the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 19 Mar 2024
| 19 Mar 2024
Contribution of expanded marine sulfur chemistry to the seasonal variability of dimethyl sulfide oxidation products and size-resolved sulfate aerosol
Qianjie Chen
Becky Alexander
Charles H. Fite
Christopher D. Holmes
Jeffrey R. Pierce
Betty Croft
Sakiko Ishino
Marine emissions of dimethyl sulfide (DMS) and the subsequent formation of its oxidation products methanesulfonic acid (MSA) and sulfuric acid (H2SO4) are well-known natural precursors of atmospheric aerosols, contributing to particle mass and cloud formation over ocean and coastal regions. Despite a long-recognized and well-studied role in the marine troposphere, DMS oxidation chemistry remains a work in progress within many current air quality and climate models, with recent advances exploring heterogeneous chemistry and uncovering previously unknown intermediate species. With the identification of additional DMS oxidation pathways and intermediate species that influence the eventual fate of DMS, it is important to understand the impact of these pathways on the overall sulfate aerosol budget and aerosol size distribution. In this work, we update and evaluate the DMS oxidation mechanism of the chemical transport model GEOS-Chem by implementing expanded DMS oxidation pathways in the model. These updates include gas- and aqueous-phase reactions, the formation of the intermediates dimethyl sulfoxide (DMSO) and methanesulfinic acid (MSIA), and cloud loss and aerosol uptake of the recently quantified intermediate hydroperoxymethyl thioformate (HPMTF). We find that this updated mechanism collectively decreases the global mean surface-layer gas-phase sulfur dioxide (SO2) mixing ratio by 40 % and enhances the sulfate aerosol () mixing ratio by 17 %. We further perform sensitivity analyses exploring the contribution of cloud loss and aerosol uptake of HPMTF to the overall sulfur budget. Comparing modeled concentrations to available observations, we find improved biases relative to previous studies. To quantify the impacts of these chemistry updates on global particle size distributions and the mass concentration, we use the TwO-Moment Aerosol Sectional (TOMAS) aerosol microphysics module coupled to GEOS-Chem and find that changes in particle formation and growth affect the size distribution of aerosol. With this new DMS-oxidation scheme, the global annual mean surface-layer number concentration of particles with diameters smaller than 80 nm decreases by 16.8 %, with cloud loss processes related to HPMTF being mostly responsible for this reduction. However, the global annual mean number of particles larger than 80 nm (corresponding to particles capable of acting as cloud condensation nuclei, CCN) increases by 3.8 %, suggesting that the new scheme promotes seasonal particle growth to these sizes.
- Article
(11688 KB) - Full-text XML
- BibTeX
- EndNote





Dimethyl sulfide (DMS: CH3SCH3) is the most abundant biological source of sulfate aerosol and has a significant influence on Earth's radiation budget and climate due to its contribution to atmospheric marine particles (Charlson et al., 1987; Fung et al., 2022). In the atmosphere, DMS reacts with hydroxyl radical (OH), nitrate radical (NO3), ozone (O3) and various halogen species (e.g., chlorine, Cl; bromine oxide, BrO), primarily forming sulfur dioxide (SO2) and methyl sulfonic acid (MSA: CH3SO3H) (Chen et al., 2018; Faloona, 2009; Hoffmann et al., 2016). These oxidation products are considered key influences on the formation and evolution of natural aerosols and clouds along with their associated climate impacts, especially in the marine boundary layer (MBL) (Carslaw et al., 2013; Sipilä et al., 2010; Schobesberger et al., 2013; Thomas et al., 2010; von Glasow and Crutzen, 2004). SO2 and MSA formed by DMS oxidation can be deposited on the Earth's surface or can further oxidize, affecting the size distribution of aerosol and cloud microphysics (Leaitch et al., 2013; Wollesen de Jonge et al., 2021). SO2 can either oxidize in the gas phase by reacting with the OH radical, forming H2SO4, which can participate in nucleation and the early growth of particles in the atmosphere, or it can be taken up by cloud droplets and undergo aqueous-phase oxidation by reacting with H2O2, O3 and O2 (catalyzed by transition metals: Mn, Fe), forming and generally only contributing to the growth of aerosol particles (Hoyle et al., 2016; Kulmala, 2003; Alexander et al., 2009). The hypohalous acids (HOBr, HOCl, HOI) also play a significant role in aqueous-phase sulfate production in the marine boundary layer (MBL) (Chen et al., 2016; Sherwen et al., 2016b). Recent studies have highlighted the importance of natural aerosols originating from DMS oxidation and their contribution to the uncertainty of aerosol radiative forcing in climate models (Carslaw et al., 2013; Fung et al., 2022; Rosati et al., 2022; Novak et al., 2021, 2022). Since DMS-derived aerosol is a major source of uncertainty when estimating the global natural aerosol burden and associated aerosol indirect radiative forcing, a more accurate representation of DMS oxidation and particle formation processes is an important step towards improved Earth system and climate modeling.
Table 1The three DMS oxidation reactions in the standard GEOS-Chem chemical mechanism.

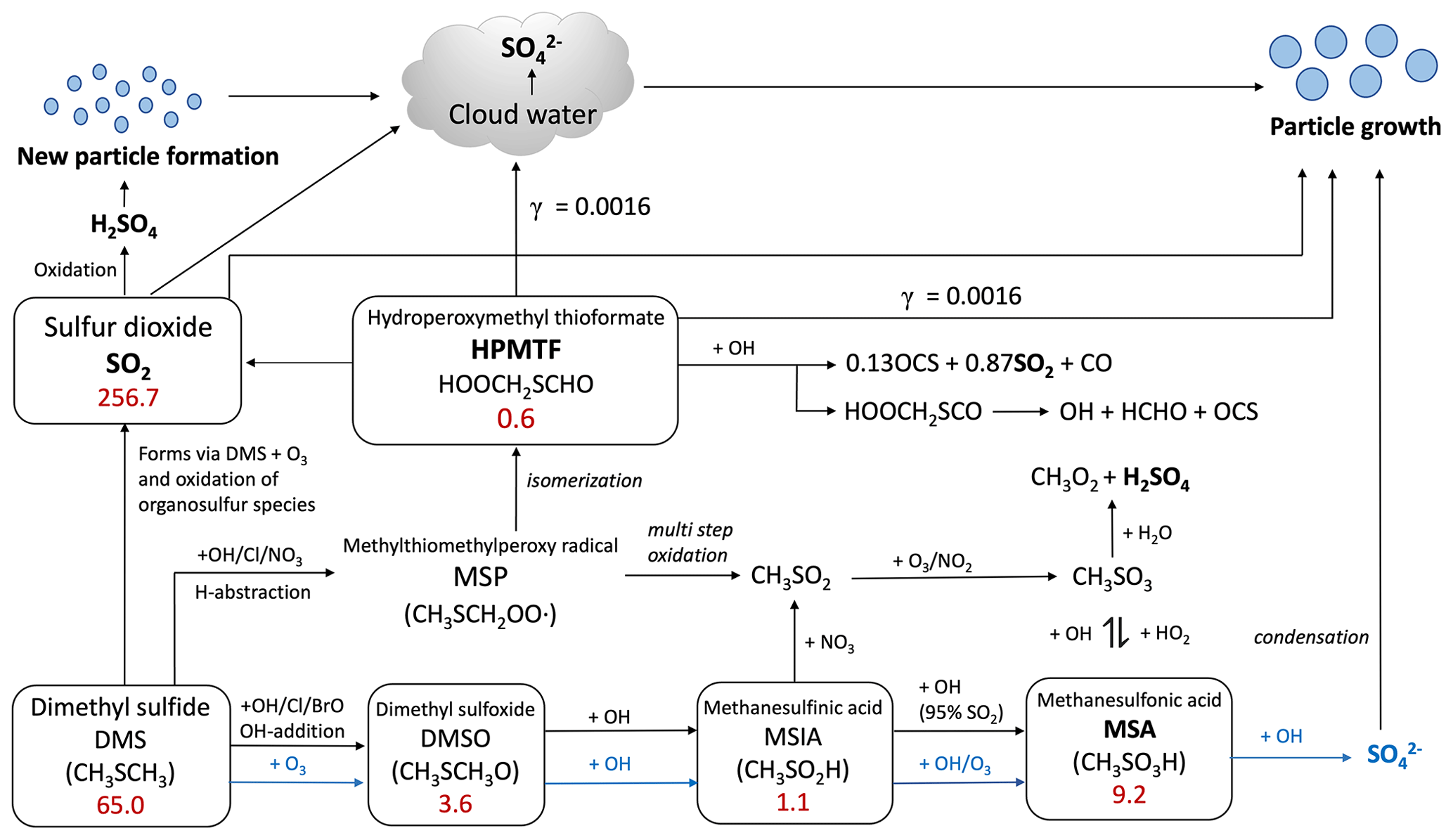
Figure 1Summary of the modified DMS oxidation mechanism used in this work (MOD simulation), showing the formation of major stable oxidation products (in bold; including the newly identified intermediate HPMTF) and their contributions to new particle formation or the growth of existing particles. The blue arrows and text represent aqueous-phase reactions. Numbers inside boxes indicate the burden in units of Gg S. γ values represent reactive uptake coefficients for the heterogeneous loss of HPMTF to cloud and aerosol. Note that SO2 formation from DMS and HPMTF involves multiple oxidation steps in this mechanism, but full pathways are simplified here for visual clarity.
Although the chemistry of DMS oxidation has been previously studied in great detail, there are still known uncertainties in and omissions from the current mechanism in current air quality and chemical transport models (Barnes et al., 2006; Fung et al., 2022; Hoffmann et al., 2016, 2021). Furthermore, while increasingly complex and experimentally validated mechanisms are under ongoing development, DMS oxidation processes in many current chemical transport models continue to be represented through simplified gas-phase reactions with the tropospheric oxidants OH and NO3 that produce the two major oxidation products SO2 and MSA in a fixed ratio, as shown in Reactions (R1)–(R3) in Table 1 (Chen et al., 2018; Chin et al., 1996; Veres et al., 2020). This type of simplified mechanism neglects the formation and loss of important intermediates such as dimethyl sulfoxide (DMSO: CH3SOCH3), methanesulfinic acid (MSIA: CH3SO2H) and the recently discovered oxidation product hydroperoxymethyl thioformate (HPMTF: HOOCH2SCHO) (Berndt et al., 2019; Veres et al., 2020; Wu et al., 2015; Khan et al., 2021).
These omissions can have major consequences for product yields of DMS oxidation, thereby affecting the aerosol burdens. For example, the OH-addition pathway of DMS forms DMSO and MSIA as the intermediates; these have been identified as a dominant source of MSA via their aqueous-phase oxidation, and a fraction of that MSA subsequently undergoes aqueous-phase oxidation to form sulfate aerosol (Chen et al., 2018; Ishino et al., 2021; Zhu et al., 2006; von Glasow and Crutzen, 2004). Previous studies have suggested that BrO is responsible for 8 %–30 % of the total DMS loss, highlighting the importance of this pathway as well (Breider et al., 2010; Boucher et al., 2003; Chen et al., 2018; Khan et al., 2016). More recent experimental and laboratory studies have confirmed the formation of methylthiomethylperoxy radicals (CH3CH2OO; abbreviated to MSP or MTMP) from the H-abstraction channel of OH oxidation, which can subsequently lead to a series of rapid intramolecular H-shift isomerization reactions, ultimately resulting in the formation of the stable intermediate HPMTF (Berndt et al., 2019; Veres et al., 2020; Vermeuel et al., 2020; Wu et al., 2015; Fung et al., 2022; Jernigan et al., 2022a). It has been reported that 30 %–46 % of the emitted DMS forms HPMTF according to different modeling studies, and this falls within the observational range from NASA Atmospheric Tomography (ATom) for ATom-3 and ATom-4 flight campaigns, where about 30 %–40 % of the DMS was oxidized to HPMTF along their flight tracks (Fung et al., 2022; Veres et al., 2020; Novak et al., 2021). Subsequent investigation of the isomerization rate and heterogeneous loss of HPMTF in cloud droplets and aerosol showed a high production rate of marine carbonyl sulfide (OCS) from the chemical loss of HPMTF, a potential precursor of stratospheric sulfate aerosol and significant inhibitor of the formation of cloud condensation nuclei (CCN) due to the resulting reduction in SO2 (Jernigan et al., 2022a). With the latest experimental findings on the heterogeneous HPMTF loss process and experimentally validated oxidation reactions for OCS formation directly from HPMTF, it is necessary to include these reactions as part of the DMS oxidation mechanism, as these will have an impact on the overall yield of SO2, thus affecting the formation probability of CCN (Jernigan et al., 2022a, b).
Considering these and other consequences of complex DMS oxidation processes, a heavily simplified oxidation scheme will necessarily neglect potentially important reaction intermediates along with their production and loss pathways, with implications for the concentration and distribution of the oxidation products, including particulate sulfate. Differing intermediate lifetimes further influence sulfur removal and transport depending on the relative dominance of pathways. Thus, the exclusion of key pathways and intermediate species can lead to errors in the representation of the spatial distributions of both gas- and particle-phase sulfur species as well as the global sulfur burden.
The DMS oxidation products sulfate and MSA play an important role in Earth's radiative budget through cloud droplet formation, and the extent of this role depends on how efficiently they can produce and grow new particles in the marine atmosphere (Thomas et al., 2010). In the gas phase, SO2 can oxidize forming H2SO4, which is a key product due to its contribution to nucleation and condensational growth, as shown in Fig. 1. SO2 oxidation through aqueous chemistry in cloud droplets does contribute to particle growth rates by providing larger aerosol particles during cloud evaporation, which act as more efficient CCN. (Kaufman and Tanré, 1994). On the other hand, MSA might participate in nucleation along with sulfuric acid in the presence of amines or ammonia (Johnson and Jen, 2023). Recent studies have highlighted the importance of aqueous-phase chemistry in the formation and loss of MSA (Boniface et al., 2000; Chen et al., 2015; Kaufman and Tanré, 1994; Kulmala et al., 2000).
Additionally, the recently identified intermediate HPMTF also has the potential for further gas-phase oxidation. Under cloud-free conditions, HPMTF can undergo gas-phase oxidation by OH, producing SO2 and eventually leading to the formation of non-sea-salt . This sulfate can contribute to aerosol formation and growth processes, with climate implications (Galí et al., 2019). Another work used direct airborne eddy covariance flux measurements to explain the chemical fate of HPMTF in the MBL and found that chemical loss due to aqueous-phase reactions in clouds is the major HPMTF removal process in cloudy conditions (Novak et al., 2021). In the same study, global model simulations showed a 35 % reduction in global annual average SO2 production from DMS and a 24 % reduction in the near-surface (0 to 3 km) global annual average SO2 concentrations over the ocean as a result of this process (Novak et al., 2021). Thus, a complete representation of cloud loss and aerosol uptake is needed to effectively evaluate the atmospheric impacts of marine DMS and their connections to cloud formation (Novak et al., 2021; Holmes et al., 2019).
To better understand the marine sulfur budget as well as the eventual formation, size distribution and seasonality of sulfate aerosol, we use the global chemical transport model GEOS-Chem, integrating previously developed mechanisms along with newly proposed pathways involving the formation and loss of the intermediates DMSO, MSIA and HPMTF. As part of this work, we further quantify the atmospheric impacts of individual reactions and mechanisms, evaluate uncertainties in the chemical mechanism, and identify the improvements necessary to better represent the impacts of DMS on atmospheric chemistry and climate more accurately. The resulting integrated scheme provides a more complete representation of marine sulfur and sulfate aerosol species in marine tropospheric environments compared to the simplified base GEOS-Chem mechanism, with improved comparisons to aircraft and surface observations. Since aerosols are a major contributor to uncertainty in climate forcing, improving oxidation and aerosol formation mechanisms by including and optimizing neglected reactions in models is a crucial step towards a more mechanistically robust representation of particle yields and sensitivities. We further perform multiple sensitivity tests to investigate how the uncertainty in the heterogeneous uptake of the newly identified HPMTF could influence DMS chemistry and tropospheric aerosol formation (Holmes et al., 2019; Novak et al., 2021). In a broader sense, our work provides a more detailed story of the heterogeneous loss, fate and ultimate impacts of DMS and its oxidation products, improving our understanding of a key ocean–atmosphere interaction in the context of global change.
The global chemical transport model GEOS-Chem v12.9.3 is used to simulate the chemistry of DMS and its oxidation products. Impacts on simulated aerosol size and number and mass concentrations are considered by coupling the TwO-Moment Aerosol Sectional (TOMAS) aerosol microphysics module with GEOS-Chem v12.9.3 (GC-TOMAS) (https://github.com/geoschem/geos-chem/tree/12.9.3, last access: 10 March 2024) (Adams and Seinfeld, 2002; Kodros and Pierce, 2017). The default GEOS-Chem chemical mechanism contains detailed HOx–NOx–VOC–O3–halogen tropospheric chemistry along with recently updated halogen chemistry and in-cloud processing (Bey et al., 2001; Holmes et al., 2019; Chen et al., 2017; Parrella et al., 2012; Schmidt et al., 2016; Wang et al., 2019). The DMS emission flux from the ocean is controlled by a gas transfer velocity which is dependent on sea surface temperature and wind speed (Johnson, 2010) and the climatology of concentrations in seawater (Lana et al., 2011; Nightingale et al., 2000). The aqueous-phase concentration of O3 in aerosols or cloud droplets is calculated assuming gas–liquid equilibrium, and the aqueous-phase concentration of OH is calculated based on [OH(aq)] = δ[OH(g)], where δ = 1 × 10−19 (Jacob et al., 2005; Chen et al., 2018).
In this study, TOMAS tracks the aerosol number and the mass of each aerosol species in 15 logarithmically sized bins, with sizes in this analysis ranging from 3 nm to 10 µm (Lee and Adams, 2012; Lee et al., 2013). All binned aerosol species undergo interactive microphysics, allowing the calculation of aerosol number budgets (Westervelt et al., 2013). The version of GC-TOMAS used here includes 47 vertical levels, a horizontal resolution of 4° × 5° and the GEOS-FP data product for meteorological inputs. Simulations are performed for 2018 with 11 months of discarded model spin-up. Nucleation is simulated via a ternary nucleation scheme involving water, sulfuric acid and ammonia, with nucleation rates scaled by 10−5 (Napari et al., 2002; Westervelt et al., 2013). In low-ammonia regions (less than 1 pptv), a binary nucleation scheme involving water and sulfuric acid is used instead (Vehkamäki et al., 2002). Previously, GC-TOMAS was used for aerosol simulations to investigate topics such as the aerosol cloud-albedo effect and cloud condensation nuclei formation (Kodros et al., 2016; Kodros and Pierce, 2017; Pierce and Adams, 2006; Westervelt et al., 2013). The aerosol species available for GC-TOMAS simulations are sulfate, aerosol water, black carbon, organic carbon, mineral dust and sea salt (Alexander et al., 2005; Bey et al., 2001; Duncan Fairlie et al., 2007; Pye et al., 2009). The wet and dry deposition schemes for aerosols and gas species are based on previous studies (Amos et al., 2012; Emerson et al., 2020; Liu et al., 2001; Wesely, 1989; Wang et al., 1998).
Table 2Overview of the mechanism for DMS oxidation via the OH-addition pathway. Note that “upd. 2006” here and in Table 4 refers to the corresponding IUPAC data sheet updated in 2006.
