the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 12 Dec 2024
| 12 Dec 2024
Formation and chemical evolution of secondary organic aerosol in two different environments: a dual-chamber study
Andreas Aktypis
Dontavious J. Sippial
Christina N. Vasilakopoulou
Angeliki Matrali
Christos Kaltsonoudis
Andrea Simonati
Marco Paglione
Matteo Rinaldi
Stefano Decesari
Spyros N. Pandis
A dual-chamber system was deployed in two different environments to study the potential of ambient air, which was directly injected into the chambers, to form secondary organic and inorganic aerosol. A total of 16 experiments took place during March 2022 in a polluted environment in the Po Valley, Italy, which is dominated by anthropogenic emissions. Another 15 experiments were conducted in the Pertouli forest, Greece, which is dominated by biogenic emissions. In both campaigns, ambient air containing highly oxidized (average O:C 0.7–0.8) aerosol was the starting point of the experiments, and its chemical evolution under the presence of OH radicals was followed. In the Po Valley secondary organic aerosol (SOA) formation was observed in all experiments but one, and the SOA formed ranged from 0.1 to 10 µg m−3. Experiments conducted under more polluted conditions (usually at night and early morning) had significantly higher SOA formation, with the concentration of the organic aerosol at the end being about 4 times higher than the initial level. Also, production of 4–230 µg m−3 of ammonium nitrate was observed in all experiments due to the high levels of ammonia in this area. The produced SOA appeared to increase as the ambient relative humidity increased, but other parameters could also be responsible for this. There was not a clear relationship between the SOA and temperature, while higher SOA production was observed when the PM1 levels in the Po Valley were high. Contrary to the Po Valley, only one experiment in the Pertouli forest resulted in the formation of detectable SOA (about 1 µg m−3). This experiment was characterized by higher ambient concentrations of both monoterpenes and isoprene. In two experiments, some SOA was formed, but its concentration dropped below detection levels after 30 min. This behavior is consistent with local formation in a chamber that was not well mixed. Although both environments have organic aerosol (OA) with O:C in the range of 0.7–0.8, the atmosphere of the two sites had very different potentials of forming SOA. In the Po Valley, the system reacts rapidly, forming large amounts of SOA, while in Pertouli the corresponding SOA formation chemistry appears to have been practically terminated before the beginning of most experiments, so there is little additional SOA formation potential left.
- Article
(2783 KB) - Full-text XML
-
Supplement
(1846 KB) - BibTeX
- EndNote





About 300 000 premature deaths are estimated to occur annually in Europe due to air pollution, the vast majority (77 %) of which are related to chronic exposure to fine particulate matter (EEA, 2023). Exposure to higher levels of atmospheric aerosols has been linked to various diseases affecting the cardiovascular and respiratory systems, as well as the brain (Pope III and Dockery, 2006). These particles are also responsible for the reduction of the atmosphere's visibility, while they play a key role in the climate system (Seinfeld and Pandis, 2016). Although the impacts of atmospheric aerosols on both climate and human health have been widely studied, large uncertainty remains about their chemical evolution in the atmosphere because their sources, composition, properties, and toxicity can vary dramatically from one environment to another.
Organic material accounts for 20 %–60 % of the total fine aerosol mass on a global scale, and it can even reach 90 % in tropical forests (Kanakidou et al., 2005). Due to its high availability and characteristics, it plays a predominant role in various physicochemical processes in the atmosphere. These include the formation of new particles and their subsequent growth (Kulmala et al., 2004; Kerminen et al., 2018), affecting climate both directly and indirectly (Seinfeld and Pandis, 2016). Organic aerosol (OA) can either be introduced in the atmosphere directly (primary organic aerosol or POA) or be formed through the oxidation of various organic vapors (secondary organic aerosol or SOA). The sources of POA include combustion processes, like biomass burning (wildfires, domestic heating, etc.), transportation, and cooking (Kanakidou et al., 2005). A higher complexity characterizes the formation of SOA, which accounts for up to 90 % of the total OA in both urban and rural environments (Huang et al., 2014; Chaturvedi et al., 2023). The gaseous precursors involved in SOA production originate from both biogenic and anthropogenic sources, interact with each other, and have different contributions to the formed SOA. The mechanisms and kinetics of SOA formation depend on ambient conditions, NOx availability, preexisting OA, the oxidant (OH, O3, or NO3) involved, etc. (Kroll and Seinfeld, 2008). Understanding how and to what extent SOA forms in the atmosphere is crucial for addressing the impact of aerosol on climate and human health. SOA can have even higher toxicity than primary particles (Ito et al., 2019).
The levels of SOA observed in the atmosphere are often underpredicted by chemical transport models as a result of our lack of understanding of the corresponding SOA formation pathways (Kanakidou et al., 2005; Volkamer et al., 2006; Robinson et al., 2007; Kroll and Seinfeld, 2008; Jimenez et al., 2009; Tsigaridis et al., 2014). While developments, such as the volatility basis set (Donahue et al., 2011) and accounting for the intermediate-volatility organic compounds (IVOCs), have significantly improved model predictions, the problem of underestimation often persists. For example, heterogeneous reactions that may affect SOA formation are not fully incorporated, even in the most recent models (Jo et al., 2024). The interactions of the oxidized precursors in the atmospheric environment could be another potential reason for model–measurement discrepancy (McFiggans et al., 2019; Schervish and Donahue, 2020; Voliotis et al., 2021; Takeuchi et al., 2022). Another explanation for this underprediction is that models rely on measurements obtained from laboratory chamber experiments that typically test the potential of individual volatile organic compounds (VOCs) to form SOA (Kroll and Seinfeld, 2008). Although these laboratory experiments provide valuable insights about the chemical pathways the carbon of a specific VOC follows during and after its oxidation, they are not always able to explain ambient observations. For instance, the SOA formed in the ambient air is usually far more oxidized than the one formed in atmospheric simulation chambers (Kroll and Seinfeld, 2008).
Biogenic volatile organic compounds (BVOCs) include isoprene (C5H8), monoterpenes (C10H16), and sesquiterpenes (C15H24) and are emitted at a higher rate on a global scale than their anthropogenic counterparts (Guenther et al., 2012; Seinfeld and Pandis, 2016). Numerous laboratory studies have investigated the oxidation of these compounds (mainly isoprene and monoterpenes) by the hydroxyl radical (OH), ozone (O3), and the nitrate radical (NO3) (Griffin et al., 1999b; Kroll et al., 2006; Qi et al., 2012; Müller et al., 2012; Wang et al., 2018a). Most of these efforts have focused on the first generation of reactions and the corresponding formation of SOA. The first-generation products, depending on their volatility and structure, can be further oxidized and produce later generations of products, a process known as chemical aging. Ng et al. (2006) suggested that several compounds containing more than one double bond, like many terpenes, continue to make a notable contribution to SOA formation, even after undergoing their initial oxidation step. For example, they observed a significant increase in SOA mass from 100 to 340 µg m−3 during β-caryophyllene ozonolysis, driven by later-generation reactions (Ng et al., 2006). Donahue et al. (2012) also reported that OH aging of biogenic SOA can enhance its concentration by a factor of 2–4. Sesquiterpenes have been found to have a notable SOA formation potential, even in later stages of oxidation (Griffin et al., 1999a; Tasoglou and Pandis, 2015; Barreira et al., 2021), but limited knowledge still exists about their detailed oxidation pathways. The complexity of understanding the aerosol aging potential increases in urban, suburban, or rural environments where anthropogenic VOCs dominate (He et al., 2021; Nault et al., 2021; Chaturvedi et al., 2023). Semivolatile (C* between 1 and 100 µg m−3) and intermediate-volatility (C* between 103 and 106 µg m−3) organic compounds (SVOCs and IVOCs, respectively) have been found to play a key role in the formation of SOA, despite their relatively low ambient concentrations compared to other VOCs. Laboratory experiments have shown that these compounds have much higher SOA yields than the VOCs with smaller molecular weight (Chan et al., 2009; Lim and Ziemann, 2009; Presto et al., 2010; Tkacik et al., 2012; Docherty et al., 2021). For example, Zhao et al. (2014) estimated that IVOCs were responsible for 30 % of the SOA formed during afternoon hours in an urban environment in the US. However, due to the experimental difficulties in measuring IVOC concentrations, there is still uncertainty about their exact role in SOA formation and especially the role of the later-generation reactions.
While investigations of the chemical evolution of specific VOCs and IVOCs within controlled chambers provide valuable information about their potential to form SOA, the corresponding experimental conditions frequently deviate from real-world atmospheric conditions. The use of ambient air as a starting point in chamber experiments can help connect ambient observations with laboratory experiments. Kaltsonoudis et al. (2019) designed a dual-chamber system to perform such experiments. Jorga et al. (2021) utilized this system to explore the nighttime chemistry of biomass burning emissions in an urban area in Greece. The system enabled them to quantify the rather high potential of the actual ambient air to form SOA, even during periods with low photochemistry. The portability of the system and its easy setup make it useful in challenging environments, such as forests.
In this study, we take advantage of the already aged aerosol in two different environments (one polluted and one more pristine inside a forest), to investigate their potential to form additional SOA, through oxidation of the existing VOCs, IVOCs, and SVOCs. We focus on oxidation by OH radicals at different NOx conditions. The dual-chamber system was initially deployed at a rural site in the Po Valley, Italy, an area that is highly polluted and dominated by anthropogenic emissions. The same system was also deployed, in Pertouli, a remote Greek forest, that is expected to be dominated by biogenic emissions. The only commonality about these two sites was the nearly identical (and relatively constant) ambient OA oxygen-to-carbon ratio (O:C), indicating highly aged aerosols in both environments. Our hypothesis is that the chemical evolution of highly aged organic aerosols differs significantly between polluted and pristine environments, despite similar initial oxidation levels of the OA. The results of all the conducted experiments are synthesized to provide insights into the SOA formation potential of the two different environments.
2.1 Description of sites and field campaigns
2.1.1 FAIRARI campaign
The Fog and Aerosol InteRAction Research Italy (FAIRARI) field campaign (November 2021–May 2022) was conducted at San Pietro Capofiume (SPC), a rural site in the southern part of the Po Valley in northern Italy (Fig. S1 in the Supplement). The main objectives of the campaign were the investigation of the interactions between aerosol and fog, the formation and chemical evolution of SOA, and the driving mechanisms of new particle formation in a polluted atmosphere. Details about the campaign can be found in the overview paper by Neuberger et al. (2024).
The greater Po Valley area is one of the most polluted environments in Europe (Decesari et al., 2014; Daellenbach et al., 2020; EEA, 2023). Its high population density, orography, and the intense industrial and agricultural activities all contribute to high levels of atmospheric pollutants and especially particulate matter (Decesari et al., 2014). During the cold period (winter and early spring) the introduction of biomass burning emissions in the atmosphere exacerbates the already poor air quality. Paglione et al. (2020) reported that SOA dominates OA, contributing at any given moment more than 65 % of the OA, with an annual average of 83 ± 16 %. The ambient OA O:C is of the order of 0.7–0.9, implying indeed aged aerosol (Paglione et al., 2020). During the cold period, the high liquid aerosol water content has also been found to participate in the aging process. The conditions in this region during the cold period (elevated concentrations of NOx, NH3, low temperature, high humidity) create an ideal environment for the formation of secondary inorganic aerosol (Squizzato et al., 2013). High efficiency in the formation of ammonium nitrate in SPC during this period has been reported (Paglione et al., 2021).
A total of 16 dual-chamber experiments (including 2 blanks, in which there was no OH production) were conducted during the FAIRARI campaign, between 2 and 17 March 2022 (Table 1).
Table 1Starting times, initial conditions, total SOA formed, SOA formation rate, and OH radicals' concentration in the perturbed chamber of each experiment conducted during the FAIRARI campaign.
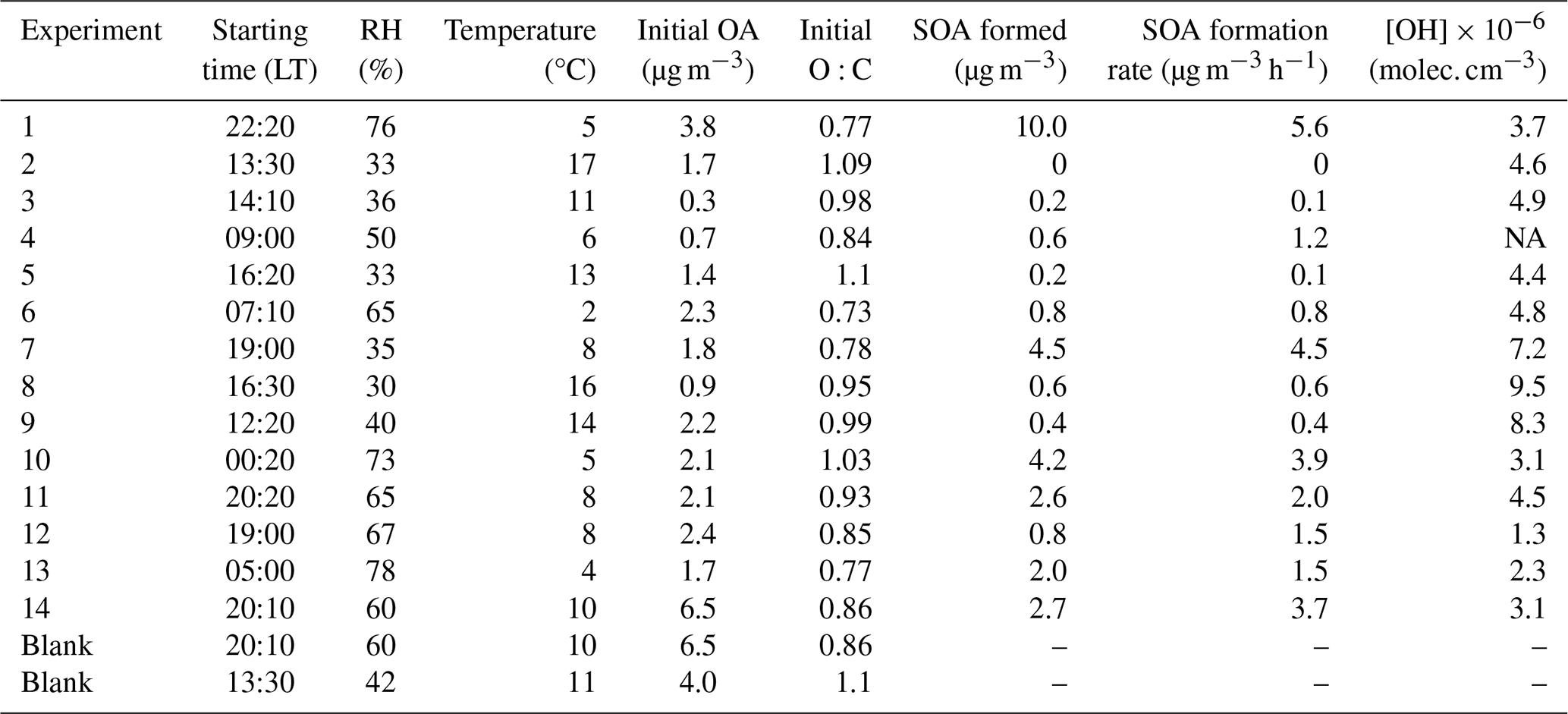
NA: not available.
2.1.2 SPRUCE-22 campaign
The Summer PeRtoUli Campaign Emissions (SPRUCE-22) field campaign was conducted during July 2022, in Pertouli, a remote mountainous forested site in Greece (Fig. S1). Its main objective was to quantify the interactions between pollutants transported over long distances and biogenic emissions in the eastern Mediterranean. It combined both chamber experiments and ambient measurements (Vasilakopoulou et al., 2023; Matrali et al., 2024). Situated at an elevation of 1300 m a.s.l., the site was positioned at the chalet atop the Pertouli ski resort (39°32′44′′ N, 21°29′55′′ E), in the middle of a spruce–fir forest. More information about the campaign can be found in Matrali et al. (2024).
The area is characterized by high biogenic emissions, especially of isoprene and monoterpenes (Harrison et al., 2001). Due to fast photochemistry during the summer, these are rapidly oxidized resulting in relatively low ambient concentrations. Positive matrix factorization (PMF) analysis revealed that the biogenic oxidized factor accounted for 23 % of the total, the rest being less-oxidized OA (37 %) and more-oxidized OA (40 %) (Vasilakopoulou et al., 2023). Detailed analysis of the latter suggested that it was mostly a result of rapid chemical aging and transport of wildfire emissions, originating hundreds of kilometers away from the site (Vasilakopoulou et al., 2023). However, biogenic SOA was also present in the less-oxidized OA, and some could even be included in the more-oxidized OA.
A total of 15 dual-chamber experiments (including 2 blanks) were conducted during the SPRUCE-22 campaign, between 7 and 25 July 2022 (Table 2).
