the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 13 Apr 2023
| 13 Apr 2023
Selective deuteration as a tool for resolving autoxidation mechanisms in α-pinene ozonolysis
Otso Peräkylä
Jonathan G. Varelas
Jingyi Luo
Runlong Cai
Yanjun Zhang
Theo Kurtén
Matthieu Riva
Matti Rissanen
Franz M. Geiger
Regan J. Thomson
Highly oxygenated organic molecules (HOMs) from α-pinene ozonolysis have been shown to be significant contributors to secondary organic aerosol (SOA), yet our mechanistic understanding of how the peroxy-radical-driven autoxidation leads to their formation in this system is still limited. The involved isomerisation reactions such as H-atom abstractions followed by O2 additions can take place on sub-second timescales in short-lived intermediates, making the process challenging to study. Similarly, while the end-products and sometimes radical intermediates can be observed using mass spectrometry, their structures remain elusive. Therefore, we propose a method utilising selective deuterations for unveiling the mechanisms of autoxidation, where the HOM products can be used to infer which C atoms have taken part in the isomerisation reactions. This relies on the fact that if a C−D bond is broken due to an abstraction by a peroxy group forming a −OOD hydroperoxide, the D atom will become labile and able to be exchanged with a hydrogen atom in water vapour (H2O), effectively leading to loss of the D atom from the molecule.
In this study, we test the applicability of this method using three differently deuterated versions of α-pinene with the newly developed chemical ionisation Orbitrap (CI-Orbitrap) mass spectrometer to inspect the oxidation products. The high mass-resolving power of the Orbitrap is critical, as it allows the unambiguous separation of molecules with a D atom (mD=2.0141) from those with two H atoms (). We found that the method worked well, and we could deduce that two of the three tested compounds had lost D atoms during oxidation, suggesting that those deuterated positions were actively involved in the autoxidation process. Surprisingly, the deuterations were not observed to decrease HOM molar yields, as would have been expected due to kinetic isotope effects. This may be an indication that the relevant H (or D) abstractions were fast enough that no competing pathways were of relevance despite slower abstraction rates of the D atom. We show that selective deuteration can be a very useful method for studying autoxidation on a molecular level and likely is not limited to the system of α-pinene ozonolysis tested here.
Please read the corrigendum first before continuing.
-
Notice on corrigendum
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
-
Article
(8160 KB)
- Corrigendum
-
Supplement
(1226 KB)
-
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(8160 KB) - Full-text XML
- Corrigendum
-
Supplement
(1226 KB) - BibTeX
- EndNote




Highly oxygenated organic molecules (HOMs) are atmospheric compounds with low to extremely low volatilities formed from volatile organic compounds (VOCs) in the atmosphere via autoxidation (Crounse et al., 2013; Ehn et al., 2014; Bianchi et al., 2019). HOMs formed from α-pinene ozonolysis have been shown to contribute significantly to secondary organic aerosol (SOA) (Ehn et al., 2014); however, a mechanistic understanding of this system is limited, despite it having been studied computationally and experimentally (Kurtén et al., 2015; Iyer et al., 2021). Autoxidation involving peroxy radicals (RO2) has been shown to be the source of the high oxygen content (Berndt et al., 2018), but the exact route or routes of isomerisation cannot be derived purely by the mass spectrometric methods (Jokinen et al., 2012; Riva et al., 2019a) typically used to detect the HOMs. In addition, the isomerisation reactions are very fast (Bianchi et al., 2019), which makes following them in real time typically not possible. One way to circumvent these limitations is to perturb the autoxidation process in a way that leads to different HOM products depending on the reaction mechanism.
Deuteration is a commonly used method for multiple applications in chemistry, including atmospheric studies (Rissanen et al., 2014; Zhang et al., 2017; Ye et al., 2018; Michelotti and Roche, 2019; Wang et al., 2020; Li et al., 2021). In deuteration, one or more hydrogen atoms in a molecule are exchanged to deuterium atoms (D). Deuteration can be a full deuteration where all hydrogen atoms are exchanged to deuterium atoms as used by Rissanen et al. (2014), or it can be done selectively, where specific hydrogen atoms are exchanged with deuterium atoms as used by Zhang et al. (2017) and Ye et al. (2018). The deuteration can have several impacts on the reactions taking place, including a decreased rate of reaction due to the higher mass of a D atom compared to a H atom, the so-called kinetic isotope effect (Laidler, 1987). Rissanen et al. (2014) used fully deuterated cyclohexene and found that the ozonolysis formed HOMs at greatly reduced yields compared to the non-deuterated cyclohexene. They also showed that D atoms were exchanged to H atoms in contact with water vapour in cases where the C−D bond was broken, and the D became attached to an oxygen through an O−D bond. In other words, the autoxidation was perturbed in predictable ways by the deuteration. Nevertheless, we expect that selective deuteration can provide substantially more information still. To our knowledge, selective deuteration has not been utilised in studying autoxidation pathways from α-pinene ozonolysis before.
In this study, we inspected α-pinene ozonolysis by simulating atmospheric conditions in a reaction chamber. We used non-deuterated as well as three selectively deuterated precursors to test how the resulting HOM spectrum might change and to gain insight into the autoxidation processes. We used a chemical ionisation Orbitrap mass spectrometer (Riva et al., 2019a) which has a high-enough resolving power to unambiguously distinguish between the different isotopes in the spectra. We compared the spectral distribution of different HOMs as well as the overall HOM yields for the different precursors. In particular, we examined the numbers of D atoms lost for the differently deuterated precursors for different HOM molecules, with the aim of assessing from which C atom abstractions take place during autoxidation. We also compare our findings to the few suggested mechanisms from earlier publications.
In this section, we first describe the selectively deuterated precursors used in this study (Sect. 2.1), after which we describe the main reaction pathways during α-pinene ozonolysis that are of relevance for this study (Sect. 2.2). Lastly, we describe how selective deuteration can be used to study the oxidation processes (Sect. 2.3).
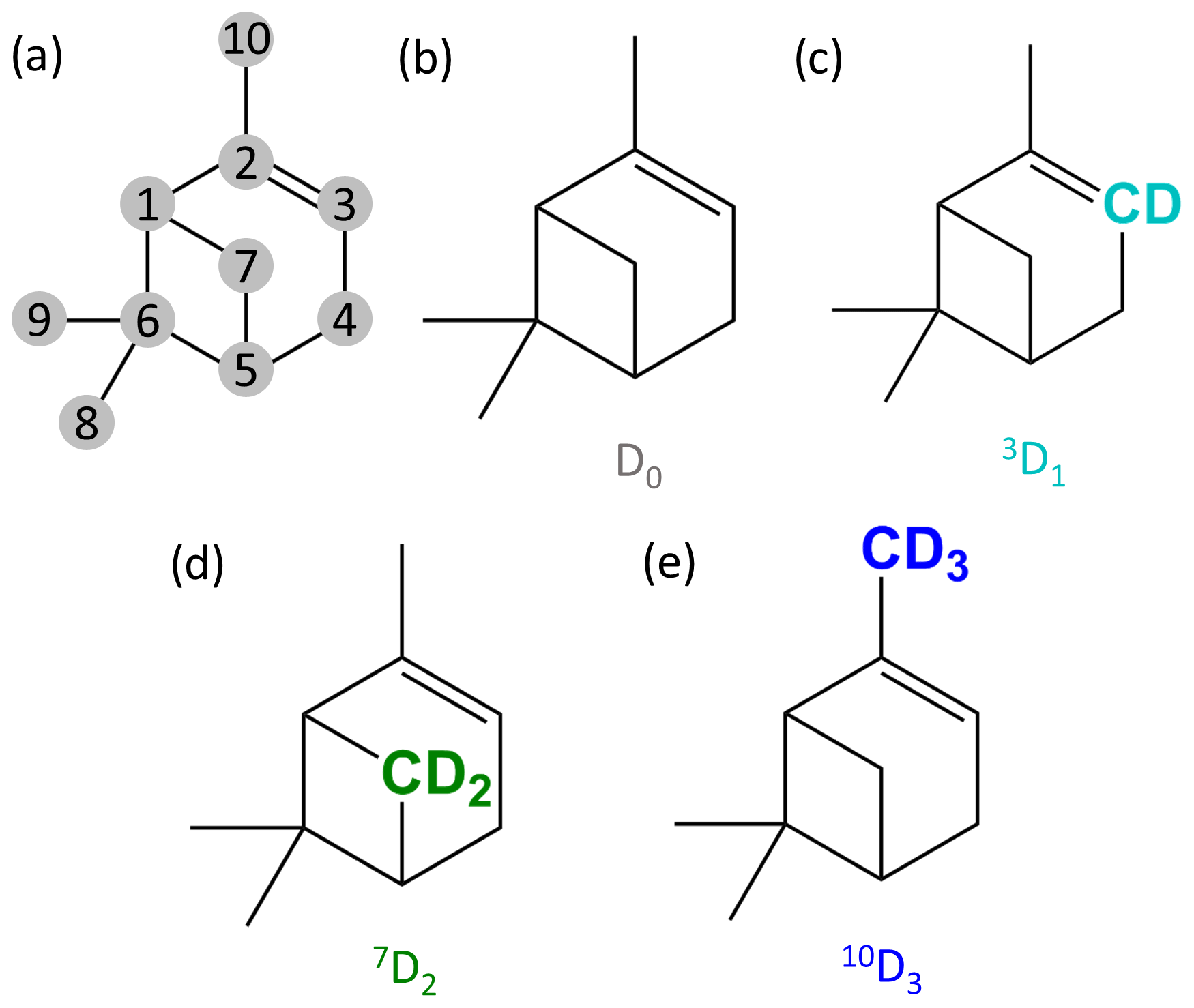
Figure 1α-pinene with numbered carbons following IUPAC numbering (a) and the molecular structures of the selectively deuterated precursors used in this study (b–e). The precursors are named based on the number of the carbon that is deuterated and the number of deuterium atoms added to the molecule. The carbons with the deuterium atoms are highlighted with colours specific to the precursors. (b) Normal α-pinene is D0, (c) α-pinene with one deuterium atom is 3D1 (cyan), (d) α-pinene with two deuterium atoms is 7D2 (green), and (e) α-pinene with three deuterium atoms is 10D3 (blue).
2.1 Selectively deuterated precursors
In this study, we used four precursors, and their chemical structures are shown in Fig. 1. The three selectively deuterated precursors were synthesised at Northwestern University, Illinois, USA. The precursors are named based on the carbon that was deuterated and the number of deuterium atoms they have: D0 is the standard α-pinene molecule without any isotopic alteration (C10H16), 3D1 is α-pinene with one deuterium atom on C-3 (C10H15D, α-pinene-3-d1; see Fig. 1), 7D2 is α-pinene with two deuterium atoms on C-7 (C10H14D2, α-pinene-7,7-d2), and 10D3 is α-pinene with three deuterium atoms on C-10 (C10H13D3, α-pinene-10,10,10-d3). The vinylic C-3 carbon and the allylic C-10 methyl group carbon are expected to undergo changes during the ozonolysis process (Jenkin et al., 1997; Kurtén et al., 2015), while the cyclobutyl-ring C-7 carbon has not to our knowledge been shown to take part in any reactions in the initial ozonolysis reaction.
Synthesis of the 10D3 and 7D2 α-pinene species was carried out according to the published literature procedures (Upshur et al., 2016, 2019), while access to the 3D1 α-pinene was accomplished through a new three-step route utilising nopinone as the starting material (see the Supplement for complete synthetic details). The purity of each compound was >95 % as determined by 1HNMR spectroscopy, and we were unable to observe residual proton resonances associated with the deuterated carbon positions for any sample, indicating complete deuterium incorporation within the detection limits of the spectrometer (see the Supplement for 1HNMR spectral data).
2.2 Autoxidation in α-pinene ozonolysis
As discussed above, autoxidation is a major source of condensable low-volatile vapours from α-pinene ozonolysis. The reaction of α-pinene with ozone starts by the attachment of ozone to the double bond, followed by bond scission and rapid isomerisations via a Criegee intermediate and vinyl hydroperoxide and OH loss, potential isomerisation, and O2 addition to form a primary peroxy radical (C10H15O4), as shown in Fig. 2a (Kurtén et al., 2015; Iyer et al., 2021). From these peroxy radicals, the process can continue with autoxidation, which is successive H shifts and oxygen additions. For pathways I to III, Kurtén et al. (2015) calculated that subsequent H shifts starting from these primary RO2 were too slow to explain observed HOM formation rates. This was primarily due to the intact cyclobutyl ring causing strain for the abstraction of hydrogen atoms across the ring. However, Iyer et al. (2021) found that an isomerisation reaction was available for the vinoxy intermediate in pathway I due to excess energy remaining from the initial ozonolysis reaction. The excess energy allows the breaking of the cyclobutyl ring (pathway IV) and subsequent rapid H shifts leading to RO2 with up to eight oxygen atoms (Fig. 2b). The O8−RO2 product remains the only experimentally and computationally supported HOM-forming pathway in this system (Rissanen et al., 2014; Iyer et al., 2021) and is thus the most promising candidate to explain many of the observed closed-shell HOMs following termination reactions.
