the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 08 Feb 2024
| 08 Feb 2024
pH regulates the formation of organosulfates and inorganic sulfate from organic peroxide reaction with dissolved SO2 in aquatic media
Xiaofan Lv
Makroni Lily
Narcisse Tsona Tchinda
Organic peroxides (OPs) are an important component of dissolved organic matter (DOM), detected in various aquatic media. Despite their unique functions as redox agents in water ecosystems, the complete mechanisms and factors controlling their transformation are not explicitly established. Here, we evaluate the pH effect on the aqueous-phase reaction of three selected OPs (methyl hydroperoxide (MHP), peracetic acid (PAA), and benzoyl peroxide (BZP)) with dissolved SO2. Results show that due to the presence of the hydroperoxyl group in their structures, MHP and PAA are susceptible to forming inorganic sulfate and organosulfate (methyl sulfate for MHP and acetyl sulfate for PAA) depending on the pH, while BZP exclusively forms organosulfate (benzoyl sulfate) in the pH range investigated. Moreover, it is seen that the ability of PAA to form inorganic sulfate relative to organosulfate is more pronounced, which is supported by a previous experimental observation. The effective rate constants of the transformation of these peroxides within the pH 1–10 and 240–340 K ranges exhibit positive pH and temperature dependencies, and BZP is seen to degrade more effectively than MHP and PAA. In addition to the pH impact, it is highlighted that the formation of organic and/or inorganic sulfate strongly depends on the nature of the substituents on the peroxy function. Namely, PAA and BZP are more reactive than MHP, which may be attributed to the electron-withdrawing effects of -C(O)R (R = -CH3 and -C6H5 for PAA and BZP, respectively) substituents that activate the peroxy function. The results further indicate that the aqueous-phase degradation of OPs can adequately drive the change in the chemical composition of DOM, both in terms of organic and inorganic sulfate mass fractions.
- Article
(3980 KB) - Full-text XML
-
Supplement
(575 KB) - BibTeX
- EndNote





Organic peroxides (OPs) are an important contributor to the atmospheric oxidation capacity, as they act as reservoirs for alkoxy radicals and organic peroxy radicals (ROx) (Ehn et al., 2014; Allen et al., 2022). Besides their well-known implication in the oxidation of biogenic volatile organic compounds and anthropogenic precursors, OPs were shown to contribute to secondary organic aerosols (SOAs) by forming an important link between sulfate and SOAs (Tkacik et al., 2012; Riva et al., 2015; Hettiyadura et al., 2017; Huang et al., 2020). Moreover, they are also known to regulate the distribution of HOx and ROx radicals through large-scale vertical and horizontal transport (Mari et al., 2000; Ravetta et al., 2001). OPs were ubiquitously detected in atmospheric media, including air and precipitation, fresh water, seawater, surface waters, and coastal environments (Sauer et al., 2001; Morgan and Jackson, 2002; Sun et al., 2021). Their sources are varied, depending on their chemical compositions and properties (Liu et al., 2014). In aqueous media, these sources include the self-reaction of photochemically formed organic radicals and partitioning from the gas phase to the particle phase (O'Sullivan et al., 2005; Sun et al., 2021; Riemer et al., 2000).
The primary sinks of OPs include SO2 oxidation in cloud and rain droplets and uptake on water surfaces and water ice, mainly forming water-soluble organic compounds and secondary sulfates (Böge et al., 2006; Hua et al., 2008). Previous studies have indeed shown that OPs are highly reactive towards dissolved SO2 in submicron aerosol (O'Sullivan et al., 1996; Liou and Storz, 2010), despite the incomplete detailed mechanisms and kinetics of their transformation. Such reactions in the aqueous phase are known to be driven by the change in solution pH. Specifically, bulk phase pH was shown to impact the rates of multiphase SO2 oxidation towards aqueous-phase aerosol formation by altering the reactions mechanisms and modifying the particle morphology, viscosity, and radiative effects (Turnock et al., 2019; Lei et al., 2022). While some of these pH impacts have been explored in detail, namely altering the aerosol radiative forcing (Turnock et al., 2019), the detailed effect on reaction mechanisms and kinetics remains not fully elucidated. The lack of comprehensive information on the pH impact on the mechanisms and kinetics of OP reactions towards SO2 hinders the complete assessment of the importance of such interactions in aquatic environments, for example in the chemical composition of dissolved organic matter (DOM).
Methyl hydroperoxide (MHP, CH3OOH), peracetic acid (PAA, CH3C(O)OOH), and benzoyl peroxide (BZP, C6H5-C(O)OO(O)C-C6H5) are typical examples of OPs both from natural and anthropogenic origins. MHP is the simplest and most abundant OP in the atmosphere and is most prevalent in the remote marine environment (Heikes et al., 1996; O'Sullivan et al., 1999). MHP is believed to form from the ozonolysis of alkenes and from biomass burning (Klippel et al., 2011; Zhang et al., 2012). PAA is one of the simplest OPs containing the carbonyl function, formed form the reaction between acetic acid and hydrogen peroxide in the presence of a strong acid catalyst. PAA is widely used as a disinfectant, sterilizing agent, oxidizer, polymerization catalyst, and sanitizer in wastewater treatment and various industrial applications and is known as a powerful degrading agent for aqueous organic micropollutants (Luukkonen et al., 2015; Zhang and Huang, 2020; Ao et al., 2021; Kiejza et al., 2021). BZP is an antiseptic much used for acne treatment and in the chemical industry (Kircik, 2013; Brammann and Mueller-Goymann, 2019). It can easily enter the human body via food intake and skin absorption, causing potential risks such as tissue damage and eventually acting as a tumor promoter (Kozan et al., 2010; Ding et al., 2019). As a strong reactive oxygen species, BZP can be reduced to benzoic acid, with the ability to inhibit the growth of some microorganisms in wheat flour and corn starch (Ding et al., 2019; Yu et al., 2022). Previous studies also mentioned the possibility of BZP thermolysis to yield benzoyloxy radicals and volatile benzene in the aqueous phase (Tu et al., 1996; Wang et al., 2023).
Despite MHP, PAA, and BZP being poorly soluble in water compared to hydrogen peroxide, for example, they are relatively present in aquatic media (Meylan and Howard, 1991; O'Sullivan et al., 1996; Sun et al., 2021). Some studies have investigated the chemistry of these OPs through particle uptake, wet and dry deposition, thermal and photochemical decomposition, and SO2 oxidation (Tan et al., 2020; Xu et al., 2021; Ignatov et al., 2011; Allen et al., 2022), yet there are still deficiencies in understanding their full chemistry in aquatic media. The current study applies quantum chemical calculation techniques to explore the aqueous-phase reactions of MHP, PAA, and BZP with dissolved SO2. The pH effect on the mechanism and kinetics of these reactions is evaluated, and their implication in altering the chemical composition of DOM is assessed.
2.1 Quantum chemical calculations
Geometry optimizations and vibrational frequency analysis of all molecules and stationary points were performed using density functional theory based on the ωB97XD/6-31G(d,p) method under the harmonic oscillator–rigid-rotor approximation (Chai and Head-Gordon, 2008; Elm, 2022). This method has been shown to adequately describe molecular clustering and reactions involving transition state (TS) configurations (Elm et al., 2017; Elm, 2019). The aqueous phase was modeled using the continuum solvation model based on the solute electron density (SMD) at the ωB97XD/6-31G(d,p) level of theory. This model, based on an implicit treatment of the solvent, is adequate for describing atmospheric processes (Ostovari et al., 2018; Xu and Coote, 2019) and was shown to outperform explicit water treatment especially in resolving for energy barriers (Chen et al., 2019). All ωB97XD/6-31G(d,p) calculations were performed using the Gaussian 09 package (Frisch et al., 2013), while single-point energy corrections on ωB97XD/6-31G(d,p) structures were calculated with the DLPNO-CCSD(T)/aug-cc-pVTZ method using Orca version 4.2.1 (Riplinger and Neese, 2013). The Gibbs free energy in the aqueous phase is calculated at standard conditions (298.15 K and 1 M molar concentration). Details are given in Sect. S1 in the Supplement.
2.2 Kinetics
The kinetic analysis was performed based on the transition state theory (Truhlar et al., 1996). The reaction proceeds through collision between initial reactants, i.e., dissolved SO2 (henceforth, simply denoted as S(IV) = SO2 • H2O + HSO SO), and the organic peroxide (OP), to form the reactant complex (RC) in equilibrium with the initial reactants, followed by a rearrangement through a TS configuration to form the product complex according to the following reaction:
The transition state theory approach to determine the bimolecular rate constant (k) of Reaction (R1) under the pseudo-steady-state approximation considers two main terms, and it is expressed as
where Keq is the equilibrium constant of the formation of RC and kreac is the unimolecular rate constant for the reaction of RC to the product complex, given, respectively, by the following equations:
where ΔGeq is the Gibbs free energy of RC formation, c0 is the standard concentration of 1 M, h is Planck's constant, kB is Boltzmann's constant, ΔG# is the Gibbs free-energy barrier separating RC from the products, R is the molar gas constant, and T is the absolute temperature.
To account for low-barrier and barrierless processes in Reaction (R1), namely the formation of the reactant complex, the contribution of molecular diffusion is considered based on the Collins–Kimball theory (Collins and Kimball, 1949), and the overall rate constant for Reaction (R1) is then expressed as
where kD is the steady-state Smoluchowski rate constant given as (Smoluchowski, 1917):
RS(IV),OP is the reaction distance between reactants S(IV) and OP, defined as the sum of their radii, RS(IV) and ROP, respectively. NA is the Avogadro number and D is the sum of reactant diffusion coefficients DS(IV) and DOP (Truhlar, 1985). The diffusion coefficient for a reactant is related to its radius in any medium of viscosity η by the Stokes–Einstein approach (Einstein, 1905). For reactants S(IV) and OP in water, the viscosity is that of water and the diffusion coefficients are calculated as
The radii were calculated with the Multiwfn software by assuming spherical reactants (Lu and Chen, 2012). It is known that the viscosity of aerosols can be affected by relative humidity, chemical composition, water uptake, and temperature (Song et al., 2021). Given that aerosols are mixtures of different proportions of organic and inorganic salts, it is obvious that the actual diffusion coefficients and overall kinetics of the studied reactions in aqueous aerosols will strongly depend on the actual aerosol viscosity.