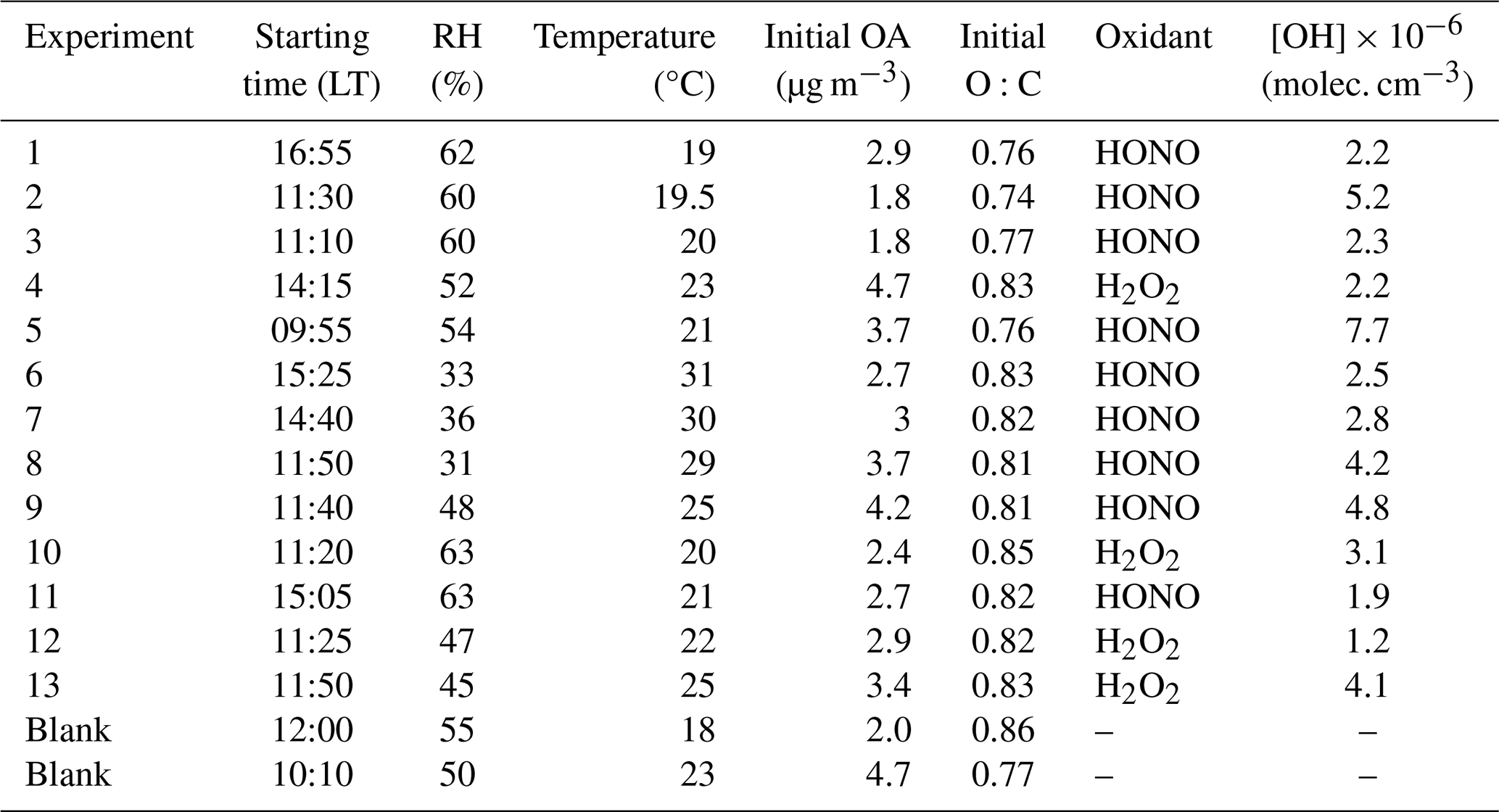
Table 2Starting times, initial conditions, and OH radicals' concentration in the perturbed chamber of each experiment conducted during the SPRUCE-22 campaign.
2.2 The dual-chamber system
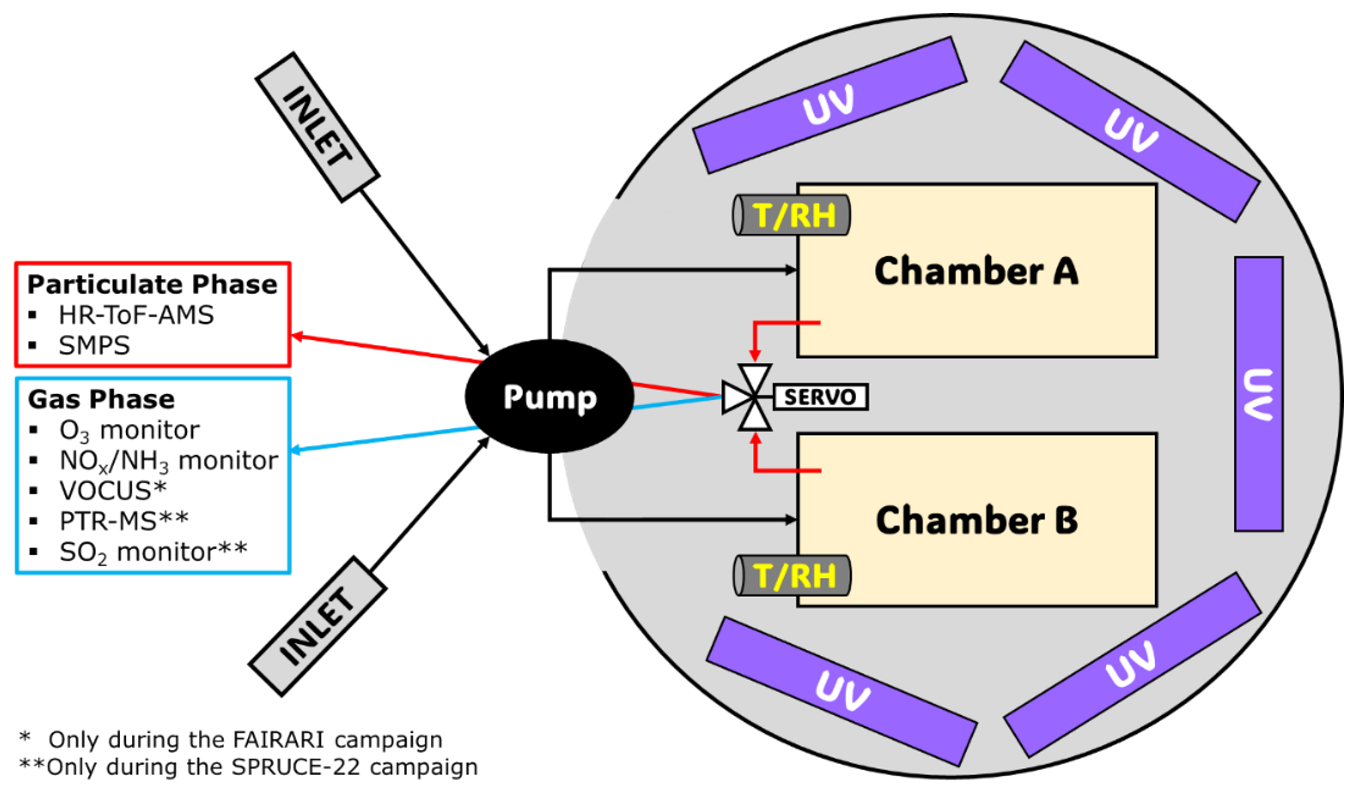
The experiments in both campaigns were conducted in the same dual-chamber system (Kaltsonoudis et al., 2019). The FORTH mobile chamber system (Fig. 1) consists of two identical PTFE chambers (1.3 m3 each), five UV light panels (a total of 60 fluorescent 36 W lamps with a broad maximum at 365 nm, yielding a of 0.1 min−1), and several instruments for the measurement of the concentration of gases and particles inside the chambers. The instruments sampled through a special port at the front side of each chamber. The two chambers and the light panels were located inside a hemispherical enclosure in order to protect them from rain, wind, etc. The top of the structure could be opened, if necessary, to expose them to natural sunlight or as a method of temperature control for the chambers. More details about the chamber setup and testing can be found in Kaltsonoudis et al. (2019). A metal-bellows pump (Senior Aerospace, model MB-302) was used to condition and fill the chambers with ambient air. Continuous measurements (1 min resolution) of temperature and relative humidity inside the chambers were performed in each experiment using two stainless-steel sensors (RH-USB, Omega).
2.3 Experimental procedure
The experimental procedure always started with the preparation of the chambers. Before each experiment, the chambers were flushed continuously with ambient air for about 2 h using the metal-bellows pump, in order to condition them and the sampling lines with ambient air that were to be used later for the actual experiment. During this period, an ionizer fan (Dr. Schneider PC, model SL-001) was used on the surface of the chamber walls for 20 min, in an effort to minimize the wall charges and thus the wall losses (Jorga et al., 2020). After this preparation procedure, the chambers were filled with ambient air while the synchronization of the different sampling instruments was performed. The concentration and chemical composition of the various gases and particles in both chambers were then characterized for 40–60 min. We measured the pollutant concentrations in both chambers using an automated servo valve that was also synchronized with the sampling instruments. D9-butanol (approximately 50–100 ppb) was injected in both chambers as an OH radical tracer to estimate from its decay the concentration of the OH radicals.
Then, nitrous acid (HONO) was bubbled in the perturbation chamber to produce OH radicals, while the second one was used as a reference. The production of HONO was performed following the work of Mellouki and Mu (2003). The line connecting the outlet of the bubbler to the perturbed chamber had a Teflon filter to prevent generated particles from getting into the chamber. The concentration of HONO inside the perturbed chamber was estimated from the NO measurements to be in the range of 0.5–1 ppm. The dissociation of HONO, together with the OH radicals, introduces high levels of NO (0.5–1 ppm) in the perturbed chamber. Nitrous acid (HONO) photolysis as a hydroxyl radical source has been used widely in many atmospheric stimulation smog chambers, and its advantages and disadvantages have been documented (Bell et al., 2023). For this work the addition of HONO was selected for most experiments to generate a high enough concentration of OH rapidly, given the limitations posed by the smaller chambers and the field conditions. The presence of high levels of NOx in the chamber means that the SOA formation takes place in the high-NOx regime. This should not be a major change in the Po Valley where the ambient atmosphere is already in this regime. Adding more NOx in this regime should have a small effect on SOA formation as far as oxidation pathways are concerned. In Pertouli the regime is changed by this addition of HONO. To investigate this effect in Pertouli we performed four experiments with H2O2 as the source of OH radicals in that forest environment. Their results were consistent with those with the HONO. The H2O2 was produced from a solution of hydrogen peroxide in water (50 %–50 %). The concentration of H2O2 was estimated, based on the concentration of the OH radicals, to be of the order of a few hundred parts per billion (ppb). The UV lights were turned on after the HONO (or the H2O2) addition, and the concentration of both gases and particles was followed for at least 2–3 more hours.
At the end of each experiment, ammonium sulfate seeds were added to both chambers to measure the size-dependent particle wall loss rate constants following the work of Wang et al. (2018b). After the photooxidation stage ended, the chambers were flushed with ambient air and were re-inflated to their approximate original volume, so that the estimated wall loss rate constants could be applicable to the corresponding experiment. Ammonium sulfate seeds (an aqueous solution of 5 g L−1 ammonium sulfate was used for their production) were then added, and their size distribution was monitored for about 2 h. Finally, the chambers were flushed again with ambient air to be ready for the next experiment.
2.4 Instrumentation
In both campaigns particle number size distributions in the range 14–700 nm were measured using a scanning mobility particle sizer (SMPS; Model 3080 Classifier), a condensation particle counter (CPC; models 3787 (FAIRARI) and 3775 (SPRUCE-22)), and a differential mobility analyzer (DMA; model 3081, TSI). A high-resolution time-of-flight aerosol mass spectrometer (HR-ToF-AMS; Aerodyne) was used to monitor the sub-micrometer particle chemical composition, mass size distribution, and organic aerosol mass spectrum. The detailed calibration procedures of the HR-ToF-AMS that were followed in the two campaigns can be found in the Supplement. Performance evaluation tests of the AMS were performed throughout both campaigns to ensure that it operated correctly by comparing its size distributions with those of an SMPS. These tests confirmed the good performance of the instrument at both low and high particle concentration levels.
A suite of gas monitors was used in both campaigns, including an O3 monitor (model 400E, Teledyne) and a NOx and NH3 analyzer (model T201, Teledyne). A Vocus chemical ionization time-of-flight (CI-ToF) mass spectrometer (Aerodyne Research Inc. and Tofwerk AG) was available during the Po Valley experiments to measure the VOCs, but due to technical issues the quantification of the measured ion signals was not possible. A quadrupole proton-transfer-reaction mass spectrometer (PTR-QMS 500, Ionicon Analytik) was used during the SPRUCE-22 campaign to measure the concentrations of volatile organic compounds. The calibration of the PTR-MS used a 15-compound VOC mixture at the beginning of the campaign, with daily background corrections through zero air. Additional information about the calibration of the PTR-MS and its response factors can be found in Matrali et al. (2024).
During the FAIRARI campaign, all measuring instruments were located inside either the FAIRARI containers or the FORTH mobile laboratory, which were both next to the chambers and had controlled temperature. During the SPRUCE-22 campaign, the instruments were all inside the FORTH mobile laboratory. Quarter-inch diameter copper (for particles) and PTFE (for gases) sampling lines were used.
Simultaneous on-site ambient measurements were utilized during the Po Valley experiments, obtained from the CNR-ISAC network and the Regional Agency for Prevention, Environment and Energy of Emilia-Romagna (ARPAE). The instruments included an SO2 (Model 43iTLE, Thermo Scientific), NOx (Model 200A, Teledyne), and NH3 (Model 201E, Teledyne) monitor. A meteorological station (Model WXT536, Vaisala Ltd) provided the various meteorological parameters in SPC. In Pertouli, ambient temperature and RH measurements were available.
2.5 Data analysis
The SeQUential Igor data RetRiEvaL (SQUIRREL) v1.57I and Peak Integration by Key Analysis (PIKA) v1.16I toolkits were used for the analysis of the HR-ToF-AMS data in Igor Pro (Wave Metrics). The improved method by Canagaratna et al. (2015) was used to calculate the various elemental ratios. The Analytic Procedure for Elemental Separation (APES) software (Aiken et al., 2007, 2008) was used for the calculation of the O:C of the produced SOA in each experiment. The default relative ionization efficiencies (RIEs) were used for NO (1.1), SO (1.2), organics (1.4), and Cl− (1.3) in both campaigns. A RIE equal to 4 in the Po Valley and 4.42 (obtained from the brute-force single-particle (BFSP) calibration) in the Pertouli campaign was used for NH. The AMS data were corrected for the collection efficiency (CE) following the work of Kostenidou et al. (2007). The method compares the mass distributions obtained from the AMS with the volume size distributions of the SMPS and calculates the CE and the corresponding density of the aerosol. For the comparison of the size distributions obtained from the two instruments, the electrical mobility diameters of the SMPS are converted to vacuum aerodynamic diameters used by the AMS, utilizing the aerosol density calculated from the algorithm. Since the Po Valley experiments were conducted in an environment dominated by ammonium nitrate, the parameterization by Middlebrook et al. (2012) was also used to estimate the CE, accounting for the ammonium nitrate fraction. The comparison of the two methods is further discussed in the results section.
Following the work of Wang et al. (2018b) the size-dependent wall loss constant was estimated for each experiment, utilizing the decay of the concentration in the different size channels of the SMPS in the wall loss experiments.
To determine the OH concentration, we used the tracer method of Barmet et al. (2012). The reaction rate coefficient of d9-butanol with the OH radicals is equal to 3.42 × 10−12 cm3 molec.−1 s−1 (Barmet et al., 2012). The concentration of d9-butanol was measured by the PTR-MS in Pertouli, with negligible interference by other organic compounds found in the atmosphere by observing changes at 66. To measure the d-9 butanol decay in the Po Valley, the Vocus instrument signal (ions s−1) was used, since the calculation of the OH radicals' concentration relies on the butanol's decay slope (Barmet et al., 2012).
The mass spectra of the organic aerosol that was initially in the two chambers were always compared to verify that there were no significant artifacts during the filling process. The mass spectra of the OA were identical (θ<5°) in the two chambers in all experiments of both campaigns. Also, a comparison of the initial PM1 and the O:C with the ambient values right before the start of each experiment was made. The O:C of the OA was practically the same inside the chambers and the ambient air. However, the PM1 inside the chambers was 30 %–50 % less than the ambient concentrations. This is mainly due to losses inside the pump used for filling the chambers and has also been observed in other similar studies (Jorga et al., 2020, 2021). Comparing ambient air with the initial in-chamber VOC concentrations, we estimate vapor losses of the order of 10 %–20 %. The different transmission efficiency of the particle and the gas phases could affect the partitioning of the organics that enter the chambers. As a quality assurance step, the AMS spectra of the ambient OA and the initial OA in the chamber were compared, and the observed changes were minimal. We estimated that these effects would change the fraction of a compound in the particulate phase by less than 20 %. This change would be relatively rapid and therefore is already included in the initial OA concentration of each experiment.
3.1 Mass concentration corrections
3.1.1 Collection efficiency (CE) correction
The AMS collection efficiency in both campaigns ranged from 0.3 to 0.9 in all experiments. During the characterization period of each experiment (before the introduction of the HONO) in the Po Valley, the CE was on average 0.35 ± 0.13, while during the SOA formation period, it increased to 0.7 ± 0.19. This significant increase can be explained by the high production of ammonium nitrate observed in every experiment. It has been found that as the fraction of ammonium nitrate in PM1 (in the current study it exceeded 75 % in most experiments) increases, the CE tends to approach unity (Middlebrook et al., 2012). The CE during each period (characterization and SOA formation) in each experiment was quite stable, and therefore an average CE was sufficient for the correction of the data of each period. The relatively low concentrations during the characterization period in some experiments resulted in noisy AMS size distributions (Fig. S2a); thus the corresponding loss corrections were more uncertain. Following the ammonium-nitrate-fraction-dependent CE parameterization developed by Middlebrook et al. (2012), the CE was found to be on average 0.5 ± 0.09 for the period before the introduction of the OH radicals and 0.87 ± 0.08 for the SOA formation period in the Po Valley experiments. Both aforementioned methods were also used for the Pertouli experiments, but in this case, there was much better agreement between them (compared to the Po Valley). The average CE found in Pertouli was 0.88 ± 0.3. While these values are reasonable, it is preferable to estimate the CE for each experiment based on the corresponding AMS and SMPS size distribution measurements. Because of the notable difference between the initial and final CE inside the perturbed chamber, an intermediate CE (average CE of before and after oxidation) was applied for the first AMS data points right after the UV illumination. This is reasonable because in this short transitional period (≈ 10 min), not enough ammonium nitrate and SOA have been formed yet.
3.1.2 Wall loss correction
About 10 experiments with ammonium sulfate seeds took place in the Po Valley and another 7 in Pertouli, usually before or after the actual experiments. The wall-loss-dependent rate constants were measured for both chambers. An example of their dependence on the particle diameter is shown in Fig. S3 for a typical experiment in the Po Valley. For particles with diameters larger than 200 nm, the rate constant was practically stable, and since the mass size distributions of the OA and the ammonium nitrate were always in the 300–800 nm size range (Fig. S4), the average wall loss rate constant in that range was used for the AMS measurements. This constant was on average 0.17 ± 0.08 h−1 for both chambers in the Po Valley and 0.22 ± 0.07 h−1 in Pertouli, while the difference between the two chambers was minor. The wall loss constant was also estimated from the decay of sulfate's concentration in the control chamber during the actual experiments. This (size independent) wall loss rate constant using this method was similar and had an average value of 0.2 h−1.
3.2 FAIRARI campaign (Po Valley)
3.2.1 Conditions during the FAIRARI campaign (Po Valley)
The period when the experiments were conducted (2–17 March 2022) was characterized by a relatively clean atmosphere for this area, with an average PM1 equal to 13.3 ± 7.9 µg m−3, ranging from 3 to 40 µg m−3. The main components of the ambient PM1 were the organics and nitrates, with an average concentration of 4.7 ± 2.2 µg m−3 and 4.0 ± 3.9 µg m−3, respectively. Ammonium (1.8 ± 1.3 µg m−3), sulfate (1.1 ± 0.5 µg m−3), and black carbon (1.5 ± 0.7 µg m−3) were also important components of the PM1. Highly oxidized aerosol was present in the ambient air during the experiment period, with an average O:C equal to 0.86 ± 0.11. NOx ranged from 5 to 35 ppb, with an average of 10 ppb during the measurement period, with an average concentration of NO2 equal to 8 ± 4.9 ppb and of NO 1.9 ± 4.2 ppb. The concentration of ammonia was also high, ranging between 5 and 20 ppb, while ozone was on average 30 ± 15 ppb, exhibiting a clear diurnal cycle peaking at 52 ppb.