The autoxidation process can be terminated via unimolecular or bimolecular reactions, and the most relevant ones are introduced below. A common unimolecular Reaction (R1) of an RO2 that has undergone autoxidation is the H abstraction from a C atom with a hydroperoxide functionality. This rapidly leads to the loss of a hydroxyl radical and the formation of a closed-shell HOM with a carbonyl functionality at said C atom (Crounse et al., 2013). The RO2 can also undergo different bimolecular termination reactions (Atkinson and Arey, 2003). In the absence of NOx, as in this study, there are two relevant reaction partners, namely HO2 and other RO2, and their main reaction products are listed below Reactions (R2)–(R6). In brief, RO2 cross-reactions can form closed-shell monomers (Reaction R2), ROOR accretion products which are commonly referred to as dimers (Reaction R3), or alkoxy radicals RO (Reaction R4), with the latter able to undergo further isomerisation or termination reactions. Reactions of RO2 with HO2 typically form hydroperoxides ROOH (Reaction R5) but can also form RO (Reaction R6), especially for complex-enough RO2 (Hasson et al., 2005; Orlando and Tyndall, 2012; Iyer et al., 2018).
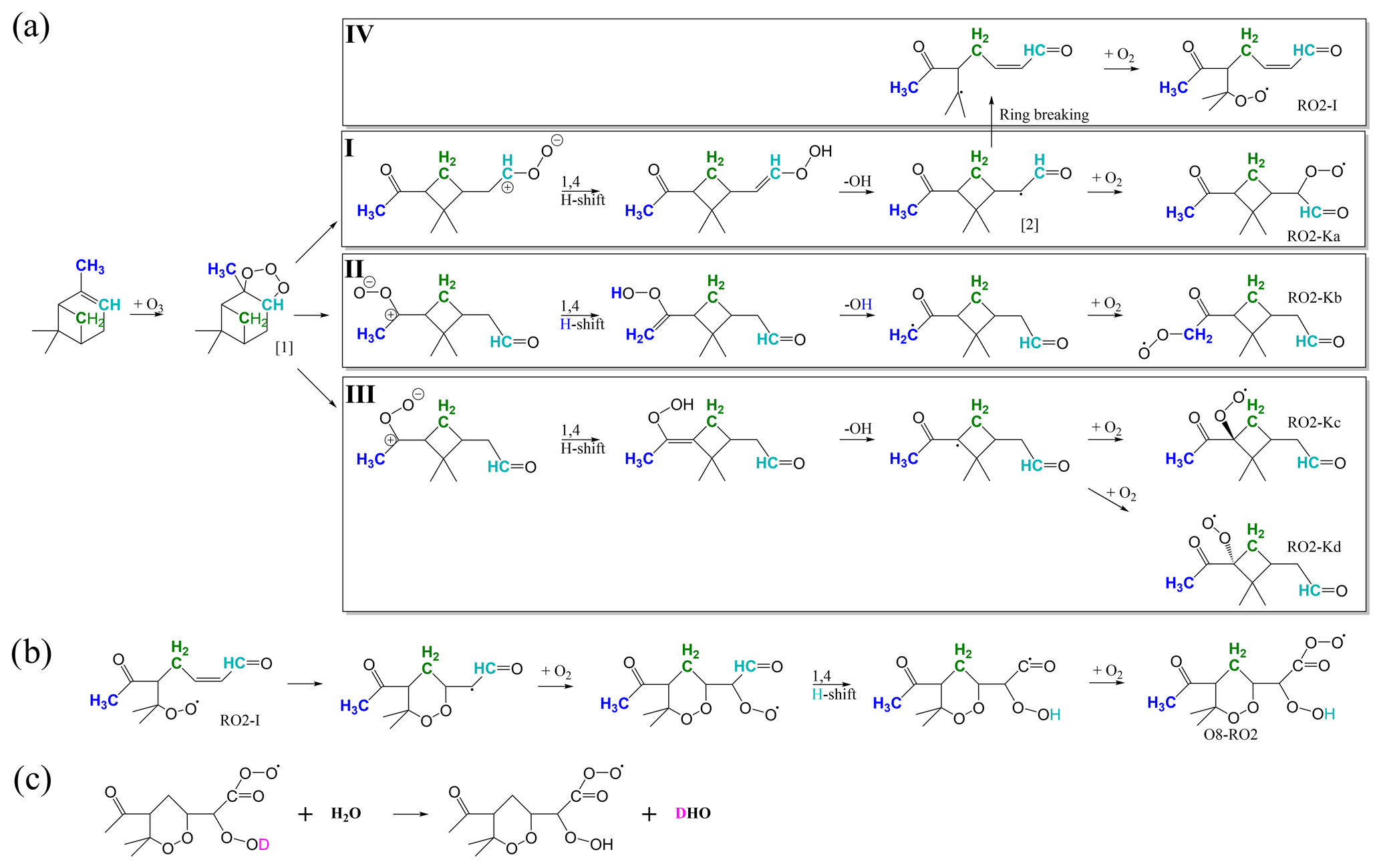
Figure 2(a) Reaction pathways for α-pinene ozonolysis up until the primary peroxy radical (RO2) from Kurtén et al. (2015) (I–III) and Iyer et al. (2021) (IV). (b) Continued reaction pathway for the IV pathway up to a peroxy radical containing eight oxygen atoms (O8−RO2) (Iyer et al., 2021). In panels (a) and (b) the carbon and hydrogen atoms corresponding to deuterated ones in the selectively deuterated precursors are coloured matching the colouring in Fig. 1; i.e. cyan corresponds to the 3D1 precursor, green corresponds to the 7D2 precursor, and blue corresponds to the 10D3 precursor. (c) Deuterium atoms are readily exchanged between HOMs and water vapour if the deuterium is in an −OD or −OOD group, resulting in HOMs without deuterium and semi-heavy water, i.e. a water molecule with one hydrogen atom and one deuterium atom instead of two hydrogen atoms (Rissanen et al., 2014).
2.3 Studying autoxidation with selective deuteration
During the oxidation and autoxidation processes, hydrogen atoms can be lost from the precursor molecule in multiple ways. If a carbon loses a hydrogen atom during an H shift, the carbon has actively participated in the autoxidation process, and we refer to this as the carbon being “active”. More specifically, when discussing the experimental results, we only consider the carbon to be active when we observe the D abstractions taking place at fractions above our uncertainty margins. While we are primarily interested in H shifts by RO2, they can also occur by RO radicals or in the Criegee intermediate step, a reaction that takes place in all cases.
When a hydrogen atom is shifted away from a carbon atom, the hydrogen atom ends up in a hydroperoxide group −OOH, except in the case of H shifts in RO, where the result is a hydroxyl group −OH. In both cases, the H atoms are now labile enough that they can be exchanged in collisions with water vapour molecules. In the special case where a deuterated carbon has been active and a D shift has taken place, the D can eventually be lost from the molecule entirely (example shown in Fig. 2c). With deuteration, we can thus observe the change in the molecular mass when the exchange has taken place and thus specify which carbon in the precursor has been active. Additionally, if a deuterated precursor produces a mass spectral signature for a given HOM molecule where part of the signal has lost a D while another has not, it is an indication of different isomers for this composition. In other words, several pathways lead to the same elemental composition but with different C atoms being active.
If the D atom was internally abstracted by the Criegee intermediate, it will be lost by ejection of the OD, as would be the case in pathway II (Fig. 2a, blue H) for the 10D3 precursor. Furthermore, deuterium atoms can be lost if the selectively deuterated carbon itself is lost through fragmentation during the HOM formation reaction.
Only two of the four pathways discussed earlier result in the precursor losing a deuterium atom. In the four pathways shown in Fig. 2a–b, the carbon and hydrogen atoms corresponding to those of the selectively deuterated precursors are highlighted with colours matching the colouring from Fig. 1. The atoms coloured with cyan correspond to the 3D1 precursor, the green ones correspond to the 7D2 precursor, and the blue ones correspond to the 10D3 precursor. In pathway II, the 10D3 precursor would lose one deuterium when OH is fragmented from the molecule, and in pathway IV with the continued steps in Fig. 2b, the deuterium in the 3D1 precursor would be shifted to a hydroperoxy group and hence readily exchanged to a hydrogen atom with water.
In addition to the deuteration impacting the molecular mass observed in the mass spectra, the H shifts themselves will become much slower since the deuterium atom being abstracted has twice the mass of a hydrogen atom. Rissanen et al. (2014) found that the HOM yields were roughly 100 times lower for ozonolysis of fully deuterated cyclohexene C6D10 compared to that of standard cyclohexene C6H10. Some of the same effects should play a part in this oxidation system, namely the kinetic isotope effect and the slowing down of quantum tunnelling. The kinetic isotope effect is caused by the lower vibrational frequencies for the C−D bond compared to the C−H bond, and it decreases the rate of the abstraction (Laidler, 1987). Furthermore, quantum mechanical tunnelling is important for the abstractions, and this causes an even larger decrease to the reaction rate (Laidler, 1987).
The exact total decrease in the reaction rate coefficients depends on the radical structure, but a typical range is a factor of 20–100 slower for D shifts versus H shifts (Crounse et al., 2011; Praske et al., 2018). However, it is important to keep in mind that the effect on the likelihood of the D shift taking place will still depend on the possible competing reactions. For example, if the H shift is 1000 times faster than any competing reaction, substituting for a D shift will barely impact the branching of the reaction, whereas with similar H-shift rates with any competing reaction, deuteration will largely shut down this pathway.
In this section, we first describe the experimental set-up and the used instruments (Sect. 3.1). Then we describe how the data were analysed (Sect. 3.2).
3.1 Experimental set-up
A schematic of the experimental set-up is shown in Fig. 3. The experiments were conducted in the 2 m3 “COALA” chamber (Teflon FEP chamber, Vector Foiltec, Germany) (Riva et al., 2019b; Peräkylä et al., 2020), a continuous stirred-tank reactor (CSTR) at the University of Helsinki, Finland. We monitored relative humidity (RH) and temperature with a temperature and humidity probe (INTERCAP® HMP60, Vaisala). RH stayed below the detection limit of the probe (under 1 %) and temperature was 26.0 ∘C within the ±1.5 ∘C range. We monitored NOx (NO and NO2) concentrations with a NO–NO2 analyser (model T200UP, Teledyne), and the concentrations were under 0.3 ppb. We generated ozone by running 5 L min−1 clean air through an ozone generator (Dasibi 1008-PC ozone generator). We ran the chamber at a stable O3 concentration ( ppb), monitoring the ozone concentration with a photometric O3 analyser (model 400, Teledyne instruments; ozone monitor in Fig. 3). The total flow through the COALA chamber was kept at 50 L min−1 by adding 45 L min−1 flow of clean air generated in a zero-air generator (AADCO, Series 737-14, Ohio, USA). As a result, we achieved an average residence time of τR≈40 min inside the chamber. With these conditions, we estimate the α-pinene loss rate [α−pinene]loss to be 23 % using the equation
where cm3 s−1 is the reaction rate coefficient for α-pinene ozonolysis at T=26 ∘C (Atkinson et al., 2006).
During an experiment, we aimed to keep the precursor concentration stable when injecting the precursor into the chamber. We ran the chamber as a “steady-state chamber”, meaning we continuously fed the precursor and O3 into the chamber throughout the experiments, aiming for stable concentrations resulting from a balance between sources (inflow) and sinks (outflow, chemical reactions, and wall losses). As such, the absolute precursor consumption varied with the amount of precursor that was fed, which often varied even across one experiment. We injected precursor into the chamber using one of two methods: an overflow set-up for small amounts of precursor and a syringe pump set-up when there was enough precursor to use a 5 µL syringe to take a sample. In both cases, the evaporated α-pinene was injected into the chamber with a small 25–150 mL min−1 N2 flow. We only had enough of one deuterated precursor (3D1) to be injected with the syringe pump set-up. With the other deuterated precursors (7D2 and 10D3), there was barely any visible liquid in the glass bottle, and thus we could not draw the precursors into the syringe to use with the syringe pump. Nevertheless, flushing N2 over the bottle did release enough precursor to provide a spectrum of oxidation products, though the overflow set-up was harder to control and produced a less stable concentration in the chamber than the syringe pump set-up. The α-pinene injection set-ups are described in more detail in the Appendix (Sect. A1).
We monitored the α-pinene ozonolysis products with a chemical ionisation Orbitrap (CI-Orbitrap). The Orbitrap (Q Exactive Plus Orbitrap, Thermo Scientific) has an ultra-high mass resolution (280 000 ), meaning it is possible to separate between two hydrogen atoms () and a deuterium atom (mD=2.0141) signal in the mass spectrum, hence enabling the use of selectively deuterated precursors. Coupled with the nitrate chemical ionisation inlet ( CI inlet) which is selective towards highly oxidised products (Hyttinen et al., 2015), the instrument has been shown to be effective at detecting HOMs (Riva et al., 2019a; Bianchi et al., 2019).
The precursor concentration was monitored using the proton-transfer-reaction time-of-flight (PTR-ToF) mass spectrometer (Ionicon Analytik GmbH, Austria; Yuan et al., 2017). However, due to instrument failure, we conducted the last experiment with the 3D1 precursor using the Vocus PTR-ToF (Tofwerk AG/Aerodyne Research, Inc.; Krechmer et al., 2018) instead of the PTR-ToF. Nonetheless, both instruments are PTR-ToF mass spectrometers that can be used to monitor VOCs such as monoterpenes (Yuan et al., 2017), and they were calibrated with the same method and hence could fulfil the experimental purposes.
We used a 10 L min−1 sample flow rate with a 60 cm-long sampling line for the CI-Orbitrap, a 1 L min−1 sampling flow rate with a roughly 2.5 m sampling line for the ozone monitor, and a 5 L min−1 sampling flow rate with a roughly 1 m sampling line that was also split to house exhaust for the PTR-ToF mass spectrometers. As a result, we had a 16 L min−1 flow for sampling, and the excess flow from the chamber was fed to the house exhaust, keeping the chamber at a slight over-pressure to ensure contaminations from the room air could not leak into the chamber.
