the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 14 Nov 2024
| 14 Nov 2024
The annual cycle and sources of relevant aerosol precursor vapors in the central Arctic during the MOSAiC expedition
Matthew Boyer
Diego Aliaga
Lauriane L. J. Quéléver
Silvia Bucci
Hélène Angot
Lubna Dada
Benjamin Heutte
Lisa Beck
Marina Duetsch
Andreas Stohl
Ivo Beck
Tiia Laurila
Nina Sarnela
Roseline C. Thakur
Branka Miljevic
Markku Kulmala
Tuukka Petäjä
Mikko Sipilä
Julia Schmale
In this study, we present and analyze the first continuous time series of relevant aerosol precursor vapors from the central Arctic (north of 80° N) during the Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition. These precursor vapors include sulfuric acid (SA), methanesulfonic acid (MSA), and iodic acid (IA). We use FLEXPART simulations, inverse modeling, sulfur dioxide (SO2) mixing ratios, and chlorophyll a (chl a) observations to interpret the seasonal variability in the vapor concentrations and identify dominant sources. Our results show that both natural and anthropogenic sources are relevant for the concentrations of SA in the Arctic, but anthropogenic sources associated with Arctic haze are the most prevalent. MSA concentrations are an order of magnitude higher during polar day than during polar night due to seasonal changes in biological activity. Peak MSA concentrations were observed in May, which corresponds with the timing of the annual peak in chl a concentrations north of 75° N. IA concentrations exhibit two distinct peaks during the year, namely a dominant peak in spring and a secondary peak in autumn, suggesting that seasonal IA concentrations depend on both solar radiation and sea ice conditions. In general, the seasonal cycles of SA, MSA, and IA in the central Arctic Ocean are related to sea ice conditions, and we expect that changes in the Arctic environment will affect the concentrations of these vapors in the future. The magnitude of these changes and the subsequent influence on aerosol processes remains uncertain, highlighting the need for continued observations of these precursor vapors in the Arctic.
- Article
(7258 KB) - Full-text XML
-
Supplement
(4191 KB) - BibTeX
- EndNote





The Arctic environment is rapidly changing as the region is warming up to 4 times faster than the rest of the planet (Rantanen et al., 2022). The central Arctic Ocean (north of 80° N), which is largely covered by sea ice, is particularly sensitive to temperature changes due to climate feedbacks involving the decline in sea ice. The Arctic atmosphere is expected to change in response to lower sea ice extent through changing atmospheric transport patterns, increasing anthropogenic activities such as ship traffic, and increasing ocean–atmosphere exchange of gases (Meier et al., 2014).
Such changes can have a direct influence on climate-relevant aerosol precursor vapors in the central Arctic north of 80° N.
Condensable vapors, either naturally emitted by the Arctic environment or transported from distant anthropogenic sources, can lead to secondary aerosol production, including new particle formation (NPF), which is estimated to be a key source of particles in the summertime Arctic atmosphere (e.g., Willis et al., 2018). Other relevant sources of particles in the high Arctic during summer could include sea spray and marine nanogels formed by bubble bursting (Bigg and Leck, 2008; Karl et al., 2013; Lawler et al., 2021; Leck and Bigg, 2005). When reaching a certain size, the newly formed particles can act as nuclei in cloud formation processes that influence the surface energy budget by scattering and absorbing incoming and outgoing radiation. NPF, which can yield high number concentrations of particles, is estimated to contribute to cloud condensation nuclei (CCN) number concentrations in the Arctic; however, these estimates remain poorly constrained. Global model estimates range up to 90 % contribution of CCN concentrations from NPF (Gordon et al., 2017), and field observations suggest that newly formed particles can increase background CCN concentrations by up to a factor of 5 (Kecorius et al., 2019). Moreover, aerosol–cloud interactions have an important, yet uncertain, role in the surface energy budget in the central Arctic Ocean, where summertime aerosol concentrations can be so low that cloud formation is CCN-limited (Mauritsen et al., 2011) due to the wet scavenging of particles during advection from the marginal ice zone into the ice pack (Leck and Svensson, 2015). Recent observations suggest that very small particles may act as CCN under such conditions during summer and early autumn in the high Arctic (Baccarini et al., 2020; Chang et al., 2022; Karlsson et al., 2022; Kecorius et al., 2019; Leaitch et al., 2016), which also agrees with modeled Aitken mode particle contribution to CCN at particle diameters < 50 nm (Bulatovic et al., 2021). The specific vapors that contribute to these climate-relevant processes in the Arctic have remained elusive, particularly over the central Arctic Ocean where data are scarce. Many of the past observations in the central Arctic Ocean are limited, either by short temporal coverage in summer/early autumn or insufficient instrumentation to quantify aerosol precursor vapor concentrations (Leck et al., 2001, 2004, 2019; Leck and Persson, 1996a; Tjernström et al., 2014). Therefore, there is a need for more detailed characterization of the seasonal variability and sources of aerosol precursor vapors in the Arctic (Kecorius et al., 2019; Schmale and Baccarini, 2021; Willis et al., 2018).
Dimethyl sulfide (DMS) is a gaseous product of phytoplankton and microalgae activities (Yoch, 2002) and the largest natural source of reduced sulfur in the atmosphere (Andreae, 1990). Specific phytoplankton species exude dimethylsulfoniopropionate (DMSP) in the water column, which can then be converted to DMS by microbial enzyme activity (Carpenter et al., 2012). A portion of the dissolved DMS subsequently outgasses from the surface ocean to the atmosphere (Stefels et al., 2007). Once emitted, DMS oxidizes in the presence of sunlight to form secondary products that act as aerosol precursors, such as methanesulfonic acid (MSA), sulfur dioxide (SO2), and sulfuric acid (SA) (Carpenter et al., 2012). In addition, there are other intermediate sulfur species that result from DMS oxidation, including dimethyl sulfoxide (DMSO), methanesulfinic acid (MSIA), and hydroperoxyl methyl thioformate (HPMTF), that readily undergo aqueous phase processing. Reactions involving these intermediate species can contribute directly to sulfur aerosol mass in the marine boundary layer and affect the yield of MSA, SO2, and SA from DMS oxidation (Hoffmann et al., 2016; Chen et al., 2018; Siegel et al., 2023; Cala et al., 2023). SO2 ultimately reaches its highest oxidation state as SA in the atmosphere, which typically requires sunlight but has also been observed to occur under conditions with very low solar radiation (Dada et al., 2020; Sipilä et al., 2021). Recent observations also suggest that direct formation of SA is possible via oxidation of DMS with the OH radical (Berndt et al., 2023), a process which has been proposed for decades (Karl et al., 2007; Lucas and Prinn, 2002). SA and MSA are important vapors for secondary aerosol formation and growth processes (Dunne et al., 2016; Gordon et al., 2017; Jokinen et al., 2018; Kirkby et al., 2011; Kulmala et al., 2013; Sipilä et al., 2021; Yan et al., 2018; Zollner et al., 2012), where SA is one of the most well-known precursors for NPF and MSA has a demonstrated role in the growth of newly formed particles (Beck et al., 2021).
The concentration of gaseous DMS and its oxidation products (SA and MSA) in both the gas and particle phases has been observed to increase at various locations across the Arctic, including during spring and summer at Ny-Ålesund, Svalbard, Norway (Beck et al., 2021); during spring at Villum Research Station, Greenland, Denmark (Nielsen et al., 2019); during spring and summer in the Canadian Arctic (Abbatt et al., 2019; Ghahremaninezhad et al., 2017); and in summer over the ice pack in the central Arctic Ocean (Kerminen and Leck, 2001; Lundén et al., 2010), which corresponds to the appearance of sunlight and phytoplankton activity in the region. Other studies suggest that DMS oxidation products affect the natural aerosol population, and potentially CCN concentrations, in the marine atmosphere (Mayer et al., 2020), over the Arctic Ocean (Ghahremaninezhad et al., 2019), and in Svalbard (Lee et al., 2020). Moreover, there is inferred evidence that decadal DMS emission trends are positive across the Arctic due to decreasing sea ice coverage (Galí et al., 2019; Kurosaki et al., 2022), where the strongest increase in emissions is observed at higher latitudes and near the marginal ice zone (Galí et al., 2021; Leck and Persson, 1996b). At the same time, observations of DMS oxidation products in the aerosol phase in the Arctic do not show a uniform trend (Schmale et al., 2022; Sharma et al., 2019), which highlights the complexity of understanding aerosol processes involving SA and MSA. As a result, DMS oxidation remains poorly represented in large-scale climate models, despite its climate relevance, and thus, direct observations of its oxidation products can help support modeling activities (Chen et al., 2018; Hoffmann et al., 2016, 2021; Quinn and Bates, 2011; Veres et al., 2020).
Anthropogenic sources of sulfur are also important in the Arctic atmosphere, particularly during the so-called “Arctic haze” phenomenon in winter (Quinn et al., 2007). During this season, SO2 and particulate sulfate emissions from anthropogenic combustion processes, ore smelters, and shipping activities are transported into the Arctic (Barrie, 1986; Corbett et al., 1999; Heidam et al., 1999; Leaitch et al., 1989; Sharma et al., 2019; Sipilä et al., 2021; Smith et al., 2001; Tunved et al., 2013). Likewise, volcanic activity represents an occasionally large natural source of atmospheric SO2 emissions (Fioletov et al., 2020). Therefore, both anthropogenic pollution and natural sources may influence the sulfur budget, and hence atmospheric SA concentrations, in the Arctic.
In addition to DMS oxidation products and SA, iodic acid (IA) has recently been identified as an important aerosol precursor gas for secondary particle formation (Allan et al., 2015; Sipilä et al., 2016). Detailed studies have identified IA to play a role in the initial steps of NPF in the Arctic atmosphere that may contribute to the CCN budget in the region (Baccarini et al., 2020; Beck et al., 2021). IA forms via atmospheric reactions with iodine, which is typically sourced from biological activities of microalgae and phytoplankton (Allan et al., 2015; Ashu-Ayem et al., 2012; O'Dowd et al., 2002). However, the mechanism of IA formation in the atmosphere is not fully resolved (Finkenzeller et al., 2023). While iodine oxoacids were thought to involve reactions between HOx (OH and HO2) and reactive iodine (Drougas and Kosmas, 2005; Khanniche et al., 2017; Plane et al., 2006), He et al. (2021) showed that iodic acid can form via the oxidation of iodine atoms (obtained by photolysis of molecular iodine) under atmospherically relevant conditions with either ozone and water or from the hydrolysis of intermediate oxygenated iodine compounds (e.g., I2O3, I2O4, and I2O5). New insights into the chemical mechanism show that IA formation can also occur at low iodine concentrations due to the catalytic recycling of iodine gas in the IA formation pathway (Finkenzeller et al., 2023). Interestingly, these reactions can proceed without light if there is a source of reactive iodine. Furthermore, recent studies have identified that IA formation follows a diurnal trend, where IA concentrations peak during periods of low solar radiation (Sipilä et al., 2016; Jokinen et al., 2018; Baccarini et al., 2021; Quéléver et al., 2022). These results may have implications for secondary aerosol formation during the polar night or during seasonal transitions in the central Arctic.
Despite knowledge from previous studies, observations of SA, MSA, and IA across the Arctic region in general are scarce. This lack of observations limits our understanding of NPF, particle growth, and aerosol size distributions to resolve climate-relevant aerosol processes in the Arctic (Beck et al., 2021; Croft et al., 2019; Willis et al., 2018; Baccarini et al., 2020). Therefore, it is necessary to obtain more measurements of atmospheric aerosol precursor vapors at high latitudes.
Herein, we present and analyze the concentrations of key aerosol precursor vapors, including MSA, SA, SO2, and IA, in the central Arctic during the Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition. This dataset represents not only the longest continuous time series of SA, MSA, IA, and SO2 in the central Arctic and the first measurements over the sea ice pack during the polar night but also one of the longest continuous measurements of these precursor vapors available in the literature. Our analysis focuses on the annual cycle in the concentrations of these aerosol precursor vapors and their primary source regions using FLEXPART (FLEXible PARTicle dispersion model) simulations, emission inventories, and inverse-modeling techniques. The results of this study are valuable for improving our understanding of these key aerosol precursor vapors and their potential sources from direct observations within the central Arctic, a region which is currently undergoing rapid changes.
2.1 The MOSAiC expedition
The MOSAiC expedition was a ship-based campaign in the Arctic Ocean on research vessel (R/V) Polarstern (Knust, 2017) between September 2019 and October 2020. The MOSAiC expedition and its objectives have been previously described in detail elsewhere (Krumpen et al., 2020; Nicolaus et al., 2022; Rabe et al., 2022; Shupe et al., 2022). The ship drifted beside an ice floe for a full year in the central Arctic Ocean and remained north of 80° N for almost the entire expedition. Refer to Fig. S1 in the Supplement for a map of the drift track and Fig. 1c for the monthly median location of Polarstern during the campaign. MOSAiC was the largest expedition to the central Arctic in history and the first with a full year of observation of various components of the Arctic environment, including during polar night. The work presented here focuses on gas phase parameters measured from the Swiss container located on board Polarstern, as described below. Refer to Shupe et al. (2022) for further description of all the atmospheric measurements and a diagram of the measurement containers onboard Polarstern during MOSAiC.
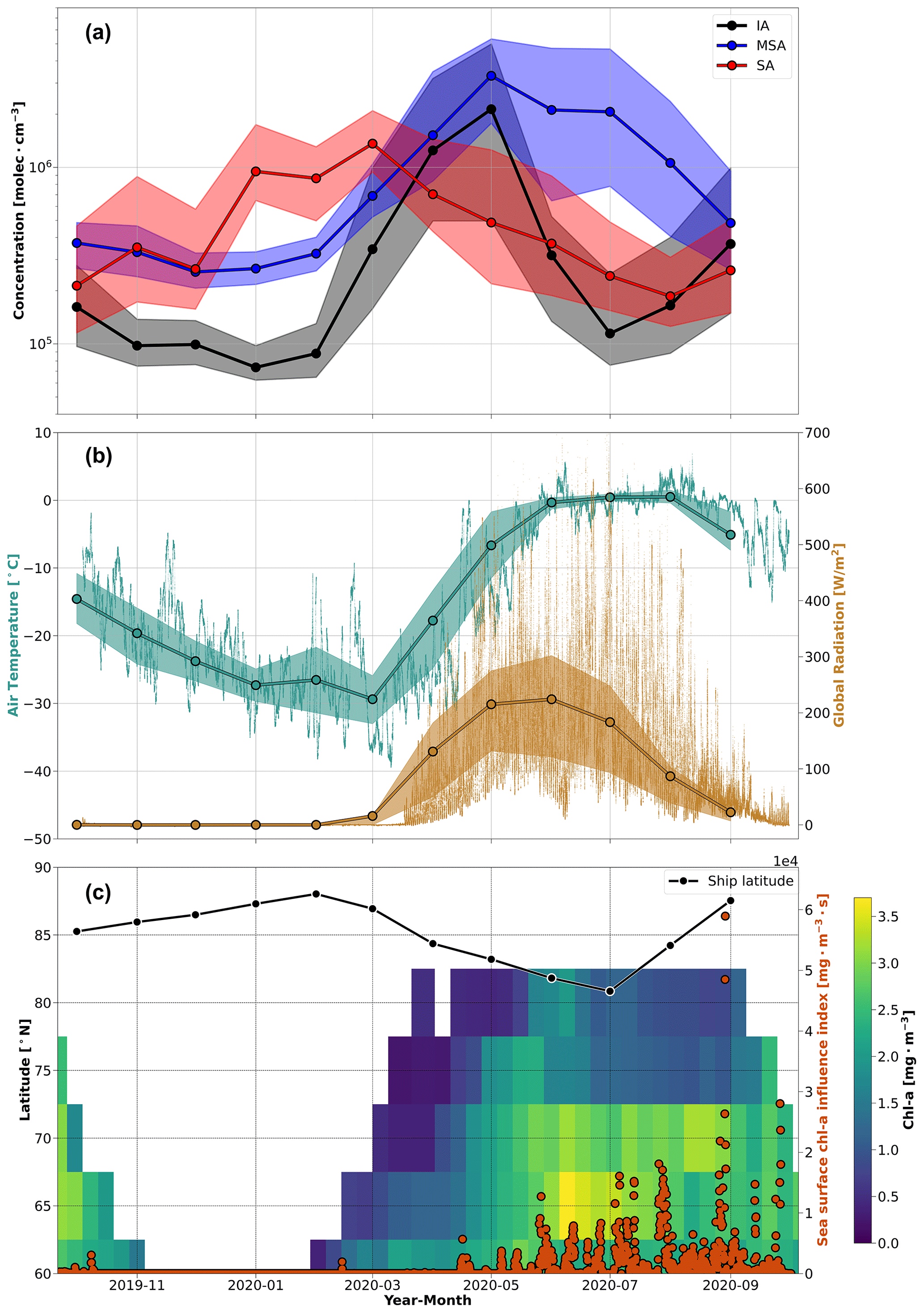
Figure 1The annual cycle of aerosol precursor vapor concentrations and related parameters during MOSAiC. (a) The monthly median vapor concentrations and the corresponding interquartile ranges of iodic acid (IA), methanesulfonic acid (MSA), and sulfuric acid (SA) are presented as the solid lines and shaded regions, respectively. Note that concentrations below the LOD for each vapor species were removed. (b) The temperature and global radiation measurements are shown at 1 min resolution with the monthly averages and interquartile ranges overlaid for context. (c) The chl a concentrations are shown as 8 d averages integrated for every 5° of latitude between 60–90° N from the Ocean Colour Climate Change Initiative (OC-CCI) satellite dataset. The color bar depicts the chl a concentrations, where the lack of color indicates missing data due to cloudiness, algorithm failure, or low-incidence sun angles in winter. The solid black line denotes the monthly median latitude of Polarstern during the campaign. The orange circles show the sea surface chl a influence index, a time series that quantifies potential air mass exposure to ocean regions with chl a derived from the convolution of the FLEXPART FES with the satellite chl a concentrations using air mass ages of 10 d, as described in Sect. 3.2.
2.2 Precursor vapors from a chemical ionization mass spectrometer
The concentrations of MSA, SA, and IA in this study were measured using a nitrate-based chemical ionization mass spectrometer (NO3-CIMS) (CI-APi-TOF; Tofwerk HTOF; Jokinen et al., 2012), using 5 s time resolution. The NO3-CIMS was located inside the Swiss container on the starboard side of the bow of Polarstern (refer to Beck et al., 2022). The NO3-CIMS sampled from a dedicated NPF inlet that was specifically designed to minimize diffusional losses (reaching a 60 + 10 L min−1 combined inlet flow shared between a neutral cluster air ion spectrometer and the NO3-CIMS, respectively). The NPF inlet was ∼ 1.3 m long, with a diameter of 10.2 cm, connected to the in. (19.05 mm) inlet tube of the CI inlet by a KNF 50 flange (see Baccarini et al., 2020, for more details). The NO3-CIMS had an individual inlet flow of 10 L min−1, which was the result of the difference between a total vacuum flow of 40 L min−1 and a sheath flow of 30 L min−1 accomplished by a set of blowers in a flow generation system (Airel OÜ). The sheath flow contained an additional flow saturated with nitric acid (HNO3) vapor at a flow rate of 5 mL min−1 that was ionized by X-ray irradiation to form nitrate ions. Background measurements were performed periodically during the campaign by placing a high-efficiency particulate air (HEPA) filter in front of the instrument inlet to quantify the limit of detection (LOD), and a calibration for SA was performed post-campaign according to the procedure described in Kürten et al. (2012). A calibration factor of 6 × 109 molec. cm−3 was obtained, and this calibration factor was then used for concentration retrieval.
The time series of SA, MSA, and IA concentrations were obtained from TofTools (Junninen et al., 2010) by integrating peaks from the high-resolution mass spectra data at 5 min averaged time resolution, normalizing the result with the sum of charger ions (NO, HNO3NO, and (HNO3)2NO), and multiplying by the calibration factor. SA was determined by peaks at mass-to-charge ratios () of 96.9601 Th (HSO) and 159.9557 Th (H2SO4NO), MSA was determined by peaks at 94.9808 Th (CH3SO) and 157.9765 Th (CH3SO3HNO), and IA was determined by peaks at 174.8898 Th (IO) and 237.8854 Th (HIO3NO). The LOD was determined for each species, according to the method discussed in He et al. (2023), as follows:
where μ is the average concentration and σ is the standard deviation during a filter measurement. The resulting LOD are 8.8 × 104, 1.5 × 105, and 5.5 × 104 molec. cm−3 for SA, MSA, and IA, respectively. The uncertainty range of the measured concentrations from the NO3-CIMS is estimated to be −50 % to +100 % (Jokinen et al., 2012), while the reported concentrations should mostly be considered low-limit values. This is due to the assumption that MSA and IA are charging at the kinetic limit, and therefore, concentrations reported herein may be underestimated (Ehn et al., 2014; Sipilä et al., 2016).
