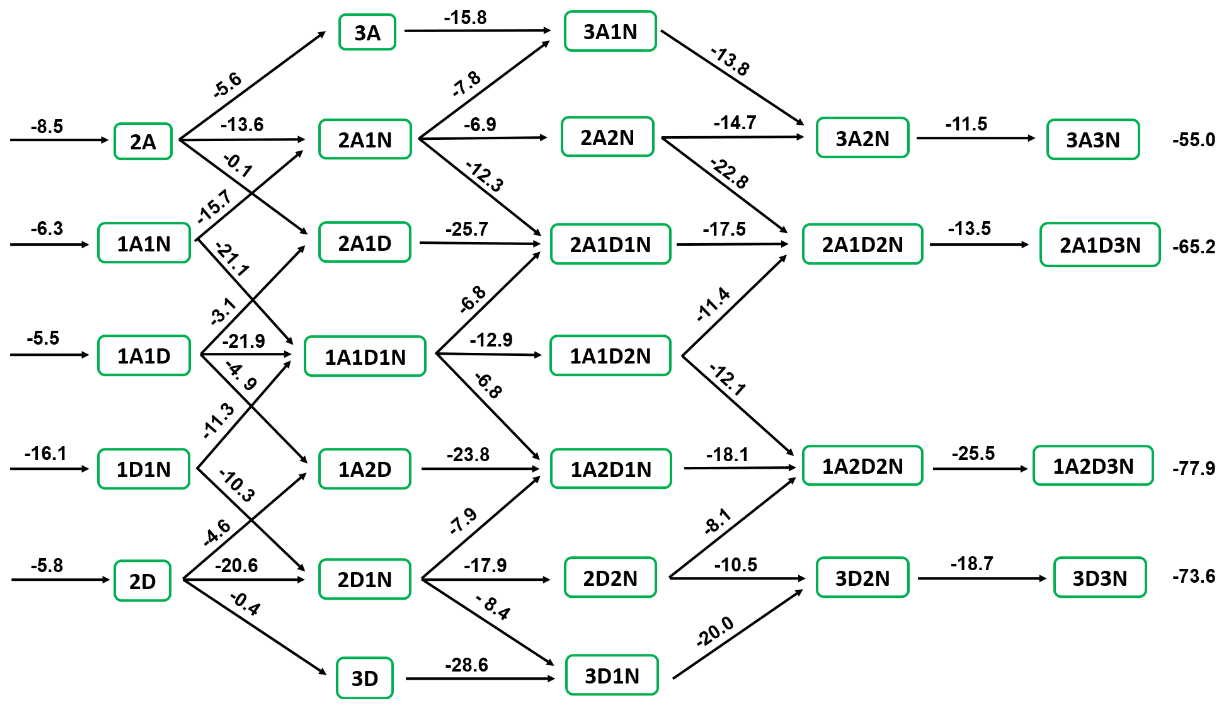
Figure 5The Gibbs free energy (kcal mol−1) diagram of (DSA)x(SA)y(A)z () clusters at 278.15 K and 1 atm. “A” refers to sulfuric acid, “D” refers to disulfuric acid, and “N” refers to ammonia.
3.3.2 Enhancement effect of DSA on NPF
From the multistep global minimum sampling technique, for (DSA)x(SA)y(A)z () molecular clusters, the 27 most stable structures in the present system have been found (Fig. S11 in the Supplement). To evaluate the thermodynamic stability of these clusters, Gibbs formation free energies (ΔG) at 278.15 K and evaporation rate coefficient (γ, s−1) for (DSA)x(SA)y(A)z () molecular clusters were calculated in Fig. 5 and Tables S11 and S12 in the Supplement, respectively. As for dimers formed by SA, A, and DSA, the ΔG of (A)1⋅(DSA)1 was −16.1 kcal mol−1, which was lowest in all dimers followed by (SA)2 (−8.5 kcal mol−1) and then (SA)1⋅(A)1 (−6.3 kcal mol−1), meanwhile, the γ of (A)1⋅(DSA)1 (1.17 × 10−3 s−1) was lower than that of (SA)2 (3.81 × 102 s−1) and (SA)1⋅(A)1 (4.19 × 104 s−1). Regarding the SA–A–DSA-based clusters, the values of ΔG and γ of SA–A–DSA-based clusters containing more DSA molecules were relatively lower than the corresponding values of other SA–A–DSA-based clusters with the same number of acid and base molecules. In the free-energy diagram for cluster formation steps of the SA–A–DSA system (Fig. 5), thermodynamic barriers were weakened mainly by the subsequential addition of the A or DSA monomer. Also, the SA–A–DSA-based growth pathway was thermodynamically favorable, with decreasing ΔG. These results indicate that DSA not only can promote the stability of SA–A–DSA-based clusters but also may synergistically participate in the nucleation process.
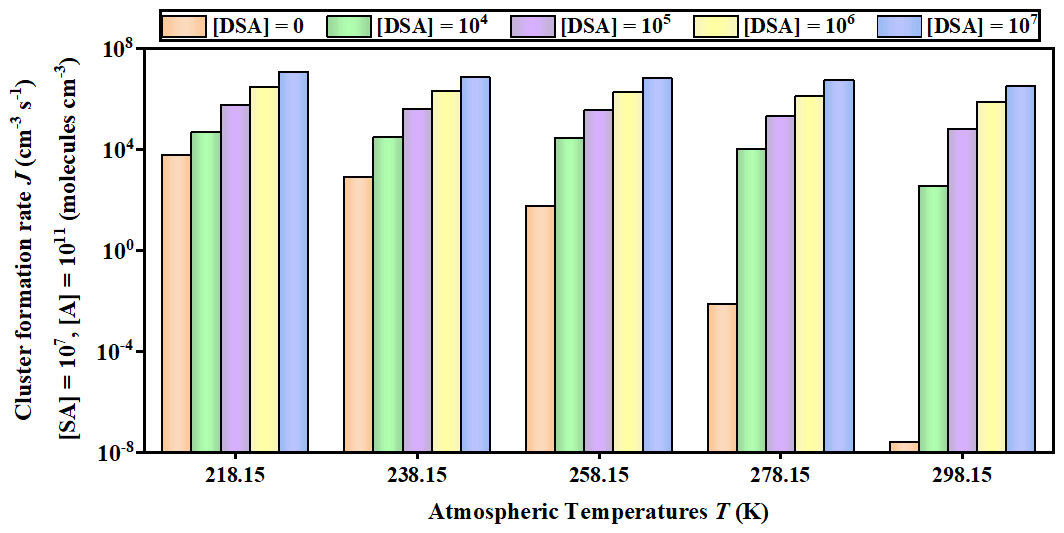
Figure 6Cluster formation rates J () against the of DSA monomer concentration (unit: ) under different temperatures (218.15, 238.15, 258.15, 278.15, and 298.15 K), where [SA] = 107 and [A] = 109 .