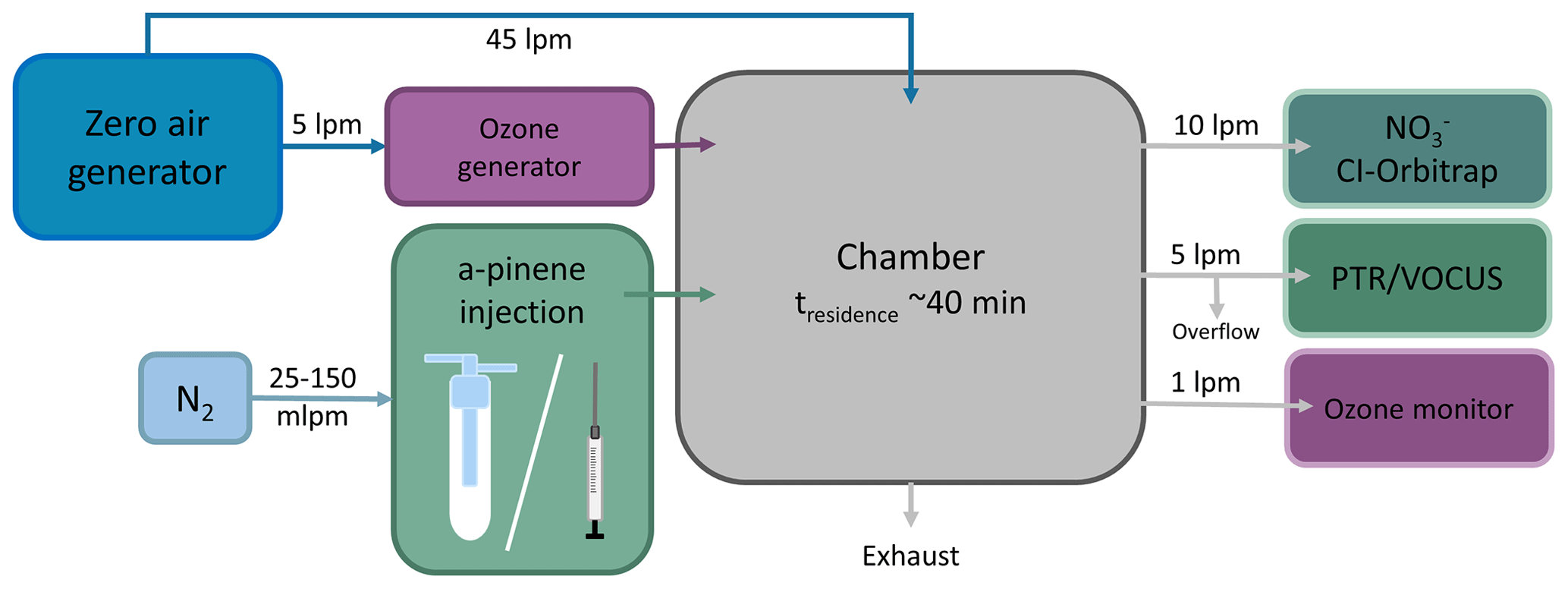
Figure 3The experimental set-up consists of a continuous stirred-tank reactor (CSTR). Additionally, there are ozone and zero-air generators, an α-pinene injection set-up, an ozone monitor, a nitrate CI-Orbitrap, and a PTR or a Vocus.
3.2 Data analysis
We preprocessed the raw data generated by the CI-Orbitrap using a new software called Orbitool (Orbitool version 1.40) developed specifically for the data analysis of time series data from Orbitrap (Cai et al., 2021). We denoised, mass calibrated, and time-averaged the data into 30 min steps using Orbitool. We then identified peaks corresponding to HOMs found in the atmosphere (Ehn et al., 2012) and exported the time series of these signals for further analysis. In this work, the term “HOM” includes all compounds with compositions and C5H6O7 for HOM monomers and for HOM dimers, dimers being ROOR′ accretion products formed from RO2 cross-reactions (Reaction R3 in Sect. 2.2). Notably, other compositions can be and are classified as HOMs (Bianchi et al., 2019), but in this study we aimed to keep the data set more compact for ease of analysis by only inspecting the most prominent and atmospherically relevant signals.
We preprocessed the PTR-ToF and Vocus-PTR raw data using tofTools, a MATLAB-based software package (Junninen et al., 2010). Additionally, we conducted sensitivity calibrations to the PTR instruments to obtain calibration factors, which we used to determine the precursor concentrations from the ion signal. The calibration factors were 15 cps ppb−1 for the PTR-ToF used for the D0, 7D2, and 10D3 precursors and 180 cps ppb−1 for the Vocus-PTR used for the 3D1 precursor.
Because the Orbitrap's sensitivity as a function of signal intensity is non-linear, we determined our instrument's sensitivity behaviour by analysing the α-pinene oxidation products and their natural isotopes following the method of Riva et al. (2020). When determining the instrument sensitivity, we excluded all isotopes that contained deuterium, because using selectively deuterated precursors distorts the signal distributions of the natural hydrogen isotopes. Based on our instrument's sensitivity behaviour, we created a correction function to correct the time series data for the non-linearity. We also used the sensitivity behaviour to determine CI-Orbitrap's detection limit, which is the signal intensity normalised with the total reagent ion signal intensity above which the signal is ≥20 % of the true intensity. The detection limit is , and it can be converted to a concentration of 104 molecules cm−3 by multiplying the signal intensity by the calibration coefficient c; however, the conversion introduces large uncertainties. As calibrations for HOMs remain limited due to a lack of suitable standards, we assumed that the calibration coefficient has the value c=1010 molecules cm−3 as an average of typically reported values (Jokinen et al., 2012; Ehn et al., 2014; Riva et al., 2019c). In order to be detected by the nitrate chemical ionisation mass spectrometer (CIMS), the molecule needs to form a very strong cluster with (Hyttinen et al., 2015), and the majority of HOMs are thus expected to be charged at the collision limit, motivating the use of one single c value for all HOMs. We estimate a large uncertainty of at least arising from the listed assumptions. However, this uncertainty is of less importance in our study, as absolute HOM concentrations are not used for any of the main conclusions of our work.
Additionally, Riva et al. (2020) found that the relative ion transmission of the Orbitrap mass spectrometer is dependent on the mass of the ion. As we did not separately determine the ion transmission of our instrument during these measurements, we corrected our data assuming the same transmission curve as Riva et al. (2020) determined for their instrument. This assumption generates some uncertainty; however, as we are mainly focused on comparing adjacent ions in the mass spectrum, the transmission is unlikely to make a large difference for the purpose of this study. In contrast, in order to assess the impact of deuteration on the total HOM formation, we did also estimate total HOM yields which could have considerable errors if an erroneous correction is used. Nevertheless, as we will show, using the correction resulted in HOM yields that better matched those of previous studies, while using no correction caused unreasonably high values. Additionally, the HOM yields already have large uncertainties because of the assumptions about the calibration coefficient.
We calculated the total HOM concentration according to
where {HOM signal} is the sum of found HOM signals, {total reagent ion signal} is the sum of the measured reagent ion monomer () and dimer () signals, and c is the calibration coefficient.
We calculated the HOM yield γ from the production and loss terms of HOMs when the concentration of HOMs [HOM] did not change significantly over time (Ehn et al., 2014):
from which we can solve for the yield γ:
where is the estimated loss rate for HOMs in the chamber dominated by the loss to the chamber walls (Peräkylä et al., 2020).
In the following, we first describe the experimental set-up and how the oxidation measurements were conducted (Sect. 4.1). We then describe how to interpret the data in the form of mass spectra (Sect. 4.2). In Sect. 4.3, we describe how the results were interpreted and what conclusions could be drawn concerning the involvement of different C atoms. Finally, in Sect. 4.4, we evaluate the effects of the deuteration on the overall HOM yields.
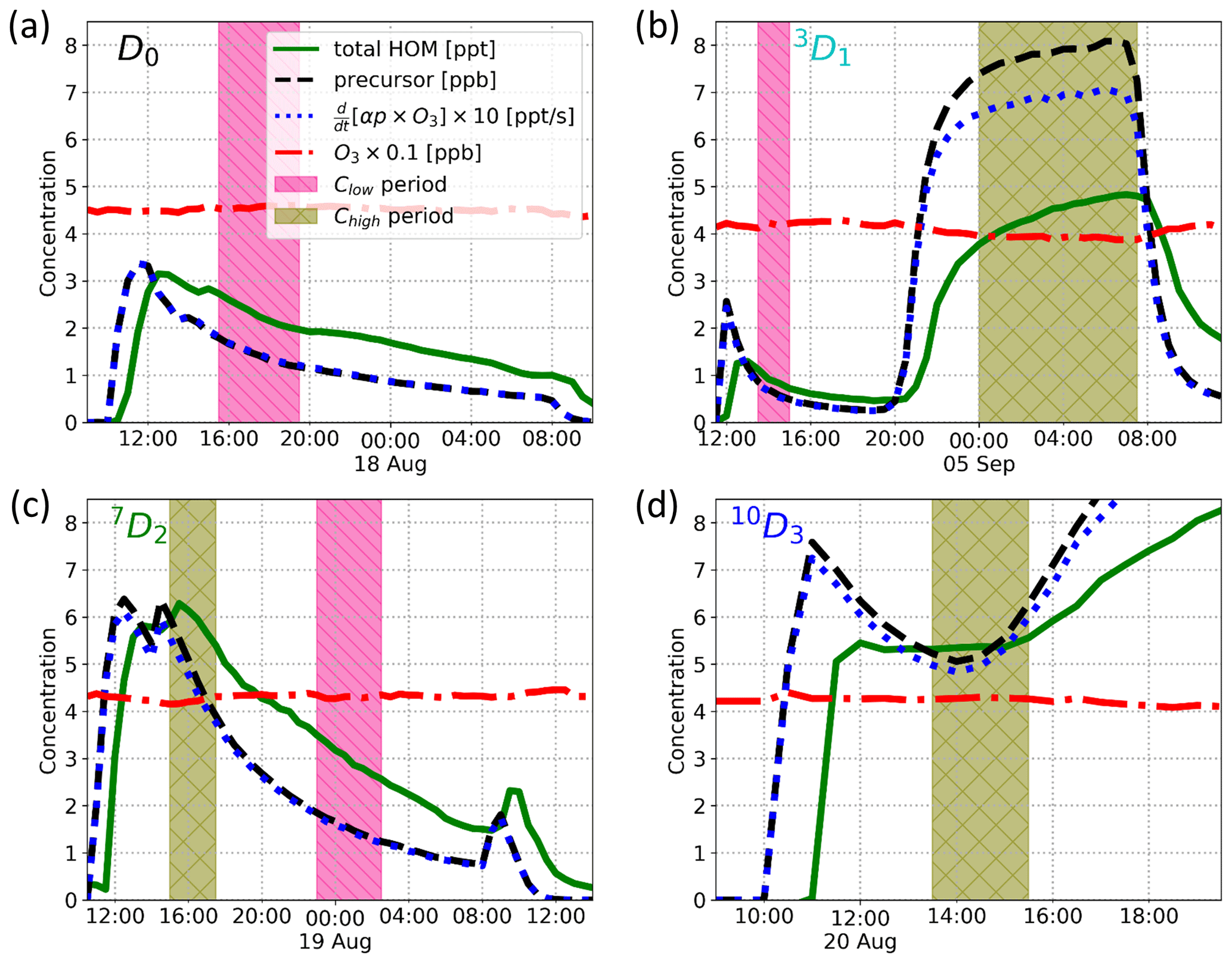
Figure 4Time series of the experiments showing total HOM concentration (green solid line), precursor concentration (black dashed line), α-pinene ozonolysis rate multiplied by 10 (blue dotted line), and ozone concentration multiplied by 0.1 (red dash-dotted line). Additionally, sample times for a low-concentration sample (Clow) and a high-concentration sample (Chigh) are shown with a magenta “\” hatched area and an olive cross-hatched area, respectively.
4.1 Overview of the experiments
We conducted four experiments with three selectively deuterated α-pinenes and standard α-pinene, i.e. one experiment per precursor, Fig. 4 showing their time series. As can be seen from Fig. 4, optimal steady-state conditions were not achieved in all cases, in particular when the injection was performed using the overflow set-up (Fig. A1a), as was the case for 7D2 and 10D3. However, we achieved a rate of change in precursor concentration of the order of 10 % h−1 or less in most cases. Thus, the conditions can be considered stable enough from the perspective of the oxidation chemistry, where HOM formation happens on timescales of seconds and HOM loss on timescales of minutes. For each experiment, we calculated average mass spectra for time ranges where the α-pinene reaction rate was comparable between the four precursors. The selected reaction rates k1[α-pinene][O3] were 0.5 and 0.015 ppt s−1, which we will refer to as high-concentration Chigh and low-concentration Clow samples, respectively. The Clow sample corresponds to precursor concentration ranges of 1.3–1.7, 0.6–1.0, and 1.4–1.8 ppb for the D0, 3D1, and 7D2 precursors, respectively. The Chigh sample corresponds to 7.6–8.0, 4.6–6.0, and 5.0–5.4 ppb for the 3D1, 7D2, 10D3 precursors.
Due to limitations in the experiments, we could only run one experiment per precursor, which led to us not achieving a low-concentration sample with the 10D3 precursor. Similarly, only after the experiment did we conclude that there was no successful Chigh measurement with the standard α-pinene. Thus, we performed the analyses with Chigh samples when comparing the changes in isotopic distributions due to deuteration, whereas we performed the yield comparisons against the standard α-pinene for the Clow for all but the 10D3 precursor.