2.3 Local pollution in the NO3-CIMS measurements
Local pollution is an important consideration in ship-based campaigns, as it affects the baseline of ambient samples (Beck et al., 2022). During MOSAiC, local pollution sources included the vessel and logistic activities surrounding the central observatory (helicopter flights, snowmobiles, etc.). Polarstern used low-sulfur diesel fuel, specifically, distillate marine fuel, which has a maximum sulfur content of 0.1 % m m−1. While drifting in the pack ice, Polarstern's main engines were turned off; however, fuel was still consumed to provide electricity and heat.
There are three possible ways in which local pollution could bias the NO3-CIMS measurements: (1) the high particulate loading in pollution plumes could increase the condensation sink and enhance the loss of condensable vapors onto particle surfaces, (2) the pollution could act as a direct source of the gas phase species, and (3) the pollution could act as a source of primary species that are subsequently oxidized to form species of interest. The secondary formation of SA from SO2 is particularly of concern, as ship stack pollution is a large source of SO2. Note that the influence of local pollution was removed from the SO2 time series data, as described in Sect. 2.4.
To evaluate the effect of local pollution on the NO3-CIMS measurements, we followed the approach of Baccarini et al. (2021) by comparing the gas phase concentrations of SA, MSA, and IA during clean and polluted periods (Fig. S2). Polluted periods, including the local pollution influence from the ship exhaust and logistical activities, were determined by applying the pollution detection algorithm developed by Beck et al. (2022) to particle concentrations from a condensation particle counter (TSI; model 3025) located in the Swiss container. This method identified that 67 % of the 5 min NO3-CIMS data were influenced by primary pollution (out of 87 506 total data points). In addition, a comparison of the particle number concentrations and aerosol precursor vapors time series during “clean” and “polluted” periods for a single day is shown in Fig. S3. For further details on timing and extent of pollution in the time series of SA, MSA, and IA, refer to Figs. S4, S5, and S6, respectively.
Overall, the comparisons presented in Figs. S2, S3, S4, S5, and S6 suggest that local pollution has minimal impact on our analysis of the seasonal cycles for all three aerosol precursor vapors during the MOSAiC expedition. While the median polluted concentrations were slightly higher for all species, we conclude that this is likely coincidental due to natural variability in the gas phase concentrations over time, given the length and extent of pollution episodes in the time series. We also expect that secondary pollution and natural variability may be seasonal in the high Arctic due to environmental changes between polar day and polar night, especially for the secondary formation of SA from SO2, which typically requires solar radiation. Moreover, the conversion of SO2 to SA occurs on the order of hours under atmospheric conditions (Lee et al., 2011); hence, the effect of the ship stack emissions may only be minimal in our measurements. The comparison of SA in Fig. S2 suggests that pollution may have been influential for a small fraction of SA concentrations higher than ∼ 107 molec. cm−3 during the year; however, the effect is minimal and, again, may be coincidental. These periods of high SA concentrations are discussed further in Sect. 3.1. Therefore, we concluded that our analyses on the seasonal cycles of SA, MSA, and IA were not affected by primary pollution in our measurements, and we did not remove any data during periods of local pollution.
2.4 Sulfur dioxide
Ambient SO2 mixing ratios, by volume, were measured in the Swiss container using a Thermo Fisher Scientific instrument (model 43i). The lower limit of detection for this SO2 instrument is 1 ppb. The measured SO2 mixing ratios were adjusted following cross-evaluation against a certified SO2 standard at the Swiss Laboratories for Materials Science and Technology (Empa), and the influence of local pollution was removed. Local pollution affected 62 % of the original SO2 time series during the campaign. For further discussion on the laboratory evaluation and pollution detection and removal in the SO2 dataset, refer to Angot et al. (2022).
2.5 Satellite ocean color data (chlorophyll a)
The 8 d averages of chlorophyll a (chl a) concentrations were downloaded from the Ocean Colour Climate Change Initiative (OC-CCI, http://www.esa-oceancolour-cci.org, last access: 10 November 2022) with a spatial resolution of 4 km. The OC-CCI is a long-term, consistent, and error-characterized dataset generated from merged normalized remote-sensing reflectance data derived from five satellite sensors: MERIS, MODIS Aqua, SeaWiFS, VIIRS, and Sentinel-3A-OLCI (Sathyendranath et al., 2019). Cloudiness, algorithm failure, or low-incidence sun angles in winter can result in missing data in the Arctic; however, overall, there are typically enough cloud-free data available to evaluate chl a in the Arctic region during summer (e.g., Becagli et al., 2016).
2.6 Source region identification using FLEXPART and inverse modeling
To infer the air mass origin, we use the Lagrangian particle model, FLEXPART v10.4 (Pisso et al., 2019), driven by ERA5 reanalysis data. Backward simulations, up to 30 d backward in time, were calculated releasing a cluster of 100 000 air tracer particles every 3 h from Polarstern's location during the MOSAiC expedition. These simulations were then used to determine the “footprint emission sensitivity” (FES), or the emission sensitivity of air masses < 100 m altitude during transport, according to the simulated “source–receptor relationship” (SRR) (Seibert and Frank, 2004). Refer to Fig. S7, taken from Boyer et al. (2023), for the FES observed during the MOSAiC expedition. The FES can then be coupled with an emission inventory to simulate concentrations at the ship, which is an established method to evaluate source regions using to the SRR (e.g., Sauvage et al., 2017). Specifically, we used the ECLIPSE v6b (Evaluating the Climate and Air Quality Impacts of Short-Lived Pollutants) emission inventory and 10 d backward air tracer simulations from FLEXPART to estimate source regions of anthropogenic sulfur from SO2 emissions, as described in Sect. 3.1. For additional details on SO2 in the emission inventory, we refer to Klimont et al. (2013).
Additional source region analyses were performed using an inverse-modeling technique (Seibert, 1998). The inverse model uses the FLEXPART SRR and the measurement time series data of SA, SO2, and MSA from the ship to identify potential source regions and estimate their emissions using the following equation:
where y is the measurement time series data, x is the source term, A is the FLEXPART SRR transport matrix, and n is the error. To simplify the model, we assumed that the source term is constant over time. In addition, we used a clustering technique to reduce the dimensionality of the FLEXPART transport matrix to 1000 groups, as described in Aliaga et al. (2021) and Faletto and Bien (2022). We also applied an elastic net regularization method (Zou and Hastie, 2005; Martinez-Camara et al., 2014, and references therein) using the following equation:
where is the updated source term with penalization applied, y is the measurement time series data, x is the source term from Eq. (2), N is the number of time steps in the time series, α is the regularization parameter that controls the overall strength of the penalty, and ρ is the mixing parameter that balances the contributions of lasso and ridge penalties (described below). A larger value of α results in more regularization and a simpler model with smaller coefficients. The ρ parameter ranges from 0 to 1. When ρ is set to 1, the elastic net model becomes equivalent to a lasso regression. When ρ is set to 0, it becomes equivalent to a ridge regression.
The objective function of the elastic net model (Eq. 3) consists of two terms. The first term is the mean squared error (MSE) loss, which quantifies the difference between the observed and predicted responses. The second term is the regularization penalty, which is a combination of the lasso penalty (L1 regularization) and the ridge penalty (L2 regularization). The lasso penalty promotes sparsity by discouraging non-zero coefficients, while the ridge penalty encourages small coefficients by penalizing large ones. The elastic net penalty is a weighted combination of both penalties and controlled by the mixing parameter α.
A common challenge in applying an elastic net model is selecting the appropriate values for α and ρ. One approach to address this issue is to use cross-validation. Cross-validation is a statistical technique used to assess the performance of machine-learning models. It involves dividing the dataset into multiple subsets, training the model on some of these subsets, and evaluating it on the remaining subsets. This process is repeated multiple times, and the average performance is computed to provide a more reliable estimate of the model's performance.
In our case, we use cross-validation to determine optimal values of α from an initial list of ρ values (0.1, 0.5, 0.9, 0.99, 0.9999, and 1). We then construct a matrix of potential solutions by multiplying the optimal values of α obtained before by the following list (L): (, , , 1, 2, and 4). This results in a matrix of 36 footprint maps, with rows determined by the ρ values and columns by L × α. Based on prior knowledge of possible source regions, we select one of the source region maps that identifies probable source regions while minimizing noisy regions. Refer to Figs. S8, S9, and S10 for the source region footprint maps for SO2, SA, and MSA obtained by the elastic net model in this study. The result is a map of potential source contributions, much like an emission inventory. The calculation of these emission fields in the inverse model used the FLEXPART FES air tracer from 1, 7, 10, and 30 d simulations backward in time, where the shorter simulations were given more weight in the estimated emissions. Using this map of estimated emissions and the FLEXPART FES, the model can simulate measured concentrations at the ship, in a similar way as using the FES coupled with the emission inventory.
Then, the inverse model identifies source region clusters, represented as polygons on a map, that are sorted by their relative contribution to the simulated concentrations. To filter noise, we only considered clusters where the annual mean source contribution was > 5 % relative to all clusters for further analyses. Adjacent clusters were grouped to simplify the interpretation of the results. Finally, we obtained a time series of simulated influence from the source region polygons, which we used to provide qualitative insights into the seasonal contributions from different source regions.
It is important to note that the inverse model assumes a constant emission rate for each identified source region and does not include chemical processing during transport, which would have to be simulated by FLEXPART rather than the inverse model. While this approach simplifies the implementation of the inverse model, it may not describe the true nature of gas phase emissions from the various source regions. Due to the constant emission limitation, the MSA inverse model simulations were carried out from March–September, the time during which we expect DMS emissions that lead to the observed maximum MSA concentrations. Since the emission of iodine species necessary for the formation of IA are highly variable, the assumption of constant emissions cannot be made. Therefore, we do not present inverse model results for IA.
2.7 Global radiation and temperature
Global radiation, or short-wave downwelling radiation, was measured using a pyranometer (Kipp & Zonen, CM11), and ambient air temperature was measured using a Vaisala HMP155. Both sensors were installed on R/V Polarstern and operated continuously during the MOSAiC campaign. The global radiation and the temperature sensors were located 34 and 29 m above the sea surface, respectively.
In this section, the annual cycle of various precursor vapors is presented and discussed. The monthly median concentrations of SA, MSA, and IA – vapors that are known to participate in aerosol formation in remote marine environments – are presented in Fig. 1a. The time series of other parameters that provide context to the gas phase species, including temperature, global radiation, the ship's latitude, and satellite observations of chl a concentrations in seawater are presented in Fig. 1b and c. We also present and discuss the measured SO2 mixing ratios in Fig. 3. These datasets offer unique insights into the seasonal variability in these vapors during the MOSAiC expedition in the high Arctic. Given the high latitude of these measurements and the changes in global radiation during the year (Fig. 1b), the seasonal cycle that we present is essentially a cycle between polar day and polar night. Herein, we give an in-depth discussion on the annual cycle of each species individually.
3.1 Sulfur dioxide and sulfuric acid: anthropogenic and biogenic sulfur species
The median SA concentration observed during the year was 5.1 × 105 molec. cm−3 (SD = 6.1 × 106 molec. cm−3), but most notably, our measurements show a large increase in SA concentrations from January through April, where the median SA concentration (avg = 9.7 × 105 molec. cm−3; SD = 9.4 × 106 molec. cm−3) is approximately twice the annual median (Fig. 1a). We also observed short-lived spikes of SA concentrations up to 1 × 108 molec. cm−3 in January and February (Fig. 2). In general, the monthly median concentrations agree with campaign observations reported from land-based sites in the Arctic (Beck et al., 2021) and sub-Arctic (Jokinen et al., 2022), and the short-lived spikes of SA in January and February are similar in magnitude to concentrations observed during local pollution events related to traffic emissions in Helsinki, Finland (Okuljar et al., 2021; Thakur et al., 2022).
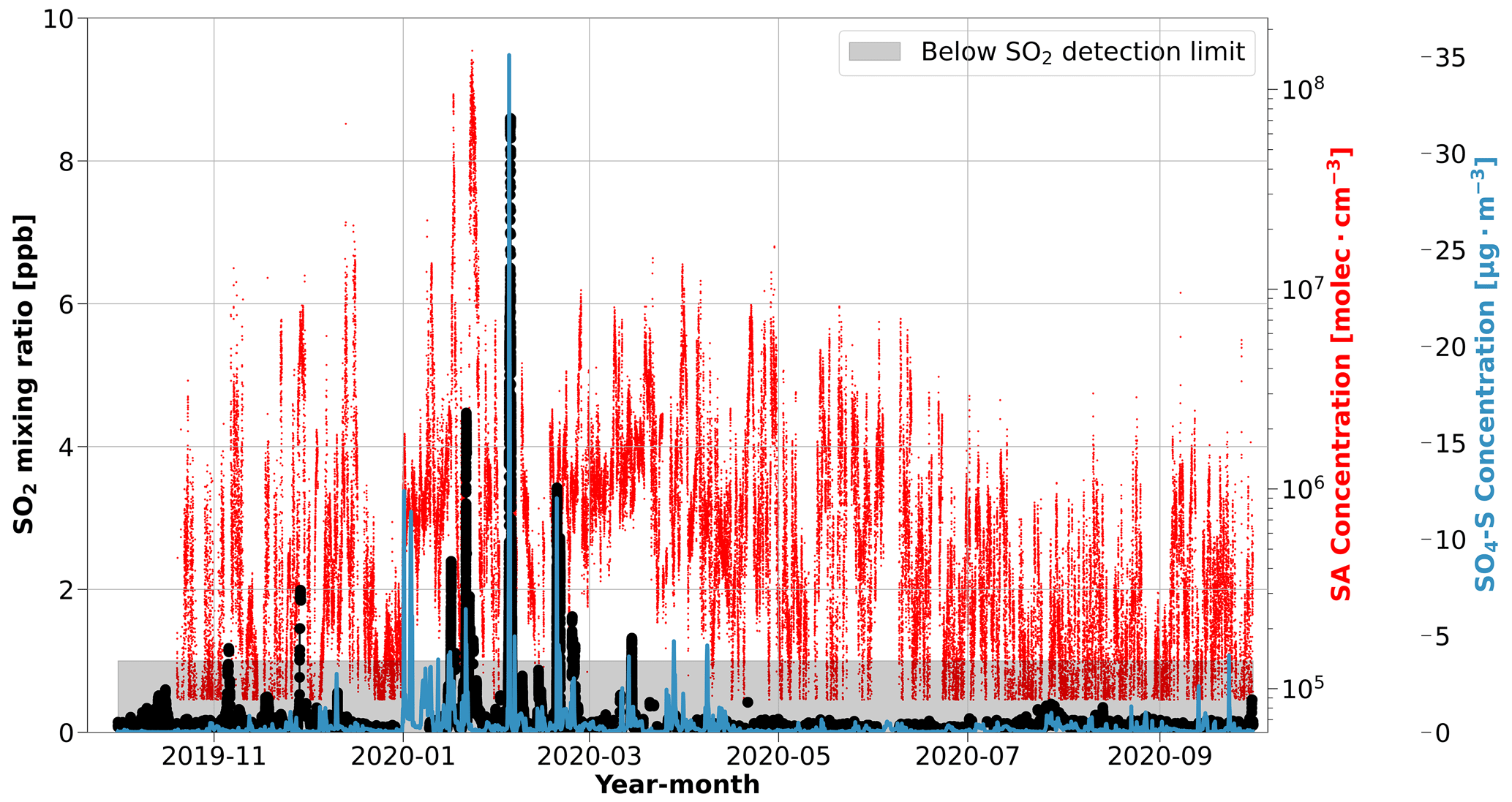
Figure 2The seasonal cycle in SO2 mixing ratios compared to SA during MOSAiC. The SO2 mixing ratio (black circles) is presented as a rolling 10 min median. The shaded gray area identifies the data below the detection limit of the SO2 instrument (1 ppb). The time series of SA concentration (red line) and the simulated mass concentration of sulfate in the aerosol phase (blue line) at the position of Polarstern from FLEXPART coupled to the ECLIPSE v6b emission inventory (SO4-S) are included to highlight the consistent behavior between the sulfur species. For the relative location of Polarstern that corresponds to these measurements, refer to Figs. 1c and S1.
The high concentrations in winter and spring (January–April) indicate a significant source of SA, even during the cold and light-limited central Arctic environment when biological processes are largely absent (Fig. 1c). The periods of high SA concentrations in winter and spring coincide with the occurrence of Arctic haze in the aerosol size distribution during MOSAiC that was driven by a strong positive phase of the Arctic Oscillation (AO) (Boyer et al., 2023; Lawrence et al., 2020), which suggests that Arctic haze, or anthropogenic pollution, is a key source of the high SA concentrations in winter and the dominant source of SA during the annual cycle. Indeed, previous analyses have identified that sulfur/sulfate is a key component of Arctic haze (e.g., Barrie, 1986; Quinn et al., 2007, 2009; Gong et al., 2010; Schmale et al., 2022).
SO2 is the primary gaseous sulfur species that, once emitted, can form SA in the atmosphere. As a result, we can use the time series of SO2 mixing ratios to provide further insight into the sources of SA in the Arctic (e.g., Dada et al., 2020). Comparing the SA data with SO2 mixing ratios leads to a similar conclusion during the winter months: Arctic haze. Throughout most of the year, the SO2 mixing ratio is below the detection limit of the instrument, which is 1 ppb. Only 0.6 % of the ship exhaust pollution filtered SO2 dataset is above the limit of detection during the entire campaign. The few occasions that we observe SO2 mixing ratios above the detection limit occurred between November–March, especially during January and February (Fig. 2). SO2 mixing ratios below the detection limit were not considered in our analysis but were included in Fig. 2 to show the continuity of the SO2 measurements during the campaign. The high SO2 mixing ratios occur during the same time of year that SA concentrations reach their highest values, which is again consistent with the timing and occurrence of Arctic haze. Further discussion of these SO2 plumes, which are associated with episodic warm- and moist-air-mass intrusion events, is given in Angot et al. (2024). It is worth noting that the photochemical conversion of SO2 to SA and the details of the sulfur chemistry is beyond the scope of this work and is not explored further.
To further evaluate the sources of SO2 and SA in our measurements, we examined source regions of anthropogenic sulfate in the aerosol phase (SO4-S), which were simulated using the ECLIPSE v6b emission inventory coupled with the FLEXPART simulations. Since the conversion of SO2 to sulfate aerosol usually occurs on the order of hours in the atmosphere (Lee et al., 2011), FLEXPART treated anthropogenic SO2 emissions as SO4-S, yielding the SO4-S-weighted influence from anthropogenic sources. As such, SO4-S is useful to interpret the sources of SO2 and infer the sources of SA, especially for distinguishing between anthropogenic and natural sources. The simulated SO4-S concentrations are included in Fig. 2 for comparison with the SA and SO2 time series. Overall, the SO4-S simulations peak during the same time of year as the measured gas phase sulfur species, especially in January and February, when we observed temporal spikes of each species (Fig. 2). The geographic regions associated with the SO4-S concentrations, determined by applying a geographic mask to the simulations, are presented in Fig. 3. Refer to Fig. S11 for a more detailed description of anthropogenic SO4-S emissions from each source region in the emission inventory. The results suggest that northern Asia dominates the anthropogenic emissions of sulfur species during January and February. This supports our conclusions; anthropogenic sulfur emissions have a strong influence in this region, as shown in the satellite-derived emission inventory of SO2 described by Liu et al. (2018). The sulfur emissions in this region predominately originate from smelters in Norilsk, Russia, that are identified as strong point source emitters (Khokhar et al., 2008), which is consistent with the SO4-S loading in the emission inventory from northern Asia in Fig. S11.