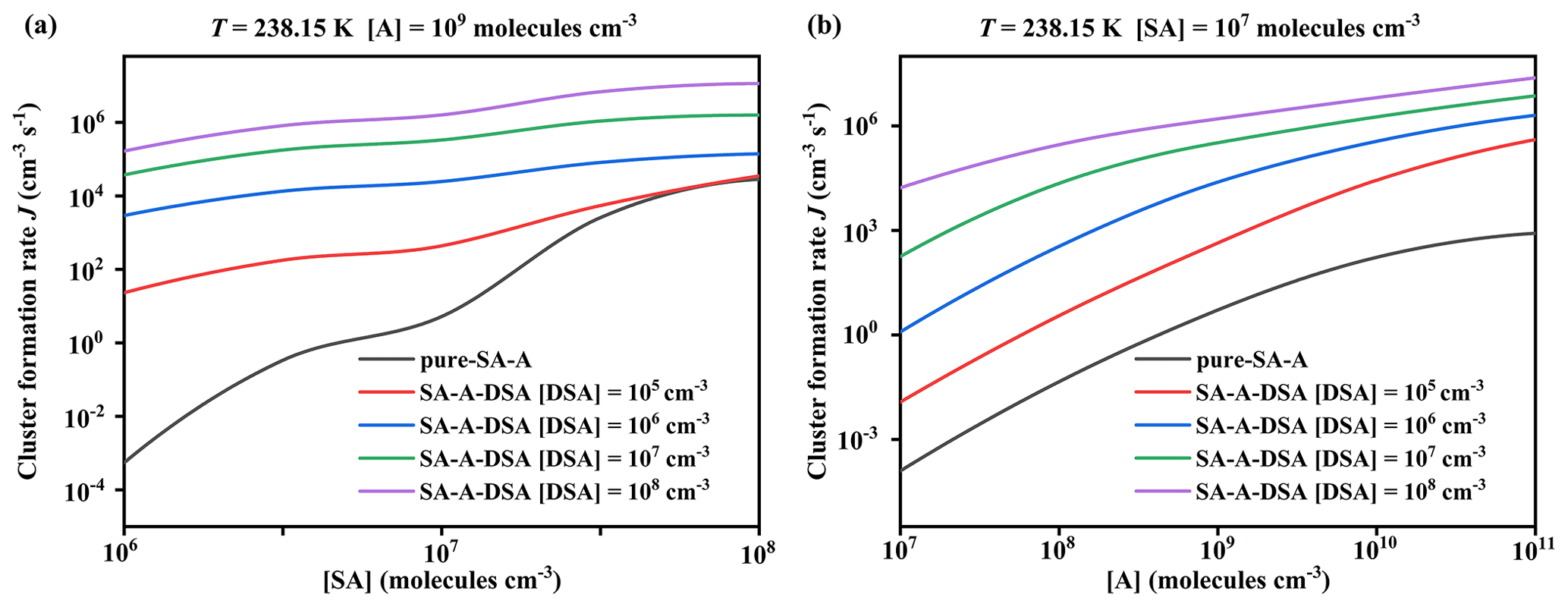
Figure 7Simulated cluster formation rates J () as a function of (a) [SA] and (b) [A], with different concentrations of disulfuric acid [DSA] of 104 (red), 105 (blue), 106 (green), 107 (purple), and 0 (black; pure SA–A), at T = 238.15 K.
The potential enhancement influence of DSA on the SA–A-based particle formation was shown in Fig. 6. The formation rate (J; ) of the SA–A–DSA-based system illustrated in Fig. 6 was negatively dependent on temperature, demonstrating that the low temperature is a key factor to accelerate cluster formation. It is noted that, at low temperatures of 218.15 K (Fig. S12 in the Supplement) and 238.15 K (Fig. S13 in the Supplement), the actual ΔG of clusters has been calculated to ensure meaningful cluster dynamics of the 3 × 3 systems, where the actual ΔG surface represented that the simulated set of clusters always included the critical cluster. In addition to temperature, the J of the SA–A–DSA-based system shown in Fig. 6 rises with the increase in [DSA]. More notably, the participation of DSA can promote J to a higher level, indicating its enhancement in SA–A nucleation. In addition, there was significantly positive dependence of the J of the SA–A–DSA-based system on both [SA] and [A] in Fig. 7 (238.15 K) and Figs. S15–S18 in the Supplement (218.15, 258.15, 278.15, and 298.15 K). This was because the higher concentration of nucleation precursors could lead to higher J. In addition, Fig. S19 in the Supplement showed the nucleation rate when the sum ([SA] + [DSA]) was kept constant. under substituted conditions was higher than that under unsubstituted conditions. These results indicated that DSA can greatly enhance the SA–A particle nucleation in the atmospheric boundary layer heavily polluted by sulfur oxide, especially at an average flight altitude of 10 km with high [DSA].
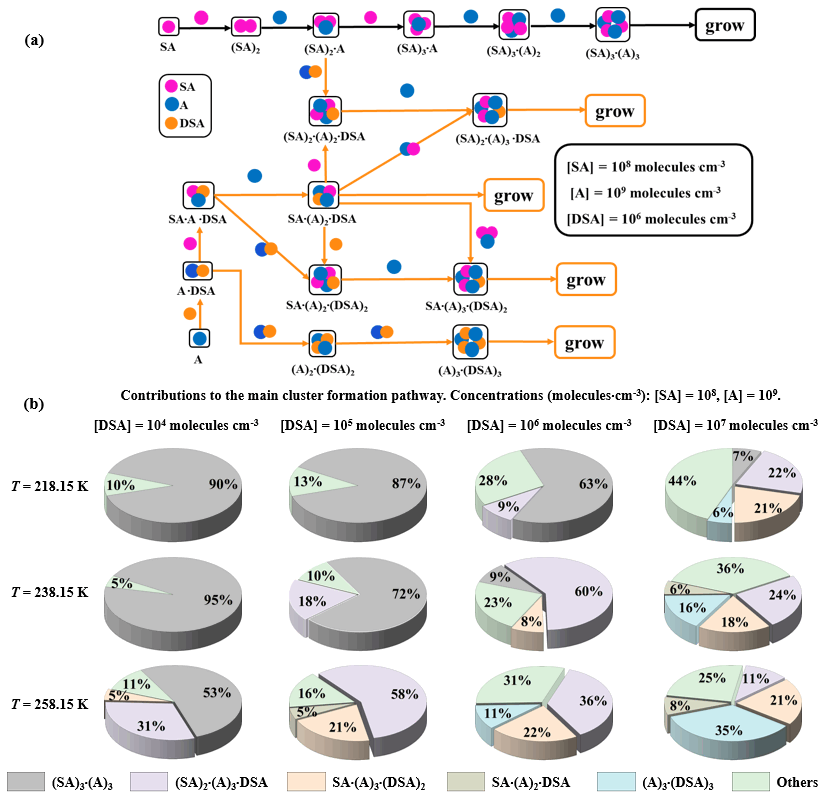
Figure 8(a) The main pathways of clusters growing out of the research system under conditions with 218.15, 238.15, and 258.15 K where [SA] = 108 , [A] = 109 , and [DSA] = 106 . (b) The contribution of different concentrations of DSA to the main cluster formation pathway at 218.15, 238.15, and 258.15 K is shown in the pie charts.