We have to consider the purity of the deuterated precursors in the chamber when analysing the data, as this is another source of uncertainty. The standard α-pinene has a natural isotope distribution where the ratio is around 0.01 %, making D-containing isotopes often fall below the detection limit of our instrument. For the selectively deuterated samples, we can inspect their purity using PTR mass-spectrometer data. As PTR instruments are known to induce isomerisation and fragmentation of the protonated molecules (Tani et al., 2003; Li et al., 2022), we expect that the observed level of deuteration in the PTR will mainly provide a lower limit for the purity. The PTR measurements suggested that over 88 % of the measured α-pinene in our chamber contained exactly the number of D atoms specified, verifying that the samples are very pure and in line with the observed purity determined using 1HNMR spectroscopy (see Sect. 2.1).
The uncertainties arising from the limited number of experiments we could conduct, the estimation of the precursor purity from PTR data, and the estimation of the calibration coefficient in addition to other uncertainty sources discussed later limit the extent to which we can draw conclusions from our data. For this reason, we opt for only broadly discussing the implications of our results and focusing on detailed mechanistic insights only when they appear unambiguous.
4.2 Interpreting the mass spectra
Interpreting the mass spectra requires some thought and is detailed below for easier reading. Each precursor can form HOM monomers with deuterium atoms up to the number the precursor initially contained. Depending on the formation reactions, the HOMs can have lost deuterium atoms during their formation. As a result, there can be HOM isotopes with the number of deuterium atoms in the molecule anywhere between the number of deuterium atoms in the precursor and zero. Similarly, HOM dimers can contain between twice the number of deuterium atoms in the precursor and zero deuterium atoms.
We show an example of the resulting mass spectra for the HOM C10H14O11 when using deuterated precursors in Fig. 5. We have gathered the measured spectra from using each deuterated precursor into one figure. In the mass spectra, the HOMs are clustered with the nitrate ion, and hence the nominal mass for C10H14O11 when using normal α-pinene (D0) is 372 Th (310+62). We omit the reagent ion from all labels for clarity.
The HOM monomers formed from the 10D3 precursor can have zero to three deuterium atoms. As one can see from Fig. 5, where the 10D3 signal is shown with blue bars, we found isotopes containing three, two, and one deuterium atom in the case of C10H14O11 and no signal for the compound with zero deuterium atoms. These three found isotopes lie on the mass spectrum 1 Th apart, at 373 Th, 374 Th, and 375 Th, their chemical formulae being C10H13DO11, C10H12D2O11, and C10H11D3O11, respectively. Furthermore, for the 10D3 precursor, the highest signal by a large margin is C10H12D2O11, the HOM that has lost one deuterium atom.
For the 7D2 precursor shown with green bars, we can find C10H12D2O11 at 374 Th, C10H13DO11 at 373 Th, and C10H14O11 at 372 Th (Fig. 5). The highest signal by a large margin is C10H12D2O11, but in contrast to the 10D3 precursor, this HOM has lost zero deuterium atoms during its formation process. Assuming the deuteration does not affect the (preference of) reaction pathways significantly, these findings suggest that in the main pathway forming C10H14O11 the C-7 carbon is inactive, whereas the C-10 carbon is active. We emphasise that we only consider the carbon to be active when we can observe the D abstractions taking place at fractions above our uncertainty margins.
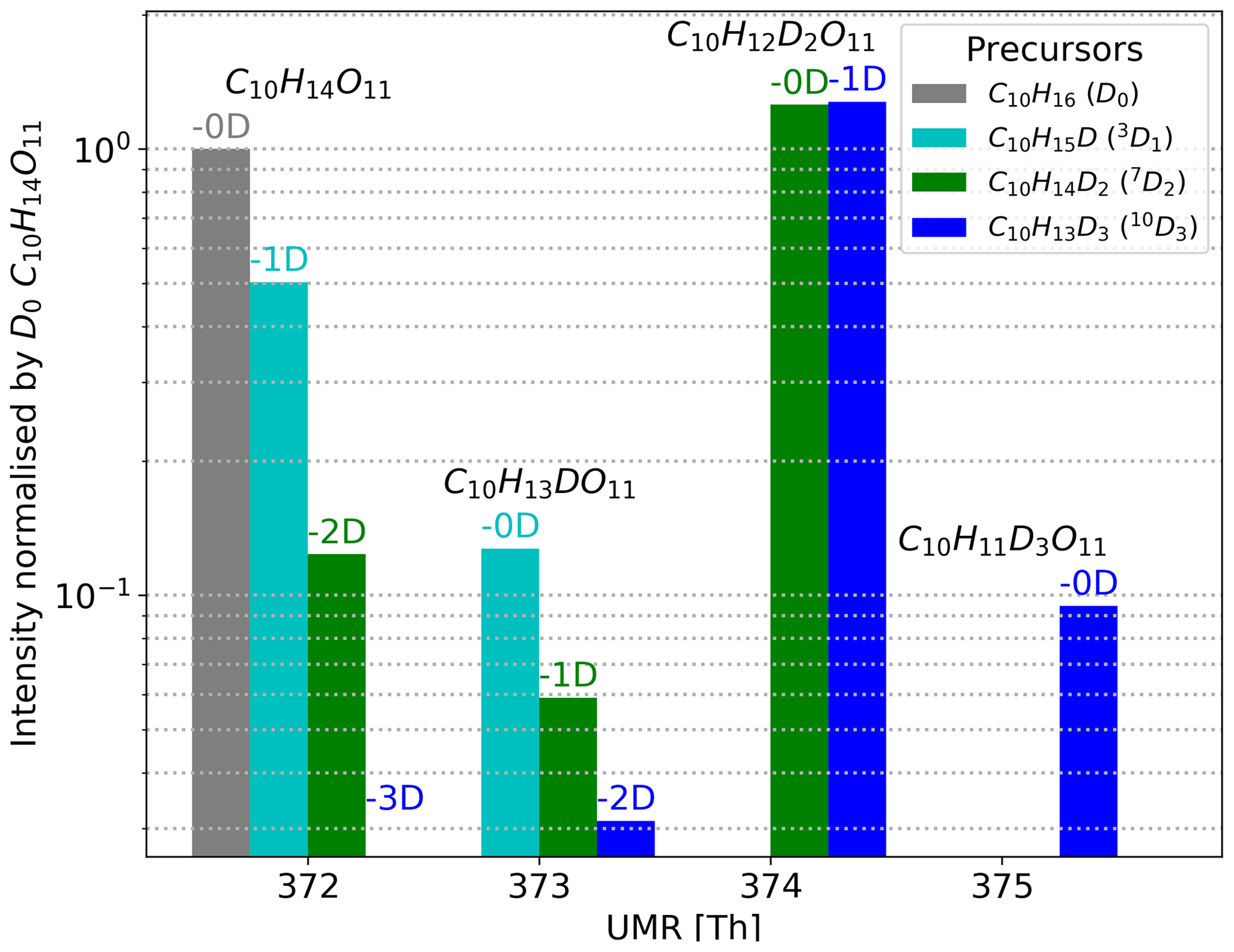
Figure 5Example spectra using C10H14O11 to show resulting HOM signals when using different selectively deuterated precursors. The nitrate cluster of C10H14O11 () is at 372 Th. The colours show which selectively deuterated α-pinene is the precursor, and the values above the bars in matching colour tell how many deuterium atoms the HOMs have lost compared to the precursor. For example, the α-pinene with three deuterium atoms (10D3, shown in blue) can form up to four different C10H14O11 isotopes in the spectrum. However, in the 10D3 precursor's case there is no signal for C10H14O11 without any deuterium atoms (−3D).
4.3 HOM isotopic distributions between deuterated precursors
Both radicals and closed-shell products can be found from the α-pinene ozonolysis HOM spectrum. Unfortunately, many of the inspected radical signals were covered under large background and oxidation product signals, making it impossible to say whether the radical signal was completely missing or just hidden under the dominant adjacent peak. For example, the radicals C10H15O8 and C10H14DO8 were typically not distinguishable due to very large signals of the closed-shell species C10H13DO8 and C10H16O8, respectively. Hence, despite the high resolution of the Orbitrap, the significant difference in signal intensities prevented us from using the radical data for further analysis in this study, instead focusing our analysis solely on the closed-shell molecules. With lower concentrations and/or shorter residence times, the radical signals would be easier to distinguish as they would account for a larger portion of all the signals (Jokinen et al., 2014; Molteni et al., 2019). We emphasise that the complication arises from the deuteration, as in normal experiments the radicals and closed-shell species would be at different integer masses and thus easily distinguishable.
We focused on compounds that Ehn et al. (2012) found to be the main HOMs in monoterpene-dominated regions like the Hyytiälä forestry field station located in the boreal forest. Due to the sheer number of different signals to look at as there are numerous H/D isotopes for every molecule, we limited the most detailed inspection to eight compounds that correspond to the most prominent signals in α-pinene ozonolysis HOM mass spectra: four HOM monomers C10H14O7, C10H14O9, C10H16O9, and C10H14O11 and four HOM dimers C19H28O11, C20H30O14, C20H30O16, and C20H30O18 (Ehn et al., 2014; Molteni et al., 2019).
By limiting our inspection to these compounds and focusing our main analysis on known ozonolysis products, we limit the effect OH reaction products have on our analysis. Firstly, earlier studies have concluded that HOM formation from OH-initiated reactions is a minor channel in α-pinene ozonolysis, only of the order of 10 % of the ozone-initiated oxidation (Ehn et al., 2014; Jokinen et al., 2014, 2015). As such, the impact of OH reactions on the data would be barely detectable, especially within our high uncertainty range. Secondly, the limited inspection compounds are formed specifically from ozone reactions (Ehn et al., 2014; Molteni et al., 2019), as the OH-derived HOMs would normally have higher hydrogen atom contents and odd numbers of oxygen atoms. Thus, our main analysis specifically focuses on known ozonolysis products, and the effect of OH reactions is further negated. Thirdly, the likelihood of multi-generation oxidation, i.e. first-generation non-HOM oxidation products reacting with OH to form HOM, is quite low in our set-up. This is because the flush-out time of the chamber is only 40 min and the timescale of the wall loss of condensable products is shorter yet (of the order of a few minutes), thus making OH reactions a relatively small sink for the oxidation products in the chamber (Peräkylä et al., 2020; Bianchi et al., 2019).
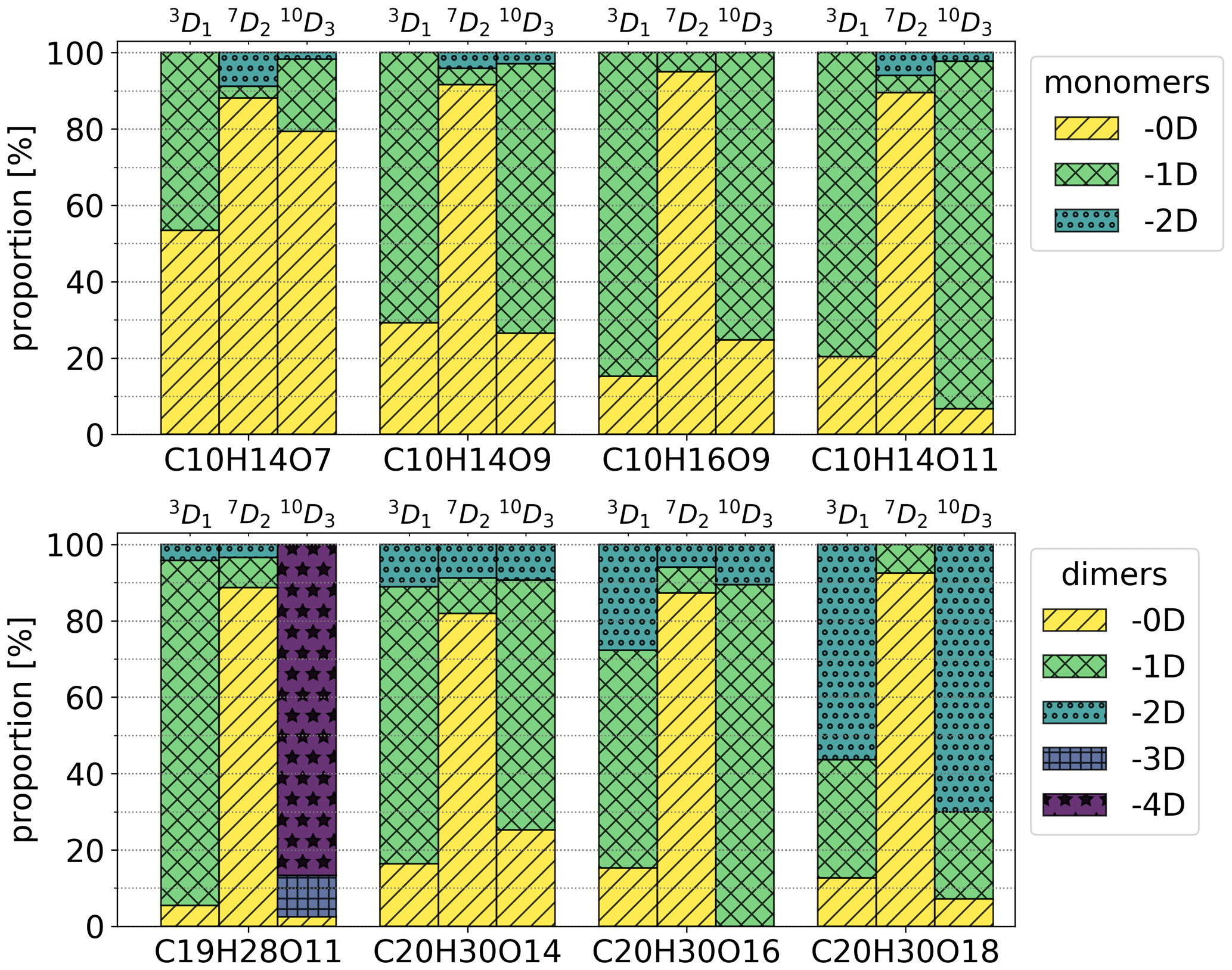
Figure 6Isotopic distributions of chosen HOMs as proportions of signals using high-concentration Chigh data. The colours show how many deuterium atoms are lost from the molecule during the formation process. For example, in the case of 3D1 precursor and C10H14O7 compounds, −0D corresponds to C10H13DO7 and −1D corresponds to C10H14O7. The entries for −3D and −4D are excluded from the upper legend as there were no corresponding compounds in the inspected monomers.