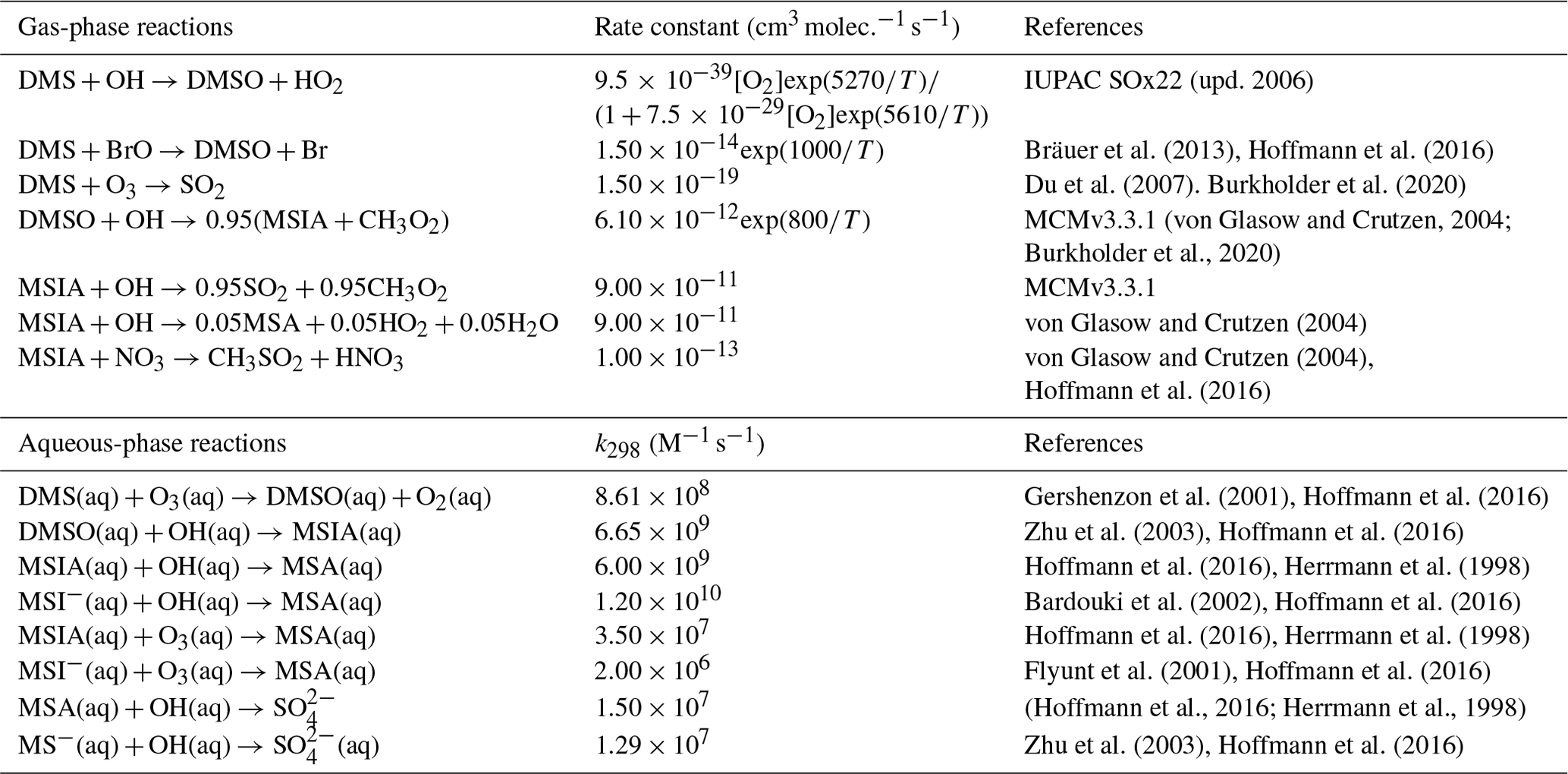
Table 3Overview of the DMS oxidation mechanism involving HPMTF formation.
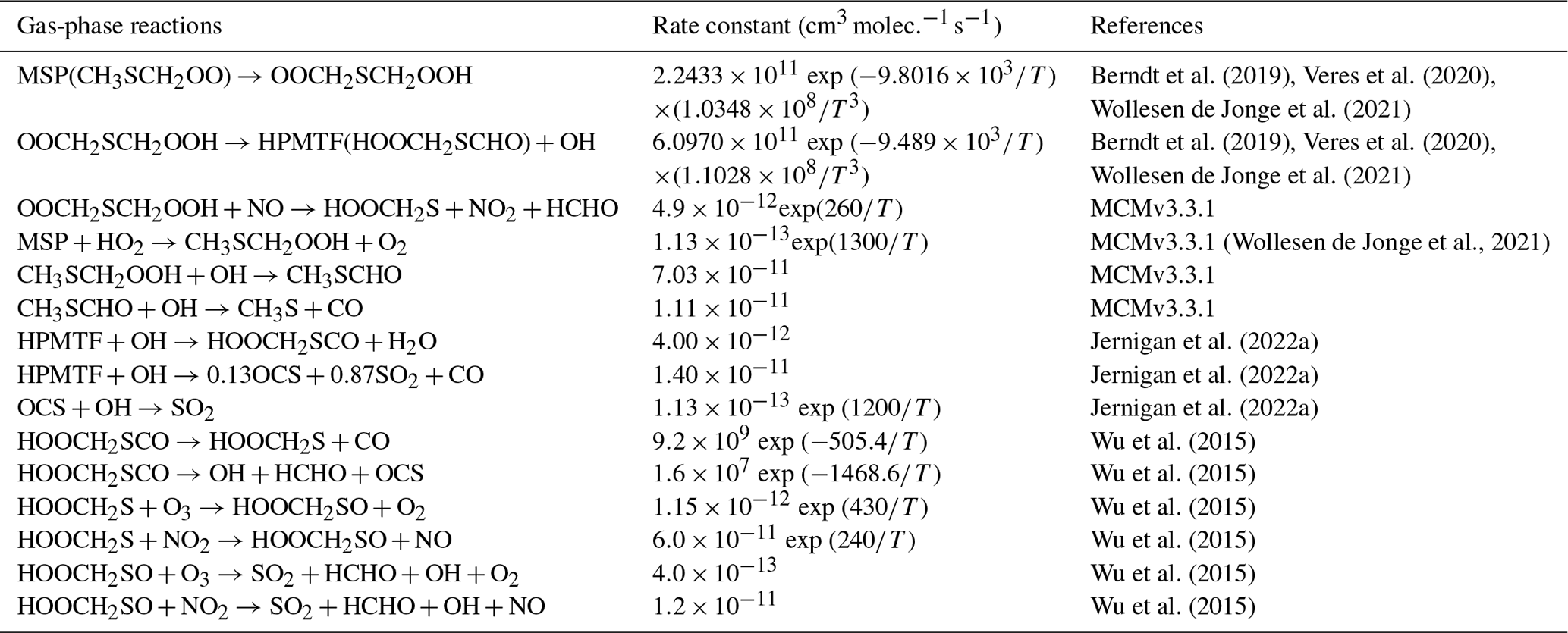
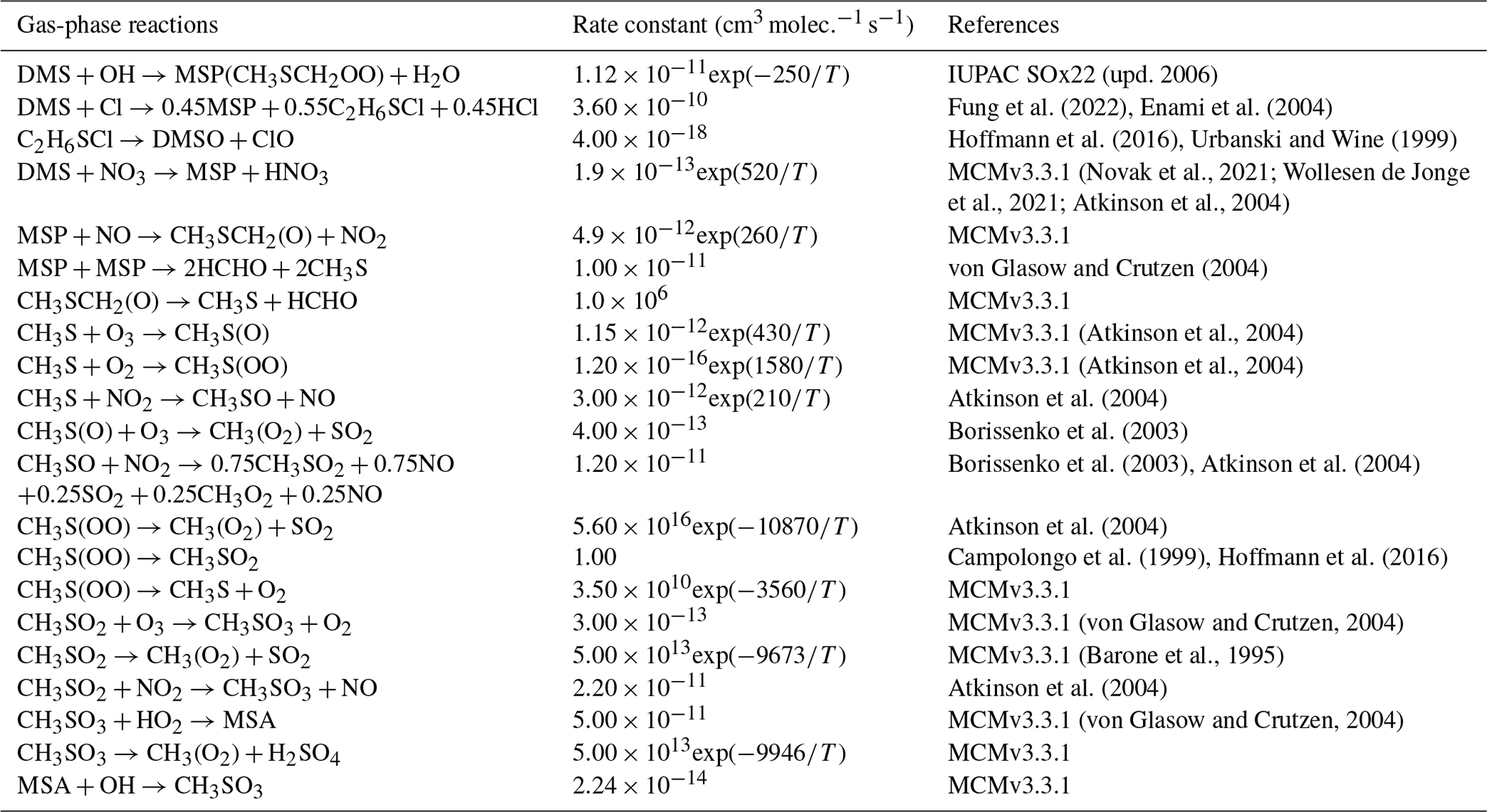
Table 4Overview of the MSA-producing branch of the H-abstraction pathway for DMS oxidation.
We refer to simulations performed using only these three DMS oxidation reactions (Table 1) as “BASE”; these involve only the direct formation of SO2 and MSA in the gas phase (Chin et al., 1996). We further implement and evaluate a custom chemical mechanism for DMS oxidation referred to as “MOD” (Tables 2–4) and representing an integration of three individual DMS oxidation mechanism updates explored previously using GEOS-Chem and CAM6-Chem. This mechanism also includes HPMTF loss to clouds and aerosols via heterogeneous chemistry, dry and wet deposition of HPMTF, along with a further improvement based on recent literature updates to chemical kinetics (Chen et al., 2018; Fung et al., 2022; Veres et al., 2020; Novak et al., 2021; Cala et al., 2023). In GC-TOMAS, we use a specific subroutine that takes the amount of sulfate produced via in-cloud oxidation and condenses it into an existing aerosol size distribution. So, the mass of sulfate produced by oxidation is apportioned to the various size bins according to the number of particles in that size bin. The microphysics of TOMAS accounts for H2SO4 formation based on the gas-phase oxidation of SO2 included in the kinetic preprocessor (KPP) equation list valid for simulation BASE. Since there are additional sources of sulfate in the integrated DMS oxidation mechanism in both the gas and aqueous phases, we made the necessary changes in the KPP code to allow H2SO4 formation by gas-phase oxidation of SO2 to be explicitly tracked. On the other hand, code changes to account for sulfate formed by heterogeneous oxidation of MSA and HPMTF (in clouds and aerosols) were performed in the GEOS-Chem microphysics module, which also handles the in-cloud oxidation of SO2 in GC version 12.9.3 (Park et al., 2004; Trivitayanurak et al., 2008).
Table 5List of mechanisms used in GEOS-Chem–TOMAS simulations.
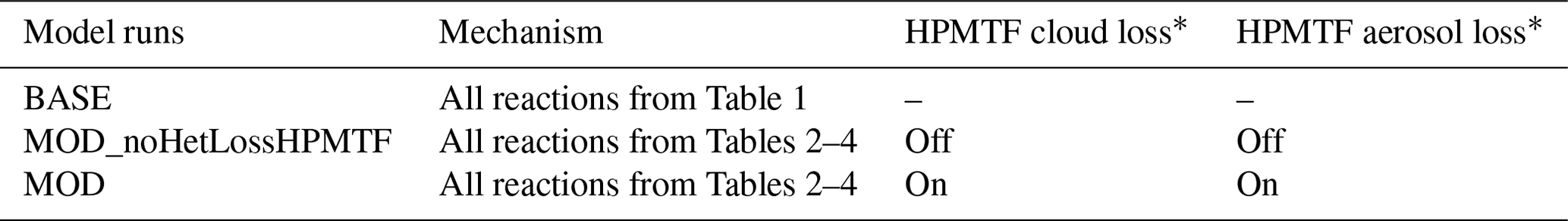
∗ The instantaneous formation of sulfate via HPMTF cloud and aerosol loss has a reactive uptake coefficient (γ) of 0.0016.
To examine the sensitivities of size-resolved aerosol formation and growth to DMS chemistry modifications, model simulations are conducted as summarized in Table 5. Output from simulations MOD and MOD_noHetLossHPMTF was then compared against simulation BASE to understand the contribution of these additional chemical reactions to the spatial pattern of the surface concentrations of major oxidation products of DMS.
As shown in Table 2, the modified DMS chemistry simulations examined here include the gas- and aqueous-phase oxidation of DMS and the intermediate products of its oxidation by OH, NO3, O3 and halogenated species, as previously explored in an older version of GEOS-Chem (Chen et al., 2018). The aqueous-phase reactions in cloud droplets and aerosols were parameterized assuming first-order loss of the gas-phase sulfur species (Chen et al., 2018). Further building upon the previous mechanism, the scheme used here also includes the formation and loss of HPMTF, previously tested in the global climate model CAM6-Chem, as shown in Table 3 (Veres et al., 2020). Table 4 presents the third piece of the mechanism: a gas-phase MSA-producing branch of the H-abstraction pathway in the DMS chemistry that bridges the other two sets of reactions (Fung et al., 2022). To avoid the addition of SO3 oxidation chemistry, we have replaced SO3 with H2SO4 following previous work on the decomposition reaction of CH3SO3 (Table 4). A similarly integrated mechanism (Tables 2–4) was previously explored using the CAM6-Chem model with a focus on radiation budget impacts, which is improved in this work through updates to rate constants and the inclusion of additional relevant reactions (Fung et al., 2022; Novak et al., 2021; Wollesen de Jonge et al., 2021; Cala et al., 2023). The newly added reactions and their respective rate constants are largely based on the Master Chemical Mechanism (MCM) v3.3.1 and the literature cited in the reference lists in Tables 2–4. We use a rate constant of 1.40 × 10−11 for HPMTF + OH, which was previously determined based on the concentrations of other known sulfur species (DMS, DMSO, SO2 and methyl thioformate (MTF); CH3SCHO, a structurally similar proxy for HPMTF) and evaluated by the box model (Jernigan et al., 2022a). An exploration of the reaction rate uncertainty for the HPMTF+OH reaction (Table 3), including both high- and low-end limits of 5.5 × 10−11 and 1.4 × 10−12 , respectively, resulted in only minor impacts on the fate of HPMTF and on sulfate formation in our simulations (Novak et al., 2021; Wu et al., 2015).
Model sensitivity simulations were also performed with (MOD simulations) and without (“MOD_noHetLossHPMTF” simulations) HPMTF heterogeneous uptake by clouds and aerosols to account for how much of the DMS-derived HPMTF eventually forms SO2 in the presence of these additional loss processes (Table 5). Previous work has shown that aerosol surface chemistry causes additional decreases in HPMTF mixing ratios, primarily over land, and that the loss of HPMTF in clouds is larger (36 %) than the losses from aerosols (15 %) when using an uptake coefficient of γ = 0.01 for both processes (Novak et al., 2021). In this work, based on recent laboratory measurements, we use a smaller uptake coefficient (γ = 0.0016) for HPMTF loss to aerosols and clouds (Table 5) (Jernigan et al., 2022b). Based on previous work, we assume that HPMTF directly produces sulfate in cloud and aerosol, though there is some uncertainty regarding the fate of HPMTF during heterogeneous loss (Zhang and Millero, 1993; Novak et al., 2021; Jernigan et al., 2022a). For the aqueous-phase reactions listed in Table 2, including the oxidation of the intermediates DMSO and MSIA in cloud droplets and aerosols, a first-order loss of the gas-phase sulfur species was assumed, following previously used parameterizations and physical parameter values (Chen et al., 2018). Alongside the gas-phase and aqueous-phase reactions relevant to the added DMS oxidation mechanism contributing to the formation of SO2 and sulfate, the default version of GC-TOMAS used here also includes in-cloud oxidation of SO2 by H2O2, O3 and O2 (catalyzed by transition metals: Mn, Fe) as well as the loss of dissolved SO2 by HOBr and HOCl, all of which are passed to TOMAS to account for sulfate production (Chen et al., 2017; Wang et al., 2021).
All simulations are conducted for the year 2018, which was chosen to match the model simulation with the dates of the NASA Atmospheric Tomography flight campaign (ATom-4) that offers observational data for HPMTF, DMS and SO2. Rate coefficients for all gas-phase sulfur reactions are obtained from the most recent JPL report and other references, while sulfur product yields for gas-phase reactions are obtained from various laboratory and modeling studies (Burkholder et al., 2020; Lucas and Prinn, 2002; Hoffmann et al., 2016; Gershenzon et al., 2001; Kowalczuk et al., 2003; Zhou et al., 2019; Jernigan et al., 2022a). The simulations included sea salt debromination except for some sensitivity tests described below (Zhu et al., 2019; Schmidt et al., 2016). In all our simulations that include MOD, DMS is advected and undergoes chemical loss and transport but does not undergo dry or wet deposition. However, dry and wet deposition of oxidation products such as DMSO, MSIA, MSA and HPMTF are included.
We note that previous work has explored the impact of MSA on aerosol growth, including modifications within TOMAS to represent this process (Hodshire et al., 2019). We do not include this process here. Future work to examine its importance in the context of the chemistry updates presented here is recommended.
3.1 Model–observation comparison
3.1.1 Surface DMS mixing ratio
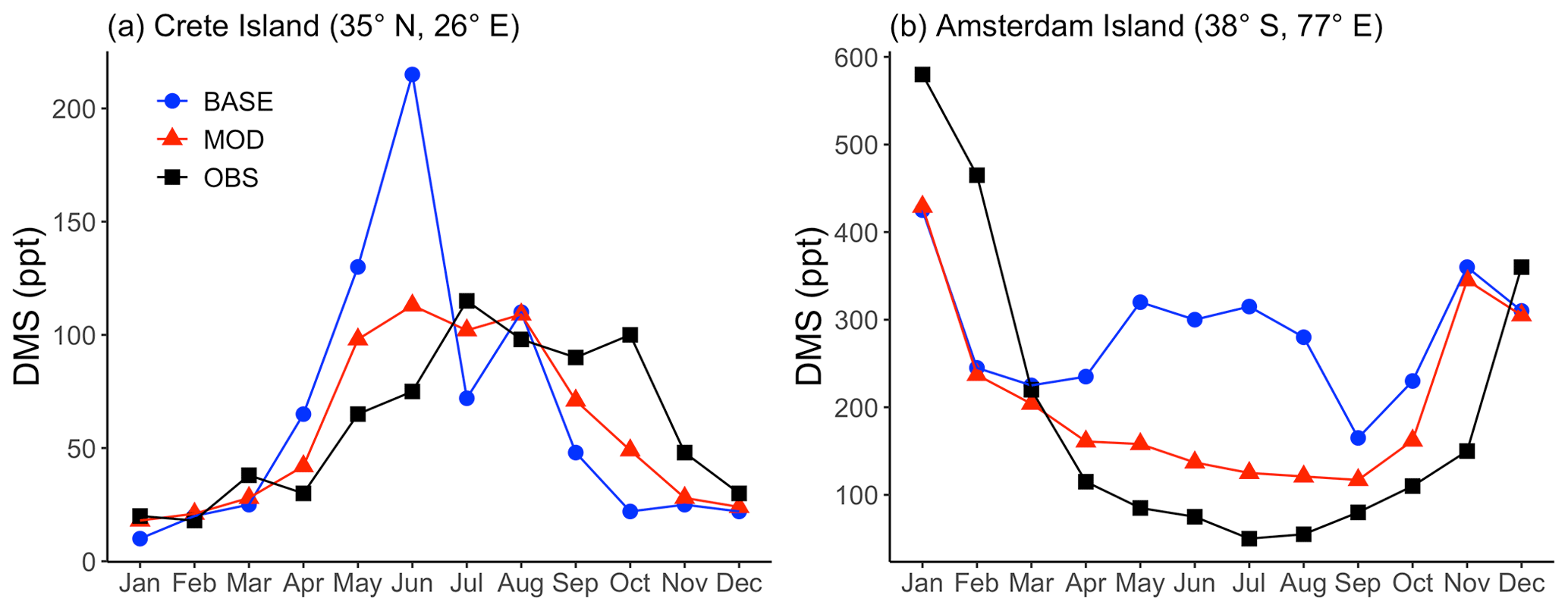
We compared the modeled DMS mixing ratio averaged for each month with the observational data collected at Crete Island (35° N, 26° E) and Amsterdam Island (37° S, 77° E) (Kouvarakis and Mihalopoulos, 2002; Chen et al., 2018; Castebrunet et al., 2009). Comparing simulations BASE and MOD, we find a closer match with DMS observations for simulations using modified DMS chemistry for both sets of observation data shown in Fig. 2. Modeled DMS mixing ratios calculated using the base chemistry show strong positive bias during the months of May and June for Crete Island. By comparison, during the same period, the modeled DMS mixing ratios calculated with the modified chemistry reduce the bias from 102 % to 42 %. Similarly, for Amsterdam Island, major overpredictions are apparent for the BASE simulation compared to MOD for the months of May–August. One reaction that may play a role in this shift is DMS + BrO, which, as indicated earlier, is responsible for a faster overall chemical loss of DMS, in particular over the Southern Hemisphere high latitudes. Besides DMS chemistry, sea surface DMS concentration is also proven to affect the modeled DMS mixing ratio (Chen et al., 2018). But the aim of this study is to investigate the chemistry of DMS oxidation, so we did not explore how the change in the DMS seawater climatology, and thus the emission of DMS, influence the surface DMS mixing ratio.