Two main cluster formation pathways, the pure SA–A-based cluster (i) and the DSA-containing cluster (ii), at different [DSA] and different temperatures (218.15, 238.15, and 258.15 K), are shown in Fig. 8a. As seen, the DSA molecule exhibited an ability to directly participate in cluster formation under high [SA] and [DSA] and median [A]. Interestingly, at different temperatures and different [DSA], the DSA molecule showed a different effect mechanism and contribution in the SA–A system. As seen in Fig. 8b and Fig. S20b in the Supplement, the cluster growth pathways were dominated by DSA-containing cluster formation under the conditions of 238.15 K ([DSA] is 106–107 ), 258.15 K ([DSA] is 105–107 ), 278.15 K ([DSA] is 104–107 ), and 298.15 K ([DSA] is 104–107 ). The cluster growth pathways were completely dominated by the DSA-containing cluster at 298.15 K, where [DSA] = 105–107 , and its contribution for growth flux out of the system reached 100 % (Fig. S22 in the Supplement). In short, on the one hand, the contribution of the DSA participation pathway has been increased with increasing temperature. On the other hand, the contribution of the pathway with participation of DSA increased with increasing [DSA], while the number of DSA molecules contained in clusters [, , , and (A)3⋅(DSA)3] that can contribute to cluster growth had a positive correlation with [DSA]. These results suggested that DSA has the ability to act as a potential contributor to SA–A-based NPF in the atmosphere at low T, low [SA], high [A], and high [DSA], and the DSA participation pathway can be dominant in the atmospheric boundary layer that is heavily polluted by sulfur oxide in the seasons of late autumn and early winter.
Table 3Gibbs free energy (ΔG, kcal mol−1) for the formation of , , , H3O+⋯NH3, H3O+⋯H2SO4, , , , H2SO4⋯NH3, , HOOCCH2COOH⋯H2SO4⋯NH3, HOCCOOSO3H⋯H2SO4⋯NH3, CH3OSO3H⋯H2SO4⋯NH3, and HOOCCH2CH(NH2)COOH⋯H2SO4⋯NH3 at 298 K.
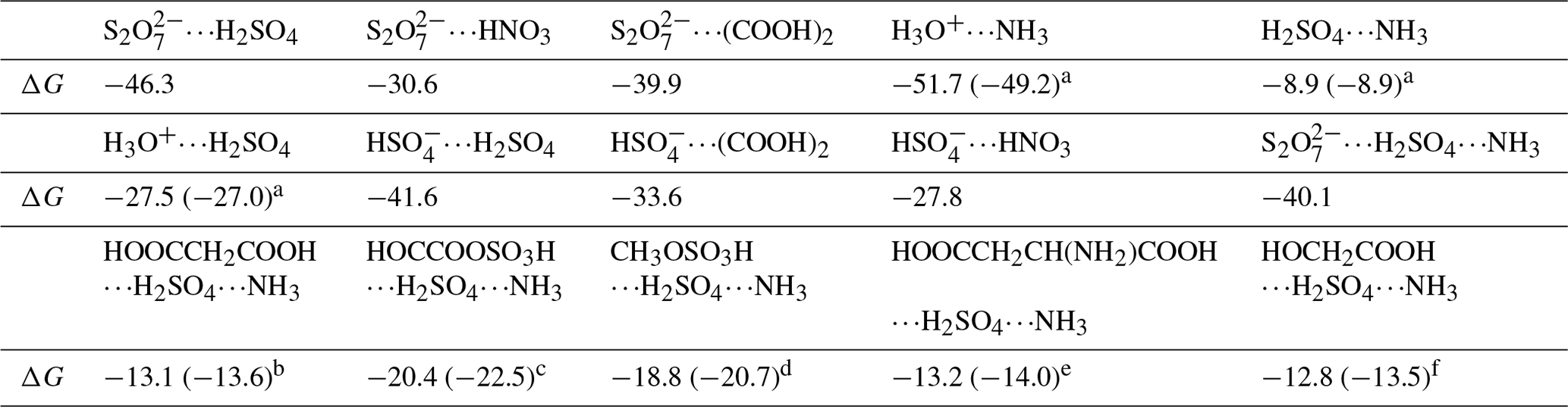
Energies are given in kcal mol−1 and calculated at the M06-2X/6-311G(2df,2pd) theoretical level. References are as follows: a Zhong et al. (2019). b Zhang et al. (2018). c Rong et al. (2020). d Gao et al. (2023). e J. Liu et al. (2021). f Zhang et al. (2017).
At the air–water interface, an important implication of the BOMD simulations was that the reaction between SO3 and SA at the air–water interface can be accomplished within a few picoseconds, whereby the interfacial water molecules played a significant role in promoting the formation of and ion pairs. Furthermore, the adsorption capacity of , H3O+, and SA− to gaseous precursors in the atmosphere was further investigated by the calculated interaction free energies. Herein, the species of SA, NH3, HNO3, and (COOH)2 have been regarded as the candidate species (Kulmala et al., 2004; Kirkby et al., 2011). Our calculated Gibbs free energies in Table 3 show that the interactions of , , , H3O+⋯NH3, H3O+⋯H2SO4, SA−⋯H2SO4, SA−⋯(COOH)2, and SA−⋯HNO3 were stronger than those of H2SO4⋯NH3 (major precursor of atmospheric aerosols), with their binding free energies enhanced by 18.6–42.8 kcal mol−1. These results reveal that interfacial , SA−, and H3O+ can attract candidate species from the gas phase to the water surface. Moreover, we evaluated whether could lead to increased particle growth in the SA–A cluster by considering geometrical structure and the formation free energies of the clusters. Compared with (SA)1(A)1(X)1 (X = HOOCCH2COOH, HOCCOOSO3H, CH3OSO3H, HOOCCH2CH(NH2)COOH and HOCH2COOH) clusters (Zhong et al., 2019; Zhang et al., 2018; Rong et al., 2020; Gao et al., 2023; J. Liu et al., 2021; Zhang et al., 2017), the number of hydrogen bonds in the cluster presented in Fig. S8 increased, and the ring of the complex was enlarged. It was demonstrated that has the highest potential to stabilize SA–A clusters and promote SA–A nucleation in these clusters due to its acidity and structural factors, such as more intermolecular hydrogen bond binding sites. Subsequently, compared to (SA)1(A)1(X)1 clusters (Table 2), the Gibbs formation free energy ΔG of cluster was lower, showing that the ion at the air–water interface has stronger nucleation ability than X in the gas phase. Therefore, we predict that at the air–water interface would lead to increased particle growth.