Isotopic distributions of the four HOM monomers and four HOM dimers for the selectively deuterated precursors are shown in Fig. 6. Figure A1 shows the same distributions for a much wider range of HOMs, but for these compounds each isotope was not checked manually, and thus the results are less reliable than the results in Fig. 6 (see the Appendix for further discussion). The isotopic distributions are shown for Chigh data. In general, for the 3D1 and 10D3 precursors, the majority of the signal is generated by isotopes that have lost one or more deuterium atoms, whereas for the 7D2 precursor the signals are predominantly from isotopes that have not lost any deuterium atoms. In fact, the signal of HOMs that have lost one or more deuterium atoms for the 7D2 precursor is only at most 10 % of the total signal. Due to the large uncertainties in the data, we cannot give an exact value, but the fraction is clearly so small that the pathways involving abstraction from this carbon are likely not significant. In other words, the vinylic carbon C-3 and allylic methyl group carbon C-10 are more active in the formation process than the cyclobutyl-ring carbon C-7.
For the more active C-3 and C-10 carbons, we can also note a clear trend for the C10H14Ox monomers and the C20H30Ox dimers, where deuterium loss increases with increasing oxygen content. This makes sense qualitatively, because in order to reach a higher O-atom content, more H shifts need to take place, and almost all potentially abstractable atoms need to have been abstracted for the most-oxygenated HOMs to form. For C-10, the monomers lost at most one D atom, while the C20 dimers lost up to two D atoms, indicating that it is rare to have more than one abstraction from the same C atom. The C19 dimer is discussed later.
Furthermore, the data for 3D1 and 10D3 precursors implicitly show that there are multiple structural isomers contributing to the different elemental compositions. If there was only one isomer, the number of lost D atoms would be identical for all molecules with a given composition, resulting in a signal of only one isotope. However, as we can see from Fig. 6 (and Fig. A3), there are signals for multiple isotopes.
We can now compare our results to those of Iyer et al. (2021), where they suggest a mechanism leading to the C10H15O8 RO2 radical shown in Fig. 2b. More specifically, we can look at the closed-shell product C10H14O7 formed from the O8−RO2 through OH loss and compare its expected behaviour based on the proposed mechanism to the behaviour observed when using selectively deuterated samples. According to the mechanism proposed by Iyer et al. (2021) (Fig. 2b), we see that, out of the deuterated positions used in this work (C-3, C-7, and C-10), only C-3 has undergone an H shift (cyan H). Hence, we would expect that the closed-shell C10H14O7 HOM would always have lost D through exchange with water (Fig. 2c) when using 3D1, while the HOM would never have lost a D atom when using 10D3 and 7D2 precursors. However, according to our observations, for the 3D1 signal of C10H14O7, roughly half still has the D attached (Fig. 6), meaning that the Iyer et al. (2021) pathway can at most explain half of the observed signals in our experiment. For the 10D3 C10H14O7, about 20 % has lost a D atom, while the other 80 % behaved according to expectations from Iyer et al. (2021). However, the Iyer et al. (2021) mechanism is not necessarily the only mechanism through which all of the 80 % of the signal is formed, as there can be other mechanisms that could explain parts of the signal.
Lastly, the C19H28O11 dimer is often the most abundant dimer measured by the nitrate CIMS from α-pinene ozonolysis (Ehn et al., 2014; Rissanen et al., 2015) and also a dominant signal in night-time atmospheric measurements (Yan et al., 2016), but no suggestions have been made concerning which C atom is lost. In Fig. 6, the signal of this dimer for 10D3 precursor has always lost at least three D atoms, providing clear evidence for C-10 being the one that is lost. This is also true more generally for C9 and C19 HOMs according to Fig. A3, where we can see a clear difference in the isotopic distributions for the 10D3 precursor; i.e. the majority of the C9 and C19 signals lost all three D atoms.
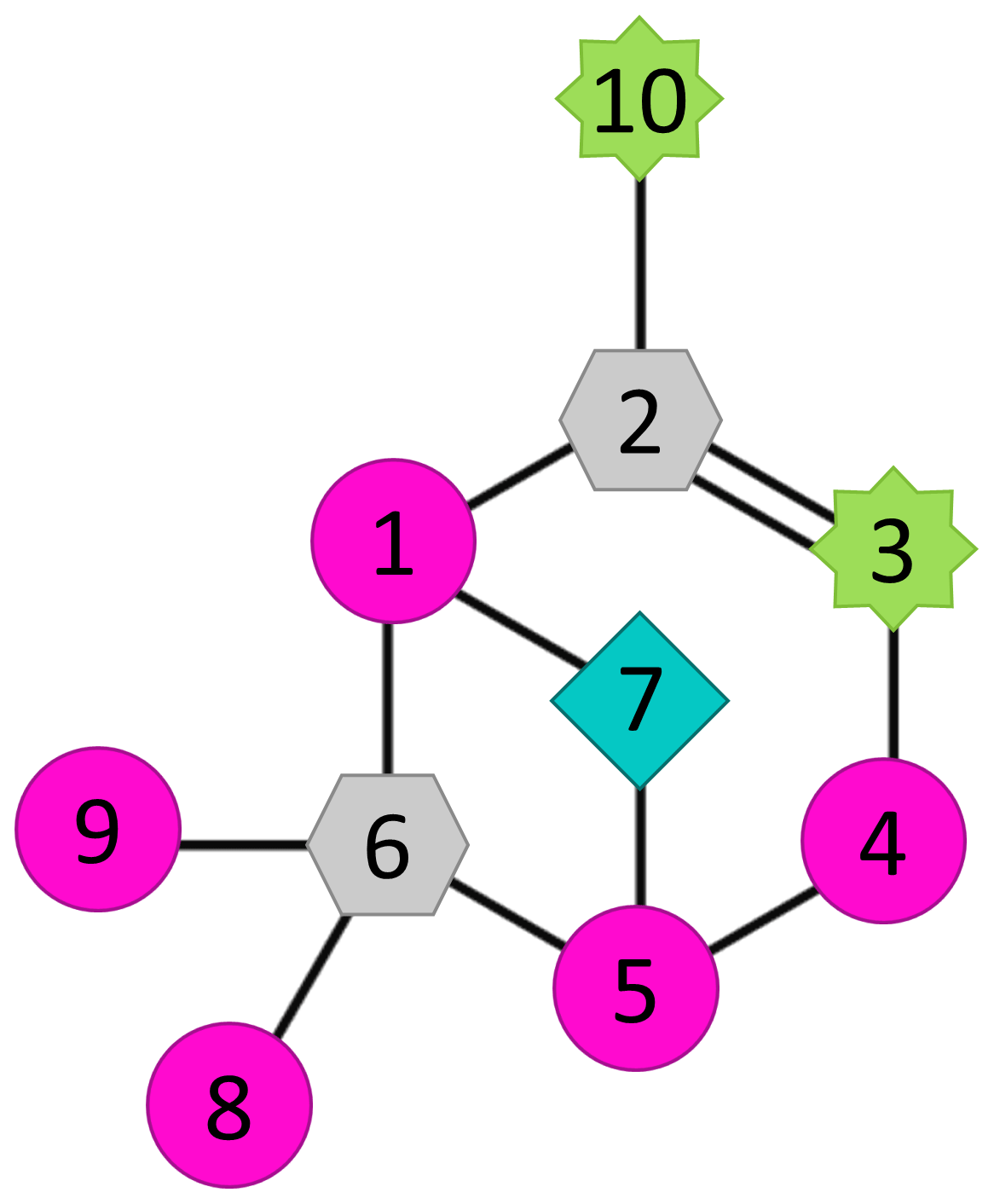
Figure 7Molecular structure of α-pinene with numbered carbons showing carbons that are active (carbons 3 and 10; green eight-point star) and inactive (carbon 7; light-blue diamond) in the formation process, carbons that were not inspected in this study (carbons 1, 4, 5, 8, and 9; magenta circle), and carbons that cannot be studied using selective deuteration (carbons 2 and 6; grey hexagon).
To summarise the findings from our approach of studying selectively deuterated α-pinene, the observed roles of different C atoms in the autoxidation process are highlighted in Fig. 7. The carbons C-3 and C-10 (green stars) are active and often lose deuterium atoms during the HOM-formation process. Additionally, the fraction of signal that had lost deuterium atoms increased with the increasing oxygen number of the HOM. In contrast, the carbon C-7 (cyan diamond) rarely lost deuterium atoms, meaning that it was mostly inactive in the HOM-formation process. As for the carbons C-2 and C-6 (grey hexagons), they can neither be active as defined in this work nor studied with selective deuteration because they are not attached to any hydrogen atoms. The carbons C-1, C-4, C-5, C-8, and C-9 were not inspected in this study, but selectively deuterated studies of these would be extremely interesting and provide crucial information for understanding the detailed autoxidation and HOM-formation mechanisms of this system.
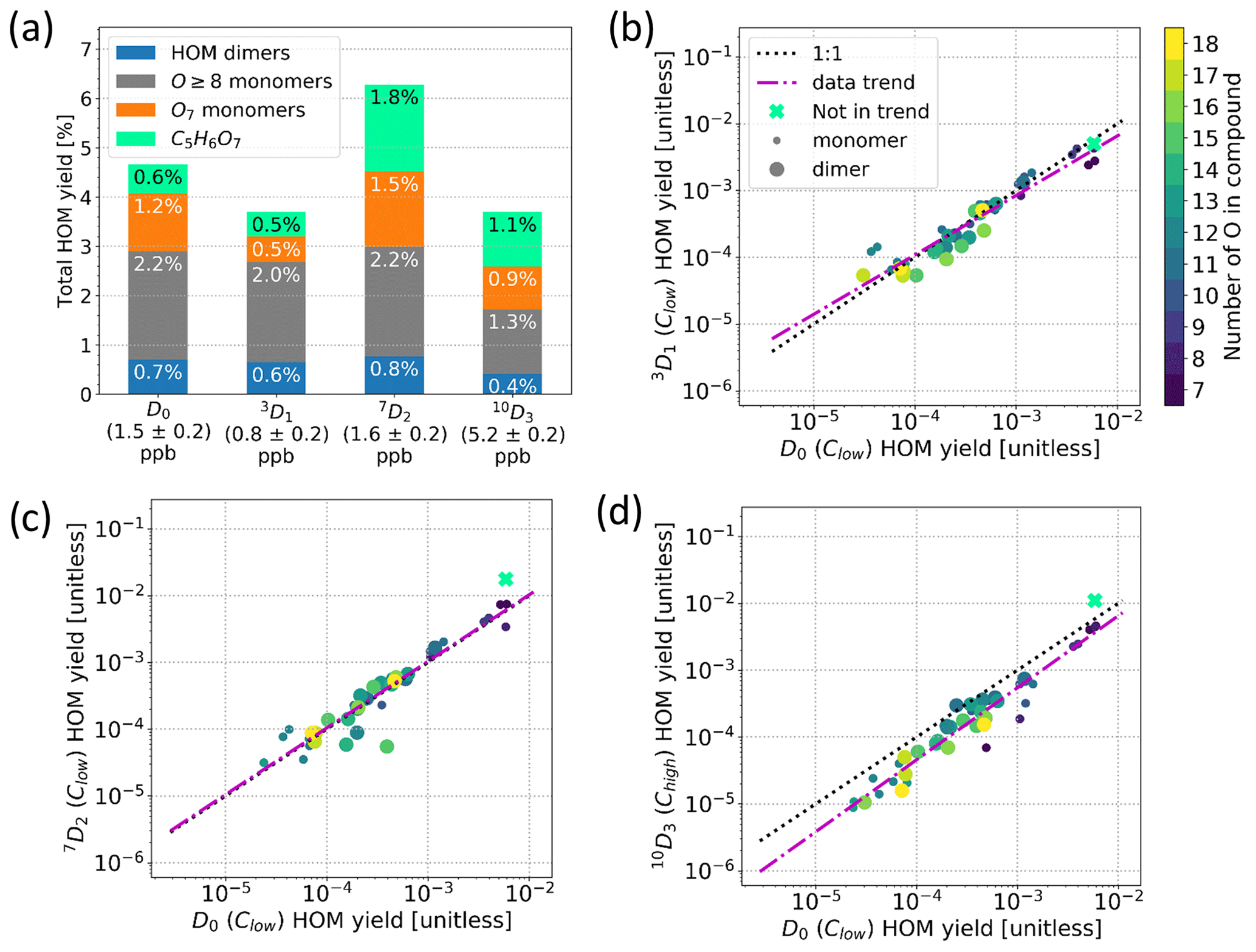
Figure 8(a) HOM yields for α-pinene samples, where yields for C5H6O7 (mint green), O7 monomers (orange), O8+ monomers (grey), and dimers (blue) are plotted separately. The corresponding precursor names and concentrations are shown on the x axis. (b–d) Comparison of HOM yields compound per compound between the D0 sample and 3D1 sample (b), 7D2 sample (c), and 10D3 sample (d), where the mint green cross corresponds to C5H6O7 and is excluded from the data trend and the colour shows the number of oxygen atoms in the compound. Clow and Chigh refer to the α-pinene concentration of the sample. Note that each point in panels (b)–(d) corresponds to the yield of the sum of all found isotopes per main compound.