The reactions with OPs were explored at three different pH ranges, according to the prevalent states of dissolved SO2: SO2 • H2O at pH < 1.81, HOSO at 1.81 < pH < 6.97, and SO at pH > 6.97. Details on the distribution of dissolved SO2 are given in the Supplement, Table S1. From this distribution, the following reactions were investigated:
3.1 MHP reaction with dissolved SO2
At a pH below 1.81, the reaction of dissolved SO2 with MHP is driven by the interaction between SO2• H2O and MHP to form SO2• H2O • MHP. The Gibbs free-energy surface of this reaction, along with the structures of all stationary states, is given in Fig. 1. The formation of SO2• H2O • MHP is relatively endergonic at 298.15 K and a standard concentration of 1 M.
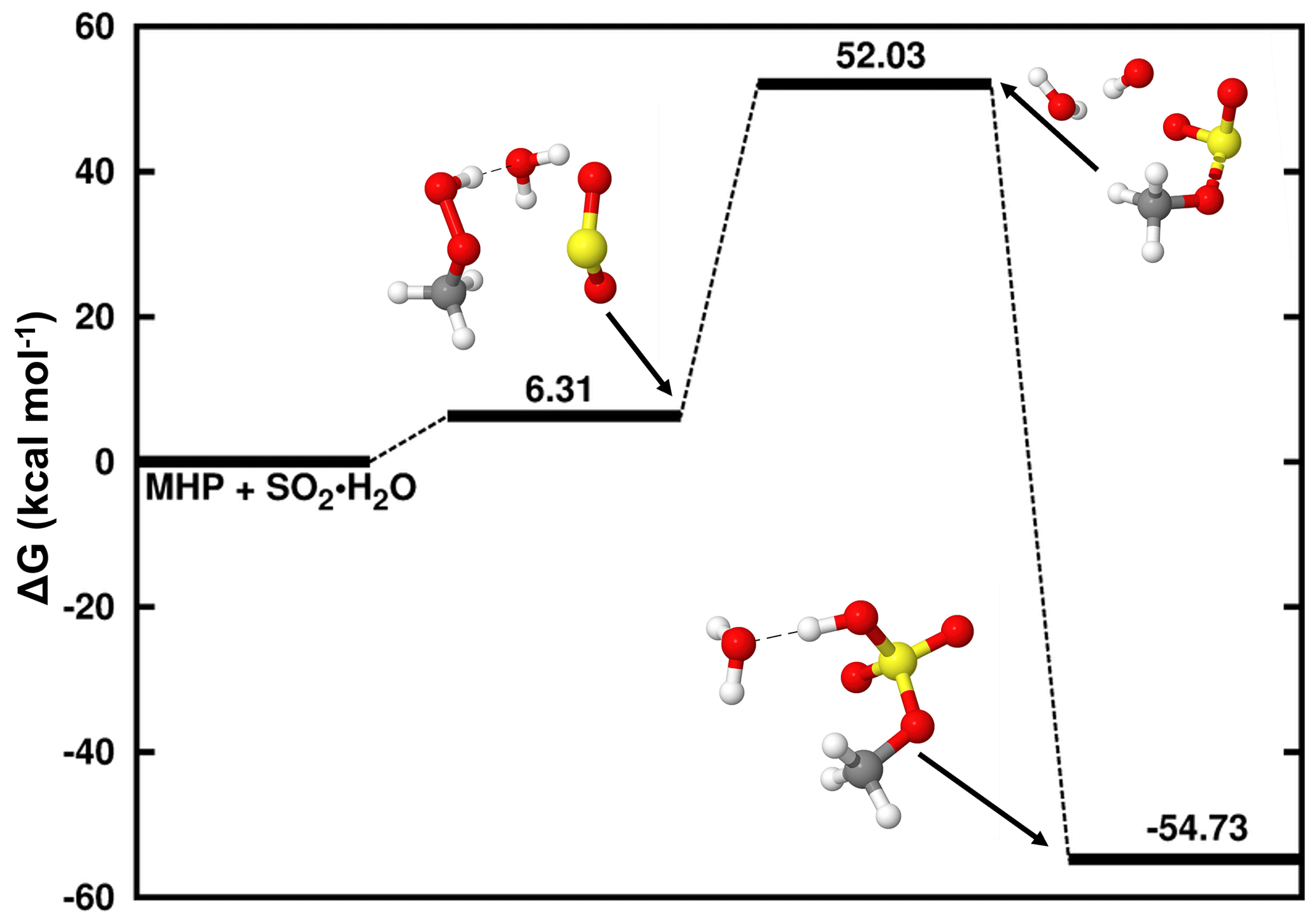
Figure 1Gibbs free-energy profile of the stationary points in the MHP + SO2• H2O reaction. Atoms' color coding is yellow for sulfur, red for oxygen, gray for carbon, and white for hydrogen.
This complex rearranges to methyl sulfate clustered to water (CH3OSO3H • H2O), through the formation of an S–O bond between the sulfur atom of SO2 and one oxygen atom of MHP. Although this product complex is formed with substantial Gibbs free-energy gain, ca. −54.73 kcal mol−1 below the reactants, the process is separated by a high energy barrier located at 45.72 kcal mol−1 above SO2• H2O • MHP. This particularly high energy barrier is associated with the difficulty of the SO2• H2O • MHP system to form two new S–O bonds prior to CH3OSO3H • H2O formation. The high energy barrier in this process is conducive to an extremely slow reaction, which occurs with a low overall rate constant of M−1 s−1 at 298.18 K. Hence, the MHP + SO2• H2O reaction forming sulfate is likely without significant relevance in natural waters.
At 1.81 < pH < 6.97, where the bisulfite ion is the prevalent state of dissolved SO2, the prevailing interaction is between MHP and HOSO, and its mechanism is slightly different from that of the MHP + SO2• H2O reaction. From the average local ionization energy-mapped (ALIE-mapped) molecular van der Waals surfaces of MHP and HOSO shown in Fig. 2, the most probable reaction sites are the oxygen atoms of MHP and the sulfur atom of HOSO. In general, based on the ALIE-mapped molecular van der Waals surfaces, the OP + S(IV) reaction is mainly driven by the interaction between the -O-O- function of OP and the sulfur atom of S(IV), while other interactions are considered to be without significance. Hence, the sulfur atom of HOSO can interact both with the oxygen atom of MHP connected to its carbon atom or that connected to its hydrogen atom, giving rise to two different configurations (RC1 and RC2, shown in Fig. 3) for MHP • HOSO. RC1 and RC2 react in different ways to form different types of sulfates according to the following reactions:
In the two TSs of Reactions (R5) and (R6), the S atom of HOSO is being connected to MHP through its CH3O and HO fragments, respectively, to form new S–O bonds. As a result, Reaction (R5) forms organic sulfate (methyl sulfate, CH3OSO), while Reaction (R6) forms inorganic sulfate (HSO). The free-energy barrier in Reaction (R5) lies 36.02 kcal mol−1 above RC1, being ∼ 10 kcal mol−1 lower than in the reaction at pH < 1.81, which also forms methyl sulfate. This is likely due to the electrostatic strain resulting from the electronic charge on HOSO, which induces a certain pronounced stability of the TS. This strain is also reflected in the stability of CH3OSO H2O, which is formed with −65.73 kcal mol−1 free-energy change.
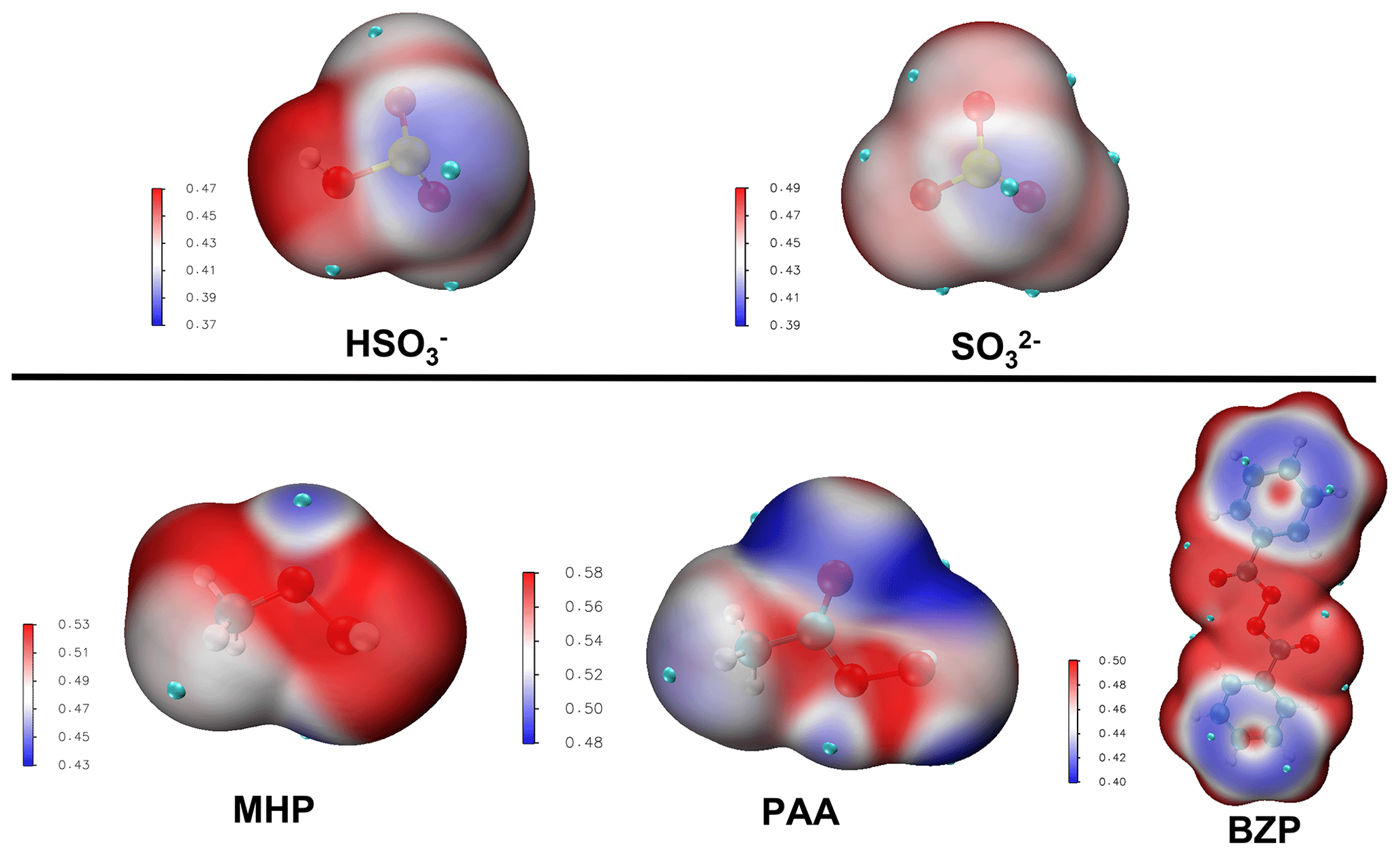
Figure 2Average local ionization energy (ALIE in a.u.) mapped molecular van der Waals surfaces of different reactants. The site possessing higher ALIE would attack the site which possesses lower ALIE, and vice versa.