In this work, we employed QC calculations, BOMD simulations, and the ACDC kinetic model to characterize the SO3–H2SO4 interaction in the gas phase and at the air–water interface to study the effect of H2S2O7 on H2SO4–NH3-based clusters. Results revealed that the energy barrier of the gas-phase SO3 + H2SO4 reaction without and with H2O was less than 2.3 kcal mol−1. Rate constants indicated that though the SO3 + H2SO4 reaction cannot compete with the H2O-assisted hydrolysis of SO3 within the temperature range of 280–320 K, its rate constant was close to the upper limits for bimolecular reactions, and H2O exerted an obvious catalytic role in promoting the reaction rate. Moreover, ACDC kinetic simulations showed that DSA has unexpected facilitate effects on the NPF process and can present a more obvious enhancement effect on SA–A-based cluster formation in the polluted atmospheric boundary layer. Of particular note is that DSA can directly participate in the SA–A-based cluster formation pathway and that the contribution of the pathway with participation of DSA increases with increasing [DSA] in regions with atmospheric pollution boundary layer of high concentrations of SO3, especially in late autumn and early winter.
At the air–water interface, H2O induced the formation of the ion pair, SA− mediated the formation of the ion pair, and the deprotonation of H2S2O7 was observed; these can occur within a few picoseconds. The formed interfacial , SA−, and H3O+ can attract candidate species (such as H2SO4, NH3, and HNO3) for particle formation from the gas phase to the water surface and thus accelerate the growth of particle. Moreover, the potential of X (X = , HOOCCH2COOH, HOCCOOSO3H, CH3OSO3H, HOOCCH2CH(NH2)COOH and HOCH2COOH) in the ternary SA–A–X cluster formation indicated that has the highest potential to stabilize SA–A clusters and promote SA–A nucleation in X.
The present work will expand our understanding of new pathways for the loss of SO3 in acidic polluted areas. Moreover, this work will also help to reveal some missing sources of NPF in metropolitan industrial regions and to understand the atmospheric organic–sulfur cycle more comprehensively.
All data presented in this study are available upon request from the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-4029-2024-supplement.
RW: methodology, validation, investigation, writing (original draft), funding acquisition. YC: writing (review), conceptualization, methodology, investigation. SC: writing (review), data computation. RL: data curation, data supplementation. YH: data curation, visualization, investigation. XG: data curation, formal analysis, investigation. TZ: writing (review and editing), project administration, funding acquisition. FS: methodology, formal analysis, funding acquisition. HL: writing (review and editing), project administration, formal analysis.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
This work was supported by the National Natural Science Foundation of China (grant nos. 22203052, 22073059, and 22006158), the Education Department of Shaanxi Provincial Government (grant no. 23JC023), the Key Cultivation Project of Shaanxi University of Technology (grant no. SLG2101), and the Special Scientific Research Project of Hanzhong City-Shaanxi University of Technology Co-construction State Key Laboratory (SXJ-2106). The authors sincerely thank Qingzhu Zhang and Fei Xu from Shandong University for their assistance in calculating the air–water interface reaction.
This research has also been supported by the Education Department of Shaanxi Provincial Government (no. 23JC023) and the Special Scientific Research Project of Hanzhong City-Shaanxi University of Technology Co-construction State Key Laboratory (SXJ-2106).
This paper was edited by Ari Laaksonen and reviewed by two anonymous referees.
Abedi, M. and Farrokhpour, H.: Acidity constants of some sulfur oxoacids in aqueous solution using CCSD and MP2 methods, Dalton T., 42, 5566–5572, https://doi.org/10.1039/C3DT33056G, 2013.
Adler, T. B., Knizia, G., and Werner, H. J.: A simple and efficient CCSD(T)-F12 approximation, J. Chem. Phys., 127, 221106, https://doi.org/10.1063/1.2817618, 2007.
Akhmatskaya, E., Apps, C., Hillier, I., Masters, A., Palmer, I., Watt, N., Vincent, M., and Whitehead, J.: Hydrolysis of SO3 and ClONO2 in water clusters: A combined experimental and theoretical study, J. Am. Chem. Soc., 93, 2775–2779, 1997.
Almeida, J., Schobesberger, S., Kürten, A., Ortega, I. K., Kupiainen-Määttä, O., Praplan, A. P., Adamov, A., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Donahue, N. M., Downard, A., Dunne, E., Duplissy, J., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Henschel, H., Jokinen, T., Junninen, H., Kajos, M., Kangasluoma, J., Keskinen, H., Kupc, A., Kurtén, T., Kvashin, A. N., Laaksonen, A., Lehtipalo, K., Leiminger, M., Leppä, J., Loukonen, V., Makhmutov, V., Mathot, S., McGrath, M. J., Nieminen, T., Olenius, T., Onnela, A., Petäjä, T., Riccobono, F., Riipinen, I., Rissanen, M., Rondo, L., Ruuskanen, T., Santos, F. D., Sarnela, N., Schallhart, S., Schnitzhofer, R., Seinfeld, J. H., Simon, M., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Tröstl, J., Tsagkogeorgas, G., Vaattovaara, P., Viisanen, Y., Virtanen, A., Vrtala, A., Wagner, P. E., Weingartner, E., Wex, H., Williamson, C., Wimmer, D., Ye, P., Yli-Juuti, T., Carslaw, K. S., Kulmala, M., Curtius, J., Baltensperger, U., Worsnop, D. R., Vehkamäki, H., and Kirkby, J.: Molecular understanding of sulphuric acid–amine particle nucleation in the atmosphere, Nature, 502, 359–363, https://doi.org/10.1038/nature12663, 2013.
Anglada, J. M., Hoffman, G. J., Slipchenko, L. V., M. Costa, M., Ruiz-Lopez, M. F., and Francisco, J. S.: Atmospheric significance of water clusters and ozone–water complexes, J. Phys. Chem. A, 117, 10381–10396, 2013.
Bandyopadhyay, B., Kumar, P., and Biswas, P.: Ammonia Catalyzed Formation of Sulfuric Acid in Troposphere: The Curious Case of a Base Promoting Acid Rain, J. Phys. Chem. A, 121, 3101–3108, https://doi.org/10.1021/acs.jpca.7b01172, 2017.
Becke, A. D.: Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 38, 3098–3100, 1988.
Bouo, F.-X., Kouamé, J., Tchétché, Y., Kré, R., Moussé, M., Assamoi, P., Cautenet, S., and Cautenet, G.: Redistribution of free tropospheric chemical species over West Africa: Radicals (OH and HO2), peroxide (H2O2) and acids (HNO3 and H2SO4), Chemosphere, 84, 1617–1629, 2011.
Calvert, J. G., Lazrus, A., Kok, G. L., Heikes, B. G., Walega, J. G., Lind, J., and Cantrell, C. A.: Chemical mechanisms of acid generation in the troposphere, Nature, 317, 27–35, 1985.