The experiments started at different hours of the day (morning, noon, afternoon, and night) (Table 1) to investigate the SOA potential at different states of the atmospheric composition. As a result, the effect of different initial meteorological conditions was also investigated. Relatively low temperatures, in the range of 2–17°C and high relative humidity (in the range of 30 %–80 %) were measured during the period that the experiments took place. The initial concentration of OA ranged from 0.3 to 6 µg m−3.
The diurnal profiles of both the meteorological parameters and the various PM1 compounds measured by the AMS can be found in Fig. S5. Elevated concentrations of all pollutants and high humidity (about 80 %) are observed during the nighttime and early morning hours, while the opposite is observed at noon.
3.2.2 Results of a typical perturbation experiment with high SOA formation
The initial PM1 inside the chambers during Exp. 1 (started at 22:20 LT on 10 March 2022) was 15 µg m−3. The organic aerosol had a concentration of 4 µg m−3, while nitrate (7 µg m−3) and ammonium (3 µg m−3) were also important fractions of the PM1 mass (Fig. 2). The rest consisted mainly of sulfate (0.5 µg m−3) and other species like chloride. The initial concentration of NO2 inside the chambers was 7 ppb and of NO 9 ppb. Ozone had a concentration of 26 ppb at the beginning of the experiment. The initial temperature in the chambers (and also the ambient air) was 5°C, and throughout the experiment it did not increase more than 2.5°C. The initial RH was 76 %, and by the end of the experiment it had decreased to 60 %.
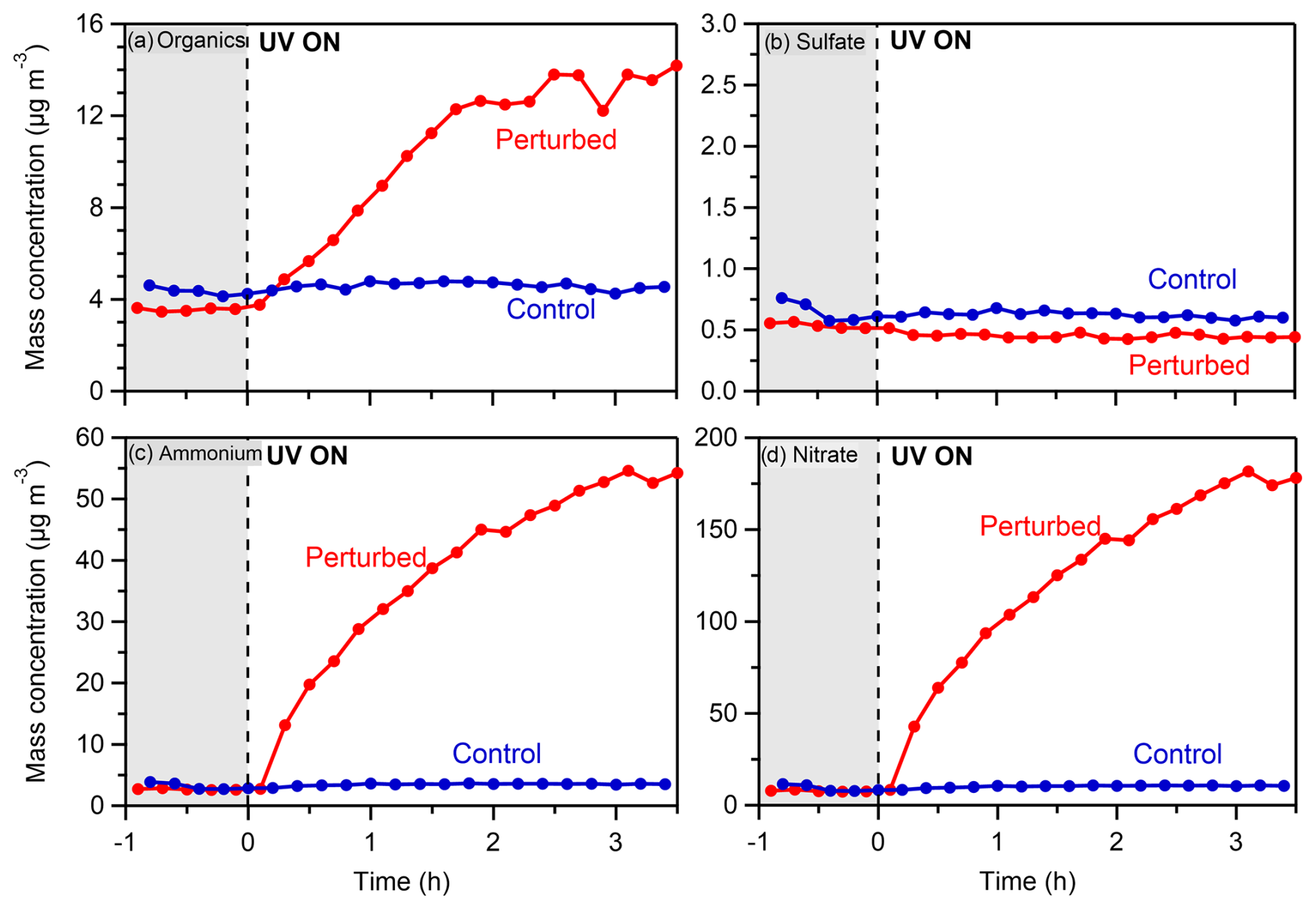
Figure 2Wall-loss- and CE-corrected mass concentrations of (a) organics, (b) sulfate, (c) ammonium, and (d) nitrate in the perturbed and the control chambers during the nighttime experiment (Exp. 1) in the Po Valley.
Following the introduction of the HONO in the perturbed chamber (average OH in this experiment was 3.7 × 106 molec. cm−3) and its illumination with UV (t= 0 h), rapid formation of SOA (Fig. 2a) was observed in the perturbed chamber. In the control, the concentration remained practically the same (within 10 % of its initial value). The concentration of OA almost quadrupled, reaching 14 µg m−3 by the end (after 3.5 h) of the experiment. The formation rate of the organic aerosol in this experiment was 5.6 µg m−3 h−1, for the first 2 h before its stabilization. No significant change in the concentration of sulfate (Fig. 2b) was observed. The SO2 was 0.12 ppb, and the expected sulfate formation based on the available OH was less than 0.05 µg m−3.
A significant increase in both the ammonium (Fig. 2c) and nitrate (Fig. 2d) concentrations was observed, indicating the production of ammonium nitrate inside the perturbed chamber. The concentration of nitrate reached 175 µg m−3 and of ammonium 54 µg m−3 by the end of the experiment. Although the NOx concentrations inside the perturbed chamber were high due to the HONO injection (NO of the order of 0.3–1 ppm), the formation of ammonium nitrate highlights the high concentration of the ambient gas-phase ammonia in San Pietro Capofiume.
Regarding the gas phase, the NO concentration in the perturbed chamber immediately increased (Fig. S6a) as expected after the introduction of the OH radicals, stabilizing at 800 ppb. The ozone concentration decreased immediately (Fig. S6b) after the HONO addition and UV illumination, from 26 to 8 ppb in the perturbed chamber, while in the control it increased by a few ppb by the end of the experiment.
3.2.3 Organic aerosol mass spectra
The initial mass spectra of both the control and the perturbed chambers, obtained from the AMS, were compared to ensure that there were no major differences in the OA. The θ angle between the two was 1°, suggesting identical OA. Notable differences (Fig. 3a) were observed between the mass spectra of the organic aerosol in the perturbed chamber before and after the SOA formation. Examining the absolute values in the mass spectra, a significant increase was observed in 18, 28 (CO+) and 44 (CO), while a notable increase was also observed at 27 (C2H), 29 (CHO+), 41 (C3H), 47 (CH3O), 48 (C), 55 (C4H and C3H3O+), and 63 (C5H). For 47 and 48, interference by NO2 containing molecules is possible. No notable decreases in the absolute values were observed. The θ angle between the two spectra was 16°, suggesting significant differences in the perturbed chamber. On the other hand, the θ angle between the two spectra in the control chamber (Fig. 3b) was 2.5°, suggesting practically identical spectra.
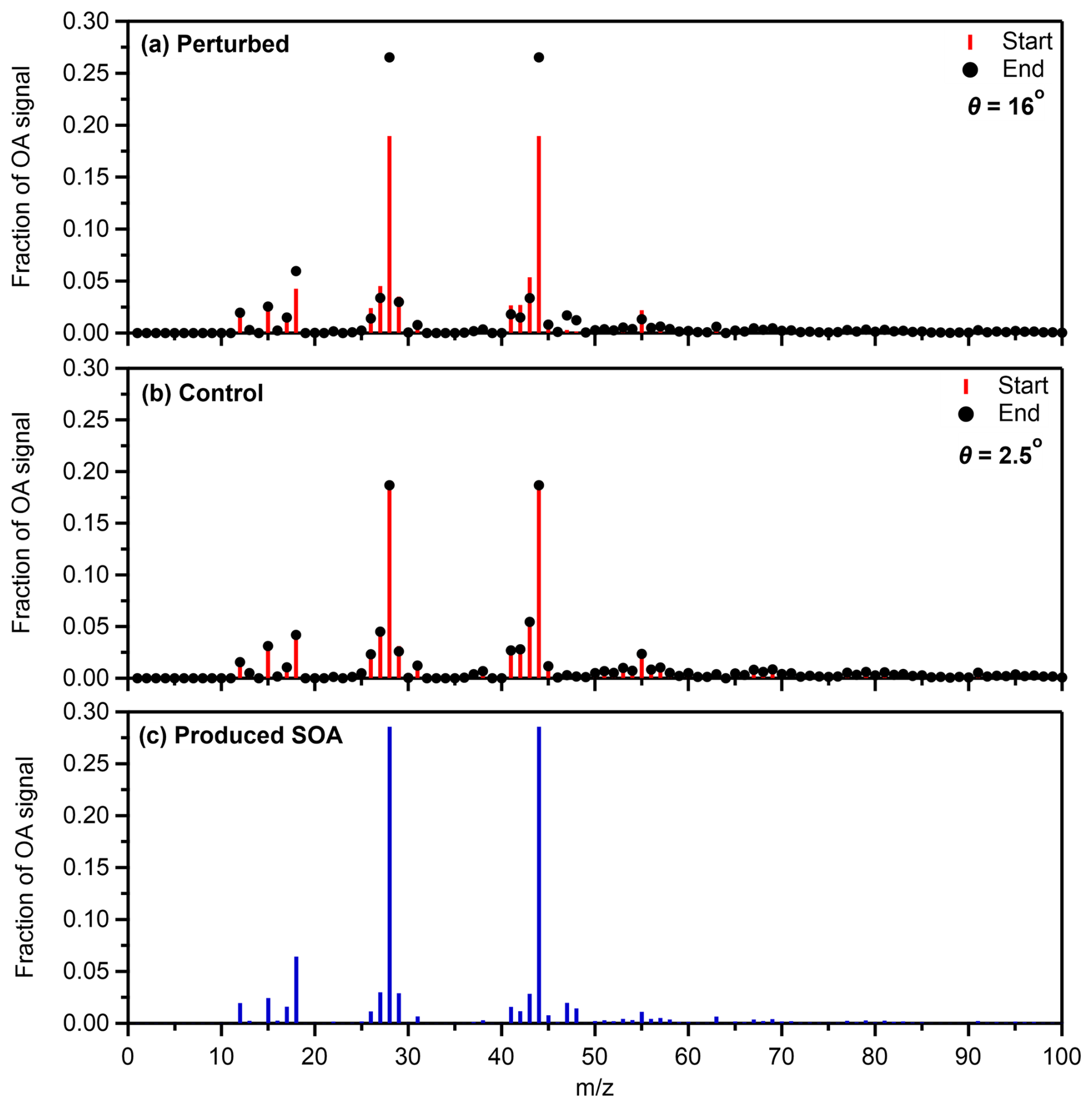
Figure 3Average fractional signal of the organic aerosol at the start and the end of Exp. 1 in the Po Valley in (a) the perturbed and (b) the control chambers. The mass spectrum of the SOA formed in the perturbed chamber during the same experiment (c) is also depicted.
The mass spectra of the produced SOA in each experiment were estimated using the method introduced by Jorga et al. (2020). The method uses the mass balance of OA before and after its oxidation, assuming that the preexisting OA remains unchanged (negligible heterogeneous reactions). The approach uses the known wall loss rate constant to calculate the mass spectra of the formed SOA. More details and an example of this calculation can be found in Jorga et al. (2020). The major values of the produced SOA mass spectrum during Exp. 1 were 18, 28, and 44 (Fig. 3c). The θ angle between the produced SOA and the initial OA in the perturbation chamber was 18.9°, suggesting significant changes in the spectrum.
The O:C ratio was initially (after the stabilization period) 0.7 in both chambers (Fig. 4), indicating an already highly oxidized aerosol. In the control chamber, the ratio remained unchanged for the first 2 h. A small decrease was observed after the second hour, and at the end of the experiment O:C was equal to 0.63. The overall stable behavior of the O:C suggests that the chemical evolution of the aerosol in the control chamber was limited, which is also confirmed by the mass spectra (Fig. 3b). On the other hand, in the perturbed chamber the O:C started increasing immediately after the production of the OH radicals and continued to do so for 1.5 h, when it stabilized at 1.05. The fact that the O:C increased by 0.35 (50 % increase) highlights the potential of the Po Valley atmosphere to produce even more-oxidized OA. The O:C of the produced SOA was equal to 1.1. This highlights the potential of this polluted atmosphere dominated by anthropogenic sources to form rapidly highly oxidized SOA.
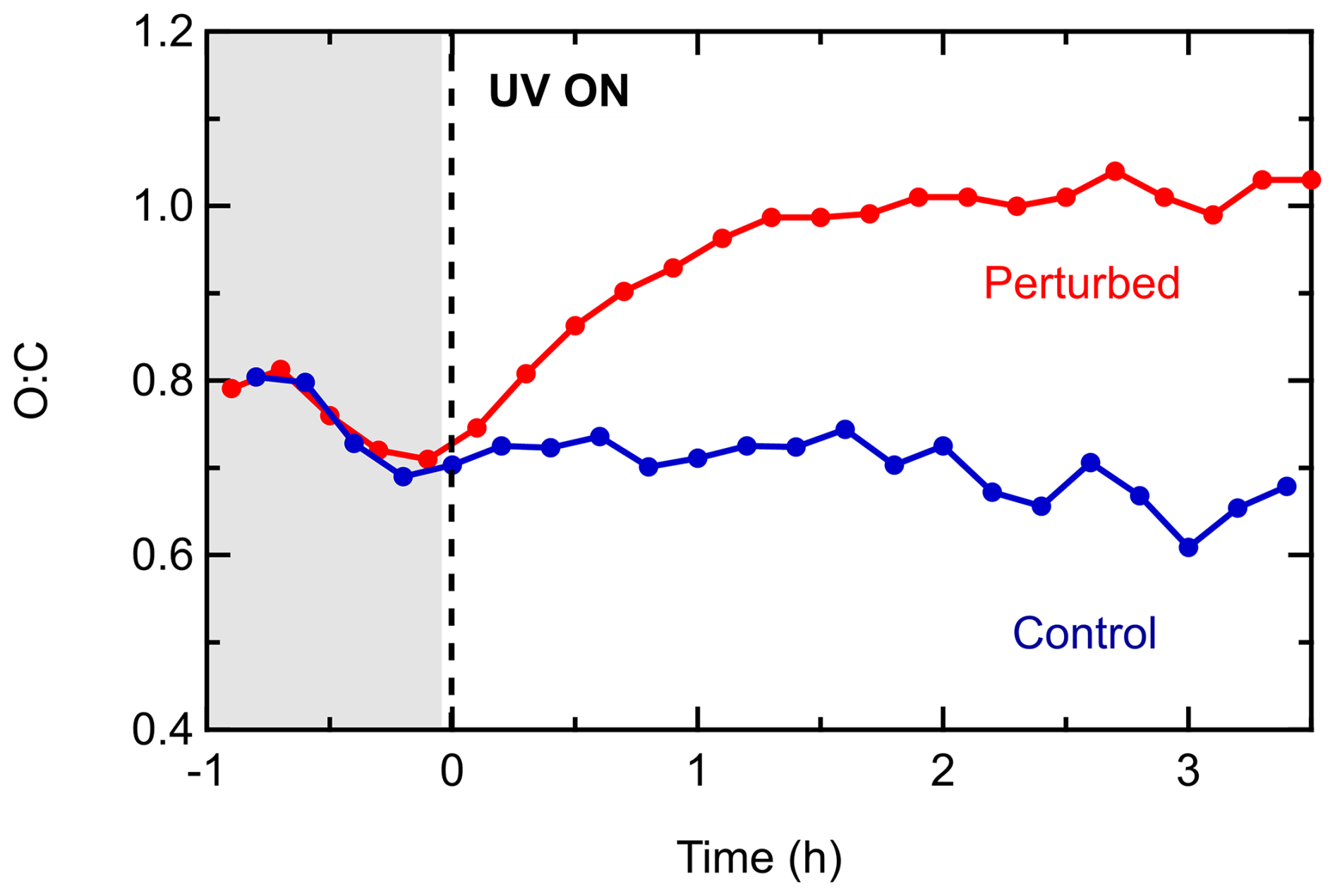
Figure 4Evolution of the O:C inside the perturbed and the control chambers during Exp. 1 in the Po Valley.
3.2.4 Results of other experiments in the Po Valley
Formation of secondary organic aerosol was observed in the perturbed chamber in all experiments but one, while ammonium nitrate was always produced. Overall, the produced SOA ranged from 0.15 to 10 µg m−3, while the formation rate ranged between 0.12 and 5.6 µg m−3 h−1 (Table 1), with an average of 1.9 ± 1.8 µg m−3 h−1. The average concentration of the OH radicals for all the experiments inside the perturbed chamber (after HONO injection and illumination with UV) was 4.3 ± 2.3 × 106 molec. cm−3 (Table 1). A clear difference in the SOA formation potential was observed between daytime and nighttime experiments (Fig. 5a). Higher potential of SOA formation was observed during the more polluted periods of the day (Fig. S5), indicating that along with the elevated PM1 levels, elevated levels of VOCs were also present during the nighttime and early morning hours, when the formation potential maximized.