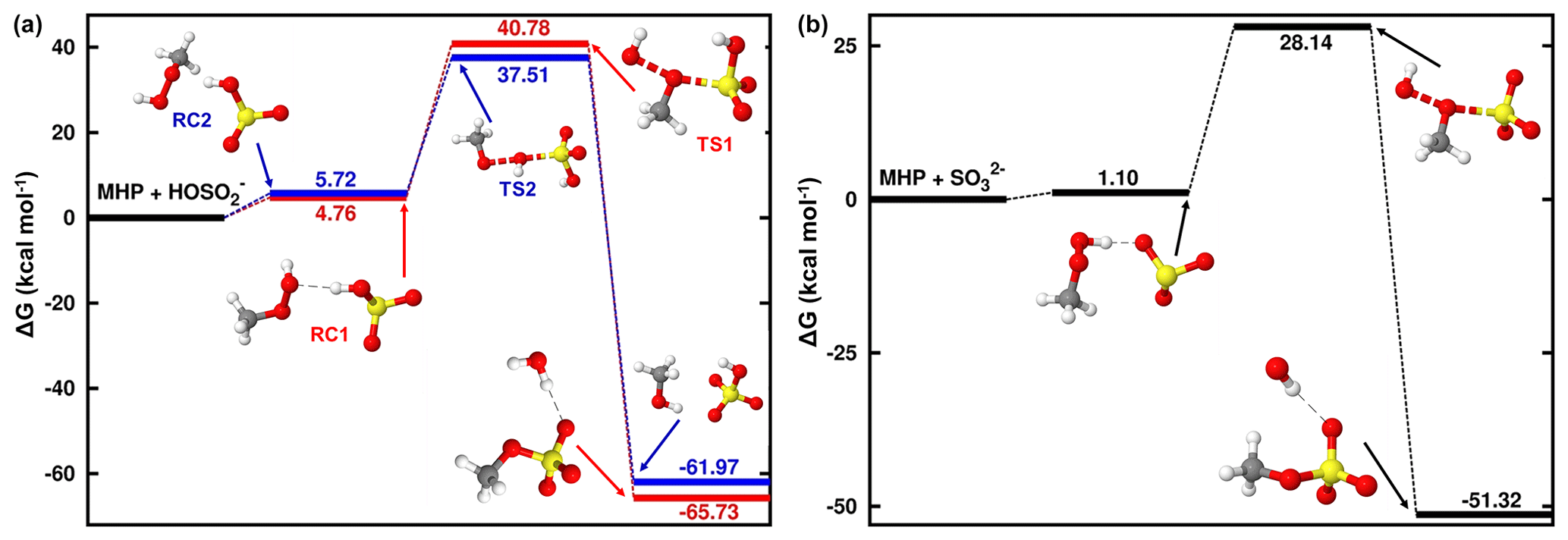
Figure 3Gibbs free-energy profile of the stationary points in the MHP + HOSO (a) and MHP+SO (b) reactions. Atoms' color coding is yellow for sulfur, red for oxygen, gray for carbon, and white for hydrogen.
Although RC1 and RC2 have almost the same formation Gibbs free energies (see Fig. 3), the free-energy barrier heights in their respective paths are 36.02 and 31.79 kcal mol−1. This shows that at 1.81 < pH < 6.97, the process to form organosulfate is energetically less favored than the process to form inorganic sulfate. Our finding is in qualitative agreement with the results of Lind et al. (1987), who found, while investigating the aqueous-phase oxidation of S(IV) by MHP over the pH range 4.0–5.2, that 73 % of inorganic sulfate was formed against 23 % of methyl sulfate. The high proportion of sulfate relative to methyl sulfate observed by Lind et al. (1987) can further be explained by the demonstrated fast hydrolysis of methyl sulfate at acidic pH (Hu et al., 2011) and its effective oxidation by OH radicals (Kwong et al., 2018) to form inorganic sulfate. The implication for this is that MHP reaction with S(IV) will contribute to aqueous aerosol mostly in terms of inorganic sulfate mass loading. This further indicates that acidic waters may highly favor inorganic sulfate from MHP relative to organosulfate. Although the presence of additional water slightly alters the barrier towards sulfate formation (see Fig. S1), the overall effect of water is not significant due to poor water clustering to MHP. Further details are given in Sect. S2.
As opposed to the bisulfite ion whose three oxygen atoms can experience different interactions from the sulfur atom, at pH > 6.97 when the ion is fully deprotonated, the three oxygen atoms of the sulfite ion (SO) are identical and only one type of product, namely organic sulfate is expected from its reaction with MHP (see Fig. 3). The reactant complex in this process is relatively more stabilized than RC1 and RC2, due to an increased electrostatic strain as the result of the presence of an additional electronic charge. The TS in this reaction lies 27.03 kcal mol−1 above the reactant complex, being nearly 9 kcal mol−1 lower than in the reaction that forms methyl sulfate in the pH range 1.81–6.97. This indicates that the organosulfate formation from the MHP + S(IV) reaction is favored at high pH.
3.2 PAA reaction with dissolved SO2
Due to the difficulty for SO2• H2O to form two new S–O bonds prior to organosulfate formation, the reaction with PAA was investigated at 1.81 < pH < 6.97 and pH > 6.97 exclusively. As expected from the ALIE-mapped molecular van der Waals surfaces of Fig. 2, the reactive sites in PAA + S(IV) reactions are the oxygen atoms in the -O-O- function of PAA and the sulfur atoms of the S(IV) entities. Regardless of the pH range, our quantum chemical calculations predict the formation of both organic and inorganic sulfates. Specifically, the sulfur atom of dissolved SO2 interacts with both oxygen atoms of the -O-O- function in PAA, and similar to the reaction with MHP, the sulfur atom attack on -O-O- through the oxygen atom connected to carbon leads to the formation of an organosulfate (acetyl sulfate), while the attack through the oxygen atom connected to hydrogen gives rise to inorganic sulfate. Energetics and optimized structures of all relevant intermediates in PAA + S(IV) reactions are given in Fig. 4.
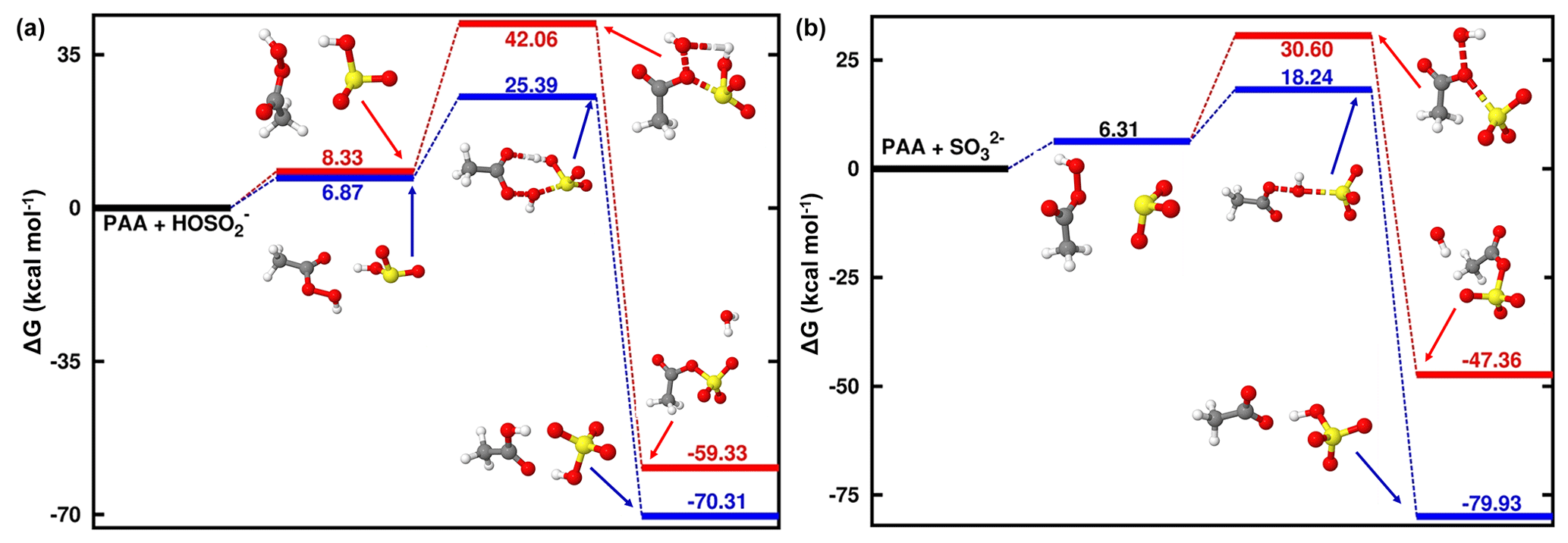
Figure 4Gibbs free-energy profile of the stationary points in the PAA + HOSO reaction (a) and PAA + SO reaction (b). Atoms' color coding is yellow for sulfur, red for oxygen, gray for carbon, and white for hydrogen.
In general, the reaction of PAA is much more favorable to the formation of inorganic sulfate than the reaction of MHP at all pH ranges, while the formation of organosulfate is substantially prevented. The transition states in the pathways of inorganic sulfate formation are located at 25.39 and 18.24 kcal mol−1 in the pH 1.81–6.97 and pH > 6.97 ranges, respectively, indicating a more favored process at basic pH than at acidic pH. The same trend is observed also in the pathways of organosulfate formation, with much higher energy barriers, in accordance with the reaction of MHP.