Cao, Y., Zhou, H., Jiang, W., Chen, C. W., and Pan, W. P.: Studies of the fate of sulfur trioxide in coal-fired utility boilers based on modified selected condensation methods, Environ. Sci. Technol., 44, 3429–3434, 2010.
Chen, L. and Bhattacharya, S.: Sulfur emission from Victorian brown coal under pyrolysis, oxy-fuel combustion and gasification conditions, Environ. Sci. Technol., 47, 1729–1734, 2013.
Chen, T. and Plummer, P. L.: Ab initio MO investigation of the gas-phase reaction sulfur trioxide + water .fwdarw. sulfuric acid, J. Phys. Chem. A, 89, 3689–3693, 1985.
Chen, X., Tao, C., Zhong, L., Gao, Y., Yao, W., and Li, S.: Theoretical study on the atmospheric reaction of SO2 with the HO2 and HO2⋅H2O complex formation HSO4 and H2SO3, Chem. Phys. Lett., 608, 272–276, 2014.
Cheng, Y., Ding, C., Wang, H., Zhang, T., Wang, R., Muthiah, B., Xu, H., Zhang, Q., and Jiang, M.: Significant influence of water molecules on the SO3 + HCl reaction in the gas phase and at the air–water interface, Phys. Chem. Chem. Phys., 25, 28885–28894, https://doi.org/10.1039/D3CP03172A, 2023.
Curtius, J., Arnold, F., and Schulte, P.: Sulfuric acid measurements in the exhaust plume of a jet aircraft in flight: Implications for the sulfuric acid formation efficiency, Geophys. Res. Lett., 29, 17-11–1714, 2002.
Elm, J., Bilde, M., and Mikkelsen, K. V.: Assessment of density functional theory in predicting structures and free energies of reaction of atmospheric prenucleation clusters, J. Chem. Theory Comput., 8, 2071–2077, 2012.
Elm, J., Bilde, M., and Mikkelsen, K. V.: Influence of nucleation precursors on the reaction kinetics of methanol with the OH radical, J. Phys. Chem. A, 117, 6695–6701, 2013.
England, G. C., Zielinska, B., Loos, K., Crane, I., and Ritter, K.: Characterizing PM2. 5 emission profiles for stationary sources: comparison of traditional and dilution sampling techniques, Fuel Process. Technol., 65, 177–188, 2000.
Finlayson-Pitts, B. J. and Pitts Jr, J. N.: Atmospheric chemistry. Fundamentals and experimental techniques, John Wiley and Sons, New York, https://doi.org/10.1029/EO068i049p01643-01, 1986.
Hratchian, H. P., Izmaylov, A. F., Bloino, J., Zheng, G., Sonnenberg, J. L., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M., Heyd, J. J., Brothers, E., Kudin, K. N., Staroverov, V. N., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Rega, N., Millam, J. M., Klene, M., Knox, J. E., Cross, J. B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R. E., Yazyev, O., Austin, A. J., Cammi, R., Pomelli, C., Ochterski, J. W., Martin, R. L., Morokuma, K., Zakrzewski, V. G., Voth, G. A., Salvador, P., Dannenberg, J. J., Dapprich, S., Daniels, A. D., Farkas, Ö., Foresman, J. B., Ortiz, J. V., Cioslowski, J., and Fox, D. J.: Gaussian 09 Revision D.01, Gaussian Inc., Wallingford, CT, http://www.gaussian.com (last access: December 2023), 2009.
Gao, J., Wang, R., Zhang, T., Liu, F., and Wang, W.: Effect of methyl hydrogen sulfate on the formation of sulfuric acid–ammonia clusters: A theoretical study, J. Chin. Chem. Soc.-Taip., 70, 689–698, https://doi.org/10.1002/jccs.202200148, 2023.
Glowacki, D. R., Liang, C.-H., Morley, C., Pilling, M. J., and Robertson, S. H.: MESMER: an open-source master equation solver for multi-energy well reactions, J. Phys. Chem. A, 116, 9545–9560, 2012.
Goedecker, S., Teter, M., and Hutter, J.: Separable dual-space Gaussian pseudopotentials, Phys. Rev. B, 54, 1703, 1996.
Gonzalez, J., Torrent-Sucarrat, M., and Anglada, J. M.: The reactions of SO3 with HO2 radical and H2O⋯HO2 radical complex. Theoretical study on the atmospheric formation of HSO5 and H2SO4, Phys. Chem. Chem. Phys., 12, 2116–2125, 2010.
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H.: A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu, J. Chem. Phys., 132, 154104, 2010.
Hartwigsen, C., Goedecker, S., and Hutter, J.: Relativistic separable dual-space Gaussian pseudopotentials from H to Rn, Phys. Rev. B, 58, 3641–3662, 1998.
Haywood, J. and Boucher, O.: Estimates of the direct and indirect radiative forcing due to tropospheric aerosols: A review, Rev. Geophys., 38, 513–543, 2000.
Hazra, M. K. and Sinha, A.: Formic acid catalyzed hydrolysis of SO3 in the gas phase: A barrierless mechanism for sulfuric acid production of potential atmospheric importance, J. Am. Chem. Soc., 133, 17444–17453, 2011.
Hofmann, M. and Schleyer, P. v. R.: Acid rain: Ab initio investigation of the H2O⋅SO3 complex and its conversion to H2SO4, J. Am. Chem. Soc., 116, 4947–4952, 1994.
Horváth, G., Horváth, I., Almousa, S. A.-D., and Telek, M.: Numerical inverse Laplace transformation using concentrated matrix exponential distributions, Perform. Evaluation, 137, 102067, https://doi.org/10.1016/j.peva.2019.102067, 2020.
Huff, A. K., Mackenzie, R. B., Smith, C. J., and Leopold, K. R.: Facile Formation of Acetic Sulfuric Anhydride: Microwave Spectrum, Internal Rotation, and Theoretical Calculations, J. Phys. Chem. A, 121, 5659–5664, https://doi.org/10.1021/acs.jpca.7b05105, 2017.
Hutter, J., Iannuzzi, M., Schiffmann, F., and VandeVondele, J.: cp2k: atomistic simulations of condensed matter systems, WIREs Comput. Mol. Sci., 4, 15–25, 2014.
Kanno, N., Tonokura, K., and Koshi, M.: Equilibrium constant of the HO2-H2O complex formation and kinetics of HO2 + HO2-H2O: Implications for tropospheric chemistry, J. Geophys. Res.-Atmos., 111, D20312, https://doi.org/10.1029/2005jd006805, 2006.
Kikuchi, R.: Environmental management of sulfur trioxide emission: impact of SO3 on human health, Environ. Manage., 27, 837–844, 2001.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011.
Knizia, G., Adler, T. B., and Werner, H.-J.: Simplified CCSD(T)-F12 methods: Theory and benchmarks, J. Chem. Phys., 130, 054104, https://doi.org/10.1063/1.3054300, 2009.