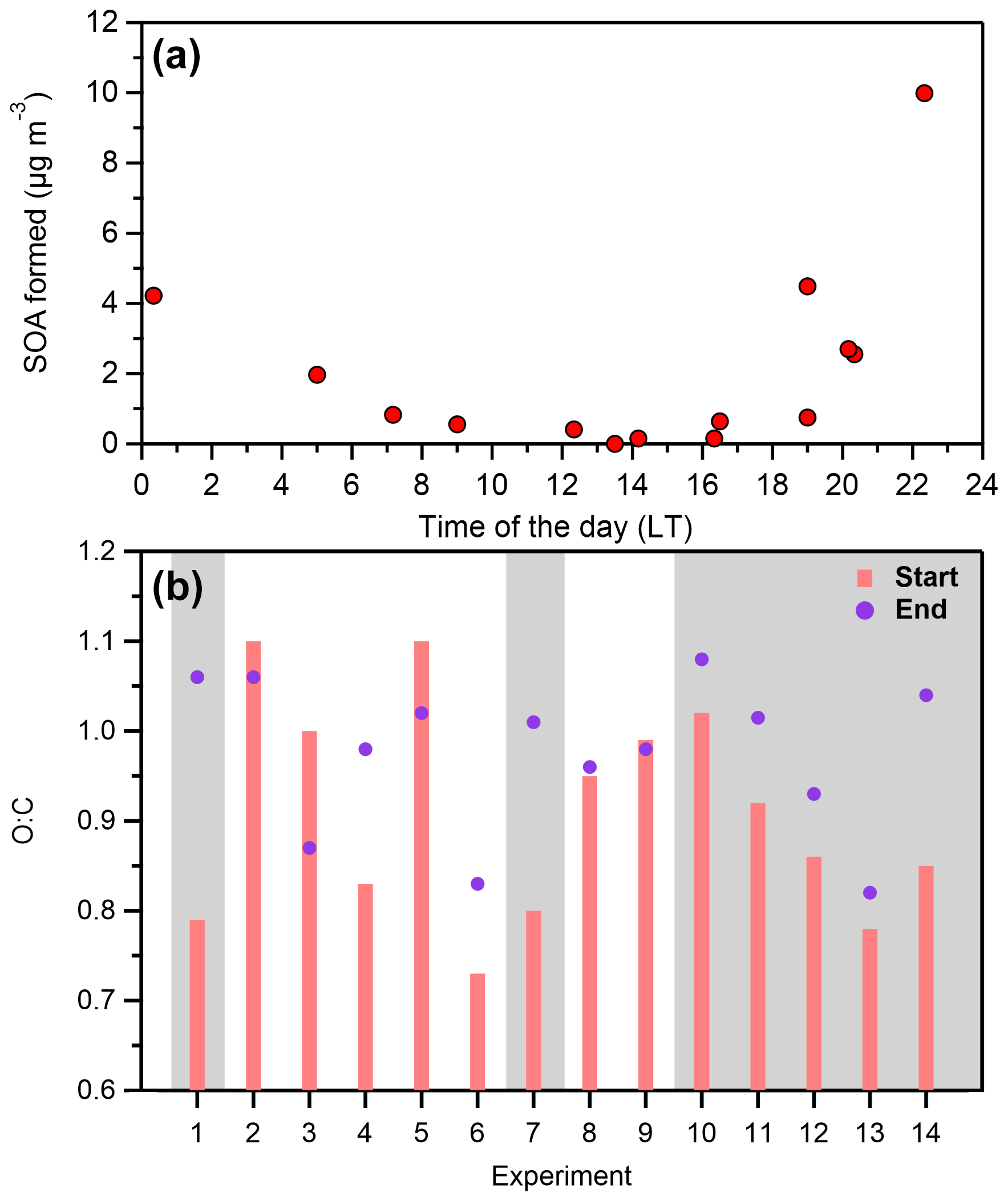
Figure 5(a) The SOA formed in each experiment in the Po Valley together with its corresponding starting time and (b) the average O:C ratio at the beginning and the end of each experiment. The grey areas denote the nighttime experiments.
Daytime experiments
Experiments that took place during the day (07:00–19:00 LT) had a range of produced SOA between 0.15 and 1 µg m−3, with an average of 0.5 µg m−3. The formation of ammonium nitrate ranged from 2.3 to 26.5 µg m−3. During all the daytime experiments a significant drop in the concentration of the formed ammonium nitrate was observed, which was higher than the corresponding decay due to the wall losses. Given that this behavior was only observed in the daytime experiments, one explanation could be that they evaporated due to the higher temperatures during these periods. In these cases, the temperature ranged from 10 to 18°C, but for a given experiment the temperature change due to the UV illumination was never higher than 3–4°C.
The experiment with no SOA formation (Exp. 2) also took place during daytime (at 13:30 LT on 7 March 2022). During this day, the ambient PM levels were low (PM1= 8 µg m−3), but even in this case, 2.3 µg m−3 of ammonium nitrate was formed (Fig. S7) inside the perturbed chamber. This day was characterized by high photochemistry and low OA, which was highly aged with an O:C of around 1.1. Given these conditions, it is expected that there were limited VOCs, IVOCs, or SVOCs available, so there was little potential for additional SOA formation. The low concentrations of pollutants in the gas phase are also consistent with the relatively low ammonium nitrate formation in this experiment. The fact that there was at least one case when no detectable SOA was formed acted as another test of our system, because it confirmed that there was no systematic artifact in the system, leading to SOA production (e.g., from the chamber walls).
The average mass spectra of the organic aerosol at the beginning and the end of each experiment were compared to quantify the chemical evolution of the aerosol in both chambers. In the control chamber the spectra remained practically constant (θ < 5°) during all experiments, except in the very low concentration experiment (Exp. 3) where the θ angle was 16° (both in the control and the perturbed chamber). At very low concentrations, interactions with the chamber walls can become significant and can affect the chemical profile of the aerosol. Comparing the mass spectra of the OA in the perturbed chamber before and after the HONO injection, a relatively small θ angle in the range of 2–9° was calculated, suggesting that no significant changes took place during the daytime experiments.
The θ angle of the produced SOA mass spectra in the various daytime experiments in the Po Valley ranged between 5.2° and 15.5°, with an average of 9.2 ± 3.9°. This indicates that the formed SOA in these cases was quite similar, with significant signals at 28, 29, 41, 43, 44, and 55 (Fig. S8a). Comparing the mass spectra of the produced SOA with the initial OA of the corresponding experiment, the θ angle ranged between 4° and 20° with an average of 11 ± 6.7°. Therefore, this later-generation SOA had some differences from the pre-existing OA.
The initial O:C during the daytime experiments ranged from 0.8 to 1.1 with an average of 1. Out of the five daytime experiments (excluding the two conducted during sunset and sunrise), the O:C only increased in one case (Exp. 4), where it was initially 0.83 and reached 0.98 in the perturbed chamber at the end of the experiment (while in the control it remained stable). In all other cases the O:C remained within 10 % of the initial value in both chambers (Fig. 5b). A decrease in the O:C was observed during the low concentration experiment (Exp. 3), which is consistent with the significant differences observed in the mass spectra of the organic aerosol. One explanation for this decrease (also observed in a few other experiments) could be that some highly oxygenated organic molecules break down into smaller molecules as they react with OH that then move to the gas phase, lowering the O:C of the remaining OA. Another explanation could be the selective loss of highly oxidized gas-phase organics to the walls due to their higher polarity, leading to evaporation of the corresponding molecules from the OA to maintain equilibrium. The O:C of the produced SOA during the daytime experiments was on average 1.05 ± 0.13, ranging from 0.83 to 1.2.
The formation of new particles was only observed in the perturbed chamber during Exp. 3. The nucleation event (Fig. S9) started immediately after the production of the OH radicals (together with a small SOA formation of the order of 0.1 µg m−3) and continued until the end of the experiment. What differentiated Exp. 3 from the others was the low ambient PM levels during that day. PM1 was equal to 3 µg m−3 (campaign minimum), resulting in a low condensation sink, which favors (when the gaseous precursors are present) the formation of new particles (Kulmala et al., 2004).
Nighttime experiments
Experiments that took place during the night (19:00–07:00 LT) had a range of produced SOA between 0.75 and 10 µg m−3, with an average of 5.8 µg m−3 (Fig. 5a). The concentration of the formed ammonium nitrate ranged from 50 to 225 µg m−3. The comparison of the organic aerosol mass spectra revealed that again in the control chamber there were minor changes (θ< 5°). In the perturbed chamber, the θ angle ranged from 4 to 16° with an average of 10°, indicating that there were more changes compared to the daytime experiments. In all nighttime experiments, a significant increase was observed mainly in the m/z values 28 and 44 as well as 47, 48, 60, and 63 and their spectra were quite similar to those in Exp. 1, described earlier.
The θ angle of the produced SOA mass spectra among the eight nighttime experiments in the Po Valley ranged between 2.4 and 19° with an average of 10.1 ± 4.9° . This indicates that the formed SOA was quite similar in the nighttime experiments as well. The comparison of the formed SOA spectra between daytime and nighttime experiments showed that they were also quite similar. The θ angle ranged from 1 to 21° and had an average of 11.6 ± 5.1°. The main fragments of the formed SOA during the nighttime experiments were 28 and 44 (Fig. S8b). Comparing the mass spectra of the produced SOA with the initial OA of the corresponding experiment, the θ angle ranged between 5.3 and 20°, with an average of 11.8 ± 5.1°.
The O:C ratio increased in all nighttime experiments (Fig. 5b) by 15 ± 9 % on average, reaching values as high as 1.05. This suggests that the oxidation cannot proceed much further than this limit in SPC, which is consistent with the ambient observations, in which the O:C never exceeded 1.1. It is important to note that the initial O:C during the night was lower (but still high) than in the daytime, with an average of 0.84 (while during the day it was close to 1). Experiments 6 and 7 were conducted close to sunrise (07:00 LT) and sunset (19:00 LT). These experiments were chemically closer to the nighttime ones, with relatively significant production of SOA and an increase in the O:C ratio. The O:C ratio of the SOA produced during the nighttime experiments was on average 1 ± 0.11, ranging from 0.77 to 1.1.
The results obtained by the nighttime experiments can be representative of the “polluted, humid conditions” that dominate the Po Valley during the fall and winter periods, even during daytime hours (Decesari et al., 2014; Paglione et al., 2020). Also, the insights from the more polluted (nighttime and early-morning periods) can be used to elucidate what the contribution of the secondary material in the total PM levels would be, following the sunrise of the next day (and the natural formation of the OH radicals).
The ammonium nitrate formed due to the HONO addition may have influenced the produced SOA in our experiments. The increase in the levels of these salts can increase the water content of the particulate phase and drive the partitioning of some water-soluble intermediate-volatility and semi-volatile organic compounds towards the particle phase. However, in most experiments (all the daytime and most of the nighttime) the concentration of the formed ammonium nitrate was of the same magnitude as the high ambient levels observed during the campaign (about 40 µg m−3), suggesting that this partitioning effect already occurs to some extent in the Po Valley. In Exp. 1, there was the highest production of ammonium nitrate (220 µg m−3). To address the magnitude of this effect, we estimated the aerosol water content using ISORROPIA-lite (Kakavas et al., 2022) after the formation of ammonium nitrate for Exp. 1 (that had the maximum ammonium nitrate levels) and found it to be 1.4 × 10−4 g m−3. At these levels of water, only compounds with a Henry's constant exceeding 107 M atm−1 would partition significantly (more than 10 % of their mass) to the particulate phase (Seinfeld and Pandis, 2016). While this partitioning could make a small contribution to the SOA formation, it is highly unlikely that it is responsible for a large fraction of the 10 µg m−3 of SOA formed in the extreme case of Exp. 1.
3.2.5 Volatile organic compounds in the Po Valley
The precursor VOCs responsible for this high potential for SOA formation in the Po Valley could not be quantified during this campaign. Anthropogenic VOCs such as toluene and other aromatic compounds and cyclic alkanes are present at higher levels in the region during the cold period (Steinbacher et al., 2005a; Decesari et al., 2014). Semi-volatile organic compounds such as polycyclic aromatic hydrocarbons (PAHs) are significant components of fine PM mass during winter (Carbone et al., 2010). Although there are no available measurements of IVOCs in the Po Valley, they are suspected to contribute to a large fraction of the SOA. Giani et al. (2019) included SVOCs and IVOCs in their model and were able to reproduce the observed SOA with a much better accuracy, indicating that these compounds are present in the atmosphere of the Po Valley. Lower levels of BVOCs have been observed in this period, with isoprene concentrations peaking during morning hours at around 2 ppb (Steinbacher et al., 2005b).
3.2.6 Dependence of the formed SOA on meteorological parameters and PM1 levels
The dependence of the formed organic and inorganic mass during each experiment on the meteorological parameters was explored to understand their role in the corresponding processes. However, because the experiments took place at different hours, and given that the examined variables vary systematically during the day (for example temperature is lower at night), it is difficult to arrive at robust conclusions about the net impact of each variable.
Higher SOA formation was observed (R2= 0.31) for the experiments that had higher relative humidity (Fig. 6a). This is also the case, even if we separate the daytime and nighttime experiments. An R2= 0.55 between RH and SOA formation was found for only the daytime experiments. Conflicting results have been found in the literature regarding the effect of RH in SOA formation. A positive correlation has been found by various studies (Healy et al., 2009; White et al., 2014; Luo et al., 2019) as well as a negative correlation (Hinks et al., 2018; Jia and Xu, 2018; Zhang et al., 2019; Lamkaddam et al., 2020).
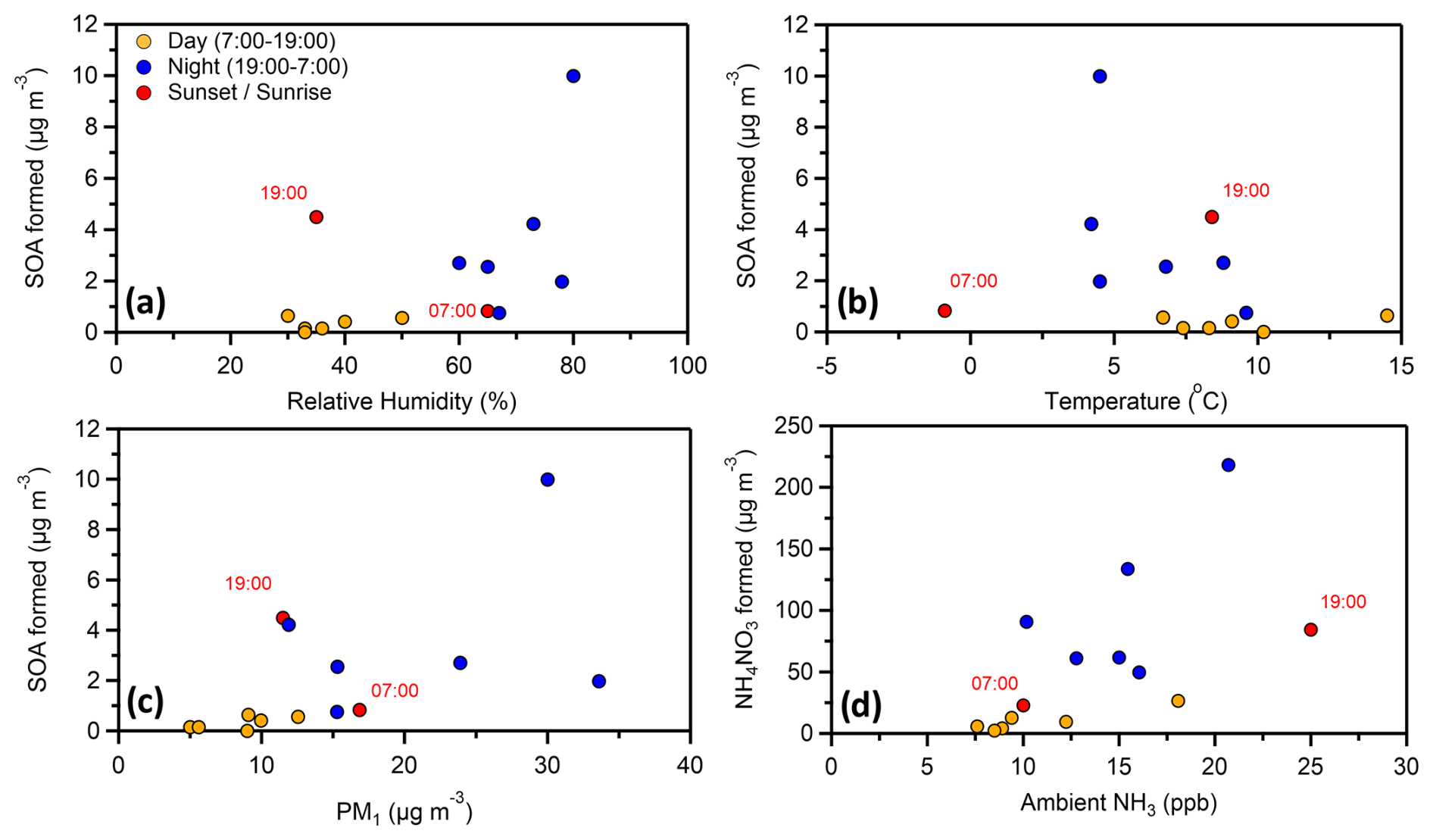
Figure 6The mass concentration of the formed SOA (raw data) in each experiment together with the ambient (a) relative humidity, (b) temperature and (c) PM1 in the beginning of the corresponding experiment. The formed mass of ammonium nitrate in each experiment is also plotted (d) against the ambient gas-phase ammonia in the beginning of the corresponding experiments.
Temperature did not appear to affect the potential of SOA formation (Fig. 6b), at least directly (R2= 0.09), implying that other parameters controlled it. The ambient PM1 was overall connected (R2= 0.33) with higher SOA formation (Fig. 6c). A more polluted atmosphere implies higher concentrations of SOA precursors and a higher mass for partitioning of the produced semi volatile compounds.
Unfortunately, measurements of VOCs and IVOCs were not available due to instrumentation problems. Overall, there are several factors that could be responsible for the higher SOA production during nighttime production. Higher availability of precursors due to the low vertical mixing could be one of the explanations. The higher RH, the lower temperature and the nighttime chemistry are other potential explanations. Quantifying the role of these effects is challenging and requires additional measurements in the future.
3.2.7 Dependence of the formed ammonium nitrate on ambient ammonia levels
A reasonable correlation (R2= 0.39) between the produced ammonium nitrate and the ambient ammonia right before the start of each experiment was found (Fig. 6d). An increasing concentration of gas-phase NH3 in the ambient air resulted in higher NH4NO3 formation, confirming that its high concentrations in the chamber were partially due to the high ammonia content in SPC. The ratio between the produced nitrate and ammonium was always around 3.3, which is close to the molar mass ratio of the corresponding compounds (3.43).