3.3 BZP reaction with dissolved SO2
Unlike MHP and PAA that possess a hydroperoxyl group and, hence, are capable of forming both inorganic and organic sulfate in its reaction with HOSO, BZP has two identical units from each side of the -O-O- group that would exhibit similar reactivities in favor of organosulfate formation exclusively. Based on the ALIE-mapped molecular van der Waals surface (Fig. 2), the oxygen atoms of the -O-O- function of BZP and the sulfur atom of HOSO are the most likely reactive sites. The mechanisms of BZP reaction with S(IV) are nearly identical at all pH above 1.81, starting with the formation of a reactant complex that rearranges through a transition state configuration to form benzoyl sulfate (organosulfate) and benzoic acid. In the pH range 1.81–6.97, the TS is located at 25.37 kcal mol−1 Gibbs free energy above the corresponding reactant complex (see Fig. 5). This is substantially decreased at pH > 6.97 with the transition state being located at 8.9 kcal mol−1 Gibbs free energy above the reactant complex, indicating a more favored process at neutral to basic pH than at acidic pH. This favored organosulfate formation at pH > 6.97 was also observed in the MHP + SO reaction, although it was less pronounced than in the BZP reaction. Wang et al. (2019) detected both benzoyl sulfate and benzoic acid as the main products of the BZP+S(IV) reaction and proposed a mechanism in which the -O-O- function of the organic peroxide reacts directly with dissolved SO2. The current study confirms their experimentally proposed mechanism for organosulfate formation, with the further highlight that this mechanism might be more dominant at a pH above 6.97.
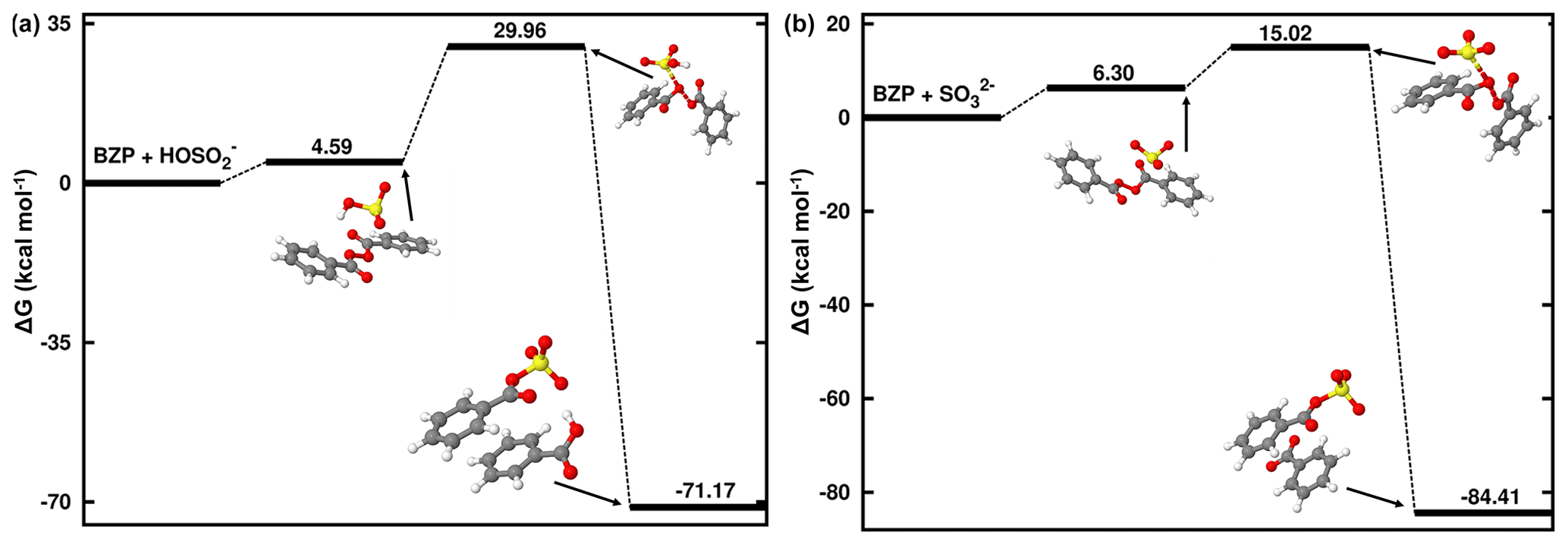
Figure 5Gibbs free-energy profile of the stationary points in the BZP + HOSO (a) and BZP+SO (b) reactions. Atoms' color coding is yellow for sulfur, red for oxygen, gray for carbon, and white for hydrogen.
3.4 Reactions with the sulfite radical ion (SO)
The importance of the sulfite radical ion (SO) to alter the atmospheric sulfur chemistry and to promote sulfate formation has been revealed in recent studies (Hung and Hoffmann, 2015; Hung et al., 2018). SO is one of the main chain carriers of the atmospheric autoxidation of SO2 commonly existing in atmospheric aquatic systems. The reactivity of SO towards organic compounds to form organosulfur compounds has been investigated in a series of previous studies (Huang et al., 2020; Liu et al., 2021). SO can readily form from the reduction of SO by NO2 according to the NO2(aq) + SO NO SO reaction catalyzed by both light and metal ions (Sapkota et al., 2015). Considering that SO and SO share similar structural properties and may coexist in some aquatic environments, SO reactions with MHP, PAA, and BZP were also investigated. While our attempt to optimize PAA + SO could not succeed, we found that SO could outperform SO in the MHP reaction, decreasing the free-energy barrier by 6.70 kcal mol−1. However, the free-energy barrier was substantially increased (by 18.31 kcal mol−1) in the BZP reaction instead. These reactivity differences between MHP and BZP towards SO can be attributed to the repulsive effects of the electron density in the benzyl rings of BZP and the spin density of the free electron in SO, which seemingly destabilizes the transition state in the reaction with BZP.
A close inspection of the configurations of different stationary states in SO reactions (Fig. 6) indicates that in the transition state of BZP reaction, the spin density is located opposite to the benzyl ring, which likely results in a strong repulsion. This obviously destabilizes the transition state, giving rise to the high energy barrier as reported above. The situation, however, is different in the transition state of the MHP reaction where the spin density is more relaxed in the configurational space of the main reactive entities.
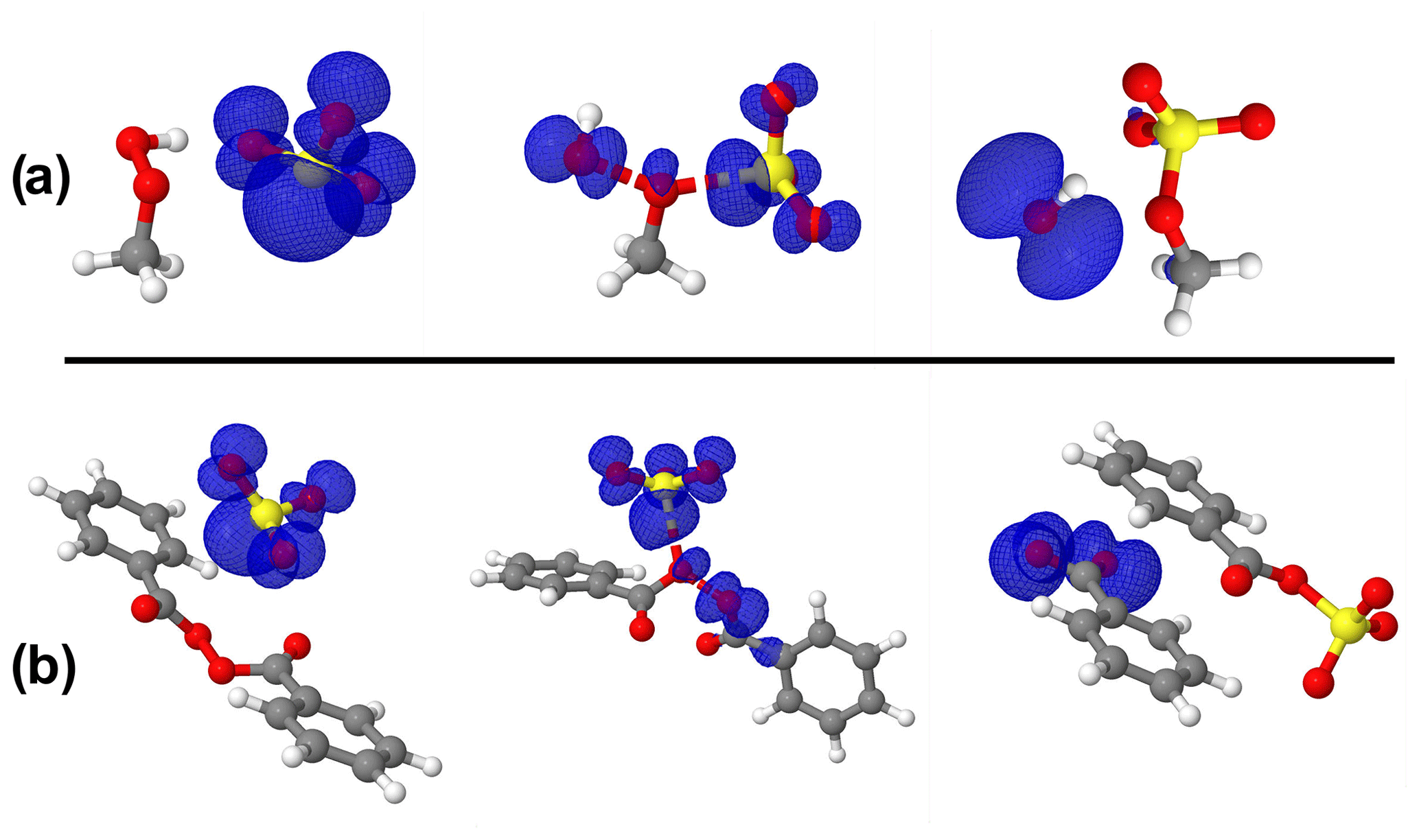
Figure 6Structures of all stationary states in the SO reactions with (a) MHP and (b) BZP, including the representation of spin density (in blue color). From left to right are reactant complex, transition state, and product complex. The spin density indicates that the extra electron initially on SO3 progressively migrates away from SO3 as the reaction proceeds. Atoms' color coding is yellow for sulfur, red for oxygen, gray for carbon, and white for hydrogen.