Kuang, C., McMurry, P. H., McCormick, A. V., and Eisele, F.: Dependence of nucleation rates on sulfuric acid vapor concentration in diverse atmospheric locations, J. Geophys. Res.-Atmos., 113, D10209, https://doi.org/10.1029/2007JD009253, 2008.
Kuczkowski, R. L., Suenram, R. D., and Lovas, F. J.: Microwave spectrum, structure, and dipole moment of sulfuric acid, J. Am. Chem. Soc., 103, 2561–2566, 1981.
Kulmala, M., Vehkamäki, H., Petäjä, T., Dal Maso, M., Lauri, A., Kerminen, V. M., Birmili, W., and McMurry, P. H.: Formation and growth rates of ultrafine atmospheric particles: a review of observations, J. Aerosol Sci., 35, 143–176, https://doi.org/10.1016/j.jaerosci.2003.10.003, 2004.
Kumar, M., Zhong, J., Francisco, J. S., and Zeng, X. C.: Criegee intermediate-hydrogen sulfide chemistry at the air/water interface, Chem. Sci., 8, 5385–5391, 2017.
Kumar, M., Zhong, J., Zeng, X. C., and Francisco, J. S.: Reaction of Criegee Intermediate with Nitric Acid at the Air–Water Interface, J. Am. Chem. Soc., 140, 4913–4921, https://doi.org/10.1021/jacs.8b01191, 2018.
Larson, L. J., Kuno, M., and Tao, F.-M.: Hydrolysis of sulfur trioxide to form sulfuric acid in small water clusters, J. Chem. Phys., 112, 8830–8838, 2000.
Lee, C., Yang, W., and Parr, R. G.: Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B, 37, 785–789, 1988.
Li, H., Zhong, J., Vehkamäki, H., Kurtén, T., Wang, W., Ge, M., Zhang, S., Li, Z., Zhang, X., Francisco, J. S., and Zeng, X. C.: Self-Catalytic Reaction of SO3 and NH3 To Produce Sulfamic Acid and Its Implication to Atmospheric Particle Formation, J. Am. Chem. Soc., 140, 11020–11028, https://doi.org/10.1021/jacs.8b04928, 2018.
Li, K., Song, X., Zhu, T., Wang, C., Sun, X., Ning, P., and Tang, L.: Mechanistic and kinetic study on the catalytic hydrolysis of COS in small clusters of sulfuric acid, Environ. Pollut., 232, 615–623, https://doi.org/10.1016/j.envpol.2017.10.004, 2018.
Li, L., Kumar, M., Zhu, C., Zhong, J., Francisco, J. S., and Zeng, X. C.: Near-barrierless ammonium bisulfate formation via a loop-structure promoted proton-transfer mechanism on the surface of water, J. Am. Chem. Soc., 138, 1816–1819, 2016.
Liu, J., Fang, S., Wang, Z., Yi, W., Tao, F. M., and Liu, J. Y.: Hydrolysis of sulfur dioxide in small clusters of sulfuric acid: Mechanistic and kinetic study, Environ. Sci. Technol., 49, 13112–13120, 2015.
Liu, J., Liu, L., Rong, H., and Zhang, X.: The potential mechanism of atmospheric new particle formation involving amino acids with multiple functional groups, Phys. Chem. Chem. Phys., 23, 10184–10195, https://doi.org/10.1039/D0CP06472F, 2021.
Liu, L., Zhong, J., Vehkamäki, H., Kurtén, T., Du, L., Zhang, X., Francisco, J. S., and Zeng, X. C.: Unexpected quenching effect on new particle formation from the atmospheric reaction of methanol with SO3, P. Natl. Acad. Sci. USA, 116, 24966–24971, 2019.
Liu, L., Yu, F., Tu, K., Yang, Z., and Zhang, X.: Influence of atmospheric conditions on the role of trifluoroacetic acid in atmospheric sulfuric acid–dimethylamine nucleation, Atmos. Chem. Phys., 21, 6221–6230, https://doi.org/10.5194/acp-21-6221-2021, 2021.
Loerting, T. and Liedl, K. R.: Toward elimination of discrepancies between theory and experiment: The rate constant of the atmospheric conversion of SO3 to H2SO4, P. Natl. Acad. Sci. USA, 97, 8874–8878, 2000.
Lohmann, U. and Feichter, J.: Global indirect aerosol effects: a review, J. Atmos. Chem. Phys., 5, 715–737, 2005.
Long, B., Long, Z. W., Wang, Y.-b., Tan, X. F., Han, Y. H., Long, C. Y., Qin, S. J., and Zhang, W. J.: Formic Acid Catalyzed Gas-Phase Reaction of H2O with SO3 and the Reverse Reaction: A Theoretical Study, ChemPhysChem, 13, 323–329, https://doi.org/10.1002/cphc.201100558, 2012.
Long, B., Chang, C. R., Long, Z. W., Wang, Y. B., Tan, X. F., and Zhang, W. J.: Nitric acid catalyzed hydrolysis of SO3 in the formation of sulfuric acid: A theoretical study, Chem. Phys. Lett., 581, 26–29, 2013a.
Long, B., Tan, X. F., Chang, C. R., Zhao, W. X., Long, Z. W., Ren, D. S., and Zhang, W. J.: Theoretical studies on gas-phase reactions of sulfuric acid catalyzed hydrolysis of formaldehyde and formaldehyde with sulfuric acid and H2SO4⋯H2O complex, J. Phys. Chem. A, 117, 5106–5116, 2013b.
Lv, G. and Sun, X. M.: The role of air–water interface in the SO3 hydration reaction, Atmos. Environ., 230, 117514, https://doi.org/10.1016/j.atmosenv.2020.117514, 2020.
Lv, G., Sun, X., Zhang, C., and Li, M.: Understanding the catalytic role of oxalic acid in SO3 hydration to form H2SO4 in the atmosphere, Atmos. Chem. Phys., 19, 2833–2844, https://doi.org/10.5194/acp-19-2833-2019, 2019.
Ma, X., Zhao, X., Ding, Z., Wang, W., Wei, Y., Xu, F., Zhang, Q., and Wang, W.: Determination of the amine-catalyzed SO3 hydrolysis mechanism in the gas phase and at the air–water interface, Chemosphere, 252, 126292, https://doi.org/10.1016/j.chemosphere.2020.126292, 2020.
Mackenzie, R. B., Dewberry, C. T., and Leopold, K. R.: Gas phase observation and microwave spectroscopic characterization of formic sulfuric anhydride, Science, 349, 58–61, 2015.
Mardirossian, N. and Head-Gordon, M.: How accurate are the Minnesota density functionals for noncovalent interactions, isomerization energies, thermochemistry, and barrier heights involving molecules composed of main-group elements?, J. Chem. Theory Comput., 12, 4303–4325, 2016.