3.2.8 Inorganic and organic secondary nitrate formation
Since the experiments in the Po Valley were conducted under high-NOx and ammonia conditions, the formation of organonitrates was investigated in more detail. The organic nitrate fraction was estimated using the method suggested by Kiendler-Scharr et al. (2016), which relies on the NO ratio from the AMS data. Before oxidation, the organic nitrates comprised 20 %–30 % of the total nitrates, yielding an organic nitrate mass concentration in the range of 0.1 to 1.6 µg m−3. This fraction appears to be close to the European average (34 %) in the ambient air, estimated by Kiendler-Scharr et al. (2016) for the same period of the year. After oxidation in the perturbed chamber, the fraction dropped to 1 %–10 %, with the lowest fractions observed during the nighttime experiments. This resulted in organic nitrate concentrations in the range of 0.3 to 4 µg m−3. These estimations suggest that while organic nitrates were produced during our experiments, their contribution to the total nitrate mass was quite small. To confirm this, we performed an ion balance of the ammonium–sulfate–nitrate system. This calculation suggested that after oxidation, practically all nitrate was inorganic. So, even if both organic nitrate estimation methods are quite uncertain, they confirm that more than 90 % of the nitrate formed was ammonium nitrate.
Increasing SOA yields in the presence of high levels of ammonia have been reported in the literature due to its interactions with the formed organics and water (Na et al., 2007; Liu et al., 2015; Horne et al., 2018; Qi et al., 2020). To gain insights into its contribution to SOA, we identified all the nitrogen-containing organic (NOC) fragments from the AMS mass spectra and calculated their mass fraction to total organics following Liu et al. (2015). The NOC mass fraction averaged was 4 % of the total signal, with little variation between initial and final conditions or across different experiments in general. The mass of the NOC ranged from 0.07 to 0.5 µg m−3 in the examined experiments. These fragments are mainly associated with organonitrates, but they can also originate from compounds produced by the interactions of organic molecules with ammonia.
3.3 SPRUCE-22 campaign (Pertouli)
3.3.1 Conditions during the SPRUCE-22 campaign
During the SPRUCE-22 campaign, the levels of ambient PM1 were comparable to those observed during the FAIRARI campaign, with an average of 12.7 ± 5.1 µg m−3, ranging between 0.7 and 27 µg m−3 (Matrali et al., 2024). Organics and sulfate were the dominant PM1 components, having average concentrations equal to 7.4 ± 3.2 and 3.8 ± 1.7 µg m−3, respectively. Ammonium (1.2 ± 1.4 µg m−3) and nitrate (0.1 ± 0.05 µg m−3) had lower concentrations, while black carbon also represented a small fraction of the PM1 (0.26 ± 0.12 µg m−3). Highly oxidized aerosol was present during the campaign, with an average O:C equal to 0.84.
Isoprene had an average concentration of 0.7 ± 1.1 ppb, while monoterpenes were 0.5 ± 0.8 ppb (Matrali et al., 2024). The diurnal profile of isoprene peaked at around 15:00 LT, reaching on average 2 ppb, while the corresponding concentration of monoterpenes was 1 ppb. The concentrations of fresh anthropogenic pollutants were low. NO2 concentration did not exceed 3.5 ppb and had an average of 1.8 ± 0.62 ppb, while the anthropogenic VOCs also had small concentrations throughout the entire campaign. For instance, toluene had an average concentration of 0.002 ± 0.05 ppb and benzene 0.004 ± 0.03 ppb. However, a peak of the order of 0.05–0.1 ppb was observed at noon, examining the diurnal profile of the anthropogenic VOCs (Matrali et al., 2024). Ozone had a relatively stable concentration with an average of 64.8 ± 8.3 ppb.
The diurnal profiles of the meteorological parameters and the concentrations of the various species in the gas or particulate phase can be found in Matrali et al. (2024).
3.3.2 Results of a perturbation experiment with SOA formation
The initial PM1 inside the chambers during Exp. 1 (took place at 17:00 LT on 10 July 2022) was 6.6 µg m−3. The organic aerosol had a concentration of 3.1 µg m−3, while sulfate was also an important fraction of the PM1 with a concentration of 2.2 µg m−3. The rest consisted mainly of nitrate (0.3 µg m−3), ammonium (0.8 µg m−3), and other species like chloride. The initial concentration of NO2 inside the chambers was 1.4 ppb and of NO 2.4 ppb. Ozone had a concentration of 39 ppb at the beginning of the experiment. The initial temperature in the chambers (and the ambient air) was 19°C, and throughout the experiment it did not increase by more than 3°C. The initial RH was 66 %, and by the end of the experiment it had decreased to 55 %.
Exp. 1 was the only experiment during the SPRUCE-22 campaign where clear formation of SOA was evident. Rapid formation of SOA was observed in the perturbed chamber (Fig. 7a), right after the introduction of the OH radicals, while the concentration of the organics in the control chamber remained constant. The concentration of the formed SOA was 1 µg m−3, which corresponds to a 27 % increase in the OA concentration, while the SOA formation rate was equal to 1.1 µg m−3 h−1. A significant production of nitrates (about 20 µg m−3) and ammonium (about 6 µg m−3) also took place in the perturbed chamber. The organic nitrates in this experiment were estimated to account for 1 % of the total nitrate mass. The sulfate concentration (Fig. 7b) fluctuated between 2–2.5 µg m−3 in the perturbed chamber both before and after the initiation of the photochemical reactions in this experiment, and overall, it remained practically constant.
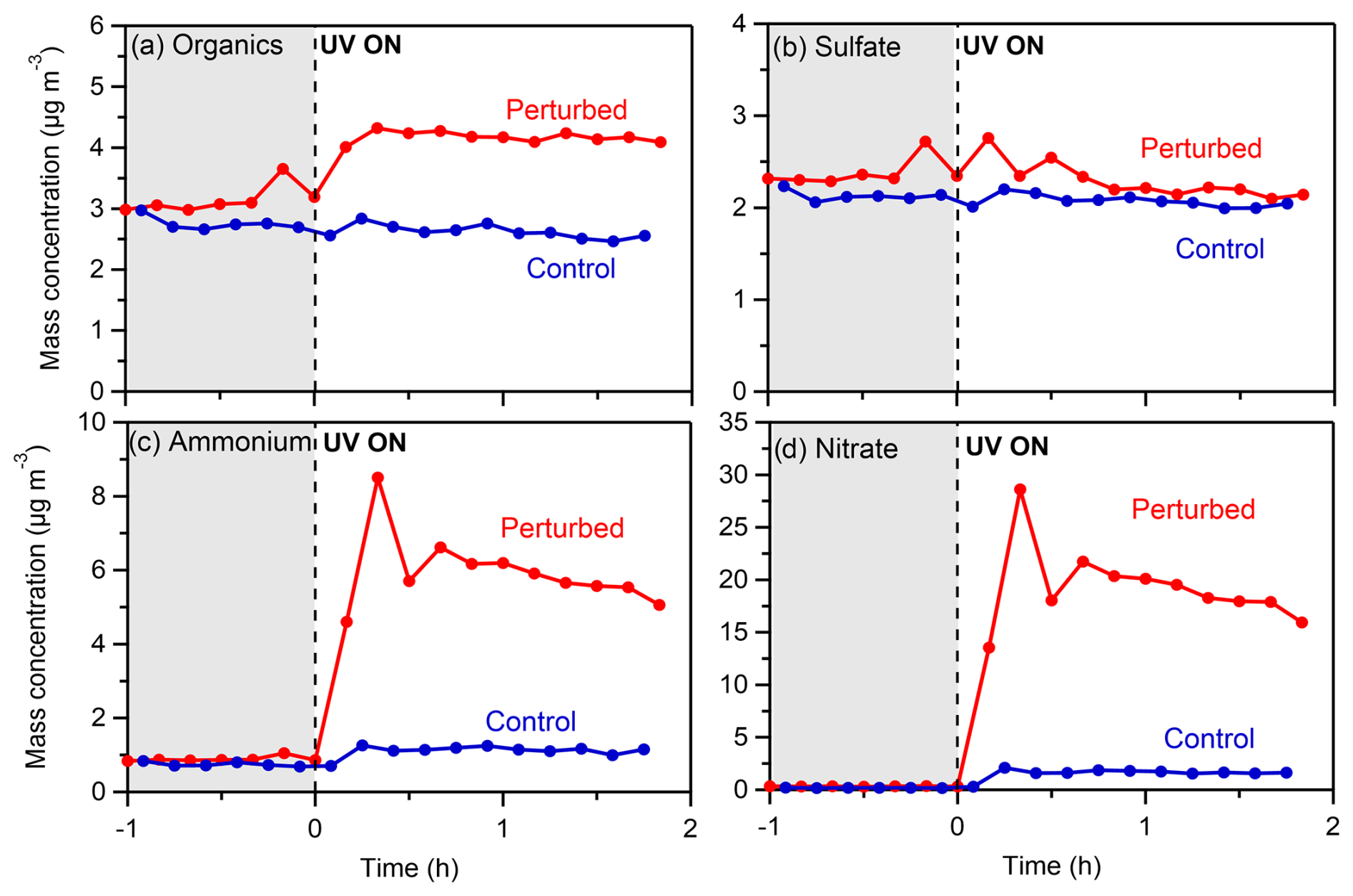
Figure 7Wall-loss- and CE-corrected mass concentrations of (a) organics, (b) sulfate, (c) ammonium, and (d) nitrate in the perturbed and the control chambers during Exp. 1 in Pertouli.
The concentration of NO reached 300 ppb (Fig. S10a) in the perturbed chamber, following the introduction of HONO. The concentration of OH radicals in the perturbed chamber was estimated to be equal to 2.2 × 106 molec. cm−3. A decrease in the concentration of O3 (Fig. S10b) from 39 to 12 ppb was observed in the perturbation chamber after the introduction of the OH radicals.
3.3.3 Organic aerosol spectra
Comparing the OA mass spectra before and after the SOA formation in the perturbed chamber, a signal increase was observed in 28 (CO+), 44 (CO), and 63 (CH3SO+ and CH3O). The θ angle between the two spectra was 8.7° (Fig. 8a), indicating similar spectra with some differences. The θ angle between the corresponding spectra in the control chamber (Fig. 8b) was 6.6°, which can be explained by the notable drop of 29.
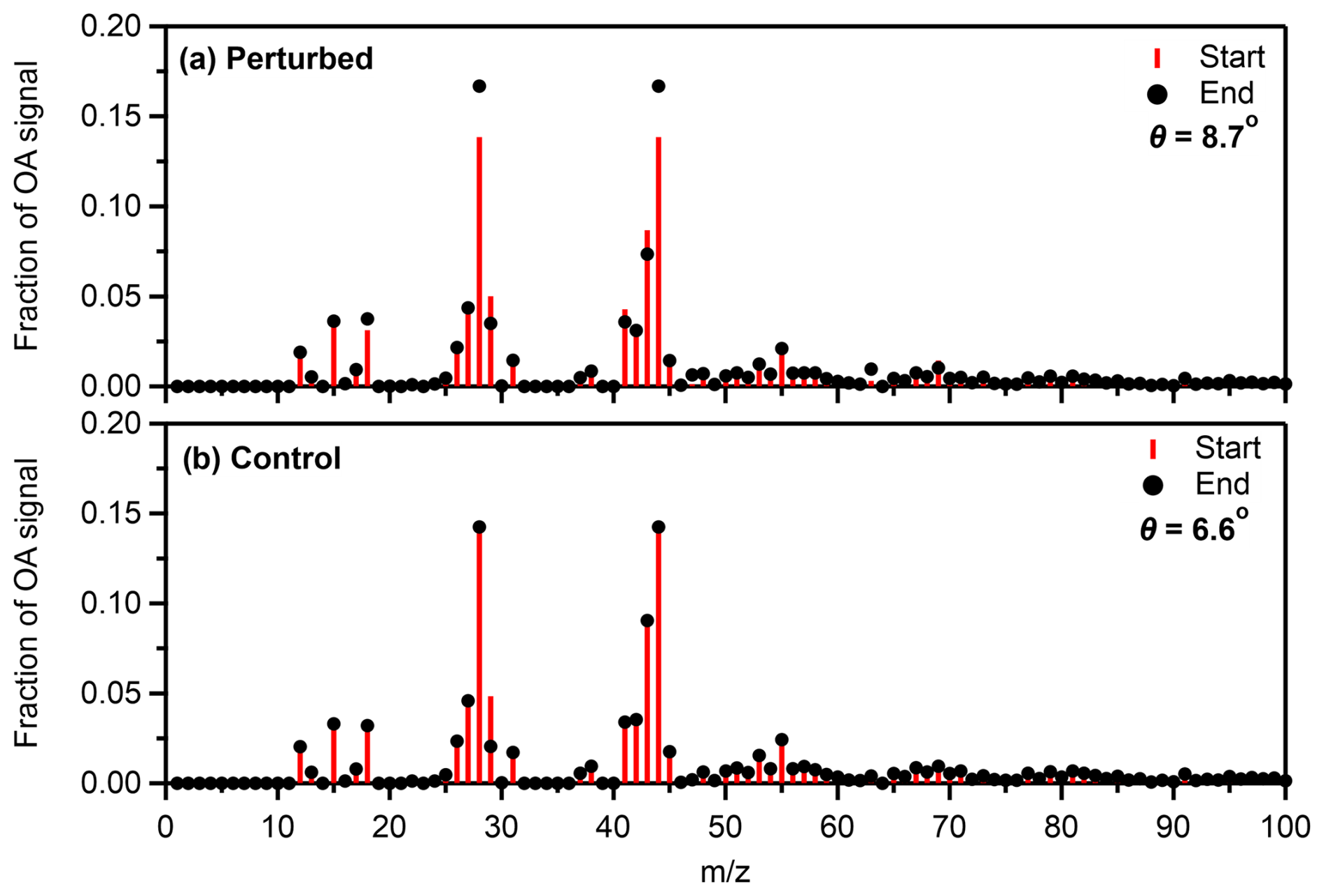
Figure 8Average fractional signal of the organic aerosol at the start and the end of Exp. 1 in Pertouli in (a) the perturbed and (b) the control chambers.
The major values of the produced SOA mass spectrum during Exp. 1 were 28 and 44 (Fig. S11), while a smaller contribution of 18, 27, 43, and 63 was also observed. The θ angle between the produced SOA and the initial OA in the perturbation chamber was 22.4°, suggesting significant changes in the spectrum in this experiment.
The O:C was initially 0.8 in both chambers, implying highly oxidized organic aerosol. A small increase of 0.04 in the O:C was observed in the perturbed chamber after the introduction of the OH radicals (Fig. 9) suggesting that the OA in this experiment had some potential to further oxidize. The O:C in the control chamber had a decreasing pattern, reaching 0.7 by the end of the experiment. This small decrease could be explained by the same points (fragmentation of some highly oxygenated molecules or their preferential loss to the chamber walls) described for the corresponding experiments in the Po Valley. The O:C of the produced SOA was about 0.9.
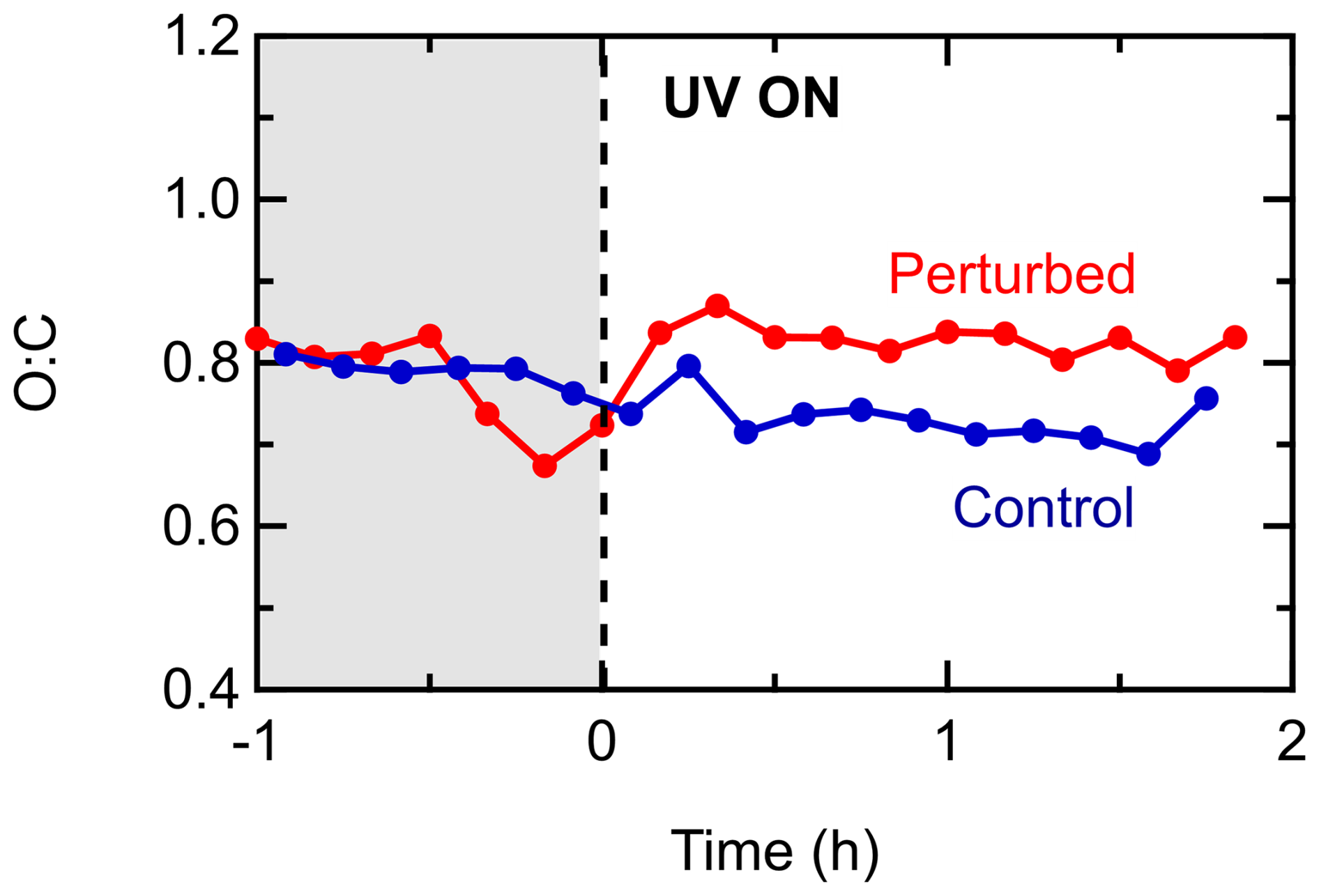
Figure 9Evolution of the O:C inside the perturbed and the control chambers during Exp. 1 in Pertouli.