3.1.2 Comparison with aircraft observations
We further evaluate the model output through a comparison with ATom-4 aircraft observations for specific days of measurement for DMS, HPMTF and SO2, as shown in Fig. 5. For this comparison, the model is sampled at the time and location of aircraft measurements by ATom-4 using the planeflight diagnostic of GEOS-Chem.
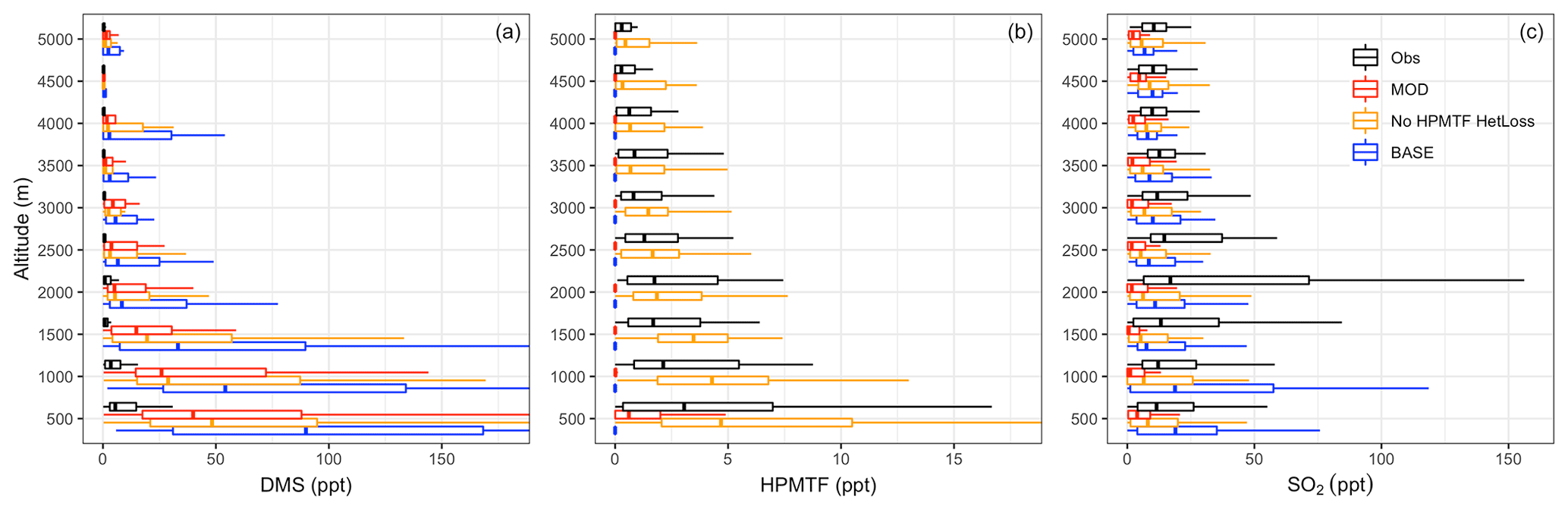
Figure 3Vertical profiles of (a) DMS, (b) HPMTF and (c) SO2 mixing ratios from ATom-4 observations (black) and the model using simulation MOD sampled along the ATom-4 flight tracks (red) binned every 500 m of flight altitude. Also shown are the modeled results obtained without heterogeneous HPMTF loss using simulation MOD_noHetLossHPMTF (yellow) and for BASE GEOS-Chem chemistry (blue). Box plot whiskers show the full range of distribution at each altitude bin. DMS observations are from whole air sampler (WAS), while HPMTF DC-8 observations are from an iodide ion chemical ionization time-of-flight mass spectrometer (CIMS). SO2 observations from the ATom-4 campaign were measured by laser-induced fluorescence (LIF).
DMS concentrations measured during ATom-4 by a whole air sampler (WAS) and modified chemistry simulation values for nearest-neighbor grid cells are shown in Fig. 3a for different altitudes. In general, the modeled DMS concentrations are significantly higher than those observed during ATom-4 missions, especially close to the surface. However, model DMS concentrations decrease more rapidly than the measurements with altitude, indicating that vertical mixing could be one of the underlying reasons for this trend. Even with this near-surface bias, simulation MOD shows, relative to BASE, greater DMS losses and a shorter DMS lifetime (from 1.5 to 0.9 d), reducing the gap between modeled and observed concentrations compared to simulation BASE. The reduction in modeled DMS is largest over the Southern Ocean (shown later in Fig. 5b), where oxidation by BrO and O3 in the aqueous phase plays the major role in reducing DMS concentration, thereby reducing the model–observation bias (Fig. 3b). The remaining model biases could be at least partially attributable to the model uncertainty in oxidant concentrations and cloud cover. The heterogeneous loss of HPMTF has a minimal impact on DMS concentration and its vertical profile.
For HPMTF, Fig. 3b shows that the observed and modeled HPMTF concentrations remain largely below 15 ppt. Agreement between observations and modeled HPMTF mixing ratios in the vertical profile (Fig. 3b) is poor for simulation MOD, even close to the surface. Removing all heterogeneous loss of HPMTF improves model comparisons across the vertical profile, though surface concentrations become overestimated (yellow line of Fig. 3b), showing a high sensitivity to cloud and aerosol loss processes. We also find that the modeled HPMTF:DMS ratios range from 0.15:1 to 0.5:1 on a daily basis in most cases when there is no heterogeneous loss of HPMTF, compared to the ratio of 0.5:1 observed during ATom-4 using the calibration maintained during measurement, implying reasonably good agreement for this value on a daily timescale (Veres et al., 2020). The SARP flight campaign data indicated much lower HPMTF:DMS ratios (< 0.2) on cloudy days, which is relatable to the modeling of HPMTF with simulation MOD (Novak et al., 2021). For simulation MOD, the modeled HPMTF:DMS ratio is 0.03:1 up to 0.5 km, after which it approaches zero with increasing altitude, indicating the need for additional work to better constrain production and loss processes of this intermediate. Our simulations indicate that cloud loss is the dominant modeled removal process of HPMTF, consistent with previous findings, while gas-phase OH oxidation plays a minor role (Novak et al., 2021). Thus, the addition of cloud uptake dramatically decreases HPMTF concentrations throughout the troposphere. Overall, this allows only 10 % of the HPMTF produced to end up as SO2, with about 89 % lost to clouds and aerosol and thus removed from the system, resulting in a net reduction in mean global SO2 of about 40 % (note that other chemical processes are involved in this reduction as well). Previous work focusing entirely on the gas-phase and heterogeneous loss of HPMTF shows a much higher bias for both DMS and HPMTF during cloudy and clear sky conditions using the same model and a condensed DMS oxidation mechanism, indicating that the addition of the gas-phase and heterogeneous oxidation of DMS – including additional intermediates such as DMSO and MSIA – further reduces model biases for HPMTF, with the overestimation of the multiphase loss for HPMTF remaining (Novak et al., 2021).
We also compared the SO2 concentrations measured during ATom-4 by laser-induced fluorescence (LIF) and simulation MOD values for nearest-neighbor grid cells, as shown in Fig. 3c for different altitudes. Modeled surface SO2 concentrations are lower than those observed during ATom-4 missions across the vertical scale shown here for simulation MOD. The greater SO2 losses result in a shorter SO2 lifetime (from 1.4 to 1.3 d) for simulation MOD relative to simulation BASE. The reduction in modeled SO2 is largest over the Southern Ocean (shown later in Fig. 7a), where heterogeneous oxidation of HPMTF is most efficient and irreversible. Also, the OH addition channel of DMS does not directly produce SO2, causing a further reduction in the concentration relative to BASE. Removing the heterogeneous loss of HPMTF increases the modeled SO2 compared to simulation MOD, with an underprediction remaining. The remaining model biases could be at least partially attributable to uncertainty in DMS oxidation processes along with other non-DMS sources that contribute high concentrations of SO2. Aside from the uncertainty in DMS emissions and oxidation, recent research in marine sulfur chemistry has shown that the emission and oxidation of underrepresented marine sulfur species such as methanethiol (CH3SH) can serve as a significant source of SO2 to the marine atmosphere, the inclusion of which could help reduce this bias (Berndt et al., 2023; Novak et al., 2022). Overall, the DMS oxidation chemistry implemented in this work reduces the model observation bias close to the surface (up to 1 km) compared to the BASE GEOS-Chem chemistry.
Besides the vertical profile shown in Fig. 3b, the global mean surface mixing ratio of HPMTF in May 2018 obtained with simulation MOD_noHetLossHPMTF is plotted in Fig. 4 and compared with the observational measurements of HPMTF made during the ATom-4 mission during the NASA DC-8 flight campaign, which sampled the daytime remote marine atmosphere over the Pacific and Atlantic oceans. The ATom-4 measurements were carried out during daytime hours between 24 April and 21 May 2018 for 21 non-continuous days.
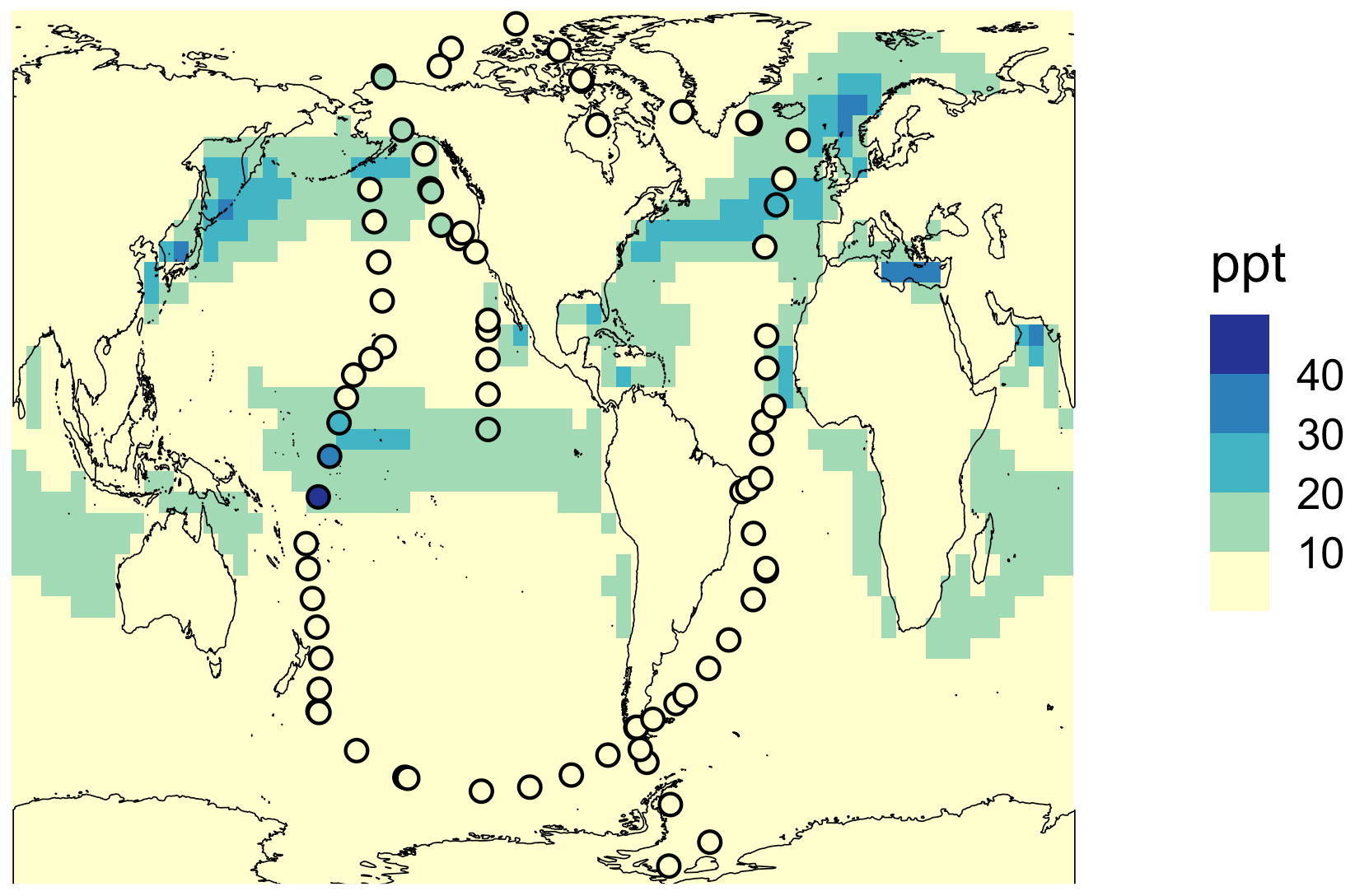
Figure 4Geographic distribution of the monthly mean surface-layer mixing ratio of HPMTF in May 2018 obtained using the simulation MOD_noHetLossHPMTF. The circles represent measurements of HPMTF made during the ATom-4 mission along NASA DC-8 flight tracks, with a limit of detection of < 1 ppt.
For this campaign, flight patterns covered vertical profiles from 0.2 to 14 km above the ocean surface. The flight leg duration was 5 min and the boundary layer altitude was 150 to 200 m above the ocean surface. Since most of these measurement days were within the month of May 2018, here we compare observations with modeled output of mean surface concentration of HPMTF for this month. With the rate of isomerization reaction used in previous work, we find that spatial patterns of monthly mean surface concentrations are generally well captured (Jernigan et al., 2022a). Overall, we find that simulation MOD_noHetLossHPMTF results in better agreement with the existing overprediction for the vertical profile (Fig. 3b) and global surface-layer HPMTF levels (Fig. 4) compared to previous modeling approaches using the CAM-chem model (Veres et al., 2020).
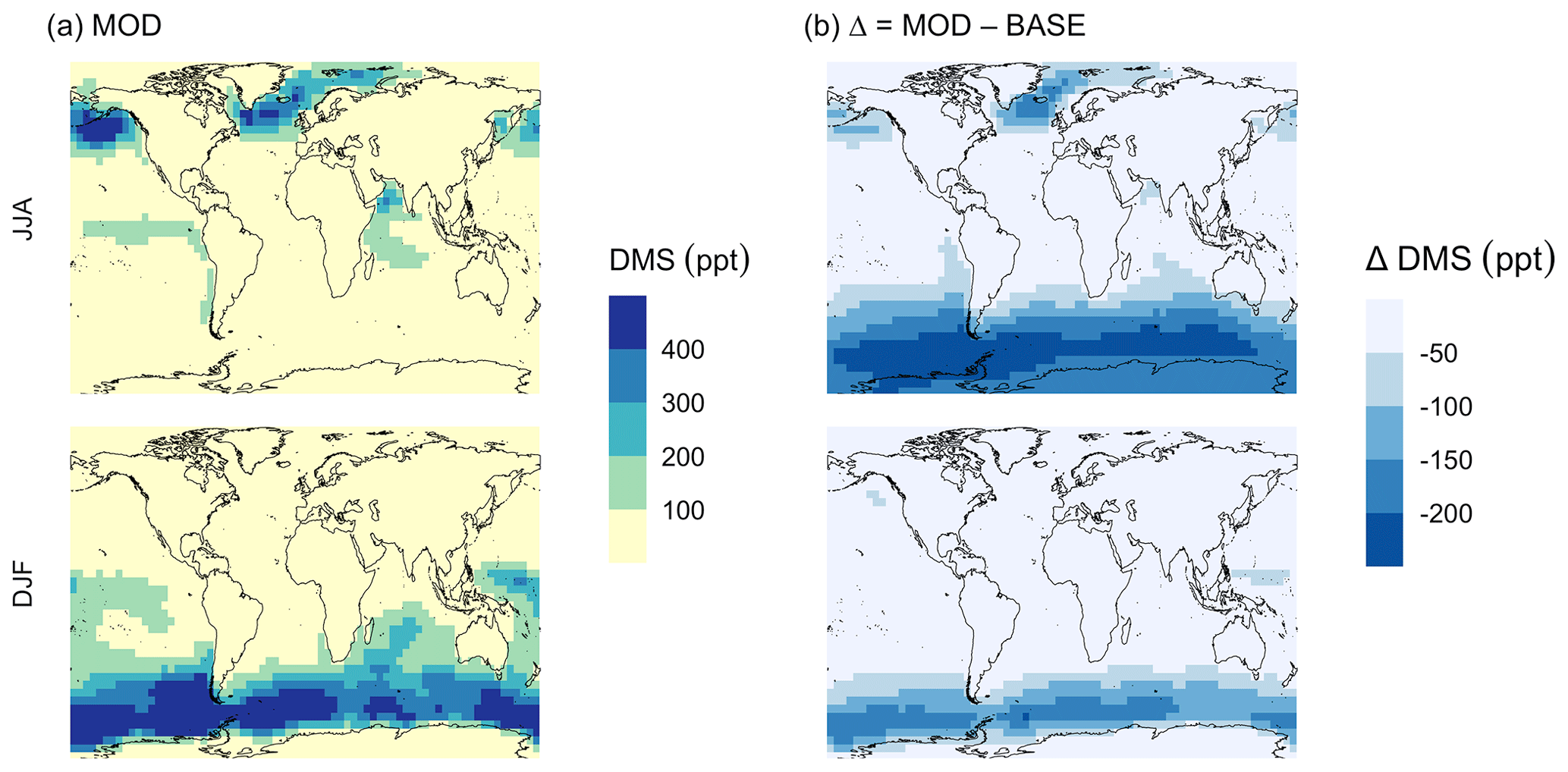
Figure 5Geographic distributions of (a) the mean surface DMS mixing ratio (ppt) for simulation MOD and (b) the difference in this parameter between simulation MOD and its baseline (Δ = MOD − BASE from GEOS-Chem simulations). Here, JJA and DJF represent June–July–August and December–January–February, respectively. The simulations are described in Table 5.
3.2 DMS burden and oxidation pathways
We find that the global burden of DMS in the MOD simulation is 65 Gg S (Table B1), 40 % lower than what we find with simulation BASE (108 Gg S). Even with this 42 % reduction, global burdens are still well within the range of 9.6–150 Gg S suggested in other studies (Faloona, 2009; Kloster et al., 2006). Figure 5a shows that surface DMS mixing ratios are highest in the North Pacific and North Atlantic oceans for June–July–August (JJA) and in the Southern Ocean during the months of December–January–February (DJF), revealing the underlying seasonality of DMS emissions. According to previous studies, the highest DMS concentrations usually occur in summer months due to higher rates of primary production in the presence of adequate solar irradiation and high temperatures for both hemispheres (Galí et al., 2018; Lana et al., 2011; Wang et al., 2020). In simulation MOD, the global mean surface-layer DMS burden was higher in the Southern Hemisphere for DJF and lower in the Northern Hemisphere for JJA, which is due to the larger ocean area in the SH than in the NH. We also find that the reactions of this expanded DMS oxidation mechanism collectively contribute to reductions in mean surface-layer DMS concentration of 58 % and 22 % compared to BASE for JJA and DJF, respectively (Fig. 5b). These reductions are due primarily to the addition of multiple new chemical loss pathways compared to BASE, which are especially impactful during the months JJA due to the elevated BrO in the SH winter, and due to higher O3 and OH concentrations in the NH summer compared to the SH summer (Zhang et al., 2018; Pound et al., 2020).
As shown in Fig. 5b, this DJF DMS reduction is seen mainly over the Southern Ocean and is largely attributable to faster chemical losses through the added reactions of DMS + BrO and DMS(aq) + O3(aq), which in earlier work was hypothesized as a possible reason for high model biases in the absence of detailed halogen chemistry (Chen et al., 2016). The global lifetime of DMS decreases from 1.5 d in the BASE simulation to 0.9 d in the MOD simulation.
These values are comparable to the range of 0.8–2.1 d reported by previous studies (Chen et al., 2018; Fung et al., 2022). The global DMS emission flux (FDMS) from the ocean to the atmosphere is 22 Tg S yr−1 and is within the range of 11–28 Tg S yr−1 simulated by GEOS-Chem and other models in previous studies (Lennartz et al., 2015; Fung et al., 2022; Chen et al., 2018; Hezel et al., 2011; Spracklen et al., 2005). Our FDMS is higher than the 18 Tg S yr−1 reported by Chen et al. (2018), who used sea surface DMS concentrations from Kettle et al. (1999); this indicates that DMS emissions vary with changes in sea surface DMS climatology. Analyzing and improving DMS emission measurements directly is not a part of this work, but we note that improved and validated inventories for DMS will certainly play a role in subsequent oxidation product comparisons. We recommend that there should be an ongoing evaluation of DMS emission inputs to complement the expanded chemical mechanism development we present here.