McGrath, M. J., Olenius, T., Ortega, I. K., Loukonen, V., Paasonen, P., Kurtén, T., Kulmala, M., and Vehkamäki, H.: Atmospheric Cluster Dynamics Code: a flexible method for solution of the birth-death equations, Atmos. Chem. Phys., 12, 2345–2355, https://doi.org/10.5194/acp-12-2345-2012, 2012.
Miller, J. A. and Klippenstein, S. J.: Master equation methods in gas phase chemical kinetics, J. Phys. Chem. A, 110, 10528–10544, 2006.
Mitsui, Y., Imada, N., Kikkawa, H., and Katagawa, A.: Study of Hg and SO3 behavior in flue gas of oxy-fuel combustion system, Int. J. Greenh. Gas Con., 5, S143–S150, 2011.
Morokuma, K. and Muguruma, C.: Ab initio molecular orbital study of the mechanism of the gas phase reaction SO3 + H2O: Importance of the second water molecule, J. Am. Chem. Soc., 116, 10316–10317, 1994.
Neese, F.: The ORCA program system, WIREs Comput. Mol. Sci., 2, 73–78, https://doi.org/10.1002/wcms.81, 2012.
Otto, A. H. and Steudel, R.: Gas-Phase Structures and Acidities of the Sulfur Oxoacids H2SnO6 (n = 2–4) and H2S2O7, Eur. J. Inorg. Chem., 2001, 3047–3054, 2001.
Pérez-Ríos, J., Ragole, S., Wang, J., and Greene, C. H.: Comparison of classical and quantal calculations of helium three-body recombination, J. Chem. Phys., 140, 044307, https://doi.org/10.1063/1.4861851, 2014.
Pöschl, U.: Atmospheric aerosols: composition, transformation, climate and health effects, Angew. Chem. Int. Edit., 44, 7520–7540, 2005.
Pöschl, U. and Shiraiwa, M.: Multiphase chemistry at the atmosphere-biosphere interface influencing climate and public health in the anthropocene, Chem. Rev., 115, 4440–4475, 2015.
Renard, J. J., Calidonna, S. E., and Henley, M. V.: Fate of ammonia in the atmosphere—a review for applicability to hazardous releases, J. Hazard. Mater., 108, 29–60, 2004.
Riipinen, I., Sihto, S.-L., Kulmala, M., Arnold, F., Dal Maso, M., Birmili, W., Saarnio, K., Teinilä, K., Kerminen, V.-M., Laaksonen, A., and Lehtinen, K. E. J.: Connections between atmospheric sulphuric acid and new particle formation during QUEST III–IV campaigns in Heidelberg and Hyytiälä, Atmos. Chem. Phys., 7, 1899–1914, https://doi.org/10.5194/acp-7-1899-2007, 2007.
Rong, H., Liu, L., Liu, J., and Zhang, X.: Glyoxylic sulfuric anhydride from the gas-phase reaction between glyoxylic acid and SO3: a potential nucleation precursor, J. Phys. Chem. A, 124, 3261–3268, 2020.
Shampine, L. F. and Reichelt, M. W.: The MATLAB ODE Suite, J. Sci. Comput., 18, 1–22, https://doi.org/10.1137/s1064827594276424, 1997.
Sihto, S.-L., Kulmala, M., Kerminen, V.-M., Dal Maso, M., Petäjä, T., Riipinen, I., Korhonen, H., Arnold, F., Janson, R., Boy, M., Laaksonen, A., and Lehtinen, K. E. J.: Atmospheric sulphuric acid and aerosol formation: implications from atmospheric measurements for nucleation and early growth mechanisms, Atmos. Chem. Phys., 6, 4079–4091, https://doi.org/10.5194/acp-6-4079-2006, 2006.
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin III, R. L., Hyvärinen, A.-P., and Lihavainen, H.: The role of sulfuric acid in atmospheric nucleation, Science, 327, 1243–1246, 2010.
Smith, C. J., Huff, A. K., Mackenzie, R. B., and Leopold, K. R.: Observation of Two Conformers of Acrylic Sulfuric Anhydride by Microwave Spectroscopy, J. Phys. Chem. A, 121, 9074–9080, https://doi.org/10.1021/acs.jpca.7b09833, 2017.
Starik, A., Savel'Ev, A., Titova, N., Loukhovitskaya, E., and Schumann, U.: Effect of aerosol precursors from gas turbine engines on the volatile sulfate aerosols and ion clusters formation in aircraft plumes, Phys. Chem. Chem. Phys., 6, 3426–3436, 2004.
Steudel, R.: Sulfuric acid from sulfur trioxide and water—a surprisingly complex reaction, Angew. Chem. Int. Edit., 34, 1313–1315, 1995.
Stewart, J.: MOPAC2016, Stewart computational chemistry, Colorado Springs, CO, OpenMOPAC, 2016.
Stewart, J. J.: Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters, J. Mol. Model., 19, 1–32, https://doi.org/10.1007/s00894-012-1667-x, 2013.
Stewart, J. J. P.: Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements, J. Mol. Model., 13, 1173–1213, https://doi.org/10.1007/s00894-007-0233-4, 2007.
Stone, D. and Rowley, D. M.: Kinetics of the gas phase HO2 self-reaction: Effects of temperature, pressure, water and methanol vapours, Phys. Chem. Chem. Phys., 7, 2156–2163, https://doi.org/10.1039/B502673C, 2005.
Tan, X. F., Long, B., Ren, D. S., Zhang, W. J., Long, Z. W., and Mitchell, E.: Atmospheric chemistry of CH3CHO: the hydrolysis of CH3CHO catalyzed by H2SO4, Phys. Chem. Chem. Phys., 20, 7701–7709, 2018.
Tilgner, A., Schaefer, T., Alexander, B., Barth, M., Collett Jr., J. L., Fahey, K. M., Nenes, A., Pye, H. O. T., Herrmann, H., and McNeill, V. F.: Acidity and the multiphase chemistry of atmospheric aqueous particles and clouds, Atmos. Chem. Phys., 21, 13483–13536, https://doi.org/10.5194/acp-21-13483-2021, 2021.
Torrent-Sucarrat, M., Francisco, J. S., and Anglada, J. M.: Sulfuric acid as autocatalyst in the formation of sulfuric acid, J. Am. Chem. Soc., 134, 20632–20644, 2012.
VandeVondele, J. and Hutter, J.: Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases, J. Chem. Phys., 127, 114105, https://doi.org/10.1063/1.2770708, 2007.
Viegas, L. P. and Varandas, A. J.: Can water be a catalyst on the HO2 + H2O + O3 reactive cluster?, Chem. Phys., 399, 17–22, 2012.
Viegas, L. P. and Varandas, A. J.: The HO2 + (H2O)n + O3 reaction: an overview and recent developments, Eur. Phys. J. D, 70, 1–9, 2016.