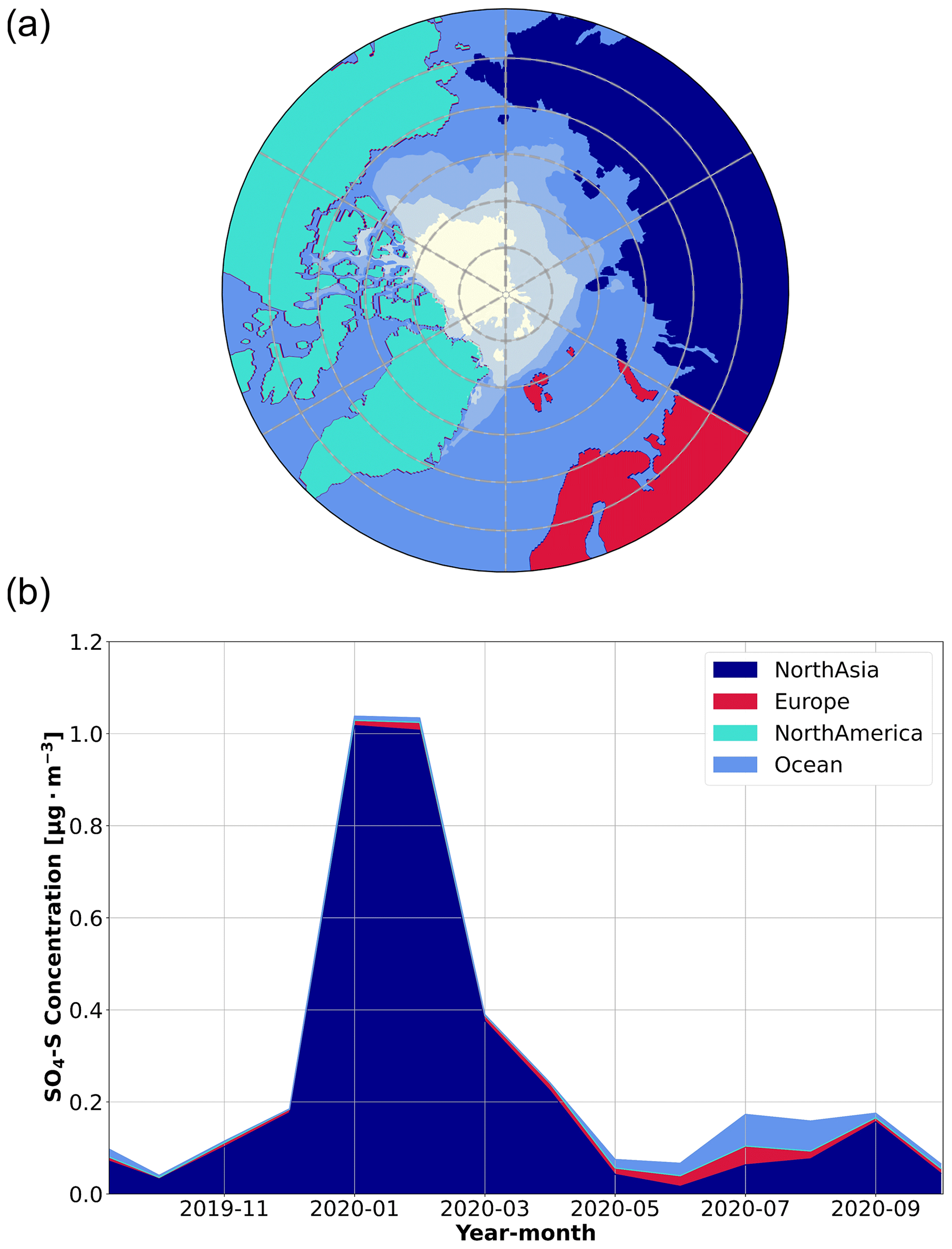
Figure 3Simulated source regions of anthropogenic SO4-S concentrations. A source region mask (a) was used to identify the contribution of each source region to the SO4-S concentrations in the aerosol phase (b), using the FLEXPART FES and the ECLIPSE v6b emission inventory. The SO4-S concentrations are presented as monthly averages according to source region. Note that while the general sea ice coverage is presented in the source region mask as the white shaded area, there are no SO4-S emissions associated with this region to show in panel (b). Emissions associated with the ocean region are due to anthropogenic emissions from ships. A description of the average SO4-S contributions from each of the regions during the entire year is provided in the Supplement to show the specific spatial distribution of the sources in the emission inventory (Fig. S11).
The inverse-modeling analysis for SO2 (Fig. 4) identified three regions as potential contributors to SO2, which we attribute to north Asia/Siberia, Europe, and the Aleutian Peninsula in Alaska. Of these three sources, north Asia/Siberia is dominant. More specifically, the dominant source region, highlighted by polygon (a) in Fig. 4, agrees well with the prevalent source of atmospheric sulfur in the emission inventory near Norilsk, Russia (Fig. S11). Therefore, these results again demonstrate that the smelter region in Norilsk, Russia, exerts a significant influence on the concentrations of SA and SO2 in the central Arctic during the Arctic haze period (Bauduin et al., 2014; Hirdman et al., 2010). These results agree with Sipilä et al. (2021), who observed that SO2 pollution from smelters in Kola Peninsula, Russia, contributes to a wintertime source of SA even during the low-light conditions of winter. The timing of our wintertime observations of SO2 and SA is also consistent with aerosol observations from the MOSAiC expedition that show an early peak in Arctic haze pollution from Eurasia during January–March 2020 that is linked to an extremely positive phase of the Arctic Oscillation index (Boyer et al., 2023; Lawrence et al., 2020). It is known that these conditions lead to high pollution in the Arctic (Eckhardt et al., 2003). Refer to Angot et al. (2024) for further analysis of these episodic pollution events associated with high-SO2 mixing ratios during the polar night on the MOSAiC expedition.
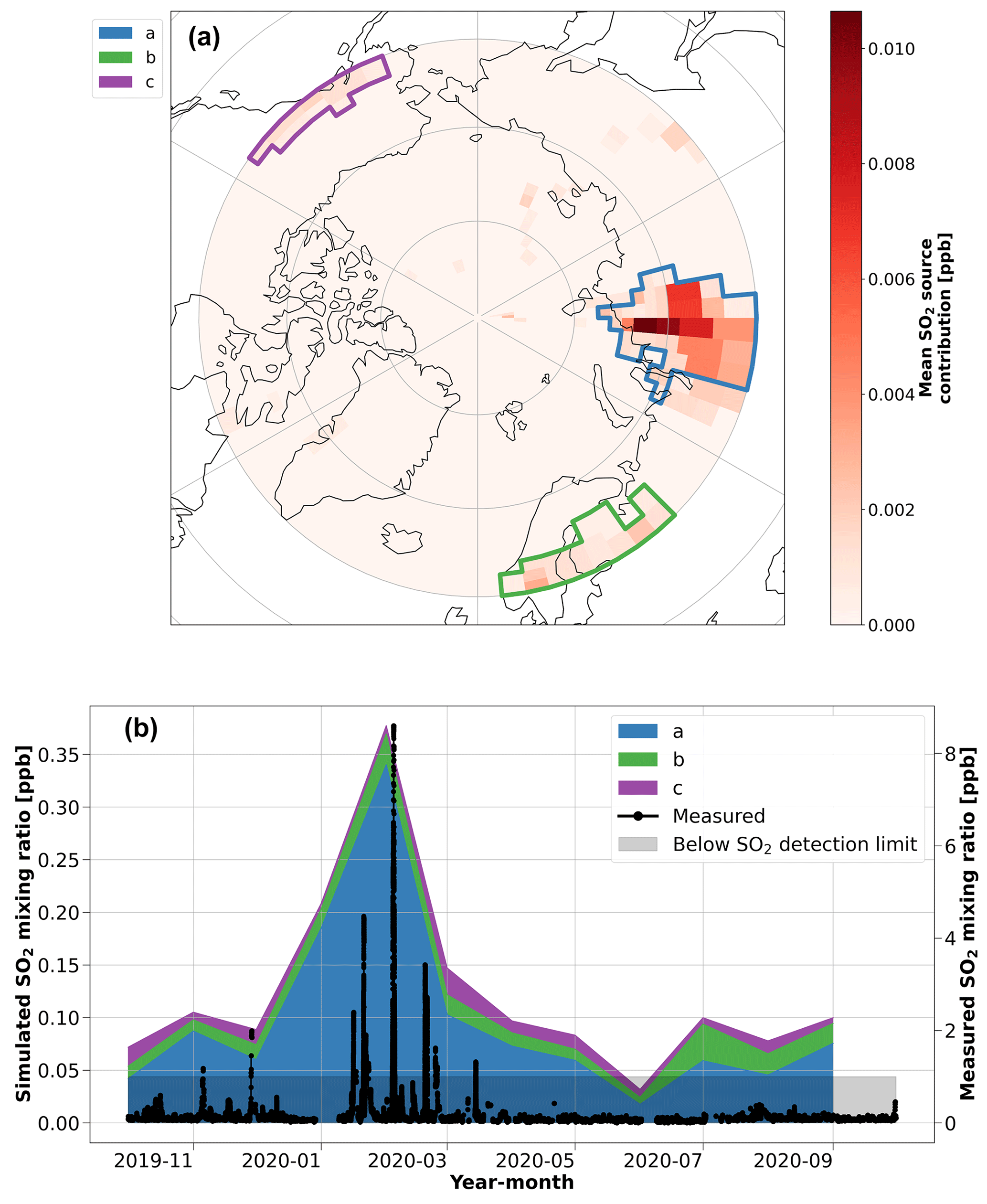
Figure 4Inverse model results for SO2. (a) The potential source regions identified using the FLEXPART air tracer data and the ambient SO2 mixing ratio in the inverse model. The red color bar shows the simulated annual mean contribution of the source regions to the SO2 mixing ratio during the year of the MOSAiC expedition. The colored polygons show the source region clusters that were identified to have a significant contribution (> 5 % annual average) on the simulated SO2 concentrations during the year, which are used in the time series analysis. (b) A time series of the monthly median SO2 mixing ratio contributions from the identified source regions, as simulated by the inverse model. The measured SO2 mixing ratio, presented as a rolling 10 min median during the year, is included on the right axis for context. Note that the shaded area shows the regions where the SO2 measurement data are below the detection limit.
Despite lower anthropogenic SO4-S influence from the north Asia sector during March and April (Fig. 3), the SA concentration remained high and even reached an annual maximum monthly median concentration of 1.36 × 106 molec. cm−3 during March (Fig. 1a). This result suggests that there is another source of the high SA observed during these months. As such, the inverse model was used to identify potential source regions for SA in March and April (Fig. 5). The inverse model results for SA also show that contributions from north Asia/Siberia (polygon a) decline in March and April; however, other source regions become more influential during this time. The more influential source regions correspond to polygons (b) and (c) in Fig. 5a, which we attribute as the Aleutian Peninsula in Alaska and the Kamchatka Peninsula in Russia, respectively, which are both regions with volcanic activity. Polygon (b) likely also includes anthropogenic emissions from oil fields in Prudhoe Bay, Alaska. These source regions are consistent with major point source emissions of SO2 (Fioletov et al., 2020) and hence SA production via photochemistry. The polygon associated with Alaska also agrees with the inverse model results for SO2 in Fig. 4, which also identifies potential contributions from the Aleutian Peninsula region during March. It is also expected that the appearance of sunlight in the central Arctic during March and April contributes to enhanced oxidation of SO2 to SA during these months compared to winter, resulting in the peak SA concentration in March.
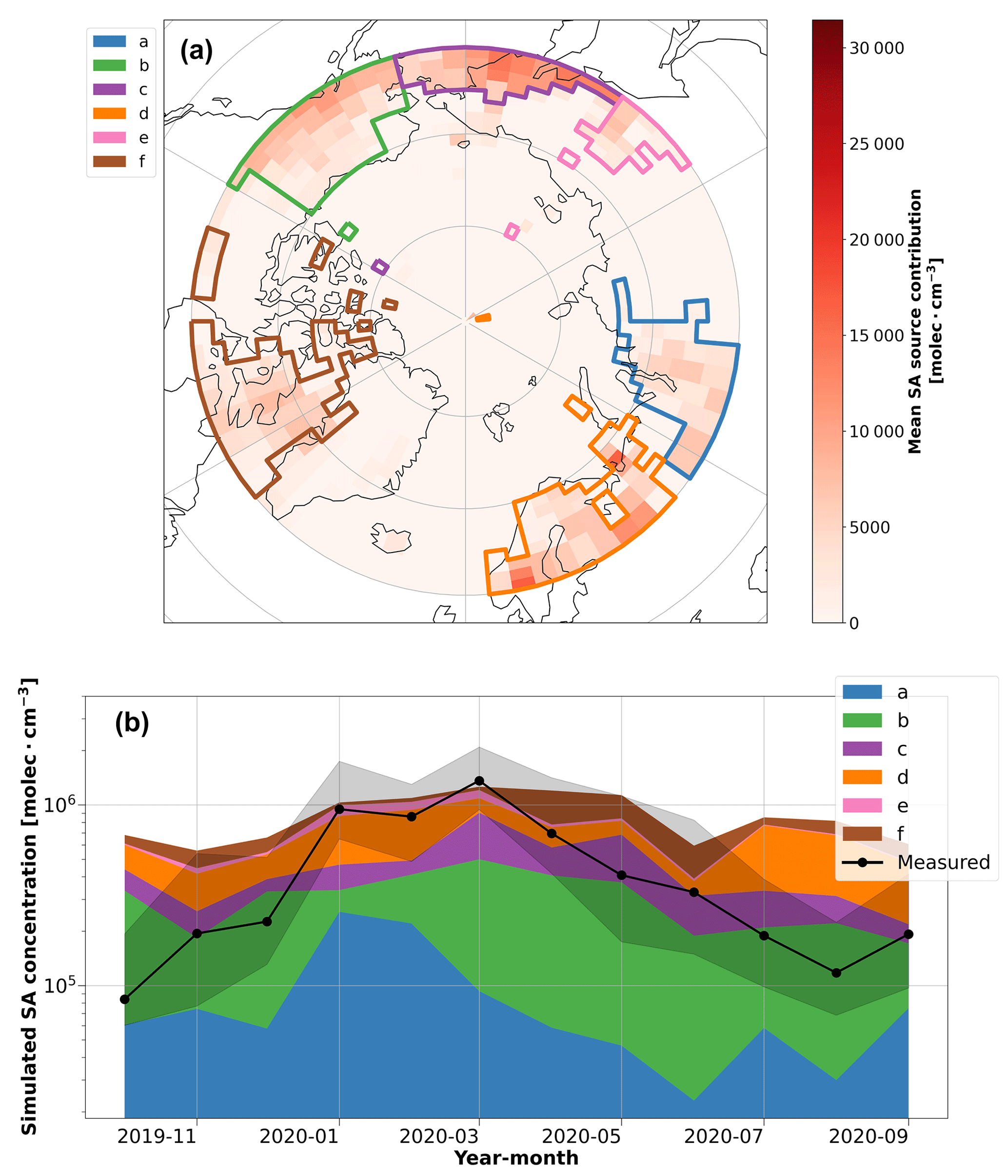
Figure 5Inverse-modeling results for SA. (a) The potential source contribution map of SA simulated by the inverse model. The red color bar shows the annual average of the potential source region contributions. The colored polygons show the source region clusters that were identified to have a significant contribution (> 5 % annual average) on the simulated SA concentrations during the year, which are used in the time series analysis. (b) The simulated SA concentration time series according to the potential source region clusters identified by the inverse model. The time series shows the potential contribution of each source region polygon to SA concentrations throughout the year. Note that the source areas here are different from those shown in Fig. 4.
SA is also a product of DMS oxidation, a process that requires biological activity and sunlight (i.e., during polar day); however, we do not observe enhanced SA concentrations in the summer. Instead, we observed that winter (i.e., polar night) and spring dominate the annual cycle of SA in the central Arctic. According to our measurements, the median SA concentration during the Arctic haze period (January–April) is between 3–4 times higher than the median SA concentration during periods of peak biological activity (May–July). This observation is contrary to typical patterns of SA behavior, where SA concentrations peak during daytime when global radiation is high (Baccarini et al., 2021; Quéléver et al., 2022). We can primarily attribute this difference in our measurements to the seasonal variation in sinks in the central Arctic. In winter and spring, Arctic haze builds up because there is little precipitation during air mass transport, leading to the accumulation of SA. In contrast, there is more precipitation during summer, which limits northward transport of anthropogenic pollution (Klonecki et al., 2003). Based on this, our results show that SA concentrations from DMS emissions in summer are small in magnitude compared to the anthropogenic sulfur sources from Arctic haze in spring. In addition, the cold air temperatures in the central Arctic during the year (Fig. 1b) suggest that DMS oxidation likely favors the production of MSA rather than SA during summer (Shen et al., 2022), which could also help explain the lack of elevated SA concentrations during periods of higher biological activity over the summertime central Arctic Ocean (Fig. 1c).
SA has a well-known role in NPF processes (Dunne et al., 2016; Gordon et al., 2017; Jokinen et al., 2018; Kulmala et al., 2013; McMurry and Friedlander, 1979), and hence, higher concentrations in the central Arctic atmosphere could have a notable impact on aerosol-related phenomena in the region, especially during summer months when SA concentrations are currently relatively low. Increased SO2 emissions from ships could become a source of SA in the central Arctic as ship traffic could become more frequent with less sea ice extent (Ferrero et al., 2016); however, it is unclear how future shipping emissions might impact SA concentrations. For instance, when primary pollution from Polarstern influenced our data, we observed SO2 concentrations exceeding 10 ppb, while only 0.6 % of the not-Polarstern-influenced SO2 data were above the detection limit of the instrument (1 ppb). The periods with data above the detection limit only occurred in the winter months, as described above. In addition, recent observations show that SO2 emissions, and subsequently SA, can lead to the formation of new particles even during low-light conditions in winter/spring in the Arctic (Sipilä et al., 2021). Our observations highlight the combined influence of both natural and anthropogenic sources of atmospheric SA during the year, where the highest concentrations occurred in winter/spring.
3.2 Methanesulfonic acid: an oxidation product from biological activity
We observed a minimum MSA concentration during the fall and winter months, particularly from October through February, with a median concentration of 2.9 × 105 molec. cm−3 (SD = 4.8 × 105 molec. cm−3) throughout this period (Fig. 1a). A clear and consistent increase was observed in the monthly MSA concentrations starting in March, which is when the global radiation starts to increase in the high latitudes of the Northern Hemisphere (Fig. 1b). The MSA concentration continued to rise after March, reaching a maximum monthly median concentration of 3.3 × 106 molec. cm−3 in May, followed by high concentrations through August, with a median concentration of 1.5 × 106 molec. cm−3 (SD = 3.0 × 106 molec. cm−3) from June through August. The concentration then decreases again in September, completing the annual cycle. In general, our observed MSA concentrations are similar in magnitude to those reported previously in Ny-Ålesund, Svalbard, Norway, especially during the annual peak in May–June (Beck et al., 2021).
Chl a, which is a well-known tracer of biological activity (e.g., Park et al., 2013), was used to evaluate the connection between DMS emissions (and subsequent formation of MSA) with biological activity in the ocean. While chl a does not provide a direct measure of DMS production (Stefels et al., 2007; Uhlig et al., 2019), we used the chl a concentrations to approximate the timing of biological activity in ocean regions between 60 and 90° N. The chl a concentrations, presented in Fig. 1c, suggest that the MSA concentrations in the central Arctic are strongly linked to biological activity from regions south of the marginal ice zone, which agrees with previous investigations (Beck et al., 2021). The annual minimum in the MSA concentrations in December and January also corresponds to the lowest chl a concentrations in the Northern Hemisphere. This period of low MSA and chl a concentrations occurs when the biological activity in the Arctic and surrounding ocean is limited due to lack of sunlight. Then, after February, the chl a concentrations start increasing at the lower latitudes, progressing northward during the spring and summer seasons. The MSA concentrations follow the same trend as chl a, reaching a maximum during May when the chl a concentration north of 75° N reaches its annual peak (Fig. 1c). The simultaneous increase in chl a and MSA concentrations is unsurprising, as gaseous DMS, and subsequently MSA, is a product of biological activity in the ocean. The comparison of the chl a and MSA time series also suggests that the MSA concentrations in the central Arctic are influenced by transport from oceanic regions further south. This is particularly clear during March and April when the MSA concentration in our measurements starts to increase despite the observation that chl a concentrations are still low at the northernmost latitudes. The increase presumably occurs due to the onset of biological activity further south combined with transport. This is also consistent with more favorable transport of air masses from southerly locations during spring (Bozem et al., 2019).
While the chl a concentrations provide insight about potential regions of biological influence, they alone cannot describe the MSA observations. The source regions of the observed air masses in the central Arctic would need to correspond with the regions of enhanced biological activity to explain the MSA measured at the ship. Therefore, we coupled the FLEXPART air tracer simulations with the oceanic chl a concentrations to calculate an index that quantifies potential air mass exposure to oceanic regions with biological influence, and hence potential DMS (MSA) emissions. The index, called the sea surface chl a influence index, was obtained by multiplying the residence time (in seconds) of the FLEXPART air tracer (based on 10 d backward simulations) below 100 m altitude with the corresponding chl a concentration maps (in mg m−3). Hence, the index is proportional to the amount of time that the air masses have spent over regions with chl a presence and to the concentrations of chl a encountered in those regions. The sea surface chl a index results are also shown in Fig. 1c, and several maps of individual trajectories are included in Fig. S12 to demonstrate how the index was calculated. As previously stated, we do not expect DMS emissions to correspond directly with chl a, especially since DMS emission are primarily associated with senescent phytoplankton cells and chl a is indicative of healthier cells, but chl a serves as a tracer for air masses with potential influence from oceanic biological activity. The index clearly indicates that the influence of ocean biology increased during the summer, which is generally consistent with the seasonal enhancement in MSA.