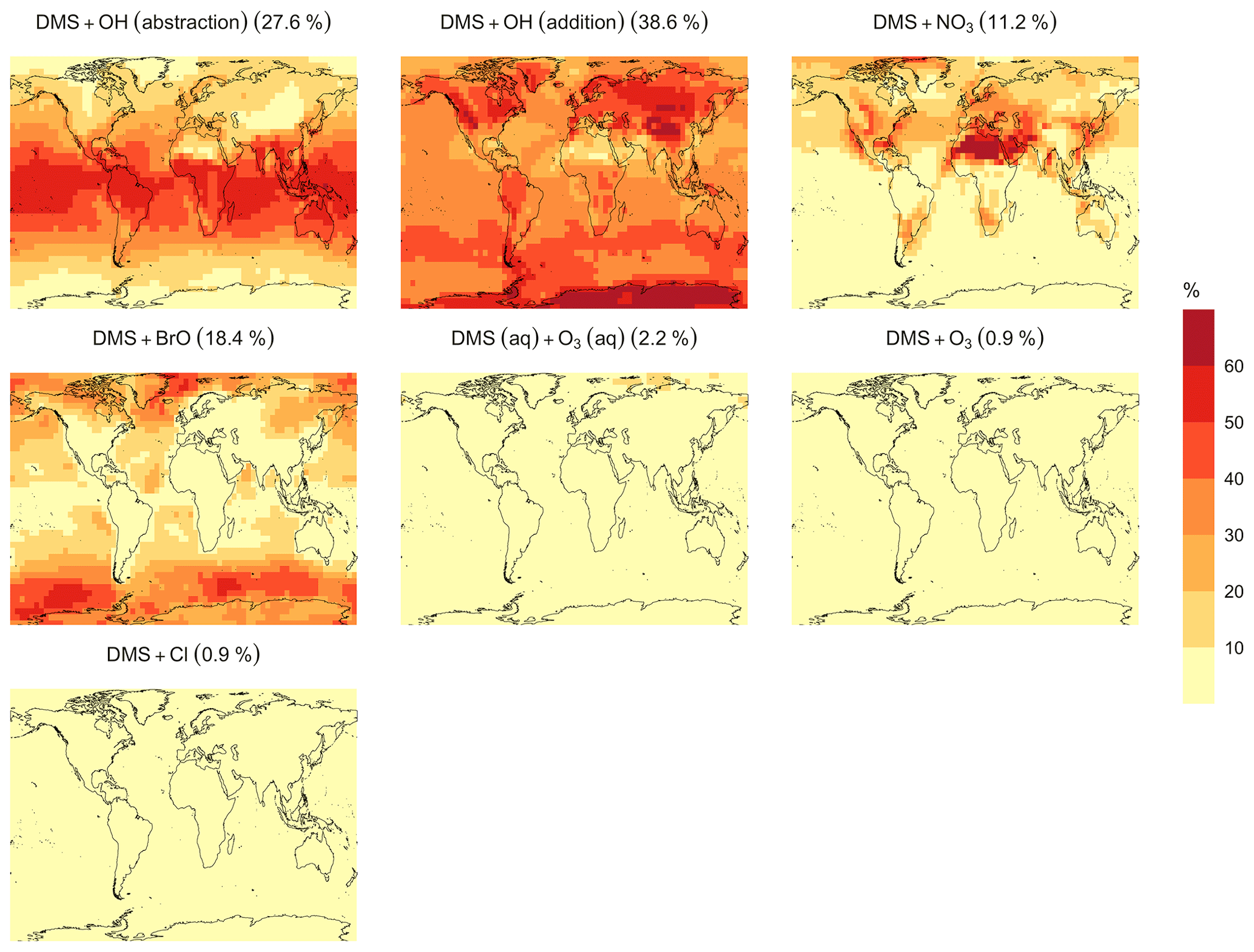
Figure 6Geographic distributions of the annual mean surface-layer fractions of total DMS oxidation (%) attributed to different tropospheric oxidants by simulation MOD (described in Table 5). Percentages in parentheses indicate the average contribution of the fraction of DMS emitted in each reaction pathway presented here to the global chemical loss.
In the BASE simulation, the chemical loss of DMS is its only sink (as opposed to dry and wet deposition), leading to the full conversion of DMS into SO2 (82.5 %) and MSA (17.5 %) (Fig. A3a). Figure 6 shows that in simulation MOD with the updated DMS oxidation scheme, DMS is mainly oxidized by OH in the gas phase, with 27.6 % of the losses proceeding via the H-abstraction channel and 38.6 % via the OH-addition pathway, which together contribute up to 66.2 % of the global average loss, with a high regional contribution over the tropical oceans via the abstraction channel, where surface OH is the highest. NO3 oxidation of DMS accounts for another 11.2 % of the global DMS chemical losses, comparable to values found in previous studies (Chen et al., 2018; Fung et al., 2022). Over the ocean, the NO3 loss pathway is strongest in the NH coastal regions due to the outflow of NOx sources from over the land, whereas values are generally less than 10 % for the SH. Oxidation by BrO is responsible for 18.4 % of the global DMS removal, which falls within the previously estimated range of 8 %–29 % (Boucher et al., 2003; Khan et al., 2016; Chen et al., 2018). Regionally, its contribution can reach 50 %–60 % over high latitudes of the Southern Hemisphere as well as to the north near the Arctic Ocean, consistent with previous box model studies based on the availability of high BrO and low OH and NO3 for those regions (Hoffmann et al., 2016). DMS + O3 accounts for 2.2 % (aqueous) and 0.9 % (gas phase) of the global surface DMS loss. The higher contribution from BrO and the lower one from O3 with this mechanism compared to some previous studies could be explained in part by the recently implemented sea salt debromination mechanism in GEOS-Chem, which results in a much higher background level of BrO as well as a lower O3 abundance, especially in the Southern Hemisphere (Boucher et al., 2003; Chen et al., 2018; Fung et al., 2022; Sherwen et al., 2016a; Wang et al., 2021). To further quantify the importance of the sea salt debromination mechanism, we perform an emissions sensitivity test by turning this emission source off while using the updated MOD chemistry (Fig. A1). As would be expected, these simulations show much lower BrO formation (as shown in Fig. A6) and resulting chemical impacts, with overall oxidation contributions comparable to previous literature (Schmidt et al., 2016; Wang et al., 2021). We find that, under this scenario, the relative contribution of BrO to the DMS loss decreases to 2.2 %, while the DMS + O3 pathway increases to 43.3 % (aqueous) and 1.4 % (gas phase) and the DMS + OH pathway increases to 31.0 % (abstraction) and 48.0 % (addition) of the global surface DMS loss (Fig. A1). The DMS loss via interaction with NO3 also increases to 2.0 % when sea salt debromination is turned off in the mechanism. The relative contributions of other oxidants remain mostly unaffected in the BrO sensitivity test.
Regionally, the fractional contribution of aqueous-phase DMS + O3 to DMS oxidation can be up to 10 %–20 % over high-latitude oceans, especially with the sea salt debromination turned off (Fig. A1). This is in the middle of the 5 %–30 % contribution to high-latitude DMS losses previously reported (Chen et al., 2018; Fung et al., 2022; von Glasow and Crutzen, 2004). The Cl oxidation reaction contributes about 0.9 % with or without sea salt debromination to the chemical removal of DMS, consistent with some previous studies (Atkinson et al., 2004; Fung et al., 2022). This does differ from other reported values, however, including those from a global model study (4 %) and box model simulations (8 %–18 %) (Chen et al., 2018; Hoffmann et al., 2016; von Glasow and Crutzen, 2004). It is worth noting that none of the studies reporting such high Cl contributions included HPMTF formation and loss. Ongoing uncertainties associated with the model–observation bias for Cl should be further resolved to allow better representation of halogenated species' contributions to DMS loss (Wang et al., 2021). Due to slower reaction kinetics and the lower fractional contribution (reported earlier) compared to BrO with DMS and the uncertainty in the surface concentrations and kinetics for photochemically generated halogenated species such as Br and IO, we did not include them in our chemical scheme (Chen et al., 2018).
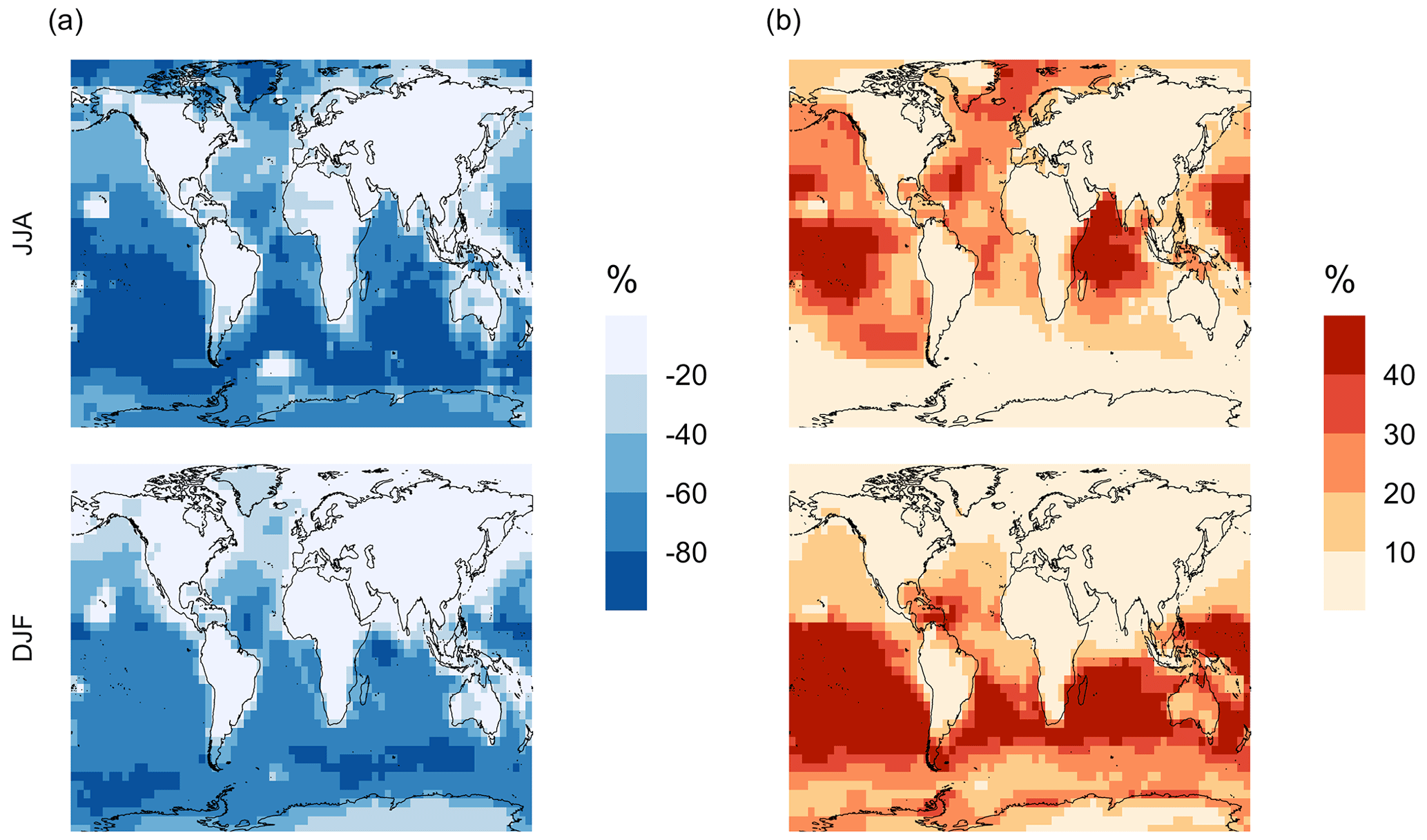
Figure 7Percent change in simulated surface-layer (a) SO2 and (b) for simulation MOD relative to BASE. Mean values for the two periods June, July and August (JJA) and December, January and February (DJF) are shown. The simulations are described in Table 5.
3.3 Implications of the extended DMS oxidation mechanism
Figure 7 shows that the MOD simulation results in a 40 % reduction of surface layer SO2 relative to BASE but a huge increase in in most regions. These changes suggest that the combination of gas-phase and aqueous-phase reactions results in a higher net yield of MSA and HPMTF and a lower net yield of gas-phase SO2. Additionally, comparison of simulation MOD to MOD_noHetLossHPMTF (Fig. A2a) shows that the loss of HPMTF in cloud droplets and aerosol reduces the global mean production of SO2 by 21.4 %, contributing to the SO2 reduction and increasing mean surface layer sulfate by 12.4 % (Fig. A2b). This reduction in SO2 is expected to reduce the availability of gas-phase sulfuric acid for new particle formation by nucleation (Clarke et al., 1998a). Total increases over the ocean, however, because the increased production from the rapid loss of MSA and HPMTF in the aqueous phase offsets the reduced oxidation of SO2 (Fig. 7b). In addition to that, reduced gas-phase sulfur species such as CH3SO3 also contribute to sulfate formation in our mechanism, as in other works (Fung et al., 2022).
Qualitatively, the regions showing the highest percent changes in SO2 are consistent with previous studies that included HPMTF chemistry and loss processes, though the extent of this reduction is much higher with the integrated mechanism used in our study (Fig. 7a) (Novak et al., 2021). The regions with the largest percent changes in SO2 reduction are those where DMS oxidation contributes most to SO2 and where HPMTF production and in-cloud oxidation of HPMTF are efficient. This spatial pattern thus helps us to identify where the production and heterogeneous loss of HPMTF and MSA are enhanced. One of the reactions that possibly contribute to delayed formation and reduction of SO2 concentration is first-generation OCS formation from OH oxidation of HPMTF. We find that the addition of cloud and aerosol loss significantly decreases the OCS production, especially in high-cloud-cover regions, as previously reported (Jernigan et al., 2022a). Even though the cloud loss of HPMTF increases the production of surface sulfate, the total global sulfate burden we calculate increases by only 6.5 % from the BASE sulfate burden of around 575 Gg S. This can be attributed to the minor contribution of DMS and its intermediate oxidation products to SO2 production compared to other non-DMS-derived sources. In addition, the production of stable intermediate oxidation products delays the conversion of SO2 to and modifies its spatial distribution in the marine environment. Thus, we should expect these aqueous-phase oxidation products to contribute to particle mass rather than increase the number of nucleated particles, as suggested in other studies (Clarke et al., 1998b; Novak et al., 2021; Williamson et al., 2019).
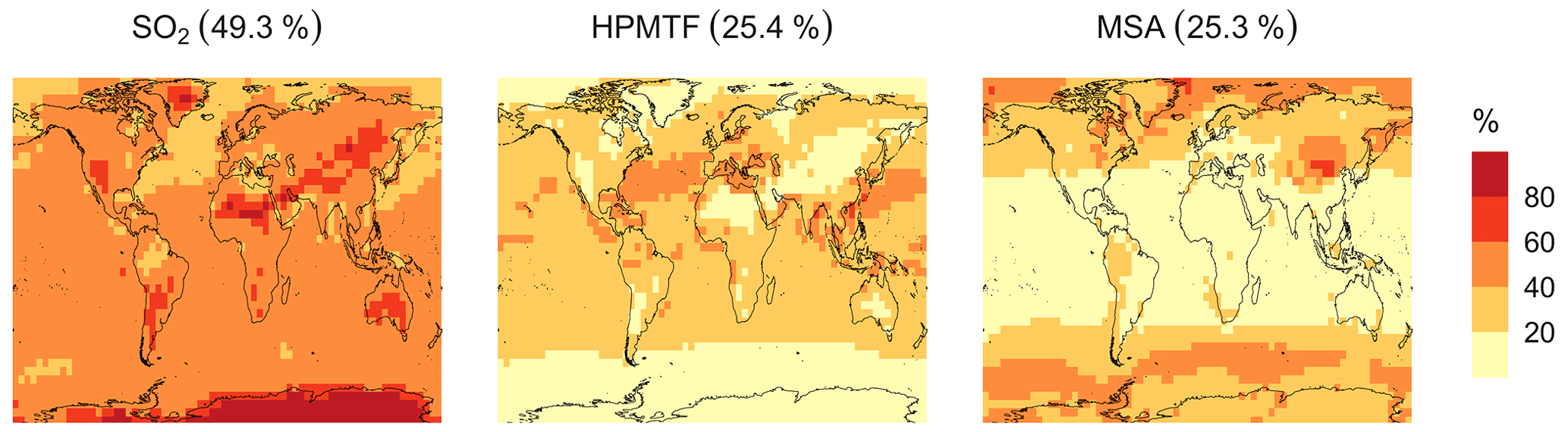
Figure 8Simulated branching ratios (in %) of the DMS oxidation mechanism calculated from the annual total production rates of the major terminal oxidation products SO2, HPMTF and MSA for simulation MOD.
The spatial distribution of product branching ratios for DMS oxidation is shown in Fig. 8. Here, 25.4 % of the annual total DMS oxidation will end up as HPMTF, while the final SO2 yield decreases to 49.3 %, compared to 82.5 % for the BASE simulation (Fig. A3a). The terminal HPMTF branch represents sulfur removed from the system by cloud and aerosol uptake of HPMTF, leading to reduced overall formation of SO2. With sea salt debromination turned off, the modified chemistry forms even more HPMTF (27.7 %) and slightly higher SO2 (51.3 %) and lowers the yield of MSA to 21.0 % (25.3 % with the sea salt debromination on), underscoring the importance of halogen chemistry for MSA production (Fig. A3b). These results are comparable with observationally constrained estimates from ATom-4 flight campaigns, where ∼ 30 %–40 % of the DMS was oxidized to HPMTF along their flight tracks, compared to 27.7 % for the full branch of HPMTF in the present work, as well as with previous modeling studies showing 33 % HPMTF formation as the terminating product (Veres et al., 2020; Fung et al., 2022). MSA is produced mostly by aqueous-phase oxidation of MSIA by O3 and OH according to the mechanism used here and has high abundance near the Southern Ocean and Antarctic belt, as reported by previous studies (Chen et al., 2018; Hoffmann et al., 2016; Fung et al., 2022). The global burden of MSA decreases dramatically, from 19 Gg S for BASE to 9.2 Gg S for simulation MOD. The higher rate of the major loss process or the lower rate of production of MSA from the aqueous phase reactions could be responsible for this reduction in the global budget (Fung et al., 2022).
3.3.1 Impact on aerosol size distributions
Based on the percent change observed in simulated surface-layer SO2 and for the modified DMS chemistry (Fig. 7), we further explore how this expanded DMS oxidation chemistry impacts modeled aerosol size distributions. Figure 9 shows the global mean surface-layer percent change in the normalized aerosol number concentration for the modified chemistry relative to the BASE simulation with and without cloud and aerosol HPMTF loss processes. The aerosol number concentration decreases for the sub-80 nm diameter size bins in both simulations, especially during the months DJF when cloud and aerosol loss pathways of HPMTF are included (the MOD case), demonstrating the negative impact of these processes on simulated new particle formation. Without these processes included (as in the MOD_noHetLossHPMTF case), percent changes are lower relative to simulation MOD but similar in terms of the directions of changes. On the other hand, the HPMTF lost to clouds and aerosols increases the simulated number of particles with diameters above 100 nm in the MOD simulation, consistent with the increase in sulfate mass concentrations shown in Fig. 7 and suggesting that heterogeneous HPMTF loss promotes simulated particle growth to diameters larger than 80–100 nm. The greater abundance of particles larger than 100 nm also acts as a condensation sink, further suppressing nucleation and growth in smaller size ranges.