3.3.4 Results of other experiments
Exp. 1 was the only experiment where clear SOA formation was observed in the perturbed chamber. The rest of the 12 experiments can be divided into two categories. The first includes experiments where formation of SOA was evident, but it could be observed for a limited amount of time (e.g., 15–30 min), and the second includes experiments where no formation of SOA (nor ammonium nitrate) was detected. The concentrations of the OH radicals inside the perturbed chamber (Table 2) were comparable to the ones in the Po Valley campaign, with an average of 3.4 ± 1.8 × 106 molec. cm−3.
An example of the first category (Exp. 2) is depicted in Fig. S12. Although the concentration of the organics increased in the perturbed chamber immediately after the production of OH radicals (reaching 2.5 µg m−3, while the OA initially was 1.9 µg m−3), it started decreasing after 15–30 min and eventually reached its original value. The same behavior was observed for nitrates and ammonium. Given that both the organic aerosol and nitrates are semi-volatile, evaporation due to increasing temperature or vapor losses to the walls is a possible explanation. The hypothesis that this increase in the concentrations was due to the change of the collection efficiency of the AMS was also tested. However, volume concentrations measured by the SMPS at the same time show an increase in concentrations in the perturbed chamber that is consistent with the increase measured by the AMS (Fig. S13). The fact that two independent instruments detected the same mass increase and decrease strongly suggests that this was not an artifact due to the AMS operation. The most probable explanation was that this bump in concentration was due to incomplete mixing of the chamber. The formation reactions took place near the inlet of the reactor; higher concentrations existed in that region (from which the instruments were sampling); and then, as the chamber was getting mixed, the concentrations decreased. Therefore, SOA was formed in these two cases, but its average concentration after the chamber was well mixed, was quite low (less than 0.1 µg m−3), and could not be distinguished from the preexisting organic aerosol inside the chamber.
The rest of the HONO experiments and all the H2O2 experiments did not show any detectable increases in organic or nitrate aerosol concentrations. An example of such an experiment (Exp. 4, conducted on 20 July 2022) is depicted in Fig. 10.
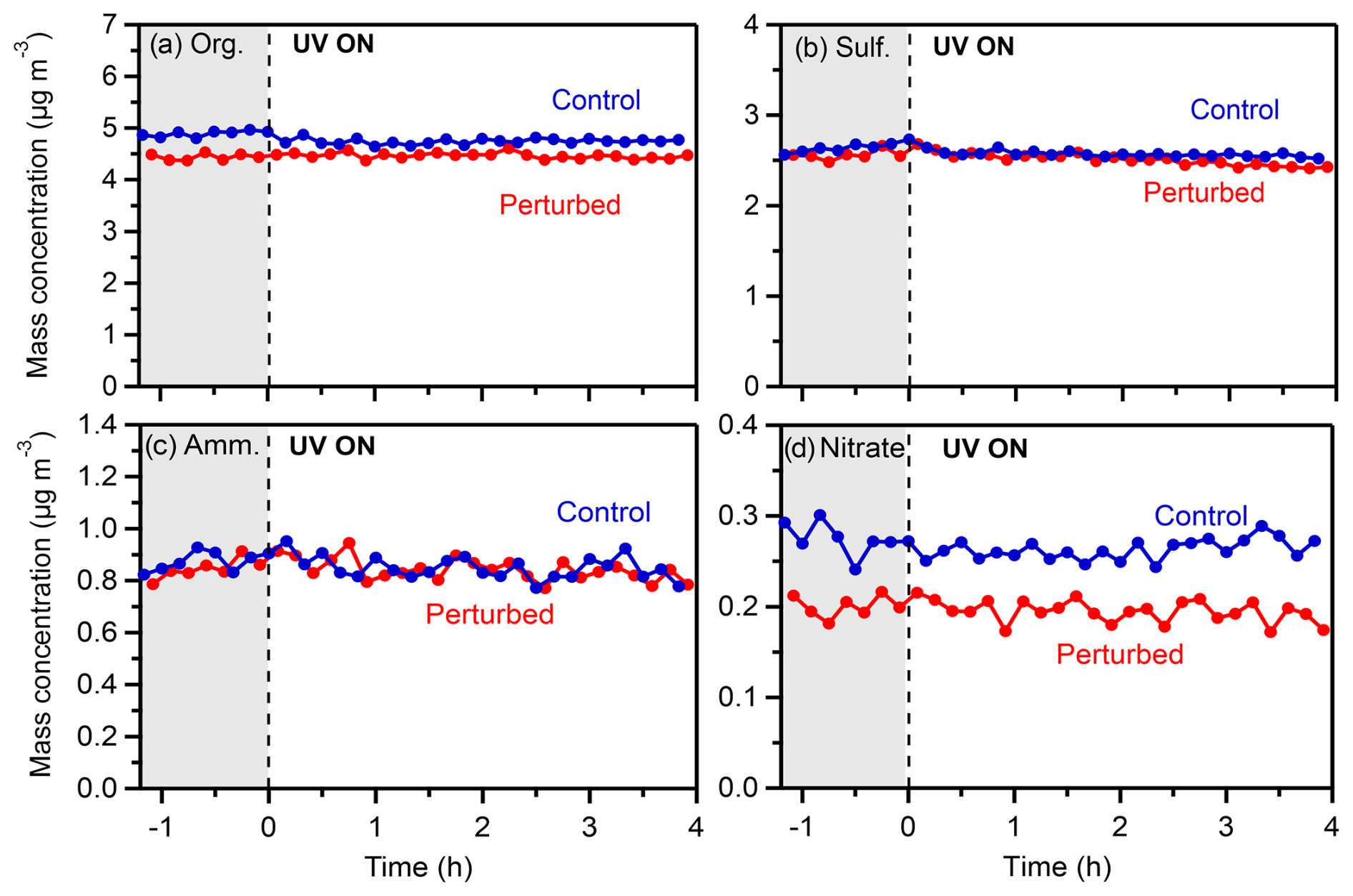
Figure 10Wall-loss-corrected mass concentrations of (a) organics, (b) sulfate, (c) ammonium, and (d) nitrate in the perturbed and the control chambers during Exp. 4 (20 July 2022) in Pertouli.
The initial O:C inside the chambers was on average 0.77 ± 0.08 and did not vary significantly from one experiment to another. The ratio remained practically stable in all experiments, excluding Exp. 1 where it increased by 0.04. During the experiments, when SOA was formed, PMF analysis (Paatero and Tapper, 1994) in the ambient air revealed that the less-oxidized organic aerosol factor (LO-OOA) was dominating, while the more-oxidized factor (MO-OOA) had low levels at the time (Vasilakopoulou et al., 2023). This indicates that the corresponding air masses were less aged. On the other hand, during the rest of the experiments, where no SOA formation was detected, the more-oxidized OA factor was dominant, suggesting that the chemistry had progressed, and additional SOA production or oxidation was slow.
Except for the higher isoprene and monoterpene concentrations mentioned earlier, the three experiments where formation of SOA was evident had the highest initial relative humidities recorded (Table 2), in the range of 60 %–63 %. These experiments were also characterized by lower temperatures (19–20°C) compared to the rest. The relatively lower temperatures during these experiments could play some role enhancing the partitioning of semi-volatile compounds to the particulate phase, leading to an increased potential of SOA formation. A smaller effect of the RH changes is expected in this range (around 60 %).
3.3.5 Volatile organic compounds in Pertouli
The available measurements of the various volatile organic compounds during the SPRUCE-22 campaign revealed that during Exp. 1 (where SOA was formed) the ambient isoprene and monoterpene concentrations were the highest recorded in all SPRUCE-22 experiments. Isoprene had a concentration of 2.4 ppb, while for monoterpenes it was 1.1 ppb. The different behavior of Exp. 1 therefore could be partially explained by the higher availability of monoterpenes and isoprene. At the same time, benzene and toluene had a concentration of 0.08 and 0.09 ppb, respectively, which was higher than the campaign averages at midday (about 0.05 ppb for both compounds). Also, the relatively lower temperature should enhance the partitioning of the products of the monoterpene reactions with OH towards the particulate phase. Notable is also the fact that this experiment took place the day after a 2 d long rainy period in Pertouli (midday of 8 July to morning of 10 July Matrali et al., 2024).
A clear dependence of the SOA formation on the BVOC availability was also observed for the two experiments with insufficient mixing that showed signs of SOA formation. The concentrations of isoprene and monoterpenes in these experiments (1.6 and 0.6 ppb for Exp. 2 and 1.3 and 0.5 ppb for Exp. 3, respectively) were elevated compared to the experiments with no SOA (Fig. S14) but still lower than Exp. 1.
During the rest of the experiments, the ambient concentrations of isoprene and monoterpenes were considerably lower (they had average concentrations of 1.2 and 0.3 ppb, respectively) than in the ones in which some formation of SOA was evident. This highlights the importance of the biogenic vapors in the formation of SOA in Pertouli.
Although anthropogenic compounds had above-average concentrations in two out of three experiments involving SOA formation, there were at least two experiments where SOA did not form, despite having even higher concentrations of benzene and toluene (e.g., 0.15 and 0.07 ppb, respectively, at the start of Experiment 7 on 23 July 2022). This suggests that anthropogenic VOCs likely played a minor role in SOA formation in Pertouli.
A more detailed analysis suggested that in Exp. 1 isoprene ( 69) decreased to almost half of its initial concentration in the first 30 min after oxidation in the perturbed chamber, while it remained practically constant in the control. A smaller decrease in monoterpenes ( 137) was found for these experiments, but this could be due to the formation of monoterpene products giving the same fragment in the PTR-MS. Methyl-vinyl-ketone and methacrolein ( 71) were identified as some of the major oxidation products in most of the performed experiments.
Overall, the negligible SOA formation observed in Pertouli is consistent with the low concentrations of VOCs in this environment. This is, to a large extent, due to the high reactivity of the biogenic precursors that are produced locally. The lack of SOA production also indicates that there was little availability of the products of these first-generation reactions or semi-volatile OA compounds, etc., that could form SOA. Finally, the fact that there was no SOA formation in this environment also in the low-NOx experiments (using H2O2 for the production of OH) suggests that the lack of production cannot be attributed just to the high-NOx environment (altered chemistry) in the corresponding experiments.
One difference between the SPRUCE-22 and the FAIRARI campaign is that no nighttime experiments took place in Pertouli, which could affect the comparability of the formed SOA levels. However, the detailed analysis of biogenic and anthropogenic VOCs during the SPRUCE-22 campaign by Matrali et al. (2024) found that the concentrations of BVOCs in Pertouli peaked on average a little after noon, when most of the experiments took place. The anthropogenic VOCs also peaked at around noon. This suggests that midday hours probably have the highest SOA formation potential in Pertouli, which is not the case in the Po Valley.
3.4 Comparison of the two campaigns
The only experiment with SOA formation in Pertouli (Exp. 1) was compared to the experiments conducted in the Po Valley. Given the high concentration of nitrates introduced in the system by the addition of HONO, we excluded the NO+ and NO signals of the AMS, in an effort to have a more direct comparison of the SOA mass spectra. There are still some fragments of organonitrates, but these had relatively weak signals and affected little the comparison of the spectra. The θ angle of the produced SOA mass spectra between Exp. 1 in Pertouli and the corresponding ones in the Po Valley was on average 11.9° ± 4.4°, ranging from 5.6 to 20°, indicating that the produced SOA had similarities in the two sites. Jimenez et al. (2009) found that the terminal oxidation of various sources often leads to similar AMS spectra that are close to the low-volatility OOA (LV-OOA) factor. A comparison between the mass spectra of the SOA in Exp. 1 (Pertouli) and the average SOA mass spectra of the daytime and nighttime experiments in the Po Valley is shown in Fig. S15.
The mass spectra of the produced SOA in the Po Valley and Pertouli (Exp. 1) were also compared to the LV-OOA (or the OOA-I) factor found in the ambient air in various European campaigns during similar periods (spring–summer). The High-Resolution AMS Spectral Database (Ulbrich et al., 2009) was utilized. The mass spectra of the produced SOA in the Po Valley were similar to the LV-OOA factor found in Rome during the DIAPASON campaign during May and June 2014 (Struckmeier et al., 2016), with an average θ angle of 11.1° ± 3.3° (ranging from 6.3 to 17.6°). The corresponding θ angles for the SV-OOA in this study were on average 46.3° ± 4.9° (ranging from 39.2 to 54.4°). The produced SOA in Exp. 1 in Pertouli was also close to the LV-OOA of this study, with a θ angle of 8.2° (while with the SV-OOA it was 49.9°).
Similarities of the produced SOA in the Po Valley were also observed with the LV-OOA from Barcelona, March 2009 (Mohr et al., 2012), with an average θ angle of 13.8° ± 4.4° (ranging from 8.2 to 21°). The corresponding value for Exp. 1 in Pertouli was 18.6° . The comparison with the SV-OOA factor of this study showed again that it was quite different with respect to the produced SOA mass spectra (the θ angles in this case were always higher than 30°). Finally, similar results were obtained comparing the experiments with the OOA factor during April 2008 found in the Po Valley (Saarikoski et al., 2012) with an average θ angle of 14° ± 2.6° for the Po Valley experiments and 15.1° for Exp. 1 in Pertouli.
Positive matrix factorization (PMF) analysis was conducted using the AMS measurements obtained from the perturbed chamber of each experiment as input, to gain insights into the composition and evolution of the OA. Two factors were identified, one that was best described as LO-OOA, dominating in the beginning of each experiment, and the MO-OOA, dominating after the HONO injection. An example of the identified PMF factors is depicted in Fig. S16 for Exp. 1 in the Po Valley. The PMF analysis also confirmed the production of highly oxidized OA in our experiments.
Laboratory studies constitute the primary source of knowledge regarding the formation of SOA from anthropogenic VOCs (Srivastava et al., 2022). One of the limitations of these studies is that laboratory-produced SOA is far less oxidized than its counterpart in the ambient air (Kroll and Seinfeld, 2008). The fact that in both campaigns, the produced OA was chemically closer to the more-oxidized factors (LV-OOA, MO-OOA, and OOAa, depending on the reference study) observed in the ambient air than in laboratory studies highlights the usefulness of such studies in understanding later-generation chemical processes. A potential limitation of this analysis, however, is the fact that the PMF factors are practically based on a relatively small set of fragments to derive the different organic sources. As the AMS heavily fragments the aerosol, it is expected that there would be some similarity between the measured SOA mass spectra and the ones observed in the ambient air across different sites. Nevertheless, the interesting result from the above comparison is the similarity of the formed SOA spectra in the two studied environments to each other and to the more-oxygenated OOA spectra reported in the literature. The differences with the less-oxidized OOA (LO-OOA) spectra are also noteworthy. This result supports the hypothesis that the MO-OOA is the result of the chemical aging of LO-OOA and that the dual-chamber system could simulate this transition.
The higher potential of SOA formation observed in the Po Valley (compared to Pertouli, where SOA was practically formed in only one experiment) suggests that the anthropogenic VOCs have a higher potential to form later-generation products, enhancing the mass of secondary material in the OA. Part of the explanation for the high SOA formation potential observed in the Po Valley could also be the contribution of less-aged biomass burning OA emissions in this area (Saarikoski et al., 2012; Paglione et al., 2020), estimated to contribute about 25 % of the OA during the FAIRARI campaign, compared to the very aged OA in Pertouli (Vasilakopoulou et al. 2023).
In this work we investigated the potential of ambient air in two different environments to form secondary organic aerosol (SOA) using a dual-chamber system.
In the Po Valley a significant mass of SOA was formed rapidly (within the first hour) following the introduction of the OH radicals in the perturbed chamber. The produced SOA ranged between 0.1 and 10 µg m−3. It was more than 4 times higher than the initial OA in some nighttime experiments. Due to the introduction of nitrous acid for the production of the hydroxyl radicals, these results correspond to high-NOx conditions, which already characterize the Po Valley during this period. Although the initial OA was already highly oxidized (O:C around 0.7–0.8) in all cases, its O:C ratio increased to a maximum of 1.05, suggesting that later-generation products with a high O:C ratio contribute much to the SOA in this environment, at least under the high-NOx conditions studied in this work. A positive correlation between the formed SOA and ambient RH and PM1 levels was observed, suggesting that there is higher potential to form SOA under humid and polluted conditions, while there was not a clear connection with temperature. The high production of ammonium nitrate was connected to the elevated concentrations of ambient ammonia. The presence of high levels of ammonium nitrate leads to an increase of the aerosol water and could thus potentially enhance the observed SOA concentrations; our estimates suggest that this should be a relatively small contribution to the produced SOA.
On the other hand, infrequent and limited formation of SOA was observed during the SPRUCE-22 campaign. In the only experiment with clear SOA production, 1 µg m−3 of organic aerosol (27 % increase) and 25 µg m−3 of ammonium nitrate were formed in the perturbed chamber. This experiment was characterized by the highest ambient concentrations of isoprene and monoterpenes (2.4 and 1.1 ppb, respectively) compared to the other experiments. The anthropogenic VOCs were higher than the campaign average in this experiment but still much lower than the BVOCs (toluene was 0.09 ppb and benzene 0.08 ppb). In two more experiments the formation of SOA was evident, but it disappeared 30 min after its formation, probably due to insufficient mixing of the chambers. The real SOA formed in these cases was probably below the detection limit after the mixing. Our results in the SPRUCE-22 campaign suggest that under the conditions of the study (summertime in southeast Europe) the chemistry of the BVOC–SOA system is quite fast and approaches its final state of the order of a few hours or less. The ambient OA is quite oxidized (initial O:C around 0.75), the VOC concentrations are quite low due to their small lifetime, and the later generations of reactions contribute little to both the SOA mass concentration and its oxidation state. Since there was not any significant SOA production, the hypothesis that the behavior of the BVOC–SOA system in this environment is consistent with the first- or second-generation reactions and that any later-generation production of SOA is of secondary importance appears to be valid.
A θ angle in the range of 5–21° was calculated comparing the mass spectra of the formed SOA in each experiment with the more-oxidized OA factors (LV-OOA or OOAa) identified by various European field campaigns. This suggests that the formed SOA in both campaigns was similar to the more-oxidized OA factors found in the ambient air, which signifies the usefulness of such experiments in bridging laboratory experiments and real-world atmospheric conditions.
Altogether, although starting from a similar oxidation state (O:C = 0.7–0.8), the atmosphere of the two sites has very different potentials of forming SOA. In the Po Valley, the chemistry rapidly proceeds to form large amounts of SOA, while in Pertouli the chemistry appears to have been terminated, even before the beginning of most experiments, so the SOA formation practically does not proceed any further.