We further examined source regions of MSA using the inverse model. The key insight obtained from the inverse model results, shown in Fig. 6a and b, is that regions south of the marginal ice zone appear to be the most influential on MSA concentrations over the central Arctic. More specifically, the inverse model identifies several oceanic regions as potential sources of MSA in our observations, where the Kara, Barents, Norwegian, and Labrador seas are the most prevalent source regions during spring and summer (polygons b, c, and d in Fig. 6a). These regions also agree well with individual trajectories from the coupled FLEXPART and chl a concentration analysis (refer to Fig. S12). Note that due to the limited domain of the FLEXPART simulations (> 60° N), source regions polygons (a) and (f) in Fig. 6a may represent the contribution of MSA transport from regions further south than the polygons depicted on the map, which could be associated with oceanic regions on the western coast of North America and the Bering Sea, respectively. Previous research has shown that the regions identified in Fig. 6a are biologically active or important sources of DMS, the precursor of MSA, from May to August (Hulswar et al., 2022; Lana et al., 2011; Leck and Persson, 1996a; Terhaar et al., 2021), which is consistent with the chl a satellite data and again highlights the importance of air mass transport from biologically active source regions further south on our MSA measurements. Our conclusions also agree with other studies that have also identified significant contributions of DMS to sulfate and MSA in the aerosol phase from biologically productive waters south of the marginal ice zone and surrounding waters (Becagli et al., 2016; Galí et al., 2021; Ghahreman et al., 2016; Ghahremaninezhad et al., 2017; Kurosaki et al., 2022; Leck and Persson, 1996b; Sharma et al., 2012), and our results further suggest that these regions are influential in MSA concentrations over the central Arctic as well.
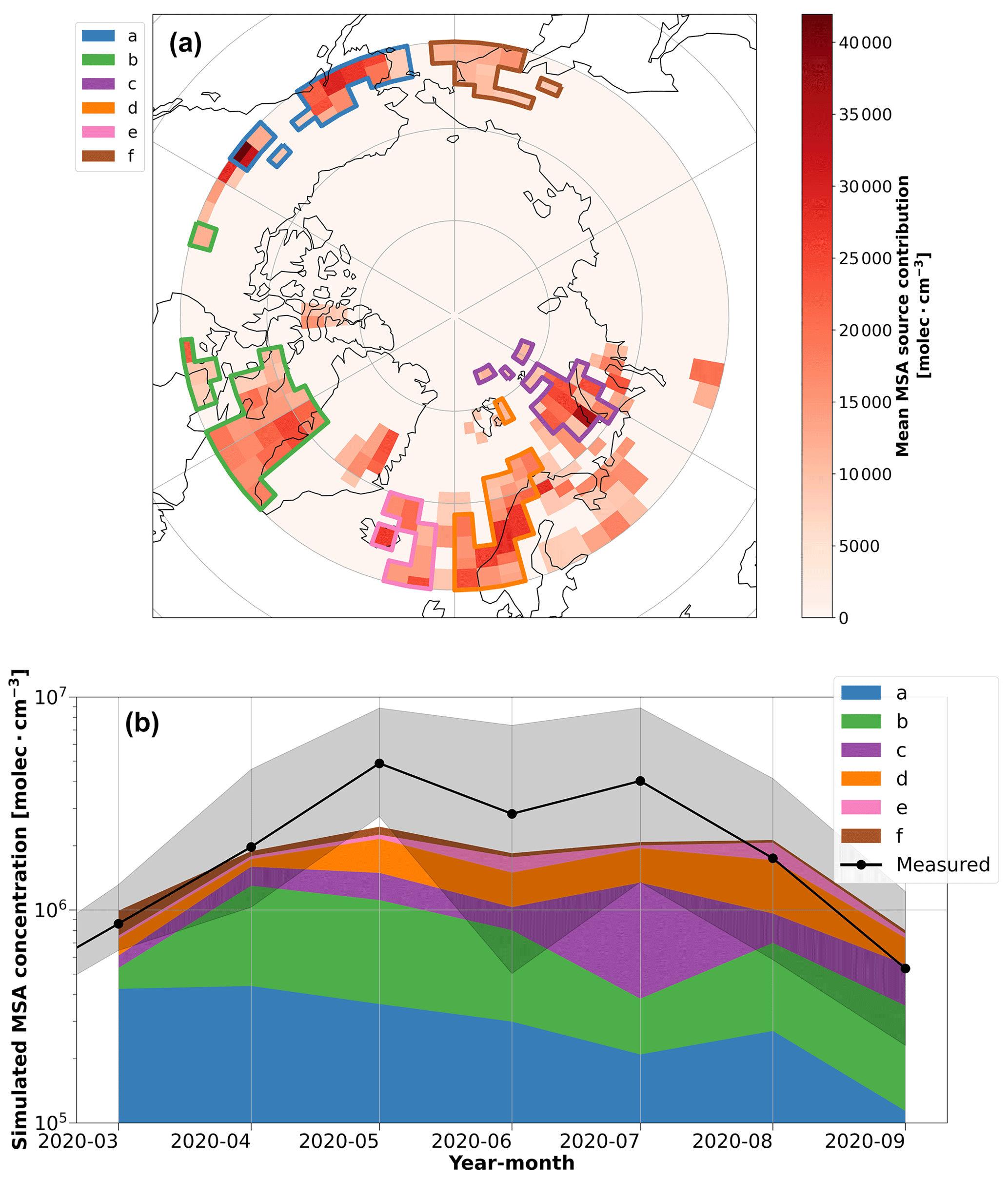
Figure 6Source regions and their contributions to MSA from the inverse model. (a) The potential source contribution map of MSA between March–September, as identified by the inverse model. The red color bar shows the annual average of the potential source region contributions. The colored polygons show the source region clusters that were identified to have a significant contribution (> 5 % annual average) on the simulated MSA concentrations during the year, which are used in the time series analysis. (b) A time series of the simulated monthly median MSA concentrations from the source region polygons identified by the inverse model. The MSA time series measured at the ship is included for context, and the shaded region shows the interquartile range. Due to the limited domain of the FLEXPART simulations (> 60° N), source regions in polygons (a) and (f) may represent the contribution of MSA transport from regions further south than the polygons depicted on the map, such as the oceanic regions on the western coast of North America and the Bering Sea, respectively.
To evaluate if transport from the regions in Fig. 6a is reasonable, we can consider the atmospheric lifetimes of MSA and DMS. Given the particle number size distributions in the central Arctic during MOSAiC (Boyer et al., 2023), we estimate the lifetime of MSA against condensation onto particle surfaces to range from ∼ 0.5 to 3 h in our observations, using the simplifying assumption that MSA condenses irreversibly onto particle surfaces. This lifetime is too short to explain the effect of transport on our MSA measurements. The lifetime of gaseous DMS in the Arctic, however, is longer. The chemical conversion of DMS in the Arctic atmosphere is limited by the presence of available oxidants, and the resulting DMS lifetime is estimated to range from 1–5 d at latitudes < 70° N and 5–20 d > 80° N (Ghahremaninezhad et al., 2019). The inverse model and FLEXPART simulations do not account for chemical processing, but from the chemical lifetime of DMS in the Arctic atmosphere, we can infer that MSA production from DMS occurs on timescales that are consistent with air mass transport into the central Arctic during summer, which is between ∼ 5–15 d (Stohl, 2006). Therefore, we conclude that transport of DMS from the regions > 60° N in Fig. 6a, followed by subsequent chemical processing during transport, could explain our MSA measurements from the central Arctic during MOSAiC, which is also consistent with the convoluted FLEXPART trajectories with the oceanic chl a concentrations on timescales between 1–5 d.
It is important to highlight the influence of transport from ocean regions south of the marginal ice zone on our observed MSA concentrations. We cannot comment on the local sulfur emissions from biological activity below the sea ice; however, there is mounting evidence that a stable meltwater layer forms on exposed leads within the sea ice during the melt season (Rabe et al., 2022; Smith et al., 2023). The meltwater layer could act as a barrier that limits ocean–atmosphere gas exchange (Nicolaus et al., 2022; Smith et al., 2023), hence limiting the release of DMS during the melt season in regions with sea ice coverage. The effect of this meltwater layer is not explored further in this study; however, its occurrence and potential implications for Arctic processes will be evaluated in future studies. There is also evidence to suggest that seawater contact with sea ice limits biological processes associated with DMS production (Uhlig et al., 2019). Therefore, as the summertime sea ice extent continues to decline, emissions of these biologically produced gases that are relevant for secondary aerosol processes could become more prevalent in the central Arctic, leading to large-scale changes in climate-relevant aerosol processes. Indeed, long-term observations of gas phase MSA concentrations in the Arctic show a strong positive association with ambient air temperature (Moffett et al., 2020), and DMS emissions are increasing across the Arctic and are expected to increase further under future scenarios with less sea ice coverage (Galí et al., 2019; Kurosaki et al., 2022).
While MSA emissions are expected to increase in the future, the subsequent changes in aerosol processes are not straightforward. Based on our observations from MOSAiC, we note that periods of high MSA concentrations coincide with the time of year when particles < 100 nm in diameter dominate the aerosol number size distribution (Boyer et al., 2023). Our results agree with previous investigations that have found that enhanced NPF and subsequent Aitken mode sulfate aerosol composition in the Arctic during summer can be directly linked to biogenic sulfur sources (i.e., MSA) (Abbatt et al., 2019; Ghahreman et al., 2016; Leaitch et al., 2013; Willis et al., 2017) caused by retreating sea ice (Dall'Osto et al., 2017). On the other hand, several other studies have observed a decrease in aerosol phase MSA in the Arctic despite increases in DMS emissions (Moffett et al., 2020; Schmale et al., 2022; Sharma et al., 2019), suggesting that higher gas phase MSA concentrations do not directly lead to increased MSA mass in particles.
The discrepancy among these observations may be due to the fate of MSA in aerosol processes in the marine atmosphere, which are complex and not yet fully understood (e.g., Hodshire et al., 2019). MSA can condense onto aerosol surfaces to grow nucleation and Aitken mode particles (Beck et al., 2021); however, the partitioning of MSA to the particle phase is linked to aerosol acidity and relative humidity (Baccarini et al., 2021; Dada et al., 2022, and references therein), which varies over time and space across the greater Arctic region (Fisher et al., 2011). Once in the aerosol phase, secondary particles containing MSA are sufficiently hygroscopic such that they may enhance CCN concentrations in the summertime Arctic atmosphere, which can experience periods of CCN-limited conditions (Mauritsen et al., 2011). As a result, particles containing condensed MSA may be effectively removed by wet deposition during regional transport in the Arctic, which is supported by recent field and modeling studies (Mahmood et al., 2019; Pernov et al., 2022). Conversely, multiphase chemistry on aerosol and cloud droplet surfaces has also been demonstrated as an important process in the formation of gas phase MSA (Baccarini et al., 2021; Hoffmann et al., 2016). Moreover, the wet deposition processes that remove biogenic-sulfur-containing aerosols may also result in periods of very low particle concentrations during summer in the Arctic, creating favorable conditions for NPF, a process that Pernov et al. (2022) reported to correspond with increased gas phase MSA concentrations in their observations from Greenland. These aerosol processes involving MSA likely vary temporally and spatially across the Arctic region, and therefore, it is necessary to continue monitoring the concentrations of MSA, in both the gas and aerosol phase, to resolve its contribution to the aerosol, CCN number concentrations, and ultimately the surface energy budget in the central Arctic as sea ice declines. Note that these aerosol processes involving MSA are beyond the scope of the seasonal analysis presented here; however, future work should aim to investigate the role of MSA in event level analyses of the mechanism of NPF and aerosol chemistry in the central Arctic region.
3.3 Iodic acid: a halogen gas phase aerosol precursor
Figure 1a shows the annual record of IA concentrations during MOSAiC. Two significant peaks in IA concentration were observed during the year, the largest of which occurred in spring followed by a secondary peak in early autumn. Peak monthly median IA concentrations of 2.1 × 106 and 3.7 × 105 molec. cm−3 were measured in May and September, respectively. In contrast, concentrations in late fall and winter were lower, with an average concentration of 9.7 × 104 molec. cm−3 (SD = 1.3 × 105 molec. cm−3) from October to February. Interestingly, there is also a low median concentration of IA during July (1.1 × 105 molec. cm−3), which is similar to the low concentrations observed during late autumn and winter. The seasonal cycle in IA concentrations correspond with the findings of Sharma et al. (2019), who observed two peaks in iodine constituents in the aerosol phase between March–May and August–September, using a long time series of aerosol filter measurements at Alert, Canada. Moreover, our IA concentrations agree well with the range of concentrations reported at both Villum Research Station, Greenland, Denmark and Ny-Ålesund, Svalbard, Norway, especially the peak observed during spring (Beck et al., 2021; Sipilä et al., 2016). The IA concentrations measured during the autumn peak also agree with the findings of Baccarini et al. (2020) during a research cruise in the central Arctic in 2018; they observed monthly median IA concentrations of 8.6 × 104 molec. cm−3 (SD = 1.2 × 106 molec. cm−3) and 6.3 × 105 molec. cm−3 (SD = 1.8 × 106 molec. cm−3) during August and September, respectively (Baccarini et al., 2020).
The early spring peak of IA concentration, the largest during the year, coincides with increasing solar radiation that starts during March in the central Arctic (Fig. 1b). There are two possible explanations for this peak in spring, from biotic and abiotic processes, both of which are driven by the appearance of solar radiation. When solar radiation increases, it boosts the photolysis of molecular iodine, as well as the biological activities of microalgae and phytoplankton, which are known sources of iodine (Allan et al., 2015; Ashu-Ayem et al., 2012; O'Dowd et al., 2002). Interestingly, the increasing IA concentrations occur during the same period of increasing MSA concentrations between March and May. As previously discussed in Sect. 3.2, increasing MSA concentrations during this time were associated with the onset of biological activity at high latitudes, which might suggest a link between IA and biological activity as well (Fig. 1c). However, the types of organisms that produce DMS (MSA) are different than those that produce iodine (IA), and the chemical mechanisms governing the production of MSA or IA differ as well. Thus, the concurrent increase in MSA and IA during spring may be coincidental and more generally associated with the appearance of solar radiation during this time of year in the high Arctic. We must also consider abiotic processes that could contribute to the spring peak in IA. The introduction of sunlight in early spring initiates heterogenous photochemistry and emission of iodine compounds from sea salts deposited on sea ice/snow surfaces by blowing snow or the upward migration of brine through the sea ice (Domine et al., 2004; Raso et al., 2017; Spolaor et al., 2019), which may proceed even with very low levels of light (He et al., 2021). Therefore, it is possible that the large increase in IA concentrations in March–May, when solar radiation returns to the Arctic, includes both biotic and abiotic processes. Sharma et al. (2019) also proposed that the iodine peaks in the aerosol constituents result from both biogenic activity and photochemical iodine processes. We are unable to resolve the contribution of these processes with the seasonal analysis presented herein.
Our observations suggest that IA concentrations are also strongly linked to seasonal changes in sea ice conditions. We observed that IA concentrations decrease during summer, possibly due to thinning of the sea ice and the reduced brine layer (Saiz-Lopez et al., 2015). It is likely that the warming of the sea ice surface in the early season promotes the emission of iodine by restructuring the brine channel network and promoting the diffusion of iodine species to the surface through increases in the volume of brine veins but only up to the point where the brine vein network breaks down due to more advanced stages of melting resulting in unfavorable conditions for the emission of reactive iodine precursors (Saiz-Lopez et al., 2007). This process suggests that abiotic processes could be important during the spring peak in IA concentrations and may also explain the low IA concentrations observed between June and August, which corresponds to the annual peak in ambient air temperatures, global radiation, and sea ice melt. Additionally, it is important to consider the parallel competitive consumption of O3 for HIO3 formation (Finkenzeller et al., 2023; He et al., 2021), as well as by other reactive halogens, such as chlorine or bromine, that are well-known actors of ozone depletion phenomena at the poles (Barrie et al., 1988; Benavent et al., 2022; Pratt, 2019). On the other hand, bromine emissions are most active in the upper layer of the snowpack that is thinning and disappearing well before the rest of the ice pack during the melting season (Custard et al., 2017).
In addition, we propose that the secondary peak in IA during the year, which occurred at the end of the summer melt season, is also associated with seasonal changes in sea ice processes and solar radiation. During this time of year, global radiation and ambient air temperature decline in the central Arctic (Fig. 1b), and the sea ice undergoes various freeze–thaw cycles. These freeze–thaw cycles could cause restructuring of the brine channel networks in a similar way as in the spring. The freezing onset during MOSAiC occurred during late August and early September, which corresponds with the timing of the secondary peak in IA concentrations. Baccarini et al. (2020) also proposed that the increase in the IA concentration that they observed in the fall was linked to elevated O3 concentrations, combined with the formation of new sea ice during the freezing onset observed in late August. Additionally, several other studies have shown that ozone enhances the emission of iodine from saline surfaces undergoing multiple freeze–thaw cycles during the autumn transition period (Abbatt et al., 2012; Carpenter et al., 2013; Halfacre et al., 2019). Moreover, the diurnal cycle of IA observed in Antarctica and over the Southern Ocean suggests that solar irradiance plays a role in atmospheric IA concentrations; higher IA concentrations were favored during periods of lower solar irradiance in the early morning and evening compared to midday (Jokinen et al., 2018; Baccarini et al., 2021). We also observed that IA reaches its maximum concentrations during the months where sunlight returns to the Arctic and freeze–thaw cycles occur. Such a result suggests that emissions from the sea ice and ocean regions are influential on the IA concentrations during seasonal transitions in sea ice processes and under conditions with low solar radiation during the seasonal transitions from polar day/night, which further supports the results of these previous studies. However, our analysis focuses on the seasonal cycle, which is not sufficient to resolve the relative contributions from these processes on IA concentrations. As such, atmospheric iodine processes, especially in the Arctic, require further investigation. A more detailed analysis of atmospheric IA formation mechanisms during the MOSAiC expedition will be given in a separate study.
IA has a demonstrated role in NPF (Allan et al., 2015; Sipilä et al., 2016). Baccarini et al. (2020) also identified that IA has an important role in NPF processes in the central Arctic, particularly during autumn when the sea ice refreezes. In contrast, another study of the mechanism of NPF at two Arctic sites identified different particle formation pathways that were dependent on season and location. The study, conducted by Beck et al. (2021), showed that IA, SA, MSA, and ammonia were all identified to play different roles in the NPF process due to changes in the surrounding environment in Greenland and Svalbard. IA-induced NPF was found to be the most relevant pathway in Greenland during spring due to its proximity to sea ice, whereas NPF proceeded with the participation of SA, MSA, and ammonia during the summer months in Greenland and Svalbard (Beck et al., 2021). During MOSAiC, the peaks in the IA concentration that we observed in spring and fall correspond with the results of both Baccarini et al. (2020) and Beck et al. (2021), showing that IA NPF occurs during seasonal transitions in sea ice processes while near the sea ice. On the other hand, previous observations show that secondary particles in the nucleation and Aitken modes dominate the aerosol size distribution throughout summer (Boyer et al., 2023; Collins et al., 2017; Croft et al., 2016; Freud et al., 2017; Pernov et al., 2022; Tunved et al., 2013), even during the months where IA concentrations are relatively low (e.g., July). This observation allows us to speculate that IA is not the only compound forming particles in the central Arctic during all seasons and that the chemistry of clusters and newly formed particles is more complex, as witnessed at land-based stations by Beck et al. (2021) (Schmale and Baccarini, 2021). The chemical mechanisms of NPF observed during MOSAiC will be evaluated in a dedicated study.
In this study, we present the annual cycles of SA, MSA, and IA measured in the central Arctic during the MOSAiC expedition. These measurements represent the first continuous annual time series of these aerosol precursor vapors ever collected over the sea ice in the central Arctic Ocean (north of 80° N), which offers new insights into their seasonal cycles.
Our results show the influence of both natural and anthropogenic sources on SA concentrations. Most notably, we show that these sources yield the highest SA concentrations in winter and spring, associated with Arctic haze and enhanced transport from continental sources further south. Comparatively, DMS oxidation from biogenic sources contributes less to SA concentrations during summer. Localized anthropogenic emissions in Siberia/northern Russia, especially from the region of Norilsk in northern Russia, contribute substantially to our observed SA concentrations during winter. Natural sources of atmospheric sulfur, including volcanically active regions, also contribute to high SA concentrations during spring when the transport of air masses from continental regions remains favorable and solar radiation increases in the Arctic region. Processes controlling SA concentrations are subject to change as the Arctic becomes more accessible as sea ice continues to decline and anthropogenic activities, such as ship traffic, become more common in the central Arctic throughout the year.
Our analyses additionally show that biological activity in the open-ocean areas south of the marginal ice zone within the Arctic region contributes to enhanced MSA concentrations, an important component of aerosol formation and growth, during late spring through summer. The timing of the annual maximum in MSA corresponds to elevated chl a concentrations north of 75° N during May and June. Transport from regions south of the marginal ice zone appear to be the primary driver of MSA concentrations in our observations over the sea ice. We do not expect that biological activity in the surface ocean within the sea ice pack is a strong source of MSA concentrations during the summer, but this may be subject to change in the future as sea ice continues to decline. MSA concentrations may increase as a result.