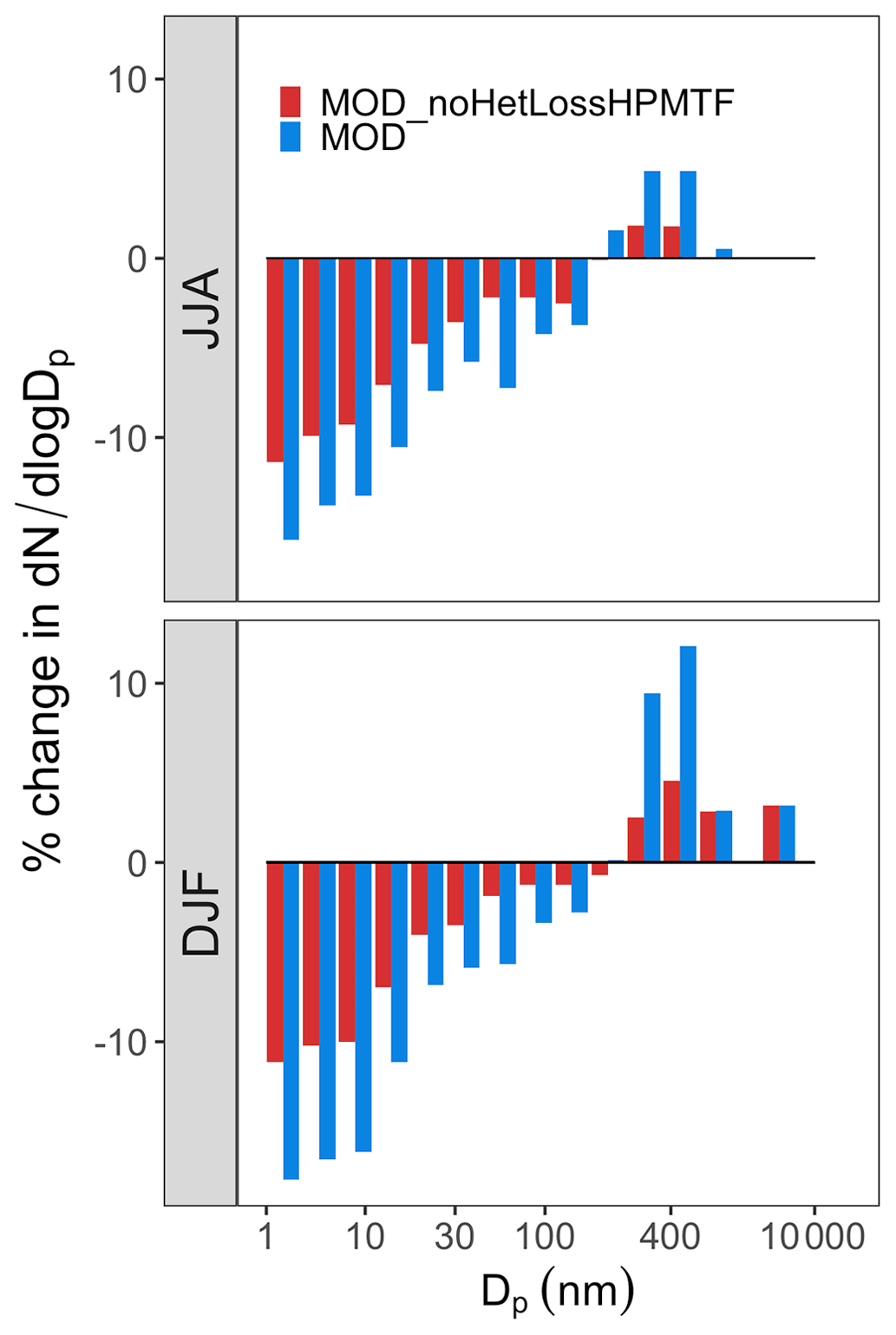
Figure 9Global mean surface-layer percent changes in normalized aerosol number concentration for different size bins with particle diameters Dp in the range of 3 nm < Dp < 10000 nm for simulations MOD and MOD_noHetLossHPMTF relative to simulation BASE. The simulations are described in Table 5.
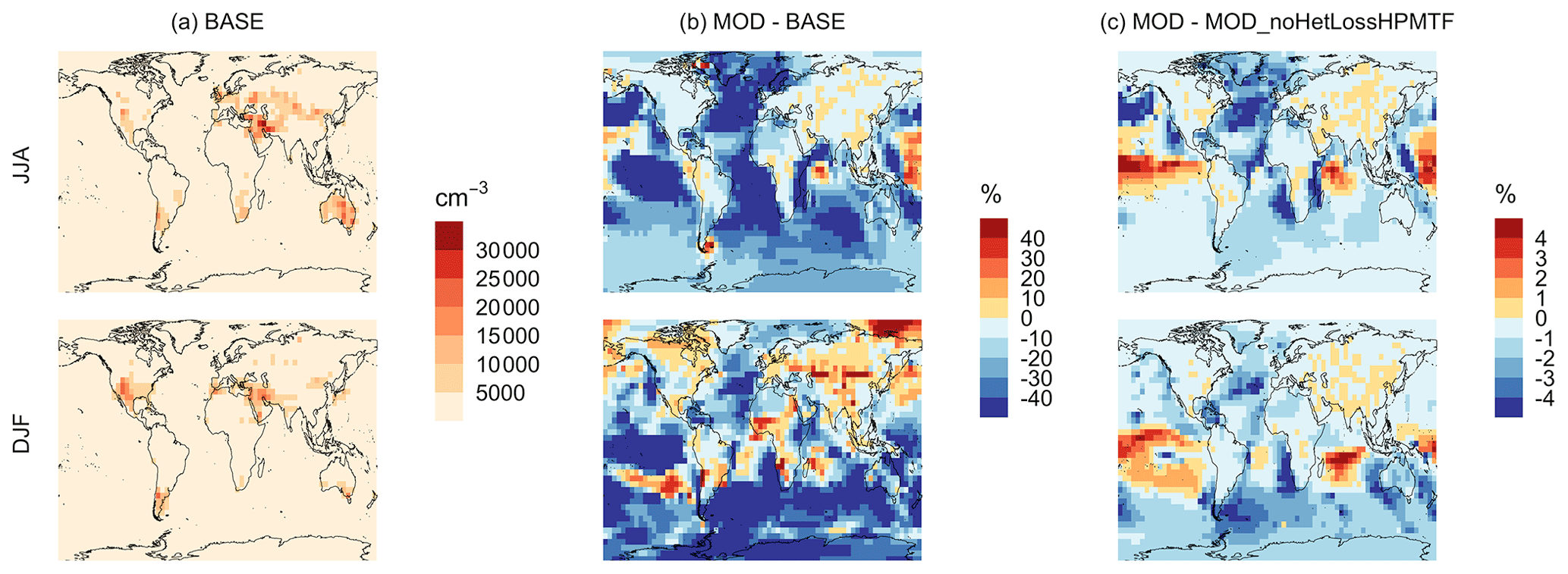
Figure 10Geographic distributions of (a) the seasonal-mean surface-layer aerosol number concentration in cm−3 (for particles with diameters between 3–80 nm) for the BASE simulation, (b) the percent difference in this parameter between MOD and BASE, and (c) the percent difference in this parameter between MOD and MOD_noHetLossHPMTF to show the role of cloud and aerosol loss of HPMTF. The top and bottom rows correspond to the months of JJA and DJF, respectively. The simulations are described in Table 5.
The geographic distributions of surface-layer aerosol number concentration for aerosol in the size range of 3–80 nm during two seasons are shown in Fig. 10. We find that global mean aerosol number concentration in this size range decreases for simulations MOD and MOD_noHetLossHPMTF relative to BASE by 16.8 % and 11.7 %, respectively. Decreases are greater for simulation MOD (Fig. 10b). Figure 10c shows the effect of HPMTF heterogeneous loss processes on the number of particles with diameters between 3–80 nm for simulation MOD relative to simulation MOD_noHetLossHPMTF. The largely negative impact of HPMTF loss to clouds and aerosols on sub-80 nm particle number is contributed to by enhanced direct sulfate formation on pre-existing particles, bypassing gas-phase SO2 formation (a precursor for new particle formation). As well, in the model, new particles grow through the condensation of H2SO4 and organics, and their growth is dependent on the condensation sink, while the loss of particle number depends on the coagulation sink. Thus, changes to the condensation and coagulation sinks and sulfuric acid production rate through the updated mechanism will also alter the growth rates of small particles (sub-80 nm) as well as their coagulation loss rates. Hence, similar to the discussion for Fig. 9, the reduction of gas-phase production of H2SO4 in MOD relative to BASE slows new-particle formation and growth, while the additional production of sulfate through aqueous chemistry on larger particles in MOD increases the coagulational scavenging of the newly formed particles. These two effects synergistically reduce the concentration of ultrafine particles in the model. The fraction of newly formed particles that can reach the size of CCN is dependent on the particle growth rates, especially for particle sizes below 10 nm, where we see the highest coagulation losses to larger particles. The sensitivity of these results to the new sea salt debromination parameterization is shown in Fig. A4, where we find a regional increase in aerosol number concentration at mid- to higher latitudes in the SH despite low BrO concentrations (Fig. A4).
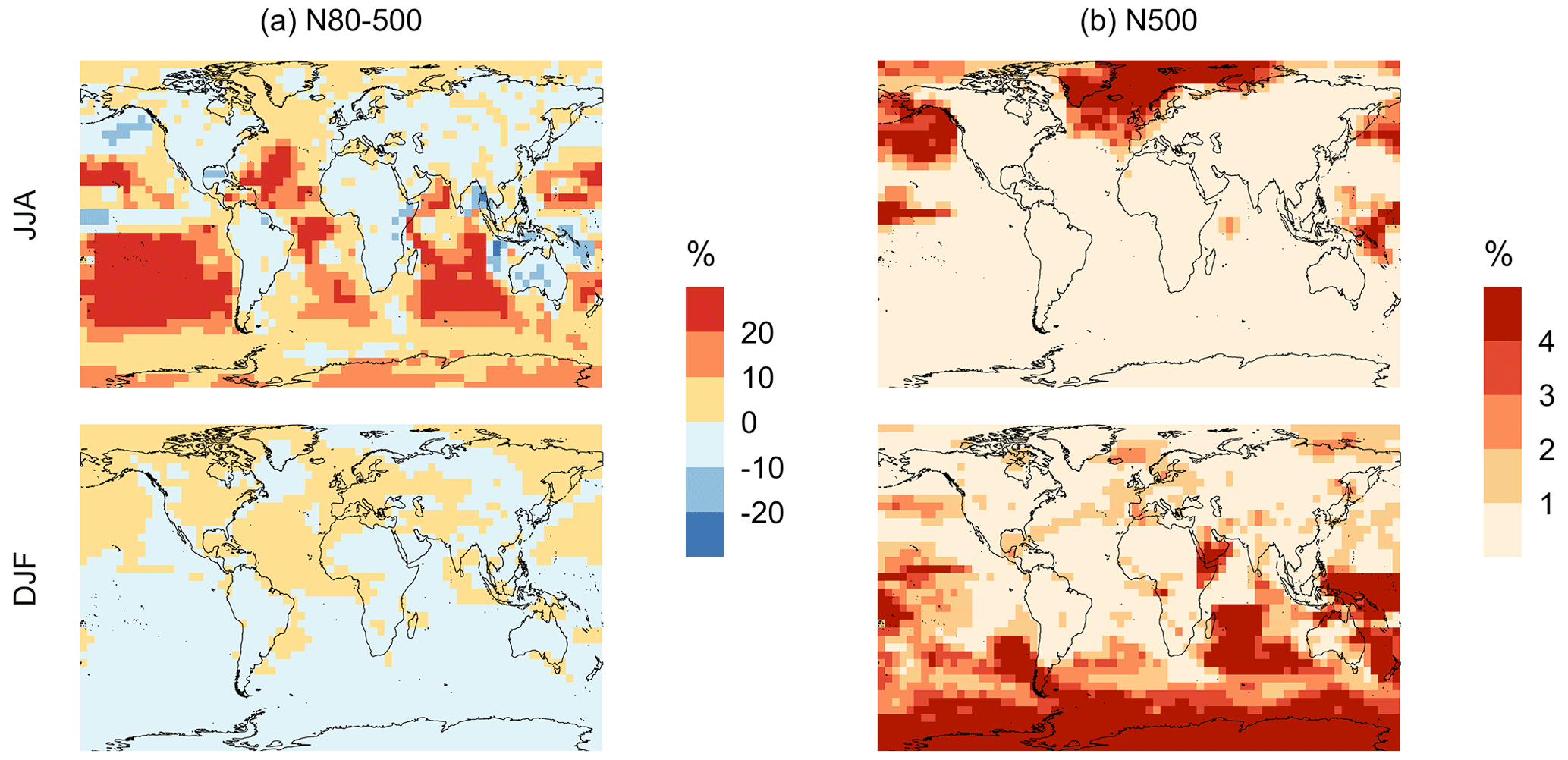
Figure 11Geographic distributions of the percent difference in seasonal-mean surface-layer aerosol number concentration (in cm−3) for simulation MOD relative to simulation BASE for diameters (a) between 80–500 nm and (b) > 500 nm. The simulations are described in Table 5.
Finally, we also analyze the impact of this expanded DMS scheme on particles larger than 80 nm (Fig. 11). We find increases of around 6.7 % for the JJA mean surface layer number concentration of aerosol with diameters between 80–500 nm, while the mean reduction is −5.4 % for DJF despite largely positive changes in the marine NH for these months (Fig. 11a). However, for the >500 nm size range (Fig. 11b), the global mean surface-layer number concentration of aerosol mostly increases, with the greatest changes occurring in the areas of peak DMS emission in both hemispheres during the summertime season. A similar trend is observed in the absence of cloud and aerosol HPMTF uptake in simulation MOD_noHetLossHPMTF (Fig. A5). Overall, the global annual mean number of particles with diameters larger than 80 nm increases by about 3.8 %.
Comparing the regional extents and directions of change in particle number concentration, we find that the net increase in particle number concentration is higher for MOD compared to MOD_noHetLossHPMTF, highlighting the importance of HPMTF loss processes to clouds and aerosols as a contributor of CCN.
In this study we have updated the default DMS oxidation scheme in the GEOS-Chem model by implementing an integrated oxidation mechanism. The new scheme includes gas-phase and aqueous-phase reactions involving DMSO, MSIA and HPMTF formation as well as newly identified HPMTF loss processes yielding considerable changes in the seasonal concentrations of major oxidation products and sulfur-derived aerosols. With this new chemistry scheme, the global annual mean surface DMS concentration decreases by 36 % relative to the BASE scheme in GEOS-Chem globally due to the presence of additional loss processes in the integrated mechanism, reducing the bias with respect to ATom-4 DMS measurement.
In this new scheme, OH, BrO, O3 and NOx species act as important sinks of DMS, contributing 66.2 %, 18.4 %, 3.1 % and 11.2 % of the global annual mean surface DMS loss and highlighting the relative importance of these loss processes in determining the surface DMS budget. We also find that at higher latitudes, gas-phase and multiphase oxidation of DMS by O3 and BrO become important in determining the budget of DMS. On the other hand, overall, OH is responsible for the largest loss of DMS via the addition and abstraction reactions relative to other sinks that contribute more from the addition reaction than the abstraction reaction. For the global distribution of simulated HPMTF, our updated scheme in GEOS-Chem provides a reduced high bias with respect to observations compared to previous studies. While emissions of BrO are uncertain in this version of GEOS-Chem, we find that the compound acts as a key sink of DMS, especially over the Southern Ocean. Overall, we find a large reduction in SO2 (40 %) and an increase in sulfate (17 %) due to the addition of heterogeneous HPMTF loss processes.
The lower SO2 obtained with the new DMS chemistry scheme contributes to a reduction of 16.8 % in the global annual mean surface-layer number concentration of particles with diameters less than 80 nm, contributed to by reductions in gas-phase precursors for new particle formation. There is a concurrent increase of 3.8 % in the global annual mean number of particles with diameters larger than 80 nm. This latter global mean particle number change varies in sign seasonally, with a 6.7 % increase for JJA and a 5.4 % decrease for DJF. This decrease is dominated by Southern Hemisphere summertime changes connected to suppressed new particle formation/growth and enhanced coagulation following additional sulfate production through aqueous chemistry. Cloud loss processes related to HPMTF make key contributions to these simulated changes through the enhancement of the aqueous-phase particle growth of those particles large enough to act as CCN.
Although the increased chemical mechanism complexity described in this work will necessarily increase the model's computational cost (MOD simulation runtimes are approximately 16 % longer), this study highlights the value of including a more realistic chemical oxidation mechanism for DMS and its stable intermediates to allow a better representation of DMS-derived aerosol in the marine atmosphere as well as its seasonal size distributions. A reduced form of this reaction mechanism including key chemical species and pathways should be able to capture the key processes with less computational impact and will be a priority in future work.
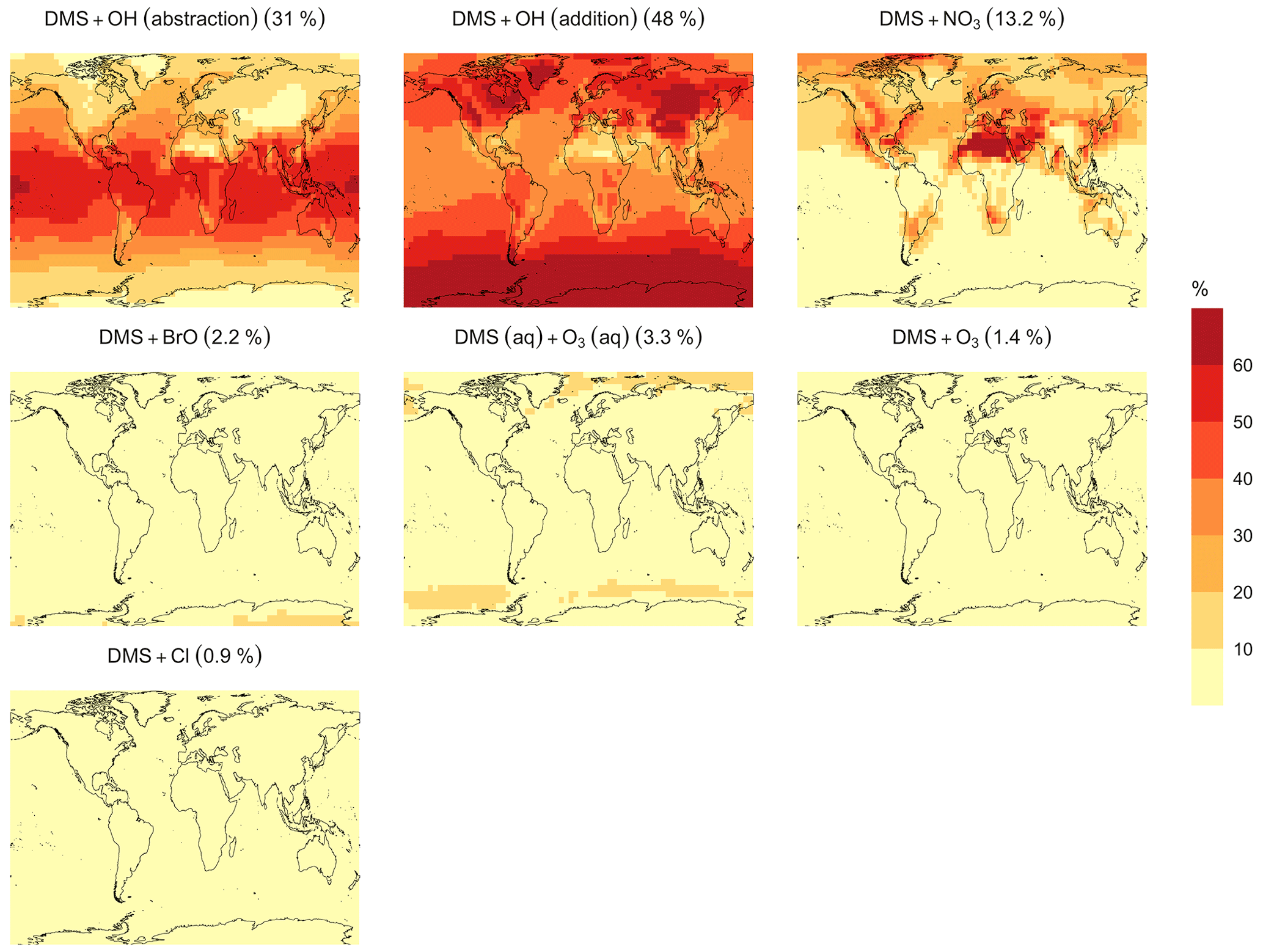
Figure A1Geographic distributions of the simulated annual mean surface-layer fractions of total DMS oxidation (in %) attributed to different tropospheric oxidants for a simulation with no sea salt debromination but which was otherwise the same as simulation MOD. Percentages in parentheses indicate the average contribution of each reaction pathway presented here to the global chemical loss of DMS. The simulations are described in Table 5.