The codes used in this work for data analysis are available upon request from the authors (spyros@chemeng.upatras.gr).
The atmospheric simulation chamber measurements are available from the authors (spyros@chemeng.upatras.gr).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-13769-2024-supplement.
AA performed the experiments in the Po Valley, analyzed the data from both campaigns, and wrote the paper; DJS performed the experiments at Pertouli and contributed to the data analysis; CNV contributed to the measurements and analyzed the AMS data; AM, CK, and AS contributed to the measurements; MP, MR, and SD helped with the planning of the Po Valley experiments, provided the AMS, and analyzed the ambient data; and SNP conceived and directed the study, synthesized the data, and edited the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
The authors also acknowledge the help of Ghislain Motos and Athanasios Nenes for the completion of this work; the Regional Agency for Prevention, Environment and Energy of Emilia-Romagna (ARPAE) for providing the ambient data; and Stockholm University for providing the Vocus instrument.
This work has received funding from the European Union's Horizon 2020 Research and Innovation program through the projects FORCeS (grant agreement no. 821205) and ATMO-ACCESS (grant agreement no. 101008004) as well as from the Chemical evolution of gas and particulate-phase organic pollutants in the atmosphere (CHEVOPIN) project of the Hellenic Foundation for Research and Innovation (HFRI; grant agreement no. 1819).
This paper was edited by Annele Virtanen and reviewed by two anonymous referees.
Aiken, A. C., DeCarlo, P. F., and Jimenez, J. L.: Elemental analysis of organic species with electron ionization high-resolution mass spectrometry, Anal. Chem., 79, 8350–8358, https://doi.org/10.1021/ac071150w, 2007.
Aiken, A. C., DeCarlo, P. F., Kroll, J. H., Worsnop, D. R., Huffman, J. A., Docherty, K. S., Ulbrich, I. M., Mohr, C., Kimmel, J. R., Sueper, D., Sun, Y., Zhang, Q., Trimborn, A., Northway, M., Ziemann, P. J., Canagaratna, M. R., Onasch, T. B., Alfarra, M. R., Prevot, A. S. H., Dommen, J., Duplissy, J., Metzger, A., Baltensperger, U., and Jimenez, J. L.: and ratios of primary, secondary, and ambient organic aerosols with high-resolution time-of-flight aerosol mass spectrometry, Environ. Sci. Technol., 42, 4478–4485, https://doi.org/10.1021/es703009q, 2008.
Barmet, P., Dommen, J., DeCarlo, P. F., Tritscher, T., Praplan, A. P., Platt, S. M., Prévôt, A. S. H., Donahue, N. M., and Baltensperger, U.: OH clock determination by proton transfer reaction mass spectrometry at an environmental chamber, Atmos. Meas. Tech., 5, 647–656, https://doi.org/10.5194/amt-5-647-2012, 2012.
Barreira, L. M. F., Ylisirniö, A., Pullinen, I., Buchholz, A., Li, Z., Lipp, H., Junninen, H., Hõrrak, U., Noe, S. M., Krasnova, A., Krasnov, D., Kask, K., Talts, E., Niinemets, Ü., Ruiz-Jimenez, J., and Schobesberger, S.: The importance of sesquiterpene oxidation products for secondary organic aerosol formation in a springtime hemiboreal forest, Atmos. Chem. Phys., 21, 11781–11800, https://doi.org/10.5194/acp-21-11781-2021, 2021.
Bell, D. M., Cirtog, M., Doussin, J.-F., Fuchs, H., Illmann, J., Muñoz, A., Patroescu-Klotz, I., Picquet-Varrault, B., Ródenas, M., and Saathoff, H.: Preparation of Experiments: Addition and In Situ Production of Trace Gases and Oxidants in the Gas Phase, in: A Practical Guide to Atmospheric Simulation Chambers, edited by: Doussin, J.-F., Fuchs, H., Kiendler-Scharr, A., Seakins, P., and Wenger, J., Springer International Publishing, Cham, 129–161, https://doi.org/10.1007/978-3-031-22277-1_4, 2023.
Canagaratna, M. R., Jimenez, J. L., Kroll, J. H., Chen, Q., Kessler, S. H., Massoli, P., Hildebrandt Ruiz, L., Fortner, E., Williams, L. R., Wilson, K. R., Surratt, J. D., Donahue, N. M., Jayne, J. T., and Worsnop, D. R.: Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 15, 253–272, https://doi.org/10.5194/acp-15-253-2015, 2015.
Carbone, C., Decesari, S., Mircea, M., Giulianelli, L., Finessi, E., Rinaldi, M., Fuzzi, S., Marinoni, A., Duchi, R., Perrino, C., Sargolini, T., Vardè, M., Sprovieri, F., Gobbi, G. P., Angelini, F., and Facchini, M. C.: Size-resolved aerosol chemical composition over the Italian Peninsula during typical summer and winter conditions, Atmos. Environ., 44, 5269–5278, https://doi.org/10.1016/j.atmosenv.2010.08.008, 2010.
Chan, A. W. H., Kautzman, K. E., Chhabra, P. S., Surratt, J. D., Chan, M. N., Crounse, J. D., Kürten, A., Wennberg, P. O., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from photooxidation of naphthalene and alkylnaphthalenes: implications for oxidation of intermediate volatility organic compounds (IVOCs), Atmos. Chem. Phys., 9, 3049–3060, https://doi.org/10.5194/acp-9-3049-2009, 2009.
Chaturvedi, S., Kumar, A., Singh, V., Chakraborty, B., Kumar, R., and Min, L.: Recent advancement in organic aerosol understanding: a review of their sources, formation, and health impacts, Water Air Soil Poll., 234, 750, https://doi.org/10.1007/s11270-023-06772-0, 2023.
Daellenbach, K. R., Uzu, G., Jiang, J., Cassagnes, L.-E., Leni, Z., Vlachou, A., Stefenelli, G., Canonaco, F., Weber, S., Segers, A., Kuenen, J. J. P., Schaap, M., Favez, O., Albinet, A., Aksoyoglu, S., Dommen, J., Baltensperger, U., Geiser, M., El Haddad, I., Jaffrezo, J.-L., and Prévôt, A. S. H.: Sources of particulate-matter air pollution and its oxidative potential in Europe, Nature, 587, 414–419, https://doi.org/10.1038/s41586-020-2902-8, 2020.
Decesari, S., Allan, J., Plass-Duelmer, C., Williams, B. J., Paglione, M., Facchini, M. C., O'Dowd, C., Harrison, R. M., Gietl, J. K., Coe, H., Giulianelli, L., Gobbi, G. P., Lanconelli, C., Carbone, C., Worsnop, D., Lambe, A. T., Ahern, A. T., Moretti, F., Tagliavini, E., Elste, T., Gilge, S., Zhang, Y., and Dall'Osto, M.: Measurements of the aerosol chemical composition and mixing state in the Po Valley using multiple spectroscopic techniques, Atmos. Chem. Phys., 14, 12109–12132, https://doi.org/10.5194/acp-14-12109-2014, 2014.
Docherty, K. S., Yaga, R., Preston, W. T., Jaoui, M., Reidel, T. P., Offenberg, J. H., Kleindienst, T. E., and Lewandowski, M.: Relative contributions of selected multigeneration products to chamber SOA formed from photooxidation of a range (C10–C17) of n-alkanes under high NOx conditions, Atmos. Environ., 244, 117976, https://doi.org/10.1016/j.atmosenv.2020.117976, 2021.
Donahue, N. M., Epstein, S. A., Pandis, S. N., and Robinson, A. L.: A two-dimensional volatility basis set: 1. organic-aerosol mixing thermodynamics, Atmos. Chem. Phys., 11, 3303–3318, https://doi.org/10.5194/acp-11-3303-2011, 2011.
Donahue, N. M., Henry, K. M., Mentel, T. F., Kiendler-Scharr, A., Spindler, C., Bohn, B., Brauers, T., Dorn, H. P., Fuchs, H., Tillmann, R., Wahner, A., Saathoff, H., Naumann, K.-H., Möhler, O., Leisner, T., Müller, L., Reinnig, M.-C., Hoffmann, T., Salo, K., Hallquist, M., Frosch, M., Bilde, M., Tritscher, T., Barmet, P., Praplan, A. P., DeCarlo, P. F., Dommen, J., Prévôt, A. S. H., and Baltensperger, U.: Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions, P. Natl. Acad. Sci. USA, 109, 13503–13508, https://doi.org/10.1073/pnas.1115186109, 2012.
EEA – European Environment Agency: Europe's air quality status 2023: https://www.eea.europa.eu/publications/europes-air-quality-status-2023/europes-air-quality-status2023 (last access: 10 December 2024), 2023.
Giani, P., Balzarini, A., Pirovano, G., Gilardoni, S., Paglione, M., Colombi, C., Gianelle, V. L., Belis, C. A., Poluzzi, V., and Lonati, G.: Influence of semi- and intermediate-volatile organic compounds (S/IVOC) parameterizations, volatility distributions and aging schemes on organic aerosol modelling in winter conditions, Atmos. Environ., 213, 11–24, https://doi.org/10.1016/j.atmosenv.2019.05.061, 2019.
Griffin, R. J., Cocker III, D. R., Seinfeld, J. H., and Dabdub, D.: Estimate of global atmospheric organic aerosol from oxidation of biogenic hydrocarbons, Geophys. Res. Lett., 26, 2721–2724, https://doi.org/10.1029/1999GL900476, 1999a.
Griffin, R. J., Cocker III, D. R., Flagan, R. C., and Seinfeld, J. H.: Organic aerosol formation from the oxidation of biogenic hydrocarbons, J. Geophys. Res.-Atmos., 104, 3555–3567, https://doi.org/10.1029/1998JD100049, 1999b.
Guenther, A. B., Jiang, X., Heald, C. L., Sakulyanontvittaya, T., Duhl, T., Emmons, L. K., and Wang, X.: The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): an extended and updated framework for modeling biogenic emissions, Geosci. Model Dev., 5, 1471–1492, https://doi.org/10.5194/gmd-5-1471-2012, 2012.
Harrison, D., Hunter, M. C., Lewis, A. C., Seakins, P. W., Bonsang, B., Gros, V., Kanakidou, M., Touaty, M., Kavouras, I., Mihalopoulos, N., Stephanou, E., Alves, C., Nunes, T., and Pio, C.: Ambient isoprene and monoterpene concentrations in a Greek fir (Abies Borisii-regis) forest. Reconciliation with emissions measurements and effects on measured OH concentrations, Atmos. Environ., 35, 4699–4711, https://doi.org/10.1016/S1352-2310(01)00091-7, 2001.
He, Q., Tomaz, S., Li, C., Zhu, M., Meidan, D., Riva, M., Laskin, A., Brown, S. S., George, C., Wang, X., and Rudich, Y.: Optical properties of secondary organic aerosol produced by nitrate radical oxidation of biogenic volatile organic compounds, Environ. Sci. Technol., 55, 2878–2889, https://doi.org/10.1021/acs.est.0c06838, 2021.
Healy, R. M., Temime, B., Kuprovskyte, K., and Wenger, J. C.: Effect of relative humidity on gas/particle partitioning and aerosol mass yield in the photooxidation of p-xylene, Environ. Sci. Technol., 43, 1884–1889, https://doi.org/10.1021/es802404z, 2009.
Hinks, M. L., Montoya-Aguilera, J., Ellison, L., Lin, P., Laskin, A., Laskin, J., Shiraiwa, M., Dabdub, D., and Nizkorodov, S. A.: Effect of relative humidity on the composition of secondary organic aerosol from the oxidation of toluene, Atmos. Chem. Phys., 18, 1643–1652, https://doi.org/10.5194/acp-18-1643-2018, 2018.
Horne, J. R., Zhu, S., Montoya-Aguilera, J., Hinks, M. L., Wingen, L. M., Nizkorodov, S. A., and Dabdub, D.: Reactive uptake of ammonia by secondary organic aerosols: Implications for air quality, Atmos. Environ., 189, 1–8, https://doi.org/10.1016/j.atmosenv.2018.06.021, 2018.
Huang, R.-J., Zhang, Y., Bozzetti, C., Ho, K.-F., Cao, J.-J., Han, Y., Daellenbach, K. R., Slowik, J. G., Platt, S. M., Canonaco, F., Zotter, P., Wolf, R., Pieber, S. M., Bruns, E. A., Crippa, M., Ciarelli, G., Piazzalunga, A., Schwikowski, M., Abbaszade, G., Schnelle-Kreis, J., Zimmermann, R., An, Z., Szidat, S., Baltensperger, U., Haddad, I. E., and Prévôt, A. S. H.: High secondary aerosol contribution to particulate pollution during haze events in China, Nature, 514, 218–222, https://doi.org/10.1038/nature13774, 2014.
Ito, T., Bekki, K., Fujitani, Y., and Hirano, S.: The toxicological analysis of secondary organic aerosol in human lung epithelial cells and macrophages, Environ. Sci. Pollut. R., 26, 22747–22755, https://doi.org/10.1007/s11356-019-05317-5, 2019.
Jia, L. and Xu, Y.: Different roles of water in secondary organic aerosol formation from toluene and isoprene, Atmos. Chem. Phys., 18, 8137–8154, https://doi.org/10.5194/acp-18-8137-2018, 2018.
Jimenez, J. L., Canagaratna, M. R., Donahue, N. M., Prevot, A. S. H., Zhang, Q., Kroll, J. H., DeCarlo, P. F., Allan, J. D., Coe, H., Ng, N. L., Aiken, A. C., Docherty, K. S., Ulbrich, I. M., Grieshop, A. P., Robinson, A. L., Duplissy, J., Smith, J. D., Wilson, K. R., Lanz, V. A., Hueglin, C., Sun, Y. L., Tian, J., Laaksonen, A., Raatikainen, T., Rautiainen, J., Vaattovaara, P., Ehn, M., Kulmala, M., Tomlinson, J. M., Collins, D. R., Cubison, M. J., E., Dunlea, J., Huffman, J. A., Onasch, T. B., Alfarra, M. R., Williams, P. I., Bower, K., Kondo, Y., Schneider, J., Drewnick, F., Borrmann, S., Weimer, S., Demerjian, K., Salcedo, D., Cottrell, L., Griffin, R., Takami, A., Miyoshi, T., Hatakeyama, S., Shimono, A., Sun, J. Y., Zhang, Y. M., Dzepina, K., Kimmel, J. R., Sueper, D., Jayne, J. T., Herndon, S. C., Trimborn, A. M., Williams, L. R., Wood, E. C., Middlebrook, A. M., Kolb, C. E., Baltensperger, U., and Worsnop, D. R.: Evolution of organic aerosols in the atmosphere, Science, 326, 1525–1529, https://doi.org/10.1126/science.1180353, 2009.
Jo, Y., Jang, M., Han, S., Madhu, A., Koo, B., Jia, Y., Yu, Z., Kim, S., and Park, J.: CAMx–UNIPAR simulation of secondary organic aerosol mass formed from multiphase reactions of hydrocarbons under the Central Valley urban atmospheres of California, Atmos. Chem. Phys., 24, 487–508, https://doi.org/10.5194/acp-24-487-2024, 2024.
Jorga, S. D., Kaltsonoudis, C., Liangou, A., and Pandis, S. N.: Measurement of formation rates of secondary Aerosol in the ambient urban atmosphere using a dual smog chamber system, Environ. Sci. Technol., 54, 1336–1343, https://doi.org/10.1021/acs.est.9b03479, 2020.
Jorga, S. D., Florou, K., Kaltsonoudis, C., Kodros, J. K., Vasilakopoulou, C., Cirtog, M., Fouqueau, A., Picquet-Varrault, B., Nenes, A., and Pandis, S. N.: Nighttime chemistry of biomass burning emissions in urban areas: A dual mobile chamber study, Atmos. Chem. Phys., 21, 15337–15349, https://doi.org/10.5194/acp-21-15337-2021, 2021.
Kakavas, S., Pandis, S. N., and Nenes, A.: ISORROPIA-Lite: A Comprehensive Atmospheric Aerosol Thermodynamics Module for Earth System Models, Tellus B, 74, 1–23, https://doi.org/10.16993/tellusb.33, 2022.
Kaltsonoudis, C., Jorga, S. D., Louvaris, E., Florou, K., and Pandis, S. N.: A portable dual-smog-chamber system for atmospheric aerosol field studies, Atmos. Meas. Tech., 12, 2733–2743, https://doi.org/10.5194/amt-12-2733-2019, 2019.
Kanakidou, M., Seinfeld, J. H., Pandis, S. N., Barnes, I., Dentener, F. J., Facchini, M. C., Van Dingenen, R., Ervens, B., Nenes, A., Nielsen, C. J., Swietlicki, E., Putaud, J. P., Balkanski, Y., Fuzzi, S., Horth, J., Moortgat, G. K., Winterhalter, R., Myhre, C. E. L., Tsigaridis, K., Vignati, E., Stephanou, E. G., and Wilson, J.: Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 5, 1053–1123, https://doi.org/10.5194/acp-5-1053-2005, 2005.
Kerminen, V.-M., Chen, X., Vakkari, V., Petäjä, T., Kulmala, M., and Bianchi, F.: Atmospheric new particle formation and growth: review of field observations, Environ. Res. Lett., 13, 103003, https://doi.org/10.1088/1748-9326/aadf3c, 2018.
Kiendler-Scharr, A., Mensah, A. A., Friese, E., Topping, D., Nemitz, E., Prevot, A. S. H., Äijälä, M., Allan, J., Canonaco, F., Canagaratna, M., Carbone, S., Crippa, M., Dall Osto, M., Day, D. A., De Carlo, P., Di Marco, C. F., Elbern, H., Eriksson, A., Freney, E., Hao, L., Herrmann, H., Hildebrandt, L., Hillamo, R., Jimenez, J. L., Laaksonen, A., McFiggans, G., Mohr, C., O'Dowd, C., Otjes, R., Ovadnevaite, J., Pandis, S. N., Poulain, L., Schlag, P., Sellegri, K., Swietlicki, E., Tiitta, P., Vermeulen, A., Wahner, A., Worsnop, D., and Wu, H.-C.: Ubiquity of organic nitrates from nighttime chemistry in the European submicron aerosol, Geophys. Res. Lett., 43, 7735–7744, https://doi.org/10.1002/2016GL069239, 2016.
Kostenidou, E., Pathak, R. K., and Pandis, S. N.: An algorithm for the calculation of secondary organic aerosol density combining AMS and SMPS data, Aerosol Sci. Tech., 41, 1002–1010, https://doi.org/10.1080/02786820701666270, 2007.