We observed peaks in atmospheric IA concentrations during seasonal transitions in spring and fall. IA has an apparent source from sea ice thawing–freezing processes during periods with low solar irradiance, which is in concordance with previous observations of IA. As there are iodine compounds sourced from marine biological activities, the spring IA peak could be partly explained by the increased production of algae (from both the sea and the ice) with the appearance of sunlight. In addition, the thinning sea ice could facilitate the exchange of iodine into the atmosphere and further reaction with O3 to form IA. Once solar radiation intensity increases and sea ice experiences more advanced stages of melting; however, the concentrations of IA decline. Therefore, we suggest that the peak IA concentrations may have biotic and abiotic origins that are strongly linked to low levels of solar radiation; however, future work is necessary to resolve the details of these processes.
Our observations provide circumstantial evidence that the current seasonal cycles of SA, MSA, IA, and SO2 in the central Arctic Ocean are linked to sea ice conditions and solar radiation due to their role in biological activity and air mass transport from southern regions. Given that sea ice is in a state of decline in the central Arctic, the concentrations of the vapors presented herein, and their influence on aerosol processes, will likely change as a result. Anthropogenic activities around the Arctic and in the central Arctic Ocean may increase as the Arctic becomes more accessible with less sea ice, which can also influence these gas phase species. The magnitude and implications of such changes on climate relevant processes remain uncertain. While our findings offer new insights that can improve climate model predictions in the remote Arctic, it is imperative to continue monitoring these aerosol precursor vapors and to further understand their role in atmospheric processes to evaluate their climate-relevant effects on aerosol formation, growth, and subsequent CCN activation in the future in the central Arctic.
All datasets used in this work that were obtained during the MOSAiC campaign are publicly available via Pangaea (https://www.pangaea.de/, last access: 15 October 2023).
Data from the Pangaea archive include the following:
-
Meteorological observations from Polarstern at https://doi.org/10.1594/PANGAEA.935221 (Schmithüsen, 2021a), https://doi.org/10.1594/PANGAEA.935222 (Schmithüsen, 2021b), https://doi.org/10.1594/ PANGAEA.935223 (Schmithüsen, 2021c), https://doi.org/10.1594/PANGAEA.935224 (Schmithüsen, 2021d), and https://doi.org/10.1594/PANGAEA.935225 (Schmithüsen, 2021e).
-
Chemical ionization mass spectrometer (NO3-CIMS) data at https://doi.org/10.1594/PANGAEA.963321 (Boyer et al., 2023b).
-
Sulfur dioxide (SO2) data at https://doi.org/10.1594/PANGAEA.944270 (Angot et al., 2022b).
-
Particle number concentration (CPC3025) data at https://doi.org/10.1594/PANGAEA.941886 (Beck et al., 2022b).
The Ocean Colour Climate Change Initiative dataset, Version 5.0, European Space Agency, is publicly available online at https://doi.org/10.5285/1dbe7a109c0244aaad713e078fd3059a (Sathyendranath et al., 2021).
An archive of the FLEXPART model output for the whole campaign can be found at https://doi.org/10.25365/phaidra.570 (Bucci, 2024). Quicklooks of the FLEXPART data are available at https://img.univie.ac.at/webdata/mosaic (last access: 23 March 2022).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-24-12595-2024-supplement.
MB led the analysis and writing process, with the assistance of LLJQ and TJ. LLJQ, IB, JS, TJ, and TL conducted the field measurements reported in this work. SB, MD, and AS performed the FLEXPART simulations. DA performed inversion modeling and source region identification. HA, LD, IB, BH, RCT, LB, NS, BM, and JS provided useful insights and assisted with the interpretation of various datasets. MK, TP, JS, and MS provided funding for the campaign and data analysis. All authors provided comments/revisions on the paper.
At least one of the (co-)authors is a member of the editorial board of Atmospheric Chemistry and Physics. The peer-review process was guided by an independent editor, and the authors also have no other competing interests to declare.
Views and opinions expressed are those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council Executive Agency. Neither the European Union nor the granting authority can be held responsible for them.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
Data reported in this work were produced as part of the international Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition with the tag MOSAiC20192020, with activities supported by the Polarstern expedition AWI_PS122_00. The authors thank the Laboratory of Atmospheric Chemistry at the Paul Scherrer Institute and ACTRIS CiGas-UHEL for their support, and we thank Janne Lampilahti, Markus Lampimäki, Tommy Chan, Katrianne Lehtipalo, Federico Bianchi, Anton Rusanen, Heikki Junninen, and the INAR technical staff for their assistance with measurement support during the campaign. We also extend our special thanks to all the personnel who made the expedition possible through the operation of the R/V Polarstern during MOSAiC in 2019–2020 (AWI_PS122_00) (Nixdorf et al., 2021).
This research has been supported by the Research Council of Finland (grant nos. 296628, 328290, 335844, 337552, and 346372), the European Research Council, HORIZON EUROPE European Research Council (grant nos. 714621 and 101076311), the EU Horizon 2020 (grant nos. 856612 and 101003826), the U.S. Department of Energy (grant no. DE-SC0022046), the Swiss Polar Institute (grant no. DIRCR-2018-004), and the Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (grant no. 188478). Julia Schmale holds the Ingvar Kamprad chair, sponsored by Ferring Pharmaceuticals, and Tuija Jokinen receives funding from the Cyprus Government.
This paper was edited by Barbara Ervens and reviewed by Karam Mansour and four anonymous referees.
Abbatt, J. P. D., Thomas, J. L., Abrahamsson, K., Boxe, C., Granfors, A., Jones, A. E., King, M. D., Saiz-Lopez, A., Shepson, P. B., Sodeau, J., Toohey, D. W., Toubin, C., von Glasow, R., Wren, S. N., and Yang, X.: Halogen activation via interactions with environmental ice and snow in the polar lower troposphere and other regions, Atmos. Chem. Phys., 12, 6237–6271, https://doi.org/10.5194/acp-12-6237-2012, 2012.
Abbatt, J. P. D., Leaitch, W. R., Aliabadi, A. A., Bertram, A. K., Blanchet, J.-P., Boivin-Rioux, A., Bozem, H., Burkart, J., Chang, R. Y. W., Charette, J., Chaubey, J. P., Christensen, R. J., Cirisan, A., Collins, D. B., Croft, B., Dionne, J., Evans, G. J., Fletcher, C. G., Galí, M., Ghahreman, R., Girard, E., Gong, W., Gosselin, M., Gourdal, M., Hanna, S. J., Hayashida, H., Herber, A. B., Hesaraki, S., Hoor, P., Huang, L., Hussherr, R., Irish, V. E., Keita, S. A., Kodros, J. K., Köllner, F., Kolonjari, F., Kunkel, D., Ladino, L. A., Law, K., Levasseur, M., Libois, Q., Liggio, J., Lizotte, M., Macdonald, K. M., Mahmood, R., Martin, R. V., Mason, R. H., Miller, L. A., Moravek, A., Mortenson, E., Mungall, E. L., Murphy, J. G., Namazi, M., Norman, A.-L., O'Neill, N. T., Pierce, J. R., Russell, L. M., Schneider, J., Schulz, H., Sharma, S., Si, M., Staebler, R. M., Steiner, N. S., Thomas, J. L., von Salzen, K., Wentzell, J. J. B., Willis, M. D., Wentworth, G. R., Xu, J.-W., and Yakobi-Hancock, J. D.: Overview paper: New insights into aerosol and climate in the Arctic, Atmos. Chem. Phys., 19, 2527–2560, https://doi.org/10.5194/acp-19-2527-2019, 2019.
Aliaga, D., Sinclair, V. A., Andrade, M., Artaxo, P., Carbone, S., Kadantsev, E., Laj, P., Wiedensohler, A., Krejci, R., and Bianchi, F.: Identifying source regions of air masses sampled at the tropical high-altitude site of Chacaltaya using WRF-FLEXPART and cluster analysis, Atmos. Chem. Phys., 21, 16453–16477, https://doi.org/10.5194/acp-21-16453-2021, 2021.
Allan, J. D., Williams, P. I., Najera, J., Whitehead, J. D., Flynn, M. J., Taylor, J. W., Liu, D., Darbyshire, E., Carpenter, L. J., Chance, R., Andrews, S. J., Hackenberg, S. C., and McFiggans, G.: Iodine observed in new particle formation events in the Arctic atmosphere during ACCACIA, Atmos. Chem. Phys., 15, 5599–5609, https://doi.org/10.5194/acp-15-5599-2015, 2015.
Andreae, M. O.: Ocean-atmosphere interactions in the global biogeochemical sulfur cycle, Mar. Chem., 30, 1–29, https://doi.org/10.1016/0304-4203(90)90059-L, 1990.
Angot, H., Blomquist, B., Howard, D., Archer, S., Bariteau, L., Beck, I., Boyer, M., Crotwell, M., Helmig, D., Hueber, J., Jacobi, H.-W., Jokinen, T., Kulmala, M., Lan, X., Laurila, T., Madronich, M., Neff, D., Petäjä, T., Posman, K., Quéléver, L., Shupe, M. D., Vimont, I., and Schmale, J.: Year-round trace gas measurements in the central Arctic during the MOSAiC expedition, Sci. Data, 9, 723, https://doi.org/10.1038/s41597-022-01769-6, 2022a.
Angot, H., Beck, I., Jokinen, T.,; Laurila, T., Quéléver, L., and Schmale, J.: Ambient air sulfur dioxide mole fractions measured in the Swiss container during MOSAiC 2019/2020, PANGAEA [data set], https://doi.pangaea.de/10.1594/PANGAEA.944270, 2022b.
Angot, H., Reveillet, M., Dada, L., Jacobi, H.-W., Archer, S., Bariteau, L., Barten, J., Beck, I., Bergner, N., Blomquist, B., Bourgeois, I., Boyer, M., Bucci, S., Creamean, J., Barry, K., Cox, C., Dahlke, S., Dütsch, M., Dumont, M., Ganzeveld, L.N., Helmig, D., Heutte, B., Howard, D., Jokinen, T., Laurila, T., Macfarlane, A. R., Pernov, J. B., Persson, O., Posman, K. M., Quéléver, L., Shupe, M. D., Stohl A., and Schmale, J.: Warm and moist air intrusions drive black carbon deposition and accelerate central Arctic snowmelt, Research Square [preprint], https://doi.org/10.21203/rs.3.rs-5197651/v1, 2024.
Ashu-Ayem, E. R., Nitschke, U., Monahan, C., Chen, J., Darby, S. B., Smith, P. D., O'Dowd, C. D., Stengel, D. B., and Venables, D. S.: Coastal Iodine Emissions. 1. Release of I 2 by Laminaria digitata in Chamber Experiments, Environ. Sci. Technol., 46, 10413–10421, https://doi.org/10.1021/es204534v, 2012.
Baccarini, A., Karlsson, L., Dommen, J., Duplessis, P., Vüllers, J., Brooks, I. M., Saiz-Lopez, A., Salter, M., Tjernström, M., Baltensperger, U., Zieger, P., and Schmale, J.: Frequent new particle formation over the high Arctic pack ice by enhanced iodine emissions, Nat. Commun., 11, 4924, https://doi.org/10.1038/s41467-020-18551-0, 2020.
Baccarini, A., Dommen, J., Lehtipalo, K., Henning, S., Modini, R. L., Gysel-Beer, M., Baltensperger, U., and Schmale, J.: Low-Volatility Vapors and New Particle Formation Over the Southern Ocean During the Antarctic Circumnavigation Expedition, J. Geophys. Res.-Atmos., 126, e2021JD035126, https://doi.org/10.1029/2021JD035126, 2021.
Barrie, L. A.: Arctic air pollution: An overview of current knowledge, Atmos. Environ., 20, 643–663, https://doi.org/10.1016/0004-6981(86)90180-0, 1986.
Barrie, L. A., Bottenheim, J. W., Schnell, R. C., Crutzen, P. J., and Rasmussen, R. A.: Ozone destruction and photochemical reactions at polar sunrise in the lower Arctic atmosphere, Nature, 334, 138–141, https://doi.org/10.1038/334138a0, 1988.
Bauduin, S., Clarisse, L., Clerbaux, C., Hurtmans, D., and Coheur, P.-F.: IASI observations of sulfur dioxide (SO2) in the boundary layer of Norilsk, J. Geophys. Res.-Atmos., 119, 4253–4263, https://doi.org/10.1002/2013JD021405, 2014.
Becagli, S., Lazzara, L., Marchese, C., Dayan, U., Ascanius, S. E., Cacciani, M., Caiazzo, L., Di Biagio, C., Di Iorio, T., di Sarra, A., Eriksen, P., Fani, F., Giardi, F., Meloni, D., Muscari, G., Pace, G., Severi, M., Traversi, R., and Udisti, R.: Relationships linking primary production, sea ice melting, and biogenic aerosol in the Arctic, Atmos. Environ., 136, 1–15, https://doi.org/10.1016/j.atmosenv.2016.04.002, 2016.
Beck, I., Angot, H., Baccarini, A., Dada, L., Quéléver, L., Jokinen, T., Laurila, T., Lampimäki, M., Bukowiecki, N., Boyer, M., Gong, X., Gysel-Beer, M., Petäjä, T., Wang, J., and Schmale, J.: Automated identification of local contamination in remote atmospheric composition time series, Atmos. Meas. Tech., 15, 4195–4224, https://doi.org/10.5194/amt-15-4195-2022, 2022a.
Beck, I., Quéléver, L., Laurila, T., Jokinen, T., and Schmale, J.: Continuous corrected particle number concentration data in 10 sec resolution, measured in the Swiss aerosol container during MOSAiC 2019/2020, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.941886, 2022b.
Beck, L. J., Sarnela, N., Junninen, H., Hoppe, C. J. M., Garmash, O., Bianchi, F., Riva, M., Rose, C., Peräkylä, O., Wimmer, D., Kausiala, O., Jokinen, T., Ahonen, L., Mikkilä, J., Hakala, J., He, X. C., Kontkanen, J., Wolf, K. K. E., Cappelletti, D., Mazzola, M., Traversi, R., Petroselli, C., Viola, A. P., Vitale, V., Lange, R., Massling, A., Nøjgaard, J. K., Krejci, R., Karlsson, L., Zieger, P., Jang, S., Lee, K., Vakkari, V., Lampilahti, J., Thakur, R. C., Leino, K., Kangasluoma, J., Duplissy, E. M., Siivola, E., Marbouti, M., Tham, Y. J., Saiz-Lopez, A., Petäjä, T., Ehn, M., Worsnop, D. R., Skov, H., Kulmala, M., Kerminen, V. M., and Sipilä, M.: Differing Mechanisms of New Particle Formation at Two Arctic Sites, Geophys. Res. Lett., 48, e2020GL091334, https://doi.org/10.1029/2020GL091334, 2021.
Benavent, N., Mahajan, A. S., Li, Q., Cuevas, C. A., Schmale, J., Angot, H., Jokinen, T., Quéléver, L. L. J., Blechschmidt, A.-M., Zilker, B., Richter, A., Serna, J. A., Garcia-Nieto, D., Fernandez, R. P., Skov, H., Dumitrascu, A., Simões Pereira, P., Abrahamsson, K., Bucci, S., Duetsch, M., Stohl, A., Beck, I., Laurila, T., Blomquist, B., Howard, D., Archer, S. D., Bariteau, L., Helmig, D., Hueber, J., Jacobi, H.-W., Posman, K., Dada, L., Daellenbach, K. R., and Saiz-Lopez, A.: Substantial contribution of iodine to Arctic ozone destruction, Nat. Geosci., 15, 770–773, https://doi.org/10.1038/s41561-022-01018-w, 2022.
Berndt, T., Hoffmann, E. H., Tilgner, A., Stratmann, F., and Herrmann, H.: Direct sulfuric acid formation from the gas-phase oxidation of reduced-sulfur compounds, Nat. Commun., 14, 4849, https://doi.org/10.1038/s41467-023-40586-2, 2023.
Bigg, E. K. and Leck, C.: The composition of fragments of bubbles bursting at the ocean surface, J. Geophys. Res.-Atmos., 113, D11209, https://doi.org/10.1029/2007JD009078, 2008.
Boyer, M., Aliaga, D., Pernov, J. B., Angot, H., Quéléver, L. L. J., Dada, L., Heutte, B., Dall'Osto, M., Beddows, D. C. S., Brasseur, Z., Beck, I., Bucci, S., Duetsch, M., Stohl, A., Laurila, T., Asmi, E., Massling, A., Thomas, D. C., Nøjgaard, J. K., Chan, T., Sharma, S., Tunved, P., Krejci, R., Hansson, H. C., Bianchi, F., Lehtipalo, K., Wiedensohler, A., Weinhold, K., Kulmala, M., Petäjä, T., Sipilä, M., Schmale, J., and Jokinen, T.: A full year of aerosol size distribution data from the central Arctic under an extreme positive Arctic Oscillation: insights from the Multidisciplinary drifting Observatory for the Study of Arctic Climate (MOSAiC) expedition, Atmos. Chem. Phys., 23, 389–415, https://doi.org/10.5194/acp-23-389-2023, 2023a.
Boyer, M., Quéléver, L., Beck, I., Laurila, T., Sarnela, N., Schmale, J., and Jokinen, T.: Ambient concentrations of aerosol precursor vapor concentrations (sulfuric acid, methanesulfonic acid, and iodic acid) in 5-minute resolution measured by a nitrate chemical ionization mass spectrometer, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.963321, 2023b.
Bozem, H., Hoor, P., Kunkel, D., Köllner, F., Schneider, J., Herber, A., Schulz, H., Leaitch, W. R., Aliabadi, A. A., Willis, M. D., Burkart, J., and Abbatt, J. P. D.: Characterization of transport regimes and the polar dome during Arctic spring and summer using in situ aircraft measurements, Atmos. Chem. Phys., 19, 15049–15071, https://doi.org/10.5194/acp-19-15049-2019, 2019.
Bucci, S.: FLEXPART airtracer trajectories for the MOSAIC campaign: Emission sensitivities below 100 m, PHAIDRA [data set], https://doi.org/10.25365/phaidra.570, 2024.
Bulatovic, I., Igel, A. L., Leck, C., Heintzenberg, J., Riipinen, I., and Ekman, A. M. L.: The importance of Aitken mode aerosol particles for cloud sustenance in the summertime high Arctic – a simulation study supported by observational data, Atmos. Chem. Phys., 21, 3871–3897, https://doi.org/10.5194/acp-21-3871-2021, 2021.
Cala, B. A., Archer-Nicholls, S., Weber, J., Abraham, N. L., Griffiths, P. T., Jacob, L., Shin, Y. M., Revell, L. E., Woodhouse, M., and Archibald, A. T.: Development, intercomparison, and evaluation of an improved mechanism for the oxidation of dimethyl sulfide in the UKCA model, Atmos. Chem. Phys., 23, 14735–14760, https://doi.org/10.5194/acp-23-14735-2023, 2023.
Carpenter, L. J., Archer, S. D., and Beale, R.: Ocean-atmosphere trace gas exchange, Chem. Soc. Rev., 41, 6473–6506, https://doi.org/10.1039/C2CS35121H, 2012.
Carpenter, L. J., MacDonald, S. M., Shaw, M. D., Kumar, R., Saunders, R. W., Parthipan, R., Wilson, J., and Plane, J. M. C.: Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine, Nat. Geosci., 6, 108–111, https://doi.org/10.1038/ngeo1687, 2013.
Chang, R. Y.-W., Abbatt, J. P. D., Boyer, M. C., Chaubey, J. P., and Collins, D. B.: Characterizing the hygroscopicity of growing particles in the Canadian Arctic summer, Atmos. Chem. Phys., 22, 8059–8071, https://doi.org/10.5194/acp-22-8059-2022, 2022.
Chen, Q., Sherwen, T., Evans, M., and Alexander, B.: DMS oxidation and sulfur aerosol formation in the marine troposphere: a focus on reactive halogen and multiphase chemistry, Atmos. Chem. Phys., 18, 13617–13637, https://doi.org/10.5194/acp-18-13617-2018, 2018.
Collins, D. B., Burkart, J., Chang, R. Y.-W., Lizotte, M., Boivin-Rioux, A., Blais, M., Mungall, E. L., Boyer, M., Irish, V. E., Massé, G., Kunkel, D., Tremblay, J.-É., Papakyriakou, T., Bertram, A. K., Bozem, H., Gosselin, M., Levasseur, M., and Abbatt, J. P. D.: Frequent ultrafine particle formation and growth in Canadian Arctic marine and coastal environments, Atmos. Chem. Phys., 17, 13119–13138, https://doi.org/10.5194/acp-17-13119-2017, 2017.
Corbett, J. J., Fischbeck, P. S., and Pandis, S. N.: Global nitrogen and sulfur inventories for oceangoing ships, J. Geophys. Res.-Atmos., 104, 3457–3470, https://doi.org/10.1029/1998JD100040, 1999.
Croft, B., Martin, R. V., Leaitch, W. R., Tunved, P., Breider, T. J., D'Andrea, S. D., and Pierce, J. R.: Processes controlling the annual cycle of Arctic aerosol number and size distributions, Atmos. Chem. Phys., 16, 3665–3682, https://doi.org/10.5194/acp-16-3665-2016, 2016.