4.4 HOM yields
Until now, we have only looked at the relative isotope distributions of specific elemental compositions from separate precursors. However, as described in Sect. 2.3, the deuteration may also slow down the autoxidation, which could result in an overall decrease in HOM yields. In this section, we inspect how selective deuterations affected the yields of different HOMs in comparison with HOM yields from standard α-pinene ozonolysis.
Following the definition of HOM in Sect. 3.2, the total HOM yields (Fig. 8a) are calculated from the sum of the signals of all found HOMs for the Clow samples, except for the 10D3 precursor for which the Chigh sample was used. Not all HOM peaks were necessarily found, especially in the lower masses and with lower signal intensities due to the high background and instrumental limitations. Nevertheless, after correcting the data for the CI-Orbitrap's relative ion transmission and non-linearity of the sensitivity as a function of signal intensity, the HOM yield of the D0 precursor is roughly 5 %, which is similar to values reported earlier for α-pinene ozonolysis experiments which ranged from 1.2 () to 7.0±3.5 % (Bianchi et al., 2019). Regardless, we again emphasise that there are large uncertainties involved in determining this absolute value, and we are mainly interested in the possible change in the HOM yields with different precursors. This comparison should be much more robust than the determination of absolute HOM yields.
We report the HOM yields grouped by their chemical compositions in Fig. 8a due to their differing behaviour. C5H6O7 is separated due to the signal being much higher than the rest of the HOM signals. The yields of HOM monomers with seven O atoms and HOM monomers with eight or more O atoms were separated due to the potentially differing volatilities between the two groups of molecules, the O7 monomers having a higher volatility (Peräkylä et al., 2020). Lastly, we give one yield value to the rest of the HOMs, namely the HOM dimers.
Surprisingly, the selective deuterations used in this study did not decrease the HOM yields significantly. We can see in Fig. 8a that the obtained total HOM yields for precursors 3D1, 7D2, and 10D3 were 3.7 %, 6.3 %, and 3.5 %, respectively. Considering the instrumental uncertainties and the differences in precursor concentrations between the experiments, we conclude that the ±20 %–40 % variability is within the uncertainty limits of our experiment. However, this change can be compared to the change of 2 orders of magnitude observed by Rissanen et al. (2014) for fully deuterated vs. non-deuterated cyclohexene, suggesting that deuteration had a minimal role, if any, in the HOM yields of our precursors.
Furthermore, the detailed spectra from the deuterated precursors were similar to that of standard α-pinene. The compound-wise comparisons of HOMs between D0 and deuterated precursors are shown in Fig. 8b–d. We calculated the compound-wise yields by using the sum of all found isotope signals corresponding to a specific HOM composition. As an example, to calculate the compound-wise yield for C10H14O11 for the 10D3 precursor, we take all the found isotope signals corresponding to C10H14O11 (blue bars in Fig. 5), sum them, and use that sum to calculate the yield for the specific compound. What is more, we limited the inspection in Fig. 8b–d to the compounds that were found in both the deuterated precursor's spectrum and the D0 precursor's spectrum. As can be seen from the figures, the data points fall very close to the 1:1 line in all cases, meaning that the total concentrations of HOMs in the deuterated precursors' spectra do not differ significantly from the concentrations of HOMs formed from the D0 precursor. Additionally, there is no clear trend that more-oxygenated HOMs would be more impacted by the deuterations, although we showed that they typically had lost more deuterium atoms. This may be an indication that the D shifts were still fast enough to outcompete other reaction pathways despite the deuteration. On the other hand, it is possible that the autoxidation could proceed through the next most competitive pathway not shut down by the deuteration and end up losing deuterium atoms later in the process, especially in the case of the more-oxidised products.
The compound-wise HOM yields for 10D3 are in general slightly lower than the yields for the D0 precursor (Fig. 8d), which is mainly caused by us comparing the high-concentration Chigh 10D3 to the low-concentration Clow D0 sample. We saw a similar decrease in HOM yields for the other two selectively deuterated precursors when using Chigh data, which may be explained by higher precursor loadings that decrease the RO2 lifetimes, thus hampering the HOM formation. In other words, comparing the Chigh 10D3 to the Clow D0 sample would only exaggerate the HOM yield decrease we expect to see from the deuteration. As the difference is still small enough, we can conclude that the decrease caused by selective deuteration is not significant for the 10D3 HOMs either, matching what was found for the other two selectively deuterated precursors.
Notably, the O7 yield in Fig. 8a is smaller for the 3D1 sample. A difference can also be seen in the mass spectra (Fig. A2); i.e. the masses 308 (C10H14O7) and 310 (C10H16O7) Th have markedly lower signals in the spectrum for 3D1 compared to the D0 spectrum. Figure A3 shows that, for C10H14,16O7, there is a clear difference in the fraction of D lost between 3D1 and 10D3, but at C10H14,16O9, the difference is gone. Likewise, the yields of HOMs with eight or more O atoms (grey bars in Fig. 8a) are very close between the different labelled compounds. From a RO2 radical perspective, it is clear that, to form C10H15O10, one necessarily has gone through C10H15O8, and as such it is surprising that the products of the less-oxidised radicals can decrease while the more-oxidised products do not. It is possible that different structures of C10H15O8 can form, and the ones that can undergo further H shifts are not hampered by deuteration, while the isomers that cannot undergo H shifts and thus will terminate to e.g. C10H14O7 are hampered. Based on the data we have, the latter option is supported; however, without data on the radicals, this finding remains highly speculative.
In this study, we tested the method of selective deuteration for assessing autoxidation mechanisms that are otherwise challenging to elucidate. We monitored the highly oxygenated oxidation products of three selectively deuterated α-pinenes to calculate the fractions of HOM that had lost D atoms. We found that, in the cases where carbons C-3 and C-10 had been deuterated, D atoms were frequently lost, indicating that HOM formation happens through H abstractions from these C atoms. Contrastingly, no significant D-atom loss was observed in the case where C-7 had been deuterated, suggesting that this C atom was rarely involved in any isomerisation reactions during autoxidation and HOM formation. Our findings not only support the existence of the channel Iyer et al. (2021) postulated, but also indicate that there are additional, so far unknown oxidation channels for HOM formation from α-pinene ozonolysis. Furthermore, we found that the used deuterations did not significantly affect the HOM yields, contrasting with our expectations based on kinetic isotope effects.
Our experiments demonstrate that selective deuteration can be a very powerful tool for identifying autoxidation mechanisms. What is more, we can identify many improvements to these initial investigations. Firstly, the interpretation of the results would be simpler if we were able to observe the radicals themselves. Even better still would be to monitor less-oxygenated radicals, which has so far been shown to be possible only using few different instruments (e.g. Berndt et al., 2018). The radical signals are more straightforward to interpret because these compounds are at an earlier stage of the oxidation processes, removing different types of branching possibilities that can take place when the inspected closed-shell products are formed. Unfortunately, due to the large background and overlapping isotope signals that possibly obfuscated some of the signals at the smaller masses, we were not able to inspect the radical signals with the experimental design we used in this study.
Secondly, for a more complete picture of the autoxidation pathways, a more comprehensive set of selectively deuterated compounds would be beneficial. Lastly, it might be possible to investigate the most prominent signals with tandem mass spectrometry (MS2) and gain further insight into their structures (Tomaz et al., 2021).
We have shown that using selectively deuterated precursors for studying the ozonolysis and oxidation of α-pinene is a viable method for mechanism development to gain insight into the autoxidation process still veiled in limited understanding of exact reaction pathways. We expect that utilising this approach and possibly other related isotope-based approaches will be the most promising route forward for understanding autoxidation propagation on a mechanistic level, both for α-pinene and other complex atmospheric VOCs of importance. As such, we are planning a continuation study with a more mechanistic orientation.
A1 α-pinene injection set-ups

Figure A1α-pinene injection set-ups used in the experiments. (a) The overflow injection set-up was a gas bubbler into which the small glass vial holding the precursor sample (and its cap) were inserted. (b) The syringe pump injection set-up consists of a syringe pump, a syringe, a metal T piece, and a septa plug.
The schematics of the α-pinene injection set-ups used in the experiments are shown in Fig. A1. The overflow injection set-up shown in Fig. A1a) consists of a gas bubbler and a small glass vial containing a very small amount (few tens of microlitres) of the precursor. The glass vial is covered with parafilm with a small hole in the film to control the rate at which the precursor is flushed out. A small flow of N2 (25–150 mL min−1) is flushed over the glass vial through the gas bubbler to carry the evaporated precursor into the chamber. Due to the very small amounts of precursor samples we had, we wanted to use all the precursor sample from the vial. Thus, we also added the cap of the glass vial inside the bubbler in case any of the precursor was on it. This set-up was used when there was not enough precursor to take a sample in a syringe, and the resulting precursor concentration in the chamber was less stable than with the syringe pump injection set-up.
The syringe pump injection set-up shown in Fig. A1b was used when there was enough precursor to take a sample with a 5 µL syringe. We used a syringe pump to inject the precursor into a stream of 0.5 L min−1 N2 flow carrying the precursor into the chamber. The precursor is injected into the stream using a small T piece, with one of its openings sealed with a septa plug through which the syringe is put and the two others connected to the N2 flow. This set-up was much more controllable than the overflow set-up; however, there was a communication problem between the pump and the controlling software which caused the pump to inject at an order of magnitude faster rate within a certain range of set injection rates. In some cases, this resulted in far-too-high precursor concentrations in the chamber, increasing the large background and prohibiting us from using those data for analysis.
A2 HOM mass spectra
Figure A2 shows the mass spectra for all the precursors used, highlighting the closely inspected compounds and their isotopes used in Fig. 6, i.e. four HOM monomers C10H14O7, C10H14O9, C10H16O9, and C10H14O11 and four HOM dimers C19H28O11, C20H30O14, C20H30O16, and C20H30O18.
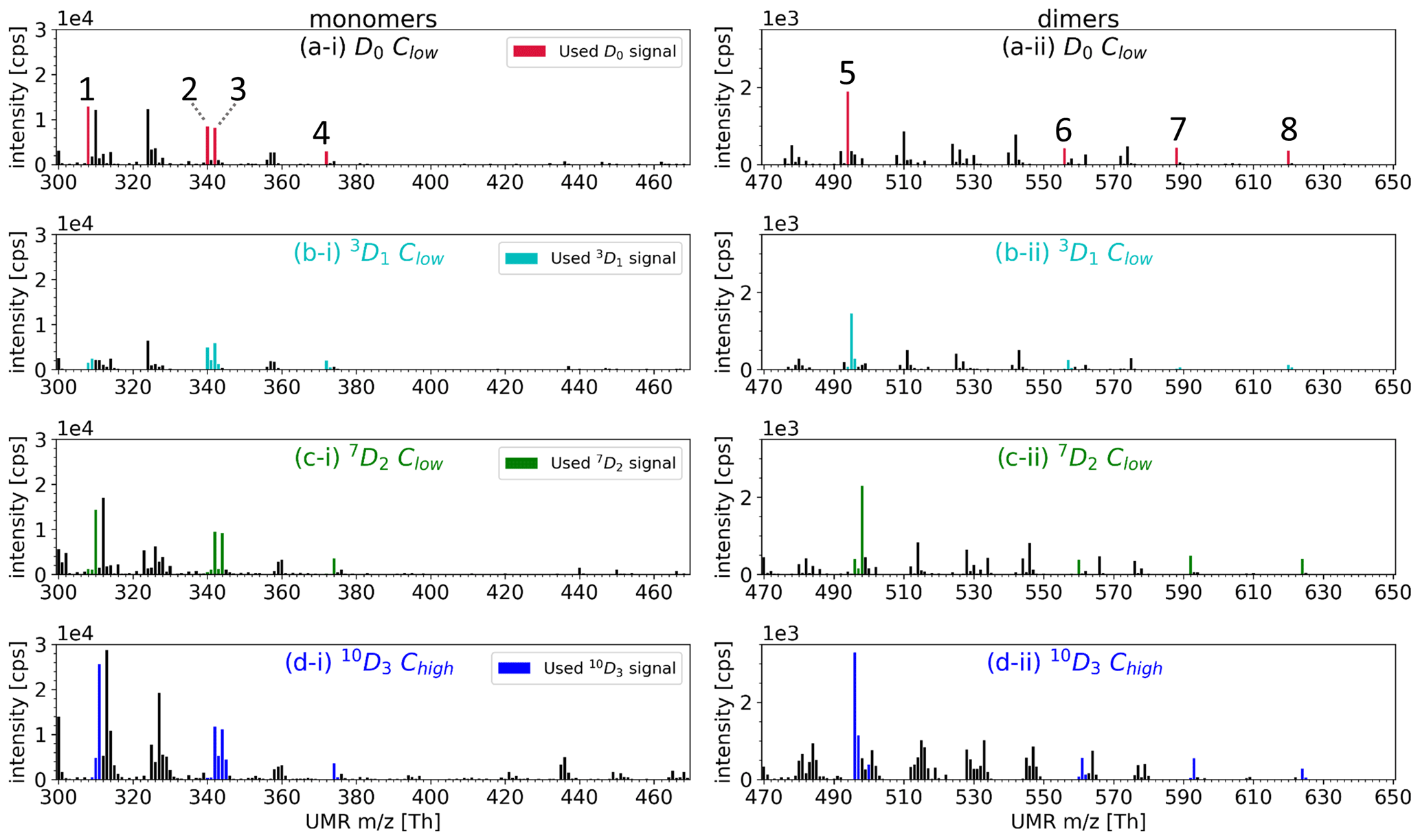
Figure A2Mass spectra of HOMs from selectively deuterated α-pinene precursors at unit mass resolution. Masses corresponding to HOM signals used on closer inspection are marked with a colour, whereas other peaks in the mass spectra are shown in black. (a-i) and (a-ii): the closer-inspection compounds are 1. C10H14O7, 2. C10H14O9, 3. C10H16O9, 4. C10H14O11, 5. C19H28O11, 6. C20H30O14, 7. C20H30O16, and 8. C20H30O18. The y scales are different between the monomer and dimer plots.