In a series of experiments, the Abbatt group suggested that in peroxide reaction with S(IV), organosulfates are formed directly from peroxide–SO2 interactions instead of SO2 being first oxidized by peroxides to SO followed by the reaction between SO and organic species (Ye et al., 2018; Wang et al., 2019, 2021). The current study is in line with the observations of the Abbatt group and further explains in detail their suggested mechanisms for sulfate formation. Our calculations indicate that the importance of the degradation pathway of OPs by reaction with S(IV) to form (organo)sulfate in the aquatic environment is strongly dependent on the pH and is especially important under acidic pH. Besides reactions with dissolved SO2, other recent studies showed that hydronium ions effectively catalyze the decomposition of organic peroxides at acidic pH (Qiu et al., 2020; Hu et al., 2020; Enami, 2021). It should be noted that in these studies, the catalytic effect of the available proton was examined on specific classes of OPs, namely hydroperoxides with α-hydroxyalkyl, α-alkoxyalkyl, and α-acyloxyalkyl substituents. The presence of the hydroperoxide group (-OOH) and the α-substituent with the oxygen atom (-O-) at the β-position in these peroxides enables the formation of a hydrogen-bonded ring structure involving the two functions and further facilitate the decomposition. Although these two requirements are not met for OPs considered in this work, the potential role of the available proton in the degradation of OPs that have no substituents at the α-position is worth thorough analysis in our future investigations.
Wang et al. (2019) raised the aerosol pH effect to be one of the fundamental issues to model aqueous-phase sulfate formation from the reactions of OPs with dissolved SO2. We explored the conditions of pH 1–10, which are dominated by different prevalent states of dissolved SO2. While pH below 1.81 is not a favorable condition for the degradations of both investigated OPs by reaction with S(IV), these reactions exhibit different mechanisms and kinetics at a pH above 1.81. Based on the mechanisms shown in Figs. 1, 3, 4, 5, and S1, the kinetics of the degradation of investigated OPs were evaluated within the ranges of pH 1–10 and 240–340 K temperature. The reaction rate at each pH is driven by specific fractional populations of the different protonated states of S(IV): δ(SO2• H2O) at pH < 1.81, δ(HOSO) at 1.81 < pH < 6.97, and δ(SO) at pH > 6.97. Details on these fractional populations are given in Table S1. At a given temperature, the effective rate constant, keff, for each reaction is calculated by weighing the overall rate constant (koverall, given by Eq. 4) of partial reactions at specific pHi over the fractional populations of S(IV) (xS(IV)) at that pH as follows:
While koverall is independent of pH and simply gives the rate constant of the OP + S(IV) reaction based on the activation energy, keff gives the rate constant of the full process, by taking into account the population of different protonated states of S(IV) according to the pH value. Atmospheric lifetimes of OPs are calculated based on the effective rate constant of Eq. (7), and considering that self-degradation is an unlikely process given the simple structure of investigated OPs relative to those of, e.g., hydroperoxides with α-hydroxyalkyl, α-alkoxyalkyl, and α-acyloxyalkyl substituents, whose self-degradations are catalyzed by the proton (Enami, 2021), these lifetimes are estimated based on S(IV) reactions exclusively.
Figure 7 shows the effects of pH and temperature on the effective rate constants while the numerical values are given in Tables 1, S2, and S3. In general, the effective rate constants of the reactions of OPs exhibit a positive temperature dependency, with the steepest increase being observed at a pH below 6.97, especially for the reaction of BZP. Above this range, the effective rate constant becomes almost insensitive to the pH. This indicates that the effect of pH change on the formation of organosulfates from reactions of OPs with dissolved S(IV) is most relevant in acidic aqueous environments.
In the pH range 1.81–6.97, the rate constant of the MHP + HOSO reaction to form methyl sulfate (Reaction R5) was estimated to be M−1 s−1 at 298.15 K, ∼ 3 orders of magnitude lower than that of the reaction to form inorganic sulfate (Reaction R6; overall rate constant of M−1 s−1) and 7 orders of magnitude higher than the overall rate constant of the MHP + SO2• H2O reaction. The branching ratios estimated from the obtained kinetics for the MHP + S(IV) reaction are 99.92 % and 0.08 % for inorganic sulfate and methyl sulfate, respectively. This favored formation of inorganic sulfate relative to methyl sulfate is in agreement with the experimental observation that inorganic sulfate is the major product while methyl sulfate is the second product in the MHP + HOSO reaction (Lind et al., 1987). Regardless of the temperature, the effective rate constant within this pH range increases by 3 orders of magnitude as the pH increases.
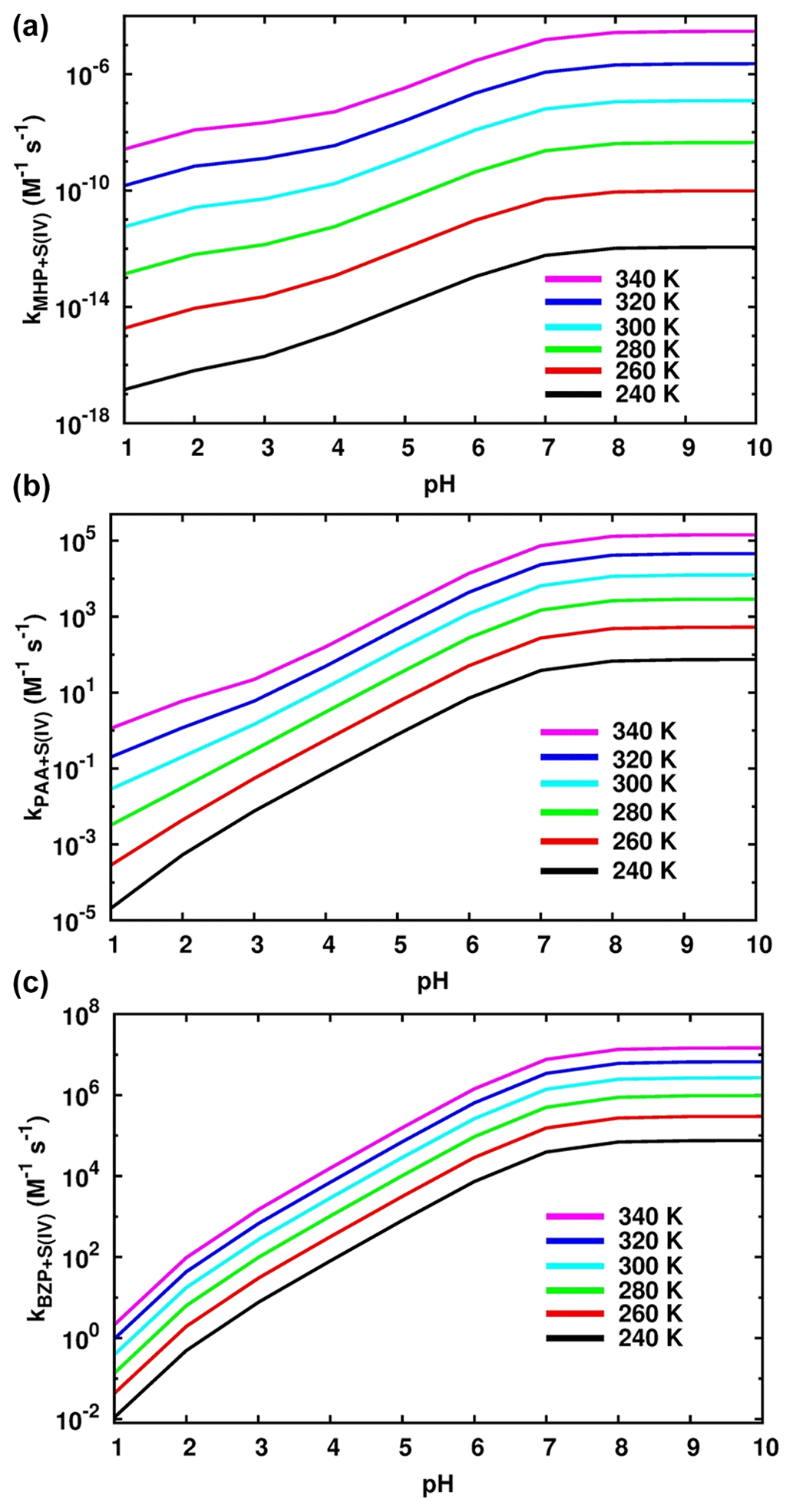
Figure 7Effective rate constants of S(IV) reaction with MHP (a), PAA (b), and BZP (c) at different values of pH and temperature.
Table 1Effective rate constants (keff, M−1 s−1) for the reactions of MHP, PAA, and BZP with S(IV) and atmospheric lifetimes (s) of corresponding OPs as a function of pH, at 298.15 K.
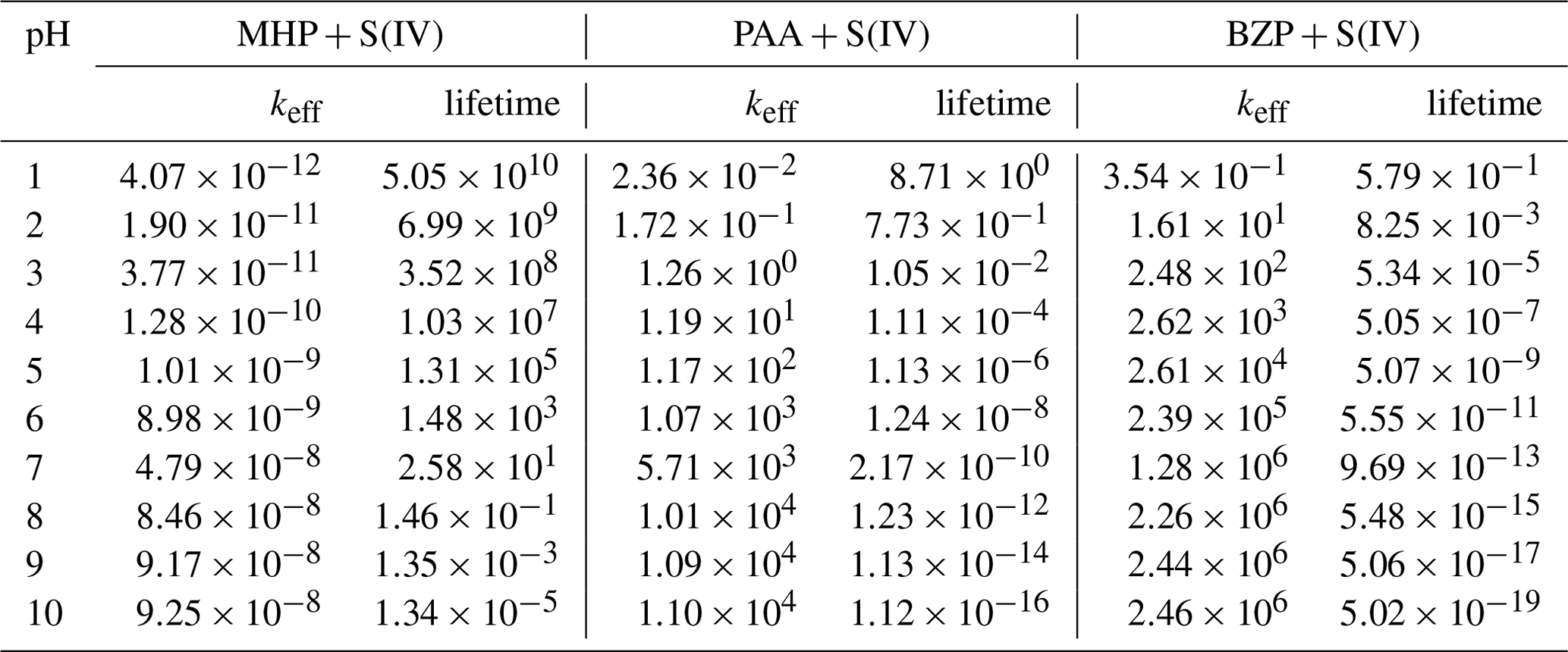
The formation of methyl sulfate at a pH above 6.97 is much more favorable than in the preceding investigated pH range, occurring with an overall rate constant of M−1 s−1 at 298.15 K. However, the effective rate constant in this range is almost insensitive to pH at all temperatures investigated (see Fig. 7). The estimated atmospheric lifetimes for MHP at 298.15 K, based on this reaction, are –5.05×1010 s in the pH range 1–10, and they are shown to decrease with increasing pH. This indicates that MHP degradation is more important in acidic waters and will alter the chemical composition of DOM by increasing the concentration of inorganic sulfate.