Croft, B., Martin, R. V., Leaitch, W. R., Burkart, J., Chang, R. Y.-W., Collins, D. B., Hayes, P. L., Hodshire, A. L., Huang, L., Kodros, J. K., Moravek, A., Mungall, E. L., Murphy, J. G., Sharma, S., Tremblay, S., Wentworth, G. R., Willis, M. D., Abbatt, J. P. D., and Pierce, J. R.: Arctic marine secondary organic aerosol contributes significantly to summertime particle size distributions in the Canadian Arctic Archipelago, Atmos. Chem. Phys., 19, 2787–2812, https://doi.org/10.5194/acp-19-2787-2019, 2019.
Custard, K. D., Raso, A. R. W., Shepson, P. B., Staebler, R. M., and Pratt, K. A.: Production and Release of Molecular Bromine and Chlorine from the Arctic Coastal Snowpack, ACS Earth Space Chem., 1, 142–151, https://doi.org/10.1021/acsearthspacechem.7b00014, 2017.
Dada, L., Ylivinkka, I., Baalbaki, R., Li, C., Guo, Y., Yan, C., Yao, L., Sarnela, N., Jokinen, T., Daellenbach, K. R., Yin, R., Deng, C., Chu, B., Nieminen, T., Wang, Y., Lin, Z., Thakur, R. C., Kontkanen, J., Stolzenburg, D., Sipilä, M., Hussein, T., Paasonen, P., Bianchi, F., Salma, I., Weidinger, T., Pikridas, M., Sciare, J., Jiang, J., Liu, Y., Petäjä, T., Kerminen, V.-M., and Kulmala, M.: Sources and sinks driving sulfuric acid concentrations in contrasting environments: implications on proxy calculations, Atmos. Chem. Phys., 20, 11747–11766, https://doi.org/10.5194/acp-20-11747-2020, 2020.
Dada, L., Angot, H., Beck, I., Baccarini, A., Quéléver, L. L. J., Boyer, M., Laurila, T., Brasseur, Z., Jozef, G., de Boer, G., Shupe, M. D., Henning, S., Bucci, S., Dütsch, M., Stohl, A., Petäjä, T., Daellenbach, K. R., Jokinen, T., and Schmale, J.: A central arctic extreme aerosol event triggered by a warm air-mass intrusion, Nat. Commun., 13, 5290, https://doi.org/10.1038/s41467-022-32872-2, 2022.
Dall'Osto, M., Beddows, D. C. S., Tunved, P., Krejci, R., Ström, J., Hansson, H.-C., Yoon, Y. J., Park, K.-T., Becagli, S., Udisti, R., Onasch, T., O'Dowd, C. D., Simó, R., and Harrison, R. M.: Arctic sea ice melt leads to atmospheric new particle formation, Sci. Rep., 7, 3318, https://doi.org/10.1038/s41598-017-03328-1, 2017.
Domine, F., Sparapani, R., Ianniello, A., and Beine, H. J.: The origin of sea salt in snow on Arctic sea ice and in coastal regions, Atmos. Chem. Phys., 4, 2259–2271, https://doi.org/10.5194/acp-4-2259-2004, 2004.
Drougas, E. and Kosmas, A. M.: Computational studies of (HIO3) isomers and the HO2 + IO reaction pathways, J. Phys. Chem. A, 109 17, 3887–92, 2005.
Dunne, E. M., Gordon, H., Kürten, A., Almeida, J., Duplissy, J., Williamson, C., Ortega, I. K., Pringle, K. J., Adamov, A., Baltensperger, U., Barmet, P., Benduhn, F., Bianchi, F., Breitenlechner, M., Clarke, A., Curtius, J., Dommen, J., Donahue, N. M., Ehrhart, S., Flagan, R. C., Franchin, A., Guida, R., Hakala, J., Hansel, A., Heinritzi, M., Jokinen, T., Kangasluoma, J., Kirkby, J., Kulmala, M., Kupc, A., Lawler, M. J., Lehtipalo, K., Makhmutov, V., Mann, G., Mathot, S., Merikanto, J., Miettinen, P., Nenes, A., Onnela, A., Rap, A., Reddington, C. L. S., Riccobono, F., Richards, N. A. D., Rissanen, M. P., Rondo, L., Sarnela, N., Schobesberger, S., Sengupta, K., Simon, M., Sipilä, M., Smith, J. N., Stozkhov, Y., Tomé, A., Tröstl, J., Wagner, P. E., Wimmer, D., Winkler, P. M., Worsnop, D. R., and Carslaw, K. S.: Global atmospheric particle formation from CERN CLOUD measurements, Science, 354, 1119–1124, https://doi.org/10.1126/science.aaf2649, 2016.
Eckhardt, S., Stohl, A., Beirle, S., Spichtinger, N., James, P., Forster, C., Junker, C., Wagner, T., Platt, U., and Jennings, S. G.: The North Atlantic Oscillation controls air pollution transport to the Arctic, Atmos. Chem. Phys., 3, 1769–1778, https://doi.org/10.5194/acp-3-1769-2003, 2003.
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Maso, M. D., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–479, https://doi.org/10.1038/nature13032, 2014.
Faletto, G. and Bien, J.: Cluster Stability Selection, arXiv [preprint], https://doi.org/10.48550/ARXIV.2201.00494, 2022.
Ferrero, L., Cappelletti, D., Busetto, M., Mazzola, M., Lupi, A., Lanconelli, C., Becagli, S., Traversi, R., Caiazzo, L., Giardi, F., Moroni, B., Crocchianti, S., Fierz, M., Močnik, G., Sangiorgi, G., Perrone, M. G., Maturilli, M., Vitale, V., Udisti, R., and Bolzacchini, E.: Vertical profiles of aerosol and black carbon in the Arctic: a seasonal phenomenology along 2 years (2011–2012) of field campaigns, Atmos. Chem. Phys., 16, 12601–12629, https://doi.org/10.5194/acp-16-12601-2016, 2016.
Finkenzeller, H., Iyer, S., He, X.-C., Simon, M., Koenig, T. K., Lee, C. F., Valiev, R., Hofbauer, V., Amorim, A., Baalbaki, R., Baccarini, A., Beck, L., Bell, D. M., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dada, L., Duplissy, J., Heinritzi, M., Kemppainen, D., Kim, C., Krechmer, J., Kürten, A., Kvashnin, A., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mauldin, R. L., Mentler, B., Müller, T., Petäjä, T., Philippov, M., Ranjithkumar, A., Rörup, B., Shen, J., Stolzenburg, D., Tauber, C., Tham, Y. J., Tomé, A., Vazquez-Pufleau, M., Wagner, A. C., Wang, D. S., Wang, M., Wang, Y., Weber, S. K., Nie, W., Wu, Y., Xiao, M., Ye, Q., Zauner-Wieczorek, M., Hansel, A., Baltensperger, U., Brioude, J., Curtius, J., Donahue, N. M., Haddad, I. E., Flagan, R. C., Kulmala, M., Kirkby, J., Sipilä, M., Worsnop, D. R., Kurten, T., Rissanen, M., and Volkamer, R.: The gas-phase formation mechanism of iodic acid as an atmospheric aerosol source, Nat. Chem., 15, 129–135, https://doi.org/10.1038/s41557-022-01067-z, 2023.
Fioletov, V., McLinden, C. A., Griffin, D., Theys, N., Loyola, D. G., Hedelt, P., Krotkov, N. A., and Li, C.: Anthropogenic and volcanic point source SO2 emissions derived from TROPOMI on board Sentinel-5 Precursor: first results, Atmos. Chem. Phys., 20, 5591–5607, https://doi.org/10.5194/acp-20-5591-2020, 2020.
Fisher, J. A., Jacob, D. J., Wang, Q., Bahreini, R., Carouge, C. C., Cubison, M. J., Dibb, J. E., Diehl, T., Jimenez, J. L., Leibensperger, E. M., Lu, Z., Meinders, M. B. J., Pye, H. O. T., Quinn, P. K., Sharma, S., Streets, D. G., Van Donkelaar, A., and Yantosca, R. M.: Sources, distribution, and acidity of sulfate–ammonium aerosol in the Arctic in winter–spring, Atmos. Environ., 45, 7301–7318, https://doi.org/10.1016/j.atmosenv.2011.08.030, 2011.
Freud, E., Krejci, R., Tunved, P., Leaitch, R., Nguyen, Q. T., Massling, A., Skov, H., and Barrie, L.: Pan-Arctic aerosol number size distributions: seasonality and transport patterns, Atmos. Chem. Phys., 17, 8101–8128, https://doi.org/10.5194/acp-17-8101-2017, 2017.
Galí, M., Devred, E., Babin, M., and Levasseur, M.: Decadal increase in Arctic dimethylsulfide emission, Proc. Natl. Acad. Sci., 116, 19311–19317, https://doi.org/10.1073/pnas.1904378116, 2019.
Galí, M., Lizotte, M., Kieber, D. J., Randelhoff, A., Hussherr, R., Xue, L., Dinasquet, J., Babin, M., Rehm, E., and Levasseur, M.: DMS emissions from the Arctic marginal ice zone, Elem. Sci. Anthr., 9, 00113, https://doi.org/10.1525/elementa.2020.00113, 2021.
Ghahreman, R., Norman, A.-L., Abbatt, J. P. D., Levasseur, M., and Thomas, J. L.: Biogenic, anthropogenic and sea salt sulfate size-segregated aerosols in the Arctic summer, Atmos. Chem. Phys., 16, 5191–5202, https://doi.org/10.5194/acp-16-5191-2016, 2016.
Ghahreman, R., Norman, A.-L., Croft, B., Martin, R. V., Pierce, J. R., Burkart, J., Rempillo, O., Bozem, H., Kunkel, D., Thomas, J. L., Aliabadi, A. A., Wentworth, G. R., Levasseur, M., Staebler, R. M., Sharma, S., and Leaitch, W. R.: Boundary layer and free-tropospheric dimethyl sulfide in the Arctic spring and summer, Atmos. Chem. Phys., 17, 8757–8770, https://doi.org/10.5194/acp-17-8757-2017, 2017.
Ghahreman, R., Gong, W., Galí, M., Norman, A.-L., Beagley, S. R., Akingunola, A., Zheng, Q., Lupu, A., Lizotte, M., Levasseur, M., and Leaitch, W. R.: Dimethyl sulfide and its role in aerosol formation and growth in the Arctic summer – a modelling study, Atmos. Chem. Phys., 19, 14455–14476, https://doi.org/10.5194/acp-19-14455-2019, 2019.
Gong, S. L., Zhao, T. L., Sharma, S., Toom-Sauntry, D., Lavoué, D., Zhang, X. B., Leaitch, W. R., and Barrie, L. A.: Identification of trends and interannual variability of sulfate and black carbon in the Canadian High Arctic: 1981–2007, J. Geophys. Res.-Atmos., 115, D07305, https://doi.org/10.1029/2009JD012943, 2010.
Gordon, H., Kirkby, J., Baltensperger, U., Bianchi, F., Breitenlechner, M., Curtius, J., Dias, A., Dommen, J., Donahue, N. M., Dunne, E. M., Duplissy, J., Ehrhart, S., Flagan, R. C., Frege, C., Fuchs, C., Hansel, A., Hoyle, C. R., Kulmala, M., Kürten, A., Lehtipalo, K., Makhmutov, V., Molteni, U., Rissanen, M. P., Stozkhov, Y., Tröstl, J., Tsagkogeorgas, G., Wagner, R., Williamson, C., Wimmer, D., Winkler, P. M., Yan, C., and Carslaw, K. S.: Causes and importance of new particle formation in the present-day and preindustrial atmospheres, J. Geophys. Res.-Atmos., 122, 8739–8760, https://doi.org/10.1002/2017JD026844, 2017.
Halfacre, J. W., Shepson, P. B., and Pratt, K. A.: pH-dependent production of molecular chlorine, bromine, and iodine from frozen saline surfaces, Atmos. Chem. Phys., 19, 4917–4931, https://doi.org/10.5194/acp-19-4917-2019, 2019.
He, X.-C., Tham, Y. J., Dada, L., Wang, M., Finkenzeller, H., Stolzenburg, D., Iyer, S., Simon, M., Kürten, A., Shen, J., Rörup, B., Rissanen, M., Schobesberger, S., Baalbaki, R., Wang, D. S., Koenig, T. K., Jokinen, T., Sarnela, N., Beck, L. J., Almeida, J., Amanatidis, S., Amorim, A., Ataei, F., Baccarini, A., Bertozzi, B., Bianchi, F., Brilke, S., Caudillo, L., Chen, D., Chiu, R., Chu, B., Dias, A., Ding, A., Dommen, J., Duplissy, J., El Haddad, I., Gonzalez Carracedo, L., Granzin, M., Hansel, A., Heinritzi, M., Hofbauer, V., Junninen, H., Kangasluoma, J., Kemppainen, D., Kim, C., Kong, W., Krechmer, J. E., Kvashin, A., Laitinen, T., Lamkaddam, H., Lee, C. P., Lehtipalo, K., Leiminger, M., Li, Z., Makhmutov, V., Manninen, H. E., Marie, G., Marten, R., Mathot, S., Mauldin, R. L., Mentler, B., Möhler, O., Müller, T., Nie, W., Onnela, A., Petäjä, T., Pfeifer, J., Philippov, M., Ranjithkumar, A., Saiz-Lopez, A., Salma, I., Scholz, W., Schuchmann, S., Schulze, B., Steiner, G., Stozhkov, Y., Tauber, C., Tomé, A., Thakur, R. C., Väisänen, O., Vazquez-Pufleau, M., Wagner, A. C., Wang, Y., Weber, S. K., Winkler, P. M., Wu, Y., Xiao, M., Yan, C., Ye, Q., Ylisirniö, A., Zauner-Wieczorek, M., Zha, Q., Zhou, P., Flagan, R. C., Curtius, J., Baltensperger, U., Kulmala, M., Kerminen, V.-M., Kurtén, T., Donahue, N. M., Volkamer, R., Kirkby, J., Worsnop, D. R., and Sipilä, M.: Role of iodine oxoacids in atmospheric aerosol nucleation, Science, 371, 589–595, https://doi.org/10.1126/science.abe0298, 2021.
He, X.-C., Shen, J., Iyer, S., Juuti, P., Zhang, J., Koirala, M., Kytökari, M. M., Worsnop, D. R., Rissanen, M., Kulmala, M., Maier, N. M., Mikkilä, J., Sipilä, M., and Kangasluoma, J.: Characterisation of gaseous iodine species detection using the multi-scheme chemical ionisation inlet 2 with bromide and nitrate chemical ionisation methods, Atmos. Meas. Tech., 16, 4461–4487, https://doi.org/10.5194/amt-16-4461-2023, 2023.
Heidam, N. Z., Wåhlin, P., and Christensen, J. H.: Tropospheric Gases and Aerosols in Northeast Greenland, J. Atmos. Sci., 56, 261–278, https://doi.org/10.1175/1520-0469(1999)056<0261:TGAAIN>2.0.CO;2, 1999.
Hirdman, D., Sodemann, H., Eckhardt, S., Burkhart, J. F., Jefferson, A., Mefford, T., Quinn, P. K., Sharma, S., Ström, J., and Stohl, A.: Source identification of short-lived air pollutants in the Arctic using statistical analysis of measurement data and particle dispersion model output, Atmos. Chem. Phys., 10, 669–693, https://doi.org/10.5194/acp-10-669-2010, 2010.
Hodshire, A. L., Campuzano-Jost, P., Kodros, J. K., Croft, B., Nault, B. A., Schroder, J. C., Jimenez, J. L., and Pierce, J. R.: The potential role of methanesulfonic acid (MSA) in aerosol formation and growth and the associated radiative forcings, Atmos. Chem. Phys., 19, 3137–3160, https://doi.org/10.5194/acp-19-3137-2019, 2019.
Hoffmann, E. H., Tilgner, A., Schrödner, R., Bräuer, P., Wolke, R., and Herrmann, H.: An advanced modeling study on the impacts and atmospheric implications of multiphase dimethyl sulfide chemistry, P. Natl. Acad. Sci. USA, 113, 11776–11781, https://doi.org/10.1073/pnas.1606320113, 2016.
Hoffmann, E. H., Heinold, B., Kubin, A., Tegen, I., and Herrmann, H.: The Importance of the Representation of DMS Oxidation in Global Chemistry-Climate Simulations, Geophys. Res. Lett., 48, e2021GL094068, https://doi.org/10.1029/2021GL094068, 2021.
Hulswar, S., Simó, R., Galí, M., Bell, T. G., Lana, A., Inamdar, S., Halloran, P. R., Manville, G., and Mahajan, A. S.: Third revision of the global surface seawater dimethyl sulfide climatology (DMS-Rev3), Earth Syst. Sci. Data, 14, 2963–2987, https://doi.org/10.5194/essd-14-2963-2022, 2022.
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012.
Jokinen, T., Sipilä, M., Kontkanen, J., Vakkari, V., Tisler, P., Duplissy, E.-M., Junninen, H., Kangasluoma, J., Manninen, H. E., Petäjä, T., Kulmala, M., Worsnop, D. R., Kirkby, J., Virkkula, A., and Kerminen, V.-M.: Ion-induced sulfuric acid-ammonia nucleation drives particle formation in coastal Antarctica, Sci. Adv., 4, eaat9744, https://doi.org/10.1126/sciadv.aat9744, 2018.
Jokinen, T., Lehtipalo, K., Thakur, R. C., Ylivinkka, I., Neitola, K., Sarnela, N., Laitinen, T., Kulmala, M., Petäjä, T., and Sipilä, M.: Measurement report: Long-term measurements of aerosol precursor concentrations in the Finnish subarctic boreal forest, Atmos. Chem. Phys., 22, 2237–2254, https://doi.org/10.5194/acp-22-2237-2022, 2022.
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010.
Karl, M., Gross, A., Leck, C., and Pirjola, L.: Intercomparison of dimethylsulfide oxidation mechanisms for the marine boundary layer: Gaseous and particulate sulfur constituents, J. Geophys. Res., 112, D15304, https://doi.org/10.1029/2006JD007914, 2007.
Karl, M., Leck, C., Coz, E., and Heintzenberg, J.: Marine nanogels as a source of atmospheric nanoparticles in the high Arctic, Geophys. Res. Lett., 40, 3738–3743, https://doi.org/10.1002/grl.50661, 2013.
Karlsson, L., Baccarini, A., Duplessis, P., Baumgardner, D., Brooks, I. M., Chang, R. Y.-W., Dada, L., Dällenbach, K. R., Heikkinen, L., Krejci, R., Leaitch, W. R., Leck, C., Partridge, D. G., Salter, M. E., Wernli, H., Wheeler, M. J., Schmale, J., and Zieger, P.: Physical and Chemical Properties of Cloud Droplet Residuals and Aerosol Particles During the Arctic Ocean 2018 Expedition, J. Geophys. Res.-Atmos., 127, e2021JD036383, https://doi.org/10.1029/2021JD036383, 2022.
Kecorius, S., Vogl, T., Paasonen, P., Lampilahti, J., Rothenberg, D., Wex, H., Zeppenfeld, S., van Pinxteren, M., Hartmann, M., Henning, S., Gong, X., Welti, A., Kulmala, M., Stratmann, F., Herrmann, H., and Wiedensohler, A.: New particle formation and its effect on cloud condensation nuclei abundance in the summer Arctic: a case study in the Fram Strait and Barents Sea, Atmos. Chem. Phys., 19, 14339–14364, https://doi.org/10.5194/acp-19-14339-2019, 2019.
Kerminen, V.-M. and Leck, C.: Sulfur chemistry over the central Arctic Ocean during the summer: Gas-to-particle transformation, J. Geophys. Res.-Atmos., 106, 32087–32099, https://doi.org/10.1029/2000JD900604, 2001.
Khanniche, S., Louis, F., Cantrel, L., and Černušák, I.: Thermochemistry of HIO2 Species and Reactivity of Iodous Acid with OH Radical: A Computational Study, ACS Earth Space Chem., 1, 39–49, https://doi.org/10.1021/acsearthspacechem.6b00010, 2017.
Khokhar, M. F., Platt, U., and Wagner, T.: Temporal trends of anthropogenic SO2 emitted by non-ferrous metal smelters in Peru and Russia estimated from Satellite observations, Atmos. Chem. Phys. Discuss., 8, 17393–17422, https://doi.org/10.5194/acpd-8-17393-2008, 2008.