Kroll, J. H. and Seinfeld, J. H.: Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere, Atmos. Environ., 42, 3593–3624, https://doi.org/10.1016/j.atmosenv.2008.01.003, 2008.
Kroll, J. H., Ng, N. L., Murphy, S. M., Flagan, R. C., and Seinfeld, J. H.: Secondary organic aerosol formation from isoprene photooxidation, Environ. Sci. Technol., 40, 1869–1877, https://doi.org/10.1021/es0524301, 2006.
Kulmala, M., Vehkamäki, H., Petäjä, T., Maso, M. D., Lauri, A., Kerminen, V. M., Birmili, W., and McMurry, P. H.: Formation and growth rates of ultrafine atmospheric particles: A review of observations, J. Aerosol Sci., 35, 143–176, https://doi.org/10.1016/j.jaerosci.2003.10.003, 2004.
Lamkaddam, H., Gratien, A., Pangui, E., David, M., Peinado, F., Polienor, J.-M., Jerome, M., Cazaunau, M., Gaimoz, C., Picquet-Varrault, B., Kourtchev, I., Kalberer, M., and Doussin, J.-F.: Role of relative humidity in the secondary organic aerosol formation from high-NOx photooxidation of long-chain alkanes: n-dodecane case study, ACS Earth Space Chem., 4, 2414–2425, https://doi.org/10.1021/acsearthspacechem.0c00265, 2020.
Lim, Y. B. and Ziemann, P. J.: Effects of molecular structure on aerosol yields from OH radical-initiated Reactions of linear, branched, and cyclic alkanes in the presence of NOx, Environ. Sci. Technol., 43, 2328–2334, https:// doi.org/10.1021/es803389s, 2009.
Liu, Y., Liggio, J., Staebler, R., and Li, S.-M.: Reactive uptake of ammonia to secondary organic aerosols: kinetics of organonitrogen formation, Atmos. Chem. Phys., 15, 13569–13584, https://doi.org/10.5194/acp-15-13569-2015, 2015.
Luo, H., Jia, L., Wan, Q., An, T., and Wang, Y.: Role of liquid water in the formation of O3 and SOA particles from 1,2,3-trimethylbenzene, Atmos. Environ., 217, 116955, https://doi.org/10.1016/j.atmosenv.2019.116955, 2019.
Matrali, A., Vasilakopoulou, C. N., Aktypis, A., Kaltsonoudis, C., Florou, K., Błaziak, A., Patoulias, D., Kostenidou, E., Błaziak, K., Seitanidi, K., Skyllakou, K., Fagault, Y., Tuna, T., Panagiotopoulos, C., Bard, E., Nenes, A., and Pandis, S. N.: Anthropogenic and biogenic pollutants in a forested environment: SPRUCE-22 campaign overview, Atmos. Environ., 334, 120722, https://doi.org/10.1016/j.atmosenv.2024.120722, 2024.
McFiggans, G., Mentel, T. F., Wildt, J., Pullinen, I., Kang, S., Kleist, E., Schmitt, S., Springer, M., Tillmann, R., Wu, C., Zhao, D., Hallquist, M., Faxon, C., Le Breton, M., Hallquist, Å. M., Simpson, D., Bergström, R., Jenkin, M. E., Ehn, M., Thornton, J. A., Alfarra, M. R., Bannan, T. J., Percival, C. J., Priestley, M., Topping, D., and Kiendler-Scharr, A.: Secondary organic aerosol reduced by mixture of atmospheric vapours, Nature, 565, 587–593, https://doi.org/10.1038/s41586-018-0871-y, 2019.
Mellouki, A. and Mu, Y.: On the atmospheric degradation of pyruvic acid in the gas phase, J. Photoch. Photobio., A, 157, 295–300, https://doi.org/10.1016/S1010-6030(03)00070-4, 2003.
Middlebrook, A. M., Bahreini, R., Jimenez, J. L., and Canagaratna, M. R.: Evaluation of composition-dependent collection efficiencies for the aerodyne aerosol mass spectrometer using field data, Aerosol Sci. Tech., 46, 258–271, https://doi.org/10.1080/02786826.2011.620041, 2012.
Mohr, C., DeCarlo, P. F., Heringa, M. F., Chirico, R., Slowik, J. G., Richter, R., Reche, C., Alastuey, A., Querol, X., Seco, R., Peñuelas, J., Jiménez, J. L., Crippa, M., Zimmermann, R., Baltensperger, U., and Prévôt, A. S. H.: Identification and quantification of organic aerosol from cooking and other sources in Barcelona using aerosol mass spectrometer data, Atmos. Chem. Phys., 12, 1649–1665, https://doi.org/10.5194/acp-12-1649-2012, 2012.
Müller, L., Reinnig, M.-C., Naumann, K. H., Saathoff, H., Mentel, T. F., Donahue, N. M., and Hoffmann, T.: Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 12, 1483–1496, https://doi.org/10.5194/acp-12-1483-2012, 2012.
Na, K., Song, C., Switzer, C., and Cocker, D. R.: Effect of ammonia on secondary organic aerosol formation from α-pinene ozonolysis in dry and humid conditions, Environ. Sci. Technol., 41, 6096–6102, https://doi.org/10.1021/es061956y, 2007.
Nault, B. A., Jo, D. S., McDonald, B. C., Campuzano-Jost, P., Day, D. A., Hu, W., Schroder, J. C., Allan, J., Blake, D. R., Canagaratna, M. R., Coe, H., Coggon, M. M., DeCarlo, P. F., Diskin, G. S., Dunmore, R., Flocke, F., Fried, A., Gilman, J. B., Gkatzelis, G., Hamilton, J. F., Hanisco, T. F., Hayes, P. L., Henze, D. K., Hodzic, A., Hopkins, J., Hu, M., Huey, L. G., Jobson, B. T., Kuster, W. C., Lewis, A., Li, M., Liao, J., Nawaz, M. O., Pollack, I. B., Peischl, J., Rappenglück, B., Reeves, C. E., Richter, D., Roberts, J. M., Ryerson, T. B., Shao, M., Sommers, J. M., Walega, J., Warneke, C., Weibring, P., Wolfe, G. M., Young, D. E., Yuan, B., Zhang, Q., de Gouw, J. A., and Jimenez, J. L.: Secondary organic aerosols from anthropogenic volatile organic compounds contribute substantially to air pollution mortality, Atmos. Chem. Phys., 21, 11201–11224, https://doi.org/10.5194/acp-21-11201-2021, 2021.
Ng, N. L., Kroll, J. H., Keywood, M. D., Bahreini, R., Varutbangkul, V., Flagan, R. C., Seinfeld, J. H., Lee, A., and Goldstein, A. H.: Contribution of first- versus second-generation products to secondary organic aerosols formed in the oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 40, 2283–2297, https://doi.org/10.1021/es052269u, 2006.
Neuberger, A., Decesari, S., Aktypis, A., Andersen, H., Baumgardner, D., Bianchi, F., Busetto, M., Cai, J., Cermak, J., Dipu, J., Ekman, A., Fuzzi, S., Gramlich, Y., Hadden, D., Haslett, S. L., Heikkinen, L., Joutsensaari, J., Kaltsonoudis, C., Kangasluoma, J., Lupi, A., Marinoni, A., Matrali, A., Mattsson, F., Mohr, M., Nenes, A., Paglione, M., Pandis, S. N., Patel, A., Riipinen, I., Rinaldi, M., Steimer, S. S., Stolzenburg, D., Sulo, J., Vasilakopoulou, C. N., and Zieger, P.: From molecules to droplets: The Fog and Aerosol InteRAction Research Italy (FAIRARI) 2021/22 campaign, B. Am. Meteorol. Soc., https://doi.org/10.1175/BAMS-D-23-0166.1, in press, 2024.
Paatero, P. and Tapper, U.: Positive matrix factorization: A non-negative factor model with optimal utilization of error estimates of data values, Environmetrics, 5, 111–126, https://doi.org/10.1002/env.3170050203, 1994.
Paglione, M., Gilardoni, S., Rinaldi, M., Decesari, S., Zanca, N., Sandrini, S., Giulianelli, L., Bacco, D., Ferrari, S., Poluzzi, V., Scotto, F., Trentini, A., Poulain, L., Herrmann, H., Wiedensohler, A., Canonaco, F., Prévôt, A. S. H., Massoli, P., Carbone, C., Facchini, M. C., and Fuzzi, S.: The impact of biomass burning and aqueous-phase processing on air quality: a multi-year source apportionment study in the Po Valley, Italy, Atmos. Chem. Phys., 20, 1233–1254, https://doi.org/10.5194/acp-20-1233-2020, 2020.
Paglione, M., Decesari, S., Rinaldi, M., Tarozzi, L., Manarini, F., Gilardoni, S., Facchini, M. C., Fuzzi, S., Bacco, D., Trentini, A., Pandis, S. N., and Nenes, A.: Historical changes in seasonal aerosol acidity in the Po Valley (Italy) as inferred from fog water and aerosol measurements, Environ. Sci. Technol., 55, 7307–7315, https://doi.org/10.1021/acs.est.1c00651, 2021.
Pope III, C. A. and Dockery, D. W.: Health Effects of Fine Particulate Air Pollution: Lines that Connect, J. Air Waste Manage., 56, 709–742, https://doi.org/10.1080/10473289.2006.10464485, 2006.
Presto, A. A., Miracolo, M. A., Donahue, N. M., and Robinson, A. L.: Secondary organic aerosol formation from high-NOx photo-oxidation of low volatility precursors: n-alkanes, Environ. Sci. Technol., 44, 2029–2034, https://doi.org/10.1021/es903712r, 2010.
Qi, L., Nakao, S., and Cocker III, D. R.: Aging of secondary organic aerosol from α-pinene ozonolysis: Roles of hydroxyl and nitrate radicals, J. Air Waste Manage., 62, 1359–1369, https://doi.org/10.1080/10962247.2012.712082, 2012.
Qi, X., Zhu, S., Zhu, C., Hu, J., Lou, S., Xu, L., Dong, J., and Cheng, P.: Smog chamber study of the effects of NOx and NH3 on the formation of secondary organic aerosols and optical properties from photo-oxidation of toluene, Sci. Total Environ., 727, 138632, https://doi.org/10.1016/j.scitotenv.2020.138632, 2020.
Robinson, A. L., Donahue, N. M., Shrivastava, M. K., Weitkamp, E. A., Sage, A. M., Grieshop, A. P., Lane, T. E., Pierce, J. R., and Pandis, S. N.: Rethinking organic aerosols: semivolatile emissions and photochemical aging, Science, 315, 1259–1262, https://doi.org/10.1126/science.1133061, 2007.
Saarikoski, S., Carbone, S., Decesari, S., Giulianelli, L., Angelini, F., Canagaratna, M., Ng, N. L., Trimborn, A., Facchini, M. C., Fuzzi, S., Hillamo, R., and Worsnop, D.: Chemical characterization of springtime submicrometer aerosol in Po Valley, Italy, Atmos. Chem. Phys., 12, 8401–8421, https://doi.org/10.5194/acp-12-8401-2012, 2012.
Schervish, M. and Donahue, N. M.: Peroxy radical chemistry and the volatility basis set, Atmos. Chem. Phys., 20, 1183–1199, https://doi.org/10.5194/acp-20-1183-2020, 2020.
Seinfeld, J. H. and Pandis, S. N.: Atmospheric Chemistry and Physics from Air Pollution to Climate Change, John Wiley, Hoboken, NJ, ISBN 978-1-118-94740-1, 2016.
Squizzato, S., Masiol, M., Brunelli, A., Pistollato, S., Tarabotti, E., Rampazzo, G., and Pavoni, B.: Factors determining the formation of secondary inorganic aerosol: a case study in the Po Valley (Italy), Atmos. Chem. Phys., 13, 1927–1939, https://doi.org/10.5194/acp-13-1927-2013, 2013.
Srivastava, D., Vu, T. V., Tong, S., Shi, Z., and Harrison, R. M.: Formation of secondary organic aerosols from anthropogenic precursors in laboratory studies, NPJ Clim. Atmos. Sci., 5, 1–30, https://doi.org/10.1038/s41612-022-00238-6, 2022.
Steinbacher, M., Dommen, J., Ordonez, C., Reimann, S., Grüebler, F. C., Staehelin, J., and Prevot, A. S. H.: Volatile Organic Compounds in the Po Basin. Part A: Anthropogenic VOCs, J. Atmos. Chem., 51, 271–291, https://doi.org/10.1007/s10874-005-3576-1, 2005a.
Steinbacher, M., Dommen, J., Ordonez, C., Reimann, S., Grüebler, F. C., Staehelin, J., Andreani-Aksoyoglu, S., and Prevot, A. S. H.: Volatile Organic Compounds in the Po Basin. Part B: Biogenic VOCs, J. Atmos. Chem., 51, 293–315, https://doi.org/10.1007/s10874-005-3577-0, 2005b.
Struckmeier, C., Drewnick, F., Fachinger, F., Gobbi, G. P., and Borrmann, S.: Atmospheric aerosols in Rome, Italy: sources, dynamics and spatial variations during two seasons, Atmos. Chem. Phys., 16, 15277–15299, https://doi.org/10.5194/acp-16-15277-2016, 2016.
Takeuchi, M., Berkemeier, T., Eris, G., and Ng, N. L.: Non-linear effects of secondary organic aerosol formation and properties in multi-precursor systems, Nat. Commun., 13, 7883, https://doi.org/10.1038/s41467-022-35546-1, 2022.
Tasoglou, A. and Pandis, S. N.: Formation and chemical aging of secondary organic aerosol during the β-caryophyllene oxidation, Atmos. Chem. Phys., 15, 6035–6046, https://doi.org/10.5194/acp-15-6035-2015, 2015.
Tkacik, D. S., Presto, A. A., Donahue, N. M., and Robinson, A. L.: Secondary organic aerosol formation from intermediate-volatility organic compounds: Cyclic, linear, and branched alkanes, Environ. Sci. Technol., 46, 8773–8781, https://doi.org/10.1021/es301112c, 2012.
Tsigaridis, K., Daskalakis, N., Kanakidou, M., Adams, P. J., Artaxo, P., Bahadur, R., Balkanski, Y., Bauer, S. E., Bellouin, N., Benedetti, A., Bergman, T., Berntsen, T. K., Beukes, J. P., Bian, H., Carslaw, K. S., Chin, M., Curci, G., Diehl, T., Easter, R. C., Ghan, S. J., Gong, S. L., Hodzic, A., Hoyle, C. R., Iversen, T., Jathar, S., Jimenez, J. L., Kaiser, J. W., Kirkevåg, A., Koch, D., Kokkola, H., Lee, Y. H., Lin, G., Liu, X., Luo, G., Ma, X., Mann, G. W., Mihalopoulos, N., Morcrette, J.-J., Müller, J.-F., Myhre, G., Myriokefalitakis, S., Ng, N. L., O'Donnell, D., Penner, J. E., Pozzoli, L., Pringle, K. J., Russell, L. M., Schulz, M., Sciare, J., Seland, Ø., Shindell, D. T., Sillman, S., Skeie, R. B., Spracklen, D., Stavrakou, T., Steenrod, S. D., Takemura, T., Tiitta, P., Tilmes, S., Tost, H., van Noije, T., van Zyl, P. G., von Salzen, K., Yu, F., Wang, Z., Wang, Z., Zaveri, R. A., Zhang, H., Zhang, K., Zhang, Q., and Zhang, X.: The AeroCom evaluation and intercomparison of organic aerosol in global models, Atmos. Chem. Phys., 14, 10845–10895, https://doi.org/10.5194/acp-14-10845-2014, 2014.
Ulbrich, I. M., Canagaratna, M. R., Zhang, Q., Worsnop, D. R., and Jimenez, J. L.: Interpretation of organic components from Positive Matrix Factorization of aerosol mass spectrometric data, Atmos. Chem. Phys., 9, 2891–2918, https://doi.org/10.5194/acp-9-2891-2009, 2009.
Vasilakopoulou, C. N., Matrali, A., Skyllakou, K., Georgopoulou, M., Aktypis, A., Florou, K., Kaltsonoudis, C., Siouti, E., Kostenidou, E., Błaziak, A., Nenes, A., Papagiannis, S., Eleftheriadis, K., Patoulias, D., Kioutsioukis, I., and Pandis, S. N.: Rapid transformation of wildfire emissions to harmful background aerosol, NPJ Clim. Atmos. Sci., 6, 1–9, https://doi.org/10.1038/s41612-023-00544-7, 2023.
Voliotis, A., Wang, Y., Shao, Y., Du, M., Bannan, T. J., Percival, C. J., Pandis, S. N., Alfarra, M. R., and McFiggans, G.: Exploring the composition and volatility of secondary organic aerosols in mixed anthropogenic and biogenic precursor systems, Atmos. Chem. Phys., 21, 14251–14273, https://doi.org/10.5194/acp-21-14251-2021, 2021.
Volkamer, R., Jimenez, J. L., San Martini, F., Dzepina, K., Zhang, Q., Salcedo, D., Molina, L. T., Worsnop, D. R., and Molina, M. J.: Secondary organic aerosol formation from anthropogenic air pollution: Rapid and higher than expected, Geophys. Res. Lett., 33, L17811, https://doi.org/10.1029/2006GL026899, 2006.
Wang, N., Kostenidou, E., Donahue, N. M., and Pandis, S. N.: Multi-generation chemical aging of α-pinene ozonolysis products by reactions with OH, Atmos. Chem. Phys., 18, 3589–3601, https://doi.org/10.5194/acp-18-3589-2018, 2018a.
Wang, N., Jorga, S. D., Pierce, J. R., Donahue, N. M., and Pandis, S. N.: Particle wall-loss correction methods in smog chamber experiments, Atmos. Meas. Tech., 11, 6577–6588, https://doi.org/10.5194/amt-11-6577-2018, 2018b.
White, S. J., Jamie, I. M., and Angove, D. E.: Chemical characterisation of semi-volatile and aerosol compounds from the photooxidation of toluene and NOx, Atmos. Environ., 83, 237–244, https://doi.org/10.1016/j.atmosenv.2013.11.023, 2014.
Zhang, Q., Xu, Y., and Jia, L.: Secondary organic aerosol formation from OH-initiated oxidation of m-xylene: effects of relative humidity on yield and chemical composition, Atmos. Chem. Phys., 19, 15007–15021, https://doi.org/10.5194/acp-19-15007-2019, 2019.
Zhao, Y., Hennigan, C. J., May, A. A., Tkacik, D. S., de Gouw, J. A., Gilman, J. B., Kuster, W. C., Borbon, A., and Robinson, A. L.: Intermediate-volatility organic compounds: A large source of secondary organic aerosol, Environ. Sci. Technol., 48, 13743–13750, https://doi.org/10.1021/es5035188, 2014.