Wayne, R. P.: Chemistry of Atmospheres. An Introduction to the Chemistry of the Atmospheres of Earth, the Planets, and Their Satellites, 3rd edn., Oxford University Press, https://doi.org/10.1021/ja004780n, 2000.
Weber, R., McMurry, P., Eisele, F., and Tanner, D.: Measurement of expected nucleation precursor species and 3–500-nm diameter particles at Mauna Loa observatory, Hawaii, J. Atmos. Sci., 52, 2242–2257, 1995.
Weber, R., Marti, J., McMurry, P., Eisele, F., Tanner, D., and Jefferson, A.: Measured atmospheric new particle formation rates: Implications for nucleation mechanisms, Chem. Eng. Commun., 151, 53–64, 1996.
Weber, R., Chen, G., Davis, D., Mauldin III, R., Tanner, D., Eisele, F., Clarke, A., Thornton, D., and Bandy, A.: Measurements of enhanced H2SO4 and 3–4 nm particles near a frontal cloud during the First Aerosol Characterization Experiment (ACE 1), J. Geophys. Res.-Atmos., 106, 24107–24117, 2001.
Yang, Y., Liu, L., Wang, H., and Zhang, X.: Molecular-Scale Mechanism of Sequential Reaction of Oxalic Acid with SO3: Potential Participator in Atmospheric Aerosol Nucleation, J. Phys. Chem. A, 125, 4200–4208, 2021.
Yao, L., Garmash, O., Bianchi, F., Zheng, J., Yan, C., Kontkanen, J., Junninen, H., Mazon, S. B., Ehn, M., Paasonen, P., Sipilä, M., Wang, M., Wang, X., Xiao, S., Chen, H., Lu, Y., Zhang, B., Wang, D., Fu, Q., Geng, F., Li, L., Wang, H., Qiao, L., Yang, X., Chen, J., Kerminen, V.-M., Petäjä, T., Worsnop, D. R., Kulmala, M., and Wang, L.: Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity, Science, 361, 278–281, https://doi.org/10.1126/science.aao4839, 2018.
Yao, L., Fan, X., Yan, C., Kurtén, T., Daellenbach, K. R., Li, C., Wang, Y., Guo, Y., Dada, L., Rissanen, M. P., Cai, J., Tham, Y. J., Zha, Q., Zhang, S., Du, W., Yu, M., Zheng, F., Zhou, Y., Kontkanen, J., Chan, T., Shen, J., Kujansuu, J. T., Kangasluoma, J., Jiang, J., Wang, L., Worsnop, D. R., Petäjä, T., Kerminen, V. M., Liu, Y., Chu, B., He, H., Kulmala, M., and Bianchi, F.: Unprecedented Ambient Sulfur Trioxide (SO3) Detection: Possible Formation Mechanism and Atmospheric Implications, Environ. Sci. Technol. Lett., 7, 809–818, https://doi.org/10.1021/acs.estlett.0c00615, 2020.
Zhao, Y. and Truhlar, D. G.: The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 120, 215–241, 2008.
Zhang, H., Kupiainen-Määttä, O., Zhang, X., Molinero, V., Zhang, Y., and Li, Z.: The enhancement mechanism of glycolic acid on the formation of atmospheric sulfuric acid–ammonia molecular clusters, J. Chem. Phys., 146, 184308, https://doi.org/10.1063/1.4982929, 2017.
Zhang, H., Li, H., Liu, L., Zhang, Y., Zhang, X., and Li, Z.: The potential role of malonic acid in the atmospheric sulfuric acid–ammonia clusters formation, Chemosphere, 203, 26–33, 2018.
Zhang, J. and Dolg, M.: ABCluster: the artificial bee colony algorithm for cluster global optimization, Phys. Chem. Chem. Phys., 17, 24173–24181, https://doi.org/10.1039/C5CP04060D, 2015.
Zhang, J. and Dolg, M.: Global optimization of clusters of rigid molecules using the artificial bee colony algorithm, Phys. Chem. Chem. Phys., 18, 3003–3010, https://doi.org/10.1039/C5CP06313B, 2016.
Zhang, R., Khalizov, A., Wang, L., Hu, M., and Xu, W.: Nucleation and growth of nanoparticles in the atmosphere, Chem. Rev., 112, 1957–2011, 2012.
Zhang, R., Wang, G., Guo, S., Zamora, M. L., Ying, Q., Lin, Y., Wang, W., Hu, M., and Wang, Y.: Formation of urban fine particulate matter, Chem. Rev., 115, 3803–3855, 2015.
Zhang, R., Shen, J., Xie, H.-B., Chen, J., and Elm, J.: The role of organic acids in new particle formation from methanesulfonic acid and methylamine, Atmos. Chem. Phys., 22, 2639–2650, https://doi.org/10.5194/acp-22-2639-2022, 2022.
Zhong, J., Zhao, Y., Li, L., Li, H., Francisco, J. S., and Zeng, X. C.: Interaction of the NH2 Radical with the Surface of a Water Droplet, J. Am. Chem. Soc., 137, 12070–12078, https://doi.org/10.1021/jacs.5b07354, 2015.
Zhong, J., Kumar, M., Zhu, C. Q., Francisco, J. S., and Zeng, X. C.: Frontispiece: Surprising Stability of Larger Criegee Intermediates on Aqueous Interfaces, Angew. Chem. Int. Edit., 56, 7740–7744, https://doi.org/10.1002/anie.201782761, 2017a.
Zhong, J., Zhu, C., Li, L., Richmond, G. L., Francisco, J. S., and Zeng, X. C.: Interaction of SO2 with the Surface of a Water Nanodroplet, J. Am. Chem. Soc., 139, 17168–17174, 2017b.
Zhong, J., Kumar, M., Francisco, J. S., and Zeng, X. C.: Insight into chemistry on cloud/aerosol water surfaces, Accounts Chem. Res., 51, 1229–1237, 2018.
Zhong, J., Li, H., Kumar, M., Liu, J., Liu, L., Zhang, X., Zeng, X. C., and Francisco, J. S.: Mechanistic Insight into the Reaction of Organic Acids with SO3 at the Air–Water Interface, Angew. Chem. Int. Edit., 131, 8439-8443, 2019.
Zhu, C., Kumar, M., Zhong, J., Li, L., Francisco, J. S., and Zeng, X. C.: New Mechanistic Pathways for Criegee-Water Chemistry at the Air/Water Interface, J. Am. Chem. Soc., 138, 11164–11169, https://doi.org/10.1021/jacs.6b04338, 2016.
Zhu, C., Kais, S., Zeng, X. C., Francisco, J. S., and Gladich, I.: Interfaces select specific stereochemical conformations: the isomerization of glyoxal at the liquid water interface, J. Am. Chem. Soc., 139, 27–30, 2017.
Zhuang, Y. and Pavlish, J. H.: Fate of hazardous air pollutants in oxygen-fired coal combustion with different flue gas recycling, Environ. Sci. Technol., 46, 4657–4665, 2012.