The kinetics of PAA + S(IV) reactions are essentially driven by the rates of inorganic sulfate formation, given the much lower energy barrier than in organosulfate formation. The rate constants for these processes at 298.15 K are and 1.10×104 M−1 s−1 for organosulfate and inorganic sulfate formation, respectively. Despite quantum chemical calculations predicting the pathways for the formation of both inorganic sulfate and organosulfate from the PAA + S(IV) reaction, the kinetics show that inorganic sulfate formation is more than 9 orders of magnitude faster than organosulfate formation in the whole pH range investigated, simply indicating that the organosulfate formation from PAA reaction is insignificant. Regardless of the pH range, the branching ratios estimated from the kinetics are 100 % for inorganic sulfate formation and 0 % for organosulfate. This is in line with the experimental observation that the reaction of PAA with dissolved SO2 almost exclusively forms inorganic sulfate (Lind et al., 1987). Lind et al. (1987) measured a rate constant of M−1 s−1 for this reaction at 291 K and pH 2.9–5.8. At the same temperature and pH 5.8, we obtained an effective rate constant of 4.20×102 M−1 s−1, in reasonable agreement with the experimental value. At pH 1–10 and 240–340 K, we obtained a positive dependency for the effective rate constant (plot and numerical values are shown in Fig. 7, Tables 1 and S3). From these kinetics, the estimated atmospheric lifetimes for PAA based on the reaction with dissolved SO2 at 298.18 K and pH 1–10 are within the range of –8.71 s. This indicates that PAA is much more reactive than MHP towards dissolved SO2 under the investigated conditions.
BZP is more reactive than MHP and PAA towards S(IV) at all pH above 1.81. Despite the mechanisms being nearly similar in the investigated pH ranges, the overall rate constants of the formation of benzoyl sulfate at 298.15 K are and 2.47×106 M−1 s−1 from BZP + HOSO and BZP + SO, respectively. The full pH and temperature dependencies of the total rate constant of the BZP + S(IV) reaction are given in Table 1 and plotted in Fig. 7. It should be noted that there is no direct formation of inorganic sulfate in BZP reactions in the pH range investigated. Sufficient evidence has been gathered about aromatic organosulfate formation connected to anthropogenic activities, namely benzyl sulfate and phenyl sulfate detected in ambient aerosols from urban sites (Kuang et al., 2016; Huang et al., 2018). These aromatic and other organosulfates derived from polycyclic aromatic hydrocarbons are believed to form from aromatics interacting with sulfur-containing species (Blair et al., 2017; Riva et al., 2015). It is likely that the investigated BZP + S(IV) mechanism in this work would explain some of such aromatic organosulfate formation mechanisms.
The calculated lifetimes for benzoyl peroxide in the investigated pH range and 298.15 K are in the range of – s (see Table 1), decreasing with increasing pH. These lifetimes are much lower than measured lifetimes of ∼ 6 d in Los Angeles from the photolysis of OPs (Krapf et al., 2016), indicating more efficient degradation in the aqueous phase. The current results highlight the strong pH impact on the degradation of BZP by S(IV). A previous study also showed that aerosol pH can substantially alter the formation of organosulfate from the reaction uptake of isoprene epoxydiols onto a mixture of ammonium sulfate and sulfuric acid particles (Lei et al., 2022).
The overall rate constants of SO reactions with MHP and BZP were determined to be and M−1 s−1, respectively, regardless of the pH. Compared to the rate constants of S(IV) reactions, the pathway for SO may also significantly contribute to degrade MHP and BZP and form organosulfate. Our results further show that sulfate formation from PAA + S(IV) and BZP + S(IV) reactions is more effective than from MHP + S(IV), and this pronounced reactivity can be attributed to electron-withdrawing effects of -C(O)R substituents (-C(O)-CH3 and -C(O)-C6H5 in PAA and BZP, respectively) that activate the -O-O- function. This finding further highlights that the specific nature of different substituents to the -O-O- function may play a determinant role in (organo)sulfate formation. It is obvious that OPs containing the hydroperoxyl function will most likely contribute to both organic and inorganic sulfate mass, whereas those not containing this function will only form organosulfates. Although OPs may not compete with, e.g., OH radicals in S(IV) oxidation, their contribution to sulfate formation may be important in low OH radical conditions. These pathways can adequately account for sulfate formation under conditions of low solar radiation such as haze events, where there is insufficient production of OH radicals. Further, the proposed mechanisms not only improve our understanding of the pH impact on the degradation efficiency but also show that different OPs would alter the chemical composition of DOM in different ways, both in terms of organic and inorganic sulfate mass fractions. Of particular importance, the investigated pathways for the degradation of OPs have direct implications for aqueous-phase sulfate aerosol formation. This provides grounds for a deeper evaluation of the pH impact of the kinetics of organic and inorganic sulfate formation from various OP+S(IV) reactions for a more complete kinetic data set. Such investigations will further guarantee a better understanding of the impacts of various sources of sulfates in aqueous-phase aerosol formation and unveil a broader impact of OPs in altering the composition of DOM in natural waters.
All data from this research can be obtained upon request by contacting the corresponding author.
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-1841-2024-supplement.
All authors contributed to the analysis and discussion of the results. NTT and LD designed the work. NTT performed all quantum chemical calculations. LD and NTT wrote the paper with contributions from all co-authors.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors would like to acknowledge the Wuxi National Centre for Supercomputing for providing the computational resources.
This research has been supported by the National Natural Science Foundation of China (grant no. 22076099) and the Youth Innovation Program of Universities in Shandong Province (grant no. 2019KJD007).
This paper was edited by Jason Surratt and reviewed by two anonymous referees.
Allen, H. M., Bates, K. H., Crounse, J. D., Kim, M. J., Teng, A. P., Ray, E. A., and Wennberg, P. O.: H2O2 and CH3OOH (MHP) in the Remote Atmosphere: 2. Physical and Chemical Controls, J. Geophys. Res.-Atmos., 127, e2021JD035702, https://doi.org/10.1029/2021JD035702, 2022.
Ao, X.-w., Eloranta, J., Huang, C.-H., Santoro, D., Sun, W.-j., Lu, Z.-d., and Li, C.: Peracetic acid-based advanced oxidation processes for decontamination and disinfection of water: A review, Water Res., 188, 116479, https://doi.org/10.1016/j.watres.2020.116479, 2021.
Blair, S. L., MacMillan, A. C., Drozd, G. T., Goldstein, A. H., Chu, R. K., Paša-Tolić, L., Shaw, J. B., Tolić, N., Lin, P., Laskin, J., Laskin, A., and Nizkorodov, S. A.: Molecular Characterization of Organosulfur Compounds in Biodiesel and Diesel Fuel Secondary Organic Aerosol, Environ. Sci. Technol., 51, 119–127, https://doi.org/10.1021/acs.est.6b03304, 2017.
Böge, O., Miao, Y., Plewka, A., and Herrmann, H.: Formation of secondary organic particle phase compounds from isoprene gas-phase oxidation products: An aerosol chamber and field study, Atmos. Environ., 40, 2501–2509, https://doi.org/10.1016/j.atmosenv.2005.12.025, 2006.
Brammann, C. and Mueller-Goymann, C. C.: Incorporation of benzoyl peroxide nanocrystals into adapalene-loaded solid lipid microparticles: Part I – Nanocrystalline benzoyl peroxide, Inter. J. Pharm., 564, 171–179, https://doi.org/10.1016/j.ijpharm.2019.04.031, 2019.
Chai, J.-D. and Head-Gordon, M.: Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections, Phys. Chem. Chem. Phys., 10, 6615–6620, https://doi.org/10.1039/B810189B, 2008.
Chen, J., Shao, Y., and Ho, J.: Are Explicit Solvent Models More Accurate than Implicit Solvent Models? A Case Study on the Menschutkin Reaction, J. Phys. Chem. A, 123, 5580–5589, https://doi.org/10.1021/acs.jpca.9b03995, 2019.
Collins, F. C. and Kimball, G. E.: Diffusion-controlled reaction rates, Journal of Colloid Science, 4, 425–437, https://doi.org/10.1016/0095-8522(49)90023-9, 1949.
Ding, H., Yuan, G., and Zhou, L.: A new water-soluble two-photon fluorescent probe for detection of trace benzoyl peroxide in wheat flour and in living cell and tissue imaging, New J. Chem., 43, 3169–3173, https://doi.org/10.1039/C8NJ06543H, 2019.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I. H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V. M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Einstein, A.: Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen, Ann. Phys.-Berlin, 322, 549–560, https://doi.org/10.1002/andp.19053220806, 1905.