Kirkby, J., Curtius, J., Almeida, J., Dunne, E., Duplissy, J., Ehrhart, S., Franchin, A., Gagné, S., Ickes, L., Kürten, A., Kupc, A., Metzger, A., Riccobono, F., Rondo, L., Schobesberger, S., Tsagkogeorgas, G., Wimmer, D., Amorim, A., Bianchi, F., Breitenlechner, M., David, A., Dommen, J., Downard, A., Ehn, M., Flagan, R. C., Haider, S., Hansel, A., Hauser, D., Jud, W., Junninen, H., Kreissl, F., Kvashin, A., Laaksonen, A., Lehtipalo, K., Lima, J., Lovejoy, E. R., Makhmutov, V., Mathot, S., Mikkilä, J., Minginette, P., Mogo, S., Nieminen, T., Onnela, A., Pereira, P., Petäjä, T., Schnitzhofer, R., Seinfeld, J. H., Sipilä, M., Stozhkov, Y., Stratmann, F., Tomé, A., Vanhanen, J., Viisanen, Y., Vrtala, A., Wagner, P. E., Walther, H., Weingartner, E., Wex, H., Winkler, P. M., Carslaw, K. S., Worsnop, D. R., Baltensperger, U., and Kulmala, M.: Role of sulphuric acid, ammonia and galactic cosmic rays in atmospheric aerosol nucleation, Nature, 476, 429–433, https://doi.org/10.1038/nature10343, 2011.
Klimont, Z., Smith, S. J., and Cofala, J.: The last decade of global anthropogenic sulfur dioxide: 2000–2011 emissions, Environ. Res. Lett., 8, 014003, https://doi.org/10.1088/1748-9326/8/1/014003, 2013.
Klonecki, A., Hess, P., Emmons, L., Smith, L., Orlando, J., and Blake, D.: Seasonal changes in the transport of pollutants into the Arctic troposphere-model study, J. Geophys. Res.-Atmos., 108, 8367, https://doi.org/10.1029/2002JD002199, 2003.
Knust, R.: Polar Research and Supply Vessel POLARSTERN Operated by the Alfred-Wegener-Institute, J. Large-Scale Res. Facil. JLSRF, 3, A119, https://doi.org/10.17815/jlsrf-3-163, 2017.
Krumpen, T., Birrien, F., Kauker, F., Rackow, T., von Albedyll, L., Angelopoulos, M., Belter, H. J., Bessonov, V., Damm, E., Dethloff, K., Haapala, J., Haas, C., Harris, C., Hendricks, S., Hoelemann, J., Hoppmann, M., Kaleschke, L., Karcher, M., Kolabutin, N., Lei, R., Lenz, J., Morgenstern, A., Nicolaus, M., Nixdorf, U., Petrovsky, T., Rabe, B., Rabenstein, L., Rex, M., Ricker, R., Rohde, J., Shimanchuk, E., Singha, S., Smolyanitsky, V., Sokolov, V., Stanton, T., Timofeeva, A., Tsamados, M., and Watkins, D.: The MOSAiC ice floe: sediment-laden survivor from the Siberian shelf, The Cryosphere, 14, 2173–2187, https://doi.org/10.5194/tc-14-2173-2020, 2020.
Kulmala, M., Kontkanen, J., Junninen, H., Lehtipalo, K., Manninen, H. E., Nieminen, T., Petäjä, T., Sipilä, M., Schobesberger, S., Rantala, P., Franchin, A., Jokinen, T., Järvinen, E., Äijälä, M., Kangasluoma, J., Hakala, J., Aalto, P. P., Paasonen, P., Mikkilä, J., Vanhanen, J., Aalto, J., Hakola, H., Makkonen, U., Ruuskanen, T., Mauldin, R. L., Duplissy, J., Vehkamäki, H., Bäck, J., Kortelainen, A., Riipinen, I., Kurtén, T., Johnston, M. V., Smith, J. N., Ehn, M., Mentel, T. F., Lehtinen, K. E. J., Laaksonen, A., Kerminen, V.-M., and Worsnop, D. R.: Direct Observations of Atmospheric Aerosol Nucleation, Science, 339, 943–946, https://doi.org/10.1126/science.1227385, 2013.
Kurosaki, Y., Matoba, S., Iizuka, Y., Fujita, K., and Shimada, R.: Increased oceanic dimethyl sulfide emissions in areas of sea ice retreat inferred from a Greenland ice core, Commun. Earth Environ., 3, 327, https://doi.org/10.1038/s43247-022-00661-w, 2022.
Kürten, A., Rondo, L., Ehrhart, S., and Curtius, J.: Calibration of a Chemical Ionization Mass Spectrometer for the Measurement of Gaseous Sulfuric Acid, J. Phys. Chem. A, 116, 6375–6386, https://doi.org/10.1021/jp212123n, 2012.
Lana, A., Bell, T. G., Simó, R., Vallina, S. M., Ballabrera-Poy, J., Kettle, A. J., Dachs, J., Bopp, L., Saltzman, E. S., Stefels, J., Johnson, J. E., and Liss, P. S.: An updated climatology of surface dimethlysulfide concentrations and emission fluxes in the global ocean, Global Biogeochem. Cy., 25, GB1004, https://doi.org/10.1029/2010GB003850, 2011.
Lawler, M. J., Saltzman, E. S., Karlsson, L., Zieger, P., Salter, M., Baccarini, A., Schmale, J., and Leck, C.: New Insights Into the Composition and Origins of Ultrafine Aerosol in the Summertime High Arctic, Geophys. Res. Lett., 48, e2021GL094395, https://doi.org/10.1029/2021GL094395, 2021.
Lawrence, Z. D., Perlwitz, J., Butler, A. H., Manney, G. L., Newman, P. A., Lee, S. H., and Nash, E. R.: The Remarkably Strong Arctic Stratospheric Polar Vortex of Winter 2020: Links to Record-Breaking Arctic Oscillation and Ozone Loss, J. Geophys. Res.-Atmos., 125, e2020JD033271, https://doi.org/10.1029/2020JD033271, 2020.
Leaitch, W. R., Hoff, R. M., and MacPherson, J. I.: Airborne and lidar measurements of aerosol and cloud particles in the troposphere over Alert Canada in April 1986, J. Atmos. Chem., 9, 187–211, 1989.
Leaitch, W. R., Sharma, S., Huang, L., Toom-Sauntry, D., Chivulescu, A., Macdonald, A. M., Von Salzen, K., Pierce, J. R., Bertram, A. K., Schroder, J. C., Shantz, N. C., Chang, R. Y.-W., and Norman, A.-L.: Dimethyl sulfide control of the clean summertime Arctic aerosol and cloud, Elem. Sci. Anthr., 1, 000017, https://doi.org/10.12952/journal.elementa.000017, 2013.
Leaitch, W. R., Korolev, A., Aliabadi, A. A., Burkart, J., Willis, M. D., Abbatt, J. P. D., Bozem, H., Hoor, P., Köllner, F., Schneider, J., Herber, A., Konrad, C., and Brauner, R.: Effects of 20–100 nm particles on liquid clouds in the clean summertime Arctic, Atmos. Chem. Phys., 16, 11107–11124, https://doi.org/10.5194/acp-16-11107-2016, 2016.
Leck, C. and Bigg, E. K.: Source and evolution of the marine aerosol – A new perspective, Geophys. Res. Lett., 32, L19803, https://doi.org/10.1029/2005GL023651, 2005.
Leck, C. and Persson, C.: Seasonal and short-term variability in dimethyl sulfide, sulfur dioxide and biogenic sulfur and sea salt aerosol particles in the arctic marine boundary layer during summer and autumn, Tellus B, 48, 272–299, https://doi.org/10.1034/j.1600-0889.48.issue2.1.x, 1996a.
Leck, C. and Persson, C.: The central Arctic Ocean as a source of dimethyl sulfide Seasonal variability in relation to biological activity, Tellus B, 48, 156–177, https://doi.org/10.3402/tellusb.v48i2.15834, 1996b.
Leck, C. and Svensson, E.: Importance of aerosol composition and mixing state for cloud droplet activation over the Arctic pack ice in summer, Atmos. Chem. Phys., 15, 2545–2568, https://doi.org/10.5194/acp-15-2545-2015, 2015.
Leck, C., Nilsson, E. D., Bigg, E. K., and Bäcklin, L.: Atmospheric program on the Arctic Ocean Expedition 1996 (AOE-96): An overview of scientific goals, experimental approach, and instruments, J. Geophys. Res.-Atmos., 106, 32051–32067, https://doi.org/10.1029/2000JD900461, 2001.
Leck, C., Tjernström, M., Matrai, P., Swietlicki, E., and Bigg, K.: Can marine micro-organisms influence melting of the Arctic pack ice?, Eos Trans. Am. Geophys. Union, 85, 25–32, https://doi.org/10.1029/2004EO030001, 2004.
Leck, C., Matrai, P., Perttu, A.-M., and Gårdfeldt, K.: Expedition report: SWEDARTIC Arctic Ocean 2018, Swedish Polar Research Secretariat, ISBN 978-91-519-3671-0, 2019.
Lee, C., Martin, R. V., Van Donkelaar, A., Lee, H., Dickerson, R. R., Hains, J. C., Krotkov, N., Richter, A., Vinnikov, K., and Schwab, J. J.: SO2 emissions and lifetimes: Estimates from inverse modeling using in situ and global, space-based (SCIAMACHY and OMI) observations, J. Geophys. Res., 116, D06304, https://doi.org/10.1029/2010JD014758, 2011.
Lee, H., Lee, K., Lunder, C. R., Krejci, R., Aas, W., Park, J., Park, K.-T., Lee, B. Y., Yoon, Y. J., and Park, K.: Atmospheric new particle formation characteristics in the Arctic as measured at Mount Zeppelin, Svalbard, from 2016 to 2018, Atmos. Chem. Phys., 20, 13425–13441, https://doi.org/10.5194/acp-20-13425-2020, 2020.
Liu, F., Choi, S., Li, C., Fioletov, V. E., McLinden, C. A., Joiner, J., Krotkov, N. A., Bian, H., Janssens-Maenhout, G., Darmenov, A. S., and da Silva, A. M.: A new global anthropogenic SO2 emission inventory for the last decade: a mosaic of satellite-derived and bottom-up emissions, Atmos. Chem. Phys., 18, 16571–16586, https://doi.org/10.5194/acp-18-16571-2018, 2018.
Lucas, D. D. and Prinn, R. G.: Mechanistic studies of dimethylsulfide oxidation products using an observationally constrained model, J. Geophys. Res.-Atmos., 107, ACH 12-1–ACH 12-26, https://doi.org/10.1029/2001JD000843, 2002.
Lundén, J., Svensson, G., Wisthaler, A., Tjernström, M., Hansel, A., and Leck, C.: The vertical distribution of atmospheric DMS in the high Arctic summer, Tellus B, 62, 160–171, https://doi.org/10.1111/j.1600-0889.2010.00458.x, 2010.
Mahmood, R., von Salzen, K., Norman, A.-L., Galí, M., and Levasseur, M.: Sensitivity of Arctic sulfate aerosol and clouds to changes in future surface seawater dimethylsulfide concentrations, Atmos. Chem. Phys., 19, 6419–6435, https://doi.org/10.5194/acp-19-6419-2019, 2019.
Martinez-Camara, M., Béjar Haro, B., Stohl, A., and Vetterli, M.: A robust method for inverse transport modeling of atmospheric emissions using blind outlier detection, Geosci. Model Dev., 7, 2303–2311, https://doi.org/10.5194/gmd-7-2303-2014, 2014.
Mauritsen, T., Sedlar, J., Tjernström, M., Leck, C., Martin, M., Shupe, M., Sjogren, S., Sierau, B., Persson, P. O. G., Brooks, I. M., and Swietlicki, E.: An Arctic CCN-limited cloud-aerosol regime, Atmos. Chem. Phys., 11, 165–173, https://doi.org/10.5194/acp-11-165-2011, 2011.
Mayer, K. J., Wang, X., Santander, M. V., Mitts, B. A., Sauer, J. S., Sultana, C. M., Cappa, C. D., and Prather, K. A.: Secondary Marine Aerosol Plays a Dominant Role over Primary Sea Spray Aerosol in Cloud Formation, ACS Cent. Sci., 6, 2259–2266, https://doi.org/10.1021/acscentsci.0c00793, 2020.
McMurry, P. H. and Friedlander, S. K.: New particle formation in the presence of an aerosol, Atmos. Environ., 13, 1635–1651, https://doi.org/10.1016/0004-6981(79)90322-6, 1979.
Meier, W. N., Hovelsrud, G. K., van Oort, B. E. H., Key, J. R., Kovacs, K. M., Michel, C., Haas, C., Granskog, M. A., Gerland, S., Perovich, D. K., Makshtas, A., and Reist, J. D.: Arctic sea ice in transformation: A review of recent observed changes and impacts on biology and human activity, Rev. Geophys., 52, 185–217, https://doi.org/10.1002/2013RG000431, 2014.
Moffett, C. E., Barrett, T. E., Liu, J., Gunsch, M. J., Upchurch, L. M., Quinn, P. K., Pratt, K. A., and Sheesley, R. J.: Long-Term Trends for Marine Sulfur Aerosol in the Alaskan Arctic and Relationships With Temperature, J. Geophys. Res.-Atmos., 125, e2020JD033225, https://doi.org/10.1029/2020JD033225, 2020.
Nicolaus, M., Perovich, D. K., Spreen, G., Granskog, M. A., von Albedyll, L., Angelopoulos, M., Anhaus, P., Arndt, S., Belter, H. J., Bessonov, V., Birnbaum, G., Brauchle, J., Calmer, R., Cardellach, E., Cheng, B., Clemens-Sewall, D., Dadic, R., Damm, E., de Boer, G., Demir, O., Dethloff, K., Divine, D. V., Fong, A. A., Fons, S., Frey, M. M., Fuchs, N., Gabarró, C., Gerland, S., Goessling, H. F., Gradinger, R., Haapala, J., Haas, C., Hamilton, J., Hannula, H.-R., Hendricks, S., Herber, A., Heuzé, C., Hoppmann, M., Høyland, K. V., Huntemann, M., Hutchings, J. K., Hwang, B., Itkin, P., Jacobi, H.-W., Jaggi, M., Jutila, A., Kaleschke, L., Katlein, C., Kolabutin, N., Krampe, D., Kristensen, S. S., Krumpen, T., Kurtz, N., Lampert, A., Lange, B. A., Lei, R., Light, B., Linhardt, F., Liston, G. E., Loose, B., Macfarlane, A. R., Mahmud, M., Matero, I. O., Maus, S., Morgenstern, A., Naderpour, R., Nandan, V., Niubom, A., Oggier, M., Oppelt, N., Pätzold, F., Perron, C., Petrovsky, T., Pirazzini, R., Polashenski, C., Rabe, B., Raphael, I. A., Regnery, J., Rex, M., Ricker, R., Riemann-Campe, K., Rinke, A., Rohde, J., Salganik, E., Scharien, R. K., Schiller, M., Schneebeli, M., Semmling, M., Shimanchuk, E., Shupe, M. D., Smith, M. M., Smolyanitsky, V., Sokolov, V., Stanton, T., Stroeve, J., Thielke, L., Timofeeva, A., Tonboe, R. T., Tavri, A., Tsamados, M., Wagner, D. N., Watkins, D., Webster, M., and Wendisch, M.: Overview of the MOSAiC expedition: Snow and sea ice, Elem. Sci. Anthr., 10, 000046, https://doi.org/10.1525/elementa.2021.000046, 2022.
Nielsen, I. E., Skov, H., Massling, A., Eriksson, A. C., Dall'Osto, M., Junninen, H., Sarnela, N., Lange, R., Collier, S., Zhang, Q., Cappa, C. D., and Nøjgaard, J. K.: Biogenic and anthropogenic sources of aerosols at the High Arctic site Villum Research Station, Atmos. Chem. Phys., 19, 10239–10256, https://doi.org/10.5194/acp-19-10239-2019, 2019.
O'Dowd, C. D., Jimenez, J. L., Bahreini, R., Flagan, R. C., Seinfeld, J. H., Hämeri, K., Pirjola, L., Kulmala, M., Jennings, S. G., and Hoffmann, T.: Marine aerosol formation from biogenic iodine emissions, Nature, 417, 632–636, https://doi.org/10.1038/nature00775, 2002.
Okuljar, M., Kuuluvainen, H., Kontkanen, J., Garmash, O., Olin, M., Niemi, J. V., Timonen, H., Kangasluoma, J., Tham, Y. J., Baalbaki, R., Sipilä, M., Salo, L., Lintusaari, H., Portin, H., Teinilä, K., Aurela, M., Dal Maso, M., Rönkkö, T., Petäjä, T., and Paasonen, P.: Measurement report: The influence of traffic and new particle formation on the size distribution of 1–800 nm particles in Helsinki – a street canyon and an urban background station comparison, Atmos. Chem. Phys., 21, 9931–9953, https://doi.org/10.5194/acp-21-9931-2021, 2021.
Park, K.-T., Lee, K., Yoon, Y.-J., Lee, H.-W., Kim, H.-C., Lee, B.-Y., Hermansen, O., Kim, T.-W., and Holmén, K.: Linking atmospheric dimethyl sulfide and the Arctic Ocean spring bloom, Geophys. Res. Lett., 40, 155–160, https://doi.org/10.1029/2012GL054560, 2013.
Pernov, J. B., Beddows, D., Thomas, D. C., Dall'Osto, M., Harrison, R. M., Schmale, J., Skov, H., and Massling, A.: Increased aerosol concentrations in the High Arctic attributable to changing atmospheric transport patterns, Npj Clim. Atmos. Sci., 5, 1–13, https://doi.org/10.1038/s41612-022-00286-y, 2022.
Pisso, I., Sollum, E., Grythe, H., Kristiansen, N. I., Cassiani, M., Eckhardt, S., Arnold, D., Morton, D., Thompson, R. L., Groot Zwaaftink, C. D., Evangeliou, N., Sodemann, H., Haimberger, L., Henne, S., Brunner, D., Burkhart, J. F., Fouilloux, A., Brioude, J., Philipp, A., Seibert, P., and Stohl, A.: The Lagrangian particle dispersion model FLEXPART version 10.4, Geosci. Model Dev., 12, 4955–4997, https://doi.org/10.5194/gmd-12-4955-2019, 2019.
Plane, J. M. C., Joseph, D. M., Allan, B. J., Ashworth, S. H., and Francisco, J. S.: An Experimental and Theoretical Study of the Reactions OIO + NO and OIO + OH, J. Phys. Chem. A, 110, 93–100, https://doi.org/10.1021/jp055364y, 2006.
Pratt, K. A.: Tropospheric Halogen Photochemistry in the Rapidly Changing Arctic, Trends Chem., 1, 545–548, https://doi.org/10.1016/J.TRECHM.2019.06.001, 2019.
Quéléver, L. L. J., Dada, L., Asmi, E., Lampilahti, J., Chan, T., Ferrara, J. E., Copes, G. E., Pérez-Fogwill, G., Barreira, L., Aurela, M., Worsnop, D. R., Jokinen, T., and Sipilä, M.: Investigation of new particle formation mechanisms and aerosol processes at Marambio Station, Antarctic Peninsula, Atmos. Chem. Phys., 22, 8417–8437, https://doi.org/10.5194/acp-22-8417-2022, 2022.
Quinn, P. K. and Bates, T. S.: The case against climate regulation via oceanic phytoplankton sulphur emissions, Nature, 480, 51–56, https://doi.org/10.1038/nature10580, 2011.
Quinn, P. K., Shaw, G., Andrews, E., Dutton, E. G., Ruoho-Airola, T., and Gong, S. L.: Arctic haze: current trends and knowledge gaps, Tellus B, 59, 99–114, https://doi.org/10.1111/j.1600-0889.2006.00236.x, 2007.
Quinn, P. K., Bates, T. S., Schulz, K., and Shaw, G. E.: Decadal trends in aerosol chemical composition at Barrow, Alaska: 1976–2008, Atmos. Chem. Phys., 9, 8883–8888, https://doi.org/10.5194/acp-9-8883-2009, 2009.
Rabe, B., Heuzé, C., Regnery, J., Aksenov, Y., Allerholt, J., Athanase, M., Bai, Y., Basque, C., Bauch, D., Baumann, T. M., Chen, D., Cole, S. T., Craw, L., Davies, A., Damm, E., Dethloff, K., Divine, D. V., Doglioni, F., Ebert, F., Fang, Y.-C., Fer, I., Fong, A. A., Gradinger, R., Granskog, M. A., Graupner, R., Haas, C., He, H., He, Y., Hoppmann, M., Janout, M., Kadko, D., Kanzow, T., Karam, S., Kawaguchi, Y., Koenig, Z., Kong, B., Krishfield, R. A., Krumpen, T., Kuhlmey, D., Kuznetsov, I., Lan, M., Laukert, G., Lei, R., Li, T., Torres-Valdés, S., Lin, L., Lin, L., Liu, H., Liu, N., Loose, B., Ma, X., McKay, R., Mallet, M., Mallett, R. D. C., Maslowski, W., Mertens, C., Mohrholz, V., Muilwijk, M., Nicolaus, M., O'Brien, J. K., Perovich, D., Ren, J., Rex, M., Ribeiro, N., Rinke, A., Schaffer, J., Schuffenhauer, I., Schulz, K., Shupe, M. D., Shaw, W., Sokolov, V., Sommerfeld, A., Spreen, G., Stanton, T., Stephens, M., Su, J., Sukhikh, N., Sundfjord, A., Thomisch, K., Tippenhauer, S., Toole, J. M., Vredenborg, M., Walter, M., Wang, H., Wang, L., Wang, Y., Wendisch, M., Zhao, J., Zhou, M., and Zhu, J.: Overview of the MOSAiC expedition: Physical oceanography, Elem. Sci. Anthr., 10, 00062, https://doi.org/10.1525/elementa.2021.00062, 2022.