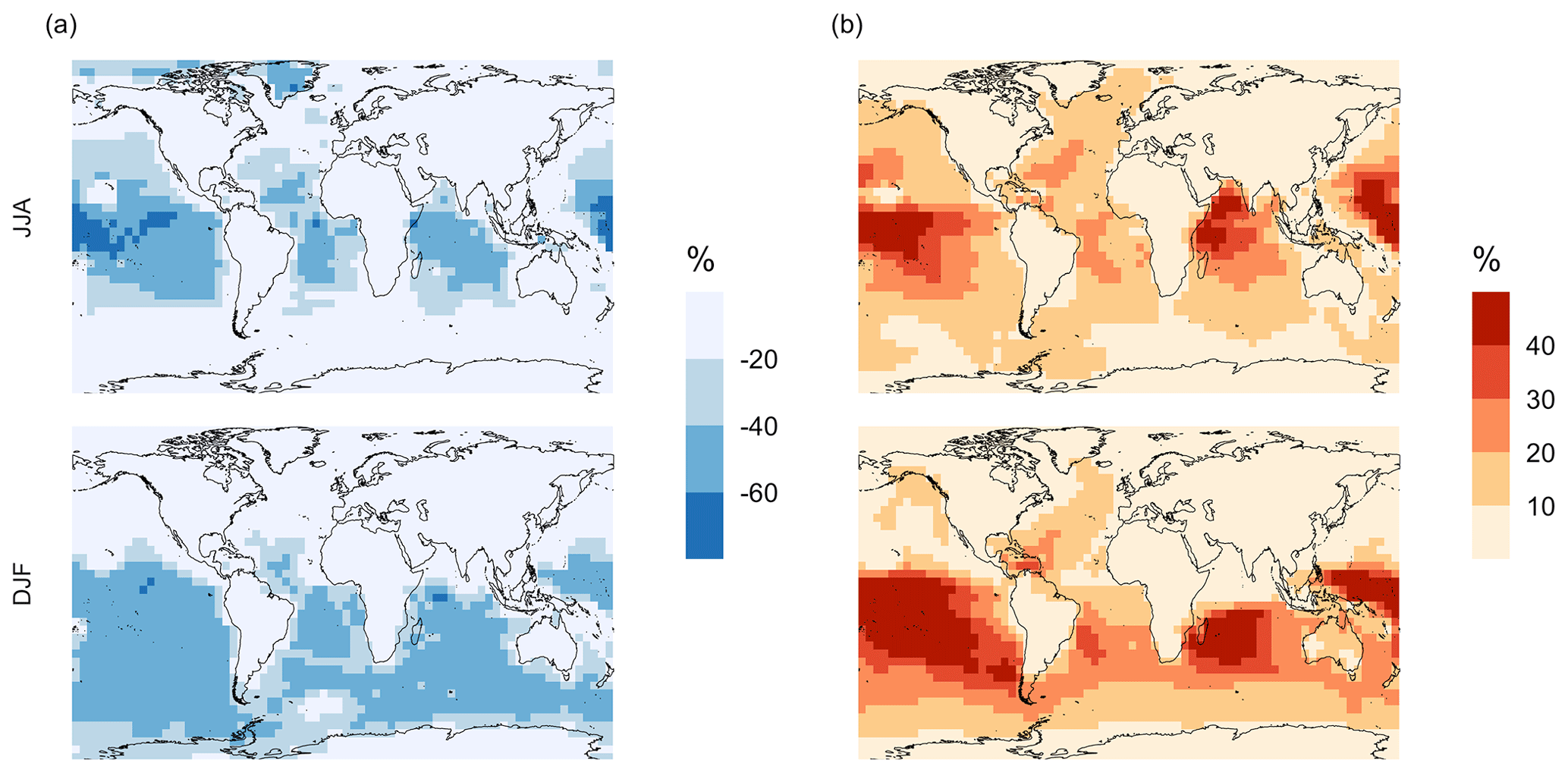
Figure A2Percent change in simulated surface-layer (a) SO2 and (b) for simulation MOD relative to MOD_noHetLossHPMTF. Mean values for the two periods June, July and August (JJA) and December, January, and February (DJF) are shown. The simulations are described in Table 5.
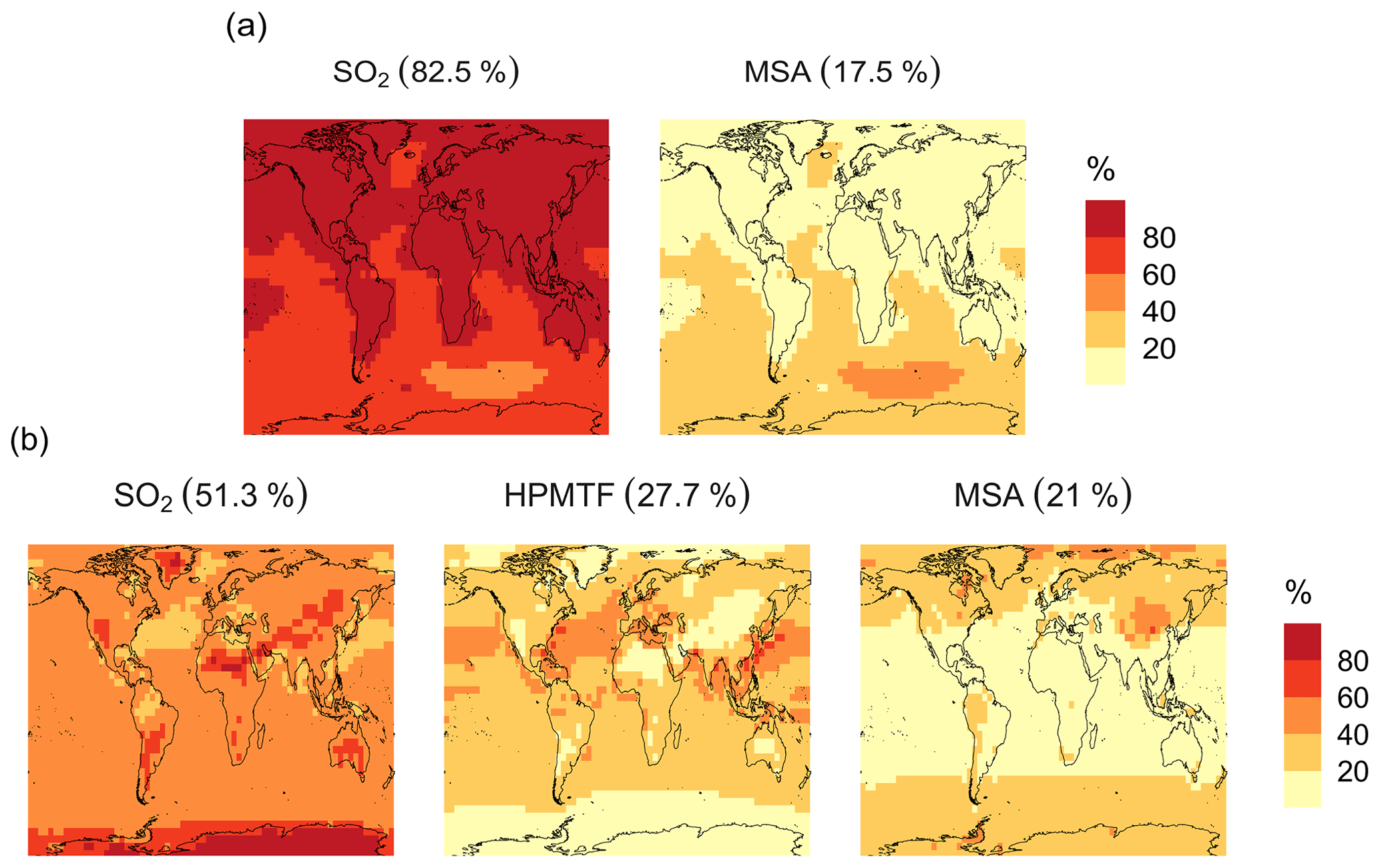
Figure A3Geographic distribution of the annual mean surface-layer branching ratios (in %) of the DMS oxidation mechanism calculated from the total production rates of the major oxidation products SO2, HPMTF and MSA for simulations similar to (a, top row) BASE and (b, bottom row) MOD except with no sea salt debromination. The simulations are described in Table 5.
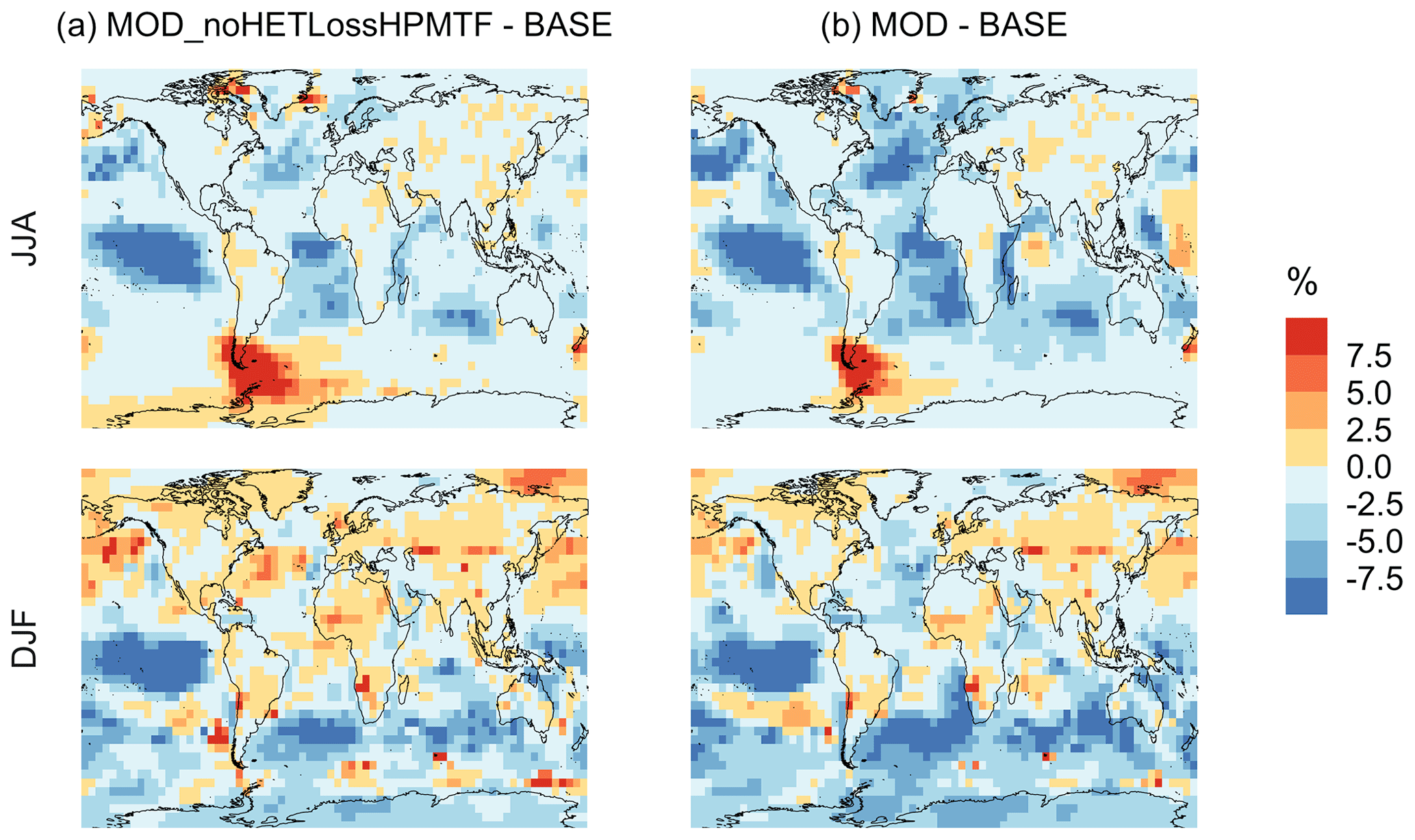
Figure A4Geographic distributions of the percent difference in seasonal-mean surface-layer aerosol number concentration (in cm−3 and for particles with diameters between 3–80 nm) between modified versions of the simulations (a) MOD_noHetLossHPMTF and BASE and (b) MOD and BASE, where the only modification made to the simulations described in Table 5 was to exclude sea salt debromination.
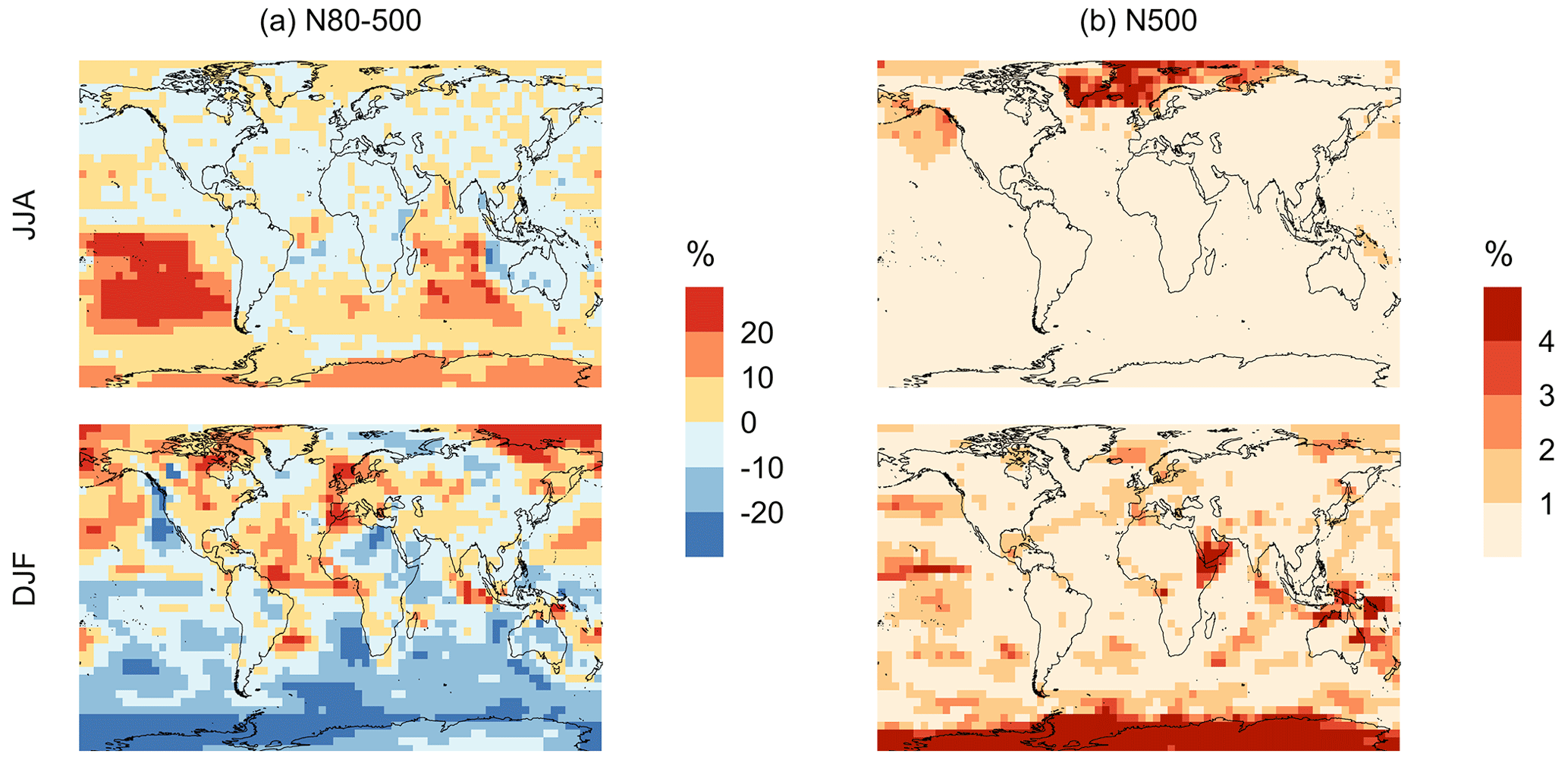
Figure A5Geographic distributions of the percent difference in seasonal-mean surface-layer aerosol number concentration in cm−3 for simulations MOD_noHetLossHPMTF relative to BASE simulations for particle diameters (a) between 80–500 nm and (b) > 500 nm. The simulations are described in Table 5.
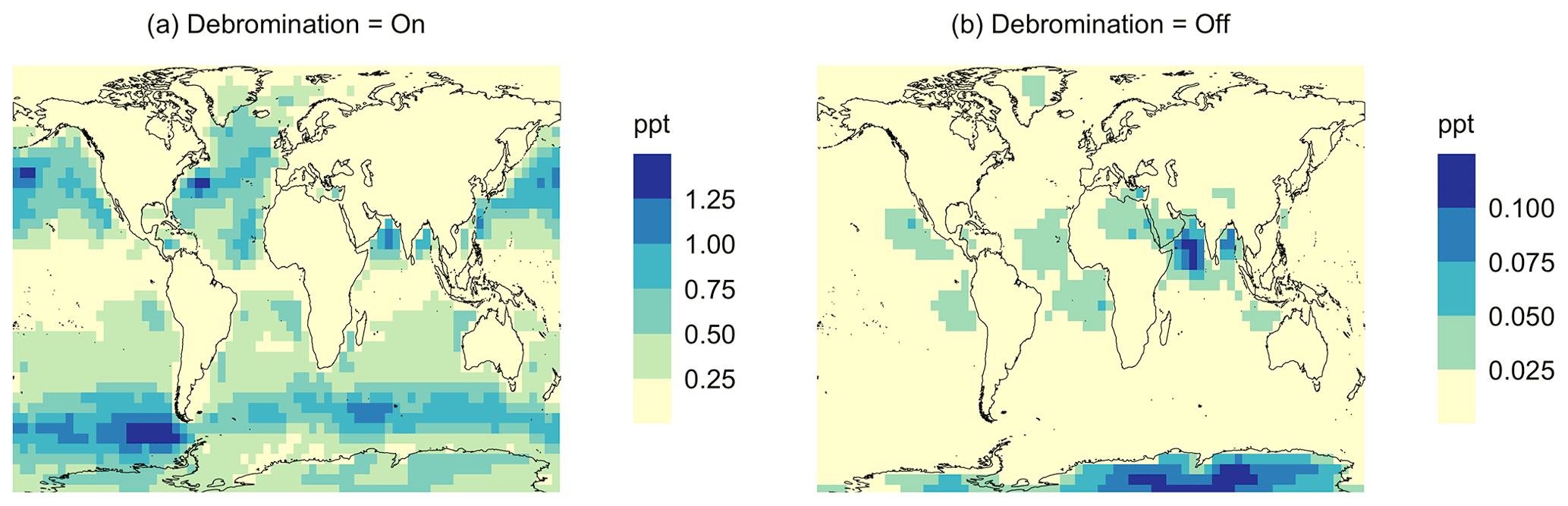
Figure A6Geographic distributions of the mean surface BrO mixing ratio (ppt) for simulation MOD (a) with sea salt debromination and (b) without sea salt debromination. The simulations are described in Table 5.
Figure A7Geographic distributions of (a) the mean surface oxidant concentration for simulation BASE and (b) the difference in mean surface oxidant concentration for MOD with respect to BASE. The simulations are described in Table 5.
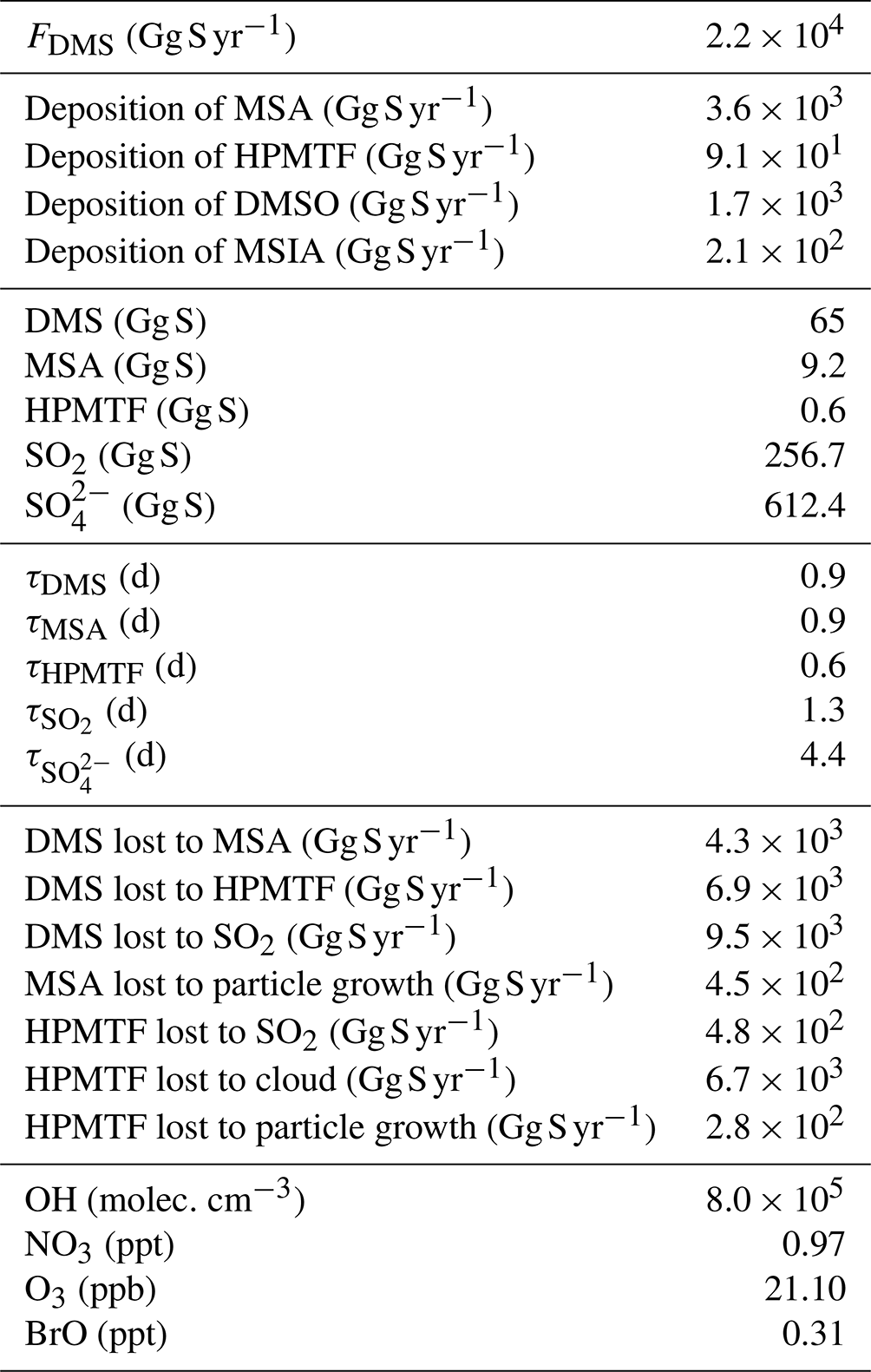
Table B1Global atmospheric flux, deposition, burdens, lifetimes of DMS and its oxidation products, chemical loss rates for specific reaction pathways, and global mean concentrations of major oxidants for simulation MOD. Note that SO2 and include natural as well as anthropogenic sources.