Elm, J.: Unexpected Growth Coordinate in Large Clusters Consisting of Sulfuric Acid and C8H12O6 Tricarboxylic Acid, J. Phys. Chem. A, 123, 3170–3175, https://doi.org/10.1021/acs.jpca.9b00428, 2019.
Elm, J.: Clusteromics III: Acid Synergy in Sulfuric Acid–Methanesulfonic Acid–Base Cluster Formation, ACS Omega, 7, 15206–15214, https://doi.org/10.1021/acsomega.2c01396, 2022.
Elm, J., Myllys, N., Olenius, T., Halonen, R., Kurtén, T., and Vehkamäki, H.: Formation of atmospheric molecular clusters consisting of sulfuric acid and C8H12O6 tricarboxylic acid, Phys. Chem. Chem. Phys., 19, 4877–4886, https://doi.org/10.1039/C6CP08127D, 2017.
Enami, S.: Fates of Organic Hydroperoxides in Atmospheric Condensed Phases, J. Phys. Chem. A, 125, 4513–4523, https://doi.org/10.1021/acs.jpca.1c01513, 2021.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Scalmani, G., Barone, V., Petersson, G. A., Nakatsuji, H., Li, X., Caricato, M., Marenich, A. V., Bloino, J., Janesko, B. G., Gomperts, R., Mennucci, B., Hratchian, H. P., Ortiz, J. V., Izmaylov, A. F., Sonnenberg, J. L., Williams, Ding, F., Lipparini, F., Egidi, F., Goings, J., Peng, B., Petrone, A., Henderson, T., Ranasinghe, D., Zakrzewski, V. G., Gao, J., Rega, N., Zheng, G., Liang, W., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Vreven, T., Throssell, K., Montgomery Jr., J. A., Peralta, J. E., Ogliaro, F., Bearpark, M. J., Heyd, J. J., Brothers, E. N., Kudin, K. N., Staroverov, V. N., Keith, T. A., Kobayashi, R., Normand, J., Raghavachari, K., Rendell, A. P., Burant, J. C., Iyengar, S. S., Tomasi, J., Cossi, M., Millam, J. M., Klene, M., Adamo, C., Cammi, R., Ochterski, J. W., Martin, R. L., Morokuma, K., Farkas, O., Foresman, J. B., and Fox, D. J.: Gaussian 09 Rev. E.01, Wallingford, CT, 2013.
Heikes, B. G., Lee, M., Bradshaw, J., Sandholm, S., Davis, D. D., Crawford, J., Rodriguez, J., Liu, S., McKeen, S., Thornton, D., Bandy, A., Gregory, G., Talbot, R., and Blake, D.: Hydrogen peroxide and methylhydroperoxide distributions related to ozone and odd hydrogen over the North Pacific in the fall of 1991, J. Geophys. Res.-Atmos., 101, 1891–1905, https://doi.org/10.1029/95JD01364, 1996.
Hettiyadura, A. P. S., Jayarathne, T., Baumann, K., Goldstein, A. H., de Gouw, J. A., Koss, A., Keutsch, F. N., Skog, K., and Stone, E. A.: Qualitative and quantitative analysis of atmospheric organosulfates in Centreville, Alabama, Atmos. Chem. Phys., 17, 1343–1359, https://doi.org/10.5194/acp-17-1343-2017, 2017.
Hu, K. S., Darer, A. I., and Elrod, M. J.: Thermodynamics and kinetics of the hydrolysis of atmospherically relevant organonitrates and organosulfates, Atmos. Chem. Phys., 11, 8307–8320, https://doi.org/10.5194/acp-11-8307-2011, 2011.
Hu, M., Chen, K., Qiu, J., Lin, Y.-H., Tonokura, K., and Enami, S.: Temperature Dependence of Aqueous-Phase Decomposition of α-Hydroxyalkyl-Hydroperoxides, J. Phys. Chem. A, 124, 10288–10295, https://doi.org/10.1021/acs.jpca.0c09862, 2020.
Hua, W., Chen, Z. M., Jie, C. Y., Kondo, Y., Hofzumahaus, A., Takegawa, N., Chang, C. C., Lu, K. D., Miyazaki, Y., Kita, K., Wang, H. L., Zhang, Y. H., and Hu, M.: Atmospheric hydrogen peroxide and organic hydroperoxides during PRIDE-PRD'06, China: their concentration, formation mechanism and contribution to secondary aerosols, Atmos. Chem. Phys., 8, 6755–6773, https://doi.org/10.5194/acp-8-6755-2008, 2008.
Huang, L., Liu, T., and Grassian, V. H.: Radical-Initiated Formation of Aromatic Organosulfates and Sulfonates in the Aqueous Phase, Environ. Sci. Technol., 54, 11857–11864, https://doi.org/10.1021/acs.est.0c05644, 2020.
Huang, R.-J., Cao, J., Chen, Y., Yang, L., Shen, J., You, Q., Wang, K., Lin, C., Xu, W., Gao, B., Li, Y., Chen, Q., Hoffmann, T., O'Dowd, C. D., Bilde, M., and Glasius, M.: Organosulfates in atmospheric aerosol: synthesis and quantitative analysis of PM2.5 from Xi'an, northwestern China, Atmos. Meas. Tech., 11, 3447–3456, https://doi.org/10.5194/amt-11-3447-2018, 2018.
Hung, H.-M., Hsu, M.-N., and Hoffmann, M. R.: Quantification of SO2 Oxidation on Interfacial Surfaces of Acidic Micro-Droplets: Implication for Ambient Sulfate Formation, Environ. Sci. Technol., 52, 9079–9086, https://doi.org/10.1021/acs.est.8b01391, 2018.
Hung, H. M. and Hoffmann, M. R.: Oxidation of Gas-Phase SO2 on the Surfaces of Acidic Microdroplets: Implications for Sulfate and Sulfate Radical Anion Formation in the Atmospheric Liquid Phase, Environ. Sci. Technol., 49, 13768–13776, https://doi.org/10.1021/acs.est.5b01658, 2015.
Ignatov, S. K., Gadzhiev, O. B., Kulikov, M. Y., Petrov, A. I., Razuvaev, A. G., Gand, M., Feigin, A. M., and Schrems, O.: Adsorption of Methyl Hydroperoxide (CH3OOH) on Water Ice. Theoretical Study with Systematic Assessment of Coordination Modes, J. Phys. Chem. C, 115, 9081–9089, https://doi.org/10.1021/jp112177x, 2011.
Kiejza, D., Kotowska, U., Polińska, W., and Karpińska, J.: Peracids - New oxidants in advanced oxidation processes: The use of peracetic acid, peroxymonosulfate, and persulfate salts in the removal of organic micropollutants of emerging concern – A review, Sci. Total Environ., 790, 148195, https://doi.org/10.1016/j.scitotenv.2021.148195, 2021.
Kircik, L. H.: The role of benzoyl peroxide in the new treatment paradigm for acne, J. Drugs Dermatol., 12, 73–76, 2013.
Klippel, T., Fischer, H., Bozem, H., Lawrence, M. G., Butler, T., Jöckel, P., Tost, H., Martinez, M., Harder, H., Regelin, E., Sander, R., Schiller, C. L., Stickler, A., and Lelieveld, J.: Distribution of hydrogen peroxide and formaldehyde over Central Europe during the HOOVER project, Atmos. Chem. Phys., 11, 4391–4410, https://doi.org/10.5194/acp-11-4391-2011, 2011.
Kozan, J. V., Silva, R. P., Serrano, S. H., Lima, A. W., and Angnes, L.: Amperometric detection of benzoyl peroxide in pharmaceutical preparations using carbon paste electrodes with peroxidases naturally immobilized on coconut fibers, Biosens. Bioelectron., 25, 1143–1148, https://doi.org/10.1016/j.bios.2009.09.044, 2010.
Krapf, M., El Haddad, I., Bruns, E. A., Molteni, U., Daellenbach, K. R., Prévôt, A. S. H., Baltensperger, U., and Dommen, J.: Labile Peroxides in Secondary Organic Aerosol, Chem, 1, 603–616, https://doi.org/10.1016/j.chempr.2016.09.007, 2016.
Kuang, B. Y., Lin, P., Hu, M., and Yu, J. Z.: Aerosol size distribution characteristics of organosulfates in the Pearl River Delta region, China, Atmos. Environ., 130, 23–35, https://doi.org/10.1016/j.atmosenv.2015.09.024, 2016.
Kwong, K. C., Chim, M. M., Davies, J. F., Wilson, K. R., and Chan, M. N.: Importance of sulfate radical anion formation and chemistry in heterogeneous OH oxidation of sodium methyl sulfate, the smallest organosulfate, Atmos. Chem. Phys., 18, 2809–2820, https://doi.org/10.5194/acp-18-2809-2018, 2018.
Lei, Z., Chen, Y., Zhang, Y., Cooke, M. E., Ledsky, I. R., Armstrong, N. C., Olson, N. E., Zhang, Z., Gold, A., Surratt, J. D., and Ault, A. P.: Initial pH Governs Secondary Organic Aerosol Phase State and Morphology after Uptake of Isoprene Epoxydiols (IEPOX), Environ. Sci. Technol., 56, 10596–10607, https://doi.org/10.1021/acs.est.2c01579, 2022.
Lind, J. A., Lazrus, A. L., and Kok, G. L.: Aqueous phase oxidation of sulfur(IV) by hydrogen peroxide, methylhydroperoxide, and peroxyacetic acid, J. Geophys. Res.-Atmos., 92, 4171–4177, https://doi.org/10.1029/JD092iD04p04171, 1987.
Liou, G.-Y. and Storz, P.: Reactive oxygen species in cancer, Free Radical Res., 44, 479–496, https://doi.org/10.3109/10715761003667554, 2010.
Liu, Q., Wang, W., Liu, Z., Wang, T., Wu, L.-Y., and Ge, M. J. R. A.: Organic hydroperoxide formation in the acid-catalyzed heterogeneous oxidation of aliphatic alcohols with hydrogen peroxide, RSC Adv., 4, 19716–19724, 2014.
Liu, W., Lv, G., Zhang, C., and Sun, X.: Mechanism of secondary organic aerosol formation from the reaction of isoprene with sulfoxy radicals, Environ. Sci. Pollut. R., 28, 42562–42569, https://doi.org/10.1007/s11356-021-13539-9, 2021.
Lu, T. and Chen, F.: Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 33, 580–592, https://doi.org/10.1002/jcc.22885, 2012.