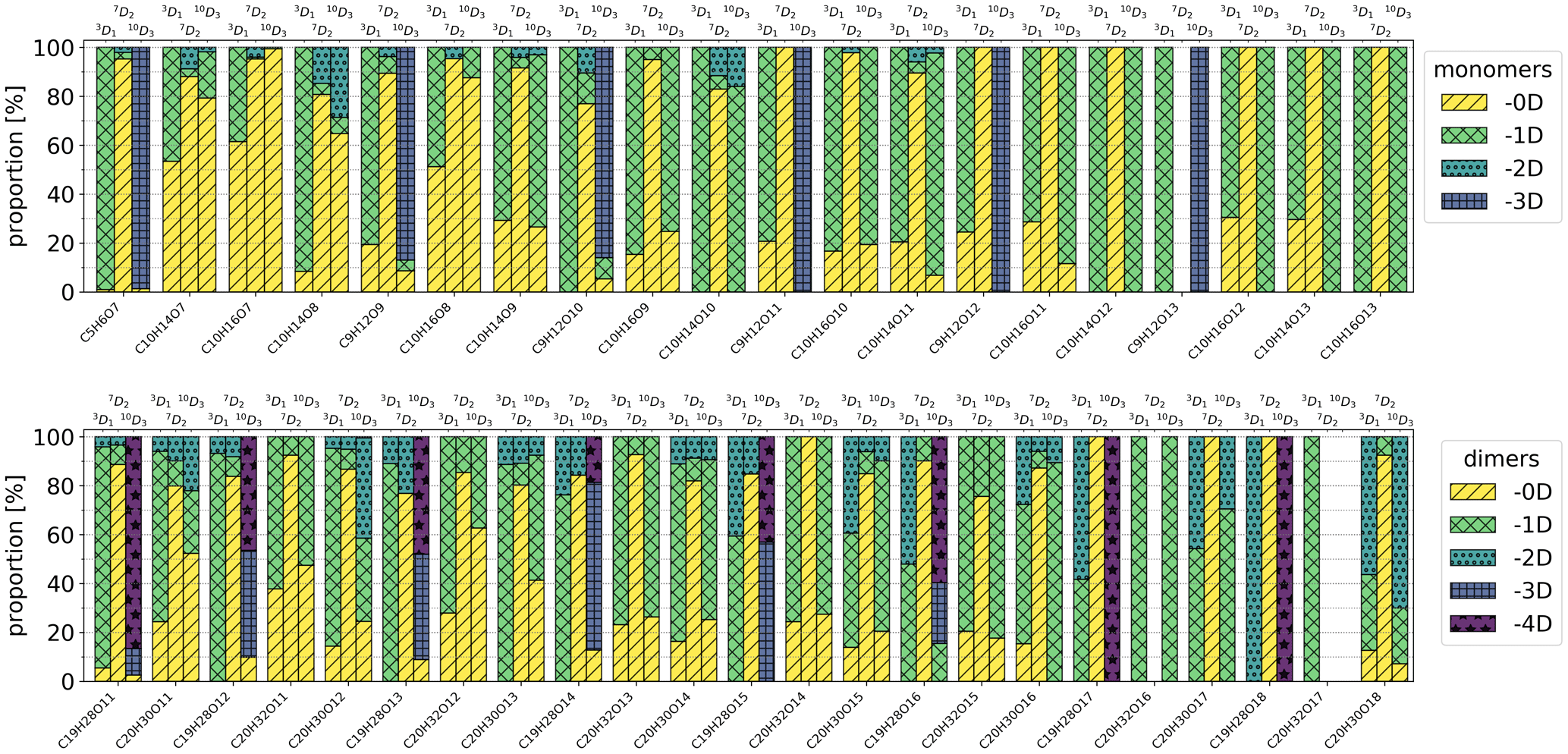
Figure A3Isotopic distributions of all inspected peaks. The colour shows how many deuterium atoms have been lost from the precursor during the formation of the HOM isotope that corresponds to the main HOM on the x axis. For example, in the case of the 3D1 precursor and the C10H14O7 compound, -0D corresponds to C10H13DO7 and -1D corresponds to C10H14O7. The entry for -4D is excluded from the upper legend as there were no corresponding compounds in the inspected monomers.

Figure A4Comparison of HOM yields compound per compound with the number of deuterium atoms lost on average shown with the colour between the D0 sample and the 3D1 sample (a), the 7D2 sample (b), and the 10D3 sample (c), where the mint green cross corresponds to C5H6O7 and is excluded from the data trend. Clow and Chigh refer to the α-pinene concentration of the sample. Note that each point corresponds to the yield of the sum of all found isotopes per main compound.
A3 HOM isotopic distributions for all found HOMs
Figure A3 shows the isotopic distributions for all found HOMs, including the compounds shown already in Fig. 6. Notably, unlike the signals for the compounds in Fig. 6, most of these signals have not been checked for overlapping signals, and hence we cannot say for sure whether there was no signal at all for some isotopes or that some of the signals were obfuscated by larger nearby signals. Additionally, since many of these signals are clearly smaller than those in Fig. 6, it is possible that only one of the isotopes' signals is above the detection limit of our instrument, which would lead to a 100 % contribution from that isotope, although others would have been almost equally high. Therefore, caution should be taken when interpreting the presented distributions.
We can see that C9 and C19 compounds have usually lost at least three deuterium atoms when using the 10D3 precursor. In other words, it is likely that the carbon lost in the formation of the C9 and C19 compounds is C-10, fragmenting with all the deuterium atoms connected to it. The behaviour differs significantly from that of C10 and C20 compounds, which have at most lost two deuterium atoms.
Additionally, when using the 3D1 precursor, the C9 compounds' signals consist mostly of the isotopes that have lost one deuterium atom, and C19 compounds mostly lost one deuterium and sometimes two deuterium atoms, meaning that the C-3 carbon is active in the formation process of these compounds. In contrast, these compounds' signals consist mostly of the isotopes that have lost zero deuterium atoms when using the 7D2 precursor, meaning that the C-7 carbon is rarely active.
Figure A3 also contains the isotopic distributions for C5H6O7, for which the carbons C-3 and C-10 have in practice always lost all of the deuterium atoms and C-7 has never lost any. This likely means that, of the five carbons lost from α-pinene when forming this compound, two of them are the C-3 and C-10 carbons.
Furthermore, we can also see the general trends depicted in Fig. 6 in Fig. A3, where C-3 and C-10 are active and C-7 is inactive in the formation process.
A4 HOM yield behaviour as a function of the average number of D atoms lost
Figure A4 shows the comparison of HOM yields compound per compound between the D0 precursor and the deuterated precursors. It is otherwise the same as Fig. 8b–d, except that the colour shows the average number of deuterium atoms lost per compound. This modification could highlight the effect of the kinetic isotope effects better; however, despite the modification, we do not see a clear effect from the kinetic isotope effects. Nonetheless, Fig. A4 highlights the differing behaviours of the precursors, i.e. how 7D2 HOMs mostly did not lose deuterium atoms, whereas 3D1 and 10D3 did (the colour scales have a larger range for the latter two precursors). Additionally, we can see that, for the 3D1 precursor, most of the HOMs have lost on average some deuterium atoms (most points are in the green-coloured region), whereas for 10D3 precursor HOMs, there are clearly some HOMs that have lost more deuterium atoms than others (some points are yellow, while others are in the blue- to green-coloured region).
Data are available upon request. Data presented in the paper are archived in Zenodo (https://doi.org/10.5281/zenodo.7756182; Meder et al., 2023).
The supplement related to this article is available online at: https://doi.org/10.5194/acp-23-4373-2023-supplement.
ME and RJT conceived of the overall project idea. JL and JGV synthesised the deuterated pinene precursors. MM and ME planned the experiments, and MM conducted the experiments, carried out the data analysis, and wrote the paper under the guidance of ME. All the authors discussed the results and commented on the paper.
The contact author has declared that none of the authors has any competing interests.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
We thank Pekka Rantala, Hannu Koskenvaara, and Petri Keronen from the technical staff at INAR/Physics at the University of Helsinki for the indispensable help and assistance during the experiments. Additionally, we thank Frans Graeffe for calibrating and running the Vocus PTR-ToF and Yihao Li for his efforts in developing the software tool Orbitool for CI-Orbitrap data analysis.
This research has been supported by the Magnus Ehrnrooth Foundation (PhD grant), the HORIZON EUROPE European Research Council (grant no. 101002728), the Academy of Finland (grant nos. 317380, 320094, and 331207), and the National Science Foundation (grant no. CHE-1607640).
Open-access funding was provided by the Helsinki University Library.
This paper was edited by Sergey A. Nizkorodov and reviewed by four anonymous referees.