The DMS observational data in Fig. 2 were obtained from the referenced papers (Kouvarakis and Mihalopoulos, 2002; Castebrunet et al., 2009). The observational data obtained during ATom-4 (Novak et al., 2021; Wollesen de Jonge et al., 2021) are published through the Distributed Active Archive Center for Biogeochemical Dynamics (DAAC) at https://doi.org/10.3334/ORNLDAAC/1921 (Veres et al., 2021) and https://doi.org/10.3334/ORNLDAAC/1890 (Rollins, 2021).
LT and WCP designed the research goals, aims and methodology and implemented the new code in GC-TOMAS. QC, BA, CHF and CDH contributed to code development. All authors provided expert advice on data analysis, interpretation and visualization. LT ran model simulations, analyzed the data, created the figures, and led the manuscript development and editing.
The contact author has declared that none of the authors has any competing interests.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
Linia Tashmim and William C. Porter gratefully acknowledge Ka Ming Fung for discussions on DMS oxidation chemistry. Betty Croft thanks Rachel Y.-W. Chang for discussions on marine aerosols.
Linia Tashmim and William C. Porter were supported by NSF (grant no. 2155192). Qianjie Chen was supported by the Hong Kong Research Grants Council (grant nos. 15223221 and 15219722). Becky Alexander was supported by NSF (grant no. AGS 2109323) and (grant no. PLR 1904128). Charles H. Fite was supported by NASA FINESST (grant no. 80NSSC19K1368). Christopher D. Holmes acknowledges funding support from NSF AGS (grant 1848372). Betty Croft gratefully acknowledges research funding supported by the Ocean Frontier Institute through an award from the Canada First Research Excellence Fund. Jeffrey R. Pierce was supported by the Atmospheric System Research (ASR) program, part of the US Department of Energy's Office of Biological and Environmental Research within the Office of Science (under grant no. DE-SC0021208). Sakiko Ishino was supported by Ferring Pharmaceuticals through the Extreme Environments Research Laboratory, École Polytechnique Fédérale de Lausanne (EPFL).
This paper was edited by Ashu Dastoor and reviewed by three anonymous referees.
Adams, P. J. and Seinfeld, J. H.: Predicting global aerosol size distributions in general circulation models, J. Geophys. Res.-Atmos., 107, AAC 4-1–AAC 4-23, https://doi.org/10.1029/2001JD001010, 2002..
Alexander, B., Park, R. J., Jacob, D. J., Li, Q. B., Yantosca, R. M., Savarino, J., Lee, C. C. W., and Thiemens, M. H.: Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes, J. Geophys. Res.-Atmos., 110, D10307, https://doi.org/10.1029/2004JD005659, 2005.
Alexander, B., Park, R. J., Jacob, D. J., and Gong, S.: Transition metal-catalyzed oxidation of atmospheric sulfur: Global implications for the sulfur budget, J. Geophys. Res.-Atmos., 114, D02309, https://doi.org/10.1029/2008JD010486, 2009.
Amos, H. M., Jacob, D. J., Holmes, C. D., Fisher, J. A., Wang, Q., Yantosca, R. M., Corbitt, E. S., Galarneau, E., Rutter, A. P., Gustin, M. S., Steffen, A., Schauer, J. J., Graydon, J. A., Louis, V. L. St., Talbot, R. W., Edgerton, E. S., Zhang, Y., and Sunderland, E. M.: Gas-particle partitioning of atmospheric Hg(II) and its effect on global mercury deposition, Atmos. Chem. Phys., 12, 591–603, https://doi.org/10.5194/acp-12-591-2012, 2012.
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., and Troe, J.: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I - gas phase reactions of Ox, HOx, NOx and SOx species, Atmos. Chem. Phys., 4, 1461–1738, https://doi.org/10.5194/acp-4-1461-2004, 2004.
Bardouki, H., da Rosa, M. B., Mihalopoulos, N., Palm, W.-U., and Zetzsch, C.: Kinetics and mechanism of the oxidation of dimethylsulfoxide (DMSO) and methanesulfinate (MSI-) by OH radicals in aqueous medium, Atmos. Environ., 36, 4627–4634, https://doi.org/10.1016/S1352-2310(02)00460-0, 2002.
Barnes, I., Hjorth, J., and Mihalopoulos, N.: Dimethyl Sulfide and Dimethyl Sulfoxide and Their Oxidation in the Atmosphere, Chem. Rev., 106, 940–975, https://doi.org/10.1021/cr020529+, 2006.
Barone, S. B., Turnipseed, A. A., and Ravishankara, A. R.: Role of adducts in the atmospheric oxidation of dimethyl sulfide, Faraday Discuss., 100, 39, https://doi.org/10.1039/fd9950000039, 1995.
Berndt, T., Scholz, W., Mentler, B., Fischer, L., Hoffmann, E. H., Tilgner, A., Hyttinen, N., Prisle, N. L., Hansel, A., and Herrmann, H.: Fast Peroxy Radical Isomerization and OH Recycling in the Reaction of OH Radicals with Dimethyl Sulfide, J. Phys. Chem. Lett., 10, 6478–6483, https://doi.org/10.1021/acs.jpclett.9b02567, 2019.
Berndt, T., Hoffmann, E. H., Tilgner, A., Stratmann, F., and Herrmann, H.: Direct sulfuric acid formation from the gas-phase oxidation of reduced-sulfur compounds, Nat. Commun., 14, 4849, https://doi.org/10.1038/s41467-023-40586-2, 2023.
Bey, I., Jacob, D. J., Yantosca, R. M., Logan, J. A., Field, B. D., Fiore, A. M., Li, Q., Liu, H. Y., Mickley, L. J., and Schultz, M. G.: Global modeling of tropospheric chemistry with assimilated meteorology: Model description and evaluation, J. Geophys. Res.-Atmos., 106, 23073–23095, https://doi.org/10.1029/2001JD000807, 2001.
Boniface, J., Shi, Q., Li, Y. Q., Cheung, J. L., Rattigan, O. V., Davidovits, P., Worsnop, D. R., Jayne, J. T., and Kolb, C. E.: Uptake of Gas-Phase SO2, H2S, and CO2 by Aqueous Solutions, J. Phys. Chem. A, 104, 7502–7510, https://doi.org/10.1021/jp000479h, 2000.
Borissenko, D., Kukui, A., Laverdet, G., and Le Bras, G.: Experimental Study of SO2 Formation in the Reactions of CH3SO Radical with NO2 and O3 in Relation with the Atmospheric Oxidation Mechanism of Dimethyl Sulfide, J. Phys. Chem. A, 107, 1155–1161, https://doi.org/10.1021/jp021701g, 2003.
Boucher, O., Moulin, C., Belviso, S., Aumont, O., Bopp, L., Cosme, E., von Kuhlmann, R., Lawrence, M. G., Pham, M., Reddy, M. S., Sciare, J., and Venkataraman, C.: DMS atmospheric concentrations and sulphate aerosol indirect radiative forcing: a sensitivity study to the DMS source representation and oxidation, Atmos. Chem. Phys., 3, 49–65, https://doi.org/10.5194/acp-3-49-2003, 2003.
Bräuer, P., Tilgner, A., Wolke, R., and Herrmann, H.: Mechanism development and modelling of tropospheric multiphase halogen chemistry: The CAPRAM Halogen Module 2.0 (HM2), J. Atmos. Chem., 70, 19–52, https://doi.org/10.1007/s10874-013-9249-6, 2013.
Breider, T. J., Chipperfield, M. P., Richards, N. a. D., Carslaw, K. S., Mann, G. W., and Spracklen, D. V.: Impact of BrO on dimethylsulfide in the remote marine boundary layer, Geophys. Res. Lett., 37, L02807, https://doi.org/10.1029/2009GL040868, 2010.
Burkholder, J. B., Sander, S. P., Abbatt, J. P. D., Barker, J. R., Cappa, C., Crounse, J. D., Dibble, T. S., Huie, R. E., Kolb, C. E., Kurylo, M. J., Orkin, V. L., Percival, C. J., Wilmouth, D. M., and Wine, P. H.: Chemical kinetics and photochemical data for use in atmospheric studies; evaluation number 19, JPL Publication: 19-5, Jet Propulsion Laboratory, Pasadena, 2020.
Cala, B. A., Archer-Nicholls, S., Weber, J., Abraham, N. L., Griffiths, P. T., Jacob, L., Shin, Y. M., Revell, L. E., Woodhouse, M., and Archibald, A. T.: Development, intercomparison, and evaluation of an improved mechanism for the oxidation of dimethyl sulfide in the UKCA model, Atmos. Chem. Phys., 23, 14735–14760, https://doi.org/10.5194/acp-23-14735-2023, 2023.
Campolongo, F., Saltelli, A., Jensen, N. R., Wilson, J., and Hjorth, J.: The Role of Multiphase Chemistry in the Oxidation of Dimethylsulphide (DMS). A Latitude Dependent Analysis, J. Atmos. Chem., 32, 327–356, https://doi.org/10.1023/A:1006154618511, 1999.
Carslaw, K. S., Lee, L. A., Reddington, C. L., Pringle, K. J., Rap, A., Forster, P. M., Mann, G. W., Spracklen, D. V., Woodhouse, M. T., Regayre, L. A., and Pierce, J. R.: Large contribution of natural aerosols to uncertainty in indirect forcing, Nature, 503, 67–71, https://doi.org/10.1038/nature12674, 2013.
Castebrunet, H., Martinerie, P., Genthon, C., and Cosme, E.: A three-dimensional model study of methanesulphonic acid to non sea salt sulphate ratio at mid and high-southern latitudes, Atmos. Chem. Phys., 9, 9449–9469, https://doi.org/10.5194/acp-9-9449-2009, 2009.
Charlson, R. J., Lovelock, J. E., Andreae, M. O., and Warren, S. G.: Oceanic phytoplankton, atmospheric sulphur, cloud albedo and climate, Nature, 326, 655–661, https://doi.org/10.1038/326655a0, 1987.
Chen, H., Ezell, M. J., Arquero, K. D., Varner, M. E., Dawson, M. L., Gerber, R. B., and Finlayson-Pitts, B. J.: New particle formation and growth from methanesulfonic acid, trimethylamine and water, Phys. Chem. Chem. Phys., 17, 13699–13709, https://doi.org/10.1039/C5CP00838G, 2015.
Chen, Q., Geng, L., Schmidt, J. A., Xie, Z., Kang, H., Dachs, J., Cole-Dai, J., Schauer, A. J., Camp, M. G., and Alexander, B.: Isotopic constraints on the role of hypohalous acids in sulfate aerosol formation in the remote marine boundary layer, Atmos. Chem. Phys., 16, 11433–11450, https://doi.org/10.5194/acp-16-11433-2016, 2016.
Chen, Q., Schmidt, J. A., Shah, V., Jaeglé, L., Sherwen, T., and Alexander, B.: Sulfate production by reactive bromine: Implications for the global sulfur and reactive bromine budgets, Geophys. Res. Lett., 44, 7069–7078, https://doi.org/10.1002/2017GL073812, 2017.
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018.
Chin, M., Jacob, D. J., Gardner, G. M., Foreman-Fowler, M. S., Spiro, P. A., and Savoie, D. L.: A global three-dimensional model of tropospheric sulfate, J. Geophys. Res.-Atmos., 101, 18667–18690, https://doi.org/10.1029/96JD01221, 1996.
Clarke, A. D., Davis, D., Kapustin, V. N., Eisele, F., Chen, G., Paluch, I., Lenschow, D., Bandy, A. R., Thornton, D., Moore, K., Mauldin, L., Tanner, D., Litchy, M., Carroll, M. A., Collins, J., and Albercook, G.: Particle Nucleation in the Tropical Boundary Layer and Its Coupling to Marine Sulfur Sources, Science, 282, 89–92, 1998a.
Clarke, A. D., Varner, J. L., Eisele, F., Mauldin, R. L., Tanner, D., and Litchy, M.: Particle production in the remote marine atmosphere: Cloud outflow and subsidence during ACE 1, J. Geophys. Res.-Atmos., 103, 16397–16409, https://doi.org/10.1029/97JD02987, 1998b.
Du, L., Xu, Y., Ge, M., Jia, L., Yao, L., and Wang, W.: Rate constant of the gas phase reaction of dimethyl sulfide (CH3SCH3) with ozone, Chem. Phys. Lett., 436, 36–40, https://doi.org/10.1016/j.cplett.2007.01.025, 2007.
Duncan Fairlie, T., Jacob, D. J., and Park, R. J.: The impact of transpacific transport of mineral dust in the United States, Atmos. Environ., 41, 1251–1266, https://doi.org/10.1016/j.atmosenv.2006.09.048, 2007.
Emerson, E. W., Hodshire, A. L., DeBolt, H. M., Bilsback, K. R., Pierce, J. R., McMeeking, G. R., and Farmer, D. K.: Revisiting particle dry deposition and its role in radiative effect estimates, P. Natl. Acad. Sci. USA, 117, 26076–26082, https://doi.org/10.1073/pnas.2014761117, 2020.
Enami, S., Nakano, Y., Hashimoto, S., Kawasaki, M., Aloisio, S., and Francisco, J. S.: Reactions of Cl Atoms with Dimethyl Sulfide: A Theoretical Calculation and an Experimental Study with Cavity Ring-Down Spectroscopy, J. Phys. Chem. A, 108, 7785–7789, https://doi.org/10.1021/jp049772y, 2004.
Faloona, I.: Sulfur processing in the marine atmospheric boundary layer: A review and critical assessment of modeling uncertainties, Atmos. Environ., 43, 2841–2854, https://doi.org/10.1016/j.atmosenv.2009.02.043, 2009.
Flyunt, R., Makogon, O., Schuchmann, M. N., Asmus, K.-D., and von Sonntag, C.: OH-Radical-induced oxidation of methanesulfinic acid. The reactions of the methanesulfonyl radical in the absence and presence of dioxygen, J. Chem. Soc. Perk. T. 2, 2001, 787–792, https://doi.org/10.1039/B009631H, 2001.
Fung, K. M., Heald, C. L., Kroll, J. H., Wang, S., Jo, D. S., Gettelman, A., Lu, Z., Liu, X., Zaveri, R. A., Apel, E. C., Blake, D. R., Jimenez, J.-L., Campuzano-Jost, P., Veres, P. R., Bates, T. S., Shilling, J. E., and Zawadowicz, M.: Exploring dimethyl sulfide (DMS) oxidation and implications for global aerosol radiative forcing, Atmos. Chem. Phys., 22, 1549–1573, https://doi.org/10.5194/acp-22-1549-2022, 2022.
Galí, M., Levasseur, M., Devred, E., Simó, R., and Babin, M.: Sea-surface dimethylsulfide (DMS) concentration from satellite data at global and regional scales, Biogeosciences, 15, 3497–3519, https://doi.org/10.5194/bg-15-3497-2018, 2018.
Galí, M., Devred, E., Babin, M., and Levasseur, M.: Decadal increase in Arctic dimethylsulfide emission, P. Natl. Acad. Sci. USA, 116, 19311–19317, https://doi.org/10.1073/pnas.1904378116, 2019.
Gershenzon, M., Davidovits, P., Jayne, J. T., Kolb, C. E., and Worsnop, D. R.: Simultaneous Uptake of DMS and Ozone on Water, J. Phys. Chem. A, 105, 7031–7036, https://doi.org/10.1021/jp010696y, 2001.
Herrmann, H., Zellner, R., Mirabel, P., Buxton, G., Salmon, A., Sehested, K., Holcman, J., and Brede, O.: Removal and interconversions of oxidants in the atmospheric aqueous phase, part 2 (RINOXA 2). Final report, Universität GH Essen, Institut für Physikalische und Theoretische Chemie, Essen, 1998.
Hezel, P. J., Alexander, B., Bitz, C. M., Steig, E. J., Holmes, C. D., Yang, X., and Sciare, J.: Modeled methanesulfonic acid (MSA) deposition in Antarctica and its relationship to sea ice, J. Geophys. Res.-Atmos., 116, D23214, https://doi.org/10.1029/2011JD016383, 2011.
Hodshire, A. L., Campuzano-Jost, P., Kodros, J. K., Croft, B., Nault, B. A., Schroder, J. C., Jimenez, J. L., and Pierce, J. R.: The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 19, 3137–3160, https://doi.org/10.5194/acp-19-3137-2019, 2019.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hoffmann, E. H., Heinold, B., Kubin, A., Tegen, I., and Herrmann, H.: The Importance of the Representation of DMS Oxidation in Global Chemistry-Climate Simulations, Geophys. Res. Lett., 48, e2021GL094068, https://doi.org/10.1029/2021GL094068, 2021.
Holmes, C. D., Bertram, T. H., Confer, K. L., Graham, K. A., Ronan, A. C., Wirks, C. K., and Shah, V.: The Role of Clouds in the Tropospheric NOx Cycle: A New Modeling Approach for Cloud Chemistry and Its Global Implications, Geophys. Res. Lett., 46, 4980–4990, https://doi.org/10.1029/2019GL081990, 2019.
Hoyle, C. R., Fuchs, C., Järvinen, E., Saathoff, H., Dias, A., El Haddad, I., Gysel, M., Coburn, S. C., Tröstl, J., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Corbin, J. C., Craven, J., Donahue, N. M., Duplissy, J., Ehrhart, S., Frege, C., Gordon, H., Höppel, N., Heinritzi, M., Kristensen, T. B., Molteni, U., Nichman, L., Pinterich, T., Prévôt, A. S. H., Simon, M., Slowik, J. G., Steiner, G., Tomé, A., Vogel, A. L., Volkamer, R., Wagner, A. C., Wagner, R., Wexler, A. S., Williamson, C., Winkler, P. M., Yan, C., Amorim, A., Dommen, J., Curtius, J., Gallagher, M. W., Flagan, R. C., Hansel, A., Kirkby, J., Kulmala, M., Möhler, O., Stratmann, F., Worsnop, D. R., and Baltensperger, U.: Aqueous phase oxidation of sulphur dioxide by ozone in cloud droplets, Atmos. Chem. Phys., 16, 1693–1712, https://doi.org/10.5194/acp-16-1693-2016, 2016.
Ishino, S., Hattori, S., Legrand, M., Chen, Q., Alexander, B., Shao, J., Huang, J., Jaeglé, L., Jourdain, B., Preunkert, S., Yamada, A., Yoshida, N., and Savarino, J.: Regional Characteristics of Atmospheric Sulfate Formation in East Antarctica Imprinted on 17O-Excess Signature, J. Geophys. Res.-Atmos., 126, e2020JD033583, https://doi.org/10.1029/2020JD033583, 2021.
Jacob, D. J., Field, B. D., Li, Q., Blake, D. R., de Gouw, J., Warneke, C., Hansel, A., Wisthaler, A., Singh, H. B., and Guenther, A.: Global budget of methanol: Constraints from atmospheric observations, J. Geophys. Res.-Atmos., 110, D08303, https://doi.org/10.1029/2004JD005172, 2005.
Jernigan, C. M., Fite, C. H., Vereecken, L., Berkelhammer, M. B., Rollins, A. W., Rickly, P. S., Novelli, A., Taraborrelli, D., Holmes, C. D., and Bertram, T. H.: Efficient Production of Carbonyl Sulfide in the Low-NOx Oxidation of Dimethyl Sulfide, Geophys. Res. Lett., 49, e2021GL096838, https://doi.org/10.1029/2021GL096838, 2022a.
Jernigan, C. M., Cappa, C. D., and Bertram, T. H.: Reactive Uptake of Hydroperoxymethyl Thioformate to Sodium Chloride and Sodium Iodide Aerosol Particles, J. Phys. Chem. A, 126, 4476–4481, https://doi.org/10.1021/acs.jpca.2c03222, 2022b.
Johnson, J. S. and Jen, C. N.: Role of Methanesulfonic Acid in Sulfuric Acid–Amine and Ammonia New Particle Formation, ACS Earth Space Chem., 7, 653–660, https://doi.org/10.1021/acsearthspacechem.3c00017, 2023.
Johnson, M. T.: A numerical scheme to calculate temperature and salinity dependent air-water transfer velocities for any gas, Ocean Sci., 6, 913–932, https://doi.org/10.5194/os-6-913-2010, 2010.
Kaufman, Y. J. and Tanré, D.: Effect of variations in super-saturation on the formation of cloud condensation nuclei, Nature, 369, 45–48, https://doi.org/10.1038/369045a0, 1994.