Rantanen, M., Karpechko, A. Y., Lipponen, A., Nordling, K., Hyvärinen, O., Ruosteenoja, K., Vihma, T., and Laaksonen, A.: The Arctic has warmed nearly four times faster than the globe since 1979, Commun. Earth Environ., 3, 1–10, https://doi.org/10.1038/s43247-022-00498-3, 2022.
Raso, A. R. W., Custard, K. D., May, N. W., Tanner, D., Newburn, M. K., Walker, L., Moore, R. J., Huey, L. G., Alexander, L., Shepson, P. B., and Pratt, K. A.: Active molecular iodine photochemistry in the Arctic, P. Natl. Acad. Sci. USA, 114, 10053–10058, https://doi.org/10.1073/pnas.1702803114, 2017.
Saiz-Lopez, A., Chance, K., Liu, X., Kurosu, T. P., and Sander, S. P.: First observations of iodine oxide from space, Geophys. Res. Lett., 34, L12812, https://doi.org/10.1029/2007GL030111, 2007.
Saiz-Lopez, A., Blaszczak-Boxe, C. S., and Carpenter, L. J.: A mechanism for biologically induced iodine emissions from sea ice, Atmos. Chem. Phys., 15, 9731–9746, https://doi.org/10.5194/acp-15-9731-2015, 2015.
Sathyendranath, S., Brewin, R. J. W., Brockmann, C., Brotas, V., Calton, B., Chuprin, A., Cipollini, P., Couto, A. B., Dingle, J., Doerffer, R., Donlon, C., Dowell, M., Farman, A., Grant, M., Groom, S., Horseman, A., Jackson, T., Krasemann, H., Lavender, S., Martinez-Vicente, V., Mazeran, C., Mélin, F., Moore, T. S., Müller, D., Regner, P., Roy, S., Steele, C. J., Steinmetz, F., Swinton, J., Taberner, M., Thompson, A., Valente, A., Zühlke, M., Brando, V. E., Feng, H., Feldman, G., Franz, B. A., Frouin, R., Gould, R. W., Hooker, S. B., Kahru, M., Kratzer, S., Mitchell, B. G., Muller-Karger, F. E., Sosik, H. M., Voss, K. J., Werdell, J., and Platt, T.: An Ocean-Colour Time Series for Use in Climate Studies: The Experience of the Ocean-Colour Climate Change Initiative (OC-CCI), Sensors, 19, 4285, https://doi.org/10.3390/s19194285, 2019.
Sathyendranath, S., Jackson, T., Brockmann, C., Brotas, V., Calton, B., Chuprin, A., Clements, O., Cipollini, P., Danne, O., Dingle, J., Donlon, C., Grant, M., Groom, S., Krasemann, H., Lavender, S., Mazeran, C., Mélin, F., Müller, D., Steinmetz, F., Valente, A., Zühlke, M., Feldman, G., Franz, B., Frouin, R., Werdell, J., and Platt, T.: ESA Ocean Colour Climate Change Initiative (Ocean_Colour_cci): Version 5.0 Data, NERC EDS Centre for Environmental Data Analysis [data set], https://doi.org/10.5285/1dbe7a109c0244aaad713e078fd3059a, 2021.
Sauvage, B., Fontaine, A., Eckhardt, S., Auby, A., Boulanger, D., Petetin, H., Paugam, R., Athier, G., Cousin, J.-M., Darras, S., Nédélec, P., Stohl, A., Turquety, S., Cammas, J.-P., and Thouret, V.: Source attribution using FLEXPART and carbon monoxide emission inventories: SOFT-IO version 1.0, Atmos. Chem. Phys., 17, 15271–15292, https://doi.org/10.5194/acp-17-15271-2017, 2017.
Schmale, J. and Baccarini, A.: Progress in Unraveling Atmospheric New Particle Formation and Growth Across the Arctic, Geophys. Res. Lett., 48, e2021GL094198, https://doi.org/10.1029/2021GL094198, 2021.
Schmale, J., Sharma, S., Decesari, S., Pernov, J., Massling, A., Hansson, H.-C., von Salzen, K., Skov, H., Andrews, E., Quinn, P. K., Upchurch, L. M., Eleftheriadis, K., Traversi, R., Gilardoni, S., Mazzola, M., Laing, J., and Hopke, P.: Pan-Arctic seasonal cycles and long-term trends of aerosol properties from 10 observatories, Atmos. Chem. Phys., 22, 3067–3096, https://doi.org/10.5194/acp-22-3067-2022, 2022.
Schmithüsen, H.: Continuous meteorological surface measurement during POLARSTERN cruise PS122/1. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.935221, 2021a.
Schmithüsen, H.: Continuous meteorological surface measurement during POLARSTERN cruise PS122/2. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.935222, 2021b.
Schmithüsen, H.: Continuous meteorological surface measurement during POLARSTERN cruise PS122/3. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.935223, 2021c.
Schmithüsen, H.: Continuous meteorological surface measurement during POLARSTERN cruise PS122/4. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.935224, 2021d.
Schmithüsen, H.: Continuous meteorological surface measurement during POLARSTERN cruise PS122/5. Alfred Wegener Institute, Helmholtz Centre for Polar and Marine Research, Bremerhaven, PANGAEA [data set], https://doi.org/10.1594/PANGAEA.935225, 2021e.
Seibert, P.: Inverse Dispersion Modelling Based on Trajectory-Derived Source-Receptor Relationships, in: Air Pollution Modeling and Its Application XII, edited by: Gryning Sven-Erikand Chaumerliac, N., Springer US, Boston, MA, 711–713, https://doi.org/10.1007/978-1-4757-9128-0_80, 1998.
Seibert, P. and Frank, A.: Source-receptor matrix calculation with a Lagrangian particle dispersion model in backward mode, Atmos. Chem. Phys., 4, 51–63, https://doi.org/10.5194/acp-4-51-2004, 2004.
Sharma, S., Chan, E., Ishizawa, M., Toom-Sauntry, D., Gong, S. L., Li, S. M., Tarasick, D. W., Leaitch, W. R., Norman, A., Quinn, P. K., Bates, T. S., Levasseur, M., Barrie, L. A., and Maenhaut, W.: Influence of transport and ocean ice extent on biogenic aerosol sulfur in the Arctic atmosphere, J. Geophys. Res.-Atmos., 117, D12209, https://doi.org/10.1029/2011JD017074, 2012.
Sharma, S., Barrie, L. a., Magnusson, E., Brattström, G., Leaitch, W. r., Steffen, A., and Landsberger, S.: A Factor and Trends Analysis of Multidecadal Lower Tropospheric Observations of Arctic Aerosol Composition, Black Carbon, Ozone, and Mercury at Alert, Canada, J. Geophys. Res.-Atmos., 124, 14133–14161, https://doi.org/10.1029/2019JD030844, 2019.
Shen, J., Scholz, W., He, X.-C., Zhou, P., Marie, G., Wang, M., Marten, R., Surdu, M., Rörup, B., Baalbaki, R., Amorim, A., Ataei, F., Bell, D. M., Bertozzi, B., Brasseur, Z., Caudillo, L., Chen, D., Chu, B., Dada, L., Duplissy, J., Finkenzeller, H., Granzin, M., Guida, R., Heinritzi, M., Hofbauer, V., Iyer, S., Kemppainen, D., Kong, W., Krechmer, J. E., Kürten, A., Lamkaddam, H., Lee, C. P., Lopez, B., Mahfouz, N. G. A., Manninen, H. E., Massabò, D., Mauldin, R. L., Mentler, B., Müller, T., Pfeifer, J., Philippov, M., Piedehierro, A. A., Roldin, P., Schobesberger, S., Simon, M., Stolzenburg, D., Tham, Y. J., Tomé, A., Umo, N. S., Wang, D., Wang, Y., Weber, S. K., Welti, A., Wollesen De Jonge, R., Wu, Y., Zauner-Wieczorek, M., Zust, F., Baltensperger, U., Curtius, J., Flagan, R. C., Hansel, A., Möhler, O., Petäjä, T., Volkamer, R., Kulmala, M., Lehtipalo, K., Rissanen, M., Kirkby, J., El-Haddad, I., Bianchi, F., Sipilä, M., Donahue, N. M., and Worsnop, D. R.: High Gas-Phase Methanesulfonic Acid Production in the OH-Initiated Oxidation of Dimethyl Sulfide at Low Temperatures, Environ. Sci. Technol., 56, 13931–13944, https://doi.org/10.1021/acs.est.2c05154, 2022.
Shupe, M. D., Rex, M., Blomquist, B., Persson, P. O. G., Schmale, J., Uttal, T., Althausen, D., Angot, H., Archer, S., Bariteau, L., Beck, I., Bilberry, J., Bucci, S., Buck, C., Boyer, M., Brasseur, Z., Brooks, I. M., Calmer, R., Cassano, J., Castro, V., Chu, D., Costa, D., Cox, C. J., Creamean, J., Crewell, S., Dahlke, S., Damm, E., de Boer, G., Deckelmann, H., Dethloff, K., Dütsch, M., Ebell, K., Ehrlich, A., Ellis, J., Engelmann, R., Fong, A. A., Frey, M. M., Gallagher, M. R., Ganzeveld, L., Gradinger, R., Graeser, J., Greenamyer, V., Griesche, H., Griffiths, S., Hamilton, J., Heinemann, G., Helmig, D., Herber, A., Heuzé, C., Hofer, J., Houchens, T., Howard, D., Inoue, J., Jacobi, H.-W., Jaiser, R., Jokinen, T., Jourdan, O., Jozef, G., King, W., Kirchgaessner, A., Klingebiel, M., Krassovski, M., Krumpen, T., Lampert, A., Landing, W., Laurila, T., Lawrence, D., Lonardi, M., Loose, B., Lüpkes, C., Maahn, M., Macke, A., Maslowski, W., Marsay, C., Maturilli, M., Mech, M., Morris, S., Moser, M., Nicolaus, M., Ortega, P., Osborn, J., Pätzold, F., Perovich, D. K., Petäjä, T., Pilz, C., Pirazzini, R., Posman, K., Powers, H., Pratt, K. A., Preußer, A., Quéléver, L., Radenz, M., Rabe, B., Rinke, A., Sachs, T., Schulz, A., Siebert, H., Silva, T., Solomon, A., Sommerfeld, A., Spreen, G., Stephens, M., Stohl, A., Svensson, G., Uin, J., Viegas,J., Voigt, C., von der Gathen, P., Wehner, B., Welker, J. M., Wendisch, M., Werner, M., Xie, Z., and Yue, F.: Overview of the MOSAiC expedition: Atmosphere, Elem. Sci. Anthr., 10, 00060, https://doi.org/10.1525/elementa.2021.00060, 2022.
Siegel, K., Gramlich, Y., Haslett, S. L., Freitas, G., Krejci, R., Zieger, P., and Mohr, C.: Arctic observations of hydroperoxymethyl thioformate (HPMTF) – seasonal behavior and relationship to other oxidation products of dimethyl sulfide at the Zeppelin Observatory, Svalbard, Atmos. Chem. Phys., 23, 7569–7587, https://doi.org/10.5194/acp-23-7569-2023, 2023.
Sipilä, M., Sarnela, N., Jokinen, T., Henschel, H., Junninen, H., Kontkanen, J., Richters, S., Kangasluoma, J., Franchin, A., Peräkylä, O., Rissanen, M. P., Ehn, M., Vehkamäki, H., Kurten, T., Berndt, T., Petäjä, T., Worsnop, D., Ceburnis, D., Kerminen, V.-M., Kulmala, M., and O'Dowd, C.: Molecular-scale evidence of aerosol particle formation via sequential addition of HIO3, Nature, 537, 532–534, https://doi.org/10.1038/nature19314, 2016.
Sipilä, M., Sarnela, N., Neitola, K., Laitinen, T., Kemppainen, D., Beck, L., Duplissy, E.-M., Kuittinen, S., Lehmusjärvi, T., Lampilahti, J., Kerminen, V.-M., Lehtipalo, K., Aalto, P. P., Keronen, P., Siivola, E., Rantala, P. A., Worsnop, D. R., Kulmala, M., Jokinen, T., and Petäjä, T.: Wintertime subarctic new particle formation from Kola Peninsula sulfur emissions, Atmos. Chem. Phys., 21, 17559–17576, https://doi.org/10.5194/acp-21-17559-2021, 2021.
Smith, M. M., Angot, H., Chamberlain, E. J., Droste, E. S., Karam, S., Muilwijk, M., Webb, A. L., Archer, S. D., Beck, I., Blomquist, B. W., Bowman, J., Boyer, M., Bozzato, D., Chierici, M., Creamean, J., D'Angelo, A., Delille, B., Fer, I., Fong, A. A., Fransson, A., Fuchs, N., Gardner, J., Granskog, M. A., Hoppe, C. J. M., Hoppema, M., Hoppmann, M., Mock, T., Muller, S., Müller, O., Nicolaus, M., Nomura, D., Petäjä, T., Salganik, E., Schmale, J., Schmidt, K., Schulz, K. M., Shupe, M. D., Stefels, J., Thielke, L., Tippenhauer, S., Ulfsbo, A., van Leeuwe, M., Webster, M., Yoshimura, M., and Zhan, L.: Thin and transient meltwater layers and false bottoms in the Arctic sea ice pack—Recent insights on these historically overlooked features, Elem. Sci. Anthr., 11, 00025, https://doi.org/10.1525/elementa.2023.00025, 2023.
Smith, S. J., Pitcher, H., and Wigley, T. M. L.: Global and regional anthropogenic sulfur dioxide emissions, Global Planet. Change, 29, 99–119, https://doi.org/10.1016/S0921-8181(00)00057-6, 2001.
Spolaor, A., Barbaro, E., Cappelletti, D., Turetta, C., Mazzola, M., Giardi, F., Björkman, M. P., Lucchetta, F., Dallo, F., Pfaffhuber, K. A., Angot, H., Dommergue, A., Maturilli, M., Saiz-Lopez, A., Barbante, C., and Cairns, W. R. L.: Diurnal cycle of iodine, bromine, and mercury concentrations in Svalbard surface snow, Atmos. Chem. Phys., 19, 13325–13339, https://doi.org/10.5194/acp-19-13325-2019, 2019.
Stefels, J., Steinke, M., Turner, S., Malin, G., and Belviso, S.: Environmental constraints on the production and removal of the climatically active gas dimethylsulphide (DMS) and implications for ecosystem modelling, Biogeochemistry, 83, 245–275, https://doi.org/10.1007/s10533-007-9091-5, 2007.
Stohl, A.: Characteristics of atmospheric transport into the Arctic troposphere, J. Geophys. Res.-Atmos., 111, D11306, https://doi.org/10.1029/2005JD006888, 2006.
Terhaar, J., Lauerwald, R., Regnier, P., Gruber, N., and Bopp, L.: Around one third of current Arctic Ocean primary production sustained by rivers and coastal erosion, Nat. Commun., 12, 169, https://doi.org/10.1038/s41467-020-20470-z, 2021.
Thakur, R. C., Dada, L., Beck, L. J., Quéléver, L. L. J., Chan, T., Marbouti, M., He, X.-C., Xavier, C., Sulo, J., Lampilahti, J., Lampimäki, M., Tham, Y. J., Sarnela, N., Lehtipalo, K., Norkko, A., Kulmala, M., Sipilä, M., and Jokinen, T.: An evaluation of new particle formation events in Helsinki during a Baltic Sea cyanobacterial summer bloom, Atmos. Chem. Phys., 22, 6365–6391, https://doi.org/10.5194/acp-22-6365-2022, 2022.
Tjernström, M., Leck, C., Birch, C. E., Bottenheim, J. W., Brooks, B. J., Brooks, I. M., Bäcklin, L., Chang, R. Y.-W., de Leeuw, G., Di Liberto, L., de la Rosa, S., Granath, E., Graus, M., Hansel, A., Heintzenberg, J., Held, A., Hind, A., Johnston, P., Knulst, J., Martin, M., Matrai, P. A., Mauritsen, T., Müller, M., Norris, S. J., Orellana, M. V., Orsini, D. A., Paatero, J., Persson, P. O. G., Gao, Q., Rauschenberg, C., Ristovski, Z., Sedlar, J., Shupe, M. D., Sierau, B., Sirevaag, A., Sjogren, S., Stetzer, O., Swietlicki, E., Szczodrak, M., Vaattovaara, P., Wahlberg, N., Westberg, M., and Wheeler, C. R.: The Arctic Summer Cloud Ocean Study (ASCOS): overview and experimental design, Atmos. Chem. Phys., 14, 2823–2869, https://doi.org/10.5194/acp-14-2823-2014, 2014.
Tunved, P., Ström, J., and Krejci, R.: Arctic aerosol life cycle: linking aerosol size distributions observed between 2000 and 2010 with air mass transport and precipitation at Zeppelin station, Ny-Ålesund, Svalbard, Atmos. Chem. Phys., 13, 3643–3660, https://doi.org/10.5194/acp-13-3643-2013, 2013.
Uhlig, C., Damm, E., Peeken, I., Krumpen, T., Rabe, B., Korhonen, M., and Ludwichowski, K.-U.: Sea Ice and Water Mass Influence Dimethylsulfide Concentrations in the Central Arctic Ocean, Front. Earth Sci., 7, 179, https://doi.org/10.3389/feart.2019.00179, 2019.
Veres, P. R., Neuman, J. A., Bertram, T. H., Assaf, E., Wolfe, G. M., Williamson, C. J., Weinzierl, B., Tilmes, S., Thompson, C. R., Thames, A. B., Schroder, J. C., Saiz-Lopez, A., Rollins, A. W., Roberts, J. M., Price, D., Peischl, J., Nault, B. A., Møller, K. H., Miller, D. O., Meinardi, S., Li, Q., Lamarque, J.-F., Kupc, A., Kjaergaard, H. G., Kinnison, D., Jimenez, J. L., Jernigan, C. M., Hornbrook, R. S., Hills, A., Dollner, M., Day, D. A., Cuevas, C. A., Campuzano-Jost, P., Burkholder, J., Bui, T. P., Brune, W. H., Brown, S. S., Brock, C. A., Bourgeois, I., Blake, D. R., Apel, E. C., and Ryerson, T. B.: Global airborne sampling reveals a previously unobserved dimethyl sulfide oxidation mechanism in the marine atmosphere, P. Natl. Acad. Sci. USA, 117, 4505–4510, https://doi.org/10.1073/pnas.1919344117, 2020.
Willis, M. D., Köllner, F., Burkart, J., Bozem, H., Thomas, J. L., Schneider, J., Aliabadi, A. A., Hoor, P. M., Schulz, H., Herber, A. B., Leaitch, W. R., and Abbatt, J. P. D.: Evidence for marine biogenic influence on summertime Arctic aerosol, Geophys. Res. Lett., 44, 6460–6470, https://doi.org/10.1002/2017GL073359, 2017.
Willis, M. D., Leaitch, W. R., and Abbatt, J. P. D.: Processes Controlling the Composition and Abundance of Arctic Aerosol, Rev. Geophys., 56, 621–671, https://doi.org/10.1029/2018RG000602, 2018.
Yan, C., Dada, L., Rose, C., Jokinen, T., Nie, W., Schobesberger, S., Junninen, H., Lehtipalo, K., Sarnela, N., Makkonen, U., Garmash, O., Wang, Y., Zha, Q., Paasonen, P., Bianchi, F., Sipilä, M., Ehn, M., Petäjä, T., Kerminen, V.-M., Worsnop, D. R., and Kulmala, M.: The role of H2SO4-NH3 anion clusters in ion-induced aerosol nucleation mechanisms in the boreal forest, Atmos. Chem. Phys., 18, 13231–13243, https://doi.org/10.5194/acp-18-13231-2018, 2018.
Yoch, D. C.: Dimethylsulfoniopropionate: its sources, role in the marine food web, and biological degradation to dimethylsulfide, Appl. Environ. Microb., 68, 5804–5815, https://doi.org/10.1128/AEM.68.12.5804-5815.2002, 2002.
Zollner, J. H., Glasoe, W. A., Panta, B., Carlson, K. K., McMurry, P. H., and Hanson, D. R.: Sulfuric acid nucleation: power dependencies, variation with relative humidity, and effect of bases, Atmos. Chem. Phys., 12, 4399–4411, https://doi.org/10.5194/acp-12-4399-2012, 2012.
Zou, H. and Hastie, T.: Regularization and Variable Selection Via the Elastic Net, J. R. Stat. Soc. B, 67, 301–320, https://doi.org/10.1111/j.1467-9868.2005.00503.x, 2005.