Atkinson, R. and Arey, J.: Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review, Atmos. Environ., 37, 197–219, https://doi.org/10.1016/S1352-2310(03)00391-1, 2003. a
Atkinson, R., Baulch, D. L., Cox, R. A., Crowley, J. N., Hampson, R. F., Hynes, R. G., Jenkin, M. E., Rossi, M. J., Troe, J., and IUPAC Subcommittee: Evaluated kinetic and photochemical data for atmospheric chemistry: Volume II – gas phase reactions of organic species, Atmos. Chem. Phys., 6, 3625–4055, https://doi.org/10.5194/acp-6-3625-2006, 2006. a
Berndt, T., Mender, B., Scholz, W., Fischer, L., Herrmann, H., Kulmala, M., and Hansel, A.: Accretion Product Formation from Ozonolysis and OH Radical Reaction of alpha-Pinene: Mechanistic Insight and the Influence of Isoprene and Ethylene, Environ. Sci. Technol., 52, 11069–11077, https://doi.org/10.1021/acs.est.8b02210, 2018. a, b
Bianchi, F., Kurtén, T., Riva, M., Mohr, C., Rissanen, M. P., Roldin, P., Berndt, T., Crounse, J. D., Wennberg, P. O., Mentel, T. F., Wildt, J., Junninen, H., Jokinen, T., Kulmala, M., Worsnop, D. R., Thornton, J. A., Donahue, N., Kjaergaard, H. G., and Ehn, M.: Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Organic Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 119, 3472–3509, https://doi.org/10.1021/acs.chemrev.8b00395, 2019. a, b, c, d, e, f
Cai, R., Li, Y., Clément, Y., Li, D., Dubois, C., Fabre, M., Besson, L., Perrier, S., George, C., Ehn, M., Huang, C., Yi, P., Ma, Y., and Riva, M.: Orbitool: a software tool for analyzing online Orbitrap mass spectrometry data, Atmos. Meas. Tech., 14, 2377–2387, https://doi.org/10.5194/amt-14-2377-2021, 2021. a
Crounse, J. D., Paulot, F., Kjaergaard, H. G., and Wennberg, P. O.: Peroxy radical isomerization in the oxidation of isoprene, Phys. Chem. Chem. Phys., 13, 13607–13613, https://doi.org/10.1039/C1CP21330J, 2011. a
Crounse, J. D., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., and Wennberg, P. O.: Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 4, 3513–3520, https://doi.org/10.1021/jz4019207, 2013. a, b
Ehn, M., Kleist, E., Junninen, H., Petäjä, T., Lönn, G., Schobesberger, S., Dal Maso, M., Trimborn, A., Kulmala, M., Worsnop, D. R., Wahner, A., Wildt, J., and Mentel, Th. F.: Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 12, 5113–5127, https://doi.org/10.5194/acp-12-5113-2012, 2012. a, b
Ehn, M., Thornton, J. A., Kleist, E., Sipilä, M., Junninen, H., Pullinen, I., Springer, M., Rubach, F., Tillmann, R., Lee, B., Lopez-Hilfiker, F., Andres, S., Acir, I.-H., Rissanen, M., Jokinen, T., Schobesberger, S., Kangasluoma, J., Kontkanen, J., Nieminen, T., Kurtén, T., Nielsen, L. B., Jørgensen, S., Kjaergaard, H. G., Canagaratna, M., Dal Maso, M., Berndt, T., Petäjä, T., Wahner, A., Kerminen, V.-M., Kulmala, M., Worsnop, D. R., Wildt, J., and Mentel, T. F.: A large source of low-volatility secondary organic aerosol, Nature, 506, 476–496, https://doi.org/10.1038/nature13032, 2014. a, b, c, d, e, f, g, h
Hasson, A. S., Kuwata, K. T., Arroyo, M. C., and Petersen, E. B.: Theoretical studies of the reaction of hydroperoxy radicals (HO2) with ethyl peroxy (CH3CH2O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals, J. Photoch. Photobio. A, 176, 218–230, https://doi.org/10.1016/j.jphotochem.2005.08.012, 2005. a
Hyttinen, N., Kupiainen-Määttä, O., Rissanen, M. P., Muuronen, M., Ehn, M., and Kurtén, T.: Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 119, 6339–6345, https://doi.org/10.1021/acs.jpca.5b01818, 26023711, 2015. a, b
Iyer, S., Reiman, H., Møller, K. H., Rissanen, M. P., Kjaergaard, H. G., and Kurtén, T.: Computational Investigation of RO2 + HO2 and RO2 + RO2 Reactions of Monoterpene Derived First-Generation Peroxy Radicals Leading to Radical Recycling, J. Phys. Chem. A, 122, 9542–9552, https://doi.org/10.1021/acs.jpca.8b09241, 2018. a
Iyer, S., Rissanen, M. P., Valiev, R., Barua, S., Krechmer, J. E., Thornton, J., Ehn, M., and Kurtén, T.: Molecular mechanism for rapid autoxidation in α-pinene ozonolysis, Nat. Commun., 12, 878, https://doi.org/10.1038/s41467-021-21172-w, 2021. a, b, c, d, e, f, g, h, i, j, k, l
Jenkin, M. E., Saunders, S. M., and Pilling, M. J.: The tropospheric degradation of volatile organic compounds: a protocol for mechanism development, Atmos. Environ., 31, 81–104, https://doi.org/10.1016/S1352-2310(96)00105-7, 1997. a
Jokinen, T., Sipilä, M., Junninen, H., Ehn, M., Lönn, G., Hakala, J., Petäjä, T., Mauldin III, R. L., Kulmala, M., and Worsnop, D. R.: Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 12, 4117–4125, https://doi.org/10.5194/acp-12-4117-2012, 2012. a, b
Jokinen, T., Sipilä, M., Richters, S., Kerminen, V.-M., Paasonen, P., Stratmann, F., Worsnop, D., Kulmala, M., Ehn, M., Herrmann, H., and Berndt, T.: Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere, Angewandte Chemie International Edition, 53, 14596–14600, https://doi.org/10.1002/anie.201408566, 2014. a, b
Jokinen, T., Berndt, T., Makkonen, R., Kerminen, V.-M., Junninen, H., Paasonen, P., Stratmann, F., Herrmann, H., Guenther, A. B., Worsnop, D. R., Kulmala, M., Ehn, M., and Sipilä, M.: Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications, P. Natl. Acad. Sci. USA, 112, 7123–7128, https://doi.org/10.1073/pnas.1423977112, 2015. a
Junninen, H., Ehn, M., Petäjä, T., Luosujärvi, L., Kotiaho, T., Kostiainen, R., Rohner, U., Gonin, M., Fuhrer, K., Kulmala, M., and Worsnop, D. R.: A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 3, 1039–1053, https://doi.org/10.5194/amt-3-1039-2010, 2010. a
Krechmer, J., Lopez-Hilfiker, F., Koss, A., Hutterli, M., Stoermer, C., Deming, B., Kimmel, J., Warneke, C., Holzinger, R., Jayne, J., Worsnop, D., Fuhrer, K., Gonin, M., and de Gouw, J.: Evaluation of a New Reagent-Ion Source and Focusing Ion-Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 90, 12011–12018, https://doi.org/10.1021/acs.analchem.8b02641, 2018. a
Kurtén, T., Rissanen, M. P., Mackeprang, K., Thornton, J. A., Hyttinen, N., Jørgensen, S., Ehn, M., and Kjaergaard, H. G.: Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in α-Pinene Ozonolysis Products, J. Phys. Chem. A, 119, 11366–11375, https://doi.org/10.1021/acs.jpca.5b08948, 2015. a, b, c, d, e
Laidler, K. J.: Chemical Kinetics, HarperCollins, ISBN 978-81-317-0972-6, 1987. a, b, c
Li, H., Almeida, T. G., Luo, Y., Zhao, J., Palm, B. B., Daub, C. D., Huang, W., Mohr, C., Krechmer, J. E., Kurtén, T., and Ehn, M.: Fragmentation inside proton-transfer-reaction-based mass spectrometers limits the detection of ROOR and ROOH peroxides, Atmos. Meas. Tech., 15, 1811–1827, https://doi.org/10.5194/amt-15-1811-2022, 2022. a
Li, L., Jakowski, J., Do, C., and Hong, K.: Deuteration and Polymers: Rich History with Great Potential, Macromolecules, 54, 3555–3584, https://doi.org/10.1021/acs.macromol.0c02284, 2021. a
Meder, M., Peräkylä, O., Varelas, J. G., Luo, J., Cai, R., Zhang, Y., Kurtén, T., Riva, M., Rissanen, M. P., Geiger, F. M., Thomson, R. J., and Ehn, M.: Data set for article “Selective deuteration as a tool for resolving autoxidation mechanisms in a-pinene ozonolysis”, Zenodo [data set], https://doi.org/10.5281/zenodo.7756182, 2023. a
Michelotti, A. and Roche, M.: 40 Years of Hydrogen-Deuterium Exchange Adjacent to Heteroatoms: A Survey, Synthesis, 51, 1319–1328, https://doi.org/10.1055/s-0037-1610405, 2019. a
Molteni, U., Simon, M., Heinritzi, M., Hoyle, C. R., Bernhammer, A.-K., Bianchi, F., Breitenlechner, M., Brilke, S., Dias, A., Duplissy, J., Frege, C., Gordon, H., Heyn, C., Jokinen, T., Kürten, A., Lehtipalo, K., Makhmutov, V., Petäjä, T., Pieber, S. M., Praplan, A. P., Schobesberger, S., Steiner, G., Stozhkov, Y., Tomé, A., Tröstl, J., Wagner, A. C., Wagner, R., Williamson, C., Yan, C., Baltensperger, U., Curtius, J., Donahue, N. M., Hansel, A., Kirkby, J., Kulmala, M., Worsnop, D. R., and Dommen, J.: Formation of Highly Oxygenated Organic Molecules from α-Pinene Ozonolysis: Chemical Characteristics, Mechanism, and Kinetic Model Development, ACS Earth Space Chem., 3, 873–883, https://doi.org/10.1021/acsearthspacechem.9b00035, 2019. a, b, c
Orlando, J. J. and Tyndall, G. S.: Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance, Chem. Soc. Rev., 41, 6294–6317, https://doi.org/10.1039/C2CS35166H, 2012. a
Peräkylä, O., Riva, M., Heikkinen, L., Quéléver, L., Roldin, P., and Ehn, M.: Experimental investigation into the volatilities of highly oxygenated organic molecules (HOMs), Atmos. Chem. Phys., 20, 649–669, https://doi.org/10.5194/acp-20-649-2020, 2020. a, b, c, d
Praske, E., Otkjær, R. V., Crounse, J. D., Hethcox, J. C., Stoltz, B. M., Kjaergaard, H. G., and Wennberg, P. O.: Atmospheric autoxidation is increasingly important in urban and suburban North America, P. Natl. Acad. Sci. USA, 115, 64–69, https://doi.org/10.1073/pnas.1715540115, 2018. a
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kangasluoma, J., Sarnela, N., Junninen, H., Jørgensen, S., Schallhart, S., Kajos, M. K., Taipale, R., Springer, M., Mentel, T. F., Ruuskanen, T., Petäjä, T., Worsnop, D. R., Kjaergaard, H. G., and Ehn, M.: The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene, J. Am. Chem. Soc., 136, 15596–15606, https://doi.org/10.1021/ja507146s, 2014. a, b, c, d, e, f, g
Rissanen, M. P., Kurtén, T., Sipilä, M., Thornton, J. A., Kausiala, O., Garmash, O., Kjaergaard, H. G., Petäjä, T., Worsnop, D. R., Ehn, M., and Kulmala, M.: Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene, J. Phys. Chem. A, 119, 4633–4650, https://doi.org/10.1021/jp510966g, 2015. a
Riva, M., Ehn, M., Li, D., Tomaz, S., Bourgain, F., Perrier, S., and George, C.: CI-Orbitrap: An Analytical Instrument To Study Atmospheric Reactive Organic Species, Anal. Chem., 91, 9419–9423, https://doi.org/10.1021/acs.analchem.9b02093, 2019a. a, b, c
Riva, M., Heikkinen, L., Bell, D. M., Peräkylä, O., Zha, Q., Schallhart, S., Rissanen, M. P., Imre, D., Petäjä, T., Thornton, J. A., Zelenyuk, A., and Ehn, M.: Chemical transformations in monoterpene-derived organic aerosol enhanced by inorganic composition, npj Climate and Atmospheric Science, 2, https://doi.org/10.1038/s41612-018-0058-0, 2019b. a
Riva, M., Rantala, P., Krechmer, J. E., Peräkylä, O., Zhang, Y., Heikkinen, L., Garmash, O., Yan, C., Kulmala, M., Worsnop, D., and Ehn, M.: Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 12, 2403–2421, https://doi.org/10.5194/amt-12-2403-2019, 2019c. a
Riva, M., Brüggemann, M., Li, D., Perrier, S., George, C., Herrmann, H., and Berndt, T.: Capability of CI-Orbitrap for Gas-Phase Analysis in Atmospheric Chemistry: A Comparison with the CI-APi-TOF Technique, Anal. Chem., 92, 8142–8150, https://doi.org/10.1021/acs.analchem.0c00111, 2020. a, b, c
Tani, A., Hayward, S., and Hewitt, C.: Measurement of monoterpenes and related compounds by proton transfer reaction-mass spectrometry (PTR-MS), Int. J. Mass Spectrom., 223–224, 561–578, https://doi.org/10.1016/S1387-3806(02)00880-1, 2003. a
Tomaz, S., Wang, D., Zabalegui, N., Li, D., Lamkaddam, H., Bachmeier, F., Vogel, A., Monge, M. E., Perrier, S., Baltensperger, U., George, C., Rissanen, M., Ehn, M., El Haddad, I., and Riva, M.: Structures and reactivity of peroxy radicals and dimeric products revealed by online tandem mass spectrometry, Nat. Commun., 12, 300, https://doi.org/10.1038/s41467-020-20532-2, 2021. a
Upshur, M. A., Chase, H. M., Strick, B. F., Ebben, C. J., Fu, L., Wang, H., Thomson, R. J., and Geiger, F. M.: Vibrational Mode Assignment of α-Pinene by Isotope Editing: One Down, Seventy-One To Go, J. Phys. Chem. A, 120, 2684–2690, https://doi.org/10.1021/acs.jpca.6b01995, 2016. a
Upshur, M. A., Vega, M. M., Bé, A. G., Chase, H. M., Zhang, Y., Tuladhar, A., Chase, Z. A., Fu, L., Ebben, C. J., Wang, Z., Martin, S. T., Geiger, F. M., and Thomson, R. J.: Synthesis and surface spectroscopy of α-pinene isotopologues and their corresponding secondary organic material, Chem. Sci., 10, 8390–8398, https://doi.org/10.1039/C9SC02399B, 2019. a
Wang, Y., Mehra, A., Krechmer, J. E., Yang, G., Hu, X., Lu, Y., Lambe, A., Canagaratna, M., Chen, J., Worsnop, D., Coe, H., and Wang, L.: Oxygenated products formed from OH-initiated reactions of trimethylbenzene: autoxidation and accretion, Atmos. Chem. Phys., 20, 9563–9579, https://doi.org/10.5194/acp-20-9563-2020, 2020. a
Yan, C., Nie, W., Äijälä, M., Rissanen, M. P., Canagaratna, M. R., Massoli, P., Junninen, H., Jokinen, T., Sarnela, N., Häme, S. A. K., Schobesberger, S., Canonaco, F., Yao, L., Prévôt, A. S. H., Petäjä, T., Kulmala, M., Sipilä, M., Worsnop, D. R., and Ehn, M.: Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 16, 12715–12731, https://doi.org/10.5194/acp-16-12715-2016, 2016. a
Ye, Q., Upshur, M. A., Robinson, E. S., Geiger, F. M., Sullivan, R. C., Thomson, R. J., and Donahue, N. M.: Following Particle-Particle Mixing in Atmospheric Secondary Organic Aerosols by Using Isotopically Labeled Terpenes, Chem, 4, 318–333, https://doi.org/10.1016/j.chempr.2017.12.008, 2018. a, b
Yuan, B., Koss, A. R., Warneke, C., Coggon, M., Sekimoto, K., and de Gouw, J. A.: Proton-Transfer-Reaction Mass Spectrometry: Applications in Atmospheric Sciences, Chem. Rev., 117, 13187–13229, https://doi.org/10.1021/acs.chemrev.7b00325, 2017. a, b
Zhang, X., Lambe, A. T., Upshur, M. A., Brooks, W. A., Gray Bé, A., Thomson, R. J., Geiger, F. M., Surratt, J. D., Zhang, Z., Gold, A., Graf, S., Cubison, M. J., Groessl, M., Jayne, J. T., Worsnop, D. R., and Canagaratna, M. R.: Highly Oxygenated Multifunctional Compounds in α-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 51, 5932–5940, https://doi.org/10.1021/acs.est.6b06588, 2017. a, b
The requested paper has a corresponding corrigendum published. Please read the corrigendum first before downloading the article.
- Article
(8160 KB) - Full-text XML
- Corrigendum
-
Supplement
(1226 KB) - BibTeX
- EndNote