Kettle, A. J., Andreae, M. O., Amouroux, D., Andreae, T. W., Bates, T. S., Berresheim, H., Bingemer, H., Boniforti, R., Curran, M. a. J., DiTullio, G. R., Helas, G., Jones, G. B., Keller, M. D., Kiene, R. P., Leck, C., Levasseur, M., Malin, G., Maspero, M., Matrai, P., McTaggart, A. R., Mihalopoulos, N., Nguyen, B. C., Novo, A., Putaud, J. P., Rapsomanikis, S., Roberts, G., Schebeske, G., Sharma, S., Simó, R., Staubes, R., Turner, S., and Uher, G.: A global database of sea surface dimethylsulfide (DMS) measurements and a procedure to predict sea surface DMS as a function of latitude, longitude, and month, Glob. Biogeochem. Cy., 13, 399–444, https://doi.org/10.1029/1999GB900004, 1999.
Khan, M. A. H., Gillespie, S. M. P., Razis, B., Xiao, P., Davies-Coleman, M. T., Percival, C. J., Derwent, R. G., Dyke, J. M., Ghosh, M. V., Lee, E. P. F., and Shallcross, D. E.: A modelling study of the atmospheric chemistry of DMS using the global model, STOCHEM-CRI, Atmos. Environ., 127, 69–79, https://doi.org/10.1016/j.atmosenv.2015.12.028, 2016.
Khan, M. A. H., Bannan, T. J., Holland, R., Shallcross, D. E., Archibald, A. T., Matthews, E., Back, A., Allan, J., Coe, H., Artaxo, P., and Percival, C. J.: Impacts of Hydroperoxymethyl Thioformate on the Global Marine Sulfur Budget, ACS Earth Space Chem., 5, 2577–2586, https://doi.org/10.1021/acsearthspacechem.1c00218, 2021.
Kloster, S., Feichter, J., Maier-Reimer, E., Six, K. D., Stier, P., and Wetzel, P.: DMS cycle in the marine ocean-atmosphere system – a global model study, Biogeosciences, 3, 29–51, https://doi.org/10.5194/bg-3-29-2006, 2006.
Kodros, J. K. and Pierce, J. R.: Important global and regional differences in aerosol cloud-albedo effect estimates between simulations with and without prognostic aerosol microphysics, J. Geophys. Res.-Atmos., 122, 4003–4018, https://doi.org/10.1002/2016JD025886, 2017.
Kodros, J. K., Cucinotta, R., Ridley, D. A., Wiedinmyer, C., and Pierce, J. R.: The aerosol radiative effects of uncontrolled combustion of domestic waste, Atmos. Chem. Phys., 16, 6771–6784, https://doi.org/10.5194/acp-16-6771-2016, 2016.
Kouvarakis, G. and Mihalopoulos, N.: Seasonal variation of dimethylsulfide in the gas phase and of methanesulfonate and non-sea-salt sulfate in the aerosols phase in the Eastern Mediterranean atmosphere, Atmos. Environ., 36, 929–938, https://doi.org/10.1016/S1352-2310(01)00511-8, 2002.
Kowalczuk, P., Cooper, W. J., Whitehead, R. F., Durako, M. J., and Sheldon, W.: Characterization of CDOM in an organic-rich river and surrounding coastal ocean in the South Atlantic Bight, Aquat. Sci., 65, 384–401, https://doi.org/10.1007/s00027-003-0678-1, 2003.
Kulmala, M.: How Particles Nucleate and Grow, Science, 302, 1000–1001, https://doi.org/10.1126/science.1090848, 2003.
Kulmala, M., Pirjola, L., and Mäkelä, J. M.: Stable sulphate clusters as a source of new atmospheric particles, Nature, 404, 66–69, https://doi.org/10.1038/35003550, 2000.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011.
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., von Salzen, K., Pierce, J. R., Bertram, A. K., Schroder, J. C., Shantz, N. C., Chang, R. Y.-W., and Norman, A.-L.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elem. Sci. Anthr., 1, 000017, https://doi.org/10.12952/journal.elementa.000017, 2013.
Lee, Y. H. and Adams, P. J.: A Fast and Efficient Version of the TwO-Moment Aerosol Sectional (TOMAS) Global Aerosol Microphysics Model, Aerosol Sci. Technol., 46, 678–689, https://doi.org/10.1080/02786826.2011.643259, 2012.
Lee, Y. H., Pierce, J. R., and Adams, P. J.: Representation of nucleation mode microphysics in a global aerosol model with sectional microphysics, Geosci. Model Dev., 6, 1221–1232, https://doi.org/10.5194/gmd-6-1221-2013, 2013.
Lennartz, S. T., Krysztofiak, G., Marandino, C. A., Sinnhuber, B.-M., Tegtmeier, S., Ziska, F., Hossaini, R., Krüger, K., Montzka, S. A., Atlas, E., Oram, D. E., Keber, T., Bönisch, H., and Quack, B.: Modelling marine emissions and atmospheric distributions of halocarbons and dimethyl sulfide: the influence of prescribed water concentration vs. prescribed emissions, Atmos. Chem. Phys., 15, 11753–11772, https://doi.org/10.5194/acp-15-11753-2015, 2015.
Liu, H., Jacob, D. J., Bey, I., and Yantosca, R. M.: Constraints from 210Pb and 7Be on wet deposition and transport in a global three-dimensional chemical tracer model driven by assimilated meteorological fields, J. Geophys. Res.-Atmos., 106, 12109–12128, https://doi.org/10.1029/2000JD900839, 2001.
Lucas, D. D. and Prinn, R. G.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res.-Atmos., 107, ACH 12-1–ACH 12-26, https://doi.org/10.1029/2001JD000843, 2002.
Napari, I., Noppel, M., Vehkamäki, H., and Kulmala, M.: Parametrization of ternary nucleation rates for H2SO4-NH3-H2O vapors, J. Geophys. Res.-Atmos., 107, AAC 6-1–AAC 6-6, https://doi.org/10.1029/2002JD002132, 2002.
Nightingale, P. D., Malin, G., Law, C. S., Watson, A. J., Liss, P. S., Liddicoat, M. I., Boutin, J., and Upstill-Goddard, R. C.: In situ evaluation of air–sea gas exchange parameterizations using novel conservative and volatile tracers, Global Biogeochem. Cy., 14, 373–387, https://doi.org/10.1029/1999GB900091, 2000.
Novak, G. A., Fite, C. H., Holmes, C. D., Veres, P. R., Neuman, J. A., Faloona, I., Thornton, J. A., Wolfe, G. M., Vermeuel, M. P., Jernigan, C. M., Peischl, J., Ryerson, T. B., Thompson, C. R., Bourgeois, I., Warneke, C., Gkatzelis, G. I., Coggon, M. M., Sekimoto, K., Bui, T. P., Dean-Day, J., Diskin, G. S., DiGangi, J. P., Nowak, J. B., Moore, R. H., Wiggins, E. B., Winstead, E. L., Robinson, C., Thornhill, K. L., Sanchez, K. J., Hall, S. R., Ullmann, K., Dollner, M., Weinzierl, B., Blake, D. R., and Bertram, T. H.: Rapid cloud removal of dimethyl sulfide oxidation products limits SO2 and cloud condensation nuclei production in the marine atmosphere, P. Natl. Acad. Sci. USA, 118, e2110472118, https://doi.org/10.1073/pnas.2110472118, 2021.
Novak, G. A., Kilgour, D. B., Jernigan, C. M., Vermeuel, M. P., and Bertram, T. H.: Oceanic emissions of dimethyl sulfide and methanethiol and their contribution to sulfur dioxide production in the marine atmosphere, Atmos. Chem. Phys., 22, 6309–6325, https://doi.org/10.5194/acp-22-6309-2022, 2022.
Park, R. J., Jacob, D. J., Field, B. D., Yantosca, R. M., and Chin, M.: Natural and transboundary pollution influences on sulfate-nitrate-ammonium aerosols in the United States: Implications for policy, J. Geophys. Res.-Atmos., 109, D15204, https://doi.org/10.1029/2003JD004473, 2004.
Parrella, J. P., Jacob, D. J., Liang, Q., Zhang, Y., Mickley, L. J., Miller, B., Evans, M. J., Yang, X., Pyle, J. A., Theys, N., and Van Roozendael, M.: Tropospheric bromine chemistry: implications for present and pre-industrial ozone and mercury, Atmos. Chem. Phys., 12, 6723–6740, https://doi.org/10.5194/acp-12-6723-2012, 2012.
Pierce, J. R. and Adams, P. J.: Global evaluation of CCN formation by direct emission of sea salt and growth of ultrafine sea salt, J. Geophys. Res.-Atmos., 111, D06203, https://doi.org/10.1029/2005JD006186, 2006.
Pound, R. J., Sherwen, T., Helmig, D., Carpenter, L. J., and Evans, M. J.: Influences of oceanic ozone deposition on tropospheric photochemistry, Atmos. Chem. Phys., 20, 4227–4239, https://doi.org/10.5194/acp-20-4227-2020, 2020.
Pye, H. O. T., Liao, H., Wu, S., Mickley, L. J., Jacob, D. J., Henze, D. K., and Seinfeld, J. H.: Effect of changes in climate and emissions on future sulfate-nitrate-ammonium aerosol levels in the United States, J. Geophys. Res.-Atmos., 114, D01205, https://doi.org/10.1029/2008JD010701, 2009.
Rollins, A. W.: ATom: Sulfur Dioxide by Laser Induced Fluorescence (LIF-SO2) for ATom-4 Campaign, ORNL DAAC, Oak Ridge, Tennessee, USA [data set], https://doi.org/10.3334/ORNLDAAC/1890, 2021.
Rosati, B., Isokääntä, S., Christiansen, S., Jensen, M. M., Moosakutty, S. P., Wollesen de Jonge, R., Massling, A., Glasius, M., Elm, J., Virtanen, A., and Bilde, M.: Hygroscopicity and CCN potential of DMS-derived aerosol particles, Atmos. Chem. Phys., 22, 13449–13466, https://doi.org/10.5194/acp-22-13449-2022, 2022.
Schmidt, J. A., Jacob, D. J., Horowitz, H. M., Hu, L., Sherwen, T., Evans, M. J., Liang, Q., Suleiman, R. M., Oram, D. E., Le Breton, M., Percival, C. J., Wang, S., Dix, B., and Volkamer, R.: Modeling the observed tropospheric BrO background: Importance of multiphase chemistry and implications for ozone, OH, and mercury, J. Geophys. Res.-Atmos., 121, 11,819-11,835, https://doi.org/10.1002/2015JD024229, 2016.
Schobesberger, S., Junninen, H., Bianchi, F., Lönn, G., Ehn, M., Lehtipalo, K., Dommen, J., Ehrhart, S., Ortega, I. K., Franchin, A., Nieminen, T., Riccobono, F., Hutterli, M., Duplissy, J., Almeida, J., Amorim, A., Breitenlechner, M., Downard, A. J., Dunne, E. M., Flagan, R. C., Kajos, M., Keskinen, H., Kirkby, J., Kupc, A., Kürten, A., Kurtén, T., Laaksonen, A., Mathot, S., Onnela, A., Praplan, A. P., Rondo, L., Santos, F. D., Schallhart, S., Schnitzhofer, R., Sipilä, M., Tomé, A., Tsagkogeorgas, G., Vehkamäki, H., Wimmer, D., Baltensperger, U., Carslaw, K. S., Curtius, J., Hansel, A., Petäjä, T., Kulmala, M., Donahue, N. M., and Worsnop, D. R.: Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules, P. Natl. Acad. Sci. USA, 110, 17223–17228, https://doi.org/10.1073/pnas.1306973110, 2013.
Sherwen, T., Schmidt, J. A., Evans, M. J., Carpenter, L. J., Großmann, K., Eastham, S. D., Jacob, D. J., Dix, B., Koenig, T. K., Sinreich, R., Ortega, I., Volkamer, R., Saiz-Lopez, A., Prados-Roman, C., Mahajan, A. S., and Ordóñez, C.: Global impacts of tropospheric halogens (Cl, Br, I) on oxidants and composition in GEOS-Chem, Atmos. Chem. Phys., 16, 12239–12271, https://doi.org/10.5194/acp-16-12239-2016, 2016a.
Sherwen, T. M., Evans, M. J., Spracklen, D. V., Carpenter, L. J., Chance, R., Baker, A. R., Schmidt, J. A., and Breider, T. J.: Global modeling of tropospheric iodine aerosol, Geophys. Res. Lett., 43, 10012–10019, https://doi.org/10.1002/2016GL070062, 2016b.
Sipilä, M., Berndt, T., Petäjä, T., Brus, D., Vanhanen, J., Stratmann, F., Patokoski, J., Mauldin, R. L., Hyvärinen, A.-P., Lihavainen, H., and Kulmala, M.: The Role of Sulfuric Acid in Atmospheric Nucleation, Science, 327, 1243–1246, https://doi.org/10.1126/science.1180315, 2010.
Spracklen, D. V., Pringle, K. J., Carslaw, K. S., Chipperfield, M. P., and Mann, G. W.: A global off-line model of size-resolved aerosol microphysics: I. Model development and prediction of aerosol properties, Atmos. Chem. Phys., 5, 2227–2252, https://doi.org/10.5194/acp-5-2227-2005, 2005.
Thomas, M. A., Suntharalingam, P., Pozzoli, L., Rast, S., Devasthale, A., Kloster, S., Feichter, J., and Lenton, T. M.: Quantification of DMS aerosol-cloud-climate interactions using the ECHAM5-HAMMOZ model in a current climate scenario, Atmos. Chem. Phys., 10, 7425–7438, https://doi.org/10.5194/acp-10-7425-2010, 2010.
Trivitayanurak, W., Adams, P. J., Spracklen, D. V., and Carslaw, K. S.: Tropospheric aerosol microphysics simulation with assimilated meteorology: model description and intermodel comparison, Atmos. Chem. Phys., 8, 3149–3168, https://doi.org/10.5194/acp-8-3149-2008, 2008.
Urbanski, S. P. and Wine, P. H.: Spectroscopic and Kinetic Study of the Cl-S(CH3)2 Adduct, J. Phys. Chem. A, 103, 10935–10944, https://doi.org/10.1021/jp992682m, 1999.
Vehkamäki, H., Kulmala, M., Napari, I., Lehtinen, K. E. J., Timmreck, C., Noppel, M., and Laaksonen, A.: An improved parameterization for sulfuric acid–water nucleation rates for tropospheric and stratospheric conditions, J. Geophys. Res.-Atmos., 107, AAC 3-1–AAC 3-10, https://doi.org/10.1029/2002JD002184, 2002.
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020.
Veres, P. R., Neuman, J. A., and Ryerson, T. B.: ATom: L2 Measurements from NOAA ToF Chemical Ionization Mass Spectrometer, Version 2, ORNL DAAC, Oak Ridge, Tennessee, USA [data set], https://doi.org/10.3334/ORNLDAAC/1921, 2021.
Vermeuel, M. P., Novak, G. A., Jernigan, C. M., and Bertram, T. H.: Diel Profile of Hydroperoxymethyl Thioformate: Evidence for Surface Deposition and Multiphase Chemistry, Environ. Sci. Technol., 54, 12521–12529, https://doi.org/10.1021/acs.est.0c04323, 2020.
von Glasow, R. and Crutzen, P. J.: Model study of multiphase DMS oxidation with a focus on halogens, Atmos. Chem. Phys., 4, 589–608, https://doi.org/10.5194/acp-4-589-2004, 2004.
Wang, W.-L., Song, G., Primeau, F., Saltzman, E. S., Bell, T. G., and Moore, J. K.: Global ocean dimethyl sulfide climatology estimated from observations and an artificial neural network, Biogeosciences, 17, 5335–5354, https://doi.org/10.5194/bg-17-5335-2020, 2020.
Wang, X., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Zhu, L., Chen, Q., Alexander, B., Sherwen, T., Evans, M. J., Lee, B. H., Haskins, J. D., Lopez-Hilfiker, F. D., Thornton, J. A., Huey, G. L., and Liao, H.: The role of chlorine in global tropospheric chemistry, Atmos. Chem. Phys., 19, 3981–4003, https://doi.org/10.5194/acp-19-3981-2019, 2019.
Wang, X., Jacob, D. J., Downs, W., Zhai, S., Zhu, L., Shah, V., Holmes, C. D., Sherwen, T., Alexander, B., Evans, M. J., Eastham, S. D., Neuman, J. A., Veres, P. R., Koenig, T. K., Volkamer, R., Huey, L. G., Bannan, T. J., Percival, C. J., Lee, B. H., and Thornton, J. A.: Global tropospheric halogen (Cl, Br, I) chemistry and its impact on oxidants, Atmos. Chem. Phys., 21, 13973–13996, https://doi.org/10.5194/acp-21-13973-2021, 2021.
Wang, Y., Jacob, D. J., and Logan, J. A.: Global simulation of tropospheric O3-NOx-hydrocarbon chemistry: 1. Model formulation, J. Geophys. Res.-Atmos., 103, 10713–10725, https://doi.org/10.1029/98JD00158, 1998.
Wesely, M. L.: Parameterization of surface resistances to gaseous dry deposition in regional-scale numerical models, Atmos. Environ. 1967, 23, 1293–1304, https://doi.org/10.1016/0004-6981(89)90153-4, 1989.
Westervelt, D. M., Pierce, J. R., Riipinen, I., Trivitayanurak, W., Hamed, A., Kulmala, M., Laaksonen, A., Decesari, S., and Adams, P. J.: Formation and growth of nucleated particles into cloud condensation nuclei: model–measurement comparison, Atmos. Chem. Phys., 13, 7645–7663, https://doi.org/10.5194/acp-13-7645-2013, 2013.
Williamson, C. J., Kupc, A., Axisa, D., Bilsback, K. R., Bui, T., Campuzano-Jost, P., Dollner, M., Froyd, K. D., Hodshire, A. L., Jimenez, J. L., Kodros, J. K., Luo, G., Murphy, D. M., Nault, B. A., Ray, E. A., Weinzierl, B., Wilson, J. C., Yu, F., Yu, P., Pierce, J. R., and Brock, C. A.: A large source of cloud condensation nuclei from new particle formation in the tropics, Nature, 574, 399–403, https://doi.org/10.1038/s41586-019-1638-9, 2019.
Wollesen de Jonge, R., Elm, J., Rosati, B., Christiansen, S., Hyttinen, N., Lüdemann, D., Bilde, M., and Roldin, P.: Secondary aerosol formation from dimethyl sulfide – improved mechanistic understanding based on smog chamber experiments and modelling, Atmos. Chem. Phys., 21, 9955–9976, https://doi.org/10.5194/acp-21-9955-2021, 2021.
Wu, R., Wang, S., and Wang, L.: New Mechanism for the Atmospheric Oxidation of Dimethyl Sulfide. The Importance of Intramolecular Hydrogen Shift in a CH3SCH2OO Radical, J. Phys. Chem. A, 119, 112–117, https://doi.org/10.1021/jp511616j, 2015.
Zhang, J.-Z. and Millero, F. J.: The products from the oxidation of H2S in seawater, Geochim. Cosmochim. Ac., 57, 1705–1718, https://doi.org/10.1016/0016-7037(93)90108-9, 1993.
Zhang, Y., Jacob, D. J., Maasakkers, J. D., Sulprizio, M. P., Sheng, J.-X., Gautam, R., and Worden, J.: Monitoring global tropospheric OH concentrations using satellite observations of atmospheric methane, Atmos. Chem. Phys., 18, 15959–15973, https://doi.org/10.5194/acp-18-15959-2018, 2018.
Zhou, Z.-X., Lujan, S. A., Burkholder, A. B., Garbacz, M. A., and Kunkel, T. A.: Roles for DNA polymerase δ in initiating and terminating leading strand DNA replication, Nat. Commun., 10, 3992, https://doi.org/10.1038/s41467-019-11995-z, 2019.
Zhu, L., Nicovich, J. M., and Wine, P. H.: Temperature-dependent kinetics studies of aqueous phase reactions of hydroxyl radicals with dimethylsulfoxide, dimethylsulfone, and methanesulfonate, Aquat. Sci., 65, 425–435, https://doi.org/10.1007/s00027-003-0673-6, 2003.
Zhu, L., Nenes, A., Wine, P. H., and Nicovich, J. M.: Effects of aqueous organosulfur chemistry on particulate methanesulfonate to non–sea salt sulfate ratios in the marine atmosphere, J. Geophys. Res.-Atmos., 111, D05316, https://doi.org/10.1029/2005JD006326, 2006.
Zhu, L., Jacob, D. J., Eastham, S. D., Sulprizio, M. P., Wang, X., Sherwen, T., Evans, M. J., Chen, Q., Alexander, B., Koenig, T. K., Volkamer, R., Huey, L. G., Le Breton, M., Bannan, T. J., and Percival, C. J.: Effect of sea salt aerosol on tropospheric bromine chemistry, Atmos. Chem. Phys., 19, 6497–6507, https://doi.org/10.5194/acp-19-6497-2019, 2019.