Luukkonen, T., Heyninck, T., Rämö, J., and Lassi, U.: Comparison of organic peracids in wastewater treatment: Disinfection, oxidation and corrosion, Water Res., 85, 275–285, https://doi.org/10.1016/j.watres.2015.08.037, 2015.
Mari, C., Jacob, D. J., and Bechtold, P.: Transport and scavenging of soluble gases in a deep convective cloud, J. Geophys. Res.-Atmos., 105, 22255–22267, https://doi.org/10.1029/2000JD900211, 2000.
Meylan, W. M. and Howard, P. H.: Bond contribution method for estimating henry's law constants, Environ. Toxicol. Chem., 10, 1283–1293, https://doi.org/10.1002/etc.5620101007, 1991.
Morgan, R. B. and Jackson, A. V.: Measurements of gas-phase hydrogen peroxide and methyl hydroperoxide in the coastal environment during the PARFORCE project, J. Geophys. Res., 107, 8109, https://doi.org/10.1029/2000JD000257, 2002.
Ostovari, H., Zahedi, E., Sarvi, I., and Shiroudi, A.: Kinetic and mechanistic insight into the formation of amphetamine using the Leuckart–Wallach reaction and interaction of the drug with GpC • CpG base-pair step of DNA: a DFT study, Monatsh. Chem., 149, 1045–1057, https://doi.org/10.1007/s00706-018-2145-7, 2018.
O'Sullivan, D. W., Lee, M., Noone, B. C., and Heikes, B. G.: Henry's Law Constant Determinations for Hydrogen Peroxide, Methyl Hydroperoxide, Hydroxymethyl Hydroperoxide, Ethyl Hydroperoxide, and Peroxyacetic Acid, J. Phys. Chem., 100, 3241–3247, https://doi.org/10.1021/jp951168n, 1996.
O'Sullivan, D. W., Heikes, B. G., Lee, M., Chang, W., Gregory, G. L., Blake, D. R., and Sachse, G. W.: Distribution of hydrogen peroxide and methylhydroperoxide over the Pacific and South Atlantic Oceans, J. Geophys. Res.-Atmos., 104, 5635–5646, https://doi.org/10.1029/98JD01250, 1999.
O'Sullivan, D. W., Neale, P. J., Coffin, R. B., Boyd, T. J., and Osburn, C. L.: Photochemical production of hydrogen peroxide and methylhydroperoxide in coastal waters, Marine Chem., 97, 14–33, https://doi.org/10.1016/j.marchem.2005.04.003, 2005.
Qiu, J., Tonokura, K., and Enami, S.: Proton-Catalyzed Decomposition of α-Hydroxyalkyl-Hydroperoxides in Water, Environ. Sci. Technol., 54, 10561–10569, https://doi.org/10.1021/acs.est.0c03438, 2020.
Ravetta, F., Jacob, D. J., Brune, W. H., Heikes, B. G., Anderson, B. E., Blake, D. R., Gregory, G. L., Sachse, G. W., Sandholm, S. T., Shetter, R. E., Singh, H. B., and Talbot, R. W.: Experimental evidence for the importance of convected methylhydroperoxide as a source of hydrogen oxide (HOx) radicals in the tropical upper troposphere, J. Geophys. Res.-Atmos., 106, 32709–32716, https://doi.org/10.1029/2001JD900009, 2001.
Riemer, D. D., Milne, P. J., Zika, R. G., and Pos, W. H.: Photoproduction of nonmethane hydrocarbons (NMHCs) in seawater, Mar. Chem., 71, 177–198, https://doi.org/10.1016/S0304-4203(00)00048-7, 2000.
Riplinger, C. and Neese, F.: An efficient and near linear scaling pair natural orbital based local coupled cluster method, J. Chem. Phys., 138, 034106, https://doi.org/10.1063/1.4773581, 2013.
Riva, M., Tomaz, S., Cui, T., Lin, Y.-H., Perraudin, E., Gold, A., Stone, E. A., Villenave, E., and Surratt, J. D.: Evidence for an Unrecognized Secondary Anthropogenic Source of Organosulfates and Sulfonates: Gas-Phase Oxidation of Polycyclic Aromatic Hydrocarbons in the Presence of Sulfate Aerosol, Environ. Sci. Technol., 49, 6654–6664, https://doi.org/10.1021/acs.est.5b00836, 2015.
Sapkota, V. N. A., Fine, N. A., and Rochelle, G. T.: NO2-Catalyzed Sulfite Oxidation, Ind. Eng. Chem. Res., 54, 4815–4822, https://doi.org/10.1021/ie504767w, 2015.
Sauer, F., Beck, J., Schuster, G., and Moortgat, G. K.: Hydrogen peroxide, organic peroxides and organic acids in a forested area during FIELDVOC'94, Chemosphere – Global Change Science, 3, 309–326, https://doi.org/10.1016/S1465-9972(01)00013-7, 2001.
Smoluchowski, M. V.: Mathematical Theory of the Kinetics of the Coagulation of Colloidal Solutions, Z. Phys. Chem., 92, 129–168, 1917.
Song, Y.-C., Lilek, J., Lee, J. B., Chan, M. N., Wu, Z., Zuend, A., and Song, M.: Viscosity and phase state of aerosol particles consisting of sucrose mixed with inorganic salts, Atmos. Chem. Phys., 21, 10215–10228, https://doi.org/10.5194/acp-21-10215-2021, 2021.
Sun, J., Ma, J., Lian, L., Yan, S., and Song, W.: Photochemical Formation of Methylhydroperoxide in Dissolved Organic Matter Solutions, Environ. Sci. Technol., 55, 1076–1087, https://doi.org/10.1021/acs.est.0c07717, 2021.
Tan, Y., Xu, Y., Shang, Y., Wang, H., Li, W., and Cao, W.: Thermal Decomposition Behavior and Thermal Hazard of Benzoyl Peroxide under Different Environmental Conditions, ChemistrySelect, 5, 5049–5054, https://doi.org/10.1002/slct.201904896, 2020.
Tkacik, D. S., Presto, A. A., Donahue, N. M., and Robinson, A. L.: Secondary Organic Aerosol Formation from Intermediate-Volatility Organic Compounds: Cyclic, Linear, and Branched Alkanes, Environ. Sci. Technol., 46, 8773–8781, https://doi.org/10.1021/es301112c, 2012.
Truhlar, D. G.: Nearly encounter-controlled reactions: The equivalence of the steady-state and diffusional viewpoints, J. Chem. Educ., 62, 104, https://doi.org/10.1021/ed062p104, 1985.
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J.: Current Status of Transition-State Theory, J. Phys. Chem., 100, 12771–12800, https://doi.org/10.1021/jp953748q, 1996.
Tu, Y.-P., Yuan, H.-S., and Liu, S.-Y.: Mass Spectrometric Evidence for the Generation of Benzyne from Benzoyl Peroxide in Thermolysis, Rapid Commun. Mass Sp., 10, 1093–1095, https://doi.org/10.1002/(SICI)1097-0231(19960715)10:9<1093::AID-RCM628>3.0.CO;2-S, 1996.
Turnock, S. T., Mann, G. W., Woodhouse, M. T., Dalvi, M., O'Connor, F. M., Carslaw, K. S., and Spracklen, D. V.: The Impact of Changes in Cloud Water pH on Aerosol Radiative Forcing, Geophys. Res. Lett., 46, 4039–4048, https://doi.org/10.1029/2019GL082067, 2019.
Wang, S., Zhou, S., Tao, Y., Tsui, W. G., Ye, J., Yu, J. Z., Murphy, J. G., McNeill, V. F., Abbatt, J. P. D., and Chan, A. W. H.: Organic Peroxides and Sulfur Dioxide in Aerosol: Source of Particulate Sulfate, Environ. Sci. Technol., 53, 10695–10704, https://doi.org/10.1021/acs.est.9b02591, 2019.
Wang, S., Liu, T., Jang, J., Abbatt, J. P. D., and Chan, A. W. H.: Heterogeneous interactions between SO2 and organic peroxides in submicron aerosol, Atmos. Chem. Phys., 21, 6647–6661, https://doi.org/10.5194/acp-21-6647-2021, 2021.
Wang, S., Zhao, Y., Chan, A. W. H., Yao, M., Chen, Z., and Abbatt, J. P. D.: Organic Peroxides in Aerosol: Key Reactive Intermediates for Multiphase Processes in the Atmosphere, Chem. Rev., 123, 1635–1679, https://doi.org/10.1021/acs.chemrev.2c00430, 2023.
Xu, L. and Coote, M. L.: Methods To Improve the Calculations of Solvation Model Density Solvation Free Energies and Associated Aqueous pKa Values: Comparison between Choosing an Optimal Theoretical Level, Solute Cavity Scaling, and Using Explicit Solvent Molecules, J. Phys. Chem. A, 123, 7430–7438, https://doi.org/10.1021/acs.jpca.9b04920, 2019.
Xu, Y., Zhang, H., Cao, W., Zhang, Y., Tan, Y., Shu, C.-M., and Luo, J.-W.: Autocatalytic decomposition properties and thermal decomposition of benzoyl peroxide, J. Therm. Anal. Calorim., 146, 2601–2611, https://doi.org/10.1007/s10973-021-10677-7, 2021.
Ye, J., Abbatt, J. P. D., and Chan, A. W. H.: Novel pathway of SO2 oxidation in the atmosphere: reactions with monoterpene ozonolysis intermediates and secondary organic aerosol, Atmos. Chem. Phys., 18, 5549–5565, https://doi.org/10.5194/acp-18-5549-2018, 2018.
Yu, L., Jiang, C., Xi, L., Zhang, X., Tong, J., Chen, Z., Chen, R., and He, H.: Colorimetric Detection of Benzoyl Peroxide in the Flour Samples Based on the Morphological Transition of Silver Nanoprisms, Food Anal. Methods, 15, 993–1000, https://doi.org/10.1007/s12161-021-02145-7, 2022.
Zhang, T. and Huang, C.-H.: Modeling the Kinetics of UV/Peracetic Acid Advanced Oxidation Process, Environ. Sci. Technol., 54, 7579–7590, https://doi.org/10.1021/acs.est.9b06826, 2020.
Zhang, X., He, S. Z., Chen, Z. M., Zhao, Y., and Hua, W.: Methyl hydroperoxide (CH3OOH) in urban, suburban and rural atmosphere: ambient concentration, budget, and contribution to the atmospheric oxidizing capacity, Atmos. Chem. Phys., 12, 8951–8962, https://doi.org/10.5194/acp-12-8951-2012, 2012.